
Home » Posts tagged 'clinical trials' (Page 7)
Tag Archives: clinical trials
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Zotarolimus, ABT 578

Zotarolimus
221877-54-9 CAS
A 179578; ABT 578; Resolute; 42-(1-Tetrazolyl)rapamycin; (42S)-42-Deoxy-42-(1H-tetrazol-1-yl)rapamycin
| Molecular Formula: C52H79N5O12 |
| Molecular Weight: 966.21 |
A tetrazole-containing Rapamycin analog as immunomodulator and useful in the treatment of restenosis and immune and autoimmune diseases.
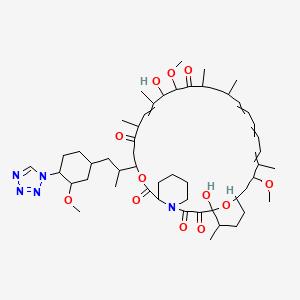
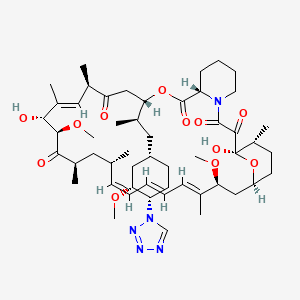
(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy-3-{(1R)-2-[(1S,3R,4S)-3-methoxy-4-(1H-tetrazol-1-yl)cyclohexyl]-1-methylethyl)-6,8,12,14,20,26-hexamethyl-4,9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-heptadecahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontine-1,5,11,28,29(6H,31H)-pentone, cas no 221877-54-9
zotarolimus in U.S. Patent Nos. 6,015,815 and 6,329,386 , and PCT Application No. WO 1999/015530
Zotarolimus (INN, codenamed ABT-578) is an immunosuppressant. It is a semi-synthetic derivative of rapamycin. It was designed for use in stents with phosphorylcholine as a carrier. Coronary stents reduce early complications and improve late clinical outcomes in patients needing interventional cardiology.[1] The first human coronary stent implantation was first performed in 1986 by Puel et al.[1][2] However, there are complications associated with stent use, development of thrombosis which impedes the efficiency of coronary stents, haemorrhagic and restenosis complications are problems associated with stents.[1]
These complications have prompted the development of drug-eluting stents. Stents are bound by a membrane consisting of polymers which not only slowly release zotarolimus and its derivatives into the surrounding tissues but also do not invoke an inflammatory response by the body.

Medtronic are using zotarolimus as the anti-proliferative agent in the polymer coating of their Endeavor and Resolute products.[3]
The inherent growth inhibitory properties of many anti-cancer agents make these drugs ideal candidates for the prevention of restenosis. However, these same properties are often associated with cytotoxicity at doses which block cell proliferation. Therefore, the unique cytostatic nature of the immunosuppressant rapamycin was the basis for the development of zotarolimus by Johnson and Johnson. Rapamycin was originally approved for the prevention of renal transplant rejection in 1999. More recently, Abbott Laboratories developed a compound from the same class, zotarolimus (formerly ABT-578), as the first cytostatic agent to be used solely for delivery from drug-eluting stents to prevent restenosis.[4]
Drug-eluting stents
Drug-eluting stents have revolutionized the field of interventional cardiology and have provided a significant innovation for preventing coronary artery restenosis. Polymer coatings that deliver anti-proliferative drugs to the vessel wall are key components of these revolutionary medical devices. The development of stents which elute the potent anti-proliferative agent, zotarolimus, from a synthetic phosphorylcholine-based polymer known for its biocompatible profile. Zotarolimus is the first drug developed specifically for local delivery from stents for the prevention of restenosis and has been tested extensively to support this indication. Clinical experience with the PC polymer is also extensive, since more than 120,000 patients have been implanted to date with stents containing this non-thrombogenic coating.[4]
Structure and properties

Zotarolimus is a analog made by substituting a tetrazole ring in place of the native hydroxyl group at position 42 in rapamycin that is isolated and purified as a natural product from fermentation. This site of modification was found to be the most tolerant position to introduce novel structural changes without impairing biologic activity. The compound is extremely lipophilic, with a very high octanol:water partition coefficient, and therefore has limited water solubility. These properties are highly advantageous for designing a drug-loaded stent containing zotarolimus in order to obtain a slow sustained release of drug from the stent directly into the wall of coronary vessels. The poor water solubility prevents rapid release into the circulation, since elution of drug from the stent will be partly dissolution rate-limited. The slow rate of release and subsequent diffusion of the molecule facilitates the maintenance of therapeutic drug levels eluting from the stent. In addition, its lipophilic character favors crossing cell membranes to inhibit neointimal proliferation of target tissue. The octanol:water partition coefficients of a number of compounds, recently obtained in a comparative study, indicate that zotarolimus is the most lipophilic of all DES drugs [4]
Stents are used to treat serious decreases in vessel or duct diameter due to a variety of diseases and conditions, especially atherosclerotic diseases, and are often used after angioplasty. While frequently used in arteries, stents are also used in other structures, including veins, bile ducts, esophagus, trachea, large bronchi, ureters, and urethras. Stents are the innovation of the English dentist Charles Stent (1845-1901).
While effective in treating deleterious lumen narrowing, vascular stents in an instance of medical irony, also risk re-creating the condition that they were used to treat. Stents can incur the development of thick endothelial tissue inside the lumen—the neointima. While the degree of development varies, the neointima can grow to occlude the vessel lumen, a type of restenosis.



Previous Syntheses of Zotarolimus
Mollison presented several methods to generate zotarolimus from sirolimus (Mollison, 2000). For example, C-40 hydroxyl of sirolimus is activated with the formation of triflate, and the triflate is then purified by column chromatography. During triflate purification, some of the activated intermediate reverts to sirolimus and its epimer, epi-sirolimus, due to presence of the water during chromatography. The purified triflate is then reacted in a second step with tetrazole to produce the 40-epi-tetrazole derivative of sirolimus, that is, zotarolimus. The crude product is then purified by column chromatography. However, even with this purification, the end product could contain sirolimus and epi-sirolimus impurities.
ISOMERS
ABT-578 [40-epi-(1-tetrazolyl)-rapamycin], known better today as zotarolimus, is a semi-synthetic macrolide triene antibiotic derived from rapamycin. Zotarolimus’ structure is shown in Formula D.

………………………

A representative procedure is shown in Scheme 1.

As shown in Scheme 1, conversion of the C-42 hydroxyl of rapamycin to a trifluoromethanesulfonate or fluorosulfonate leaving group provided A. Displacement of the leaving group with tetrazole in the presence of a hindered, non-nucleophilic base, such as 2,6-lutidine, or, preferably, diisopropylethyl amine provided epimers B and C, which were separated and purified by flash column chromatography.
Synthetic Methods
The foregoing may be better understood by reference to the following examples which illustrate the methods by which the compounds of the invention may be prepared and are not intended to limit the scope of the invention as defined in the appended claims.
Example 1 42-Epi-(tetrazolyl)-rapamycin (less polar isomer) Example 1AA solution of rapamycin (100 mg, 0.11 mmol) in dichloromethane (0.6 mL) at −78° C. under a nitrogen atmosphere was treated sequentially with 2,6-lutidine (53 uL, 0.46 mmol, 4.3 eq.) and trifluoromethanesulfonic anhydride (37 uL, 0.22 mmol), and stirred thereafter for 15 minutes, warmed to room temperature and eluted through a pad of silica gel (6 mL) with diethyl ether. Fractions containing the triflate were pooled and concentrated to provide the designated compound as an amber foam.
Example 1B 42-Epi-(tetrazolyl)-rapamycin (less polar isomer)A solution of Example 1A in isopropyl acetate (0.3 mL) was treated sequentially with diisopropylethylamine (87 L, 0.5 mmol) and 1H-tetrazole (35 mg, 0.5 mmol), and thereafter stirred for 18 hours. This mixture was partitioned between water (10 mL) and ether (10 mL). The organics were washed with brine (10 mL) and dried (Na2SO4). Concentration of the organics provided a sticky yellow solid which was purified by chromatography on silica gel (3.5 g, 70-230 mesh) eluting with hexane (10 mL), hexane:ether (4:1(10 mL), 3:1(10 mL), 2:1(10 mL), 1:1(10 mL)), ether (30 mL), hexane:acetone (1:1(30 mL)). One of the isomers was collected in the ether fractions.
MS (ESI) m/e 966 (M)−;
Example 2 42-Epi-(tetrazolyl)-rapamycin (more polar isomer) Example 2A 42-Epi-(tetrazolyl)-rapamycin (more polar isomer)Collection of the slower moving band from the chromatography column using the hexane:acetone (1:1) mobile phase in Example 1B provided the designated compound.
MS (ESI) m/e 966 (M)−.
………………………………………………….

sirolimus (commercially available or produced as described ((Paiva et al., 1991; Sehgal et al., 1975; Vezina et al., 1975) is dissolved in DCM:toluene (such as 1:2) 100. The reaction mixture is concentrated to dryness 105, and the azeo-drying process 105 is repeated 1-5 times more, more preferably 2-4 times, most preferably twice, preferably with DCM:toluene. The resulting foamy solid is dissolved in IPAc 110, and then 2,6-Lutidine is added 115. The solution is cooled to −30° C. 115. Triflic anhydride is then slowly added to the solution 115. After stirring the reaction mixture, the solution is filtered under nitrogen. The recovered salts 120 are washed with IPAc 125.

To the salts is added 1-H-tetrazole and DIEA 130. The reaction mixture is stirred at room temperature (e.g., 22-25° C.) 135and then concentrated. The crude reaction mixture is purified, using for example, a silica gel column and using, e.g., 1:1 THF:heptane to elute 140. The fractions are monitored for the N-1 isomer (which elutes more slowly than the N-2 isomer), pooled and concentrated, forming an oil. The oil is dissolved in minimum DCM and the solution loaded on a silica gel column packed in, for example, 65:35 heptane:acetone 145. The column is eluted with, for example, 65:35 heptane:acetone, the fractions monitored for the pure product, pooled and concentrated 150.


The purified product is then dissolved in t-BME, and then n-heptane is slowly added to form a precipitate while vigorously stirring the solution 150. The precipitated solids are stirred at 5-10° C., filtered, washed again with heptane, and dried on the funnel with nitrogen. The product is dissolved in acetone and treated with BHT 155. The solution is concentrated, dissolved in acetone, and then concentrated to dryness. The product is then dried under vacuum at 47° C. 160.
EXAMPLES
Example 1 Dichloromethane-Toluene Isopropylacetate One-Pot Process with Filtration (1)
In this example, zotarolimus was prepared from rapamycin in a one-pot process using dichloromethane, toluene and isopropylacetate; the preparation was then purified, concentrated, and dried. The purified product was then characterized by its 1H, 13C NMR resonances from COSY, ROESY, TOCSY, HSQC, and HMBC spectra.
Rapamycin (10 g) was dissolved in dichloromethane (DCM, 25 ml) and toluene (50 ml). The reaction mixture was concentrated to dryness. This azeo-drying process was repeated twice with DCM/toluene. The foamy solid was dissolved in isopropylacetate (IPAc, 65 ml), and 2,6-Lutidine (3.2 ml) was added. The solution was cooled to −30° C. acetonitrile-dry ice bath, and triflic anhydride (2.8 ml) was added slowly in 10 minutes. The reaction mixture was stirred for 30 minutes, and then filtered under nitrogen atmosphere. The salts were washed with IPAc (10 ml). 1-H-tetrazole (2.3 g), followed by diisopropylethylamine (DIEA, 7.4 ml) were added. The reaction mixture was stirred for 6 hours at room temperature, and then concentrated. The crude reaction mixture was purified on a silica gel column (350 g) eluting with 1:1 THF/heptane. The fractions containing product that eluted later (predominantly N-1 isomer) were collected and concentrated. The concentrated oil was dissolved in minimum DCM and loaded on a silica gel column packed in 65:35 heptane:acetone. The column was eluted with 65:35 heptane:acetone, and fractions containing pure product were concentrated.
The purified product was then dissolved in t-butylmethyl ether (t-BME, 13.5 g), and n-heptane (53 g) was added slowly with vigorous stirring. The precipitated solids were stirred at 5-10° C. for 2 hours, filtered, washed with heptane and dried on the funnel with nitrogen to give 3.2 g wet product. The solids (1.0 g) were dissolved in acetone (10 ml) and treated with 2,6-di-tert-butyl-4-ethylphenol (DEP, 0.2%). The solution was concentrated, dissolved in acetone (10 ml) and concentrated to dryness. The product was dried under vacuum for 18 hours at 47° C., yielding 0.83 g of zotarolimus. The product was characterized by its 1H, 13C NMR resonances from its COSY, ROESY, TOCSY, HSQC, and HMBC spectra.
1H-NMR (DMSO-d6, position in bracket): ppm 0.73 (Me, 43); 0.81 (Me, 49); 0.84 (Me, 46); 0.89 (Me, 48); 0.98 (Me, 45); 1.41, 1.05 (CH2, 24); 1.18, 1.10 (CH2, 36); 1.52 (CH, 37); 1.53 (CH2, 12 & 42); 1.59, 1.30 (CH2, 5); 1.41, 1.67 (CH2, 4); 1.11, 1.73 (CH2, 38); 1.21, 1.83 (CH2, 15); 1.21, 1.83 (CH2, 13); 1.62 (Me, 44); 1.73 (Me, 47); 1.76 (CH, 35); 1.60, 2.09 (CH2, 3); 1.93, 2.21 (CH2, 41); 2.05 (CH, 11); 2.22 (CH, 23); 2.47 (CH, 25); 2.40, 2.77 (CH2, 33); 3.06 (OCH3, 50); 3.16 (OCH3, 51); 3.22, 3.44 (CH2, 6); 3.29 (OCH2, 52); 3.29 (CH, 31); 3.60 (CH, 39), 3.62 (CH, 16); 3.89 (CH, 27); 4.01 (CH, 14); 4.02 (CH, 28); 4.95 (CH, 2); 5.02 (CH, 34); 5.10 (═CH, 30); 5.17 (CH, 40); 5.24 (OH, 28); 5.46 (═CH, 22); 6.09 (═CH, 18); 6.15 (═CH, 21); 6.21 (═CH, 20); 6.42 (═CH, 19); 6.42 (OH, 10), 9.30 (CH, 53).
13C NMR (DMSO-d6, position in bracket): ppm 10.4 (Me, 44); 13.1 (Me, 47); 13.6 (Me, 46); 14.5 (Me, 49); 15.5 (Me, 43 & 48); 20.3 (CH2, 4); 21.6 (Me, 45); 24.4 (CH2, 4); 26.2 (CH2, 12); 26.4 (CH2, 3); 26.8 (CH2, 41); 27.2 (CH2, 42); 29.6 (CH2, 13); 31.6 (CH2, 38), 31.7 (CH, 37); 32.9 (CH, 35); 34.8 (CH, 11); 35.2 (CH, 23); 38.2 (CH2, 36); 39.1 (CH, 25); 39.4 (CH2, 33); 39.6 (CH2, 24), 40.0 (CH2, 15); 43.4 (CH2, 6); 45.2 (CH, 31); 50.6 (CH, 2); 55.4 (OCH3, 50); 55.8 (OCH3, 52); 57.0 (OCH3, 52); 55.9 (CH, 40); 66.2 (CH, 14); 73.4 (CH, 34); 75.6 (CH, 28); 77.4 (CH, 39); 82.3 (CH, 16); 85.7 (CH, 27); 99.0 (CH, 10); 125.3 (═CH, 30); 127.0 (═CH, 18 & 19); 130.4 (═CH, 21); 132.2 (═CH, 20); 137.2 (═CMe, 29); 137.7 (═CMe, 17); 139.2 (═CH, 22); 144.6 (CH, 53); 167.0 (C═O, 8); 169.1 (C═O, 1); 199.0 (C═O, 9); 207.5 (C═O, 32); 210.7 (C═O, 26).
Example 2 Dichloromethane-Isopropylacetate One-Pot Process (2)
In this example, zotarolimus was prepared from rapamycin in a one-pot process using dichloromethane and isopropylacetate. The compound was then purified, concentrated, and dried.
Rapamycin (10 g) was dissolved in dichloromethane (DCM, 100 g). 2,6-Lutidine (2.92 g) was added. The solution was cooled to −30° C. in acetonitrile-dry ice bath, and triflic anhydride (4.62 g) was added slowly in 10 minutes. The reaction mixture was stirred for 20 minutes, and then warmed to 10° C. within 15 minutes. The reaction solution was then concentrated. The residue was dissolved in IPAc (55 g). 1-H-tetrazole (2.68 g), followed by diisopropylethylamine (DIEA, 7.08 g) were then added. The reaction mixture was stirred for 6 hours at room temperature and then concentrated. The crude reaction mixture was purified on a silica gel column (360 g), eluting with 1:1 THF:heptane. The fractions containing product that eluted later (principally N-1) were collected and concentrated. The concentrated oil was dissolved in minimum DCM and loaded on a silica gel column (180 g) that was packed in 65:35 heptane:acetone. The column was then eluted with 65:35 heptane:acetone, and fractions containing pure product were concentrated.
The purified product was dissolved in t-butylmethyl ether (t-BME, 23 g) and added slowly to n-heptane (80 g) with vigorous stirring. The precipitated solids were stirred at 5-10° C. for not longer than 1 hour, filtered, washed with heptane and dried on the funnel with nitrogen. BHT (0.015 g) was added to the solids. The solids were dissolved in acetone (20 g), passed through a filter, and concentrated. The residue was treated with acetone two times (20 g), and concentrated each time to dryness. The product was then dried under vacuum for 18 h at not more than 50° C. to give 2.9 g of zotarolimus.
Example 3 Dichloromethane One Pot Process (3)
In this example, zotarolimus was prepared from rapamycin in a one-pot process using dichloromethane. The compound was then purified, concentrated, and dried as described in Example 2.
Rapamycin (7.5 g) was dissolved in DCM (30 g). 2,6-Lutidine (1.76 g) was added. The solution was cooled to −30° C. in acetonitrile-dry ice bath, and triflic anhydride (2.89 g) was added slowly in 10 minutes. The reaction mixture was stirred for 20 minutes, and then assayed for the presence of rapamycin to determine consumption in the reaction. 1-H-tetrazole (1.44 g), followed by DIEA (5.29 g) was added. The reaction mixture was stirred for 6 hours at room temperature, and then directly loaded on a silica gel (270 g) column prepared in 1:1 THF:n-heptane (v/v). The crude reaction mixture was purified with 1:1 THF:n-heptane. The fractions containing product that elute later were collected and concentrated. The concentrated solids were dissolved in minimum DCM and loaded on a silica gel column (135 g) packed in 70:30 n-heptane:acetone. The column was eluted with 70:30 n-heptane:acetone, and fractions containing pure product, as identified by thin-layer chromatography (TLC), were concentrated.
The purified product was dissolved in t-BME (9 g), and added slowly to n-heptane (36 g) with vigorous stirring at 10±10° C. The precipitated solids were stirred at 5-10° C. for not longer than 1 hour, filtered, washed with n-heptane and dried on the funnel with nitrogen. BHT (0.006 g) was added to the solids. The solids were dissolved in acetone (20 g), passed through a filter, and concentrated. The residue was treated with acetone twice (20 g each) and concentrated each time to dryness. The product was dried under vacuum for not longer than 18 hours at not more than 50° C. to give 2.5 g of zotarolimus.
The above process, when carried out with rapamycin presence of 2,6-di-tert-butylpyridine or 2,4,6-collidine (2,3,5-trimethylpyridine) as a non-nucleophilic in step 1a gave zotarolimus of acceptable purity, but a lower yield.
Example 4 High-Pressure Liquid Chromatography HPLC Purification of Zotarolimus Prepared by the One-Pot Synthesis Method
In this example, zotarolimus was made from rapamycin using a one-pot synthesis method of the invention (using DCM), and then subjected to an additional round of purification using HPLC.
Rapamycin (3.75 g) was dissolved in dichloromethane (DCM, 15 g). 2,6-Lutidine (0.88 g) was then added. The solution was cooled to −30° C. in acetonitrile-dry ice bath, and triflic anhydride (1.45 g) was added slowly in 10 minutes. The reaction mixture was stirred for 20 minutes, and then 1-H-tetrazole (0.72 g), followed by DIEA (2.65 g) was added. The reaction mixture was stirred for 6 hours at 25° C., and then directly loaded on a silica gel (115 g) column prepared in 70:30 n-heptane:acetone. The crude reaction mixture was purified with 70:30 n-heptane:acetone. The fractions containing product were collected, and concentrated.
The concentrated solids were dissolved in acetonitrile-water and loaded on a C-18 TechniKrom column (5 cm×25 cm), and eluted with 64:36 acetonitrile-water containing 0.1% BHT. Fractions were analyzed by reverse phase (RP)—HPLC, and product fractions pooled and concentrated to remove acetonitrile. The product was extracted with ethyl acetate or isopropyl acetate, dried (sodium sulfate) and concentrated.
The purified product was dissolved in t-BME (4.5 g), and added slowly to n-heptane (18 g) with vigorous stirring at −10° C. The precipitated solids were stirred at 5-10° C. for not longer than 1 hour, filtered, washed with n-heptane and dried on the funnel with nitrogen. BHT (0.005 g) was added to the solids. The solids were dissolved in acetone (20 g), passed through a filter, and concentrated. The residue was treated with acetone twice (20 g), and concentrated each time to dryness. The product was dried under vacuum for not longer than 18 hours at not more than 50° C. to give 1.2 g of high quality zotarolimus.

- Braunwald E, Zipes D, Libby P, ed. (2001). Heart diseases: a textbook of cardiovascular disease (6th ed.). Philadelphia: Saunders Elsevier.
- Sigwart, U; Puel, J; Mirkovitch, V; Joffre, F; Kappenberger, L (1987). “Intravascular stents to prevent occlusion and restenosis after transluminal angioplasty”. The New England journal of medicine 316 (12): 701–6. doi:10.1056/NEJM198703193161201. PMID 2950322.
- “Medtronic Receives FDA Approval for Endeavor Zotarolimus-Eluting Coronary Stent System”.
- Burke, Sandra E.; Kuntz, Richard E.; Schwartz, Lewis B. (2006). “Zotarolimus (ABT-578) eluting stents”. Advanced Drug Delivery Reviews 58 (3): 437–46.doi:10.1016/j.addr.2006.01.021. PMID 16581153.
- Heitman, J; Movva, NR; Hall, MN (1991). “Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast”. Science 253 (5022): 905–9. PMID 1715094.

The FDA has approved the zotarolimus-eluting stent (Medtronic).
|
3-7-2012
|
ASSAY FOR IMMUNOSUPPRESSANT DRUGS
|
|
|
3-7-2012
|
ONE POT SYNTHESIS OF TETRAZOLE DERIVATIVES OF RAPAMYCIN
|
|
|
7-15-2011
|
NON-DENATURING LYSIS REAGENT
|
|
|
4-22-2011
|
IMMUNOSUPPRESSANT DRUG EXTRACTION REAGENT FOR IMMUNOASSAYS
|
|
|
3-30-2011
|
NON-DENATURING LYSIS REAGENT
|
|
|
10-27-2010
|
METHODS OF MANUFACTURING CRYSTALLINE FORMS OF RAPAMYCIN ANALOGS
|
|
|
10-13-2010
|
CRYSTALLINE FORMS OF RAPAMYCIN ANALOGS
|
|
|
4-21-2010
|
One pot synthesis of tetrazole derivatives of rapamycin
|
|
|
10-16-2009
|
Heparin Prodrugs and Drug Delivery Stents Formed Therefrom
|
|
|
2-20-2009
|
Medical Devices Containing Rapamycin Analogs
|
|
2-20-2009
|
Medical Devices Containing Rapamycin Analogs
|
|
|
11-26-2008
|
Medical devices containing rapamycin analogs
|
|
|
11-21-2008
|
CASCADE SYSTEM
|
|
|
9-5-2008
|
Method Of Treating Disorders Using Compositions Comprising Zotarolimus And Paclitaxel
|
|
|
7-25-2008
|
Medical Devices Containing Rapamycin Analogs
|
|
|
7-16-2008
|
Methods of administering tetrazole-containing rapamycin analogs with other therapeutic substances using medical devices
|
|
|
6-27-2008
|
Medical Devices Containing Rapamycin Analogs
|
|
|
9-8-2006
|
COMPOSITIONS AND METHODS OF ADMINISTERING RAPAMYCIN ANALOGS USING MEDICAL DEVICES FOR LONG-TERM EFFICACY
|
| WO2001087372A1 * | Apr 25, 2001 | Nov 22, 2001 | Cordis Corp | Drug combinations useful for prevention of restenosis |
| EP1826211A1 * | Feb 20, 2007 | Aug 29, 2007 | Cordis Corporation | Isomers and 42-Epimers of rapamycin alkyl ether analogs, methods of making and using the same |
| US5151413 * | Nov 6, 1991 | Sep 29, 1992 | American Home Products Corporation | Rapamycin acetals as immunosuppressant and antifungal agents |
| US5362718 * | Apr 18, 1994 | Nov 8, 1994 | American Home Products Corporation | Rapamycin hydroxyesters |
| US7193078 | Mar 1, 2005 | Mar 20, 2007 | Terumo Kabushiki Kaisha | Process for production of O-alkylated rapamycin derivatives |
| US7220755 | Nov 12, 2003 | May 22, 2007 | Biosensors International Group, Ltd. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US7279571 | Dec 1, 2005 | Oct 9, 2007 | Teva Gyógyszergyár Zártkörüen Müködö Részvénytársaság | Methods of preparing pimecrolimus |
| US7812155 | Nov 26, 2007 | Oct 12, 2010 | Terumo Kabushiki Kaisha | Process for preparing an o-alkylated rapamycin derivative and o-alkylated rapamycin derivative |
| US7872122 | May 8, 2009 | Jan 18, 2011 | Chunghwa Chemical Synthesis & Biotech Co., Ltd. | Process for making Biolimus A9 |
| US20050101624 * | Nov 12, 2003 | May 12, 2005 | Betts Ronald E. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20090209572 | Nov 19, 2008 | Aug 20, 2009 | Biotica Technology Limited | 36-Des(3-Methoxy-4-Hydroxycyclohexyl) 36-(3-Hydroxycycloheptyl) Derivatives of Rapamycin for the Treatment of Cancer and Other Disorders |
| US20100204466 | Feb 23, 2010 | Aug 12, 2010 | Abbott Laboratories | One pot synthesis of tetrazole derivatives of rapamycin |
| US20100249415 | Mar 29, 2010 | Sep 30, 2010 | Kwang-Chung Lee | Process for preparation of temsirolimus |
READ
ANONYMOUS: “Randomised comparison of zotarolimus eluting and sirolimus-eluting stents in patients with coronary artery disease (ENDEAVOUR III)” JOURNAL OF AMERICAN COLLEGE OF CARDIOLOGY, vol. 46, no. 11, 6 December 2005 (2005-12-06), pages CS5-CS6, XP009089338
Dr. Reddy’s Announces the Launch of Decitabine for Injection
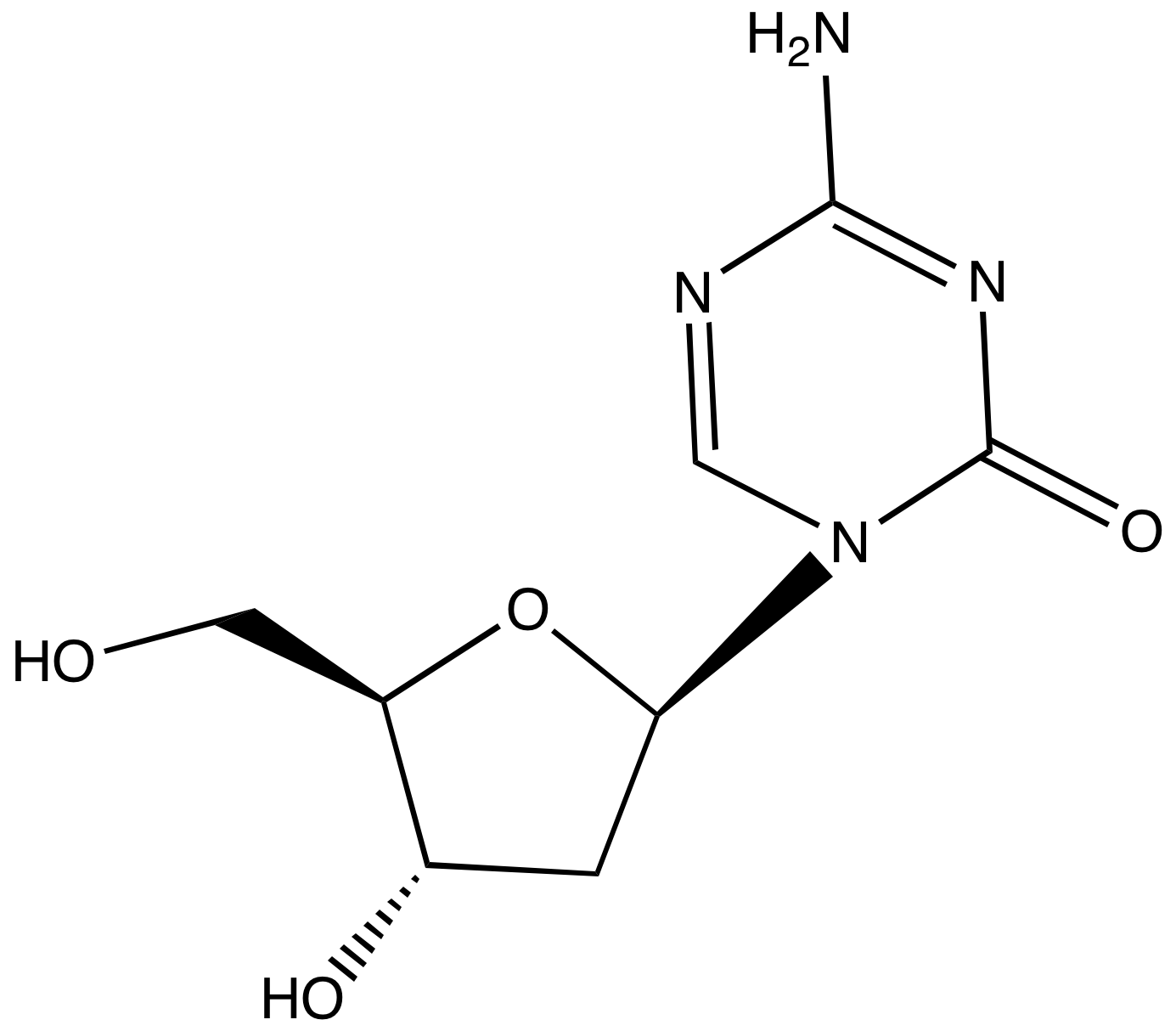
Decitabine
Hyderabad, India, July 12, 2013 — Dr. Reddy’s Laboratories announced today that it has launched Decitabine for Injection (50mg) a therapeutic equivalent generic version of Dacogen (Decitabine for Injection) in the US market on July 11, 2013, following the approval by the United States Food & Drug Administration (USFDA) of Dr. Reddy’s ANDA for Decitabine for Injection.
The Dacogen brand has U.S. sales of approximately $260 Million MAT for the most recent twelve months ending in July 2013 according to IMS Health*.
Dr. Reddy’s Decitabine for Injection 50 mg is available as a single dose vial.
About Dr. Reddy’s

Dr. Reddy’s Laboratories Ltd. (NYSE: RDY) is an integrated global pharmaceutical company, committed to providing affordable and innovative medicines for healthier lives. Through its three businesses – Pharmaceutical Services and Active Ingredients, Global Generics and Proprietary Products – Dr. Reddy’s offers a portfolio of products and services including APIs, custom pharmaceutical services, generics, biosimilars, differentiated formulations and NCEs. Therapeutic focus is on gastro-intestinal, cardiovascular, diabetology, oncology, pain management, anti-infective and pediatrics. Major markets include India, USA, Russia and CIS, Germany, UK, Venezuela, S. Africa, Romania, and New Zealand. For more information, log on to: http://www.drreddys.com.
Dacogen® is a registered trademark used by Eisai Inc. under license from Astex Pharmaceuticals, Inc
Decitabine (trade name Dacogen), or 5-aza-2′-deoxycytidine, is a drug for the treatment of myelodysplastic syndromes, a class of conditions where certain blood cells are dysfunctional, and for acute myeloid leukemia (AML).[1] Chemically, it is a cytidine analog.
Decitabine is a hypomethylating agent.[2][3] It hypomethylates DNA by inhibiting DNA methyltransferase.
It functions in a similar manner to azacitidine, although decitabine can only be incorporated into DNA strands while azacitidine can be incorporated into both DNA and RNA chains.

Clinical uses
Decitabine is indicated for the treatment of myelodysplastic syndromes (MDS) including previously treated and untreated, de novo and secondary MDS of all French-American-British subtypes (refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia) and Intermediate-1, Intermediate-2, and High-Risk International Prognostic Scoring System groups. In patients with renal insufficiency, Batty and colleagues reported the first case series on the feasibility of therapy with hypomethylating agents in patients with renal insufficiency.[4]
Chemical synthesis
Decitabine can be synthesized from a benzoyl-protected chlorosugar:[5] 
- “EC Approves Marketing Authorization Of DACOGEN For Acute Myeloid Leukemia”. 2012-09-28. Retrieved 28 September 2012.
- Kantarjian H, Issa JP, Rosenfeld CS, et al. (April 2006). “Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study”. Cancer 106 (8): 1794–803. doi:10.1002/cncr.21792. PMID 16532500.
- Kantarjian HM, O’Brien S, Cortes J, et al. (August 2003). “Results of decitabine (5-aza-2’deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia”. Cancer 98 (3): 522–8. doi:10.1002/cncr.11543. PMID 12879469.
- Ravandi, F.; Cortés, J. E.; O’Brien, S.; Pierce, S.; Garcia-Manero, G.; McCue, D.; Santos, F. P. S.; Jabbour, E. et al. (2010). “Feasibility of Therapy with Hypomethylating Agents in Patients with Renal Insufficiency”. Clinical Lymphoma, Myeloma & Leukemia 10 (3): 205–210. doi:10.3816/CLML.2010.n.032. PMID 20511166.
|displayauthors=suggested (help) edit - Piml, J.; Sorm, F. (1964). Coll. Czech. Chem. Commun. 29: 2576.

Greek Herbs- Fennel (saunf)

………………………………………………
……………………………………………….
History of Fennel
Ancient Greeks and Indian cultures used fennel for cooking and as part of traditional herbal medicine. The Greeks and Indians traditionally combined fennel with other herbs to make home remedies for the relief of gastrointestinal problems such as acidity and indigestion.
Fennel Composition
The essential oil of fennel contains approximately 5 percent limonene, 50 to 80 percent anethole and 5 percent fenchone. Additionally, the oil contains trace amounts of a-pinene, estragole, b-pinene, safrole, b-myrcene, camphene and p-cymene. The seeds from the fennel plant also contain fiber and complex carbohydrates. Fennel contains nutrients including vitamin B-3, magnesium, molybdenum, copper, phosphorus, iron, calcium, manganese, vitamin C, folate and potassium.
Fennel Uses
As a health supplement, fennel can help to prevent gas, support digestion and function as an expectorant that can help to relieve minor respiratory problems such as mucus. Fennel also contains anti-inflammatory properties when used externally. The leaves from the fennel plant can facilitate the healing of wounds and burns. The root of the fennel plant is diuretic and can help treat urine infections. Fennel also contains a combination of phytonutrients including the flavonoids rutin, quercitin and kaempferol. Fennel also has antioxidant properties and as a dietary fiber, it can help lower your cholesterol levels.
Fennel Supplements
Health supplement manufacturers offer fennel supplements in powdered form. As a supplement, manufacturers recommend taking 1 to 4 g per day of the powdered fennel supplement. The Food and Drug Administration, however, has not established a recommended dose for fennel powder. There are no known side effects of consuming fennel powder supplements, although you should speak with your doctor prior to using fennel powder if you are attempting to treat a specific medical condition.
The bulb, foliage, and seeds of the fennel plant are widely used in many of the culinary traditions of the world. The small flowers of wild fennel (mistakenly known in America as fennel “pollen” ) are the most potent form of fennel, but also the most expensive.Dried fennel seed is an aromatic, anise-flavoured spice, brown or green in colour when fresh, slowly turning a dull grey as the seed ages. For cooking, green seeds are optimal. The leaves are delicately flavoured and similar in shape to those of dill. The bulb is a crisp vegetable that can be sautéed, stewed, braised, grilled, or eaten raw. They are used for garnishes and to add flavor to salads. They are also added to sauces and served with pudding. The leaves used in soups and fish sauce and sometimes eaten raw as salad.
Fennel seeds are sometimes confused with those of anise, which are similar in taste and appearance, though smaller. Fennel is also used as a flavouring in some natural toothpastes. The seeds are used in cookery and sweet desserts.
Many cultures in India, Pakistan, Afghanistan, Iran and the Middle East use fennel seed in their cookery. It is one of the most important spices in Kashmiri Pandit and Gujarati cooking. It is an essential ingredient of the Assamese/Bengali/Oriya spice mixture panch phoron and in Chinese five-spice powders. In many parts of India and Pakistan, roasted fennel seeds are consumed as mukhwas, an after-meal digestive and breath freshener. Fennel leaves are used as leafy green vegetables either by themselves or mixed with other vegetables, cooked to be served and consumed as part of a meal, in some parts of India. In Syria and Lebanon, it is used to make a special kind of egg omelette (along with onions, and flour) called ijjeh.
Many egg, fish, and other dishes employ fresh or dried fennel leaves. Florence fennel is a key ingredient in some Italian and German salads, often tossed with chicory and avocado, or it can be braised and served as a warm side dish. It may be blanched or marinated, or cooked in risotto.
In Spain the stems of the fennel plant are used in the preparation of pickled eggplants, “berenjenas de Almagro”.
Medicinal uses

Fennel (Foeniculum vulgare) essential oil in clear glass vial
Fennel contains anethole, which can explain some of its medical effects: It, or its polymers, act as phytoestrogens.
The essence of fennel can be used as a safe and effective herbal drug for primary dysmenorrhea, but could have lower potency than mefenamic acid at the current study level.
Intestinal tract
Fennel is widely employed as a carminative, both in humans and in veterinary medicine (e.g., dogs), to treat flatulence by encouraging the expulsion of intestinal gas. Anethole is responsible for the carminative action.
Mrs. Eencher Herbal states:
On account of its carminative properties, fennel is chiefly used medicinally with purgatives to allay their side effects, and for this purpose forms one of the ingredients of the well-known compound liquorice powder. Fennel water has properties similar to those of anise and dill water: mixed with sodium bicarbonate and syrup, these waters constitute the domestic ‘gripe water‘ used to correct the flatulence of infants. Volatile oil of fennel has these properties in concentration. Commercial preparations of fennel are widely available as alternative treatment for baby colic. Fennel tea, also employed as a carminative, is made by pouring boiling water on a teaspoonful of bruised fennel seeds.
Fennel can be made into a syrup to treat babies with colic (formerly thought to be due to digestive upset), but long-term ingestion of fennel preparations by babies is a known cause of thelarche.
Eyes
In the Indian subcontinent, fennel seeds are also eaten raw, sometimes with some sweetener, as they are said to improve eyesight. Ancient Romans regarded fennel as the herb of sight.Root extracts were often used in tonics to clear cloudy eyes. Extracts of fennel seed have been shown in animal studies to have a potential use in the treatment of glaucoma.
Blood and urine
Fennel may be an effective diuretic and a potential drug for treatment of hypertension.
Breastmilk
There are historical anecdotes that fennel is a galactagogue,improving the milk supply of a breastfeeding mother. This use, although not supported by direct evidence, is sometimes justified by the fact that fennel is a source of phytoestrogens, which promote growth of breast tissue. However, normal lactation does not involve growth of breast tissue. A single case report of fennel tea ingested by a breastfeeding mother resulted in neurotoxicity for the newborn child.
Other uses
Syrup prepared from fennel juice was formerly given for chronic coughs. It is one of the plants which is said to be disliked by fleas, and powdered fennel has the effect of driving away fleas from kennels and stables.
References
- “Herbs That Work: The Scientific Evidence of Their Healing Powers”; David Armstrong
- “The Encyclopedia of Herbs: A Comprehensive Reference to Herbs of Flavor and Fragrance”; Arthur O. Tucker and Thomas DeBaggio; 2009
- “Pocket Guide to Herbal Remedies”; Lane P. Johnson; 2002
- “Encyclopedia of Natural Medicine”; Michael Murray and Joseph Pizzorno; 1997
seeds
DRUG DISCOVERY PRESENTATION BY DR ANTHONY CRASTO

Please wait and allow slideshare to load…………..
Nanotechnology, its applications in medicine, pharmaceuticals,drug developments

Nanotechnology can be defined as a technology which deals with manipulation, study, and designing and developing particles, bio-molecules of the size more than 1 nm and less than 100 nanometer, with the intention of modification enhancement or lowering a particular property of a molecule or a particle, which can be used in developing a device or molecule

.Nanotechnology involves developing materials or devices in the size range of 1 nm to 100 nanometer. At this scale quantum mechanical effects have very important implications in the quantum realm; nanotechnology controls the properties of material on an atomic level.

A serious cause of concern about nanotechnology is its safety and hazardous effects on environment and health, nanomaterial is required to be handled with special care and requires special methods for its disposal.
Drugs that use nanotechnology are also required to qualify for its effectiveness and safety, safety studies are very important factors as so far there is not enough data of drugs developed using nanotechnology and tested for safety.

Nanostructures provide this surface with superhydrophobicity, which lets water droplets roll down the inclined plane.
In pharmaceuticals nanotechnology has wide applications some of which are given below.
1. Targeting a drug to a particular tissue, to, enhancing absorption of a drug molecule in a particular tissue
2. To reduce degradation of a drug and enhance bioavailability and reduce untoward toxic effect of a drug molecule.
3. To enhance the microbial stability of a product
4. In cosmetics zinc oxide nanoparticles are used to increase its antimicrobial properties , and titanium dioxide nano particles effectively block UV rays in both cases concentrations required are very low compared to conventional use.
5. Nanoemulsions for increasing the absorption of a drug molecules.
6. To develop molecules as tracer marker compound to identify the toxic and untoward effects or spilage

Graphical representation of a rotaxane, useful as a molecular switch.
Antineoplastic drugs bring about their anticancer action by inhibiting cancerour cells growth by virtue of alkylation of nucleotides in cancerous cells or by inhibition of folic acid uptake by cancerous cells or by inhibiting cell division by binding with tubulin and microtubulin in a cancerous cells, it is likely that these drug are also absorbed in to normal tissues, leading to untoward serious cytotoxic effects , like kidney damage and nerve damage in chemotherapy with cisplatin, a drug of choice in most of anticancer chemotherapies.


This DNA tetrahedron is an artificially designed nanostructure of the type made in the field of DNA nanotechnology. Each edge of the tetrahedron is a 20 base pair DNA double helix, and each vertex is a three-arm junction.

This device transfers energy from nano-thin layers of quantum wells to nanocrystals above them, causing the nanocrystals to emit visible light.
A team of scientists from the Massachusetts Institute of Technology and Brigham and Women’s Hospital conducted study. They stored an prodrug of cisplatin (which is used in most of cancer chemotherapies) within nanoparticles which they developed to target a specific protein in cancerous cells in prostate gland.
After these prodrug loaded nanoparticles were absorbed by cancerous cells the prodrug was released in to the cancerous cells and was converted in to an active form . The team demonstrated that these prodrug carrying nanoparticles were able to kill cancer cells in culture more efficiently than the drug alone.

Study was conducted by researchers, led by Dr. Omid Farokhzad and Dr. Stephen Lippard, to study nanoparticle drug delivery system for an effective and safer option for chemotherapy in living animals. Their research work is published in Proceedings of the National Academy of Sciences, in Jan 2011 issue of the journal, the study was funded in part by NIH’s National Cancer Institute (NCI) and National Institute for Biomedical Imaging and Bioengineering (NIBIB).
By applying this drug delivery by nanoparticles they were able to shrink tumors in mice with smaller doses of the drug to reduce harmful side effects. Only 30% of the dose of prodrug of cisplatin was required to diminish the tumor by using the drug carrying nanoparticles, than that of standard dose of cisplatin as such.
Researchers initially studied different doses of nanoparticle bound drug in rats and mice, both the types of animals maintained their body weight and survived at higher doses of the drug when drug was delivered using nanoparticles than when injected without nanoparticles. It was also found that the kidney damage was less in rats which received the nanoparticle bound drug.
Also it was found that binding nanoparticles provided greater stability of cisplatin prodrug in blood stream than that of injected alone , after one hour about 77 % of prodrug was found in blood stream when it was delivered using nanoparticles compared to only 16% available drug in case of drug delivered without nanoparticles, cispaltin is very unstable drug and remains in blood for very short time , which calls for more dose to get the desired effect.
Transdermal drug delivery system new requirements for quality and for regulatory submissions
US FDA issued new guidelines for Transdermal drug delivery system and related drug delivery systems.
US FDA stated in its new guidelines on transdermal drug delivery system and related drug delivery systems that the initial drug load concentration has tremendous potential for impacting quality of product its safety and efficacy and it has great potential for drug abuse.
There are many advantages and disadvantages of transdermal drug delivery system TDDS , like a drug can be administered without pain to patient, patients to like the dosage form greatly as they wont feel as they are on medication, and a constant plasma drug concentration can be easily achieved for a drug for a longer period of time without giving the untoward effect of initial higher plasma level of a drug as in case of conventional dosage forms, also drug escape the first pass metabolism through transdermal drug delivery , some a critical drugs which are known to save life are also administered as Transdermal patches for example nitroglycerin in congestive cardiac diseases.
There are some serious effects observed in resent time, like accidental high dose of a drug up on accidental sticking on handling or accidental contact with skin which has lead individual serious and to fatal conditions some times a life threatening one.
The fatal untoward effects are also seen in health care providers which accidentally handled the patches and got drug dose from remaining drug load from the used transdermal drug delivery patch.
The important factor.
The drug concentration which is required to be loaded on to a Transdermal drug delivery or related drug delivery systems is very high than that of the actual drug being absorbed and required to be achieved in to plasma of a patient.

Nanotechnology cancer treatments would use gold particles to carry anticancer drugs straight to the cancer. Learn about nanotechnology cancer treatments.
US FDA guidelines for Transdermal drug delivery patches and related drug delivery systems
In order to finally achieve consistent low residual drug with the desired quality of the Transdermal drug delivery systems ,
1. US FDA requires a drug manufacturers to submit the initial loaded drug concentration in the transdermal drug delivery patch and related drug delivery systems , be provided in the application for investigational new drug applications (INDs), new drug applications (NDAs), abbreviated new drug applications (ANDAs), and supplemental new drug applications (sNDAs) for TDDS, TMDS, and topical patch products.
2.US FDA now requires that the all justifications for initial drug load or concentration should be included in the application.
3.It also states that a proper scientific risk based approach must be taken to minimize the drug residue in the system so that a lowest possible concentration remains in the system.
4.The amount of residual drug in the transdemanl drug delivery system must not exceed than those already approved by FDA .
5. US FDA also requires that the information of product and process development and how the final formulation is justified should be given in the common technical document (CTD) formatted application in section for Pharmaceutical Development.
US FDA has put emphasis on following points
1.) Quality By Design Concept
2.) Minimizing Residual Drug
The transdermal drug delivery patches and related products , be developed with the intention of giving efficacy and safety as well,
The quality by design concept basically requires a formulator to plan for a desired quality, quality of a drug can be best achieved when it is planed than when it monitored.
Planing of quality of a drug product through logical application of past findings and research data and chemistry of drug molecule and exceipients being used, to achieve minimum drug load and this can lead to achieve minimum residual drug in transdermal drug delivery systems after use. Which will ensure that the abuse potential of the transdermal drug delivery systems are taken care of.

Buckminsterfullerene C60, also known as the buckyball, is a representative member of the carbon structures known as fullerenes. Members of the fullerene family are a major subject of research falling under the nanotechnology umbrella.
Nanotechnology and Cancer
Nanotechnology is one of the most popular areas of scientific research, especially with regard to medical applications. We’ve already discussed some of the new detection methods that should bring about cheaper, faster and less invasive cancer diagnoses. But once the diagnosis occurs, there’s still the prospect of surgery, chemotherapy or radiation treatment to destroy the cancer. Unfortunately, these treatments can carry serious side effects. Chemotherapy can cause a variety of ailments, including hair loss, digestive problems, nausea, lack of energy and mouth ulcers.
But nanotechnologists think they have an answer for treatment as well, and it comes in the form of targeted drug therapies. If scientists can load their cancer-detecting gold nanoparticles with anticancer drugs, they could attack the cancer exactly where it lives. Such a treatment means fewer side effects and less medication used. Nanoparticles also carry the potential for targeted and time-release drugs. A potent dose of drugs could be delivered to a specific area but engineered to release over a planned period to ensure maximum effectiveness and the patient’s safety.
These treatments aim to take advantage of the power of nanotechnology and the voracious tendencies of cancer cells, which feast on everything in sight, including drug-laden nanoparticles. One experiment of this type used modified bacteria cells that were 20 percent the size of normal cells. These cells were equipped with antibodies that latched onto cancer cells before releasing the anticancer drugs they contained.
Another used nanoparticles as a companion to other treatments. These particles were sucked up by cancer cells and the cells were then heated with a magnetic field to weaken them. The weakened cancer cells were then much more susceptible to chemotherapy.
It may sound odd, but the dye in your blue jeans or your ballpoint pen has also been paired with gold nanoparticles to fight cancer. This dye, known as phthalocyanine, reacts with light. The nanoparticles take the dye directly to cancer cells while normal cells reject the dye. Once the particles are inside, scientists “activate” them with light to destroy the cancer. Similar therapies have existed to treat skin cancers with light-activated dye, but scientists are now working to use nanoparticles and dye to treat tumors deep in the body.
From manufacturing to medicine to many types of scientific research, nanoparticles are now rather common, but some scientists have voiced concerns about their negative health effects. Nanoparticles’ small size allows them to infiltrate almost anywhere. That’s great for cancer treatment but potentially harmful to healthy cells and DNA. There are also questions about how to dispose of nanoparticles used in manufacturing or other processes. Special disposal techniques are needed to prevent harmful particles from ending up in the water supply or in the general environment, where they’d be impossible to track.
Gold nanoparticles are a popular choice for medical research, diagnostic testing and cancer treatment, but there are numerous types of nanoparticles in use and in development. Bill Hammack, a professor of chemical engineering at the University of Illinois, warned that nanoparticles are “technologically sweet” [Source: Marketplace]. In other words, scientists are so wrapped up in what they can do, they’re not asking if they should do it. The Food and Drug Administration has a task force on nanotechnology, but as of yet, the government has exerted little oversight or regulation.
robotics

A mechanical white blood cell attacks bacteria. The bacteria cannot develop immunity to mechanical devices as it would towards a drug
Nanotechnology, perhaps, has been most popularly recognized for it’s applications in robotics. Nano-robotics, although having many applications in other areas (such as particle manipulation and, has the most useful and variety of uses in medical fields.
Drugs have been shown to be effective during treatment and so has surgery. However, both are only temporary. We do not have much control over the drugs that have entered our body. As mentioned in the “Applications in Drugs and Therapeutics” page, nanotechnology can play an important role by being used for designing drug delivery systems.
Nanorobots, once fully developed, will be more effective than drugs. This is because nanobots cab always be present in the body, fighting off pathogens such as viruses and tumors. Nanorobots will not require any additional treatment and will become relatively cheap after development.
Some of the potential applications for nano-robotics in medicine include early diagnosis and targeted drug delivery for cancer, biomedical instrumentation, surgery, pharmacokinetics, monitoring of diabetes, and health care. Medical nanotechnology in the future will use nanorobots injected into the patient to perform treatments at cellular levels
Some other possible applications using medical nanorobots are as follows:
· To cure skin diseases, a cream containing nanorobots may be used. This cream would remove the right amounts of dead skin cells, remove excess oils which may cause oily skin, insert missing oils, apply the specifically right amounts of natural moisturizing compounds. Dermatological problems would thus be avoided or removed.
· A mouthwash full of water and smart nanorobots could identify and destroy pathogenic bacteria, particles of food, plaque, or tartar, while allowing the harmless flora of the mouth to flourish. Being suspended in liquid and able to swim about, devices would be able to reach surfaces beyond reach of toothbrush bristles or the floss fibers. As short-lifetime medical nano-devices, the bots could be built to last only a few minutes in the body before falling apart into materials of the sort found in foods (such as fibers and other organic compounds). This would not cause any toxic harmful effects in the body, and there would be no need for toothbrushes.
· Medical nanodevices could augment the immune system by finding and disabling unwanted bacteria and viruses. When an invader is identified, it can be punctured, letting its contents spill out and ending its effectiveness. If the contents were known to be hazardous by themselves, then the immune machine could hold on to it long enough to dismantle it more completely. With even more innovation, pathogens could be broken down into simple substances such as oxygen and extra cellular material which can be used for benefit of the body!
· Devices working in the bloodstream could nibble away at arteriosclerotic deposits, widening the affected blood vessels. Various nano-devices could restore the strength of the arteries and veins. With such applications, many heart attacks would be prevented.
More Background on Nanotechnology:
| Nanotechnology Basics – For students and other learners | |
| Managing Magic – A brief overview of the challenges posed by advanced nanotechnology | |
| Nanotechnology on an Upward Slope – An online PowerPoint presentation | |
| Turn on the Nanotech High Beams – An essay published by Future Brief | |
| Nano Simulation – A way to visualize what is meant by molecular manufacturing | |
| Debating the Future of Nanotechnology – Perspective from the Foresight Institute | |
| Safe Utilization of Advanced Nanotechnology – One of the founding papers of CRN | |
| 5-Minute Nanosystems– A quick summary of Eric Drexler’s foundational work on nanotechnology | |
| Nanotechnology Press Kit – Compiled and published by Nanotechnology Now |
Quad Pill for HIV Appears Safe in Renal Disease

Published: Jul 7, 2013
KUALA LUMPUR — HIV patients with mild to moderate renal impairment appear to tolerate treatment with a combination tablet that contains drugs known to impact kidney function, a phase III, open-label, two-cohort study found.
The treatment group receiving the four-drug combination of elvitegravir, cobicistat, tenofovir DF, and emtricitabine, branded as Stribild
GSK Duchenne drug gets ‘breakthrough’ status

The US Food and Drug Administration has granted breakthrough therapy designation to GlaxoSmithKline’s drisapersen for the potential treatment of patients with Duchenne muscular dystrophy. read all at
http://www.pharmatimes.com/Article/13-06-28/GSK_Duchenne_drug_gets_breakthrough_status.aspx
Drisapersen (also known as PRO051 and GSK2402968 ) is an experimental drug under development by Prosensa for the treatment of Duchenne muscular dystrophy.
The compound is in a Phase III trial which is anticipated to complete by the end of 2013.
- ^ “PRO051/GSK2402968”. Prosensa. Retrieved 29 October 2012
- ^ “A Clinical Study to Assess the Efficacy and Safety of GSK2402968 in Subjects With Duchenne Muscular Dystrophy (DMD114044)”. ClinicalTrials.gov. Retrieved 29 October 2012.

Amgen In Focus

Amgen In Focus
Seeking Alpha
According to Amgen, they have 45 drugs in development from Phase 1 to Phase 3. Conversely, Gilead has 32 drugs in development and Pfizer has 64. Meanwhile, Gilead only has 8 drugs in Phase 3, Pfizer has 25, and Amgen has 14. 7 of those Phase 3 …
http://seekingalpha.com/article/1510002-amgen-in-focus?source=google_news
Amgen has the second deepest pipeline of drugs of the three large cap biotechs. According to Amgen, they have 45 drugs in development from Phase 1 to Phase 3. Conversely, Gilead has 32 drugs in development and Pfizer has 64. Meanwhile, Gilead only has 8 drugs in Phase 3, Pfizer has 25, and Amgen has 14. 7 of those Phase 3 drugs are focused on cancer treatments for Amgen, more than either Pfizer or Gilead. Keep in mind that 12.4 million people learn they have cancer each year, while 7.6 million people lose that battle each year. The CDC predicts that the global number of cancer related deaths will increase by 80% by 2030. It doesn’t take a rocket scientist to know that cancer treating drugs presents the largest opportunity for any drug maker considering those statistics. Amgen has the inside track versus Gilead and Pfizer as far as quantity of drugs in late stage development.
Modality
Pharmathene Will Proceed With Anthrax Vaccine Test
May 31, 2013 — PharmAthene Inc. said Thursday that the Food and Drug Administration will allow it to proceed with new clinical trials of its experimental anthrax vaccine SparVax.
http://www.pharmalive.com/pharmathene-will-proceed-with-anthrax-vaccine-test

