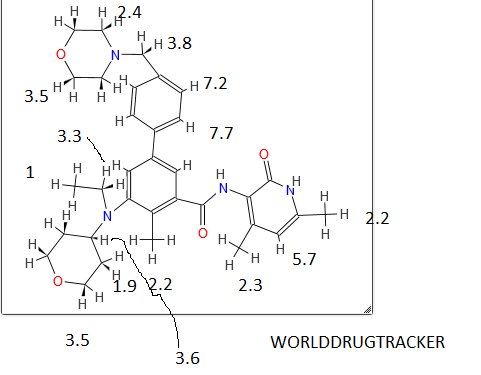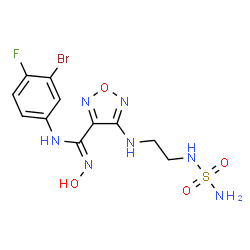
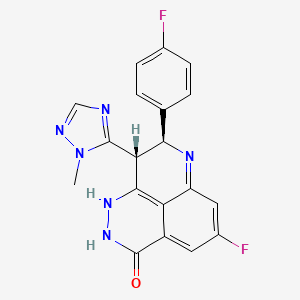
Epacadostat
(Z)-N-(3-bromo-4-fluorophenyl)-N’-hydroxy-4-[2-(sulfamoylamino)ethylamino]-1,2,5-oxadiazole-3-carboxamidine
1,2,5-Oxadiazole-3-carboximidamide, 4-[[2-[(aminosulfonyl)amino]ethyl]amino]-N-(3-bromo-4-fluorophenyl)-N’-hydroxy-
1204669-58-8
INCB024360
N-(3-Brom-4-fluorphenyl)-N’-hydroxy-4-{[2-(sulfamoylamino)ethyl]amino}-1,2,5-oxadiazol-3-carboximidamid
UNII 71596A9R13
(Z)-N-(3-bromo-4-fluorophenyl)-N’-hydroxy-4-(2-(sulfamoylamino)ethylamino)-1,2,5-oxadiazole-3-carboximidamide
1,2,5-Oxadiazole-3-carboximidamide, 4-[[2-[(aminosulfonyl)amino]ethyl]amino]-N’-(3-bromo-4-fluorophenyl)-N-hydroxy-
Molecular Formula, C11H13BrFN7O4S
Average mass438.233 Da
cas 1204669-58-8 (or 1204669-37-3)
| Synonym: |
IDO1 inhibitor INCB024360
indoleamine-2,3-dioxygenase inhibitor INCB024360 |
| Code name: |
INCB 024360
INCB024360 |
| Chemical structure: |
1,2,5-Oxadiazole-3-carboximidamide, 4-((2-((Aminosulfonyl)amino)ethyl)amino)-N-(3-bromo-4-fluorophenyl)-N’-hydroxy-, (C(Z))- |
| Company |
Incyte Corp. |
| Description |
Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor |
| Molecular Target |
Indoleamine 2,3-dioxygenase 1 (IDO1) |
| Mechanism of Action |
Indoleamine 2,3-dioxygenase (INDO) inhibitor |
| Therapeutic Modality |
Small molecule |


- OriginatorIncyte Corporation
- DeveloperFred Hutchinson Cancer Research Center; Incyte Corporation; Merck AG
- ClassAmides; Antineoplastics; Imides; Oxadiazoles; Small molecules
-
- Phase IIFallopian tube cancer; Malignant melanoma; Non-small cell lung cancer; Ovarian cancer; Peritoneal cancer; Solid tumours
Most Recent Events
- 15 Jan 2016Phase-II clinical trials in Solid tumours (Combination therapy, Late-stage disease, Second-line therapy or greater) in USA (PO)
- 11 Jan 2016Phase-II clinical trials in Non-small cell lung cancer (Combination therapy, Late-stage disease, Second-line therapy or greater) in USA (PO)
- 11 Jan 2016The US FDA and Health Canada approve IND application and Clinical Trial Application, respectively, for a phase Ib trial in Ovarian cancer (Combination therapy, Recurrent, Second-line therapy or greater)
In 2016, orphan drug designation was assigned to the compound in the US. for the treatment of stage IIB-IV melanoma
EpacadostatAn orally available hydroxyamidine and inhibitor of indoleamine 2,3-dioxygenase (IDO1), with potential immunomodulating and antineoplastic activities. epacadostat targets and binds to IDO1, an enzyme responsible for the oxidation of tryptophan into kynurenine. By inhibiting IDO1 and decreasing kynurenine in tumor cells, epacadostat increases and restores the proliferation and activation of various immune cells, including dendritic cells (DCs), NK cells, and T-lymphocytes, as well as interferon (IFN) production, and a reduction in tumor-associated regulatory T cells (Tregs). Activation of the immune system, which is suppressed in many cancers, may inhibit the growth of IDO1-expressing tumor cells. IDO1 is overexpressed by a variety of tumor cell types and DCsINCB24360 (epacadostat), An Agent For Cancer Immunotherapy
Incyte and Merck Expand Clinical Collaboration to Include Phase 3 Study Investigating the Combination of Epacadostat with Keytruda® (pembrolizumab) as First-line Treatment for Advanced Melanoma
Pivotal study to evaluate Incyte’s IDO1 inhibitor in combination with Merck’s anti-PD-1 therapy in patients with advanced or metastatic melanoma
WILMINGTON, Del. and KENILWORTH, N.J. — October 13, 2015 — Incyte Corporation (Nasdaq: INCY) and Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced the expansion of the companies’ ongoing clinical collaboration to include a Phase 3 study evaluating the combination of epacadostat, Incyte’s investigational selective IDO1 inhibitor, with Keytruda® (pembrolizumab), Merck’s anti-PD-1 therapy, as first-line treatment for patients with advanced or metastatic melanoma. The Phase 3 study, which is expected to begin in the first half of 2016, will be co-funded by Incyte and Merck.
“We are very pleased to expand our collaboration with Merck and to move the clinical development program for epacadostat in combination with Keytruda into Phase 3,” said Hervé Hoppenot, President and Chief Executive Officer of Incyte. “We believe the combination of these two immunotherapies shows promise and, if successfully developed, may help to improve clinical outcomes for patients with metastatic melanoma.”
“The initiation of this large Phase 3 study with Incyte in the first-line advanced melanoma treatment setting is an important addition to our robust immunotherapy clinical development program for Keytruda,” said Dr. Roger Dansey, senior vice president and therapeutic area head, oncology late-stage development, Merck Research Laboratories. “We continue to explore the benefit that Keytruda brings to patients suffering from advanced melanoma when used alone, and we are pleased to be able to add this important combination study with epacadostat to our Keytruda development program.”
Under the terms of the agreement Incyte and Merck have also agreed, for a period of two years, not to initiate new pivotal studies of an IDO1 inhibitor in combination with a PD-1/PD-L1 antagonist as first-line therapy in advanced or metastatic melanoma with any third party. During this time, the companies will each offer the other the opportunity to collaborate on any new pivotal study involving an IDO1 inhibitor in combination with a PD-1/PD-L1 antagonist for types of melanoma and lines of therapy outside of the current collaboration agreement.
The agreement is between Incyte and certain subsidiaries and Merck through its subsidiaries.
Epacadostat and Keytruda are part of a class of cancer treatments known as immunotherapies that are designed to enhance the body’s own defenses in fighting cancer; the two therapies target distinct regulatory components of the immune system. IDO1 is an immunosuppressive enzyme that has been shown to induce regulatory T cell generation and activation, and allow tumors to escape immune surveillance. Keytruda is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. Preclinical evidence suggests that the combination of these two agents may lead to an enhanced anti-tumor immune response compared with either agent alone.
Safety and efficacy data from the ongoing Phase 1/2 study evaluating the combination of epacadostat with Keytruda in patients with advanced malignancies is scheduled to be highlighted as a late-breaking oral presentation (Abstract #142) at the upcoming Society for Immunotherapy of Cancer 30th Anniversary Annual Meeting & Associated Programs, November 4–8, 2015 at the Gaylord National Resort & Convention Center in National Harbor, MD.
Metastatic Melanoma
Melanoma, the most serious form of skin cancer, strikes adults of all ages and accounts for approximately five percent of all new cases of cancer in the United States each year. The number of new cases of melanoma continues to rise by almost three percent each year which translates to 76,000 new cases yearly in the U.S. alone.[i] The 5-year survival rate for late-stage or metastatic disease is 15 percent.[ii]
About Epacadostat (INCB024360)
Indoleamine 2,3-dioxygenase 1 (IDO1) is an immunosuppressive enzyme that has been shown to induce regulatory T cell generation and activation, and allow tumors to escape immune surveillance. Epacadostat is an orally bioavailable small molecule inhibitor of IDO1 that has nanomolar potency in both biochemical and cellular assays and has demonstrated potent activity in enhancing T lymphocyte, dendritic cell and natural killer cell responses in vitro, with a high degree of selectivity. Epacadostat has shown proof-of-concept clinical data in patients with unresectable or metastatic melanoma in combination with the CTLA-4 inhibitor ipilimumab, and is currently in four proof-of-concept clinical trials with PD-1 and PD-L1 immune checkpoint inhibitors in a variety of cancer histologies.

PATENT
WO 2014066834
https://www.google.com/patents/WO2014066834A1?cl=en
EXAMPLE 1
4-({2-[(Aminosulfonyl)amino]ethyl}amino)- V-(3-bromo-4-fluorophenyl)- V -hydroxy- l,2,5-oxadiazole-3-carboximidamide
Step 1: 4-Amino-N’-hydroxy-l,2,5-oxadiazole-3-carboximidamide
[00184] Malononitrile (320.5 g, 5 mol) was added to water (7 L) preheated to 45 °C and stirred for 5 min. The resulting solution was cooled in an ice bath and sodium nitrite (380 g, 5.5 mol) was added. When the temperature reached 10 °C, 6 N hydrochloric acid (55 mL) was added. A mild exothermic reaction ensued with the temperature reaching 16 °C. After 15 min the cold bath was removed and the reaction mixture was stirred for 1.5 hrs at 16-18 °C. The reaction mixture was cooled to 13 °C and 50% aqueous hydroxylamine (990 g, 15 mol) was added all at once. The temperature rose to 26 °C. When the exothermic reaction subsided the cold bath was removed and stirring was continued for 1 hr at 26-27 °C, then it was slowly brought to reflux. Reflux was maintained for 2 hrs and then the reaction mixture was allowed to cool overnight. The reaction mixture was stirred in an ice bath and 6 N hydrochloric acid (800 mL) was added in portions over 40 min to pH 7.0. Stirring was continued in the ice bath at 5 °C. The precipitate was collected by filtration, washed well with water and dried in a vacuum oven (50 °C) to give the desired product (644 g, 90%). LCMS for C3H6N5O2
(M+H)+: m/z = 144.0. 13C MR (75 MHz, CD3OD): δ 156.0, 145.9, 141.3. Step 2: 4-Amino-N-hydroxy-l,2,5-oxadiazole-3-carboximidoyl chloride [00185] 4-Amino-N,-hydroxy-l ,2,5-oxadiazole-3-carboximidamide (422 g, 2.95 mol) was added to a mixture of water (5.9 L), acetic acid (3 L) and 6 Ν hydrochloric acid (1.475 L, 3 eq.) and this suspension was stirred at 42 – 45 °C until complete solution was achieved. Sodium chloride (518 g, 3 eq.) was added and this solution was stirred in an ice/water/methanol bath. A solution of sodium nitrite (199.5 g, 0.98 eq.) in water (700 mL) was added over 3.5 hrs while maintaining the temperature below 0 °C. After complete addition stirring was continued in the ice bath for 1.5 hrs and then the reaction mixture was allowed to warm to 15 °C. The precipitate was collected by filtration, washed well with water, taken in ethyl acetate (3.4 L), treated with anhydrous sodium sulfate (500 g) and stirred for 1 hr. This suspension was filtered through sodium sulfate (200 g) and the filtrate was concentrated on a rotary evaporator. The residue was dissolved in methyl i-butyl ether (5.5 L), treated with charcoal (40 g), stirred for 40 min and filtered through Celite. The solvent was removed in a rotary evaporator and the resulting product was dried in a vacuum oven (45 °C) to give the desired product (256 g, 53.4%). LCMS for C3H4CIN4O2 (M+H)+: m/z = 162.9. 13C NMR (100 MHz, CD3OD): 5 155.8, 143.4, 129.7.
Step 3: 4-Amino-N’-hydroxy-N-(2-methoxyethyl)-l,2,5-oxadiazole-3-carboximidamide [00186] 4-Amino-N-hydroxy-l ,2,5-oxadiazole-3-carboximidoyl chloride (200.0 g, 1.23 mol) was mixed with ethyl acetate (1.2 L). At 0-5 °C 2-methoxyethylamine [Aldrich, product # 143693] (119.0 mL, 1.35 mol) was added in one portion while stirring. The reaction temperature rose to 41 °C. The reaction was cooled to 0 – 5 °C. Triethylamine (258 mL, 1.84 mol) was added. After stirring 5 min, LCMS indicated reaction completion. The reaction solution was washed with water (500 mL) and brine (500 mL), dried over sodium sulfate, and concentrated to give the desired product (294 g, 1 19%) as a crude dark oil.
LCMS for C6Hi2 503 (M+H)+: m/z = 202.3. 1H NMR (400 MHz, DMSO- ): δ 10.65 (s, 1 H), 6.27 (s, 2 H), 6.10 (t, J = 6.5 Hz, 1 H), 3.50 (m, 2 H), 3.35 (d, J = 5.8 Hz, 2 H), 3.08 (s, 3 H).
Step 4: N’-Hydroxy-4-[(2-methoxyethyl)amino]-l,2,5-oxadiazole-3-carboximidamide
[00187] 4-Amino-N-hydroxy-N-(2-methoxyethyl)-l,2,5-oxadiazole-3- carboximidamide (248.0 g, 1.23 mol) was mixed with water (1 L). Potassium hydroxide (210 g, 3.7 mol) was added. The reaction was refluxed at 100 °C overnight (15 hours). TLC with 50% ethyl acetate (containing 1% ammonium hydroxide) in hexane indicated reaction completed (product Rf = 0.6, starting material Rf = 0.5). LCMS also indicated reaction completion. The reaction was cooled to room temperature and extracted with ethyl acetate (3 x 1 L). The combined ethyl acetate solution was dried over sodium sulfate and concentrated to give the desired product (201 g, 81%) as a crude off-white solid. LCMS for C6H12N5O3 (M+H)+: m/z = 202.3 LH NMR (400 MHz, OMSO-d6): δ 10.54 (s, 1 H), 6.22 (s, 2 H), 6.15 (t, J = 5.8 Hz, 1 H), 3.45 (t, J= 5.3 Hz, 2 H), 3.35 (m, 2 H), 3.22 (s, 3 H). Step 5: N-Hydroxy-4-[(2-methoxyethyl)amino]-l,2,5-oxadiazole-3-carboximidoyl chloride
[00188] At room temperature N’-hydroxy-4-[(2-methoxyethyl)amino]- 1 ,2,5- oxadiazole-3-carboximidamide (50.0 g, 0.226 mol) was dissolved in 6.0 M hydrochloric acid aqueous solution (250 mL, 1.5 mol). Sodium chloride (39.5 g, 0.676 mol) was added followed by water (250 mL) and ethyl acetate (250 mL). At 3-5 °C a previously prepared aqueous solution (100 mL) of sodium nitrite (15.0 g, 0.217 mol) was added slowly over 1 hr. The reaction was stirred at 3 – 8 °C for 2 hours and then room temperature over the weekend. LCMS indicated reaction completed. The reaction solution was extracted with ethyl acetate (2 x 200 mL). The combined ethyl acetate solution was dried over sodium sulfate and concentrated to give the desired product (49.9 g, 126%) as a crude white solid. LCMS for
C6HioClN403 (M+H)+: m/z = 221.0. !H NMR (400 MHz, DMSO-d6): δ 13.43 (s, 1 H), 5.85 (t, J= 5.6 Hz, 1 H), 3.50 (t, J= 5.6 Hz, 2 H), 3.37(dd, J= 10.8, 5.6 Hz, 2 H), 3.25 (s, 3 H).
Step 6 : N-(3-Bromo-4-fluorophenyl)-N’-hydroxy-4- [(2-methoxyethyl)amino] – 1 ,2,5- oxadiazole-3-carboximidamide [00189] N-Hydroxy-4-[(2-methoxyethyl)amino]- 1 ,2,5-oxadiazole-3-carboximidoyl chloride (46.0 g, 0.208 mol) was mixed with water (300 mL). The mixture was heated to 60 °C. 3-Bromo-4-fluoroaniline [Oakwood products, product # 013091] (43.6 g, 0.229 mol) was added and stirred for 10 min. A warm sodium bicarbonate (26.3 g, 0.313 mol) solution (300 mL water) was added over 15 min. The reaction was stirred at 60 °C for 20 min. LCMS indicated reaction completion. The reaction solution was cooled to room temperature and extracted with ethyl acetate (2 x 300 mL). The combined ethyl acetate solution was dried over sodium sulfate and concentrated to give the desired product (76.7 g, 98%) as a crude brown solid. LCMS for Ci2Hi4BrF503 (M+H)+: m/z = 374.0, 376.0. 1H NMR (400 MHz, DMSO- tf): δ 11.55 (s, 1 H), 8.85 (s, 1 H), 7.16 (t, J= 8.8 Hz, 1 H), 7.08 (dd, J= 6.1, 2.7 Hz, 1 H), 6.75 (m, 1 H), 6.14 (t, J= 5.8 Hz, 1 H), 3.48 (t, J = 5.2 Hz, 2 H), 3.35 (dd, J= 10.8, 5.6 Hz, 2 H), 3.22 (s, 3 H).
Step 7: 4-(3-Bromo-4-fluorophenyl)-3-{4- [(2-methoxyethyl)amino]-l,2,5-oxadiazol-3- yl}-l,2,4-oxadiazol-5(4H)-one
[00190] A mixture of N-(3-bromo-4-fluorophenyl)-N’-hydroxy-4-[(2- methoxyethyl)amino]-l,2,5-oxadiazole-3-carboximidamide (76.5 g, 0.204 mol), 1,1 ‘- carbonyldiimidazole (49.7 g, 0.307 mol), and ethyl acetate (720 mL) was heated to 60 °C and stirred for 20 min. LCMS indicated reaction completed. The reaction was cooled to room temperature, washed with 1 N HC1 (2 x 750 mL), dried over sodium sulfate, and concentrated to give the desired product (80.4 g, 98%) as a crude brown solid. LCMS for
(M+H)+: m/z = 400.0, 402.0. 1H NMR (400 MHz, DMSO-c½): δ 7.94 (t, J = 8.2 Hz, 1 H), 7.72 (dd, J = 9.1, 2.3 Hz, 1 H), 7.42 (m, 1 H), 6.42 (t, J= 5.7 Hz, 1 H), 3.46 (t, J = 5.4 Hz, 2 H), 3.36 (t, J= 5.8 Hz, 2 H), 3.26 (s, 3 H).
Step 8: 4-(3-Bromo-4-fluorophenyl)-3-{4-[(2-hydroxyethyl)amino]-l,2,5-oxadiazol-3- yl}-l,2,4-oxadiazol-5(4H)-one
[00191] 4-(3-Bromo-4-fluoroplienyl)-3-{4-[(2-metlioxyethyl)amino]-l,2,5-oxadiazol- 3-yl}-l,2,4-oxadiazol-5(4H)-one (78.4 g, 0.196 mol) was dissolved in dichloromethane (600 mL). At -67 °C boron tribromide (37 mL, 0.392 mol) was added over 15 min. The reaction was warmed up to -10 °C in 30 min. LCMS indicated reaction completed. The reaction was stirred at room temperature for 1 hour. At 0 – 5 °C the reaction was slowly quenched with saturated sodium bicarbonate solution (1.5 L) over 30 min. The reaction temperature rose to 25 °C. The reaction was extracted with ethyl acetate (2 x 500 mL, first extraction organic layer is on the bottom and second extraction organic lager is on the top). The combined organic layers were dried over sodium sulfate and concentrated to give the desired product (75 g, 99%) as a crude brown solid. LCMS for Ci2HioBrFN504 (M+H)+: m/z = 386.0, 388.0.
1H NMR (400 MHz, DMSO-^): δ 8.08 (dd, J = 6.2, 2.5 Hz, 1 H), 7.70 (m, 1 H), 7.68 (t, J = 8.7 Hz, 1 H), 6.33 (t, J = 5.6 Hz, 1 H), 4.85 (t, J= 5.0 Hz, 1 H), 3.56 (dd, J= 10.6, 5.6 Hz, 2 H), 3.29 (dd, J= 11.5, 5.9 Hz, 2 H).
Step 9 : 2-({4- [4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro- 1 ,2,4-oxadiazol-3-yl] – l,2,5-oxadiazol-3-yl}amino)ethyl methanesulfonate
[00192] To a solution of 4-(3-bromo-4-fluorophenyl)-3-{4-[(2-hydroxyethyl)amino]- l,2,5-oxadiazol-3-yl}-l,2,4-oxadiazol-5(4H)-one (1.5 kg, 3.9 mol, containing also some of the corresponding bromo-compound) in ethyl acetate (12 L) was added methanesulfonyl chloride (185 mL, 2.4 mol) dropwise over 1 h at room temperature. Triethylamine (325 mL, 2.3 mol) was added dropwise over 45 min, during which time the reaction temperature increased to 35 °C. After 2 h, the reaction mixture was washed with water (5 L), brine (1 L), dried over sodium sulfate, combined with 3 more reactions of the same size, and the solvents removed in vacuo to afford the desired product (7600 g, quantitative yield) as a tan solid. LCMS for C HnBrFNsOeS a (M+Na)+: m/z = 485.9, 487.9. !H NMR (400 MHz, DMSO- d6): δ 8.08 (dd, J = 6.2, 2.5 Hz, 1 H), 7.72 (m, 1 H), 7.58 (t, J = 8.7 Hz, 1 H), 6.75 (t, J = 5.9 Hz, 1 H), 4.36 (t, J = 5.3 Hz, 2 H), 3.58 (dd, J = 11.2, 5.6 Hz, 2 H), 3.18 (s, 3 H).
Step 10: 3-{4-[(2-Azidoethyl)amino]-l,2,5-oxadiazol-3-yl}-4-(3-bromo-4-fluorophenyl)- l,2,4-oxadiazol-5(4H)-one
To a solution of 2-({4-[4-(3-bromo-4-f uorophenyl)-5-oxo-4,5-dihydro-l ,2,4- oxadiazol-3-yl]-l ,2,5-oxadiazol-3-yl}amino)ethyl methanesulfonate (2.13 kg, 4.6 mol, containing also some of the corresponding bromo-compound) in dimethylformamide (4 L) stirring in a 22 L flask was added sodium azide (380 g, 5.84 mol). The reaction was heated at 50 °C for 6 h, poured into ice/water (8 L), and extracted with 1 : 1 ethyl acetate:heptane (20 L). The organic layer was washed with water (5 L) and brine (5 L), and the solvents removed in vacuo to afford the desired product (1464 g, 77%) as a tan solid. LCMS for CnHgBrFNsOs a
(M+Na)+: m/z = 433.0, 435.0. !H NMR (400 MHz, DMSO-J6): δ 8.08 (dd, J = 6.2, 2.5 Hz, 1 H), 7.72 (m, 1 H), 7.58 (t, J= 8.7 Hz, 1 H), 6.75 (t, J = 5.7 Hz, 1 H), 3.54 (t, J = 5.3 Hz, 2 H), 3.45 (dd, J= 1 1.1 , 5.2 Hz, 2 H).
Step 11: 3-{4-[(2-Aminoethyl)amino]-l,2,5-oxadiazol-3-yl}-4-(3-bromo-4-fluorophenyl)-
1.2.4- oxadiazol-5(4H)-one hydrochloride
[00194] Sodium iodide (1080 g, 7.2 mol) was added to 3-{4-[(2-azidoethyl)amino]-
1.2.5- oxadiazol-3-yl}-4-(3-bromo-4-fluorophenyl)-l ,2,4-oxadiazol-5(4H)-one (500 g, 1.22 mol) in methanol (6 L). The mixture was allowed to stir for 30 min during which time a mild exotherm was observed. Chlorotrimethylsilane (930 mL, 7.33 mol) was added as a solution in methanol (1 L) dropwise at a rate so that the temperature did not exceed 35 °C, and the reaction was allowed to stir for 3.5 h at ambient temperature. The reaction was neutralized with 33 wt% solution of sodium thiosulfate pentahydrate in water (-1.5 L), diluted with water (4 L), and the pH adjusted to 9 carefully with solid potassium carbonate (250 g – added in small portions: watch foaming). Di-ieri-butyl dicarbonate (318 g, 1.45 mol) was added and the reaction was allowed to stir at room temperature. Additional potassium carbonate (200 g) was added in 50 g portions over 4 h to ensure that the pH was still at or above 9. After stirring at room temperature overnight, the solid was filtered, triturated with water (2 L), and then MTBE (1.5 L). A total of 11 runs were performed (5.5 kg, 13.38 mol). The combined solids were triturated with 1 : 1 THF:dichloromethane (24 L, 4 runs in a 20 L rotary evaporator flask, 50 °C, 1 h), filtered, and washed with dichloromethane (3 L each run) to afford an off- white solid. The crude material was dissolved at 55 °C tetrahydrofuran (5 mL/g), treated with decolorizing carbon (2 wt%) and silica gel (2 wt%), and filtered hot through celite to afford the product as an off-white solid (5122 g). The combined MTBE, THF, and dichloromethane filtrates were concentrated in vacuo and chromatographed (2 kg silica gel, heptane with a 0-100% ethyl acetate gradient, 30 L) to afford more product (262 g). The combined solids were dried to a constant weight in a convection oven (5385 g, 83%).
In a 22 L flask was charged hydrogen chloride (4 N solution in 1 ,4-dioxane, 4 L, 16 mol). tert-Butyl [2-({4-[4-(3-bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-l ,2,4- oxadiazol-3-yl]-l ,2,5-oxadiazol-3-yl}amino)ethyl]carbamate (2315 g, 4.77 mol) was added as a solid in portions over 10 min. The slurry was stirred at room temperature and gradually became a thick paste that could not be stirred. After sitting overnight at room temperature, the paste was slurried in ethyl acetate (10 L), filtered, re-slurried in ethyl acetate (5 L), filtered, and dried to a constant weight to afford the desired product as a white solid (combined with other runs, 5 kg starting material charged, 41 13 g, 95%). LCMS for
Ci2HnBrFN603 (M+H)+: m/z = 384.9, 386.9. 1H NMR (400 MHz, DMSO-^): δ 8.12 (m, 4 H), 7.76 (m, 1 H), 7.58 (t, J = 8.7 Hz, 1 H), 6.78 (t, J = 6.1 Hz, 1 H), 3.51 (dd, J = 1 1.8, 6.1 Hz, 2 H), 3.02 (m, 2 H).
Step 12: tert-Butyl ({[2-({4-[4-(3-bromo-4-nuorophenyl)-5-oxo-4,5-dihydro-l,2,4- oxadiazol-3-yl]-l,2,5-oxadiazol-3-yl}amino)ethyl]amino}sulfonyl)carbamate
A 5 L round bottom flask was charged with chlorosulfonyl isocyanate [Aldrich, product # 142662] (149 mL, 1.72 mol) and dichloromethane (1.5 L) and cooled using an ice bath to 2 °C. teri-Butanol (162 mL, 1.73 mol) in dichloromethane (200 mL) was added dropwise at a rate so that the temperature did not exceed 10 °C. The resulting solution was stirred at room temperature for 30-60 min to provide tert-bvAy\ [chlorosulfonyl]carbamate.
A 22 L flask was charged with 3- {4-[(2-aminoethyl)amino]- 1 ,2,5-oxadiazol-3- yl}-4-(3-bromo-4-fluorophenyl)-l,2,4-oxadiazol-5(4H)-one hydrochloride (661 g, 1.57 mol) and 8.5 L dichloromethane. After cooling to -15 °C with an ice/salt bath, the solution oi tert- Vmtvl i Vi 1 r>rosulfonyl]carbamate (prepared as above) was added at a rate so that the temperature did not exceed -10 °C (addition time 7 min). After stirring for 10 min, triethylamine (1085 mL, 7.78 mol) was added at a rate so that the temperature did not exceed -5 °C (addition time 10 min). The cold bath was removed, the reaction was allowed to warm to 10 °C, split into two portions, and neutralized with 10% cone HC1 (4.5 L each portion). Each portion was transferred to a 50 L separatory funnel and diluted with ethyl acetate to completely dissolve the white solid (-25 L). The layers were separated, and the organic layer was washed with water (5 L), brine (5 L), and the solvents removed in vacuo to afford an off- white solid. The solid was triturated with MTBE (2 x 1.5 L) and dried to a constant weight to afford a white solid. A total of 4113 g starting material was processed in this manner (5409 g, 98%). 1H NMR (400 MHz, DMSO-^): δ 10.90 (s, 1 H), 8.08 (dd, J = 6.2, 2.5 Hz, 1 H), 7.72 (m, 1 H), 7.59 (t, J = 8.6 Hz, 1 H), 6.58 (t, J = 5.7 Hz, 1 H), 3.38 (dd, J= 12.7, 6.2 Hz, 2 H), 3.10 (dd, J= 12.1 , 5.9 Hz, 2 H), 1.41 (s, 9 H).
Step 13: N-[2-({4-[4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-l,2,4-oxadiazol-3-yl]- l,2,5-oxadiazol-3-yl}amino)ethyl]sulfamide
[00198] To a 22 L flask containing 98:2 trifluoroacetic acid:water (8.9 L) was added tert-bvXyl ({[2-({4-[4-(3-bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-l,2,4-oxadiazol-3-yl]- l,2,5-oxadiazol-3-yl}amino)ethyl]amino}sulfonyl)carbamate (1931 g, 3.42 mol) in portions over 10 minutes. The resulting mixture was stirred at room temperature for 1.5 h, the solvents removed in vacuo, and chased with dichloromethane (2 L). The resulting solid was treated a second time with fresh 98:2 trifluoroacetic acid:water (8.9 L), heated for 1 h at 40- 50 °C, the solvents removed in vacuo, and chased with dichloromethane (3 x 2 L). The resulting white solid was dried in a vacuum drying oven at 50 °C overnight. A total of 5409 g was processed in this manner (4990 g, quant, yield). LCMS for C12H12BrFN705S (M+H)+: m/z = 463.9, 465.9. 1H NMR (400 MHz, DMSO- ): δ 8.08 (dd, J = 6.2, 2.5 Hz, 1 H), 7.72 (m, 1 H), 7.59 (t, J= 8.7 Hz, 1 H), 6.67 (t, J = 5.9 Hz, 1H), 6.52 (t, J= 6.0 Hz, 1 H), 3.38 (dd, J = 12.7, 6.3 Hz, 2 H), 3.11 (dd, J = 12.3, 6.3 Hz). Step 14: 4-({2-[(Aminosulfonyl)amino]ethyl}amino)-N-(3-bromo-4-fluorophenyl)-N’- hydroxy-l,2,5-oxadiazole-3-carboximidamide
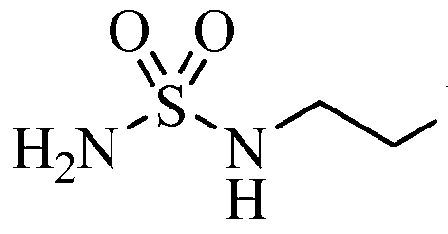
[00199] To a crude mixture of N-[2-({4-[4-(3-bromo-4-fluorophenyl)-5-oxo-4,5- dihydro-l,2,4-oxadiazol-3-yl]-l,2,5-oxadiazol-3-yl}amino)ethyl]sulfamide (2.4 mol) containing residual amounts of trifluoroacetic acid stirring in a 22 L flask was added THF (5 L). The resulting solution was cooled to 0 °C using an ice bath and 2 N NaOH (4 L) was added at a rate so that the temperature did not exceed 10 °C. After stirring at ambient temperature for 3 h (LCMS indicated no starting material remained), the pH was adjusted to 3-4 with concentrated HC1 (-500 mL). The THF was removed in vacuo, and the resulting mixture was extracted with ethyl acetate (15 L). The organic layer was washed with water (5 L), brine (5 L), and the solvents removed in vacuo to afford a solid. The solid was triturated with MTBE (2 x 2 L), combined with three other reactions of the same size, and dried overnight in a convection oven to afford a white solid (3535 g). The solid was recrystallized (3 x 22 L flasks, 2:1 watenethanol, 14.1 L each flask) and dried in a 50 °C convection oven to a constant weight to furnish the title compound as an off-white solid (3290 g, 78%). LCMS for CnHnBrF yC S (M+H)+: m/z = 437.9, 439.9. i NMR (400 MHz, DMSO-J^): δ 11.51 (s, 1 H), 8.90 (s, 1 H), 7.17 (t, J= 8.8 Hz, 1 H), 7.11 (dd, J= 6.1, 2.7 Hz, 1 H), 6.76 (m, 1 H), 6.71 (t, J = 6.0 Hz, 1 H), 6.59 (s, 2 H), 6.23 (t, J= 6.1 Hz, 1 H), 3.35 (dd, J= 10.9, 7.0 Hz, 2 H), 3.10 (dd, J= 12.1, 6.2 Hz, 2 H).
PATENT
WO 2010005958
https://www.google.com/patents/WO2010005958A2?cl=en
EXAMPLES Example 1
4-({2-[(Aminosulfonyl)amino]ethyl}amino)-7V-(3-bromo-4-fluorophenyl)-iV’-hydroxy- l,2,5-oxadiazole-3-carboximidamide
Step A: 4-Amino-N’-hydroxy-l,2,5-oxadiazole-3-carboximidamide

Malononitrile [Aldrich, product # M1407] (320.5 g, 5 mol) was added to water (7 L) preheated to 45 0C and stirred for 5 min. The resulting solution was cooled in an ice bath and sodium nitrite (380 g, 5.5 mol) was added. When the temperature reached 10 0C, 6 N hydrochloric acid (55 mL) was added. A mild exothermic reaction ensued with the temperature reaching 16 0C. After 15 min the cold bath was removed and the reaction mixture was stirred for 1.5 hrs at 16-18 0C. The reaction mixture was cooled to 13 0C and 50% aqueous hydroxylamine (990 g, 15 mol) was added all at once. The temperature rose to 26 0C. When the exothermic reaction subsided the cold bath was removed and stirring was continued for 1 hr at 26-270C, then it was slowly brought to reflux. Reflux was maintained for 2 hrs and then the reaction mixture was allowed to cool overnight. The reaction mixture was stirred in an ice bath and 6 N hydrochloric acid (800 mL) was added in portions over 40 min to pH 7.0. Stirring was continued in the ice bath at 5 0C. The precipitate was collected by filtration, washed well with water and dried in a vacuum oven (50 0C) to give the desired product (644 g, 90%). LCMS for C3H6N5O2 (M+H)+: m/z = 144.0. 13C NMR (75 MHz, CD3OD): δ 156.0, 145.9, 141.3. Step B: 4-Amino-N-hydroxy-l,2,5-oxadiazole-3-carboximidoyl chloride

4-Amino-N’-hydroxy-l,2,5-oxadiazole-3-carboximidamide (422 g, 2.95 mol) was added to a mixture of water (5.9 L), acetic acid (3 L) and 6 Ν hydrochloric acid (1.475 L, 3 eq.) and this suspension was stirred at 42 – 45 0C until complete solution was achieved. Sodium chloride (518 g, 3 eq.) was added and this solution was stirred in an ice/water/methanol bath. A solution of sodium nitrite (199.5 g, 0.98 eq.) in water (700 mL) was added over 3.5 hrs while maintaining the temperature below 0 0C. After complete addition stirring was continued in the ice bath for 1.5 hrs and then the reaction mixture was allowed to warm to 15 0C. The precipitate was collected by filtration, washed well with water, taken in ethyl acetate (3.4 L), treated with anhydrous sodium sulfate (500 g) and stirred for 1 hr. This suspension was filtered through sodium sulfate (200 g) and the filtrate was concentrated on a rotary evaporator. The residue was dissolved in methyl f-butyl ether (5.5 L), treated with charcoal (40 g), stirred for 40 min and filtered through Celite. The solvent was removed in a rotary evaporator and the resulting product was dried in a vacuum oven (45 0C) to give the desired product (256 g, 53.4%). LCMS for C3H4ClN4O2(M+H)+: m/z = 162.9. 13c NMR (100 MHz, CD3OD): δ 155.8, 143.4, 129.7.
Step C: 4-Amino-N’-hydroxy-N-(2-methoxyethyl)- 1 ,2,5-oxadiazole-3-carboximidamide
4-Amino-N-hydroxy-l,2,5-oxadiazole-3-carboximidoyl chloride (200.0 g, 1.23 mol) was mixed with ethyl acetate (1.2 L). At 0-50C 2-methoxyethylamine [Aldrich, product # 143693] (119.0 mL, 1.35 mol) was added in one portion while stirring. The reaction temperature rose to 41 0C. The reaction was cooled to 0 – 5 °C. Triethylamine (258 mL, 1.84 mol) was added. After stirring 5 min, LCMS indicated reaction completion. The reaction solution was washed with water (500 mL) and brine (500 mL), dried over sodium sulfate, and concentrated to give the desired product (294 g, 119%) as a crude dark oil. LCMS for C6Hi2N5O3 (M+H)+: m/z = 202.3. 1H NMR (400 MHz, DMSO-J6): δ 10.65 (s, 1 H), 6.27 (s, 2 H), 6.10 (t, J= 6.5 Hz, 1 H), 3.50 (m, 2 H), 3.35 (d, J= 5.8 Hz, 2 H), 3.08 (s, 3 H).
Step D: N’-Hydroxy-4-[(2-methoxyethyl)amino]-l ,2,5-oxadiazole-3-carboximidamide
4-Amino-N’-hydroxy-N-(2-methoxyethyl)-l,2,5-oxadiazole-3-carboximidaniide (248.0 g, 1.23 mol) was mixed with water (1 L). Potassium hydroxide (210 g, 3.7 mol) was added. The reaction was refluxed at 100 0C overnight (15 hours). TLC with 50% ethyl acetate (containing 1% ammonium hydroxide) in hexane indicated reaction completed (product Rf= 0.6, starting material Rf = 0.5). LCMS also indicated reaction completion. The reaction was cooled to room temperature and extracted with ethyl acetate (3 x 1 L). The combined ethyl acetate solution was dried over sodium sulfate and concentrated to give the desired product (201 g, 81%) as a crude off-white solid. LCMS for C6H12N5O3 (M+H)+: m/z = 202.3 1H NMR (400 MHz, DMSO-Gk): δ 10.54 (s, 1 H), 6.22 (s, 2 H), 6.15 (t, J= 5.8 Hz, 1 H), 3.45 (t, J= 5.3 Hz, 2 H), 3.35 (m, 2 H), 3.22 (s, 3 H).
Step E: N-Hydroxy-4-[(2-methoxyethyl)amino]-l,2,5-oxadiazole-3-carboximidoyl chloride
Ν. ,Ν O
At room temperature N’-hydroxy-4-[(2-methoxyethyl)amino]-l,2,5-oxadiazole-3- carboximidamide (50.0 g, 0.226 mol) was dissolved in 6.0 M hydrochloric acid aqueous solution (250 mL, 1.5 mol). Sodium chloride (39.5 g, 0.676 mol) was added followed by water (250 mL) and ethyl acetate (250 mL). At 3-5 0C a previously prepared aqueous solution (100 mL) of sodium nitrite (15.0 g, 0.217 mol) was added slowly over 1 hr. The reaction was stirred at 3 – 8 0C for 2 hours and then room temperature over the weekend. LCMS indicated reaction completed. The reaction solution was extracted with ethyl acetate (2 x 200 mL). The combined ethyl acetate solution was dried over sodium sulfate and concentrated to give the desired product (49.9 g, 126%) as a crude white solid. LCMS for C6Hi0ClN4O3 (M+H)+: m/z = 221.0. 1H NMR (400 MHz, DMSO-J6): δ 13.43 (s, 1 H), 5.85 (t, J= 5.6 Hz, 1 H), 3.50 (t, J= 5.6 Hz, 2 H), 3.37(dd, J= 10.8, 5.6 Hz, 2 H), 3.25 (s, 3 H).
Step F: N-(3-Bromo-4-fluorophenyl)-N’-hydroxy-4-[(2-methoxyethyl)amino]- 1 ,2,5- oxadiazole-3 -carboximidamide

N-Hydroxy-4-[(2-methoxyethyl)amino]-l,2,5-oxadiazole-3-carboximidoyl chloride (46.0 g, 0.208 mol) was mixed with water (300 mL). The mixture was heated to 60 °C. 3-Bromo-4- fluoroaniline [Oakwood products, product # 013091] (43.6 g, 0.229 mol) was added and stirred for 10 nrn‘n. A warm sodium bicarbonate (26.3 g, 0.313 mol) solution (300 mL water) was added over 15 min. The reaction was stirred at 60 0C for 20 min. LCMS indicated reaction completion. The reaction solution was cooled to room temperature and extracted with ethyl acetate (2 x 300 mL). The combined ethyl acetate solution was dried over sodium sulfate and concentrated to give the desired product (76.7 g, 98%) as a crude brown solid. LCMS for Ci2Hi4BrFN5O3 (M+H)+: m/z = 374.0, 376.0. 1H NMR (400 MHz, DMSO-J6): δ 11.55 (s, 1 H), 8.85 (s, 1 H), 7.16 (t, J= 8.8 Hz, 1 H), 7.08 (dd, J= 6.1, 2.7 Hz, 1 H), 6.75 (m, 1 H), 6.14 (t, J= 5.8 Hz, 1 H), 3.48 (t, J= 5.2 Hz, 2 H), 3.35 (dd, J= 10.8, 5.6 Hz, 2 H), 3.22 (s, 3 H).
Step G: 4-(3-Bromo-4-fluorophenyl)-3-{4-[(2-methoxyethyl)amino]-l,2,5-oxadiazol-3-yl}- 1 ,2,4-oxadiazol-5(4H)-one
A mixture of N-(3-bromo-4-fluorophenyl)-N’-hydroxy-4-[(2-methoxyethyl)amino]-l,2,5- oxadiazole-3-carboximidamide (76.5 g, 0.204 mol), l,r-carbonyldiimidazole (49.7 g, 0.307 mol), and ethyl acetate (720 mL) was heated to 60 0C and stirred for 20 min. LCMS indicated reaction completed. The reaction was cooled to room temperature, washed with 1 Ν HCl (2 x 750 mL), dried over sodium sulfate, and concentrated to give the desired product (80.4 g, 98%) as a crude brown solid. LCMS for C13H12BrFN5O4 (M+H)+: m/z = 400.0, 402.0. 1H NMR (400 MHz, OMSO-d6): δ 7.94 (t, J= 8.2 Hz, 1 H), 7.72 (dd, J= 9.1, 2.3 Hz, 1 H), 7.42 (m, 1 H), 6.42 (t, J= 5.7 Hz, 1 H), 3.46 (t, J= 5.4 Hz, 2 H), 3.36 (t, J= 5.8 Hz, 2 H), 3.26 (s, 3 H).
Step H: 4-(3-Bromo-4-fluorophenyl)-3-{4-[(2-liydroxyethyl)amino]-l,2,5-oxadiazol-3-yl}- 1 ,2,4-oxadiazol-5(4H)-one

4-(3-Bromo-4-fluorophenyl)-3-{4-[(2-methoxyetliyl)amino]-l,2,5-oxadiazol-3-yl}-l,2,4- oxadiazol-5(4H)-one (78.4 g, 0.196 mol) was dissolved in dichloromethane (600 mL). At -67 0C boron tribromide (37 mL, 0.392 mol) was added over 15 min. The reaction was warmed up to -10 0C in 30 min. LCMS indicated reaction completed. The reaction was stirred at room temperature for 1 hour. At 0 – 5 0C the reaction was slowly quenched with saturated sodium bicarbonate solution (1.5 L) over 30 min. The reaction temperature rose to 25 0C. The reaction was extracted with ethyl acetate (2 x 500 mL, first extraction organic layer is on the bottom and second extraction organic lager is on the top). The combined organic layers were dried over sodium sulfate and concentrated to give the desired product (75 g, 99%) as a crude brown solid. LCMS for C12H10BrFN5O4 (M+H)+: m/z = 386.0, 388.0. 1H NMR (400 MHz, DMSO-^6): δ 8.08 (dd, J= 6.2, 2.5 Hz, 1 H), 7.70 (m, 1 H), 7.68 (t, J= 8.7 Hz, 1 H), 6.33 (t, J= 5.6 Hz, 1 H), 4.85 (t, J= 5.0 Hz, 1 H), 3.56 (dd, J= 10.6, 5.6 Hz, 2 H), 3.29 (dd, J= 11.5, 5.9 Hz, 2 H).
Step I: 2-({4-[4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-l,2,4-oxadiazol-3-yl]-l,2,5- oxadiazol-3-yl}amino)ethyl methanesulfonate
To a solution of 4-(3-bromo-4-fluorophenyl)-3-{4-[(2-hydroxyethyl)amino]-l,2,5-oxadiazol- 3-yl}-l,2,4-oxadiazol-5(4H)-one (1.5 kg, 3.9 mol, containing also some of the corresponding bromo-compound) in ethyl acetate (12 L) was added methanesulfonyl chloride (185 mL, 2.4 mol) dropwise over 1 h at room temperature. Triethylamine (325 mL, 2.3 mol) was added dropwise over 45 min, during which time the reaction temperature increased to 35 0C. After 2 h, the reaction mixture was washed with water (5 L), brine (I L), dried over sodium sulfate, combined with 3 more reactions of the same size, and the solvents removed in vacuo to afford the desired product (7600 g, quantitative yield) as a tan solid. LCMS for
Ci3HnBrFN5O6SNa (M+Na)+: m/z = 485.9, 487.9. 1H NMR (400 MHz, DMSCW6): δ 8.08 (dd, J= 6.2, 2.5 Hz, 1 H), 7.72 (m, 1 H), 7.58 (t, J= 8.7 Hz, 1 H), 6.75 (t, J- 5.9 Hz, 1 H), 4.36 (t, J= 5.3 Hz, 2 H), 3.58 (dd, J= 11.2, 5.6 Hz, 2 H), 3.18 (s, 3 H).
Step J: 3-{4-[(2-Azidoethyl)amino]-l,2,5-oxadiazol-3-yl}-4-(3-bromo-4-fluorophenyl)- 1 ,2,4-oxadiazol-5(4H)-one
To a solution of 2-({4-[4-(3-bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-l,2,4-oxadiazol-3-yl]- l,2,5-oxadiazol-3-yl}amino)ethyl methanesulfonate (2.13 kg, 4.6 mol, containing also some of the corresponding bromo-compound) in dimethylformamide (4 L) stirring in a 22 L flask was added sodium azide (380 g, 5.84 mol). The reaction was heated at 500C for 6 h, poured into ice/water (8 L), and extracted with 1 : 1 ethyl acetate:heptane (20 L). The organic layer was washed with water (5 L) and brine (5 L), and the solvents removed in vacuo to afford the desired product (1464 g, 77%) as a tan solid. LCMS for C12H8BrFN8O3Na (M+Na)+: m/z =
433.0, 435.0. 1H NMR (400 MHz, DMSO-*/*): δ 8.08 (dd, J= 6.2, 2.5 Hz, 1 H), 7.72 (m, 1 H), 7.58 (t, J= 8.7 Hz, 1 H), 6.75 (t, J= 5.7 Hz, 1 H), 3.54 (t, J= 5.3 Hz, 2 H), 3.45 (dd, J= 11.1, 5.2 Hz, 2 H).
Step K: 3-{4-[(2-Aminoethyl)amino]-l,2,5-oxadiazol-3-yl}-4-(3-bromo-4-fluorophenyl)- 1 ,2,4-oxadiazol-5(4H)-one hydrochloride

Sodium iodide (1080 g, 7.2 mol) was added to 3-{4-[(2-azidoethyl)amino]-l,2,5-oxadiazol-3- yl}-4-(3-bromo-4-fluorophenyl)-l,2,4-oxadiazol-5(4H)-one (500 g, 1.22 mol) in methanol (6 L). The mixture was allowed to stir for 30 min during which time a mild exotherm was observed. Chlorotrimethylsilane (930 mL, 7.33 mol) was added as a solution in methanol (1 L) dropwise at a rate so that the temperature did not exceed 35 0C, and the reaction was allowed to stir for 3.5 h at ambient temperature. The reaction was neutralized with 33 wt% solution of sodium thiosulfate pentahydrate in water (~1.5 L), diluted with water (4 L), and the pΗ adjusted to 9 carefully with solid potassium carbonate (250 g – added in small portions: watch foaming). Di-fe/t-butyl dicarbonate (318 g, 1.45 mol) was added and the reaction was allowed to stir at room temperature. Additional potassium carbonate (200 g) was added in 50 g portions over 4 h to ensure that the pΗ was still at or above 9. After stirring at room temperature overnight, the solid was filtered, triturated with water (2 L), and then MTBE (1.5 L). A total of 11 runs were performed (5.5 kg, 13.38 mol). The combined solids were triturated with 1 : 1 TΗF:dichloromethane (24 L, 4 runs in a 20 L rotary evaporator flask, 50 0C, 1 h), filtered, and washed with dichloromethane (3 L each run) to afford an off- white solid. The crude material was dissolved at 55 0C tetrahydrofuran (5 mL/g), treated with decolorizing carbon (2 wt%) and silica gel (2 wt%), and filtered hot through celite to afford the product as an off-white solid (5122 g). The combined MTBE, THF, and dichloromethane filtrates were concentrated in vacuo and chromatographed (2 kg silica gel, heptane with a 0-100% ethyl acetate gradient, 30 L) to afford more product (262 g). The combined solids were dried to a constant weight in a convection oven (5385 g, 83%).
In a 22 L flask was charged hydrogen chloride (4 N solution in 1,4-dioxane, 4 L, 16 mol). fert-Butyl [2-({4-[4-(3-bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-l,2,4-oxadiazol-3-yl]- l,2,5-oxadiazol-3-yl}amino)ethyl]carbamate (2315 g, 4.77 mol) was added as a solid in portions over 10 min. The slurry was stirred at room temperature and gradually became a thick paste that could not be stirred. After sitting overnight at room temperature, the paste was slurried in ethyl acetate (10 L), filtered, re-slurried in ethyl acetate (5 L), filtered, and dried to a constant weight to afford the desired product as a white solid (combined with other runs, 5 kg starting material charged, 4113 g, 95%). LCMS for C12HnBrFN6O3 (M+H)+: m/z
= 384.9, 386.9. 1H NMR (400 MHz, DMSO-J6): δ 8.12 (m, 4 H), 7.76 (m, 1 H), 7.58 (t, J= 8.7 Hz, 1 H), 6.78 (t, J= 6.1 Hz, 1 H), 3.51 (dd, J= 11.8, 6.1 Hz, 2 H), 3.02 (m, 2 H).
Step L: tert-Butyl ({[2-({4-[4-(3-bromo-4-fluorophenyl)-5-oxo-4,5-diliydro-l,2,4-oxadiazol- 3-yl]-l,2,5-oxadiazol-3-yl}amino)ethyl]amino}sulfonyl)carbamate
A 5 L round bottom flask was charged with chlorosulfonyl isocyanate [Aldrich, product #
142662] (149 mL, 1.72 mol) and dichloromethane (1.5 L) and cooled using an ice bath to 2 0C. tert-Butanol (162 mL, 1.73 mol) in dichloromethane (200 mL) was added dropwise at a rate so that the temperature did not exceed 10 0C. The resulting solution was stirred at room temperature for 30-60 min to provide tert-butyl [chlorosulfonyljcarbamate.
A 22 L flask was charged with 3-{4-[(2-aminoethyl)amino]-l,2,5-oxadiazol-3-yl}-4-(3- bromo-4-fluorophenyl)-l,2,4-oxadiazol-5(4H)-one hydrochloride (661 g, 1.57 mol) and 8.5 L dichloromethane. After cooling to -15 0C with an ice/salt bath, the solution of tert-butyl [chlorosulfonyl]carbamate (prepared as above) was added at a rate so that the temperature did not exceed -10 0C (addition time 7 min). After stirring for 10 min, triethylamine (1085 mL, 7.78 mol) was added at a rate so that the temperature did not exceed -5 0C (addition time 10 min). The cold bath was removed, the reaction was allowed to warm to 10 0C, split into two portions, and neutralized with 10% cone HCl (4.5 L each portion). Each portion was transferred to a 50 L separatory funnel and diluted with ethyl acetate to completely dissolve the white solid (~25 L). The layers were separated, and the organic layer was washed with water (5 L), brine (5 L), and the solvents removed in vacuo to afford an off-white solid. The solid was triturated with MTBE (2 x 1.5 L) and dried to a constant weight to afford a white solid. A total of 4113 g starting material was processed in this manner (5409 g, 98%). *Η NMR (400 MHz, OMSO-d6): δ 10.90 (s, 1 H), 8.08 (dd, J= 6.2, 2.5 Hz, 1 H), 7.72 (m, 1 H), 7.59 (t, J= 8.6 Hz, 1 H), 6.58 (t, J= 5.7 Hz, 1 H), 3.38 (dd, J= 12.7, 6.2 Hz, 2 H), 3.10 (dd, J = 12.1, 5.9 Hz, 2 H), 1.41 (s, 9 H). Step M: N-[2-({4-[4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dmydro-l ,2,4-oxadiazol-3-yl]- l,2,5-oxadiazol-3-yl}amino)ethyl]sulfamide

To a 22 L flask containing 98:2 trifluoroacetic acid:water (8.9 L) was added tert-butyl ({[2- ({4-[4-(3-bromo-4-fluorophenyl)-5-oxo-4,5-diliydro-l,2,4-oxadiazol-3-yl]-l,2,5-oxadiazol-3- yl}amino)ethyl]amino}sulfonyl)carbamate (1931 g, 3.42 mol) in portions over 10 minutes. The resulting mixture was stirred at room temperature for 1.5 h, the solvents removed in vacuo, and chased with dichloromethane (2 L). The resulting solid was treated a second time with fresh 98:2 trifluoroacetic acid:water (8.9 L), heated for 1 h at 40-50 0C, the solvents removed in vacuo, and chased with dichloromethane (3 x 2 L). The resulting white solid was dried in a vacuum drying oven at 50 0C overnight. A total of 5409 g was processed in this manner (4990 g, quant, yield). LCMS for C]2H12BrFN7O5S (M+H)+: m/z = 463.9, 465.9.
1H NMR (400 MHz, OM$>O-d6): δ 8.08 (dd, J= 6.2, 2.5 Hz, 1 H), 7.72 (m, 1 H), 7.59 (t, J= 8.7 Hz, 1 H), 6.67 (t, J= 5.9 Hz, IH), 6.52 (t, J= 6.0 Hz, 1 H), 3.38 (dd, J= 12.7, 6.3 Hz, 2 H), 3.11 (dd, J= 12.3, 6.3 Hz).
Step N: 4-( {2-[(Aminosulfonyl)amino]ethyl} amino)-N-(3-bromo-4-fluorophenyl)-N- hydroxy-l,2,5-oxadiazole-3-carboximidamide
To a crude mixture of N-[2-({4-[4-(3-bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-l,2,4- oxadiazol-3-yl]-l,2,5-oxadiazol-3-yl}amino)ethyl]sulfamide (2.4 mol) containing residual amounts of trifluoroacetic acid stirring in a 22 L flask was added THF (5 L). The resulting solution was cooled to 0 °C using an ice bath and 2 Ν NaOH (4 L) was added at a rate so that the temperature did not exceed 10 0C. After stirring at ambient temperature for 3 h (LCMS indicated no starting material remained), the pH was adjusted to 3-4 with concentrated HCl (-500 mL). The THF was removed in vacuo, and the resulting mixture was extracted with ethyl acetate (15 L). The organic layer was washed with water (5 L), brine (5 L), and the solvents removed in vacuo to afford a solid. The solid was triturated with MTBE (2 x 2 L), combined with three other reactions of the same size, and dried overnight in a convection oven to afford a white solid (3535 g). The solid was recrystallized (3 x 22 L flasks, 2: 1 water: ethanol, 14.1 L each flask) and dried in a 50 0C convection oven to a constant weight to furnish the title compound as an off-white solid (3290 g, 78%). LCMS for CnH14BrFN7O4S (M+H)+: m/z = 437.9, 439.9. 1H NMR (400 MHz, DMSO-J6): δ 11.51 (s, 1 H), 8.90 (s, 1 H), 7.17 (t, J= 8.8 Hz, 1 H), 7.11 (dd, J= 6.1, 2.7 Hz, 1 H), 6.76 (m, 1 H), 6.71 (t, J= 6.0 Hz, 1 H), 6.59 (s, 2 H), 6.23 (t, J= 6.1 Hz, 1 H), 3.35 (dd, J= 10.9, 7.0 Hz, 2 H), 3.10 (dd, J= 12.1, 6.2 Hz, 2 H).
The final product was an anhydrous crystalline solid. The water content was determined to be less than 0.1% by Karl Fischer titration.
CLIP
Incyte’s Andrew P. Combs presented the company’s clinical candidate for cancer immunotherapy. The basic tenet of this burgeoning field is that the human body’s immune system is a tremendous resource for fighting disease; scientists just need to figure out how to unleash it. One target that’s proven to be particularly attractive for this purpose in recent years is indoleamine-2,3-dioxygenase-1, or IDO1 (C&EN, April 6, page 10).
IDO1 plays a role in signaling the immune system to stand down from attacking foreign bodies it might otherwise go after, such as fetuses. Tumors also produce IDO1 to evade the immune system, so molecules that can inhibit this enzyme could bring the full force of the body’s defenses to bear on these deadly invaders.
Incyte’s search for an IDO1 inhibitor began with a high-throughput screen, which led to a proof-of-concept compound. But the compound had poor oral bioavailability. What’s more, the molecule and its analogs underwent glucuronidation during its metabolism: Enzymes tacked on a glucuronic acid group to the structure’s amidoxime, which was key to its activity.
The chemists reasoned they could block this metabolism by sterically hindering that position. Making such molecules proved to be more difficult than they expected. But then they unearthed a Latvian paper from 1993 that gave them the synthetic method they needed to make the series of compounds that would lead to their clinical candidate INCB24360 (epacadostat).
With its furazan core, as well as its amidoxime, bromide, and sulfuric diamide functional groups, INCB24360 is something of an odd duck, Combs acknowledged. “Some of you in the audience may be looking at this and saying, ‘That molecule does not look like something I would bring forward or maybe even make,’ ” he said, noting that the structure breaks many medicinal chemistry rules. “We’re a data-centric company, and we followed the data, not the rules,” Combs told C&EN.
The compound has completed Phase I clinical trials and is now being used in collaborative studies with several other pharmaceutical companies that combine INCB24360 with other cancer immunotherapy agents.
TEAMWORK
Incyte’s team (from left): Andrew Combs, Dilip Modi, Joe Glenn, Brent Douty, Padmaja Polam, Brian Wayland, Rick Sparks, Wenyu Zhu, and Eddy Yue.
Credit: Incyte
| WO2007113648A2 * |
Mar 26, 2007 |
Oct 11, 2007 |
Pfizer Products Inc. |
Ctla4 antibody combination therapy |
| US20070185165 * |
Dec 19, 2006 |
Aug 9, 2007 |
Combs Andrew P |
N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| US20100055111 * |
Feb 14, 2008 |
Mar 4, 2010 |
Med. College Of Georgia Research Institute, Inc. |
Indoleamine 2,3-dioxygenase, pd-1/pd-l pathways, and ctla4 pathways in the activation of regulatory t cells |
| US20120058079 * |
Nov 11, 2011 |
Mar 8, 2012 |
Incyte Corporation, A Delaware Corporation |
1,2,5-Oxadiazoles as Inhibitors of Indoleamine 2,3-Dioxygenase |
REFERENCES
1: Vacchelli E, Aranda F, Eggermont A, Sautès-Fridman C, Tartour E, Kennedy EP, Platten M, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology. 2014 Dec 15;3(10):e957994. eCollection 2014 Nov. Review. PubMed PMID: 25941578; PubMed Central PMCID: PMC4292223.
2: Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, Leffet L, Hansbury MJ, Thomas B, Rupar M, Waeltz P, Bowman KJ, Polam P, Sparks RB, Yue EW, Li Y, Wynn R, Fridman JS, Burn TC, Combs AP, Newton RC, Scherle PA. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010 Apr 29;115(17):3520-30. doi: 10.1182/blood-2009-09-246124. Epub 2010 Mar 2. PubMed PMID: 20197554.
3: Koblish HK, Hansbury MJ, Bowman KJ, Yang G, Neilan CL, Haley PJ, Burn TC, Waeltz P, Sparks RB, Yue EW, Combs AP, Scherle PA, Vaddi K, Fridman JS. Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and the growth of IDO-expressing tumors. Mol Cancer Ther. 2010 Feb;9(2):489-98. doi: 10.1158/1535-7163.MCT-09-0628. Epub 2010 Feb 2. PubMed PMID: 20124451.
//////////1204669-58-8 , INCB024360, INCB24360, epacadostat, PHASE 2, CANCER, orphan drug designation
Fc1ccc(cc1Br)N/C(=N\O)c2nonc2NCCNS(N)(=O)=O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

























 67
67 66
66



















































































.JPG)





































 .
.
 .
.