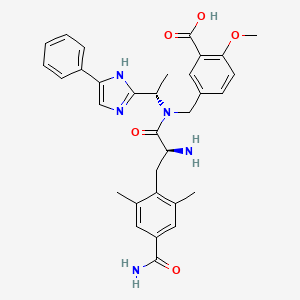
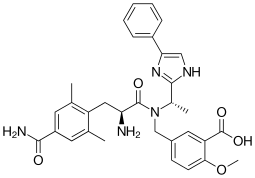
Eluxadoline
- Molecular FormulaC32H35N5O5
- Average mass569.651 Da
5-({[(2S)-2-amino-3-(4-carbamoyl-2,6-dimethylphenyl)propanoyl][(1S)-1-(4-phenyl-1H-imidazol-2-yl)ethyl]amino}methyl)-2-methoxybenzoic acid
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
864821-90-9 CAS
JNJ-27018966
Molecular Formula: C32H35N5O5
Molecular Weight: 569.6508
Agents for Irritable Bowel Syndrome, mu-Opioid Agonists, delta-Opioid Antagonists
Eluxadoline
Trade Name: Viberzi®
Research Code: JNJ-27018966, JNJ27018966, JNJ 27018966
Chemical Name: 5 – [[[(2S) -2-amino-3- [4- (aminocarbonyl) -2,6-dimethylphenyl ] -1- oxopropyl] [(1S) -1- (4-phenyl-1H-imidazol-2-yl) ethyl] amino] methyl] -2-methoxybenzoic acid
MOA: mu opioid receptor agonist, Indication: Irritable bowel syndrome with diarrhea (IBS-D)
Approval Date: May 27, 2015 (US)
Originator: Furiex Pharmaceuticals Inc ( Furiex acquired Eluxadoline from Janssen in 2011 )
Developer: Forest Laboratories Inc. (acquired by Actavis PLC in 2014 )
Eluxadoline, sold under the brand names Viberzi ( vy-BUR-zee) in the US and Truberzi in Europe,[2] is a medication taken by mouth for the treatment of diarrhea and abdominal pain in individuals with diarrhea-predominant irritable bowel syndrome (IBS-D).[3]It was approved for use in the United States in 2015.[4] The drug originated from Janssen Pharmaceutica and was developed by Actavis.
Contraindications
This drug is contraindicated in case of having:
Adverse effects
Common adverse effects are constipation and nausea, but rates of discontinuation due to constipation were low for both eluxadoline and placebo. Rare adverse effects: fatigue, bronchitis, viral gastroenteritis. Rare serious adverse effects include pancreatitis with a general incidence of 0.3% – higher incidence with 100 mg dose (0.3%) than with 75 mg dose (0.2%).[6] The risk is even greater in those who do not have a gall bladder and the medication is not recommended in this group.[7]
In March 2017, the U.S. Food and Drug Administration issued a safety alert for eluxadoline concerning an increased risk of serious pancreatitis in patients without a gallbladder.[8] An FDA review found that in such patients, spasm of the sphincter of Oddi may lead to severe pancreatitis.[9] The FDA reported that in some cases symptoms have occurred with just one or two doses at the recommended dosage for patients without a gallbladder (75 mg).[9] Of two deaths associated with eluxadoline reported up to February 2017, both occurred in patients without a gallbladder.[8]
Interactions
Elevated concentrations of eluxadoline were observed with co-administration of inhibitors of the transporter protein OATP1B1, such as:
Also, concurrent use of other drugs that cause constipation is not preferred, such as:
Eluxadoline increases the concentrations of drugs which are OATP1B1 and BCRP substrates. Also, co-administration of eluxadoline with rosuvastatin may increase the risk of rhabdomyolysis.[1]
Pharmacology
Mechanism of action
Eluxadoline is a μ- and κ-opioid receptor agonist and δ-opioid receptor antagonist [11] that acts locally in the enteric nervous system, possibly decreasing adverse effects on the central nervous system.[12][13]
Pharmacokinetics
In the in vitro studies, eluxadoline was found to be transported by OAT3 (SLC22A8), OATP1B1 (SLCO1B1) and BSEP (ABCB11) at the highest concentrations tested (400 ng/ml which is 162-fold larger than the observed Cmax of the highest therapeutic dose of 100 mg). However, it was not to be transported by OCT1 POU2F1, OAT1 Organic anion transporter 1, OCT2, OATP1B3 (SLCO1B3), P-gp (P-glycoprotein), or BCRP (ABCG2).
Multidrug resistance-associated protein 2 (MRP2)-vesicular accumulation of eluxadoline was observed, indicating that the drug is a substrate of MRP2. Eluxadoline was not found to inhibit BCRP-, BSEP-, MRP2-, OCT1-, OCT2-, OAT1-, OAT3-, or OATP1B3-mediated transport of probe substrates but inhibited the transport of probe substrates of OATP1B1 and P-gp. Also in the in vitro studies, it was observed that eluxadoline is an in vivo substrate of OATP1B1, OAT3, and MRP2. Finally, no inhibition or induction of cytochrome P450enzymes was observed.[14]
Following a 100 mg dose of eluxadoline, the Cmax was about 2 to 4 ng/ml and AUC was 12-22 ng.h/ml. Eluxadoline has linear pharmacokinetics with no accumulation upon repeated twice daily dosing. Taking eluxadoline with high fat meal decreased the Cmax by 50% and AUC by 60%.[1]
Chemistry
Synthesis
The synthesis of eluxadoline was extensively discussed in the patent No. WO2006099060 A2, with the title : “Process for the preparation of opioid modulators” which was published in Sept. 2006[15]
A CLIP
5 JAN 2014
Furiex Pharmaceuticals Inc. more than doubled in its best day of trading after its experimental drug alleviated diarrhea and abdominal pain caused by irritable bowel syndrome in two studies.
The drug eluxadoline met targets for improvements in stool consistency and abdominal pain that were developed in conjunction with U.S. and European regulators, the company said today. Furiex will apply for approval in June, Chairman Fred Eshelman said in an investor call today. He estimated annual sales of $750 million to $1 billion.
“By our math, it looks like a pretty doggone good market,” Eshelman said on the call, noting that there is only one currently approved drug available in the U.S. for the condition.
Diarrhea-predominant irritable bowel syndrome is a chronic disorder that affects about 28 million patients in the U.S. and Europe, Furiex said in the statement.Furiex said it would apply by mid-year for U.S. approval of the drug, eluxadoline, to treat diarrhea-predominant irritable bowel syndrome (IBS-d), a debilitating bowel disorder that affects about 28 million people in the United States and major European markets.
Furiex said it expected to seek European approval in early 2015.
“We believe that there are a lot of patients out there who need this drug. There is a huge unmet need,” Furiex Chief Medical Officer June Almenoff said in a telephone interview.
Currently approved drugs for IBS address constipation associated with the disorder, but there are few options for diarrhea predominant IBS.
Furiex founder and chairman Fred Eshelman said he believes the drug has the potential for blockbuster sales, which he defined as annual sales of between $750 million and $1 billion.
Eluxadoline was tested at two doses against a placebo over the course of 12 weeks to meet requirements by the U.S. Food and Drug Administration, and for 26 weeks for European health regulators, in Phase III studies involving 2,428 patients, Furiex said.
For the combined goal of improvement in abdominal pain and stool consistency for at least half the days in the study, eluxadoline achieved a statistically significant improvement at the 100 milligram and 75 mg doses through 12 weeks in both studies.
On the 26-week measure, the higher dose succeeded in both studies but the lower dose missed statistical significance in one of the two trials, according to initial results released by the company.
The success appeared to be driven by the percentage of patients reporting improvements in diarrhea, which ranged from 30 percent to 37 percent versus 22 percent and 20.9 percent for the placebo groups.
When the composite goal was broken into its two components, researchers found a numerical improvement in pain response rates that did not achieve statistical significance.
The drug appeared to be safe and well-tolerated in both studies, Furiex said. The most commonly reported side effects were constipation and nausea.
The company plans to present a far more detailed analysis of the late stage studies at an upcoming medical meeting.
“We’re very excited about the path ahead and about how this can transform patients’ lives,” Almenoff said.
Mu Delta is a locally active mu opioid receptor agonist and delta opioid receptor antagonist in phase III clinical evaluation at Furiex Pharmaceuticals for the oral treatment of diarrheal predominant irritable bowel syndrome (d-IBS).
The product candidate holds an advantage over currently marketed products for this indication because it acts locally on the enteric nervous system, possibly decreasing adverse effects on the central nervous system. In 2011, fast track designation was assigned in the U.S. for the treatment of d-IBS. In 2011, Mu Delta was licensed to Furiex Pharmaceuticals by Janssen for the treatment of d-IBS, granting an option to Furiex to continue development and commercialization following phase II proof of concept studies.
The opioid receptors were identified in the mid-1970’s, and were quickly categorized into three sub-sets of receptors (mu, delta and kappa). More recently the original three types of receptors have been further divided into sub-types. Also known is that the family of opioid receptors are members of the G-protein coupled receptor (GPCR) super-family. More physiologically pertinent are the well established facts that opioid receptors are found throughout the central and peripheral nervous system of many mammalian species, including humans, and that modulation of the respective receptors can elicit numerous, albeit different, biological effects, both desirable and undesirable (D. S. Fries, “Analgesics”, inPrinciples of Medicinal Chemistry, 4th ed.; W. O. Foye, T. L. Lemke, and D. A. Williams, Eds.; Williams and Wilkins: Baltimore, Md., 1995; pp. 247-269; J. V. Aldrich, “Analgesics”, Burger’s Medicinal Chemistry and Drug Discovery, 5thEdition, Volume 3: Therapeutic Agents, John Wiley & Sons, Inc., 1996, pp. 321-441). In the most current literature, the likelihood of heterodimerization of the sub-classes of opioid receptors has been reported, with respective physiological responses yet undetermined (Pierre J. M. Riviere and Jean-Louis Junien, “Opioid receptors: Targets for new gastrointestinal drug development”, Drug Development 2000, pp. 203-238).
A couple biological effects identified for opioid modulators have led to many useful medicinal agents. Most significant are the many centrally acting mu opioid agonist modulators marketed as analgesic agents to attenuate pain (e.g., morphine), as well as peripherally acting mu agonists to regulate motility (e.g., loperamide). Currently, clinical studies are continuing to evaluate medicinal utility of selective delta, mu, and kappa modulators, as well as compounds possessing combined sub-type modulation. It is envisioned such explorations may lead to agents with new utilities, or agents with minimized adverse side effects relative to currently available agents (examples of side effects for morphine includes constipation, respiratory depression, and addiction potential). Some new GI areas where selective or mixed opioid modulators are currently being evaluated includes potential treatment for various diarrheic syndromes, motility disorders (post-operative ileus, constipation), and visceral pain (post operative pain, irritable bowel syndrome, and inflammatory bowel disorders) (Pierre J. M. Riviere and Jean-Louis Junien, “Opioid receptors: Targets for new gastrointestinal drug development” Drug Development, 2000, pp. 203-238).
Around the same time the opioid receptors were identified, the enkephalins were identified as a set of endogenous opioid ligands (D. S. Fries, “Analgesics”, inPrinciples of Medicinal Chemistry, 4th ed.; W. O. Foye; T. L. Lemke, and D. A. Williams, Eds.; Williams and Wilkins: Baltimore, Md., 1995; pp. 247-269). Schiller discovered that truncating the original pentapeptide enkephalins to simplified dipeptides yielded a series of compounds that maintained opioid activity (Schiller, P. WO 96/06855). However one potential drawback cited for such compounds is the likelihood of their inherent instability (P. W. Schiller et al., Int. J. Pept. Protein Res. 1993, 41 (3), pp. 313-316).
More recently, a series of opioid pseudopeptides containing heteroaromatic or heteroaliphatic nuclei were disclosed, however this series is reported showing a different functional profile than that described in the Schiller works. (L. H. Lazarus et al., Peptides 2000, 21, pp. 1663-1671).
Most recently, works around morphine related structures were reported by Wentland, et al, where carboxamido morphine derivatives and it’s analogs were prepared (M. P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, pp. 1717-1721; M. P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, pp. 623-626). Wentland found that substitution for the phenol moiety of the morphine related structures with a primary carboxamide led anywhere from equal activities up to 40 fold reduced activities, depending on the opioid receptor and the carboxamide. It was also revealed that any additional N-substitutions on the carboxamide significantly diminished the desired binding activity.
Compounds of the present invention have not been previously disclosed and are believed to provide advantages over related compounds by providing improved pharmacological profiles.
Opioid receptor modulators, agonists or antagonists are useful in the treatment and prevention of various mammalian disease states, for example pain and gastrointestinal disorders such as diarrheic syndromes, motility disorders including post-operative ileus and constipation, and visceral pain including post-operative pain, irritable bowel syndrome and inflammatory bowel disorders.
It is an object of the present invention to provide opioid receptor modulators. It is a further object of the invention to provide opioid receptor agonists and opioid receptor antagonists. It is an object of the present invention to provide opioid receptor ligands that are selective for each type of opioid receptor, mu, delta and kappa. It is a further object of the present invention to provide opioid receptor ligands that modulate two or three opioid receptor types, mu, delta and kappa, simultaneously.
It is an object of the invention to provide certain instant compounds that are also useful as intermediates in preparing new opioid receptor modulators. It is also an object of the invention to provide a method of treating or ameliorating a condition mediated by an opioid receptor. And, it is an object of the invention to provide a useful pharmaceutical composition comprising a compound of the present invention useful as an opioid receptor modulator.
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1 h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid is an opoid receptor modulator (mu receptor agonist and delta receptor antagonist) and may be useful for treating irritable bowel syndrome, pain or other opioid receptor disorders.
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid and methods of making this molecule are disclosed in
US application 2005/02033143. Example 9 of US application 2005/02033143 makes the hydrochloride salt of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
Applicants have discovered a process of making the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid and two novel crystals of this zwitterion. In Applicant’s hands, these novel crystals provide improved properties and can be purified at higher purity. Applicant’s new process results in improved and less costly process manufacturing conditions than the procedure disclosed in US application 2005/02033143.
FIG. 6 is the molecular structure of the zwitterion 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.

US7994206
SYNTHESIS OF 5-formyl-2- methoxy-benzoic acid methyl ester
WO2002022612A1
Example 8: 2-Methoxy-5-formylbenzoic acid
Lithium hydroxide (1.04g, 0.043mol, 3eq) in water (lOmL) was added to a stirred solution of methyl 2-methoxy-5-formylbenzoate (2.8g, 0.014mol, leq) in a mixture of tetrahydrofuran (30mL) and methanol (20mL). The solution was stirred overnight, acidified to pH 1 with 10% HCl and the organic solvents removed in vacuo. The aqueous solution was extracted with ethyl acetate (lOOmL) and the organic solution washed with brine (lOOmL), then extracted with saturated aqueous sodium bicarbonate (3 x lOOmL). The basic solution was washed with ethyl acetate (lOOmL), then acidified to pH 1 with 10% HCl and back extracted with dichloromethane (3 x lOOmL). The organic solution was dried over sodium sulfate and evaporated in vacuo to give a cream coloured powder (2.01g, 77%). 1H NMR (CDC13) δ 9.99 (s, IH, O=C- H), 4.14 (s, 3H, CH3).
ANALOGOUS METHOD TO PREPARE..2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester

USE 5-formyl-2- methoxy-benzoic acid methyl ester for 3,4- dimethoxybenzaldehyde, TO GET 2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester
Example 4
(3,4-Dimethoxy-benzyl)-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amine
NOTE THIS IS NOT THE COMPD….IT IS REF FOR AN ANALOGOUS PROCEDURE
A solution of 1-(4-phenyl-1 W-imidazol-2-yl)-ethylamine (0.061 g, 0.33 mmol) of Example 3, and 0.55 g (0.33 mmol) of 3,4-dimethoxybenzaldehyde in 5 ml_ of anhydrous methanol was stirred at room temperature for 1 h and then cooled to about 0-100C in an ice bath for 1 h. The reaction was treated carefully with 0.019 g (0.49 mmol) of sodium borohydride in one portion and maintained at about 0-100C for 21 h. Cold 2M aqueous HCI was added dropwise (30 drops), the mixture was stirred for 5 min, and then partially concentrated in vacuo unheated. The residual material was taken up in EtOAc to yield a suspension that was treated with 5 ml_ of cold 3M aqueous NaOH and stirred vigorously until clear. The phases were separated and the aqueous layer was extracted three times additional with EtOAc. The combined extracts were dried over MgSO4, filtered, and concentrated to yield (3,4-dimethoxy- benzyl)-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amine as a light yellow oil (HPLC: 87% @ 254nm and 66% @ 214 nm).
MS (ES+) (relative intensity): 338.1 (100) (M+1)
This sample was of sufficient quality to use in the next reaction without further purification.
SYNTHESIS
WO2006099060A2
In an embodiment, the present invention is directed to processes for the preparation of the compound of formula (IV)
also known as, 5-({[2-amino-3-(4-carbamoyl-2,5-dimethyl-phenyl)- propionyl]-[1 -(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy- benzoic acid
Example 1
(S)-2-ferf-Butoxycarbonylamino-3-(4-carbamoyl-2.6-dimethyl-phenyl)- propionic acid
STEP A: Trifluoromethanesulfonic acid 4-bromo-3,5-dimethyl-phenyl ester
To a cooled (0°C) solution of 4-bromo-3,5-dimethylphenol (3.05 g, 15.2 mmol) in pyridine (8 ml_) was added trifluoromethanesulfonic anhydride (5.0 g, 17.7 mmol) dropwise. After completion of addition, the resulting mixture was stirred at 0°C for 15 min, and then at room temperature overnight. The reaction was quenched by addition of water, and then extracted with EtOAc. The organic extracts were washed sequentially with water, 2N HCI (2x ), brine, and then dried over MgSO4. Filtration and evaporation to dryness yielded compound 1 b as a colorless oil.
1H NMR (300 MHz, CDCI3): δ 2.45 (6H, s), 7.00 (2H, s).
Step B: 4-Bromo-3,5-dimethylbenzoic acid
Into a solution of compound 1 b (6.57 g, 19.7 mmol) in DMF (65 ml_) were added K2CO3 (13.1 g, 94.7 mmol), Pd(OAc)2 (0.44 g, 1.97 mmol) and 1 ,1′-bis(diphenylphosphino)ferrocene (2.29 g, 4.14 mmol). The resulting mixture was bubbled in gaseous CO for 10 min and was heated to 60°C for 7.5h with a CO(9) balloon. The cooled mixture was partitioned between aqueous NaHCO3 and EtOAc, and filtered. The aqueous phase was separated, acidified with aqueous 6N HCI, extracted with EtOAc, and then dried over Na2SO4. Filtration and concentration of the filtrate yielded crude compound 1c as a brown residue, which was used in the next step without further purification. STEP C: Method A: 4-Bromo-3,5-dimethyl-benzamide
Into a suspension of compound 1c in DCM (40 ml_) was added SOCI2 (3.1 rnL, 42 mmol) and the mixture was heated at reflux for 2 h. Upon removal of the solvent by evaporation, the residue was dissolved in DCM (40 ml_) and then ammonium hydroxide (28% NH3 in water, 2.8 ml_) was added. The reaction mixture was heated at 5O0C for 2 h and concentrated. The residue was diluted with H2O, extracted with EtOAc, and the organic portion was dried over Na2SO4. After filtration and evaporation, the residue was purified by flash column chramotagraphy (eluent: EtOAc) to yield compound 1 d as an off-white solid.
1H NMR (300 MHz, CD3CN): δ 2.45 (6H, s), 5.94 (1 H, br s), 6.71 (1 H, br s), 7.57 (2H, s)
MS(ES+)(relative intensity): 228.0 (100%) (M+1).
Step C: Method B: 4-Bromo-3,5-dimethyl-benzamide
A mixture of compound 1 b (3.33 g, 10 mmol), PdCI2 (0.053 g, 0.3 mmol), hexamethyldisilazane (HMDS, 8.4 ml_, 40 mmol), and DPPP (0.12 g, 0.3 mmol) was bubbled with a gaseous CO for 5 min and then stirred in a CO balloon at 80°C for 4 h. To the reaction mixture was added MeOH (5 ml_). The reaction mixture was stirred for 10 min, diluted with 2N H2SO4 (200 ml_), and then extracted with EtOAc. The EtOAc extract was washed with saturated aqueous NaHCO3, brine, and then dried over Na2SO4. Filtration and evaporation of the resultant filtrate yielded a residue, which was purified by flash column chromatography (eluent: EtOAc) to yield compound 1d as a white solid.
Step D: 2-terf-Butoxycarbonylaminoacrylic acid methyl ester
To a suspension of /V-Boc-serine methyl ester (Compound 1e, 2.19 g, 10 mmol) and EDCI (2.01 g, 10.5 mmol) in DCM (70 ml_) was added CuCI (1.04 g, 10.5 mmol). The reaction mixture was stirred at room temperature for 72 h. Upon removal of the solvent, the residue was diluted with EtOAc, washed sequentially with water and brine and then dried over MgSO4. The crude product was purified by flash column chromatography (eluent: EtOAc:hexane ~1 :4) to yield compound 1f as a colorless oil.
1H NMR (300 MHz, CDCI3): δ 1.49 (9H, s), 3.83 (3H, s), 5.73 (1 H, d, J = 1.5 Hz), 6.16 (1 H1 S), 7.02 (1 H, s).
STEP E: (2)-2-fert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)acrylic acid methyl ester
A flask charged with compound 1d (0.46 g, 2.0 mmol), compound 1f (0.80 g, 4.0 mmol), tri-o-tolylphosphine (0.098 g, 0.32 mmol) and DMF (8 ml_) was purged with N2(g) 3 times. After the addition of tris(dibenzylideneacetone)dipalladium (0) (0.074 g, 0.08 mmol) and TEA (0.31 ml_, 2.2 mol), the reaction mixture was heated at 110°C for 24 h. At that time, the reaction was quenched by addition of water, and then extracted with EtOAc. The organic phase was washed with 1 N HCI, saturated aqueous NaHCO3, brine, and dried over MgSO4. The mixture was concentrated to a residue, which was purified by flash column chromatography (eluent: EtOAc:hexane~1 :1 to EtOAc only) to yield compound 1g as a white solid.
1H NMR (300 MHz, CD3OD): δ 1.36 (9H, s), 2.26 (6H, s), 3.83 (3H, s), 7.10 (1 H, s), 7.56 (2H, s); 13C NMR (75 MHz, DMSO-d6): δ 17.6, 25.7, 50.2, 78.7, 124.9, 126.4,
128.3, 131.2, 135.2, 135.5, 152.8, 164.3, 169.6;
MS (ES+) (relative intensity): 349.1 (38%)(M+1).
STEP F: (S)-2-ferf-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid methyl ester
Into a reactor charged with a solution of compound 1g (0.56 g, 1.6 mmol) in degassed MeOH (80 mL) was added [Rh(COd)(H1R-DIPAMP)J+BF4 “ under a stream of argon. The reactor was sealed and flushed with H2, stirred at 6O0C under 1000 psi of H2 for 14 days. The crude product was purified by flash column chromatography (eluent: EtOAc:hexane ~1 :1) to yield compound 1 h as a white solid. ee: >99%; 1H NMR (300 MHz, CDCI3): δ 1.36 (9H, s), 2.39 (6H, s), 3.11 (2H, J = 7.2 Hz), 3.65 (3H, s), 4.53-4.56 (1 H, m), 5.12 (1 H, d, J = 8.7 Hz), 5.65 (1 H, br s), 6.09 (1 H, br s), 7.46 (2H, s);
MS(ES+) (relative intensity): 250.9 (100) (M-BoC)+.
STEP G: (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid
Into an ice-cooled solution of compound “I h (0.22 g, 0.63 mmol) in THF (3.5 ml_) was added an aqueous LiOH solution (1 N, 3.5 ml_) and the reaction mixture stirred at 0°C. Upon completion of the reaction, the reaction mixture was concentrated and the aqueous phase was neutralized with cooled aqueous 1 N HCI at 0°C, and then extracted with EtOAc. The combined extracts were dried over Na2SO4 overnight. Filtration and evaporation of the filtrate to dryness yielded compound 1j as a white solid. 1H NMR (300 MHz, DMSO-cfe): δ 1.30 (9H, s), 2.32 (6H, s), 2.95(1 H, dd,
J= 8.8, 13.9 Hz), 3.10 (1 H, dd, J= 6.2, 14.0 Hz), 4.02-4.12 (1 H, m), 7.18-7.23 (2H, m), 7.48 (2H1 s), 7.80 (1 H, s);
MS(ES+) (relative intensity): 236.9 (6) (M-BoC)+.
Example 5
5-((r2-Amino-3-(4-carbamoyl-2.6-dimethyl-phenyl)-propionvn-n-(4-phenyl- 1 H-imidazol-2-yl)-ethvπ-aminol-methyl)-2-methoxy-benzoic acid
STEP A. 2-Methoxy-5-{[1-(4-phenyl-1 W-imidazol-2-yl)-ethylamino]-methyl}- benzoic acid methyl ester
Using the procedures described for Example 4, substituting 5-formyl-2- methoxy-benzoic acid methyl ester (WO 02/22612) for 3,4- dimethoxybenzaldehyde, 2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester was prepared.
STEP B. 5-({[2-ferf-ButoxycarbonylmethyI-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl]-[1 -(4-phenyl-1 H-imidazoI-2-yl)-ethyl]-amino}-methyl)-2- methoxy-benzoic acid methyl ester
Using the procedure of Example 3 for the conversion of Cpd 3d to Cpd 3e, substituting 2-methoxy-5-{[1-(4-phenyl-1 /-/-imidazol-2-yl)-ethylamino]- methylj-benzoic acid methyl ester for Cpd 3d and substituting 2-tert- Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionic acid for 2- tø/t-Butoxycarbonylamino-3-(4-hydroxy-2,6-dimethyl-phenyl)-propionic acid, Cpd 5a was prepared.
STEP C. 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl]-[1 -(4-phenyl-1 W-imidazol-2-yl)-ethyl]-amino}-methyl)-2- methoxy-benzoic acid
5-({[2-tørf-Butoxycarbonylmethyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)- propionyl]-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy- benzoic acid methyl ester was dissolved in an ice-chilled (0-10°C), mixed solvent system of THF (10 ml_) and MeOH (5 ml_). A LiOH H2O/water suspension (2.48 M; 3.77 ml_) was added dropwise, then the reaction was allowed to warm to room temperature and stirred overnight. The resulting mixture was cooled in an ice bath and the basic solution was neutralized with 2N citric acid until slightly acidic. The mixture was concentrated under reduced pressure to remove the volatile materials, after which time the remaining aqueous phase was extracted with EtOAc (3 x 26 ml_). These combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure to yield a pale yellowish white solid. This crude material was dissolved in a 10% MeOH/CH2CI2 solution and adsorbed onto 30 g of silica. The adsorbed material was divided and chromatographed on an ISCO normal phase column over two runs, using a 40 g Redi-Sep column for both runs. The solvent system was a gradient MeOHZCH2CI2 system as follows: Initial 100% CH2CI2, 98%-92% over 40 min; 90% over 12 min, and then 88% over 13 min. The desired product eluted cleanly between 44-61 min. The desired fractions were combined and concentrated under reduced pressure to yield 5-({[2-terf- butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4- phenyl-1 /-/-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid, Cpd 5b, as a white solid.
STEP D. 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1 – (4-phenyl-1 W-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A portion of Cpd 5b (0.27g, 0.41 mmol) was dissolved in EtOAc (39 ml_)/THF (5 ml_), filtered, and subsequently treated with gaseous HCI for 15 min. After completion of the HCI addition, the reaction was slowly warmed to room temperature and a solid precipitate formed. After 5 h the reaction appeared >97% complete by LC (@214nm; 2.56 min.). The stirring was continued over 3 d, then the solid was collected and rinsed with a small amount of EtOAc. The resulting solid was dried under high vacuum under refluxing toluene for 2.5 h to yield Cpd 5c as a white solid di-HCI salt.
Example 2
Racemic 2-terf-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethvl- phenvD-propionic acid
STEP A: Racemic 2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)propionic acid methyl ester
To a reactor charged with a solution of compound 1g (0.68 g, 1.95 mmol) in MeOH (80 mL) was added 10% Pd-C (0.5 g). The reactor was connected to a hydrogenator and shaken under 51 psi of H2 overnight. The mixture was filtered through a pad of Celite and the filtrate was concentrated to dryness to yield compound 2a as a white solid.
The 1H NMR spectrum was identical to that of (S)-2-tert- butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)propionic acid methyl ester, compound 1 h.
STEP B: Racemic 2-terf-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)propionic acid
Following the procedure described for Example 1 , STEP G (preparation of (S)-2-teAt-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid), compound 2b – racemic 2-te/?-butoxycarbonylamino-3- (4-carbamoyl-2,6-dimethyl-phenyl)propionic acid – was prepared.
POLYMORPHS
US8609865
Example 1 Preparation of the zwitterion of 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A 1 L three-necked round-bottomed flask equipped with a mechanical stirrer, addition funnel and a thermocouple was charged without agitation. 34.2 g of 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid (see Example 9 of US 2005/0203143), 340 ml of acetone, and 17 ml of 204 mmolar concentrated HCl were combined in the flask. The stirring was started and the resulting slurry formed a clear solution. This solution was heated to 45° C. under vigorous stirring and aged at this temperature for a period of two hours. After the completion, the reaction mass was cooled to ambient temperature and the supernatant was removed by suction. The vessel along with the residue was rinsed with 20 ml of acetone and then removed as previously. 170 ml of water was added and the reaction mass and was aged under stirring until a homogeneus solution resulted. This solution was then added over a period of ˜½ hr to a solution of 90 ml of 1N NaOH and water. The pH was adjusted to 6.5-7.0 accordingly. The resulting slurry was aged for about 2 hrs at ambient temperature, cooled to 10-15° C., aged at that temperature for about 1 hr, and then filtered. The solid was washed with 10 ml water, air-dried for a period of 4 to 5 hrs, and then placed in a vacuum oven at 50-55° C. until the water content was less than 3%.
Example 2 Preparation of the Form α Crystal
The Form α crystal can be prepared by storing the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid at 0-25% relative humidity for 3 days. Representative PXRD, TGA, and DSC data are shown in FIGS. 1-3 respectively.
Example 3 Preparation of the Form β crystal
The Form β crystal can be prepared by storing the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid at greater than 60% relative humidity for 3 days. Representative PXRD, TGA, and DSC data are shown in FIGS. 1, 4, and 5 respectively.
SYNTHESIS
US20050203143
Example 9 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A. 2-Methoxy-5{[1-(4-phenyl-1 H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester.
Using the procedures described for Example 3, substituting 5-formyl-2-methoxy-benzoic acid methyl ester (WO 02/22612) for 3,4-dimethoxybenzaldehyde, 2-methoxy-5-{[1-(4-phenyl-1H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester was prepared.
B. 5-({[2-tert-Butoxycarbonyl methyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid methyl ester.
Using the procedure of Example 1 for the conversion of Cpd 1d to Cpd 1e, substituting 2-methoxy-5-{[1-(4-phenyl-1H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester for Cpd 1 d and substituting 2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl-propionic acid of Example 8 for 2-tert-Butoxycarbonylamino-3-(4-hydroxy-2,6-dimethyl-phenyl)-propionic acid, Cpd 9a was prepared.
C. 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[11-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
5-({[2-tert-Butoxycarbonyl methyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid methyl ester was dissolved in an ice-chilled (0-10° C.), mixed solvent system of THF (10 mL) and MeOH (5 mL). A LiOH.H2O/water suspension (2.48 M; 3.77 mL) was added dropwise, then the reaction was allowed to warm to room temperature and stirred overnight. The resulting mixture was cooled in an ice bath and the basic solution was neutralized with 2N citric acid until slightly acidic. The mixture was concentrated under reduced pressure to remove the volatile materials, after which time the remaining aqueous phase was extracted with EtOAc (3×26 mL). These combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure to give 2.26 g (146% of theory) of pale yellowish white solid. This crude material was dissolved in a 10% MeOH/CH2Cl2 solution and adsorbed onto 30 g of silica. The adsorbed material was divided and chromatographed on an ISCO normal phase column over two runs, using a 40 g Redi-Sep column for both runs. The solvent system was a gradient MeOH/CH2Cl2 system as follows: Initial 100% CH2Cl2, 98%-92% over 40 min; 90% over 12 min, and then 88% over 13 min. The desired product eluted cleanly between 44-61 min. The desired fractions were combined and concentrated under reduced pressure to yield 1.74 g (113% of theory) of 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid, Cpd 9b, as a white solid.
D. 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
A portion of Cpd 9b (0.27g, 0.41 mmol) was dissolved in EtOAc (39 mL)/THF (5 mL), filtered, and subsequently treated with gaseous HCl for 15 min. After completion of the HCl addition, the reaction was slowly warmed to room temperature and a solid precipitate formed. After 5 h the reaction appeared >97% complete by LC (@214 nm; 2.56 min.). The stirring was continued over 3 d, then the solid was collected and rinsed with a small amount of EtOAc. The resulting solid was dried under high vacuum under refluxing toluene for 2.5 h to yield 0.19 g (71%) of desired Cpd 9c as a white solid di-HCl salt.
Example 8 (S)-2-tert-Butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester

A. (S)-2-tert-Butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester. Into a cool solution of Boc-L-(2,6-diMe)Tyr-OMe (7.0 g, 21.6 mmol; Sources: Chiramer or RSP AminoAcidAnalogues) and N-phenyltrifluoromethanesulfonimide (7.9 g, 22.0 mmol) in dichloromethane (60 mL) was added triethylamine (3.25 mL, 23.3 mmol). The resulting solution was stirred at 0° C. for 1 h and slowly warmed to rt. Upon completion, the reaction was quenched by addition of water. The separated organic phase was washed with 1 N NaOH aqueous solution, water and dried over Na2SO4 overnight. After filtration and evaporation, the residue was purified by flash column chromatography (eluent: EtOAc-hexane: 3:7) to give the desired product (9.74 g, 99%) as a clear oil; 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.39 (6H, s), 3.06 (2H, d, J=7.7 Hz), 3.64 (3H, s), 4.51-4.59 (1H, m), 5.12 (1H, d, J=8.5 Hz), 6.92 (2H, s); MS (ES+) (relative intensity): 355.8 (100) (M−Boc)+.
B. (S)4-(2-tert-Butoxycarbonylamino-2-methoxycarbonylethyl)-3,5-dimethylbenzoic acid. To a suspension of (S)-2-tert-butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester (9.68 g, 21.3 mmol), K2CO3 (14.1 g, 0.102 mol), Pd(OAc)2 (0.48 g, 2.13 mmol) and 1,1′-bis(diphenylphosphino)ferrocene (2.56 g, 4.47 mmol) in DMF (48 mL) was bubbled in gaseous CO for 15 min. The mixture was heated to 60° C. for 8 h with a CO balloon. The cool mixture was partitioned between NaHCO3 and EtOAc, and filtered. The aqueous layer was separated, acidified with 10% citric acid aqueous solution, extracted with EtOAc, and finally dried over Na2SO4. Filtration and concentration of the filtrate resulted in a residue. The residue was recrystallized from EtOAc-hexanes to afford the desired product (7.05 g, 94%); 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.42 (6H, s), 3.14 (2H, J=7.4 Hz), 3.65 (3H, s), 4.57-4.59 (1H, m), 5.14 (1H, d, J=8.6 Hz), 7.75 (2H, s); MS(ES+) (relative intensity): 251.9 (100) (M−Boc)+.
C. (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid methyl ester. Into a stirring solution of (S)-4-(2-tert-butoxycarbonylamino-2-methoxycarbonylethyl)-3,5-dimethyl benzoic acid (3.00 g, 8.54 mmol), PyBOP (6.68 g, 12.8 mmol) and HOBt (1.74 g, 12.8 mmol) in DMF (36 mL) was added DIPEA (5.96 mL, 34.2 mmol) and NH4Cl (0.92 g, 17.1 mmol). The resulting mixture was stirred at rt for 40 min before being partitioned between aqueous NH4Cl solution and EtOAc. The separated organic phase was washed sequentially with 2N citric acid aqueous solution, saturated aqueous NaHCO3 solution, and brine, then dried over Na2SO4 overnight. After filtration and concentration, the residue was purified by flash column chromatography (eluent: EtOAc) to give the product. (3.00 g, 100%); 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.39 (6H, s), 3.11 (2H, J=7.2 Hz), 3.65 (3H, s), 4.53-4.56 (1H, m), 5.12 (1H, d, J=8.7 Hz), 5.65 (1H, brs), 6.09 (1H, br s), 7.46 (2H, s); MS(ES+) (relative intensity): 250.9 (100) (M−Boc)+.
D. (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid. Into an ice-cooled solution of methyl ester from Step C (2.99 g, 8.54 mmol) in THF (50 mL) was added an aqueous LiOH solution (1N, 50 mL) and stirred at 0° C. Upon consumption of the starting materials, the organic solvents were removed and the aqueous phase was neutralized with cooled 1N HCl at 0° C., and extracted with EtOAc, and dried over Na2SO4 overnight. Filtration and evaporation to dryness led to the title acid (S)-2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid (2.51 g, 87%); 1H NMR (300 MHz, DMSO-d6): δ 1.30 (9H, s), 2.32 (6H, s), 2.95 (1H, dd, J=8.8, 13.9 Hz), 3.10 (1H, dd, J=6.2, 14.0 Hz), 4.02-4.12 (1H, m), 7.18-7.23 (2H, m), 7.48 (2H, s), 7.80 (1H, s); MS(ES+) (relative intensity): 236.9 (6) (M−Boc)+.
PAPER
Bioorg Med Chem Lett. 2012 Jul 15;22(14):4869-72.
PATENTS
1.WO 2005090315
2..WO 2006099060
3.WO 2009009480
4. WO 2010062590
5.US 2011263868 *
Patent
https://patentscope2.wipo.int/search/de/detail.jsf;jsessionid=17DB1184234A30C42C287EBFB95A7EF3?docId=WO2018198101&tab=PCTDESCRIPTION&office=&prevFilter=%26fq%3DOF%3AWO&sortOption=Ver%C3%83%C2%B6ffentlichungsdatum+ab&queryString=&recNum=9351&maxRec=3410922
Eluxadoline chemically is 5-[[[(25)-2-amino-3-[4-(aminocarbonyl)-2, 6-dimethylphenyl] – 1 -oxopropyl] [( 15)- 1 -(4-phenyl- lH-imidazol-2-yl)ethyl] amino] methyl] -2-methoxybenzoic acid, represented by Formula I.

Formula I
Eluxadoline is a mu-opioid receptor agonist, indicated in adults for the treatment of irritable bowel syndrome with diarrhea (IBS-D).
U.S. Patent No. 7,741 ,356 describes a process for the preparation of eluxadoline. U.S. Patent Nos. 7,629,488 and 8,710,256 describe processes for the preparation of intermediates of eluxadoline.
PCT Publication No. WO2009/009480 purportedly discloses forms alpha and beta crystals of eluxadoline and processes thereof. PCT Publication No. WO2009/009480 discloses that form alpha crystals can be prepared by storing the zwitterion of eluxadoline at 0-25% relative humidity (RH) for 3 days and form beta crystals can be prepared by storing the zwitterion of eluxadoline at greater than 60% RH for 3 days.
PCT Publication No. WO2017/015606 purportedly discloses amorphous form, crystalline forms I, II, III and IV, and processes for their preparation and a process for the preparation of form alpha crystal of eluxadoline
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018138274
Eluxadoline is the INN denomination assigned to the compound having lUPAC name 5-({[(2S)-2-amino-3-(4-carbamoyl-2,6-dimethylphenyl)propanoyl][(1 S)-1 -(4-phenyl-1 /-/-imidazol-2-yl)ethyl]amino}methyl)-2-methoxybenzoic acid and the formula reported below:

Eluxadoline is a μ- and κ-opioid receptor agonist and δ-opioid receptor antagonist that acts locally in the enteric nervous system. The drug, administered orally, is active locally in the intestine and is able to control gastrointestinal function (Gl) and at the same time to reduce the pain and mitigate the effect of constipation. Its use has been approved for the treatment of diarrhea and abdominal pain in individuals with diarrhea-predominant irritable bowel syndrome (IBS-D).
The family of compounds to which eluxadoline belongs is disclosed in patent application WO 2005/090315 A1 , while patent application WO 2006/099060 A2 is directed to processes for the preparation of these compounds.
As generally known, any active principle may exist under amorphous or different crystalline forms (polymorphs), either as pure compound or in forms in which, in the structure of the crystal, are present molecules of water (hydrates) or of another solvent (solvates); besides, in case of hydrates and solvates, the ratio between the number of molecules of active principle and molecules of water or solvent may vary, giving rise to different solid forms of the compound.
Different salts and solid-state forms of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid-state forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favourable direction, or improving stability (polymorphic and/or chemical) and shelf-life. These variations in the properties of different salts and solid-state forms may also offer improvements to the final dosage form, for instance, if they serve to improve bioavailability. Different salts, solid-state forms and solvates of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which, in turn, may provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
While not intending to be bound by any theory, certain solid forms are characterized by physical properties, e.g., stability, solubility and dissolution rate, appropriate for pharmaceutical and therapeutic dosage forms. Moreover, while not wishing to be bound by any theory, certain solid forms are characterized by physical properties (e.g., density, compressibility, hardness, morphology, cleavage, stickiness, solubility, water uptake, electrical properties, thermal behaviour, solid-state reactivity, physical stability, and chemical stability) affecting particular processes (e.g., yield, filtration, washing, drying, milling, mixing, tableting, flowability, dissolution, formulation, and lyophilization) which make certain solid forms suitable for the manufacture of a solid dosage form. Such properties can be determined using analytical chemical techniques, including solid-state analytical techniques (e.g., X-ray diffraction, microscopy, spectroscopy and thermal analysis), as described herein and known in the art.
For these reasons, chemical compounds useful in the pharmaceutical field are systematically screened looking for the physical form(s) that present an improved set of production, storage and handling properties, and which result in an improved administration to the patients.
Patent application WO 2009/009480 A2 discloses two crystalline forms of eluxadoline, referred to in the document respectively as Form a and Form β. Form a is characterized by an X-ray powder diffraction pattern having the main peaks at about 10.2°, 1 1.3°, 1 1.8°, 14.0°, 14.3°, 14.7°, 16.1 ° and 18.3° 2Θ, while Form β is characterized by an X-ray powder diffraction pattern having the main peaks at about 1 1.0°, 12.4°, 14.9°, 15.2°, 22.1 °, 25.6°, 27.4°, and 30.4° 2Θ.
Patent application WO 2017/015606 A1 discloses several crystalline forms of eluxadoline, referred to therein as Form I, Form II, Form III, and Form IV. Form I is characterized by an X-ray powder diffraction pattern having peaks at about 6.4°, 7.5°, 9.1 °, 10.0°, and 13.0° 2Θ. Form II is characterized by an X-ray powder diffraction profile having peaks at about 7.2°, 1 1 .6°, 12.1 °, 12.7° and 16.9° 2Θ. Form III is characterized by an X-ray powder diffraction pattern having peaks at about 9.3°, 10.2°, 1 1 .5°, 13.3° and 21.8° 2Θ. Form IV is characterized by an X-ray powder diffraction profile having peaks at about 9.3°, 10.2°, 1 1.5°, 13.3° and 21 .8° 2Θ.
However, no information is provided in any of these documents about any useful
properties from the standpoint of the pharmaceutical industry, neither regarding ease of handling of the forms in the production of formulations nor regarding the storage stability (polymorphic and/or chemical) of eluxadoline when prepared in one of these crystalline forms.
An object of the present invention is the provision of a novel process for the preparation of a polymorphic form a’ of eluxadoline (as defined hereinbelow) which, surprisingly, is polymorphically and chemically stable. Since this polymorphic form represents a valuable product, it is an object that upscaling of this process, in order to meet the needs of industrial-scale production, should be easily accomplishable. It is a further object of the present invention that said novel process should produce high-purity products which must contain as low an amount of possibly harmful compounds as possible.
Surprisingly, it was found that new solvate forms ε of eluxadoline allow for the realization of this process and, thus, of the new polymorphically and chemically stable crystalline form α’. It was found that in terms of the starting material from which the solvate forms ε of eluxadoline can be produced, they are extremely flexible.
Further, it was found that the reaction conditions necessary to produce these solvate forms are highly advantageous in terms of energy consumption in combination with the chemical nature of the solvents used
High Performance Liquid Chromatography-Ultraviolet Detection (HPLC-UV):
Chemical stability tests were performed using the following HPLC method
Column: XBridge C8 150 X 4.6 mm, 3.5 μηι
Mobile Phase A: 0.1 % (V/V) phosphoric acid aqueous solution
Mobile Phase B: Acetonitrile
Diluent: 1 :1 (V/V) Mixture of Mobile Phases A and B
Flow Rate: 1.3 mL/min
Runtime: 35 min
Column Temperature 30 °C
Autosampler Temperature: Ambient
Injection Volume: 5 μΙ_
Detection: 210 nm
Sample concentration: 0.4 mg/mL
Gradient Program:

PATENT
WO 2018020450
https://patents.google.com/patent/WO2018020450A2/en
Example 1
Preparation of Eluxadoline
Step 1- Preparation of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[l- (4-phenyl- lH-imidazol-2-yl)-ethyl]-amino} -methyl)-2-methoxy-benzoic acid
To a stirred solution of 1 -(4-phenyl- lH-imidazole-2-yl)-ethyl amine (20 gm) and 5-formyl-2-methoxy-benzoic acid methyl ester (20 gm) in methanol was added catalytic amount of acetic acid (3 ml). The reaction mixture was cooled at 5°C-10°C and sodium borohydride (4 gm) was added. The reaction mixture was further stirred for 2-3 hours at room temperature. The resultant mixture was diluted with water and then partially concentrated. To this mixture was added 2N HCl solution followed by addition of dichloromethane. The phases were separated and the pH (9-1 1) of aqueous layer was adjusted using 2N NaOH solution; which was further extracted with dichloromethane. The combined organic layers were concentrated under vacuum to afford titled compound as oil (yield: 40 gm).
Step 2- Preparation of 5-({[2-tert-butoxycarbonylmethyl-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl] – [ 1 -(4-phenyl- 1 H-imidazol-2-yl)-ethyl] -amino} -methyl)-2-methoxy- benzoic acid methyl ester

To a stirring mixture of 2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl-propionic acid (100 gm), l-ethyl-3-(3- dimethylaminopropyl)carbodiimide hydrochloride (159.4 gm) and 1- hydroxybenzotriazole (45.4 gm) in dimethylformamide (80 ml) & dichloromethane (1920 ml) was added 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[l-(4- phenyl-lH-imidazol-2-yl)-ethyl] -amino }-methyl)-2-methoxy-benzoic acid (step 1 product, 146.6 gm). The resulting mixture was stirred at room temperature for overnight and further diluted with water. The separated organic phase was washed sequentially with aqueous Na2C03 solution, IN HCl solution, water and brine. After concentration, the residue was further dissolved in DCM. The resultant solution was washed sequentially with water & IN HCl solution and then concentrated under vacuum to afford titled compound (yield: 145 gm).
Step 3- Preparation of methyl 5-((2-amino-3-(4-carbamoyl-2,6-dimethylphenyl)-N-(l-(4- phenyl- 1 H-imidazol-2-yl)ethyl) enzoate
To a stirred solution of 5-({[2-tert-butoxycarbonylmethyl-3-(4-carbamoyl-2,6- dimethyl-phenyl)-propionyl] – [ 1 -(4-phenyl- 1 H-imidazol-2-yl)-ethyl] -amino } -methyl)-2- methoxy-benzoic acid methyl ester (step -2 product, 20 gm) in THF (80 ml) was added Cone. HCl solution (30 ml). The reaction mixture was heated at 40°C-45°C. After completion of reaction, the mixture was concentrated and resultant residue was diluted with water. The pH (9-10) was adjusted using 3N NaOH solution; and resultant stick mass was dissolved in methanol. The resultant solution was concentrated under vacuum to afford titled compound (yield: 18.1 gm).
Step 4- Preparation of Eluxadoline
Into an ice cooled solution of methyl 5-((2-amino-3-(4-carbamoyl-2,6- dimethylphenyl)-N-( 1 -(4-phenyl- 1 H-imidazol-2-yl)ethyl)propanamido)methyl)-2- methoxybenzoate (step 3 product, 15 gm) in methanol was added an aqueous lithium hydroxide (3.23 gm in 30 ml water) and resultant mixture was heated at 40°C-45°C. After completion of reaction, mixture was concentrated and further diluted with water. The pH (6-7) was adjusted using 2N citric acid and resultant residue was dissolved in methanol. The resultant solution was added slowly to the acetone and stirring was continued for overnight. The solid precipitated was filtered, washed with acetone and dried to obtain an amorphous form of titled compound (Yield: 3.50 gm).
Example 2
Preparation of Eluxadoline
Step 1 : Preparation of 5-( {[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl] – [ 1 -(4-phenyl- 1 W-imidazol-2-yl)-ethyl] -amino } -methyl)-2-methoxy- benzoic acid
Into an ice cooled solution of 5-({[2-tert-butoxycarbonylmethyl-3-(4-carbamoyl- 2,6-dimethyl-phenyl)-propionyl] – [ 1 -(4-phenyl- 1 H-imidazol-2-yl)-ethyl] -amino } -methyl)- 2-methoxy-benzoic acid methyl ester (160 gm) in methanol (800 ml) was added an aqueous solution of lithium hydroxide (29.46 gm in 350 ml water) and resultant mixture was stirred at room temperature for overnight. After completion of reaction, mixture was partially concentrated and further diluted with water. The pH (4-5) was adjusted using 2N citric acid and further stirred for 60 min. The solid precipitated was filtered, washed with water and dried to obtain titled compound (yield: 140 gm). Step 2: Preparation of Eluxadoline
To a stirred solution of 5-( {[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)-propionyl] – [ 1 -(4-phenyl- 1 H-imidazol-2-yl)-ethyl] -amino } -methyl)-2- methoxy-benzoic acid (step-1 product, 100 gm) in acetone (1200 ml) was added Cone. HC1 solution (50 ml). The reaction mixture was heated at 40°C-45°C. After completion of reaction, the supernatant solution was decanted; resultant residue was rinsed with acetone and further dissolved in water. The pH (6-7) was adjusted using IN NaOH solution and the precipitated was filtered, washed with water and dried to obtain an amorphous form of eluxadoline (yield: 72 gm).
Example 3
Preparation of Eluxadoline
To a stirred solution of 5-( {[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)-propionyl] – [ 1 -(4-phenyl- 1 -imidazol-2-yl)-ethyl] -amino } -methyl)-2- methoxy-benzoic acid (50 gm) in dichloromethane (250 ml) were added solution of Cone. HC1 (50 ml) and water (50 ml). The reaction mixture was heated at 35°C-40°C and further stirred for 10-20 minutes. Tetrahydrofuran (50 ml) & Cone. HC1 (20 ml) were added to the sticky mass and reaction mixture was heated at 40°C for 2 hours. After completion of reaction, the mixture was diluted with water. The pH (6-7) was adjusted using 4N NaOH solution and the obtained sticky mass was dissolved in methanol. The resultant solution was concentrated under vacuum to afford eluxadoline (yield: 16 gm).
Example 4
Preparation of amorphous form of eluxadoline Eluxadoline (1 gm) was dissolved in methanol (20 ml) at 25-30°C. Water (60 ml) was added to the resultant solution and stirred for 15-20 minutes. The resultant slurry was filtered, washed with water and further dried to obtain amorphous form of eluxadoline (Yield: 0.80 gm).
Example 5
Preparation of amorphous form of eluxadoline
Eluxadoline (1 gm) was dissolved in methanol (20 ml) at 25-30°C. Acetone (80 ml) was added to the resultant solution and stirred for 15-20 minutes. The resultant slurry was filtered, washed with acetone and further dried to obtain amorphous form of eluxadoline (Yield: 0.80 gm).
Example 6
Preparation of Form L of eluxadoline
Eluxadoline (1 gm) was charged into flask containing acetonitrile (60 ml) and slurried for 24 hours to 25 hours at 50°C. The resultant solid was filtered, and dried to obtain titled compound (Yield: 0.80 gm).
clip
Eluxadoline (Viberzi)
Eluxadoline, originally developed by Janssen and currently marketed by Allergan (formerly Actavis), was approved in May 2015 by the FDA for the treatment of diarrhea-predominant irritable bowel syndrome (IBS-D).
(60)Eluxadoline, an orally dosed agent, employs a unique mechanism for IBS-D treatment, as it functions simultaneously as a μ- and κ-opioid receptor agonist and a δ-opioid receptor antagonist,
(61) leading to a first-in-class therapy for treatment of IBS-D. Specifically, in animal studies, eluxadoline was found to interact with opioid receptors in the gut, inhibiting neurogenically mediated secretion and reducing intestinal contractility.
(62)Additionally, the treatment led to a decrease in stress-induced acceleration of upper GI transit without causing rebound constipation,
(60-62) earning its mark as a first-line therapeutic treatment for IBS-D. In two phase III clinical trials of over 2400 patients with IBS-D, patients taking eluxadoline showed a greater improvement toward the end point (≥30% improvement from their baseline IBS-D score on at least 50% of days treated with eluxadoline) compared to patients treated with placebo.
(63)The synthesis of eluxadoline begins with preparation of advanced coupling component
85, which could be completed via a four-step route from commercially available
N-Boc-protected aminoester
83 (Scheme 15).
(64) Triflate formation using
N-phenyltrifluoromethanesulfinimide in DCM under basic conditions led to nearly quantitative yield of the desired triflate, which was subjected to a carbonylation reaction to yield aryl acid
84 in 94% yield. Employing NH
4Cl as a source of ammonia, amidation of
84 took place in the presence of PyBOP/HOBt and DIPEA in DMF. Finally, acid
85 was revealed upon methyl ester saponification with aqueous LiOH in THF. This sequence provided
85 without purification ,and this acid could be used directly as applied in Scheme 16.
(64)Scheme 15. Synthesis of Eluxadoline Intermediate 85
Scheme 16. Synthesis of Eluxadoline (XII)
With coupling component
85 in hand, the synthesis of eluxadoline proceeds as described in Scheme 16 and initiated from a HOBt and EDC·HCl-mediated coupling of commercial
N-Cbz-
l-alanine (
86) with commercial 2-amino acetophenone hydrochloride (
87) to provide intermediate
88in 83% yield.
(64, 65) Addition of NH
4OAc and AcOH to a suspension of
88 in refluxing xylenes furnished the desired imidazole in excellent yield (95%). Submission of this
N-Cbz-imidazole to hydrogenation conditions (H
2, Pd/C, MeOH) enabled liberation of the free amine to access
89 in quantitative yield following filtration and concentration. From intermediate
89, reductive amination with commercially available aryl aldehyde
90 under standard conditions (NaBH
4, MeOH) followed by subsequent coupling of the corresponding crude amine with acid
85 using HOBt/EDC·HCl enabled formation of the carbon framework of eluxadoline (
91). Saponification of the ester within
91 with LiOH in MeOH/THF yielded the corresponding acid in quantitative yield. Immediate subjection of this intermediate to acidic conditions (HCl in EtOAc/THF) led to
N-Boc cleavage and isolation of eluxadoline (
XII) as the bis-HCl salt in 71% yield, requiring no further purification.
(64, 65) It should be noted that since this initial report, additional details for the isolation of eluxadoline in high purity in various crystal forms and as a zwitterion have been reported,
(66) although most reported routes described isolation of this drug in its HCl salt form.
(64, 65)
-
60.Garnock-Jones, K. P. Eluxadoline: First Global Approval Drugs 2015, 75, 1305– 1310 DOI: 10.1007/s40265-015-0436-4
-
61.Davenport, J. M.; Covington, P.; Bonifacio, L.; McIntyre, G.; Venitz, J. Effect of Uptake Transporters OAT3 and OATP1B1 and Efflux Transporter MRP2 on the Pharmacokinetics of Eluxadoline J. Clin. Pharmacol.2015, 55, 534– 542 DOI: 10.1002/jcph.442
-
62.Wade, P. R.; Palmer, J. M.; McKenney, S.; Kenigs, V.; Chevalier, K.; Moore, B. A.; Mabus, J. R.;Saunders, P. R.; Wallace, N. H.; Schneider, C. R.; Kimball, E. S.; Breslin, H. J.; He, W.; Hornby, P. J.Modulation of Gastrointestinal Function by MuDelta, a Mixed μ Opioid Receptor Agonist/δ Opioid Receptor Antagonist Br. J. Pharmacol. 2012, 167, 1111– 1125 DOI: 10.1111/j.1476-5381.2012.02068.x
-
63.Lembo, A. J.; Lacy, B. E.; Zuckerman, M. J.; Schey, R.; Dove, L. S.; Andrae, D. A.; Davenport, J. M.;McIntyre, G.; Lopez, R.; Turner, L.; Covington, P. S. Eluxadoline for Irritable Bowel Syndrome with DiarrheaN. Engl. J. Med. 2016, 374, 242– 253 DOI: 10.1056/NEJMoa1505180
-
64.Breslin, H. J.; Cai, C.; He, W.; Kavash, R. W. Preparation of Imidazole Derivatives as Opioid Receptor Modulators. WO 20050203143A1, 2005.
-
65.cai, C.; He, W. Process for the Preparation of Amino Acid Derivatives as Opioid Modulators. WO 2006099060A1,
2006.
|
|
12-24-2010
|
NOVEL COMPOUNDS AS OPIOID RECEPTOR MODULATORS
|
|
|
8-32-2010
|
Compounds as opioid receptor modulators
|
|
|
6-23-2010
|
Compounds as opioid receptor modulators
|
|
|
2-12-2010
|
PROCESS FOR THE PREPARATION OF OPIOD MODULATORS
|
|
|
12-9-2009
|
Process for the preparation of opioid modulators
|
| US7629488 * |
Mar 6, 2006 |
Dec 8, 2009 |
Janssen Pharmaceutica N.V. |
Process for the preparation of opioid modulators |
| US7741356 * |
Mar 14, 2005 |
Jun 22, 2010 |
Janssen Pharmaceutica N.V. |
Compounds as opioid receptor modulators |
| US7786158 * |
Oct 24, 2007 |
Aug 31, 2010 |
Janssen Pharmaceutica N.V. |
Compounds as opioid receptor modulators |
| US7994206 |
Jul 7, 2008 |
Aug 9, 2011 |
Janssen Pharmaceutica, N.V. |
Crystals and process of making 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid |
| CN1950342A |
Mar 14, 2005 |
Apr 18, 2007 |
詹森药业有限公司 |
Novel compounds as opioid receptor modulators |
References
- ^ Jump up to:a b c “Viberzi (eluxadoline) Tablets, for Oral Use, CIV. Full Prescribing Information”. Actavis Pharma, Inc. Parsippany, NJ 07054 USA. Retrieved 26 December 2015.
- ^ “Truberzi”. European Medicines Agency. 29 September 2016.
- ^ Fragkos, Konstantinos C (2017-09-25). “Spotlight on eluxadoline for the treatment of patients with irritable bowel syndrome with diarrhea”. Clinical and Experimental Gastroenterology. 10: 229–240. doi:10.2147/ceg.s123621.
- ^ “FDA approves two therapies to treat IBS-D”. http://www.fda.gov. Retrieved 2015-06-01.
- ^ “Viberzi Information from Drugs.com”. http://www.drugs.com. Retrieved 2015-06-01.
- ^ Limbo AJ, et al. Eluxadoline in Irritable Bowel Syndrome with Diarrhea. NEJM 2016;374:242-53
- ^ Commissioner, Office of the (15 March 2017). “Safety Alerts for Human Medical Products – Viberzi (eluxadoline): Drug Safety Communication – Increased Risk of Serious Pancreatitis In Patients Without A Gallbladder”. http://www.fda.gov. Retrieved 19 March 2017.
- ^ Jump up to:a b Brooks, Megan (March 2017). “FDA: Avoid IBS Drug Viberzi in Patients With No Gallbladder”. http://www.medscape.com. Retrieved 2017-09-18.
- ^ Jump up to:a b Commissioner, Office of the. “Safety Alerts for Human Medical Products – Viberzi (eluxadoline): Drug Safety Communication – Increased Risk of Serious Pancreatitis In Patients Without A Gallbladder”. http://www.fda.gov. Retrieved 2017-09-18.
- ^ “bismuth subsalicylate”. reference.medscape.com. Retrieved 2016-05-10.
- ^ Levy-Cooperman, N; McIntyre, G; Bonifacio, L; McDonnell, M; Davenport, JM; Covington, PS; Dove, LS; Sellers, EM (December 2016). “Abuse Potential and Pharmacodynamic Characteristics of Oral and Intranasal Eluxadoline, a Mixed μ- and κ-Opioid Receptor Agonist and δ-Opioid Receptor Antagonist”. The Journal of Pharmacology and Experimental Therapeutics. 359 (3): 471–481. doi:10.1124/jpet.116.236547. PMC 5118645. PMID 27647873.
- ^ “Actavis Announces FDA Acceptance for Filing of NDA for Eluxadoline”. http://www.drugs.com. Retrieved 2015-06-01.
- ^ “FDA Approves Viberzi (eluxadoline) for Irritable Bowel Syndrome with Diarrhea (IBS-D) in Adults”. http://www.drugs.com. Retrieved 2015-06-01.
- ^ Davenport, J. Michael; Covington, Paul; Bonifacio, Laura; McIntyre, Gail; Venitz, Jürgen (2015). “Effect of uptake transporters OAT3 and OATP1B1 and efflux transporter MRP2 on the pharmacokinetics of eluxadoline”. The Journal of Clinical Pharmacology. 55 (5): 534–542. doi:10.1002/jcph.442. ISSN 0091-2700. PMC 4402028.
- ^ [1], Process of the Preparation of Opioid modulators.
The active ingredient in VIBERZI is eluxadoline, a mu-opioid receptor agonist.
The full chemical name is 5-[[[(2S)-2-amino-3-[4-(aminocarbonyl)-2,6-dimethylphenyl]-1- oxopropyl][(1S)-1-(4-phenyl-1H-imidazol-2-yl)ethyl]amino]methyl]-2-methoxybenzoic acid.
Eluxadoline has a molecular weight of 569.65 and a molecular formula of C32H35N5O5. The chemical structure of eluxadoline is:
VIBERZI is available as 75 mg and 100 mg tablets for oral administration. In addition to the active ingredient, eluxadoline, each tablet contains the following inactive ingredients: silicified microcrystalline cellulose, colloidal silica, crospovidone, mannitol, magnesium stearate, and Opadry II (partially hydrolyzed polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, iron oxide yellow, and iron oxide red).
//////////////////JNJ-27018966, iberzi, элуксадолин ,إيلوكسادولين ,艾沙多林 ,ELUXADOLINE, FDA 2015, エルクサドリン,
CC1=CC(=CC(=C1CC(C(=O)N(CC2=CC(=C(C=C2)OC)C(=O)O)C(C)C3=NC=C(N3)C4=CC=CC=C4)N)C)C(=O)N

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
















































































 2a is the drug
2a is the drug