WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Join me on Facebook
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
Googleplus
MYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Tiabendazole (INN, BAN), thiabendazole (AAN, USAN), TBZ (and the trade names Mintezol, Tresaderm, and Arbotect) is a preservative[1]
2-Substituted benzimidazole first introduced in 1962. It is active against a variety of nematodes and is the drug of choice for strongyloidiasis. It has CNS side effects and hepatototoxic potential. (From Smith and Reynard, Textbook of Pharmacology, 1992, p919)
Percent Composition: C 59.68%, H 3.51%, N 20.88%, S 15.93%
Literature References: Prepd by the reaction of 4-thiazolecarboxamide with o-phenylenediamine in polyphosphoric acid: H. D. Brown et al.,J. Am. Chem. Soc.83, 1764 (1961); L. H. Sarett, H. D. Brown, US3017415 (1962 to Merck & Co.). Synthesis of labeled thiabendazole: D. J. Tocco et al.,J. Med. Chem.7, 399 (1964). Alternate route of synthesis: V. J. Grenda et al.,J. Org. Chem.30, 259 (1965). Anthelmintic props: H. D. Brown et al.,loc. cit.; K. C. Kates et al.,J. Parasitol.57, 356 (1971). Fungicidal props: H. J. Robinson et al.,J. Invest. Dermatol.42, 479 (1966). Systemic props in plants: D. C. Erwin et al.,Phytopathology58,860 (1968). Toxicity: H. J. Robinson et al.,Toxicol. Appl. Pharmacol.7, 53 (1965). Residue analysis: IUPAC Appl. Chem. Div., Pure Appl. Chem.52, 2567 (1980). Comprehensive description: V. K. Kapoor, Anal. Profiles Drug Subs.16, 611-639 (1986).
Properties: Colorless crystals, mp 304-305°. uv max (methanol): 298 nm (e 23330). Fluorescence max in acid soln: 370 nm (310 nm excitation). Max soly in water at pH 2.2: 3.84%. Soluble in DMF, DMSO. Slightly soluble in alcohols, esters, chlorinated hydrocarbons. LD50 in mice, rats, rabbits (g/kg): 3.6, 3.1, >3.8 orally (Robinson).
Melting point: mp 304-305°
Absorption maximum: uv max (methanol): 298 nm (e 23330)
Use: Fungicide for spoilage control of citrus fruit; for treatment and prevention of Dutch elm disease in trees; for control of fungal diseases of seed potatoes.
Therap-Cat: Anthelmintic (Nematodes).
Therap-Cat-Vet: Anthelmintic, fungicide.
Keywords: Anthelmintic (Nematodes).
Thiabendazole, 2-(4′-thiazolyl)-benzimidazole (TBZ) (I) is an important anthelmintic and fungicidal agent widely used in pharmaceutical, agriculture and food industry. Owing to the commercial importance of thiabendazole, the various synthetic routes are disclosed in the literature for preparing this pharmacologically and fungicidally active compound.
The various literature discloses the synthesis of thiabendazole by using aniline, 4-cyanothiazole and hydrogen chloride in polychlorobenzene such as dichloro- or a trichlorobenzene solvent under high pressure reaction conditions to obtain N-phenyl-(thiazole-4-amidine)-hydrochloride (amidine hydrochloride). This amidine hydrochloride is then treated with hypohalites such as sodium or potassium hypochlorite, sodium hypobromite and calcium hypochlorite in presence of base such as alkali or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, calcium hydroxide; or an alkali metal carbonate or bicarbonate such sodium carbonate, sodium bicarbonate to obtain thiabendazole.
NMR
The US patent no. US 3,274,208 discloses the process for preparation of amidine hydrochloride by reacting 4-cynothiazole and aniline in presence of aluminum chloride at 180 °C. The amidine hydrochloride is purified by acid base treatment.
The US patent no. US 3,299,081 (henceforth patent ‘081) discloses the process for preparation of N-phenyl-(thiazole-4-amidine)-hydrochloride (amidine hydrochloride) and thiabendazole by heating together 4-cyanothiazole and aniline hydrochloride and purging of excess dry hydrogen chloride gas under pressure (15 psig) reaction condition in a 1,2-dichlorobenzene solvent at 135 to 140 °C using closed reactor. The amidine hydrochloride is isolated by filtration and it is then cyclized to N-chloro-N’-phenyl-(thiazole-4-amidine) intermediate by reaction with sodium hypochlorite in water-methanol solvent, further the intermediate is then converted to thiabendazole by treatment with potassium hydroxide in ethanol. The preferred embodiment of the said patent discloses the use of excess hydrogen chloride in a polychlorobenzene medium to achieve higher yields of amidine hydrochloride. The reaction with gas under pressure is exothermic, so the reaction is unsafe.
As per the background of the patent ‘081, the prior art processes were disclosed that the N-aryl amidines could be prepared by reacting together a nitrile and an aromatic amine in the presence of a metal catalyst such as aluminum chloride or zinc chloride. The process involved the use of a metallic halide as an additional substance in the reaction mixture with the result that metal complexes are obtained which have to be decomposed and the metal removed before pure amidine compounds can be recovered. It was also known to prepare N-aryl amidines by reacting the nitrile and the aromatic amine hydrochloride in a solvent such as ether in the absence of metallic halide. The process referred to affords only poor yields of the desired amidine. Hence, neither of these methods are entirely satisfactory.
13C NMR
The US patent no. US 3,299,082 discloses the process for preparation of N-phenyl-(thiazole-4-amidine)-hydrochloride (amidine hydrochloride) by reacting aniline and 4-cyanothiazole in in the presence of a Friedel Crafts type catalyst such as aluminum chloride at temperature 180 °C. The amidine hydrochloride is reacted with hydroxylamine hydrochloride, in presence of base such as sodium bicarbonate and water as solvent to obtain N-phenyl-(thiazole-4-hydroxyamidine) which is then treated with alkyl or aryl sulfonyl halide such methane sulfonyl chloride in the presence of a base such as pyridine to obtain thiabendazole.
The US patent no. US 3,325,506 discloses the process for preparation of thiabendazole by reacting amidine hydrochloride with hypohalites such as sodium or potassium hypochlorite, sodium hypobromite and calcium hypochlorite in presence of base such as alkali or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, calcium hydroxide; or an alkali metal carbonate or bicarbonate such sodium carbonate, sodium bicarbonate in water or mixtures of water and organic solvents to obtain thiabendazole.
The significance of by-products from reactions in process development work arises from the need to control or eliminate their formation which might affect product cost, process safety, product purity and environmental health. Very few reactions go to 100% completion in the desired sense. Even when conversion is 100% selectivity is not 100%. Most reactions are accompanied by by-products which arise as a direct consequence of a primary synthetic step including work-up and isolation and as a result of various types of side reactions. By-products from the latter type also include tars, polymeric materials, and coloring matters. The level of some by-products from side reactions depends frequently on the batch size.
MASS
In the pharmaceutical industry, an impurity is considered as any other inorganic or organic material, or residual solvents other than the drug substances, or ingredients, arise out of synthesis or unwanted chemicals that remains with APIs. Organic impurities are those substances which are formed in the drug substance during the process of synthesis of drug product or even formed during the storage of drug product. This type of impurity includes-intermediate, starting material, degradation product, reagents, ligands, catalyst and by product. Inorganic impurities present mainly include heavy metals, residual solvents, inorganic salts, filter aids, charcoal, reagent, ligands and catalyst.
Impurity profiling includes identification, structure elucidation and quantitative determination of impurities and degradation products in bulk drug materials and pharmaceutical formulations. Impurity profiling has gained importance in modern pharmaceutical analysis since an unidentified, potentially toxic impurities are hazardous to health and the presence of unwanted impurities may influence bioavailability, safety and efficacy of APIs. Now days, not only purity profile but also impurity profile has become mandatory according to various regulatory authorities. The International Conference on Harmonization (ICH) has published guidelines on impurities in new drug substances, products, and residual solvents.
IR
The prior art processes for preparing thiabendazole suffer from inherent drawbacks and inconveniences, such as low yields, additional reaction steps, high-pressure and unsafe reaction conditions. Moreover, the prior art processes for preparation of thiabendazole are end up with surplus level of potential impurities such as 4-chloro thiabendazole (V) or 5-chloro thiabendazole (VI). Also, the prior processes are silent about these impurities. Since, the strict regulations of the regulatory authorities pertaining to the presence of impurities in the active ingredient, it is highly essential to align the research inline with the guidelines of the regulatory authorities in accordance to appropriate regulations and limits to register and commercialize the product in respective countries.
(V) (VI)
Hence, with objective of developing the short process, more direct and less expensive methods, significant improvement in the art for preparation of thiabendazole with controlled level of 4-chloro thiabendazole or 5-chloro thiabendazole impurities, residual solvents (methanol, benzene) and heavy metals (selenium, cobalt, molybdenum), the inventors of the instant invention are motivated to pursue the research to synthesize thiabendazole in under atmospheric conditions with high yield and high chemical purity for agricultural and pharmaceutical use.
Genes responsible for the maintenance of cell walls in yeast have been shown to be responsible for angiogenesis in vertebrates. Tiabendazole serves to block angiogenesis in both frog embryos and human cells. It has also been shown to serve as a vascular disrupting agent to reduce newly established blood vessels. Tiabendazole has been shown to effectively do this in certain cancer cells.[5]
Pharmacodynamics
TBZ works by inhibition of the mitochondrial, helminth-specific enzyme, fumarate reductase, with possible interaction with endogenous quinone.[6]
Other
Medicinally, thiabendazole is also a chelating agent, which means it is used medicinally to bind metals in cases of metal poisoning, such as lead, mercury, or antimony poisoning.
In dogs and cats, thiabendazole is used to treat ear infections.
Thiabendazole is also used as a food additive,[7][8] a preservative with E number E233 (INS number 233). For example, it is applied to bananas to ensure freshness, and is a common ingredient in the waxes applied to the skins of citrus fruits. It is not approved as a food additive in the EU,[9] Australia and New Zealand.[10]
Safety
The substance appears to have a slight toxicity in higher doses, with effects such as liver and intestinal disorders at high exposure in test animals (just below LD50 level).[citation needed] Some reproductive disorders and decreasing weaning weight have been observed, also at high exposure. Effects on humans from use as a drug include nausea, vomiting, loss of appetite, diarrhea, dizziness, drowsiness, or headache; very rarely also ringing in the ears, vision changes, stomach pain, yellowing eyes and skin, dark urine, fever, fatigue, increased thirst and change in the amount of urine occur.[citation needed] Carcinogenic effects have been shown at higher doses.[11]
Intermediate arylamidine2 is prepared by the dry HCl catalyzed addition of aniline to the nitrile function of 4-cyanothiazole (1). Amidine (2) is then converted to its N-chloro analog 3by means of NaOCl. On base treatment, this apparently undergoes a nitrene insertion reaction (4) to produce thiabendazole (5). Note the direction of the arrow is from the benzene to the nitrene since the nitrene is an electrophilic species.
Alternative route of synthesis: 4-thiazolecarboxamide with o-phenylenediamine in polyphosphoric acid.[13]
The present invention relates to an improved process for preparing thiabendazole of formula (I) with high yield, high purity, in economical and commercially viable manner for agricultural and pharmaceutical use.
Process for preparing thiabendazole with higher yield, purity, in an economical and commercially viable manner. Thiabendazole is an important anthelmintic and fungicidal agent widely used in pharmaceutical, agriculture and food industry. Represents the first filing from the Hikal Ltd and the inventors on thiabendazole.
The structural details of the 4-chloro thiabendazole (V) and 5-chloro thiabendazole (VI) impurities are as follow.
1. 4-Chloro thiabendazole:
(a) FT-IR study: The FT-IR spectrum was recorded in the KBr pellet using ABB FTLA-2000 FT-IR Spectrometer. The IR data is tabulated below.
Frequency (cm“1) Assignment (s)
1576.37 C=C stretching
1309.16 C-N stretching
3073.38 N-H stretching
(b) NMR spectral data:
NMR experiment was carried out on 400 MHz Bruker spectrometer using DMSO as solvent. The chemical shifts are reported on the δ scale in ppm relative DMSO at 2.5 ppm. The 1H spectra displayed in respectively. The NMR assignment of 4-chloro thiabendazole is shown below.
Proton assignments of 4-Chloro thiabendazole:
s-singlet, d-doublet, t -triplet, q- quartet, dd-doublet of doublet, br-broad, m-multiplet.
2. 5-Chloro thiabendazole:
(a) FT-IR study:
The FT-IR spectrum was recorded in the KBr pellet using ABB FTLA- 2000 Spectrometer. The IR data is tabulated below.
(b) NMR spectral data:
NMR experiment was carried out on 400 MHz Bruker spectrometer using DMSO-d6 as solvent. The chemical shifts are reported on the δ scale in ppm relative DMSO-d6 at 2.50
ppm. The 1H spectra displayed in respectively. The NMR assignment of 5-chloro thiabendazole is shown below.
Example 1: Preparation of amidine hydrochloride (IV)
To the 4-neck, 1 lit RBF, fixed with thermo pocket, condenser and hydrogen chloride (HC1) gas inlet, 100 g (0.908 moles, 1.0 eq) of 4-cyanothiazole, 386 (3.86 V) ml of 1,2-dichlorobenzene and 86.02 (0.924 moles, 1.02 eq) g of aniline were charged. The reaction mass was heated to 55 to 60 °C and hydrogen chloride (HC1) gas was purged till exotherm ceased. Then the temperature of the reaction mass was raised to 135 to 140 °C and again dry HC1 gas was purged till 4-cyanothiazole was reduced to less than 0.2 % (w/w) analyzed by HPLC. The reaction mass was cooled to 45 to 50° C and 500 mL of water was charged and the reaction mass was stirred for half an hour. The pH of the reaction mass was adjusted between 3 to 5 using caustic lye. The reaction mass was filtered through hyflo bed, and bed was washed with 50 (0.5 V) mL of water. The organic layer was separated, and the aqueous layer was charged back to the RBF. 20 g of activated charcoal was added in aqueous layer under stirring at 45 to 50 °C. The reaction mass was heated to 55 to 60 °C and maintained under stirring for 1.0 hour. The reaction mass was filtered through the hyflo bed under
vacuum, and bed was washed with 50 mL of hot water and suck dried till no more filtrate collected. 300-400 mL of water was distilled from the aqueous layer at 55 °C under 50 m bar of vacuum. Then the reaction mass was cooled to 0 to 5 °C and maintained under stirring for 1 hour. The obtain amidine hydrochloride was filtered by using Buckner funnel and suck dried till no more filtrate collected from it. The wet cake was dried under vacuum at 55 to 60 °C to get 189 g (86.83% yield, HPLC purity 99.85%) of amidine hydrochloride.
Example 2: Preparation of thiabendazole (I)
The 5 lit RBF was fixed with over head stirrer, thermo pocket, condenser and addition funnel. 185 g (0.772 moles, 1.0 eq.) of amidine hydrochloride and 1536 mL (7.33V) of water were charged. The reaction mass was cooled to 0 to 5 °C. 1233 mL of methanol was added to the mass and the pH of the reaction mass was adjusted between 9 to 10 by using 5N sodium carbonate solution. The reaction mass was warmed to 10 to 15 °C and 415.35 g (12.57 % w/w, 0.91 eq.) sodium hypochlorite was slowly added by maintaining temperature between 10 to 15 °C. The reaction mass was stirred at same temperature for half an hour. Then the reaction mass was heated to 60 to 65 °C and 46.15 g (12.57 % w/w, 0.1 eq) sodium hypochlorite was added. The reaction mass was stirred at 60 to 65 °C for 1.0 hour and the reaction mass was cooled to 30 to 40 °C. The reaction mass was filtered, the bed was washed with 925 mL of water (5.0 V) and suck dried for 10 minutes to get 238 g (152 g on dry basis, 97.82 % yield, HPLC purity 99.77%) of thiabendazole.
Example 3: Purification of thiabendazole (I)
The 5 lit RBF was fixed with over head stirrer, thermo pocket, condenser and addition funnel. 224 g of wet crude thiabendazole (145 g on dry basis) was charged at 25 to 30 °C. 2392 mL (16.5 V) of water was charged and the reaction mass was heated to 75 to 80 °C. The pH of the reaction mass was adjusted between 1 to 2 by adding concentrated hydrochloride. Then 21.75 g (15 %, w/w) activated charcoal was added and the reaction mass was stirred for 1.0 hour at 75 to 80 °C. The reaction mass was filtered through hyflo bed and the bed was washed with 1445 mL (1.0 V) of hot water. The aqueous layer was charged back to clean RBF and cooled to 0 to 5 °C and stirred for 10 hours. The solid was filtered and suck dried under vacuum to get 224 g wet cake of thiabendazole hydrochloride (135 g on dry basis).
1261 niL (10 V w.r.t dry thiabendazole hydrochloride) was charged and then 224 g wet cake of thiabendazole hydrochloride was added. The reaction mass was heated to 70 to 80 °C and maintained under stirring for half an hour to get clear solution. The pH of the reaction mass was adjusted to 7 to 8 by using liquor ammonia. The reaction mass was cooled to 25 to 30 °C and stirred for 1.0 hour. The reaction mass was filtered, and the wet cake was slurry washed twice with 1350 mL (10V x 2 times). Then the bed was washed with 675 mL (5.0 V) water. The solid was dried under vacuum at 60 to 70 °C to afford 119 g (79.33% yield, HPLC purity 99.96%) of pure thiabendazole.
CLIP
Fig. 5 Raman spectrum of solid thiabendazole, and SERS spectra of ethanol – water solutions on a re-used 3 m m thick Au woodpile array. Spurious bands from impurities are marked with asterisks.
CLIP
Fig. 6 (A) Proton NMR spectrum of thiabendazole in DMSO-d 6 solution. (B) Plots of normalized selective relaxation rate enhancements of H1/ H2, H14, and H12. [TBZ] ¼ 2 Â 10 À3 mol L À1 , [DNA] ¼ 1, 2, 5, 10, 20 Â 10 À5 mol L À1 , pH ¼ 7.4, T ¼ 298 K. (C) Equilibrium constant of the TBZ-DNA system. [DNA] ¼ 2 Â 10 À5 mol L À1 , [TBZ] ¼ 2, 2.5, 3, 3.5, 4 Â 10 À3 mol L À1 , pH ¼ 7.4, T ¼ 298 K.
CLIP
Thiabendazole has been prepared by heating thiazole-4-carboxamide and benzene-1,2-diamine in polyphosphoric acid (Scheme 13) (1961JA(83)1764). An alternative synthesis involves 4-carboxythiazole (CA 162 590253 (2015), CA 62 90958 (1964)) or 4-cyanothiazole (CA 130 110264 (1996), CA 121 57510 (1994)) as starting materials. A different approach to the synthesis of thiabendazole has been described starting from N-arylamidines; in the presence of sodium hypochlorite and a base, N-arylamidine hydrochlorides are transformed to benzimidazoles via formation of N-chloroamidine intermediate followed by ring closure in a stepwise or concerted mechanism (1965JOC(30)259).
A recent report (1) from workers at Chonnam National University (Gwangju, Korea) describes a benzimidazole synthesis which:
produces good product yields (40-98%, for about 30 examples)
and proceeds in one pot from three readily available components: sodium azide, an aldehyde, and 2-haloanilines
shows good functional group tolerance(nitro-, ester-, chloro-, and various heterocyclic functionalities on the aldehyde or haloaniline component).
The Benzimidazole Synthesis of Lee and coworkers (1)
Naturally, there are many established ways to synthesize benzimidazoles, which are important substances used in the design of bioactive substances (2). Recent work has sought to address specific drawbacks associated with these methods, which can include harsh reaction conditions and complicated product mixtures.
Further developments have focused on the use of 2-haloacetanilides, 2-haloarylamidines, arylamino oximes, and N-arylbenzimidamides (3). This work notable due to the useful anthelmintic properties. Anthelmintic agents work to kill or repel intestinal worms. A review (3) discusses the synthesis of benzimidazoles, and cites the breakthrough discovery of thiabendazole by researchers at Merck in 1961. Thiabendazole was found to have potent broad spectrum activity against gastrointestinal parasites.
Early thiabendazole synthesis (3)
The initial synthesis of thiabendazole occured via dehydrative cyclization of 1,2 diaminobenzenze in polyphosphoric acid (PPA). The commercialized process involved the conversion of N-arylamidines using hypochlorite (4). Although this process can be performed in ‘one-pot’ fashion it is more typically performed in two steps.
The ‘one-pot’ benzimidazole synthesis described by Lee et. Al. is showcased by its ability to produce thiabendazole in one step, from readily available starting materials (2-haloanilines, thiazole-4-carboxaldehyde) – in 97% yield.
Their work builds on the report of Driver and coworkers (5) that showed that benzimidazoles could be had from 2-azidoanilines in good yield. Indeed, Lee proposes a mechanism that produces an azidoaldimine intermediate, which foregoes the multistep preparation of 2-azidoaniline starting materials.
One proposed mechanistic pathway is shown, with the following steps:
initial in situ formation of an aldimine, via addition of aniline to an aldehyde;
Ar-X insertion of the copper catalyst;
Cu-azide association, with transfer of azide to the aromatic ring;
loss of nitrogen with concomitant ring formation, and catalyst regeneration
One mechanistic explanation proposed by Lee and coworkers (1).
In developing their method, they investigated a number of factors:
Solvent. DMSO outperformed other polar solvents (NMP, DMF, DMAc). Less polar solvents failed (toluene, diglyme).
Source of Copper catalyst. The oxidation state of copper was not a factor, as Cu(I) and Cu(II) salts showed similar performance.
Ligand Evaluation. Ligand selection was not a large factor. Several were tested; ultimately TMEDA was selected.
Substituents on the aniline / pyridyl component. Base sensitive substituents were tolerated (benzoate ester) and 3-Cl groups were fine. The sensitivity to a broad range of substituents (the usual EWD- and ED-groups) was not rigorously determined
Nature of the haloaniline. Although both bromo- and iodoaniline examples were given, the predominance of iodoaniline examples suggests it was prefered by the authors for unstated reasons.
Reactivity of various aldehyde reactants. Aldehydes of varying classes were evaluated. Yields from aromatic substrates bearing ED groups(benzaldehyde, 4-Cl benzaldehyde, 4-methoxybenzaldehyde) produced the highest product yields. Aliphatic aldehydes produced noticeably lower yields, with the curious exception of pivaldehyde. Several heterocyclic aldehydes (2- furyl- and 2-thionylaldehyde were tested and provided good results.
A synopsis of the Lee Procedure follows:
CuCl (0.1 mmol), haloaniline (2.0 mmol), TMEDA (0.1 mmol), NaN3 (4.0 mmol), aldehyde (2.4 mmol) were combined in DMSO mL), The mixture was heated at 120 C for 12 hours. After cooling to room temperature the mixture was poured onto EtOAc (50 mL), washed with brine (25 mL) and water (25 mL). The organic phase was dried over Mg2SO4, and the residue from evaporation was purified by column chromatography (1:1 hexane / EtOAc mobile phase).
Artie McKim.
(1) Kim, Y.; Kumar, M.R.; Park, N.; Heo, Y.; Lee, S. J. Org. Chem. 2011, 76, 9577-9583.
(2) Tumulty, D.; Cao, K.; Homes, C.P. Org Lett. 2001, 3, 83.; Wu, Z. Rea. P.; Wickham, G.; Tetrahedron Lett. 2000, 41, 9871.; Chari, M.A.; Shobha, P.S.D.; Mukkanti, K. J. Heterocycl. Chem.2010, 47, 153.
(3) Townsend, L.B.; Wise, D.S. Parasitology Today 6, 4 (1990) 107-112.
(4) Grenda, V. J.; Jones, R.E; Gal,G.; Sletzinger J. Org Chem. 30 (1965), 259-261.
(5) Shen, M.; Driver, T.G. Org Lett. 2008, 10, 3367.
^Gilman, A.G., T.W. Rall, A.S. Nies and P. Taylor (eds.). Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 8th ed. New York, NY. Pergamon Press, 1990., p. 970
^Rosenblum, C (March 1977). “Non-Drug-Related Residues in Tracer Studies”. Journal of Toxicology and Environmental Health. 2 (4): 803–14. doi:10.1080/15287397709529480. PMID853540.
^Sax, N.I. Dangerous Properties of Industrial Materials. Vol 1-3 7th ed. New York, NY: Van Nostrand Reinhold, 1989., p. 3251
^Setzinger, Meyer; Painfield, North; Gaines, Water A.; Grenda, Victor J. (1965). “Novel Preparation of Benzimidazoles from N-Arylamidines. New Synthesis of Thiabendazole1”. The Journal of Organic Chemistry. 30: 259–261. doi:10.1021/jo01012a061.
^Brown, H. D.; Matzuk, A. R.; Ilves, I. R.; Peterson, L. H.; Harris, S. A.; Sarett, L. H.; Egerton, J. R.; Yakstis, J. J.; Campbell, W. C.; Cuckler, A. C. (1961). “Antiparasitic Drugs. Iv. 2-(4′-Thiazolyl)-Benzimidazole, A New Anthelmintic”. Journal of the American Chemical Society. 83 (7): 1764–1765. doi:10.1021/ja01468a052.
^Tocco, D. J.; Buhs, R. P.; Brown, H. D.; Matzuk, A. R.; Mertel, H. E.; Harman, R. E.; Trenner, N. R. (1964). “The Metabolic Fate of Thiabendazole in Sheep1”. Journal of Medicinal Chemistry. 7 (4): 399–405. doi:10.1021/jm00334a002.
^Hoff, D. R.; Fisher, M. H.; Bochis, R. J.; Lusi, A.; Waksmunski, F.; Egerton, J. R.; Yakstis, J. J.; Cuckler, A. C.; Campbell, W. C. (1970). “A new broad-spectrum anthelmintic: 2-(4-Thiazolyl)-5-isopropoxycarbonylamino-benzimidazole”. Experientia. 26 (5): 550–551. doi:10.1007/BF01898506.
^Chronicles of Drug Discovery, Book 1, pp 239-256.
Elapegademase is a PEGylated recombinant adenosine deaminase. It can be defined molecularly as a genetically modified bovine adenosine deaminase with a modification in cysteine 74 for serine and with about 13 methoxy polyethylene glycol chains bound via carbonyl group in alanine and lysine residues.[4] Elapegademase is generated in E. coli, developed by Leadiant Biosciences and FDA approved on October 5, 2018.[1, 5]
Indication
Elapegademase is approved for the treatment of adenosine deaminase severe combined immune deficiency (ADA-SCID) in pediatric and adult patients.[1] This condition was previously treated by the use of pegamedase bovine as part of an enzyme replacement therapy.[2]
ADA-SCID is a genetically inherited disorder that is very rare and characterized by a deficiency in the adenosine deaminase enzyme. The patients suffering from this disease often present a compromised immune system. This condition is characterized by very low levels of white blood cells and immunoglobulin levels which results in severe and recurring infections.[3]
Pharmacodynamics
In clinical trials, elapegademase was shown to increase adenosine deaminase activity while reducing the concentrations of toxic metabolites which are the hallmark of ADA-SCID. As well, it was shown to improve the total lymphocyte count.[6]
Mechanism of action
The ADA-SCID is caused by the presence of mutations in the ADA gene which is responsible for the synthesis of adenosine deaminase. This enzyme is found throughout the body but it is mainly active in lymphocytes. The normal function of adenosine deaminase is to eliminate deoxyadenosine, created when DNA is degraded, by converting it into deoxyinosine. This degradation process is very important as deoxyadenosine is cytotoxic, especially for lymphocytes. Immature lymphocytes are particularly vulnerable as deoxyadenosine kills them before maturation making them unable to produce their immune function.[3]
Therefore, based on the causes of ADA-SCID, elapegademase works by supplementing the levels of adenosine deaminase. Being a recombinant and an E. coli-produced molecule, the use of this drug eliminates the need to source the enzyme from animals, as it was used previously.[1]
Absorption
Elapegademase is administered intramuscularly and the reported Tmax, Cmax and AUC are approximately 60 hours, 240 mmol.h/L and 33000 hr.mmol/L as reported during a week.[Label]
Volume of distribution
This pharmacokinetic property has not been fully studied.
Protein binding
This pharmacokinetic property is not significant as the main effect is in the blood cells.
Metabolism
Metabolism studies have not been performed but it is thought to be degraded by proteases to small peptides and individual amino acids.
Route of elimination
This pharmacokinetic property has not been fully studied.
Half life
This pharmacokinetic property has not been fully studied.
Clearance
This pharmacokinetic property has not been fully studied.
Toxicity
As elapegademase is a therapeutic protein, there is a potential risk of immunogenicity.
There are no studies related to overdose but the highest weekly prescribed dose in clinical trials was 0.4 mg/kg. In nonclinical studies, a dosage of 1.8 fold of the clinical dose produced a slight increase in the activated partial thromboplastin time.[Label]
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
Ogeda (formerly Euroscreen ) is developing fezolinetant, an NK3 antagonist, for treating endometriosis, benign prostate hyperplasia, polycystic ovary syndrome, uterine fibroids and hot flashes. In November 2018, drug was listed under phase II development for PCOS, uterine fibroids and hot flashes in company’s pipeline. In October 2018, the company was proceeding to phase III study preparation, and regulatory filings were expected in 2021 or later .
Unlike GnRH modulators, but similarly to estrogens, NK3 receptor antagonists including fezolinetant and MLE-4901 (also known as AZD-4901, formerly AZD-2624) have been found to alleviate hot flashes in menopausal women.[6][7] This would seem to be independent of their actions on the hypothalamic–pituitary–gonadal axis and hence on sex hormone production.[6][7] NK3 receptor antagonists are anticipated as a useful clinical alternative to estrogens for management of hot flashes, but with potentially reduced risks and side effects.[6][7]
PATENT
WO2011121137
hold protection in most of the EU states until 2031 and expire in the US in 2031.
PATENT
US 20170095472
PATENT
WO2016146712
PATENT
WO-2019012033
Novel deuterated analogs of fezolinetant , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating pain, convulsion, obesity, inflammatory disease including irritable bowel syndrome, emesis, asthma, cough, urinary incontinence, reproduction disorders, testicular cancer and breast cancer. Further claims are processes for the preparation of fezolinetant. claiming use of NK3R antagonist eg fezolinetant, for treating pathological excess body fat or prevention of obesity.
Fezolinetant was developed as selective antagonist of NK-3 receptor and is useful as therapeutic compound, particularly in the treatment and/or prevention of sex-hormone dependent diseases. Fezolinetant corresponds to (R)-(4-fluorophenyl)-(8-methyl-3-(3-memyl-l,2,4-miacMazol-5-yl)-5,6-dmy(ko-[l,2,4]trizolo[4,3-a]pyrazin-7(8H)-yl)methanone and is described in WO2014/154895.
Drug-drug interactions are the most common type of drug interactions. They can decrease how well the medications works, may cause serious unexpected side effects, or even increase the blood level and possible toxicity of a certain drug.
Drug interaction may occur by pharmacokinetic interaction, during which one drug affects another drug’s absorption, distribution, metabolism, or excretion. Regarding metabolism, it should be noted that drugs are usually eliminated from the body as either the unchanged drug or as a metabolite. Enzymes in the liver, usually the cytochrome P450s (CYPs) enzymes, are often responsible for metabolizing drugs. Therefore, determining the CYP profile of a drug is of high relevancy to determine if it will affect the activity of CYPs and thus if it may lead to drug-drug interactions.The five most relevant CYPs for drug-drug interaction are CYP3A4, 2C9, 2C19, 1A2 and 2D6, among which isoforms 3A4, 2C9 and 2C19 are the major ones. The less a drug inhibits these CYPs, the less drug-drug interactions would be expected.
Therefore, it is important to provide drugs that present the safest CYP profile in order to minimize as much as possible the potential risks of drug-drug interactions.Even if fezolinetant possesses a good CYP profile, providing analogs of fezolinetant with a further improved CYP profile would be valuable for patients.
In a completely unexpected way, the Applicant evidenced that deuteration of fezolinetant provides a further improved CYP profile, especially on isoforms CYP 2C9 and 2C19. This was evidenced for the deuterated form (R)-(4-fluorophenyl)-(8-methyl-3-(3-(memyl-d.?)-l,2,4-miacttazol-5-y ^yl)methanone, hereafter referred to as “deuterated fezolinetant”.
Importantly, deuterated fezolinetant retains the biological activity of fezolinetant as well as its lipophilic efficiency.
Deuterated fezolinetant also presents the advantage to enable improvement of the in vivo half -life of the drug. For example, half -life is increased by a factor 2 in castrated monkeys, compared to fezolinetant.
Synthetic scheme
Deuterated fezolinetant may be synthesized using the methodology described following schemes (Part A and Part B):
Part A: Preparation of deuterated key intermediate (ii)
Part B: Synthesis of deuterated fezolinetant using intermediate (ii)
Synthesis of deuterated fezolinetant was performed through key intermediate (ii). Part A corresponds to the synthesis of intermediate (ii). Part B leads to deuterated fezolinetant (d3-fezolinetant), using intermediate (ii), using procedures adapted from WO2014/154895.
Experimental details
Part A – Step 1): Formation of d3-acetamide (b)
To i¾-acetic acid (a) (10 g, 1 equiv.) in DCM (100 mL) CDI (25.3 g, 1 equiv.) was added and the resultant mixture stirred at RT for 30 min, thereupon ammonia gas was bubbled through the reaction mixture for 40 min at 0-5 °C. Thereafter the bubbling was stopped, the mixture was filtered and the filtrate was evaporated under reduced pressure to give 30.95 g crude product that was purified using flash chromatography on silica to furnish 6.65 g (yield: 73 %) deuterated acetamide (b) was obtained (GC (column RTX-1301 30 m x 0.32 mm x 0.5 μπι) Rt 7.4 min, 98 %).
Part A – Step 2): Ring closure leading to compound (c)
<¾-Acetamide (b) (3.3 g, 1 equiv.) and chlorocarbonylsulfenyl chloride (CCSC) (8.4 g, 1.2 equiv.) were combined in 1,2-dichloroethane (63 mL), and refluxed for 4.5 h. CCSC can be prepared as per the procedure described in Adeppa et al. (Synth. Commun., 2012, Vol. 42, pp. 714-721). The volatiles were then removed to obtain 6.60 g (102 % yield) oxathiazolone (c) product as a yellow oil. The product was analyzed by GC (Rt= 7.8 min, 97 ). 13C NMR (CDC13): 16.0, 158.7, 174.4 ppm.
Part A – Step 3): formation of compound (d)
To oxathiazolone (c) (6.6 g, 1 equiv) in rn-xylene (231 mL) methyl cyanoformate (14.70 g, 3.2 equiv.) was added. The mixture was stirred at 130 °C for 19 h and thereafter the volatiles removed under reduced pressure at 50 °C to obtain 4.53 g brown oil (yield: 51 %). The product (d) was analyzed by GC (Rt = 11.8 min, 81 %) and mass spectrometry (M+H = 162).
Part A – Step 4): formation of intermediate (ii)
The ester (d) obtained above (3.65 g, lequiv.) was dissolved in ethanol (45 mL). The undissolved material was filtered off then hydrazine hydrate (2.3 mL, 1.15 equiv. 55w/w in H20) was added to the stirred solution. Thick suspension formed in minutes, the suspension was stirred for 45 min, filtered and washed with EtOH (3 mL) to furnish intermediate (ii) a pale yellow solid (2.43 g, 55 % yield). Mass spectrometry (M+H = 162, M+Na = 184); ¾ NMR (cfe-DMSO): 4.79 ppm (br s, 2H), 10.55 ppm (br s, 1H); 13C NMR (fife-DMSO): 17.4 ppm, 155.6 ppm, 173.4 ppm, 183.0 ppm.
Part B – Step a): formation of compound (iii)
Intermediate (i) was prepared as described in WO2014/154895.
Intermediate (ii) (490 mg, 3.04 mmol) and compound (i) (1.0 g (87 mol 1.3 content), 2.97 mmol) were taken up in MeOH and the reaction mixture was stirred at a temperature ranging from 55°C to 70°C for a period of time ranging from 6 hours to 8 hours. The reaction was deemed complete by TLC. The reaction mixture was evaporated and the crude product was purified by flash chromatography on silica in DCM : MeOH eluent to afford 1.13 g (97 % yield) of compound (iii) as a yellow oil. JH NMR (CDC13): δ (ppm) 7.26 (d, 1H), 6.48-6.49 (2H), 4.50 (m, 1H), 4.30 (m, 1H), 4.09 (m, 1H), 3.94 (d, 1H), 3.80 (s, 6H), 3.61 (d, 1H), 3.22 (m, 1H), 2.75 (m, 1H), 1.72 (d, 3H); Mass spectrometry (M+H = 390, 2M+Na = 801). Chiral LC (column: Chiralpak IC, 250 x 4.6 mm – eluent: MTBE MeOH DEA 98/2/0.1) 99.84 .
Part B – Step b): deprotection leading to compound (iv)
Intermediate (iii) prepared above (1.05 g, 2.7 mmol) was dissolved in DCM and washed with aq. NaOH. The organic phase was dried, then TFA (1.56 mL, 2.3 g, 7.5 equiv.) was added at RT. The resulting solution was stirred at RT for 2 h. The reaction was monitored by TLC. After completion of the reaction water was added to the reaction mixture, and the precipitate filtered and washed with water. The phases were separated, the pH of the aq. phase was adjusted to pH 13 by addition of 20 % aq. NaOH. NaCl was then added to the aqueous solution that was then extracted with DCM. The organic phase was evaporated under reduced pressure to give 504 mg of compound (iv) (78 % yield). ¾ NMR (cfe-DMSO): δ (ppm) 4.42 (m, 1H), 4.10 (m, 2H), 3.0 (m, 1H), 2.82 (m, 1H), 1.46 (d, 3H). 13C NMR (rf6-DMSO): δ (ppm) 174.8, 173.4, 156.2, 145.0, 48.1, 45.7, 40.7, 19.1. Mass spectrometry (M+H = 240, 2M+Na = 501).
Part B – Step c): acylation and recrystallization to form deuterated fezolinetant
Intermediate (iv) (450 mg, 1.88 mmol) was dissolved in DCM, then sat. aq. NaHC03 was added and the mixture was stirred for 30 min. To this mixture 4-fluorobenzoyl chloride (v) (220 1 equiv.) was added dropwise at RT. The reaction was stirred for a period of time ranging from about 20 min to overnight at RT and reaction progress monitored by TLC. After completion the phases were separated, the organic phase was washed with water, dried over MgS04, filtered and evaporated under reduced pressure to give 745 mg crude <i3-fezolinetant (110 % yield). The crude product was purified by flash chromatography using MeOH : DCM together with a second batch, then
crystallized (EtOH H20) before final analysis. ¾ NMR (d6-DMSO): δ (ppm) 7.60 (m, 2H), 7.33 (m, 2H), 5.73 (m, 1H), 4.68 (dd, 1H), 4.31 (m, 1H), 4.06 (m, 1H), 3.65 (m, 1H), 1.61 (d, 3H). 13C NMR (d6-DMSO): δ (ppm) 174.4, 173.5, 168.7, 163.7, 161.8, 154.1, 144.9, 131.6, 129.5, 115.5, 44.7, 18.7. Isotopic purity based on an intense molecular ion observed at m/z = 362.2 Da is estimated as approximately 100 % isotopic purity. Chiral purity (LC) (column: Chiralpak IC, 250 x 4.6 mm – eluent: n-hexane/EtOH DEA 80/20/0.1) >99.9 %. A single crystal X-ray structure of the deuterated fezolinetant final product was obtained (Figure 1) that confirmed the structure of the compound as well as the stereochemistry.
^ Jump up to:abFraser GL, Ramael S, Hoveyda HR, Gheyle L, Combalbert J (2016). “The NK3 Receptor Antagonist ESN364 Suppresses Sex Hormones in Men and Women”. J. Clin. Endocrinol. Metab. 101 (2): 417–26. doi:10.1210/jc.2015-3621. PMID26653113.
^ Jump up to:abFraser GL, Hoveyda HR, Clarke IJ, Ramaswamy S, Plant TM, Rose C, Millar RP (2015). “The NK3 Receptor Antagonist ESN364 Interrupts Pulsatile LH Secretion and Moderates Levels of Ovarian Hormones Throughout the Menstrual Cycle”. Endocrinology. 156 (11): 4214–25. doi:10.1210/en.2015-1409. PMID26305889.
NOVEL N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS, PHARMACEUTICAL COMPOSITION, METHODS FOR USE IN NK-3 RECEPTOR-MEDIATED DISORDERS
NOVEL N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS, PHARMACEUTICAL COMPOSITION, METHODS FOR USE IN NK-3 RECEPTOR-MEDIATED DISORDERS
NOVEL N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS, PHARMACEUTICAL COMPOSITION, METHODS FOR USE IN NK-3 RECEPTOR-MEDIATED DISORDERS
////////////////Fezolinetant, ESN-364, фезолинетант, فيزولينيتانت, 非唑奈坦, Phase II, Hot flashes, Polycystic ovary syndrome, Uterine leiomyoma, Euroscreen, Ogeda, FDA 2023, APPROVALS 2023, Veozah
Smiles
C[C@H]1N(CCn2c1nnc2c3nc(C)ns3)C(=O)c4ccc(F)cc4
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
Asparaginase (Escherichia coli isoenzyme II), conjugate with alpha-(((2,5-dioxo-1-pyrrolidinyl)oxy)carbonyl)-omega-methoxypoly(oxy-1,2-ethanediyl)
List Acronyms
Peptide
Calaspargase pegol
calaspargase pegol-mknl
EZN-2285
Used to treat acute lymphoblastic leukemia., Antineoplastic
BAX-2303
SC-PEG E. Coli L-asparaginase
SHP-663
Calaspargase pegol-mknl (trade name Asparlas) is a drug for the treatment of acute lymphoblastic leukemia (ALL). It is approved by the Food and Drug Administration for use in the United States as a component of a multi-agent chemotherapeutic regimen for ALL in pediatric and young adult patients aged 1 month to 21 years.[1]
Calaspargase pegol was first approved in 2018 in the U.S. as part of a multi-agent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia.
In 2008, orphan drug designation was assigned in the E.U.
Calaspargase pegol is an engineered protein consisting of the E. coli-derived enzyme L-asparaginase II conjugated with succinimidyl carbonate monomethoxypolyethylene glycol (pegol).[2] The L-asparaginase portion hydrolyzesL-asparagine to L-aspartic acid depriving the tumor cell of the L-asparagine it needs for survival.[2] The conjugation with the pegol group increases the half-life of the drug making it longer acting.
Asparaginase is an important agent used to treat acute lymphoblastic leukemia (ALL) [1]. Asparagine is incorporated into most proteins, and the synthesis of proteins is stopped when asparagine is absent, which inhibits RNA and DNA synthesis, resulting in a halt in cellular proliferation. This forms the basis of asparaginase treatment in ALL [1], [2], [6].
Calaspargase pegol, also known as asparlas, is an asparagine specific enzyme which is indicated as a part of a multi-agent chemotherapy regimen for the treatment of ALL [3]. The asparagine specific enzyme is derived from Escherichia coli, as a conjugate of L-asparaginase (L-asparagine amidohydrolase) and monomethoxypolyethylene glycol (mPEG) with a succinimidyl carbonate (SC) linker to create a stable molecule which increases the half-life and decreases the dosing frequency [Label], [1].
Calaspargase pegol, by Shire pharmaceuticals, was approved by the FDA on December 20, 2018 for acute lymphoblastic anemia (ALL) [3].
Indication
This drug is is an asparagine specific enzyme indicated as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia in pediatric and young adult patients age 1 month to 21 years [Label].
The pharmacokinetics of calaspargase pegol were examined when given in combination with multiagent chemotherapy in 124 patients with B-cell lineage ALL [3]. The FDA approval of this drug was based on the achievement and maintenance of nadir serum asparaginase activity above the level of 0.1 U/mL when administering calaspargase, 2500 U/m2 intravenously, at 3-week intervals.
The effect of this drug is believed to occur by selective killing of leukemic cells due to depletion of plasma L-asparagine. Leukemic cells with low expression of asparagine synthetase are less capable of producing L-asparagine, and therefore rely on exogenous L-asparagine for survival [Label]. When asparagine is depleted, tumor cells cannot proliferate [6].
During remission induction, one dose of SC-PEG (2500 IU/m2) results in a sustained therapeutic serum asparaginase activity (SAA) without excessive toxicity or marked differences in the proportion of patients with low end-induction minimum residual disease (MRD) [5].
Pharmacodynamic (PD) response was studied through measurement of plasma and cerebrospinal fluid (CSF) asparagine concentrations with an LC-MS/MS assay (liquid chromatography–mass spectrometry). Asparagine concentration in plasma was sustained below the assay limit of quantification for more than 18 days after one dose of calaspargase pegol, 2,500 U/m2, during the induction phase of treatment. Average cerebrospinal asparagine concentrations decreased from a pretreatment concentration of 0.8 μg/mL (N=10) to 0.2 μg/mL on Day 4 (N=37) and stayed decreased at 0.2 μg/mL (N=35) 25 days after the administration of one of 2,500 U/m2 in the induction phase [Label].
Mechanism of action
L-asparaginase (the main component of this drug) is an enzyme that catalyzes the conversion of the amino acid L-asparagine into both aspartic acid and ammonia [Label], [2]. This process depletes malignant cells of their required asparagine. The depletion of asparagine then blocks protein synthesis and tumor cell proliferation, especially in the G1 phase of the cell cycle. As a result, tumor cell death occurs. Asparagine is important in protein synthesis in acute lymphoblastic leukemia (ALL) cells which, unlike normal cells, cannot produce this amino acid due to lack of the enzyme asparagine synthase[2], [Label].
Pegylation decreases enzyme antigenicity and increases its half-life. Succinimidyl carbamate (SC) is used as a PEG linker to facilitate attachment to asparaginase and enhances the stability of the formulation [4], [1]. SC-PEG urethane linkages formed with lysine groups are more hydrolytically stable [2].
Toxicity
Pancreatitis, hepatotoxicity, hemorrhage, and thrombosis have been observed with calaspargase pegol use [Label].
Pancreatitis: Discontinue this drug in patients with pancreatitis, and monitor blood glucose.
Hepatotoxicity: Hepatic function should be tested regularly, and trough levels of this drug should be measured during the recovery phase of the drug cycle [Label].
Hemorrhage or Thrombosis: Discontinue this drug in serious or life-threatening hemorrhage or thrombosis. In cases of hemorrhage, identify the cause of hemorrhage and treat appropriately. Administer anticoagulant therapy as indicated in thrombotic events [Label].
A note on hypersensitivity:
Observe the patient for 1 hour after administration of calaspargase pegol for possible hypersensitivity [Label]. In cases of previous hypersensitivity to this drug, discontinue this drug immediately.
Lactation: Advise women not to breastfeed while taking this drug [Label].
Pregnancy: There are no available data on the use of calaspargase pegol in pregnant women to confirm a risk of drug-associated major birth defects and miscarriage. Published literature studies in pregnant animals suggest asparagine depletion can cause harm to the animal offspring. It is therefore advisable to inform women of childbearing age of this risk. The background risk of major birth defects and miscarriage for humans is unknown at this time [Label].
Pregnancy testing should occur before initiating treatment. Advise females of reproductive potential to avoid becoming pregnant while taking this drug. Females should use effective contraceptive methods, including a barrier methods, during treatment and for at least 3 months after the last dose. There is a risk for an interaction between calaspargase pegol and oral contraceptives. The concurrent use of this drug with oral contraceptives should be avoided. Other non-oral contraceptive methods should be used in women of childbearing potential [Label].
References
Angiolillo AL, Schore RJ, Devidas M, Borowitz MJ, Carroll AJ, Gastier-Foster JM, Heerema NA, Keilani T, Lane AR, Loh ML, Reaman GH, Adamson PC, Wood B, Wood C, Zheng HW, Raetz EA, Winick NJ, Carroll WL, Hunger SP: Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: results from Children’s Oncology Group Study AALL07P4. J Clin Oncol. 2014 Dec 1;32(34):3874-82. doi: 10.1200/JCO.2014.55.5763. Epub 2014 Oct 27. [PubMed:25348002]
Appel IM, Kazemier KM, Boos J, Lanvers C, Huijmans J, Veerman AJ, van Wering E, den Boer ML, Pieters R: Pharmacokinetic, pharmacodynamic and intracellular effects of PEG-asparaginase in newly diagnosed childhood acute lymphoblastic leukemia: results from a single agent window study. Leukemia. 2008 Sep;22(9):1665-79. doi: 10.1038/leu.2008.165. Epub 2008 Jun 26. [PubMed:18580955]
Blood Journal: Randomized Study of Pegaspargase (SS-PEG) and Calaspargase Pegol (SPC-PEG) in Pediatric Patients with Newly Diagnosed Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma: Results of DFCI ALL Consortium Protocol 11-001 [Link]
Angiolillo AL, Schore RJ, Devidas M, Borowitz MJ, Carroll AJ, Gastier-Foster JM, Heerema NA, Keilani T, Lane AR, Loh ML, Reaman GH, Adamson PC, Wood B, Wood C, Zheng HW, Raetz EA, Winick NJ, Carroll WL, Hunger SP: Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: results from Children’s Oncology Group Study AALL07P4. J Clin Oncol. 2014 Dec 1;32(34):3874-82. doi: 10.1200/JCO.2014.55.5763. Epub 2014 Oct 27. [PubMed:25348002]
Appel IM, Kazemier KM, Boos J, Lanvers C, Huijmans J, Veerman AJ, van Wering E, den Boer ML, Pieters R: Pharmacokinetic, pharmacodynamic and intracellular effects of PEG-asparaginase in newly diagnosed childhood acute lymphoblastic leukemia: results from a single agent window study. Leukemia. 2008 Sep;22(9):1665-79. doi: 10.1038/leu.2008.165. Epub 2008 Jun 26. [PubMed:18580955]
Blood Journal: Randomized Study of Pegaspargase (SS-PEG) and Calaspargase Pegol (SPC-PEG) in Pediatric Patients with Newly Diagnosed Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma: Results of DFCI ALL Consortium Protocol 11-001 [Link]
5′-(O-(2-hydroxy-3-((1-oxohexadecyl)amino)propyl)phosphorothioate)-d(3′-amino-3′-deoxy-p-thio)(t-a-g-g-g-t-t-a-g-a-c-a-a), sodium salt (13)
DNA, d(3′-amino-3′-deoxy-p-thio)(T-A-G-G-G-T-T-A-G-A-C-A-A), 5′-(o-(2-hydroxy-3-((1-oxohexadecyl)amino)propyl) hydrogen phosphorothioate), sodium salt (1:13)
UNII-2AW48LAZ4I, Antineoplastic
In 2014, Geron entered into an exclusive worldwide license and collaboration agreement with Janssen Biotech for the treatment of hematologic cancers. However, in 2018, the agreement was terminated and Geron regained global rights to the product.
In 2015, imetelstat was granted orphan drug status in the U.S. for the treatment of myelodysplastic syndrome, as well as in both the U.S. and the E.U. for the treatment of myelofibrosis. In 2017, fast track designation was received in the U.S. for the treatment of adult patients with transfusion-dependent anemia due to low or intermediate-1 risk myelodysplastic syndromes (MDS) who are non-del(5q) and who are refractory or resistant to treatment with an erythropoiesis stimulating agent (ESA).
Imetelstat Sodium is the sodium salt of imetelstat, a synthetic lipid-conjugated, 13-mer oligonucleotide N3′ P5′-thio-phosphoramidate with potential antineoplastic activity. Complementary to the template region of telomerase RNA (hTR), imetelstat acts as a competitive enzyme inhibitor that binds and blocks the active site of the enzyme (a telomerase template antagonist), a mechanism of action which differs from that for the antisense oligonucleotide-mediated inhibition of telomerase activity through telomerase mRNA binding. Inhibition of telomerase activity in tumor cells by imetelstat results in telomere shortening, which leads to cell cycle arrest or apoptosis.
Imetelstat sodium, a lipid-based conjugate of Geron’s first-generation anticancer drug, GRN-163, is in phase III clinical trials at Geron for the treatment of myelodysplastic syndrome, as well as in phase II for the treatment of myelofibrosis.
Geron is developing imetelstat, a lipid-conjugated 13-mer thiophosphoramidate oligonucleotide and the lead in a series of telomerase inhibitors, for treating hematological malignancies, primarily myelofibrosis.
Imetelstat, a first-in-class telomerase inhibitor and our sole product candidate, is being developed for the potential treatment of hematologic myeloid malignancies. Imetelstat is currently in two clinical trials being conducted by Janssen under the terms of an exclusive worldwide collaboration and license agreement.
Originally known as GRN163L, imetelstat sodium (imetelstat) is a 13-mer N3’—P5’ thio-phosphoramidate (NPS) oligonucleotide that has a covalently bound 5’ palmitoyl (C16) lipid group. The proprietary nucleic acid backbone provides resistance to the effect of cellular nucleases, thus conferring improved stability in plasma and tissues, as well as significantly improved binding affinity to its target. The lipid group enhances cell permeability to increase potency and improve pharmacokinetic and pharmacodynamic properties. The compound has a long residence time in bone marrow, spleen and liver. Imetelstat binds with high affinity to the template region of the RNA component of telomerase, resulting in direct, competitive inhibition of telomerase enzymatic activity, rather than elicit its effect through an antisense inhibition of protein translation. Imetelstat is administered by intravenous infusion.
Preclinical Studies with Imetelstat
A series of preclinical efficacy studies of imetelstat have been conducted by Geron scientists and academic collaborators. These data showed that imetelstat:
Inhibits telomerase activity, and can shorten telomeres.
Inhibits the proliferation of a wide variety of tumor types, including solid and hematologic, in cell culture systems and rodent xenograft models of human cancers, impacting the growth of primary tumors and reducing metastases.
Inhibits the proliferation of malignant progenitor cells from hematologic cancers, such as multiple myeloma, myeloproliferative neoplasms and acute myelogenous leukemia.
Has additive or synergistic anti-tumor effect in a variety of cell culture systems and xenograft models when administered in combination with approved anti-cancer therapies, including radiation, conventional chemotherapies and targeted agents.
Clinical Experience with Imetelstat
Over 500 patients have been enrolled and treated in imetelstat clinical trials.
PHASE 1
Six clinical trials evaluated the safety, tolerability, pharmacokinetics and pharmacodynamics both as a single agent and in combination with standard therapies in patients with solid tumors and hematologic malignancies:
Single agent studies of imetelstat were in patients with advanced solid tumors, multiple myeloma and chronic lymphoproliferative diseases. Combination studies with imetelstat were with bortezomib in patients with relapsed or refractory multiple myeloma, with paclitaxel and bevacizumab in patients with metastatic breast cancer, and with carboplatin and paclitaxel in patients with advanced non-small cell lung cancer (NSCLC).
Doses ranging from 0.5 mg/kg to 11.7 mg/kg were tested in a variety of dosing schedules ranging from weekly to once every 28 days.
The human pharmacokinetic profile was characterized in clinical trials of patients with solid tumors and chronic lymphoproliferative diseases. Single-dose kinetics showed dose-dependent increases in exposure with a plasma half-life (t1/2) ranging from 4-5 hours. Residence time in bone marrow is long (0.19-0.51 µM observed at 41-45 hours post 7.5 mg/kg dose).
Telomerase inhibition was observed in various tissues where the enzymes’s activity was measurable.
PHASE 2
Imetelstat was studied in two randomized clinical trials, two single arm proof-of-concept studies and an investigator sponsored pilot study:
Randomized trials were in combination with paclitaxel in patients with metastatic breast cancer and as maintenance treatment following a platinum-containing chemotherapy regimen in patients with NSCLC.
Single arm studies were as a single agent or in combination with lenalidomide in patients with multiple myeloma and as a single agent in essential thrombocythemia (ET) or polycythemia vera (PV).
The safety profile of imetelstat across the Phase 1 and 2 trials has been generally consistent. Reported adverse events (AEs) and laboratory investigations associated with imetelstat administration included cytopenias, transient prolonged activated partial thromboplastin time (aPTT; assessed only in Phase 1 trials), gastrointestinal symptoms, constitutional symptoms, hepatic biochemistry abnormalities, and infusion reactions. Dose limiting toxicities include thrombocytopenia and neutropenia.
A Focus on Hematologic Myeloid Malignancies
Early clinical data from the Phase 2 clinical trial in ET and the investigator sponsored pilot study in MF suggest imetelstat may have disease-modifying activity by suppressing the proliferation of malignant progenitor cell clones for the underlying diseases, and potentially allowing recovery of normal hematopoiesis in patients with hematologic myeloid malignancies.
Results from these trials were published in the New England Journal of Medicine:
Imetelstat is currently being tested in two clinical trials: IMbark, a Phase 2 trial in myelofibrosis (MF), and IMerge, a Phase 2/3 trial in myelodysplastic syndromes (MDS).
IMbark
IMbark is the ongoing Phase 2 clinical trial to evaluate two doses of imetelstat in intermediate-2 or high-risk MF patients who are refractory to or have relapsed after treatment with a JAK inhibitor.
Internal data reviews were completed in September 2016, April 2017 and March 2018. The safety profile was consistent with prior clinical trials of imetelstat in hematologic malignancies, and no new safety signals were identified. The data supported 9.4 mg/kg as an appropriate starting dose in the trial, but an insufficient number of patients met the protocol defined interim efficacy criteria and new patient enrollment was suspended in October 2016. As of January 2018, median follow up was approximately 19 months, and median overall survival had not been reached in either dosing arm. In March 2018, the trial was closed to new patient enrollment. Patients who remain in the treatment phase of the trial may continue to receive imetelstat, and until the protocol-specified primary analysis, all safety and efficacy assessments are being conducted as planned in the protocol, including following patients, to the extent possible, until death, to enable an assessment of overall survival.
IMerge
IMerge is the ongoing two-part Phase 2/3 clinical trial of imetelstat in red blood cell (RBC) transfusion-dependent patients with lower risk MDS who are refractory or resistant to treatment with an erythropoiesis stimulating agent (ESA). Part 1 is a Phase 2, open-label, single-arm trial of imetelstat administered as a single agent by intravenous infusion, and is ongoing. Part 2 is designed to be a Phase 3, randomized, controlled trial, and has not been initiated.
The data showed that among the subset of 13 patients who had not received prior treatment with either lenalidomide or a hypomethylating agent (HMA) and did not have a deletion 5q chromosomal abnormality (non-del(5q)), 54% achieved RBC transfusion-independence (TI) lasting at least 8 weeks, including 31% who achieved a 24-week RBC-TI. In the overall trial population, the rates of 8- and 24-week RBC-TI were 38% and 16%, respectively. Cytopenias, particularly neutropenia and thrombocytopenia, were the most frequently reported adverse events, which were predictable, manageable and reversible.
Based on the preliminary data from the 13-patient subset, Janssen expanded Part 1 of IMerge to enroll approximately 20 additional patients who were naïve to lenalidomide and HMA treatment and non-del(5q) to increase the experience and confirm the benefit-risk profile of imetelstat in this refined target patient population
Patients of acute myeloid leukemia (AML) have limited treatment options at diagnosis; treatment typically takes the form of chemotherapy to quickly reduce the leukemic cell burden. Invasive leukapheresis procedures to remove large numbers of leukocytes (normal and diseased) may be applied in parallel to chemotherapy to temporarily lower tumor cell burden. Induction phase chemotherapy can be successful but, most healthy cells residing in patient bone marrow are also killed, causing illness and requiring additional palliative therapy to ward off infection and raise leukocyte counts. Additional rounds of chemotherapy can be used in an attempt to keep patients in remission; but relapse is common.
[0005] Telomerase is present in over 90% of tumors across all cancer types; and is lacking in normal, healthy tissues. Imetelstat sodium is a novel, first-in-class telomerase inhibitor that is a covalently-lipidated 13-mer oligonucleotide (shown below) complimentary to the human telomerase RNA (hTR) template region. Imetelstat sodium does not function through an anti-sense mechanism and therefore lacks the side effects commonly observed with such therapies. Imetelstat sodium is the sodium salt of imetelstat (shown below):
Imetelstat sodium
Unless otherwise indicated or clear from the context, references below to imetelstat also include salts thereof. As mentioned above, imetelstat sodium in particular is the sodium salt of imetelstat.
[0006] ABT-199/venetoclax (trade name Venclexta) is an FDA approved Bcl-2 inhibitor for use in chronic lymphocytic leukemia (CLL) patients with dell7p who are relapsed/refractory. ABT-199 is also known as ABT 199, GDC0199, GDC-0199 or RG7601. The chemical name for ABT-199 is 4-[4-[[2-(4-chlorophenyl)-4,4-dimethylcyclohexen-l-yl]methyl]piperazin-l-yl]-N-[3-nitro-4-(oxan-4-ylmethylamino)phenyl]sulfonyl-2-(lH-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (Cas No. 1257044-40-8). Unless otherwise indicated or clear from the context, references below to ABT-199 also include pharmaceutically acceptable salts thereof. Specifically in the Examples however, ABT-199 was used in the free base form.
[0007] ABT-199, shown below in the free base form, is highly specific to Bcl-2, unlike other first generation inhibitors which show affinity for related Bel family members and induce greater side effects. Inhibition of Bcl-2 blocks the pro-apoptotic signals caused by damage to or abnormalities within cellular DNA and ultimately leads to programmed cell death in treated cells via the caspase cascade and apoptosis through the intrinsic pathway.
Improved process for preparing imetelstat . claiming use of a combination comprising a telomerase inhibitor, specifically imetelstat sodium and a Bcl-2 inhibitor, specifically ABT-199 for treating hematological cancer such as acute myeloid leukemia, essential thrombocythemia and polycythemia vera, specifically acute myeloid leukemia.
Imetelstat (SEQ ID NO: 1 ) is a N3′- P5′ thiophosphoramidate oligonucleotide covalently linked to a palmitoyl lipid moiety and has been described in WO-2005/023994 as compound (1 F). The sodium salt of imetelstat acts as a potent and specific telomerase inhibitor and can be used to treat telomerase-mediated disorders, e.g. cancer, including disorders such as myelofibrosis (MF), myelodysplastic syndromes (MDS) and acute myelogenous leukemia (AML).
The structure of imetelstat sodium is shown below :
The structure of imetelstat can also be represented as shown below
imetelstat
The LPT group represents the palmitoyi lipid that is covalently linked to the N3′- P5′ thiophosphor-amidate oligonucleotide. The base sequence of the thirteen nucleotides is as follows :
TAGGGTTAGACAA and is represented by the bases B1 to B13. The -NH-P(=S)(OH)-and -0-P(=S)(OH)- groups of the structure can occur in a salt form. It is understood that salt forms of a subject compound are encompassed by the structures depicted herein, even if not specifically indicated.
Imetelstat sodium can also be represented as follows
o H
LPT = CH3-(CH2)i4-C-N-CH2-(CHOH)-CH2-
The -NH-P(=S)(OH)- group and the thymine, adenine, guanine and cytosine bases can occur in other tautomeric arrangements then used in the figures of the description. It is understood that all tautomeric forms of a subject compound are encompassed by a structure where one possible tautomeric form of the compound is described, even if not specifically indicated.
Prior art
The synthetic scheme used in WO-2005/023994 to prepare imetelstat as compound (1 F) is described in Scheme 1 and Scheme 2. The synthesis of this oligonucleotide is achieved using the solid-phase phosphoramidite methodology with all reactions taking place on solid-phase support. The synthesis of imetelstat is carried out on controlled pore glass (LCAA-CPG) loaded with
3-palmitoylamido-1-0-(4, 4′-dimethoxytrityl)-2-0-succinyl propanediol. The oligonucleotide is assembled from the 5′ to the 3′ terminus by the addition of protected nucleoside 5′-phosphor-amidites with the assistance of an activator. Each elongation cycle consists of 4 distinct, highly controlled steps : deprotection, amidite coupling, sulfurization and a capping step.
Scheme 1 : imetelstat synthetic scheme cycle 1
3. Sulfurization
In Scheme 1 the solid-phase supported synthesis starts with removal of the acid-labile 4,4-dimethoxy-trityl (DMT) protecting group from the palmitoylamidopropanediol linked to the solid-phase support. The first phosphoramidite nucleotide is coupled to the support followed by sulfurization of the phosphor using a 0.1 M solution of phenylacetyl disulfide (PADS) in a mixture of acetonitrile and 2,6-lutidine (1 : 1 ratio). Then a capping step is applied to prevent any unreacted solid-phase support starting material from coupling with a phosphoramidite nucleotide in the following reaction cycles. Capping is done using an 18:1 :1 mixture of THF / isobutyric anhydride / 2,6-lutidine.
After the first cycle on the solid-phase support, chain elongation is achieved by reaction of the 3′-amino group of the support-bound oligonucleotide with an excess of a solution of the protected nucleotide phosphoramidite monomer corresponding to the next required nucleotide in the sequence as depicted in Scheme 2.
Scheme 2 : imetelstat synthetic scheme cycle 2-13
In Scheme 2 the first cycle is depicted of the chain elongation process which is achieved by deprotection of the 3′-amino group of the support-bound oligonucleotide (a), followed by a coupling reaction of the 3′-amino group of the support-bound oligonucleotide (b) with an excess of a solution of a 5′-phosphoramidite monomer corresponding to the next required nucleotide in the sequence of imetelstat. The coupling reaction is followed by sulfurization of the phosphor of the support-bound oligonucleotide (c) and a capping step (see Scheme 3) to prevent any unreacted solid-phase support starting material (b) from coupling with a 5′-phosphoramidite nucleotide in the following reaction cycles. The reaction cycle of Scheme 2 is repeated 12 times before the solid-phase support-bound oligonucleotide is treated with a 1 :1 mixture of ethanol and concentrated ammonia, followed by HPLC purification to obtain imetelstat.
Scheme 3
The capping step using an 18:1 : 1 mixture of THF / isobutyric anhydride / 2,6-lutidine is done to convert after the coupling step any remaining solid-phase support bound oligonucleotide (b) with a primary 3′-amino group into oligonucleotide (e) with a protected (or ‘capped’) 3′-amino group in order to prevent the primary 3′-amino group from coupling with a phosphoramidite nucleotide in the next reaction cycles.
WO-01/18015 discloses in Example 3 with SEQ ID No. 2 a N3’^P5′ thiophosphoramidate oligonucleotide and a process for preparing this oligonucleotide encompassing a capping step.
Herbert B-S et al. discusses the lipid modification of GRN163 (Oncogene (2005) 24, 5262-5268).
Makiko Horie et al. discusses the synthesis and properties of 2′-0,4′-C-ethylene-bridged nucleic acid oligonucleotides targeted to human telomerase RNA subunit (Nucleic Acids Symposium Series (2005) 49, 171-172).
Description of the invention
The coupling reaction in the solid-phase support bound process disclosed in WO-01/18015 and WO-2005/023994 include a capping step to prevent any unreacted primary 3′ amino groups on the support-bound oligonucleotide from reacting during subsequent cycles.
It has now surprisingly been found that the use of a capping step as described in the prior art is superfluous and that imetelstat can be prepared using a 3-step cycle without an additional capping step with nearly identical yield and purity compared to the prior art 4-step cycle that uses a specific capping step. Eliminating the capping step from each cycle benefits the overall process by reducing the number of cycle steps by 22% (from 54 to 42 steps) and consequent reduction of process time. Also, the solvent consumption is reduced due to the reduction of cycle steps which makes for a greener process.
Wherever the term “capping step” is used throughout this text, it is intended to define an additional chemical process step wherein the primary free 3′-amino group on the solid-phase support bound oligonucleotide is converted into a substituted secondary or tertiary 3′-amino group that is not capable of participating in the coupling reaction with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylamino-phosphoramidite monomer in the ensuing coupling step.
In one embodiment, the present invention relates to a method of synthesizing an oligonucleotide N3′ – P5′ thiophosphoramidate of formula
imetelstat
the method comprises of
a) providing a first 3′-amino protected nucleotide attached to a solid-phase support of formula (A) wherein PG is an acid-labile protecting group;
b) deprotecting the protected 3′-amino group to form a free 3′-amino group;
c) reacting the free 3′-amino group with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N- diisopropylaminophosphoramidite monomer of formula (B n) wherein n = 2 to form an internucleoside N3′- P5′-phosphoramidite linkage;
mer (B’n)
d) sulfurization of the internucleoside phosphoramidite group using an acyl disulfide to form a N3′- P5′ thiophosphoramidate;
e) repeating 1 1 times in successive order the deprotection step b), the coupling step c) with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylamino-phosphoramidite monomer of formula (B n) wherein the protected nucleoside base B’ in monomer (B n) is successively the protected nucleobase B3 to B13 in the respective 1 1 coupling steps, and the sulfurization step d);
f) removing the acid-labile protecting group PG; and
g) cleaving and deprotecting imetelstat from the solid-phase support;
characterized in that no additional capping step is performed in any of the reaction steps a) to e).
In one embodiment, the present invention relates to a method of synthesizing the N3′ – P5′
thiophosphoramidate oligonucleotide imetelstat of formula
imetelstat
the method comprises of
a) providing a first 3′-amino protected nucleotide attached to a solid-phase support of formula (A) wherein PG is an acid-labile protecting group;
b) deprotecting the protected 3′-amino group to form a free 3′-amino group;
c) reacting the free 3′-amino group with a protected 3′-aminonucleoside-5′-0-cyanoethyl- Ν,Ν-diisopropylaminophosphoramidite monomer of formula (B n), wherein B n with n = 2 is protected A, to form an internucleoside N3′- P5′-phosphoramidite linkage;
mer
d) sulfurization of the internucleoside phosphoramidite group using an acyl disulfide to form a N3′- P5′ thiophosphoramidate;
e) repeating 1 1 times in successive order the deprotection step b), the coupling step c) with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylamino-phosphoramidite monomer of formula (B n) wherein the nucleoside base B’ of monomer (B n) is protected B except when B is thymine, and wherein Bn is successively nucleobase B3 to B13 in the respective 1 1 coupling steps, and the sulfurization step d);
f) removing the acid-labile protecting group PG; and
g) deprotecting and cleaving imetelstat from the solid-phase support;
characterized in that no additional capping step is performed in any of the reaction steps a) to e).
In one embodiment, the present invention relates to a method of synthesizing the N3′ – P5′
thiophosphoramidate oligonucleotide imetelstat of formula
imetelstat
thymine
adenine
guanine
cytosine
9 H
LPT =CH3-(CH2)i4-C-N-CH2-(CHOH)-CH2-
the method comprises of
a) providing a first protected 3′-amino nucleotide attached to a solid-phase support of formula (A) wherein PG is an acid-labile protecting group;
b) deprotecting the PG-protected 3′-amino nucleotide to form a free 3′-amino nucleotide of formula (A’);
c) coupling the free 3′-amino nucleotide with a protected 3′-aminonucleoside-5′-0- cyanoethyl-N,N-diisopropylaminophosphoramidite monomer (B n), wherein B nwith n = 2 is protected A, to form an internucleoside N3′- P5′-phosphoramidite linkage;
monomer (B’n)
d) sulfurizing the N3′- P5′-phosphoramidite linkage using an acyl disulfide to form an internucleoside N3′- P5′ thiophosphoramidate linkage;
e) repeating 1 1 times in successive order:
the deprotecting step b);
the coupling step c) with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N- diisopropylamino-phosphoramidite monomer (B n) wherein the nucleoside base B’ of monomer (B n) is protected B except when B is thymine, and wherein Bn is successively nucleobase B3 to B13 in the respective 1 1 coupling steps; and
the sulfurizing step d);
to produce a protected N3′ – P5′ thiophosphoramidate oligonucleotide imetelstat attached to the solid-phase support;
f) removing the 3′-terminal acid-labile protecting group PG from the protected N3′ – P5′ thiophosphoramidate oligonucleotide imetelstat; and
g) deprotecting and cleaving the protected N3′ – P5′ thiophosphoramidate oligonucleotide imetelstat from the solid-phase support to produce imetelstat;
characterized in that no additional capping step is performed in any of the reaction steps a) to e).
A wide variety of solid-phase supports may be used with the invention, including but not limited to, such as microparticles made of controlled pore glass (CPG), highly cross-linked polystyrene, hybrid controlled pore glass loaded with cross-linked polystyrene supports, acrylic copolymers, cellulose, nylon, dextran, latex, polyacrolein, and the like.
The 3′-amino protected nucleotide attached to a solid-phase support of formula (A)
can be prepared as disclosed in WO-2005/023994 wherein a controlled pore glass support loaded with 3-palmitoylamido-1-0-(4, 4′-dimethoxytrityl)-2-0-succinyl propanediol has been coupled with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylaminophosphoramidite monomer of formula (B^ )
monomer (B’-| ) wherein B’-| = T
wherein PG is an acid-labile protecting group. Suitable acid-labile 3′-amino protecting groups PG are, but not limited to, e.g. triphenylmethyl (i.e. trityl or Tr), p-anisyldiphenylmethyl (i.e. mono-methoxytrityl or MMT), and di-p-anisylphenylmethyl (i.e. dimethoxytrityl or DMT).
The protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylaminophosphoramidite monomers of formula (B n) have a 3′-amino protecting group PG which is an acid-labile group, such as triphenylmethyl (i.e. trityl or Tr), p-anisyldiphenylmethyl (i.e. monomethoxytrityl or MMT), or di-p-anisylphenylmethyl (i.e. dimethoxytrityl or DMT). Furthermore the nucleoside base B’ is protected with a base-labile protecting group (except for thymine).
ed A ed C ed A ed A
B’s = protected A G = guanine
B’g = protected G C = cytosine
The nucleotide monomers and B’2 to B’13 are used successively in the 13 coupling steps starting from the provision of a solid-phase support loaded with 3-palmitoylamido-1-0-(4, 4′-dimethoxytrityl)-2-0-succinyl propanediol and coupled to nucleotide monomer and the following cycle of 12 deprotection, coupling, and sulfurization reactions wherein the nucleotide monomers B’2 to B -I 3 are used.
The 3′-amino protecting group PG can be removed by treatment with an acidic solution such as e.g. dichloroacetic acid in dichloromethane or toluene.
The nucleoside base B’ in the protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropyl-aminophosphoramidite monomers of formula (B n) is protected with a base-labile protecting group which is removed in step g). Suitable base-labile protecting groups for the nucleoside base adenine, cytosine or guanine are e.g. acyl groups such as acetyl, benzoyl, isobutyryl, dimethyl-formamidinyl, or dibenzylformamidinyl. Under the reaction conditions used in oligonucleotide synthesis the thymine nucleoside base does not require protection. Such protected 3′- amino-nucleoside-5′-0-cyanoethyl-N,N-diisopropylaminophosphoramidite monomers of formula (B N) having a 3′-amino protected with an acid-labile group protecting group PG and a nucleoside base B’ protected with a base-labile protecting group are commercially available or can be prepared as described in WO-2006/014387.
The coupling step c) is performed by adding a solution of protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylaminophosphoramidite monomer of formula (BN) and a solution of an activator (or a solution containing the phosphoramidite monomer (BN) and the activator) to the reaction vessel containing the free amino group of an (oligo)nucleotide covalently attached to a solid support. The mixture is then mixed by such methods as mechanically vortexing, sparging with an inert gas, etc. Alternately, the solution(s) of monomer and activator can be made to flow through a reaction vessel (or column) containing the solid-phase supported (oligo)nucleotide with a free 3′-amino group. The monomer and the activator either can be premixed, mixed in the valve-block of a suitable synthesizer, mixed in a pre-activation vessel and preequilibrated if desired, or they can be added separately to the reaction vessel.
Examples of activators for use in the invention are, but not limited to, tetrazole, 5-(ethylthio)-1 H-tetrazole, 5-(4-nitro-phenyl)tetrazole, 5-(2-thienyl)-1 H-tetrazole, triazole, pyridinium chloride, and the like. Suitable solvents are acetonitrile, tetrahydrofuran, dichloromethane, and the like. In practice acetonitrile is a commonly used solvent for oligonucleotide synthesis.
The sulfurization agent for use in step d) is an acyl disulfide dissolved in a solvent. Art know acyl disulfides are e.g. dibenzoyl disulphide, bis(phenylacetyl) disulfide (PADS), bis(4-methoxybenzoyl) disulphide, bis(4-methylbenzoyl) disulphide, bis(4-nitrobenzoyl) disulphide and bis(4-chlorobenzoyl) disulfide.
Phenylacetyl disulfide (PADS) is a commonly used agent for sulfurization reactions that it is best ‘aged’ in a basic solution to obtain optimal sulfurization activity (Scotson J.L. et al., Org. Biomol. Chem., vol. 14, 10840 – 10847, 2016). A suitable solvent for PADS is e.g. a mixture of a basic solvent such as e.g. 3-picoline or 2,6-lutidine with a co-solvent such as acetonitrile, toluene, 1-methyl-pyrrolidinone or tetrahydrofuran. The amount of the basic solvent to the amount of the co-solvent can be any ratio including a 1 :1 ratio. Depending upon the phosphite ester to be converted into its corresponding thiophospate, both ‘fresh’ and ‘aged’ PADS can be used however ‘aged’ PADS has been shown to improve the rate and efficiency of sulfurization. ‘Aged’ PADS solutions are freshly prepared PADS solutions that were maintained some time before usage in the sulfurization reaction. Aging times can vary from a few hours to 48 hours and the skilled person can determine the optimal aging time by analysing the sulfurization reaction for yield and purity.
For the preparation of imetelstat in accordance with the present invention, a PADS solution in a mixture of acetonitrile and 2,6-lutidine, preferably in a 1 :1 ratio, with an aging time of 4 to 14 hours is used. It has been found that when 2,6-lutidine is used, limiting the amount of 2,3,5-collidine (which is often found as an impurity in 2,6-lutidine) below 0.1 % improves the efficiency of sulfurization and less undesirable phosphor oxidation is observed.
In step g) imetelstat is deprotected and cleaved from the solid-phase support. Deprotection includes the removal of the β-cyanoethyl groups and the base-labile protecting groups on the nucleotide bases. This can be done by treatment with a basic solution such as a diethylamine (DEA) solution in acetonitrile, followed by treatment with aqueous ammonia dissolved in an alcohol such as ethanol.
The reaction steps a) to f) of the present invention are carried out in the temperature range of 10°C to 40°C. More preferably, these reactions are carried out at a controlled temperature ranging from 15°C to 30°C. In particular reaction step b) of the present invention is carried out in the temperature range of 15°C to 30°C; more in particular 17°C to 27°C. In particular reaction step d) of the present invention is carried out in the temperature range of 17°C to 25°C; more in particular 18°C to 22°C; even more in particular 19°C. The step g) wherein imetelstat is deprotected and cleaved from the solid-phase support is carried out at a temperature ranging from 30°C to 60°C. Depending upon the equipment and the specific reaction conditions used, the optimal reaction temperature for each step a) to g) within the above stated ranges can be determined by the skilled person.
After each step in the elongation cycle, the solid-phase support is rinsed with a solvent, for instance acetonitrile, in preparation for the next reaction.
After step g), crude imetelstat is obtained in its ammonium salt form which is then purified by a preparative reversed phase high performance liquid chromatography (RP-HPLC) by using either polymeric or silica based resins to get purified imetelstat in triethyl amine form. An excess of a sodium salt is added, and then the solution is desalted by diafiltration thereby yielding imetelstat sodium which is then lyophilized to remove water.
Experimental part
‘Room temperature’ or ‘ambient temperature’ typically is between 21-25 °C.
Experiment 1 (no capping step)
All the reagents and starting material solutions were prepared including 3% dichloroacetic acid (DCA) in toluene, 0.5 M 5-(ethylthio)-1 H-tetrazole in acetonitrile, 0.15 M of all 4 nucleotide monomers of formula (B n) in acetonitrile, 0.2 M phenyl acetyl disulfide (PADS) in a 1 :1 mixture of acetonitrile and 2,6-lutidine and 20% DEA (diethylamine) in acetonitrile.
The oligonucleotide synthesis was performed in the direction of 5′ to 3′ utilizing a repetitive synthesis cycle consisting of detritylation followed by coupling, and sulfurization performed at ambient temperature.
A column (diameter : 3.5 cm) was packed with a solid-support loaded with 3-palmitoylamido-1-0- (4, 4′-dimethoxytrityl)-2-0-succinyl propanediol (3.5 mmol based on a capacity of 400 μιηοΙ/g) that was coupled with the nucleotide monomer B Detritylation was achieved using 3% dichloroacetic acid (DCA) in toluene (amount is between 6.5 and 13.4 column volumes in each detritylation step) and the solid-support bound nucleotide was washed with acetonitrile (amount: 5 column volumes). Coupling with the next nucleotide monomer of formula (B n) was achieved by pumping a solution of 0.5 M 5-(ethylthio)-1 H-tetrazole in acetonitrile and 0.15 M of the next nucleotide monomer of formula (B n) in the sequence, dissolved in acetonitrile, through the column. The column was washed with acetonitrile (amount : 2 column volumes). Then sulfurization was performed by
pumping a solution of 0.2 M phenyl acetyl disulfide (PADS) in a 1 :1 mixture of acetonitrile and 2,6-lutidine mixture through the column followed by washing the column with acetonitrile (amount : 5 column volumes).
The synthesis cycle of detritylation, coupling with the next nucleotide monomer of formula (B n) and sulfurization was repeated 12 times, followed by detritylation using 3% dichloroacetic acid (DCA) in toluene (amount is between 6.5 and 13.4 column volumes).
Upon completion of the synthesis cycle, the crude oligonucleotide on the solid-support support was treated with a diethylamine (DEA) solution followed by treatment with ammonium hydroxide solution: ethanol (3: 1 volume ratio) at a temperature of 55°C. The reaction mixture was aged for
4 to 24 hours at 55°C, cooled to room temperature, and slurry was filtered to remove the polymeric support. The solution comprising imetelstat in its ammonium form was subjected to the HPLC analysis procedure of Experiment 3.
Experiment 2 (with capping step)
All the reagents and starting material solutions were prepared including 3% dichloroacetic acid (DCA) in toluene, 0.5 M 5-(ethylthio)-1 H-tetrazole in acetonitrile, 0.15 M of all 4 nucleotide monomers of formula (B n) in acetonitrile, 0.2 M phenyl acetyl disulfide (PADS) in a 1 :1 mixture of acetonitrile and 2,6-lutidine mixture, 20% N-methylimidazole (NMI) in acetonitrile as capping agent A, isobutryic anhydride in a 1 :1 mixture of acetonitrile and 2,6-lutidine mixture as capping agent B and 20% DEA in acetonitrile.
The oligonucleotide synthesis was performed in the direction of 5′ to 3′ utilizing a repetitive synthesis cycle consisting of detritylation followed by coupling, and sulfurization performed at ambient temperature.
A column (diameter : 3.5 cm) was packed with a solid-support loaded with 3-palmitoylamido-1-0-(4, 4′-dimethoxytrityl)-2-0-succinyl propanediol (3.5 mmol based on a capacity of 400 μιηοΙ/g) that was coupled with the nucleotide monomer B Detritylation was achieved using 3% dichloroacetic acid (DCA) in toluene (amount is between 6.5 and 13.4 column volumes in each detritylation step) and the solid-support bound nucleotide was washed with acetonitrile (amount : 5 column volumes). Coupling with the next nucleotide monomer of formula (B n) was achieved by pumping a solution of 0.5 M 5-(ethylthio)-1 H-tetrazole in acetonitrile and 0.15 M of the next nucleotide monomer of formula (B n) in the sequence, dissolved in acetonitrile, through the column. The column was washed with acetonitrile (amount : 2 column volumes). Then sulfurization was performed by pumping a solution of 0.2 M phenyl acetyl disulfide (PADS) in a 1 :1 mixture of acetonitrile and 2,6-lutidine mixture through the column followed by washing the column with acetonitrile (amount :
5 column volumes).
The sulfurization was followed by a capping step. Each capping in a given cycle used 37-47 equivalents (eq.) of the capping agent NMI, and 9-1 1 equivalents of the capping agent B isobutryic anhydride (IBA), and 1 .4-1.8 equivalents of 2,6 lutidine. Capping agents A and B were pumped through the column with separate pumps at different ratios such as 50:50, 35:65, 65:35.
The synthesis cycle of detritylation, coupling with the next nucleotide monomer of formula (B n) and sulfurization, and capping step was repeated 12 times, followed by detritylation using 3% dichloroacetic acid (DCA) in toluene (amount is between 6.5 and 13.4 column volumes).
Upon completion of the synthesis cycle, the crude oligonucleotide on the solid-support support was treated with a diethylamine (DEA) solution followed by treatment with ammonium hydroxide solution: ethanol (3: 1 volume ratio) at a temperature of 55°C. The reaction mixture was aged for 4 to 24 hours at 55°C, cooled to room temperature, and slurry was filtered to remove the polymeric support. The solution comprising imetelstat in its ammonium form was subjected to the HPLC analysis procedure of Experiment 3.
Experiment 3 : comparision of no-capping vs. capping
Imetelstat obtained in Experiment 1 and Experiment 2 was analysed by HPLC. The amount of the desired full length oligonucleotide having 13 nucleotides was determined and listed in the Table below for Experiment 1 and Experiment 2. Also, the total amount of shortmer, specifically the 12mer, was determined and listed in the Table below for Experiment 1 and Experiment 2.
HPLC analysis method :
column type: Kromasil C18, 3.5 μιτι particle size, 4.6 X 150 mm
eluent:
A: 14.4 mM TEA/386 mM HFIP (hexafluoroisopropanol) /100 ppm(w/v) Na2EDTA in water B: 50% MeOH, 50% EtOH containing 5% IPA
Gradient :
Step Run time (minutes) %B
1 0 10
2 5 10
3 12 26 (linear)
4 35 45 (linear)
5 40 50 (linear)
6 42 50
7 44 10 (linear)
8 50 10
Table : capping vs. no-capping experiments (Experiment 1 was run twice and results are listed as Experiment 1a and 1 b).
The HPLC analysis of Experiment 1 and Experiment 2 demonstrates that yield and purity are comparable for the no-capping experiment vs. the capping experiment.
Main peak % includes Full length oligonucleotide + PO impurities + depurinated impurities.
PO impurities are impurities including one or more oxophosphoramidate internucleoside linkages instead of thiophosphoramidate internucleoside linkages.
Solvent use and reaction time
0.45 L of acetonitrile/mmol is used to prepare capping agent A and capping agent B reagents which corresponds to approximately 25 % of the overall acetonitrile use during the preparation of the reagents. Since each chemical reaction step is followed by a solvent wash, after each capping step too, a solvent wash takes place which is equivalent to about 40 column volumes of the solvent. Considering that about 212 column volumes of the solvent wash is done for a given synthesis run, about 19 % of the wash solvent is used for the capping steps. Each capping step takes between 3 – 6 minutes. This corresponds to about 8 % of the overall synthesis time including the 13 cycles and DEA treatment.
Experiment 4 (detritylation temperature)
The detritylation temperature has an impact in terms of controlling n-1 and depurinated impurities. The temperature of the deblocking solution at the entrance of the synthesizer was chosen between 17.5 and 27 °C (at 3.5 mmol scale) and the selected temperature was kept the same for all detritylation steps. The acetonitrile washing was also kept at the same temperature of the deblocking solution. The % depurinated impurities increased linearly with temperature while n-1 was higher at lower temperatures.
Temperature n-1 % Depurinated Impurity %
17.5 10.7 5.3
19 7.6 6.4
22 5.4 8.7
25 6.1 10.8
27 5.3 12.3
Experiment 5 (sulfurization step temperature)
In the experiments below, the temperature (RT means room temperature) of the PADS solution used in the sulfurization reactions was tested for the % of less favourable PO impurities (these are impurities where phosphor oxidation occurred instead of sulfurization). Lower temperature results in lower PO %.
SEQ ID NO:1 – imetelstat and imetelstat sodium
5′-R-TAGGGTTAGACAA-NH2-3′
wherein R represents palmitoyl [(CH2)1 CH3] amide is conjugated through an aminoglycerol linker to the 5′-thiophosphate group of an N3′ – P5′ thiophosphoramidate (NPS) -linked oligonucleotide.
///////////IMETELSTAT, GRN163L, PHASE 3, orphan drug, FAST TRACK
Romosozumab was originally discovered by Chiroscience,[2] which was acquired by Celltech (now owned by UCB).[3] Celltech entered in a partnership with Amgen in 2002 for the product’s development.[4]
In 2016 results from 12 months of a clinical study were reported.[5]
Some results from the FRAME[6] and ARCH clinical studies were reported on in 2017.[7]
Japan’s Ministry of Health, Labor and Welfare has granted a marketing authorization for romosozumab (EVENITY)for the treatment of osteoporosis in patients at high risk of fracture. Developed by Amgen and UCB, romosozumab is a humanized IgG2 monoclonal antibody that targets sclerostin. The approval in Japan is based on results from the Phase 3 FRAME and BRIDGE studies, which included 7,180 postmenopausal women with osteoporosis and 245 men with osteoporosis, respectively.
A biologics license application (BLA) for romosozumab as a treatment of osteoporosis in postmenopausal women at high risk for fracture was submitted to the U.S. Food and Drug Administration (FDA) in July 2016, but additional safety and efficacy data was requested in the FDA’s complete response letter, as announced by Amgen and UCB in July 2017. In July 2018, Amgen and UCB announced that the BLA had been resubmitted. In addition to data from early-stage clinical studies, the original BLA included data from the Phase 3 FRAME study. The resubmitted BLA includes results from the more recent Phase 3 ARCH study, an alendronate-active comparator trial including 4,093 postmenopausal women with osteoporosis who experienced a fracture, and the Phase 3 BRIDGE study. The FDA’s Bone, Reproductive and Urologic Drugs Advisory Committee is scheduled to review data supporting the BLA for romosozumab at a meeting on January 16, 2019.
The European Medicines Agency is also currently reviewing a marketing application for romosozumab.
Commercial production of cell culture-derived products (for example, protein-based products, such as monoclonal antibodies (mAbs)), requires optimization of cell culture parameters in order for the cells to produce enough product to meet clinical and commercial demands. However, when cell culture parameters are optimized for improving productivity of a protein product, it is also necessary to maintain desired quality specifications of the product such as glycosylation profile, aggregate levels, charge heterogeneity, and amino acid sequence integrity (Li, et al., 2010 , mAbs., 2(5):466-477).
For instance, an increase of over 20% volumetric titer results in a significant improvement in large-scale monoclonal antibody production economics. Additionally, the ability to control the glycan forms of proteins produced in cell culture is important. Glycan species have been shown to significantly influence pharmacokinetics (PK) and pharmacodynamics (PD) of therapeutic proteins such as mAbs. Moreover, the ability to modulate the relative percentage of various glycan species can have drastic results over the behavior of a protein in vivo. For example, increased mannose-5-N-acetylglycosamine-2 (“Man5”) and other high-mannose glycan species have been shown to decrease mAb in vivo half-life (Liu, 2015 , J Pharm Sci., 104(6):1866-84; Goetze et al., 2011 , Glycobiology, 21(7):949-59; and Kanda et al. 2007 , Glycobiology, 17(1):104-18). On the other hand, glycosylated mAbs with mannose-3-N-acetylglycosamine-4 (“G0”) glycan species have been shown to impact antibody dependent cellular cytotoxicity (ADCC).
Bioreactors have been successfully utilized for the cell-based production of therapeutic proteins using fed-batch, immobilized, perfusion and continuous modes. Strategies, such as the use of temperature, media formulation, including the addition of growth inhibitors, autocrine factors or cyclic mononucleotides, and hyperstimulation by osmolarity stress, have been used to enhance protein production by cells in culture. To the extent that they have worked at all, these approaches have shown only marginal success.
As such, there is a particular need for improved compositions for use in cell culture for the production of medically or industrially useful products, such as antibodies. Ideally, such compositions and methods for utilizing the same would result in higher titers, modulated (e.g. decreased) high and low molecular weight species, as well as a more favorable glycosylation profile of the derived products in cell culture.
Throughout this specification, various patents, patent applications and other types of publications (e.g., journal articles, electronic database entries, etc.) are referenced. The disclosure of all patents, patent applications, and other publications cited herein are hereby incorporated by reference in their entirety for all purposes.
Gemigliptin L-tartrate sesquihydrate was approved by Korean food and Drug Administration on June 27, 2012. It was developed and marketed as Zemiglo® by LG Life Sciences in KR.
Gemigliptin L-tartrate sesquihydrate is a dipeptidyl peptidase-4 inhibitor indicated for the treatment of type 2 diabetes mellitus.
Zemiglo® is available as tablet for oral use, containing 50 mg of free Gemigliptin. The recommended dose is 50 mg once daily taken regardless of meals.
Originator LG Life Sciences
Developer LG Chem; Sanofi
Class Antihyperglycaemics; Piperidines; Pyrimidines; Small molecules
Mechanism of Action CD26 antigen inhibitors
Marketed Type 2 diabetes mellitus
Phase II/III Acute kidney injury
24 Jun 2018 Biomarkers information updated
05 Apr 2018 LG Chem initiates enrolment in a phase I trial for Type-2 diabetes mellitus (Combination therapy) in South Korea (NCT03565458)
08 Feb 2018 LG Life Sciences completes the phase III ZEUS II trial in Type-2 diabetes mellitus (Adjunctive treatment) in South Korea (PO) (NCT02831361)
Research Code:LC-15-0444
Trade Name:Zemiglo®
MOA:Dipeptidyl peptidase-4 (DPP-4) inhibitor
Indication:Type 2 diabetes
Status:Approved 2012-06-27 korea
Company:LG Life Sciences (Originator)
Sales:ATC Code:A10BH06
Mechanism of Action
● Gemigliptin is a selective DPP-4 inhibitor[4-7].
● DPP-4 inhibition IC50=16 nM.
● Selectivity compared against DPP8/9 >3000 fold.
● Gemigliptin bound to DPP-4 enzyme with a Ki=15.2 nM.
In Vivo Efficacy
Minimum effective dose of gemigliptin in animal models:
● HbA1c reduce: DIO mice: 3 mg/kg/day for 4 weeks.
Absorption
● The oral bioavailability of gemigliptin in the rats, dogs and monkeys are species-dependent with the values of 94 %, 73 %, and 26 %, respectively.
● Gemigliptin is rapidly absorbed after single oral dose administration, with Tmax occurring 0.3 to 0.5 hr postdose in rats and dogs.
● Half-life of gemigliptin is moderate in rats, dogs, monkeys (3.6 – 5.8 hrs) and long in humans (30.8 hrs), clearance of gemigliptin ranged from 0.6 L/hr/kg (32 % of liver blood flow) in dogs to 4.1 L/hr/kg (123 % of liver blood flow).
● Volume of distribution of gemigliptin is greater than body water volume, occurring 3.7 to 14 L/kg, which suggested extensive extravascular distribution.
Distribution
● [14C] Gemigliptin and metabolites were not extensively bound to plasma proteins.
● Following oral administration of gemigliptin, [14C] gemigliptin-derived radioactivity was widely distributed to most tissues and organs in rats.
● In most tissues and organs, the total radioactivity decreased with time and was almost entirely eliminated 24 hrs after administration.
● The highest concentrations were observed in the tissues of the alimentary (the small intestine and large intestine) and the excretory or metabolic (the kidney, pancreas and liver) systems.
● Accumulation of the drug was not observed in most of the tissues or organs tested but the time course of radioactivity in testis showed some possibility of drug accumulation.
Metabolism
● Following oral administration of 10 mg/kg [14C] gemigliptin to male rats: The major circulating metabolites were LC15-0516 (dehydrated), LC15-0635 and LC15-0636 (hydroxylated), but the majority of the radioactivity in plasma was associated with the parent compound. The metabolite profile in urine was similar to that in plasma, but the profile in bile was somewhat different from that in urine or plasma. The major metabolic pathway was hydroxylation.
● Following oral administration of 50 mg [14C] gemigliptin to healthy male subjects: Gemigliptin was the most abundant component accounting for 67.2%-100% of plasma radioactivity. Unchanged gemigliptin accounted for 44.8%-67.2% of urinary radioactivity and 27.7%-51.8% of fecal radioactivity. LC15-0636 was the most abundant metabolite in plasma, accounting for 9.1%–17.5 % of plasma radioactivity. LC15-0516 and LC15-0635 were not detected in plasma samples. LC15-0636 was the only human metabolite with systemic exposure more than 10% of total drug-related exposure.
● CYP3A4 was identified as the dominant CYP isozyme converting gemigliptin to LC15-0636 in recombinant CYP/FMO enzymes.
Gemigliptin was initially developed solely by LG Life Sciences. In 2010, Double-Crane Pharmaceutical Co. (DCPC) joined with LGLS to co-develop the final compound and collaborate on the marketing of the drug in China. LGLS also announced in November 2010 that NOBEL Ilac has been granted rights to develop and commercialize gemigliptin in Turkey.
A New Drug Application (NDA) for gemigliptin in the treatment of type 2 diabetes was submitted to the Korea Food & Drug Administration (KFDA) in July 2011. Then on June 27, 2012, the KFDA has approved the manufacture and distribution of LG Life Sciences’ diabetes treatment, Zemiglo, the main substance of which is gemigliptin. LG Life Sciences signed a licensing agreement with multinational pharmaceutical companies such as Sanofi (Paris, France) and Stendhal (Mexico City, Mexico) for 104 countries. Currently, gemigliptin has been approved in 11 countries such as India, Columbia, Costa Rica, Panama, and Ecuador, and several clinical studies are in progress in Russia, Mexico, and Thailand.
History
The NDA for gemigliptin was submitted to KFDA in July, 2011 and it was approved on June 27, 2012. By the end of 2012, gemigliptin will be marketed in Korea as Zemiglo which is the fifth new DPP-4 inhibitor diabetes treatment in the world. Sanofi-Synthelabo India Private Limited announced the launch of drug for type 2 diabetes patients in India: Zemiglo (gemigliptin) on Juty 19, 2016. Zemiglo is a once daily, oral tablet. As per the International Diabetes Federation Diabetes Atlas 2015, India is home to the second largest number of adults living with diabetes worldwide, after China, with 69.1 million patients and expected to rise to 1401 million in 2040. India is the largest contributor to South East Asia regional mortality, with 1 million deaths attributable to diabetes. These statistics reveal how diabetes is fast gaining the status of a potential epidemic in India and establishes the need for treatment compliance and effective control through diet, exercise and drugs for long-term positive effects in disease management.
Mechanism of action
DPP-4 is a serine protease located on the cell surfaces throughout the body. In plasma, DPP-4 enzyme rapidly inactivates incretins including GLP-1 and GIP which are produced in the intestine depending on the blood glucose level and contribute to the physiological regulation of glucose homeostatis. Active GLP-1 and GIP increase the production and release of insulin by pancreatinc beta cells. GLP-1 also reduces the secretion of glucacon by pancreatic alpha cells, thereby resulting in a decreased hepatic glucose production. However these incretins are rapidly cleaved by DPP-4 and their effects last only for a few minutes. DPP-4 inhibitors block the cleavage of the gliptins and thus lead to an increasee insulin level and a reduced glucagon level in a glucose-dependent way. This results in a decrease of fasting and postprandial glycemia, as well as HbA1c levels.[2]
Preclinical studies
Gemigliptin is a competitive, reversible DPP-4 inhibitor (Ki = 7.25 ± 0.67 nM) with excellent selectivity over other critical human proteases such as DPP-2, DPP-8, DPP-9, elastase, trypsin, urokinase and cathepsin G. The kinetics of DPP-4 inhibition by gemigliptin was characterized by a fast association and a slow dissociation rate compared to sitagliptin (fast on and fast off rate) or vildagliptin (slow on and slow off rate). Gemigliptin was rapidly absorbed after single oral dosing and the compound was eliminated with a half-life of 3.6 h, 5.2 h, and 5.4 h in the rat, dog, and monkey, respectively.
The bioavailability of gemigliptin in the rat, dog, and monkey was species-dependent with the values of 94%, 73%, and 26%, respectively. Following the oral administration of gemigliptin in the rat, dog and monkey, about 80% inhibition of plasma DPP-4 activity were observed at the plasma levels of 18 nM, 14 nM and 4 nM, respectively.
In a diet-induced obesity model, gemigliptin reduced glucose excursion during OGTT in a dose dependent manner with the minimum effective dose of 0.3 mg/kg and enhanced glucose-stimulated plasma GLP-1 increase in a dose dependent manner reaching the maximum effect at the dose of 1 mg/kg.
Following 4 week oral repeat dosing in the DIO mice, gemigliptin reduced significantly HbA1c with the minimum effective dose of 3 mg/kg. In the beagle dog, gemigliptin significantly enhanced active GLP-1, decreased glucagon, and reduced glucose excursion during OGTT following a single dosing.
Studies on animals suggest its positive effect on hepatic and renal fibrosis .[3][4] Data on human patients are still inconclusive .[5]
Clinical studies
Monotherapy
The efficacy and safety of gemigliptin monotherapy were evaluated in two double-blind placebo controlled studies and one double-blind active-controlled study. A phase II study (study identifier: LG-DPCL002) of gemigliptin was conducted in a randomized, double-blind, placebo-controlled, parallel group design with three doses of 50, 100, and 200 mg qd for the purpose of finding a dose responsiveness and an optimal dose in patients with T2DM. The mean changes of HbA1c at week 12 from the baseline were –0.98%, –0.74%, –0.78% (when adjusted with placebo data, –0.92%, –0.68%, and –0.72%) at 50, 100, and 200 mg, respectively. Among the effective doses obtained from the phase II study in patients with T2DM, the 50 mg dose showed a similar efficacy as the 100 and 200 mg doses, within the maximum safety margin. Similar findings were reported from two phase III studies. Patients were randomized to receive gemigliptin, either a 50 mg qd (n=90) or a placebo (n=92) for 24 weeks (study identifier: LG-DPCL005; ClinicalTrials.gov registration number: NCT01601990). The placebo-subtracted changes from baseline in HbA1c were reported to be −0.71% (95% confidence interval [CI], −1.04 to −0.37) with gemigliptin 50 mg. In addition, a 28-week open-label extension study was designed to evaluate the long-term safety and efficacy of gemigliptin. Among 165 patients who consented to participate in the extension period of study LG-DPCL005, 158 patients (96%) completed their treatments for 52 weeks. All patients were switched to or continued their treatments only with gemigliptin 50 mg qd during the extension period. A further decrease in HbA1c was observed in the continued treatment with gemigliptin 50 mg in this extension period, and the mean change from baseline at 52 weeks (–0.87%) was still clinically and statistically significant (full analysis set analysis, P<0.0001). In another double-blind, active-controlled, phase III trial (study identifier: LG-DPCL011), eligible patients with HbA1c greater than 7.5% were randomized to receive gemigliptin 50 mg qd with metformin slow release (SR) qd (n=141), gemigliptin 50 mg qd (n=142), or metformin SR qd (n=150) for 24 weeks. After 24 weeks, the reduction from the baseline in HbA1c was –1.24% for gemigliptin monotherapy.
Initial combination therapy with metformin
In this randomized, double-blind, active-controlled, phase III trial (study identifier: LG-DPCL011, INICOM study; ClinicalTrials.gov registration number: NCT01787396), eligible patients with an HbA1c greater than 7.5% were randomized to gemigliptin 50 mg qd+metformin SR qd (n=141), gemigliptin 50 mg qd (n=142), or metformin SR qd (n=150). From weeks 2 to 6, metformin SR was uptitrated incrementally from 500 to 2,000 mg/day maximum in the gemigliptin/metformin and metformin groups. The mean daily doses of metformin at week 24 were 1,699 and 1,868 mg for the gemigliptin/metformin group and the metformin group, respectively. Mean change in HbA1c from baseline was –2.06% for gemigliptin/metformin group versus –1.24% for the gemigliptin group and –1.47% for the metformin group, respectively (P<0.0001 for all comparisons of combination therapy vs. monotherapy). The differences in proportions achieving an HbA1c <7% or <6.5% were also statistically significant (P<0.0001) between the combination therapy and the respective monotherapy groups.
Add-on to metformin
A 24-week, multinational, randomized, double-blind, active-controlled study (study identifier: LG-DPCL006; ClinicalTrials.gov registration number: NCT01602003) was designed to assess the efficacy and safety of gemigliptin 50 mg compared to the active control (sitagliptin) added to ongoing metformin therapy in patients with T2DM inadequately controlled with metformin alone (HbA1c, 7% to 11%). After 24 weeks, the reduction from baseline for HbA1c was 0.81% for gemigliptin 25 mg twice a day (bid) and 0.77% for gemigliptin 50 mg qd, and the differences in the least square mean changes from baseline between groups (each group of gemigliptin-sitagliptin group) were −0.011% in gemigliptin 25 mg bid and 0.004% in gemigliptin 50 mg qd. The proportion of patients achieving an HbA1c <7% at week 24 (gemigliptin 25 mg bid group, 50%; gemigliptin 50 mg qd group, 54.07%) was comparable to the results with sitagliptin 100 mg qd (48.87%). The efficacy of lowering HbA1c in the gemigliptin group was generally consistent across the subgroups based on age (<65 or ≥65 years), gender, duration of T2DM (5, >5 to 10, or >10 years), and baseline body mass index (BMI, <25 or ≥25 kg/m2). In addition, gemigliptin groups led to a significantly greater inhibition of plasma DPP-4 compared to sitagliptin. This study was extended by 28 weeks in order to evaluate the long-term efficacy and safety of gemigliptin. All treatment groups showed clinically and statistically (P<0.0001) significant improvement in glycemic control from baseline after 52 weeks. The reduction from the baseline in HbA1c was –1.06 (95% CI, –1.28 to –0.85) in the patients who continued to receive gemigliptin 50 mg qd.
Add-on to metformin and glimepiride
In this multicenter, randomized, double-blind, phase III study (study identifier: LG-DPCL010, TROICA study; ClinicalTrials.gov registration number: NCT01990469), eligible patients with inadequate glycemic control (7%≤HbA1c≤11%) were randomized to gemigliptin 50 mg qd (n=109) or placebo (n= 110). The baseline demographics were similar between groups (age, 60.9 years; BMI, 24.9 kg/m2; duration of T2DM, 12.9 years), with mean±standard deviation (SD) baseline HbA1c of 8.12%± 0.82% in the gemigliptin group and 8.15%±0.89% in the placebo group. At week 24, the adjusted mean±standard error change for HbA1c with gemigliptin was –0.88%±0.17% (change with placebo –0.01%±0.18%; difference –0.87%±0.12%; 95% CI, –1.09 to –0.64; P<0.0001).
Add-on therapy in patients with renal impairment
RI in T2DM limits the usable medications for lowering glucose level and requires frequent monitoring of renal function. Gemigliptin has balanced elimination between urinary/fecal excretion and hepatic metabolism; therefore, it does not require dose adjustment in patient with moderate to severe RI. This study evaluated the efficacy and safety of gemigliptin in T2DM patients with moderate to severe RI. This randomized, double-blind, parallel group, phase IIIb study (study identifier: LG-DPCL015, GUARD study; ClinicalTrials.gov registration number: NCT01968044) was composed of a 12-week, placebo controlled period, followed by a 40-week, double-blind active controlled extension period (placebo switched to linagliptin). A total of 132 patients with moderate or severe RI were randomized to receive gemigliptin (n=66) or placebo (n=66). Insulin was used as predominant background therapy (63.1%). At week 12, the placebo-adjusted mean change in HbA1c from the baseline was –1.20% (95% CI, –1.53 to –0.87; P<0.0001). A similar profile was also observed in other glycemic control parameters (fasting plasma glucose, glycated albumin, and fructosamine).
Effects on glycemic variability
Glycemic variability and chronic sustained hyperglycemia are the main components of dysglycemia in diabetes. The previous studies suggested that different pharmacodynamic profiles between DPP-4 inhibitors have been associated with the different effects on glycemic variability. In this study, a multicenter, randomized, active-controlled, parallel group, open-label, exploratory study was designed to evaluate the efficacy on glycemic variability and safety of initial combination therapy of gemigliptin 50 mg qd versus sitagliptin 100 mg qd, or glimepiride 2 mg qd with metformin in patients with T2DM (study identifier: LG-DPCL012, STABLE study; ClinicalTrials.gov registration number: NCT01890629). The mean amplitude of glycemic excursions (MAGE) and SD of glucose were used for assessing glucose fluctuations from the baseline after 12 weeks of treatment. At 12 weeks, MAGE was significantly lower in the DPP-4 inhibitor groups (gemigliptin and sitagliptin) than in the glimepiride group (–43.1, –38.3, and –21.7 mg/dL, respectively). Furthermore, the SD of mean glucose was significantly lower in patients with gemigliptin when compared with sitagliptin (P=0.023) and glimepiride (P=0.0058).
Ongoing studies
Several clinical studies in LG Life Sciences are actively underway to assess the efficacy and safety as an add-on combination therapy with insulin (with or without metformin) (ClinicalTrials.gov registration number: NCT02831361), to evaluate the efficacy and safety of gemigliptin-rosuvastatin fixed-dose combination in patients with T2DM and dyslipidemia in phase III clinical trials (ClinicalTrials.gov registration number: NCT02126358), and to evaluate the efficacy and safety of gemigliptin compared with vildagliptin in Russian patients with T2DM (ClinicalTrials.gov registration number: NCT02343926).
Key Characteristics
·Gemigliptin is a reversible, potent, selective, competitive, and long-acting inhibitor of DPP-4.
·Gemigliptin is orally administered 50 mg once daily either as monotherapy or in combination with other drugs. It can be taken with or without food.
·No dose adjustment is recommended for patients with renal or hepatic impairment.
·Gemigliptin shows a low propensity of drug interactions with metformin, pioglitazone, glimepiride, CYP3A4 inhibitors, rosuvastatin, or irbesartan, and dose adjustment of gemigliptin is not required for the patients who are concomitantly receiving these drugs.
·Gemigliptin decreases the mean level of HbA1c from baseline by 1.24% in monotherapy and 0.8% in add-on therapy with metformin. For gemigliptin as an initial combination with metformin, the mean reduction from baseline in HbA1c was 2.8%. In head-to-head comparisons, the mean reduction from baseline in HbA1c was 0.8% for gemigliptin with metformin and 0.8% for sitagliptin with metformin, hence the efficacy of gemigliptin is found to be comparable to sitagliptin.
·Gemigliptin was shown to be more effective in reduction of glycemic variability than glimepiride and sitagliptin with metformin as an initial combination therapy for drug naïve patients with T2DM.
·Gemigliptin is generally well tolerated in controlled clinical studies as monotherapy and as part of combination therapy. The incidences of AEs are generally similar to those of placebo and active control groups.
The present invention relates to the following formula 1- {(2S) -2- amino-I shown 4- [2,4-bis (trifluoromethyl) _5,8_ dihydro-pyrido [3,4-d ] pyrimidin -7 (6H) – yl] -4-oxo – 1.5 hydrate butyl} -5,5-difluoropiperidin-2-one (hereinafter, referred to as “compound I”) and its tartrate Preparation.
Preparation of Hydrate (Form I) of Example 1 I tartrate salt of the compound of embodiment [0072]
[0073]
[0074] The compound 2 was dissolved in about 1.87Kg 9L of ethanol.Was added 0.94Kg at O~10 ° C SOCl2, and then stirred while maintaining a low temperature.After concentration under reduced pressure, the concentrate was dissolved in MTBE 11.2L (MTBE), and the resulting mixture was adjusted to pH7~8 with ION NaOH solution.After the layers were separated, and the aqueous layer was extracted with MTBE about 3.7L and 3.7L extracted twice with MTBE, and then concentrated under reduced pressure.The resulting brown turbid solution was dissolved in 12L of ethanol, to which was added 0.47kg of dissolved water of about 1.5L of L- tartaric acid, and then stirred for I hour.The resulting crystalline slurry was filtered, washed with water and ethanol: washing (18), and then dried to obtain the title compound 1.13kg (yield 97.5%) of.
2.99 (m, 2Η), 3.11 (bt, 1H), 3.21 (bt, 1Η), 3.50 – 3.55 (m, 1Η), 3.72 – 3.91 (m, 5Η), 3.98 (t, J = 5.2Hz, 1Η) , 4.38 (s, 2Η), 4.97 -. 5.00 (m, 2Η) [0076] Example 2 compound I tartrate embodiment 1.5 hydrate (Form I) was recrystallized from water
[0077] obtained from Example 1 50g of compound I was added 250~500ml tartrate dissolved in water and water, while with ION NaOH solution was adjusted to pH6~7.Was dissolved in 23.5ml of water was added 11.7g of L- tartaric acid, and shown in Table I below, with changes in temperature, stirring speed and stirring time to obtain crystals.Then, the crystals were filtered and dried to obtain Form I.Stirring rate of change in 50~400rpm range, the temperature change in the range of 5~32 ° C.The volume of water for the recrystallization, stirring rate, temperature and mixing time shown in Table I below.
Preparation of 1.5 hydrate of tartrate salt of Compound 1 (crystal form I)
1.87 kg of the compound 2 was dissolved in about 9 L of ethanol. 0.94 kg of SOCl2was added at 0~10℃ and then stirred while maintaining low temperature. After concentrating under reduced pressure, the concentrate was dissolved in 11.2 L of MTBE (methyl t-butyl ether), and the resulting mixture was adjusted with 10 N NaOH solution to pH 7~8. After separating the layers, the aqueous layer was extracted with about 3.7 L of MTBE and twice with 3.7 L of MTBE, and then concentrated under reduced pressure. The resulting brown turbid solution was dissolved in 12 L of ethanol, 0.47 kg of L-tartaric acid dissolved in about 1.5 L of water was added thereto, and then stirred for 1 hour. The resulting crystalline slurry was filtered, washed with water and ethanol (1:8), and then dried to obtain 1.13 kg (yield 97.5%) of the title compound.
Recrystallization of 1.5 hydrate of tartrate salt of Compound 1 (crystal form I) from water
50 g of tartrate salt of Compound 1 obtained from Example 1 was added to 250~500 ml of water, and dissolved in water while adjusting the solution with 10 N NaOH to pH 6~7. 11.7 g of L-tartaric acid dissolved in 23.5 ml of water was added, and crystals were obtained with varying the temperature, stirring rate and stirring time as shown in the following Table 1. Then, the crystals were filtered and dried to obtain the crystal form I. The stirring rate was varied in the range of 50~400 rpm, and the temperature was varied in the range of 5~32℃. The volume of water used for recrystallization, the stirring rate, temperature and stirring time are represented in the following Table 1.
Table 1
Conditions for HPLC analysis
Column: Atlantis dC18 (4.6 mm I.D x 250 mm L, Particle Size 5㎛, Waters)
Column Temperature: 10℃
Mobile phase:
Mobile phase A: MeCN/TFA = 100/0.1 (v/v)
Mobile phase B: H2O/TFA = 100/0.1 (v/v)
Gradient condition:
Flow rate: 0.7 ml/min.
Detection: 256 nm, UV
Injection volume: 10㎕
Total analysis time: 55 min.
The results of the stability for the crystal form I and the crystal form II are shown in the following Table 4.
Table 4
As shown in Table 4, it could be confirmed that upon keeping the crystal form I and the crystal form II at 40±2℃, 75±5% RH or 60±2℃, 5±5% RH they exhibit a superior stability up to 8 weeks. However, according to the result of XRD analysis the crystal form I did not show any change up to 8 weeks, but the crystal form II was converted into the crystal form I at 8 week under the condition of 40℃/75% RH (see Figure 16).
Gemigliptin, a novel dipeptidyl peptidase 4 inhibitor: first new anti-diabetic drug in the history of Korean pharmaceutical industry
Abstract
Gemigliptin, a potent, selective and long-acting DPP 4 inhibitor was developed by LG Life Sciences and approved for use in patients with type 2 diabetes mellitus by the Korean Food and Drug Administration in June 2012 under the trade name Zemiglo®. Clinical pharmacokinetic and pharmacodynamic data suggest the efficacy and once daily dosing of gemigliptin. In clinical phase III studies, gemigliptin was efficacious as either monotherapy or combination therapy (add-on to metformin) and well tolerated in patients with type 2 diabetes. Further development of combination therapy is on-going.
The discovery of a series of non-peptide factor Xa (FXa) inhibitors incorporating 3-(S)-amino-2-pyrrolidinone as a central template is described. After identifying compound 4, improvements in in vitro potency involved modifications of the liphophilic group and optimizing the angle of presentation of the amidine group to the S1 pocket of FXa. These studies ultimately led to compound RPR120844, a potent inhibitor of FXa (Ki = 7 nM) which shows selectivity for FXa over trypsin, thrombin, and several fibrinolytic serine proteinases. RPR120844 is an effective anticoagulant in both the rat model of FeCl2-induced carotid artery thrombosis and the rabbit model of jugular vein thrombus formation.
-difluoro-2-oxpiperidin-l-yl)methyl]-3-oxpropyl}carbamate obtained in
PREPARATION 143 was used. [1962] 1K NMR (CD3OD) δ 5.05-4.92 (2H, m), 3.98-3.91 (2H, m), 3.85-3.79 (2H, m),
3.70-3.59 (2H, m), 3.54-3.48 (IH, m), 3.36-3.33 (2H, m), 3.24 (IH, bra), 3.14 (IH, bra), 2.83-2.76 (IH, m), 2.72-2.53 (3H, m), 2.43-2.34 (2H, m) [1963] Mass (m/e) 490 (M+l)
[1966] 14 mg of the title compound was obtained in a yield of 17% at the same manner as in PREPARATION 45, except that 43.7 mg (0.138 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-[2(S)-2-methyl-5-oxomoφholin-4-yl]-butanoic acid obtained in PREPARATION 55 and 42.5 mg (0.138 mmol) of 2,4-bis(trifluoromethyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine hydrochloric acid salt (product of PREPARATION 127) were used.
[1967] 1K NMR (CDCl3) δ 5.85-5.83 (IH, m), 5.09-4.92 (IH, m), 4.95-4.78 (IH, m),
4.23-4.08 (3H, m), 4.04-3.76 (3H, m), 3.73-3.66 (IH, m), 3.46-3.38 (IH, m), 3.36-3.21 (2H, m), 3.18-3.10 (2H, m), 2.96-2.81 (IH, m), 2.61-2.50 (IH, m), 1.43-1.41 (9H, m), 1.28-1.24 (3H, m)
PREPARATION 1: Synthesis of diethyl 2,2-difluoropentanedioate
To a solution of ethyl bromodifluoroacetate (33.2 g) in tetrahydrofuran (94.0 g) was added ethyl acrylate (8.2 g) and copper powder (10.9 g). After heating to 50℃, TMEDA (9.5 g) was added dropwise and the reaction mixture was then stirred for 3 hours at the same temperature. Upon disappearance of ethyl acrylate as the starting material, to the reaction solution was added methyl t-butyl ether (MTBE, 73.7 g) followed by addition of 10% aqueous ammonium chloride solution (49.8 g) dropwise, and the mixture was then stirred for 30 minutes. The remaining copper residue was removed by filtration through a celite, and methyl t-butyl ether (MTBE, 66.3 g) was added to separate the layers. The separated organic layer was washed successively with 10% aqueous NH4Cl solution (66.3 g) and 3 N aqueous hydrochloric acid solution (99.6 g) in order and then distilled under reduced pressure to obtain 55.0 g of the desired title compound.
PREPARATION 2: Synthesis of ethyl 4,4-difluoro-5-hydroxypentanoate
14.8 g of the compound obtained from the above Preparation 1 was diluted with ethanol (20.4 g) and tetrahydrofuran (69.1 g) and then cooled to 0℃. To this solution was slowly added sodium borohydride (NaBH4, 3.5 g) stepwise while keeping the internal temperature below 30℃. After confirming completion of the reaction by 1H NMR, the reaction solution was cooled to the temperature of 10℃ and 10% aqueous ammonium chloride solution (77.7 g) was slowly added. The remaining boron compound was filtered through celite, and the filtrate was distilled under reduced pressure to remove tetrahydrofuran. Then, ethyl acetate (105.2 g) was added to separate the layers, and the organic layer was distilled under reduced pressure to obtain 10.8 g of the title compound.
EXAMPLE 1: Synthesis of ethyl 4,4-difluoro-5-{[(trifluoromethyl)sulfonyl]oxy}- pentanoate
To the solution of 10.8 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (100.2 g) was added pyridine (7.0 g), and then the mixture was cooled to -5.0℃. After completion of cooling, trifluoromethane sulfonic acid anhydride (20.1 g) was slowly added dropwise while keeping the reaction temperature below 6.3℃. After stirring the reaction solution for 30 minutes, 1.5 N hydrochloric acid solution was added dropwise at 0℃ to separate the layers. The aqueous layer as separated was back-extracted twice with dichloromethane (33.4 g), and the extracts were combined with the organic layer separated from the above and then distilled under reduced pressure to obtain 19.7 g of the title compound as a yellow oil.
EXAMPLE 2-1: Synthesis of ethyl 4,4-difluoro-5-{[(nonafluorobutyl)sulfonyl]- oxy}pentanoate
To the solution of 100.0 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (300.0 ml) was added pyridine (65.7 g), and the mixture was then cooled to -10.0℃. After completion of cooling, nonafluorobutanesulfonic anhydride (477.4 g) was slowly added dropwise. After stirring the reaction solution for 3 hours, 1.0 N hydrochloric acid solution (300.0 ml) was added dropwise to separate the layers. The aqueous layer as separated was back extracted once with dichloromethane (500.0 ml), and the extracts were combined with the organic layer separated from the above and then distilled under reduced pressure to obtain 177.5 g of the title compound.
EXAMPLE 2-2: Synthesis of ethyl 4,4-difluoro-5-{[(nonafluorobutyl)sulfonyl]- oxy}pentanoate
To the solution of 500.0 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (1000.0 ml) was added triethylamine (389.0 g), and the mixture was then cooled to 0℃. After completion of cooling, perfluorobutanesulfonyl chloride (948.80 g) was slowly added dropwise. The reaction solution was stirred for 3 hours at room temperature, distilled under reduced pressure, dissolved in methyl t-butyl ether (MTBE, 3000.0 ml) and then washed three times with water. The organic layer thus obtained was dehydrated with magnesium sulfate, filtered through a celite and then distilled under reduced pressure to obtain 960.0 g of the title compound.
EXAMPLE 3: Synthesis of methyl (2S)-2-[(tert-butoxycarbonyl)amino]-4-oxo- pentanoate
To 25.0 g of the starting material, (3S)-3-[(t-butoxycarbonyl)amino]-4-oxo- pentanoic acid, was added t-butanol (96.9 g) followed by the addition of Boc2O (25.4 g) and dimethylaminopyridine (DMAP, 62.0 g, 0.5 mol%) at room temperature, and the reaction mixture was then stirred for 23 hours at 40℃. Upon completion of the reaction, ethylene dichloride (62.3 g) in t-butanol was added, and the mixture was then distilled under reduced pressure to obtain 30.7 g of the title compound.
EXAMPLE 4: Synthesis of tert-butyl (3S)-3-[(tert-butoxycarbonyl)amino]-4-hydroxy- butanoate
30.7 g of the compound obtained from the above Example 3 was dissolved in ethanol (112.3 g) and, after lowering the internal temperature to 10.5℃ sodium borohydride (NaBH4, 5.7 g) was slowly added dropwise. This reaction solution was stirred while maintaining the temperature below 22℃. After confirming completion of the reaction by 1H NMR and TLC, to the reaction solution was slowly added 3.0 N hydrochloric acid solution (30.7 g) dropwise at the internal temperature of 10℃ followed by addition of diluted 0.2% hydrochloric acid solution (100.0 g). The reaction solution was adjusted to pH 3~4 with addition of 9.0% aqueous hydrochloric acid solution, and then back-extracted twice with ethyl acetate (100.0 g) and toluene (44.0 g). The organic layer thus obtained was distilled under reduced pressure to obtain 25.1 g of the title compound.
EXAMPLE 5: tert-butyl (3S)-[(tert-butoxycarbonyl)amino]-4-[(methylsulfonyl)oxy]- butanoate
To 25.1 g of the compound obtained from the above Example 4 was added dichloromethane (133.0 g) and triethylamine (148.0 g), and the mixture was then cooled to 0℃. To this reaction solution was slowly added methanesulfonyl chloride (11.8 g) diluted with dichloromethane (39.9 g) dropwise for 50 minutes while maintaining the internal temperature below 12℃. After completion of the reaction, the reaction solution was washed with 0.5 N aqueous hydrochloric acid solution (120.0 g) and water (100.4 g), and then distilled under reduced pressure to obtain 31.5 g of the title compound.
EXAMPLE 6: Synthesis of tert-butyl (3S)-4-azido-3-[(tert-butoxycarbonyl)amino]- butanoate
Sodium azide (NaN3, 11.6 g) was diluted with dimethylacetamide (DMAc, 260.0 g). After elevating the internal temperature to 80℃, a solution of 31.5 g of the compound, as obtained from the above Example 5, diluted with dimethylacetamide (DMAc, 45.0 g) was added thereto. The reaction proceeded at 80℃ for 2 hours. To the reaction solution were added toluene (251.0 g) and water (320.0 g) to separate the layers. The organic layer thus obtained was distilled under reduced pressure to obtain 24.0 g of the title compound.
EXAMPLE 7: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- butanoate
To 21.0 g of the compound obtained from the above Example 6 was added tetrahydrofuran (93.3 g) followed by the addition of triphenylphosphine (PPh3, 21.0 g) at 40℃, the mixture was stirred for 2 hours at the same temperature, and water (3.8 g) was then added thereto. The reaction solution was distilled under reduced pressure, and the resulting triphenylphosphine oxide solid was diluted with toluene (26.0 g) and n-hexane (41.0 g), and then filtered off. The filtrate was adjusted to pH 2~3 with 1.0 N aqueous hydrochloric acid solution (110.0 g) and then subjected to separation of the layers. To remove any residual triphenylphosphine oxide solid, the aqueous layer obtained above was washed with dichloromethane (100.0 g) and then adjusted to pH 8~9 with 28% aqueous ammonia solution (7.6 g). The aqueous solution thus obtained was extracted with dichloromethane (100.0 g) and distilled under reduced pressure to obtain 8.5 g of the title compound as a white solid.
EXAMPLE 8: Synthesis of N,N-dibenzyl-L-N(Boc)-aspartamide 4-tert-butyl ester
N-Boc-L-aspartic acid 4-t-butyl ester (29.0 g, 0.10 mol) was added to THF (200 ml). After cooling to temperature below -5℃, to the reaction solution was added isobutylchloroformate (13.0 ml, 0.10 mol) followed by addition of N-methyl morpholine (12.0 ml, 0.10 mol) dropwise, and the reaction mixture was stirred for over 30 minutes. To the reaction mixture was added dropwise dibenzylamine (21.1 ml, 0.11 mol), and the mixture was then stirred for over 3 hours and monitored for the reaction progress by TLC (EtOAc: Hexane=1:4). Upon completion of the reaction, the reaction solution was stirred with addition of ethyl acetate (300.0 mL) and 1 N hydrochloric acid to separate the layers, and distilled under reduced pressure to precipitate a solid. The solid was filtered and washed with ethyl acetate (100 ml), and then the washings were concentrated by distillation again under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (41.7 g, 0.89 mol).
Mass (ESI, m/z): 491 (M+Na), 469 (M+H), 413 (M-55).
EXAMPLE 9: Synthesis of N, N-diallyl-L-N(Boc)-aspartamide 4-tert-butyl ester
L-N(Boc)-aspartic acid 4-t-butyl ester (5.00 g, 17.3 mol) was added to THF (50 ml). After cooling to temperature below -5℃, to the reaction solution was added isobutylchloroformate (2.26 ml, 17.3 mol) followed by addition of N-methyl morpholine (1.90 ml, 17.3 mol) dropwise, and the reaction mixture was stirred for over 30 minutes. To the reaction mixture was added dropwise diallylamine (2.35 ml, 19.0 mol), and the mixture was then stirred for over 3 hours and monitored for the reaction progress by TLC (EtOAc: Hexane=1:4). Upon completion of the reaction, the reaction solution was stirred with addition of ethyl acetate (60 ml) and 1 N hydrochloric acid and, after separating the layers, concentrated by distillation under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (6.0 g, 16.3 mol).
Mass (ESI, m/z): 391 (M+Na), 369 (M+H), 313 (M-55).
EXAMPLE 10: Synthesis of N,N-dibenzyl-4-amino-3(S)-N(Boc)-aminobutanoic acid 4-tert-butyl ester
10.0 g of the compound obtained from the above Example 8, Ru3(CO)12 (136 mg, 1mol%), and diphenylsilane (19.7 ml, 106.7 mmol) were added to tetrahydrofuran (50 ml), and the reaction solution was stirred under reflux for over 40 hours. The reaction solution was extracted with ethyl acetate (200 ml) and concentrated by distillation under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (4.7 g, 10.5 mmol).
EXAMPLE 11: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- 4-oxobutanoate
360.0 g of the starting material, N-Boc-Asp(O-t-Bu)OH, together with Boc2O (353.0 g) and ammonium bicarbonate (NH4HCO3, 123.9 g) was added to dimethylformamide (1174.6 g), and pyridine (61.0 g) was added dropwise thereto at room temperature, and the reaction mixture was then stirred for about 3 hours. Upon completion of the reaction, water (1440 ml) and toluene (1800 ml) were added to the reaction solution and stirred for 30 minutes to separate the layers. The organic layer thus obtained was distilled under reduced pressure to remove t-butanol and toluene to obtain the title compound, which was directly used in the next reaction.
EXAMPLE 12: Synthesis of (S)-tert-butyl 3-(tert-butoxycarbonylamino)-3-cyanopropanoate
To the compound obtained from Example 11 was added dimethylformamide (1019.5 g) followed by addition of cyanuric chloride (112.0 g) dropwise for 1.5 hours at temperature below 25℃. The reaction solution was stirred for one hour at room temperature, and then 0.1 N aqueous sodium hydroxide solution (1850.0 g) and toluene (1860 ml) were added thereto to separate the layers. The organic layer thus obtained was washed once again with water (700 ml) and then distilled under reduced pressure to obtain 318.3 g of the title compound.
EXAMPLE 13: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- butanoate
To 212.1 g of the compound obtained from the above Example 12 was added acetic acid (4000 ml) followed by addition of 20 wt% Pd(OH)2 (1.1 g) at 40℃. The mixture was stirred for 8 hours while keeping the internal temperature below 45℃ and 3 atmospheric pressure of hydrogen. Upon completion of the reaction, the reaction solution was distilled under reduced pressure to remove acetic acid, diluted with toluene (640 L) and then filtered through a celite. To the filtrate was added 0.25 N aqueous hydrochloric acid solution (1060 ml) to separate the layers. The aqueous layer thus obtained was basified with aqueous ammonia solution (543.1 g) and then extracted with methyl t-butyl ether (MTBE, 1000 ml). The organic layer thus obtained was distilled under reduced pressure to obtain 185.0 g of the title compound.
EXAMPLE 14: Synthesis of 3-t-butoxycarbonylamino-4-(5,5-difluoro-2-oxo- piperidin-1-yl)-butyric acid t-butyl ester
Triethylamine (13.2 g) was added to 16.0 g of the compound obtained from the above Example 1 or 2-1 or 2-2, and 14.1 g of the compound obtained from the above Example 7 or 13, and the mixture was then stirred for 21 hours at 40℃. Then, dichloromethane (154.8 g) and acetic acid (18.3 g) were added, and the mixture was stirred for 5 hours at room temperature. To the resulting reaction solution was added 0.5 N aqueous hydrochloric acid solution (116.8 g) and then, the mixture was stirred for 30 minutes to separate the layers. The organic layer thus obtained was distilled under reduced pressure to obtain 23.6 g of the title compound.
EXAMPLE 15: Synthesis of 3-t-butoxycarbonylamino-4-(5,5-difluoro-2-oxo- piperidin-1-yl)-butyric acid
23.6 g of the compound obtained from the above Example 14 was added to dichloromethane (20.0 g) followed by addition of H3PO4 (30.0 g), and the mixture was stirred for 16 hours at room temperature. After confirming the detachment of all of t-butyl group and t-butyloxycarbonyl group, the reaction solution was adjusted to pH 7.0~8.0 with 10 N aqueous hydrogen peroxide, and Boc2O (16.0 g) was added thereto. After completion of the addition, 10 N aqueous hydrogen peroxide was used to maintain the pH of the reaction solution at 8.0~9.0. After stirring for 3 hours, the resulting sodium phosphate was filtered off, and the filtrate was then adjusted to pH 2.0~3.0 with 3.0 N aqueous hydrochloric acid solution. The resulting solid was filtered and dried under nitrogen to obtain 14.5 g of the title compound.
For the title compound resulting from the above, its enantiomeric isomers―i.e. S-form and R-form―were measured by HPLC (high-performance liquid chromatography), and an excess of the enantiomeric isomers (S vs. R form) (enantiomeric excess; ee) was then calculated as being ee > 99%. On the other hand, in case of the Comparative Example prepared according to the prior method based on WO 06/104356, as described below, the excess (ee) of enantiomeric isomers (S vs. R form) was 80%. From this, it can be identified that the compound of formula (2) having an optically high purity could be obtained according to the method of the present invention.
COMPARATIVE EXAMPLE 1: Synthesis of 3-t-butoxycarbonylamino-4-(5,5- difluoro-2-oxo-piperidin-1-yl)-butyric acid t-butyl ester
COMPARATIVE EXAMPLE 1-1: Synthesis of methyl 5-amino-4,4-difluoro- pentanoate HCl
To 10.0 g of the compound obtained from Example 1 was added 40 ml of anhydrous ammonia solution (7 M solution in methanol), and the mixture was stirred for 3 hours. The reaction solution was distilled and 30 ml of hydrochloric acid solution saturated with methanol was added dropwise thereto. The reaction mixture was stirred at room temperature and then distilled to obtain 7.2 g of the title compound as a white solid.
COMPARATIVE EXAMPLE 1-2: Synthesis of 3-t-butoxycarbonylamino-4-(5,5- difluoro-2-oxo-piperidin-1-yl)-butyric acid t-butyl ester
To the solution of the compound (1.93 g), as obtained from the above Example 4, dissolved in dichloromethane (20.0 g) and H2O (4.0 g) were added NaBr (0.8 g) and TEMPO (11 mg, 1 mol%). To this reaction solution was slowly added a solution of 5% NaOCl (11.5 g) and NaHCO3 (1.7 g) dissolved in H2O (12.0 g) dropwise for about 2 hours while maintaining the temperature below 5℃. Upon completion of dropwise addition, the reaction solution was stirred for 30 minutes to separate the layers. To the organic layer thus obtained was added the compound (1.6 g) obtained from the above Comparative Example 1-1. After stirring for 15 minutes at room temperature, NaBH(OAc)3 (2.23 g) was added to the reaction solution. After stirring for about 19 hours, 10% aqueous NaHCO3 solution (20.0 g) and 0.5 N aqueous hydrochloric acid solution (20.0 g) were added dropwise to the reaction solution to separate the layers. The organic layer thus obtained was dehydrated under anhydrous MgSO4 to obtain 2.0 g (yield 73%) of the same title compound as Example 14, as a yellow solid. For the title compound resulting from the above, its enantiomeric isomers―i.e., S-form and R-form―were measured by HPLC (high-performance liquid chromatography), and an excess (ee) of the enantiomeric isomers (S vs. R form) was then calculated as being ee = 80%.
PAPER
Gemigliptin is a prolyl-specific dipeptidyl aminopeptidase IV (DPP IV, DPP-4, CD26) inhibitor
approved for the treatment of type 2 diabetes mellitus by the Korean Food and Drug Administration in
2012. Gemigliptin was discovered and developed by LG Life Sciences81 and is now the sixth DPP-4
inhibitor approved for the treatment of type 2 diabetes.82 At the time this review was prepared, there
were no publications describing the discovery strategy and preclinical data that led to the advancement
of gemigliptin to the clinic. Additionally, the synthesis of the drug has only been described in the patent
literature.83-85
The molecule was prepared via a convergent route and the synthesis of the dihydropyridopyrimidine
fragment is described in Scheme 11.85 Commercial N-Boc-3-piperidone (71) was treated with lithium
hexamethyldisilazane (LHMDS) followed by ethyl trifluoroacetate to effect a Claisen condensation,
producing diketone 72 in 81% yield. Cyclization of 72 with 2,2,2-trifluoroacetamide (73) gave bistrifluoromethyl
dihydropyridopyrimidine 74 in 23% yield. Removal of the Boc protecting group
efficiently provided amine 75 in 96% yield.
SCHEME 11
The synthesis of the carbon skeleton of the difluoropyridone fragment 80 is described in Scheme
12.84 1,4-Addition of ethyl bromodifluoroacetate (76) to ethyl acrylate (77) in the presence of copper powder and tetramethylethylenediamine (TMEDA) gave diester 78, which was selectively reduced with
sodium borohydride (NaBH4) to give alcohol 79 in 90% overall yield for the two-step procedure.
Alcohol 79 was then treated with perfluorobutanesulfonyl chloride and triethylamine to give activated
alcohol 80 in 75% yield.
87 in 51% yield. Removal of the Boc group with thionyl chloride in ethanol followed by neutralization
with aqueous sodium hydroxide and salt formation with L-tartaric acid provided gemigliptin L-tartrate
hydrate (X) in 97.5% yield.83
The completion of the synthesis of gemigliptin is described in Scheme 13.83, 84 Boc-L-aspartic acid
4-tert-butyl ester (81) was treated with ammonium bicarbonate and pyridine in the presence of di-tertbutyl
dicarbonate to give formamide 82. Dehydration of 82 to give nitrile 83 was accomplished through
reaction with cyanuric chloride in 95% overall yield for the two-step sequence. Hydrogenation of 83 in
the presence of Pearlman’s catalyst provided butyl amine 84. Alkylation of 84 with activated alcohol 80
in triethylamine followed by cyclization in acetic acid afforded difluoropyridone 85. Acidic hydrolysis
of the ester proceeded with concomitant removal of the Boc protecting group, and was followed by
reprotection of the amine with di-tert-butyl dicarbonate to give acid 86 in 84% overall yield for the
three-step procedure in >97% ee. Coupling of 86 with fragment 75 in the presence of
hydroxybenzotriazole (HOBT) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) gave amide
81. Kim, S.-H.; Lee, S.-H.; Yim, H.-J. Arch. Pharmacal Res. 2013, 36, 1185. 82. Juillerat-Jeanneret, L. J. Med. Chem. 2014, 57, 2197. 83. Park, K. S.; Yun, J. M.; Kim, B. C.; Kim, K. Y.; Lee, J. H. WO Patent 2012060590A2, 2012. 84. Kim, B. C.; Kim, K. Y.; Lee, H. B.; An, J. E.; Lee, K. W. WO Patent 2012030106A2, 2012. 85. Lee, C.-S.; Koh, J. S.; Koo, K. D.; Kim, G. T.; Kim, K.-H.; Hong, S. Y.; Kim, S.; Kim, M.-J.; Yim, H. J.; Lim, D.; Kim, H. J.; Han, H. O.; Bu, S. C.; Kwon, O. H.; Kim, S. H.; Hur, G.-C.; Kim, J. Y.; Yeom, Z.-H.; Yeo, D.-J. WO Patent 2006104356A1, 2006.
PAPER
Gemigliptin, (LC15-0444, LG Life Sciences)
Gemigliptin is a sitagliptin analogue discovered by LG Life sciences Ltd, Korea via the derivatization of the compounds. It is potent and long acting DPP-IV inhibitor with high selectivity profile (3000-fold) against isoenzymes. The binding mode of gemigliptin is not reported, but expected as sitagliptin due to structural similarity. It inhibited more than 80% of DPP-IV activity and exhibited the bioavailability of 94% in rats. It also showed the lowering of
blood glucose and elevating of GLP-1 levels in dose-dependent manner in the diet-induced obese mice. Gemigliptin displayed a noteworthy lowering in HbA1c level (0.77%) at a dose of 3.0 mg/kg.[57] It is approved by Korean FDA in June 2012 for the treatment of T2DM.[58]
Synthesis of gemigliptin involved the preparation of two key intermediates dihydropyrido[3,4-d]pyrimidine moiety 88 and β-amino acid moiety 92. Compound 86 was prepared by generating enolate from compound 85 using LHMDS and adding trifluoroacetate.
Compound 86 gave the 87 in reflux condition which after Boc deprotection afforded key amine intermediate 88. The β-amino derivative 91 was synthesized by cyclization reaction between 89 and 90, which on benzyl deprotection using Pd/C gave desired β-amino intermediate 92.
Coupling of this intermediate with 88 using EDC/HOBt followed by Boc deprotection offered
gemigliptin 94 via 93 (Scheme 13).[59,60]
[57] S. J. Yang, K. W. Min, S. K. Gupta, J. Y. Park, V. K. Shivane, S. U. Pitale, P. K.
Agarwal, A. Sosale, P. Gandhi, M. Dharmalingam, V. Mohan, U. Mahesh, D. M. Kim, Y.
S. Kim, J. A. Kim, P. K. Kim, and S. H. Baik, Diabetes, Obes. Metab., 2013, 15, 410–
416.
[58] S. H. Kim, S. H. Lee, and H. J. Yim, Arch. Pharm. Res., 2013, 36 (10), 1185-1188. [59] C. S. Lee, J. S. Koh, K. D. Koo, G. T. Kim, K. H. Kim, S. Y. Hong, S. Kim, M. J. Kim, H. J. Yim, D. Lim, H. J. Kim, H. O. Han, S. C. Bu, O. H. Kwon, S. H. Kim, G. C. Hur, J. Y. Kim, Z. H. Yeom, D. J. Yeo, WO 2006/104356 A1, 2006. [60] K. S. Park, J. M. Yun, B. C. Kim, Y. U. Kim, J. H. Lee, WO 2012/060590, 2012.
CN101151265A *2005-04-012008-03-26株式会社Lg生命科学Dipeptidyl peptidase-iv inhibiting compounds, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent
WO2004007468A1 *2002-07-152004-01-22Merck & Co., Inc.Piperidino pyrimidine dipeptidyl peptidase inhibitors for the treatment of diabetes
WO2004069162A3 *2003-01-312005-05-19Wallace T Ashton3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes
Reference:
[1]. J. Med. Chem., Ahead of Print.
[2]. Clinical therapeutics2008, 30, 1817-1830.
[3]. Int. J. Res. Dev. Pharm. Life Sci. 2013, 2, 602-610, 609 pp.
[4]. Xenobiotica; the fate of foreign compounds in biological systems 2014, 44, 627-634.
[5]. Poster presented at the annual meeting of American Diabetes Association, 2008, San Francisco: CA.
^Lim KS, Kim JR, Choi YJ, Shin KH, Kim KP, Hong JH, Cho JY, Shin HS, Yu KS, Shin SG, Kwon OH, Hwang DM, Kim JA, Jang IJ (October 2008). “Pharmacokinetics, pharmacodynamics, and tolerability of the dipeptidyl peptidase IV inhibitor LC15-0444 in healthy Korean men: a dose-block-randomized, double-blind, placebo-controlled, ascending single-dose, Phase I study”. Clin Ther. 30 (10): 1817–30. doi:10.1016/j.clinthera.2008.10.013. PMID19014837.
^Kaji K (Mar 2014). “Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats”. J Gastroenterol.. 49 (3): 481–91. doi:10.1007/s00535-013-0783-4. PMID23475323.
^Min HS (Jun 2014). “Dipeptidyl peptidase IV inhibitor protects against renal interstitial fibrosis in a mouse model of ureteral obstruction”. Lab. Invest. 94 (5): 598–607. doi:10.1038/labinvest.2014.50. PMID24687121.
Rhee EJ, Lee WY, Yoon KH, Yoo SJ, Lee IK, Baik SH, Kim YK, Lee MK, Park KS, Park JY, Cha BS, Lee HW, Min KW, Bae HY, Kim MJ, Kim JA, Kim DK, Kim SW (December 2010). “A multicenter, randomized, placebo-controlled, double-blind phase II trial evaluating the optimal dose, efficacy and safety of LC 15-0444 in patients with type 2 diabetes”. Diabetes Obes Metab. 12 (12): 1113–1119. doi:10.1111/j.1463-1326.2010.01303.x. PMID20977584.
////////////////gemigliptin, LC15-0444, KOREA 2012, Gemigliptin tartrate sesquihydrate, GLIPTIN, ゲミグリプチン , LG Chem, Sanofi DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
In February 2013, EC approval in all EU member states was granted for the reduction of alcohol consumption in adults with alcohol dependence
Nalmefene hydrochloride dihydrate is a white or almost white crystalline powder. The chemical name is 17-(Cyclopropylmethyl)-4,5-α-epoxy-6-methylene-morphinan-3,14-diol hydrochloride dihydrate, has the following molecular formula C21H25NO3 ⋅ HCl ⋅ 2 H2O
Nalmefene hydrochloride dihydrate is very soluble in water and is not hygroscopic. Nalmefene hydrochloride dihydrate is a chiral compound, containing 4 asymmetric carbon atoms. Only one crystal form of Nalmefene hydrochloride dihydrate has been identified. Nalmefene hydrochloride dihydrate does not melt, but becomes amorphous after dehydration.
The structure of nalmefene hydrochloride dihydrate was demonstrated by elemental analysis, IR, UV/Vis, 1 H-NMR and 13C-NMR spectroscopy as well as MS spectrometry. Its crystal structure was analysed by X-ray diffraction and specific optical rotation was determined. It has been shown that no polymorphic forms were observed.
PATENTS AND GENERICS
The original product patent was based on US 03814768 which expired in 1991. However, a number of patents cover formulations and use. Lundbeck and Biotie have a family based on WO 2010063292 which claims novel crystal forms and hydrate salts, in particular Nalmefene hydrochloride dihydrate, and their use in alcohol dependence. There are European and US patents granted on this EP 02300479 will expire December 2029 and US-08530495 will expire August 2030.
Nalmefene hydrochloride was approved by the U.S. Food and Drug Administration (FDA) on Apr 17, 1995. It was developed and marketed asRevex® by Baxterin in the US.
Nalmefene is an opioid receptor antagonist. It acts as a silent antagonist of the μ-opioid receptor and as a partial agonist of the κ-opioid receptor, it also possesses affinity for the δ-opioid receptor. Revex® is indicated for the complete or partial reversal of opioid drug effects, including respiratory depression, induced by either natural or synthetic opioids. It is also indicated in the management of known or suspected opioid overdose.
Revex® is available as a sterile solution for intravenous, intramuscular and subcutaneous administration in two concentrations, containing 100 μg or 1.0 mg of nalmefene free base per mL. The recommended dose is initiating at 0.25 μg/kg followed by 0.25 μg/kg incremental doses at 2-5 minute intervals for reversal of postoperative opioid depression, stopping as soon as the desired degree of opioid reversal is obtained.
Nalmefene (trade name Selincro), originally known as nalmetrene, is an opioid antagonist used primarily in the management of alcohol dependence. It has also been investigated for the treatment of other addictions such as pathological gambling.[1]
Nalmefene is an opiate derivative similar in both structure and activity to the opioid antagonist naltrexone. Advantages of nalmefene relative to naltrexone include longer half-life, greater oral bioavailability and no observed dose-dependent liver toxicity.[2]
As with other drugs of this type, nalmefene may precipitate acute withdrawal symptoms in patients who are dependent on opioid drugs, or more rarely when used post-operatively, to counteract the effects of strong opioids used in surgery.
Medical uses
Opioid overdose
Intravenous doses of nalmefene have been shown effective at counteracting the respiratory depression produced by opioid overdose.[3]
This is not the usual application for this drug, for two reasons:
The half-life of nalmefene is longer than that of naloxone. One might have thought this would make it useful for treating overdose involving long-acting opioids: it would require less frequent dosing, and hence reduce the likelihood of renarcotization as the antagonist wears off. But, in fact, the use of nalmefene is not recommended in such situations. Unfortunately, opioid-dependent patients may go home and use excessive doses of opioids in order to overcome nalmefene’s opioid blockade and to relieve the discomfort of opioid withdrawal. Such large doses of opioids may be fatal. This is why naloxone (a shorter-acting drug) is normally a better choice for overdose reversal.[4]
In addition, injectable nalmefene is no longer available on the market.
When nalmefene is used to treat an opioid overdose, doses of nalmefene greater than 1.5 mg do not appear to give any greater benefit than doses of only 1.5 mg.
Alcohol dependence
Nalmefene is used in Europe to reduce alcohol dependence[5] and NICE recommends the use of nalmefene to reduce alcohol consumption in combination with psychological support for people who drink heavily.[6]
Based on a meta analysis, the usefulness of nalmefene for alcohol dependence is unclear.[7] Nalmefene, in combination with psychosocial management, may decrease the amount of alcohol drunk by people who are alcohol dependent.[7][8] The medication may also be taken “as needed”, when a person feels the urge to consume alcohol.[8]
Side effects
The following adverse effects have been reported with nalmefene:
Nalmefene is structurally related to naltrexone and differs from it by substitution of the ketone group at the 6-position of naltrexone with a methylene group (CH2). It binds to the MOR with similar affinity relative to naltrexone, but binds “somewhat more avidly” to the KOR and DOR in comparison.[10][13]
Pharmacokinetics
Nalmefene is extensively metabolized in the liver, mainly by conjugation with glucuronic acid and also by N-dealkylation. Less than 5% of the dose is excreted unchanged. The glucuronide metabolite is entirely inactive, while the N-dealkylated metabolite has minimal pharmacological activity.[citation needed]
Chemistry
Nalmefene is a derivative of naltrexone and was first reported in 1975.[17]
Society and culture
United States
In the US, immediate-release injectable nalmefene was approved in 1995 as an antidote for opioid overdose. It was sold under the trade name Revex. The product was discontinued by its manufacturer around 2008.[18][19] Perhaps, due to its price, it never sold well. (See § Opioid overdose, above.)
Nalmefene in pill form, which is used to treat alcohol dependence and other addictive behaviors, has never been sold in the United States.[2]
Europe
Lundbeck has licensed nalmefene from Biotie Therapies and performed clinical trials with nalmefene for treatment of alcohol dependence.[20] In 2011 they submitted an application for their drug termed Selincro to the European Medicines Agency.[21] The drug was approved for use in the EU in March 2013.[22] and in October 2013 Scotland became the first country in the EU to prescribe the drug for alcohol dependence.[23] England followed Scotland by offering the substance as a treatment for problem drinking in October 2014.[24] In November 2014 nalmefene was appraised and approved as a treatment supplied by Britain’s National Health Service (NHS) for reducing alcohol consumption in people with alcohol dependence.[25]
Research
Nalmefene is a partial agonist of the κ-opioid receptor and may be useful to treat cocaine addiction.[26]
SYN
Nalmefene (CAS NO.: 55096-26-9), with its systematic name of Morphinan-3,14-diol, 17-(cyclopropylmethyl)-4,5-epoxy-6-methylene-, (5alpha)-, could be produced through many synthetic methods.
Following is one of the synthesis routes:
By a Wittig reaction at naltrexone (I) with triphenylmethylphosphonium bromide (II) in DMSO in the presence of NaH as base.
Nalmefene (trade name Selincro), originally known as nalmetrene, is an opioid receptorantagonist developed in the early 1970s,[1] and used primarily in the management of alcohol dependence, and also has been investigated for the treatment of other addictions such as pathological gambling and addiction to shopping.
Nalmefene is an opiate derivative similar in both structure and activity to the opiate antagonist naltrexone. Advantages of nalmefene relative to naltrexone include longer half-life, greater oral bioavailability and no observed dose-dependent liver toxicity. As with other drugs of this type, nalmefene can precipitate acute withdrawal symptoms in patients who are dependent on opioid drugs, or more rarely when used post-operatively to counteract the effects of strong opioids used in surgery.
Nalmefene differs from naltrexone by substitution of the ketone group at the 6-position of naltrexone with a methylene group (CH2), which considerably increases binding affinity to the μ-opioid receptor. Nalmefene also has high affinity for the other opioid receptors, and is known as a “universal antagonist” for its ability to block all three.
In clinical trials using this drug, doses used for treating alcoholism were in the range of 20–80 mg per day, orally.[2] The doses tested for treating pathological gambling were between 25–100 mg per day.[3] In both trials, there was little difference in efficacy between the lower and higher dosage regimes, and the lower dose (20 and 25 mg, respectively) was the best tolerated, with similar therapeutic efficacy to the higher doses and less side effects. Nalmefene is thus around twice as potent as naltrexone when used for the treatment of addictions.
Intravenous doses of nalmefene at between 0.5 to 1 milligram have been shown effective at counteracting the respiratory depression produced by opiate overdose,[4] although this is not the usual application for this drug as naloxone is less expensive.
Doses of nalmefene greater than 1.5 mg do not appear to give any greater benefit in this application. Nalmefene’s longer half-life might however make it useful for treating overdose involving longer acting opioids such as methadone, as it would require less frequent dosing and hence reduce the likelihood of renarcotization as the antagonist wears off.
Nalmefene is extensively metabolised in the liver, mainly by conjugation with glucuronic acid and also by N-dealkylation. Less than 5% of the dose is excreted unchanged. The glucuronide metabolite is entirely inactive, while the N-dealkylated metabolite has minimal pharmacological activity.
Lundbeck has licensed the drug from Biotie Therapies and performed clinical trials with nalmefene for treatment of alcohol dependence.[5] In 2011 they submitted an application for their drug termed Selincro to the European Medicines Agency.[6] It has not been available on the US market since at least August 2008.[citation needed]
Soluble in water up to 130 mg/mL, soluble in chloroform up to 0.13 mg/mL
pKa 7.6
Distribution half-life: 41 minutes
Nalmefene is a known opioid receptor antagonist which can inhibit pharmacological effects of both administered opioid agonists and endogenous agonists deriving from the opioid system. The clinical usefulness of nalmefene as antagonist comes from its ability to promptly (and selectively) reverse the effects of these opioid agonists, including the frequently observed depressions in the central nervous system and the respiratory system.
Nalmefene has primarily been developed as the hydrochloride salt for use in the management of alcohol dependency, where it has shown good effect in doses of 10 to 40 mg taken when the patient experiences a craving for alcohol (Karhuvaara et al, Alcohol. Clin. Exp. Res., (2007), Vol. 31 No. 7. pp 1179-1187). Additionally, nalmefene has also been investigated for the treatment of other addictions such as pathological gambling and addiction to shopping. In testing the drug in these developmental programs, nalmefene has been used, for example, in the form of parental solution (Revex™).
Nalmefene is an opiate derivative quite similar in structure to the opiate antagonist naltrexone. Advantages of nalmefene compared to naltrexone include longer half- life, greater oral bioavailability and no observed dose-dependent liver toxicity. Nalmefene differs structurally from naltrexone in that the ketone group at the 6- position of naltrexone is replaced by a methylene (CH2) group, which considerably increases binding affinity to the μ-opioid receptor. Nalmefene also has high affinity for the other opioid receptors (K and δ receptors) and is known as a “universal antagonist” as a result of its ability to block all three receptor types.
Nalmefene can be produced from naltrexone by the Wittig reaction. The Wittig reaction is a well known method within the art for the synthetic preparation of olefins (Georg Wittig, Ulrich Schόllkopf (1954). “Uber Triphenyl-phosphin- methylene ah olefinbildende Reagenzien I”. Chemische Berichte 87: 1318), and has been widely used in organic synthesis.
The procedure in the Wittig reaction can be divided into two steps. In the first step, a phosphorus ylide is prepared by treating a suitable phosphonium salt with a base. In the second step the ylide is reacted with a substrate containing a carbonyl group to give the desired alkene.
The preparation of nalmefene by the Wittig reaction has previously been disclosed by Hahn and Fishman (J. Med. Chem. 1975, 18, 259-262). In their method, naltrexone is reacted with the ylide methylene triphenylphosphorane, which is prepared by treating methyl triphenylphosphonium bromide with sodium hydride (NaH) in DMSO. An excess of about 60 equivalents of the ylide is employed in the preparation of nalmefene by this procedure.
For industrial application purposes, the method disclosed by Hahn and Fishman has the disadvantage of using a large excess of ylide, such that very large amounts phosphorus by-products have to be removed before nalmefene can be obtained in pure form. Furthermore, the NaH used to prepare the ylide is difficult to handle on an industrial scale as it is highly flammable. The use of NaH in DMSO is also well known by the skilled person to give rise to unwanted runaway reactions. The Wittig reaction procedure described by Hahn and Fishman gives nalmefene in the form of the free base. The free base is finally isolated by chromatography, which may be not ideal for industrial applications.
US 4,535,157 also describes the preparation of nalmefene by use of the Wittig reaction. In the method disclosed therein the preparation of the ylide methylene triphenylphosphorane is carried out by using tetrahydrofuran (THF) as solvent and potassium tert-butoxidc (KO-t-Bu) as base. About 3 equivalents of the ylide are employed in the described procedure.
Although the procedure disclosed in US 4,535,157 avoids the use of NaH and a large amount of ylide, the method still has some drawbacks which limit its applicability on an industrial scale. In particular, the use of THF as solvent in a Wittig reaction is disadvantageous because of the water miscibility of THF. During the aqueous work-up much of the end product (nalmefene) may be lost in the aqueous phases unless multiple re-extractions are performed with a solvent which is not miscible with water.
Furthermore, in the method described in US 4,535,157, multiple purification steps are carried out in order to remove phosphine oxide by-products of the Wittig reaction. These purification steps require huge amounts of solvents, which is both uneconomical and labor extensive requiring when running the reaction on an industrial scale. As in the case of the Wittig reaction procedure described by Hahn and Fishman (see above) the Wittig reaction procedure disclosed in US 4,535,157 also yields nalmefene as the free base, such that an additional step is required to prepare the final pharmaceutical salt form, i.e. the hydrochloride, from the isolated nalmefene base.
US 4,751,307 also describes the preparation of nalmefene by use of the Wittig reaction. Disclosed is a method wherein the synthesis is performed using anisole (methoxybenzene) as solvent and KO-t-Bu as base. About 4 equivalents of the ylide methylene triphenylphosphorane were employed in this reaction. The product was isolated by extraction in water at acidic pHs and then precipitating at basic pHs giving nalmefene as base.
Even though the isolation procedure for nalmefene as free base is simplified, it still has some disadvantages. The inventors of the present invention repeated the method disclosed in US 4,751,307 and found that the removal of phosphine oxide by-products was not efficient. These impurities co-precipitate with the nalmefene during basifϊcation, yielding a product still contaminated with phosphorus byproducts and having, as a consequence, a low chemical purity, as illustrated in example 2 herein.
There is therefore a need within the field to improve the method of producing nalmefene by the Wittig reaction. In particular, there is a need for a method that is readily applicable on a large industrial scale and which avoids the use of water- miscible solvents, such as THF, in the Wittig reaction, and permits easy isolation of nalmefene in a pure form suitable for its transformation to the final pharmaceutical salt form.
present invention the Wittig reaction may be performed by mixing a methyltriphenylphosphonium salt with 2- methyltetrahydrofuran (MTHF) and a suitable base to afford the ylide methylene triphenylphosphorane :
Methyltriphenylphosphonium salt Methylene triphenylphosphorane Yhde
The preformed ylide is subsequently reacted ‘in situ’ with naltrexone to give nalmefene and triphenylphosphine oxide (TPPO):
Naltrexone Yhde Nalmefene TPPO
Example 1 Methyltriphenylphosphonium bromide (MTPPB, 25.8 Kg) was suspended in 2- methyltetrahydrofuran (MTHF, 56 litres). Keeping the temperature in the range 20-250C, KO-t-Bu (8.8 kg) was charged in portions under inert atmosphere in one hour. The suspension turned yellow and was stirred further for two hours. An anhydrous solution of naltrexone (8.0 Kg) in MTHF (32 litres) was then added over a period of one hour at 20-250C. The suspension was maintained under stirring for a few hours to complete the reaction. The mixture was then treated with a solution of ammonium chloride (4.2 Kg) in water (30.4 litres) and then further diluted with water (30.4 litres). The phases were separated, the lower aqueous phase was discarded and the organic phase was washed twice with water (16 litres). The organic phase was concentrated to residue under vacuum and then diluted with dichloromethane (40 litres) to give a clear solution. Concentrated aqueous hydrochloric acid (HCl 37%, 2 litres) was added over one hour at 20- 250C. The suspension was stirred for at least three hours at the same temperature, and then filtered and washed with dichloromethane (8 litres) and then with acetone (16 litres). The solid was then re-suspended in dichloromethane (32 litres) at 20-250C for a few hours and then filtered and washed with dichloromethane (16 litres), affording 9.20 Kg of nalmefene hydrochloride, corresponding to 7.76 kg of nalmefene hydrochloride (99.7% pure by HPLC). Molar yield 89%.
HPLC Chromatographic conditions
Column: Zorbax Eclipse XDB C-18, 5 μm, 150 x 4.6 mm or equivalent Mobile Phase A: Acetonitrile / Buffer pH = 2.3 10 / 90
Buffer: Dissolve 1.1 g of Sodium Octansulfonate in 1 L of water. Adjust the pH to 2.3 with diluted
H3PO4. Column Temperature: 35°C
Detector: UV at 230 nm
Flow: 1.2 ml/min
Injection volume: 10 μl
Time of Analysis: 55 minutes
Example 2
The procedure described in US 4,751,307 was repeated, starting from 1Og of naltrexone and yielding 8.5g of nalmefene. The isolated product showed the presence of phosphine oxides by-products above 15% molar as judged by 1HNMR.
Example 3.
Methyltriphenylphosphonium bromide (MTPPB, 112.9g) was suspended in 2- methyltetrahydrofuran (MTHF, 245 ml). Keeping the temperature in the range 20- 25°C, KO-t-Bu (38.7 g) was charged in portions under inert atmosphere in one hour. The suspension was stirred for two hours. An anhydrous solution of naltrexone (35 g) in MTHF (144 ml) was then added over a period of one hour at 20-250C. The suspension was maintained under stirring overnight. The mixture was then treated with a solution of glacial acetic acid (17.7 g) in MTHF. Water was then added and the pH was adjusted to 9-10. The phases were separated, the lower aqueous phase was discarded and the organic phase was washed twice with water. The organic phase was concentrated to residue under vacuum and then diluted with dichloromethane (175 ml) to give a clear solution. Concentrated aqueous hydrochloric acid (HCl 37%, 10. Ig) was added over one hour at 20- 25°C. The suspension was stirred and then filtered and washed with dichloromethane and acetone. The product was dried affording 38.1g of Nalmefene HCl. Example 4
Example 3 was repeated but the Wittig reaction mixture after olefmation completeness was treated with acetone and then with an aqueous solution of ammonium chloride. After phase separation, washings, distillation and dilution with dichloromethane, the product was precipitated as hydrochloride salt using HCl 37%. The solid was filtered and dried affording 37.6 g of Nalmefene HCl.
Example 5 Preparation of Nalmefene HCl dihydrate from Nalmefene HCl Nalmefene HCl (7.67 Kg, purity 99.37%, assay 93.9%) and water (8.6 litres) were charged into a suitable reactor. The suspension was heated up to 800C until the substrate completely dissolved. Vacuum was then applied to remove organic solvents. The resulting solution was filtered through a 0.65 μm cartridge and then diluted with water (2.1 litres) that has been used to rinse the reactor and pipelines. The solution was cooled down to 500C and 7 g of Nalmefene HCl dihydrate seeding material was added. The mixture was cooled to 0-50C over one hour with vigorous stirring and then maintained under stirring for one additional hour. The solid was filtered of and washed with acetone. The wet product was dried at 25°C under vacuum to provide 5.4 Kg of Nalmefene HCl dihydrate (purity 99.89%, KF 8.3% , yield 69%).
Lundbeck’s novel alcohol dependency drug has been endorsed by the National Institute for Health and Care Excellence (NICE) for use in Britain’s state health service.
A structural analog of Naltrexone (N285780) with opiate antagonist activity used in pharmaceutical treatment of alcoholism. Other pharmacological applications of this compound aim to reduce food cravings, drug abuse and pulmonary disease in affected individuals. Used as an opioid-induced tranquilizer on large animals in the veterinary industry. Narcotic antagonist.
Lundbeck will be celebrating news that European regulators have issued a green light for Selincro, making it the first therapy approved for the reduction of alcohol consumption in dependent adults.
Selincro (nalmefene) is a unique dual-acting opioid system modulator that acts on the brain’s motivational system, which is dysregulated in patients with alcohol dependence.
The once daily pill has been developed to be taken on days when an alcoholic feels at greater risk of having a drink, in a strategy that aims to reduce – rather than stop – alcohol consumption, which some experts believe is a more realistic goal.
Clinical trials of the drug have shown that it can reduce alcohol consumption by approximately 60% after six months treatment, equating to an average reduction of nearly one bottle of wine per day.
In March last year, data was published from two Phase III trials, ESENSE 1 and ESENSE 2, showing that the mean number of heavy drinking days decreased from 19 to 7 days/month and 20 to 7 days/month, while TAC fell from 85 to 43g/day and from 93 to 30g/day at month six. However, the placebo effect was also strong in the studies.
According to Anders Gersel Pedersen, Executive Vice President and Head of Research & Development at Lundbeck, Selincro “represents the first major innovation in the treatment of alcohol dependence in many years,” and he added that its approval “is exciting news for the many patients with alcohol dependence who otherwise may not seek treatment”.
Alcohol dependence is considered a major public health concern, and yet it is both underdiagnosed and undertreated, highlighting the urgent need for better management of the condition.
In Europe, more than 90% of the 14 million patients with alcohol dependence are not receiving treatment, but research suggests that treating just 40% of these would save 11,700 lives each year.
The Danish firm said it expects to launch Selincro in its first markets in mid-2013, and that it will provide the drug as part of “a new treatment concept that includes continuous psychosocial support focused on the reduction of alcohol consumption and treatment adherence”.
Nalmefene (Revex), originally known as nalmetrene, is an opioid receptor antagonistdeveloped in the early 1970s, and used primarily in the management of alcoholdependence, and also has been investigated for the treatment of other addictions such aspathological gambling and addiction to shopping.
Nalmefene is an opiate derivative similar in both structure and activity to the opiate antagonist naltrexone. Advantages of nalmefene relative to naltrexone include longer half-life, greater oral bioavailability and no observed dose-dependent liver toxicity. As with other drugs of this type, nalmefene can precipitate acute withdrawal symptoms in patients who are dependent on opioid drugs, or more rarely when used post-operatively to counteract the effects of strong opioids used in surgery.
Nalmefene differs from naltrexone by substitution of the ketone group at the 6-position of naltrexone with a methylene group (CH2), which considerably increases binding affinity to the μ-opioid receptor. Nalmefene also has high affinity for the other opioid receptors, and is known as a “universal antagonist” for its ability to block all three.
US patent 3814768, Jack Fishman et al, “6-METHYLENE-6-DESOXY DIHYDRO MORPHINE AND CODEINE DERIVATIVES AND PHARMACEUTICALLY ACCEPTABLE SALTS”, published 1971-11-26, issued 1974-06-04
Sihuan Pharmaceutical Holdings Group Ltd a leading pharmaceutical company with the largest cardio-cerebral vascular drug franchise in China’s prescription market, announced that the new Category 3.1 drug, the Nalmefene Hydrochloride Injection received a new drug certificate (H20120078) and approval for production (2012S00818) from the State Food and Drug Administration. Nalmefene Hydrochloride is yet another generic drug for which the Company has received approval for production following the Roxatidine Acetate Hydrochloridefor Injection. It will be manufactured by Beijing Sihuan Pharmaceutical Co., Ltd., a wholly-owned manufacturing subsidiary of the Company.
Nalmefene hydrochloride is a next generation opioid (opium) receptor inhibitor following Naloxone and Naltrexone. The injection formulation of Naloxone hydrochloride was invented by Ohmeda Pharmaceuticals and was approved by the US Food and Drug Administration (FDA) in 1995. The clinical uses of Nalmefene hydrochloride include anti-shock, neuroprotection, treatment for acute morphine poisoning, drug relapse prevention, recovery from the after-effects of anesthesia such as respiratory and nerve center depression and the treatment of unconsciousness persons.
The drug is also effective for treating heart failure and spinal cord injuries, for cerebral protection, etc. Multi-centre, randomized, blind, and positive-controlled clinical research of Nalmefene hydrochloride of Sihuan Pharmaceutical were performed by the Peking University First Hospital, the First Affiliated Hospital of China Medical University, Xijing Hospital (The First Affiliated Hospital of the Fourth Military Medical College) and Qingdao Municipal Hospital.
Compared to Naloxone, Nalmefene demonstrates longer curative effects and fewer adverse reactions. With its high bioavailability, biological activities and biofilm penetration ability, it helps to regulate respiration, circulation, digestion, and the endocrine and nervous systems. It is becoming a substitute for Naloxone, and has been included in Part B of the National Medicine Catalogue. At present, the size of the Nalmefene hydrochloride market in China is approximately RMB1 billion. As a substitution for Naloxone hydrochloride, Nalmefene hydrochloride has enormous market potential.
Diseases of the central nervous system (CNS) are common in China, which has an immense patient base. Due to the rapid pace of modern life, accelerated urbanisation and mental stress, the demand for CNS medicines has seen rapid growth in recent years given the rising number of patients. According to IMS, the size of the CNS drug market now exceeds RMB 23 billion. With the CNS drug market expected to reach RMB 100 billion in 2020, the Group sees great potential and strong growth prospects in the market.Dr. Che Fengsheng, Chairman and CEO of Sihuan Pharmaceutical, said, “Nalmefene Hydrochloride has shown better characteristics for treatment and higher clinical value than Naloxone. Its market demonstrates great potential to expand. Leveraging Sihuan Pharmaceutical’s strong marketing capabilities and extensive sales and distribution network, we believe that our market share for Nalmefene Hydrochloride will see rapid growth, which will strengthen our position in drugs for the treatment of major diseases of the central nervous system. Together with other new products, this will in turn enhance the continuous development and growth of Sihuan Pharmaceutical in China’s prescription drug market and create value for the shareholders and the Company.”
REVEX (nalmefene hydrochloride injection), an opioid antagonist, is a 6-methylene analogue of naltrexone. The chemical structure is shown below:
Molecular Formula: C21H25NO3•HCl
Molecular Weight: 375.9, CAS # 58895-64-0
Chemical Name: 17-(Cyclopropylmethyl)-4,5a-epoxy-6-methylenemorphinan-3,14-diol, hydrochloride salt.
Nalmefene hydrochloride is a white to off-white crystalline powder which is freely soluble in water up to 130 mg/mL and slightly soluble in chloroform up to 0.13 mg/mL, with a pKa of 7.6.
REVEX is available as a sterile solution for intravenous, intramuscular, and subcutaneous administration in two concentrations, containing 100 µg or 1.0 mg of nalmefene free base per mL. The 100 µg/mL concentration contains 110.8 µg of nalmefene hydrochloride and the 1.0 mg/mL concentration contains 1.108 mg of nalmefene hydrochloride per mL. Both concentrations contain 9.0 mg of sodium chloride per mL and the pH is adjusted to 3.9 with hydrochloric acid.
Concentrations and dosages of REVEX are expressed as the free base equivalent of nalmefene
////////////////////JF-1, NIH-10365, ORF-11676, SRD-174, JAPAN 2019, FDA 1995, Nalmefene hydrochloride dihydrate, ナルメフェン塩酸塩水和物 , Nalmefene, ema 2013, china, 2013, Lu-AA36143
Class Antihyperglycaemics; Antihypertensives; Pyrroles; Small molecules; Sulfones
Mechanism of Action Mineralocorticoid receptor antagonists
Registered Hypertension
Phase III Diabetic nephropathies
No development reported Cardiovascular disorders; Heart failure
09 Jan 2019 Registered for Hypertension in Japan (PO) – First global approval
27 Nov 2018 Daiichi Sankyo completes a phase III trial in Diabetic nephropathies in Japan (PO) (JapicCTI-173696)
08 Jun 2018 Efficacy and adverse events data from the phase III ESAX-HTN trial in Essential hypertension presented 28th European Meeting on Hypertension and Cardiovascular Protection (ESH-2018)
CS 3150, angiotensin II receptor antagonist, for the treatment or prevention of such hypertension and heart disease similar to olmesartan , losartan, candesartan , valsartan, irbesartan, telmisartan, eprosartan,
Cas name 1H-Pyrrole-3-carboxamide, 1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-, (5S)-
Esaxerenone 1632006-28-0, FDA approved new drug will be a big potential drug. Original Route of Synthesis …
Esaxerenone, also known as CS-3150, XL-550, is a nonsteroidal antimineralocorticoid which was discovered by Exelixis and is now under development by Daiichi Sankyo Company for the treatment of hypertension, essential hypertension, hyperaldosteronism, and diabetic nephropathies. It acts as a highly selective silent antagonist of the mineralocorticoid receptor (MR), the receptor for aldosterone, with greater than 1,000-fold selectivity for this receptor over other steroid hormone receptors, and 4-fold and 76-fold higher affinity for the MR relative to the existing antimineralocorticoids spironolactone and eplerenone.
01 Jan 2015Daiichi Sankyo initiates a phase IIb trial for Diabetic nephropathies in Japan (NCT02345057)
01 Jan 2015Daiichi Sankyo initiates a phase IIb trial for Hypertension in Japan (NCT02345044)
01 May 2013Phase-II clinical trials in Diabetic nephropathies in Japan (PO)
Currently, angiotensin II receptor antagonists and calcium antagonists are widely used as a medicament for the treatment or prevention of such hypertension or heart disease.
Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.
Renin – angiotensin II receptor antagonists are inhibitors of angiotensin system is particularly effective in renin-dependent hypertension, and show a protective effect against cardiovascular and renal failure. Also, the calcium antagonists, and by the function of the calcium channel antagonizes (inhibits), since it has a natriuretic action in addition to the vasodilating action, is effective for hypertension fluid retention properties (renin-independent) .
Therefore, the MR antagonist, when combined angiotensin II receptor antagonists or calcium antagonists, it is possible to suppress the genesis of multiple hypertension simultaneously, therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology is expected to exhibit.
Also, diuretics are widely used as a medicament for the treatment or prevention of such hypertension or heart disease. Diuretic agent is effective in the treatment of hypertension from its diuretic effect. Therefore, if used in combination MR antagonists and diuretics, the diuretic effect of diuretics, it is possible to suppress the genesis of multiple blood pressure at the same time, shows a therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology it is expected.
1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (hereinafter, compound ( I)) is, it is disclosed in Patent Documents 1 and 2, hypertension, for the treatment of such diabetic nephropathy are known to be useful.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Useful as a mineralocorticoid receptor (MR) antagonist, for treating hypertension, cardiac failure and diabetic nephropathy. It is likely to be CS-3150, a non-steroidal MR antagonist, being developed by Daiichi Sankyo (formerly Sankyo), under license from Exelixis, for treating hypertension and diabetic nephropathy (phase 2 clinical, as of March 2015). In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month (NCT02345057).
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.
Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.
Daiichi Sankyo (formerly Sankyo), under license from Exelixis, is developing CS-3150 (XL-550), a non-steroidal mineralocorticoid receptor (MR) antagonist, for the potential oral treatment of hypertension and diabetic nephropathy, microalbuminuria , By October 2012, phase II development had begun ; in May 2014, the drug was listed as being in phase IIb development . In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month. At that time, the trial was expected to complete in March 2017 .
Exelixis, following its acquisition of X-Ceptor Therapeutics in October 2004 , was investigating the agent for the potential treatment of metabolic disorders and cardiovascular diseases, such as hypertension and congestive heart failure . In September 2004, Exelixis expected to file an IND in 2006. However, it appears that the company had fully outlicensed the agent to Sankyo since March 2006 .
Description
Small molecule antagonist of the mineralocorticoid receptor (MR)
In January 2015, a multi-center, placebo-controlled, randomized, 5-parallel group, double-blind, phase II trial (JapicCTI-152774; NCT02345057; CS3150-B-J204) was planned to be initiated later that month in Japan, in patients with type 2 diabetes mellitus and microalbuminuria, to assess the efficacy and safety of different doses of CS-3150 compared to placebo. At that time, the trial was expected to complete in March 2017; later that month, the trial was initiated in the Japan
By October 2012, phase II development had begun in patients with essential hypertension
By January 2011, phase I trials had commenced in Japan
After methyl 4-methyl-5-[2-(trifluoromethyl) phenyl]-1H-pyrrole-3-carboxylate was obtained by the method described in Example 16 of WO 2006/012642 , the following reaction was performed using this compound as a raw material.
Methyl 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate (1.4 g, 4.9 mmol) was dissolved in methanol (12 mL), and a 5 M aqueous sodium hydroxide solution (10 mL) was added thereto, and the resulting mixture was heated under reflux for 3 hours. After the mixture was cooled to room temperature, formic acid (5 mL) was added thereto to stop the reaction. After the mixture was concentrated under reduced pressure, water (10 mL) was added thereto to suspend the resulting residue. The precipitated solid was collected by filtration and washed 3 times with water. The obtained solid was dried under reduced pressure, whereby 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylic acid (1.1 g, 83%) was obtained as a solid. The thus obtained solid was suspended in dichloromethane (10 mL), oxalyl chloride (0.86 mL, 10 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 2 hours. After the mixture was concentrated under reduced pressure, the residue was dissolved in tetrahydrofuran (10 mL), and 4-(methylsulfonyl)aniline hydrochloride (1.0 g, 4.9 mmol) and N,N-diisopropylethylamine (2.8 mL, 16 mmol) were sequentially added to the solution, and the resulting mixture was heated under reflux for 18 hours. After the mixture was cooled to room temperature, the solvent was distilled off under reduced pressure, and acetonitrile (10 mL) and 3 M hydrochloric acid (100 mL) were added to the residue. A precipitated solid was triturated, collected by filtration and washed with water, and then, dried under reduced pressure, whereby 4-methyl-N-[4-(methylsulfonyl) phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (1.4 g, 89%) was obtained as a solid. 1H-NMR (400 MHz, DMSO-d6) δ11.34 (1H, brs,), 9.89 (1H, s), 7.97 (2H, d, J = 6.6 Hz), 7.87-7.81 (3H, m), 7.73 (1H, t, J = 7.4 Hz), 7.65-7.61 (2H, m), 7.44 (1H, d, J = 7.8 Hz), 3.15 (3H, s), 2.01 (3H, s).
Sodium hydride (0.12 g, 3 mmol, 60% dispersion in mineral oil) was dissolved in N,N-dimethylformamide (1.5 mL), and 4-methyl -N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (0.47 g, 1.1 mmol) was added thereto, and then, the resulting mixture was stirred at room temperature for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (0.14 g, 1.2 mmol) was added thereto, and the resulting mixture was stirred at room temperature. After 1 hour, sodium hydride (40 mg, 1.0 mmol, oily, 60%) was added thereto again, and the resulting mixture was stirred for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (12 mg, 0.11 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 1 hour. After the mixture was concentrated under reduced pressure, methanol (5 mL) was added to the residue and insoluble substances were removed by filtration, and the filtrate was concentrated again. To the residue, tetrahydrofuran (2 mL) and 6 M hydrochloric acid (2 mL) were added, and the resulting mixture was stirred at 60°C for 16 hours. The reaction was cooled to room temperature, and then dissolved in ethyl acetate, and washed with water and saturated saline. The organic layer was dried over anhydrous sodium sulfate and filtered. Then, the filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate), whereby the objective compound (0.25 g, 48%) was obtained. 1H-NMR (400 MHz, CDCl3) δ: 7.89-7.79 (m, 6H), 7.66-7.58 (m, 2H), 7.49 (s, 1H), 7.36 (d, 1H, J = 7.4Hz), 3.81-3.63 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1246.
Anal. calcd for C22H21F3N2O4S: C, 56.65; H, 4.54; N, 6.01; F, 12.22; S, 6.87. found: C, 56.39; H, 4.58; N, 5.99; F, 12.72; S, 6.92.
(Example 4)
Optical Resolution of Compound of Example 3
Resolution was performed 4 times in the same manner as in Example 2, whereby 74 mg of Isomer C was obtained as a solid from a fraction containing Isomer C (tR = 10 min), and 71 mg of Isomer D was obtained as a solid from a fraction containing Isomer D (tR = 11 min).
1- [2- (trifluoromethyl) phenyl] propan-1-one 75 g (370 mmol) in t- butyl methyl ether (750 mL), and I was added bromine 1.18 g (7.4 mmol). After confirming that the stirred bromine color about 30 minutes at 15 ~ 30 ℃ disappears, cooled to 0 ~ 5 ℃, was stirred with bromine 59.13 g (370 mmol) while keeping the 0 ~ 10 ℃. After stirring for about 2.5 hours, was added while maintaining 10 w / v% aqueous potassium carbonate solution (300 mL) to 0 ~ 25 ℃, was further added sodium sulfite (7.5 g), was heated to 20 ~ 30 ℃. The solution was separated, washed in the resulting organic layer was added water (225 mL), to give t- butyl methyl ether solution of the title compound and the organic layer was concentrated under reduced pressure (225 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.91 (3H, D, J = 4.0 Hz), 4.97 (1H, Q, J = 6.7 Hz), 7.60 ~ 7.74 (4H, M).
2-bromo-1- [2- (trifluoromethyl) phenyl] propan-1 / t- butyl methyl ether solution (220 mL) in dimethylacetamide (367 mL), ethyl cyanoacetate obtained in Example 1 53.39 g (472 mmol), potassium carbonate 60.26 g (436 mmol) were sequentially added, and the mixture was stirred and heated to 45 ~ 55 ℃. After stirring for about 2 hours, 20 is cooled to ~ 30 ℃, water (734 mL) and then extracted by addition of toluene (367 mL), washed by adding water (513 mL) was carried out in the organic layer (2 times implementation). The resulting organic layer was concentrated under reduced pressure to obtain a toluene solution of the title compound (220 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.33 ~ 1.38 (6H, M), 3.80 ~ 3.93 (2H, M), 4.28 ~ 4.33 (2H, M), 7.58 ~ 7.79 (4H, M).
The 20 ~ 30 ℃ 2-cyano-3-methyl-4-oxo-4 was obtained [2- (trifluoromethyl) phenyl] butanoate in toluene (217 mL) by the method of Example 2 ethyl acetate (362 mL) Te, after the addition of thionyl chloride 42.59 g (358 mmol), cooled to -10 ~ 5 ℃, was blown hydrochloric acid gas 52.21 g (1432 mmol), further concentrated sulfuric acid 17.83 g (179 mmol) was added, and the mixture was stirred with hot 15 ~ 30 ℃. After stirring for about 20 hours, added ethyl acetate (1086 mL), warmed to 30 ~ 40 ℃, after the addition of water (362 mL), and the layers were separated. after it separated organic layer water (362 mL) was added for liquid separation, and further 5w / v% was added for liquid separation aqueous sodium hydrogen carbonate solution (362 mL).
Subsequently the organic layer was concentrated under reduced pressure, the mixture was concentrated under reduced pressure further added toluene (579 mL), was added toluene (72 mL), and cooled to 0 ~ 5 ℃. After stirring for about 2 hours, the precipitated crystals were filtered, and washed the crystals with toluene which was cooled to 0 ~ 5 ℃ (217 mL). The resulting wet goods crystals were dried under reduced pressure at 40 ℃, the title compound was obtained (97.55 g, 82.1% yield).
Example obtained by the production method of the three 2-chloro-4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylate 97.32 g (293 mmol) in ethanol (662 mL), tetrahydrofuran (117 mL), water (49 mL), sodium formate 25.91 g (381 mmol) and 5% palladium – carbon catalyst (water content 52.1%, 10.16 g) was added at room temperature, heated to 55 ~ 65 ℃ the mixture was stirred. After stirring for about 1 hour, cooled to 40 ℃ less, tetrahydrofuran (97 mL) and filter aid (KC- flock, Nippon Paper Industries) 4.87 g was added, the catalyst was filtered and the residue using ethanol (389 mL) was washed. The combined ethanol solution was used for washing the filtrate after concentration under reduced pressure, and with the addition of water (778 mL) was stirred for 0.5 hours at 20 ~ 30 ℃. The precipitated crystals were filtered, and washed the crystals with ethanol / water = 7/8 solution was mixed with (292 mL). The resulting wet goods crystals were dried under reduced pressure at 40 ℃, the title compound was obtained (86.23 g, 98.9% yield).
N to the fourth embodiment of the manufacturing method by the resulting 4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylate 65.15 g (219 mmol), N- dimethylacetamide ( 261 mL), ethylene carbonate 28.95 g (328.7 mmol), 4- dimethylaminopyridine 2.68 g (21.9 mmol) were sequentially added at room temperature, and heated to 105 ~ 120 ℃, and the mixture was stirred. After stirring for about 10 hours, toluene was cooled to 20 ~ 30 ℃ (1303 mL), and the organic layer was extracted by adding water (326 mL). Subsequently, was washed by adding water (326 mL) to the organic layer (three times). The resulting organic layer was concentrated under reduced pressure, ethanol (652 mL) was added, and was further concentrated under reduced pressure, ethanol (130 mL) was added to obtain an ethanol solution of the title compound (326 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.35 (3H, t, J = 7.1 Hz), 1.84 (1H, Broad singlet), 2.00 (3H, s), 3.63 ~ 3.77 (4H, M), 4.27 (2H , m), 7.35 ~ 7.79 (5H, m).
Obtained by the method of Example 5 (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid ethyl / ethanol (321 mL) solution in water (128.6 mL), was added at room temperature sodium hydroxide 21.4 g (519 mmol), and stirred with heating to 65 ~ 78 ℃. After stirring for about 6 hours, cooled to 20 ~ 30 ℃, after the addition of water (193 mL), and was adjusted to pH 5.5 ~ 6.5, while maintaining the 20 ~ 30 ℃ using 6 N hydrochloric acid. was added as seed crystals to the pH adjustment by a liquid (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 6.4 mg , even I was added to water (193mL). Then cooled to 0 ~ 5 ℃, again, adjusted to pH 3 ~ 4 with concentrated hydrochloric acid and stirred for about 1 hour. Then, filtered crystals are precipitated, and washed the crystals with 20% ethanol water is cooled to 0 ~ 5 ℃ (93 mL). The resulting wet product crystals were dried under reduced pressure at 40 ℃, to give the title compound (64.32 g, 95.0% yield). 1 H NMR (400 MHz, DMSO-D 6 ) delta: 1.87 (3H, s), 3.38 ~ 3.68 (4H, M), 7.43 ~ 7.89 (5H, M).
(Example 7)
(S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
(7-1) (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
obtained by the method of Example 6 the (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 50.00 g (160 mmol), N, N- dimethylacetamide (25 mL), ethyl acetate (85 mL) was added and dissolved at room temperature (solution 1).
Quinine 31.05 g (96 mmol) in N, N- dimethylacetamide (25 mL), ethyl acetate (350 mL), was heated in water (15 mL) 65 ~ 70 ℃ was added, was added dropwise a solution 1. After about 1 hour stirring the mixture at 65 ~ 70 ℃, and slowly cooled to 0 ~ 5 ℃ (cooling rate standard: about 0.3 ℃ / min), and stirred at that temperature for about 0.5 hours. The crystals were filtered, 5 ℃ using ethyl acetate (100 mL) which was cooled to below are washed crystals, the resulting wet product crystals was obtained and dried under reduced pressure to give the title compound 43.66 g at 40 ℃ (Yield 42.9%). Furthermore, the diastereomeric excess of the obtained salt was 98.3% de. 1 H NMR (400 MHz, DMSO-D 6 ) delta: 1.30 ~ 2.20 (10H, M), 2.41 ~ 2.49 (2H, M), 2.85 ~ 3.49 (6H, M), 3.65 ~ 3.66 (1H, M), 3.88 (3H, s), 4.82 (1H, broad singlet), 4.92 ~ 5.00 (2H, m), 5.23 ~ 5.25 (1H, m), 5.60 (1H, br), 5.80 ~ 6.00 (1H, m), 7.36 ~ 7.92 (9H, M), 8.67 (1H, D, J = 4.6 Hz) (7-2) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3 diastereomeric excess of the carboxylic acid quinine salt HPLC measurements (% de) that the title compound of about 10 mg was collected, and the 10 mL was diluted with 50v / v% aqueous acetonitrile me was used as a sample solution.
Column: DAICEL CHIRALPAK IC-3 (4.6 mmI.D. × 250 mm, 3 μm)
mobile phase A: 0.02mol / L phosphorus vinegar buffer solution (pH 3)
mobile phase B: acetonitrile
solution sending of mobile phase: mobile phase A and I indicates the mixing ratio of mobile phase B in Table 1 below.
[Table 1]
Detection: UV 237 nm
flow rate: about 0.8 mL / min
column temperature: 30 ℃ constant temperature in the vicinity of
measuring time: about 20 min
Injection volume: 5 μL
diastereomeric excess (% de), the title compound (retention time about 12 min), was calculated by the following equation using a peak area ratio of R-isomer (retention time of about 13 min).
% De = {[(the title compound (S body) peak area ratio) – (R body peak area ratio)] ÷ [(the title compound (S body) peak area ratio) + (R body peak area ratio)]} × 100
(Example 8)
(S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxamide (Compound (A))
(8-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3 – carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 40.00 g (63 mmol) in ethyl acetate (400 mL), was added 2N aqueous hydrochloric acid (100 mL) was stirred at room temperature and separated . The resulting organic layer was concentrated under reduced pressure (120 mL), and added ethyl acetate (200 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (120 mL).
(8-2) N – {[4- (methylsulfonyl) phenyl] amino} oxamic acid 2 – ((S) -3- methyl-4 – {[4- (methylsulfonyl) phenyl] carbamoyl} -2- [ 2- (trifluoromethyl) phenyl] -1H- pyrrol-1-yl) ethyl
ethyl acetate (240 mL), was mixed tetrahydrofuran (80 mL) and oxalyl chloride 20.72 g (163 mmol), and cooled to 10 ~ 15 ℃ was. Then the resulting solution was added while keeping the 10 ~ 15 ℃ Example (8-1) and stirred for about 1 hour by heating to 15 ~ 20 ℃. After stirring, acetonitrile (120 mL) and pyridine 2.46 g (31 mmol) was added and the reaction mixture was concentrated under reduced pressure (120 mL), acetonitrile (200 mL) was added and further concentrated under reduced pressure (120 mL).
After completion concentration under reduced pressure, acetonitrile (200 mL) was added and cooled to 10 ~ 15 ℃ (reaction 1).
Acetonitrile (240mL), pyridine 12.39 g (157 mmol), 4- were successively added (methylsulfonyl) aniline 26.85 g (157 mmol), the reaction solution 1 was added while maintaining the 10 ~ 15 ℃, the 20 ~ 25 ℃ and the mixture was stirred and heated to about 1 hour.
The resulting reaction solution in acetonitrile (40 mL), 2 N hydrochloric acid water (120 mL), was added sodium chloride (10.0 g) was stirred, and the layers were separated. Again, 2N aqueous hydrochloric acid to the organic layer (120 mL), was added sodium chloride (10.0 g) was stirred, and the layers were separated. After filtering the resulting organic layer was concentrated under reduced pressure (400 mL). Water (360 mL) was added to the concentrated liquid, after about 1 hour stirring, the crystals were filtered, washed with 50v / v% aqueous acetonitrile (120 mL), wet product of the title compound (undried product, 62.02 g) and obtained. 1 H NMR (500 MHz, DMSO-D 6 ) delta: 1.94 (s, 3H), 3.19 (s, 3H), 3.20 (s, 3H), 3.81 (t, 1H), 4.12 (t, 1H), 4.45 ( t, 2H, J = 5.81 Hz), 7.62 (t, 1H, J = 4.39 Hz), 7.74 (t, 2H, J = 3.68 Hz), 7.86 (dd, 3H), 7.92 (dd, 3H, J = 6.94 , 2.13 Hz), 7.97 (DD, 2H, J = 6.80, 1.98 Hz), 8.02 (DD, 2H), 10.03 (s, 1H), 11.19 (s, 1H)
(8-3) (S)-1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (Compound (A)) ( the resulting wet product crystals 8-2), t- butyl methyl ether (200 mL), acetonitrile (40 mL), 48w / w potassium hydroxide aqueous solution (16 g) and water (200 mL) was added, I was stirred for about 2 hours at 25 ~ 35 ℃. After stirring, and the mixture is separated, the resulting organic layer was concentrated under reduced pressure (120 mL), ethanol (240 mL) was added and further concentrated under reduced pressure (120 mL). After completion concentration under reduced pressure, ethanol (36 mL), and heated in water (12 mL) was added 35 ~ 45 ℃, while maintaining the 35 ~ 45 ℃ was added dropwise water (280 mL), and was crystallized crystals. After cooling the crystal exudates to room temperature, I was filtered crystal. Then washed with crystals 30v / v% aqueous ethanol solution (80 mL), where it was dried under reduced pressure at 40 ℃, the title compound was obtained in crystalline (26.26 g, 89.7% yield). Moreover, the enantiomers of the resulting crystals was 0.3%.
(8-4) (S)-1-(2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3- HPLC method for measuring the amount enantiomer carboxamide (%) and collected the title compound of about 10 mg is, what was the 10 mL was diluted with 50v / v% aqueous acetonitrile to obtain a sample solution.
(12-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H – pyrrole-3-carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 10.00 g (16 mmol) in t- butyl methyl ether (90 mL), water (10 mL) 36w / w% aqueous hydrochloric acid ( 5 mL) was added and stirring at room temperature and separated. The resulting organic layer was concentrated under reduced pressure (30 mL), was added ethyl acetate (50 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (30 mL).
(12-2) N – {[4- (methylsulfonyl) phenyl] amino} oxamic acid 2 – ((S) -3- methyl-4 – {[4- (methylsulfonyl) phenyl] carbamoyl} -2- [ 2- (trifluoromethyl) phenyl] -1H- pyrrol-1-yl) ethyl
ethyl acetate (50 mL), was mixed with tetrahydrofuran (20 mL) and oxalyl chloride 5.18 g (41 mmol), and cooled to 0 ~ 5 ℃ was.Then the resulting solution was added in Examples while maintaining the 0 ~ 5 ℃ (12-1), and the mixture was stirred for 6 hours at 0 ~ 10 ℃. After stirring, acetonitrile (30 mL) and pyridine 0.62 g (8 mmol) was added and the reaction mixture was concentrated under reduced pressure (30 mL), acetonitrile (50 mL) was added, and further concentrated under reduced pressure (30 mL).
After concentration under reduced pressure end, is added acetonitrile (10 mL) and oxalyl chloride 0.10 g (1 mmol), and cooled to 0 ~ 5 ℃ (reaction 1).
Acetonitrile (30mL), pyridine 3.15 g (40 mmol), 4- were successively added (methylsulfonyl) aniline 6.71 g (39 mmol), the reaction solution 1 was added while maintaining the 10 ~ 15 ℃, the 20 ~ 25 ℃ and the mixture was stirred and heated to about 1 hour.
Insolubles from the resulting reaction solution was filtered, washed with acetonitrile (10 mL), and stirred for about 2 hours the addition of water (15 mL), followed by dropwise addition of water (75 mL) over about 1 hour . After about 1 hour stirring the suspension was filtered crystals were washed with 50v / v% aqueous acetonitrile (20 mL), wet product of the title compound (undried product, 15.78 g) to give a. 1 H NMR (500 MHz, DMSO-D 6 ) delta: 1.94 (s, 3H), 3.19 (s, 3H), 3.20 (s, 3H), 3.81 (t, 1H), 4.12 (t, 1H), 4.45 ( t, 2H, J = 5.81 Hz), 7.62 (t, 1H, J = 4.39 Hz), 7.74 (t, 2H, J = 3.68 Hz), 7.86 (dd, 3H), 7.92 (dd, 3H, J = 6.94 , 2.13 Hz), 7.97 (DD, 2H, J = 6.80, 1.98 Hz), 8.02 (DD, 2H), 10.03 (s, 1H), 11.19 (s, 1H)
(12-3) (S)-1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (Compound (A)) ( the resulting wet product crystals 12-2), t- butyl methyl ether (50 mL), acetonitrile (10 mL), 48w / w potassium hydroxide aqueous solution (4 g) and water (50 mL) was added, 15 I was about 2 hours of stirring at ~ 25 ℃. After stirring, and the mixture is separated, the resulting organic layer was concentrated under reduced pressure (30 mL), was added ethanol (60 mL), was further concentrated under reduced pressure (30 mL). After completion concentration under reduced pressure, ethanol (14 mL), after addition of water (20 mL), was added a seed crystal, and was crystallized crystals. After dropwise over about 1 hour water (50 mL), and about 1 hour stirring, and crystals were filtered off. Then washed with crystals 30v / v% aqueous ethanol solution (10 mL), where it was dried under reduced pressure at 40 ℃, the title compound was obtained in crystal (6.36 g, 87.0% yield). Moreover, the enantiomers of the resulting crystals was 0.05%. Enantiomers amount, I was measured by the method of (Example 8-4). 1 H NMR (400 MHz, CDCl 3 ) delta: 1.74 (1H, Broad singlet), 2.08 (3H, s), 3.04 (3H, s), 3.63 ~ 3.80 (4H, M), 7.36 (1H, D, J = 7.2 Hz), 7.48 (1H, s), 7.58 ~ 7.67 (2H, m), 7.77 ~ 7.90 (6H, m).
Angiotensin II receptor 桔抗 agent used as the component (A), olmesartan medoxomil, olmesartan cilexetil, losartan, candesartan cilexetil, valsartan, biphenyl tetrazole compounds such as irbesartan, biphenyl carboxylic acid compounds such as telmisartan, eprosartan, agile Sultan, and the like, preferably, a biphenyl tetrazole compound, more preferably, olmesartan medoxomil, is losartan, candesartan cilexetil, valsartan or irbesartan, particularly preferred are olmesartan medoxomil, losartan or candesartan cilexetil, Most preferably, it is olmesartan medoxomil.
Olmesartan medoxomil, JP-A-5-78328, US Patent No. 5,616,599
is described in Japanese or the like, its chemical name is (5-methyl-2-oxo-1,3-dioxolen-4-yl ) methyl 4- (1-hydroxy-1-methylethyl) -2-propyl-1 – in [2 ‘(1H- tetrazol-5-yl) biphenyl-4-ylmethyl] imidazole-5-carboxylate, Yes, olmesartan medoxomil of the present application includes its pharmacologically acceptable salt.
OLMESARTAN
Losartan (DUP-753) is, JP 63-23868, is described in US Patent No. 5,138,069 JP like, and its chemical name is 2-butyl-4-chloro-1- [2 ‘ – The (1H- tetrazol-5-yl) biphenyl-4-ylmethyl] -1H- is imidazol-5-methanol, application of losartan includes its pharmacologically acceptable salt (losartan potassium salt, etc.).
LOSARTAN
Candesartan cilexetil, JP-A-4-364171, EP-459136 JP, is described in US Patent No. 5,354,766 JP like, and its chemical name is 1- (cyclohexyloxycarbonyloxy) ethyl-2 ethoxy-1- [2 ‘one (1H- tetrazol-5-yl) -4-Bife~eniru ylmethyl] -1H- benzimidazole-7-carboxylate is a salt application of candesartan cilexetil, which is a pharmacologically acceptable encompasses.
Valsartan (CGP-48933), the JP-A-4-159718, are described in EP-433983 JP-like, and its chemical name, (S) -N- valeryl -N- [2 ‘- (1H- tetrazol – It is a 5-yl) biphenyl-4-ylmethyl) valine, valsartan of the present application includes its pharmacologically acceptable ester or a pharmacologically acceptable salt thereof.
Irbesartan (SR-47436), the Japanese Patent Publication No. Hei 4-506222, is described in JP WO91-14679 publication, etc., its chemical name, 2-N–butyl-4-spiro cyclopentane-1- [2′ The (tetrazol-5-yl) biphenyl-4-ylmethyl] -2-imidazoline-5-one, irbesartan of the present application includes its pharmacologically acceptable salts.
Eprosartan (SKB-108566) is described in US Patent No. 5,185,351 JP etc., the chemical name, 3- [1- (4-carboxyphenyl-methyl) -2-n- butyl – imidazol-5-yl] The 2-thienyl – methyl-2-propenoic acid, present in eprosartan, the carboxylic acid derivatives, pharmacologically acceptable ester or a pharmacologically acceptable salt of a carboxylic acid derivative (eprosartan mesylate, encompasses etc.).
Telmisartan (BIBR-277) is described in US Patent No. 5,591,762 JP like, and its chemical name is 4 ‘- [[4 Mechiru 6- (1-methyl-2-benzimidazolyl) -2 – is a propyl-1-benzimidazolyl] methyl] -2-biphenylcarboxylic acid, telmisartan of the present application includes its carboxylic acid derivative, a pharmacologically acceptable ester or a pharmacologically acceptable salt thereof of carboxylic acid derivatives .
Agile Sultan, is described in Patent Publication No. 05-271228 flat JP, US Patent No. 5,243,054 JP like, and its chemical name is 2-ethoxy-1 {[2 ‘- (5-oxo-4,5-dihydro 1,2,4-oxadiazole-3-yl) biphenyl-4-yl] methyl} -1H- benzo [d] imidazole-7-carboxylic acid (2-Ethoxy-1 {[2 ‘- (5- oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) biphenyl-4-yl] is a methyl} -1H-benzo [d] imidazole-7-carboxylic acid).
Class Analgesics; Antineoplastics; Ketones; Pyrimidines; Small molecules
Mechanism of Action LHRH receptor antagonists
Preregistration Uterine leiomyoma
Phase III Pain; Prostate cancer
No development reported Solid tumours
08 Nov 2018 Myovant announces intention to submit NDA for Uterine leiomyoma in Q3 of 2019
08 Nov 2018 Myovant Sciences completes enrollment in the phase III LIBERTY 1 trial for Uterine leiomyoma (Combination therapy) in USA (PO)(NCT03049735)
25 Oct 2018 Myovant Sciences completes enrolment in its phase III HERO trial for Prostate cancer (Late-stage disease) in Denmark, Australia, Austria, Belgium, Canada, United Kingdom, USA, Japan, Taiwan, Sweden, Spain, Slovakia, New Zealand, Netherlands, South Korea, Germany, France and Finland (PO) (NCT03085095)
Relugolix has been used in trials studying the treatment of Endometriosis, Prostate Cancer, Uterine Fibroids, and Androgen Deprivation Treatment-naïve Nonmetastatic Prostate Cancer.
A single oral administration of relugolix at a dose of 3 mg/kg has been found to suppress luteinizing hormone (LH) levels for more than 24 hours in castratedcynomolgus monkeys, indicating a long duration of action.[6] The drug (80–160 mg/day) has been found to reduce testosterone levels to sustained castrate levels in men with once-daily administration.[8] Lower dosages (10–40 mg/day) are being studied in the treatment of endometriosis and uterine fibroids to achieve partial sex hormone suppression.[4] The reasoning behind partial suppression for these conditions is to reduce the incidence and severity of menopausal symptoms such as hot flushes and to avoid bone mineral density changes caused by estrogen deficiency that can eventually lead to osteoporosis.[4][9]
History
Relugolix was first described in 2004.[10][6] It superseded sufugolix, which was developed by the same group.[6]
Society and culture
Generic names
Relugolix is the generic name of the drug and its INN and USAN.[11] It is also known by its developmental code names RVT-601 and TAK-385.[1][11]
SYN
Journal of Medicinal Chemistry, 54(14), 4998-5012; 2011
Production of N-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxy-3-pyridazinyl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-N’-methoxyurea
The similar reaction as described in Example 4 by using the compound (100 mg, 0.164 mmol) obtained in Reference Example 54 and methyl iodide (0.010 ml, 0.164 mmol) gave the title compound (17.3 mg, 17 %) as colorless crystals. 1 H-NMR(CDCl3) δ: 2.15 (6H, s), 3.6-3.8 (2H, m), 3.82 (3H, s), 4.18 (3H, s), 5.35 (2H, s), 6.92 (2H, t, J = 8.2 Hz), 7.12 (1H, d, J = 8.8 Hz), 7.2-7.65 (7H, m), 7.69 (1H, s).
PAPER
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
Method for the production of TAK-385 or its salt and crystals starting from 6-(4-aminophenyl)-1-(2,6-difluorobenzyl)-5-dimethylaminomethyl-3-(6-methoxypyridazin-3-yl) thieno[2,3-d] pyrimidine-2,4 (1H,3H)-dione or its salt. Takeda Pharmaceutical is developing relugolix (TAK-385), an oral LHRH receptor antagonist analog of sufugolix, for the treatment of endometriosis and uterine fibroids. As of April 2014, the drug is in Phase 2 trails. See WO2010026993 claiming method for improving the oral absorption and stability of tetrahydro-thieno[2,3-d]pyrimidin-6-yl]-phenyl)-N’-methoxy urea derivatives.
Endometriosis is a common estrogen-dependent gynecological diseases, often occurs in women during their childbearing years, and its mechanism is unclear. Complex and difficult to diagnose the cause of the symptoms of endometriosis is unknown, serious block to the discovery of effective therapies. Currently, endometriosis primarily by laparoscopy diagnosis, and treatment by surgery, or pill, or progesterone receptor agonists of GnRH reduce estrogen levels to control.
Currently the high incidence of endometriosis, Datamonitor 2009 year data show that only two countries, India and China, the number of female patients suffering from endometriosis had more than 68 million (31,288,000 India, China 3753.5 million) passengers, while the national prevalence of the number seven major markets have more than 17 million. Datamonitor expects 2009 to 2018, endometriosis market from 2009 to $ 764 million (US $ 596 billion and the EU $ 117 million, Japan US $ 051 million) in 2018 increased to US $ 1.156 billion (US 8.44 billion dollars, 206 million US dollars the European Union, Japan $ 106 million), while the Chinese market will have more room for growth.
Gonadotropin-releasing hormone (Gonadoliberin; gonadotropin releasing hormone; GnRH), also known as luteinizing hormone releasing hormone (LHRH), is synthesized by neuroendocrine cells of the hypothalamus hormones decapeptide (pGlu-His-Trp-Ser-Tyr-Gly- Leu-Arg-Pro-Gly-NH2), a central regulator of reproductive endocrine system. Which conveys the circulatory system through hypothalamus-pituitary portal to the pituitary, bind to the cells of the anterior pituitary GnRH receptor, such as gonadotropin luteinizing hormone (Luteinizing Hormone, LH) and FSH (Follicle-Stimulating Hormone, FSH ) secretion and release, regulation of normal development and corpus luteum of the ovary, hypothalamic – pituitary – gonadal axis plays an important role. GnRH receptors capable of activating the G protein coupled calcium phosphatidylinositol second messenger system exert their regulatory role, and LH is adjusted to produce steroids, FSH regulating development of the male and female follicle spermatogenesis.
LH and FSH are released into the circulation, and combined with the ovaries or testes specific cell receptors, stimulating the production of steroids. The presence of sex steroids, diseases such as endometriosis, uterine fibroids, prostate cancer and exacerbations, to be given long-acting GnRH receptor agonists and antagonists for treatment control peptides.
Peptide GnRH receptor antagonists include linear peptides (US 5,171,835) GnRH-derived, cyclic hexapeptide derivatives (US 2002/0065309), a bicyclic peptide derivative (Journal of Medicinal Chemistry, 1993; 36: 3265-73), etc. ; and GnRH receptor peptide agonists include leuprolide (leuprorelin, pGlu-His-Trp-Ser-Tyr-d-Leu-Leu-Arg-Pro-NHEt). However, there are many problems including oral absorbability, dosage form, dose volume, drug stability, sustained action, and metabolic stability of the peptide-type compound to be resolved. But the main reason small molecule GnRH receptor antagonists of peptide-based therapy is superior to the existing method is that small molecule GnRH receptor antagonist may be orally administered directly, convenient. Studies have shown that small molecule antagonists of endometriosis, precocious puberty, prostate cancer and other hormone-dependent diseases having a significant effect.
GnRH receptor agonist mediated indirect mechanisms of tumor suppression by long-term effects on the hypothalamic – pituitary – gonadal axis, leading to pituitary gonadotropins (FSH, LH) is reduced, thereby reducing the secretion of sex hormones and indirectly inhibit growth of tumor cells. And a GnRH receptor antagonist directly to inhibit the release of the pituitary gonadotropins, thereby inhibiting tumor cell growth.
Given the limitations of peptide GnRH receptor antagonists, non-peptide GnRH receptor antagonists have been proposed and into the development, clinical trials and launch phase, such as Elagolix (NBI-56418, or also known as ABT-620) is a Abbott and Neurocrine Biosciences Inc company co-developed small molecule GnRH receptor antagonist, is currently in phase III clinical stage, mainly used in the treatment of endometriosis (III phase) and uterine fibroids (II period). June 2012, data released results of a Phase II clinical endometrial endometriosis Houston, the 94th annual meeting of the Endocrine Society: 131 accepts elagolix (150 or 250mg qd), leuprorelin depot (3.75mg sc in, once a month, female patients with endometriosis endometrium 12 weeks) or placebo treatment, elagolix treatment groups in patients with serum hormone estrogen compared to leuprorelin therapy group and the placebo group was significantly reduced. At the same time, elagolix safety and tolerability have been well verified.
Relugolix also known as TAK-385, is a GnRH by the Japanese Takada Pharmaceutical company developed an oral small molecule receptor antagonist, for the treatment of endometriosis, uterine fibroids and prostate. 2011 entered endometriosis and uterine fibroids clinical phase II study, carried out a clinical study of prostate cancer in the same year.
It disclosed a series of current small molecule GnRH receptor antagonists including patent WO2006096785, WO2010026993, WO2011076687, WO2012175514 like.
Despite the large number of interesting studies have been conducted in this field, there remains a need to continue research and development of more effective small molecule GnRH receptor antagonists, the present invention provides a novel GnRH receptor antagonist structure, and found to have such a structure compounds having good activity, reproductive endocrine system effective to treat the disease.
Production of N-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxy-3-pyridazinyl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-N′-methoxyurea
The similar reaction as described in Example 4 by using the compound (100 mg, 0.164 mmol) obtained in Reference Example 54 and methyl iodide (0.010 ml, 0.164 mmol) gave the title compound (17.3 mg, 17%) as colorless crystals.
Discovery of TAK-385, a thieno[2,3-d]pyrimidine-2,4-dione derivative, as a potent and orally bioavailable nonpeptide antagonist of gonadotropin releasing hormone (GnRH) receptor
238th ACS Natl Meet (August 16-20, Washington) 2009, Abst MEDI 386
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
^Goenka L, George M, Sen M (June 2017). “A peek into the drug development scenario of endometriosis – A systematic review”. Biomed. Pharmacother. 90: 575–585. doi:10.1016/j.biopha.2017.03.092. PMID28407578.
^Dellis A, Papatsoris A (October 2017). “Therapeutic outcomes of the LHRH antagonists”. Expert Rev Pharmacoecon Outcomes Res. 17 (5): 481–488. doi:10.1080/14737167.2017.1375855. PMID28870102.
^ Jump up to:abcStreuli I, de Ziegler D, Borghese B, Santulli P, Batteux F, Chapron C (March 2012). “New treatment strategies and emerging drugs in endometriosis”. Expert Opin Emerg Drugs. doi:10.1517/14728214.2012.668885. PMID22439891.
^Elancheran, R.; Maruthanila, V. L.; Ramanathan, M.; Kabilan, S.; Devi, R.; Kunnumakara, A.; Kotoky, Jibon (2015). “Recent discoveries and developments of androgen receptor based therapy for prostate cancer”. Med. Chem. Commun. 6 (5): 746–768. doi:10.1039/C4MD00416G. ISSN2040-2503.
^ Jump up to:abcdefMiwa K, Hitaka T, Imada T, Sasaki S, Yoshimatsu M, Kusaka M, Tanaka A, Nakata D, Furuya S, Endo S, Hamamura K, Kitazaki T (July 2011). “Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor”. J. Med. Chem. 54 (14): 4998–5012. doi:10.1021/jm200216q. PMID21657270.
^ Jump up to:abcNakata D, Masaki T, Tanaka A, Yoshimatsu M, Akinaga Y, Asada M, Sasada R, Takeyama M, Miwa K, Watanabe T, Kusaka M (January 2014). “Suppression of the hypothalamic-pituitary-gonadal axis by TAK-385 (relugolix), a novel, investigational, orally active, small molecule gonadotropin-releasing hormone (GnRH) antagonist: studies in human GnRH receptor knock-in mice”. Eur. J. Pharmacol. 723: 167–74. doi:10.1016/j.ejphar.2013.12.001. PMID24333551.
Alizulatide vixocianine CAS 2924859-51-6 MF C115H145N17O25S, 2,197.55 L-Serine, N-[6-[2-[7-[1,3-dihydro-1,1-dimethyl-3-(4-sulfobutyl)-2H-benz[e]indol-2-ylidene]-1,3,5-heptatrien-1-yl]-1,1-dimethyl-1H-benz[e]indolio]-1-oxohexyl]-L-α-glutamyl-L-α-glutamyl-L-α-aspartyl-3-cyclohexyl-L-alanyl-L-phenylalanyl-D-seryl-D-arginyl-L-tyrosyl-L-leucyl-L-tryptophyl-, inner salt 4-[2-[7-[3-[6-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2R)-1-[[(2R)-5-carbamimidamido-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(1S)-1-carboxy-2-hydroxyethyl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-1-oxopentan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-cyclohexyl-1-oxopropan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-6-oxohexyl]-1,1-dimethylbenzo[e]indol-3-ium-2-yl]hepta-2,4,6-trienylidene]-1,1-dimethylbenzo[e]indol-3-yl]butane-1-sulfonate diagnostic imaging agent, 8M3Q8XZ6MJ Alizulatide vixocianine is a polypeptide that can be discovered…
Limnetrelvir CAS 2923500-04-1 MF C27H23F4N5O4 MW 557.50 N-[(3R)-1-[4-cyano-2-(morpholine-4-carbonyl)-6-(trifluoromethyl)phenyl]pyrrolidin-3-yl]-8-fluoro-2-oxo-1H-quinoline-4-carboxamide N-{(3R)-1-[4-cyano-2-(morpholine-4-carbonyl)-6-(trifluoromethyl)phenyl]pyrrolidin-3-yl}-8-fluoro-2-oxo1,2-dihydroquinoline-4-carboxamideantiviral, ABBV-903, ABBV 903, 4TPS988XGG Limnetrelvir (ABBV-903) is a MPro inhibitor. Limnetrelvir could be used in antiviral research. SYN https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=4D458E543A941751578F87216FA39801.wapp1nA?docId=WO2024081351&_cid=P10-MJQJMM-46773-1#detailMainForm:MyTabViewId:PCTDESCRIPTION Example…
Iscartrelvir CAS 2921711-74-0 MF 2921711-74-0, 526.4 g/mol N-{(1S,2R)-2-[4-bromo-2-(methylcarbamoyl)-6-nitroanilino]cyclohexyl}isoquinoline-4-carboxamideantiviral, WU-04, WU 04, W2LTV65R5E Iscartrelvir is an investigational new drug developed by the Westlake University for the treatment of COVID-19. It targets the SARS-CoV-2 3CL…
Zopocianine CAS 2206660-94-6, NA SALT 2206660-95-7 MF C74H93N7O27S4, 1,640.83 L-Tyrosine, N-[[[(1S)-1,3-dicarboxypropyl]amino]carbonyl]-L-g-glutamyl-3-[2-(2-aminoethoxy)ethoxy]propanoyl-L-phenylalanyl-O-[6-[2-[1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene]ethylidene]-2-[2-[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-3H-indolium-2-yl]ethenyl]-1-cyclohexen-1-yl]-, inner salt N-{[(1S)-1,3-dicarboxypropyl]carbamoyl}-L-γ-glutamyl3-[2-(2-aminoethoxy)ethoxy]propanoyl-L-phenylalanylO-[(6Ξ)-2-{(1Ξ)-2-[3,3-dimethyl-1-(4-sulfobutyl)-5-sulfonato-3H-indol-1-ium-2-yl]ethen-1-yl}-6-{(2Ξ)-2-[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2Hindol-2-ylidene]ethylidene}cyclohex-1-en-1-yl]-Ltyrosinediagnostic imaging agent, UD9V5S9M7A, OTL 0078, OTL 78 AS ON OCT2025 4.511…
Zomiradomide CAS 2655656-99-6 MF C45H48F3N7O6S MW871.97 antineoplastic, IRAK degrader-1, AQ5UXV5646 Zomiradomide is an orally active PROTAC degrader for IRAK4 (DC50=6 nM), thereby inhibiting the NF-κB signaling pathway. Zomiradomide acts also as…
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO



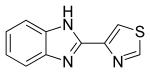

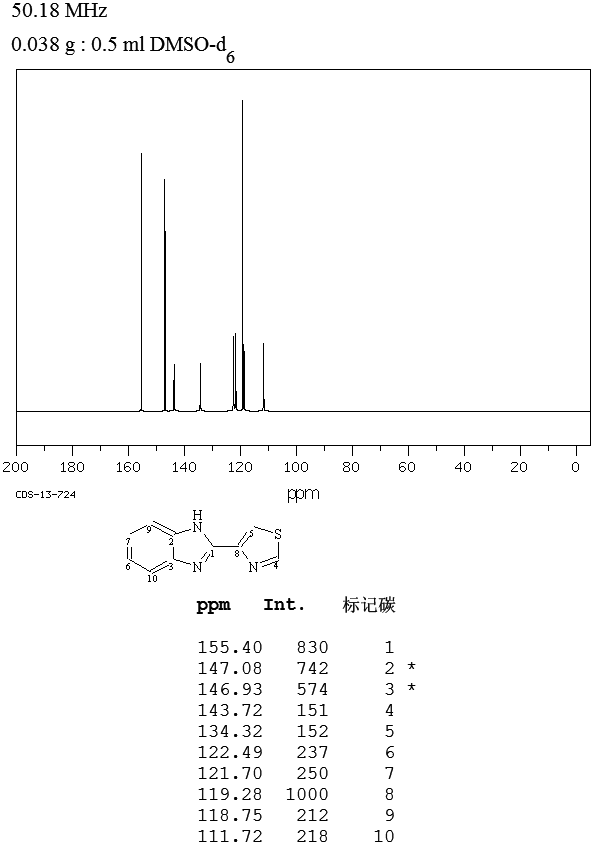
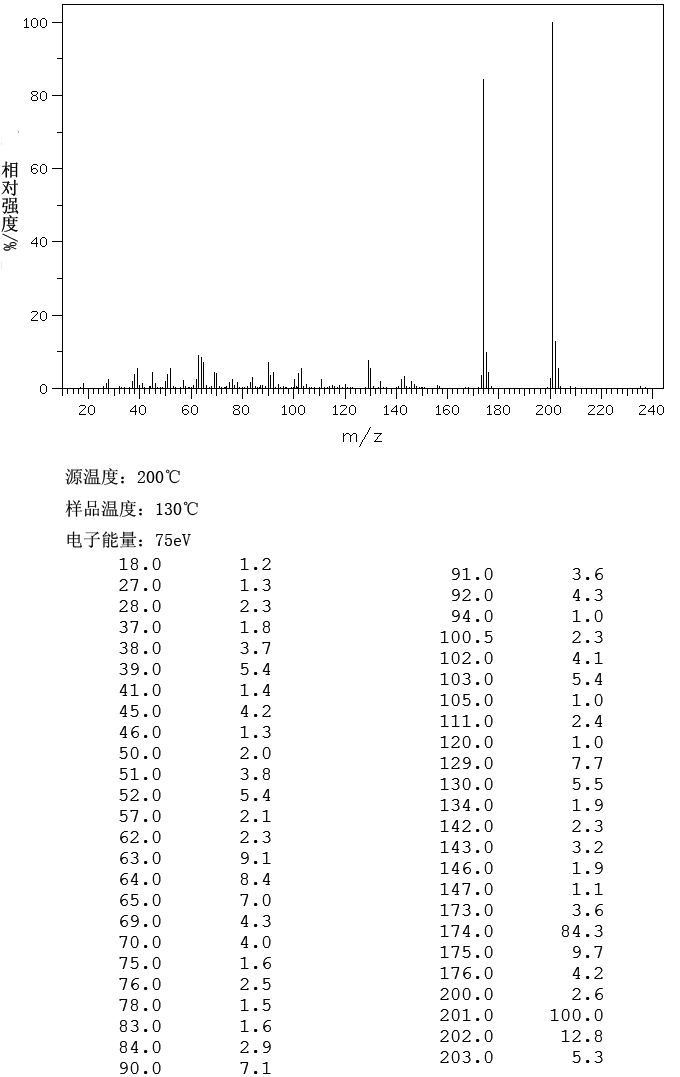








 One mechanistic explanation proposed by Lee and coworkers (1).
One mechanistic explanation proposed by Lee and coworkers (1).






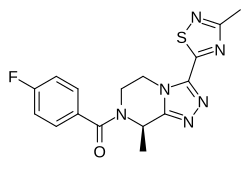









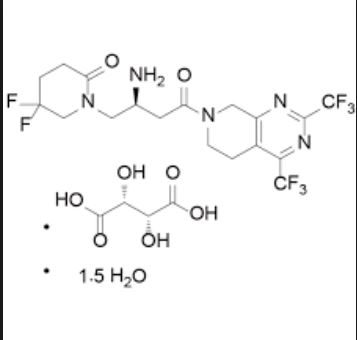








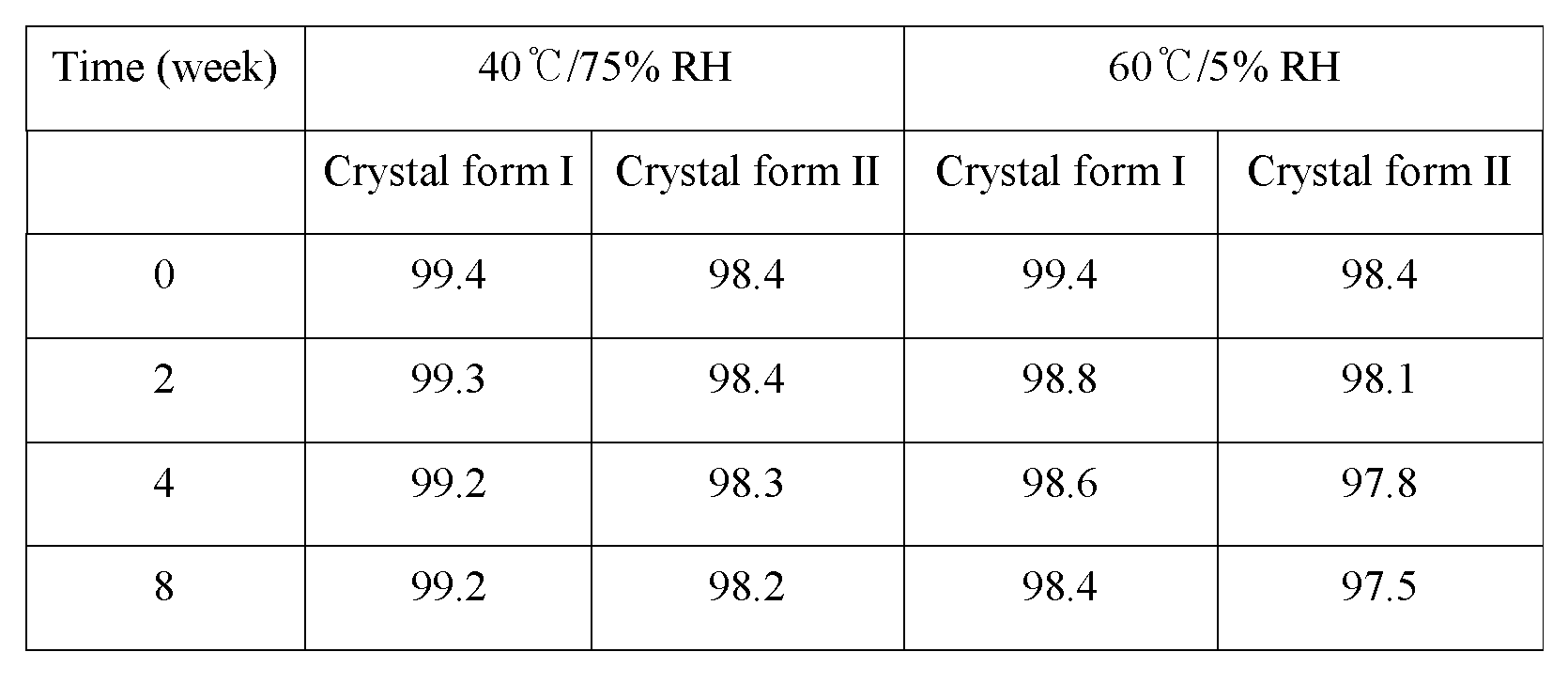





















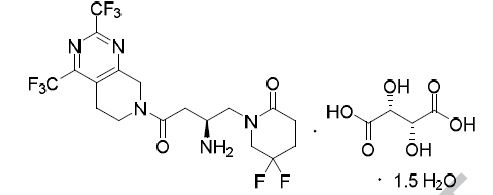




.svg)
 LIONEL MY SON
LIONEL MY SON














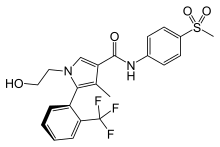
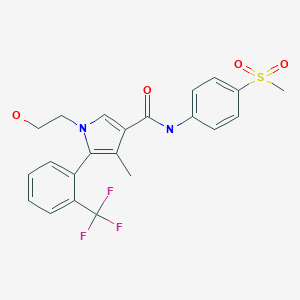
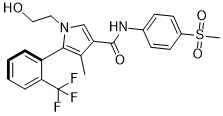





 OLMESARTAN
OLMESARTAN


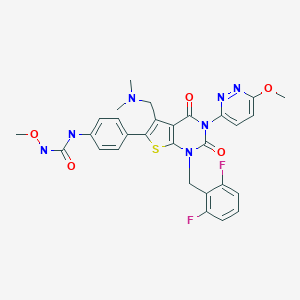






 tak 385
tak 385




{kind=link}