
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

MITAPIVAT
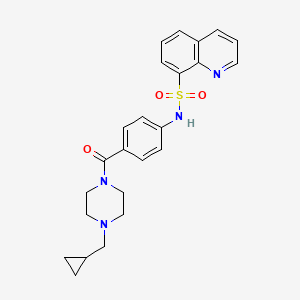

MITAPIVAT
CAS 1260075-17-9
MF C24H26N4O3S
MW 450.55
UPDATE…. FDA APPROVE 2/17/2022 To treat hemolytic anemia in pyruvate kinase deficiency, Pyrukynd
8-Quinolinesulfonamide, N-[4-[[4-(cyclopropylmethyl)-1-piperazinyl]carbonyl]phenyl]-
N-[4-[[4-(Cyclopropylmethyl)-1-piperazinyl]carbonyl]phenyl]-8-quinolinesulfonamide
- AG-348
- Originator Agios Pharmaceuticals
- Class Antianaemics; Piperazines; Quinolines; Small molecules; Sulfonamides
- Mechanism of Action Pyruvate kinase stimulants
- Orphan Drug Status Yes – Inborn error metabolic disorders
- New Molecular Entity Yes
- Phase III Inborn error metabolic disorders
- Phase II Thalassaemia
- 27 Feb 2019 Agios Pharmaceuticals plans a phase III trial for Inborn error metabolic disorders (Pyruvate kinase deficiency) (Treatment-experienced) in the US, Brazil, Canada, Czech Republic, Denmark, France, Germany, Ireland, Italy, Japan, South Korea, Netherlands, Portugal, Spain, Switzerland, Thailand, Turkey and United Kingdom in March 2019 (NCT03853798) (EudraCT2018-003459-39)
- 11 Dec 2018 Phase-II clinical trials in Thalassaemia in Canada (PO) (NCT03692052)
- 29 Aug 2018 Chemical structure information added
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Mitapivat sulfate | N4JTA67V3O | Not Available | Not applicable |
Mitapivat sulfate
CAS#: 2151847-10-6 (sulfate hydrate)
Chemical Formula: C48H60N8O13S3
Exact Mass: 1052.3442
Molecular Weight: 1053.23
Elemental Analysis: C, 54.74; H, 5.74; N, 10.64; O, 19.75; S, 9.13
N-(4-(4-(cyclopropylmethyl)piperazine-1-carbonyl)phenyl)quinoline-8-sulfonamide hemisulfate trihydrate
Related CAS #: 2151847-10-6 (sulfate hydrate) 1260075-17-9 (free) 2329710-91-8 (sulfate)
Mitapivat, sold under the brand name Pyrukynd, is a medication used to treat hemolytic anemia.[1] It is taken as the sulfate hydrate salt by mouth.[1]
Mitapivat is a pyruvate kinase activator.[1]
Mitapivat was approved for medical use in the United States in February 2022.[1][2][3]
Activator of pyruvate kinase isoenzyme M2 (PKM2), an enzyme involved in glycolysis. Since all tumor cells exclusively express the embryonic M2 isoform of PK, it is hypothesized that PKM2 is a potential target for cancer therapy. Modulation of PKM2 might also be effective in the treatment of obesity, diabetes, autoimmune conditions, and antiproliferation-dependent diseases.
Agios Pharmaceuticals is developing AG-348 (in phase 3 , in June 2019), an oral small-molecule allosteric activator of the red blood cell-specific form of pyruvate kinase (PK-R), for treating PK deficiency and non-transfusion-dependent thalassemia.
Mitapivat is a novel, first-in-class pyruvate kinase activator. It works to increase the activity of erythrocyte pyruvate kinase, a key enzyme involved in the survival of red blood cells. Defects in the pyruvate kinase enzyme in various red blood cells disorders lead to the lack of energy production for red blood cells, leading to lifelong premature destruction of red blood cells or chronic hemolytic anemia.1
On February 17, 2022, the FDA approved mitapivat as the first disease-modifying treatment for hemolytic anemia in adults with pyruvate kinase (PK) deficiency, a rare, inherited disorder leading to lifelong hemolytic anemia.6 Mitapivat has also been investigated in other hereditary red blood cell disorders associated with hemolytic anemia, such as sickle cell disease and alpha- and beta-thalassemia.1
SYN
WO 20100331307

CAS 59878-57-8 TO CAS 57184-25-5
Eisai Co., Ltd., EP1508570, Lithium aluminium hydride (770 mg, 20.3 mmol) was suspended in tetrahydrofuran (150 mL), 1-(cyclopropylcarbonyl)piperazine (1.56 g, 10.1 mmol) was gradually added thereto, and the reaction mixture was heated under reflux for 30 minutes. The reaction mixture was cooled to room temperature, and 0.8 mL of water, 0.8 mL of a 15percent aqueous solution of sodium hydroxide and 2.3 mL of water were seque ntially gradually added thereto. The precipitated insoluble matter was removed by filtration through Celite, and the filtrate was evaporated to give the title compound (1.40g) as a colorless oil. The product was used for the synthesis of (8E,12E,14E)-7-((4-cyclopropylmethylpiperazin-1-yl)carbonyl)oxy-3,6,16,21-tetrahydroxy-6,10,12,16,20-pentamethyl-18,19-epoxytricosa-8,12,14-trien-11-olide (the co mpound of Example 27) without further purification.1H-NMR Spectrum (CDCl3,400MHz) delta(ppm): 0.09-0.15(2H,m), 0.48-0.56(2H,m),0.82-0.93(1H,m),2.25(2H,d,J=7.2Hz) 2.48-2.65(4H,m),2.90-2.99(4H,m).
CAS 91-22-5 TO CAD 18704-37-5
chlorosulfonic acid;
Russian Journal of Organic Chemistry, vol. 36, 6, (2000), p. 851 – 853
NMR
US2010/331307
dimethylsulfoxide-d6, 1H
1H NMR (400 MHz, DMSO-d6) δ: 1.2 (t, 2H), 1.3 (t, 2H), 1.31-1.35 (m, 1H), 2.40 (s, 2H), 3.68 (br s, 4H), 3.4-3.6 (m, 4H), 7.06 (m, 6H), 7.25-7.42 (m, 3H), 9.18 (s, 1H) 10.4 (s, 1H)
1H NMR (400 MHz, DMSO-d6) δ: 0.04-0.45 (m, 2H), 0.61-0.66 (m, 2H), 1.4-1.6 (m, 1H), 2.21-2.38 (m, 4H), 2.61 (d, 2H), 3.31-3.61 (br s, 4H), 6.94-7.06 (m, 4H), 7.40 (d, 2H), 7.56-7.63 (m, 2H), 8.28 (d, 1H), 9.18 (s, 1H), 10.4 (s, 1H)
SYN
SYN
J. Med. Chem. 2024, 67, 4376−4418
Mitapivat (Pyrukynd). Mitapivat (9), developed by Agios Pharmaceuticals, Inc., was approved in the EU and US as an oral treatment of hemolytic anemia and pyruvate kinase deficiency (PKD), a rare, inherited condition associated with chronic hemolytic anemia. 66 Previous clinical management of PKD has been limited to only supportive therapies associated with short- and long-term risks, including red cell transfusions and splenectomy. 67 Mitapivat is a first-in-class activator of erythrocyte pyruvate kinase and has been shown to increase hemoglobin levels in patients who were not receiving regular red cell transfusions. In addition to its approved treatment of PKD, it is also currently in clinical studies for the treatment of sickle cell disease and thalassemia. 66 68
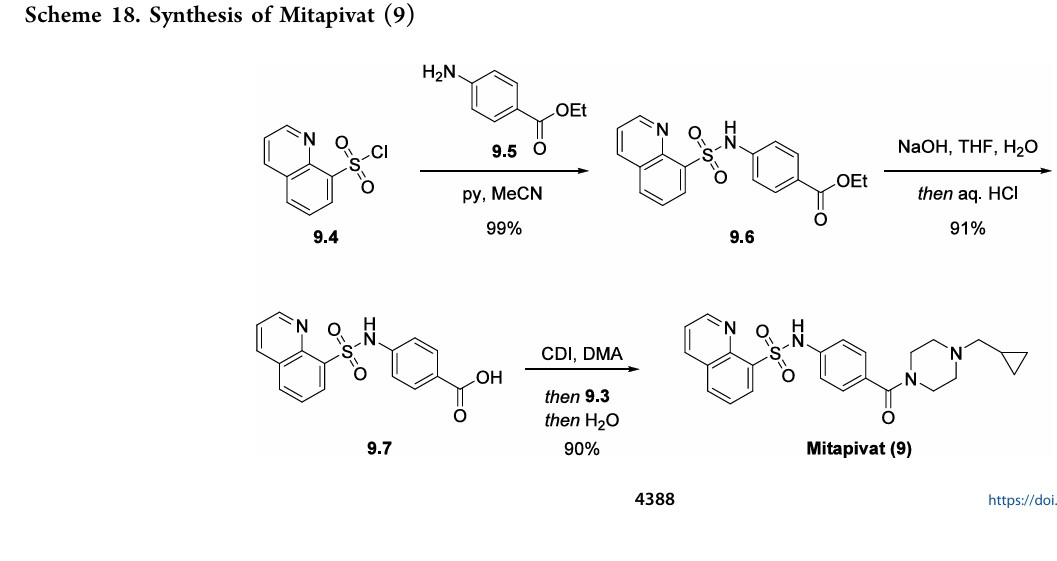
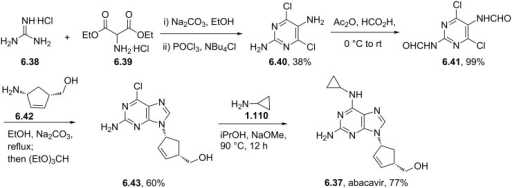
As compared to the linear medicinal chemistry route utilized for analog synthesis, Agios Pharmaceuticals, Inc. developed a convergent, high-yielding route to mitapivat depicted in Scheme 17 and Scheme 18.
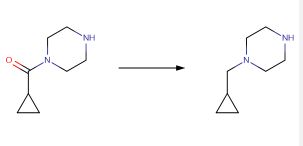
69 Piperazine 9.3 was synthesized in two steps from tert-butyl piperazine-1-carboxylate (9.1) in
92% yield (Scheme 17) beginning with a reductive amination in the presence of excess cyclopropanecarbaldehyde (9.2) to give the desired tertiary amine. This was carried forward as a
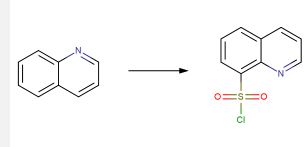
solution into the N-Boc deprotection, precipitating the piperazine as bis-HCl salt 9.3. The second module of mitapivat, sulfonamide 9.7, was prepared in two steps beginning with the reaction of sulfonyl chloride 9.4 with aniline 9.5 to afford ester 9.6 in 99% yield (Scheme 18). Saponification of ester 9.6 under basic
conditions provided carboxylic acid 9.7 in 91% yield. Efficient coupling was demonstrated on the kilogram scale by activation of carboxylic acid 9.7 with CDI, followed by addition of piperazine 9.3. Finally, addition of water to the reaction mixture induced crystallization of the product, providing mitapivat in 90% isolated yield.
(66) Pilo, F.; Angelucci, E. Mitapivat for sickle cell disease and
thalassemia. Drugs Today (Barc) 2023, 59, 125−134.
(67) Al-Samkari, H.; Galacteros, F.; Glenthoej, A.; Rothman, J. A.;
Andres, O.; Grace, R. F.; Morado-Arias, M.; Layton, D. M.; Onodera,
K.; Verhovsek, M.; et al. Mitapivat versus placebo for pyruvate kinase
deficiency. N. Engl. J. Med. 2022, 386, 1432−1442.
(68) Salituro, F. G.; Saunders, J. O.; Yan, S. Preparation of
aroylpiperazines and related compounds as pyruvate kinase M2
modulators useful in treatment of cancer. U.S. Patent US
20100331307, 2010.
(69) Sizemore, J. P.; Guo, L.; Mirmehrabi, M.; Su, Y. Preparation of
amorphous and crystalline forms of N-(4-(4-(cyclopropylmethyl)
piperazine-1-carbonyl)phenyl)quinoline-8-sulfonamide useful for
treatment of pyruvate kinase associated disorders. WO 2019104134,
2019.

Development Overview
Introduction
Mitapivat (designated AG 348), an orally available, first-in-class, small molecule stimulator of pyruvate kinase (PK), is being developed by Agios Pharmaceuticals for the treatment of pyruvate kinase deficiency (Inborn error metabolic disorders in development table) and thalassemia. Mitapivat is designed to activate the wild-type (normal) and mutated PK-R (the isoform of pyruvate kinase that is present in erythrocytes), in order to correct the defects in red cell glycolysis found within mutant cells. Clinical development is underway for inborn error metabolic disorders in the US, Spain and Denmark and for Thalassaemia in Canada.
Mitapivat emerged from Agios’ research programme focussed on the discovery of small molecule therapeutics for inborn metabolic disorders [see Adis Insight Drug Profile 800036791].
Key Development Milestones
In June 2018, Agios Pharmaceuticals initiated the phase III ACTIVATE trial to evaluate the efficacy and safety of orally administered mitapivat as compared with placebo in participants with pyruvate kinase deficiency (PKD), who are not regularly receiving blood transfusions (NCT03548220; AG348-C-006). The randomised, double-blind, placebo-controlled global trial intends to enrol 80 patients in the US, Canada, Denmark, France, Germany, Italy, Japan, South Korea, Netherlands, Poland, Portugal, Spain, Switzerland, Thailand and United Kingdom. The study design has two parts. Part 1 is a dose optimisation period where patients start at 5mg of mitapivat or placebo twice daily, with the flexibility to titrate up to 20mg or 50mg twice daily over a three month period to establish their individual optimal dose, as measured by maximum increase in hemoglobin levels. After the dose optimisation period, patients will receive their optimal dose for an additional three months in part 2. The primary endpoint of the study is the proportion of patients who achieve at least a 1.5 g/dL increase in haemoglobin sustained over multiple visits in part 2 of the trial
In February 2018, Agios Pharmaceuticals initiated the phase III ACTIVATE-T trial to assess the efficacy and safety of mitapivat in regularly transfused adult subjects with pyruvate kinase deficiency (Inborn error metabolism disorders in development table) (EudraCT2017-003803-22; AG348-C-007). The open label trial will enrol approximately 20 patients in Denmark and Spain and will expand to Canada, France, Italy, Japan, the Netherlands, the UK and the US
In December 2018, Agios Pharmaceuticals initiated a phase II study to assess the safety, efficacy, pharmacokinetics and pharmacodynamics of mitapivat (50mg and 100mg) for the treatment of patients with non-transfusion-dependent thalassemia (AG348-C-010; EudraCT2018-002217-35; NCT03692052). This study will include a 24-week core period followed by a 2-year extension period for eligible participants. The open-label trial intends to enrol approximately 17 patients. Enrolment has been initiated in Canada and may expand to the US and the UK
Agios Pharmaceuticals, in June 2015 initiated the phase II DRIVE PK trial to evaluate the safety, efficacy, pharmacokinetics and pharmacodynamics of mitapivat in adult transfusion-independent patients with pyruvate kinase deficiency (Inborn error metabolism disorders in development table) (AG348-C-003; NCT02476916). The trial will include two arms with 25 patients each. The patients in the first arm will receive 50mg twice daily, and the patients in the second arm will receive 300mg twice daily. The study will include a six-month dosing period with the opportunity for continued treatment beyond six months based on safety and clinical activity. The open-label, randomised trial completed enrolment of targeted 52 patients in the US, in November 2016. Preliminary data from the trial was presented at the 21st Congress of the European Haematology Association (EHA-2016). Updated results were presented by Agios at the 58th Annual Meeting and Exposition of the American Society of Haematology in December 2016. Based on results of the DRIVE PK trial, Agios plans to develop a registration path for mitapivat. Updated data from the trial was presented at the 22nd Congress of the European Haematology Association (EHA-2017)
In June 2018, Agios Pharmaceuticals completed a phase I trial in healthy male volunteers to assess the absorption, distribution, metabolism, excretion and absolute bioavailability of AG 348 (AG348-C-009; NCT03703505). Radiolabelled analytes of AG 348 ([14C]AG 348 and [13C6]AG 348) were administered in a single oral and intravenous dose on day 1. The open label trial was initiated in May 2018 and enrolled 8 volunteers in the US
In November 2017, Agios Pharmaceuticals completed a phase I trial that evaluated the relative bioavailability and safety of the mitapivat tablet and capsule formulations after single-dose administration in healthy adults (AG348-C-005; NCT03397329). The open-label trial enrolled 26 subjects in the US and was initiated in October 2017
In October 2017, Agios Pharmaceuticals completed a phase I trial that evaluated the pharmacokinetics, safety and effect on QTc interval of mitapivat in healthy volunteers (AG348-C-004; NCT03250598). This single-dose, open-label trial was initiated in August 2017 and enrolled 60 volunteers in the US
In November 2014, Agios completed a randomised, double-blind, placebo-controlled phase I trial that assessed the safety, pharmacokinetics and pharmacodynamics of multiple escalating doses of mitapivat in healthy volunteers (MAD; AG-348MAD; AG348-C-002; NCT02149966). Mitapivat was dosed daily for 14 days. The trial recruited 48 subjects in the US. In June 2015, positive results from the trial were presented at the 20th congress of the European Haematology Association (EHA-2015). Mitapivat showed a favourable pharmacokinetic profile with rapid absorption, low to moderate variability and a dose-proportional increase in exposure following multiple doses and serum hormone changes consistent with reversible aromatase inhibition were also observed
Agios Pharmaceuticals completed a randomised, double-blind, placebo-controlled phase I clinical trial of mitapivat in August 2014 (AG-348 SAD; AG348-C-001; NCT02108106). The study evaluated the safety, pharmacokinetics and pharmacodynamics of single escalating doses of the agent in healthy volunteers. Potential metabolic biomarkers were also explored. The trial enrolled 48 participants in the US
Patent Information
As of January 2018, Agios Pharmaceuticals owned approximately six issued US patents, 65 issued foreign patents, five pending US patent applications and 55 pending foreign patent applications in a number of jurisdictions directed to PK deficiency programme, including mitapivat (AG 348). The patents are valid till at least 2030
- Route of administrationPO
- FormulationTablet, unspecified
- ClassAntianaemics, Piperazines, Quinolines, Small molecules, Sulfonamides
- Mechanism of ActionPyruvate kinase stimulants
- WHO ATC codeA16A-X (Various alimentary tract and metabolism products)B03 (Antianemic Preparations)B06A (Other Hematological Agents)
- EPhMRA codeA16A (Other Alimentary Tract and Metabolism Products)B3 (Anti-Anaemic Preparations)B6 (All Other Haematological Agents)
- Chemical nameN-[4-[4-(cyclopropylmethyl)piperazine-1-carbonyl]phenyl]quinoline-8-sulfonamide
- Molecular formulaC24 H26 N4 O3 S
References
-
Agios Reports First Quarter 2017 Financial Results.
Media Release -
Agios Announces Initiation of Global Phase 3 Trial (ACTIVATE) of AG-348 in Adults with Pyruvate Kinase Deficiency Who Are Not Regularly Transfused.
Media Release -
A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of AG-348 in Not Regularly Transfused Adult Subjects With Pyruvate Kinase Deficiency
ctiprofile -
Agios Provides Business Update on Discovery Research Strategy and Pipeline, Progress on Clinical Programs, Commercial Launch Preparations and Reports First Quarter 2018 Financial Results at Investor Day.
Media Release -
An Open-Label Study To Evaluate the Efficacy and Safety of AG-348 in Regularly Transfused Adult Subjects With Pyruvate Kinase (PK) Deficiency
ctiprofile -
A Phase 2, Open-label, Multicenter Study to Determine the Efficacy, Safety, Pharmacokinetics, and Pharmacodynamics of AG-348 in Adult Subjects With Non-transfusion-dependent Thalassemia
ctiprofile -
Agios Announces Key Upcoming Milestones to Support Evolution to a Commercial Stage Biopharmaceutical Company in 2017.
Media Release -
Agios to Present Clinical and Preclinical Data at the 20th Congress of the European Hematology Association.
Media Release -
Agios Announces Updated Data from Fully Enrolled DRIVE PK Study Demonstrating AG-348s Potential as the First Disease-modifying Treatment for Patients with Pyruvate Kinase Deficiency.
Media Release -
Agios Announces New Data from AG-348 and AG-519 Demonstrating Potential for First Disease-modifying Treatment for Patients with PK Deficiency.
Media Release -
Agios Provides Update on PKR Program.
Media Release -
AG-348 Achieves Proof-of-Concept in Ongoing Phase 2 DRIVE-PK Study and Demonstrates Rapid and Sustained Hemoglobin Increases in Adults with Pyruvate Kinase Deficiency.
Media Release -
Agios Reports New, Final Data from Phase 1 Multiple Ascending Dose (MAD) Study in Healthy Volunteers for AG-348, an Investigational Medicine for Pyruvate Kinase (PK) Deficiency.
Media Release -
Grace RF, Layton DM, Galacteros F, Rose C, Barcellini W, Morton DH, et al. Results Update from the DRIVE PK Study: Effects of AG-348, a Pyruvate Kinase Activator, in Patients with Pyruvate Kinase Deficiency. ASH-Hem-2017 2017; abstr. 2194.
Available from: URL: https://ash.confex.com/ash/2017/webprogram/Paper102236.html -
A Phase 2, Open Label, Randomized, Dose Ranging, Safety, Efficacy, Pharmacokinetic and Pharmacodynamic Study of AG-348 in Adult Patients With Pyruvate Kinase Deficiency
ctiprofile -
A Phase I, Open-label Study to Evaluate the Absorption, Distribution, Metabolism, and Excretion and to Assess the Absolute Bioavailability of AG-348 in Healthy Male Subjects Following Administration of a Single Oral Dose of [14C]AG-348 and Concomitant Single Intravenous Microdose of [13C6]AG-348
ctiprofile -
A Phase 1, Randomized, Open-Label, Two-Period Crossover Study Evaluating the Relative Bioavailability and Safety of the AG-348 Tablet and Capsule Formulations After Single-Dose Administration in Healthy Adults
ctiprofile -
A Phase 1, Single-Dose, Open-Label Study to Characterize and Compare the Pharmacokinetics, Safety, and Effect on QTc Interval of AG-348 in Healthy Subjects of Japanese Origin and Healthy Subjects of Non-Asian Origin
ctiprofile -
Agios Pharmaceuticals Initiates Multiple Ascending Dose Trial in Healthy Volunteers of AG-348 for the Potential Treatment of PK Deficiency, a Rare, Hemolytic Anemia.
Media Release -
A Phase 1, Randomized, Double-Blind, Placebo-Controlled, Multiple Ascending Dose, Safety, Pharmacokinetic, and Pharmacodynamic Study of Orally Administered AG-348 in Healthy Volunteers
ctiprofile -
Agios Initiates Phase 1 Study of AG-348, a First-in-class PKR Activator, for Pyruvate Kinase Deficiency.
Media Release -
A Phase I, Randomized, Double-Blind, Placebo-Controlled, Single Ascending Dose, Safety, Pharmacokinetic and Pharmacodynamic Study of Orally Administered AG-348 in Healthy Volunteers
ctiprofile -
Agios Pharmaceuticals Reports First Quarter 2014 Financial Results.
Media Release -
Agios Pharmaceuticals Reports Third Quarter 2013 Financial Results.
Media Release -
Agios Pharmaceuticals to Present Preclinical Research at the 2013 American Society of Hematology Annual Meeting.
Media Release -
Agios Presents Preclinical Data from Lead Programs at American Society of Hematology Annual Meeting.
Media Release -
Agios Pharmaceuticals Form 10-K, February 2018. Internet-Doc 2018;.
Available from: URL: https://www.sec.gov/Archives/edgar/data/1439222/000143922218000004/agio-123117x10k.htm -
Agios Outlines Key 2018 Priorities Expanding Clinical and Research Programs to Drive Long Term Value.
Media Release -
Grace RF, Layton DM, Galacteros F, Rose C, Barcellini W, Morton DH, et al. Effects of Ag-348, a Pyruvate Kinase Activator, in Patients with Pyruvate Kinase Deficiency: Updated Results from the Drive Pk Study. EHA-2017 2017; abstr. S451.
Available from: URL: https://learningcenter.ehaweb.org/eha/2017/22nd/181738/rachael.f.grace.effects.of.ag-348.a.pyruvate.kinase.activator.in.patients.with.html?f=m3e1181l15534 -
Agios Presents Updated Data from DRIVE PK Study Demonstrating AG-348 is Well-Tolerated and Results in Clinically Relevant, Rapid and Sustained Hemoglobin Increases in Patients with Pyruvate Kinase Deficiency.
Media Release
Medical uses
Mitapivat is indicated for the treatment of hemolytic anemia in adults with pyruvate kinase deficiency.[1][3]Pharmacology
Mechanism of action
Mitapivat binds to and activates pyruvate kinase, thereby enhancing glycolytic pathway activity, improving adenosine triphosphate (ATP) levels and reducing 2,3-diphosphoglycerate (2,3-DPG) levels.[4] Mutations in pyruvate kinase cause deficiency in pyruvate kinase which prevents adequate red blood cell (RBC) glycolysis, leading to a buildup of the upstream glycolytic intermediate 2,3-DPG and deficiency in the pyruvate kinase product ATP.[4][5]Society and culture
Names
Mitapivat is the international nonproprietary name (INN).[6]References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/216196s000lbl.pdf
- ^ “Agios Announces FDA Approval of Pyrukynd (mitapivat) as First Disease-Modifying Therapy for Hemolytic Anemia in Adults with Pyruvate Kinase Deficiency” (Press release). Agios Pharmaceuticals. 17 February 2022. Retrieved 19 February 2022 – via GlobeNewswire.
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/216196Orig1s000ltr.pdf
- ^ Jump up to:a b “Mitapivat (Code C157039)”. NCI Thesaurus. 31 January 2022. Retrieved 19 February 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “PK-R allosteric activator AG-348”. NCI Drug Dictionary. National Cancer Institute. Retrieved 19 February 2022. This article incorporates text from this source, which is in the public domain.
- ^ World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 78”. WHO Drug Information. 31 (3): 539. hdl:10665/330961.
Further reading
- Kung C, Hixon J, Kosinski PA, Cianchetta G, Histen G, Chen Y, et al. (September 2017). “AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency”. Blood. 130 (11): 1347–1356. doi:10.1182/blood-2016-11-753525. PMC 5609468. PMID 28760888.
- Rab MA, Van Oirschot BA, Kosinski PA, Hixon J, Johnson K, Chubukov V, et al. (January 2021). “AG-348 (Mitapivat), an allosteric activator of red blood cell pyruvate kinase, increases enzymatic activity, protein stability, and ATP levels over a broad range of PKLR genotypes”. Haematologica. 106 (1): 238–249. doi:10.3324/haematol.2019.238865. PMC 7776327. PMID 31974203.
External links
- “Mitapivat sulfate”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03548220 for “A Study to Evaluate Efficacy and Safety of AG-348 in Not Regularly Transfused Adult Participants With Pyruvate Kinase Deficiency (PKD)” at ClinicalTrials.gov
- Clinical trial number NCT03559699 for “A Study Evaluating the Efficacy and Safety of AG-348 in Regularly Transfused Adult Participants With Pyruvate Kinase Deficiency (PKD)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Pyrukynd |
| Other names | AG-348, Mitapivat sulfate (USAN US) |
| License data | |
| Routes of administration | By mouth |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| DrugBank |
|
| ChemSpider | |
| UNII |
|
| KEGG |
|
| ChEMBL |
|
| Chemical and physical data | |
| Formula | C24H26N4O3S |
| Molar mass | 450.56 g·mol−1 |
| 3D model (JSmol) | |
ABACAVIR
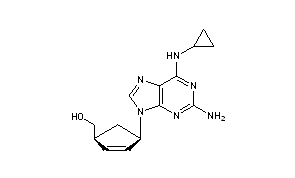
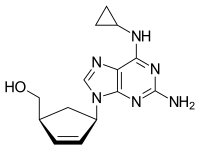
Abacavir (ABC) is a medication used to prevent and treat HIV/AIDS.[1][2] Similar to other nucleoside analog reverse-transcriptase inhibitors (NRTIs), abacavir is used together with other HIV medications, and is not recommended by itself.[3] It is taken by mouth as a tablet or solution and may be used in children over the age of three months.[1][4]
Abacavir is generally well tolerated.[4] Common side effects include vomiting, trouble sleeping, fever, and feeling tired.[1] More severe side effects include hypersensitivity, liver damage, and lactic acidosis.[1] Genetic testing can indicate whether a person is at higher risk of developing hypersensitivity.[1] Symptoms of hypersensitivity include rash, vomiting, and shortness of breath.[4] Abacavir is in the NRTI class of medications, which work by blocking reverse transcriptase, an enzyme needed for HIV virus replication.[5] Within the NRTI class, abacavir is a carbocyclic nucleoside.[1]
Abacavir was patented in 1988 and approved for use in the United States in 1998.[6][7] It is on the World Health Organization’s List of Essential Medicines, the most effective and safe medicines needed in a health system.[8] It is available as a generic medication.[1] The wholesale cost in the developing world as of 2014 is between US$0.36 and US$0.83 per day.[9] As of 2016 the wholesale cost for a typical month of medication in the United States is US$70.50.[10] Commonly, abacavir is sold together with other HIV medications, such as abacavir/lamivudine/zidovudine, abacavir/dolutegravir/lamivudine, and abacavir/lamivudine.[4][5]
Medical uses
_300mg.jpg)
Two Abacavir 300mg tablets
Abacavir tablets and oral solution, in combination with other antiretroviral agents, are indicated for the treatment of HIV-1 infection.
Abacavir should always be used in combination with other antiretroviral agents. Abacavir should not be added as a single agent when antiretroviral regimens are changed due to loss of virologic response.
Side effects
Common adverse reactions include nausea, headache, fatigue, vomiting, diarrhea, loss of appetite and trouble sleeping. Rare but serious side effects include hypersensitivity reaction or rash, elevated AST and ALT, depression, anxiety, fever/chills, URI, lactic acidosis, hypertriglyceridemia, and lipodystrophy.[11]
People with liver disease should be cautious about using abacavir because it can aggravate the condition. Signs of liver problems include nausea and vomiting, abdominal pain, dark-colored urine and yellowing of the skin or whites of the eyes. The use of nucleosidedrugs such as abacavir can very rarely cause lactic acidosis. Signs of lactic acidosis include fast or irregular heartbeat, unusual muscle pain, fatigue, difficulty breathing and stomach pain with nausea and vomiting.[12] Abacavir can also lead to immune reconstitution inflammatory syndrome, a change in body fat as well as an increased risk of heart attack.
Resistance to abacavir has developed in laboratory versions of HIV which are also resistant to other HIV-specific antiretrovirals such as lamivudine, didanosine, and zalcitabine. HIV strains that are resistant to protease inhibitors are not likely to be resistant to abacavir.
Abacavir is contraindicated for use in infants under 3 months of age.
Little is known about the effects of Abacavir overdose. Overdose victims should be taken to a hospital emergency room for treatment.
Hypersensitivity syndrome
Hypersensitivity to abacavir is strongly associated with a specific allele at the human leukocyte antigen B locus namely HLA-B*5701.[13][14][15] There is an association between the prevalence of HLA-B*5701 and ancestry. The prevalence of the allele is estimated to be 3.4 to 5.8 percent on average in populations of European ancestry, 17.6 percent in Indian Americans, 3.0 percent in Hispanic Americans, and 1.2 percent in Chinese Americans.[16][17] There is significant variability in the prevalence of HLA-B*5701 among African populations. In African Americans, the prevalence is estimated to be 1.0 percent on average, 0 percent in the Yorubafrom Nigeria, 3.3 percent in the Luhya from Kenya, and 13.6 percent in the Masai from Kenya, although the average values are derived from highly variable frequencies within sample groups.[18]
Common symptoms of abacavir hypersensitivity syndrome include fever, malaise, nausea, and diarrhea. Some patients may also develop a skin rash.[19] Symptoms of AHS typically manifest within six weeks of treatment using abacavir, although they may be confused with symptoms of HIV, immune reconstitution syndrome, hypersensitivity syndromes associated with other drugs, or infection.[20] The U.S. Food and Drug Administration (FDA) released an alert concerning abacavir and abacavir-containing medications on July 24, 2008,[21] and the FDA-approved drug label for abacavir recommends pre-therapy screening for the HLA-B*5701 allele and the use of alternative therapy in subjects with this allele.[22] Additionally, both the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group recommend use of an alternative therapy in individuals with the HLA-B*5701 allele.[23][24]

Skin-patch testing may also be used to determine whether an individual will experience a hypersensitivity reaction to abacavir, although some patients susceptible to developing AHS may not react to the patch test.[25]
The development of suspected hypersensitivity reactions to abacavir requires immediate and permanent discontinuation of abacavir therapy in all patients, including patients who do not possess the HLA-B*5701 allele. On March 1, 2011, the FDA informed the public about an ongoing safety review of abacavir and a possible increased risk of heart attack associated with the drug. A meta-analysis of 26 studies conducted by the FDA, however, did not find any association between abacavir use and heart attack [26][27]
Immunopathogenesis
The mechanism underlying abacavir hypersensitivity syndrome is related to the change in the HLA-B*5701 protein product. Abacavir binds with high specificity to the HLA-B*5701 protein, changing the shape and chemistry of the antigen-binding cleft. This results in a change in immunological tolerance and the subsequent activation of abacavir-specific cytotoxic T cells, which produce a systemic reaction known as abacavir hypersensitivity syndrome.[28]
Interaction
Abacavir, and in general NRTIs, do not undergo hepatic metabolism and therefore have very limited (to none) interaction with the CYP enzymes and drugs that effect these enzymes. That being said there are still few interactions that can affect the absorption or the availability of abacavir. Below are few of the common established drug and food interaction that can take place during abacavir co-administration:
- Protease inhibitors such as tipranavir or ritonovir may decrease the serum concentration of abacavir through induction of glucuronidation. Abacavir is metabolized by both alcohol dehydrogenase and glucuronidation.[29][30]
- Ethanol may result in increased levels of abacavir through the inhibition of alcohol dehydrogenase. Abacavir is metabolized by both alcohol dehydrogenase and glucuronidation.[29][31]
- Methadone may diminish the therapeutic effect of Abacavir. Abacavir may decrease the serum concentration of Methadone.[32][33]
- Orlistat may decrease the serum concentration of antiretroviral drugs. The mechanism of this interaction is not fully established but it is suspected that it is due to the decreased absorption of abacavir by orlistat.[34]
- Cabozantinib: Drugs from the MPR2 inhibitor (Multidrug resistance-associated protein 2 inhibitors) family such as abacavir could increase the serum concentration of Cabozantinib.[35]
Mechanism of action
Abacavir is a nucleoside reverse transcriptase inhibitor that inhibits viral replication. It is a guanosine analogue that is phosphorylated to carbovir triphosphate (CBV-TP). CBV-TP competes with the viral molecules and is incorporated into the viral DNA. Once CBV-TP is integrated into the viral DNA, transcription and HIV reverse transcriptase is inhibited.[36]
Pharmacokinetics
Abacavir is given orally and is rapidly absorbed with a high bioavailability of 83%. Solution and tablet have comparable concentrations and bioavailability. Abacavir can be taken with or without food.
Abacavir can cross the blood-brain barrier. Abacavir is metabolized primarily through the enzymes alcohol dehydrogenase and glucuronyl transferase to an inactive carboxylate and glucuronide metabolites. It has a half-life of approximately 1.5-2.0 hours. If a person has liver failure, abacavir’s half life is increased by 58%.
Abacavir is eliminated via excretion in the urine (83%) and feces (16%). It is unclear whether abacavir can be removed by hemodialysis or peritoneal dialysis.[36]
History
Robert Vince and Susan Daluge along with Mei Hua, a visiting scientist from China, developed the medication in the ’80s.[37][38][39]
Abacavir was approved by the Food and Drug Administration (FDA) on December 18, 1998, and is thus the fifteenth approved antiretroviral drug in the United States. Its patent expired in the United States on 2009-12-26.
Synthesis

Abacavir synthesis:[40]
References











ABOVE From internet free resources
- Crimmins, M. T.; King, B. W. (1996). “An Efficient Asymmetric Approach to Carbocyclic Nucleosides: Asymmetric Synthesis of 1592U89, a Potent Inhibitor of HIV Reverse Transcriptase”. The Journal of Organic Chemistry. 61 (13): 4192–4193. doi:10.1021/jo960708p. PMID 11667311.

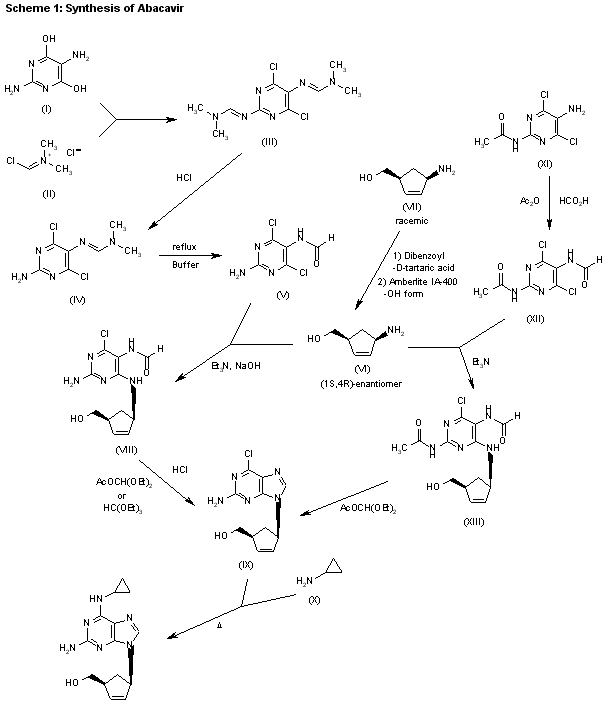
| AU 8937025; EP 0349242; JP 1990045486; JP 1999139976; US 5034394; US 5089500
|
SYN 2
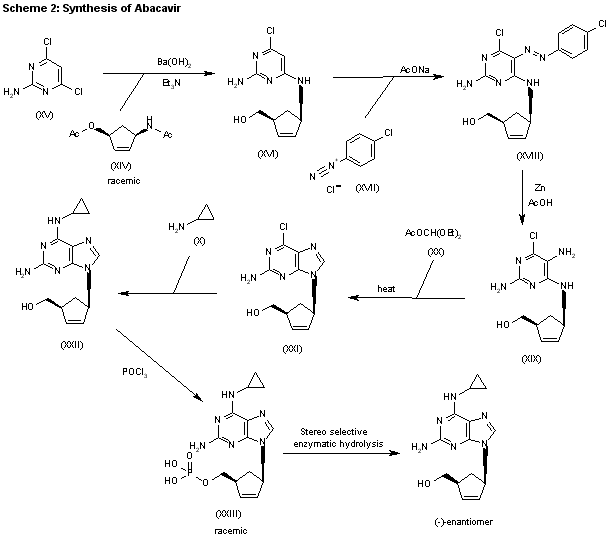
The condensation of (?-cis-4-acetamido-2-cyclopentenylmethyl acetate (XIV) with 2-amino-4,6-dichloropyrimidine (XV) by means of Ba(OH)2 and triethylamine in refluxing butanol gives the expected condensation product (XVI), which is treated with 4-chlorophenyldiazonium chloride (XVII) in water/acetic acid to yield the corresponding azo-compound (XVIII). The reduction of (XVIII) with Zn/acetic acid in ethanol affords the diamine (XIX), which is cyclized with refluxing diethoxymethyl acetate (XX) to afford the corresponding purine (XXI). The reaction of (XXI) with cyclopropylamine (X) in refluxing ethanol affords racemic abacavir (XXII), which is phosphorylated with POCl3 giving the racemic 4′-O-phosphate (XXIII). Finally, this compound is submitted to stereoselective enzymatic dephosphorylation using snake venom 5′-nucleotidase (EC 3.1.3.5) from Crotalus atrox yielding the (-)-enantiomer, abacavir.
SYN 3
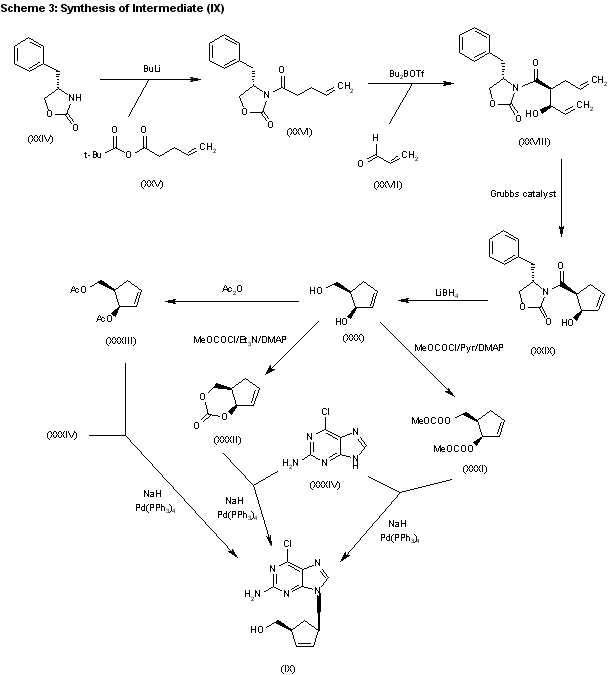
The acylation of 4(S)-benzyloxazolidin-2-one (XXIV) with 4-pentenoyl pivaloyl anhydride (XXV) by means of NaH in THF gives 4(S)-benzyl-3-(4-pentenoyl)oxazolidin-2-one (XXVI), which is submitted to a diastereoselective syn aldol condensation with acrolein (XXVII), using dibutylboron triflate as catalyst, affording the aldol (XXVIII). The cyclization of (XXVIII) by means of the Grubbs catalyst in dichloromethane yields the cyclopentenol (XXIX), which is reduced with LiBH4 in THF/methanol to give the key intermediate 5(R)-(hydroxymethyl)-2-cyclopenten-1(R)-ol (XXX). The reaction of (XXX) with methyl chloroformate/pyridine/DMAP or methyl chloroformate/triethylamine/DMAP or acetic anhydride gives the diols (XXXI), (XXXII) and (XXXIII), respectively, each of which coupled with 2-amino-6-chloropurine (XXXIV) in the presence of NaH and palladium tetrakis(triphenylphosphine) in THF/DMSO, affords the purine intermediate (IX) already reported.
SYN
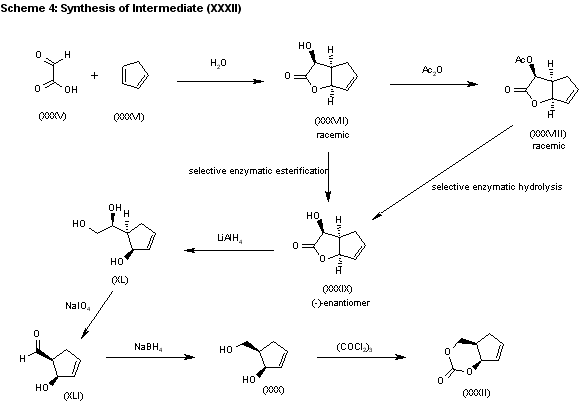
The water promoted condensation of glyoxylic acid (XXXV) with cyclopentadiene (XXXVI) gives the racemic cis-hydroxylactone (XXXVII), which is acetylated with acetic anhydride to the acetate (XXXVIII). The selective enzymatic hydrolysis of (XXXVIII) with Pseudomonas fluorescens lipase yields the pre (-)-enantiomer (XXXIX), which is reduced with LiAlH4 in refluxing THF, affording triol (XL). The oxidation of the vicinal glycol of (XL) with NaIO4 in ethyl ether/water yields the hydroxyaldehyde (XLI), which is reduced with NaBH4 in ethanol to give the key intermediate 5(R)-(hydroxymethyl)-2-cyclopenten-1(R)-ol (XXX). This compound, by reaction with triphosgene and triethylamine in dichloromethane, results in the cyclic carbonate intermediate (XXXII), already reported.
SYN
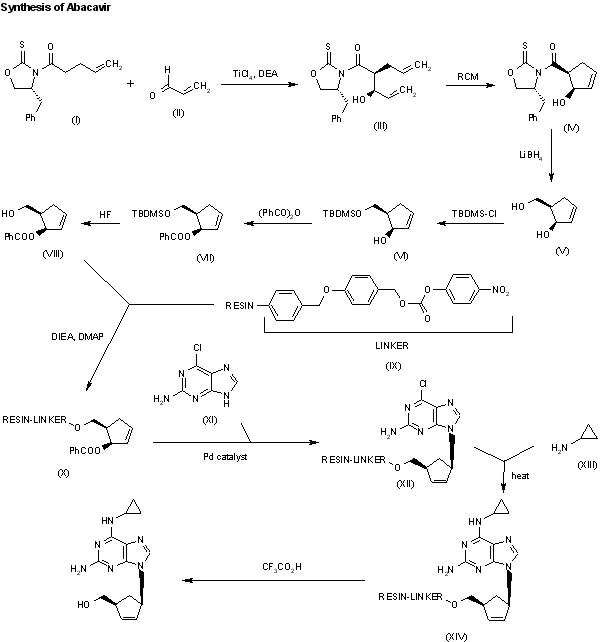
A new solid phase synthesis of abacavir has been reported: Condensation of the chiral 4(R)-benzyl-3-(4-pentenoyl)oxazolidin-2-thione (I) with acrolein (II) by means of TiCl4 and DIEA gives the adduct (III), which was transformed into the chiral cyclopentene (IV) by catalytic ring-closing metathesis. The reductive removal of the chiral auxiliary with LiBH4 affords the chiral diol (V), which is selectively silylated with TBDMSCl providing the primary silyl ether (VI). Acylation of the secondary alcohol of (VI) with benzoic anhydride gives the benzoate (VII), which is desilylated with HF in acetonitrile yielding the allylic benzoate (VIII). Benzoate (VIII) is condensed with a p-nitrophenyl Wang carbonate resin (IX) by means of DIEA and DMAP affording the solid phase resin (X) which is condensed with 2-amino-6-chloropurine (XI) by means of a Pd catalyst furnishing the adduct (XII). Thermal condensation of (XII) with cyclopropylamine (XIII) yields the diaminopurine resin (XIV) which, after cleavage from the resin by a treatment with TFA in dichloromethane, gives directly abacavir.
SYN
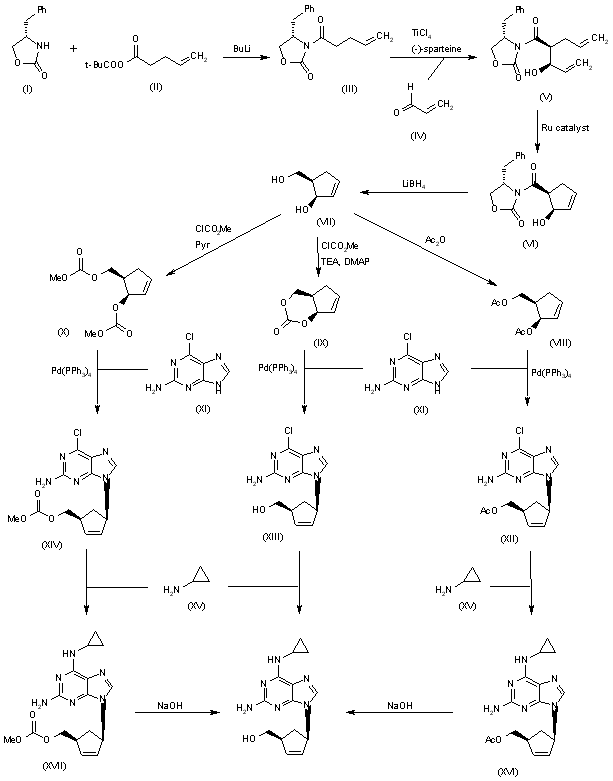
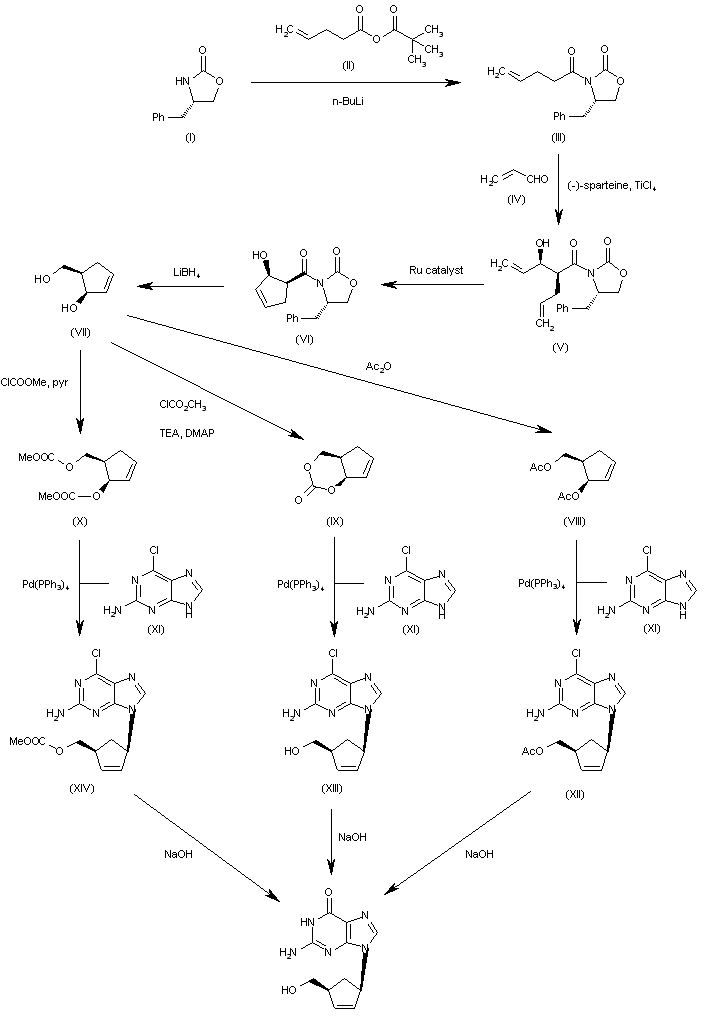 The condensation of the chiral oxazolidinone (I) with the pentenoic anhydride (II) by means of n-BuLi in THF gives the N-pentenoyloxazolidinone (III), which is condensed with acrolein (IV) catalyzed by TiCl4 and (-)-spartein in dichloromethane, yielding the chiral adduct (V). The ring-closing metathesis of (V) by means of a Ru catalyst in dichloromethane affords the chiral cyclopentenol derivative (VI), which is reduced to the (R,R)-5-(hydroxymethyl)-2-cyclopenten-1-ol (VII) by means of LiBH4 in THF. The reaction of diol (VII) with Ac2O; with methyl chloroformate, TEA and DMAP; or with ethyl chloroformate and pyridine gives the diacetate (VIII), the cyclic carbonate (IX) or the dicarbonate (X), respectively. The condensation of (VIII), (IX) or (X) with 2-amino-6-chloropurine (XI) by means of Pd(PPh3)4 yields the carbocyclic purines (XII), (XIII) or (XIV), respectively. Finally, these compounds are hydrolyzed with aqueous NaOH to the target carbocyclic guanine.
The condensation of the chiral oxazolidinone (I) with the pentenoic anhydride (II) by means of n-BuLi in THF gives the N-pentenoyloxazolidinone (III), which is condensed with acrolein (IV) catalyzed by TiCl4 and (-)-spartein in dichloromethane, yielding the chiral adduct (V). The ring-closing metathesis of (V) by means of a Ru catalyst in dichloromethane affords the chiral cyclopentenol derivative (VI), which is reduced to the (R,R)-5-(hydroxymethyl)-2-cyclopenten-1-ol (VII) by means of LiBH4 in THF. The reaction of diol (VII) with Ac2O; with methyl chloroformate, TEA and DMAP; or with ethyl chloroformate and pyridine gives the diacetate (VIII), the cyclic carbonate (IX) or the dicarbonate (X), respectively. The condensation of (VIII), (IX) or (X) with 2-amino-6-chloropurine (XI) by means of Pd(PPh3)4 yields the carbocyclic purines (XII), (XIII) or (XIV), respectively. Finally, these compounds are hydrolyzed with aqueous NaOH to the target carbocyclic guanine.
SYN
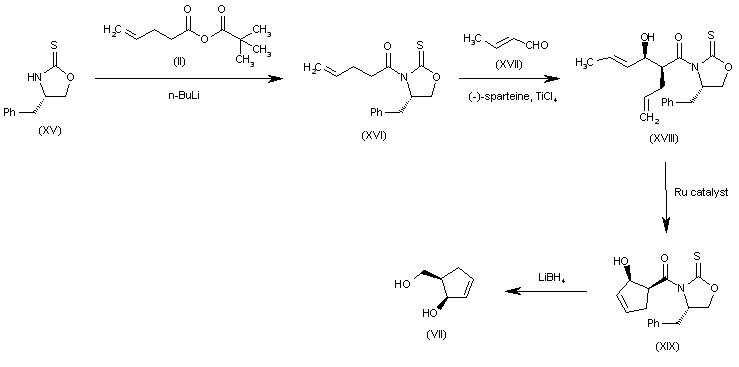
Alternatively, the (R,R)-5-(hydroxymethyl)-2-cyclopenten-1-ol (VII) can also be obtained as follows: The condensation of the chiral oxazolidinethione (XV) with the pentenoic anhydride (II) by means of n-BuLi in THF gives the N-pentenoyloxazolidinethione (XVI), which is condensed with crotonaldehyde (XVII) catalyzed by TiCl4 and (-)-spartein in dichloromethane, yielding the chiral adduct (XVIII). The ring-closing metathesis of (XVIII) by means of a Ru catalyst in dichloromethane affords the chiral cyclopentenol derivative (XIX), which is reduced to the target diol (VII) by means of LiBH4 in THF.
SYN
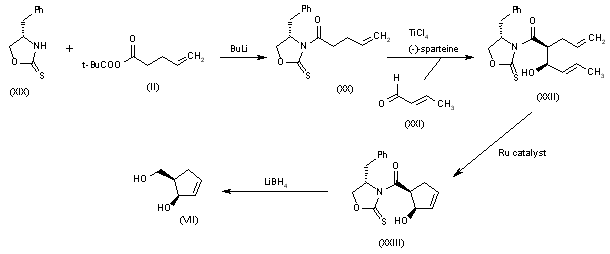
An efficient asymmetric synthesis of abacavir has been reported: Acylation of the chiral oxazolidinone (I) with the mixed anhydride (II) by means of BuLi in THF gives the N-pentenoyloxazolidinone (III), which by condensation with acrolein (IV) catalyzed by TiCl4 and (?-spartein in dichloromethane yields the chiral adduct (V). The ring-closing metathesis of adduct (V) by means of the ruthenium catalyst (Cy3P)Cl2Ru=CHPh in dichloromethane affords the chiral cyclopentenol (VI), which is reduced to 5(R)-(hydroxymethyl)-2-cyclopenten-1(R)-ol (VII) by means of LiBH4 in THF. Reaction of diol (VII) with a) Ac2O, TEA and DMAP, b) methyl chloroformate, TEA and DMAP or c) methyl chloroformate, pyridine and DMAP gives a) the diacetate (VIII), b) the cyclic carbonate (IX) or c) the dicarbonate (X), respectively. The condensation of diacetate (VIII), cyclic carbonate (IX) or dicarbonate (X) with 2-amino-6-chloropurine (XI) by means of Pd(PPh3)4 yields the carbocyclic purines (XII), (XIII) or (XIV), respectively. Treatment of these chloro-purines (XII), (XIII) and (XIV) with cyclopropylamine (XV) in hot DMSO provides the corresponding cyclopropylaminopurine carbonate (XVI), abacavir or cyclopropylaminopurine acetate (XVII), respectively. Finally, the protecting groups of purines (XVI) and (XVII) are hydrolyzed with aqueous NaOH.
SYN
Alternatively, 5(R)-(hydroxymethyl)-2-cyclopenten-1(R)-ol (VII) can also be obtained as follows: Acylation of the chiral oxazolidinethione (XIX) with the mixed anhydride (II) by means of BuLi in THF gives the N-pentenoyl-oxazolidinethione (XX), which by condensation with crotonaldehyde (XXI) catalyzed by TiCl4 and (?-spartein in dichloromethane yields the chiral adduct (XXII). The ring-closing metathesis of (XXII) by means of the ruthenium catalyst in dichloromethane affords the chiral cyclopentenol derivative (XXIII), which is reduced to the target diol (VII) by means of LiBH4 in THF.
SYN
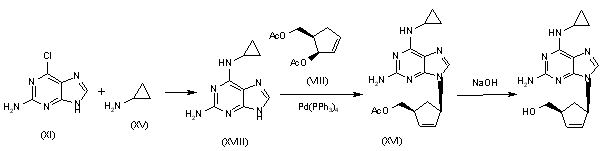
Alternatively, 2-amino-6-chloropurine (XI) is treated with cyclopropylamine (XV) in hot DMSO to give 2-amino-6-(cyclopropylamino)purine (XVIII), which is condensed with the chiral diacetate (VIII) by means of Pd(PPh3)4 to yield the carbocyclic purine acetate (XVI). Finally, purine (XVI) is deprotected by hydrolysis with aqueous NaOH.
CLIP

https://www.sciencedirect.com/science/article/pii/S0960894X15007581
CLIP
CLIP
CLIP
Production of Abacavir
030-8 1.0g (0.0053mol), in the reaction flask was added cesium carbonate 1.75 g (0.0054 mol) and dry DMSO 50ml, stirred under N2 protection, the temperature was raised to 60 °C and stirred at this temperature for 2 h the mixture wascooled to room temperature, then add tetrakis (triphenylphosphine) combined palladium (TTP) [0.85 (0.00074mol)] and compound 030-5 [0.79g (0.0034 mol), DMSO (10 ml) solution was stirred and heated to 65 °C held 65 °C and stirred reaction 2.25h. The you can get the mixture containing compounds 030-9.
To the mixture was added methanol 100ml and K2CO3 is 2.10g, the mixture reaction was stirred for 45min at 40 °C, a solid precipitate which was filtered through a Celite layer and the filtrate was evaporated to a small volume under vacuum at 90 °C, and the remaining gum pounding mill was extracted with dichloromethane (100ml * 2) to give a brown solid residue was purified by silica gel (Merck 9385) column chromatography [eluent: dichloromethane / methanol (volume ratio 9:1)] to give a yellow foam was 030 0.26 g, yield 26.8.

CLIP
https://pubs.rsc.org/en/content/articlehtml/2012/ra/c2ra20842c
References
- ^ Jump up to:a b c d e f g h “Abacavir Sulfate”. The American Society of Health-System Pharmacists. Archived from the original on 8 September 2017. Retrieved 31 July 2015.
- ^ “Drug Name Abbreviations Adult and Adolescent ARV Guidelines”. AIDSinfo. Archived from the original on 2016-11-09. Retrieved 2016-11-08.
- ^ “What Not to Use Adult and Adolescent ARV Guidelines”. AIDSinfo. Archived from the original on 2016-11-09. Retrieved 2016-11-08.
- ^ Jump up to:a b c d Yuen, GJ; Weller, S; Pakes, GE (2008). “A review of the pharmacokinetics of abacavir”. Clinical Pharmacokinetics. 47 (6): 351–71. doi:10.2165/00003088-200847060-00001. PMID 18479171.
- ^ Jump up to:a b “Nucleoside reverse transcriptase inhibitors (NRTIs or ‘nukes’) – HIV/AIDS”. http://www.hiv.va.gov. Archived from the original on 2016-11-09. Retrieved 2016-11-08.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 505. ISBN 9783527607495. Archived from the original on 2017-09-08.
- ^ Kane, Brigid M. (2008). HIV/AIDS Treatment Drugs. Infobase Publishing. p. 56. ISBN 9781438102078. Archived from the original on 2017-09-08.
- ^ “WHO Model List of Essential Medicines (19th List)” (PDF). World Health Organization. April 2015. Archived (PDF) from the original on 13 December 2016. Retrieved 8 December 2016.
- ^ “International Drug Price Indicator Guide”. ERC. Retrieved 20 November 2016.
- ^ “NADAC as of 2016-12-07 | Data.Medicaid.gov”. Centers for Medicare and Medicaid Services. Archived from the original on 21 December 2016. Retrieved 12 December 2016.
- ^ “Abacavir Adverse Reactions”. Epocrates Online.
- ^ “Abacavir | Dosage, Side Effects | AIDSinfo”. AIDSinfo. Archived from the original on 2017-03-06. Retrieved 2016-11-08.
- ^ Mallal, S., Phillips, E., Carosi, G.; et al. (2008). “HLA-B*5701 screening for hypersensitivity to abacavir”. New England Journal of Medicine. 358 (6): 568–579. doi:10.1056/nejmoa0706135. PMID 18256392.
- ^ Rauch, A., Nolan, D., Martin, A.; et al. (2006). “Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study”. Clinical Infectious Diseases. 43 (1): 99–102. doi:10.1086/504874. PMID 16758424.
- ^ Dean, Laura (2012), Pratt, Victoria; McLeod, Howard; Rubinstein, Wendy; Dean, Laura (eds.), “Abacavir Therapy and HLA-B*57:01 Genotype”, Medical Genetics Summaries, National Center for Biotechnology Information (US), PMID 28520363, retrieved 2019-01-14
- ^ Heatherington; et al. (2002). “Genetic variations in HLA-B region and hypersensitivity reactions to abacavir”. Lancet. 359 (9312): 1121–1122. doi:10.1016/s0140-6736(02)08158-8. PMID 11943262.
- ^ Mallal; et al. (2002). “Association between presence of HLA*B5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir”. Lancet. 359(9308): 727–732. doi:10.1016/s0140-6736(02)07873-x. PMID 11888582.
- ^ Rotimi, C. N.; Jorde, L. B. (2010). “Ancestry and disease in the age of genomic medicine”. New England Journal of Medicine. 363 (16): 1551–1558. doi:10.1056/nejmra0911564. PMID 20942671.
- ^ Phillips, E., Mallal, S. (2009). “Successful translation of pharmacogenetics into the clinic”. Molecular Diagnosis & Therapy. 13: 1–9. doi:10.1007/bf03256308.
- ^ Phillips, E., Mallal S. (2007). “Drug hypersensitivity in HIV”. Current Opinion in Allergy and Clinical Immunology. 7 (4): 324–330. doi:10.1097/aci.0b013e32825ea68a. PMID 17620824.
- ^ “Postmarket Drug Safety Information for Patients and Providers – Information for Healthcare Professionals: Abacavir (marketed as Ziagen) and Abacavir-Containing Medications”. Center for Drug Evaluation and Research at the US FDA. Archived from the original on 2013-12-11. Retrieved 2013-11-29.
- ^ “Archived copy”. Archived from the original on 2014-08-08. Retrieved 2014-07-31.
- ^ Swen JJ, Nijenhuis M, de Boer A, et al. (May 2011). “Pharmacogenetics: from bench to byte–an update of guidelines”. Clin Pharmacol Ther. 89 (5): 662–73. doi:10.1038/clpt.2011.34. PMID 21412232.
- ^ Martin MA, Hoffman JM, Freimuth RR, et al. (May 2014). “Clinical Pharmacogenetics Implementation Consortium Guidelines for HLA-B Genotype and Abacavir Dosing: 2014 update”. Clin Pharmacol Ther. 95 (5): 499–500. doi:10.1038/clpt.2014.38. PMC 3994233. PMID 24561393.
- ^ Shear, N.H., Milpied, B., Bruynzeel, D. P.; et al. (2008). “A review of drug patch testing and implications for HIV clinicians”. AIDS. 22 (9): 999–1007. doi:10.1097/qad.0b013e3282f7cb60. PMID 18520343.
- ^ “FDA Alert: Abacavir – Ongoing Safety Review: Possible Increased Risk of Heart Attack”. Drugs.com. Archived from the original on 2013-12-10. Retrieved 2013-11-29.
- ^ Ding X, Andraca-Carrera E, Cooper C, et al. (December 2012). “No association of abacavir use with myocardial infarction: findings of an FDA meta-analysis”. J Acquir Immune Defic Syndr. 61 (4): 441–7. doi:10.1097/QAI.0b013e31826f993c. PMID 22932321.
- ^ Illing, PT; et al. (2012). “Immune self-reactivity triggered by drug-modified HLA-peptide repertoire”. Nature. 486 (7404): 554–8. doi:10.1038/nature11147. PMID 22722860.
- ^ Jump up to:a b Prescribing information. Ziagen (abacavir). Research Triangle Park, NC: GlaxoSmithKline, July 2002
- ^ Vourvahis, M; Kashuba, AD (2007). “Mechanisms of Pharmacokinetic and Pharmacodynamic Drug Interactions Associated with Ritonavir-Enhanced Tipranavir”. Pharmacotherapy. 27 (6): 888–909. doi:10.1592/phco.27.6.888. PMID 17542771.
- ^ McDowell, JA; Chittick, GE; Stevens, CP; et al. (2000). ““, “Pharmacokinetic Interaction of Abacavir (1592U89) and Ethanol in Human Immunodeficiency Virus-Infected Adults”. Antimicrob Agents Chemother. 44 (6): 1686–90. doi:10.1128/aac.44.6.1686-1690.2000. PMC 89933. PMID 10817729.
- ^ Berenguer, J; Perez-Elias, MJ; Bellon, JM; et al. (2006). “Effectiveness and safety of abacavir, lamivudine, and zidovudine in antiretroviral therapy-naive HIV-infected patients: results from a large multicenter observational cohort”. J Acquir Immune Defic Syndr. 41 (2): 154–159. doi:10.1097/01.qai.0000194231.08207.8a. PMID 16394846.
- ^ Dolophine(methadone) [prescribing information]. Columbus, OH: Roxane Laboratories, Inc.; March 2015.
- ^ Gervasoni, C; Cattaneo, D; Di Cristo, V; et al. (2016). “Orlistat: weight lost at cost of HIV rebound”. J Antimicrob Chemother. 71 (6): 1739–1741. doi:10.1093/jac/dkw033. PMID 26945709.
- ^ Cometriq (cabozantinib) [prescribing information]. South San Francisco, CA: Exelixis, Inc.; May 2016.
- ^ Jump up to:a b Product Information: ZIAGEN(R) oral tablets, oral solution, abacavir sulfate oral tablets, oral solution. ViiV Healthcare (per Manufacturer), Research Triangle Park, NC, 2015.
- ^ “Dr. Robert Vince – 2010 Inductee”. Minnesota Inventors Hall of Fame. Minnesota Inventors Hall of Fame. Archived from the original on 15 February 2016. Retrieved 10 February 2016.
- ^ “Robert Vince, PhD (faculty listing)”. University of Minnesota. University of Minnesota. Archived from the original on 2016-02-17.
- ^ Daluge SM, Good SS, Faletto MB, Miller WH, St Clair MH, Boone LR, Tisdale M, Parry NR, Reardon JE, Dornsife RE, Averett DR (May 1997). “1592U89, a novel carbocyclic nucleoside analog with potent, selective anti-human immunodeficiency virus activity”. Antimicrobial Agents and Chemotherapy. 41 (5): 1082–1093. doi:10.1128/AAC.41.5.1082. PMC 163855. PMID 9145874.
- ^ Crimmins, M. T.; King, B. W. (1996). “An Efficient Asymmetric Approach to Carbocyclic Nucleosides: Asymmetric Synthesis of 1592U89, a Potent Inhibitor of HIV Reverse Transcriptase”. The Journal of Organic Chemistry. 61 (13): 4192–4193. doi:10.1021/jo960708p. PMID 11667311.
External links
- Full Prescribing Information
- Abacavir pathway on PharmGKB
- Abacavir dosing guidelines from the Clinical Pharmacogenetics Implementation Consortium (CPIC)
- Abacavir dosing guidelines from the Dutch Pharmacogenetics Working Group (DPWG)
References
-
- Crimmins, M.T. et al.: J. Org. Chem. (JOCEAH) 61 4192 (1996).
- b Olivo, H.F. et al.: J. Chem. Soc., Perkin Trans. 1 (JCPRB4) 1998, 391.
- US 5 089 500 (Burroughs Wellcome; 18.2.1992; GB-prior. 27.6.1988).
- a EP 434 450 (Wellcome Found.; 26.6.1991; appl. 21.12.1990; USA-prior. 22.12.1989).
- EP 1 857 458 (Solmag; appl. 5.5.2006).
- aa EP 424 064 (Enzymatix; appl. 24.4.1991; GB-prior. 16.10.1989).
- US 6 340 587 (SmithKline Beecham; 22.1.2002; appl. 20.8.1998; GB-prior. 22.8.1997).
- c US 5 034 394 (Welcome Found.; 23.7.1991; appl. 22.12.1989; GB-prior. 27.6.1988).
- d WO 9 924 431 (Glaxo; appl. 12.11.1998; WO-prior. 12.11.1997).
-
Alternative syntheses:
- EP 878 548 (Lonza; appl. 13.5.1998; CH-prior. 13.5.1997).
-
Preparation of chloropyrimidine intermediate V:
- US 6 448 403 (SmithKline Beecham; 10.9.2002; appl. 3.2.1995; GB-prior. 4.2.1994).
-
Condensation of pyrimidines with cyclopentylamine IV:
- Vince, R.; Hua, M.: J. Med. Chem. (JMCMAR) 33 (1), 17 (1990).
- Grumam, A. et al.: Tetrahedron Lett. (TELEAY) 36 (42), 7767 (1995).
- EP 349 242 (Wellcome Found.; appl. 26.6.1989; GB-prior. 27.6.1988).
- EP 366 385 (Wellcome Found.; appl. 23.10.1989; GB-prior. 24.10.1988).
- US 6 646 125 (SmithKline Beecham; 11.11.2003; appl. 14.10.1998; GB-prior. 14.10.1997).
- JP 1 022 853 (Asahi Glass Co.; appl. 17.7.1987).
-
Alternative preparation of 4-amino-2-cyclopentene-1-methanol:
- EP 926 131 (Lonza; appl. 24.11.1998; CH-prior. 27.11.1997).
- WO 9 745 529 (Lonza; appl. 30.5.1997; CH-prior. 30.5.1996).
- WO 9 910 519 (Glaxo; 4.3.1999; GB-prior. 20.8.1998).
- WO 9 824 741 (Glaxo; 11.6.1998; GB-prior. 7.12.1996).
- WO 2 001 017 952 (Chirotech; 15.3.2001; GB-prior. 9.9.1999).
-
Abacavir hemisulfate salt:
- US 6 294 540 (Glaxo Wellcome; 25.9.2001; appl. 14.5.1998; GB-prior. 17.5.1997).
-
Abacavir succinate as antiviral agent:
- WO 9 606 844 (Wellcome; 7.3.1996; appl. 25.8.1995; GB-prior. 26.8.1994).
-
Pharmaceutical formulations:
- US 6 641 843 (SmithKline Beecham; 4.11.2003; appl. 4.2.1999; GB-prior. 6.2.1998).
-
Synergistic combinations for treatment of HIV infection:
- WO 9 630 025 (Wellcome; 3.10.1996; appl. 28.3.1996; GB-prior. 30.3.1995).
 |
|

Chemical structure of abacavir
|
|
| Clinical data | |
|---|---|
| Pronunciation | /əˈbækəvɪər/ ( |
| Trade names | Ziagen, others[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a699012 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth (solution or tablets) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 83% |
| Metabolism | Liver |
| Elimination half-life | 1.54 ± 0.63 h |
| Excretion | Kidney (1.2% abacavir, 30% 5′-carboxylic acid metabolite, 36% 5′-glucuronide metabolite, 15% unidentified minor metabolites). Fecal (16%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| NIAID ChemDB | |
| ECHA InfoCard | 100.149.341 |
| Chemical and physical data | |
| Formula | C14H18N6O |
| Molar mass | 286.332 g/mol g·mol−1 |
| 3D model (JSmol) | |
| Melting point | 165 °C (329 °F) |
 |
|

Chemical structure of abacavir
|
{(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]cyclopent-2-en-1-yl}methanol
(-)-cis-4-[2-Amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
(1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol
Abacavir
Abacavir (ABC) is a powerful nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. [Wikipedia] Chemically, it is a synthetic carbocyclic nucleoside and is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. In vivo, abacavir sulfate dissociates to its free base, abacavir.
Abacavir (ABC) ![]() i/ʌ.bæk.ʌ.vɪər/ is a nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. It is available under the trade name Ziagen (ViiV Healthcare) and in the combination formulations Trizivir (abacavir, zidovudine andlamivudine) and Kivexa/Epzicom (abacavir and lamivudine). It has been well tolerated: the main side effect is hypersensitivity, which can be severe, and in rare cases, fatal. Genetic testing can indicate whether an individual will be hypersensitive; over 90% of patients can safely take abacavir. However, in a separate study, the risk of heart attack increased by nearly 90%.[1]
i/ʌ.bæk.ʌ.vɪər/ is a nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. It is available under the trade name Ziagen (ViiV Healthcare) and in the combination formulations Trizivir (abacavir, zidovudine andlamivudine) and Kivexa/Epzicom (abacavir and lamivudine). It has been well tolerated: the main side effect is hypersensitivity, which can be severe, and in rare cases, fatal. Genetic testing can indicate whether an individual will be hypersensitive; over 90% of patients can safely take abacavir. However, in a separate study, the risk of heart attack increased by nearly 90%.[1]
Viral strains that are resistant to zidovudine (AZT) or lamivudine (3TC) are generally sensitive to abacavir (ABC), whereas some strains that are resistant to AZT and 3TC are not as sensitive to abacavir.
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basic health system.[2]
Abacavir is a nucleoside reverse transcriptase inhibitor (NRTI) with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Abacavir is phosphorylated to active metabolites that compete for incorporation into viral DNA. They inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis. The concentration of drug necessary to effect viral replication by 50 percent (EC50) ranged from 3.7 to 5.8 μM (1 μM = 0.28 mcg/mL) and 0.07 to 1.0 μM against HIV-1IIIB and HIV-1BaL, respectively, and was 0.26 ± 0.18 μM against 8 clinical isolates. Abacavir had synergistic activity in cell culture in combination with the nucleoside reverse transcriptase inhibitor (NRTI) zidovudine, the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine, and the protease inhibitor (PI) amprenavir; and additive activity in combination with the NRTIs didanosine, emtricitabine, lamivudine, stavudine, tenofovir, and zalcitabine.
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | J05AF06 | C 14 H 18 N 6 O | 286.34 g / mol | 136470-78-5 |
| succinate | J05AF06 | C 14 H 18 N 6 O · C 4 H 6 O | 356.43 g / mol | 168146-84-7 |
| sulfate | J05AF06 | C 14 H 18 N 6 O · 1 / 2H 2 SO 4 | 670.76 g / mol | 188062-50-2 |
| Systematic (IUPAC) name | |
|---|---|
| {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]cyclopent-2-en-1-yl}methanol | |
| Clinical data | |
| Trade names | Ziagen |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699012 |
| Pregnancy cat. | B3 (AU) C (US) |
| Legal status | POM (UK) ℞-only (US) |
| Routes | Oral (solution or tablets) |
| Pharmacokinetic data | |
| Bioavailability | 83% |
| Metabolism | Hepatic |
| Half-life | 1.54 ± 0.63 h |
| Excretion | Renal (1.2% abacavir, 30% 5′-carboxylic acid metabolite, 36% 5′-glucuronide metabolite, 15% unidentified minor metabolites). Fecal (16%) |
| Identifiers | |
| CAS number | 136470-78-5 |
| ATC code | J05AF06 |
| PubChem | CID 441300 |
| DrugBank | DB01048 |
| ChemSpider | 390063 |
| UNII | WR2TIP26VS |
| KEGG | D07057 |
| ChEBI | CHEBI:421707 |
| ChEMBL | CHEMBL1380 |
| NIAID ChemDB | 028596 |
| Chemical data | |
| Formula | C14H18N6O |
| Mol. mass | 286.332 g/mol |
Abacavir is a carbocyclic synthetic nucleoside analogue and an antiviral agent. Intracellularly, abacavir is converted by cellular enzymes to the active metabolite carbovir triphosphate, an analogue of deoxyguanosine-5′-triphosphate (dGTP). Carbovir triphosphate inhibits the activity of HIV-1 reverse transcriptase (RT) both by competing with the natural substrate dGTP and by its incorporation into viral DNA. Viral DNA growth is terminated because the incorporated nucleotide lacks a 3′-OH group, which is needed to form the 5′ to 3′ phosphodiester linkage essential for DNA chain elongation.
Application
-
an antiviral agent, is used in the treatment of AIDS
-
ingibitor convertibility transkriptazы
Classes of substances
-
Adenine (6-aminopurines)
-
Aminoalcohols
-
Cyclopentenes and cyclopentadienes
-
Tsyklopropanы
-
-
-
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2289753 | 2007-01-23 | 2018-05-14 |
| Canada | 1340589 | 1999-06-08 | 2016-06-08 |
| Canada | 2216634 | 2004-07-20 | 2016-03-28 |
| United States | 6641843 | 2000-02-04 | 2020-02-04 |
| United States | 5089500 | 1992-12-26 | 2009-12-26 |
PATENT
US5034394
Synthesis pathway
Abacavir, (-) cis-[4-[2-amino-6-cyclopropylamino)-9H-purin-9-yl]-2-cyclopenten-yl]-1 – methanol, a carbocyclic nucleoside which possesses a 2,3-dehydrocyclopentene ring, is referred to in United States Patent 5,034,394 as a reverse transcriptase inhibitor. Recently, a general synthetic strategy for the preparation of this type of compound and intermediates was reported [Crimmins, et. al., J. Org. Chem., 61 , 4192-4193 (1996) and 65, 8499-8509-4193 (2000)].
-
-
Abacavir is the International Nonproprietary Name (INN) of {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol and CAS No. 136470-78-5. Abacavir and therapeutically acceptable salts thereof, in particular the hemisulfate salt, are well-known as potent selective inhibitors of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
-
The structure of abacavir corresponds to formula (I):
-

-
EP 434450-A discloses certain 9-substituted-2-aminopurines including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
-
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring.
-
According to the teachings of EP 434450-A , the abacavir base is finally isolated by trituration using acetonitrile (ACN) or by chromatography, and subsequently it can be transformed to a salt of abacavir by reaction with the corresponding acid. Such isolation methods (trituration and chromatography) usually are limited to laboratory scale because they are not appropriate for industrial use. Furthermore, the isolation of the abacavir base by trituration using acetonitrile gives a gummy solid (Example 7) and the isolation by chromatography (eluted from methanol/ethyl acetate) yields a solid foam (Example 19 or 28).
-
Other documents also describe the isolation of abacavir by trituration or chromatography, but always a gummy solid or solid foam is obtained (cf. WO9921861 and EP741710 ), which would be difficult to operate on industrial scale.
-
WO9852949 describes the preparation of abacavir which is isolated from acetone. According to this document the manufacture of the abacavir free base produces an amorphous solid which traps solvents and is, therefore, unsuitable for large scale purification, or for formulation, without additional purification procedures (cf. page 1 of WO 9852949 ). In this document, it is proposed the use of a salt of abacavir, in particular the hemisulfate salt which shows improved physical properties regarding the abacavir base known in the art. Said properties allow the manufacture of the salt on industrial scale, and in particular its use for the preparation of pharmaceutical formulations.
-
However, the preparation of a salt of abacavir involves an extra processing step of preparing the salt, increasing the cost and the time to manufacture the compound. Generally, the abacavir free base is the precursor compound for the preparation of the salt. Thus, depending on the preparation process used for the preparation of the salt, the isolation step of the abacavir free base must also be done.

………………………………
http://www.google.co.in/patents/US5034394
EXAMPLE 21(-)-cis-4-[2-Amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
The title compound of Example 7, (2.00 g, 6.50 mmol) was dissolved in 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (Aldrich, 20 mL). Phosphoryl chloride (2.28 mL, 24.0 mmol) was added to the stirred, cooled (-10° C.) solution. After 3 minutes, cold water (80 mL) was added. The solution was extracted with chloroform (3×80 mL). The aqueous layer was diluted with ethanol (400 mL) and the pH adjusted to 6 with saturated aqueous NaOH. The precipitated inorganic salts were filtered off. The filtrate was further diluted with ethanol to a volume of 1 liter and the pH adjusted to 8 with additional NaOH. The resulting precipitate was filtered and dried to give the 5′-monophosphate of (±)-cis-4-[2-amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol as white powder (4.0 mmoles, 62% quantitated by UV absorbance); HPLC analysis as in Example 17 shows one peak. This racemic 5′ -monophosphate was dissolved in water (200 mL) and snake venom 5′-nucleotidase (EC 3.1.3.5) from Crotalus atrox (5,000 IU, Sigma) was added. After incubation at 37° C. for 10 days, HPLC analysis as in Example 17 showed that 50% of the starting nucleotide had been dephosphorylated to the nucleoside. These were separated on a 5×14 cm column of DEAE Sephadex A25 (Pharmacia) which had been preequilibrated with 50 mM ammonium bicarbonate. Title compound was eluted with 2 liters of 50 mM ammonium bicarbonate. Evaporation of water gave white powder which was dissolved in methanol, adsorbed on silica gel, and applied to a silica gel column. Title compound was eluted with methanol:chloroform/1:9 as a colorless glass. An acetonitrile solution was evaporated to give white solid foam, dried at 0.3 mm Hg over P2 O5 ; 649 mg (72% from racemate); 1 H-NMR in DMSO-d6 and mass spectrum identical with those of the racemate (title compound of Example 7); [α]20 D -48.0°, [α]20 436 -97.1°, [α]20 365 -149° (c=0.14, methanol).
Anal. Calcd. for C15 H20 N6 O.0.10CH3 CN: C, 59.96; H, 6.72; N, 28.06. Found: C, 59.93; H, 6.76; N, 28.03.
Continued elution of the Sephadex column with 2 liters of 100 mM ammonium bicarbonate and then with 2 liters of 200 mM ammonium bicarbonate gave 5′-monophosphate (see Example 22) which was stable to 5′-nucleotidase.
…………………………………………
| Синтез a) |
|---|
        |
| Синтез b) |
     |
| Preparation c) |
    |
| Synthesis d) |
 |

An enantiopure β-lactam with a suitably disposed electron withdrawing group on nitrogen, participated in a π-allylpalladium mediated reaction with 2,6-dichloropurine tetrabutylammonium salt to afford an advanced cis-1,4-substituted cyclopentenoid with both high regio- and stereoselectivity. This advanced intermediate was successfully manipulated to the total synthesis of (−)-Abacavir.

http://pubs.rsc.org/en/content/articlelanding/2012/ob/c2ob06775g#!divAbstract
………………………………….
http://www.google.com.ar/patents/EP2085397A1?cl=en
Example 1: Preparation of crystalline Form I of abacavir base using methanol as solvent
-
[0026]Abacavir (1.00 g, containing about 17% of dichloromethane) was dissolved in refluxing methanol (2.2 mL). The solution was slowly cooled to – 5 °C and, the resulting suspension, was kept at that temperature overnight under gentle stirring. The mixture was filtered off and dried under vacuum (7-10 mbar) at 40 °C for 4 hours to give a white solid (0.55 g, 66% yield, < 5000 ppm of methanol). The PXRD analysis gave the diffractogram shown in FIG. 1.
……………………………………..
http://www.google.com/patents/WO2008037760A1?cl=en
Abacavir, is the International Nonproprietary Name (INN) of {(1 S,4R)-4-[2- amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol, and CAS No. 136470-78-5. Abacavir sulfate is a potent selective inhibitor of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
The structure of abacavir hemisulfate salt corresponds to formula (I):

(I)
EP 434450-A discloses certain 9-substituted-2-aminopuhnes including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring. Pyrimidine compounds which have been identified as being useful as intermediates of said preparation processes include N-2-acylated abacavir intermediates such as N-{6- (cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H-purin- 2-yl}acetamide or N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-
(hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide. The removal of the amino protective group of these compounds using acidic conditions is known in the art. According to Example 28 of EP 434450-A, the amino protective group of the N-{6-(cyclopropylamino)-9-[(1 R,4S)-4- (hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide is removed by stirring with 1 N hydrochloric acid for 2 days at room temperature. The abacavir base, after adjusting the pH to 7.0 and evaporation of the solvent, is finally isolated by trituration and chromatography. Then, it is transformed by reaction with an acid to the corresponding salt of abacavir. The main disadvantages of this method are: (i) the use of a strongly corrosive mineral acid to remove the amino protective group; (ii) the need of a high dilution rate; (iii) a long reaction time to complete the reaction; (iv) the need of isolating the free abacavir; and (v) a complicated chromatographic purification process.
Thus, despite the teaching of this prior art document, the research of new deprotection processes of a N-acylated {(1 S,4R)-4-[2-amino-6- (cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is still an active field, since the industrial exploitation of the known process is difficult, as it has pointed out above. Thus, the provision of a new process for the removal of the amino protective group of a N-acylated {(1 S,4R)-4-[2-amino-6-
(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is desirable.
Example 1 : Preparation of abacavir hemisulfate
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (6.56 g, 18.40 mmol) was slurried in a mixture of isopropanol (32.8 ml) and 10% solution of NaOH (36.1 ml, 92.0 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (32.8 ml) was added. The layers were separated and H2SO4 96% (0.61 ml, 11.03 mmol) was added dropwise to the organic layer. This mixture was cooled to 0-50C and the resulting slurry filtered off.
The solid was dried under vacuum at 40 0C. Abacavir hemisulfate (5.98 g, 97%) was obtained as a white powder.
Example 6: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.0 g, 2.80 mmol) was slurried in a mixture of isopropanol (2 ml) and 10% solution of NaOH (1.1 ml, 2.80 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (2 ml) was added. The aqueous layer was discarded, the organic phase was cooled to 0-5 0C and the resulting slurry filtered off. The solid was dried under vacuum at 400C. Abacavir (0.62 g, 77%) was obtained as a white powder.
Example 7: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetone. Abacavir (0.47 g, 47%) was obtained as a white powder.
Example 8: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetonitrile. Abacavir (0.43 g, 43%) was obtained as a white powder.
Example 9: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in ethyl acetate (150 ml) to afford abacavir (7.2 g, 90%).
Example 10: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in acetone (300 ml) to afford abacavir (7.0 g, 88%).
…………………………………
http://www.google.com/patents/WO2004089952A1?cl=en
Abacavir of formula (1) :

or (1 S,4R)-4-[2-Amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1 – methanol and its salts are nucleoside reverse transcriptase inhibitors. Abacavir sulfate is a nucleoside reverse transcriptase inhibitor and used in the treatment of human immunodeficiency virus infection. Abacavir sulfate and related compounds and their therapeutic uses are disclosed in US 5,034,394.
Crystalline forms of abacavir sulfate have not been reported in the literature. Moreover, the processes described in the literature do not produce abacavir sulfate in a stable, well-defined and reproducible crystalline form. It has now been discovered that abacavir sulfate can be prepared in three stable, well-defined and consistently reproducible crystalline forms.
Example 1
Abacavir free base (3.0 gm, obtained by the process described in example 21 of US 5,034,394) is dissolved in ethyl acetate (15 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 3 hours at 20°C and filtered to give 3.0 gm of form I abacavir sulfate. Example 2 Abacavir free base (3.0 gm) is dissolved in acetone (20 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 6 hours at 25°C and filtered to give 2.8 gm of form I abacavir sulfate.
Example 3 Abacavir free base (3.0 gm) is dissolved in acetonitrile (15 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 2 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 4 Abacavir free base (3.0 gm) is dissolved in methyl tert-butyl ether (25 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 1 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 5 Abacavir free base (3.0 gm) is dissolved in methanol (15 ml) and sulfuric acid (0.3 ml) is added to the solution. The contents then are cooled to 0°C and diisopropyl ether (15 ml) is added. The reaction mass is stirred for 2 hours at about 25°C and the separated solid is filtered to give 3.0 gm of form III abacavir sulfate
…………………………….
http://www.google.com.ar/patents/WO1999021861A1?cl=en
The present invention relates to a new process for the preparation of the chiral nucleoside analogue (1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol (compound of Formula (I)).
The compound of formula (I) is described as having potent activity against human immunodeficiency virus (HIV) and hepatitis B virus (HBV) in EPO34450.

Results presented at the 34th Interscience Conference on Antimicrobial Agents and Chemotherapy (October 4-7, 1994) demonstrate that the compound of formula I has significant activity against HIV comparable to, and if not better than, some current anti HIV drugs, such as zidovudine and didanosine.
Currently the compound of Formula (I) is undergoing clinical investigation to determine its safety and efficacy in humans. Therefore, there exists at the present time a need to supply large quantities of this compound for use in clinical trials.
Current routes of synthesising the compound of formula (I) involve multiple steps and are relatively expensive. It will be noted that the compound has two centres of asymmetry and it is essential that any route produces the compound of formula (I) substantially free of the corresponding enantiomer, preferably the compound of formula (I) is greater than 95% w/w free of the corresponding enantiomer.
Processes proposed for the preparation of the compound of formula (I) generally start from a pyrimidine compound, coupling with a 4-amino-2-cyclopentene-1- methanol analogue, cyciisation to form the imidazole ring and then introduction of the cyclopropylamine group into the 6 position of the purine, such routes include those suggested in EPO434450 and WO9521161. Essentially both routes disclosed in the two prior patent applications involve the following steps:-
(i) coupling (1S, 4R)-4-amino-2-cyclopentene-1 -methanol to N-(4,6-dichloro-5- formamido-2-pyrimidinyl) acetamide or a similar analogue thereof, for example N- (2-amino-4,6-dichloro-5-pyrimidinyl) formamide;
(ii) ring closure of the resultant compound to form the intermediate (1 S, 4R)-4- (2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1 -methanol;
(iii) substituting the halo group by a cyclopropylamino group on the 6 position of the purine ring.
The above routes are multi-step processes. By reducing the number of processing steps significant cost savings can be achieved due to the length of time to manufacture the compound being shortened and the waste streams minimised.
An alternative process suggested in the prior art involves the direct coupling of carbocyclic ribose analogues to the N atom on the 9 position of 2-amino-6-chloro purine. For example WO91/15490 discloses a single step process for the formation of the (1S, 4R)- 4-(2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1- methanol intermediate by reacting (1S, 4R)-4-hydroxy-2-cyclopentene-1 -methanol, in which the allylic hydroxyl group has been activated as an ester or carbonate and the other hydroxyl group has a blocking group attached (for example 1 ,4- bis- methylcarbonate) with 2-amino-6-chloropurine.
However we have found that when synthesising (1S, 4R)-4-(2-amino-6-chloro-9H- purin-9-yl)-2-cyclopentene-1- methanol by this route a significant amount of an N- 7 isomer is formed (i.e. coupling has occurred to the nitrogen at the 7- position of the purine ring) compared to the N-9 isomer desired. Further steps are therefore required to convert the N-7 product to the N-9 product, or alternatively removing the N-7 product, adding significantly to the cost. We have found that by using a transition metal catalysed process for the direct coupling of a compound of formula (II) or (III),

Example 1 (1 S. 4R)-4-[2-Amino-6-(cvclopropylamino)-9H purin-9-vπ-2-cvclopentene-1 – methanol
Triphenylphosphine (14mg) was added, under nitrogen, to a mixture of (1S.4R)- 4-hydroxy-2-cyclopentene -1 -methanol bis(methylcarbonate) (91 mg), 2-amino-6- (cyclopropylamino) purine (90mg), tris(dibenzylideneacetone)dipalladium (12mg) and dry DMF (2ml) and the resulting solution stirred at room temperature for 40 min.
The DMF was removed at 60° in vacuo and the residue partitioned between ethyl acetate (25ml.) and 20% sodium chloride solution (10ml.). The ethyl acetate solution was washed with 20% sodium chloride (2x12ml.) and with saturated sodium chloride solution, then dried (MgSO4) and the solvent removed in vacuo.
The residue was dissolved in methanol (10ml.), potassium carbonate (17mg) added and the mixture stirred under nitrogen for 15h.
The solvent was removed in vacuo and the residue chromatographed on silica gel
(Merck 9385), eluting with dichloromethane-methanol [(95:5) increasing to (90:10)] to give the title compound (53mg) as a cream foam.
δ(DMSO-d6): 7.60 (s.1 H); 7.27 (s,1 H); 6.10 (dt,1 H); 5.86 (dt, 1 H); 5.81 (s,2H); 5.39 (m,1H); 4.75 (t,1H); 3.44 (t,2H); 3.03 (m, 1H): 2.86 (m,1H);2.60 (m,1H); 1.58 (dt, 1 H); 0.65 (m, 2H); 0.57 (m,2H).
TLC SiO2/CHCI3-MeOH (4:1 ) Rf 0.38; det. UN., KMnO4
Trade Names
| Page | Trade name | Manufacturer |
|---|---|---|
| Germany | Kiveksa | GlaxoSmithKline |
| Trizivir | -»- | |
| Ziagen | -»- | |
| France | Kiveksa | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| United Kingdom | Kiveksa | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| Italy | Trizivir | -»- |
| Ziagen | -»- | |
| Japan | Épzikom | -»- |
| Ziagen | -»- | |
| USA | Épzikom | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| Ukraine | Virol | Ranbaksi Laboratories Limited, India |
| Ziagen | GlaksoSmitKlyayn Inc.., Canada | |
| Abamun | Tsipla Ltd, India | |
| Abacavir sulfate | Aurobindo Pharma Limited, India |
Formulations
-
Oral solution 20 mg / ml;
-
Tablets of 300 mg (as the sulfate);
-
Trizivir tablets 300 mg – abacavir in fixed combination with 150 mg of lamivudine and 300 mg zidovudine
ZIAGEN is the brand name for abacavir sulfate, a synthetic carbocyclic nucleoside analogue with inhibitory activity against HIV-1. The chemical name of abacavir sulfate is (1S,cis)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol sulfate (salt) (2:1). Abacavir sulfate is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. It has a molecular formula of (C14H18N6O)2•H2SO4 and a molecular weight of 670.76 daltons. It has the following structural formula:
 |
Abacavir sulfate is a white to off-white solid with a solubility of approximately 77 mg/mL in distilled water at 25°C. It has an octanol/water (pH 7.1 to 7.3) partition coefficient (log P) of approximately 1.20 at 25°C.
ZIAGEN Tablets are for oral administration. Each tablet contains abacavir sulfate equivalent to 300 mg of abacavir as active ingredient and the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The tablets are coated with a film that is made of hypromellose, polysorbate 80, synthetic yellow iron oxide, titanium dioxide, and triacetin.
ZIAGEN Oral Solution is for oral administration. Each milliliter (1 mL) of ZIAGEN Oral Solution contains abacavir sulfate equivalent to 20 mg of abacavir (i.e., 20 mg/mL) as active ingredient and the following inactive ingredients: artificial strawberry and banana flavors, citric acid (anhydrous), methylparaben and propylparaben (added as preservatives), propylene glycol, saccharin sodium, sodium citrate (dihydrate), sorbitol solution, and water.
In vivo, abacavir sulfate dissociates to its free base, abacavir. All dosages for ZIAGEN are expressed in terms of abacavir.
History
Abacavir was approved by the Food and Drug Administration (FDA) on December 18, 1998 and is thus the fifteenth approved antiretroviral drug in the United States. Its patent expired in the United States on 2009-12-26.
Links
-
US 5 089 500 (Burroughs Wellcome; 18.2.1992; GB-prior. 27.6.1988).
-
Synthesis a)
-
EP 434 450 (Wellcome Found .; 26.6.1991; appl. 21.12.1990; prior-USA. 22.12.1989).
-
Crimmins, MT et al .: J. Org. Chem. (JOCEAH) 61 4192 (1996).
-
EP 1 857 458 (Solmag; appl. 5.5.2006).
-
EP 424 064 (Enzymatix; appl. 24.4.1991; GB -prior. 16.10.1989).
-
U.S. 6 340 587 (Beecham SMITHKLINE; 22.1.2002; appl. 20.8.1998; GB -prior. 22.8.1997).
-
-
Синтез b)
-
Olivo, HF et al .: J. Chem. Soc., Perkin Trans. 1 (JCPRB4) 1998, 391.
-
-
Preparation c)
-
U.S. 5 034 394 (Wellcome Found .; 23.7.1991; appl. 22.12.1989; GB -prior. 27.6.1988).
-
-
Synthesis d)
-
WO 9 924 431 (Glaxo; appl. 12.11.1998; WO-prior. 12.11.1997).
-
| WO2008037760A1 * | Sep 27, 2007 | Apr 3, 2008 | Esteve Quimica Sa | Process for the preparation of abacavir |
| EP1905772A1 * | Sep 28, 2006 | Apr 2, 2008 | Esteve Quimica, S.A. | Process for the preparation of abacavir |
| US8183370 | Sep 27, 2007 | May 22, 2012 | Esteve Quimica, Sa | Process for the preparation of abacavir |
| EP0434450A2 | 21 Dec 1990 | 26 Jun 1991 | The Wellcome Foundation Limited | Therapeutic nucleosides |
| EP0741710A1 | 3 Feb 1995 | 13 Nov 1996 | The Wellcome Foundation Limited | Chloropyrimide intermediates |
| WO1998052949A1 | 14 May 1998 | 26 Nov 1998 | Glaxo Group Ltd | Carbocyclic nucleoside hemisulfate and its use in treating viral infections |
| WO1999021861A1 | 24 Oct 1997 | 6 May 1999 | Glaxo Group Ltd | Process for preparing a chiral nucleoside analogue |
| WO1999039691A2 * | 4 Feb 1999 | 12 Aug 1999 | Brooks Nikki Thoennes | Pharmaceutical compositions |
| WO2008037760A1 * | 27 Sep 2007 | 3 Apr 2008 | Esteve Quimica Sa | Process for the preparation of abacavir |
References
- Jump up^ SFGate.com
- Jump up^ “WHO Model List of EssentialMedicines”. World Health Organization. October 2013. Retrieved 22 April 2014.
- Jump up^ https://online.epocrates.com/noFrame/showPage.do?method=drugs&MonographId=2043&ActiveSectionId=5
- Jump up^ Mallal, S., Phillips, E., Carosi, G. et al. (2008). “HLA-B*5701 screening for hypersensitivity to abacavir”. New England Journal of Medicine 358: 568–579.doi:10.1056/nejmoa0706135.
- Jump up^ Rauch, A., Nolan, D., Martin, A. et al. (2006). “Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study”. Clinical Infectious Diseases 43: 99–102. doi:10.1086/504874.
- Jump up^ Heatherington et al. (2002). “Genetic variations in HLA-B region and hypersensitivity reactions to abacavir”. Lancet 359: 1121–1122.
- Jump up^ Mallal et al. (2002). “Association between presence of HLA*B5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir”. Lancet359: 727–732. doi:10.1016/s0140-6736(02)07873-x.
- Jump up^ Rotimi, C.N.; Jorde, L.B. (2010). “Ancestry and disease in the age of genomic medicine”. New England Journal of Medicine 363: 1551–1558.
- Jump up^ Phillips, E., Mallal, S. (2009). “Successful translation of pharmacogenetics into the clinic”. Molecular Diagnosis & Therapy 13: 1–9. doi:10.1007/bf03256308.
- Jump up^ Phillips, E., Mallal S. (2007). “Drug hypersensitivity in HIV”. Current Opinion in Allergy and Clinical Immunology 7: 324–330. doi:10.1097/aci.0b013e32825ea68a.
- Jump up^http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm123927.htmAccessed November 29, 2013.
- Jump up^ http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=ca73b519-015a-436d-aa3c-af53492825a1
- Jump up^ Martin MA, Hoffman JM, Freimuth RR et al. (May 2014). “Clinical Pharmacogenetics Implementation Consortium Guidelines for HLA-B Genotype and Abacavir Dosing: 2014 update”. Clin Pharmacol Ther. 95 (5): 499–500. doi:10.1038/clpt.2014.38.PMC 3994233. PMID 24561393.
- Jump up^ Swen JJ, Nijenhuis M, de Boer A et al. (May 2011). “Pharmacogenetics: from bench to byte–an update of guidelines”. Clin Pharmacol Ther. 89 (5): 662–73.doi:10.1038/clpt.2011.34. PMID 21412232.
- Jump up^ Shear, N.H., Milpied, B., Bruynzeel, D.P. et al. (2008). “A review of drug patch testing and implications for HIV clinicians”. AIDS 22: 999–1007.doi:10.1097/qad.0b013e3282f7cb60.
- Jump up^ http://www.drugs.com/fda/abacavir-ongoing-safety-review-possible-increased-risk-heart-attack-12914.html Accessed November 29, 2013.
- Jump up^ Ding X, Andraca-Carrera E, Cooper C et al. (December 2012). “No association of abacavir use with myocardial infarction: findings of an FDA meta-analysis”. J Acquir Immune Defic Syndr. 61 (4): 441–7. doi:10.1097/QAI.0b013e31826f993c.PMID 22932321.
- Illing PT et al. 2012, Nature, doi:10.1038/nature11147
External links
- Full Prescribing Information
- Abacavir pathway on PharmGKB
- Abacavir dosing guidelines from the Clinical Pharmacogenetics Implementation Consortium (CPIC)
- Abacavir dosing guidelines from the Dutch Pharmacogenetics Working Group (DPWG)
EXTRA INFO
How to obtain carbocyclic nucleosides?
Carbocyclic nucleosides are synthetically the most challenging class of nucleosides, requiring multi-step and often elaborate synthetic pathways to introduce the necessary stereochemistry. There are two main strategies for the preparation of carbocyclic nucleosides. In the linear approach a cyclopentylamine is used as starting material and the heterocycle is built in a stepwise manner (see Scheme 1).
Scheme 1: Linear approach for the synthesis of abacavir.[5]

The more flexible strategy is a convergent approach: a functionalized carbocyclic moiety is condensed with a heterocycle rapidly leading to a variety of carbocyclic nucleosides. Initially, we started our syntheses from cyclopentadiene 1 that is deprotonated and alkylated with benzyloxymethyl chloride to give the diene 2. This material is converted by a hydroboration into cyclopentenol 3 or isomerized into two thermodynamically more stable cyclopentadienes 4a,b. With the protection and another hydroboration step to 5 we gain access to an enantiomerically pure precursor for the synthesis of a variety of carbocyclic 2’-deoxynucleosides e.g.:carba-dT, carba-dA or carba-BVDU.[6] The isomeric dienes 4a,b were hydroborated to the racemic carbocyclic moiety 6.

Scheme 2: Convergent approach for the synthesis of carba-dT.
The asymmetric synthesis route and the racemic route above are short and efficient ways to diverse carbocyclic D- or L-nucleosides (Scheme 2). Different heterocycles can be condensed to these precursors leading to carbocyclic purine- and pyrimidine-nucleosides. Beside α- and β-nucleosides, carbocyclic epi– andiso-nucleosides in the 2’-deoxyxylose form were accessable.[7]
What else is possible? The racemic cyclopentenol 6 can be coupled by a modified Mitsunobu-reaction.Moreover, this strategy offers the possibility of synthesizing new carbocyclic nucleosides by functionalizing the double bond before or after introduction of the nucleobase (scheme 3).[8]

Scheme 3: Functionalized carbocyclic nucleosides based on cyclopentenol 6.
Other interesting carbocyclic precursors like cyclopentenol 7 can be used to synthesize several classes of carbocyclic nucleoside analogues, e.g.: 2’,3’-dideoxy-2’,3’-didehydro nucleosides (d4-nucleosides), 2’,3’-dideoxynucleosides (ddNs), ribonucleosides, bicyclic nucleosides or even 2’-fluoro-nucleosides.

Scheme 4: Functionalized carbocyclic thymidine analogues based on cyclopentenol 7.
[1] V. E. Marquez, T. Ben-Kasus, J. J. Barchi, K. M. Green, M .C. Nicklaus, R. Agbaria, J. Am. Chem. Soc.2004,126, 543.
[2] A. D. Borthwick, K. Biggadike, Tetrahedron 1992, 48, 571.
[3] H. Bricaud, P. Herdewijn, E. De Clercq, Biochem. Pharmacol. 1983, 3583.
[4] P. L. Boyer, B. C. Vu, Z. Ambrose, J. G. Julias, S. Warnecke, C. Liao, C. Meier, V. E. Marquez, S. H. Hughes, J. Med. Chem. 2009, 52, 5356.
[5] S. M. Daluge, M. T. Martin, B. R. Sickles, D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids 2000,19, 297.
[6] O. R. Ludek, C. Meier, Synthesis 2003, 2101.
[7] O. R. Ludek, T. Kraemer, J. Balzarini, C. Meier, Synthesis 2006, 1313.
[8] M. Mahler, B. Reichardt, P. Hartjen, J. van Lunzen, C. Meier, Chem. Eur. J. 2012, 18, 11046-11062.
FDA approves new treatment for hospital-acquired and ventilator-associated bacterial pneumonia
The U.S. Food and Drug Administration today approved a new indication for the previously FDA-approved drug, Zerbaxa (ceftolozane and tazobactam) for the treatment of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia (HABP/VABP) in patients 18 years and older. The FDA initially approved Zerbaxa in 2014 to treat complicated intra-abdominal infections and for complicated urinary tract infections.
“A key global challenge we face as a public health agency is addressing the threat of antimicrobial-resistant infections,” said FDA Principal Deputy Commissioner Amy Abernethy, M.D., Ph.D. “Hospital-acquired and ventilator-associated bacterial pneumonia are serious infections that can result in death in some patients. New therapies to treat these infections are important to …
- June 03, 2019
The U.S. Food and Drug Administration today approved a new indication for the previously FDA-approved drug, Zerbaxa (ceftolozane and tazobactam) for the treatment of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia (HABP/VABP) in patients 18 years and older. The FDA initially approved Zerbaxa in 2014to treat complicated intra-abdominal infections and for complicated urinary tract infections.
“A key global challenge we face as a public health agency is addressing the threat of antimicrobial-resistant infections,” said FDA Principal Deputy Commissioner Amy Abernethy, M.D., Ph.D. “Hospital-acquired and ventilator-associated bacterial pneumonia are serious infections that can result in death in some patients. New therapies to treat these infections are important to meet patient needs because of increasing antimicrobial resistance. That’s why, among our other efforts to address antimicrobial resistance, we’re focused on facilitating the development of safe and effective new treatments to give patients more options to fight life-threatening infections.”
HABP/VABP occur in patients in hospitals or other health care facilities and can be caused by a variety of bacteria. According to data from the U.S. Centers for Disease Control and Prevention, HABP and VABP are currently the second most common type of hospital-acquired infection in the United States, and are a significant issue in patients in the intensive care unit (ICU).
The safety and efficacy of Zerbaxa for the treatment of HABP/VABP, administered via injection, was demonstrated in a multinational, double-blind study that compared Zerbaxa to another antibacterial drug in 726 adult patients hospitalized with HABP/VABP. The study showed that mortality and cure rates were similar between Zerbaxa and the comparator treatment.
The most common adverse reactions observed in the HABP/VABP trial among patients treated with Zerbaxa were elevated liver enzyme levels, renal impairment or failure, and diarrhea.
Zerbaxa should not be used in patients with known serious hypersensitivity to components of Zerbaxa, as well as hypersensitivity to piperacillin/tazobactam or other members of the beta lactam class of antibacterial drugs.
Zerbaxa received FDA’s Qualified Infectious Disease Product (QIDP) designation for the treatment of HABP/VABP. The QIDP designation is given to antibacterial and antifungal drug products intended to treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act. As part of QIDP designation, the Zerbaxa marketing application for the HABP/VABP indication was granted Priority Review under which the FDA’s goal is to take action on an application within an expedited time frame.
The FDA granted the approval of Zerbaxa for the treatment of HABP/VABP to Merck & Co., Inc.
//////////////ceftolozane, tazobactam, FDA 2019, Zerbaxa, HABP/VABP, Merck , Qualified Infectious Disease Product, (QIDP), Priority Review
Onasemnogene abeparvovec オナセムノジーンアベパルボベック
Onasemnogene abeparvovec
オナセムノジーンアベパルボベック
DNA (synthetic adeno-associated virus 9 vector scAAV9.CB.hSMN human survivor motor neuron protein-specifying)
Zolgensma
FDA 2019/5/24 APPROVED
CAS: 1922968-73-7
AVXS-101
Spinal muscular atrophy treatment
Treatment of Spinal Muscular Atrophy (SMA) Type 1
Gene therapy product
![]()
Onasemnogene abeparvovec, sold under the trade name Zolgensma, is a gene therapy medication used to treat spinal muscular atrophy (SMA).
SMA is a neuromuscular disorder caused by a mutation in the SMN1 gene, which in turn reduces the amount of SMN protein necessary for survival of motor neurons. Onasemnogene abeparvovec is a biologic drug consisting of AAV9 virus capsids that have been deprived of the original viral DNA and instead contain a SMN1 transgene along with promoters. The drug is administered intravenously or intrathecally. Upon administration, the AAV9 viral vector delivers the SMN1 transgene to cell nuclei where the transgene begins encoding SMN protein, thus addressing the root cause of the disease. Since motor neurons do not divide, it is thought that a single dose of the drug will have a lifelong effect.[1]
The medication was developed by a US biotechnology company AveXis, a subsidiary of Novartis,[2] based on an earlier discovery by French researchers.[3] The intravenous formulation was approved in May 2019 in the United States for use in children under 2 years.[4]It carries a list price of US$ 2.125 million per dose (one-time treatment), making it the most expensive medication in the world as of 2019.[5]
Terminology
Onasemnogene abeparvovec is the international nonproprietary name (INN) and US adopted name (USAN).[6] It was previously known under compound name AVXS-101.
FDA approves a gene therapy that is the most expensive drug in the world
FDA on Friday approved onasemnogene abeparvovec-xioi (Zolgensma—AveXis), a one-time gene therapy for the treatment of spinal muscular atrophy (SMA).
FDA on Friday approved onasemnogene abeparvovec-xioi (Zolgensma—AveXis), a one-time gene therapy for the treatment of spinal muscular atrophy (SMA). The ultrarare disease affects infants. In announcing the approval, Novartis—which acquired AveXis last year—also disclosed the price of the drug, $2.1 million. The company noted that it would provide rebates to insurance companies if the drug is not successful, though it did not offer details about what would be considered failure. Novartis also said it will set up 5-year payment plans for states, small insurance firms, and self-insured employers. Another drug, nusinersen (Spinraza—Biogen) is already available for the treatment of SMA; however, that drug must continue to be injected into patients’ spines throughout their lives, at a cost of $750,000 in the first year and $375,000 a year after that. “Patients with SMA now have another treatment option to minimize the progression of SMA and improve survival,” said Peter Marks, director of FDA’s Center for Biologics Evaluation and Research.
References
- ^ “Novartis announces FDA filing acceptance and Priority Review of AVXS-101, a one-time treatment designed to address the genetic root cause of SMA Type 1 | Novartis”. Novartis. Retrieved 2018-12-04.
- ^ “Novartis successfully completes acquisition of AveXis, Inc. | Novartis”. Novartis. Retrieved 2018-10-06.
- ^ “AveXis receives FDA approval for Zolgensma®, the first gene therapy for paediatric patients with SMA”. SMA Europe. 2015-05-25. Retrieved 2019-05-25.
- ^ “FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality”. FDA. 2019-05-24. Retrieved 2019-05-24.
- ^ Reuters (2019-05-25). “$2.1m Novartis gene therapy to become world’s most expensive drug”. The Guardian. ISSN 0261-3077. Retrieved 2019-05-25.
- ^ “Onasemnogene abeparvovec – AveXis – AdisInsight”. adisinsight.springer.com. Retrieved 2018-10-06.
| Clinical data | |
|---|---|
| Trade names | Zolgensma |
| Synonyms | AVXS-101 |
| License data | |
| Routes of administration |
Intravascular |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Duration of action | lifetime (?) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| KEGG | |
/////////Onasemnogene abeparvovec, Zolgensma, FDA 2019, オナセムノジーンアベパルボベック ,Spinal muscular atrophy, Gene therapy product, AVXS-101
FDA approves first PI3K inhibitor Piqray (alpelisib) for breast cancer

FDA approves first PI3K inhibitor for breast cancer
syn https://newdrugapprovals.org/2018/06/25/alpelisib-byl-719/
Today, the U.S. Food and Drug Administration approved Piqray (alpelisib) tablets, to be used in combination with the FDA-approved endocrine therapy fulvestrant, to treat postmenopausal women, and men, with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced or metastatic breast cancer (as detected by an FDA-approved test) following progression on or after an endocrine-based regimen.
The FDA also approved the companion diagnostic test, therascreen PIK3CA RGQ PCR Kit, to detect the PIK3CA mutation in a tissue and/or a liquid biopsy. Patients who are negative by
- May 24, 2019
Today, the U.S. Food and Drug Administration approved Piqray (alpelisib) tablets, to be used in combination with the FDA-approved endocrine therapy fulvestrant, to treat postmenopausal women, and men, with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced or metastatic breast cancer (as detected by an FDA-approved test) following progression on or after an endocrine-based regimen.
The FDA also approved the companion diagnostic test, therascreen PIK3CA RGQ PCR Kit, to detect the PIK3CA mutation in a tissue and/or a liquid biopsy. Patients who are negative by the therascreen test using the liquid biopsy should undergo tumor biopsy for PIK3CA mutation testing.
“Piqray is the first PI3K inhibitor to demonstrate a clinically meaningful benefit in treating patients with this type of breast cancer. The ability to target treatment to a patient’s specific genetic mutation or biomarker is becoming increasingly common in cancer treatment, and companion diagnostic tests assist oncologists in selecting patients who may benefit from these targeted treatments,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “For this approval, we employed some of our newer regulatory tools to streamline reviews without compromising the quality of our assessment. This drug is the first novel drug approved under the Real-Time Oncology Review pilot program. We also used the updated Assessment Aid, a multidisciplinary review template that helps focus our written review on critical thinking and consistency and reduces time spent on administrative tasks.”
Metastatic breast cancer is breast cancer that has spread beyond the breast to other organs in the body (most often the bones, lungs, liver or brain). When breast cancer is hormone-receptor positive, patients may be treated with anti-hormonal treatment (also called endocrine therapy), alone or in combination with other medicines, or chemotherapy.
The efficacy of Piqray was studied in the SOLAR-1 trial, a randomized trial of 572 postmenopausal women and men with HR-positive, HER2-negative, advanced or metastatic breast cancer whose cancer had progressed while on or after receiving an aromatase inhibitor. Results from the trial showed the addition of Piqray to fulvestrant significantly prolonged progression- free survival (median of 11 months vs. 5.7 months) in patients whose tumors had a PIK3CA mutation.
Common side effects of Piqray are high blood sugar levels, increase in creatinine, diarrhea, rash, decrease in lymphocyte count in the blood, elevated liver enzymes, nausea, fatigue, low red blood cell count, increase in lipase (enzymes released by the pancreas), decreased appetite, stomatitis, vomiting, weight loss, low calcium levels, aPTT prolonged (blood clotting taking longer to occur than it should), and hair loss.
Health care professionals are advised to monitor patients taking Piqray for severe hypersensitivity reactions (intolerance). Patients are warned of potentially severe skin reactions (rashes that may result in peeling and blistering of skin or mucous membranes like the lips and gums). Health care professionals are advised not to initiate treatment in patients with a history of severe skin reactions such as Stevens-Johnson Syndrome, erythema multiforme, or toxic epidermal necrolysis. Patients on Piqray have reported severe hyperglycemia (high blood sugar), and the safety of Piqray in patients with Type 1 or uncontrolled Type 2 diabetes has not been established. Before initiating treatment with Piqray, health care professionals are advised to check fasting glucose and HbA1c, and to optimize glycemic control. Patients should be monitored for pneumonitis/interstitial lung disease (inflammation of lung tissue) and diarrhea during treatment. Piqray must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Piqray is the first new drug application (NDA) for a new molecular entity approved under the Real-Time Oncology Review (RTOR) pilot program, which permits the FDA to begin analyzing key efficacy and safety datasets prior to the official submission of an application, allowing the review team to begin their review and communicate with the applicant earlier. Piqray also used the updated Assessment Aid (AAid), a multidisciplinary review template intended to focus the FDA’s written review on critical thinking and consistency and reduce time spent on administrative tasks. With these two pilot programs, today’s approval of Piqray comes approximately three months ahead of the Prescription Drug User Fee Act (PDUFA) VI deadline of August 18, 2019.
The FDA granted this application Priority Review designation. The FDA granted approval of Piqray to Novartis. The FDA granted approval of the therascreen PIK3CA RGQ PCR Kit to QIAGEN Manchester, Ltd.
//////////////FDA, PI3K inhibitor, breast cancer, fda 2019, Piqray, alpelisib, therascreen PIK3CA RGQ PCR Kit, QIAGEN Manchester, Priority Review, BYL719, BYL 719
CT 1812
CT-1812
Elayta
Condition(s): Alzheimer’s Disease
U.S. FDA Status: Alzheimer’s Disease (Phase 2)
Company: Cognition Therapeutics Inc.
CAS: 1802632-22-9
Chemical Formula: C24H33NO4S
Molecular Weight: 431.591
2-(tert-butoxy)-4-(3-methyl-3-(5-(methylsulfonyl)isoindolin-2-yl)butyl)phenol
Phenol, 4-[3-[1,3-dihydro-5-(methylsulfonyl)-2H-isoindol-2-yl]-3-methylbutyl]-2-(1,1-dimethylethoxy)-
- Originator Cognition Therapeutics
- Class Antidementias; Neuroprotectants; Nootropics; Small molecules
- Mechanism of Action Sigma-2 receptor antagonists
- Phase II Alzheimer’s disease
- Phase I Cognition disorders
- 21 Feb 2019 Cognition Therapeutics receives patent for a composition of matter patent covering Elayta™ in Europe
- 19 Feb 2019 Pharmacokinetics and adverse events data from a phase I trial in Cognition disorders released by Cognition Therapeutics
- 22 Oct 2018 CTP push 289675: Updated KDM, forwarded USA line from PI/II to PII
CT-1812 is a first-in-class, orally available sigma-2/PGRMC1 antagonist (alpha beta oligomer receptor antagonist), is being developed by Cognition. sCT-1812 is a novel therapeutic candidate for Alzheimer’s disease
SYN

BACKGROUND
CT1812 is a small-molecule antagonist of the sigma2 receptor, also known as the progesterone receptor membrane component 1. The rationale behind this therapeutic approach is that ligands for the sigma2/PGRMC1 receptor will compete with oligomeric Aβ binding to this receptor and thus interfere with Aβ-induced synaptic toxicity. CT1812 grew out of screening programs at Cognition Therapeutics. Company scientists have reported that compounds in this series not only block binding of a range of different Aβ species to this receptor but also displace it when applied after Aβ has bound (Dec 2014 conference news).
The structure of CT1812 has not been disclosed, but similar compounds in the series have been reported to enter the brain, occupy up to 80 percent of sigma2/PGRMC1 receptors, and restore behavioral deficits in APP transgenic mice (Izzo et al., 2014; Izzo et al., 2014).
FINDINGS
From September 2015 to May 2016, Cognition Therapeutics ran a Phase 1 trial in 80 healthy volunteers aged 18 to 75 in Melbourne, Australia; target enrollment was originally listed as 114. Single-ascending-dose administration was followed by multiple ascending doses given once daily for two weeks. The dose range in this trial spanned 10 to 650 mg; if this would not generate data to set a maximum tolerated dose, doses up to 1,350 mg were to be tried. Outcome measures included safety, tolerability, plasma pharmacokinetics, and CSF CT1812 concentration. At the 2016 and 2017 AAIC conferences, company scientists reported that single doses up to 1,120 mg were given, as were multiple doses of up to 840 mg in young and up to 560 mg in elderly volunteers. The drug was reported to be well-tolerated, with suitable pharmacokinetics, sufficient brain penetrance and target exposure, and minimal drug-drug interactions affecting cytochrome P450 activity (Catalano et al., 2016; Catalano et al., 2017).
From September 2016 to August 2017, Cognition Therapeutics ran a Phase 1/2 trial at four sites in Australia, enrolling 19 participants with mild to moderate Alzheimer’s disease supported by a recent MRI. It compared a four-week course of 90, 280, or 560 mg of CT 1812 to placebo, taken once daily, on safety and tolerability parameters. At the subsequent CTAD conference, Elayta was reported to have been generally safe and well tolerated, though there were four cases of lymphocytopenia. Exploratory measures such as ADAS-Cog14, verbal or category fluency tests recorded no difference between groups, but exploratory biomarker analyses yielded possible signals of synapse protection (Dec 2017 conference news).
In April 2018, a Phase 1/2 study started enrolling 21 people whose mild to moderate AD was confirmed by amyloid PET or CSF testing. Conducted at Yale University School of Medicine and dubbed COG0105 or SPARC, this trial will compare a six-month course of 100 or 300 mg of Elayta, or placebo. The primary outcome is cognition as assessed by the Alzheimer’s Disease Clinical Study Activities of Daily Living (ADCS-ADL), but the trial will also use the investigational PET tracer UCB-J, which binds to the synaptic vesicle glycoprotein 2A, in an attempt to monitor synapse density before and after treatment (see company press release; Jul 2016 news).
In summer 2018, a Phase 1b target engagement study at the University of Pennsylvania will start enrolling 18 people whose mild to moderate AD is confirmed by amyloid PET. Called COG0104 or SNAP, it will compare single injections of 90, 280, or 560 mg of Elayta or placebo for their ability to displace Aβ oligomers and clear them into the CSF, as measured by a CSF Aβ oligomer assay.
Also in summer 2018, a Phase 2 multi-center study is expected to begin enrolling 24 people with mild to moderate AD as confirmed by amyloid PET for a six-month course of 100 or 300 mg of Elayta, or placebo. As of May 22, 2018, this trial lists CT1812 pharmacodynamic effects on CSF biomarkers, specifically as assessed by CSF neurogranin levels, as primary outcome.
For all trials of this compound, see clinicaltrials.gov.
PATENT
WO 2015116923
https://patents.google.com/patent/WO2015116923A1
There are only five medications currently FDA-approved for the treatment of Alzheimer’s Disease (AD). Four are cholinesterase inhibitors: tacrine (COGNEX®; Sciele), donepezil (ARICEPT®; Pfizer), rivastigmine (EXELON®; Novartis), and galantamine (RAZADYNE®; Ortho-McNeil-Janssen). Donepezil, rivastigmine, and galantamine are successors to tacrine, a first generation compound rarely prescribed because of the potential for hepatotoxicity; they are roughly equally efficacious at providing symptomatic improvement of cognition and function at all stages of AD. The fifth approved medication is memantine (NAMENDA®; Forest), a low-affinity, use dependent N-methyl-D-aspartate glutamate receptor antagonist that offers similar benefits, but only in moderate to severe AD. The clinical effects of these compounds are small and impermanent, and currently available data are inconclusive to support their use as disease modifying agents. See, e.g., Kerchner et al, 2010, Bapineuzumab, Expert Opin Biol Ther., 10(7): 1121-1130. Clearly, alternative approaches to treatment of AD are required.
[004] Certain isoindoline compounds are provided that act as sigma-2 receptor functional antagonists and inhibit the deleterious effects of soluble Αβ oligomers. In some embodiments, isoindoline sigma-2 receptor antagonist compounds and compositions are used to treat or prevent synaptic dysfunction in a subject.
Example 21 illustrates representative preparation of 2-(Tert-butoxy)-
4-(3-methyl-3-(5-(methylsulfonyl)isoindolin-2-yl)butyl)phenol, Example Compound 62, as shown in Scheme 17.


10 Compound 62
[0534] Scheme 17: Procedure for preparation of 2-(Tert-butoxy)-4-(3- methyl-3-(5-(methylsulfonyl)isoindolin-2-yl)butyl)phenol, Example Compound 62.
[0535] Preparation of compound l(Scheme 17): To a glass pressure -bottle at -30 °C containing a mixture of catechol (50.0 g, 454 mmol, 1.0 eq), concentrated sulfuric acid (0.3 mL) in dichloromethane (200 mL), isobutene (152.6 g, 2.72 mol, 6.0 eq) was condensed. After sealing the pressure-bottle with a threaded Teflon cap tipped with a Teflon-protected rubber O-ring, the mixture was heated at 35 °C for 3 h until a clear solution was obtained. After cooling (-30 °C), triethylamine (1.5 mL, 10.8 mmol) was added and the mixture was concentrated. The residue was suspended in 0.5 M NaOH (1 L) and stirred for 10 min. The dark-green colored solution was washed with petroleum ether (2x 100 mL) and the washing layers were reextracted with 0.5 M NaOH (3x 100 mL). The combined aqueous layers were brought to pH 7-8 with 2 N HCl (400 mL), and extracted with ethyl acetate (2* 1 L), dried over sodium sulfate and concentrated to afford product 1 (67.7 g, 90%) as a colorless oil, which was used directly for the next step reaction without further purification. TLC: PE/EA = 50/1 ; Rf (Catechol) = 0.1 ; Rf (Compound 1) = 0.6.
[0536] Preparation of compound 2 (Scheme 17): To a stirred solution of compound 1 (1 12.2 g, 676 mmol, 1.2 eq) and potassium iodide (1 12.2 g, 676 mmol, 1.0 eq) in methanol (2 L) at 0 °C was slowly added sodium hydroxide (27.0 g, 676 mmol, 1.0 eq), followed with aqueous sodium chlorite (7% aq., 718.8 mL, 710 mmol, 1.05 eq) dropwise over 3 h while keeping the reaction below 0 °C. The mixture was stirred at 0 °C for another 30 min and neutralized by adding 2 N HCl at 0 °C till pH 7, extracted with DCM (2 x 1 L). The organic layers were dried over sodium sulfate and concentrated to afford product 2 (179.8 g, 91%). TLC: PE/EA = 50/1; Rf(Compound 1) = 0.6 ; Rf (Compound 2) = 0.6.
[0537] Preparation of compound 3(Scheme 17): To a stirred solution of compound 2 (179.8 g, 616 mmol, 1.0 eq) and triethylamine (186.6 g, 1.85 mol, 3.0 eq) in dichloromethane (2 L) at 0 °C was slowly added acetyl chloride (53.2 g, 677 mmol, 1.1 eq). The mixture was stirred at 0 °C for another 30 min, and warmed up to rt, and stirred at rt for 3 h, water (1 L) was added into the reaction mixture and the organic layer was washed with brine, dried over sodium sulfate and concentrated to afford product 3 (206 g, 100%), which was used directly to the next step without further purification. TLC: PE/EA = 50/1; Rf (Compound 2) = 0.6; Rf (Compound 3) = 0.5.
[0538] Preparation of compound 4 (Scheme 17): To a stirred solution of compound 3 (206 g, 616 mmol, 1.0 eq) in triethylamine (4.0 L) was added 2- methylbut-3-yn-2-amine (102.5 g, 1.23 mol, 2.0 eq), Pd(PPh3)2Cl2 (15.1 g, 18.5 mmol, 0.03 eq) and copper(I) iodide (5.9 g, 31 mmol, 0.05 eq) and resulting mixture was stirred at rt for 17 h. The solvent was removed under reduced pressure and the crude product was purified by silica gel chromatography to afford the title compound 4 (132.7 g, 74%). TLC: PE/EA = 1/1; Rf (Compound 3) = 0.9; Rf (Compound 4) = 0.3. [0539] Preparation of compound 5(Scheme 17): To a stirred solution of compound 4 (104.5 g, 0.36 mol) in ethanol (1.5 L) was added Pd/C (10% wt, 10.5 g). The mixture was stirred under hydrogen (balloon) overnight, and filtered. The filtrate was evaporated to dryness to afford compound 5 (106.3 g, 100%), which was used directly to the next step without further purification. TLC: PE/EA = 1/1; Rf(Compound 4) = 0.3 ; Rf (Compound 5) = 0.3.
[0540] Preparation of compound 6 (Scheme 17): To a solution of o-xylene
(115.7 g, 1.09 mol, 1.0 eq) in chloroform (1.0 L) at 0 °C was added C1S03H (254 g, 2.18 mol, 2.0 eq) dropwise. After the addition, the reaction mixture was stirred at room temperature for 2 days, and poured into ice. The crude mixture was extracted with dichloromethane (3 x 1.0 L). The organic layers were combined, dried over anhydrous sodium sulfate, concentrated to afford the crude compound 6 (161.5 g, 80%) as a white solid, which was used directly to the next step without further purification. TLC: PE/EA = 5/1; Rf (Compound 6) = 0.7.
[0541] General procedure for the preparation of compound 7 (Scheme
17): To a stirred solution of compound 6 (161.5 g, 0.87 mol, 1.0 eq) in saturated sodium sulfite solution (273 g, 2.17 mol, 2.5 eq, in 2.0 L of water) was added dropwise 32% NaOH (69.4 g, 1.73 mol, 2.0 eq) till the solution reached pH 9. After stirring at rt overnight, the reaction mixture was acidified with cone. HC1 in ice- cooling bath till pH 1. The precipitate was filtered, and washed with ice-water (2x), dried in vacuo to afford the crude product 7 (131 g, 88%), which was used directly for next step without further purification. TLC: PE/EA = 5/1; Rf (Compound 6) = 0.7; Rf (Compound 7) = 0.6.
[0542] Preparation of compound 8 (Scheme 17): To a stirred solution of compound 7 (130 g, 0.76 mol, 1.0 eq) and potassium carbonate (211 g, 1.53 mol, 2.0 eq) in DMF (300 mL) was added iodomethane (96 mL, 1.53 mol, 2.0 eq). The reaction was stirred at 40 °C overnight. The reaction mixture was evaporated to dryness, extracted with ethyl acetate. The organic layers were washed with water and brine, dried over sodium sulfate and concentrated, purified by flash column chromatography (PE: EA,10: 1 ~ 5: 1) to afford compound 8 (85.2 g, 61%). TLC: PE/EA = 5/1; Rf (Compound 7) = 0.6; Rf (Compound 8) = 0.3. [0543] Preparation of compound 9 (Scheme 17):To a stirred solution of compound 8 (78.2 g, 424 mmol, 1.0 eq) in 1 ,2-dichloroethane (1.2 L), were added N-bromosuccinimide (166 g, 934 mmol, 2.2 eq) and AIBN (6.9 g, 42.4 mmol, 0.1 eq). The reaction was stirred at reflux overnight. The reaction was diluted with water and dichloromethane. The organic layer was collected, and dried over sodium sulfate and concentrated, purified by flash column chromatography to afford compound 9, which was further recrystallized from hot methanol to afford the pure product 8 (75 g, 52%). TLC: PE/EA = 5/1; Rf (Compound 8) = 0.3; Rf (Compound 9) = 0.2.
[0544] Preparation of compound 10 (Scheme 17):To a stirred solution of compound 5 (46 g, 157 mmol, 1.0 eq) and compound 9 (53.5 g, 157 mmol, 1.0 eq) in THF (460 mL) was added triethylamine (47.7 g, 472 mmol, 3.0 eq). The reaction was stirred at 40 °C overnight, filtered and the filtrate was evaporated to dryness and purified by flash column chromatography to afford compound 10 (45 g, 63%). TLC: PE/EA = 1/1; Rf (Compound 5) = 0.3; Rf (Compound 9) = 1.0; Rf (Compound 10) = 0.4.
[0545] Preparation of Compound 62 (Scheme 17):To a stirred solution of compound 10 (45 g, 98.4 mmol) in methanol (300 mL) was added sodium methoxide (844 mg, 15.6 mmol, 0.16 eq) in one portion. The solution was stirred at rt overnight. Water (250 mL) was added dropwise into the reaction mixture over 1 h, the mixture was stirred at rt for 2 h, and filtered. The white solid was collected and dried on vacuum overnight to afford pure example Compound 62 base (38 g, 89%>). TLC: PE/EA = 1/1; Rf (Compound 10) = 0.4; Rf (Compound 62) = 0.4; ESI-MS: 432 (M+l)+; 1H NMR (400 MHz, CDC13) δ 7.80-7.78 (m, 2H). 7.40-7.38 (m, 1H), 6.87-6.79 (m, 3H), 5.58 (s, 1H), 4.11 (s, 4H), 3.05 (s, 3H), 2.61-2.57 (m, 2H), 1.76- 1.72 (m, 2H), 1.48 (s, 9H), 1.18 (s, 6H). Example 22: Preparation of (2-(4-(4-Hydroxy-3-methoxyphenyl)-2- methylbutan-2-yl)isoindolin-4-yl)(piperazin-l-yl)methanone,
REFERENCES
1: Grundman M, Morgan R, Lickliter JD, Schneider LS, DeKosky S, Izzo NJ,
Guttendorf R, Higgin M, Pribyl J, Mozzoni K, Safferstein H, Catalano SM. A phase
1 clinical trial of the sigma-2 receptor complex allosteric antagonist CT1812, a
novel therapeutic candidate for Alzheimer’s disease. Alzheimers Dement (N Y).
2019 Jan 23;5:20-26. doi: 10.1016/j.trci.2018.11.001. eCollection 2019. PubMed
PMID: 30723776; PubMed Central PMCID: PMC6352291.
Paper Citations
- Catalano S, Grundman M, Schneider LS, DeKosky S, Morgan R, Higgin M, Pribyl J, Mozzoni K, Izzo NJ, Safferstein H, Lickliter J. A Two-Part, Double-Blind, Placebo-Controlled, Phase 1 Study of the Safety and Pharmacokinetics of Single and Multiple Ascending Doses of Ct1812 in Healthy Volunteers. Alzheimer’s & Dementia, July 2016, Volume 12, Issue 7, Supplement
- Catalano S, Grundman M, Schneider LS, DeKosky S, Morgan R, Guttendorf R, Higgin M, Pribyl J, Mozzoni K, Izzo NJ, Safferstein H. A Phase 1 Safety Trial of the aβ Oligomer Receptor Antagonist CT1812. Alzheimer’s & Dementia, July 2017, Volume 13, Issue 7
- Izzo NJ, Staniszewski A, To L, Fa M, Teich AF, Saeed F, Wostein H, Walko T 3rd, Vaswani A, Wardius M, Syed Z, Ravenscroft J, Mozzoni K, Silky C, Rehak C, Yurko R, Finn P, Look G, Rishton G, Safferstein H, Miller M, Johanson C, Stopa E, Windisch M, Hutter-Paier B, Shamloo M, Arancio O, LeVine H 3rd, Catalano SM. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS One. 2014;9(11):e111898. Epub 2014 Nov 12 PubMed.
- Izzo NJ, Xu J, Zeng C, Kirk MJ, Mozzoni K, Silky C, Rehak C, Yurko R, Look G, Rishton G, Safferstein H, Cruchaga C, Goate A, Cahill MA, Arancio O, Mach RH, Craven R, Head E, LeVine H 3rd, Spires-Jones TL, Catalano SM. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS One. 2014;9(11):e111899. Epub 2014 Nov 12PubMed.
/////CT-1812, CT 1812, CT1812, Alzheimers , Cognition Therapeutics, Elayta, phase 2, Cognition disorders
OC1=CC=C(CCC(C)(N2CC3=C(C=C(S(=O)(C)=O)C=C3)C2)C)C=C1OC(C)(C)C
Dipivefrine, дипивефрин , ديبيفيفرين , 地匹福林 , ジピベフリン
Dipivefrine
- Molecular FormulaC19H29NO5
- Average mass351.437 Da

Dipivefrine (INN) or dipivefrin (USAN), trade name Propine among others, is a prodrug of epinephrine, and is used to treat open-angle glaucoma.[1][2] It is available as a 0.1% ophthalmic solution. It is no longer available in the United States.[3]
Dipivefrin is a prodrug with little or no pharmacologically activity until it is hydrolyzed into epinephrine inside the human eye. The liberated epinephrine, an adrenergic agonist, appears to exert its action by stimulating α -and/or β2-adrenergic receptors, leading to a decrease in aqueous production and an enhancement of outflow facility. The dipivefrin prodrug delivery system is a more efficient way of delivering the therapeutic effects of epinephrine, with fewer side effects than are associated with conventional epinephrine therapy. Dipivefrin is used as initial therapy for the control of intraocular pressure in chronic open-angle glaucoma.

Contraindications
Use in narrow-angle glaucoma may be dangerous because it could make the eye susceptible to an attack of angle closure,[2] causing an increase in pressure and pain, and possibly loss of vision.
Side effects
The most common side effects of dipivefrine are burning, stinging and other irritations of the eye. Possible, but uncommon, side effects are those of epinephrine: tachycardia (fast heartbeat), hypertension (high blood pressure) and arrhythmias (irregular heartbeat).[2]
Pharmacology
Dipivefrine penetrates the cornea and is then hydrolysed to epinephrine by esterase enzymes. It increases outflow of the aqueous humour and also reduces its formation (mediated by its action on α1 and α2 receptors), thus reducing pressure inside the eye. It also increases the conductivity of trabecular filtering cells (a β2 receptor mediated action). It is preferred to epinephrine because it is longer acting, more consistent in its action and better tolerated.[1]
Patent
https://patents.google.com/patent/CN102153485A/en

Example 1 [0023] Embodiment
[0024] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 5 ° C, was added 666g (6. 58mol) of triethylamine, and then was added dropwise 784g (6. 5mol) pivaloyl chloride was added dropwise and stirring was continued after the pool. Filtered off with suction, the filtrate by rotary evaporation; to give 990g yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 96.2%. [0025] The 35mol) N- methyl amine section, 370g (3. 66mol) of triethylamine, 25g (0. 15mol) KI, 3L DMF was added 4-neck flask of the IOL. Cooled to 0 ° C, was added dropwise 990g (2. 8mol) 4- (2- chloroacetyl) -I, DMF solution tank 2-phenyl pivalate ester. At room temperature was stirred for 4h.
[0026] suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 923. Og. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 96.5%.
[0027] Take 625g (1. 422mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 65g (1.71mol) of sodium borohydride. At room temperature was stirred for 4h. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 552. 5g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.2%.
[0028] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 13g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0029] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0030] After the solution was washed with methanol hydrochloride salt to give an off-white solid 119. 9g, dipivefrin i.e., the content of 98.9%.
[0031] m.p. 161 ~162 ° C;
[0032] 1H NMR (CDCl3) δ: 1. 35 (s, 18Η), 2 68 (s, 3Η), 3 07-3 13 (m, 2Η), 5 36-5 39 (m….. , 1H),
[0033] 7. 06-7. 30 (m, 3H), 8. 61 (s, 1H), 9. 48 (s, 1H)
Dipivefrin prepared: Example 2 [0034] Embodiment
[0035] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 10 ° C, was added 666g (6. 58mol) of triethylamine, and then dropwise 78½ (6. 5mol) pivaloyl chloride was added dropwise and stirring was continued after the pool. Filtered off with suction, the filtrate by rotary evaporation; 978. 2g to give yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 96. 2% o
[0036] The 35mol) N- methyl amine section, 370g (3. 66mol) of triethylamine, 25g (0. 15mol) KI, 3L DMF was added 4-neck flask of the IOL. Cooled to O0C, dropwise 978. 2g (2. 77mol) 4- (2- chloroacetyl) of DMF solution tank Laid-1,2-phenyl valerate. At room temperature was stirred for 4h.
[0037] suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 910. 2g. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 96.3%.
[0038] Take 625g (1. 422mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 97g (l. SOmol) potassium borohydride. Stirred cell at room temperature. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 532. 7g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.0%.
[0039] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 15g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0040] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0041] After the solution was washed with methanol hydrochloride salt to give an off-white solid was 112. 8g, i.e., dipivefrin, content 98.6%.
3 [0042] Example 2: Preparation of dipivefrin
[0043] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 5 ° C, was added 897g (6. 5mol) of potassium carbonate, and then drops was added 784g (6. 5mol) pivaloyl chloride addition was completed stirring was continued Syndrome. Filtered off with suction, the filtrate by rotary evaporation; to give 900g yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 95.6%.
[0044] A 526g (4. 35mol) N_ methylbenzylamine, 414g (3. Omol) of potassium carbonate, 25g (0. 15mol) KI, 3L DMF force Λ IOL of four port flask. Cooled to O0C, was added dropwise 900g (2. 55mol) 4- (2- chloroacetyl) of DMF solution of 1,2-Shan Laid phenyl valerate. It was stirred at room temperature Mi.
[0045] The suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 820g. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 95.6%.
[0046] Take 625g (1. 42mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 65g (1.71mol) of sodium borohydride. Stirred cell at room temperature. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 512. 5g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.0%.
[0047] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 16g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0048] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0049] After the solution was washed with methanol hydrochloride salt to give an off-white solid was 109. 8g, i.e., dipivefrin, content 98.5%.
SYN
SYN
2-chloro-3′,4′-dihydroxyacetophenone, 99-40-1
3′,4′-dihydroxy-2-methylaminoacetophenone, 99-45-6
2,2-dimethylpropanoic acid 4-[(methylamino)acetyl]-1,2-phenylene ester, 52245-00-8
Pivaloyl chloride, 3282-30-2
Trimethylacetyl chloride, 3282-30-2

1-(3,4-dipivaloyloxyphenyl)-2-(benzylmethylamino)ethan-1-one, 42146-03-2
SPECTROSCOPY

infrared spectral assignments for dipiveh hydrochloride
Wavelength (cm-1) Assignment
3255,2804,2475, 2397 RflHz+-NH stretch
2974-2875 sp3 C-H stretch
1273, 1258-1163 C-0-C stretch
3600-3400 0-H stretch
phenyl ester C=O stretch 1761
aromatic C-C stretch 1614, 1595, 1562, 1504
sp3 C-H bending and scissoring 1481, 1461, 1441, 1397
tert-butyl C-H bending1368, 1332
secondary alcohol C-0 stretch 1 124- 1028
out-of-plane bending for 1,substituted benzene ring 3,4 891,842

Ultraviolet absorption of dipivefrin hydrochloride
E (176, 1 cm)
Solvent 210 nm 264 Nn 270 nm
Acetonitrile 267.3 14.8 13.4
Ethanol 246.8 14.5 13.1
pH 3 Buffer 266.7 12.4 10.4
pH 7 Buffer 257.6 10.8 8.9
Water 278.0 18.0 16.2




References
- ^ Jump up to:a b KD Tripari. Essentials of Medical Pharmacology (5 ed.). Jaypee Brothers Medical Publishers(P) Ltd. p. 88. ISBN 81-8061-187-6.
- ^ Jump up to:a b c Dipivefrin FDA Professional Drug Information.
- ^ Zhang L, Weizer JS, Musch DC (2017). “Perioperative medications for preventing temporarily increased intraocular pressure after laser trabeculoplasty”. Cochrane Database Syst Rev. 2: CD010746. doi:10.1002/14651858.CD010746.pub2. PMC 5477062. PMID 28231380.
-
- Hussain, A.; Truelove, J.E.: J. Pharm. Sci. (JPMSAE) 65, 1510 (1976).
- US 3 839 584.
- a DOS 2 343 657 (Interx Res. Corp.; appl. 30.8.1973; USA-prior. 31.8.1972).
- US 3 809 714 (Interx; 7.5.1974; prior. 31.8.1972) also racemate resolution.
- b DOS 2 152 058 (Klinge; appl. 19.10.1971).
 |
|
| Clinical data | |
|---|---|
| Trade names | Propine, Pivalephrine |
| Synonyms | Dipivefrin |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a686005 |
| Pregnancy category |
|
| Routes of administration |
Eye drops |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C19H29NO5 |
| Molar mass | 351.437 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////дипивефрин , ديبيفيفرين , 地匹福林 , Dipivefrine, antiglaucoma, GENERIC, ジピベフリン
Etosalamide, этосаламид , إيتوسالاميد , 依托柳胺 ,
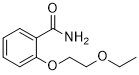
Cas 15302-15-5
Chemical Formula: C11H15NO3
Molecular Weight: 209.245
o-(2-Ethoxyethoxy)benzamide
Etosalamide, also known as Ethosalamide, is an antipyretic and analgesics agent
SYN

OR

CAS:592-55-2, 2-Bromoethyl ethyl ether
Cas, 611-20-1, 2-Hydroxybenzonitrile

PATENT
DE 1013643
PATENT
GB 774635
PATENT
US2822391
78 – 79 MP
PAPER
Journal of Chemical and Engineering Data (1962), 7, 265-6
70 – 71.5 MP
PATENT
WO 2004003198
US 20100226943
/////////Etosalamide, этосаламид , إيتوسالاميد , 依托柳胺 , ethosalamide
O=C(N)C1=CC=CC=C1OCCOCC
FDA approves first treatment Ruzurgi (amifampridine) for children with Lambert-Eaton myasthenic syndrome, a rare autoimmune disorder

FDA approves first treatment Ruzurgi (amifampridine) for children with Lambert-Eaton myasthenic syndrome, a rare autoimmune disorder
The U.S. Food and Drug Administration today approved Ruzurgi (amifampridine) tablets for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in patients 6 to less than 17 years of age. This is the first FDA approval of a treatment specifically for pediatric patients with LEMS. The only other treatment approved for LEMS is only approved for use in adults.
“We continue to be committed to facilitating the development and approval of treatments for rare diseases, particularly those in children,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This approval will provide a much-needed treatment option for pediatric patients with LEMS who have significant weakness and fatigue that can often cause great difficulties with daily activities.”
LEMS is a rare autoimmune disorder that affects the connection between nerves and muscles and causes weakness and other symptoms in affected patients. In people with LEMS, the body’s own immune system attacks the neuromuscular junction (the connection between nerves and muscles) and disrupts the ability of nerve cells to send signals to muscle cells. LEMS may be associated with …
- May 06, 2019
The U.S. Food and Drug Administration today approved Ruzurgi (amifampridine) tablets for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in patients 6 to less than 17 years of age. This is the first FDA approval of a treatment specifically for pediatric patients with LEMS. The only other treatment approved for LEMS is only approved for use in adults.
“We continue to be committed to facilitating the development and approval of treatments for rare diseases, particularly those in children,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This approval will provide a much-needed treatment option for pediatric patients with LEMS who have significant weakness and fatigue that can often cause great difficulties with daily activities.”
LEMS is a rare autoimmune disorder that affects the connection between nerves and muscles and causes weakness and other symptoms in affected patients. In people with LEMS, the body’s own immune system attacks the neuromuscular junction (the connection between nerves and muscles) and disrupts the ability of nerve cells to send signals to muscle cells. LEMS may be associated with other autoimmune diseases, but more commonly occurs in patients with cancer such as small cell lung cancer, where its onset precedes or coincides with the diagnosis of cancer. LEMS can occur at any age. The prevalence of LEMS specifically in pediatric patients is not known, but the overall prevalence of LEMS is estimated to be three per million individuals worldwide.
Use of Ruzurgi in patients 6 to less than 17 years of age is supported by evidence from adequate and well-controlled studies of the drug in adults with LEMS, pharmacokinetic data in adult patients, pharmacokinetic modeling and simulation to identify the dosing regimen in pediatric patients and safety data from pediatric patients 6 to less than 17 years of age.
The effectiveness of Ruzurgi for the treatment of LEMS was established by a randomized, double-blind, placebo-controlled withdrawal study of 32 adult patients in which patients were taking Ruzurgi for at least three months prior to entering the study. The study compared patients continuing on Ruzurgi to patients switched to placebo. Effectiveness was measured by the degree of change in a test that assessed the time it took the patient to rise from a chair, walk three meters, and return to the chair for three consecutive laps without pause. The patients that continued on Ruzurgi experienced less impairment than those on placebo. Effectiveness was also measured with a self-assessment scale for LEMS-related weakness that evaluated the feeling of weakening or strengthening. The scores indicated greater perceived weakening in the patients switched to placebo.
The most common side effects experienced by pediatric and adult patients taking Ruzurgi were burning or prickling sensation (paresthesia), abdominal pain, indigestion, dizziness and nausea. Side effects reported in pediatric patients were similar to those seen in adult patients. Seizures have been observed in patients without a history of seizures. Patients should inform their health care professional immediately if they have signs of hypersensitivity reactions such as rash, hives, itching, fever, swelling or trouble breathing.
The FDA granted this application Priority Review and Fast Track designations. Ruzurgi also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Ruzurgi to Jacobus Pharmaceutical Company, Inc.
/////////////////FDA 2019, Ruzurgi, amifampridine, Lambert-Eaton myasthenic syndrome, LEMS, RARE DISEASES, CHILDREN, Jacobus Pharmaceutical Company, Priority Review, Fast Track designations, Orphan Drug designation
Beperminogene perplasmid, ベペルミノゲンペルプラスミド
Beperminogene perplasmid
ベペルミノゲンペルプラスミド
HGF plasmid
- DNA (human hepatocyte growth factor plasmid pVAX1 cDNA)
- DNA (plasmid pVAX1HGF/MGBI)
- AMG-0001
DS-992
Nucleic Acid Sequence
Sequence Length: 51811342 a 1223 c 1314 g 1302 t
APPROVED, japan 2019, Collategene, 2019/3/29
Antiparkinsonian, Angiogenesis inducing agent
CAS: 627861-07-8
- Originator AnGes MG
- Developer AnGes MG; Osaka University Hospital
- Class Antiparkinsonians; Gene therapies; Ischaemic heart disorder therapies; Vascular disorder therapies
- Mechanism of Action Angiogenesis inducing agents; Gene transference; Hepatocyte growth factor expression stimulants
- Available For Licensing Yes – Ischaemic heart disorders; Lymphoedema; Parkinson’s disease
- Registered Peripheral arterial disorders
- Phase I/II Lymphoedema
- No development reported Arteriosclerosis obliterans; Ischaemic heart disorders; Parkinson’s disease; Thromboangiitis obliterans
- 26 Mar 2019 Registered for Peripheral arterial disorders in Japan (IM)
- 21 Feb 2019 The Pharmaceutical Affairs and Food Sanitation Council recommends conditional and time-limited approval of beperminogene perplasmid for the improvement of ulcers associated with chronic peripheral arterial disease
- 21 Feb 2019 AnGes plans a clinical study to assess the efficacy of beperminogene perplasmid in improvement of pain at rest in chronic peripheral arterial disorders
- In 2010, the product received fast track designation in the U.S. for the treatment of critical limb ischemia
HGF Plasmid (Beperminogene Perplasmid)Critical Limb Ischemia (Arteriosclerosis Obliterans & Buerger’s Disease) AMG0001 Injection, JAPAN AND US ALLIANCE Mitsubishi Tanabe Pharma
PATENT
WO 2017126488
US 20170283446
Expert Review of Cardiovascular Therapy (2014), 12(10), 1145-1156.
////////////Beperminogene perplasmid, japan 2019, ベペルミノゲンペルプラスミド , AnGes MG, Osaka University Hospital, Critical Limb Ischemia, Arteriosclerosis Obliterans, Buerger’s Disease, AMG0001, AMG-0001, DS-992 , HGF plasmid , fast track designation
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

