
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

GRAPIPRANT

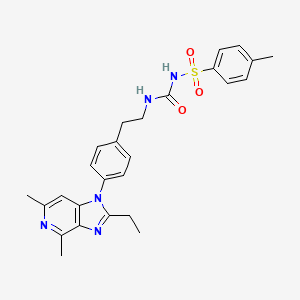

GRAPIPRANT
- Molecular FormulaC26H29N5O3S
- Average mass491.605 Da
CAS 415903-37-6
UNII-J9F5ZPH7NB, CJ 023423, CJ-023423,
Phase II, Arrys Therapeutics, CANCER,
PAIN, AskAt Phase II,
- N-[[[2-[4-(2-Ethyl-4,6-dimethyl-1H-imidazo[4,5-c]pyridin-1-yl)phenyl]ethyl]amino]carbonyl]-4-methylbenzenesulfonamide
- 1-[2-[4-(2-Ethyl-4,6-dimethylimidazo[4,5-c]pyridin-1-yl)phenyl]ethyl]-3-(4-methylphenyl)sulfonylurea
- 2-Ethyl-4,6-dimethyl-1-[4-[2-[[[[(4-methylphenyl)sulfonyl]amino]carbonyl]amino]ethyl]phenyl]-1H-imidazo[4,5-c]pyridine
- AAT 007
- CJ 023423
- Grapiprant
- MR 10A7
- RQ 00000007
- RQ 7
Synonyms and Mappings
- 415903-37-6
- GRAPIPRANT [GREEN BOOK]
- CJ-023
- GRAPIPRANT [INN]
- GRAPIPRANT [WHO-DD]
- MR-10A7
- AAT-007
- MR10A7
- RQ-00000007
- RQ-7
- GRAPIPRANT [USAN]
- GRAPIPRANT
- 2-ETHYL-4,6-DIMETHYL-1-(4-(2-(((((4-METHYLPHENYL)SULFONYL)AMINO)CARBONYL)AMINO)ETHYL)PHENYL)-1H-IMIDAZO(4,5-C)PYRIDINE
- N-(((2-(4-(2-ETHYL-4,6-DIMETHYL-1H-IMIDAZO(4,5-C)PYRIDIN-1-YL)PHENYL)ETHYL)AMINO)CARBONYL)-4-METHYLBENZENESULFONAMIDE
- CJ 023423
- BENZENESULFONAMIDE, N-(((2-(4-(2-ETHYL-4,6-DIMETHYL-1H-IMIDAZO(4,5-C)PYRIDIN-1-YL)PHENYL)ETHYL)AMINO)CARBONYL)-4-METHYL-
- CJ-023,423
- N-(((2-(4-(2-ETHYL-4,6-DIMETHYL-1H-IMIDAZO(4,5-C)PYRIDIN-1-YL)PHENYL)ETHYL)AMINO)CARBONYL)-4-METHYL-BENZENESULFONAMIDE
- CJ-023423
SYN

Arrys Therapeutics (under license from AskAt ) and affiliate Ikena Oncology (formerly known as Kyn Therapeutics ) are developing ARY-007 , an oral formulation of grapiprant, for treating cancers; in December 2019, preliminary data were expected in 2020
Grapiprant (trade name Galliprant) is a small molecule drug that belongs in the piprant class. This analgesic and anti-inflammatory drug is primarily used as a pain relief for mild to moderate inflammation related to osteoarthritis in dogs. Grapiprant has been approved by the FDA’s Center for Veterinary Medicine and was categorized as a non-cyclooxygenase inhibiting non-steroidal anti-inflammatory drug (NSAID) in March 2016.[1]
Preclinical studies also indicate that grapiprant is not only efficacious as a acute pain but also in chronic pain relief and inflammation drug. The effect of the drug is directly proportional to the dosage and its effects were comparable to human medication such as rofecoxib and piroxicam.[2]
Grapiprant, a prostanoid EP4 receptor antagonist, is in phase II clinical trials at AskAt for the treatment of chronic pain. Phase I/II clinical trials are ongoing at Arrys Therapeutics in combination with pembrolizumab for the treatment of patients with microsatellite stable colorectal cancer and in patients with advanced or metastatic PD-1/L1 refractory non-small cell lung cancer (NSCLC).
Grapiprant is also used in humans, and was researched to be used as a pain control and inflammation associated with osteoarthritis. The effect of grapiprant could be explained through the function of prostaglandin E2, in which acts as a pro-inflammatory mediator of redness of the skin, edema and pain which are the typical signs of inflammation. The effect of PGE2 stems from its action through the four prostaglandin receptor subgroups EP1, EP2, EP3 and EP4, in which the prostaglandin EP4 receptor acts as the main intermediary of the prostaglandin-E2-driven inflammation. Grapiprant is widely accepted in veterinary medicine due to its specific and targeted approach to pain management in dogs. The serum concentration of grapiprant is increased when used in conjunction with other drugs such as acetaminophen, albendazole, and alitretinoin.
Common side effects are intestinal related effects such as mild diarrhea, appetite loss, and vomiting.[3] Additionally, it is found that it might lead to reduced tear production due to it being a sulfa-based medication and also reduced albumin levels.
Grapiprant, a prostanoid EP4 receptor antagonist, is in phase II clinical trials at AskAt for the treatment of chronic pain. Phase I/II clinical trials are ongoing at Arrys Therapeutics in combination with pembrolizumab for the treatment of patients with microsatellite stable colorectal cancer and in patients with advanced or metastatic PD-1/L1 refractory non-small cell lung cancer (NSCLC).
Medical uses
Grapiprant is used once a day as an oral pain relief for dogs with inflammation-related osteoarthritis. It is a non-steroidal anti-inflammatory (NSAID) that functions as a targeted action to treat osteoarthritis pain and inflammation in dogs.
Mechanism of action
Grapiprant acts as a specific antagonist that binds and blocks the prostaglandin EP4 receptor, one out of the four prostaglandin E2 (PGE2) receptor subgroups. The EP4 receptor then mediates the prostaglandin-E2-elicited response to pain, and hence grapiprant was proven to be effective in the decrease of pain in several inflammatory pain models of rats. It was also proven to be effective in reducing osteoarthritis-related pain in humans, which serves as a proof for its mechanism of action. The approximate calculation for canine efficacy dose is between the range of 1.3 and 1.7 mg/kg, in conjunction with a methylcellulose suspending agent. Based on the calculations from the comparisons of binding affinity of grapiprant to the EP4 receptors of dogs, rats, and humans, the study of plasma and serum protein binding determinations, the effective doses determined in inflammation pain models of rats, and human-related clinical studies, it is evaluated that Grapiprant should be administered just once a day. The approved dose of the commercial Grapiprant tablet by the FDA for the pain relief and inflammation associated with osteoarthritis to dogs is reported to be 2 mg/kg a day.[4]
Absorption
Studies in animals such as horses have shown the presence of Grapiprant in serum 72 hours with a concentration >0.005 ng/ml after the initial administration of a dose of 2 mg/kg. Grapiprant is expeditiously absorbed and the reported serum concentration was reported to be 31.9 ng/ml in an amount of time of 1.5 hours. The actual body exposure to grapiprant after administration of one dose was shown to be 2000 ng.hr/ml. The degree and rate at which grapiprant is absorbed into the body, presents a mean bioavailability of 39%. A significant reduction in the bioavailability, concentration time and maximal concentration were reported to have occurred after food intake.[1] And thus, grapiprant is usually not administered with food as it will not be as efficient.[5]
Distribution
The volume of distribution in cat studies was reported to be 918 ml/kg.[1]
Route of elimination
Following an oral administration, the majority of the dose was metabolized within the first 72 hours. Equine studies have shown that grapiprant is present in urine 96 hours after the first administration of a dose of 2 mg/kg and has a concentration >0.005 ng/ml. From the excreted dose conducted in horses, it is found that 55%, 15% and 19% of the orally-administered dose was excreted in bile, urine, and faeces respectively.[1]
Toxicity
Safety studies conducted on grapiprant have demonstrated that it generally possesses an exceptional safety profile and a wide safety margin in veterinary studies.[6] In animal studies, a research on 2.5-12 times overdose was conducted for grapiprant and the study resulted in soft-blobs and mucous-filled faeces, occasional bloody stools and emesis.
PATENT
WO-2020014465
Novel crystalline forms of grapiprant and their salts eg HCl (designated as Form A), useful for inhibiting prostaglandin EP4 receptor activity and treating cancers.
Prostaglandins are mediators of pain, fever and other symptoms associated with inflammation. Prostaglandin E2 (PGE2) is the predominant eicosanoid detected in inflammation conditions. In addition, it is also involved in various physiological and/or pathological conditions such as hyperalgesia, uterine contraction, digestive peristalsis, awakeness, suppression of gastric acid secretion, blood pressure, platelet function, bone metabolism, angiogenesis or the like.
[0003] Four PGE2 receptor subtypes (EP1, EP2, EP3 and EP4) displaying different pharmacological properties exist. The EP4 subtype, a Gs-coupled receptor, stimulates cAMP production as well as PI3K and GSK3P signaling, and is distributed in a wide variety of tissue suggesting a major role in PGE2-mediated biological events. Various EP4 inhibitors have been described previously, for example, in WO 2002/032900, WO 2005/021508, EiS 6,710,054, and US 7,238,714, the contents of which are incorporated herein by reference in their entireties.
[0004] Accordingly, there is a need for treating, preventing, and/or reducing severity of a proliferative disorder associated with prostaglandin EP4 receptor activity. The present invention addresses such a need.
It has now been found that compounds of the present invention, and compositions thereof, are useful for treating, preventing, and/or reducing severity of a proliferative disorder associated with prostaglandin EP4 receptor activity. In general, salt forms and co-crystal forms, and pharmaceutically acceptable compositions thereof, are useful for treating or lessening the severity of proliferative disorders associated with prostaglandin EP4 receptor activity, as described in detail herein. Such compounds are represented by the chemical structure below, denoted as compound A (also known as grapiprant):
A
or a pharmaceutically acceptable salt thereof.
United States Patent 7,960,407, filed March 1, 2006 and issued June 14, 2011 (“the ‘407 patent,” the entirety of which is hereby incorporated herein by reference), describes certain EP4 inhibitor compounds. Such compounds include compound A:
or a pharmaceutically acceptable salt thereof.
[0037] Compound A, N-[({2-[4-(2-Ethyl-4,6-dimethyl-lH-imidazo[4,5-c]pyridin-l-yl) phenyl]ethyl}amino)carbonyl]-4-methylbenzenesulfonamide, is described in detail in the ‘407
patent, including its synthetic route. The ‘407 patent also discloses a variety of physical forms of compound A.
[0038] It would be desirable to provide a solid form of compound A (e.g., as a co-crystal thereof or salt thereof) that imparts characteristics such as improved aqueous solubility, stability and ease of formulation. Accordingly, the present invention provides both co-crystal forms and salt forms of compound A:
A.
PATENT
WO 2002032900
PATENT
WO 2002032422
Family members of the product case ( WO0232422 ) of grapiprant have protection in most of the EU states until October 2021 and expire in the US in October 15, 2021.
PATENT
WO 2003086371
PATENT
WO2020014445 covering combinations of grapiprant and an immuno-oncology agent.
WO 2005102389
WO 2006095268
US 7960407
US 20190314390
References
- ^ Jump up to:a b c d “Grapiprant”. http://www.drugbank.ca. Retrieved 2019-05-15.
- ^ PubChem. “Grapiprant”. pubchem.ncbi.nlm.nih.gov. Retrieved 2019-05-15.
- ^ Paul Pion, D. V. M.; Spadafori, Gina (2017-08-08). “Veterinary Partner”. VIN.com.
- ^ Nagahisa, A.; Okumura, T. (2017). “Pharmacology of grapiprant, a novel EP4 antagonist: receptor binding, efficacy in a rodent postoperative pain model, and a dose estimation for controlling pain in dogs”. Journal of Veterinary Pharmacology and Therapeutics. 40 (3): 285–292. doi:10.1111/jvp.12349. ISSN 1365-2885. PMID 27597397.
- ^ Paul Pion, D. V. M.; Spadafori, Gina (2017-08-08). “Veterinary Partner”. VIN.com.
- ^ Kirkby Shaw, Kristin; Rausch-Derra, Lesley C.; Rhodes, Linda (February 2016). “Grapiprant: an EP4 prostaglandin receptor antagonist and novel therapy for pain and inflammation”. Veterinary Medicine and Science. 2 (1): 3–9. doi:10.1002/vms3.13. ISSN 2053-1095. PMC 5645826. PMID 29067176.
 |
|
| Clinical data | |
|---|---|
| Trade names | Galliprant |
| Routes of administration |
Oral |
| ATCvet code | |
| Pharmacokinetic data | |
| Bioavailability | 6.6 L/kg, high volume of distribution |
| Elimination half-life | 5.86 hours in horses |
| Excretion | Urine |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C26H29N5O3S |
| Molar mass | 491.61 g·mol−1 |
| 3D model (JSmol) | |
//////////////GRAPIPRANT, 415903-37-6, UNII-J9F5ZPH7NB, CJ 023423, CJ-023423, RQ-00000007, MR10A7, Galliprant, Phase II, Arrys Therapeutics, CANCER, PAIN, AskAt
CCC1=NC2=C(N1C3=CC=C(C=C3)CCNC(=O)NS(=O)(=O)C4=CC=C(C=C4)C)C=C(N=C2C)C
Brilliant blue G , ブリリアントブルーG ,
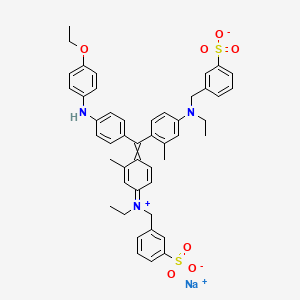
Brilliant blue G
FDA 2019, 12/20/2019, TISSUEBLUE, New Drug Application (NDA): 209569
Company: DUTCH OPHTHALMIC, PRIORITY; Orphan
OPQ recommends APPROVAL of NDA 209569 for commercialization of TissueBlue (Brilliant Blue G Ophthalmic Solution), 0.025%
Neuroprotectant
sodium;3-[[4-[[4-(4-ethoxyanilino)phenyl]-[4-[ethyl-[(3-sulfonatophenyl)methyl]azaniumylidene]-2-methylcyclohexa-2,5-dien-1-ylidene]methyl]-N-ethyl-3-methylanilino]methyl]benzenesulfonate
| Formula |
C47H48N3O7S2. Na
|
|---|---|
| CAS |
6104-58-1
|
| Mol weight |
854.0197
|
ブリリアントブルーG, C.I. Acid Blue 90
UNII-M1ZRX790SI
M1ZRX790SI
6104-58-1
Brilliant Blue G
Derma Cyanine G
SYN


////////////Brilliant blue G , ブリリアントブルーG , C.I. Acid Blue 90, FDA 2019, PRIORITY, Orphan
CCN(CC1=CC(=CC=C1)S(=O)(=O)[O-])C2=CC(=C(C=C2)C(=C3C=CC(=[N+](CC)CC4=CC(=CC=C4)S(=O)(=O)[O-])C=C3C)C5=CC=C(C=C5)NC6=CC=C(C=C6)OCC)C.[Na+]
- Benzenemethanaminium, N-[4-[[4-[(4-ethoxyphenyl)amino]phenyl][4-[ethyl[(3-sulfophenyl)methyl]amino]-2-methylphenyl]methylene]-3-methyl-2,5-cyclohexadien-1-ylidene]-N-ethyl-3-sulfo-, hydroxide, inner salt, monosodium salt
- Benzenemethanaminium, N-[4-[[4-[(4-ethoxyphenyl)amino]phenyl][4-[ethyl[(3-sulfophenyl)methyl]amino]-2-methylphenyl]methylene]-3-methyl-2,5-cyclohexadien-1-ylidene]-N-ethyl-3-sulfo-, inner salt, monosodium salt (9CI)
- Brilliant Indocyanine G (6CI)
- C.I. Acid Blue 90 (7CI)
- C.I. Acid Blue 90, monosodium salt (8CI)
- Acid Blue 90
- Acid Blue G 4061
- Acid Blue PG
- Acid Bright Blue G
- Acid Brilliant Blue G
- Acid Brilliant Cyanine G
- Acidine Sky Blue G
- Amacid Brilliant Cyanine G
- Anadurm Cyanine A-G
- BBG
- Benzyl Cyanine G
- Biosafe Coomassie Stain
- Boomassie blue silver
- Brilliant Acid Blue G
- Brilliant Acid Blue GI
- Brilliant Acid Blue J
- Brilliant Acid Cyanine G
- Brilliant Blue G
- Brilliant Blue G 250
- Brilliant Blue J
- Brilliant Indocyanine GA-CF
- Bucacid Brilliant Indocyanine G
- C.I. 42655
- CBB-G 250
- Colocid Brilliant Blue EG
- Coomassie Blue G
- Coomassie Blue G 250
- Coomassie Brilliant Blue G
- Coomassie Brilliant Blue G 250
- Coomassie G 250
- Cyanine G
- Daiwa Acid Blue 300
- Derma Cyanine G
- Derma Cyanine GN 360
- Dycosweak Acid Brilliant Blue G
- Eriosin Brilliant Cyanine G
- Fenazo Blue XXFG
- Impero Azure G
- Kayanol Cyanine G
- Lerui Acid Brilliant Blue G
- Milling Brilliant Blue 2J
- NSC 328382
- Optanol Cyanine G
- Orient Water Blue 105
- Orient Water Blue 105S
- Polar Blue G
- Polar Blue G 01
- Polycor Blue G
- Sandolan Cyanine N-G
- Sellaset Blue B
- Serva Blue G
- Serva Blue G 250
- Silk Fast Cyanine G
- Simacid Blue G 350
- Sumitomo Brilliant Indocyanine G
- Supranol Cyanin G
- Supranol Cyanine G
- TissueBlue
- Triacid Fast Cyanine G
- Water Blue 105
- Water Blue 105S
- Water Blue 150
- Xylene Brilliant Cyanine G
Fluorodopa F 18, フルオロドパ (18F), флуородопа (18F) , فلورودوبا (18F) , 氟[18F]多巴 ,
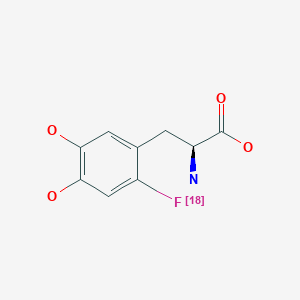
Fluorodopa F 18
2019/10/10, fda 2019,
| Formula |
C9H10FNO4
|
|---|---|
| Cas |
92812-82-3
|
| Mol weight |
215.1784
|
Diagnostic aid (brain imaging), Radioactive agent, for use in positron emission tomography (PET)
CAS 92812-82-3
フルオロドパ (18F)
Fluorodopa, also known as FDOPA, is a fluorinated form of L-DOPA primarily synthesized as its fluorine-18isotopologue for use as a radiotracer in positron emission tomography (PET).[1] Fluorodopa PET scanning is a valid method for assessing the functional state of the nigrostriatal dopaminergic pathway. It is particularly useful for studies requiring repeated measures such as examinations of the course of a disease and the effect of treatment
In October 2019, Fluorodopa was approved in the United States for the visual detection of certain nerve cells in adult patients with suspected Parkinsonian Syndromes (PS).[2][3]
The U.S. Food and Drug Administration (FDA) approved Fluorodopa F 18 based on evidence from one clinical trial of 56 patients with suspected PS.[2] The trial was conducted at one clinical site in the United States.[2]
PAPER
A one-pot two-step synthesis of 6-[18F]fluoro-L-DOPA ([18F]FDOPA) has been developed involving Cu-mediated radiofluorination of a pinacol boronate ester precursor. The method is fully automated, provides [18F]FDOPA in good activity yield (104 ± 16 mCi, 6 ± 1%), excellent radiochemical purity (>99%) and high molar activity (3799 ± 2087 Ci mmol−1), n = 3, and has been validated to produce the radiotracer for human use.
![Graphical abstract: One-pot synthesis of high molar activity 6-[18F]fluoro-l-DOPA by Cu-mediated fluorination of a BPin precursor](https://pubs.rsc.org/en/Image/Get?imageInfo.ImageType=GA&imageInfo.ImageIdentifier.ManuscriptID=C9OB01758E&imageInfo.ImageIdentifier.Year=2019 "Graphical abstract")

PATENT
KR 2019061368

The present invention relates to an L-dopa precursor compd., a method for producing the same, and a method for producing 18F-labeled L-dopa using the same. The method of prepg. 18F-labeled L-dopa I using the L-dopa precursor II [A = halogen-(un)substituted alkyl; W, X, Y = independently protecting group] can improve the labeling efficiency of 18F. After the labeling reaction, sepn. and purifn. steps of the product can be carried out continuously and it can be performed with on-column labeling (a method of labeling through the column). The final product I, 18 F-labeled L-dopa, can be obtained at a high yield relative to conventional methods. Further, it has an advantage that it is easy to apply various methods such as bead labeling.
PAPER
Science (Washington, DC, United States) (2019), 364(6446), 1170-1174.

PAPER
European Journal of Organic Chemistry (2018), 2018(48), 7058-7065.
PATENT
WO 2018115353
CN 107311877
References
- ^ Deng WP, Wong KA, Kirk KL (June 2002). “Convenient syntheses of 2-, 5- and 6-fluoro- and 2,6-difluoro-L-DOPA”. Tetrahedron: Asymmetry. 13 (11): 1135–1140. doi:10.1016/S0957-4166(02)00321-X.
- ^ Jump up to:a b c “Drug Trials Snapshots: Fluorodopa F 18”. U.S. Food and Drug Administration (FDA). 27 November 2019. Archived from the original on 27 November 2019. Retrieved 27 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Drug Approval Package: Fluorodopa F18”. U.S. Food and Drug Administration (FDA). 20 November 2019. Archived from the original on 27 November 2019. Retrieved 26 November 2019. This article incorporates text from this source, which is in the public domain.
 |
|
| Clinical data | |
|---|---|
| Other names | 6-fluoro-L-DOPA, FDOPA |
| License data |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| ChemSpider | |
| UNII | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C9H10FNO4 |
| Molar mass | 215.18 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////////////Fluorodopa F 18, フルオロドパ (18F), FDA 2019, флуородопа (18F) , فلورودوبا (18F) , 氟[18F]多巴 , radio labelled
N[C@@H](CC1=CC(O)=C(O)C=C1[18F])C(O)=O
Enfortumab vedotin

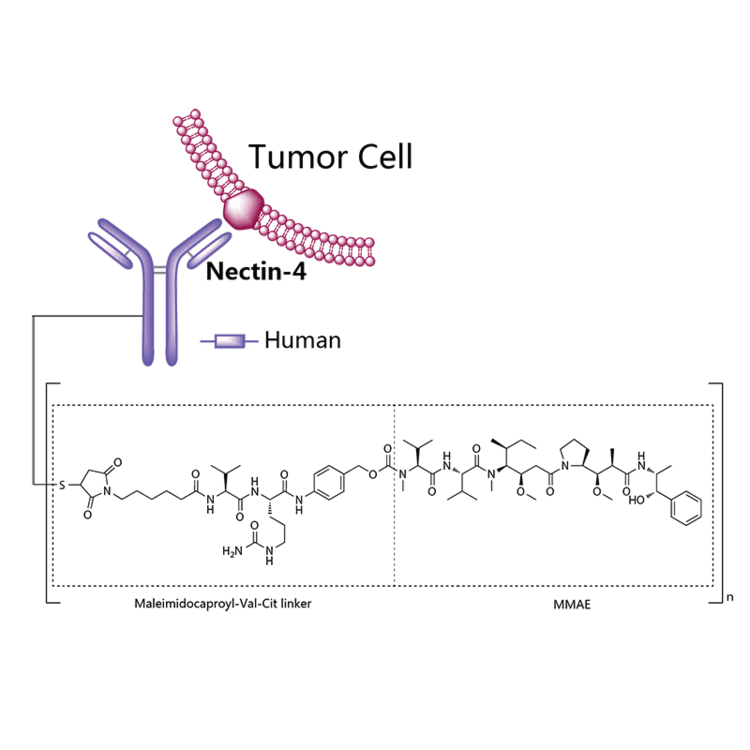

Enfortumab vedotin
| Formula |
C6642H10284N1742O2063S46
|
|---|---|
| Cas |
1346452-25-2
|
| Mol weight |
149022.148
|
AGS-22M6E, enfortumab vedotin-ejfv
Fda approved 2019/12/18, Padcev
Antineoplastic, Nectin-4 antibody, Tubulin polymerization inhibitor, Urothelial cancer
エンホルツマブベドチン (遺伝子組換え);
protein Based Therapies, Monoclonal antibody, mAb,
UNII DLE8519RWM
Other Names
- AGS 22CE
- AGS 22M6E
- AGS 22ME
- Enfortumab vedotin
- Enfortumab vedotin-ejfv
- Immunoglobulin G1 (human monoclonal AGS-22M6 γ1-chain), disulfide with human monoclonal AGS-22M6 κ-chain, dimer, tetrakis(thioether) with N-[[[4-[[N-[6-(3-mercapto-2,5-dioxo-1-pyrrolidinyl)-1-oxohexyl]-L-valyl-N5-(aminocarbonyl)-L-ornithyl]amino]phenyl]methoxy]carbonyl]-N-methyl-L-valyl-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-hydroxy-1-methyl-2-phenylethyl]amino]-1-methoxy-2-methyl-3-oxopropyl]-1-pyrrolidinyl]-2-methoxy-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-L-valinamide
- Padcev
Protein Sequence
Sequence Length: 1322, 447, 447, 214, 214multichain; modified (modifications unspecified)
Enfortumab vedotin is an antibody-drug conjugate used in the treatment of patients with advanced, treatment-resistant urothelial cancers.3 It is comprised of a fully human monoclonal antibody targeted against Nectin-4 and a microtubule-disrupting chemotherapeutic agent, monomethyl auristatin E (MMAE), joined by a protease-cleavable link.3 It is similar to brentuximab vedotin, another antibody conjugated with MMAE that targets CD-30 instead of Nectin-4.
The clinical development of enfortumab vedotin was the result of a collaboration between Astellas Pharma and Seattle Genetics2 and it was first approved for use in the United States in December 2019 under the brand name PadcevTM.3
The most common side effects for patients taking enfortumab vedotin were fatigue, peripheral neuropathy (nerve damage resulting in tingling or numbness), decreased appetite, rash, alopecia (hair loss), nausea, altered taste, diarrhea, dry eye, pruritis (itching) and dry skin. [4]Enfortumab vedotin[1] (AGS-22M6E) is an antibody-drug conjugate[2] designed for the treatment of cancer expressing Nectin-4.[3]Enfortumab refers to the monoclonal antibody part, and vedotin refers to the payload drug (MMAE) and the linker.
The fully humanized antibody was created by scientists at Agensys (part of Astellas) using Xenomice from Amgen; the linker technology holding the antibody and the toxin together was provided by and licensed from Seattle Genetics.[5]
Results of a phase I clinical trial were reported in 2016.[2]
In December 2019, enfortumab vedotin-ejfv was approved in the United States for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.[4]
Enfortumab vedotin was approved based on the results of a clinical trial that enrolled 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.[4] The overall response rate, reflecting the percentage of patients who had a certain amount of tumor shrinkage, was 44%, with 12% having a complete response and 32% having a partial response.[4] The median duration of response was 7.6 months.[4]
The application for enfortumab vedotin-ejfv was granted accelerated approval, priority review designation, and breakthrough therapydesignation.[4] The U.S. Food and Drug Administration (FDA) granted the approval of Padcev to Astellas Pharma US Inc.[4]
Indication
Enfortumab vedotin is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor, and a platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced, or metastatic setting.3
Associated Conditions
Pharmacodynamics
Enfortumab vedotin is an anti-cancer agent that destroys tumor cells by inhibiting their ability to replicate.3 Patients with moderate to severe hepatic impairment should not use enfortumab vedotin – though it has not been studied in this population, other MMAE-containing antibody-drug conjugates have demonstrated increased rates of adverse effects in patients with moderate-severe hepatic impairment.3 Enfortumab vedotin may also cause significant hyperglycemia leading, in some cases, to diabetic ketoacidosis, and should not be administered to patients with a blood glucose level >250 mg/dl.3
Mechanism of action
Enfortumab vedotin is an antibody-drug conjugate comprised of multiple components.3 It contains a fully human monoclonal antibody directed against Nectin-4, an extracellular adhesion protein which is highly expressed in urothelial cancers,1 attached to a chemotherapeutic microtubule-disrupting agent, monomethyl auristatin E (MMAE). These two components are joined via a protease-cleavable linker. Enfortumab vedotin binds to cells expressing Nectin-4 and the resulting enfortumab-Nectin-4 complex is internalized into the cell. Once inside the cell, MMAE is released from enfortumab vedotin via proteolytic cleavage and goes on to disrupt the microtubule network within the cell, arresting the cell cycle and ultimately inducing apoptosis.3
PATENT
WO 2016176089
WO 2016138034
WO 2017186928
WO 2017180587
WO 2017200492
US 20170056504
PAPER
Cancer Research (2016), 76(10), 3003-3013.
General References
- Hanna KS: Clinical Overview of Enfortumab Vedotin in the Management of Locally Advanced or Metastatic Urothelial Carcinoma. Drugs. 2019 Dec 10. pii: 10.1007/s40265-019-01241-7. doi: 10.1007/s40265-019-01241-7. [PubMed:31823332]
- McGregor BA, Sonpavde G: Enfortumab Vedotin, a fully human monoclonal antibody against Nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial Carcinoma. Expert Opin Investig Drugs. 2019 Oct;28(10):821-826. doi: 10.1080/13543784.2019.1667332. Epub 2019 Sep 17. [PubMed:31526130]
- FDA Approved Drug Products: Padcev (enfortumab vedotin-ejfv) for IV injection [Link]
References
- ^ World Health Organization (2013). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 109”(PDF). WHO Drug Information. 27 (2).
- ^ Jump up to:a b Seattle Genetics and Agensys, an Affiliate of Astellas, Highlight Promising Enfortumab Vedotin (ASG-22ME) and ASG-15ME Phase 1 Data in Metastatic Urothelial Cancer at 2016 ESMO Congress. Oct 2016
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Enfortumab Vedotin, American Medical Association.
- ^ Jump up to:a b c d e f g “FDA approves new type of therapy to treat advanced urothelial cancer”. U.S. Food and Drug Administration (FDA) (Press release). 18 December 2019. Archived from the original on 19 December 2019. Retrieved 18 December 2019. This article incorporates text from this source, which is in the public domain.
- ^ Challita-Eid PM, Satpayev D, Yang P, et al. (May 2016). “Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models”. Cancer Research. 76 (10): 3003–13. doi:10.1158/0008-5472.can-15-1313. PMID 27013195.
External links
- “Enfortumab vedotin”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Nectin-4 |
| Clinical data | |
| Trade names | Padcev |
| Other names | AGS-22M6E, AGS-22CE, enfortumab vedotin-ejfv |
| License data | |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChemSID | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C6642H10284N1742O2063S46 |
| Molar mass | 149.0 kg/mol g·mol−1 |
PADCEV™
(enfortumab vedotin-ejfv) for Injection, for Intravenous Use
DESCRIPTION
Enfortumab vedotin-ejfv is a Nectin-4 directed antibody-drug conjugate (ADC) comprised of a fully human anti-Nectin-4 IgG1 kappa monoclonal antibody (AGS-22C3) conjugated to the small molecule microtubule disrupting agent, monomethyl auristatin E (MMAE) via a protease-cleavable maleimidocaproyl valine-citrulline (vc) linker (SGD-1006). Conjugation takes place on cysteine residues that comprise the interchain disulfide bonds of the antibody to yield a product with a drug-to-antibody ratio of approximately 3.8:1. The molecular weight is approximately 152 kDa.
Figure 1: Structural Formula
|
Approximately 4 molecules of MMAE are attached to each antibody molecule. Enfortumab vedotin-ejfv is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells and the small molecule components are produced by chemical synthesis.
PADCEV (enfortumab vedotin-ejfv) for injection is provided as a sterile, preservative-free, white to off-white lyophilized powder in single-dose vials for intravenous use. PADCEV is supplied as a 20 mg per vial and a 30 mg per vial and requires reconstitution with Sterile Water for Injection, USP, (2.3 mL and 3.3 mL, respectively) resulting in a clear to slightly opalescent, colorless to slightly yellow solution with a final concentration of 10 mg/mL [see DOSAGE AND ADMINISTRATION]. After reconstitution, each vial allows the withdrawal of 2 mL (20 mg) and 3 mL (30 mg). Each mL of reconstituted solution contains 10 mg of enfortumab vedotin-ejfv, histidine (1.4 mg), histidine hydrochloride monohydrate (2.31 mg), polysorbate 20 (0.2 mg) and trehalose dihydrate (55 mg) with a pH of 6.0.
///////////////Enfortumab vedotin, AGS-22M6E, エンホルツマブベドチン (遺伝子組換え) , protein Based Therapies, Monoclonal antibody, mAb, FDA 2019
[*]SC1CC(=O)N(CCCCCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(=O)N)C(=O)Nc2ccc(COC(=O)N(C)[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@@H](CC(=O)N3CCC[C@H]3[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c4ccccc4)OC)cc2)C1=O
RESMETIROM
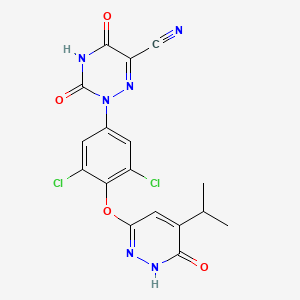
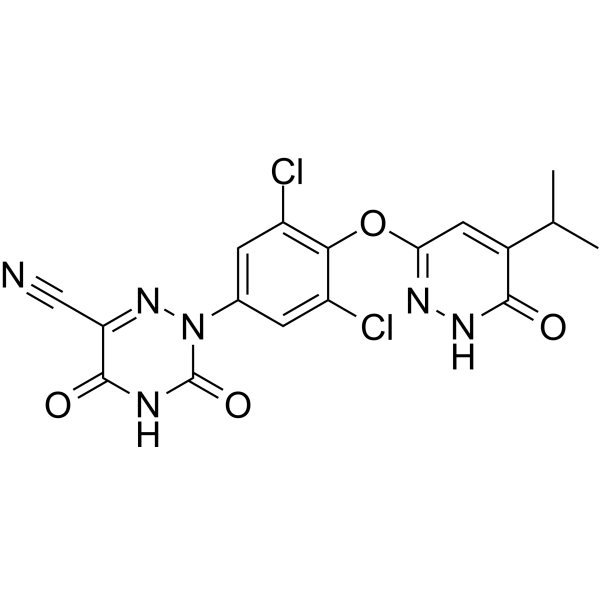
RESMETIROM
| C17H12Cl2N6O4 |
435.2 g/mol
MGL-3196
CAS 920509-32-6, Resmetirom, VIA-3196, UNII-RE0V0T1ES0
FDA APPROVED 3/14/2024, To treat noncirrhotic non-alcoholic steatohepatitis with moderate to advanced liver scarring
Press Release
Phase III, Non-alcoholic fatty liver disease (NAFLD)
2-[3,5-dichloro-4-[(6-oxo-5-propan-2-yl-1H-pyridazin-3-yl)oxy]phenyl]-3,5-dioxo-1,2,4-triazine-6-carbonitrile
2-(3,5-DICHLORO-4-((5-ISOPROPYL-6-OXO-1,6-DIHYDROPYRIDAZIN-3-YL)OXY)PHENYL)-3,5-DIOXO-2,3,4,5-TETRAHYDRO-(1,2,4)TRIAZINE-6-CARBONITRILE
1,2,4-TRIAZINE-6-CARBONITRILE, 2-(3,5-DICHLORO-4-((1,6-DIHYDRO-5-(1-METHYLETHYL)-6-OXO-3-PYRIDAZINYL)OXY)PHENYL)-2,3,4,5-TETRAHYDRO-3,5-DIOXO-
Madrigal Pharmaceuticals , following the merger between Synta and Madrigal Pharmaceuticals (pre-merger) (following the acquisition of VIA Pharmaceuticals ‘ assets (originally under license from Roche )), is developing resmetirom (MGL-3196, VIA-3196), the lead from oral capsule formulation thyroid hormone receptor (THR) beta agonists, cholesterol and triglyceride modulators, for the use in the treatment of metabolic disorders including hypercholesterolemia and other dyslipidemias, and non-alcoholic steatohepatitis.
MGL-3196 is a first-in-class, orally administered, small-molecule, liver-directed, THR β-selective agonist. Preclinical, toxicology and Phase 1 clinical data suggest MGL-3196 has an attractive, differentiated profile as a potential treatment for non-alcoholic steatohepatitis (NASH) and dyslipidemias. THR-β selectivity also enhances the safety profile of MGL-3196, compared to non-selective agents. MGL-3196 has shown no suppression of the central thyroid axis, no THR-α effects on heart rate or bone, and no elevation of liver enzymes. These characteristics make MGL-3196 among the most promising molecules in development in this therapeutic area. MGL-3196 is in a Phase 2 clinical trial for the treatment of non-alcoholic steatohepatitis (NASH).
PATENT
WO-2020010068
Novel crystalline salt of resmetirom as thyroid hormone receptor agonists useful for treating obesity, hyperlipidemia, hypercholesterolemia and diabetes. Appears to be the first filing from the assignee and the inventors on this compound,
Thyroid hormones are critical for normal growth and development and for maintaining metabolic homeostasis (Paul M. Yen, Physiological reviews, Vol. 81(3): pp. 1097-1126 (2001)). Circulating levels of thyroid hormones are tightly regulated by feedback mechanisms in the hypothalamus/pituitary/thyroid (HPT) axis. Thyroid dysfunction leading to hypothyroidism or hyperthyroidism clearly demonstrates that thyroid hormones exert profound effects on cardiac function, body weight, metabolism, metabolic rate, body temperature, cholesterol, bone, muscle and behavior.
[0005] The biological activity of thyroid hormones is mediated by thyroid hormone receptors (TRs or THRs) (M. A. Lazar, Endocrine Reviews, Vol. 14: pp. 348-399 (1993)). TRs belong to the superfamily known as nuclear receptors. TRs form heterodimers with the retinoid receptor that act as ligand-inducible transcription factors. TRs have a ligand binding domain, a DNA binding domain, and an amino terminal domain, and regulate gene expression through interactions with DNA response elements and with various nuclear co-activators and co repressors. The thyroid hormone receptors are derived from two separate genes, a and b. These distinct gene products produce multiple forms of their respective receptors through differential RNA processing. The major thyroid receptor isoforms are aΐ, a2, bΐ, and b2. Thyroid hormone receptors aΐ, bΐ, and b2 bind thyroid hormone. It has been shown that the thyroid hormone receptor subtypes can differ in their contribution to particular biological responses. Recent studies suggest that TIIb 1 plays an important role in regulating TRH (thyrotropin releasing hormone) and on regulating thyroid hormone actions in the liver. T11b2 plays an important role in the regulation of TSH (thyroid stimulating hormone) (Abel et. al, J. Clin. Invest., Vol 104: pp. 291-300 (1999)). TIIb 1 plays an important role in regulating heart rate (B. Gloss et. al. Endocrinology, Vol. 142: pp. 544-550 (2001); C. Johansson et. al, Am. J. Physiol., Vol. 275: pp. R640-R646 (1998)).
[0006] Efforts have been made to synthesize thyroid hormone analogs which exhibit increased thyroid hormone receptor beta selectivity and/or tissue selective action. Such thyroid hormone mimetics may yield desirable reductions in body weight, lipids, cholesterol, and lipoproteins, with reduced impact on cardiovascular function or normal function of the hypothalamus/pituitary/thyroid axis (see, e.g., Joharapurkar et al, J. Med. Chem, 2012, 55 (12), pp 5649-5675). The development of thyroid hormone analogs which avoid the undesirable effects of hyperthyroidism and hypothyroidism while maintaining the beneficial effects of thyroid hormones would open new avenues of treatment for patients with metabolic disease such as obesity, hyperlipidemia, hypercholesterolemia, diabetes and other disorders and diseases such as liver steatosis and NASH, atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer, thyroid diseases, a resistance to thyroid hormone (RTH) syndrome, and related disorders and diseases.
PATENT
WO2018075650
In one embodiment, the metabolite of Compound A comprises a compound
having the following structure:
(“Ml”).
PATENT
WO 2007009913
PATENT
WO 2014043706
https://patents.google.com/patent/WO2014043706A1/en
Example 3: Preparation of (Z)-ethyl (2-cyano-2-(2-(3,5-dichloro-4-((5-isopropyl-6- oxo- l,6-dihydropyridazin-3-yl)oxy)phenyl)hydrazono)acetyl)carbamate (Int. 8)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet was charged with Int. 7 (75.0 g, 0.239 mol, 1 wt), acetic acid (600 mL, 8 vol), water (150 mL, 2 vol), and concentrated HC1 (71.3 mL, 0.95 vol). The resulting thin slurry was cooled to 6 °C and a solution of NaN02 (16.8 g, 0.243 mol, 1.02 equiv) in water (37.5 mL, 0.5 vol) was added over a period of 10 min while maintaining a batch temperature below 10 °C. After an additional 10 min of agitation between 5-10 °C, HPLC analysis showed complete conversion of Int. 7 to the diazonium intermediate. A solution of NaOAc (54.5 g, 0.664 mol, 2.78 equiv) in water (225 mL, 3 vol) was added over a period of 6 min while maintaining a batch temperature below 10 °C. N-cyanoacetylurethane (37.9 g, 0.243 mol, 1.02 equiv) was immediately added, the cooling was removed, and the batch naturally warmed to 8 °C over 35 min. HPLC analysis showed complete consumption of the diazonium intermediate and the reaction was deemed complete. The batch warmed naturally to 21 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (375 mL, 5 vol) twice. The collected orange solid was dried in a 35 °C vacuum oven for 64 h to provide crude Int. 8 (104.8 g, 91%).
A I L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, and N2 inlet/outlet was charged with crude Int. 8 (104.4 g, 1 wt) and acetic acid (522 mL, 5 vol). The resulting slurry was heated to 50 °C and held at that temperature for 1.5 h. The batch cooled naturally to 25 °C over 2 h and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (522 mL, 5 vol) and the cake conditioned under vacuum for 1.75 h. The light orange solid was dried to constant weight in a 40 °C vacuum oven to provide 89.9 g (78% from Int. 7) of the desired product. 1H NMR (DMSO) was consistent with the assigned structure.
Example 4: Preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, N2 inlet/outlet, and reflux condenser was charged with Int. 8 (89.3 g, 0.185 mol, 1 wt), DMAC (446 mL, 5 vol), and KOAc (20.0 g, 0.204 mol, 1.1 equiv). The mixture was heated to 120 °C and held at that temperature for 2 h. HPLC analysis showed complete conversion to Compound A. The batch temperature was adjusted to 18 °C over 1 h, and acetic acid (22.3 mL, 0.25 vol) was added. The batch temperature was adjusted to 8 °C, and water (714 mL, 8 vol) was added over 1 h; an orange slurry formed. The batch was filtered through Sharkskin filter paper and the cake was allowed to condition overnight under N2 without vacuum for convenience. A premixed solution of 1 : 1 acetone/water (445 mL, 5 vol) was charged to the flask and added to the cake as a rinse with vacuum applied. After 2 h of conditioning the cake under vacuum, it was transferred to a clean 1 L, three-neck, round- bottom flask equipped with overhead stirring, a thermocouple, and N2inlet/outlet. Ethanol (357 mL, 4 vol) and acetone (357 mL, 4 vol) were charged and the resulting slurry was heated to 60 °C; dissolution occurred. Water (890 mL, 10 vol) was added over a period of 90 min while maintaining a batch temperature between 55-60 °C. The resulting slurry was allowed to cool to 25 °C and filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with a solution of 1:1 EtOH/water (446 mL, 5 vol). The cake was conditioned overnight under N2 without vacuum for convenience. The cracks in the cake were smoothed and vacuum applied. The cake was washed with water (179 mL, 2 vol) and dried in a 45 °C vacuum oven to a constant weight of 70.5 g (87%, crude Compound A). HPLC analysis showed a purity of 94.8%.
A 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser was charged with crude Compound A (70.0 g) and MIBK (350 mL, 5 vol). The orange slurry was heated to 50 °C and held at that temperature for 2 h. The batch cooled naturally to 23 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with MIBK (35 mL, 0.5 vol) twice. The collected solids were dried in a 45 °C vacuum oven to a constant weight of 58.5 g (84%). This solid was charged to a 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser. Ethanol (290 mL, 5 vol) was added and the slurry was heated to reflux. After 3.5 h at reflux, XRPD showed the solid was consistent with Form I, and heating was removed. Upon reaching 25 °C, the batch was filtered through filter paper, and the reactor and cake were washed sequentially with EtOH (174 mL, 3 vol). The tan solid Compound A was dried in a 40 °C vacuum oven to a constant weight of 50.4 g (87%, 64% from Int. 8). HPLC analysis showed a purity of 99.1%. 1H NMR (DMSO) was consistent with the assigned structure.
Example 5: Scaled up preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A larger scale batch of Compound A was synthesized according to the scheme below. The conditions in the scheme below are similar to those described in Examples 1-4 above.

6A

Compound A
Synthesis of 4: A 50 L jacketed glass vessel (purged with N2) was charged with 3,6- dichloropyridazine (2.00 kg), 4-amino-2,6-dichlorophenol (2.44 kg) and N,N- dimethylacetamide (10.0 L). The batch was vacuum (26 inHg) / nitrogen (1 PSIG) purged 3 times. Cesium carbonate (5.03 kg) was added and the batch temperature was adjusted from 22.3 °C to 65.0 °C over 3.5 hours. The batch was held at 65.0 °C for 20 hours. At this point,
NMR analysis indicated 3.34% 3.6-dichloropyridazine relative to 2. The batch temperature was adjusted to 21.5 °C and ethyl acetate (4.00 L) was added to the batch. The batch was agitated for 10 minutes and then filtered through a 18″ Nutsche filter equipped with polypropylene filter cloth. The filtration took 15 minutes. Ethyl acetate (5.34 L) was charged to the vessel and transferred to the filter as a rinse. The batch was then manually re- suspended in the filter before re-applying vacuum. This process was repeated 2 more times and the filter cake was conditioned for 10 minutes. The filtrate was charged to a 100-L vessel that contained (16.0 L) of a previously prepared 15% sodium chloride in H20. The batch was agitated for 5 minutes and then allowed to separate for 35 minutes. The interface was not visible, so the calculated 23 L of the lower aqueous phase was removed. 16.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 6 minutes and then allowed to separate for 7 minutes. The interface was visible at -19 L and the lower aqueous phase was removed. 17.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 7 minutes and then allowed to separate for 11 minutes. The lower aqueous phase was removed. The vessel was set up for vacuum distillation and the batch was concentrated from 17.0 L to 8.0 L over 2 hours 20 minutes with the batch temperature kept around 21 °C. Benzoic anhydride (3.19 kg) and acetic acid (18.0 L) were charged to the vessel. The vessel was set up for vacuum distillation and the batch was concentrated from 28.0 L to 12.0 L over 2 days (overnight hold at 20 °C) with the batch temperature kept between 20 and 55 °C. At this point, JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.015. Acetic acid (4.0 L) was charged to the batch and the batch was distilled to 12 L. JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.0036. Acetic acid (20.0 L) was charged to the batch and the batch temperature was adjusted to 70.0 °C. The batch was sampled for HPLC analysis and 2 was 0.16%. Sodium acetate (2,20 kg) was added to the batch and the batch temperature was adjusted from 72.4 °C to 110.0 °C. After 18.5 hours, HPLC analysis indicated no Int. B detected. The batch temperature was adjusted from 111.3 to 74.7 °C and DI water (30.0 L) was added to the batch over 2 hours. The batch temperature was adjusted to 20 .5 °C and then filtered using a 24″ Haselloy Nutsche filter equipped with polypropylene filter cloth. A previously prepared solution of 1:1 acetic acid in DI H20 (10.0 L) was charged to the vessel and agitated for 5 minutes. The wash was transferred to the filter and the batch was then manually re- suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged to the vessel and then transferred to the filter. The batch was manually re-suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged directly to the filter and the batch was then manually re-suspended in the filter before re-applying vacuum. The filter cake was allowed to condition for 18 hours to give 14.4 kg of 4. HPLC analysis indicated a purity of 93.7%. This wet cake was carried forward into the purification. A 100 L jacketed glass vessel (purged with N2) was charged with crude 4 (wet cake 14.42 kg), acetic acid (48.8 L) and the agitator was started. DI H20 (1.74 L) was charged. The batch (a slurry) temperature was adjusted from 18.1 to 100.1 °C over 4.25 hours. The batch was held at 100.1 to 106.1 °C for 1 hour and then adjusted to 73.1 °C. DI H20 (28.0 L) was added to the batch over 1 hour keeping the batch temperature between 73.1 and 70.3 °C. The batch temperature was adjusted further from 70.3 °C to 25.0 °C overnight. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with polypropylene filter cloth. The filtration took 13 minutes. A solution of DI H20 (9.00 L) and acetic acid (11.0 L) was prepared and added to the 100 L vessel. The mixture was agitated for 5 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 6 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 9 minutes and then transferred to the filter cake. The batch was allowed to condition for 3 days and then transferred to drying trays for vacuum oven drying. After 3 days at 50 °C and 28’7Hg, the batch gave a 74% yield (3.7 kg) of4 as an off-white solid. The JH NMR spectrum was consistent with the assigned structure, HPLC analysis indicated a purity of 98.87% and KF analysis indicated 0.14% H20. Synthesis of Int. 7: A 100-L jacketed glass vessel (purged with N2) was charged with tetrahydrofuran (44.4 L). The agitator was started (125 RPM) and 4 (3.67 kg) was charged followed by lithium chloride (1.26 kg). The batch temperature was observed to be 26.7 ° C and was an amber solution. Isopropenylmagnesium bromide 1.64 molar solution in 2-methyl THF (21.29 kg) was added over 2 ½ hours keeping the batch between 24.3 and 33.6 °C. The batch was agitated at 24.5 °C for 17 hours at which point HPLC analysis indicated 9% 4. A 2nd 100-L jacketed glass vessel (purged with N2) was charged with 3N hydrogen chloride (18.3 L). The batch was transferred to the vessel containing the 3N HC1 over 25 minutes keeping the batch temperature between 20 and 46 °C. A bi-phasic solution was observed. The quenched batch was transferred back to the 1st 100-L vessel to quench the small amount of residue left behind. THF (2.00 L) was used as a rinse. The batch temperature was observed to be 40.9 ° C and was agitated at 318 RPM for 45 minutes. The batch temperature was adjusted to 21.8 ° C and the layers were allowed to separate. The separation took 10 minutes. The lower aqueous phase was removed (-26.0 L). A solution of sodium chloride (1.56 kg) in DI water (14.0 L) was prepared and added to the batch. This was agitated at 318 RPM for 10 minutes and agitator was stopped. The separation took 3 minutes. The lower aqueous phase was removed (-16.0 L). The batch was vacuum distilled from 58.0 L to 18.4 L using ~24’7Hg and a jacket temperature of 50 to 55 °C. A solution of potassium hydroxide (2.30 kg) in DI water (20.7 L) was prepared in a 72-L round bottom flask. The vessel was set up for atmospheric distillation using 2 distillation heads and the batch was transferred to the 72-L vessel. THF (0.75 L) was used as a rinse. The batch volume was -41.0 L, the temperature was adjusted to 64.1 °C and distillation started with the aid of a N2 sweep. Heating was continued to drive the batch temperature to 85.4 °C while distilling at which point the 72-L vessel was set up for reflux (batch volume was about 28.0 L at the end of the distillation). The batch was held at 85 °C for 13 hours at which point HPLC analysis indicated 0.3% compound 6A. Heating was stopped and the batch was transferred to a 100-L jacketed glass vessel. Solids were observed. The batch temperature was adjusted from 70.6 °C to 56.7 °C. A previously prepared solution of sodium hydrogen carbonate (2.82 kg) in DI water (35.0 L) was added over 80 minutes keeping the batch temperature between 56.7 and 46.7 °C. The batch pH at the end of the addition was 9.8. The batch was held at
46.7 to 49.0 °C for 40 minutes and then cooled to 25.0 °C. The batch was filtered using a 18″ stainless steel Nutsche filter. DI water (18.4 L) was charged to the vessel and transferred to the filter. The filter cake was manually re-suspended in the filter and then the liquors were removed. This process was repeated once more and the filter cake was 3″ thick. The filter cake was conditioned on the filter for 3 days, was transferred to drying trays and dried in a vacuum oven at 45 °C to provide 2.93 kg Int. 7 (95% yield) with an HPLC purity of 87.6%.
Synthesis of Int. 8: A 100 L jacketed glass vessel (purged with N2 and plumbed to a caustic scrubber) was charged with acidic acid (13.0 L). Int. 7 (2.85 kg) was charged to the vessel and the agitator was started. N-Cyanoacetylurethane (1.56 kg) and DI water (5.70 L) were charged to the vessel. The batch temperature was adjusted from 17.0 °C to 5.5 °C and a thin slurry was observed. At this point 37% hydrogen chloride (2.70 L) was added over 10 minutes keeping the batch temperature between 4.8 °C and 8.8 °C. A previously prepared solution of sodium nitrite (638 g) in DI water (1.42 L) was added over 26 minutes keeping the batch temperature between 5.8 °C and 8.7 °C. A brown gas was observed in the vessel head space during the addition. HPLC analysis indicated no Int. 7 detected. At this point a previously prepared solution of sodium acetate (2.07 kg) in DI water (8.50 L) was added over 47 minutes keeping the batch temperature between 5.5 °C and 9.5 °C. After the addition, a thin layer of orange residue was observed on the vessel wall just above the level of the batch. The batch temperature was adjusted from 9.4 °C to 24.5 °C and held at 25 °C (+ 5 °C) for 12 hours. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with tight-weave polypropylene filter cloth. The filtration took 30 minutes. The vessel was rinsed with 14.3 L of a 1 : 1 acidic acid / DI water. The orange residue on the reactor washed away with the rinse. The rinse was transferred to the filter where the batch was manually re-suspended. Vacuum was re-applied to remove the wash. A 2nd 1 : 1 acidic acid / DI water wash was performed as above and the batch was conditioned on the filter for 26 hours. HPLC analysis of the wet filter cake indicated purity was 90.4%. The batch was dried to a constant weight of 3.97 kg (91% yield) in a vacuum oven at 45 °C and 287Hg. Preparation of Compound A DMAC Solvate
A 100 L, jacketed, glass vessel purged with N2 was charged with Int. 8 (3.90 kg) and potassium acetate (875 g). N,N-dimethylacetamide (DMAC, 18.3 L) was charged to the vessel and the agitator was started. The batch temperature was adjusted to 115 °C over 2 h. After 2 h at 115 °C, the batch was sampled and HPLC analysis indicated 0.27% Int. 8 remained. The batch temperature was adjusted to 25.0 °C overnight. Acetic acid (975 mL) was added to the batch and the batch was agitated further for 3 h. The batch was transferred to a carboy and the vessel was rinsed clean with 800 mL of DMAC. The batch was transferred back to the 100 L vessel using vacuum through a 10 μιη in-line filter and a DMAC rinse (1.15 L) was used. The filtration was fast at the beginning but slow at the end, plugging up the filter. The batch temperature was adjusted to 11.1 °C and DI water (35.1 L) was added over 2 h 20 min, keeping the batch temperature between 5-15 °C. The batch was held for 1 h and filtered, using an 18″ Nutsche filter equipped with tight-weave
polypropylene cloth. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel, cooled to 10 °C, and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg to give 89% yield (3.77 kg) of Compound A DMAC solvate as an orange/tan solid. The 1H NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated 0.49% H20. XRPD indicated the expected form, i.e., Compound A DMAC solvate. Thermogravimetric analysis (TGA) indicated 16% weight loss. HPLC analysis indicated a purity of 93.67%.
Preparation of Crude Compound A
A 100 L, jacketed, glass vessel purged with N2 was charged with Compound A
DMAC solvate (3.75 kg) and ethanol (15.0 L). The agitator was started and acetone (15.0 L) was added. The batch temperature was adjusted from 10.6 °C to 60.0 °C over 1 h. At this point, the batch was in solution. DI water was added to the batch over 1.5 h, keeping the batch temperature at 60 + 5 °C. The batch was held at 60 + 5 °C for 1 h and cooled to 23.5 °C. An 18″ Nutsche filter equipped with tight-weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg for five days to give a 94% yield (2.90 kg) of Compound A as a powdery tan solid. The NMR spectrum is consistent with the assigned structure and Karl Fischer analysis indicated 6.6% H20. XRPD indicated the expected form of dihydrate. TGA indicated 6.7% weight loss. HPLC analysis indicated a purity of 96.4% (AUC).
Purification of Crude Compound A
A 50 L, jacketed, glass vessel purged with N2 was charged with Compound A crude
(2.90 kg) and methyl isobutyl ketone (14.5 L). The agitator was started and the batch temperature was adjusted from 20.2 °C to 50.4 °C over 1.5 h. The batch was held at 50 °C (+ 5 °C) for 1 h and cooled to 20-25 °C. The batch was held at 20-25 °C for 2.5 h. An 18″ Nutsche filter equipped with tight- weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 20 min. Methyl isobutyl ketone (MIBK, 1.45 L) was charged to the vessel and transferred to the filter cake. The cake was manually resuspended and the liquors were pulled through with vacuum. Methyl isobutyl ketone (2.90 L) was charged to the filter cake and the cake was manually resuspended. The liquors were pulled through with vacuum and the cake was conditioned with vacuum and nitrogen for 15 h. The filter cake dried into a tan, hard 18″ x 1 ½” disc. This was manually broken up and run through coffee grinders to give a 76% yield (2.72 kg) of MGL-3196 MIBK solvate as a tan, powdery solid. No oven drying was necessary. The NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated <0.1 % H20. XRPD indicated the expected form MIBK solvate. TGA indicated 17.3% weight loss. HPLC analysis indicated a purity of 98.5%.
Example 6: Conversion of Compound A to Form I
Purified Compound A (4802 g) as a 1:1 MIBK solvate which was obtained from Int. 8 as described in Example 5 above was added into a jacketed, 100 L reactor along with 24 liters of ethanol. The resulting slurry was heated to 80 + 5 °C (reflux) over 1 h 25 min; the mixture was stirred at that temperature for 4 h 25 min. Analysis of the filtered solids at 2 h 55 min indicated that the form conversion was complete, with the XRPD spectra conforming to Form I. The mixture was cooled to 20 + 5 °C over 45 min and stirred at that temperature for 15 min. The slurry was filtered and the filter cake was washed twice with prefiltered ethanol (2 x 4.8 L). The wet cake (4.28 kg) was dried under vacuum at 40 + 5 °C for 118 h to afford 3390 g of Compound A form I.
PAPER
Journal of Medicinal Chemistry (2014), 57(10), 3912-3923
https://pubs.acs.org/doi/abs/10.1021/jm4019299
The beneficial effects of thyroid hormone (TH) on lipid levels are primarily due to its action at the thyroid hormone receptor β (THR-β) in the liver, while adverse effects, including cardiac effects, are mediated by thyroid hormone receptor α (THR-α). A pyridazinone series has been identified that is significantly more THR-β selective than earlier analogues. Optimization of this series by the addition of a cyanoazauracil substituent improved both the potency and selectivity and led to MGL-3196 (53), which is 28-fold selective for THR-β over THR-α in a functional assay. Compound 53 showed outstanding safety in a rat heart model and was efficacious in a preclinical model at doses that showed no impact on the central thyroid axis. In reported studies in healthy volunteers, 53 exhibited an excellent safety profile and decreased LDL cholesterol (LDL-C) and triglycerides (TG) at once daily oral doses of 50 mg or higher given for 2 weeks.

//////////////RESMETIROM , MGL-3196, VIA-3196, UNII-RE0V0T1ES0, Phase III
CC(C)C1=CC(=NNC1=O)OC2=C(C=C(C=C2Cl)N3C(=O)NC(=O)C(=N3)C#N)Cl
Avapritinib, アバプリチニブ , авапритиниб , أفابريتينيب ,

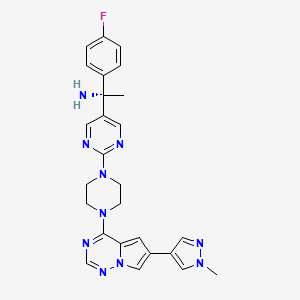
Avapritinib
BLU-285, BLU285
Antineoplastic, Tyrosine kinase inhibitor
アバプリチニブ
(1S)-1-(4-fluorophenyl)-1-[2-[4-[6-(1-methylpyrazol-4-yl)pyrrolo[2,1-f][1,2,4]triazin-4-yl]piperazin-1-yl]pyrimidin-5-yl]ethanamine
| Formula |
C26H27FN10
|
|---|---|
| CAS |
1703793-34-3
|
| Mol weight |
498.558
|
| No. | Drug Name | Active Ingredient | Approval Date | FDA-approved use on approval date* |
|---|---|---|---|---|
| 1. | Ayvakit | avapritinib | 1/9/2020 | To treat adults with unresectable or metastatic gastrointestinal stromal tumor (GIST) |
PRIORITY; Orphan, NDA 212608
Avapritinib, sold under the brand name Ayvakit, is a medication used for the treatment of tumors due to one specific rare mutation: It is specifically intended for adults with unresectable or metastatic ( y) gastrointestinal stromal tumor (GIST) that harbor a platelet-derived growth factor receptor alpha (PDGFRA) exon 18 mutation.[1]
Common side effects are edema (swelling), nausea, fatigue/asthenia (abnormal physical weakness or lack of energy), cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation (secretion of tears), abdominal pain, constipation, rash. and dizziness.[1]
Ayvakit is a kinase inhibitor.[1]
History
The U.S. Food and Drug Administration (FDA) approved avapritinib in January 2020.[1] The application for avapritinib was granted fast track designation, breakthrough therapy designation, and orphan drug designation.[1] The FDA granted approval of Ayvakit to Blueprint Medicines Corporation.[1]
Avapritinib was approved based on the results from the Phase I NAVIGATOR[2][3] clinical trial involving 43 patients with GIST harboring a PDGFRA exon 18 mutation, including 38 subjects with PDGFRA D842V mutation.[1] Subjects received avapritinib 300 mg or 400 mg orally once daily until disease progression or they experienced unacceptable toxicity.[1] The recommended dose was determined to be 300 mg once daily.[1] The trial measured how many subjects experienced complete or partial shrinkage (by a certain amount) of their tumors during treatment (overall response rate).[1] For subjects harboring a PDGFRA exon 18 mutation, the overall response rate was 84%, with 7% having a complete response and 77% having a partial response.[1] For the subgroup of subjects with PDGFRA D842V mutations, the overall response rate was 89%, with 8% having a complete response and 82% having a partial response.[1] While the median duration of response was not reached, 61% of the responding subjects with exon 18 mutations had a response lasting six months or longer (31% of subjects with an ongoing response were followed for less than six months).[1]
PATENT
WO 2015057873
https://patents.google.com/patent/WO2015057873A1/en
Example 7: Synthesis of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4- yl)pyrrolo[2, 1 -f\ [ 1 ,2,4] triazin-4-yl)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine and (S)- 1 – (4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine (Compounds 43 and 44)


Step 1 : Synthesis of (4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l- f] [ 1 ,2,4] triazin-4-yl)piperazin- 1 -yl)pyrimidin-5-yl)methanone:

4-Chloro-6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/] [l,2,4]triazine (180 mg, 0.770 mmol), (4-fluorophenyl)(2-(piperazin-l-yl)pyrimidin-5-yl)methanone, HC1 (265 mg, 0.821 mmol) and DIPEA (0.40 mL, 2.290 mmol) were stirred in 1,4-dioxane (4 mL) at room temperature for 18 hours. Saturated ammonium chloride was added and the products extracted into DCM (x2). The combined organic extracts were dried over Na2S04, filtered through Celite eluting with DCM, and the filtrate concentrated in vacuo. Purification of the residue by MPLC (25- 100% EtOAc-DCM) gave (4-fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methanone (160 mg, 0.331 mmol, 43 % yield) as an off-white solid. MS (ES+) C25H22FN90 requires: 483, found: 484 [M + H]+.
Step 2: Synthesis of (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-p razol-4-yl)p rrolo[2, l- ] [l,2,4]triazin-4- l)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide:

(S)-2-Methylpropane-2-sulfinamide (110 mg, 0.908 mmol), (4-fluorophenyl)(2-(4-(6-(l- methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5- yl)methanone (158 mg, 0.327 mmol) and ethyl orthotitanate (0.15 mL, 0.715 mmol) were stirred in THF (3.2 mL) at 70 °C for 18 hours. Room temperature was attained, water was added, and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0- 10% MeOH-EtOAc) gave (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)methylene)-2- methylpropane-2-sulfinamide (192 mg, 0.327 mmol, 100 % yield) as an orange solid. MS (ES+) C29H3iFN10OS requires: 586, found: 587 [M + H]+.
Step 3: Synthesis of (lS’)-N-(l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- l)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-

(lS’,Z)-N-((4-Fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide (190 mg, 0.324 mmol) was taken up in THF (3 mL) and cooled to 0 °C. Methylmagnesium bromide (3 M solution in diethyl ether, 0.50 mL, 1.500 mmol) was added and the resulting mixture stirred at 0 °C for 45 minutes. Additional methylmagnesium bromide (3 M solution in diethyl ether, 0.10 mL, 0.300 mmol) was added and stirring at 0 °C continued for 20 minutes. Saturated ammonium chloride was added and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0-10% MeOH-EtOAc) gave (lS’)-N-(l-(4-fluorophenyl)-l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol, 61.5 % yield) as a yellow solid (mixture of diastereoisomers). MS (ES+) C3oH35FN10OS requires: 602, found: 603 [M + H]+. Step 4: Synthesis of l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l- f\ [ 1 ,2,4] triazin-4- l)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine:

(S)-N- ( 1 – (4-Fluorophenyl)- 1 -(2- (4- (6-( 1 -methyl- 1 H-pyrazol-4-yl)pyrrolo [2,1- /] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol) was stirred in 4 M HCl in 1,4-dioxane (1.5 mL)/MeOH (1.5 mL) at room temperature for 1 hour. The solvent was removed in vacuo and the residue triturated in EtOAc to give l-(4-fluorophenyl)- l-(2-(4-(6-(l -methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4- yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine, HCl (110 mg, 0.206 mmol, 103 % yield) as a pale yellow solid. MS (ES+) C26H27FN10requires: 498, found: 482 [M- 17 + H]+, 499 [M + H]+.
Step 5: Chiral separation of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine and (5)-1-(4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin- 1 -yl)pyrimidin- -yl)ethanamine:

The enantiomers of racemic l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (94 mg, 0.189 mmol) were separated by chiral SFC to give (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH- pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine (34.4 mg, 0.069 mmol, 73.2 % yield) and (lS,)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (32.1 mg, 0.064 mmol, 68.3 % yield). The absolute stereochemistry was assigned randomly. MS (ES+)
C26H27FN10 requires: 498, found: 499 [M + H]+.
References
- ^ Jump up to:a b c d e f g h i j k l m “FDA approves the first targeted therapy to treat a rare mutation in patients with gastrointestinal stromal tumors”. U.S. Food and Drug Administration (FDA) (Press release). 9 January 2020. Archived from the original on 11 January 2020. Retrieved 9 January 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Blueprint Medicines Announces FDA Approval of AYVAKIT (avapritinib) for the Treatment of Adults with Unresectable or Metastatic PDGFRA Exon 18 Mutant Gastrointestinal Stromal Tumor”. Blueprint Medicines Corporation (Press release). 9 January 2020. Archived from the original on 11 January 2020. Retrieved 9 January 2020.
- ^ “Blueprint Medicines Announces Updated NAVIGATOR Trial Results in Patients with Advanced Gastrointestinal Stromal Tumors Supporting Development of Avapritinib Across All Lines of Therapy”. Blueprint Medicines Corporation (Press release). 15 November 2018. Archived from the original on 10 January 2020. Retrieved 9 January 2020.
Further reading
- Wu CP, Lusvarghi S, Wang JC, et al. (July 2019). “Avapritinib: A Selective Inhibitor of KIT and PDGFRα that Reverses ABCB1 and ABCG2-Mediated Multidrug Resistance in Cancer Cell Lines”. Mol. Pharm. 16 (7): 3040–3052. doi:10.1021/acs.molpharmaceut.9b00274. PMID 31117741.
- Gebreyohannes YK, Wozniak A, Zhai ME, et al. (January 2019). “Robust Activity of Avapritinib, Potent and Highly Selective Inhibitor of Mutated KIT, in Patient-derived Xenograft Models of Gastrointestinal Stromal Tumors”. Clin. Cancer Res. 25 (2): 609–618. doi:10.1158/1078-0432.CCR-18-1858. PMID 30274985.
External links
- “Avapritinib”. Drug Information Portal. U.S. National Library of Medicine (NLM).
| Clinical data | |
|---|---|
| Trade names | Ayvakit |
| Other names | BLU-285, BLU285 |
| License data | |
| Routes of administration |
By mouth |
| Drug class | Antineoplastic agents |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C26H27FN10 |
| Molar mass | 498.570 g·mol−1 |
| 3D model (JSmol) | |
///////Avapritinib, 2020 APPROVALS, PRIORITY, Orphan, BLU-285, BLU285, FDA 2020, Ayvakit, アバプリチニブ , авапритиниб , أفابريتينيب ,
TERIPARATIDE, テリパラチド , терипаратид , تيريباراتيد , 特立帕肽 ,
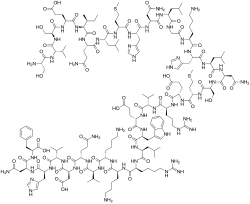
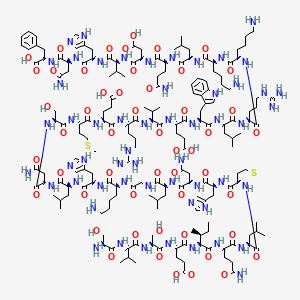


TERIPARATIDE
テリパラチド;
- PTH 1-34
- LY 333334 / LY-333334 / LY333334 / ZT-034
|
Ser Val Ser Glu Ile Gln Leu Met His Asn Leu Gly Lys His Leu Asn
Ser Met Glu Arg Val Glu Trp Leu Arg Lys Lys Leu Gln Asp Val His Asn Phe-OH |
|
| Type |
Peptide
|
|---|
| Formula |
C181H291N55O51S2
|
|---|---|
| CAS |
52232-67-4
99294-94-7 (acetate)
|
| Mol weight |
4117.7151
|
(4S)-4-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S,3S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-3-hydroxypropanoyl]amino]-3-methylbutanoyl]amino]-3-hydroxypropanoyl]amino]-4-carboxybutanoyl]amino]-3-methylpentanoyl]amino]-5-oxopentanoyl]amino]-4-methylpentanoyl]amino]-4-methylsulfanylbutanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-oxobutanoyl]amino]-4-methylpentanoyl]amino]acetyl]amino]hexanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-3-hydroxypropanoyl]amino]-4-methylsulfanylbutanoyl]amino]-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-4-amino-1-[[(1S)-1-carboxy-2-phenylethyl]amino]-1,4-dioxobutan-2-yl]amino]-3-(1H-imidazol-5-yl)-1-oxopropan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-1-oxohexan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-5-oxopentanoic acid
| SVG Image |
 |
|---|---|
| IUPAC Condensed | H-Ser-Val-Ser-Glu-Ile-Gln-Leu-Met-His-Asn-Leu-Gly-Lys-His-Leu-Asn-Ser-Met-Glu-Arg-Val-Glu-Trp-Leu-Arg-Lys-Lys-Leu-Gln-Asp-Val-His-Asn-Phe-OH |
| Sequence | SVSEIQLMHNLGKHLNSMERVEWLRKKLQDVHNF |
| PLN | H-SVSEIQLMHNLGKHLNSMERVEWLRKKLQDVHNF-OH |
| HELM | PEPTIDE1{S.V.S.E.I.Q.L.M.H.N.L.G.K.H.L.N.S.M.E.R.V.E.W.L.R.K.K.L.Q.D.V.H.N.F}$$$$ |
| IUPAC | L-seryl-L-valyl-L-seryl-L-alpha-glutamyl-L-isoleucyl-L-glutaminyl-L-leucyl-L-methionyl-L-histidyl-L-asparagyl-L-leucyl-glycyl-L-lysyl-L-histidyl-L-leucyl-L-asparagyl-L-seryl-L-methionyl-L-alpha-glutamyl-L-arginyl-L-valyl-L-alpha-glutamyl-L-tryptophyl-L-leucyl-L-arginyl-L-lysyl-L-lysyl-L-leucyl-L-glutaminyl-L-alpha-aspartyl-L-valyl-L-histidyl-L-asparagyl-L-phenylalanine |
Other Names
- L-Seryl-L-valyl-L-seryl-L-α-glutamyl-L-isoleucyl-L-glutaminyl-L-leucyl-L-methionyl-L-histidyl-L-asparaginyl-L-leucylglycyl-L-lysyl-L-histidyl-L-leucyl-L-asparaginyl-L-seryl-L-methionyl-L-α-glutamyl-L-arginyl-L-valyl-L-α-glutamyl-L-tryptophyl-L-leucyl-L-arginyl-L-lysyl-L-lysyl-L-leucyl-L-glutaminyl-L-α-aspartyl-L-valyl-L-histidyl-L-asparaginyl-L-phenylalanine
- (1-34)-Human parathormone
- (1-34)-Human parathyroid hormone
- 1-34-Human PTH
- 1-34-Parathormone (human)
- 11: PN: WO0039278 SEQID: 17 unclaimed protein
- 14: PN: WO0181415 SEQID: 16 claimed protein
- 15: PN: WO0123521 SEQID: 19 claimed protein
- 1: PN: EP2905289 SEQID: 1 claimed protein
- 1: PN: WO0198348 SEQID: 13 claimed protein
- 1: PN: WO2011071480 SEQID: 14 claimed protein
- 225: PN: US20090175821 SEQID: 272 claimed protein
- 22: PN: US6110892 SEQID: 22 unclaimed protein
- 2: PN: US20100261199 SEQID: 4 claimed protein
- 31: PN: US20070099831 PAGE: 7 claimed protein
- 32: PN: WO2008068487 SEQID: 32 claimed protein
- 5: PN: WO2008033473 SEQID: 4 claimed protein
- 692: PN: WO2004005342 PAGE: 46 claimed protein
- 69: PN: US20050009742 PAGE: 20 claimed sequence
- 7: PN: WO0031137 SEQID: 8 unclaimed protein
- 7: PN: WO0040611 PAGE: 1 claimed protein
- 93: PN: WO0069900 SEQID: 272 unclaimed protein
- Forsteo
- Forteo
- HPTH-(1-34)
- Human PTH(1-34)
- Human parathormone(1-34)
- Human parathyroid hormone-(1-34)
- LY 333334
- Osteotide
- Parathar
- Parathormone (human)
- Teriparatide
- ZT 034
Product Ingredients
| INGREDIENT | UNII | CAS | |
|---|---|---|---|
| Teriparatide acetate | 9959P4V12N | 99294-94-7 |
Teriparatide is a form of parathyroid hormone consisting of the first (N-terminus) 34 amino acids, which is the bioactive portion of the hormone. It is an effective anabolic (promoting bone formation) agent[2] used in the treatment of some forms of osteoporosis.[3] It is also occasionally used off-label to speed fracture healing. Teriparatide is identical to a portion of human parathyroid hormone (PTH) and intermittent use activates osteoblasts more than osteoclasts, which leads to an overall increase in bone.
Recombinant teriparatide is sold by Eli Lilly and Company under the brand name Forteo/Forsteo. A synthetic teriparatide from Teva Generics has been authorised for marketing in European territories[4]. Biosimilar product from Gedeon Richter plc has been authorised in Europe[5]. On October 4, 2019 the US FDA approved a recombinant teriparatide product, PF708, from Pfenex Inc. PF708 is the first FDA approved proposed therapeutic equivalent candidate to Forteo.
Teriparatide (recombinant human parathyroid hormone) is a potent anabolic agent used in the treatment of osteoporosis. It is manufactured and marketed by Eli Lilly and Company.
Teriparatide is a recombinant form of parathyroid hormone. It is an effective anabolic (i.e., bone growing) agent used in the treatment of some forms of osteoporosis. It is also occasionally used off-label to speed fracture healing. Teriparatide is identical to a portion of human parathyroid hormone (PTH) and intermittent use activates osteoblasts more than osteoclasts, which leads to an overall increase in bone. Teriparatide is sold by Eli Lilly and Company under the brand name Forteo.
Indication
For the treatment of osteoporosis in men and postmenopausal women who are at high risk for having a fracture. Also used to increase bone mass in men with primary or hypogonadal osteoporosis who are at high risk for fracture.
Associated Conditions
Pharmacodynamics
Clinical trials indicate that teriparatide increases predominantly trabecular bone in the lumbar spine and femoral neck; it has less significant effects at cortical sites. The combination of teriparatide with antiresorptive agents is not more effective than teriparatide monotherapy. The most common adverse effects associated with teriparatide include injection-site pain, nausea, headaches, leg cramps, and dizziness. After a maximum of two years of teriparatide therapy, the drug should be discontinued and antiresorptive therapy begun to maintain bone mineral density.
Mechanism of action
Teriparatide is the portion of human parathyroid hormone (PTH), amino acid sequence 1 through 34 of the complete molecule which contains amino acid sequence 1 to 84. Endogenous PTH is the primary regulator of calcium and phosphate metabolism in bone and kidney. Daily injections of teriparatide stimulates new bone formation leading to increased bone mineral density.
Medical uses
Teriparatide has been FDA-approved since 2002.[6] It is effective in growing bone (e.g., 8% increase in bone density in the spine after one year)[7] and reducing the risk of fragility fractures.[6][8] When studied, teriparatide only showed bone mineral density (BMD) improvement during the first 18 months of use. Teriparatide should only be used for a period of 2 years maximum. After 2 years, another agent such a bisphosphonate or denosumab should be used in cases of osteoporosis. [9]
Teriparatide cuts the risk of hip fracture by more than half but does not reduce the risk of arm or wrist fracture.[10]
Other
Teriparatide can be used off-label to speed fracture repair and treat fracture nonunions.[11] It has been reported to have been successfully used to heal fracture nonunions.[12] Generally, due to HIPAA regulations, it is not publicized when American athletes receive this treatment to improve fracture recovery.[11] But an Italian football player, Francesco Totti, was given teriparatide after a tibia/fibula fracture, and he unexpectedly recovered in time for the 2006 World Cup.[11] It has been reported that Mark Mulder used it to recover from a hip fracture Oakland A’s for the 2003 MLB playoffs[13] and Terrell Owens to recover from an ankle fracture before the 2005 Super Bowl.[13]
Administration
Teriparatide is administered by injection once a day in the thigh or abdomen.
Contraindications
Teriparatide should not be prescribed for people who are at increased risks for osteosarcoma. This includes those with Paget’s Diseaseof bone or unexplained elevations of serum alkaline phosphate, open epiphysis, or prior radiation therapy involving the skeleton. In the animal studies and in one human case report, it was found to potentially be associated with developing osteosarcoma in test subjects after over 2 years of use. [14]
Patients should not start teriparatide until any vitamin D deficiency is corrected. [15]
Adverse effects
Adverse effects of teriparatide include headache, nausea, dizziness, and limb pain.[6] Teriparatide has a theoretical risk of osteosarcoma, which was found in rat studies but not confirmed in humans.[2] This may be because unlike humans, rat bones grow for their entire life.[2] The tumors found in the rat studies were located on the end of the bones which grew after the injections began.[15]After nine years on the market, there were only two cases of osteosarcoma reported.[7] This risk was considered by the FDA as “extremely rare” (1 in 100,000 people)[6] and is only slightly more than the incidence in the population over 60 years old (0.4 in 100,000).[6]
Mechanism of action
Teriparatide is a portion of human parathyroid hormone (PTH), amino acid sequence 1 through 34, of the complete molecule (containing 84 amino acids). Endogenous PTH is the primary regulator of calcium and phosphate metabolism in bone and kidney. PTH increases serum calcium, partially accomplishing this by increasing bone resorption. Thus, chronically elevated PTH will deplete bone stores. However, intermittent exposure to PTH will activate osteoblasts more than osteoclasts. Thus, once-daily injections of teriparatide have a net effect of stimulating new bone formation leading to increased bone mineral density.[16][17][18]
Teriparatide is the first FDA approved agent for the treatment of osteoporosis that stimulates new bone formation.[19]
FDA approval
Teriparatide was approved by the Food and Drug Administration (FDA) on 26 November 2002, for the treatment of osteoporosis in men and postmenopausal women who are at high risk for having a fracture. The drug is also approved to increase bone mass in men with primary or hypogonadal osteoporosis who are at high risk for fracture.
Combined teriparatide and denosumab
Combined teriparatide and denosumab increased BMD more than either agent alone and more than has been reported with approved therapies. Combination treatment might, therefore, be useful to treat patients at high risk of fracture by increasing BMD. However, there is no evidence of fracture rate reduction in patients taking a teriparatide and denosumab combination. Moreover, the combination therapy group showed a significant decrease in their bone formation marker, indicating that denosumab, an antiresorptive agent, might actually counteract the effect of teriparatide, a bone formation anabolic agent, in bone formation. [20]
PATENT
KR 2011291
WO 2019077432
CN 109897099
CN 109879955
CN 109879954
CN 108373499
PATENT
WO-2020000555
Process for preparing teriparatide as parathyroid hormone receptor agonist, useful for treating osteoporosis in menopausal women. Appears to be the first filing from the assignee and the inventors on this compound, however, this invention was previously seen as a Chinese national filing published in 12/2013. Daiichi Sankyo , through its subsidiary Asubio Pharma , was developing SUN-E-3001 , a nasally administered recombinant human parathyroid hormone, for the treatment of osteoporosis.
Teriparatide is a 1-34 fragment of human parathyroid hormone, which has the same biological activity as human parathyroid hormone. Hypogonadous osteoporosis and osteoporosis in menopausal women have great market prospects.
In patent CN201410262511, a pseudoproline dipeptide Fmoc-Asn (Trt) -Ser (ψ Me, Me Pro) -OH is used instead of the two amino acids at the original 16-17 positions for coupling one by one, and the final cleavage yields teriparatide. This method adopts the method of feeding pseudoproline dipeptide to avoid the generation of oxidative impurities, but it cannot avoid a variety of missing peptides due to the excessively long peptide chain. At the same time, the pseudoproline dipeptide is expensive and difficult to obtain.
References
- ^ http://www.minsa.gob.pa/sites/default/files/alertas/nota_seguridad_teriparatida.pdf
- ^ Jump up to:a b c Riek AE and Towler DA (2011). “The pharmacological management of osteoporosis”. Missouri Medicine. 108 (2): 118–23. PMC 3597219. PMID 21568234.
- ^ Saag KG, Shane E, Boonen S, et al. (November 2007). “Teriparatide or alendronate in glucocorticoid-induced osteoporosis”. The New England Journal of Medicine. 357 (20): 2028–39. doi:10.1056/NEJMoa071408. PMID 18003959.
- ^ BfArM (2017-05-08). “PUBLIC ASSESSMENT REPORT – Decentralised Procedure – Teriparatid-ratiopharm 20 µg / 80ml, Solution for injection” (PDF).
- ^ “Summary of the European public assessment report (EPAR) for Terrosa”. Retrieved 2019-08-14.
- ^ Jump up to:a b c d e Rizzoli, R.; Reginster, J. Y.; Boonen, S.; Bréart, G. R.; Diez-Perez, A.; Felsenberg, D.; Kaufman, J. M.; Kanis, J. A.; Cooper, C. (2011). “Adverse Reactions and Drug–Drug Interactions in the Management of Women with Postmenopausal Osteoporosis”. Calcified Tissue International. 89 (2): 91–104. doi:10.1007/s00223-011-9499-8. PMC 3135835. PMID 21637997.
- ^ Jump up to:a b Kawai, M.; Mödder, U. I.; Khosla, S.; Rosen, C. J. (2011). “Emerging therapeutic opportunities for skeletal restoration”. Nature Reviews Drug Discovery. 10 (2): 141–156. doi:10.1038/nrd3299. PMC 3135105. PMID 21283108.
- ^ Murad, M. H.; Drake, M. T.; Mullan, R. J.; Mauck, K. F.; Stuart, L. M.; Lane, M. A.; Abu Elnour, N. O.; Erwin, P. J.; Hazem, A.; Puhan, M. A.; Li, T.; Montori, V. M. (2012). “Comparative Effectiveness of Drug Treatments to Prevent Fragility Fractures: A Systematic Review and Network Meta-Analysis”. Journal of Clinical Endocrinology & Metabolism. 97(6): 1871–1880. doi:10.1210/jc.2011-3060. PMID 22466336.
- ^ O’Connor KM. Evaluation and Treatment of Osteoporosis. Med Clin N Am. 2016; 100:807-26
- ^ Díez-Pérez A, Marin F, Eriksen EF, Kendler DL, Krege JH, Delgado-Rodríguez M (September 2018). “Effects of teriparatide on hip and upper limb fractures in patients with osteoporosis: A systematic review and meta-analysis”. Bone. 120: 1–8. doi:10.1016/j.bone.2018.09.020. PMID 30268814.
- ^ Jump up to:a b c Bruce Jancin (2011-12-12). “Accelerating Fracture Healing With Teriparatide”. Internal Medicine News Digital Network. Retrieved 2013-09-20.
- ^ Giannotti, S.; Bottai, V.; Dell’Osso, G.; Pini, E.; De Paola, G.; Bugelli, G.; Guido, G. (2013). “Current medical treatment strategies concerning fracture healing”. Clinical Cases in Mineral and Bone Metabolism. 10 (2): 116–120. PMC 3796998. PMID 24133528.
- ^ Jump up to:a b William L. Carroll (2005). “Chapter 1: Defining the Issue”. The Juice: The Real Story of Baseball’s Drug Problems. ISBN 1-56663-668-X. Retrieved 2013-09-23.
- ^ Harper KD, Krege JH, Marcus R, et al. Osteosarcoma and teriparatide? J Bone Miner Res 2007;22(2):334
- ^ Jump up to:a b https://www.drugs.com/pro/forteo.html
- ^ Bauer, E; Aub, JC; Albright, F (1929). “Studies of calcium and phosphorus metabolism: V. Study of the bone trabeculae as a readily available reserve supply of calcium”. J Exp Med. 49 (1): 145–162. doi:10.1084/jem.49.1.145. PMC 2131520. PMID 19869533.
- ^ Selye, H (1932). “On the stimulation of new bone formation with parathyroid extract and irradiated ergosterol”. Endocrinology. 16 (5): 547–558. doi:10.1210/endo-16-5-547.
- ^ Dempster, D. W.; Cosman, F.; Parisien, M.; Shen, V.; Lindsay, R. (1993). “Anabolic actions of parathyroid hormone on bone”. Endocrine Reviews. 14 (6): 690–709. doi:10.1210/edrv-14-6-690. PMID 8119233.
- ^ Fortéo: teriparatide (rDNA origin) injection Archived 2009-12-27 at the Wayback Machine
- ^ Tsai, Joy N; Uihlein, Alexander V; Lee, Hang; Kumbhani, Ruchit; Siwila-Sackman, Erica; McKay, Elizabeth A; Burnett-Bowie, Sherri-Ann M; Neer, Robert M; Leder, Benjamin Z (2013). “Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: The DATA study randomised trial”. The Lancet. 382 (9886): 1694–1700. doi:10.1016/S0140-6736(13)60856-9. PMC 4010689. PMID 24517156.
External links
 |
|
| Clinical data | |
|---|---|
| Trade names | Forteo/Forsteo, Teribone[1] |
| AHFS/Drugs.com | Monograph |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Subcutaneous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 95% |
| Metabolism | Hepatic (nonspecific proteolysis) |
| Elimination half-life | Subcutaneous: 1 hour |
| Excretion | Renal (metabolites) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.168.733 |
| Chemical and physical data | |
| Formula | C181H291N55O51S2 |
| Molar mass | 4117.72 g/mol g·mol−1 |
| 3D model (JSmol) | |
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| CA2314313 | No | 2005-02-08 | 2018-12-08 |  |
| CA2325371 | No | 2004-08-17 | 2019-08-19 | |
| US6770623 | No | 2004-08-03 | 2018-12-08 |  |
| US6977077 | No | 2005-12-20 | 2019-08-19 | |
| US7163684 | No | 2007-01-16 | 2019-08-19 | |
| US7351414 | No | 2008-04-01 | 2019-08-19 | |
| US7550434 | No | 2009-06-23 | 2018-12-08 | |
| US7144861 | No | 2006-12-05 | 2018-12-08 | |
| US7517334 | No | 2009-04-14 | 2025-03-25 | |
FORTEO (teriparatide [rDNA origin] injection) contains recombinant human parathyroid hormone (1- 34), and is also called rhPTH (1-34). It has an identical sequence to the 34 N-terminal amino acids(the biologically active region) of the 84-amino acid human parathyroid hormone.
Teriparatide has a molecular weight of 4117.8 daltons and its amino acid sequence is shown below:
 |
Teriparatide (rDNA origin) is manufactured using a strain of Escherichia coli modified by recombinant DNA technology. FORTEO is supplied as a sterile, colorless, clear, isotonic solution in a glass cartridge which is pre-assembled into a disposable delivery device (pen) for subcutaneous injection. Each prefilled delivery device is filled with 2.7 mL to deliver 2.4 mL. Each mL contains 250 mcg teriparatide (corrected for acetate, chloride, and water content), 0.41 mg glacial acetic acid, 0.1 mg sodium acetate (anhydrous), 45.4 mg mannitol, 3 mg Metacresol, and Water for Injection. In addition, hydrochloric acid solution 10% and/or sodium hydroxide solution 10% may have been added to adjust the product to pH 4.
Each cartridge, pre-assembled into a delivery device, delivers 20 mcg of teriparatide per dose each day for up to 28 days.
REFERENCES
1: Lindsay R, Krege JH, Marin F, Jin L, Stepan JJ. Teriparatide for osteoporosis: importance of the full course. Osteoporos Int. 2016 Feb 22. [Epub ahead of print] Review. PubMed PMID: 26902094.
2: Im GI, Lee SH. Effect of Teriparatide on Healing of Atypical Femoral Fractures: A Systemic Review. J Bone Metab. 2015 Nov;22(4):183-9. doi: 10.11005/jbm.2015.22.4.183. Epub 2015 Nov 30. Review. PubMed PMID: 26713309; PubMed Central PMCID: PMC4691592.
3: Babu S, Sandiford NA, Vrahas M. Use of Teriparatide to improve fracture healing: What is the evidence? World J Orthop. 2015 Jul 18;6(6):457-61. doi: 10.5312/wjo.v6.i6.457. eCollection 2015 Jul 18. Review. PubMed PMID: 26191492; PubMed Central PMCID: PMC4501931.
4: Lecoultre J, Stoll D, Chevalley F, Lamy O. [Improvement of fracture healing with teriparatide: series of 22 cases and review of the literature]. Rev Med Suisse. 2015 Mar 18;11(466):663-7. Review. French. PubMed PMID: 25962228.
5: Sugiyama T, Torio T, Sato T, Matsumoto M, Kim YT, Oda H. Improvement of skeletal fragility by teriparatide in adult osteoporosis patients: a novel mechanostat-based hypothesis for bone quality. Front Endocrinol (Lausanne). 2015 Jan 30;6:6. doi: 10.3389/fendo.2015.00006. eCollection 2015. Review. PubMed PMID: 25688232; PubMed Central PMCID: PMC4311704.
6: Wheeler AL, Tien PC, Grunfeld C, Schafer AL. Teriparatide treatment of osteoporosis in an HIV-infected man: a case report and literature review. AIDS. 2015 Jan 14;29(2):245-6. doi: 10.1097/QAD.0000000000000529. Review. PubMed PMID: 25532609; PubMed Central PMCID: PMC4438749.
7: Campbell EJ, Campbell GM, Hanley DA. The effect of parathyroid hormone and teriparatide on fracture healing. Expert Opin Biol Ther. 2015 Jan;15(1):119-29. doi: 10.1517/14712598.2015.977249. Epub 2014 Nov 3. Review. PubMed PMID: 25363308.
8: Yamamoto M, Sugimoto T. [Glucocorticoid and Bone. Beneficial effect of teriparatide on fracture risk as well as bone mineral density in patients with glucocorticoid-induced osteoporosis]. Clin Calcium. 2014 Sep;24(9):1379-85. doi: CliCa140913791385. Review. Japanese. PubMed PMID: 25177011.
9: Chen JF, Yang KH, Zhang ZL, Chang HC, Chen Y, Sowa H, Gürbüz S. A systematic review on the use of daily subcutaneous administration of teriparatide for treatment of patients with osteoporosis at high risk for fracture in Asia. Osteoporos Int. 2015 Jan;26(1):11-28. doi: 10.1007/s00198-014-2838-7. Epub 2014 Aug 20. Review. PubMed PMID: 25138261.
10: Eriksen EF, Keaveny TM, Gallagher ER, Krege JH. Literature review: The effects of teriparatide therapy at the hip in patients with osteoporosis. Bone. 2014 Oct;67:246-56. doi: 10.1016/j.bone.2014.07.014. Epub 2014 Jul 15. Review. PubMed PMID: 25053463.
11: Meier C, Lamy O, Krieg MA, Mellinghoff HU, Felder M, Ferrari S, Rizzoli R. The role of teriparatide in sequential and combination therapy of osteoporosis. Swiss Med Wkly. 2014 Jun 4;144:w13952. doi: 10.4414/smw.2014.13952. eCollection 2014. Review. PubMed PMID: 24896070.
12: Krege JH, Lane NE, Harris JM, Miller PD. PINP as a biological response marker during teriparatide treatment for osteoporosis. Osteoporos Int. 2014 Sep;25(9):2159-71. doi: 10.1007/s00198-014-2646-0. Epub 2014 Mar 6. Review. PubMed PMID: 24599274; PubMed Central PMCID: PMC4134485.
13: Nakano T. [Once-weekly teriparatide treatment on osteoporosis]. Clin Calcium. 2014 Jan;24(1):100-5. doi: CliCa1401100105. Review. Japanese. PubMed PMID: 24369286.
14: Yano S, Sugimoto T. [Daily subcutaneous injection of teriparatide : the progress and current issues]. Clin Calcium. 2014 Jan;24(1):35-43. doi: CliCa14013543. Review. Japanese. PubMed PMID: 24369278.
15: Lewiecki EM, Miller PD, Harris ST, Bauer DC, Davison KS, Dian L, Hanley DA, McClung MR, Yuen CK, Kendler DL. Understanding and communicating the benefits and risks of denosumab, raloxifene, and teriparatide for the treatment of osteoporosis. J Clin Densitom. 2014 Oct-Dec;17(4):490-5. doi: 10.1016/j.jocd.2013.09.018. Epub 2013 Oct 25. Review. PubMed PMID: 24206867.
16: Delivanis DA, Bhargava A, Luthra P. Subungual exostosis in an osteoporotic patient treated with teriparatide. Endocr Pract. 2013 Sep-Oct;19(5):e115-7. doi: 10.4158/EP13040.CR. Review. PubMed PMID: 23757619.
17: Borges JL, Freitas A, Bilezikian JP. Accelerated fracture healing with teriparatide. Arq Bras Endocrinol Metabol. 2013 Mar;57(2):153-6. Review. PubMed PMID: 23525295.
18: Thumbigere-Math V, Gopalakrishnan R, Michalowicz BS. Teriparatide therapy for bisphosphonate-related osteonecrosis of the jaw: a case report and narrative review. Northwest Dent. 2013 Jan-Feb;92(1):12-8. Review. PubMed PMID: 23516715.
19: Lamy O. [Bone anabolic treatment with Teriparatide]. Ther Umsch. 2012 Mar;69(3):187-91. doi: 10.1024/0040-5930/a000272. Review. German. PubMed PMID: 22403112.
20: Narváez J, Narváez JA, Gómez-Vaquero C, Nolla JM. Lack of response to teriparatide therapy for bisphosphonate-associated osteonecrosis of the jaw. Osteoporos Int. 2013 Feb;24(2):731-3. doi: 10.1007/s00198-012-1918-9. Epub 2012 Mar 8. Review. PubMed PMID: 22398853.
/////TERIPARATIDE, テリパラチド , терипаратид , تيريباراتيد , 特立帕肽 , PTH 1-34, LY 333334, LY-333334, LY333334, ZT-034, 52232-67-4, PEPTIDES
Coblopasvir

Coblopasvir
CAS: 1312608-46-0
Chemical Formula: C41H50N8O8
Molecular Weight: 782.89
methyl {(2S)-1-[(2S)-2-(4-{4-[7-(2-[(2S)-1-{(2S)-2- [(methoxycarbonyl)amino]-3-methylbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl)-2H-1,3-benzodioxol-4-yl]phenyl}-1Himidazol-2-yl)pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl}carbamate
hepatitis C virus infection
KW-136
Coblopasvir is an antiviral drug candidate.
Coblopasvir dihydrochloride

CAS 1966138-53-3
| C41 H50 N8 O8 . 2 Cl H | |
| Molecular Weight | 855.806 |
| PHASE 3 | Beijing Kawin Technology Share-Holding |
Beijing Kawin Technology Share-Holding, in collaboration with Beijing Fu Rui Tiancheng Biotechnology and Ginkgo Pharma , is developing coblopasvir as an oral capsule formulation of dihydrochloride salt (KW-136), for treating hepatitis C virus infection. In June 2018, an NDA was filed in China by Beijing Kawin Technology and Sichuan Qingmu Pharmaceutical . In August 2018, the application was granted Priority Review in China . Also, Beijing Kawin is investigating a tablet formulation of coblopasvir dihydrochloride.
PATENT
WO2011075607 , claiming substituted heterocyclic derivatives as HCV replication inhibitors useful for treating HCV infection and liver fibrosis, assigned to Beijing Kawin Technology Share-Holding Co Ltd and InterMune Inc ,
PATENT
CN 108675998
PATENT
WO-2020001089
Novel crystalline and amorphous forms of methyl carbamate compound, particularly coblopasvir dihydrochloride , (designated as Forms H) processes for their preparation, compositions and combinations comprising them are claimed. Also claim is an article or kit comprising a container and a package insert, wherein the container contains coblopasvir dihydrochloride.

///////////////Coblopasvir , KW-136, hepatitis C virus infection, CHINA, Beijing Kawin Technology, NDA, Phase III
O=C(OC)N[C@@H](C(C)C)C(N1[C@H](C2=NC(C3=CC=C(C4=C5OCOC5=C(C6=CNC([C@H]7N(C([C@@H](NC(OC)=O)C(C)C)=O)CCC7)=N6)C=C4)C=C3)=CN2)CCC1)=O
ADAFOSBUVIR, адафосбувир , أدافوسبوفير ,


ADAFOSBUVIR
AL335; ALS-335; JNJ-64146212 , D11364
Propan-2-yl N-((P5’S)-4′-fluoro-2′-C-methyl-p-o-phenyl- 5′-uridylyl)-L-alaninate
propan-2-yl (2S)-2-{[(S)-{[(2S,3S,4R,5R)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-2-fluoro-3,4-dihydroxy-4-methyloxolan-2-yl]methoxy}(phenoxy)phosphoryl]amino}propanoate
Isopropyl (2S)-2-{[(S)-{[(2S,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl)-2-fluoro-3,4-dihydroxy-4-methyltetrahydro-2-furanyl]methoxy}(phenoxy)phosphoryl]amino}propanoate (non-preferred name
Propan-2-yl N-((P5’S)-4′-fluoro-2′-C-methyl-p-o-phenyl- 5′-uridylyl)-L-alaninate
545.5 g/mol, C22H29FN3O10P
CAS Registry Number 1613589-09-5
Adafosbuvir is under investigation in clinical trial NCT02894905 (A Study to Evaluate the Effect of Renal Impairment on the Pharmacokinetics of AL-335).
- Originator Alios BioPharma
- Developer Alios BioPharma; Janssen
- Class Antivirals; Pyrimidine nucleotides; Uracil nucleotides
- Mechanism of Action Hepatitis C virus NS 5 protein inhibitors
- Phase II Hepatitis C
- 28 Oct 2019 No recent reports of development identified for phase-I development in Hepatitis-C(In volunteers) in USA (PO)
- 28 Sep 2018 No recent reports of development identified for phase-I development in Hepatitis-C in France (PO)
- 28 Sep 2018 No recent reports of development identified for phase-I development in Hepatitis-C in Georgia (PO)
Adafosbuvir (AL 335), a monophosphate prodrug, is being developed by Alios BioPharma (a subsidiary of Johnson & Johnson) for the treatment of hepatitis C virus (HCV) infections. Adafosbuvir acts a uridine-based nucleotide analogue polymerase inhibitor. Clinical development is underway in New Zealand, Japan, the UK, the US, France, Georgia, Mauritius and Moldova.
Adafosbuvir has emerged from the company’s research programme focused on developing anti-viral nucleotides for the treatment of HCV infections , In November 2014, Alios BioPharma was acquired by Johnson & Johnson As at September 2018, no recent reports of development had been identified for phase-I development in Hepatitis-C in France (PO), Georgia (PO).
As at October 2019, no recent reports of development had been identified for phase-I development in Hepatitis-C (In volunteers) in USA (PO).
useful for the treatment of hepatitis C viral infections, assignaed to Janssen Pharmaceuticals Inc and Achillion Pharmaceuticals Inc . Janssen Pharmaceuticals, following Johnson & Johnson’s acquisition of Alios , was developing adafosbuvir, a uridine (pyrimidine) nucleotide analog, from a series of back-up compounds, that acts by inhibiting HCV NS5B polymerase, for the potential treatment of HCV infection.
As of December 2019, AL-335 dose increased from 400 to 800 mg qd in the presence of reduced simeprevir and odalasvlr doses increased ALS-022227 less than dose proportionally. However, this effect was minimal in the absence of slmeprevir [1973148]. Also, the company was also developing JNJ-4178 , a triple combination of adafosbuvir, odalasvir and simeprevir for the same indication.

McGuigan phosphoramidate nucleotide prodrugs. (a) Sofosbuvir (GS-7977) (Sp isomer), (b) BMS-986094 (Rp and Sp isomer mixture), (c) Adafosbuvir (AL-335) (Sp isomer), (d) ACH-3422*, and (e) MIV-802* (Sp isomer)

Figure 3. Clinical and preclinical 30,50-CPO prodrug. (a) GS-0938 (Rp isomer) and (b) IDX19368 (Sp isomer).

PAPER
Journal of Medicinal Chemistry (2019), 62(9), 4555-4570.
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.9b00143
We report the synthesis and biological evaluation of a series of 4′-fluoro-2′-C-substituted uridines. Triphosphates of the uridine analogues exhibited a potent inhibition of hepatitis C virus (HCV) NS5B polymerase with IC50values as low as 27 nM. In an HCV subgenomic replicon assay, the phosphoramidate prodrugs of these uridine analogues demonstrated a very potent activity with EC50 values as low as 20 nM. A lead compound AL-335(53) demonstrated high levels of the nucleoside triphosphate in vitro in primary human hepatocytes and Huh-7 cells as well as in dog liver following a single oral dose. Compound 53 was selected for the clinical development where it showed promising results in phase 1 and 2 trials.

PATENT
WO 2014209979
WO2014100505
Family members of the product case of adafosbuvir, WO2014100505 , expire in the US in December 2033.
PATENT
US 20150368286
WO 2015054465
PATENT
WO2017059147 ( US20170087174 ), claiming combination comprising simeprevir , odalasvir and AL-335
PATENT
WO-2019237297
Process for preparing AL-335 (also known as adafosbuvir) and its intermediates. AL-355 is a nucleoside inhibitor of NS3B polymerase, which plays an important role in the replication of the hepatitis C virus.



Compound 4 may be prepared in accordance with the procedures described in international patent application WO 2015/200216. Compound 4 (1.0 equiv) was then dissolved in THF (10 L/kg) and cooled down to -25℃. iPrMgCl (2M in THF) was added slowly over one hour and the resulting mixture was stirred for one hour. The Compound 3 solution previously made (see above) was then added dropwise at -25℃ and the mixture was stirred for 5h at that temperature before being warmed to -5℃ and stirred for 10 additional hours at that temperature. Once the reaction was complete, the reaction was warmed up to 5℃ and an aqueous solution of NH 4Cl (5L/kg -9 w/w%) was added slowly over 30 minutes. After phase separation, the organic layer was washed with aqueous NaHCO 3 solution (5L/kg -10 w/w%) and twice with aqueous NaCl solution (5L/kg -10 w/w%) . After solvent switch to acetonitrile, the reaction was assayed and stored under nitrogen and used as such in the next step.
/////////////ADAFOSBUVIR, AL335, ALS-335, JNJ-64146212, Alios BioPharma, Janssen, hepatitis C viral infections, D11364, адафосбувир , أدافوسبوفير , PHASE 2
CC(C)OC(=O)[C@H](C)N[P@](=O)(OC[C@@]1(F)O[C@@H](N2C=CC(=O)NC2=O)[C@](C)(O)[C@@H]1O)OC1=CC=CC=C1
Cefiderocol, セフィデロコル , цефидерокол , سيفيديروكول , 头孢德罗 ,

Cefiderocol
セフィデロコル;
| Formula |
C30H34ClN7O10S2
|
|---|---|
| CAS |
1225208-94-5
|
| Mol weight |
752.2149
|
Antibacterial, Cell wall biosynthesis inhibitor, enicillin binding protein, Siderophore cephalosporin
Fetroja (TN)
FDA, Cefiderocol, APPROVED, 2019/11/14
(6R,7R)-7-{[(2Z)-2-(2-Amino-1,3-thiazol-4-yl)-2-{[(2-carboxy-2-propanyl)oxy]imino}acetyl]amino}-3-[(1-{2-[(2-chloro-3,4-dihydroxybenzoyl)amino]ethyl}-1-pyrrolidiniumyl)methyl]-8-oxo-5-thia-1-azabicycl o[4.2.0]oct-2-ene-2-carboxylate
S-649266, GSK 2696266D
Cefiderocol, sold under the brand name Fetroja, is an antibiotic used to treat complicated urinary tract infections when no other options are available.[2] It is indicated for the treatment of multi-drug-resistant Gram-negative bacteria including Pseudomonas aeruginosa.[3][4][5] It is given by injection into a vein.[6]
It is in the cephalosporin family of medications.[2][7] Cefiderocol was approved for medical use in the United States on November 14, 2019.[2][8]
Cefiderocol, also known as S-649266, is a potent siderophore cephalosporin antibiotic with a catechol moiety on the 3-position side chain. S-649266 shows potent in vitro activity against the non-fermenting Gram-negative bacteria Acinetobacter baumannii, Pseudomonas aeruginosa and Stenotrophomonas maltophilia, including MDR strains such as carbapenem-resistant A. baumannii and metallo-β-lactamase-producing P. aeruginosa. S-649266 showed potent in vitro activities against A. baumannii producing carbapenemases such as OXA-type β-lactamases, and P. aeruginosa producing metallo-β-lactamases such as IMP type and VIM type. FDA approved this drug in 11/14/2019 To treat patients with complicated urinary tract infections who have limited or no alternative treatment options
Medical uses
Cefiderocol is used to treat adults with complicated urinary tract infections, including kidney infections caused by susceptible Gram-negative microorganisms, who have limited or no alternative treatment options.[2][7]
Mechanism of action
Its mechanism of entry into bacterial cells is by binding to iron, which is actively transported into the bacterial cells along with the cefiderocol.[6][9][10][11][12] It is in a medication class known as siderophores,[6][7] and was the first siderophore antibiotic to be approved by the U.S. Food and Drug Administration (FDA).[13] It bypasses the bacterial porin channels by using the bacteria’s own iron-transport system for being transported in.[14]
History
In 2019, cefiderocol was approved in the United States as an antibacterial drug for treatment of adults 18 years of age or older with complicated urinary tract infections (cUTI), including kidney infections caused by susceptible Gram-negative microorganisms, who have limited or no alternative treatment options.[2][8]
The safety and effectiveness of cefiderocol was demonstrated in a study of 448 patients with cUTIs.[2] Of the patients who were administered cefiderocol, 72.6% had resolution of symptoms and eradication of the bacteria approximately seven days after completing treatment, compared with 54.6% in patients who received an alternative antibiotic.[2] The clinical response rates were similar between the two treatment groups.[2]
Labeling for cefiderocol includes a warning regarding the higher all-cause mortality rate observed in cefiderocol-treated patients compared to those treated with other antibiotics in a trial in critically ill patients with multidrug-resistant Gram-negative bacterial infections.[2] The cause of the increase in mortality has not been established.[2] Some of the deaths were a result of worsening or complications of infection, or underlying co-morbidities.[2] The higher all-cause mortality rate was observed in patients treated for hospital-acquired/ventilator-associated pneumonia (i.e.nosocomial pneumonia), bloodstream infections, or sepsis.[2] The safety and efficacy of cefiderocol has not been established for the treatment of these types of infections.[2]
Cefiderocol received a Qualified Infectious Disease Product designation from the U.S. Food and Drug Administration (FDA) and was granted priority review.[2] The FDA granted approval of Fetroja, on November 14, 2019, to Shionogi & Co., Ltd.[2]
PATENT
WO 2010050468
WO 2016035845
WO 2016035847
PATENT
WO 2017216765,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017216765&tab=PCTDESCRIPTION
Bacterial infections continue to remain one of the major causes contributing towards human diseases. One of the key challenges in treatment of bacterial infections is the ability of bacteria to develop resistance to one or more antibacterial agents over time. Examples of such bacteria that have developed resistance to typical antibacterial agents include: Penicillin-resistant Streptococcus pneumoniae, Vancomycin-resistant Enterococci, and Methicillin-resistant Staphylococcus aureus. The problem of emerging drug-resistance in bacteria is often tackled by switching to newer antibacterial agents, which can be more expensive and sometimes more toxic. Additionally, this may not be a permanent solution as the bacteria often develop resistance to the newer antibacterial agents as well in due course. In general, bacteria are particularly efficient in developing resistance, because of their ability to multiply very rapidly and pass on the resistance genes as they replicate. Therefore, there is a need for development of newer ways to treat infections that are becoming resistant to known therapies and methods.
Surprisingly, it has been found that the compositions comprising a compound of Formula (I) or a pharmaceutically acceptable salt thereof and at least one beta-lactamase inhibitor or a pharmaceutically acceptable salt thereof, exhibit synergistic antibacterial activity, even against resistant bacterial strains.
Formula (I)
Example 1
Synthesis of Compound of formula (I)
Step-1: Preparation of intermediate (1):
To the clear solution of (Z)-2[(2-tert-butoxycarbonyl amino-thiazol-4-yl)-carboxy-methyleneaminooxy]2-methyl-propionic acid tert-butyl ester (30 gm, 69.93 mmol) in N,N-dimethyl acetamide (300 ml) was charged triethylamine (17.68 ml, 125.87 mmol) under stirring. The reaction mixture was cooled to -15°C. Methane sulfonyl chloride (12.01 gm, 104. 89 mmol) was charged to this cooled reaction mixture via addition funnel while maintaining temperature at about -15°C. The reaction mixture was stirred for 30 minutes at -15°C after the addition. To the reaction mixture was charged (6 ?,75)-4-methoxybenzyl-7-amino-3-chloromethyl-8-oxo-5-thia-l-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylate hydrochloride salt (28.25 gm, 69.93 mmol) along with N-methyl morpholine (15.5 ml, 139.86 mmol). The reaction mixture was stirred further for 1 hour at -15°C and the reaction progress was monitored using TLC. After completion of reaction, ethyl acetate (1.2 L) was charged followed by IN aqueous hydrochloric acid (1.2 L) under stirring and cooling was removed to warm up reaction mixture to room temperature. Layers were separated and organic layer was washed with saturated aqueous sodium bicarbonate solution (500 ml) followed by brine (500 ml). Organic layer was dried over sodium sulphate and was evaporated under vacuum to provide a crude mass. It was purified using silica gel column chromatography (60-120 mesh, 30% ethyl acetate in hexane) to provide 38 gm of intermediate (1).
Analysis:
1H NMR (CDCls) δ ppm: 8.29 (br s, 1H), 8.17 (d, 1H), 7.35 (d, 2H), 7.31 (s, 1H), 6.91 (d, 2H), 6.21 (dd, 1H), 5.23 (dd, 2H), 5.05 (d, 1H), 4.55 (d, 1H), 4.46 (d, 1H), 3.82 (s, 3H), 3.65 (d, 1H), 3.48 (d, 1H), 1.62 (s, 3H), 1.59 (s, 3H), 1.53 (s, 9H), 1.45 (s, 9H).
Step-2: Preparation of intermediate (2):
The solution of intermediate 1 (45 gm, 57.76 mmol) in dichloro methane (450 ml) was cooled to about -40°C and m-chloroperbenzoic acid (18 gm, 57.76 mmol) was added in three lots at -40°C under stirring. The mixture was stirred for 30 minutes and allowed to warm at -20°C. As TLC showed complete conversion, 5% aqueous sodium thiosulfate solution (1.2 L) was added at -15°C under stirring. The mixture was allowed to warm at room temperature and was charged with ethyl acetate (1.5 L) and stirred for 30 minutes and layers were separated. Organic layer was washed with saturated aqueous sodium bicarbonate solution (1 L) followed by brine (500 ml).
Organic layer was dried over sodium sulphate and evaporated under vacuum to provide 46 gm of intermediate (2).
Analysis:
1H NMR (CDC13) δ ppm: 8.48 (br s, 1H), 7.89 (d, 1H), 7.34 (d, 2H), 7.29 (s, 1H), 6.92 (d, 2H), 6.21 (dd, 1H), 5.27 (dd, 2H), 5.04 (br d, 1H), 4.58 (d, 1H), 4.23 (d, 1H), 3.83 (s, 3H), 3.82 (d, 1H), 3.43 (d, 1H), 1.60 (s, 3H), 1.58 (s, 3H), 1.53 (9H)1.42 (s, 9H).
Step-3: Preparation of intermediate (3):
Part-1: To the clear solution of intermediate 2 (35 gm, 44.02 mmol) in tetrahydrofuran (350 ml) was charged potassium iodide (14.61 gm, 88.05 mmol) under stirring at 25°C. The suspension was stirred for 5 hours at the same temperature and the reaction was monitored using mass spectroscopy. After completion of the reaction ethyl acetate (600 ml) was added to the reaction mixture followed by 5% aqueous sodium thiosulphate (600 ml). The reaction mixture was stirred for 15 minutes and layers were separated. Organic layer was washed with demineralised water (500 ml) followed by brine (500 ml). Organic layer was dried over sodium sulphate and evaporated to dryness under vacuum to provide 38 gm of corresponding iodo-methyl intermediate.
Part-2: To the iodo-methyl intermediate obtained (37.24 gm, 41.98 mmol) in N,N-dimethylformamide (35 ml) was added 2-chloro-3,4-di-(4-methoxybenzyloxy)-N-(pyrrolidin-l-ylethyl)-benzamide (22 gm, 42.98 mmol). The thick mass was stirred at 25°C for 15 hours and the reaction was monitored using mass spectroscopy. Potassium iodide (48.78 gm, 293.8 mmol) was charged to the reaction mass under stirring at 25 °C. The reaction mixture was cooled to -40°C and acetyl chloride (12 ml, 167.9 mmol) was added. After completion of the reaction ethyl acetate (1.2 L) followed by demineralised water (1.2 L) was added to the reaction mass at 0°C. Layers were separated and organic layer was washed with demineralised water (500 ml) followed by brine (500 ml). Organic layer was dried over sodium sulphate and was evaporated to dryness under vacuum to obtain quaternary intermediate (3) as iodide salt.
Step-4: Preparation compound of Formula (I):
Compound (3) (30 gm, 21.5 mmol) was dissolved in dichloro methane (300 ml) and anisole (30 gm, mmol) was added under stirring at 25°C. The mixture was cooled to -40° C and 2M aluminium chloride solution in nitromethane (150 ml) was added over 45 minutes at -40°C. As addition was completed reaction mixture was stirred for 1 hour at 0°C. To the reaction mixture 2M aqueous hydrochloric acid (750 ml) and acetonitrile (750 ml) were added and the stirring was
continued for 15 minutes. Di-isopropyl ether (1.5 L) was charged to the reaction mixture and the reaction mass was stirred for 15 minutes at 25°C, and the layers were separated. Aqueous layer was washed with additional di-isopropyl ether (500 ml). HP-21 resin (150 gm) was charged to the aqueous layer. The aqueous layer along with resin was loaded on a resin HP-21 column. The column was eluted with demineralised water till pH of eluent became neutral. Then the column was eluted with 10% acetonitrile in water mixture. Finally the column was eluted with 20% acetonitrile in water mixture. Evaporation of required fractions below 40°C under vacuum provided 5.5 gm of crude compound (I). The crude compound (I) was purified by dissolving in acetonitrile (200 ml) and demineralised water (200 ml) mixture followed by addition of HP-21 resin (200 gm).The slurry thus obtained was loaded on HP-21 resin column. The column was eluted first with demineralised water (3 L) followed by 10% acetonitrile in water mixture (2 L) then followed by 20% acetonitrile in water mixture till complete pure compound from the column is eluted. Pure fractions were collected and lyophilized under vacuum to provide titled compound (I) in pure form.
Analysis:
1H NMR (DMSO d6) δ ppm: 12.5 (br s, 2H), 9.42 (br s, 1H), 8.41 (br t, 1H), 7.28 (br s, 3H), 6.78 (s, 2H), 6.73 (s, 1H), 5.73 (dd, 1H), 5.15 (d, 1H), 5.08 (br d, 1H), 3.71-3.91 (m, 4H), 3.21-3.60 (m, 7H), 1.95-2.19 (m, 4H)1.76 (s, 3H), 1.44 (s, 3H).
HPLC purity: 90.80%
PATENT
WO 2019093450
Prior art documents
Non-patent literature
Non-patent Document 2: The Lancet Infrction diseases, 13 (9), 785-796,2013
Non-patent Document 3: Antimicrobial Agents and Chemotherapy, 61 (3), 1-11, 2017
PAPER
European Journal of Medicinal Chemistry (2018), 155, 847-868
References
- ^ Katsube, T.; Echols, R.; Arjona Ferreira, J. C.; et al. (2017). “Cefiderocol, a Siderophore Cephalosporin for Gram‐Negative Bacterial Infections: Pharmacokinetics and Safety in Subjects With Renal Impairment”. Journal of Clinical Pharmacology. 57 (5): 584–591. doi:10.1002/jcph.841. PMC 5412848. PMID 27874971.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves new antibacterial drug to treat complicated urinary tract infections as part of ongoing efforts to address antimicrobial resistance”. U.S. Food and Drug Administration (FDA) (Press release). 14 November 2019. Archived from the original on 16 November 2019. Retrieved 15 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ Choi, Justin J; McCarthy, Matthew W. (24 January 2018). “Cefiderocol: a novel siderophore cephalosporin”. Expert Opinion on Investigational Drugs. 27 (2): 193–197. doi:10.1080/13543784.2018.1426745. PMID 29318906.
- ^ Aoki, Toshiaki; Yoshizawa, Hidenori; Yamawaki, Kenji; et al. (15 July 2018). “Cefiderocol (S-649266), A new siderophore cephalosporin exhibiting potent activities against Pseudomonas aeruginosa and other gram-negative pathogens including multi-drug resistant bacteria: Structure activity relationship”. European Journal of Medicinal Chemistry. 155: 847–868. doi:10.1016/j.ejmech.2018.06.014. ISSN 1768-3254. PMID 29960205.
- ^ Portsmouth, Simon; van Veenhuyzen, David; Echols, Roger; et al. (25 October 2018). “Cefiderocol versus imipenem-cilastatin for the treatment of complicated urinary tract infections caused by Gram-negative uropathogens: a phase 2, randomised, double-blind, non-inferiority trial”. The Lancet Infectious Diseases. 0 (12): 1319–1328. doi:10.1016/S1473-3099(18)30554-1. ISSN 1473-3099. PMID 30509675.
- ^ Jump up to:a b c “Fetroja (cefiderocol) for injection, for intravenous use full prescribing information”(PDF). November 2019. Retrieved 17 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c Zhanel GG, Golden AR, Zelenitsky S, et al. (February 2019). “Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli”. Drugs. 79 (3): 271–289. doi:10.1007/s40265-019-1055-2. PMID 30712199.
- ^ Jump up to:a b “Cefiderocol New Drug Application”. U.S. Food and Drug Administration (FDA). Archived from the original on 4 December 2019. Retrieved 22 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ Sato T, Yamawaki K (November 2019). “Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin”. Clin. Infect. Dis. 69 (Supplement_7): S538–S543. doi:10.1093/cid/ciz826. PMC 6853759. PMID 31724047.
- ^ Matthews-King A (26 October 2018). “Antibiotic ‘Trojan horse’ could defeat superbugs causing global medical crisis, trial finds”. The Independent. Retrieved 26 October 2018.
- ^ Newey S (26 October 2018). “New ‘Trojan horse’ drug proves effective against antibiotic resistant bacteria”. The Telegraph. ISSN 0307-1235. Retrieved 26 October 2018.
- ^ Simpson DH, Scott P (2017). “Antimicrobial Metallodrugs”. In Lo K (ed.). Inorganic and Organometallic Transition Metal Complexes with Biological Molecules and Living Cells. Elsevier. ISBN 9780128038871.
- ^ Saisho, Yutaka; Katsube, Takayuki; White, Scott; et al. (March 2018). “Pharmacokinetics, Safety, and Tolerability of Cefiderocol, a Novel Siderophore Cephalosporin for Gram-Negative Bacteria, in Healthy Subjects” (PDF). Antimicrobial Agents and Chemotherapy. 62 (3): 1–12. doi:10.1128/AAC.02163-17. PMC 5826143. PMID 29311072. Retrieved 22 November 2019.
- ^ Ito A, Nishikawa T, Matsumoto S, et al. (December 2016). “Siderophore Cephalosporin Cefiderocol Utilizes Ferric Iron Transporter Systems for Antibacterial Activity against Pseudomonas aeruginosa”. Antimicrobial Agents and Chemotherapy. 60 (12): 7396–7401. doi:10.1128/AAC.01405-16. PMC 5119021. PMID 27736756.
External links
- “Cefiderocol”. Drug Information Portal. U.S. National Library of Medicine.
ADDITIONAL INFORMATION
S-649266 shows potent in vitro activity against the non-fermenting Gram-negative bacteria Acinetobacter baumannii, Pseudomonas aeruginosa and Stenotrophomonas maltophilia, including MDR strains such as carbapenem-resistant A. baumannii and metallo-β-lactamase-producing P. aeruginosa. MIC90s of S-649266 for A. baumannii, P. aeruginosa and S. maltophilia were 2, 1 and 0.5 mg/L, respectively, whereas MIC90s of meropenem were >16 mg/L. S-649266 showed potent in vitro activities against A. baumannii producing carbapenemases such as OXA-type β-lactamases, and P. aeruginosa producing metallo-β-lactamases such as IMP type and VIM type. MIC90 values for these A. baumannii strains and P. aeruginosa strains were 8 and 4 mg/L, respectively.
REFERENCES
1: Yamano Y. In Vitro Activity of Cefiderocol Against a Broad Range of Clinically Important Gram-negative Bacteria. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S544-S551. doi: 10.1093/cid/ciz827. PubMed PMID: 31724049; PubMed Central PMCID: PMC6853761.
2: Echols R, Ariyasu M, Nagata TD. Pathogen-focused Clinical Development to Address Unmet Medical Need: Cefiderocol Targeting Carbapenem Resistance. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S559-S564. doi: 10.1093/cid/ciz829. PubMed PMID: 31724048; PubMed Central PMCID: PMC6853756.
3: Sato T, Yamawaki K. Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S538-S543. doi: 10.1093/cid/ciz826. PubMed PMID: 31724047; PubMed Central PMCID: PMC6853759.
4: Bonomo RA. Cefiderocol: A Novel Siderophore Cephalosporin Defeating Carbapenem-resistant Pathogens. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S519-S520. doi: 10.1093/cid/ciz823. PubMed PMID: 31724046; PubMed Central PMCID: PMC6853757.
5: Katsube T, Echols R, Wajima T. Pharmacokinetic and Pharmacodynamic Profiles of Cefiderocol, a Novel Siderophore Cephalosporin. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S552-S558. doi: 10.1093/cid/ciz828. PubMed PMID: 31724042; PubMed Central PMCID: PMC6853762.
6: Kidd JM, Abdelraouf K, Nicolau DP. Efficacy of Humanized Cefiderocol Exposure is Unaltered by Host Iron Overload in the Thigh Infection Model. Antimicrob Agents Chemother. 2019 Oct 28. pii: AAC.01767-19. doi: 10.1128/AAC.01767-19. [Epub ahead of print] PubMed PMID: 31658966.
7: Chen IH, Kidd JM, Abdelraouf K, Nicolau DP. Comparative In Vivo Antibacterial Activity of Human-Simulated Exposures of Cefiderocol and Ceftazidime against Stenotrophomonas maltophilia in the Murine Thigh Model. Antimicrob Agents Chemother. 2019 Oct 7. pii: AAC.01558-19. doi: 10.1128/AAC.01558-19. [Epub ahead of print] PubMed PMID: 31591126.
8: Stevens RW, Clancy M. Compassionate Use of Cefiderocol in the Treatment of an Intraabdominal Infection Due to Multidrug-Resistant Pseudomonas aeruginosa: A Case Report. Pharmacotherapy. 2019 Nov;39(11):1113-1118. doi: 10.1002/phar.2334. Epub 2019 Oct 22. PubMed PMID: 31550054.
9: Sanabria C, Migoya E, Mason JW, Stanworth SH, Katsube T, Machida M, Narukawa Y, Den Nagata T. Effect of Cefiderocol, a Siderophore Cephalosporin, on QT/QTc Interval in Healthy Adult Subjects. Clin Ther. 2019 Sep;41(9):1724-1736.e4. doi: 10.1016/j.clinthera.2019.07.006. Epub 2019 Aug 1. PubMed PMID: 31378318.
10: Trecarichi EM, Quirino A, Scaglione V, Longhini F, Garofalo E, Bruni A, Biamonte E, Lionello R, Serapide F, Mazzitelli M, Marascio N, Matera G, Liberto MC, Navalesi P, Torti C; IMAGES Group . Successful treatment with cefiderocol for compassionate use in a critically ill patient with XDR Acinetobacter baumannii and KPC-producing Klebsiella pneumoniae: a case report. J Antimicrob Chemother. 2019 Nov 1;74(11):3399-3401. doi: 10.1093/jac/dkz318. PubMed PMID: 31369095.
11: Nakamura R, Ito-Horiyama T, Takemura M, Toba S, Matsumoto S, Ikehara T, Tsuji M, Sato T, Yamano Y. In Vivo Pharmacodynamic Study of Cefiderocol, a Novel Parenteral Siderophore Cephalosporin, in Murine Thigh and Lung Infection Models. Antimicrob Agents Chemother. 2019 Aug 23;63(9). pii: e02031-18. doi: 10.1128/AAC.02031-18. Print 2019 Sep. PubMed PMID: 31262762; PubMed Central PMCID: PMC6709502.
12: Katsube T, Saisho Y, Shimada J, Furuie H. Intrapulmonary pharmacokinetics of cefiderocol, a novel siderophore cephalosporin, in healthy adult subjects. J Antimicrob Chemother. 2019 Jul 1;74(7):1971-1974. doi: 10.1093/jac/dkz123. PubMed PMID: 31220260; PubMed Central PMCID: PMC6587409.
13: Jean SS, Hsueh SC, Lee WS, Hsueh PR. Cefiderocol: a promising antibiotic against multidrug-resistant Gram-negative bacteria. Expert Rev Anti Infect Ther. 2019 May;17(5):307-309. doi: 10.1080/14787210.2019.1612240. Epub 2019 May 6. PubMed PMID: 31055983.
14: Hackel MA, Tsuji M, Yamano Y, Echols R, Karlowsky JA, Sahm DF. Reproducibility of broth microdilution MICs for the novel siderophore cephalosporin, cefiderocol, determined using iron-depleted cation-adjusted Mueller-Hinton broth. Diagn Microbiol Infect Dis. 2019 Aug;94(4):321-325. doi: 10.1016/j.diagmicrobio.2019.03.003. Epub 2019 Mar 23. PubMed PMID: 31029489.
15: Miyazaki S, Katsube T, Shen H, Tomek C, Narukawa Y. Metabolism, Excretion, and Pharmacokinetics of [(14) C]-Cefiderocol (S-649266), a Siderophore Cephalosporin, in Healthy Subjects Following Intravenous Administration. J Clin Pharmacol. 2019 Jul;59(7):958-967. doi: 10.1002/jcph.1386. Epub 2019 Feb 7. PubMed PMID: 30730562; PubMed Central PMCID: PMC6593826.
16: Zhanel GG, Golden AR, Zelenitsky S, Wiebe K, Lawrence CK, Adam HJ, Idowu T, Domalaon R, Schweizer F, Zhanel MA, Lagacé-Wiens PRS, Walkty AJ, Noreddin A, Lynch Iii JP, Karlowsky JA. Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli. Drugs. 2019 Feb;79(3):271-289. doi: 10.1007/s40265-019-1055-2. Review. PubMed PMID: 30712199.
17: Huttner A. Cefiderocol for treatment of complicated urinary tract infections – Author’s reply. Lancet Infect Dis. 2019 Jan;19(1):24-25. doi: 10.1016/S1473-3099(18)30728-X. PubMed PMID: 30587291.
18: Portsmouth S, Echols R, Den Nagata T. Cefiderocol for treatment of complicated urinary tract infections. Lancet Infect Dis. 2019 Jan;19(1):23-24. doi: 10.1016/S1473-3099(18)30721-7. PubMed PMID: 30587290.
19: Wagenlehner FME, Naber KG. Cefiderocol for treatment of complicated urinary tract infections. Lancet Infect Dis. 2019 Jan;19(1):22-23. doi: 10.1016/S1473-3099(18)30722-9. PubMed PMID: 30587289.
20: Portsmouth S, van Veenhuyzen D, Echols R, Machida M, Ferreira JCA, Ariyasu M, Tenke P, Nagata TD. Cefiderocol versus imipenem-cilastatin for the treatment of complicated urinary tract infections caused by Gram-negative uropathogens: a phase 2, randomised, double-blind, non-inferiority trial. Lancet Infect Dis. 2018 Dec;18(12):1319-1328. doi: 10.1016/S1473-3099(18)30554-1. Epub 2018 Oct 25. PubMed PMID: 30509675.
 |
|
| Clinical data | |
|---|---|
| Trade names | Fetroja |
| Routes of administration |
Intravenous infusion |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 56–58%[1] |
| Elimination half-life | 2.8 hours |
| Excretion | mainly renal (60–70% unchanged) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C30H34ClN7O10S2 |
| Molar mass | 752.21 g·mol−1 |
| 3D model (JSmol) | |
////////////Cefiderocol, セフィデロコル , FDA 2019, цефидерокол , سيفيديروكول , 头孢德罗 , S-649266, GSK 2696266D
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}