
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Patent Battle Over Generic Drug Trade in India
Patent Battle Over Generic Drug Trade in India
India’s Patent rules for drug trade is grabbing global eyeballs ever since we acceded to WTO agreement in 2005. Recent court rulings have again raised a major debate on Indian rules join in our discussions about technology and business in today’s world failing to protect IPR of original inventors.
Source: Life Science India
You can also check out our premium service offerings for Mobile Applications, Social Media & Cyber Laws and Pharmaceuticals, Biotechnology, Food & Healthcare We regularly update our Facebook page and share similar stories on Twitter. Do have a look at our fresh tweets, news items and articles, and join in our discussions about technology and business in today’s world.
View original post 139 more words
Patent Searching in India| Patent Searcher in India | India Patent – Patent Guide for Inventors
Medical Devices: Technology, Emerging Sectors, Patent Trends and Roadblocks
Bio Corp Legal denotes premium service offerings offered by Tech Corp Legal LLP, an international law firm in India focusing on technology laws and business intelligence services.
We provide IP business services in the following domains:
- Pharma
- Biotech
- Medical Diagnostics
- Cell Based Research
- Cell & Gene Therapy
- Food Tech
- Peptides
- Biological Manufacturing Chemical based technologies
- Healthcare
- Drug Approval
- Life Sciences & related industries.
Online Patent Searching Tools
View original post 517 more words
Japanese Patent Applications can be translated to English by EPO’s Automatic Machine Translation
Japanese Patent Applications can be translated to English by EPO’s Automatic Machine Translation

Today EPO and the Japan Patent Office (JPO) announced the launch of the Japanese-English component of the EPO’s automatic translation service Patent Translate. This is a very good news for patent applicants and inventors across the globe as they can now easily translate the patent published in Japanese language. Numerous Japanese patent specifications available via the EPO’s global patent database Espacenet (www.epo.org/espacenet) can now be translated into English at zero cost to the applicant.
Recently, EPO and Google have been working together for sometime now to provide translation service optimised for patent specifications. Currently, patent translate covers translations between English and 15 other languages, namely Chinese, Danish, Dutch, Finnish, French, German, Greek, Hungarian, Italian, Japanese, Norwegian, Polish, Portuguese, Spanish and Swedish. In the time to come more languages will be added to the database.
Patent Translate
Patent Translate…
View original post 744 more words
FDA has granted tentative approval to India-based Strides Arcolab’s HIV drug emtricitabine and tenofovir disoproxil fumarate tablets, 200 mg/300 mg.

chief executive and vice chairman Arun Kumar –Strides arcolab

Deepak Vaidya, chaiman
Headquartered in India, Strides Arcolab is a pharmaceutical company with a key focus on the development and manufacture of IP-led niche products, particularly sterile injectables. It is among the world’s largest manufacturers of soft gelatin capsules. With 14 world-class manufacturing facilities, an innovative R&D hub and a marketing network in 70 countries, Strides is well positioned to meet the demands of the global pharmaceutical industry and has partnered with several of the world’s leading pharmaceutical companies.
2 August 2013
Strides Arcolab’s HIV drug, which is generic version of Gilead Sciences’ Truvada, gets tentative approval from the US FDA.
Tentative approval implies that the drug has met all standards but cannot be marketed in the US due to existing patent protections Good news for HIV AIDS patients
FDA has granted tentative approval to India-based Strides Arcolab’s HIV drug emtricitabine and tenofovir disoproxil fumarate tablets, 200 mg/300 mg. The drug is a generic version of Truvada, 200 mg/300 mg tablets, which is manufactured by Gilead Sciences.


The product is indicated for use in combination with other antiretroviral agents for the treatment of human immunodeficiency virus (HIV)-1


………………
………….
…………..
EMTRICITABINE

credit-chemdrug


Theravance Announces FDA Advisory Committee to Review ANORO(TM) ELLIPTA(TM) (UMEC/VI) for COPD
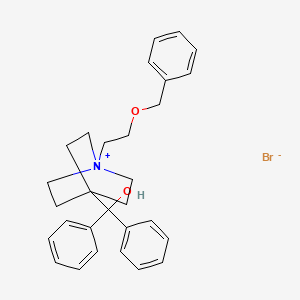
umeclidinium bromide

vilanterol
Theravance Announces FDA Advisory Committee to Review ANORO(TM) ELLIPTA(TM) (UMEC/VI) for COPD
SOUTH SAN FRANCISCO, CA — 08/01/13 –Theravance, Inc. today announced that on September 10, 2013, the U.S. Food and Drug Administration’s Pulmonary-Allergy Drugs Advisory Committee will discuss the new molecular entity New Drug Application (NDA) 203975 for umeclidinium bromide and vilanterol dry powder for inhalation (proposed trade name ANORO™ ELLIPTA™), sponsored by Glaxo Group (d/b/a GSK) for the long-term, once-daily, maintenance treatment of airflow obstruction in patients with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema. The advanced display of the Federal Register notice on the advisory committee can be found at:http://www.ofr.gov/OFRUpload/OFRData/2013-18633_PI.pdf
UMEC/VI is a combination of two investigational bronchodilator molecules — GSK573719 or umeclidinium bromide (UMEC), a long-acting muscarinic antagonist (LAMA) and vilanterol (VI), a long-acting beta2 agonist (LABA), administered using the ELLIPTA™ inhaler. The Prescription Drug User Fee Act (PDUFA) goal date is December 18, 2013. UMEC/VI is in development under the LABA collaboration agreement between Glaxo Group Limited and Theravance, Inc.
About Theravance
Theravance is a biopharmaceutical company with a pipeline of internally discovered product candidates and strategic collaborations with pharmaceutical companies. Theravance is focused on the discovery, development and commercialization of small molecule medicines across a number of therapeutic areas including respiratory disease, bacterial infections, and central nervous system (CNS)/pain. Theravance’s key programs include: RELVAR™ ELLIPTA™ or BREO™ ELLIPTA™ (FF/VI), ANORO™ ELLIPTA™ (UMEC/VI) and MABA (Bifunctional Muscarinic Antagonist-Beta2 Agonist), each partnered with GlaxoSmithKline plc, and its oral Peripheral Mu Opioid Receptor Antagonist program. By leveraging its proprietary insight of multivalency to drug discovery, Theravance is pursuing a best-in-class strategy designed to discover superior medicines in areas of significant unmet medical need. For more information, please visit Theravance’s web site atwww.theravance.com.
THERAVANCE®, the Theravance logo, and MEDICINES THAT MAKE A DIFFERENCE® are registered trademarks of Theravance, Inc.
RELVAR™, BREO™, ANORO™ and ELLIPTA™ are trademarks of the GlaxoSmithKline group of companies. The use of the brand names ANORO™ and RELVAR™ has not yet been approved by any regulatory authority.
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

