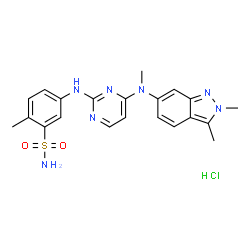
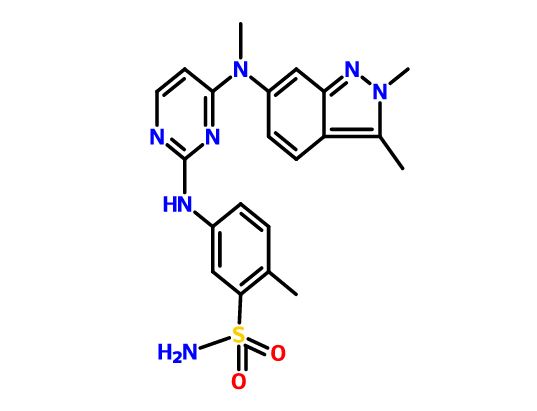
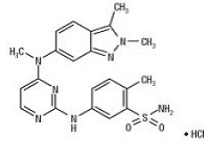
Pazopanib
パゾパニブ塩酸塩
Пазопаниба Гидрохлорид
5-[[4-[(2,3-Dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methylbenzolsulfonamide
Pazopanib is a small molecule inhibitor of multiple protein tyrosine kinases with potential antineoplastic activity. It is developed by GlaxoSmithKline and was FDA approved on October 19, 2009.
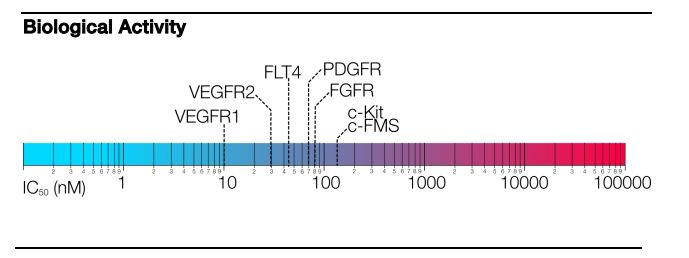
Pazopanib is a potent and selective multi-targeted receptor tyrosine kinase inhibitor of VEGFR-1, VEGFR-2, VEGFR-3,
PDGFR-a/b, and c-kit that blocks tumor growth and inhibits angiogenesis. It was approved for renal cell carcinoma by the U.S. Food and Drug Administration in 2009 and is marketed under the trade name Votrient by the drug’s manufacturer, GlaxoSmithKline.
GW 786034
M.Wt: 437.53
C21H23N7O2S
Pazopanib CAS No.: 444731-52-6
CAS No.: 635702-64-6 (PAZOPANIB HYDROCHLORIDE)
Pazopanib Hydrochloride
CAS No.: 635702-64-6 (PAZOPANIB HYDROCHLORIDE)
- MFC21H24ClN7O2S
- MW473.979
GW786034;Votrient;Armala;GW 786034;GW-786034
GW786034GW786034, VOTRIENT
5-({4-[(2,3-Dimethyl-2H-indazol-6-yl)(methyl)amino]-2-pyrimidinyl}amino)-2-methylbenzenesulfonamide hydrochloride (1:1)
Antineoplastic; Tyrosine Kinase Inhibitors, Protein Kinase Inhibitors; Renal Cell Carcinoma Therpay; Soft Tissue Sarcoma Therapy
パゾパニブ塩酸塩
Pazopanib Hydrochloride

C21H23N7O2S▪HCl : 473.98
[635702-64-6] |
Pazopanib (trade name Votrient) is a potent and selective multi-targeted receptor tyrosine kinase inhibitor that blocks tumour growth and inhibits angiogenesis. It has been approved for renal cell carcinoma and soft tissue sarcoma by numerous regulatory administrations worldwide.[2][3][4][5]
Pazopanib (Votrient®; GlaxoSmithKline, Brentford, U.K.) is currently approved by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency for the treatment of patients with metastatic renal cell carcinoma (mRCC)
Medical uses
It is approved by numerous regulatory administrations worldwide (including the FDA (19 October 2009), EMA (14 June 2010), MHRA(14 June 2010) and TGA (30 June 2010)) for use as a treatment for advanced/metastatic renal cell carcinoma and advanced soft tissue sarcomas.[1][2][3][4][5] In Australia and New Zealand, it is subsidised under the PBS and by Pharmac respectively, under a number of conditions, including:[6][7]
- The medication is used to treat clear cell variant renal cell carcinoma.
- The treatment phase is continuing treatment beyond 3-months.
- The patient has been issued an authority prescription for pazopanib
- The patient must have stable or responding disease according to the Response Evaluation Criteria In Solid Tumours (RECIST)
- This treatment must be the sole tyrosine kinase inhibitor subsidised for this condition.
It has also demonstrated initial therapeutic properties in patients with ovarian and non-small cell lung cancer,[8] though plans to apply to the EMA for a variation to include advanced ovarian cancer have been withdrawn and a license will not be sought in any country.[9][10]


SYNTHESIS
Pazopanib hydrochloride drug substance is manufactured by Glaxo Wellcome Manufacturing Pte. Limited, Jurong, Singapore
NDA 22-465 was submitted by GlaxoSmithKline (GSK) for VOTRIENT™ (pazopanib), an immediate release tablet for oral administration containing either 200 mg or 400 mg of pazopanib free base (GW786034X) as the hydrochloride salt (GW786034B). Pazopanib is a new molecular entity and is submitted for review pursuant to Section 505(b)(1) of the Food, Drug and Cosmetic Act. Reference is made to one active Investigational New Drug application, IND 65,747.
Quality by Design (QbD) approach and risk management to increase their understanding of the process and drug substance properties. A number of Critical Quality Attributes (CQAs) were identified. These are: Identity by IR, Chloride Identity, Crystalline Form, Content by HPLC, Drug-related Impurities (including named impurities and genotoxic (b) (4) (b) (4) (b) (4) (b) (4) Executive Summary Section CMC Review #1 Page 9 of 262 CMC REVIEW OF NDA 22-465 impurities content), Residue on Ignition, Particle Size, Residual Solvents, Water Content by Karl Fischer, Description, Pd Content, and Heavy Metals.
CQA are mainly controlled by controlling starting material attributes, intermediate attributes (e.g. specifications of GW786034 quality process parameters, and by following the manufacturing process. A risk based method (e.g. failure mode and effects analysis (FMEA)) was used to identify the Quality Critical Process Parameters (QCPPs), Quality Process Parameters (QPPs), CQAs and Quality Attributes (QAs) for the pazopanib hydrochloride manufacturing process. The inputs to the FMEA were knowledge gained through the work to develop the impurity fate map, spiking and purging studies, the Design of Experiments (DOE) work to establish potential QCPPs/QPPs, one factor at a time experiments to establish parameter proven acceptable ranges, and 6 years of plant experience preparing over batches of pazopanib hydrochloride in 3 plants throughout the GSK network. It was concluded from this risk assessment that there are no QCPP but a few QPP in the drug substance (DS) manufacturing process. Stages 1 and 2 had no QPP, the few were only present in stages 3 and 4. All the QPP were scale invariant. A combination of multivariate DOE and univariate experimentation was used to determine the Proven acceptable Ranges (PAR) for the variables. The risks for combining univariate and multivariate experimentation were found to be minimal, on the basis of outcome from the robustness study. For this study, all the process parameters were all set at the lower limit of the PARs to create a worst-case scenario for impurity purging. Neither new impurities nor elevated levels of known impurities were detected. This data demonstrated that multivariate interactions will not lead to elevated levels of impurities.
Drug Product Pazopanib Tablets, 200 mg and 400 mg are film-coated IR oral tablets. The two strengths contain 216.7 mg and 433.4 mg pazopanib hydrochloride, respectively, which are equivalent to 200 mg and 400 mg pazopanib (free base), respectively. Excipients in the tablet core are: microcrystalline cellulose, sodium starch glycolate, povidone, and magnesium stearate. Pazopanib Tablets, 200 mg are modified capsule-shaped, gray film-coated tablets, one side plain and the opposite side debossed with an identifying code of ‘GS JT’. Pazopanib Tablets, 400 mg are modified capsule-shaped, yellow film-coated tablets, one side plain and the opposite side debossed with an identifying code of ‘GS UHL’. The tablets are manufactured at Glaxo Operations UK Limited, Priory Street, Ware, Hertfordshire SG12 0DJ, United Kingdom. Primary packaging of tablets will be performed by either Glaxo Operations UK Limited, Priory Street, Ware, Hertfordshire SG12 0DJ, United Kingdom or GlaxoSmithKline Inc, 1011 North Arendell Avenue, Zebulon, North Carolina 27597, USA.
Pazopanib hydrochloride is a new molecular entity of Biopharmaceutics Classification System (BCS) Class 2 (poor solubility, high permeability) and a crystalline solid. Its solubility in pH 1.1 is 0.65 mg/mL. The conjugate acids of the basic nitrogens have the following acidity constants: pKa – pK1 = 2.1 (indazole), pK2 = 6.4 (pyrimidine), pK3 = 10.2 (sulfonamide).


“Synthetic approaches to the 2009 new drugs”
Kevin K.-C. Liua, Subas M. Sakyab, Christopher J. O’Donnellb, Andrew C. Flickb, Jin Lic,
Bioorganic & Medicinal Chemistry, Volume 19, Issue 3, Pages 1136–1154
“An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals”., Beilstein J. Org. Chem., 2011, 7, 442–495.


PATENT
https://www.google.com/patents/WO2015068175A2?cl=en
Pazopanib is marketed as hydrochloride salt by Glaxoshiithkline under the trade name VOTRIENT® is tyrosine kinase inhibitor and indicated for the treatment of patients with advanced renal cell carcinoma (RCC) and treatment of patients with advanced soft tissue sarcoma (STS) who have received prior chemotherapy.
U.S. Patent No (s). US 7105530 (“the ‘530 patent”), US7262203 (“the ‘203 patent”) and US8114885 (“the ‘885 patent”) discloses a variety of pyrimidineamines and their derivatives such as Pazopanib, processes for their preparation, pharmaceutical compositions comprising the derivatives, and method of use thereof.
The process disclosed in the ‘530 patent is schematically represented as follows:

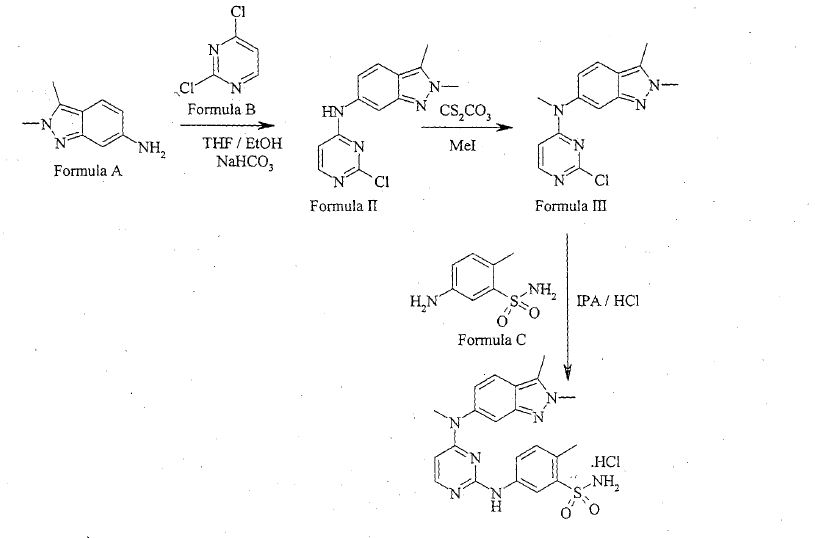
Patent publication No. WO 2011/050159 (“the Ί59 publication”) disclosed process for preparation of Pazopanib hydrochloride, which involves condensation of 2,3-dimethyl-2H-indazol-6-amine of Formula A and 2,4-dicMoropyrimidine of Formula B in a solvent like industrial methylated sprit and specific reaction conditions like, in presence of a base, sodium bicarbonate having a particle size distribution of > 250μηι or 50 to 150μηι selected to ensure that the pH of the reaction mixture is less than 7 for the reaction time period not more than 300 min to obtain N-(2-c oropyrimidin-4-yl)- 2,3-dimethyl-2H-indazol-6-amine of Formula II. The compound of Formula Π was methylated in presence of a methylating agent in an organic solvent like dimethylformamide by using specific reaction conditions like, in presence of a base i.e. Potassium carbonate having a particle size distribution D99 of > 300μηι or D99 of < 200μηι selected to ensure that the reaction time needs to reduce the starting material to less than 2% in less than 8 his to obtain N-(2-cMoropyrimidin-4-yl)-N-2,3-trimemyl-2H-mdazol-6-amine of Formula III. The resultant methylated compound was condensed with 5-amino-2-methylbenzenesulfonamide of Formula C in presence of 4M HC1 and methanol to yield Pazopanib hydrochloride.
WO Ί59 publication disclosed that use of sodium bicarbonate with specific particle size distribution of > 250μπι or 50 to 150μηι is key element in condensation of compound of Formula A and Formula B to niinimize the formation of Impurity of Formula 1 within

WO Ί59 publication also disclosed that use of potassium carbonate with specific particle size distribution D99 of > 300μπι or D99 of < 200μηι is key element in methylation of compound of Formula II to reduce the formation of Impurities of Formula 2, Formula 3 and Formula 4 within the range of about 0.05-3%.

Patent publication No. WO 2012/073254 (“the ‘254 publication”) disclosed a process for preparation of pazopanib hydrochloride, which involves condensation of 2,4-dicMoropyrimidine of Formula B with 5-amino-2-methylbenzenesulfonamide of Formula C in presence of a base like sodium bicarbonate and a solvent like ethanol to yield 5-(4-chloropyrimidm-2-yl-ammo)-2-memylbenzenesulfonamide The resultant compound was condensed with N-2,3-1rimethyl-2H-indazole-6-amine of Formula D in an alcoholic solvent like ethanol. WO ‘254 publication also discloses process for purification of pazopanib hydrochloride from alcoholic solvent and water. The process disclosed in the ‘254 publication is schematically represented as follows: 
Patent publication No. IN 2505/CHE/2011 disclosed a process for preparation of pazopanib, which involves condensation of 2,3-dimethyl-2H-indazol-6-amine of Formula A and 2,4-dichloropyrimidine of Formula B in presence of sodium bicarbonate and a phase transfer catalyst like tetrabutyl ammonium bromide in a solvent like methanol to obtain N-(2-chloropyrimidin-4-yl)-2,3 -dimethyl -2H-indazol-6-amine of Formula II. The resultant compound was methylated in presence of methyl iodide, potassium carbonate in a solvent like dimethylformamide to obtain compound of Formula III. The obtained Formula III was condensed with 5-amino-2-methylbenzenesulfonamide of Formula C in presence of dimethylformamide and concentrated HC1 to yield pazopanib hydrochloride.
Patent publication No. CN 103373989 (“the ‘989 publication”) disclosed a process for preparation of Pazopanib intermediate of Formula III by condensation of N-2,3-trimethyl-2H-indazole-6-amine of Formula D with 2,4-dicWoropyrimidine of Formula B in
Patent publication No. WO 2014/97152 (“the Ί52 publication”) disclosed a process for preparation of Pazopanib hydrochloride starting from 2,3-dimethyl-6-nitro-2H-indazole.
The processes for preparation of pazopanib described in the above literature have certain drawbacks as it involves: a) use of specific predefined particles of bases like sodium bicarbonate and potassium carbonate, which involves additional process steps like milling, grinding etc, b) use of expensive phase transfer catalysts and c) multiple steps making the process quite expensive, particularly on large scale.
European Medicines Agency (EMA) public assessment report disclosed that pazopanib hydrochloride is a white to slightly yellow, non-hygroscopic, crystalline substance and the manufacturing process consistently produces pazopanib hydrochloride Form 1. However, the EMEA does not describe any particular characterization data for the disclosed polymorph Form 1.
PCT Publication No. WO 2011/058179 (“the Ί79 publication”) discloses pazopanib base crystalline Forms such as Form-I and Form-II and a process for its preparation; also disclosed characterization data of Form-I and Form-II by XRD, IR and melting point. –
PCT Publication No. WO 2011/069053 (“the ‘053 publication”) discloses crystalline pazopanib base and crystalline pazopanib hydrochloride Forms such as Form-II, Form-Ill, Form-TV, Form-V, Form- VI, Form- VIII, Form-IX, Form-X, Form-XI, Form-XII, Form-XIII, Form-A, Form-G and also discloses crystalline Pazopanib dihydrochloride Forms such as Form-I, Form-XIV, Form-XV. The crystalline Forms reported in the PCT publication characterized by its XRD pattern.
IN Publication No. 3023/CHE/2010 discloses crystalline pazopanib dihydrochloride Form-I and crystalline pazopanib mono hydrochloride, process for it preparation and characterization by XRD of the same.
IN Publication No. 1535/CHE/2012 discloses crystalline pazopanib hydrochloride Form-SP and a process for its preparation; also disclosed characterization data, of Form-SP by XRD, DSC and TR.
PCT Publication No (s): WO 2007/143483, WO 2007/064753, WO 2006/20564 and WO 2005/105094 as well as US Publication No. US 2008/0293691 disclose anhydrous and hydrated Forms of pazopanib hydrochloride and their process for preparation thereof. ‘
IP. Com journal disclosure Number IPCOM000207426D discloses crystalline Form of pazopanib hydrochloride Form-R, which is characterized by XRD pattern.
Further, IP.Com journal disclosure Number IPCOM000193076D discloses crystalline Forms of N-(2-cUoropyrirnidin-4-yl)-N-2,3-trimethyl-2H-indazol-6-amine of
Formula III such as Form I and Form II along with characteristic data of XRD pattern
PATENT
https://www.google.com/patents/WO2012073254A1?cl=en
Examples
Example 1:
Preparation of 5-(4-chloropyrimidin-2ylamino)-2-methyIbenzenesulfonamide
To a mixture of 5-amino-2-methylbenzenesulfonamide (20 gm) in ethanol (208 ml) and tetrahydrofuran (52 ml) was added 2,4-dichloropryrimidine (44 gm) and sodium bicarbonate (36 gm) at room temperature. The contents were heated to 70 to 75°C and maintained for 13 hours. The reaction mass was then cooled to 10°C and maintained for 2 hours. The reaction mass was filtered and the solvent was distilled off under vacuum at below 50 to 55°C to obtain a residual mass. To the residual mass was added ethyl acetate (100 ml) and stirred for 1 hour, filtered. The solid obtained was dried to give 15.5 gm of 5-(4-chloropyrimidin-2ylamino)-2-methylbenzenesulfonamide. Example 2:
Preparation of N,2,3-trimethyI-2H-indazol-6-amine
Sodium methoxide (19 gm) was dissolved in methanol (610 ml) and then added 2,3-dimethyl-2H-indazol-6-amine (13 gm). The reaction mixture was stirred for 15 minutes and then added paraformaldehyde (3.9 gm). The contents were heated to 60°C and stirred for 10 hours. The reaction mass was then cooled to room temperature and maintained for 4 hours 30 minutes. Sodium borohydride (2.8 gm) was added to the reaction mass slowly at room temperature and then heated to reflux. The reaction mass was maintained for 2 hours at reflux and then cooled to room temperature. The reaction mass was stirred for 14 hours at room temperature and then added sodium hydroxide solution (1M, 100 ml). The pH of the reaction mass was adjusted to 8.0 to 8.5 with hydrochloric acid solution (40 ml) and then added ethyl acetate (400 ml). Then the layers were separated and the aqueous layer was extracted with ethyl acetate. The organic layer was dried with sodium sulfate and treated with carbon. The combined organic layers were washed with sodium chloride solution and dried with sodium sulfate. The organic layer was treated with carbon and filtered through hi-flow bed. The solvent was distilled off under vacuum at below 50°C to obtain a residual mass. To the residual mass was added diisopropyl ether (75 ml) and stirred for 1 hour, filtered. The solid obtained was dried to give 10 gm of N,2,3-trimethyl-2H-indazol-6-amine.
Example 3:
Preparation of pazopanib hydrochloride
5-(4-Chloropyrimidin-2ylamino)-2-methylbenzenesulfonamide (17 gm) as obtained in example 1, N,2,3-trimethyl-2H-indazol-6-amine (10 gm) as obtained in example 2 and ethanol (166 ml) were added at room temperature and then heated to reflux. The reaction mass was maintained for 3 hours at reflux and then added concentrated hydrochloric acid (1 ml). The reaction mass was maintained for 10 hours at reflux and then cooled to room temperature. The separated solid was filtered and dried to obtain 17 gm of pazopanib hydrochloride (HPLC Purity: 97.5%). Example 4:
Purification of pazopanib hydrochloride
Pazopanib hydrochloride (5 gm; HPLC Purity: 97.5%) as obtained in example 3 was dissolved in a mixture of methanol (100 ml) and water (10 ml) at room temperature and then heated to reflux. The reaction mass was maintained for 30 minutes at reflux and filtered. The filtrate obtained was cooled to room temperature and maintained for 2 hours at room temperature. The solid obtained was collected by filtration and dried to obtain 3.5 gm of pazopanib hydrochloride (HPLC Purity: 99.9%). Example 5:
Purification of pazopanib hydrochloride
Pazopanib hydrochloride (22 gm; HPLC Purity: 98%), methanol (528 ml), water (55 ml) and concentrated hydrochloric acid (0.2 ml) were added at room temperature. The contents were heated to reflux and maintained for 30 minutes, filtered. Take the filtrate and the solvent was distilled off under vacuum to obtain a residual mass. The residual mass was then cooled to room temperature and stirred for 30 minutes at room temperature. The contents were further cooled to 0 to 5°C, stirred for 1 hour and filtered. The solid obtained was dried to give 19 gm of pazopanib hydrochloride (HPLC Purity: 99.85%).
Example 6:
Purification of pazopanib hydrochloride
Pazopanib hydrochloride (10 gm; HPLC Purity: 96%), methanol (250 ml), water (25 ml) and concentrated hydrochloric acid (0.1 ml) were added at room temperature. The contents were heated to reflux and maintained for 30 minutes, filtered. The filtrate obtained was then cooled to room temperature and stirred for 30 minutes at room temperature. The contents further cooled to 0 to 10°C and stirred for 1 hour. The separated solid was filtered and dried to obtain 6.6 gm of pazopanib hydrochloride (HPLC Purity: 99.8%).
Example 7: Purification of pazopanib hydrochloride
Pazopanib hydrochloride (22 gm; HPLC Purity: 97%) was dissolved in a mixture of isopropanol (132 ml) and water (20 ml) at room temperature and then heated to reflux. The reaction mass was maintained for 1 hour at reflux and then cooled to room temperature. The reaction mass was stirred for 1 hour at room temperature and filtered. The solid obtained was dried to give 18 gm of pazopanib hydrochloride (HPLC Purity: 99.8%).
Paper
http://pubs.acs.org/doi/full/10.1021/op400139z
Assessment of Predictivity of Semiquantitative Risk Assessment Tool: Pazopanib Hydrochloride Genotoxic Impurities
† GlaxoSmithKline, Park Road, Ware, Hertfordshire, United Kingdom SG12 0DP
‡ GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
Org. Process Res. Dev., 2013, 17 (8), pp 1036–1041
DOI: 10.1021/op400139z
Publication Date (Web): July 02, 2013
Copyright © 2013 American Chemical Society
Abstract
The recently developed semiquantitative assessment tool for the evaluation of carryover potential of mutagenic impurities (MIs) into the final API was applied to the five identified MIs within pazopanib hydrochloride (dimethyl sulfate (DMS) and compounds II, III, VI, and VIII). The theoretical and predicted purge factors were compared. The tool accurately predicted the purging capacity for the most reactive MI, DMS, giving a theoretical purge factor of 30000 versus an actual value of 29411 (for spiking at stage 1). For the other less reactive MIs, both measured and predicted values agreed reasonably well, and the high values for the purging factors were indicative of an effective process capability that could significantly reduce observed MI levels. The only exception was for compound VI, where although the measured and theoretical purge factors were in agreement, they were significantly lower (<200) than for the other MIs. In this case, a strategy was implemented including a requirement for control of this MI on API specification. The purge-factor assessment tool has the potential to play a key role in GRA (genotoxic risk assessment) processes and subsequent regulatory submissions. This tool could provide regulators with additional confidence to accept these purging arguments without resorting to testing. This could potentially significantly reduce the analytical testing burden for early clinical candidates.


PATENT
WO 2011058179
The compound 5-(4-(N-(2,3-dimethyl-2H-indazole-6-yl)-N-methylamino)pyrimidine-2- ylamino)-2-methylbenzenesulfonamide, also known as Pazopanib, is useful in the treatment of disorders associated with inappropriate or pathological angiogenesis, such as cancer, in mammals. Pazopanib has the following formula (I):
H CH,
NH2
In WO 02/059110 the preparation of 5-(4-(N-(2,3-dimethyl-2H-indazole-6-yl)-N- methylamino)pyrimidine-2-ylamino)-2-methylbenzenesulfonamide hydrochloride as well as the uses of this compound have been disclosed. In particular, this compound is an inhibitor of tyrosine kinase enzymes, namely vascular endothelial growth factor receptors, and can be used for the treatment and/or prevention of diseases which are associated with tyrosine kinase enzymes such as vascular endothelial growth factor receptors, such as cancer, particularly breast cancer and colon cancer.
Alternative methods for the preparation of 5-(4-(N-(2,3-dimethyl-2H-indazole-6-yl)-N- methylamino)pyrimidine-2-ylamino)-2-methylbenzenesulfonamide hydrochloride are disclosed in WO 03/106416.
In WO 2007/064752 the use of Pazopanib for the treatment of age related macula degeneration is disclosed. WO 2007/064753 further discloses Pazopanib for the treatment of various types of cancer, e.g. brain cancer, glioblastoma multiforme, neuroendocrine cancer, prostate cancer, myeloma, lung cancer, liver cancer, gallbladder cancer or skin cancer.
Typically Pazopanib is administered orally, as this route provides great comfort and convenience of dosing. Although the hydrochloride form of Pazopanib is known in the art, as described above, this form is not optimal in regard to bioavailability, inter-patient variability, and safety. Further, the known form of Pazopanib hydrochloride is not optimal with regard to mechanical and chemical stability, which is in particular necessary for manufacturing tablets, as well as not optimal in regard to flow properties, compressibility, dissolution rate. Additionally, it is at least to some extent hygroscopic and shows electrostatic charging. These properties constitute disadvantages in the preparation of pharmaceutical compositions
PAPER
Synthesis and biological evaluation of novel pazopanib derivatives as antitumor agents
- doi:10.1016/j.bmcl.2014.01.003
Abstract
A series of novel pazopanib derivatives, 7a–m, were designed and synthesized by modification of terminal benzene and indazole rings in pazopanib. The structures of all the synthesized compounds were confirmed by 1H NMR and MS. Their inhibitory activity against VEGFR-2, PDGFR-α and c-kit tyrosine kinases were evaluated. All the compounds exhibited definite kinase inhibition, in which compound 7l was most potent with IC50 values of 12 nM against VEGFR-2. Furthermore, compounds 7c, 7d and 7mdemonstrated comparable inhibitory activity against three tyrosine kinases to pazopanib, and compound 7f showed superior inhibitory effects than that of pazopanib.

Figure 1.
Chemical structure of pazopanib.
Patent
https://www.google.co.in/patents/WO2002059110A1?cl=en
Example 69
5-({4-[(2,3-dimethyl-2r/-indazol-6-yl)(methyl)amino]pyrimidin-2-yl}amino)-2- methylbenzenesulfonamide
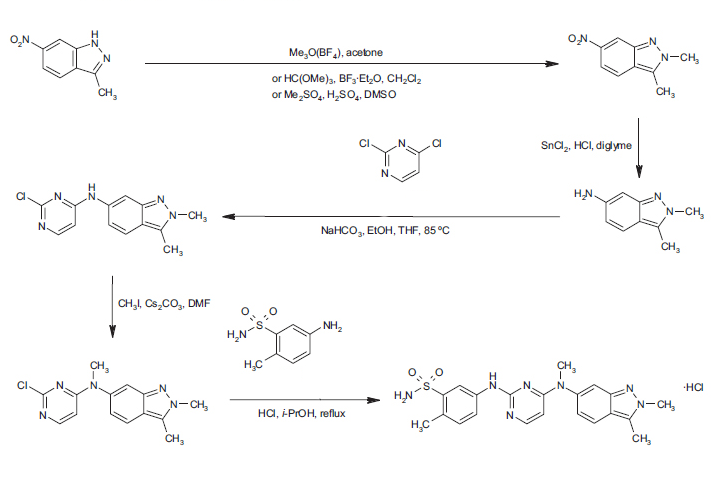
To a solution of Intermediate Example 13 (200 mg, 0.695 mmol) and 5-amino-2- ethylbenzenesulfonamide (129.4 mg, 0.695 mmol) in isopropanol (6 ml) was added 4 drops of cone. HCI. The mixture was heated to reflux overnight. The mixture was cooled to rt and diluted with ether (6 ml). Precipitate was collected via filtration and washed with ether. HCI salt of 5-({4-[(2,3-dimethyl-2H-indazol-6-yl)(methyl)amino]-pyrimidin-2- yl}amino)-2-methylbenzenesulfonamide was isolated as an off-white solid. Y\ NMR (400 MHz, deDMSO+NaHCOa) δ 9.50 (br s, 1 H), 8.55 (br s, 1 H), 7.81 (d, J = 6.2 Hz, 1 H), 7.75 (d, J = 8.7 Hz, 1 H), 7.69 (m, 1 H), 7.43 (s, 1 H), 7.23 (s, 2H), 7.15 (d, J = 8.4 Hz, 1 H), 6.86 (m, 1 H), 5.74 (d, J = 6.1 Hz, 1 H), 4.04 (s, 3H), 3.48 (s, 3H), 2.61 (s, 3H), 2.48 (s, 3H). MS (ES+, m/z) 438 (M+H).
Example 13
Preparation of Λ/-(2-chloropyrimidin-4-yl)-Λ/,2,3-trimethyl-2r/-indazol-6-amine
To a stirred solution of the Intermediate 12 (7.37 g) in DMF (50 ml) was added CS2CO3 (7.44 g, 2 eqv.) and Mel (1.84 ml, 1.1 eqv.) at room temperature. Mixture was stirred at rt for overnight The reaction mixture was poured into ice-water bath, and the precipitate was collected via filtration and washed with water. The precipitate was air- dried to afford Λ/-(2-chloropyrimidin-4-yl)-Λ/,2,3-trimethyl-2r/-indazol-6-amine as an off-white solid (6.43 g, 83%). ‘H NMR (400 MHz, dsDMSO) δ 7.94 (d, J = 6.0 Hz, 1 H), 7.80 (d, J = 7.0 Hz, 1 H), 7.50 (d, J = 1.0 Hz, 1 H), 6.88 (m, 1 H), 6.24 (d, J = 6.2 Hz, 1 H), 4.06 (s, 3H), 3.42 (s, 3H), 2.62 (s, 3H). MS (ES+, m/z) 288 (M+H).
Intermediate Example 12 Preparation of Λ/-(2-chloropyrimidin-4-yl)-2,3-dimethyl-2H-indazol-6-amine
to a stirred solution of Intermediate Example 11 (2.97 g, .015 mol) and NaHCOs (5.05 g, .06 mol) in THF (15 mL) and ethanol (60 mL) was added 2,4-dichloropyrimidine (6.70 g, .045 mol) at room temperature. After the reaction was stirred for four hours at 85 °C, the suspension was cooled to rt, filtered and washed thoroughly with ethyl acetate. The filtrate was concentrated under reduced pressure, and the resulting solid was triturated with ethyl acetate to yield 3.84 g (89 % yield) of Λ/-(2-chloropyrimidin-4-yl)- 2,3-dimethyl-2tf-indazol-6-amine. 1H NMR (400 MHz, deDMSO) δ 7.28 (d, J = 9.0 Hz, 1 H), 6.42 (d, J = 8.8 Hz, 1 H), 6.37 (s, 1 H), 5.18 (br s, 1 H), 3.84 (s, 3H), 2.43 (s, 3H). MS (ES+, m/z) 274 (M+H).
Intermediate Example 11
Preparation of 2,3-dimethyl-2r/-indazol-6-amine
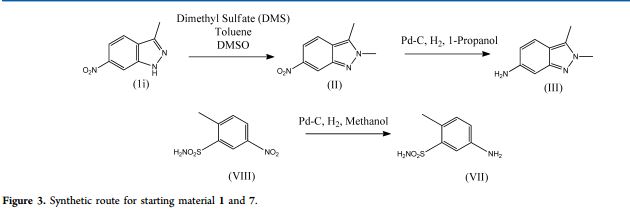
To a stirred solution of 18.5 g (0.11 mol) of 3-methyl-6-nitro- 7W-indazole in 350 ml acetone, at room temperature, was added 20 g (0.14 mol) of trimethyloxonium tetraflouroborate. After the solution was allowed to stir under argon for 3 hours, the solvent was removed under reduced pressure. To the resulting solid was added saturated aqueous NaHC03 (600 ml) and a 4:1 mixture of chloroform-isopropanol (200 ml), and the mixture was agitated and the layers were separated. The aqueous phase was washed with additional chloroform: isopropanol (4 x 200 ml) and the combined organic phase was dried (Na2S04). Filtration and removal of solvent gave a tan solid. The solid was washed with ether (200 ml) to afford 2,3-dimethyl-6-nitro-2r/-indazole as a yellow solid (15.85 g, 73 o/o). 1H NMR (300 MHz, dβDMSO) δ 8.51 (s, I H), 7.94 (d, J = 9.1 Hz, 1 H), 7.73 (d, J = 8.9 Hz, 1 H), 4.14 (s, 3H), 2.67 (s, 3H). MS (ES+, m/z) 192 (M+H).
To a stirred solution of 2,3-dimethyl-6-nitro-2V-indazole (1.13 g) in 2- methoxyethyl ether (12 ml), at 0 °C, was added a solution of 4.48 g of tin(ll) chloride in 8.9 ml of concentrated HCI dropwise over 5 min. After the addition was complete, the ice bath was removed and the solution was allowed to stir for an additional 30 min. Approximately 40 ml of diethyl ether was added to reaction, resulting in precipitate formation. The resulting precipitate was isolated by filtration and washed with diethyl ether, and afforded a yellow solid (1.1 g, 95 %), the HCI salt 2,3-dimethyl-2/7-indazol-6- amine. 1H NMR (300 MHz, deDMSO) δ 7.77 (d, J = 8.9 Hz, 1 H), 7.18 (s, 1 H), 7.88 (m, 1 H), 4.04 (s, 3H), 2.61 (s, 3H). MS (ES+, m/z) 162 (M+H).
CLIP
innovareacademics.in/journals/index.php/ijpps/article/download/10558/4196
CLIP FROM
https://ayurajan.blogspot.in/2014/12/pazopanib.html
WO2003106416A2 (same appears in Drug Future 2006, 31, 7, 585-589)
Pazopanib synthesis: J Med Chem 2008, 51, 4632-4640 (same appears in Beilstein J Org Chem 2011, 7, 442–495)
CLIP
LINK



PAPER
http://www.eurekaselect.com/97375
10.2174/157017812800233714
A Novel Practical Synthesis of Pazopanib: An Anticancer Drug
Author(s): YiCheng Mei, BaoWei Yang, Wei Chen, DanDan Huang, Ying Li, Xin Deng, BaoMing Liu, JingJie Wang, Hai Qian and WenLong Huang
Affiliation: Center of Drug Discovery, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing, Jiangsu 210009, P.R. China.
Abstract:
This paper reports a novel approach to synthesize pazopanib. In our synthetic route, the potently mutagenic alkylating agents such as dimethyl sulfate and methyl iodide are avoided. A novel regioselective methylation of the 2- position of 3-methyl-6-nitro-1H-indazole was reported. This novel route is one step shorter than the previously reported route.
PATENT
https://www.google.com/patents/WO2014097152A1?cl=en
Pazopanib is a tyrosine kinase inhibitor of Formula la.
Formula la
Pazopanib is marketed as the hydrochloride salt, with the chemical name 5-[[4- [(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2- methylbenzenesulfonamide monohydrochloride, having the structure as depicted in Formula I:
Formula I
U.S. Patent No. 7,105,530 provides a process for the preparation of a hydrochloride salt of a compound of Formula II
Formula II involving the reduction of 2,3-dimethyl-6-nitro-2H-indazole with tin (II) chloride in concentrated hydrochloric acid in the presence of 2-methoxyethyl ether at 0°C. It also describes the preparation of a compound of Formula III
Formula III involving the reaction of a hydrochloride salt of compound of Formula II with 2,4- dichloropyrimidine in the presence of a base and solvent mixture of
tetrahydrofuran/ethanol followed by stirring for 4 hours at 85°C.
PCT Publication No. WO 2007/064752 provides a process for the preparation of a compound of Formula II comprising reducing 2,3-dimethyl-6-nitro-2H-indazole with 10% Palladium-carbon (50% wet) in the presence of methanol, followed by the addition of ammonium formate at a rate that ensures the reaction temperature is maintained at or between 25°C and 30°C. It also discloses the preparation of a compound of Formula III comprising heating the compound of Formula II with sodium bicarbonate in presence of tetrahydrofuran and ethanol at or between 75°C and 80°C followed by cooling to 20°C to
25°C.
The present invention provides a process for the preparation of a compound of Formula II which offers recycling of the Raney nickel catalyst used in the process, and an easy filtration work-up procedure. Further, the present invention offers selective reduction under mild conditions that is economical to use at an industrial scale.
The present invention also provides a process for the preparation of compound of Formula III which avoids the use of two or more solvents, and additionally, also circumvents heating and cooling procedures during the reaction. The aforesaid advantages yield a compound of Formula III with a lesser amount of N-(4-chloropyrimidin-2-yl)-2,3- dimethyl-2H-indazol-6-amine (CPDMI) impurity.
The compounds of Formula II and Formula III prepared by the present invention yield a compound of Formula la or its salts in comparable yield and suitable purity required for medicinal preparations.
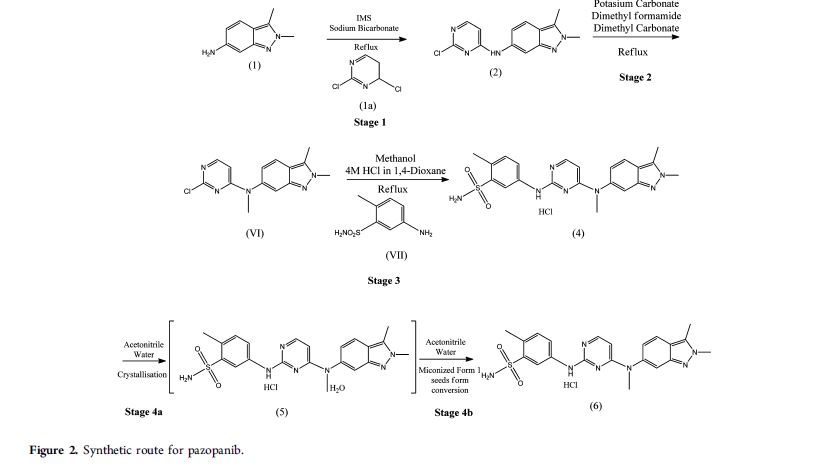
EXAMPLES
Step 1: Synthesis of 2,3-dimcthyl-6-nitro-2H-indazole
Example 1 :
Trimethyloxonium tetrafluoroborate (125.2 g, 0.85 mol) was added to a stirred suspension of 3-methyl-6-nitro-indazole (100 g, 0.56 mol) in ethyl acetate (2000 mL) over a period of 4 hours in four equal lots at 1 hour time intervals. The reaction mixture was stirred at 25 °C to 30°C for 16 hours. The solvent was recovered under reduced pressure. A saturated sodium bicarbonate solution (3240 mL) was added to the mixture slowly, and the reaction mixture was extracted with 4: 1 mixture of dichloromethane:isopropyl alcohol (1080 mL x 5). The solvent was recovered under reduced pressure. Methyl fert-butyl ether (800 mL) was added to the residue, and the reaction mixture was stirred for 30 minutes at 45 °C to 50°C. The reaction mixture was cooled to 25 °C to 30°C and was stirred at this temperature for 30 minutes. The solid was filtered, washed with methyl tert- butyl ether (100 mL x 2), and dried in an air oven at 50°C for 12 hours to afford 2,3- dimethyl-6-nitro-2H-indazole as a yellow solid.
Yield: 82.4% w/w
Step 2: Synthesis of 2,3-dimethyl-2H-indazol-6-amine
Example 2a:
Raney nickel ( 12.50 g) was added to a suspension of 2,3-dimethyl-6-nitro-2H- indazole (50 g, 0.26 mol) in methanol (500 mL). The reaction mixture was stirred in an autoclave under hydrogen pressure of 3.5 kg/cm2 – 4.0 kg/cm2 at 25°C to 30°C for 5 hours. Further, the reaction mixture was filtered through a hyflo bed, and the catalyst was washed with methanol (100 mL x 2). The filtrates were combined, and the solvent was recovered completely. «-Heptane (250 mL) and dichloromethane (50 mL) were added to the residue, and the reaction mixture was stirred for 1 hour at 25°C to 30°C. The solid was collected by filtration, washed with n-heptane (50 mL x 2), and dried under vacuum at 40°C to 45 °C to afford 2,3-dimethyl-2H-indazol-6-amine as a light brown solid.
Yield: 95% w/w
Example 2b:
Raney nickel (21.25 g) was added to a suspension of 2,3-dimethyl-6-nitro-2H- indazole (85 g, 0.45 mol) in methanol (850 mL). The reaction mixture was stirred in an autoclave under hydrogen pressure of 3.5 kg/cm2 – 4.0 kg/cm2 at 25°C to 30°C for 5 hours. Further, the reaction mixture was filtered through a hyflo bed, and the catalyst was washed with methanol (85 mL x 3). The filtrates were combined, and the solvent was recovered up to the volume of 850 mL. The 2,3-dimethyl-2H-indazol-6-amine in methanol was used as such in the next step. Step 3: Synthesis of N-(2-chloropyrimidin-4-yl)-2,3-dimethyl-2H-indazol-6-amine
Example 3 :
Sodium bicarbonate ( 112 g, 1.34 mol) was added to a stirred solution of 2,3- dimethyl-2H-indazol-6-amine (as obtained from step 2; Examples 2a and 2b) in methanol. 2,4-Dichloropyrimidine (99.35 g, 0.67 mol) was added to the reaction mixture followed by stirring of the reaction mixture for 24 hours at 25°C to 30°C. De-ionized water (850 mL) was added to the reaction mixture followed by stirring of the reaction mixture at 25 °C to 30°C for 1 hour. The solid was filtered. The wet solid was washed with de-ionized water (170 mL x 2) to obtain a wet material. De-ionized water (850 mL) was added to the wet material to obtain a slurry, and the slurry was stirred at 25°C to 30°C for 30 minutes. The solid was filtered, then washed with de-ionized water (170 mL x 2). The wet material obtained was treated with ethyl acetate (340 mL) to obtain a slurry. The slurry was stirred at 35°C to 40°C for 30 minutes and then cooled to 0°C to 5°C. The slurry was further stirred at 0°C to 5°C for 30 minutes. The solid was collected by filtration, then washed with cold ethyl acetate (170 mL x 2). The solid was dried in an air oven at 50°C for 16 hours to afford N-(2-chloropyrimidin-4-yl)-2,3 -dimethyl -2H-indazol-6-amine as an off- white solid.
Yield: 86.7% w/w
Step 4: Synthesis of pazopanib hydrochloride
Example 4a: Synthesis of N-(2-Chloropyrimidin-4-yl)-N.2.3-trimethyl-2H-indazol-6- amine
Cesium carbonate (238 g, 0.73 mol) and iodomethane (57 g, 0.40 mol) were added to a stirred suspension of N-(2-chloropyrimidin-4-yl)-2,3-dimethyl-2H-indazol-6-amine (lOOg, 0.37 mol) in N,N-dimethylformamide (300 mL) at 25°C to 30°C. The reaction mixture was further stirred at 25 °C to 30°C for 6 hours followed by cooling of the reaction mixture to 0°C to 5°C. De-ionized water (300 mL) was added drop-wise to the reaction mixture, then the reaction mixture was stirred at 5°C to 10°C for 30 minutes. The solid was collected by filtration, and washed with de-ionized water (100 mL x 2). The wet material so obtained was dried in an air oven at 50°C for 12 hours to obtain the title compound.
Yield: 90.4% w/w Example 4b: Synthesis of pazopanib hydrochloride
To a suspension of N-(2-chloropyrimidin-4-yl)-N-2,3-trimethyl-2H-indazol-6- amine (90 g, 0.312 mol) and 5-amino-2-methyl benzene sulfonamide (64.07 g, 0.344 mol) in isopropyl alcohol (900 mL) was added 4M hydrochloric acid solution in isopropyl alcohol (1.56 mL, 6.25 mol). The reaction mixture was heated to reflux temperature for 10 hours to 12 hours. The reaction mixture was cooled to 25°C. The reaction mixture was further stirred at 25°C to 30°C for 30 minutes, then the solid was filtered. The wet solid was washed with isopropyl alcohol (180 mL x 2), and then dried under vacuum at 45 °C to 50°C for 12 hours to afford the hydrochloride salt of 5-({4-[(2,3-dimethyl-21-I-indazol-6- yl)(methyl) amino] pyrimidin-2-yl} amino-Z-methylbenzene sulfonamide as a light brown solid.
Yield: 97% w/w
PATENT
https://www.google.com/patents/CN104557881A?cl=en
pazopanib hydrochloride monohydrate prepared:
(1) chemical reaction formula
(2) Operation process
In the reaction flask pazopanib hydrochloride crude 100g, 700ml of acetonitrile was added under stirring and purified water 200ml, feeding is completed, begin heating to 75~80 ° C, until clear solvent filtration system, slowly dropped 10~20 ° C, keep stirring lh, filtered, and the filter cake washed with purified water and acetonitrile respectively, drained and the filter cake was dried at 60 ° C blast 5h, have pazopanib hydrochloride monohydrate solid 84g, yield 80.9%. For example, crystalline form pazopanib hydrochloride prepared the following examples.
pazopanib hydrochloride polymorph of preparation:
Example 1:
In the reaction flask pazopanib hydrochloride monohydrate 8. 0g, ethanol 50ml and purified water 0.Iml (the volume of water accounted for a mixed solution of 2% of the total volume of the square, ethanol – water mixture total volume was 6.26 times pazopanib hydrochloride monohydrate quality), heated to 75 ° C, stirred at reflux for about 5h, after cooling to 10~20 ° C, keep stirring lh, the filter cake washed with ethanol, then blast drying at 105 ° C 5h, to obtain ultrafine powder solid 6. 8g, yield of 81. 9%, HPLC purity was 99.8%, as measured crystal X- ray powder diffraction pattern of FIG. 1 the basic consistent, as measured with a DSC thermogram consistent FIG. 2, the particle size distribution measurement is basically the same as Fig 3 (D90 <10ym).
CLIP
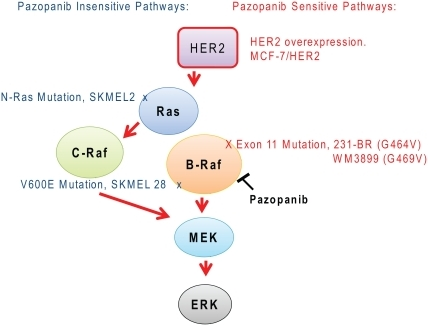
Pazopanib is a highly bio-available, multi- tyrosine kinase inhibitor of vascular endothelial growth factor receptor (VEGFR)-l, -2, -3, platelet-derived factor receptor (PDGFR) -α, -β, cytokine receptor (cKit), interleukin-2 receptor inducible T-cell kinase (Itk), leukocyte-specific protein tyrosine kinase (Lck), and transmembrane glycoprotein receptor tyrosine kinase (c-Fms). Pazopanib was recently approved by the Food and Drug Administration (FDA) for the treatment of patients with advanced renal cell carcinoma; thus adding to the other FDA-approved VEGF pathway inhibitors, sunitinib, bevacizumab (in combination with interferon) and sorafinib for this same indication.
Processes by which pazopanib and its intermediates can be synthesized have been described in US Patent No. 7,105,530 as well as in the published PCT application WO03/106416.
U.S. patent no. 7,105,530 disclosed pyrimidineamines and their derivatives thereof. These compounds are antineoplastic agents, and are useful in the treatment of various cancers and renal cell carcinoma. Among them pazopanib hydrochloride, chemically 5-[4-[N-(2,3-Dimethyl-2H-indazol-6-yl)-N-methylamino]pyrimidin-2- ylamino]-2-methylbenzenesulfonamide hydrochloride. Pazopanib hydrochloride is represented by the following structure:
Pazopanib hydrochloride is a potent and selective multi-targeted receptor tyrosine kinase inhibitor of VEGFR (Vascular endothelial growth factor receptors)- 1, VEGFR-2, VEGFR-3, PDGFR (Platelet-derived growth factor receptors )-a/p, and c-kit that blocks tumor growth and inhibits angiogenesis. It has been approved for renal cell carcinoma by the U.S. Food and Drug Administration. Pazopanib hydrochloride may also be active in ovarian cancer and soft tissue sarcoma. Pazopanib hydrochloride also appears effective in the treatment of non-small cell lung carcinoma. Pazopanib hydrochloride is marketed under the brand name Votrient® by Glaxosmithkline in the form of tablet.
Processes for the preparation of pazopanib hydrochloride and related compounds were disclosed in U.S. patent no. 7,105,530 and U.S. patent no. 7,262,203.
According to U.S. patent no. 7,105,530, pazopanib hydrochloride can be prepared by reacting the N-(2-chloropyrimidin-4-yl)-N,2,3-trimethyl-2H-indazol-6-amine with 5- amino-2-methylbenzenesulfonamide in the presence of hydrochloric acid in isopropanol and ether.
U.S. patent application publication no. 2006/0252943 disclosed a process for the preparation of pazopanib hydrochloride. According to this patent, pazopanib hydrochloride can be prepared by reacting the N-(2-chloropyrimidin-4-yl)-N,2,3- trimethyl-2H-indazol-6-amine with 5-amino-2-methylbenzenesulfonamide in the presence of hydrochloric acid in ethanol or methanol or tetrahydrofuran or acetonitrile and dioxane.

Drugs of the Future, 2006, 31 (7): 585-589
WO0306416
CLIP
Marcus Baumann

, Ian R. Baxendale
, Steven V. Ley
and Nikzad Nikbin
Innovative Technology Centre, Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW Cambridge, UK
Corresponding author email
Editor-in-Chief: J. Clayden
Beilstein J. Org. Chem. 2011, 7, 442–495.
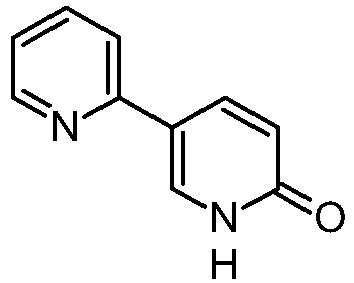
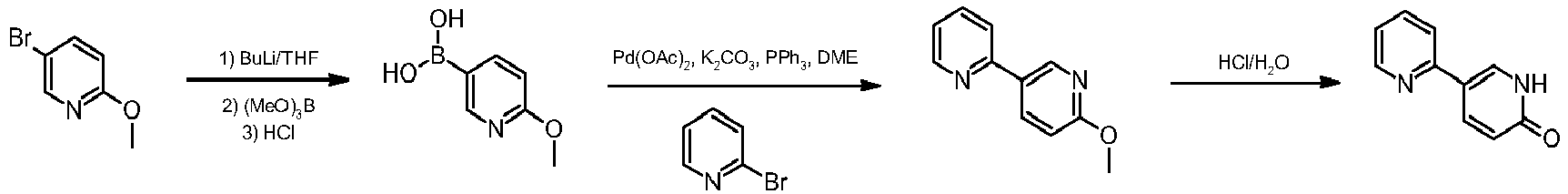
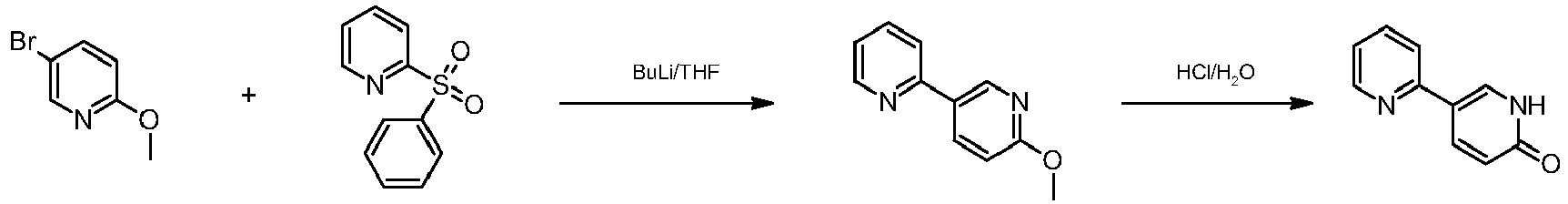
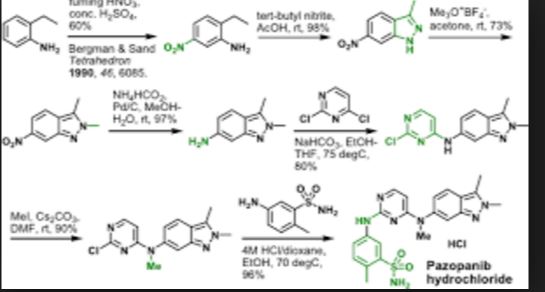
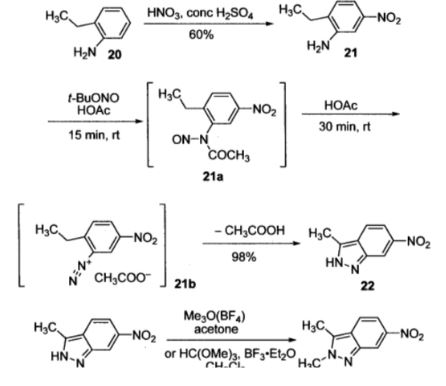
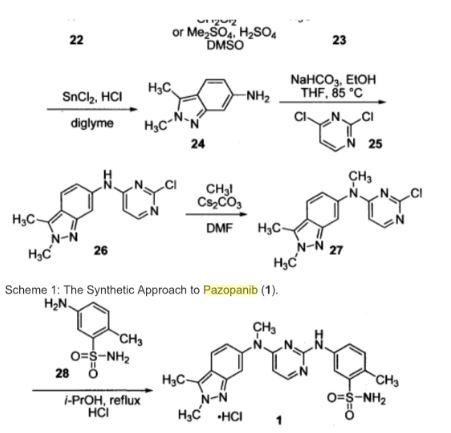
Pazopanib (246, Votrient) is a new potent multi-target tyrosine kinase inhibitor for various human cancer cell lines. Pazopanib is considered a promising replacement treatment to imatinib and sunitinib and was approved for renal cell carcinoma by the FDA in late 2009. The indazole system is built up via diazotisation and spontaneous cyclisation of 2-ethyl-5-nitroaniline (247) using tert-butyl nitrite. The resulting indazole structure 249 can be methylated entirely regioselectively with either Meerwein’s salt, trimethyl orthoformate or dimethyl sulfate. A tin-mediated reduction of the nitro group unmasks the aniline which undergoes nucleophilic aromatic substitution to introduce the pyrimidine system with the formation of 253. Methylation of the secondary amine function with methyl iodide prior to a second SNAr reaction with a sulfonamide-derived aniline affords pazopanib .
Synthesis of pazopanib.
- Pandite, A. N.; Whitehead, B. F.; Ho, P. T. C.; Suttle, A. B. Cancer Treatment Method. WO Patent 2007/064753, June 7, 2007.
- Harris, P. A.; Boloor, A.; Cheung, M.; Kumar, R.; Crosby, R. M.; Davis-Ward, R. G.; Epperly, A. H.; Hinkle, K. W.; Hunter, R. N., III; Johnson, J. H.; Knick, V. B.; Laudeman, C. P.; Luttrell, D. K.; Mook, R. A.; Nolte, R. T.; Rudolph, S. K.; Szewczyk, J. R.; Truesdale, A. T.; Veal, J. M.; Wang, L.; Stafford, J. A. J. Med. Chem.2008,51,4632–4640. doi:10.1021/jm800566m
CLIP

Pazopanib hydrochloride (Votrient)
Pazopanib is a potent and selective multi-targeted receptor tyrosine kinase inhibitor of VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-a/b, and c-kit that blocks tumor growth and inhibits angiogenesis. It was approved for renal cell carcinoma by the U.S. Food
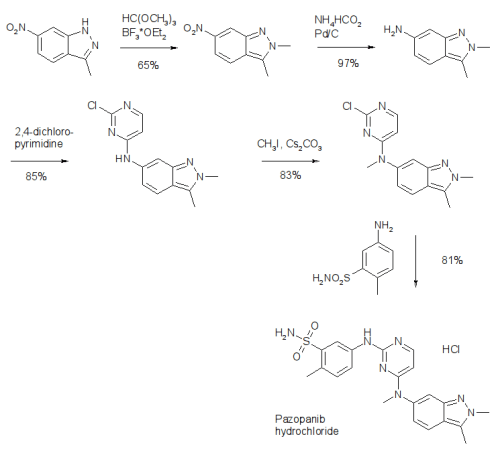
and Drug Administration in 2009 and is marketed under the trade name Votrient by the drug’s manufacturer, GlaxoSmithKline. The
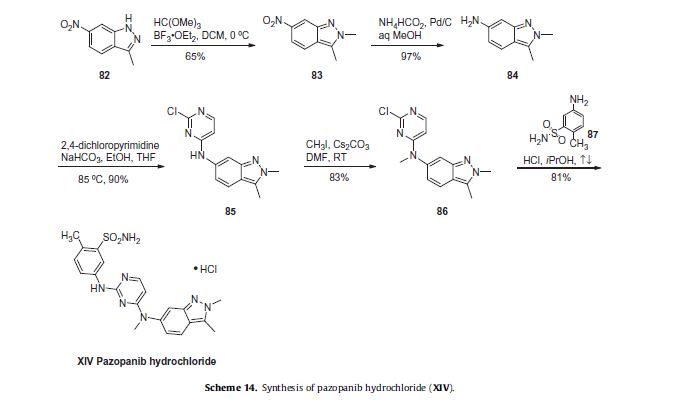
synthesis of pazopanib begins with methylation of 3-methyl-6-nitroindazole (82) with trimethyl orthoformate in the presence of BF3OEt to give indazole 83 in 65% yield (Scheme 14).65 Reduction of the nitro group was achieved via transfer hydrogenation to give 84 in 97% yield, and this was followed by coupling the aniline with 2,4-dichloropyrimidine in a THF-ethanol mixture at elevated
temperature to provide diarylamine 85 in 90% yield. The aniline nitrogen was then methylated using methyl iodide to give 86 in
83% yield prior to coupling with 5-amino-2-methylbenzenesulfonamide (87) and salt formation using an alcoholic solution of
HCl to furnish pazopanib hydrochloride (XIV) in 81% yield.

FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 3 |
| Patent |
7262203 |
| Expiration |
Dec 19, 2021 |
| Applicant |
NOVARTIS PHARMS CORP |
| Drug Application |
- N022465 (Discontinued Drug: VOTRIENT. Ingredients: PAZOPANIB HYDROCHLORIDE)
- N022465 (Prescription Drug: VOTRIENT. Ingredients: PAZOPANIB HYDROCHLORIDE)
|
| FDA Orange Book Patents: 2 of 3 |
| Patent |
8114885 |
| Expiration |
Dec 19, 2021 |
| Applicant |
NOVARTIS PHARMS CORP |
| Drug Application |
- N022465 (Discontinued Drug: VOTRIENT. Ingredients: PAZOPANIB HYDROCHLORIDE)
- N022465 (Prescription Drug: VOTRIENT. Ingredients: PAZOPANIB HYDROCHLORIDE)
|
| FDA Orange Book Patents: 3 of 3 |
| Patent |
7105530 |
| Expiration |
Oct 19, 2023 |
| Applicant |
NOVARTIS PHARMS CORP |
| Drug Application |
- N022465 (Discontinued Drug: VOTRIENT. Ingredients: PAZOPANIB HYDROCHLORIDE)
- N022465 (Prescription Drug: VOTRIENT. Ingredients: PAZOPANIB HYDROCHLORIDE)
|
VOTRIENT (pazopanib) is a tyrosine kinase inhibitor (TKI). Pazopanib is presented as the hydrochloride salt, with the chemical name 5-[[4-[(2,3-dimethyl-2H-indazol-6- yl)methylamino]-2-pyrimidinyl]amino]-2-methylbenzenesulfonamide monohydrochloride. It has the molecular formula C21H23N7O2S•HCl and a molecular weight of 473.99. Pazopanib hydrochloride has the following chemical structure:
Pazopanib hydrochloride is a white to slightly yellow solid. It is very slightly soluble at pH 1 and practically insoluble above pH 4 in aqueous media.
Tablets of VOTRIENT are for oral administration. Each 200 mg tablet of VOTRIENT contains 216.7 mg of pazopanib hydrochloride, equivalent to 200 mg of pazopanib free base. The inactive ingredients of VOTRIENT are:Tablet Core: Magnesium stearate, microcrystalline cellulose, povidone, sodium starch glycolate. Coating: Gray film-coat: Hypromellose, iron oxide black, macrogol/polyethylene glycol 400 (PEG 400), polysorbate 80, titanium dioxide.
- FierceBiotech. 2008-09-15. Retrieved 2010-08-10.
|
Country
|
Patent Number
|
Approved
|
Expires (estimated)
|
| United States |
7105530 |
2009-10-19 |
2023-10-19 |
| United States |
7262203 |
2009-10-19 |
2021-12-19 |
| United States |
8114885 |
2009-10-19 |
2021-12-19 |
|
JUNE 4 2013 old article cut paste
GlaxoSmithKline’s (GSK) Votrient (pazopanib) has met the primary objective of a statistically significant improvement in the time to disease progression or death that is the progression-free survival (PFS) against placebo in Phase III ovarian cancer..
http://clinicaltrials.pharmaceutical-business-review.com/news/gsks-votrient-meets-primary-objective-in-phase-iii-ovarian-cancer-trial-030613

Pazopanib shrinks lung cancers before surgery
FORMULATION
https://www.google.co.in/patents/US20140255505
Pazopanib is an angiogenesis inhibitor targeting vascular endothelial growth factor receptors (VEGFR)-1, -2, and -3, platelet-derived growth factor receptors (PDGFR)-α/-β, and c-Kit. The hydrochloride salt of pazopanib (5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methylbenzenesulfonamide) is marketed by GlaxoSmithKline as Votrient®, which is approved in the United States and other countries for the treatment of renal cell carcinoma (RCC).
Votrient® is currently prescribed to adults in the form of 200 mg tablets for oral administration, with each 200 mg tablet containing an amount of pazopanib hydrochloride equivalent to 200 mg of pazopanib free base.
Though the current tablets are acceptable of use in adults, the tablets are not preferred for use in potential future use for administering pazopanib to children. In pediatric populations, it is often desired that drug be available as a powder for reconstitution to an oral suspension. Manufacture of such a powder requires dry blending of various excipients with the active substance to provide good flow properties and content uniformity of the powder blend.
Several additional challenges exist concerning the use of pazopanib in a pediatric formulation. For instance, the nature of the drug substance favors conversion from the hydrochloride salt to the free base and hydrate forms in an aqueous environment such that standard formulations fail to provide adequate suspension stability at long term storage conditions of 25° C./65% RH or room temperature. Further, the drug has been found to have a bitter taste and, therefore, taste masking is critical.It is desired to invent a pediatric formulation of pazopanib hydrochloride suitable for administration to a pediatric population
References
- “Votrient (pazopanib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 27 January 2014.
- “VOTRIENT (pazopanib hydrochloride) tablet, film coated [GlaxoSmithKline LLC]”(PDF). DailyMed. GlaxoSmithKline LLC. November 2013. Retrieved 27 January 2014.
- “Votrient : EPAR – Product Information” (PDF). European Medicines Agency. Glaxo Group Ltd. 23 January 2014. Retrieved 27 January 2014.
- “Votrient 200 mg and 400 mg film coated tablets – Summary of Product Characteristics (SPC)”. electronic Medicines Compendium. GlaxoSmithKline UK. 20 December 2013. Retrieved 27 January 2014.
- “PRODUCT INFORMATION VOTRIENT® TABLETS” (PDF). TGA eBusiness Services. GlaxoSmithKline Australia Pty Ltd. 25 March 2013. Retrieved 27 January 2014.
- “Pharmaceutical Benefits Scheme (PBS) – Pazopanib”. Pharmaceutical Benefits Scheme. Australian Government. Retrieved 27 January 2014.
- “Pazopanib – Online Pharmaceutical Schedule”. Pharmaceutical Management Agency. Retrieved 9 June 2015.
- ^ “Pazopanib shows encouraging activity in several tumour types, including soft tissue sarcoma and ovarian cancer”. FierceBiotech. 2008-09-15. Retrieved 2010-08-10.
- “GSK pulls bid to extend use of kidney drug to ovarian cancer”. Reuters. 31 March 2014. Retrieved 7 April 2014.
- “Regulatory update: Votrient (pazopanib) as maintenance therapy for advanced ovarian cancer in the EU”. GlaxoSmithKline. 31 March 2014. Retrieved 7 April 2014.
- Zivi, A; Cerbone, L; Recine, F; Sternberg, CN (September 2012). “Safety and tolerability of pazopanib in the treatment of renal cell carcinoma”. Expert Opinion on Drug Safety. 11 (5): 851–859. doi:10.1517/14740338.2012.712108. PMID 22861374.
- Khurana V, Minocha M, Pal D, Mitra AK (March 2014). “Role of OATP-1B1 and/or OATP-1B3 in hepatic disposition of tyrosine kinase inhibitors.”. Drug Metabol Drug Interact. 0 (0): 1–11. doi:10.1515/dmdi-2013-0062. PMID 24643910.
- Khurana V, Minocha M, Pal D, Mitra AK (May 2014). “Inhibition of OATP-1B1 and OATP-1B3 by tyrosine kinase inhibitors.”. Drug Metabol Drug Interact. 0 (0): 1–11.doi:10.1515/dmdi-2014-0014. PMID 24807167.
- Verweij, J; Sleijfer, S (May 2013). “Pazopanib, a new therapy for metastatic soft tissue sarcoma”. Expert Opinion on Pharmacotherapy. 14 (7): 929–935.doi:10.1517/14656566.2013.780030. PMID 23488774.
- Schöffski, P (June 2012). “Pazopanib in the treatment of soft tissue sarcoma”. Expert Review of Anticancer Therapy. 12 (6): 711–723. doi:10.1586/era.12.41.PMID 22716487.
- Pick, AM; Nystrom, KK (March 2012). “Pazopanib for the treatment of metastatic renal cell carcinoma”. Clinical Therapeutics. 34 (3): 511–520.doi:10.1016/j.clinthera.2012.01.014. PMID 22341567.
- Rimel, BJ (April 2015). “Antiangiogenesis agents in ovarian cancer”. Contemporary Oncology. 7 (2): 16–19. PMID 21638926.
| WO2003106416A2 * |
Jun 17, 2003 |
Dec 24, 2003 |
Smithkline Beecham Corporation |
Chemical process |
| WO2005054212A2 * |
Nov 26, 2004 |
Jun 16, 2005 |
Ciba Specialty Chemicals Holding Inc. |
Electroluminescent device |
| WO2007064752A2 |
Nov 29, 2006 |
Jun 7, 2007 |
Smithkline Beecham Corporation |
Treatment of ocular neovascular disorders such as macular degeneration, angiod streaks, uveitis and macular edema |
| WO2011069053A1 |
Dec 3, 2010 |
Jun 9, 2011 |
Teva Pharmaceutical Industries Ltd. |
Process for the preparation of pazopanip hcl and crystalline forms of pazopanib hcl |
| WO2012051659A1 * |
Oct 20, 2011 |
Apr 26, 2012 |
Biota Scientific Management Pty Ltd |
Viral polymerase inhibitors |
| US7105530 |
Dec 19, 2001 |
Sep 12, 2006 |
Smithkline Beecham Corporation |
Pyrimidineamines as angiogenesis modulators |
| US20060252943 * |
Jun 17, 2003 |
Nov 9, 2006 |
Amogh Boloor |
Chemical process |
| US20080269170 * |
Jan 9, 2008 |
Oct 30, 2008 |
Sanofi-Aventis |
Novel 2,4-Dianilinopyrimidine Derivatives, the Preparation Thereof, Their Use as Medicaments, Pharmaceutical Compositions and, in Particular, as IKK Inhibitors |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2014085373A1 * |
Nov 26, 2013 |
Jun 5, 2014 |
Glaxosmithkline Llc |
Combination |
| CN103232443A * |
Feb 1, 2013 |
Aug 7, 2013 |
天津药物研究院 |
Indazole derivative crystal and its preparation method and use |
| CN104557881A * |
Dec 30, 2014 |
Apr 29, 2015 |
山东博迈康药物研究有限公司 |
Preparation method of pazopanib hydrochloride crystal form |
Boloor, A.; et. al. Pyrimidineamines as angiogenesis modulators. US7105530B2
2. Boloor, A.; Harris, P. A.; et. al. Discovery of 5-[[4-[(2,3-Dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a Novel and Potent Vascular Endothelial Growth Factor Receptor Inhibitor. J Med Chem 2008, 51(15), 4632–4640.
3. Bhanushali, D. S.; et. al. Compositions and processes. WO2011050159A1
4. Boloor, A.; et. al. Chemical Process. WO2003106416A2
5. Pandite, A. M.; et. al. Cancer treatment method. WO2007064753A2

////////////PAZOPANIB, GW786034, Votrient, Armala, GW 786034, GW-786034, GW786034GW786034, VOTRIENT, Pazopanib hydrochloride, FDA 2009, Antineoplastic, Tyrosine Kinase Inhibitors, Protein Kinase Inhibitors, Renal Cell Carcinoma Therpay, Soft Tissue Sarcoma Therapy, パゾパニブ塩酸塩 , Пазопаниба Гидрохлорид
O=S(=O)(N)c1c(ccc(c1)Nc2nccc(n2)N(c4ccc3c(nn(c3C)C)c4)C)C

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO







































![[1860-5397-7-57-i33]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i33.png)
![[1860-5397-7-57-i34]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i34.png)


























![[1860-5397-7-57-i50]](http://beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i50.png?scale=2.0&max-width=1024&background=FFFFFF)






![[1860-5397-7-57-i20]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i20.png)
![[1860-5397-7-57-i21]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i21.png)
![[1860-5397-7-57-i22]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i22.png)
![[1860-5397-7-57-i23]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i23.png)