
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

TEDIGLUTIDE ..Glucagon-like peptide 2 (GLP-2) analog; protects small intestinal stem cells from radiation damage.


TEDUGLUTIDE
Glucagon-like peptide 2 (GLP-2) analog; protects small intestinal stem cells from radiation damage.
Gattex (teduglutide) is a recombinant analog of human glucagon-like peptide 2 for the treatment of adults with short bowel syndrome.
- (Gly2)GLP-2
- ALX 0600
- ALX-0600
- Gattex
- Gly(2)-GLP-2
- Teduglutide
- UNII-7M19191IKG
[Gly2]hGLP-2, [Gly2]-hGLP-2, ALX-0600,
Gattex, Revestive
| CAS number | 197922-42-2 |
|---|
L-histidylglycyl-L-α-aspartylglycyl-L-seryl-L-phenylalanyl-L-seryl-L-α-aspartyl-L-α-glutamyl-L-methionyl-L-asparaginyl-L-threonyl-L-isoleucyl-L-leucyl-L-α-aspartyl-L-asparaginyl-L-leucyl-L-alanyl-L-alanyl-L-arginyl-L-α-aspartyl-L-phenylalanyl-L-isoleucyl-L-asparaginyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-glutaminyl-L-threonyl-L-lysyl-L-isoleucyl-L-threonyl-L-aspartic acid
| Formula | C164H252N44O55S |
|---|---|
| Mol. mass | 3752.082 g/mol |
Gattex, ALX-0600, (Gly2)GLP-2, Gly(2)-GLP-2, ALX 0600, [Gly2]GLP-2, Glucagon-like peptide II (2-glycine) (human), UNII-7M19191IKG
LAUNCHED 2013, NPS Pharmaceuticals
APPROVAL FDA
Company: NPS Pharmaceuticals, Inc.
Date of Approval: December 21, 2012 FDA
NDA 203441
POWDER; SUBCUTANEOUS GATTEX
U-1320=TREATMENT OF ADULT PATIENTS WITH SHORT BOWEL SYNDROME WHO ARE DEPENDENT ON PARENTERAL SUPPORT
| Patent No | Patent Expiry Date | Patent use code |
|---|---|---|
| 5789379 | Apr 14, 2015 | U-1320 |
| 7056886 | Sep 18, 2022 | U-1320 |
| 7847061 | Nov 1, 2025 | U-1320 |
| Exclusivity Code | Exclusivity_Date |
|---|---|
| ORPHAN DRUG EXCLUSIVITY | Dec 21, 2019 |
| NEW CHEMICAL ENTITY | Dec 21, 2017 |
SEE FDA
http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203441Orig1s000lbl.pdf
CLINICAL TRIALS
http://clinicaltrials.gov/search/intervention=Teduglutide+OR+ALX-0600
The active ingredient in GATTEX (teduglutide [rDNA origin]) for injection is teduglutide (rDNA origin), which is a 33 amino acid glucagon-like peptide-2 (GLP-2) analog manufactured using a strain of Escherichia coli modified byrecombinant DNA technology. The chemical name of teduglutide is L-histidyl-L-glycyl-L-aspartyl-L-glycyl-L-seryl-L-phenylalanyl-L-seryl-L-aspartyl-L-glutamyl-L-methionyl-L-asparaginyl-L-threonyl-L-isoleucyl-L-leucyl-L-aspartyl-L-asparaginyl-L-leucyl-L-alanyl-L-alanyl-L-arginyl-L-aspartyl-L-phenylalanyl-L-isoleucyl-L-asparaginyl-L-tryptophanyl-L-leucyl-L-isoleucyl-L-glutaminyl-L-threonyl-L-lysyl-L-isoleucyl-L-threonyl-L-aspartic acid. The structural formula is:
Figure 1: Structural formula of teduglutide

Teduglutide has a molecular weight of 3752 Daltons. Teduglutide drug substance is a clear, colorless to light-straw–colored liquid.
Each single-use vial of GATTEX contains 5 mg of teduglutide as a white lyophilized powder for solution for subcutaneous injection. In addition to the active pharmaceutical ingredient (teduglutide), each vial of GATTEX contains 3.88 mg L-histidine, 15 mg mannitol, 0.644 mg monobasic sodium phosphate monohydrate, 3.434 mg dibasic sodium phosphate heptahydrate as excipients. No preservatives are present.
At the time of administration the lyophilized powder is reconstituted with 0.5 mL of Sterile Water for Injection, which is provided in a prefilled syringe. A 10 mg/mL sterile solution is obtained after reconstitution. Up to 0.38 mL of the reconstituted solution which contains 3.8 mg of teduglutide can be withdrawn for subcutaneous injection upon reconstitution.
Teduglutide (brand names Gattex and Revestive) is a 36-membered polypeptide andglucagon-like peptide-2 analog that is used for the treatment of short bowel syndrome. It works by promoting mucosal growth and possibly restoring gastric emptying and secretion.[1] In Europe it is marketed under the brand Revestive by Nycomed. It was approved by the United States under the name Gattex on December 21, 2012.
Teduglutide is a proprietary analogue of glucagon-like peptide 2 (GLP-2) which was approved in the U.S. in December 2012 for the once-daily treatment of short-bowel syndrome in adults who are dependent on parenteral support. Commercial launch took place in 2013.The product was filed for approval in the E.U. in 2011 by Nycomed for this indication. In June 2012, a positive opinion was received in the E.U. and final approval was assigned in September 2012.
At NPS Pharmaceuticals, the compound is in phase III clinical development for this indication in pediatric patients and in phase II clinical studies for the treatment of Crohn’s disease. Preclinical studies are also ongoing at the company for the treatment of chemotherapy-induced enterocolitis and for the prevention and treatment of necrotizing enterocolitis (NEC) in preterm infants.
Teduglutide has been found to induce intestinal hyperplasia, reduce apoptosis and inflammation and improve cell barrier integrity in animal models. In 2001, orphan drug designation was assigned to teduglutide for the treatment of short-bowel syndrome.
In 2007, the compound was licensed to Nycomed for development and commercialization outside the U.S., Canada and Mexico for the treatment of gastrointestinal disorders. In 2012, the product was licensed to Neopharm by NPS Pharmaceuticals in Israel for development and commercialization for the treatment of gastrointestinal disorders.
The estimated prevalence of short bowel syndrome (SBS) patients with non-malignant disease requiring home parenteral nutrition (HPN) is at least 40 per million of the U.S. population. SBS usually results from surgical resection of some or most of the small intestine for conditions such as Crohn’s disease, mesenteric infarction, volvulus, trauma, congenital anomalies, and multiple strictures due to adhesions or radiation. Surgical resection may also include resection of all or part of the colon. SBS patients suffer from malabsorption that may lead to malnutrition, dehydration and weight loss. Some patients can maintain their protein and energy balance through hyperphagia; more rarely they can sustain fluid and electrolyte requirements to become independent from parenteral fluid.
Although long-term parenteral nutrition (PN) is life saving in patients with intestinal failure, it is expensive, impairs quality of life and is associated with serious complications such as catheter sepsis, venous occlusions and liver failure. Treatments that amplify absolute intestinal absorption, and eliminate or minimize the need for PN have great potential significance to SBS patients.
The endogenous meal-stimulated hormone, glucagon-like peptide-2 (GLP-2), raises considerable interest for SBS patients. GLP-2 functions to slow gastric emptying, reduce gastric secretions, increase intestinal blood-flow and stimulate growth of the small and large intestine. In animal studies, GLP-2 administration induces mucosal epithelial proliferation in the stomach and small and large intestine by stimulation of crypt cell proliferation and inhibition of enterocyte apoptosis.
SBS patients with end-jejunostomy and no colon have low basal GLP-2 levels and limited meal-stimulated GLP-2 secretion due to removal of GLP-2 secreting L-cells, which are located primarily in the terminal ileum and colon. This GLP-2 deficiency results in a minimal adaptive response following resection and could explain the gastric hypersecretion, rapid intestinal transit and lack of intestinal adaptation observed in these SBS patients.
Jeppesen et al. (Gastroenterology 2001; 120:806-815) have described positive benefit in an open-label study using pharmacologic doses of native GLP-2 in SBS jejunostomy patients. There was significant improvement in intestinal wet weight absorption and a more modest improvement in energy absorption that led to an increase in body weight, lean body mass and a rise in urinary creatinine excretion.
In contrast, SBS patients with colon-in-continuity have elevated basal endogenous GLP-2 levels resulting in an adaptive response to resection characterized by improved wet weight gain and energy absorption. The potential for added benefit of pharmacologic doses of GLP-2 receptor agonists in these patients is not obvious and has not been studied.

TEDUGLUTIDE
- Jeppesen PB (May 2012). “Teduglutide, a novel glucagon-like peptide 2 analog, in the treatment of patients with short bowel syndrome”. Therap Adv Gastroenterol 5 (3): 159–71. doi:10.1177/1756283X11436318. PMC 3342570. PMID 22570676.
- US 2013157954
- WO 2006050244
- WO 2005021022
- US 6586399
- WO 2002066062
- US 6297214
- US 2001021767
- WO 2001041779
- WO 1999058144
- WO 1998052600
Gattex Approved By FDA For Short Bowel Syndrome
 Gattex (teduglutide) has been approved by the U.S. Food and Drug Administration to be used in patients that have short bowel syndrome and require parenteral nutrition.
Gattex (teduglutide) has been approved by the U.S. Food and Drug Administration to be used in patients that have short bowel syndrome and require parenteral nutrition.
The drug, once it is in the market, will compete against two others that have been approved by the FDA for this type of patient population. Those two medications are Nutrestore (glutamine) and Zorbtive (Somatropin).
Short bowel syndrome comes on following the removal surgically of part of the large or small intestine or part of both. Patients who are affected must have parenteral nutrition due to the poor absorption they have of nutrients and fluids. Teduglutide is injected one time each day and improves the absorption making it less important to have nutrition assistance.
The advisory committee for the FDA voted unanimously in October to recommend the drug’s approval after seeing the results from a pair of clinical trials that showed the advantage teduglutide had over just a placebo in at least a reduction of 20% in the amount of parenteral nutrition at 6 months.
During the first clinical trial, 46% of the patients that took the drug saw a level of reduction, which was compared to only 6% who had taken only a placebo. In the other study, the figure increased to 63%, while the placebo rated was up to 30%
The side effects most common found in those who use teduglutide during the trials included nausea, reactions around the injection site, abdominal pain abdominal distension and headaches.
………..
| US5789379 | Jun 28, 1996 | Aug 4, 1998 | 1149336 Ontario Inc. | Glucagon-like peptide-2 analogs |
| US6077949 | Apr 24, 1997 | Jun 20, 2000 | Allelix Biopharmaceuticals, Inc. | Cloned glucagon-like peptide 2 receptors |
| US6184201 * | Apr 8, 1997 | Feb 6, 2001 | Nps Allelix Corp. | Intestinotrophic glucagon-like peptide-2 analogs |
| US7411039 | Oct 14, 2003 | Aug 12, 2008 | Novo Nordisk A/S | GLP-2 compounds, formulations, and uses thereof |
| EP1231219A1 | Apr 11, 1997 | Aug 14, 2002 | 1149336 Ontario Inc. | GLucagon-like peptide-2 analogs |
| WO1997039031A1 | Apr 11, 1997 | Oct 23, 1997 | Allelix Biopharma | Glucagon-like peptide-2 analogs |
| WO1997039091A1 | Apr 16, 1997 | Oct 23, 1997 | Burckett St Laurent James Char | Mid-chain branched surfactants |
| WO2002066511A2 | Feb 15, 2002 | Aug 29, 2002 | Conjuchem Inc | Long lasting glucagon-like peptide 2 (glp-2) for the treatment of gastrointestinal diseases and disorders |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
FDA grants fast-track status to Peregrine’s bavituximab for lung cancer treatment
FDA grants fast-track status to Peregrine’s bavituximab for lung cancer treatment
The US Food and Drug Administration (FDA) has granted fast-track designation for Peregrine Pharmaceuticals’ lead investigational immunotherapy ‘bavituximab’ to treat patients with second-line non-small cell lung cancer (NSCLC).

AZASETRON..a selective 5-HT3 receptor antagonist
AZASETRON, NAZASETRON
N-(1-azabicyclo[2.2.2]octan-8-yl)-6-chloro-4-methyl-3-oxo-1,4-benzoxazine-8-carboxamide
6-chloro-3,4-dihydro-4-methyl-3-oxo-N-(3-quinuclidinyl)-2H-1,4-benzoxazine-8-carboxamide
123039-99-6 , 123040-69-7 (free base)
141922-90-9 HYD SALT
Y-25130 (hydrochloride)LAUNCHED 1994 AS HCL SALT FORM SEROTONE
a selective 5-HT3 receptor antagonist , ANTIEMETIC
- UNII-77HC7URR9Z
Mitsubishi Tanabe Pharma, JAPAN TOBACCO (Originator)
FOR..Nausea and Vomiting, Treatment of
Prokinetic Agents

Azasetron is an antiemetic which acts as a 5-HT3 receptor antagonist.
Chemical and Pharmaceutical Bulletin, 1992 , vol. 40, 3 p. 624 – 630, ENTRY 15 , MP 305 OF HCL SALT
Biological and Pharmaceutical Bulletin, 2006 , vol. 29, 9 p. 1931 – 1935, AS MERCK 903
US 4892872…
CN 101786963…
WO 1996001630..
WO 2006006595..
WO 2007134077..
CN 102526740, CN 102451166, US 4892872, JP 2008297277
US5773436 A1, WO2006/119295 A2, EP1336602 A1, US4892872 A1,
US2003/18008 A1, US2002/147197 A1, US4892872



The nitration of 5-chloro-2-hydroxybenzoic acid methyl ester (I) with nitric acid in sulfuric acid gives 5-chloro-2-hydroxy-3-nitrobenzoic acid methyl ester (II), which is reduced with Fe and NH4Cl in water yielding 3-amino-5-chloro-2-hydroxybenzoic acid methyl ester (III). The cyclization of (III) with chloroacetyl chloride (IV) by means of NaHCO3 in CHCl3 – water affords 6-chloro-3-oxo-3,4-dihydro-2H-1,4-benzoxazine-8-carboxylic acid methyl ester (V), which is methylated with methyl iodide and K2CO3 in DMF affording the corresponding 4-methyl derivative (VI). Hydrolysis of (VI) with ethanolic NaOH gives the corresponding acid (VII), which by treatment with refluxing SOCl2 is converted into its acyl chloride (VIII). Finally, this compound is condensed with 3-aminoquinuclidine (IX) by means of N-methylmorpholine (NMM) in CHCl3.
………………
SYNTHESIS
Azasetron hydrochloride (Azasetron hydrochloride) is a secondary cancer drugs, mainly synthetic route is as follows
[0003]

[0004] In EP 0313393; JP 1989207290; JP 1990005415; US 4892872; Eur.Pat.Appl., 313393,, Chem pharm Bull, 1992,40 (3) :624-630 reported the synthesis of intermediates III is with ammonium chloride, iron as the reducing agent, the nitro substrate was dissolved in toluene and added dropwise to a reduction of the media inside, the study found that this approach has a number of disadvantages, notably the reaction can not be completely, while post-processing is very troublesome, purity of the product obtained is poor, and difficult purification. Although the “Chinese Medicinal Chemistry” magazine, 10 (2) ,138-140; 2000, the author hydrochloride instead of ammonium chloride, but still need the nitro group was dissolved in toluene was added dropwise, in fact, the nitro compound in toluene the solubility is limited, and in the process will precipitate dropwise addition, there are also incomplete reaction, impurities, and complicated post-treatment problems.
[0005] In the “China Pharmaceutical Industry” magazine 34 (3) ,214-215; 2003 in order to restore hydrosulfite as reductant,
Azasetron preparation of intermediates, the steps are as follows:
[0015] (I) A mixture of 3 – nitro-5 – chloro-salicylate added glacial acetic acid was stirred for 30-40 minutes, add water, temperature 30 ~ 100 ° C, adding iron powder, 2.5-3 hour plus finished, the addition was completed, at a temperature of 30 ~ 100 ° C the reaction for 5 to 10 hours; give 3 – amino-5-chloro-salicylate mixture;
[0016] The mass ratio of the added material is 3 – nitro-5 – chloro-salicylate: acetic acid: water: iron = 1: 5 ~ 10: 5 ~ 10: 0.5 ~ I;
[0017] (2) obtained in the step ⑴ added to a mixture of glacial acetic acid, glacial acetic acid added in the amount of step ⑴ same amount of glacial acetic acid, 70 ° C was stirred for 30-35 minutes, filtered through celite to remove the iron sludge to obtain filter cake; this procedure to that step (I) the resulting product was dissolved in glacial acetic acid solvent sufficient, in order to facilitate post-processing; while removing impurities.
[0018] (3) The filter cake was washed with an organic solvent, the combined filtrate and stirred for 10-15 minutes, allowed to stand for 30-35 minutes, the organic layer was separated; aqueous phase was extracted once again with an organic solvent, the combined organic solution was washed three times , mention made three – amino -5 chloro methyl salicylate (intermediate III) sulphate solution.
[0019] (4) of step (3) of 3 – amino-5-chloro-salicylate was added a saturated NaHCO3 solution, saturated NaHCO3 solution and the amount of acetic acid in step ⑴ same volume, cooling to _1 ° C ~ 0 ° C, control the temperature -1 ° C_5 ° C was added dropwise acetyl chloride, drop Bi. Reaction 2-3 hours. To give 3 – (2 – chloro-acetylamino)-5-chloro-mixed solution of methyl salicylate;
[0020] The amount of chloroacetyl chloride is added in step ⑴ source material 3 – nitro-5 – chloro molar ratio of methyl salicylate
I: 0.9-0.95.
[0021] (5) obtained in the step ⑷ 3 – (2 – chloro-acetylamino) methyl salicylate _5 chloride mixture was heated to 30-40 ° C, stirred for 1-1.5 hours; organic layer was separated, water phase extracted once again with an organic solvent, the combined organic layer was washed with water three times, separated and the organic layer is directly subjected to atmospheric distillation, the organic solvent is distilled off, distillation was complete, methanol or ethanol as the crystallization solvent, 80 ° C under stirring for 2.5 hours under reflux , to give 3 – (2 – chloro-acetylamino) _5 chlorine in alcohol solution of methyl salicylate. This step is intended to 3 – (2 – chloro-acetylamino)-5-chloro methyl salicylate purification.
[0022] (6) in the step (5) cooling to room temperature, an alcoholic solution, crystallization, centrifugation, and washed with ethanol crystal; 80 ° C drying in needle-like crystals 3 – (2 – chloro-acetylamino) _5 Chlorine methyl salicylate (intermediate IV).
……………………………………………………
USEFUL PATENTS
|
6-31-1998
|
Use of serotonin antagonists for treating fibromyalgia
|
|
|
11-28-1997
|
COMPOSITIONS COMPRISING CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND TAXOTERE
|
|
|
11-28-1997
|
DHA-PHARMACEUTICAL AGENT CONJUGATES DHA-PHARMACEUTICAL AGENT CONJUGATES
|
|
|
11-28-1997
|
CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND PACLITAXEL
|
|
|
3-28-1997
|
5-HT3 RECEPTOR ANTAGONISTS FOR DYSKINESIA
|
|
|
2-19-1997
|
Nasally administrable compositions
|
|
|
1-26-1996
|
PREVENTIVE OR REMEDY FOR IRRITABLE BOWEL SYNDROME OR DIARRHEA
|
|
|
3-17-1994
|
Benzoxazine compounds and pharmaceutical use thereof.
|
|
|
1-10-1990
|
Benzoxazine compounds and pharmaceutical use thereof
|
|
12-17-1999
|
MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT3 RECEPTORS MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT>3< RECEPTORS MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT3 RECEPTORS
|
|
|
11-17-1999
|
Use of serotonin antagonists for treating fibromyalgia
|
|
|
11-5-1999
|
CNRE BINDING FACTORS AND USES THEREOF
|
|
|
7-23-1999
|
TRANSGLUTAMINASE LINKAGE OF AGENTS TO TISSUE TRANSGLUTAMINASE LINKAGE OF AGENTS TO TISSUE
|
|
|
7-7-1999
|
Taxane compounds and compositions
|
|
|
6-25-1999
|
ORAL DELIVERY FORMULATION
|
|
|
4-16-1999
|
MEDICAMENTS MEDICAMENTS
|
|
|
2-5-1999
|
CHEMOTHERAPY SYNERGISTIC AGENT
|
|
|
10-30-1998
|
USE OF 5HT3 ANTAGONISTS FOR PROMOTING INTESTINAL LAVAGE
|
|
|
8-19-1998
|
DHA-pharmaceutical agent conjugates of taxanes
|
|
8-7-2009
|
ALISKIREN MODULATION OF NEUROGENESIS
|
|
|
7-3-2003
|
USE OF 5HT3 ANTAGONISTS FOR PROMOTING INTESTINAL LAVAGE
|
|
|
9-18-2002
|
Inhibition of emetic effect of metformin with 5-HT3 receptor antagonists
|
|
|
11-8-2001
|
CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND PACLITAXEL
|
|
|
6-14-2001
|
Nasally administrable compositions
|
|
|
12-22-2000
|
RECEPTOR AGONISTS AND ANTAGONISTS COMPOUND FOR USE AS A MEDICAMENT FOR TREATMENT OF DISORDERS INVOLVING BRONCHOCONTRACTION COMPOUND FOR USE AS A MEDICAMENT FOR TREATMENT OF DISORDERS INVOLVING BRONCHOCONTRACTION
|
|
|
12-15-2000
|
PHARMACEUTICAL COMPOSITION FOR INTRANASAL USE OF ACTIVE SUBSTANCES THAT ARE INSOLUBLE AND/OR HARDLY SOLUBLE IN WATER
|
|
|
11-31-2000
|
ANTI-TUMOR COMPRISING BOROPROLINE COMPOUNDS
|
|
|
11-30-2000
|
TRANSGLUTAMINASE LINKAGE OF AGENTS TO TISSUE
|
|
11-17-2000
|
FATTY ACID-N-SUBSTITUTED INDOL-3-GLYOXYL-AMIDE COMPOSITIONS AND USES THEREOF
|
|
|
10-12-2000
|
COMPOSITIONS COMPRISING CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND TAXOTERE
|
|
|
9-15-2000
|
FATTY ACID-ANTICANCER CONJUGATES AND USES THEREOF FATTY ACID-ANTICANCER CONJUGATES AND USES THEREOF
|
|
|
7-20-2000
|
$g(b)2-ADRENERGIC RECEPTOR AGONISTS $g(b)2-ADRENERGIC RECEPTOR AGONISTS
|
|
|
7-7-2000
|
USE OF CD40 ENGAGEMENT TO ALTER T CELL RECEPTOR USAGE USE OF CD40 ENGAGEMENT TO ALTER T CELL RECEPTOR USAGE USE OF CD40 ENGAGEMENT TO ALTER T CELL RECEPTOR USAGE
|
|
|
6-28-2000
|
Taxanes
|
|
|
5-32-2000
|
$g(b)2-ADRENERGIC RECEPTOR AGONISTS
|
|
|
1-14-2000
|
NOVEL INDOLOCARBAZOLE DERIVATIVES USEFUL FOR THE TREATMENT OF NEURODEGENERATIVE DISEASES AND CANCER
|
|
|
1-12-2000
|
Indolocarbazole derivatives useful for the treatment of neurodegenerative diseases and cancer
|
|
|
12-30-1999
|
METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS
|
………..AZASETRON
AZASETRON
NMR OF HCL SALT
http://file.selleckchem.com/downloads/nmr/S210601-Azasetron-hydrochloride-NMR-Selleck.pdf
…………….
SYNTHESIS
EXAMPLE 15
To a solution of 3.0 g of 3-aminoquinuclidine and 3.0 g of N-methylmorpholine in 60 ml of chloroform is added 6.2 g of 6-chloro-3,4-dihydro-4-methyl-3-oxo-2H-1,4-benzoxazine-8-carboxylic acid chloride under cooling and stirring followed by stirring for 2 hours. The resultant solution is washed with water, aqueous sodium hydrogen carbonate and then water, and dried over magnesium sulfate. After the solvent is distilled off under reduced pressure, the residue is recrystallized from ethanol-isopropyl ether and treated with ethanolic hydrochloric acid to give 6-chloro-3,4-dihydro-4-methyl-3-oxo-N-(3-quinuclidinyl)-2H-1,4-benzoxazine-8-carboxamide hydrochloride, melting at 281
………..
SYNTHESIS AND + AND – ISOMERS
EXAMPLE 36
A solution of 6 g of 6-chloro-3,4-dihydro-4-methyl-3-oxo-N-(3-quinuclidinyl)-2H-1,4-benzoxazine-8-carboxamide and 2.7 g of D-(-)-tartaric acid in 100 ml of methanol and 200 ml of ethanol is allowed to stand. The precipitated crystals are collected by filtration and recrystallized from methanol repeatedly to give the R-(+) isomer, [α].sub.D.sup.25 =+36.72 (c=1, chloroform), melting at 177 with ethanolic hydrochloric acid, and then the precipitated crystals are collected by filtration and dried to give the R-(+) isomer hydrochloride, [α].sub.D.sup.25 =+1.4 (c=1, water), melting at 309
By using L-(+)-tartaric acid in a similar manner, the R-(-) optical isomer, [α].sub.D.sup.25.5 =-36.76 (c=1, chloroform), melting at 176 [α].sub.D.sup.25.5 =-1.2 (c=1, water), melts at 310
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
Aeterna Zentaris Submits New Drug Application to FDA for Macimorelin Acetate (AEZS-130) for Evaluation of AGHD

Macimorelin
CAS 381231-18-1
Chemical Formula: C26H30N6O3
Exact Mass: 474.23794
Molecular Weight: 474.55480
Elemental Analysis: C, 65.80; H, 6.37; N, 17.71; O, 10.11

945212-59-9 (Macimorelin acetate)
AEZS-130
ARD-07
D-87875
EP-01572
EP-1572
JMV-1843
USAN (ab-26)
MACIMORELIN ACETATE
THERAPEUTIC CLAIM
Diagnostic agent for adult growth hormone deficiency (AGHD)
CHEMICAL NAMES
1. D-Tryptophanamide, 2-methylalanyl-N-[(1R)-1-(formylamino)-2-(1H-indol-3-yl)ethyl]-, acetate (1:1)
2. N2-(2-amino-2-methylpropanoyl-N1-[(1R)-1-formamido-2-(1H-indol-3-yl)ethyl]- D-tryptophanamide acetate
MOLECULAR FORMULA
C26H30N6O3.C2H4O2
MOLECULAR WEIGHT
534.6
SPONSOR
Aeterna Zentaris GmbH
CODE DESIGNATIONS
D-87575, EP 1572, ARD 07
CAS REGISTRY NUMBER
945212-59-9
Macimorelin (also known as AEZS-130, EP-1572) is a novel synthetic small molecule, acting as a ghrelin agonist, that is orally active and stimulates the secretion of growth hormone (GH). Based on results of Phase 1 studies, AEZS-130 has potential applications for the treatment of cachexia, a condition frequently associated with severe chronic diseases such as cancer, chronic obstructive pulmonary disease and AIDS. In addition to the therapeutic application, a Phase 3 trial with AEZS-130 as a…
View original post 2,777 more words
World Drug Tracker: SOVAPREVIR in phase II clinical trials at Achillion for the oral treatment of naive patients with chronic hepatitis C virus genotype 1
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
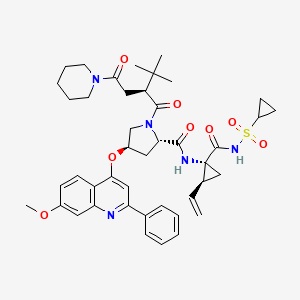
OVAPREVIR
ethenylcyclopropyl]-1-[(2S)-3,3-dimethyl-1-oxo-2-[2-oxo-2-(1-piperidinyl)ethyl]butyl]-4-
[(7-methoxy-2-phenyl-4-quinolinyl)oxy]-, (2S,4R)-
3,3-dimethyl-2-[2-oxo-2-(piperidin-1-yl)ethyl]butanoyl}-4-[(7-methoxy-2-phenylquinolin-
4-yl)oxy]pyrrolidine-2-carboxamide
- ACH-0141625
- Sovaprevir
- UNII-2ND9V3MN6O
THE CURRENT SAFETY DATABASE FOR SOVAPREVIR INCLUDES MORE THAN 560 SUBJECTS DOSED TO DATE AND DEMONSTRATES THAT SOVAPREVIR IS WELL TOLERATED IN THESE SUBJECTS.








WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
NETUPITANT

NETUPITANT
- Ro 67-3189/000
- UNII-7732P08TIR
- Ro-67-3189
- Netupitant, an NK-1 antagonist is under development for the treatment of overactive bladder. HELSINN GROUP
CAS: 290297-26-6
290296-54-7 (di HCl)
U.S. Pat. Nos. 6,303,790, 6,531,597, 6,297,375 and 6,479,483, 6,719,996 and 6,593,472 to Hoffmann La Roche(originator).
IUPAC/Chemical name:
2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide
Chemical Formula: C30H32F6N4O
Exact Mass: 578.24803
Molecular Weight: 578.59
Elemental Analysis: C, 62.28; H, 5.57; F, 19.70; N, 9.68; O, 2.77
Netupitant is another selective NKi receptor antagonist under development by Helsinn Healthcare, having the formula 2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethyl-N-[4-(2- methylphenyl)-6-(4-methylpiperazin- l-yl)pyridin-3-yl]propanamide, or Benzeneacetamide, N,a,a-trimethyl-N-[4-(2-methylphenyl)-6-(4-methyl-l-piperazinyl)-3-pyridinyl]-3,5- bis(trifluoromethyl)-, and the below chemical structure:

Netupitant is a tachykinin NK-1 antagonist which had been in phase III clinical trials at Helsinn for the prophylaxis of chemotherapy-induced nausea and vomiting and in phase II clinical studies for the treatment of overactive bladder. However, no recent development has been reported for this research.
NK-1 receptor antagonists work by blocking the action of neurokinin-1 (Substance P), a naturally-occurring neurotransmitter in the brain that causes emesis. Netupitant was originally developed at Roche. In June 2005, Helsinn and Roche signed a licensing agreement granting Helsinn worldwide rights to the drug candidate.
Methods of synthesizing and formulating netupitant and its prodrugs are described in U.S. Patent Nos. 6,297,375, 6,719,996 and 6,593,472 to Hoffmann La Roche.
Netupitant is a highly selective NK1 receptor antagonist, which is thought to work by blocking the action of substance P, an endogenous neurotransmitter contained in high concentrations in the vomiting center of the brainstem that can stimulate the vomiting reflex. Netupitant is currently under phase III trials.
Chemotherapy is one of the treatment options utilized by oncologists in treating different types of cancers. Nausea and vomiting are the most common side-effects experienced by cancer patients when administered with chemotherapy. Netupitant-palonosetron, which is currently in Phase III trials helps in preventing CINV. The blockage of P/NK1 receptors by Netupitant in the central nervous system inhibits the binding of endogenous tachykinin neuropeptide substance and this result in preventing the chemotherapy-induced nausea and vomiting. Moreover, Palonosetron helps in the blockage of serotonin at 5-hydroxytryptamine type 3 (5-HT3) receptors and it also helps in the chemotherapy-induced nausea and vomiting.

Netupitant-Palonosetron FDC is estimated to answer significant unmet needs of the CINV market post its launch that is expected to be commercialized in 2014, as it would overcome the problems associated with current treatment with 5-HT3 receptor antagonists. Similar to Emend, Netupitant-Palonosetron FDC would gain considerable patient pool after its estimated launch in 2014, and subsequently match the patient share of Aloxi by 2018. Netupitant-Palonosetron FDC sales are expected to reach an estimated USD 515.0 million USD by 2018. FDC combination of 5-HT3 receptor antagonist and neurokinin-1 (NK1) receptor antagonist have shown better efficacy results in Phase II clinical trials for CINV patients and would thus lead to high uptake due to shifting physician and patient preference pattern towards better treatment for CINV.

Neurokinin 1 receptor antagonists are being developed for the treatment of a number of physiological disorders associated with an excess or imbalance of tachykinin, in particular substance P. Examples of conditions in which substance P has been implicated include disorders of the central nervous system such as anxiety, depression and psychosis (WO 95/16679, WO 95/18124 and WO 95/23798).
The neurokinin-1 receptor antagonists are further useful for the treatment of motion sickness and for treatment induced vomiting. The New England Journal of Medicine, Vol. 340, No. 3 190-195, 1999 has been described the reduction of cisplatin-induced emesis by a selective neurokinin-l-receptor antagonist. US5,972,938 describes a method for treating a psychoimmunologic or a psychosomatic disorder by administration of a tachykinin receptor, such as NK-1 receptor antagonist.
With the development of the 5-HT3 antagonist in the early 1990s, there emerged new strategies in the medical community to better control nausea and vomiting caused by various medical procedures, including chemotherapy (CINV), surgery (PONV), and radiation therapy (RINV). When added to steroids such as dexamethasone, several 5-HT3 antagonists have been demonstrated to significantly improve the standard of life for patients undergoing emetogenic medical procedures. Examples of 5-HT3 antagonists include ondansetron, marketed by
GlaxoSmithKline, and palonosetron, developed by Helsinn Healthcare.
Netupitant is another selective NKi receptor antagonist under development by Helsinn Healthcare, having the formula 2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethyl-N-[4-(2- methylphenyl)-6-(4-methylpiperazin- l-yl)pyridin-3-yl]propanamide, or Benzeneacetamide, N,a,a-trimethyl-N-[4-(2-methylphenyl)-6-(4-methyl-l-piperazinyl)-3-pyridinyl]-3,5- bis(trifluoromethyl)-, and the below chemical structure:
Methods of synthesizing and formulating netupitant and its prodrugs are described in U.S. Patent Nos. 6,297,375, 6,719,996 and 6,593,472 to Hoffmann La Roche.
Other representative NKi antagonists include ZD4974 (developed by AstraZeneca), CGP49823 (developed by Ciba-Geigy), Lanepitant and LY686017 (developed by Eli Lilly), FK888 (developed by Fujisawa), Vofopitant, Vestipitant and Orvepitant (developed by
GlaxoSmithKline), Befetupitant (developed by Hoffmann-La Roche), Rl 16031 (developed by Janssen), L-733060 and L-736281 (developed by Merck), TKA731, NKP608 and DNK333 (developed by Novartis), CP-96345, CP-99994, CP- 122721, CJ-17493, CJ-11974 and CJ-11972 (developed by Pfizer), RP67580 and Dapitant (developed by Rhone-Poulenc Rorer),
Nolpitantium and SSR240600 (developed by Sanofi-Aventis), SCH388714 and Rolapitant (developed by Schering-Plough), TAK637 (developed by Takeda), HSP117 (developed by Hisamitsu), KRP103 (developed by Kyorin Pharm) and SLV317 (developed by Solvay).
Chemical structures of the above-mentioned NKi antagonists are shown below and discussion of those compounds as well as other NKi antagonists is present in Expert Opin. Ther. Patents (2010) 20(8), pp 1019- 1045 by Huang et al.
………………………………………………
WO 2013057554
WO 2011061622
WO 2010119347
WO 2003006016
WO 2006002860///
WO 2002085458
US 2002091265…….
…………………………………………………..
http://pubs.acs.org/doi/full/10.1021/jo0523666
…………………………………………..
https://www.google.co.in/patents/US6297375
(2-(3,5-bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide) which has the formula Ib

and to pharmaceutically acceptable acid addition salts thereof.
The compound of formula Ib and its salts is also characterized by valuable therapeutic properties as a highly selective antagonist of the Neurokinin 1 (NK-1, substance P) The present compound of formula lb and its pharmaceutically acceptable salts can be prepared by methods known in the art, for example, by processes described below, which process comprises
a) reacting the compound of formula

with the compound of formula

to the compound of formula



EXAMPLE 14
2-(3,5-Bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide hydrochloride (1:2)
a) 1-Methyl-4-(5-nitro-pyridin-2-yl)-piperazine
To a solution of 20 g (126 mmol) of 2-chloro-5-nitropyridine in 200 ml tetrahydrofuran were added dropwise 35 ml (315 mmol) 1-methylpiperazine within 10 min. The reaction mixture was refluxed for additional 1.5 h. After cooling to room temperature, the solvent was removed in vacuo and the residue was re-dissolved in 200 ml ethyl acetate. The organic phase was washed with 200 ml 1 N sodium bicarbonate solution, dried (magnesium sulfate) and evaporated to give 27.9 g (quantitative) of the title compound as a yellow solid.
MS m/e (%):223 (M+H+, 100).
b)2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-propionamide
To a solution of 27.9 g (125.5 mmol) of 1-methyl-4-(5-nitro-pyridin-2-yl)-piperazine in 400 ml methanol were added 2.6 g of 10% of palladium on activated charcoal. The reaction mixture was hydrogenated (room temperature to ca. 45° C., 1 bar) until the theoretical amount of hydrogen was taken up (about 2 h). The catalyst was filtered off and was washed twice with 100 ml portions of methanol. The filtrate was evaporated in vacuo to give 28 g of a purple oil which consisted to ca. 90% of the desired aniline derivative according to analysis by thin layer chromatography.
This crude product was dissolved in a mixture of 400 ml tetrahydrofuran and 100 ml diethyl ether. After cooling to 0° C., 30 ml (215 mmol) of triethylamine were added in one portion. Stirring was continued while 26 g (215 mmol) of pivaloyl chloride were added dropwise within a period of 10 min. The ice bath was removed and the reaction mixture was stirred for 1 h at room temperature. Then, the solvent was removed in vacuo and the residue was suspended in 200 ml 1 N sodium bicarbonate solution. The product was extracted three times with 200 ml portions of dichloromethane, dried (sodium sulfate) and purified by flash chromatography to give 30 g (86%) of the title compound as pink crystals.
MS m/e (%):277 (M+H+, 100).
c) N-[4-Iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide
A solution of 30 g (108 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-propionamide and 58 ml (380 mmol) N,N,N′,N′-tetramethylethylenediamine under argon in 650 ml tetrahydrofuran was cooled in a dry ice bath to −78° C. Within lh, 239 ml (380 mmol) of a 1.6 N n-butyllithium solution in hexane were added dropwise. The reaction mixture was allowed to warm up to −30° C. overnight. After cooling again to −78° C., 43.6 g (170 mmol) iodine dissolved in 60 ml tetrahydrofuran were added dropwise during 15 min. The dry ice bath was replaced by an ice bath and a solution of 90 g (363 mmol) sodium thiosulfate pentahydrate in 250 ml water were added within 10 min when the temperature of the reaction mixture had reached 0° C. Then, 1000 ml diethyl ether were added and the organic layer was separated. The aqueous layer was extracted twice with 500 ml dichloromethane and the combined organic layers were dried (magnesium sulfate) and evaporated. Flash chromatography gave 18.5 g (42%) of the title compound as a light brown oil which crystallized upon standing at room temperature.
MS m/e (%): 403 (M+H+, 100).
d) 2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide
A mixture of 54 g (134 mmol) N-[4-iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide, 420 ml toluene, 150 ml 2 N sodium carbonate solution, 4.63 g (3.9 mmol) tetrakis(triphenylphosphine)palladium(0) and 20.16 g (147 mmol) o-tolylboronic acid was heated under argon at 80° C. for 12 h. After cooling to room temperature, the aqueous phase was separated and washed twice with toluene. The combined organic layers were washed with 50 ml brine, dried (sodium sulfate), evaporated and dried in vacuo to yield 49 g (quantitative) of the title compound as a brown oil.
MS m/e (%): 367 (M+H+, 100).
e) 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine
A suspension of 56 g (152 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide in 1300 ml 3 N hydrochloric acid solution was heated to 90-95° C. overnight. The reaction mixture was cooled to room temperature, washed with three 500 ml portions diethyl ether and filtered over celite. The filtrate was diluted with 500 ml water and was adjusted to pH 7-8 by addition of 28% sodium hydroxide solution under ice cooling. The product was extracted with four 1000 ml portions of dichloromethane. The combined organic layers were washed with 500 ml brine, dried (magnesium sulfate) and evaporated to give 35 g (82%) of the title compound as a light brown oil.
MS m/e (%):283 (M+H+, 100).
f) Methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine
A solution of 35 g (124 mmol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine in 270 ml trimethyl orthoformate and 8 drops trifluoroacetic acid was heated for 3 h at 130° C. The reaction mixture was evaporated and dried in vacuo for 30 min. The residual oil was dissolved in 100 ml tetrahydrofuran and was added dropwise under ice cooling to 9.4 g (248 mmol) lithium aluminum hydride in 300 ml tetrahydrofuran. The reaction mixture was stirred for lh at room temperature, cooled to 0° C. again and acidified (pH 1-2) by addition of 28% hydrochloric acid solution. After stirring for 5 min, 28% sodium hydroxide solution was added to reach pH 10. The solution was filtered over celite, evaporated and purified by flash chromatography to give 23.6 g (64%) of the title compound as a light brown oil.
MS m/e (%):297 (M+H+, 100).
g) 2-(3,5-Bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide
A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane. The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of the title compound as white crystals. M.p. 155-157° C.
MS m/e (%): 579 (M+H+, 100).
h)2-(3,5-Bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide hydrochloride (1:2)
To a solution of 31.6 g (54.6 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide in 250 ml diethyl ether were added under ice cooling 60 ml 3 N hydrochloric acid solution in diethyl ether. After stirring for 15 min at 0° C., the suspension was evaporated to dryness, re-suspended in 100 ml diethyl ether, filtered and dried in vacuo to give 34.8 g (98%) of the title compound as white crystals. M.p. 235-238° C.
MS m/e (%): 579 (M+H+, 100).
……………………………….
2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (Netupitant)

Other general procedures of preparing similar compounds to intermediate 1 of Scheme 1 are also disclosed in U.S. Pat. Nos. 6,303,790, 6,531,597, 6,297,375 and 6,479,483, the entirety of which are incorporated herein by reference.
Synthesis of methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine

Step 1:
13.0 g (82.5 mMol) 6-Chloro-nicotinic acid in 65 ml THF were cooled to 0° C. and 206.3 ml (206.3 mMol) o-tolylmagnesium chloride solution (1M in THF) were added over 45 minutes. The solution obtained was further stirred 3 hours at 0° C. and overnight at room temperature. It was cooled to −60° C. and 103.8 ml (1.8 Mol) acetic acid were added, followed by 35 ml THF and 44.24 g (165 mMol) manganese(III) acetate dihydrate. After 30 minutes at −60° C. and one hour at room temperature, the reaction mixture was filtered and THF removed under reduced pressure. The residue was partitioned between water and dichloromethane and extracted. The crude product was filtered on silica gel (eluent: ethyl acetate/toluene/formic acid 20:75:5) then partitioned between 200 ml aqueous half-saturated sodium carbonate solution and 100 ml dichloromethane. The organic phase was washed with 50 ml aqueous half-saturated sodium carbonate solution, The combined aqueous phases were acidified with 25 ml aqueous HCl 25% and extracted with dichloromethane. The organic extracts were dried (Na2SO4) and concentrated under reduced pressure to yield 10.4 g (51%) of 6-chloro-4-o-tolyl-nicotinic acid as a yellow foam. MS (ISN): 246 (M−H, 100), 202 (M-CO2H, 85), 166 (36).
Step 2:
To a solution of 8.0 g (32.3 mMol) 6-chloro-4-o-tolyl-nicotinic acid in 48.0 ml THF were added 3.1 ml (42.0 mMol) thionylchloride and 143 .mu.l (1.8 mMol) DMF. After 2 hours at 50° C., the reaction mixture was cooled to room temperature and added to a solution of 72.5 ml aqueous ammonium hydroxide 25% and 96 ml water cooled to 0″C. After 30 minutes at 0° C., THF was removed under reduced pressure and the aqueous layer was extracted with ethyl acetate. Removal of the solvent yielded 7.8 g (98%) 6-chloro-4-o-tolyl-nicotinamide as a beige crystalline foam. MS (ISP): 247 (M+H30 , 100).
Step 3:
1.0 g (4.05 mMol) 6-Chloro-4-o-tolyl-nicotinamidein 9.0 ml 1-methyl-piperazine was heated to 100° C. for 2 hours. The excess N-methyl-piperazine was removed under high vacuum and the residue was filtered on silica gel (eluent: dichloromethane) to yield 1.2 g (95%) 6-(4-methyl-piperazin-1yl)-4-o-tolyl-nicotinamide as a light yellow crystalline foam. MS (ISP): 311 (M+H+, 100), 254 (62).
Step 4:
A solution of 0.2 g (0.6 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 1.0 ml methanol was added to a solution of 103 mg (2.6 mMol) sodium hydroxide in 1.47 ml (3.2 mMol) NaOCl (13%) and heated for 2 hours at 70° C. After removal of methanol, the aqueous layer was extracted with ethyl acetate. The combined. organic extracts were dried (Na2SO4), concentrated under reduced pressure and the residue filtered on silica gel (eluent: dichloromethane/methanol 4:1) to yield 100 mg (70%) 6-(4-methyl-piperazine-1-yl)-4o-tolyl-pyridin-3-ylamine as a brown resin. MS (ISP): 283 (M+H+, 100), 226 (42).
Step 5:
2.15 ml (11.6 mMol) Sodium methoxide in methanol were added over 30 minutes to a suspension of 0.85 g (4.6 mMol) N-bromosuccinimide in 5.0 ml dichloromethane cooled to −5° C. The reaction mixture was stirred 16 hours at −5° C. Still at this temperature, a solution of 1.0 g (3.1 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 5.0 ml methanol was added over 20 minutes and stirred for 5 hours. 7.1 ml (7.1 mMol) Aqueous HCl 1N and 20 ml dichloromethane were added. The phases were separated and the organic phase was washed with deionized water. The aqueous phases were extracted with dichloromethane, brought to pH=8 with aqueous NaOH 1N and further extracted with dichloromethane. The latter organic, extracts were combined, dried (Na2SO4) and concentrated to yield 1.08 g (quant.) [6-(4-methyl-piperazin-1yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester as a grey foam. MS (ISP): 341 (M+H+, 100), 284 (35).
Step 6:
A solution of 0.5 g (1.4 mMol) [6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester in 3.0 ml dichloromethane was added over 10 minutes to a solution of 1.98 ml (6.9 mMol) Red-Al.RTM. (70% in toluene) and 2.5 ml toluene (exothermic, cool with a water bath to avoid temperature to go >50° C.). The reaction mixture was stirred 2 hours at 50° C. in CH2Cl2, extracted with ethyl acetate and cooled to 0° C. 4 ml Aqueous NaOH 1N were carefully (exothermic) added over 15 minutes, followed by 20 ml ethyl acetate. The phases were separated and the aqueous phase was extracted with ethyl acetate. The combined organic extracts were washed with deionized water and brine, dried (Na2SO4) and concentrated under reduced pressure to yield 0.37 g (89%) methyl-[6-4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine as an orange resin. MS (ISP): 297 (M+H+, 100).
Synthesis of 2-(3,5-bis-Trifluoromethyl-phenyl)-2-methyl-propionyl Chloride

15.0 g (50 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionic acid were dissolved in 127.5 ml dichloromethane in the presence of 0.75 ml DMF. 8.76 ml (2 eq.) Oxalyl chloride were added and after 4.5 hours, the solution was rotary evaporated to dryness. 9 ml Toluene were added and the resulting solution was again rotary evaporated, then dried under high vacuum yielding 16.25 g (quant.) of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride as a yellow oil of 86% purity according to HPLC analysis. NMR (250 MHz, CDCl3): 7.86 (br s, 1H); 7.77, (br s, 2H, 3 Harom); 1.77 (s, 6H, 2 CH3).
Synthesis of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (Netupitant)

A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane, The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1yl)-4-(o-tolyl)pyridin-3yl)propanamide as white crystals. M.P. 155-157° C.; MS m/e (%): 579 (M+H+, 100).
…………………………………..
http://www.google.com/patents/US20130231315
N OXIDE SYNTHESIS
Synthesis of 5-(2-(3,5-bis(trifluoromethyl)phenyl-N,2-dimethylpropanamido)2-(4-methylpiperazin-1yl)-4-(o-tolyl)pyridine 1-oxide

Step 1:
The solution of 6-chloropyridin-3-amine (115 g, 0.898 mol) and (Boc)2O (215.4 g, 0.988 mol) in 900 mL of dioxane was refluxed overnight. The resulting solution was poured into 1500 mL of water. The resulting solid was collected, washed with water and re-crystallized from EtOAc to afford 160 g tert-butyl (6-chloropyridin-3yl)carbamate as a white solid (Yield: 78.2%).
Step 2:
To the solution of tert-butyl (6-chloropyridin-3-yl)carbamate (160 g, 0.7 mol) in 1 L of anhydrous THF was added n-BuLi (600 mL, L5 ml) at −78° C. under N2 atmosphere. After the addition was finished, the solution was stirred at −78° C. for 30 min, and the solution of I2 (177.68 g, 0.7 mol) in 800 mL of anhydrous THF was added. Then the solution was stirred at −78° C. for 4 hrs, TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and purified by flash chromatography to afford 80 g of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate as a yellow solid (32.3%).
Step 3:
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate (61 g, 0.172 mol) in 300 of anhydrous THF was added 60% NaH (7.6 g, 0.189 mol) at 0° C. under N2 atmosphere. After the addition was finished, the solution was stirred for 30 min, and then the solution of MeI (26.92 g, 0.189 mol) in 100 mL of dry THF was added. Then the solution was stirred at 0° C. for 3 hrs. TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and concentrated to afford 63 g of crude tert-butyl (6-chloro-4-iodopyridin-3-yl)methyl)carbamate used into the following de-protection without the further purification.
Step 4:
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)(methyl)carbamate (62.5 g, 0.172 mol) in 500 mL of anhydrous DCM was added 180 mL of TFA. Then the solution was stirred at room temperature for 4 hrs. Concentrated to remove the solvent, and purified by flash chromatography to afford 45.1 g 6-chloro-4-iodo-N-methylpyridin-3-amine as a yellow solid (Yield: 97.3%).
Step 5:
To the solution of 6-chloro-4-iodo-N-methylpyridin-3-amine (40.3 g, 0.15 mol) and 2-methylbenzene boric acid (24.5 g, 0.18 mol) in 600 mL of anhydrous toluene was added 400 mL of 2 N aq. Na2CO3 solution, Pd(OAc)2 (3.36 g, 15 mmol) and PPh3(7.87 g, 0.03 mmol), The solution was stirred at 100° C. for 2 hrs. Cooled to room temperature, and diluted with water. EtOAc was added to extract twice. The combined organic phases were washed with water and brine consecutively, dried over Na2SO4, concentrated and purified by flash chromatography to afford 19 g 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine as a white solid (Yield: 54.6%).
Step 6:
To the solution of 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine (18.87 g, 81.3 mmol) and DMAP (29.8 g, 243.9 mmol) in 200 mL of anhydrous toluene was added the solution of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride (28.5 g, 89.4 mmol) in toluene under N2 atmosphere. The solution was heated at 120° C. for 23 hrs. Cooled to room temperature, poured into 1 L of 5% aq. NaHCO3 solution, and extracted with EtOAc twice. The combined organic phases were washed by water and brine consecutively, dried. over Na2SO4, filtered and purified by flash chromatography to afford 35 g 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(4-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide as a white solid (Yield: 83.9%).
Step 7:
To the solution of 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(o-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide (5.14 g, 10 mmol) in 60 mL of DCM was added m-CPBA (6.92 g, 40 mmol) at 0° C. under N2 atmosphere. Then the solution was stirred overnight at room temperature. 1 N aq. NaOH solution was added to wash twice for removing the excess m-CPBA. and a side product. The organic phase was washed by brine, dried over Na2SO4, filtered and concentrated to afford 5.11 g of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl-N,2-dimethylpropanamido)-2-chloro-4(o-tolyl)pyridine 1-oxide as a white solid (Yield: 96.4%).
Step 8:
To the solution of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-chloro-4-(o-tolyl)pyridine 1-oxide (5.1 g, 9.62 mmol) in 80 mL of n-BuOH was added N-methylpiperazine (7.41 g, 74.1 mmol) under N2 atmosphere. Then the solution was stirred at 80° C. overnight. Concentrated and purified by flash chromatography to afford 4.98 g 5-(2-(3,5-bis(trifluoromethyl)phenyl-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide as a white solid (Yield: 87.2%), 1HNMR (CDCl3, 400 MHz) δ 8.15 (s, 1H), 7.93 (s, 1H), 7.78 (s, 2H), 7.38 (m, 2H), 7.28 (m, 1H), 7.17 (m, 1H), 7.07 (s, 1H), 5.50 (s, 3H), 2.72 (d, J=4.4 Hz, 4H), 2.57 (m, 3H), 2.40 (s, 3H), 2.23 (s, 3H), 1.45-1.20 (m, 6H).
………………………………….
https://www.google.co.in/patents/US6479483


EXAMPLE 14 2-(3,5-bis-Trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperan-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide Hydrochloride (1:2)
a) 1-Methyl-4-(5-nitro-pyridin-2-yl)-piperazine
To a solution of 20 g (126 mmol) of 2-chloro-5-nitropyridine in 200 ml tetrahydrofuran were added dropwise 35 ml (315 mmol) 1-methylpiperazine within 10 min. The reaction mixture was refluxed for additional 1.5 h. After cooling to room temperature, the solvent was removed in vacuo and the residue was re-dissolved in 200 ml ethyl acetate. The organic phase was washed with 200 ml 1 N sodium bicarbonate solution, dried (magnesium sulfate) and evaporated to give 27.9 g (quantitative) of the title compound as a yellow solid.
MS m/e (%): 223 (M+H+, 100).
b) 2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl-propionamide
To a solution of 27.9 g (125.5 mmol) of 1-methyl-4-(5-nitro-pyridin-2-yl)-piperazine in 400 ml methanol were added 2.6 g of 10% of palladium on activated charcoal. The reaction mixture was hydrogenated (room temperature to ca. 45° C., 1 bar) until the theoretical amount of hydrogen was taken up (about 2 h). The catalyst was filtered off and was washed twice with 100 ml portions of methanol. The filtrate was evaporated in vacuo to give 28 g of a purple oil which consisted to ca. 90% of the desired aniline derivative according to analysis by thin layer chromatography.
This crude product was dissolved in a mixture of 400 ml tetrahydrofuran and 100 ml diethyl ether. After cooling to 0° C., 30 ml (215 mmol) of triethylamine were added in one portion. Stirring was continued while 26 g (215 mmol) of pivaloyl chloride were added dropwise within a period of 10 min. The ice bath was removed and the reaction mixture was stirred for 1 h at room temperature. Then, the solvent was removed in vacuo and the residue was suspended in 200 ml 1 N sodium bicarbonate solution. The product was extracted three times with 200 ml portions of dichloromethane, dried (sodium sulfate) and purified by flash chromatography to give 30 g (86%) of the title compound as pink crystals.
MS m/e (%): 277 (M+H+, 100).
c) N-[4-Iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide
A solution of 30 g (108 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-propionamide and 58 ml (380 mmol) N,N,N′,N′-tetramethylethylenediamine under argon in 650 ml tetrahydrofuran was cooled in a dry ice bath to −78° C. Within 1 h, 239 ml (380 mmol) of a 1.6 N n-butyllithium solution in hexane were added dropwise. The reaction mixture was allowed to warm up to −30° C. overnight. After cooling again to −78° C., 43.6 g (170 mmol) iodine dissolved in 60 ml tetrahydrofuran were added dropwise during 15 min. The dry ice bath was replaced by an ice bath and a solution of 90 g (363 mmol) sodium thiosulfate pentahydrate in 250 ml water were added within 10 min when the temperature of the reaction mixture had reached 0° C. Then, 1000 ml diethyl ether were added and the organic layer was separated. The aqueous layer was extracted twice with 500 ml dichloromethane and the combined organic layers were dried (magnesium sulfate) and evaporated. Flash chromatography gave 18.5 g (42%) of the tide compound as a light brown oil which crystallized upon standing at room temperature.
MS m/e (%): 403 (M+H+, 100).
d) 2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide
A mixture of 54 g (134 mmol) N-[4-iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide, 420 ml toluene, 150 ml 2 N sodium carbonate solution, 4.63 g (3.9 mmol) tetrakis(triphenylphosphine)palladium(0) and 20.16 g (147 mmol) o-tolylboronic acid was heated under argon at 80° C. for 12 h. After cooling to room temperature, the aqueous phase was separated and washed twice with toluene. The combined organic layers were washed with 50 ml brine, dried (sodium sulfate), evaporated and dried in vacuo to yield 49 g (quantitative) of the title compound as a brown oil.
MS m/e (%): 367 (M+H+, 100).
e) 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine
A suspension of 56 g (152 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide in 1300 ml 3 N hydrochloric acid solution was heated to 90-95° C. overnight. The reaction mixture was cooled to room temperature, washed with three 500 ml portions diethyl ether and filtered over celite. The filtrate was diluted with 500 ml water and was adjusted to pH 7-8 by addition of 28% sodium hydroxide solution under ice cooling. The product was extracted with four 1000 ml portions of dichloromethane. The combined organic layers were washed with 500 ml brine, dried (magnesium sulfate) and evaporated to give 35 g (82%) of the title compound as a light brown oil.
MS m/e (%):283 (M+H+, 100).
f) Methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine
A solution of 35 g (124 mmol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine in 270 ml trimethyl orthoformate and 8 drops trifluoroacetic acid was heated for 3 h at 130° C. The reaction mixture was evaporated and dried in vacuo for 30 min. The residual oil was dissolved in 100 ml tetrahydrofuran and was added dropwise under ice cooling to 9.4 g (248 mmol) lithium aluminum hydride in 300 ml tetrahydrofuran. The reaction mixture was stirred for 1 h at room temperature, cooled to 0° C. again and acidified (pH 1-2) by addition of 28% hydrochloric acid solution. After stirring for 5 min, 28% sodium hydroxide solution was added to reach pH 10. The solution was filtered over celite, evaporated and purified by flash chromatography to give 23.6 g (64%) of the title compound as a light brown oil.
MS m/e (%): 297 (M+H+, 100).
g) 2-(3,5-bis-Trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide
A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane. The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of the title compound as white crystals. M.p. 155-157° C.
MS m/e (%): 579 (M+H+, 100).
h) 2-(3,5-bis-Trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide Hydrochloride (1:2)
To a solution of 31.6 g (54.6 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide in 250 ml diethyl ether were added under ice cooling 60 ml 3 N hydrochloric acid solution in diethyl ether. After stirring for 15 min at 0° C., the suspension was evaporated to dryness, re-suspended in 100 ml diethyl ether, filtered and dried in vacuo to give 34.8 g (98%) of the title compound as white crystals. M.p. 235-238° C.
MS m/e (%): 579 (M+H+, 100).
…………………………….
Research and development of an efficient process for the construction of the 2,4,5-substituted pyridines of NK-1 receptor antagonists
Org Process Res Dev 2006, 10(6): 1157
Navari RM.
Drugs. 2013 Mar;73(3):249-62. doi: 10.1007/s40265-013-0019-1. Review.
-
Hoffmann-Emery F, Hilpert H, Scalone M, Waldmeier P.
J Org Chem. 2006 Mar 3;71(5):2000-8.
Hoffmann T, Bös M, Stadler H, Schnider P, Hunkeler W, Godel T, Galley G, Ballard TM, Higgins GA, Poli SM, Sleight AJ.
Bioorg Med Chem Lett. 2006 Mar 1;16(5):1362-5. Epub 2005 Dec 5.
http://www.sciencedirect.com/science/article/pii/S0960894X05014824
…………………………………….……………………………………………………….
| US6897226 * | 9 Jul 2003 | 24 May 2005 | Hoffmann-La Roche Inc. | NK-1 receptor active amine oxide prodrugs |
| US7211579 * | 15 Mar 2006 | 1 May 2007 | Hoffmann-La Roche Inc. | NK-1 receptor antagonists |
| US8426450 | 23 May 2012 | 23 Apr 2013 | Helsinn Healthcare Sa | Substituted 4-phenyl pyridines having anti-emetic effect |
| WO2011061622A1 | 18 Nov 2010 | 26 May 2011 | Helsinn Healthcare S.A. | Compositions for treating centrally mediated nausea and vomiting |
| WO2013057554A2 | 10 Oct 2012 | 25 Apr 2013 | Helsinn Healthcare Sa | Therapeutic combinations of netupitant and palonosetron |
| US8426450 | 23 May 2012 | 23 Apr 2013 | Helsinn Healthcare Sa | Substituted 4-phenyl pyridines having anti-emetic effect |
| WO2011061622A1 | 18 Nov 2010 | 26 May 2011 | Helsinn Healthcare S.A. | Compositions for treating centrally mediated nausea and vomiting |
| WO2013057554A2 | 10 Oct 2012 | 25 Apr 2013 | Helsinn Healthcare Sa | Therapeutic combinations of netupitant and palonosetron |
……………………………………………………………………………………….

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007
DR ANTHONY MELVIN CRASTO
ORGANIC SPECTROSCOPY

