
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Orphan Drugs: Global Regulatory Events
This Blog Post discusses recent global non-US regulatory events for orphan drugs.
I – Europe
At a January meeting, the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) gives the following opinions for three orphan drugs :
• Positive recommendation for Bayer’s Adempas (Riociguat) for the treatment of Chronic Thromboembolic Pulmonary Hypertension (CTEPH) and Pulmonary Arterial Hypertension (PAH)
• Negative opinion for AB Science’s Masiviera (Masitinib) which is intended for the treatment of non resectable locally advanced or metastatic pancreatic cancer
• Negative opinion for PTC Therapeutics’ Translarna (Ataluren) which is intended for the treatment of Duchenne Muscular Dystrophy.
Also, EMA’s Committee for Orphan Medicinal Products (COMP) issues 15 positive opinions for ODD at their January meeting (Reference Blog Post). These ODDs are to be presented to the European Commission (EC) for final approval. If the EC approves these ODDs, the drugs receive ODD in the…
View original post 230 more words
UDENAFIL …The Eastern Viagra (like)

UDENAFIL
An oral phosphodiesterase 5 inhibitor used for the treatment of erectile dysfunction.
268203-93-6 CAS NO
LAUNCHED 2005 MEZZION DA-8159 ME-3113 Udzire Zydena MEZZION …INNOVATOR
POWERPOINT PRESENTATION BY INNOVATOR.. CLICK HERE
| Synonyms: | Zydena;Udenafi;Da-8159;Da 8159;Udenafil;Udenafil(DA 8159,Zydena);5-(2-Propyloxy-5-(1-methyl-2-pyrollidinylethylamidosulfonyl)phenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo(4,3-D)pyrimidine-7-one;5-[2-propyloxy-5-[2-(1-Methyl-2-pyrrolidinyl)ethylaMinosulfonyl]phenyl]-1-Methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyriMidine-7-one;5-[2-propyloxy-5-(2-(1-Methylpyrrolidin-2-yl)ethylaMinosulphonyl)phenyl]-1-Methyl-3-propyl-6,7-dihydro-1H-pyrazolo(4,3-d)pyriMidin-7-one;3-(6,7-Dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methyl-2-pyrrolidinyl)ethyl]-4-propoxybenzenesulfonamide |
| Molecular Formula: | C25H36N6O3S2 |
| Formula Weight: | 516.66 |
3-(1-methyl-7-oxo-3-propyl-4,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide
(5- [2-propyloxy-5- (1- methyl-2-pyrolidinylethylamidosulfonyl) phenyl] -1-methyl- propyl-1, β-dihydro-7H-pyrazolo (4 , 3-d) pyrimidin-7-one)
A pyrazolo-pyrimidinone similar to sildenafil; phosphodiesterase type 5 inhibitor. Udenafil is a new phosphodiesterase type 5 (PDE5) inhibitor used to treat erectile dysfunction (ED). It has been approved in South Korea and will be marketed under the brand name Zydena.
It is not yet approved for use in the U.S., E.U., or Canada. Udenafil (Zydena®) is also a potent and selective PDE5i developed by Dong-A Pharmaceutical Company in Korea (Kim et al., 2008; Han et al., 2010).
It has not yet been approved by FDA or the European Medicines Agency (EMEA) and was only approved by the Korean Food and Drug Administration (KFDA), being currently used in Korea and Russia (Alwaal et al., 2011; Cho et al., 2012).
- DA 8159
- DA-8159
- Udenafil
- UNII-L5IB4XLY36
- Zydena
Udenafil is a drug used in urology to treat erectile dysfunction. It belongs to a class of drugs called PDE5 inhibitor, which many other erectile dysfunction drugs such as sildenafil, tadalafil, and vardenafil also belong to. It was developed by Dong-A Pharmaceutical Co., Ltd. and is marketed under the trade name Zydena™.[2] With a T max of 1.0-1.5 h and a T 1/2 of 11-13 h (a relatively rapid onset and a long duration of action), both on-demand and once-daily use of udenafil have been reported.[3] Typical doses are 100 and 200 mg. It is not approved for use in the United States by theU.S. Food and Drug Administration. Udenafil (DA-8159), a pyrazolopyramidinone derivative that acts as a phosphodiesterase 5 (PDE5) inhibitor, was launched by Dong-A Pharmtech (currently Mezzion Pharma) in late 2005 in Korea for the oral treatment of erectile dysfunction (ED). The company is currently conducting phase III clinical trials in the U.S. for this indication.
Dong-A Pharmatech is conducting phase III clinical trials for the treatment of patients with portal hypertension resulting from liver disease and for the treatment of benign prostatic hyperplasia (BPH). Phase II/III clinical studies at Dong-A Pharmatech for the treatment of secondary Raynaud phenomenon have been completed. Meiji Seika Pharma is developing the compound in phase I clinical trials for the treatment of BPH in Japan.
Phosphodiesterases regulate the tissue concentration of cyclic guanosine monophosphate (cGMP), which in turn triggers smooth muscle relaxation, allowing blood to flow into the penis and resulting in erection. PDE5 is the most abundant phosphodiesterase in the human corpus cavernosum, and as such its inhibition by DA-8159 enhances erectile function by increasing the concentration of cGMP. Results from phase I studies indicate that udenafil has a unique pharmacokinetic profile with a relatively rapid onset and sufficiently long duration to make it effective for up to 24 hours. In 2009, the compound was licensed to Warner Chilcott (acquired by Actavis in 2013) by Dong-A Pharmatech for development and marketing in the U.S. for the oral treatment of erectile dysfunction.
In 2011, udenafil was licensed to Meiji Seika Pharma by Dong-A ST in Japan for the treatment of benign prostatic hyperplasia. Udenafil is a potent novel phosphodiesterase-5 inhibitor approved for use in Korea. Udenafil has unique properties, with a T max of 1.0–1.5 h and a T 1/2 of 11–13 h (a relatively rapid onset and a long duration of action). Therefore, both on-demand and once-daily use of udenafil have been reported. Udenafil’s efficacy and tolerability have been evaluated in several studies, and recent and continuing studies have demonstrated udenafil’s promise in both dosing regimens. Presently, tadalafil is the only FDA-approved drug for daily dosing, but udenafil can be used as a once-daily dose for erectile dysfunction patients who cannot tolerate tadalafil due to phosphodiesterase subtype selectivity. Udenafil as an on-demand or once-daily dose is effective and tolerable, but more studies are needed in patients of other ethnicities and with comorbid conditions such as diabetes mellitus, hypertension, and benign prostate hyperplasia.
Erectile dysfunction (ED) is defined as the inability to achieve and maintain a sufficient erection to permit satisfactory intercourse [Montorsi et al. 2010]. Numerous strategies have been used to overcome ED. Therapies for ED include intracavernosal injection, vacuum erection devices, intraurethral suppositories, penile prosthesis surgery and oral phosphodiesterase-5 (PDE5) inhibitors [Dinsmore and Evans, 1999]. Oral PDE5-inhibitor medications have revolutionized the treatment of ED. Men prefer oral medications as the first-line therapeutic option in the absence of a specific contraindication to their use [Ding et al. 2012].
There are currently four PDE5 inhibitors (sildenafil, vardenafil, tadalafil, and avanafil) approved worldwide for the treatment of male erectile dysfunction, with two other agents (udenafil and mirodenafil) currently approved only in Korea [Bell and Palmer, 2011]. The choice of PDE5 inhibitor for each patient should be determined after physician and patient discuss the characteristics of different drugs and the individual patient’s sexual habits, preferences, and expectations [Hatzimouratidis et al. 2010]. There are two types of treatment usage of PDE5 inhibitors according to their pharmacological characteristics. On-demand treatment of ED with PDE5 inhibitors allows the patient to have intercourse within 1 hour, but can remove spontaneity from sexual activity and be burdensome to patients and their partners [Hanson-Divers et al. 1998]. Once-daily dosing of a PDE5 inhibitor is an alternative for couples that prefer spontaneous sexual activities.
A new oral selective PDE5 inhibitor, udenafil (Zydena, Dong-A, Seoul, Korea), has recently been developed for the treatment of ED. Udenafil is a novel pyrazolopyrimidinone compound developed by Dong-A Pharmaceutical Co., Ltd (Seoul, Korea) for the treatment of ED which has the same mechanism of action as sildenafil [Kim et al. 2008]. Udenafil is rapidly absorbed, reaching peak plasma concentrations at 0.8–1.3 h, then declining monoexponentially with a terminal half-life (T 1/2) between 7.3 and 12.1 hours, giving it the unique pharmacokinetics of both relatively rapid onset and long duration [Salem et al. 2006]. Thus, both on-demand treatment and once-daily dosing have been reported in the literature. The purpose of this review is to evaluate the efficacy and tolerability of udenafil for patients with ED according to the currently available literature.
Udenafil” refers to the chemical compound, 3-(1-methyl-7-oxo-3-propyl-4,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide and has the following chemical formula:

More information about udenafil can be found at Kouvelas D. et al., (2009) Curr Pharm Des, 15(30):3464-75. Udenafil is marketed under the trade name Zydena® but not approved for use in the United States. TRADE NAME IN INDIA – UDEZIRE Erectile dysfunction (ED) is an inability to achieve or sustain an erection suitablefor sexual intercourse.
Sexologists say at least 10% men in India may have to use these drugs at some point. Studies have shown that 40% of men up to the age of 40 years have ED andit goesup 70% by 60 years. The commonly prescribed drugs for the disorder in India are sildenafil(Viagra) and tadalafil,which belong to a category called phosphodiesterasetype5 drugs.
Now, Zydus, a pharmaceutical company, has got exclusive permission to sell udenafil. It’s not always that the release of a drug is celebrated by many, particularly men. A drug that was released in India last week is the recent in the list of drugs that has a cure for erectile dysfunction. The manufacturers say udenafil, which will be marketed under the brand name Udezire, will be long-acting, but with minimal side effects. Erectile dysfunction (ED) is an inability to achieve or sustain an erection suitable for sexual intercourse. Sexologists say at least 10% men in India may have to use these drugs at some point. Studies have shown that 40% of men up to the age of 40 years have ED and it goes up 70% by 60 years

UDENAFIL
……………………
INTRODUCTION
Udenafil (Zydena®) is a therapeutic agent hypothesized to improve erectile function endpoints through interaction with the phosphodiesterase type 5 (PDE5) enzyme. As such, udenafil belongs to the class of such agents that includes tadalafil (Clalis®), sildenafil (Viagra®), and vardenafil (Levitra®). These agents are purported to promote erectile response through inhibition of PDE5, the predominant PDE within the penis, which leads to higher intracellular levels of cyclic guanylate cyclase (cGMP). cGMP is a second messenger for the smooth-muscle relaxing effects of nitric oxide within the penis. The various agents differ in pharmacology primarily based on 1) onset and duration of action and 2) selectivity profiles vs. other PDEs. All three marketed agents have proven remarkably safe. These agents should not be taken by patients with unstable cardiovascular disease. Udenafil has been shown to exhibit greater selectivity against the known PDE homologues, than other PDE5 inhibitors. Udenafil is comparable to tadalafil in many respects, such as duration of action and high selectivity for PDE6, but udenafil has greater selectivity for PDE11 than tadalafil.
Tadalafil, with a half life of 17.5 hours, has a much longer duration of action and improved exercise tolerance than either sildenfail or vardenafil, which have half lives of 4-5 hours. Consequently, tadalafil is associated with less planning or pressure to have sexual intercourse after dosing. Dissociation of the sexual activity from the time of dosing is associated with higher rates of patient and partner satisfaction. In prospective, randomized crossover clinical studies, patients preferred tadalafil over sildenafil by margins ranging from 7:3 to 9:1. Sildenafil and vardenafil both modulate PDE6 at higher rate than tadalafil. PDE6 modulation has been associated with chromatopsia. The side effects of chromatopsia, such as sensitivity to light and blurred vision, are therefore higher in patients taking sildenafil or vardenafil, about 2-3%, than patients taking tadalafil, about <0.1%. Tadalafil is less selective than sildenafil and vardenafil for PDE5 and for PDE11a. Activity at PDE11a is suspected to have a causal relationship with myalgia and testicular toxicity. The selectivity profile for udenafil is similar to sildenafil, which should impart greater safety for this agent.

The benefits and shortcomings of these drugs have been reviewed. Some of these shortcomings can be traced to metabolism-related phenomena. Udenafil is converted in vivo by oxidative and conjugative degradation to multiple metabolites. Phase I metabolism leads to demethylation of the pyrazole, hydroxylation of the pyrazole propyl group, and dealkylation alpha to the sulfonamide nitrogen to afford an active metabolite. Because udenafil is metabolized primarily by cytochrome P450 subtype 3A4 (CYP3A4), exposure to udenafil can influence polypharmacy. For example, CYP3A4 inhibitors such as HIV protease inhibitors, azole antifungals, and erythromycin can lead to higher than otherwise expected blood levels of udenafil. Conversely, co-administration of CYP3A4 inducers such as rifampin can decrease the otherwise expected blood levels of udenafil. Thus, the polypharmacy of udenafil is necessarily complex and has potential for adverse events. In addition, there may be increased inter-patient variability in response to polypharmacy.
Analogs of udenafil as described herein have the potential to alleviate the problems associated with the commercially available PDE5 inhibitors while maintaining or improving efficacy. It is believed that the reduction in CYP3A4 clearance of udenafil analogs will be expected to increase the proportion of clearance via mechanisms less susceptible to polypharmaceutical complications. In addition, analogs of udenafil having an attenuated rate of oxidative metabolism will have an increased half-life, further augmenting their advantages vs. tadalafil, sildenafil and vardenafil. Potentially, a single dose of an udalafil analog, described herein, having an increased half-life may provide therapeutic coverage for an entire weekend or beyond while increasing safety parameters by reducing the likelihood of drug-drug interactions and by increasing safety as a result of the increased selectivity.

The compounds of formula 1 may contain asymmetric centers and thus they can exist as enantiomers. The present invention includes both mixtures and separate individual isomers . Male erectile dysfunction is one of the most common sexual dysfunctions in men. Although erectile dysfunction can be primarily psychogenic in origin, it often accompanies chronic illnesses, such as diabetes mellitus, heart disease, hypertension, and a variety of neurological diseases. Its prevalence is strongly related to age, with a estimated prevalence of 2% at age 40 years rising to 25-30% by age of 65. Although no data are available on the prevalence of erectile dysfunction in men aged over 75, it is probably over 50%. Various treatment options for erectile dysfunction are available, such as counseling, hormonal therapy, self-injection or transurethral application of vasodilator agents, vacuum devices, prosthesis implantation, and venous/arterial surgery. However, these therapeutic options have several limitations such as side effects, high-cost and low efficacy.
Therefore it has called for research efforts to develop new, high effective and simple to use treatment methods, potentially oral medication. Recently, sildenafil has been developed as a therapeutic agent for male erectile dysfunction by oral administration. Sildenafil is the first in a new class of drugs known as inhibiting phosphodiesterase-5 enzyme distributed specifically in corpus cavernosal tissues and induces relaxation of the corpus cavernosal smooth muscle cells, so that blood flow to the penis is enhanced, leading to an erection.
Sildenafil has shown a response rate of around 80% in men with erectile dysfunction of organic cause. On the other hand, USP 3,939,161 discloses that 1 , 3 -dimethyl -lH-pyrazolopyrimidinone derivatives exhibit anticonvulsant and sedative activiity, and also exhibit anti-inflammatory activity and gastric antisecretory activity; EP 201,188 discloses that 5-substituted pyrazolopyrimidinone derivatives have effects of antagonizing adenosine receptor and of inhibiting phosphodiesterase enzymes and can be used for the treatment of cardiovascular disorders such as heart failure or cardiac insufficiency; EP 463,756, EP 526,004, WO 93/6,104 and WO 93/7,149 disclose that pyrazolopyrimidinone derivatives which inhibit c-GMP phosphodiesterase more selectively than c-AMP phosphodiesterase have efficacy on cardiovascular disorders such as angina pectoris, hypertension, heart failure, atherosclerosis, chronic asthma, etc.; and WO 94/28,902, WO 96/16,644, WO 94/16,657 and WO 98/49,166 disclose that the known inhibitors of c-GMP phosphodiesterase including the pyrazolopyrimidinone derivatives of the above mentioned patents can be used for the treatment of male erectile dysfunction Since sildenafil has been developed, various compounds for inhibiting phosphodiesterase-5 have been reported.
Among them, pyrazolopyrimidinone compounds of formula 1 (KR Pat. No. 99-49384) were reported having better potency than that of sildenafil, based on the mechanism of inhibiting phosphodiesterase-5 and having better selectivity over phosphodiesterase-6 distributed in retina and phosphodiesterase-3 distributed in heart to reduce the side effects. Further, the pyrazolopyrimidinone compounds of formula 1 were said to be improved the solubility and the metabolism in the liver, which are very important factor affecting the rate of the absorption when administered orally.
The KR patent No. 99-49384 also disclosed a process for preparing the pyrazolopyrimidinone compounds of formula , comprising the steps of: a) reacting chlorosulfonated alkoxy bonzoic acid with a primary amine to obtain sulfonamide-substituted benzoic acid; b) reacting the obtained sulfonamide-substituted benzoic acid with pyrazolamine in the presence of activating reagent of carboxylic group or coupling agent of carboxylic group with amine group to obtain corresponding amide compound; and, c) performing an intramolecular cyclization of the obtained amide compound to obtain the pyrazolopyrimidinone compound of formula 1. This reaction is represented in scheme 1 Scheme 1

…………………..
SYNTHESIS
The present invention provides an agent comprising a pyrazolopyrimidinone compound (5- [2-propyloxy-5- (1- methyl-2-pyrolidinylethylamidosulfonyl) phenyl] -1-methyl- propyl-1, 6-dihydro-7H-pyrazolo (4, 3-d) pyrimidin-7-one) expressed as formula 1 as an effective ingredient for preventing and treating benign prostatic hyperplasia (BPH) . Formula 1

The pyrazolopyrimidinone compound represented as formula 1 is one of the PDE-5 inhibitors and has characteristics in that it has a strong inhibitive activity and an excellent selectivity for PDE-5; it is readily absorbed as its solubility is improved; it has a good bioavailability and a large volume of distribution; and it has an in vivo half-life longer three times than sildenafil or vardenafil, a drug of the same mechanism. Physicochemical properties of the pyrazolopyrimidinone compound of formula 1 are as follows: it is hardly dissolved in water; however, it is readily dissolved in acetic acid, methanol, chloroform and the like; and it is a white or pale yellow powder, not a hydrate or a solvate, having a melting point of 158 to 161 “Q and having pKal and pKa2 of about 6.5 and 12.5, respectively. The pyrazolopyrimidinone compound represented as formula 1 is prepared via a synthetic process consisting of roughly three steps. The inventors of the present invention have disclosed a method for preparing the same in WO2000/027847 (Corresponding Korean Patent No.0353014), which will now be described roughly as follows. First, in the first step, 4- [2-propyloxy-5- (chlorosulfonyl) benzamido] -l-methyl-3-propyl-5-carbamoyl pyrazole is prepared.
For such preparation, a specified amount of 4- [2-propyloxybenzamido] -l-methyl-3-propyl-5- carbamoyl pyrazole is added to a specified amount of chlorosulfonic acid cooled to 0 °Q then, the resultant mixture is stirred, filtered, washed and dried to obtain 4- [2-propyloxy-5- (chlorosulfonyl) benzamido] -l-methyl-3- propyl-5-carbomoyl pyrazole. In the second step, from the pyrazole compound prepared in the first step, 4- [2-propyloxy-5- ( l-methyl-2- pyrolidinylethylamidosulfonyl) benzamido] -l-methyl-3- propyl-5-carbomoyl pyrazole is prepared. For such preparation, a specified amount of 2- (2-aminoethyl) -1- methyl pyrolidine is added in dichloromethane solution of the specified amount of 4- [2-propyloxy-5- (chlorosulfonyl) benzamido] -l-methyl-3-propyl-5-carbamoyl pyrazole prepared in the first step to be stirred. Then, the reactant solution is diluted with dichloromethane. The organic layer is washed, dried, concentrated and filtered to obtain 4- [2-propyloxy-5- (l-methyl-2- pyrolidinylethylamidosulfonyl) benzamido] -l-methyl-3- propyl-5-carbomoyl pyrazole is obtained.
Last, in the third step, the pyrazolopyrimidinone compound of the present invention (5- [2-propyloxy-5- (1- methyl-2-pyrolidinylethylamidosulfonyl) phenyl] -1-methyl- propyl-1, β-dihydro-7H-pyrazolo (4 , 3-d) pyrimidin-7-one) is prepared from the compound obtained in the second step. For such preparation, the specified amount of pyrazole compound prepared in the second step is dissolved in t- butanol . A specified amount of potassium t-butoxide is added in the resultant solution and, then, reflux-stirred for a predetermined time. After the resultant solution is cooled, diluted, washed and dried, distillation under reduced pressure, solvolysis and silica gel column chromatography are carried out, thus obtaining a specified amount of pure pyrazolopyrimidinone compound of the present invention
. …………………………..
SYNTHESIS WO2000027848A1
REACTION SCHEME 2

The process for preparation according to the present invention comprises the steps of : 1) reacting the chlorosulfonated compound of formula ( 2 ) and primary amine (3_) under the condition of suitable temperature and suitable solvent to give sulfonamide (4.) (step 1) ; 2) reacting the carboxylic acid (4.) prepared in step 1 and pyrazoleamine (5) to give an amide (6.) by the known method preparing amide from carboxylic acid and amine (step 2) ; and 3) cyclizing the amide (6.) prepared in step 2 to give the desired compound of formula 1 by the known cyclization method used for preparation of pyrimidinone (step 3) .
In step 1, a little excess of 2 equivalents of amine may be used, or a little excess of 1 equivalent of amine and 1 equivalent of acid scavenger such as tertiary amine are may be used together. The reaction temperature is preferred below 20 °C. The known method preparing amide from carboxylic acid and amine in step 2 is the process, for example, in which carboxyl group is transformed into activated acid chloride or acid anhydride by using thionyl chloride, pivaloyl chloride, trichlorobenzoyl chloride, carbonyldiimidazole, diphenylphosphinic chloride, etc. and followed by reacting with amine group, or the process using coupling agents such as DCC (1,3-dicyclo hexylcarbodiimide) or EEDQ (N-ethoxycarbonyl -2 -ethoxy- 1, 3-dihydroquinoline) .
The cyclization process in step 3 may be carried out in the presence of a suitable base and a suitable solvent. Preferred bases which are employed in step 3 are metal alkoxides; metal salts of ammonia; amine; hydrides of alkali metal or alkaline earth metal; hydroxides; carbonates; bicarbonates ; and bicyclic amidines such as DBU (1 , 8-diazabicyclo [5.4.0] undec -7-ene) and DBΝ (1 , 5-diazabicyclo [4.3.0] non-5-ene) . Preferred solvents which are employed in step 3 are alcohols such as methanol, ethanol, isopropanol, t-butanol, etc.; ethers such as tetrahydrofuran, dimethoxyethane, dioxane, etc.; aromatic – hydrocarbons such as benzene, toluene, xylene, chlorobenzene, etc.; acetonitrile; dimethylsulfoxide; dimethylformamide; N-methylpyrrolidin-2 -one ; and pyridine.
SEE ENTRY no 68
5- [2-propyloxy-5- ( 1-methyl-2-pyrrolidinylethyl amidosulfonyl) phenyl] -l-methyl-3 -propyl-1 , 6-dihydro-7 H-pyrazolo (4 , 3-d) yrimidin-7-one (compound of example68) 
ACCORDING TO ME ENTRY IS 68 ANY ERROR, amcrasto@gmail.com
Synthesis WO2001098304A1
The present invention relates to a process for preparing pyrazolopyrimidinone derivatives of formula 1 and pharmaceutically acceptable salts thereof which have an efficacy on impotence, comprising the steps of chlorosulfonation of pyrazolamide compounds of formula 2, followed by amination with a primary amine and intramolecular cyclization. Formula 1

Formula 2

The compounds of formula 1 may exist in tautomeric equilibrium as shown below.

The compounds of formula 1 may also contain asymmetric centers and thus they can exist as enantiomers. The present invention includes both racemic mixture and separate individual enantiomers. Scheme 2

……………………………….
SYNTHESIS WO2010013925A2
INTERMEDIATES
4-[2-propyloxy benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole
CHLOROSULPHONIC ACID
4-[2-propyloxy-5-(chlorosulfonyl)benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole
2-(2-aminoethyl)-l-methylpyrrolidine 4-[2-propyloxy-5-(l-methyl-2-pyrrolidinylethyl amido- sulfonyl)benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole
potassium t-butoxide
3, 5-[2-propyloxy-5-(l-methyl-2-pyrrolidinylethyl amido- sulfonyl)phenyl]-l-methyl-3-propyl-l,6-dihydro-7H-pyrazolo(4,3-d)pyrimidin-7-one UDENAFIL
The present invention provides a pharmacological compound containing 5- [2-propyloxy-5-( 1 -methyl-2-pyrolidinylethylamidosulphonyl)phenyl] – 1 -methyl-prop yl- 1 ,6-dihydro-7H-pyrazolo(4,3-d)pyrimidin-7-one, a pyrazolopyrimidinone compound, represented by the following Chemical Formula 1 or pharmaceutically acceptable salts thereof, as an active ingredient for prevention and treatment of respiratory diseases. [14] [Chemical Formula 1]

………………
SYNTHESIS
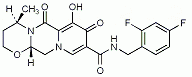
EXAMPLE 2 3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methyl-pyrrolidin-2-yl)-ethyl]-4-propoxy-benzenesulfonamide

Step 1

2,4-Dioxo-heptanoic acid methyl ester: Sodium (25.3 g, 1.1 mol) was proportionally added to ethanol (350 mL) at ambient temperature with vigorous stirring, and the solution was cooled to 0° C. Pentan-2-one (86 g, 1.0 mol) and diethyl oxalate (146 g, 1.0 mol) were added sequentially at 0° C., and stirring was continued for 1 hour at 0° C., and overnight at ambient temperature. The solvent was removed under reduced pressure, diethyl ether (200 mL) and cold dilute hydrochloric acid (500 mL) were added. Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound (141 g, 76%). 1H-NMR (300 MHz, CDCl3) δ 14.51 (broad s, 1H), 6.37 (s, 1H), 4.35 (q, 2H, J=6.6 Hz), 2.47 (t, 2H, J=7.2 Hz), 1.76-1.66 (m, 2H), 1.38 (t, 3H, J=7.2 Hz), 0.97 (t, 3H, J=7.5 Hz); GC-MS: 186 (M)+, 113 (M-73)+
Step 2

5-Propyl-2H-pyrazole-3-carboxylic acid ethyl ester: Hydrazine hydrate (41.4 g, 827 mmol) was slowly added to a solution of 2,4-dioxo-heptanoic acid methyl ester (140 g, 753 mmol) in 280 mL of acetic acid at 0° C. The mixture was heated to reflux for 8 hours and cooled. The solvent was removed under reduced pressure; the residue was diluted with diethyl ether (300 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound as a white solid (131 g, 96%). 1H NMR (300 MHz, CDCl3) δ 9.27 (broad s, 1H), 6.61 (s, 1H), 4.37 (q, 2H, J=7.2 Hz), 2.68 (t, 2H, J=7.5 Hz), 1.75-1.62 (m, 2H), 1.37 (t, 3H, J=6.6 Hz), 0.96 (t, 3H, J=7.2 Hz); LC-MS: m/z=183 (MH)+;
Step 3

2-Methyl-5-propyl-2H-pyrazole-3-carboxylic acid ethyl ester: A mixture of 5-propyl-2H-pyrazole-3-carboxylic acid ethyl ester (32.8 g, 180 mmol) and dimethyl sulfate (24.9 g, 198 mmol) was heated at 90° C. for 3 hours. The reaction was cooled and diluted with dichloromethane (200 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield a crude residue which was purified by flash chromatography on silica gel to give the title compound as a colorless oil (23 g, 65%). 1H NMR (300 MHz, CDCl3) δ 6.59 (s, 1H), 4.37 (q, 2H, J=7.2 Hz), 2.58 (t, 2H, J=7.2 Hz), 1.76-1.64 (m, 2H), 1.40 (t, 3H, J=6.6 Hz), 1.01 (t, 3H, J=7.2 Hz), 4.40 (q, 2H), 3.89 (s, 3H), 2.59 (t, 2H), 1.69 (2H), 1.37 (t, 3H), 1.01 (t, 3H); LC-MS: m/z=197 (MH)+.
Step 4

2-Methyl-5-propyl-2H-pyrazole-3-carboxylic acid: 2-methyl-5-propyl-2H-pyrazole-3-carboxylic acid ethyl ester (29.4 g, 150 mmol) was suspended in 6N sodium hydroxide (120 mL, 720 mmol) and heated to 80° C. for 2 hours, cooled, diluted with water (100 mL) and acidified with 5N hydrochloric acid (200 mL) to give a precipitate which was filtered off and dried to give the title compound as a white solid (24.2 g, 96%). 1H NMR (300 MHz, CDCl3) δ 6.76 (s, 1H), 4.17 (s, 3H), 2.63 (t, 2H, J=7.2 Hz), 1.70-1.68 (m, 2H), 0.98 (t, 3H, J=7.2 Hz); LC-MS: m/z=169 (M+H)+;
Step 5

2-Methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxylic acid: A solution of 2-methyl-5-propyl-2H-pyrazole-3-carboxylic acid (22 g, 131 mmol) in concentrated sulfuric acid (98%, 85 mL) was heated to 50° C. and treated with a mixture of fuming nitric acid (95%, 7.7 mL) and concentrated sulfuric acid (98%, 18 mL), while keeping the reaction temperature between 50 and 55° C. The reaction mixture was kept for 8 hours at 50° C., cooled to ambient temperature, and slowly added to cold water (600 mL, 4° C.), keeping the temperature below 25° C. The precipitate was collected by filtration, and dried below 80° C. to give the title compound as a white solid (25 g, 90%). 1H NMR (300 MHz, CDCl3) δ 4.25 (s, 3H), 2.92 (t, 2H, J=7.5 Hz), 1.77-1.70 (m, 2H), 1.03 (t, 3H, J=7.2 Hz); LC-MS: m/z=214 (M+H)+
Step 6

2-Methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxamide: To a suspension of 2-methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxylic acid (17.0 g, 79.8 mmol) in dry toluene (85 mL) was added a catalytic quantity of dimethylformamide (0.6 mL). The mixture was heated to 50° C. and thionyl chloride (17.1 g, 143.7 mmol) was added over 30 minutes. The reaction was stirred and heated at 55-60° C. for 6 hours. The solvent was removed, dry toluene (80 mL) was added and the mixture was cooled to 20° C. and cold (5° C.) concentrated ammonium hydroxide (100 mL) was added. The precipitate was filtered, washed with water and dried to give the title compound as an off-white solid (14.8 g, 87%). LC-MS: m/z=213 (M+H)+, 235 (M+Na)+.
Step 7

4-Amino-2-methyl-5-propyl-2H-pyrazole-3-carboxamide: To a suspension of 2-methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxamide (14.7 g, 69.3 mmol) in ethyl acetate (130 mL), was added 10% palladium on carbon (3.3 g). The mixture was reacted at 50° C. and 4 atm hydrogen pressure overnight. The reaction mixture was cooled, and the catalyst was filtered off and washed with ethyl acetate and dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to give the title compound (13.8 g, 98%). 1H NMR (300 MHz, CDCl3) δ 4.12 (s, 3H), 2.84 (s, 2H), 2.55 (t, 2H, J=7.2 Hz), 1.71-1.61 (m, 2H), 0.99 (t, 3H, J=7.2 Hz); LC-MS: m/z=183 (MH)+
Step 8

2-Methyl-4-(2-propoxybenzoylamino)-5-propyl-2H-pyrazole-3-carboxamide: A solution of 2-propoxybenzoic acid (13.7 g, 76.1 mmol) and thionyl chloride (36.2 g, 304.4 mmol) in dry dichloromethane (80 mL) was heated for 3 hours at reflux. The solvent and excess thionyl chloride were distilled off under reduced pressure. The residue was taken up in dry dichloromethane (60 mL) and reacted with a solution of 4-amino-2-methyl-5-propyl-2H-pyrazole-3-carboxamide (12.6 g, 69.2 mmol), dry triethylamine (7 g, 69.2 mmol) and 4-(N,N-dimethylamino)pyridine (84.5 mg, 0.7 mmol) in dry dichloromethane (200 mL) at 0° C. Stirring was maintained for 1 hour, and the reaction mixture was successively washed with water (150 mL), saturated aqueous sodium carbonate solution (200 mL) and saturated brine (200 mL). The organic layer was dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated to about 60 mL, and then hexane (150 mL) was added to give precipitate product as a white solid (22 g, 92%). 1H NMR (300 MHz, CDCl3) δ 9.47 (s, 1H), 8.28 (d, 1H, J=7.8 Hz), 7.87 (br.s, 1H), 7.57-7.52 (m, 1H), 7.16-7.05 (m, 2H), 5.53 (s, 1H), 4.20 (t, 2H, J=6.6 Hz), 4.09 (s, 3H), 2.54 (t, 2H, J=7.5 Hz), 1.97-1.85 (m, 2H), 1.69-1.26 (m, 2H), 1.07 (t, 3H, J=7.2 Hz), 0.95 (t, 3H, J=7.5 Hz). LC-MS: m/z=345 (M+H)+
Step 9

3-(5-Carbamoyl-1-methyl-3-propyl-1H-pyrazol-4-ylcarbamoyl)-4-propxy-benzenesulfonyl chloride: 2-Methyl-4-(2-propoxybenzoylamino)-5-propyl-2H-pyrazole-3-carboxamide (20 g, 58.1 mmol) was added to chlorosulfonic acid (81.3 g, 698 mmol) at 0° C. and the reaction was warmed to ambient temperature and stirred for 2 hours. The reaction mixture was poured into ice water (800 g) and mechanically stirred for 1 hour to give a white solid, which was filtered and washed with water. Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound (8 g, 31%). 1H NMR (300 MHz, CDCl3) δ 9.19 (s, 1H), 8.97 (s, 1H), 8.19 (t, 1H, J=8.9 Hz), 7.56 (br. s, 1H), 4.35 (t, 2H, J=6.6 Hz), 4.07 (s, 3H), 2.53 (t, 2H, J=7.5 Hz), 2.06-1.94 (m, 2H), 1.78-1.60 (m, 2H), 1.18 (t, 3H, J=7.5 Hz), 0.95 (t, 3H, J=7.2 Hz); LC-MS: m/z=443.1 (M+H)+
Step 10

2-Methyl-4-{5-[2-(1-methyl-pyrrolidin-2-yl)-ethylsulfamoyl]-2-propoxy-benzoylamino}-5-propyl-2H-pyrazole-3-carboxamide: To a solution of 3-(5-carbamoyl-1-methyl-3-propyl-1H-pyrazol-4-ylcarbamoyl)-4-propoxy-benzenesulfonyl chloride (2.12 g, 4.8 mmol) and dry triethylamine (0.5 g, 4.8 mmol) in dichloromethane (20 mL), was added 2-(2-aminoethyl)-1-methylpyrrolidine (0.6 g, 4.8 mmol) at 0° C. The reaction was warmed to ambient temperature, stirred for 1 hour at ambient temperature, and diluted with dichloromethane (40 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound (2.2 g) which was used directly in the next step. LC-MS: m/z=535 (M+H)+
Step 11

3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methyl-pyrrolidin-2-yl)-ethyl]-4-propoxy-benzenesulfonamide: Potassium tert-butoxide (0.9 g, 8.0 mmol) was added to a solution of crude 2-methyl-4-{5-[2-(1-methyl-pyrrolidin-2-yl)-ethylsulfamoyl]-2-propoxy-benzoylamino}-5-propyl-2H-pyrazole-3-carboxamide (2.14 g, 4.0 mmol) in dry tert-butanol (50 mL), and the mixture was heated to reflux for 8 hours. The reaction mixture was cooled to ambient temperature and diluted with ethyl acetate (300 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield a crude residue which was purified by flash chromatography to give the title compound (1.1 g, 53%).
1H NMR (300 MHz, CDCl3) δ 10.90 (broad s, 1H), 8.93 (s, 1H), 7.96 (d, 1H, J=8.7 Hz), 7.15 (d, 1H, J=8.7 Hz), 4.28-4.24 (m, 3H), 4.24 (s, 2H), 3.13 (t, 3H, J=6.9 Hz), 2.93 (t, 3H, J=7.8 Hz), 2.56 (s, 1H), 2.40 (s, 3H), 2.26-2.24 (m, 1H), 2.10-1.99 (m, 2H), 1.89-1.80 (m, 4H), 1.67 (s, 3H, J=7.2 Hz), 1.56-1.52 (m, 1H), 1.22 (t, 3H, J=7.5 Hz), 1.03 (t, 3H, J=7.2 Hz);
LC-MS: m/z=517 (MH)+
…………………….
References
- Udenafil Information
- Zydena (udenafil) product information page. Dong-A Pharmaceutical. Retrieved on April 13, 2009.
- Udenafil: efficacy and tolerability in the management of erectile dysfunction.
- http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3607490/
- British Journal of Pharmacology, 2008 , vol. 153, 7 PG. 1568 – 1578
- Arzneimittel-Forschung/Drug Research, 2009 , vol. 59, 12 pg. 641 – 646
- Chemical and Pharmaceutical Bulletin, 2011 , vol. 59, 9 PG. 1083 – 1088
- WO2010/13925 A2, …
- US2010/173915 A1
- WO2010/95849 A2,
- WO2007/114534 A1, …..
- Life Sciences, 2004 , vol. 75, 9 pg. 1075 – 1083 …………..mp 162 – 164 °C
- US2008/194529 A1,
-
WO2008100886A1 * Feb 12, 2008 Aug 21, 2008 Auspex Pharmaceuticals Inc Preparation and use of deuterated udenafil analogues as highly selective pde5 modulators for the treatment of erectile dysfunction -
US6333330 * Oct 22, 1999 Dec 25, 2001 Pfizer Inc. Pyrazolopyrimidinone CGMP PDE5 inhibitors for the treatment of sexual dysfunction US20040029891 * Sep 2, 2003 Feb 12, 2004 Pfizer Inc. Use of PDE5 inhibitors in the treatment of polycystic ovary syndrome -
WO1993006104A1 * Sep 4, 1992 Apr 1, 1993 Pfizer Pyrazolopyrimidinone antianginal agents WO1994028902A1 * May 13, 1994 Dec 22, 1994 Peter Ellis Pyrazolopyrimidinones for the treatment of impotence WO1996016657A1 * Oct 16, 1995 Jun 6, 1996 Simon Fraser Campbell Bicyclic heterocyclic compounds for the treatment of impotence WO1998049166A1 * Apr 10, 1998 Nov 5, 1998 Mark Edward Bunnage PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3′,5′-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION EP0463756A1 * Jun 7, 1991 Jan 2, 1992 Pfizer Limited Pyrazolopyrimidinone antianginal agents -
WO1993006104A1 * Sep 4, 1992 Apr 1, 1993 Pfizer Pyrazolopyrimidinone antianginal agents WO1998049166A1 * Apr 10, 1998 Nov 5, 1998 Mark Edward Bunnage PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3′,5′-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION WO2000027848A1 * Nov 10, 1999 May 18, 2000 Byoung Ok Ahn Pyrazolopyrimidinone derivatives for the treatment of impotence EP0463756A1 * Jun 7, 1991 Jan 2, 1992 Pfizer Limited Pyrazolopyrimidinone antianginal agents -
WO2004108726A1 May 14, 2004 Dec 16, 2004 Tianjin North Pharma Sci Tech 2-SUBSTITUTED PHENYL-5,7-DIALKYL-3,7-DIHYDROPYRROLE [2,3-d] PYRIMIDINE-4-ONE DERIVATIVES, THE PREPARATION AND THE PHARMACEUTICAL USE THEREOF US7741483 Mar 6, 2008 Jun 22, 2010 Yangtze River Pharmaceutical (Group) Co., Ltd. Process for making substituted pyrrolo[2,3-d]pyrimidine derivatives as inhibitors of phosphodiesterase 5

Dolutegravir approved by the EU Commission (synthesis included in this post)

Dolutegravir
2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
(4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Trade Name:Tivicay
Synonym:GSK1349572, S-349572, GSK572
Date of Approval: August 12, 2013 (US)
Indication:HIV infection
Drug class: Integrase strand transfer inhibitor
Company: ViiV Healthcare,GlaxoSmithKline
INNOVATOR …ViiV Healthcare
CAS number: 1051375-16-6
MF:C20H19F2N3O5
MW:419.4
Chemical Name: (4R,12aS)-N-[(2,4-difluorophenyl)methyl]-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a- hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Patent: US8129385
Patent expiration date: Oct 5, 2027
PCT patent application: W02006116764
Dolutegravir (DTG, GSK1349572) is an integrase inhibitor being developed for the treatment of human immunodeficiency virus (HIV)-1 infection by GlaxoSmithKline (GSK) on behalf of Shionogi-ViiV Healthcare LLC. DTG is metabolized primarily by uridine diphosphate glucuronyltransferase (UGT)1A1, with a minor role of cytochrome P450 (CYP)3A, and with renal elimination of unchanged drug being extremely low (< 1% of the dose).
The European Commission has on 21 January 2014 Dolutegravir (Tivicay, ViiV) permit as part of combination therapy for the treatment of HIV-infected persons over the age of 12 years.Dolutegravir (Tivicay, ViiV) is an integrase inhibitor, in combination with other antiretroviral drugs in adults and adolescents can be used from 12 years for the treatment of HIV infection.
Source: Communication from the European Commission
Dolutegravir[1] is a FDA-approved drug[2] for the treatment of HIV infection. Dolutegravir is an integrase inhibitor. Known as S/GSK1349572 or just “572” the drug is marketed as Tivicay[3] by GlaxoSmithKline (GSK). In February, 2013 the Food and Drug Administration announced that it would fast track dolutegravir’s approval process.[4] On August 13, 2013, dolutegravir was approved by the FDA. On November 4, 2013, dolutegravir was approved by Health Canada.[5]
The oral HIV integrase inhibitor S-349572 was originated by Shionogi-GlaxoSmithKline and Shionogi-ViiV Healthcare. In 2013, the product was approved and launched in the U.S. for the treatment of HIV-1 in adults and children aged 12 years and older, in combination with other antiretroviral agents. A positive opinion was received in the E.U for this indication and, in 2014, approval was attained in Europe for this indication. Registration is pending in Japan.
In 2013, orphan drug designation in Japan was assigned to the compound.
Dolutegravir is approved for use in a broad population of HIV-infected patients. It can be used to treat HIV-infected adults who have never taken HIV therapy (treatment-naïve) and HIV-infected adults who have previously taken HIV therapy (treatment-experienced), including those who have been treated with other integrase strand transfer inhibitors. Tivicay is also approved for children ages 12 years and older weighing at least 40 kilograms (kg) who are treatment-naïve or treatment-experienced but have not previously taken other integrase strand transfer inhibitors.[6]
Dolutegravir has also been compared head-to-head with a preferred regimen from the DHHS guidelines in each of the three classes (i.e. 1.) nuc + non-nuc, 2.) nuc + boosted PI, and 3.) nuc + integrase inhibitor).
SPRING-2 compared dolutegravir to another integrase inhibitor, raltegravir, with both coformulated with a choice of TDF/FTC orABC/3TC. After 48 weeks of treatment 88% of those on dolutegravir had less than 50 copies of HIV per mL compared to 85% in the raltegravir group, thus demonstrating non-inferiority.[9]
The FLAMINGO study has been presented at scientific meetings but as of early 2014 has not yet been published. It is an open-label trial of dolutegravir versus darunavir boosted with ritonavir. In this trial 90% of those on dolutegravir based regimens had viral loads < 50 at 48 weeks compared to 83% in the darunavir/r.[10] This 7% difference was statistically significant for superiority of the dolutegravir based regimens.
Another trial comparing dolutegravir to efavirenz, SINGLE, was the first trial to show statistical superiority to an efavirenz/FTC/TDF coformulated regimen for treatment naive patients.[11] After 48 weeks of treatment, 88% of the dolutegravir group had HIV RNA levels < 50 copies / mL versus 81% of the efavirenz group. This has led one commentator to predict that it may replace efavirenz as the first line choice for initial therapy as it can also be formulated in one pill, once-a-day regimens.[12]
Doultegravir has also been studied in patients who have been on previous antiretroviral medications. The VIKING trial looked at patients who had known resistance to the first generation integrase inhibitor raltegravir. After 24 weeks 41% of patients on 50mg dolutegravir once daily and 75% of patients on 50mg twice daily (both along with an optimized background regimen) achieved an HIV RNA viral load of < 50 copies per mL. This demonstrated that there was little clinical cross-resistance between the two integrase inhibitors. [13]
Dolutegravir (also known as S/GSK1349572), a second-generation integrase inhibitor under development by GlaxoSmithKline and its Japanese partner Shionogi for the treatment of HIV infection, was given priority review status from the US Food and Drug Administration (FDA) in February, 2013.
GlaxoSmithKline marketed the first HIV drug Retrovir in 1987 before losing out to Gilead Sciences Inc. (GILD) as the world’s biggest maker of AIDS medicines. The virus became resistant to Retrovir when given on its own, leading to the development of therapeutic cocktails.
The new once-daily drug Dolutegravir, which belongs to a novel class known as integrase inhibitors that block the virus causing AIDS from entering cells, is owned by ViiV Healthcare, a joint venture focused on HIV in which GSK is the largest shareholder.
Raltegravir (brand name Isentress) received approval by the U.S. Food and Drug Administration (FDA) on 12 October 2007, the first of a new class of HIV drugs, the integrase inhibitors, to receive such approval. it is a potent and well tolerated antiviral agent. However, it has the limitations of twice-daily dosing and a relatively modest genetic barrier to the development of resistance, prompting the search for agents with once-daily dosing.
Elvitegravir, approved by the FDA on August 27, 2012 as part of theelvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine fixed-dose combination pill (Quad pill, brand name Stribild) has the benefit of being part of a one-pill, once-daily regimen, but suffers from extensive cross-resistance with raltegravir.
Gilead’s Atripla (Emtricitabine/Tenofovir/efavirenz), approved in 2006 with loss of patent protection in 20121, is the top-selling HIV treatment. The $3.2 billion medicine combines three drugs in one pill, two compounds that make up Gilead’s Truvada (Emtricitabine/Tenofovir) and Bristol- Myers Squibb Co.’s Sustiva (Efavirenz).
A three-drug combination containing dolutegravir and ViiV’s older two-in-one treatment Epzicom(Abacavir/Lamivudine, marketed outside US as Kivexa) proved better than Gilead’s market-leading Atripla in a clinical trial released in July, 2012 (See the Full Conference Report Here), suggesting it may supplant the world’s best-selling AIDS medicine as the preferred front-line therapy. In the latest Phase III study, after 48 weeks of treatment, 88% of patients taking the dolutegravir-based regimen had reduced viral levels to the goal compared with 81% of patients taking Atripla. More patients taking Atripla dropped out of the study because of adverse events compared with those taking dolutegravir — 10% versus just 2% — which was the main driver of the difference in efficacy. The result was the second positive final-stage clinical read-out for dolutegravir, following encouraging results against U.S. company Merck & Co’s rival Isentress in April, 2012 (See the Conference Abstract Here)..
Dolutegravir is viewed by analysts as a potential multibillion-dollar-a-year seller, as its once-daily dosing is likely to be attractive to patients. The FDA is scheduled to issue a decision on the drug’s approval by August 17。
TIVICAY contains dolutegravir, as dolutegravir sodium, an HIV INSTI. The chemical name of dolutegravir sodium is sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate. The empirical formula is C20H18F2N3NaO5 and the molecular weight is 441.36 g/mol. It has the following structural formula:
 |
Dolutegravir sodium is a white to light yellow powder and is slightly soluble in water.
Each film-coated tablet of TIVICAY for oral administration contains 52.6 mg of dolutegravir sodium, which is equivalent to 50 mg dolutegravir free acid, and the following inactive ingredients: D-mannitol, microcrystalline cellulose, povidone K29/32, sodium starch glycolate, and sodium stearyl fumarate. The tablet film-coating contains the inactive ingredients iron oxide yellow, macrogol/PEG, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
……………………………………
INTRODUCTION
Among viruses, human immunodeficiency virus (HIV), a kind of retrovirus, is known to cause acquired immunodeficiency syndrome (AIDS). The therapeutic agent for AIDS is mainly selected from a group of reverse transcriptase inhibitors (e.g., AZT, 3TC) and protease inhibitors (e.g., Indinavir), but they are proved to be accompanied by side effects such as nephropathy and the emergence of resistant viruses. Thus, the development of anti-HIV agents having the other mechanism of action has been desired.
On the other hand, a combination therapy is reported to be efficient in treatment for AIDS because of the frequent emergence of the resistant mutant. Reverse transcriptase inhibitors and protease inhibitors are clinically used as an anti-HIV agent, however agents having the same mechanism of action often exhibit cross-resistance or only an additional activity. Therefore, anti-HIV agents having the other mechanism of action are desired.
Under the circumstances above, an HIV integrase inhibitor has been focused on as an anti-HIV agent having a novel mechanism of action (Ref: Patent Documents 1 and 2). As an anti-HIV agent having such a mechanism of action, known are carbamoyl-substituted hydroxypyrimidinone derivative (Ref: Patent Documents 3 and 4) and carbamoyl-substituted hydroxypyrrolidione derivative (Ref: Patent Document 5). Further, a patent application concerning carbamoyl-substituted hydroxypyridone derivative has been filed (Ref: Patent Document 6, Example 8).
Other known carbamoylpyridone derivatives include 5-alkoxypyridine-3-carboxamide derivatives and γ-pyrone-3-carboxamide derivatives, which are a plant growth inhibitor or herbicide (Ref: Patent Documents 7-9).
Other HIV integrase inhibitors include N-containing condensed cyclic compounds (Ref: Patent Document 10).
- [Patent Document 1] WO03/0166275
- [Patent Document 2] WO2004/024693
- [Patent Document 3] WO03/035076
- [Patent Document 4] WO03/035076
- [Patent Document 5] WO2004/004657
- [Patent Document 6] JP Patent Application 2003-32772
- [Patent Document 7] JP Patent Publication 1990-108668
- [Patent Document 8] JP Patent Publication 1990-108683
- [Patent Document 9] JP Patent Publication 1990-96506
- [Patent Document 10] WO2005/016927
-
Patent Document 1 describes compounds (I) and (II), which are useful as anti-HIV drugs and shown by formulae:

-
This document describes the following reaction formula as a method of producing compound (I).


-
Furthermore, Patent Documents 2 to 6 describe the following reaction formula as an improved method of producing compound (I).


-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
- [PATENT DOCUMENTS]
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
-



…………………………………………
Dolutegravir synthesis (EP2602260, 2013). LiHMDS as the non-nucleophilic strong base pulling compound 1 carbonyl group proton alpha position with an acid chloride after 2 and ring closure reaction to obtain 3 , 3 via primary amine 4 ring opening ring closure to obtain 5 , NBS the bromine under acidic conditions to obtain aldehyde acetal becomes 6 , 6 of the aldehyde and amino alcohols 7 and turn off the condensation reaction obtained by the ring 8 , alkaline hydrolysis 8 of bromine into a hydroxyl group and hydrolyzable ester obtained 9 after the 10 occurred acid condensation Dolutegravir.
………………………………………………………
Synthesis of Dolutegravir (S/GSK1349572, GSK1349572)

………………………
SYNTHESIS

2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS) ………..dolutegravir
PATENT US8129385

Desired isomer
Example Z-1
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

a)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0.14 mL, 1.74 mmol) and 10 drops of glacial acetic acid. The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added to the mixture and the solvents removed in vacuo and the material was purified via silica gel chromatography (2% CH3OH/CH2Cl2 gradient elution) to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 92%) as a glass. 1H NMR (CDCl3) δ 10.38 (m, 1H), 8.42 (s, 1H), 7.54-7.53 (m, 2H), 7.37-7.24 (m, 4H), 6.83-6.76 (m, 2H), 5.40 (d, J=10.0 Hz, 1H), 5.22 (d, J=10.0 Hz, 1H), 5.16 (dd, J=9.6, 6.0 Hz, 1H), 4.62 (m, 2H), 4.41 (m, 1H), 4.33-4.30 (m, 2H), 3.84 (dd, J=12.0, 10.0 Hz, 1H), 3.63 (dd, J=8.4, 7.2 Hz, 1H), 1.37 (d, J=6.0 Hz, 3H); ES+MS: 496 (M+1).
b)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt. To a solution of (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture was filtered through Celite with methanol and dichloromethane.
The filtrate was concentrated in vacuo to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide , DOLUTEGRAVIR as a pink tinted white solid (278 mg, 86%).
1H NMR (CDCl3) δ 11.47 (m, 1H), 10.29 (m, 1H), 8.32 (s, 1H), 7.36 (m, 1H), 6.82 (m, 2H), 5.31 (dd, J=9.6, 3.6 Hz, 1H), 4.65 (m, 2H), 4.47-4.38 (m, 3H), 3.93 (dd, J=12.0, 10.0 Hz, 1H), 3.75 (m, 1H), 1.49 (d, J=5.6 Hz, 3H); ES+ MS: 406 (M+1).
DOLUTEGRAVIR NA SALT
The above material (278 mg, 0.66 mmol) was taken up in ethanol (10 mL) and treated with 1 N sodium hydroxide (aq) (0.66 ml, 0.66 mmol). The resulting suspension was stirred at room temperature for 30 min. Ether was added and the liquids were collected to provide the sodium salt of the title compound as a white powder (291 mg, 99%). 1H NMR (DMSO-d6) δ 10.68 (m, 1H), 7.90 (s, 1H), 7.35 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.20 (m, 1H), 4.58 (m, 1H), 4.49 (m, 2H), 4.22 (m, 2H), 3.74 (dd, J=11.2, 10.4 Hz, 1H), 3.58 (m, 1H), 1.25 (d, J=4.4 Hz, 3H).
UNDESIRED ISOMER
Example Z-9
(3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

The title compound was made in two steps using a similar process to that described in example Z-1. 16a (510 mg, 1.08 mmol) and (25)-2-amino-1-propanol (0.17 mL, 2.17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give (3S,11aR)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500 mg, 93%). This material was hydrogenated in a second step as described in example Z-1 to give (3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (386 mg, 94%) as a tinted white solid. 1H NMR (CDCl3) δ 11.46 (m, 1H), 10.28 (m, 1H), 8.32 (s, 1H), 7.35 (m, 1H), 6.80 (m, 2H), 5.30 (dd, J=10.0, 4.0 Hz, 1H), 4.63 (m, 2H), 4.48-4.37 (m, 3H), 3.91 (dd, J=12.0, 10.0 Hz, 1H), 3.73 (m, 1H), 1.48 (d, J=6.0 Hz, 3H); ES+ MS: 406 (M+1). This material (385 mg, 0.95 mmol) was treated with sodium hydroxide (0.95 mL, 1.0 M, 0.95 mmol) in ethanol (15 mL) as described in example Z-1 to provide its corresponding sodium salt (381 mg, 94%) as a white solid. 1H NMR (DMSO-d6) δ 10.66 (m, 1H), 7.93 (s, 1H), 7.33 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.19 (m, 1H), 4.59 (m, 1H), 4.48 (m, 2H), 4.22 (m, 2H), 3.75 (m, 1 H), 3.57 (m, 1H), 1.24 (d, J=5.6 Hz, 3H).
SYNTHESIS OF INTERMEDIATES

IN ABOVE SCHEME SYNTHESIS UPTO COMPD 9 MAY BE USEFUL IN SYNTHESIS BUT READERS DISCRETION IS SOUGHT IN THIS ?????????????????
1) Maltol 1 (189 g, 1.5 mol) was dissolved in dimethylformamide (1890 ml), and benzyl bromide (184 ml, 1.5 mol) was added. After the solution was stirred at 80° C. for 15 minutes, potassium carbonate (228 g, 1.65 mol) was added, and the mixture was stirred for 1 hour. After the reaction solution was cooled to room temperature, an inorganic salt was filtered, and the filtrate was distilled off under reduced pressure. To the again precipitated inorganic salt was added tetrahydrofuran (1000 ml), this was filtered, and the filtrate was distilled off under reduced pressure to obtain the crude product (329 g, >100%) of 3-benzyloxy-2-methyl-pyran-4-one 2 as a brown oil.
NMR (CDCl3) δ: 2.09 (3H, s), 5.15 (2H, s), 6.36 (1H, d, J=5.6 Hz), 7.29-7.41 (5H, m), 7.60 (1H, d, J=5.6 Hz).
2) The compound 2 (162.2 g, 750 mmol) was dissolved in ethanol (487 ml), and aqueous ammonia (28%, 974 ml) and a 6N aqueous sodium hydroxide solution (150 ml, 900 mmol) were added. After the reaction solution was stirred at 90° C. for 1 hour, this was cooled to under ice-cooling, and ammonium chloride (58 g, 1080 mmol) was added. To the reaction solution was added chloroform, this was extracted, and the organic layer was washed with an aqueous saturated sodium bicarbonate solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, isopropyl alcohol and diethyl ether were added to the residue, and precipitated crystals were filtered to obtain 3-benzyloxy-2-methyl-1H-pyridine-4-one 3 (69.1 g, 43%) as a pale yellow crystal.
NMR (DMSO-d6) δ: 2.05 (3H, s), 5.04 (2H, s), 6.14 (1H, d, J=7.0 Hz), 7.31-7.42 (5H, m), 7.46 (1H, d, J=7.2 Hz), 11.29 (1H, brs).
3) The above compound 3 (129 g, 699 mmol) was suspended in acetonitrile (1300 ml), and N-bromosuccinic acid imide (117 g, 659 mmol) was added, followed by stirring at room temperature for 90 minutes. Precipitated crystals were filtered, and washed with acetonitrile and diethyl ether to obtain 3-benzyloxy-5-bromo-2-methyl-pyridine-4-ol 4 (154 g, 88%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 5.04 (2H, s), 7.32-7.42 (5H, m), 8.03 (1H, d, J=5.5 Hz), 11.82 (1H, brs).
4) To a solution of the compound 4 (88 g, 300 mmol), palladium acetate (13.4 g, 60 mmol) and 1,3-bis(diphenylphosphino)propane (30.8 g, 516 mmol) in dimethylformamide (660 ml) were added methanol (264 ml) and triethylamine (210 ml, 1.5 mol) at room temperature. The interior of a reaction vessel was replaced with carbon monoxide, and the material was stirred at room temperature for 30 minutes, and stirred at 80 degree for 18 hours. A vessel to which ethyl acetate (1500 ml), an aqueous saturated ammonium chloride solution (1500 ml) and water (1500 ml) had been added was stirred under ice-cooling, and the reaction solution was added thereto. Precipitates were filtered, and washed with water (300 ml), ethyl acetate (300 ml) and diethyl ether (300 ml) to obtain 5-benzyloxy-4-hydroxy-6-methyl-nicotinic acid methyl ester 5 (44.9 g, 55%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 3.72 (3H, s), 5.02 (2H, s), 7.33-7.42 (5H, m), 8.07 (1H, s).
5) After a solution of the compound 5 (19.1 g, 70 mmol) in acetic anhydride (134 ml) was stirred at 130° C. for 40 minutes, the solvent was distilled off under reduced pressure to obtain 4-acetoxy-5-benzyloxy-6-methyl-nicotinic acid methyl ester 6 (19.9 g, 90%) as a flesh colored crystal.
NMR (CDCl3) δ: 2.29 (3H, s), 2.52 (3H, s), 3.89 (3H, s), 4.98 (2H, s), 7.36-7.41 (5H, m), 8.85 (1H, s).
6) To a solution of the compound 6 (46.2 g, 147 mmol) in chloroform (370 ml) was added metachloroperbenzoic acid (65%) (42.8 g, 161 mmol) in portions under ice-cooling, and this was stirred at room temperature for 90 minutes. To the reaction solution was added a 10% aqueous potassium carbonate solution, and this was stirred for 10 minutes, followed by extraction with chloroform. The organic layer was washed with successively with a 10% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under induced pressure, and the residue was washed with diisopropyl ether to obtain 4-acetoxy-5-benzyloxy-6-methyl-1-oxy-nicotinic acid methyl ester 7 (42.6 g, 87%) as a colorless crystal.
NMR (CDCl3) δ: 2.30 (3H, s), 2.41 (3H, s), 3.90 (3H, s), 5.02 (2H, s), 7.37-7.39 (5H, m), 8.70 (1H, s).
7) To acetic anhydride (500 ml) which had been heated to stir at 130° C. was added the compound 7 (42.6 g, 129 mmol) over 2 minutes, and this was stirred for 20 minutes. The solvent was distilled off under reduced pressure to obtain 4-acetoxy-6-acetoxymethyl-5-benzyloxy-nicotinic acid methyl ester 8 (49.6 g, >100%) as a black oil.
NMR (CDCl3) δ: 2.10 (3H, s), 2.28 (3H, s), 3.91 (3H, s), 5.07 (2H, s), 5.20 (2H, s), 7.35-7.41 (5H, m), 8.94 (1H, s).
8) To a solution of the compound 8 (46.8 g, 125 mmol) in methanol (140 ml) was added a 2N aqueous sodium hydroxide solution (376 ml) under ice-cooling, and this was stirred at 50° C. for 40 minutes. To the reaction solution were added diethyl ether and 2N hydrochloric acid under ice-cooling, and precipitated crystals were filtered. Resulting crystals were washed with water and diethyl ether to obtain 5-benzyloxy-4-hydroxy-6-hydroxymethyl-nicotinic acid 9 (23.3 g, 68%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.49 (2H, s), 5.19 (2H, s), 5.85 (1H, brs), 7.14-7.20 (2H, m), 7.33-7.43 (7H, m), 8.30 (1H, s), 10.73 (1H, t, J=5.8 Hz), 11.96 (1H, brs).
9) To a solution of the compound 9 (131 g, 475 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (219 g, 1140 mmol) and 1-hydroxybenzotriazole (128 g, 950 mmol) in dimethylformamide (1300 ml) was added 4-fluorobenzylamine (109 ml, 950 mmol), and this was stirred at 80° C. for 1.5 hours. After the reaction solution was cooled to room temperature, hydrochloric acid was added, followed by extraction with ethyl acetate. The extract was washed with a 5% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure to obtain a mixture (175 g) of 10 and 11. the resulting mixture was dissolved in acetic acid (1050 ml) and water (1050 ml), and zinc (31.1 g, 475 mmol) was added, followed by heating to reflux for 1 hour. After the reaction solution was cooled to room temperature, a 10% aqueous potassium carbonate solution was added, followed by extraction with ethyl acetate. The extract was washed with an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. After the solvent was distilled off under reduced pressure, this was washed with diethyl ether to obtain 5-benzyloxy-N-(4-fluoro-benzyl)-4-hydroxy-6-hydroxymethyl-nicotinic acid amide 10 (107 g, 59%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.45 (2H, d, J=4.3 Hz), 4.52 (2H, d, J=5.8 Hz), 5.09 (2H, s), 6.01 (1H, brs), 7.36-7.43 (5H, m), 8.31 (1H, s), 12.63 (1H, brs).
………………..
SYNTHESIS
- Example 3

3H IS DOLUTEGRAVIR
Step 1
-
N,N-dimethylformamide dimethyl acetal (4.9 ml, 36.5 mmol) was added dropwise to compound 3A (5.0 g, 30.4 mmol) under cooling at 0°C. After stirring at 0°C for 1 hour, 100 ml of ethyl acetate was added to the reaction solution, and the organic layer was washed with a 0.5 N aqueous hydrochloric acid solution (50 ml). The aqueous layer was separated, followed by extraction with ethyl acetate (50 ml). The organic layers were combined, washed with a saturated aqueous solution of sodium bicarbonate and saturated saline in this order, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 1:1 (v/v) → ethyl acetate) to obtain 4.49 g (yield: 67%) of compound 3B as an oil.
1H-NMR (CDCl3)δ:1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Step 2
-
Lithium hexamethyldisilazide (1.0 M solution in toluene, 49 ml, 49.0 mmol) was diluted with tetrahydrofuran (44 ml). A tetrahydrofuran (10 ml) solution of compound 3B (4.49 g, 20.4 mmol) was added dropwise thereto under cooling at -78°C, and a tetrahydrofuran (10 ml) solution of ethyl oxalyl chloride (3.35 g, 24.5 mmol) was then added dropwise to the mixture. The mixture was stirred at -78°C for 2 hours and then heated to 0°C. 2 N hydrochloric acid was added to the reaction solution, and the mixture was stirred for 20 minutes, followed by extraction with ethyl acetate (200 ml x 2). The organic layer was washed with a saturated aqueous solution of sodium bicarbonate and saturated saline and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 7:3 → 5:5 → 0:10 (v/v)) to obtain 1.77 g (yield: 31%) of compound 3C as a white solid.
1H-NMR (CDCl3)δ:1.36-1.46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Step 3
-
Aminoacetaldehyde dimethyl acetal (0.13 ml, 1.20 mmol) was added to an ethanol (6 ml) solution of compound 3C (300 mg, 1.09 mmol) at 0°C, and the mixture was stirred at 0°C for 1.5 hours, then at room temperature for 18 hours, and at 60°C for 4 hours. The solvent in the reaction solution was distilled off under reduced pressure, and the obtained residue was then purified by silica gel column chromatography (n-hexane-ethyl acetate: 5:5 → 0:10 (v/v)) to obtain 252 mg (yield: 64%) of compound 3D as an oil.
1H-NMR (CDCl3)δ:1.36-1.47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Step 4
-
62% H2SO4 (892 mg, 5.64 mmol) was added to a formic acid (10 ml) solution of compound 3D (1.02 g, 2.82 mmol), and the mixture was stirred at room temperature for 16 hours. The formic acid was distilled off under reduced pressure. To the residue, methylene chloride was added, and the mixture was pH-adjusted to 6.6 by the addition of a saturated aqueous solution of sodium bicarbonate. The methylene chloride layer was separated, while the aqueous layer was subjected to extraction with methylene chloride. The methylene chloride layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 531.8 mg of compound 3E as a yellow oil.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Step 5
-
Methanol (0.20 ml, 5.0 mmol), (R)-3-amino-butan-1-ol (179 mg, 2.0 mmol), and acetic acid (0.096 ml, 1.70 mmol) were added to a toluene (5 ml) solution of compound 3E (531 mg, 1.68 mmol), and the mixture was heated to reflux for 4 hours. The reaction solution was cooled to room temperature, then diluted with chloroform, and then washed with a saturated aqueous solution of sodium bicarbonate. The aqueous layer was subjected to extraction with chloroform. The chloroform layers were combined, washed with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (chloroform-methanol: 100:0 → 90:10) to obtain 309.4 mg of compound 3F as a brown oil.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
Step 6
-
Potassium trimethylsilanolate (333 mg, 2.34 mmol) was added to a 1,2-dimethoxyethane (2 ml) solution of compound 3F (159 mg, 0.47 mmol), and the mixture was stirred at room temperature for 7 hours. 1 N hydrochloric acid and saturated saline were added to the reaction solution, followed by extraction with chloroform. The chloroform layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 34.4 mg (yield: 25%) of compound 3G as an orange powder.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H,m), 4.06-4.10 (2H, m), 4.31 (1H, dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz), 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
Step 7
-
Compound 3G (16 mg, 0.054 mmol) and 2,4-difluorobenzylamine (17 mg, 0.12 mmol) were dissolved in N,N-dimethylformamide (1 ml). To the solution, N,N,N’,N’-tetramethyl-O-(7-aza-benzotriazol-1-yl)uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol) and N-methylmorpholine (0.031 ml, 0.28 mmol) were added, and the mixture was stirred at room temperature for 16 hours. 2,4-difluorobenzylamine (17 mg, 0.12 mmol), HATU (64 mg, 0.17 mmol), and N-methylmorpholine (0.037 ml, 0.34 mmol) were further added thereto, and the mixture was stirred at room temperature for additional 16 hours. 0.5 N hydrochloric acid was added to the reaction solution, followed by extraction with ethyl acetate. The ethyl acetate layers were combined, washed with 0.5 N hydrochloric acid and then with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by preparative high-performance liquid chromatography to obtain 12.5 mg (yield: 55%) of compound 3H as an orange solid.
- DOLUTEGRAVIR
-
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s).
ISOMERS OF DOLUTEGRAVIR
- Reference Example 1

Step 1
-
Acetic acid (180 mg, 3.00 mmol) was added to a toluene (90 ml) solution of compound A-1 (4.39 g, 9.33 mmol) and (R)-3-aminobutan-1-ol (998 mg, 11.2 mmol), and the mixture was stirred at 50°C for 90 minutes. The reaction solution was allowed to cool to room temperature and then poured to a saturated aqueous solution of sodium bicarbonate. The organic layer was separated, while the aqueous layer was subjected to extraction three times with ethyl acetate. The combined extracts were washed with saturated saline and then dried over sodium sulfate. The solvent was distilled off to obtain 4.29 g of crude product A-2.
Step 2
-
The crude product A-2 obtained in the preceding step was dissolved in ethanol (40 ml). To the solution, a 2 N aqueous sodium hydroxide solution (20 ml) was added at room temperature, and the mixture was stirred at the same temperature for 2 hours. The reaction solution was neutralized to pH 7 using a 2 N aqueous hydrochloric acid solution. The solvent was directly distilled off. The obtained crude product A-3 was subjected to azeotropy with toluene (100 ml) and used in the next step without being purified.
Step 3
-
HOBt (1.65 g, 12.2 mmol) and WSC HCl (2.34 g, 12.2 mmol) were added at room temperature to a DMF (100 ml) solution of the crude product A-3 obtained in the preceding step, and the mixture was stirred at the same temperature for 15 hours. Water was added to the reaction solution, followed by extraction three times with ethyl acetate. The combined extracts were washed with water three times and then dried over sodium sulfate. The solvent was distilled off, and the obtained oil was subjected to silica gel column chromatography for purification. Elution was performed first with n-hexane-ethyl acetate (3:7, v/v) and then with only ethyl acetate. The fraction of interest was concentrated, and the obtained oil was then dissolved in ethyl acetate. The solution was crystallized with diisopropyl ether as a poor solvent. The obtained crystals were collected by filtration and dissolved again in ethyl acetate. The solution was recrystallized to obtain 1.84 g of compound A-4.
1HNMR (CDCl3) δ: 1.49 (3H, d, J = 6.6 Hz), 1.88-1.96 (1H, m), 2.13-2.26 (1H, m), 3.90-4.17 (4H, m), 4.42-4.47 (1H, m), 4.63 (2H, d, J = 6.0 Hz), 5.12-5.17 (1H, m), 5.17 (1H, d, J = 9.9 Hz), 5.33 (1H, d, J = 9.9 Hz), 6.77-6.87 (2H, m), 7.27-7.42 (4H, m), 7.59-7.62 (2H, m), 8.35 (1H, s), 10.41 (1H, t, J = 5.7 Hz).
Step 4
-
The compound A-4 was subjected to the hydroxy deprotection reaction described in Step F of the paragraph [0088] to obtain compound A-5.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).
- Reference Example 2
-

-
Compound A-1 was reacted with (S)-3-aminobutan-1-ol in Step 1. Compound B-5 was obtained in the same way as in Reference Example 1.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).

……………..

ENTRY 68
………………………….
WO 2010068262
…………………………
WO 2010068253
…………………………………
WO 2011119566
…………………………..
Synthesis
Example 3

I was under cooling added dropwise at 0 ℃ (4.9 ml, 36.5 mmol) and N, N-dimethylformamide dimethyl acetal (5.0 g, 30.4 mmol) in the first step compound 3A. After stirring for 1 hour at 0 ℃, ethyl acetate was added to 100ml, the reaction mixture was washed with 0.5N aqueous hydrochloric acid (50 ml). Was extracted with ethyl acetate (50ml) and solution was separated and the aqueous layer. The organic layers were combined, washed successively with saturated aqueous sodium bicarbonate solution and saturated brine, and then dried over anhydrous sodium sulfate. After the solvent was distilled off, silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) → ethyl acetate 1:1) to an oil (67% yield) of Compound 3B 4.49 g I got a thing.
1 H-NMR (CDCl 3) δ: 1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Diluted with tetrahydrofuran (44 ml) (1.0M toluene solution, 49 ml, 49.0 mmol) the second step lithium hexamethyldisilazide, under cooling at -78 ℃, compound 3B (4.49 g, 20.4 mmol) in this After dropwise tetrahydrofuran (10 ml) was added dropwise tetrahydrofuran (3.35 g, 24.5 mmol) of ethyl oxalyl chloride and (10 ml) solution. After stirring for 2 hours at -78 ℃, I was warmed to 0 ℃. After washing (200 ml x 2), saturated aqueous sodium bicarbonate solution and the organic layer with saturated brine After stirring for 20 minutes, extracted with ethyl acetate by adding 2N hydrochloric acid, the reaction solution was dried over anhydrous sodium sulfate. After removal of the solvent, silica gel column chromatography and the residue obtained – was purified (n-hexane (v / v) ethyl acetate 7:3 → 5:5 → 0:10), compound 3C 1.77 g (yield I as a white solid 31%).
1 H-NMR (CDCl 3) δ :1.36-1 .46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Was added at 0 ℃ (0.13 ml, 1.20 mmol) the aminoacetaldehyde dimethyl acetal ethanol (300 mg, 1.09 mmol) of the third step compound 3C to (6 ml) solution, 1 hour and 30 minutes at 0 ℃, 18 hours at room temperature , then I was stirred for 4 hours at 60 ℃. After the solvent was evaporated under reduced pressure and the reaction mixture by silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) ethyl acetate 5:5 → 0:10), compound 3D 252 mg (yield: I got as an oil 64%) rate.
1 H-NMR (CDCl 3) δ :1.36-1 .47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Was added (892 mg, 5.64 mmol) and 2 SO 4 62-H% formic acid (1.02 g, 2.82 mmol) in a fourth step the compound for 3D (10 ml) solution was stirred at room temperature for 16 hours. Methylene chloride was added to the residue Shi distilled off under reduced pressure and formic acid was adjusted to pH = 6.6 by addition of saturated aqueous sodium bicarbonate. The solution was separated methylene chloride layer was extracted with methylene chloride and the aqueous layer. I was dried over anhydrous sodium sulfate combined methylene chloride layers. The solvent was then distilled off and was obtained as a yellow oil 531.8 mg compound 3E.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Amino – – butane – 1 – ol (179 mg, 2.0 mmol), methanol (0.20 ml, 5.0 mmol), (R) -3 toluene (531 mg, 1.68 mmol) in the fifth step to compound 3E (5 ml) solution was added (0.096 ml, 1.70 mmol) acetic acid was heated under reflux for 4 hours. After dilution with chloroform, cooled to room temperature, the reaction mixture was washed with a saturated aqueous sodium bicarbonate solution, and the aqueous layer was extracted with chloroform. After washing with saturated brine combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was then distilled off, silica gel column chromatography and the residue obtained – and (chloroform methanol 100:0 → 90:10), was obtained as a brown oil 309.4 mg compound 3F.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz ), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
1,2 (159 mg, 0.47 mmol) in the sixth step compound 3F – was added (333 mg, 2.34 mmol) and potassium trimethylsilanolate dimethoxyethane (2 ml) solution was stirred for 7 hours at room temperature. Brine was added to the 1N-hydrochloric acid to the reaction mixture, followed by extraction with chloroform. The combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was removed by distillation, and I as an orange powder (25% yield) of compound 3G 34.4 mg.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H, m), 4.06-4.10 (2H, m), 4.31 (1H , dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz) , 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
2,4 (16 mg, 0.054 mmol) and the seventh step compound 3G – was dissolved in N, N-dimethylformamide (1 ml) (17 mg, 0.12 mmol) difluorobenzyl amine, N, N, N ‘, N was added (0.031 ml, 0.28 mmol) and N-methylmorpholine uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol), and ‘- tetramethyl-O-(yl 7 – aza – – benzo triazolopyrimidine -1) I was stirred at room temperature for 16 h. 2,4 – was added (0.037 ml, 0.34 mmol) and N-methylmorpholine (64 mg, 0.17 mmol) and (17 mg, 0.12 mmol), HATU difluorobenzylamine, and the mixture was stirred for 16 hours at room temperature. I was extracted with ethyl acetate addition of 0.5N-hydrochloric acid to the reaction mixture. 0.5N-hydrochloric acid and then was washed with saturated brine, and dried over anhydrous sodium sulfate and combined ethyl acetate layer. The solvent was then distilled off, and purified by preparative high performance liquid chromatography residue was obtained as an orange solid (55% yield) of compound 3H 12.5 mg.
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s)
References
- [1] American Medical Association (AMA), STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL (Dolutegravir) Accessed 3 December 2011.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- “U.S. FDA approves GlaxoSmithKline’s HIV drug Tivicay”. Reuters. 12 August 2013. Retrieved 13 February 2013.
- “GSK wins priority status for new HIV drug in U.S”. Reuters. 16 February 2013. Retrieved 18 February 2013.
- “ViiV Healthcare receives approval for Tivicay™ (dolutegravir) in Canada for the treatment of HIV”. Retrieved 11 November 2013.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- U.S. FDA approves GlaxoSmithKline’s HIV drug Tivicay http://www.reuters.com/article/2013/08/12/us-glaxosmithkline-hivdrug-idUSBRE97B0WU20130812 Mon Aug 12, 2013 6:40pm EDT
- “Dolutegravir Prescribing Information”. Retrieved 3 January 2014.
- Raffi, F; Jaeger, H; Quiros-Roldan, E; Albrecht, H; Belonosova, E; Gatell, JM; Baril, JG; Domingo, P; Brennan, C; Almond, S; Min, S; extended SPRING-2 Study, Group (Nov 2013). “Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial.”. The Lancet infectious diseases 13 (11): 927–35. PMID 24074642.
- http://www.natap.org/2013/ICAAC/ICAAC_24.htm
- Walmsley, Sharon L.; Antela, Antonio; Clumeck, Nathan; Duiculescu, Dan; Eberhard, Andrea; Gutiérrez, Felix; Hocqueloux, Laurent; Maggiolo, Franco; Sandkovsky, Uriel; Granier, Catherine; Pappa, Keith; Wynne, Brian; Min, Sherene; Nichols, Garrett (7 November 2013). “Dolutegravir plus Abacavir–Lamivudine for the Treatment of HIV-1 Infection”. New England Journal of Medicine 369 (19): 1807–1818. doi:10.1056/NEJMoa1215541.
- Sax, Paul. “SINGLE Study Underscores Waning of the Efavirenz Era — But Probably Just in the USA – See more at:http://blogs.jwatch.org/hiv-id-observations/index.php/single-study-underscores-waning-of-the-efavirenz-era-but-probably-just-in-the-usa/2013/11/06/#sthash.A39SderN.dpuf”. Retrieved 19 December 2013.
- Eron, JJ; Clotet, B; Durant, J; Katlama, C; Kumar, P; Lazzarin, A; Poizot-Martin, I; Richmond, G; Soriano, V; Ait-Khaled, M; Fujiwara, T; Huang, J; Min, S; Vavro, C; Yeo, J; VIKING Study, Group (2013 Mar 1). “Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study.”. The Journal of infectious diseases 207 (5): 740–8. PMID 23225901.
-
WO2010011812A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds WO2010011819A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds -
-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
-
…………………
Sources:
Johns, Brian Alvin; Kawasuji, Takashi; Taishi, Teruhiko; Taoda, Yoshiyuki ; Polycyclic carbamoylpyridone derivative having HIV integrase inhibitory activity and their preparation; PCT Int. Appl., WO2006116764, 02 Nov 2006
Johns, Brian Alvin; Weatherhead, Jason Gordon;Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection; PCT Int. Appl., WO2010011812, 28 Jan 2010
Johns, Brian Alvin; Weatherhead, Jason Gordon; Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection;PCT Int. Appl., WO2010011819, 28 Jan 2010
Yoshida, Hiroshi; Taoda, Yoshiyuki; Johns, Brian Alvin; Synthesis of fused tricyclic carbamoylpyridone HIV integrase inhibitors and intermediates;PCT Int. Appl.,WO2010068253, 17 Jun 2010
Johns, Brian Alvin; Duan, Maosheng; Hakogi, Toshikazu;Processes and intermediates for fused tricyclic carbamoylpyridone HIV integrase inhibitors;PCT Int. Appl., WO2010068262, 17 Jun 2010
Sumino, Yukihito; Okamoto, Kazuya; Masui, Moriyasu; Yamada, Daisuke; Ikarashi, Fumiya;Preparation of compounds having HIV integrase inhibitory activity; PCT Int. Appl.,WO2012018065, 09 Feb 2012
Kawasuji, Takashi; Johns, Brian A.;Discovery of dolutegravir and S/GSK1265744: Carbamoyl pyridone HIV-1 integrase inhibitors;Abstracts, 64th Southeast Regional Meeting of the American Chemical Society, Raleigh, NC, United States, November 14-17 (2012), SERM-176.
Kawasuji, Takashi; Johns, Brian A.; Yoshida, Hiroshi; Weatherhead, Jason G.; Akiyama, Toshiyuki; Taishi, Teruhiko; Taoda, Yoshiyuki; Mikamiyama-Iwata, Minako; Murai, Hitoshi; Kiyama, Ryuichi; Fuji, Masahiro; Tanimoto, Norihiko; Yoshinaga, Tomokazu; Seki, Takahiro; Kobayashi, Masanori; Sato, Akihiko; Garvey, Edward P.; Fujiwara, Tamio; Carbamoyl Pyridone HIV-1 Integrase Inhibitors. 2. Bi- and Tricyclic Derivatives Result in Superior Antiviral and Pharmacokinetic Profiles;Journal of Medicinal Chemistry (2013), 56(3), 1124-1135
Walmsley S et al. Dolutegravir (DTG; S/GSK1349572) + abacavir/lamivudine once daily statistically superior to tenofovir/emtricitabine/efavirenz: 48-week results – SINGLE (ING114467). 52nd ICAAC, 9-12 September 2012, San Francisco. Abstract H-556b.
http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=e1c18d5b-830f-4b4e-8671-35bcfb20eed5&cKey=af219b7d-2171-46b2-91ef-b8049552c9e5&mKey=%7b6B114A1D-85A4-4054-A83B-04D8B9B8749F%7d
http://www.natap.org/2012/ICAAC/ICAAC_06.htm
http://i-base.info/htb/20381
Raffi F et al. Once-daily dolutegravir (DTG; S/GSK1349572) is non-inferior to raltegravir (RAL) in antiretroviral-naive adults: 48 week results from SPRING-2 (ING113086). 19th International AIDS Conference. 22-27 July 2012, Washington. Late breaker oral presentation THLBB04.
http://pag.aids2012.org/abstracts.aspx?aid=20990
National Institutes of Health (U.S.). A trial comparing GSK1349572 50 mg plus abacavir/lamivudine once daily to Atripla (also called the SINGLE trial). Available from:http://clinicaltrials.gov/ct2/show/NCT01263015.
Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in combination therapy exhibits rapid and sustained antiviral response in ARV-naïve adults: 96-week results from SPRING-1 (ING112276) (Abstract 102LB). Paper presented at: 19th Conference on Retroviruses and Opportunistic Infections; 2012 March 5–8; Seattle, WA. Available from:http://www.retroconference.org/2012b/Abstracts/45432.html
AVANAFIL …..A PDE5 inhibitor.

AVANAFIL
A phosphodiesterase (PDE5) inhibitor, used to treat erectile dysfunction.

Avanafil is a new phosphodiesterase-5 inhibitor that is faster acting and more selective than other drugs belonging to the same class. Chemically, it is a derivative of pyrimidine and is only available as the S-enantiomer. FDA approved on April 27, 2012.
CAS RN: 330784-47-9
4-{[(3-chloro-4-methoxyphenyl)methyl]amino}-2-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide
| (S)-2-(2-Hydroxymethyl-1-pyrrolidinyl)-4-(3-chloro-4-methoxybenzylamino)-5-[(2-pyrimidinylmethyl)carbamoyl]pyrimidine |
| 4-[[(3-Chloro-4-methoxyphenyl)methyl]amino]-2-[(2S)-2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide |
| TA 1790 |
Molecular Formular: C23H26ClN7O3
Molecular Mass: 483.95064
- Stendra
- TA 1790
- TA-1790
- UNII-DR5S136IVO
- NDA 202276
INNOVATOR — VIVUS
APPROVED FDA 27/4/2-12
| Patent No | Patent Expiry | patent use code |
|---|---|---|
| 6656935 | Sep 13, 2020 | U-155 |
| 7501409 | May 5, 2023 |
U 155… TREATMENT OF ERECTILE DYSFUNCTION
| Exclusivity Code | Exclusivity_Date |
|---|---|
| NCE | Apr 27, 2017 |
Stendra (avanafil) was given the green light by the US Food and Drug Administration 27/4/2012, but there has been no launch yet as Vivus has been seeking a partner. The latest data should be attractive to potential suitors and could help Stendra take on other phosphodiesterase type 5 (PDE5) inhibitors, notably Pfizer’s Viagra (sildenafil) but also Eli Lilly’s Cialis (tadalafil) and Bayer’s Levitra (vardenafil).
read all at
http://www.pharmatimes.com/Article/13-06-20/Vivus_ED_drug_gets_to_work_in_less_than_15_mins.aspx
STENDRA (avanafil) is a selective inhibitor of cGMP-specific PDE5.
。

Avanafil is designated chemically as (S)-4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2pyrimidinylmethyl)-5-pyrimidinecarboxamide and has the following structural formula:
 |
Avanafil occurs as white crystalline powder, molecular formula C23H26ClN7O3 and molecular weight of 483.95 and is slightly soluble in ethanol, practically insoluble in water, soluble in 0.1 mol/L hydrochloric acid. STENDRA, for oral administration, is supplied as oval, pale yellow tablets containing 50 mg, 100 mg, or 200 mg avanafil debossed with dosage strengths. In addition to the active ingredient, avanafil, each tablet contains the following inactive ingredients: mannitol, fumaric acid, hydroxypropylcellulose, low substituted hydroxypropylcellulose, calcium carbonate, magnesium stearate, and ferric oxide yellow.
AVANAFIL

Avanafil is a PDE5 inhibitor approved for erectile dysfunction by FDA on April 27, 2012 [1] and by EMA on June 21, 2013.[2] Avanafil is known by the trademark names Stendra and Spedra and was developed by Vivus Inc. In July 2013 Vivus announced partnership with Menarini Group, which will commercialise and promote Spedra in over 40 European countries plus Australia and New Zealand.[3] Avanafil acts by inhibiting a specificphosphodiesterase type 5 enzyme which is found in various body tissues, but primarily in the corpus cavernosum penis, as well as the retina. Other similar drugs are sildenafil, tadalafil and vardenafil. The advantage of avanafil is that it has very fast onset of action compared with other PDE5 inhibitors. It is absorbed quickly, reaching a maximum concentration in about 30–45 minutes.[4] About two-thirds of the participants were able to engage in sexual activity within 15 minutes.[4]
Avanafil is a highly selective PDE5 inhibitor that is a competitive antagonist of cyclic guanosine monophosphate. Specifically, avanafil has a high ratio of inhibiting PDE5 as compared with other PDE subtypes allowing for the drug to be used for ED while minimizing adverse effects. Absorption occurs quickly following oral administration with a median Tmax of 30 to 45 minutes and a terminal elimination half-life of 5 hours. Additionally, it is predominantly metabolized by cytochrome P450 3A4. As such, avanafil should not be co-administered with strong cytochrome P450 3A4 inhibitors. Dosage adjustments are not warranted based on renal function, hepatic function, age or gender. Five clinical trials suggest that avanafil 100 and 200 mg doses are effective in improving the Sexual Encounter Profile and the Erectile Function Domain scores among men as part of the International Index of Erectile Function. A network meta-analysis comparing the PDE5 inhibitors revealed avanafil was less effective on Global Assessment Questionnaire question 1 while safety data indicated no major differences among the different PDE5 inhibitors. The most common adverse effects reported from the clinical trials associated with avanafil were headache, flushing, nasal congestion, nasopharyngitis, sinusitis, and dyspepsia.
A “phosphodiesterase type 5 inhibitor” or “PDE5 inhibitor” refers to an agent that blocks the degradative action of phosphodiesterase type 5 on cyclic GMP in the arterial wall smooth muscle within the lungs and in the smooth muscle cells lining the blood vessels supplying the corpus cavernosum of the penis. PDE5 inhibitors are used for the treatment of pulmonary hypertension and in the treatment of erectile dysfunction. Examples of PDE5 inhibitors include, without limitation, tadalafil, avanafil, lodenafil, mirodenafil, sildenafil citrate, vardenafil and udenafil and pharmaceutically acceptable salts thereof.
“Avanafil” refers to the chemical compound 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide, and its pharmaceutically acceptable salts. Avanafil is described in Limin M. et al., (2010) Expert Opin Investig Drugs, 19(11):1427-37. Avanafil has the following chemical formula:

Avanafil is being developed for erectile dysfunction. Avanafil currently has no trademarked term associated with it but it is being developed by Vivus Inc.
…………………………………
DESCRIPTION IN A PATENT
EXAMPLE 92-145
The corresponding starting compounds are treated in a similar manner to give the compounds as listed in the following Table 7.
| TABLE 7 |
 |
 |
 |
Amorphous MS(m/z): 484(MH+) |
ENTRY 98 IS AVANAFIL
…………………………………………………….
The invention discloses a preparation method of Avanafil (Avanafil, I), which comprises the following steps: carrying out a substitution reaction on 6-amino-1, 2-dihydro pyrimidine-2-keto-5-carboxylic acid ethyl ester (XII) and 3-chloro-4-methoxy benzyl chloride (XIII) so as to obtain 6-(3-chloro-4-methoxy benzyl amino)-1, 2-dihydro pyrimidine-2-keto-5-carboxylic acid ethyl ester (IXV); carrying out condensation on the compound (IXV) and S-hydroxymethyl pyrrolidine (II) so as to generate 4-[(3-chloro-4-methoxy benzyl) amino]-2-[2-(hydroxymethyl)-1-pyrrole alkyl] pyrimidine-5-carboxylic acid ethyl ester (XI); and carrying out hydrolysis on the compound (XI) and then carrying out an acylation reaction on the compound (XI) and the compound (XI) so as to obtain Avanafil (I). The preparation method is simple in process, economic and environmental-friendly, suitable for the requirements of industrialization amplification.
……………………………………………………
The invention discloses a method for preparing avanafil (Avanafil, I). The method comprises the steps of taking cytosine as an initial material; and orderly carrying out replacement, halogen addition and condensation reaction on a side chain 3-chlorine-4-methoxy benzyl halide (III), N-(2-methylpyrimidine) formamide (IV) and S-hydroxymethyl pyrrolidine (II), so as to obtain a target product avanafil (I). The preparation method is available in material, concise in technology, economic and environment-friendly, and suitable for the demands of industrial amplification.
…………………………………………………….
SYNTHESIS
Avanafil can be synthesized from a benzylamine derivative and a pyrimidine derivative REF 5:Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709

- ………………………………………………………
- SYNTHESIS
- A cutting that phenanthrene by a methylthio urea ( a ) and ethoxy methylene malonate ( 2 ) cyclization of 3 , chloride, phosphorus oxychloride get 4 , 4 with benzyl amine 5 occurred SNAr the reaction product after oxidation with mCPBA 6 . In pyrimidine, if the 2 – and 4 – positions are active simultaneously the same leaving group in the case, SNAr reaction occurs preferentially at 4 – position, but does not guarantee the 2 – side reaction does not occur. Here is an activity of the poor leaving group sulfide spans 2 – bit, and a good leaving group active chlorine occupy four – position, thus ensuring a high regioselectivity of the reaction. 4 – position after completion of the reaction, then the 2 – position of the group activation, where sulfide sulfoxide better than the leaving group. Amino alcohols 7 and 6 recurrence SNAr reaction 8 , 8 after alkaline hydrolysis and acid alpha amidation get that phenanthrene.
AVANAFIL
- …………………………….

-
- FDA approves Stendra for erectile dysfunction” (Press release). Food and Drug Administration (FDA). April 27, 2012.
- http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002581/human_med_001661.jsp&mid=WC0b01ac058001d124
- http://ir.vivus.com/releasedetail.cfm?releaseid=775706
- Kyle, Jeffery; Brown, Dana (2013). “Avanafil for Erectile Dysfunction”. Annals of Pharmacotherapy (Sage Publishing). doi:10.1177/1060028013501989. Retrieved 28 September 2013.
- Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709
-
- Peterson CA. Hemodynamic effect of avanafil and glyceryl trinitrate coadministration. , Drugs Context , Volume 2013 , 2013 Feb 26
- Gur S. The Effect of Intracavernosal Avanafil, a Newer Phosphodiesterase-5 Inhibitor, on Neonatal Type 2 Diabetic Rats With Erectile Dysfunction. , Urology , 2013 Dec 9
- Hill JK. Avanafil for erectile dysfunction. , Ann Pharmacother , Volume 47 , Issue 10 , 2013 Oct
- Sanford M. Avanafil: a review of its use in patients with erectile dysfunction. , Drugs Aging , Volume 30 , Issue 10 , 2013 Oct
- Hellstrom WJ. PDE5 inhibitors: considerations for preference and long-term adherence. , Int J Clin Pract , Volume 67 , Issue 8 , 2013 Aug
- Aversa A. An update on pharmacological treatment of erectile dysfunction with phosphodiesterase type 5 inhibitors. , Expert Opin Pharmacother , Volume 14 , Issue 10 , 2013 Jul
- Oelke M. Phosphodiesterase inhibitors in clinical urology. , Expert Rev Clin Pharmacol , Volume 6 , Issue 3 , 2013 May
- Kukreja RC. Sildenafil and cardioprotection. , Curr Pharm Des , Volume 19 , Issue 39 , 2013
- Day WW. An open-label, long-term evaluation of the safety, efficacy and tolerability of avanafil in male patients with mild to severe erectile dysfunction. , Int J Clin Pract, Volume 67 , Issue 4 , 2013 Apr
- Tang J. Comparative effectiveness and safety of oral phosphodiesterase type 5 inhibitors for erectile dysfunction: a systematic review and network meta-analysis. , Eur Urol , Volume 63 , Issue 5 , 2013 May
United States APPROVED 6656935 2012-04-27 EXPIRY 2020-09-13 United States 7501409 2012-04-27 2023-05-05 - Faster-Working Erectile Dysfunction Drug?. CBS News. November 24, 2009.
- Vivus says men taking avanafil were more likely to be ready for sex within 15 minutes. The Gaea Times. January 11, 2010.
- “Avanafil is the New Player in The Erectile Dysfunction Field”. June 28, 2011.
-
- • Hatzimouratidis, K., et al.: Drugs, 68, 231 (2008)
-
4-20-2011Tablets quickly disintegrated in oral cavity7-16-2010Combination treatment for diabetes mellitus8-28-2009Roflumilast for the Treatment of Pulmonary Hypertension1-32-2008Cyclic compounds
US5242391 Oct 30, 1991 Sep 7, 1993 ALZA Corporation Urethral insert for treatment of erectile dysfunction US5474535 Jul 19, 1993 Dec 12, 1995 Vivus, Inc. Dosage and inserter for treatment of erectile dysfunction US5773020 Oct 28, 1997 Jun 30, 1998 Vivus, Inc. Treatment of erectile dysfunction US6656935 Aug 10, 2001 Dec 2, 2003 Tanabe Seiyaku Co., Ltd. Aromatic nitrogen-containing 6-membered cyclic compounds Update nov 2015
NEW PATENT WO 2015177807

WO 2015177807
Suryakant Shivaji Pol; Nitin Sharadchandra Pradhan; Shashikant Balu Padwal; Vihar Raghunath Telange; Nitn Shankar Bondre
Wanbury ltd

The present invention relates to a novel compound of Formula (II), and its use in preparation of Avanafil, [Formula should be inserted here] wherein R is -OH, -CI or -OR1 and R1 is C1 to C3 alkyl group
It having been developed and launched by VIVUS and JW Pharmaceutical, under license from Mitsubishi Tanabe Pharma, and Auxilium Pharmaceuticals, for treating ED.
A process for preparation of Avanafil was first disclosed in US 6,797,709 (depicted in Scheme I), wherein 4-chloro-5-ethoxycarbonyl-2-methylthio-pyrimidine is coupled with 3-chloro-4-methoxybenzylamine in presence of triethylamine to provide compound of Formula (A), which on oxidization provides a sulfonyl compound of Formula (B). Said compound of Formula (B) is reacted with L-prolinol and exert compound of Formula (C). The resulting compound of Formula (C) undergoes column chromatographic purification and crystallization, while further subjected to hydrolysis to obtain compound of Formula (D). The compound of Formula (D) is coupled with 2-aminomethylpyrimidine to obtain Avanafil of Formula (I). The final product obtained is purified by column chromatography. The need to purify the intermediate compound of Formula (C) and final product, by column chromatography makes this process cumbersome, time consuming and unviable for large scale production thereby contributing to main disadvantages of the process.
Scheme I

Formula (A)m-CPBA/chloroform

Formula (C) Formula (B)
NaOH/DMSO

Formula (D) Formula (I)CN 103254179, discloses a process for preparation of Avanafi, wherein 3-chloro-4-methoxybenzylhalide is coupled with cytosine to result compound of Formula (E), later on condensation with L-prolinol yields 4-[(3-chloro-4-methoxy benzyl)amino-2-(2-hydroxymethyl)-l -pyrrolinyl]pyrimidine of Formula (F). The compound of Formula (F) is then condensed with N-(2-pyrimidylmethyl)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme II
Scheme II

Formula (F) Formula (I)
CN 103254180 describes an alternate process for preparation of Avanafil of Formula (I), wherein a substitution reaction on 6-amino-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester and 3-chloro-4-methoxybenzylchloride provides 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester of Formula (G) which on condensation with L-prolinoI generates 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid ethyl ester of Formula (H). The compound of Formula (H) is then hydrolysed and coupled with N-(2-pyrimidylmethyI)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme III
Scheme III

Formula (H) Formula (Γ)
In all the prior art discussed above, chiral compound L-prolinol is coupled in molecule in earlier steps of synthesis. This approach seems to be less feasible for large scale production; the insertion of L-prolinol in early stage may need to exert number of purifications for intermediates. Further the main shortcoming in such process is that the chirality of molecule is disturbed by inserting L-prolinol in early stages because there are number of operations in line in process to obtain the target compound.
CN 103483323, discloses a synthetic method for preparation of avanafil, wherein amidation of pyrimidine-5-carbonyl chlorides with 2-(aminomethyl)pyrimidine at temperature ranging from -10 to 5°C resulted an amide (intermediates A); which underwent condensation with 3-chloro-4-methoxybenzylamine at the temperature ranging from 0 -3°C to give 4-[(3-chloro-4-methoxybenzyl)amino]-5-
pyrimidinecarboxamides (intermediates B), which further on condensation with L-prolinol gave avanafil. The disadvantage of this process is the need to maintain the reaction temperature in range of – 10 to 5°C which adds up to cost of process and makes the process complicated. The process is depicted in Scheme IV.
Scheme IV

Intermediate (A)

wherein, R’ & R2 are independently, hydrogen, halogen, alkoxy, alkoxyalkyl, cyno group, amino group
Hence, to overcome shortcomings of prior art the inventors of present invention have skillfully designed a process with novel intermediate which concomitantly result Avanafil compound of Formula (I), substantially free from impurities. Further this invention encompass L-proline in last stage of molecule in order to avoid the number of purifications of intermediate which relent the economic significances by taking into account yield of each stage.
Object of the invention
1. The main object of the invention is to provide a novel compound of Formula
(ID-
2. Another object of present invention is to provide a process for preparation of a novel compound of Formula (II).
3. Yet another object of present invention is to provide a process for preparation of Avanafil of Formula (I), in high yield and purity using a novel compound of Formula (II).
4. Yet another object of the present invention to provide simple, economic and industrially scalable process for the preparation of Avanafil o Formula (I).
Summary of the invention
According to an aspect of present invention, there is provided a novel compound of Formula (II).

Formula (II)
wherein R is -OH, -CI or -OR and R is Q to C3 alkyl group
The invention will be specifically described below with reference to Examples but it should not be construed that the scope of the invention is limited thereto. Since the starting compound was produced by a modified method from that described in prior art, it will be described as Referential Example 1 to 3. Here synthesis routes of Referential Example 1 to 3 and Example 1 to 10 are illustrated below in Scheme (V).
Scheme (V)

Formula (I) Referential Examples
Referential Example 1 – Preparation of ethyl 4-[(3-chloro-4-methoxybenzyl)amino]-2-(methyl sulfanyl)pyrimidine-5-carboxylate
To 600ml of methylene dichloride was added l OOg of ethyl 4-chloro-2-(methylsulfanyl) pyrimidine-5-carboxylate and 91.2g of 3-chloro-4-methoxybenzylamine. The reaction mixture was stirred and 500ml of water, 48g of sodium carbonate and Ig of tetra-butylammonium bromide were added to it. The reaction mixture was then maintained overnight at 25-30°C. After completion of reaction, methylene dichloride layer was separated, washed with water and evaporated to obtain 145g of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate having 95% of HPLC purity.
Above reaction can also be carried out using ammonia or triethylamine in same reaction conditions and parameters, in place of sodium carbonate.
Referential Example 2 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid
To 600ml of methanol was added l OOg of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate (Referential Example 1) and an aqueous solution of sodium hydroxide (15g of NaOH in 140ml of water). The reaction mixture was heated to reflux temperature. After completion of reaction, the pH of mixture was adjusted to 1 -2 using concentrated hydrochloric acid followed by stirring the mixture for 1 hour at 10-15°C. The solid product obtained was filtered, washed sequentially with water and methanol, and dried overnight at 70-75°C to get 87g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid.
Referential Example 3 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III)
To a mixture of 400ml of toluene and 0.5ml of dimethyl formamide was added 50g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid (Referential Example 2) and 70g of thionyl chloride, and the reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain yellow solid mass. The solid mass thus obtained, was cooled to 15-20°C followed by addition of 1 75ml of methylene dichloride, 36. l g of 2-amino methyl pyrimidine mesylate and 35.55g of triaethylamine. The reaction mixture was stirred overnight at 25-30°C. After completion of reaction, methylene dichloride was distilled out to get residue. The residue was washed sequentially with 2.5% sodium carbonate solution and water. The residue was then treated with methanol to obtain 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) having HPLC purity of more than 95% (yield: 80%)
Referential Example 4 – Preparation of 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[(2S)-2-(hydroxymethyl)-l -pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide (Avanafil)
Step i)
To 200ml of dichloromethane was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyI)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting dichloromethane layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the dichloromethane layer obtained in step i), was added 2.57g of triethylamine followed by slow addition of 125ml solution of L-prolinol in dichloromethane (2.46g of L-prolinol in 125ml of dichlromethane). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water followed by evaporation of dichloromethane to obtain an oily mass. The oily mass thus obtained was treated with methanol to yield 8g of Avanafil.
Examples
Example 1 : Preparation of Compound of Formula (II) (wherein R is -OH)
Step i)
To 200ml of methylene dichloride was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting methylene dichloride layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the methylene dichloride layer comprising compound of Formula (IV) obtained in step i), was added 5g of triethylamine followed by slow addition of 125ml solution of L-proline in methylene dichloride (2.8g of L-proline in 125ml of methylene dichloride). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water and 5% sodium carbonate solution, followed by evaporation of methylene dichloride to obtain an oily mass. The oily mass obtained was stripped with 50ml acetone to yield 9g of compound of Formula (II) having HPLC purity 98%.
Example 2: Preparation of Compound of Formula (II) (wherein R is -OC2H5)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filtrate containing product. The ethanol in filtrate is completely distilled out to isolate 10.45g of esterified compound of Formula (II).
Example 3 : Preparation of Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of toluene and 0.5ml of dimethylformamide was added 50g of compound of Formula (II) obtained in example 1 , and 70g of thionyl chloride. The reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain 50.5g of oily carboxylic acid chloride compound of Formula (II).
Example 4: Preparation of Avanafil of Formula (I)
In an inert atmosphere, a solution of 30g of compound of Formula (II) obtained in example 1 or 2, in 150 ml of tetrahydrofuran was dropwise added to 180ml of suspension of 1.0M lithium aluminium hydride solution in tetrahydrofuran, The reaction mixture was refluxed for 5 hours. After completion of reaction, the mixture was cooled in ice-bath and saturated aqueous solution of sodium sulfate was added to decompose excess of lithium aluminium hydride. The mixture was then diluted with 200ml of methylene dichloride and thus formed organic layer was separated. The organic layer was washed with water (3 χ 100 ml), dried over MgS04 and concentrated to collect crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 22.8g of Avanafil of Formula (I) having HPLC purity of 99.20%.
Example 5 : Preparation of Avanafil of Formula (I)
To a mixture of 1.3g sodium borohydride, 1 ml methanesulfonic acid and 50ml ethanol was added l Og of compound of Formula (II) obtained in example 1 or 2, and the mixture was stirred at 25-30°C for 5 hours. After completion of reaction, 100ml water was added and the mixture was extracted with 1 00ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with ethyl acetate at 45- 50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 6 to Example 8
The procedure is carried out as in example 5 except for instead of methanesulfonic acid other reducing agents are used in combination with sodium borohydride. The results are given in Table I
Table I

Example 9: Preparation of Avanafil of Formula (I)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filterate containing product. To the fi Iterate was added 1.2g of sodium borohydride and 2.6g of lithium bromide, and the mixture was stirred for 5 hours. After complete conversion of ester to final product, l OOml water was added and the mixture was extracted with 100ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with 25ml ethyl acetate at 45-50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7.5g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 10: Preparation of Avanafil of Formula (I) from Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of tetrahydrofuran and 50g of carboxylic acid chloride compound of Formula (II) obtained in example 3, was added 12g sodium borohydride at 0-5°C. After completion of reaction, water was added to reaction mixture to decompose excess of sodium borohydride present. The reaction mixture was then concentrated and a solution of 30g of potassium hydroxide in 200 ml of water was added. The mixture was heated to 60-70°C and maintained for 15-18 hours. The mixture was then cooled to 25-30°C and 500 ml of methylene dichloride was added. The organic layer thus formed, was separated and evaporated to yield crude Avanafil
of Formula (I) which was then subjected to purification using methanol as solvent to obtain 40g of Avanafil of Formula (I) having HPLC purity of 99.01%.

Mr. K. Chandran Wholetime Director & Vice Chairman EXTRAS
A “phosphodiesterase type 5 inhibitor” or “PDE5 inhibitor” refers to an agent that blocks the degradative action of phosphodiesterase type 5 on cyclic GMP in the arterial wall smooth muscle within the lungs and in the smooth muscle cells lining the blood vessels supplying the corpus cavernosum of the penis. PDE5 inhibitors are used for the treatment of pulmonary hypertension and in the treatment of erectile dysfunction. Examples of PDE5 inhibitors include, without limitation, tadalafil, avanafil, lodenafil, mirodenafil, sildenafil citrate, vardenafil and udenafil and pharmaceutically acceptable salts thereof. In one aspect, the PDE5 inhibitor is tadalafil.
“Tadalafil” or “TAD” is described in U.S. Pat. Nos. 5,859,006 and 6,821,975. It refers to the chemical compound, (6R-trans)-6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-methyl-pyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione and has the following chemical formula:

Tadalafil is currently marketed in pill form for treating erectile dysfunction (ED) under the trade name Cialis® and under the trade name Adcirca® for the treatment of PAH.
“Avanafil” refers to the chemical compound 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide, and its pharmaceutically acceptable salts. Avanafil is described in Limin M. et al., (2010) Expert Opin Investig Drugs, 19(11):1427-37. Avanafil has the following chemical formula:

Avanafil is being developed for erectile dysfunction. Avanafil currently has no trademarked term associated with it but it is being developed by Vivus Inc.
“Lodenafil” refers to the chemical compound, bis-(2-{4-[4-ethoxy-3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-benzenesulfonyl]piperazin-1-yl}-ethyl)carbonate and has the following chemical formula:

More information about lodenafil is available at Toque H A et al., (2008) European Journal of Pharmacology, 591(1-3):189-95. Lodenafil is manufactured by Cristália Produtos Químicose Farmacêuticos in Brazil and sold there under the brand-name Helleva®. It has undergone Phase III clinical trials, but is not yet approved for use in the United States by the U.S. FDA.
“Mirodenafil” refers to the chemical compound, 5-Ethyl-3,5-dihydro-2-[5-([4-(2-hydroxyethyl)-1-piperazinyl]sulfonyl)-2-propoxyphenyl]-7-propyl-4H-pyrrolo[3,2-d]pyrimidin-4-one and has the following chemical formula:

More information about mirodenafil can be found at Paick J S et al., (2008) The Journal of Sexual Medicine, 5 (11): 2672-80. Mirodenafil is not currently approved for use in the United States but clinical trials are being conducted.
“Sildenafil citrate,” marketed under the name Viagra®, is described in U.S. Pat. No. 5,250,534. It refers to 1-[4-ethoxy-3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)phenylsulfonyl]-4-methylpiperazine and has the following chemical formula:

Sildenafil citrate, sold as Viagra®, Revatio® and under various other trade names, is indicated to treat erectile dysfunction and PAH.
“Vardenafil” refers to the chemical compound, 4-[2-Ethoxy-5-(4-ethylpiperazin-1-yl)sulfonyl-phenyl]-9-methyl-7-propyl-3,5,6,8-tetrazabicyclo[4.3.0]nona-3,7,9-trien-2-one and has the following chemical formula:

Vardenafil is described in U.S. Pat. Nos. 6,362,178 and 7,696,206. Vardenafil is marketed under the trade name Levitra® for treating erectile dysfunction.
“Udenafil” refers to the chemical compound, 3-(1-methyl-7-oxo-3-propyl-4,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide and has the following chemical formula:

More information about udenafil can be found at Kouvelas D. et al., (2009) Curr Pharm Des, 15(30):3464-75. Udenafil is marketed under the trade name Zydena® but not approved for use in the United States.



THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family






VORINOSTAT

Vorinostat, MK0683
CAS 149647-78-9
Zolinza, SAHA, suberoylanilide hydroxamic acid, Suberanilohydroxamic acid, N-hydroxy-N’-phenyloctanediamide
US patent 5369108, PDT PATENT
For the treatment of cutaneous manifestations in patients with cutaneous T-cell lymphoma who have progressive, persistent or recurrent disease on or following two systemic therapies. Inhibits histone deacetylase I & 3.
- CCRIS 8456
- HSDB 7930
- M344
- N-Hydroxy-N’-phenyloctanediamide
- SAHA
- SAHA cpd
- Suberanilohydroxamic acid
- suberoylanilide hydroxamic acid
- UNII-58IFB293JI
- MK0683
| Average: 264.3202 Monoisotopic: 264.147392516 |
|
| Chemical Formula | C14H20N2O3 |
|---|
| N-hydroxy-N‘-phenyl-octanediamide | |
|---|---|
| Trade names | Zolinza, 100 MG, CAPSULE, ORAL |
| ZOLINZA (VORINOSTAT) [Merck Sharp & Dohme Corp.] | |
| MedlinePlus | a607050 |
| Licence data | US FDA:link |
| LAUNCHED 2006 MERCKhttp://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021991s002lbl.pdf | |
| Legal status | ℞-only (US) |
| Routes | Oral |
| Pharmacokinetic data | |
| Protein binding | 71% |
| Metabolism | Hepatic glucuronidation andoxidation CYP system not involved |
| Half-life | 2 hours |
| Excretion | Renal (negligible) |
| Identifiers | |
| CAS number | 149647-78-9 |
| ATC code | L01XX38 |
| Chemical data | |
| Formula | C14H20N2O3 |
| Mol. mass | 264.32 g/mol |
CLINICAL TRIALS..http://clinicaltrials.gov/search/intervention=Vorinostat
Vorinostat (rINN) also known as suberanilohydroxamic acid (suberoyl+anilide+hydroxamic acid abbreviated as SAHA) is a member of a larger class of compounds that inhibit histone deacetylases (HDAC). Histone deacetylase inhibitors (HDI) have a broad spectrum of epigenetic activities.
Vorinostat is marketed under the name Zolinza for the treatment of cutaneous T cell lymphoma (CTCL) when the disease persists, gets worse, or comes back during or after treatment with other medicines.[1] The compound was developed by Columbia University chemist, Ronald Breslow.
Vorinostat was the first histone deacetylase inhibitor[2] approved by the U.S. Food and Drug Administration (FDA) for the treatment of CTCL on October 6, 2006. It is manufactured by Patheon, Inc., in Mississauga, Ontario, Canada, for Merck & Co., Inc., White House Station, New Jersey.[3]
ZOLINZA contains vorinostat, which is described chemically as N-hydroxy-N’-phenyloctanediamide. The empirical formula is C14H20N2O3. The molecular weight is 264.32 and the structural formula is:
 |
Vorinostat is a white to light orange powder. It is very slightly soluble in water, slightly soluble in ethanol, isopropanol and acetone, freely soluble in dimethyl sulfoxide and insoluble in methylene chloride. It has no chiral centers and is non-hygroscopic. The differential scanning calorimetry ranged from 161.7 (endotherm) to 163.9°C. The pH of saturated water solutions of vorinostat drug substance was 6.6. The pKa of vorinostat was determined to be 9.2.
Each 100 mg ZOLINZA capsule for oral administration contains 100 mg vorinostat and the following inactive ingredients: microcrystalline cellulose, sodium croscarmellose and magnesium stearate. The capsule shell excipients are titanium dioxide, gelatin and sodium lauryl sulfate.
Vorinostat has been shown to bind to the active site of histone deacetylases and act as a chelator for Zinc ions also found in the active site of histone deacetylases [4] Vorinostat’s inhibition of histone deacetylases results in the accumulation of acetylated histones and acetylated proteins, including transcription factors crucial for the expression of genes needed to induce cell differentiation. [4]
SAHA inhibits class I and class II HDACs at nanomolar concentrations and arrests cell growth in a wide variety of transformed cells in culture at 2.5-5.0 µM. This compound efficiently suppressed MES-SA cell growth at a low dosage (3 µM) already after 24 hours treatment. Decrease of cell survival was even more pronounced after prolonged treatment and reached 9% and 2% after 48 and 72 hours of treatment, respectively. Colony forming capability of MES-SA cells treated with 3 µM vorinostat for 24 and 48 hours was significantly diminished and blocked after 72 hours.

Vorinostat has also been used to treat Sézary syndrome, another type of lymphoma closely related to CTCL.[5]
A recent study suggested that vorinostat also possesses some activity against recurrent glioblastoma multiforme, resulting in a median overall survival of 5.7 months (compared to 4 – 4.4 months in earlier studies).[6] Further brain tumor trials are planned in which vorinostat will be combined with other drugs.
Including vorinostat in treatment of advanced non-small-cell lung cancer (NSCLC) showed improved response rates and increased median progression free survival and overall survival (although the survival improvements were not significant at the P=0.05 level).[7]
It has given encouraging results in a phase II trial for myelodysplastic syndromes in combination with Idarubicin and Cytarabine.[8]
Vorinostat is an interesting target for scientists interested in eradicating HIV from infected persons.[9] Vorinostat was recently shown to have both in vitro and in vivo effects against latently HIV infected T-cells.[10][11]
Vorinostat, represented by structural formula (I) and chemically named as N-hydroxy-N’- phenyl-octanediamide or suberoylanilide hydroxamic acid (SAElA), is a member of a larger class of compounds that inhibit histone deacetylases (HDAC). Histone deacetylase inhibitors (HDI) have a broad spectrum of epigenetic activities and vorinostat is marketed, under the brand name Zolinza®, for the treatment of a type of skin cancer called cutaneous T-cell lymphoma (CTCL). Vorinostat is approved to be used when the disease persists, gets worse, or comes back during or after treatment with other medicines. Vorinostat has also been used to treat Sέzary’s disease and, in addition, possesses some activity against recurrent glioblastoma multiforme.

Vorinostat was first described in US patent 5369108, wherein four different synthetic routes for the preparation of vorinostat are disclosed (Schemes 1 to 4).
The single step process illustrated in Scheme 1 involves coupling of the diacid chloride of suberic acid with aniline and hydiOxylamine hydrochloride. However, the yield of this reaction is only 15-30%.

Scheme 1
The multistep process illustrated in Scheme 2 begins with the monomethyl ester of suberic acid, which undergoes conversion to the corresponding acid chloride. Further coupling with aniline gives the methyl ester of suberanilic acid. Hydrolysis of the ester and further coupling with benzyl protected hydroxylamine gives benzyl protected vorinostat which on deprotection gives vorinostat.
HO. (CH2J6 OMe . ,OOMM e
O O



Scheme 2
In addition to the disadvantage of being a five-step process with overall yields reported as 35-65%, this process suffers from further disadvantages such as the use of the expensive monomethyl ester of suberic acid.

Scheme 3
The two step process illustrated in Scheme 3 involves coupling of the diacid chloride of suberic acid with aniline and O-benzyl hydroxylamine and then deprotection. However, the overall yield of this reaction is only 20-35%.

Scheme 4
The process illustrated in Scheme 4 is similar to that illustrated in Scheme 3, with the exception that O-trimethylsilyl hydroxylamine was used instead of O-benzyl hydroxylamine. The overall yield of this reaction is reported as 20-33%.
Another process for the preparation of vorinostat has been reported in J. Med. Chem.,
1995, vol. 38(8), pages 1411-1413. The reported process, illustrated in Scheme 5, begins with the conversion of suberic acid to suberanilic acid by a high temperature melt reaction.
Suberanilic acid is further converted to the corresponding methyl ester using Dowex resin and the methyl ester of suberanilic acid thus formed is converted to vorinostat by treatment with hydroxylamine hydrochloride. However, this process employs high temperatures (1900C) in the preparation of vorinostat which adds to the inefficiency and high processing costs on commercial scale. The high temperatures also increase the likelihood of impurities being formed during manufacture and safety concerns. The overall yield reported was a poor 35%.

MeOH, Dowex, 22 hours


Scheme 5
Another process for the preparation of vorinostat has been reported in OPPI Briefs, 2001, vol. 33(4), pages 391-394. The reported process, illustrated in Scheme 6, involves conversion of suberic acid to suberic anhydride, which on treatment with aniline gives suberanilic acid. Coupling of this suberanilic acid with ethyl chloroformate gives a mixed anhydride which upon treatment with hydroxylamine gives vorinostat in an overall yield of 58%. In the first step, there is competition between the formation of suberic anhydride and the linear anhydride and consequently isolation of pure suberic anhydride from the reaction mixture is very difficult. This process step is also hindered by the formation of process impurities and competitive reactions. In the second step, there is formation of dianilide by reaction of two moles of aniline with the linear anhydride. In the third step, suberanilic acid is an inconvenient by-product as the suberanilic acid is converted to a mixed anhydride with ethyl chloroformate, which is highly unstable and is converted back into suberanilic acid. Consequently, it is very difficult to obtain pure vorinostat from the reaction mixture. Although the reported yield was claimed to be 58%, when repeated a yield of only 38% was obtained.

Scheme 6
A further process for the preparation of vorinostat has been reported in J. Med. Chem., 2005, vol. 48(15), pages 5047-5051. The reported process, illustrated in Scheme 7, involves conversion of monomethyl suberate to monomethyl suberanilic acid, followed by coupling with hydroxylamine hydrochloride to afford vorinostat in an overall yield of 79%. However, the process uses the expensive monomethyl ester of suberic acid as starting material.
HOBt, DCC, DMF, RT, 4 hours



CLIP
Vorinostat (ZolinzaTM) Vorinostat, a histone deacetylase (HDAC) inhibitor from Merck, was approved for the treatment of cutaneous T-cell lymphoma (CTCL), a type of non-Hodgkin’s lymphoma.
Vorinostat was shown to inhibit HDAC1, HDAC2, HDAC3 and HDAC6 at nanomolar concentrations. HDAC inhibitors are potent differentiating agents toward a variety of neoplasms, including leukemia and breast and prostate cancers [58].
Commercially available monomethyl ester 125 wasVorinostat (ZolinzaTM) Vorinostat, a histone deacetylase (HDAC) inhibitor from Merck, was approved for the treatment of cutaneous T-cell lymphoma (CTCL), a type of non-Hodgkin’s lymphoma.
Vorinostat was shown to inhibit HDAC1, HDAC2, HDAC3 and HDAC6 at nanomolar concentrations. HDAC inhibitors are potent differentiating agents toward a variety of neoplasms, including leukemia and breast and prostate cancers [58].
Commercially available monomethyl ester 125 was reacted with aniline in the presence of DCC and HOBt in DMF to give amide 127 in 89%yield [59] (Scheme 16).
Methyl ester amide 127 was then reacted with hydroxylamine HCl salt and potassium hydroxide in methanol to give vorinostat(XVI) in 90% yield.
[58] Breslow, R.; Marks, P.A.; Rifkind, R. A.; Jursic, B. WO9307148,2003.
[59] Gediya, L. K.; Chopra, P.; Purushottamachar, P.; Maheshwari, N.;Njar, V. C. O. J. Med. Chem., 2005, 48, 5047.
PATENT
VORINOSTAT
http://www.google.com/patents/EP2349985A2
A preferred embodiment of the first aspect of the present invention is illustrated in Scheme

suberic acid subefanilic acid NH2OHHCl, CDI

suberoylanilide hydroxamic acid (T)
Scheme 8
Optionally, an activating agent can be used in step (a) and/ or step (b) to afford products with high yields and purity. Preferably, the activating agent is selected from cyanuric chloride, cyanuric fluoride, catecholborane, or a mixture thereof. The activating agent is preferably used in combination with the coupling agent. A preferred embodiment of the process according to the first aspect of the present invention comprises the following steps:
(i) taking a mixture of THF, CDI and DCC;
(ii) adding suberic acid; (iii) adding aniline in THF to the solution from step (ii);
(iv) stirring at 25-30°C;
(v) filtering off the solid dicyclohexyl urea formed in the reaction;
(vi) concentrating the filtrate in vacuo;
(vii) adding a solution of KOH in water; (vϋi) filtering off the solid by-product;
(ix) heating the filtrate;
(x) adding aq. HCl;
(xi) isolating suberanilic acid;
(xii) mixing the suberanilic acid and CDI in DMF; (xiii) adding hydroxylamine hydrochloride as solid to the mixture from step (xii);
(xiv) isolating vorinostat from the mixture obtained in step (xiii);
(xv) adding acetonitrile and aq. ammonia to the vorinostat from step (xiv);
(xvi) heating the mixture;
(xvii) cooling the mixture to 20-27°C; and (xvϋi) isolating pure vorinostat from the mixture obtained in step (xvii).
Preferably, by utilising the same organic solvent in steps (a) and (b), pure vorinostat can be obtained without isolation of any synthetic intermediate^).
A preferred embodiment of the second aspect of the present invention is illustrated in Scheme 9.

suberic acid N-hydtoxy-7-carboxy-heptanamide

Example 1
Stage 1 : Conversion of suberic acid to suberanilic acid
A mixture of CDI (0.5eq) and DCC (0.8eq) in THF (15 vol) was stirred for 1 hour at 25- 3O0C. Suberic acid (leq) and aniline (leq) in THF (1 vol) was added and the mixture stirred for a further 16-20 hours. The solid by-product was removed by filtration and the filtrate was concentrated in vacuo at 5O0C. The solid residue obtained was treated with a solution of KOH (2eq) in water (10 vol) and stirred for 30 minutes at 25-300C and any solid byproduct formed was removed by filtration. The filtrate obtained was heated at 6O0C for 3-4 hours and cooled to 200C before addition of an aqueous solution of HCl (17.5%, 3 vol). The mixture was stirred for 30 minutes and the solid filtered, washed with water (2×5 vol) and dried under vacuum at 60-650C. Molar Yield = 60-65% Purity by HPLC = 99.5%
Stage 2: Conversion of suberanilic acid to crude vorinostat The suberanilic acid (leq) obtained in stage 1 was dissolved in DMF (5 vol) and CDI (2eq) was added at 25-3O0C and maintained for 30 minutes under stirring. Hydroxylamine hydrochloride (4eq) was added and stirring continued for 30 minutes. Water (25 vol) was then added and the mixture stirred for 2 hours. The precipitated solid was filtered, washed with water (2×5 vol) and dried under vacuum at 500C. Molar Yield = 70-75% Purity by HPLC = 99% Stage 3: Purification of crude vorinostat
Aqueous ammonia (2.5 vol) was added to the crude vorinostat (leq) in acetonitrile (15 vol) at 25-30°C. The mixture was then maintained at 55-60°C for 1 hour before being cooled to 20-25°C and being stirred for a further hour. The resulting solid was filtered, washed with acetonitrile (2×0.5 vol) and dried under vacuum at 45-5O0C for 5 hours. Molar Yield = 55-60% Purity by HPLC > 99.8%
Example 2
Stage 1 : Conversion of suberic acid to crude vorinostat
A mixture of CDI (0.5eq) and DCC (0.8eq) in THF (15 vol) was stirred for 1 hour at 25- 30°C. Suberic acid (leq) and hydroxylamine (leq) in THF (1 vol) was added and the mixture stirred for a further 1 hour. Then CDI (0.5eq), DCC (0.8eq) and aniline (leq) were added to the mixture and the mixture was stirred for a further 16-20 hours. The solid byproduct was removed by filtration and the filtrate was concentrated in vacuo at 50°C to obtain crude vorinostat. Molar Yield = 55-60% Purity by HPLC > 95.8%
Stage 2: Purification of crude vorinostat
Aqueous ammonia (2.5 vol) was added to the crude vorinostat (leq) in acetonitrile (15 vol) at 25-3O0C. The mixture was then maintained at 55-600C for 1 hour before being cooled to 20-250C and being stirred for a further hour. The resulting solid was filtered, washed with acetonitrile (2×0.5 vol) and dried under vacuum at 45-500C for 5 hours. Molar Yield = 35-40% Purity by HPLC > 99.8%
PATENT
SYNTHESIS
Scheme V. – –

Vorinostat
Suberic acid (l.Oeq) was dissolved in tetrahydrofuran (15vol) and the clear solution was chilled to 0-5°C. Methyl chloro formate (l.leq) and triethylamine (1.1 eq) were added to the solution at the same temperature and the mixture was stirred for 15 minutes. The triethylamine.HCl salt formed was filtered off, then aniline (leq) was added to the reaction mixture at 0-50C and stirring was continued for 15 minutes. Methyl chloroformate (l.leq) and triethylamine (l.leq) were added to the clear solution and stirring was continued for a further 15 minutes at 0-5°C. This chilled reaction mixture was added to a freshly prepared hydroxylamine solution in methanol (*see below) chilled to 0-5°C and stirred for 15 minutes at 0-5°C. The solvent was removed under vacuum at 40°C and the residue obtained was taken in methylene dichloride and the organic solution was washed with water and dried over anhydrous sodium sulfate. Methylene dichloride was removed under vacuum at 40°C and acetonitrile was added to the residue. This mixture was stirred for 15 minutes before the solid was filtered under vacuum and dried under vacuum at 60°C to afford the product as a white solid. Molar yield = 35-41%; HPLC purity = 99.90%.
VORINOSTAT
1H-NMR (DMSO-d6): 1.27 (m, 4H, 2 x -CH2-), 1.53 (m, 4H, 2 x -CH2-), 1.94 (t, J = 7.3 Hz, 2H, -CH2-), 2.29 (t, J = 7.4 Hz, 2H, -CH2-), 7.03 (t, J = 7.35 Hz, IH, aromatic para position), 7.27 (t, J = 7.90 Hz, 2H, aromatic meta position), 7.58 (t, J = 7.65 Hz, 2H, aromatic ortho position), 8.66 (s, IH, -OH, D2O exchangeable), 9.85 (s, IH, amide -NH-, D2O exchangeable), 10.33 (s, IH, -NH-OH, D2O exchangeable).
13C-NMR (DMSO-d6): 25.04 (2C, 2 x -CH2-), 28.43 (2C, 2 x -CH2-), 32.24 (1C, -CH2-), 36.34 (1C, -CH2-), 119.01 (2C, Ar-C), 122.96 (1C, Ar-C), 128.68 (2C, Ar-C), 139.24 (1C, Ar- C, =CNH-), 169.23 (1C, -CO-), 171.50 (1C, -CO-).
*Preparation of hydroxylamine solution:
Potassium hydroxide (l.leq) was added to methanol (8vol) and the solution was chilled to 0-5°C. Similarly hydroxylamine hydrochloride (l.leq) was added to methanol (8vol) and chilled to 0-5°C. The chilled amine solution was added to the chilled alkali solution and stirred for 15 minutes at 0-50C. The white potassium chloride salt was filtered off and the filtrate was used as such.
SPECTRAL DATA AND SYNTHESIS
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
for structures see above link
Suberoylanilide hydroxamic acid (26, SAHA, vorinostat).
Suberic acid monomethyl ester (23) (15.09 g, 80.2 mmol) and DMF (0.10 mL) in anhydrous
DCM (300 mL) was added SOCl2 (34.6 mL, 0.481 mol), and the reaction mixture was refluxed for 3
h. The mixture was then concentrated. Toluene (300 mL) was added to the residue and evaporated
to afford crude acid chloride 24. Crude 24 was dissolved in DCM (240 mL), and followed by
addition of aniline (7.3 mL, 80.2 mmol) and Et3N (16.9 mL, 0.120 mol). The reaction mixture was
stirred for 90 min at room temp. The course of reaction was monitored by TLC (30% EtOAc in
hexanes) and LC–MS. DCM was removed, and ethyl acetate (500 mL) was added to dissolve the
residue. The organic layer was washed with aqueous NaHCO3 (500 mL × 2), 1 N HCl (400 mL × 2),
water, dried (Na2SO4), and evaporated to dryness under reduced pressure. The residue was purified
by vacuum liquid chromatography (silica, 20% EtOAc in hexanes) to afford compound 25as white crystalline solids (20.15 g, 96 %). NaOMe in MeOH solution (5.4 M, 106 mL, 0.573 mol) was added to a solution of compound 25 (10.05 g, 38.2 mmol) and NH2OH·HCl (26.54 g, 0.382 mol) in
dry MeOH (375 mL). The reaction mixture was stirred for 40 min at room temp. The reaction was
quenched by adding of 1 N HCl to pH 7–8. MeOH was removed under reduced pressure and water
(1 L) was added to the residue. The precipitated solid was filtered and washed with water (300 mL)
and EtOAc (150 mL) to afford crude 26 which was further purified by recrystallization. MeOH (200
mL) was added to crude 26 (5 g) and warmed to dissolve all solids. The MeOH solution was filtered,
and deionized water (400 mL) was added to the filtrate, the resulting solution was placed at 4 oC
overnight. Crystals obtained were filtered and washed with deionized water (100 mL) to afford pure
26 (vorinostat, SAHA) as off-white crystals. Overall yield: 80–85% from compound 23. Compound
26,
LC–MS m/z 265.1 ([M + H]+).
1H NMR (DMSO-d6) 10.35 (1H, s), 9.86 (1H, s), 8.68 (1H, s),
7.58 (2H, d, J = 7.6 Hz), 7.28 (2H, t, J = 7.5 Hz), 7.02 (1H, t, J = 7.4 Hz), 2.29 (2H, t, J = 7.4 Hz),
1.94 (2H, t, J = 7.4 Hz), 1.57 (2H, m), 1.49 (2H, m), 1.33 – 1.20 (2H, m); 13C NMR (DMSO-d6)
171.2, 169.1, 139.3, 128.6, 122.9, 119.0, 36.3, 32.2, 28.4, 28.3, 25.0. Anal. (C10H20N2O3) C, H, N.
CLIP

Suberic acid monomethyl ester (23) (15.09 g, 80.2 mmol) and DMF (0.10 mL) in anhydrous DCM (300 mL) was added SOCl2 (34.6 mL, 0.481 mol), and the reaction mixture was refluxed for 3 h. The mixture was then concentrated. Toluene (300 mL) was added to the residue and evaporated to afford crude acid chloride 24. Crude 24 was dissolved in DCM (240 mL), and followed by addition of aniline (7.3 mL, 80.2 mmol) and Et3N (16.9 mL, 0.120 mol). The reaction mixture was stirred for 90 min at room temp. The course of reaction was monitored by TLC (30% EtOAc in hexanes) and LC–MS. DCM was removed, and ethyl acetate (500 mL) was added to dissolve the residue. The organic layer was washed with aqueous NaHCO3 (500 mL × 2), 1 N HCl (400 mL ×2), water, dried (Na2SO4), and evaporated to dryness under reduced pressure. The residue was purified by vacuum liquid chromatography (silica, 20% EtOAc in hexanes) to afford compound 25 as white crystalline solids (20.15 g, 96 %). NaOMe in MeOH solution (5.4 M, 106 mL, 0.573 mol) was added to a solution of compound 25 (10.05 g, 38.2 mmol) and NH2OH·HCl (26.54 g, 0.382 mol) in dry MeOH (375 mL). The reaction mixture was stirred for 40 min at room temp. The reaction was quenched by adding of 1 N HCl to pH 7–8. MeOH was removed under reduced pressure and water (1 L) was added to the residue. The precipitated solid was filtered and washed with water (300 mL) and EtOAc (150 mL) to afford crude 26 which was further purified by recrystallization. MeOH (200 mL) was added to crude 26 (5 g) and warmed to dissolve all solids. The MeOH solution was filtered, S37 and deionized water (400 mL) was added to the filtrate, the resulting solution was placed at 4 oC overnight. Crystals obtained were filtered and washed with deionized water (100 mL) to afford pure 26 (vorinostat, SAHA) as off-white crystals. Overall yield: 80–85% from compound 23.
. Compound 26,
LC–MS m/z 265.1 ([M + H] + ).
1H NMR (DMSO-d6) 10.35 (1H, s), 9.86 (1H, s), 8.68 (1H, s), 7.58 (2H, d, J = 7.6 Hz), 7.28 (2H, t, J = 7.5 Hz), 7.02 (1H, t, J = 7.4 Hz), 2.29 (2H, t, J = 7.4 Hz), 1.94 (2H, t, J = 7.4 Hz), 1.57 (2H, m), 1.49 (2H, m), 1.33 – 1.20 (2H, m);
13C NMR (DMSO-d6) 171.2, 169.1, 139.3, 128.6, 122.9, 119.0, 36.3, 32.2, 28.4, 28.3, 25.0.
Anal. (C10H20N2O3) C, H, N.


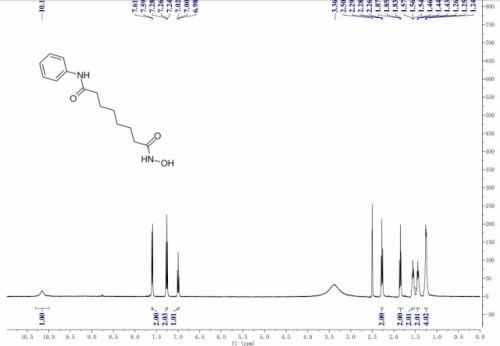
References
- “ZOLINZA, Merck’s Investigational Medicine for Advanced Cutaneous T-Cell Lymphoma (CTCL), To Receive Priority Review from U.S. Food and Drug Administration” (Press release). Merck & Co. June 7, 2006. Retrieved 2006-10-06.
- HDAC Inhibitors Base (vorinostat)
- “FDA Approves New Drug for Skin Cancer, Zolinza” (Press release). Food and Drug Administration. October 6, 2006. Retrieved 2006-10-06.
- Richon, Victoria. “Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor”. British Journal of Cancer. Retrieved 3 May 2012.
- Cuneo A, Castoldi. “Mycosis fungoides/Sezary’s syndrome”. Retrieved 2008-02-15.
- “Vorinostat shows anti-cancer activity in recurrent gliomas” (Press release). Mayo Clinic. June 3, 2007. Retrieved 2007-06-03.
- http://www.rtmagazine.com/reuters_article.asp?id=20091209clin013.html Dec 2009. URL dead Jan 2012
- “Zolinza, Idarubicin, Cytarabine Combination Yields High Response Rates In MDS Patients (ASH 2011)”.
- “Study of the Effect of Vorinostat on HIV RNA Expression in the Resting CD4+ T Cells of HIV+ Pts on Stable ART”. ClinicalTrials.gov. 2011-03-21.
- Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM (2009). “Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid.”. AIDS Res Hum Retroviruses 25 (2): 207–12. doi:10.1089/aid.2008.0191. PMC 2853863. PMID 19239360.
- Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J et al. (2009). “Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells.”. J Biol Chem 284 (11): 6782–9.doi:10.1074/jbc.M807898200. PMC 2652322. PMID 19136668.
- Vorinostat bound to proteins in the PDB
- J. Med. Chem.,1995, vol. 38(8), pages 1411-1413.
- A new simple and high-yield synthesis of suberoylanilide hydroxamic acid and its inhibitory effect alone or in combination with retinoids on proliferation of human prostate cancer cells
J Med Chem 2005, 48(15): 5047 - A new facile and expeditious synthesis of N-hydroxy-N’-phenyloctanediamide, a potent inducer of terminal cytodifferentiation
Org Prep Proced Int 2001, 33(4): 391 - US patent 5369108, PDT PATENT
- WO2007/22408………
- WO 1993007148
- CN 102344392
| United States | 7456219 | APPROVAL 2006-11-14 | EXPIRY 2026-11-14 |
| United States | 6087367 | 1994-10-04 | 2011-10-04 |
| Canada | 2120619 | 2006-11-21 | 2012-10-05 |
| Patent | Patent Expiry | pat use code |
|---|---|---|
| 7399787 | Feb 9, 2025 | U-892 |
| 7456219 | Mar 11, 2027 | |
| 7652069 | Mar 4, 2023 | |
| 7732490 | Mar 4, 2023 | U-892 |
| 7851509 | Feb 21, 2024 | U-892 |
| 8067472 | Mar 4, 2023 | U-892 |
| 8093295 | May 16, 2026 | |
| 8101663 | Mar 4, 2023 | U-892 |
| RE38506 | Nov 29, 2013 |
U 892 =TREATMENT OF CUTANEOUS MANIFESTATIONS IN PATIENTS WTIH CUTANEOUS T-CELL LYMPHOMA (CTCL)
| Exclusivity Code | Exclusivity_Date |
|---|---|
| ODE | Oct 6, 2013 |
| WO2009098515A1 * | Feb 6, 2009 | Aug 13, 2009 | Generics Uk Ltd | Novel process for the preparation of vorinostat |
Marks, P.A., Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotech 25(1) 84-90 (2007). DOI: 10.1038/nbt1272
Takashi Kumagai, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. International Journal of Cancer. 2007 Aug 1;121(3):656-65. DOI: 10.1002/ijc.22558
Hrzenjak A, et al. Histone deacetylase inhibitor vorinostat suppresses the growth of uterine sarcomas in vitro and in vivo. Mol Cancer. 2010 Mar 4;9:49. DOI: 10.1186/1476-4598-9-49
………………………………………………………………………………………
|

| US7148257 | Aug 26, 2003 | Dec 12, 2006 | Merck Hdac Research, Llc | Methods of treating mesothelioma with suberoylanilide hydroxamic acid |
| US7375137 | Mar 28, 2006 | May 20, 2008 | Merck Hdac Research, Llc | Methods of treating cancer with HDAC inhibitors |
| US7399787 | Jul 9, 2003 | Jul 15, 2008 | Merck Hdac Research, Llc | Methods of treating cancer with HDAC inhibitors |
| US7456219 | Jun 19, 2003 | Nov 25, 2008 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7652069 | Oct 30, 2007 | Jan 26, 2010 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7732490 | Sep 11, 2007 | Jun 8, 2010 | Merck Hdac Research, Llc | Methods of treating cancer |
| US7847122 | Mar 18, 2008 | Dec 7, 2010 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7851509 | Mar 18, 2008 | Dec 14, 2010 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7879865 | Nov 18, 2005 | Feb 1, 2011 | Sloan-Kettering Institute For Cancer Research | Treatment of cancer of the brain using histone deacetylase inhibitors |
| US7998957 | Feb 6, 2008 | Aug 16, 2011 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicylcoheptenes, their preparation and use |
| US8058268 | Jul 29, 2009 | Nov 15, 2011 | Lixte Biotechnology, Inc. | Neuroprotective agents for the prevention and treatment of neurodegenerative diseases |
| US8067472 | Apr 23, 2010 | Nov 29, 2011 | Merck Hdac Research, Llc | Methods of treating Hodgkin’s and non-Hodgkin’s lymphoma |
| US8088951 | Nov 30, 2007 | Jan 3, 2012 | Massachusetts Institute Of Technology | Epigenetic mechanisms re-establish access to long-term memory after neuronal loss |
| US8093295 | May 16, 2006 | Jan 10, 2012 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing the same |
| US8101663 | Dec 7, 2009 | Jan 24, 2012 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US8143445 | Oct 1, 2008 | Mar 27, 2012 | Lixte Biotechnology, Inc. | HDAC inhibitors |
| US8227473 | Jul 17, 2009 | Jul 24, 2012 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicycloheptenes, their preparation and use |
| US8288440 * | Jan 13, 2010 | Oct 16, 2012 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US8329719 | Aug 1, 2011 | Dec 11, 2012 | Lixte Biotechnology, Inc. | Neuroprotective agents for the prevention and treatment of neurodegenerative diseases |
| US8426444 | Jun 30, 2011 | Apr 23, 2013 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicycloheptenes, their preparation and use |
| US8450372 * | Jan 13, 2010 | May 28, 2013 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US8455688 | Mar 21, 2012 | Jun 4, 2013 | Lixte Biotechnology, Inc. | HDAC inhibitors |
| US8541458 | Jun 11, 2012 | Sep 24, 2013 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicycloheptenes, their preparation and use |
| US8563615 | Nov 1, 2010 | Oct 22, 2013 | Massachusetts Institute Of Technology | Use of CI-994 and dinaline for the treatment of memory/cognition and anxiety disorders |
| US20100112046 * | Jan 13, 2010 | May 6, 2010 | Jeannie Chow Wong | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US20100113829 * | Jan 13, 2010 | May 6, 2010 | Cote Aaron S | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US20100119596 * | Jan 13, 2010 | May 13, 2010 | Jeannie Chow Wong | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US20110263712 * | Oct 14, 2009 | Oct 27, 2011 | Generics (Uk) Limited | Process for the preparation of vorinostat |
| US20110313044 * | Jun 16, 2011 | Dec 22, 2011 | Urquima S.A. | Polymorphs of Suberoylanilide Hydroxamic Acid |
| EP2079304A1 * | Sep 24, 2007 | Jul 22, 2009 | Merck & Co., Inc. | Amine base salts of saha and polymorphs thereof |
| EP2229941A1 * | May 16, 2006 | Sep 22, 2010 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| EP2292221A2 * | May 16, 2006 | Mar 9, 2011 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| WO2006127319A2 * | May 16, 2006 | Nov 30, 2006 | Merck & Co Inc | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| WO2006127321A2 * | May 16, 2006 | Nov 30, 2006 | Merck & Co Inc | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| WO2008039421A2 * | Sep 24, 2007 | Apr 3, 2008 | Arlene E Mckeown | Pharmaceutical compositions of hdac inhibitors and chelatable metal compounds, and metal-hdac inhibitor chelate complexes |
| WO2008042146A1 * | Sep 24, 2007 | Apr 10, 2008 | Arlene E Mckeown | Amine base salts of saha and polymorphs thereof |
| WO2008097654A1 * | Feb 8, 2008 | Aug 14, 2008 | Nancie M Archin | Methods of using saha for treating hiv infection |
| WO2009020565A1 * | Aug 1, 2008 | Feb 12, 2009 | Lixte Biotechnology Inc | Use of phosphatases to treat neuroblastomas and medulloblastomas |
| WO2010061220A2 * | Nov 25, 2009 | Jun 3, 2010 | Generics [Uk] Limited | Novel processes and pure polymorphs |
EXTRAS
MS-275 (Entinostat); CI-994 (Tacedinaline); BML-210; M344; MGCD0103 (Mocetinostat); PXD101 (Belinostat); LBH-589 (Panobinostat); Tubastatin A; Scriptaid; NSC 3852; NCH 51; HNHA; BML-281; CBHA; Salermide; Pimelic Diphenylamide; ITF2357 (Givinostat); PCI-24781; APHA Compound 8; Droxinostat; SB939.
SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
//////////////149647-78-9, MK0683, VORINOSTAT, Zolinza
ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1
PANOBINOSTAT

Panobinostat
HDAC inhibitors, orphan drug
cas 404950-80-7
2E)-N-hydroxy-3-[4-({[2-(2-methyl-1H-indol-3-yl)ethyl]amino}methyl)phenyl]acrylamide
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (alternatively, N-hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide)
Molecular Formula: C21H23N3O2 Molecular Weight: 349.42622
- Faridak
- LBH 589
- LBH589
- Panobinostat
- UNII-9647FM7Y3Z
A hydroxamic acid analog histone deacetylase inhibitor from Novartis.
NOVARTIS, innovator
Histone deacetylase inhibitors
Is currently being examined in cutaneous T-cell lymphoma, CML and breast cancer.
clinical trials click here phase 3
DRUG SUBSTANCE–LACTATE AS IN http://www.google.com/patents/US7989639 SEE EG 31

Panobinostat (LBH-589) is an experimental drug developed by Novartis for the treatment of various cancers. It is a hydroxamic acid[1] and acts as a non-selective histone deacetylase inhibitor (HDAC inhibitor).[2]
panobinostat
Panobinostat is a cinnamic hydroxamic acid analogue with potential antineoplastic activity. Panobinostat selectively inhibits histone deacetylase (HDAC), inducing hyperacetylation of core histone proteins, which may result in modulation of cell cycle protein expression, cell cycle arrest in the G2/M phase and apoptosis. In addition, this agent appears to modulate the expression of angiogenesis-related genes, such as hypoxia-inducible factor-1alpha (HIF-1a) and vascular endothelial growth factor (VEGF), thus impairing endothelial cell chemotaxis and invasion. HDAC is an enzyme that deacetylates chromatin histone proteins. Check for
As of August 2012, it is being tested against Hodgkin’s Lymphoma, cutaneous T cell lymphoma (CTCL)[3] and other types of malignant disease in Phase III clinical trials, against myelodysplastic syndromes, breast cancer and prostate cancer in Phase II trials, and against chronic myelomonocytic leukemia (CMML) in a Phase I trial.[4][5]
Panobinostat is a histone deacetylase (HDAC) inhibitor which was filed for approval in the U.S. in 2010 for the oral treatment of relapsed/refractory classical Hodgkin’s lymphoma in adult patients. The company is conducting phase II/III clinical trials for the oral treatment of multiple myeloma, chronic myeloid leukemia and myelodysplasia. Phase II trials are also in progress for the treatment of primary myelofibrosis, post-polycythemia Vera, post-essential thrombocytopenia, Waldenstrom’s macroglobulinemia, recurrent glioblastoma (GBM) and for the treatment of pancreatic cancer progressing on gemcitabine therapy. Additional trials are under way for the treatment of hematological neoplasms, prostate cancer, colorectal cancer, renal cell carcinoma, non-small cell lung cancer (NSCLC), malignant mesothelioma, acute lymphoblastic leukemia, acute myeloid leukemia, head and neck cancer and gastrointestinal neuroendocrine tumors. Early clinical studies are also ongoing for the treatment of HER2 positive metastatic breast cancer. Additionally, phase II clinical trials are ongoing at Novartis as well as Neurological Surgery for the treatment of recurrent malignant gliomas as are phase I/II initiated for the treatment of acute graft versus host disease. The National Cancer Institute had been conducting early clinical trials for the treatment of metastatic hepatocellular carcinoma; however, these trials were terminated due to observed dose-limiting toxicity. In 2009, Novartis terminated its program to develop panobinostat for the treatment of cutaneous T-cell lymphoma. A program for the treatment of small cell lung cancer was terminated in 2012. Phase I clinical trials are ongoing for the treatment of metastatic and/or malignant melanoma and for the treatment of sickle cell anemia. The University of Virginia is conducting phase I clinical trials for the treatment of newly diagnosed and recurrent chordoma in combination with imatinib. Novartis is evaluating panobinostat for its potential to re-activate HIV transcription in latently infected CD4+ T-cells among HIV-infected patients on stable antiretroviral therapy.
Mechanistic evaluations revealed that panobinostat-mediated tumor suppression involved blocking cell-cycle progression and gene transcription induced by the interleukin IL-2 promoter, accompanied by an upregulation of p21, p53 and p57, and subsequent cell death resulted from the stimulation of caspase-dependent and -independent apoptotic pathways and an increase in the mitochondrial outer membrane permeability. In 2007, the compound received orphan drug designation in the U.S. for the treatment of cutaneous T-cell lymphoma and in 2009 and 2010, orphan drug designation was received in the U.S. and the E.U., respectively, for the treatment of Hodgkin’s lymphoma. This designation was also assigned in 2012 in the U.S. and the E.U. for the treatment of multiple myeloma.
Cardiovascular disease is the leading cause of morbidity and mortality in the western world and during the last decades it has also become a rapidly increasing problem in developing countries. An estimated 80 million American adults (one in three) have one or more expressions of cardiovascular disease (CVD) such as hypertension, coronary heart disease, heart failure, or stroke. Mortality data show that CVD was the underlying cause of death in 35% of all deaths in 2005 in the United States, with the majority related to myocardial infarction, stroke, or complications thereof. The vast majority of patients suffering acute cardiovascular events have prior exposure to at least one major risk factor such as cigarette smoking, abnormal blood lipid levels, hypertension, diabetes, abdominal obesity, and low-grade inflammation.
Pathophysiologically, the major events of myocardial infarction and ischemic stroke are caused by a sudden arrest of nutritive blood supply due to a blood clot formation within the lumen of the arterial blood vessel. In most cases, formation of the thrombus is precipitated by rupture of a vulnerable atherosclerotic plaque, which exposes chemical agents that activate platelets and the plasma coagulation system. The activated platelets form a platelet plug that is armed by coagulation-generated fibrin to form a biood clot that expands within the vessel lumen until it obstructs or blocks blood flow, which results in hypoxic tissue damage (so-called infarction). Thus, thrombotic cardiovascular events occur as a result of two distinct processes, i.e. a slowly progressing long-term vascular atherosclerosis of the vessel wall, on the one hand, and a sudden acute clot formation that rapidly causes flow arrest, on the other. This invention solely relates to the latter process.
Recently, inflammation has been recognized as an important risk factor for thrombotic events. Vascular inflammation is a characteristic feature of the atherosclerotic vessel wall, and inflammatory activity is a strong determinant of the susceptibility of the atherosclerotic plaque to rupture and initiate intravascular clotting. Also, autoimmune conditions with systemic inflammation, such as rheumatoid arthritis, systemic lupus erythematosus and different forms of vasculitides, markedly increase the risk of myocardial infarction and stroke.
Traditional approaches to prevent and treat cardiovascular events are either targeted 1) to slow down the progression of the underlying atherosclerotic process, 2) to prevent clot formation in case of a plaque rupture, or 3) to direct removal of an acute thrombotic flow obstruction. In brief, antiatherosclerotic treatment aims at modulating the impact of general risk factors and includes dietary recommendations, weight loss, physical exercise, smoking cessation, cholesterol- and blood pressure treatment etc. Prevention of clot formation mainly relies on the use of antiplatelet drugs that inhibit platelet activation and/or aggregation, but also in some cases includes thromboembolic prevention with oral anticoagulants such as warfarin. Post-hoc treatment of acute atherothrombotic events requires either direct pharmacological lysis of the clot by thrombolytic agents such as recombinant tissue-type plasminogen activator or percutaneous mechanical dilation of the obstructed vessel.
Despite the fact that multiple-target antiatherosclerotic therapy and clot prevention by antiplatelet agents have lowered the incidence of myocardial infarction and ischemic stroke, such events still remain a major population health problem. This shows that in patients with cardiovascular risk factors these prophylactic measures are insufficient to completely prevent the occurrence of atherothrombotic events.
Likewise, thrombotic conditions on the venous side of the circulation, as well as embolic complications thereof such as pulmonary embolism, still cause substantial morbidity and mortality. Venous thrombosis has a different clinical presentation and the relative importance of platelet activation versus plasma coagulation are somewhat different with an preponderance for the latter in venous thrombosis, However, despite these differences, the major underlying mechanisms that cause thrombotic vessel occlusions are similar to those operating on the arterial circulation. Although unrelated to atherosclerosis as such, the risk of venous thrombosis is related to general cardiovascular risk factors such as inflammation and metabolic aberrations.

Panobinostat can be synthesized as follows: Reduction of 2-methylindole-3-glyoxylamide (I) with LiAlH4 affords 2-methyltryptamine (II). 4-Formylcinnamic acid (III) is esterified with methanolic HCl, and the resulting aldehyde ester (IV) is reductively aminated with 2-methyltryptamine (II) in the presence of NaBH3CN (1) or NaBH4 (2) to give (V). The title hydroxamic acid is then obtained by treatment of ester (V) with aqueous hydroxylamine under basic conditions.
Panobinostat is currently being used in a Phase I/II clinical trial that aims at curing AIDS in patients on highly active antiretroviral therapy (HAART). In this technique panobinostat is used to drive the HI virus’s DNA out of the patient’s DNA, in the expectation that the patient’s immune system in combination with HAART will destroy it.[6][7]
 panobinostat
panobinostat
Panobinostat has been found to synergistically act with sirolimus to kill pancreatic cancer cells in the laboratory in a Mayo Clinic study. In the study, investigators found that this combination destroyed up to 65 percent of cultured pancreatic tumor cells. The finding is significant because the three cell lines studied were all resistant to the effects of chemotherapy – as are many pancreatic tumors.[8]
Panobinostat has also been found to significantly increase in vitro the survival of motor neuron (SMN) protein levels in cells of patients suffering fromspinal muscular atrophy.[9]
Panobinostat was able to selectively target triple negative breast cancer (TNBC) cells by inducing hyperacetylation and cell cycle arrest at the G2-M DNA damage checkpoint; partially reversing the morphological changes characteristic of breast cancer cells.[10]
Panobinostat, along with other HDAC inhibitors, is also being studied for potential to induce virus HIV-1 expression in latently infected cells and disrupt latency. These resting cells are not recognized by the immune system as harboring the virus and do not respond to antiretroviral drugs.[11]
Panobinostat inhibits multiple histone deacetylase enzymes, a mechanism leading to apoptosis of malignant cells via multiple pathways.[1]
The compound N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (alternatively, N-hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide) has the formula

as described in WO 02/22577. Valuable pharmacological properties are attributed to this compound; thus, it can be used, for example, as a histone deacetylase inhibitor useful in therapy for diseases which respond to inhibition of histone deacetylase activity. WO 02/22577 does not disclose any specific salts or salt hydrates or solvates of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide.
The compounds described above are often used in the form of a pharmaceutically acceptable salt. Pharmaceutically acceptable salts include, when appropriate, pharmaceutically acceptable base addition salts and acid addition salts, for example, metal salts, such as alkali and alkaline earth metal salts, ammonium salts, organic amine addition salts, and amino acid addition salts, and sulfonate salts. Acid addition salts include inorganic acid addition salts such as hydrochloride, sulfate and phosphate, and organic acid addition salts such as alkyl sulfonate, arylsulfonate, acetate, maleate, fumarate, tartrate, citrate and lactate. Examples of metal salts are alkali metal salts, such as lithium salt, sodium salt and potassium salt, alkaline earth metal salts such as magnesium salt and calcium salt, aluminum salt, and zinc salt. Examples of ammonium salts are ammonium salt and tetramethylammonium salt. Examples of organic amine addition salts are salts with morpholine and piperidine. Examples of amino acid addition salts are salts with glycine, phenylalanine, glutamic acid and lysine. Sulfonate salts include mesylate, tosylate and benzene sulfonic acid salts.
……………………………..
GENERAL METHOD OF SYNTHESIS
ADD YOUR METHYL AT RIGHT PLACE
As is evident to those skilled in the art, the many of the deacetylase inhibitor compounds of the present invention contain asymmetric carbon atoms. It should be understood, therefore, that the individual stereoisomers are contemplated as being included within the scope of this invention.
The hydroxamate compounds of the present invention can be produced by known organic synthesis methods. For example, the hydroxamate compounds can be produced by reacting methyl 4-formyl cinnamate with tryptamine and then converting the reactant to the hydroxamate compounds. As an example, methyl 4-formyl cinnamate 2, is prepared by acid catalyzed esterification of 4-formylcinnamic acid 3 (Bull. Chem. Soc. Jpn. 1995; 68:2355-2362). An alternate preparation of methyl 4-formyl cinnamate 2 is by a Pd- catalyzed coupling of methyl acrylate 4 with 4-bromobenzaldehyde 5.
CHO

Additional starting materials can be prepared from 4-carboxybenzaldehyde 6, and an exemplary method is illustrated for the preparation of aldehyde 9, shown below. The carboxylic acid in 4-carboxybenzaldehyde 6 can be protected as a silyl ester (e.g., the t- butyldimethylsilyl ester) by treatment with a silyl chloride (e.g., f-butyldimethylsilyl chloride) and a base (e.g. triethylamine) in an appropriate solvent (e.g., dichloromethane). The resulting silyl ester 7 can undergo an olefination reaction (e.g., a Horner-Emmons olefination) with a phosphonate ester (e.g., triethyl 2-phosphonopropionate) in the presence of a base (e.g., sodium hydride) in an appropriate solvent (e.g., tetrahydrofuran (THF)). Treatment of the resulting diester with acid (e.g., aqueous hydrochloric acid) results in the hydrolysis of the silyl ester providing acid 8. Selective reduction of the carboxylic acid of 8 using, for example, borane-dimethylsuflide complex in a solvent (e.g., THF) provides an intermediate alcohol. This intermediate alcohol could be oxidized to aldehyde 9 by a number of known methods, including, but not limited to, Swern oxidation, Dess-Martin periodinane oxidation, Moffatt oxidation and the like.

The aldehyde starting materials 2 or 9 can be reductively aminated to provide secondary or tertiary amines. This is illustrated by the reaction of methyl 4-formyl cinnamate 2 with tryptamine 10 using sodium triacetoxyborohydride (NaBH(OAc)3) as the reducing agent in dichloroethane (DCE) as solvent to provide amine 11. Other reducing agents can be used, e.g., sodium borohydride (NaBH ) and sodium cyanoborohydride (NaBH3CN), in other solvents or solvent mixtures in the presence or absence of acid catalysts (e.g., acetic acid and trifluoroacetic acid). Amine 11 can be converted directly to hydroxamic acid 12 by treatment with 50% aqueous hydroxylamine in a suitable solvent (e.g., THF in the presence of a base, e.g., NaOH). Other methods of hydroxamate formation are known and include reaction of an ester with hydroxylamine hydrochloride and a base (e.g., sodium hydroxide or sodium methoxide) in a suitable solvent or solvent mixture (e.g., methanol, ethanol or methanol/THF).

NOTE ….METHYL SUBSTITUENT ON 10 WILL GIVE YOU PANOBINOSTAT

……………………………….
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide
lactate
(34, panobinostat, LBH589)
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
for str see above link
α-methyl-β-(β-bromoethyl)indole (29) was made according to method reported by Grandberg et al.(2. Grandberg, I. I.; Kost, A. N.; Terent’ev, A. P. Reactions of hydrazine derivatives. XVII. New synthesis of α-methyltryptophol. Zhurnal Obshchei Khimii 1957, 27, 3342–3345. )
The bromide 29 was converted to amine 30 by using similar method used by Sletzinger et al.(3. Sletzinger, M.; Ruyle, W. V.; Waiter, A. G. (Merck & Co., Inc.). Preparation of tryptamine
derivatives. U.S. Patent US 2,995,566, Aug 8, 1961.)
To a 500 mL flask, crude 2-methyltryptamine 30 (HPLC purity 75%, 1.74 g, 7.29 mmol) and 3-(4-
formyl-phenyl)-acrylic acid methyl ester 31 (HPLC purity 84%, 1.65 g, 7.28 mmol) were added,
followed by DCM (100 mL) and MeOH (30 mL). The clear solution was stirred at room temp for 30
min, then NaBH3CN (0.439 g, 6.99 mmol) was added in small portions. The reaction mixture was
stirred at room temp overnight. After removal of the solvents, the residue was diluted with DCM and
added saturated NaHCO3 aqueous solution, extracted with DCM twice. The DCM layer was dried
and concentrated, and the resulting residue was purified by flash chromatography (silica, 0–10%
MeOH in DCM) to afford 33 as orange solid (1.52 g, 60%). LC–MS m/z 349.2 ([M + H]+). 33 was
converted to hydroxamic acid 34 according to procedure D (Experimental Section), and the freebase
34 was treated with 1 equiv of lactic acid in MeOH–water (7:3) to form lactic acid salt which was
further recrystallized in MeOH–EtOAc to afford the lactic acid salt of 34as pale yellow solid. LC–MS m/z 350.2 ([M + H − lactate]+).
= DELTA
1H NMR (DMSO-d6) 10.72 (s, 1H, NH), 7.54 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 16 Hz, 1H), 7.43 (d, J = 7.8 Hz, 2H), 7.38 (d, J = 7.6 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 6.97 (td, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d, J = 7.8Hz, 2H), 7.01 (t, J = 7.4, 0.9 Hz, 1H), 6.91 (td, J = 7.4, 0.9 Hz, 1H), 6.47 (d, J = 15.2 Hz, 1H), 3.94(q, J = 6.8 Hz, 1H, lactate CH), 3.92 (s, 2H), 2.88 and 2.81 (m, each, 4H, AB system, CH2CH2),2.31 (s, 3H), 1.21 (d, J = 6.8 Hz, 3H).;
13C NMR (DMSO-d6) 176.7 (lactate C=O), 162.7, 139.0,
137.9, 135.2, 134.0, 132.1, 129.1, 128.1, 127.4, 119.9, 119.0, 118.1, 117.2, 110.4, 107.0, 66.0, 51.3,
48.5, 22.9, 20.7, 11.2.
…………………………………………..
PANOBINOSTAT DRUG SUBSTANCE SYNTHESIS AND DATA
http://www.google.com/patents/US7989639

A flow diagram for the synthesis of LBH589 lactate is provided in FIG. A. A nomenclature reference index of the intermediates is provided below in the Nomenclature Reference Index:
| Nomenclature reference index | |
| Compound | Chemical name |
| 1 | 4-Bromo-benzaldehyde |
| 2 | Methyl acrylate |
| 3 | (2E)-3-(formylphenyl)-2-propenoic acid, methyl ester |
| 4 | 3-[4-[[[2-(2-Methyl-1H-indol-3- |
| yl)ethyl]amino]methyl]phenyl]-2- | |
| propenoic acid, methyl ester, monohydrochloride | |
| 5 | (2E)-N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3- |
| yl)ethyl]amino]methyl]phenyl]-2-propenamide | |
| 6 | 2-hydroxypropanoic acid, compd. with 2(E)-N- |
| hydroxy-3-[4-[[[2-(2-methyl-1H- | |
| indol-3-yl)ethyl]amino]methyl]phenyl]-2-propenamide | |
| Z3a | 2-Methyl-1H-indole-3-ethanamine |
| Z3b | 5-Chloro-2-pentanone |
| Z3c | Phenylhydrazine |
The manufacture of LBH589 lactate (6) drug substance is via a convergent synthesis; the point of convergence is the condensation of indole-amine Z3a with aldehyde 3.
The synthesis of indole-amine Z3a involves reaction of 5-chloro-2 pentanone (Z3b) with phenylhydrazine (Z3c) in ethanol at reflux (variation of Fischer indole synthesis).
Product isolation is by an extractive work-up followed by crystallization. Preparation of aldehyde 3 is by palladium catalyzed vinylation (Heck-type reaction; Pd(OAc)2/P(o-Tol)3/Bu3N in refluxing CH3CN) of 4-bromo-benzyladehyde (1) with methyl acrylate (2) with product isolation via precipitation from dilute HCl solution. Intermediates Z3a and 3 are then condensed to an imine intermediate, which is reduced using sodium borohydride in methanol below 0° C. (reductive amination). The product indole-ester 4, isolated by precipitation from dilute HCl, is recrystallized from methanol/water, if necessary. The indole ester 4 is converted to crude LBH589 free base 5 via reaction with hydroxylamine and sodium hydroxide in water/methanol below 0° C. The crude LBH589 free base 5 is then purified by recrystallization from hot ethanol/water, if necessary. LBH589 free base 5 is treated with 85% aqueous racemic lactic acid and water at ambient temperature. After seeding, the mixture is heated to approximately 65° C., stirred at this temperature and slowly cooled to 45-50° C. The resulting slurry is filtered and washed with water and dried to afford LBH589 lactate (6).
If necessary the LBH589 lactate 6 may be recrystallised once again from water in the presence of 30 mol % racemic lactic acid. Finally the LBH589 lactate is delumped to give the drug substance. If a rework of the LBH589 lactate drug substance 6 is required, the LBH589 lactate salt is treated with sodium hydroxide in ethanol/water to liberate the LBH589 free base 5 followed by lactate salt formation and delumping as described above.
All starting materials, reagents and solvents used in the synthesis of LBH589 lactate are tested according to internal specifications or are purchased from established suppliers against a certificate of analysis.
EXAMPLE 7 Formation of Monohydrate Lactate Salt
About 40 to 50 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base was suspended in 1 ml of a solvent as listed in Table 7. A stoichiometric amount of lactic acid was subsequently added to the suspension. The mixture was stirred at ambient temperature and when a clear solution formed, stirring continued at 4° C. Solids were collected by filtration and analyzed by XRPD, TGA and 1H-NMR.
| TABLE 7 | |||||
| LOD, % | |||||
| Physical | Crystallinity | (Tdesolvation) | |||
| Solvent | T, ° C. | Appear. | and Form | Tdecomposit. | 1H-NMR |
| IPA | 4 | FFP | excellent | 4.3 (79.3) | — |
| HA | 156.3 | ||||
| Acetone | 4 | FFP | excellent | 4.5 (77.8) | 4.18 (Hbz) |
| HA | 149.5 | ||||
The salt forming reaction in isopropyl alcohol and acetone at 4° C. produced a stoichiometric (1:1) lactate salt, a monohydrate. The salt is crystalline, begins to dehydrate above 77° C., and decomposes above 150° C.
EXAMPLE 18 Formation of Anhydrous Lactate Salt
DL-lactic acid (4.0 g, 85% solution in water, corresponding to 3.4 g pure DL-lactic acid) is diluted with water (27.2 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution is allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base (10.0 g) is placed in a 4-necked reaction flask with mechanical stirrer. Demineralized water (110.5 g) is added, and the suspension is heated to 65° C. (inner temperature) within 30 minutes. The DL-lactic acid solution is added to this suspension during 30 min at 65° C. During the addition of the lactate salt solution, the suspension converted into a solution. The addition funnel is rinsed with demineralized water (9.1 g), and the solution is stirred at 65° C. for an additional 30 minutes. The solution is cooled down to 45° C. (inner temperature) and seed crystals (10 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate monohydrate) are added at this temperature. The suspension is cooled down to 33° C. and is stirred for additional 20 hours at this temperature. The suspension is re-heated to 65° C., stirred for 1 hour at this temperature and is cooled to 33° C. within 1 hour. After additional stirring for 3 hours at 33° C., the product is isolated by filtration, and the filter cake is washed with demineralized water (2×20 g). The wet filter-cake is dried in vacuo at 50° C. to obtain the anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt as a crystalline product. The product is identical to the monohydrate salt (form HA) in HPLC and in 1H-NMR, with the exception of the integrals of water signals in the 1H-NMR spectra.
In additional salt formation experiments carried out according to the procedure described above, the product solution was filtered at 65° C. before cooling to 45° C., seeding and crystallization. In all cases, form A (anhydrate form) was obtained as product.
EXAMPLE 19 Formation of Anhydrous Lactate Salt
DL-lactic acid (2.0 g, 85% solution in water, corresponding to 1.7 g pure DL-lactic acid) is diluted with water (13.6 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution was allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base (5.0 g) is placed in a 4-necked reaction flask with mechanical stirrer. Demineralized water (54.85 g) is added, and the suspension is heated to 48° C. (inner temperature) within 30 minutes. The DL-lactic acid solution is added to this suspension during 30 minutes at 48° C. A solution is formed. Seed crystals are added (as a suspension of 5 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form A, in 0.25 g of water) and stirring is continued for 2 additional hours at 48° C. The temperature is raised to 65° C. (inner temperature) within 30 minutes, and the suspension is stirred for additional 2.5 hours at this temperature. Then the temperature is cooled down to 48° C. within 2 hours, and stirring is continued at this temperature for additional 22 hours. The product is isolated by filtration and the filter cake is washed with demineralized water (2×10 g). The wet filter-cake is dried in vacuo at 50° C. to obtain anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A) as a crystalline product.
EXAMPLE 20 Conversion of Monohydrate Lactate Salt to Anhydrous Lactate Salt
DL-lactic acid (0.59 g, 85% solution in water, corresponding to 0.5 g pure DL-lactic acid) is diluted with water (4.1 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution is allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
10 g of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt monohydrate is placed in a 4-necked reaction flask. Water (110.9 g) is added, followed by the addition of the lactic acid solution. The addition funnel of the lactic acid is rinsed with water (15.65 g). The suspension is heated to 82° C. (inner temperature) to obtain a solution. The solution is stirred for 15 minutes at 82° C. and is hot filtered into another reaction flask to obtain a clear solution. The temperature is cooled down to 50° C., and seed crystals are added (as a suspension of 10 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form, in 0.5 g of water). The temperature is cooled down to 33° C. and stirring is continued for additional 19 hours at this temperature. The formed suspension is heated again to 65° C. (inner temperature) within 45 minutes, stirred at 65° C. for 1 hour and cooled down to 33° C. within 1 hour. After stirring at 33° C. for additional 3 hours, the product is isolated by filtration and the wet filter cake is washed with water (50 g). The product is dried in vacuo at 50° C. to obtain crystalline anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A).
EXAMPLE 21 Formation of Anhydrous Lactate Salt
DL-lactic acid (8.0 g, 85% solution in water, corresponding to 6.8 g pure DL-lactic acid) was diluted with water (54.4 g), and the solution was heated to 90° C. (inner temperature) for 15 hours. The solution was allowed to cool down to room temperature and was used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (20 g) is placed in a 1 L glass reactor, and ethanol/water (209.4 g of a 1:1 w/w mixture) is added. The light yellow suspension is heated to 60° C. (inner temperature) within 30 minutes, and the lactic acid solution is added during 30 minutes at this temperature. The addition funnel is rinsed with water (10 g). The solution is cooled to 38° C. within 2 hours, and seed crystals (20 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form) are added at 38° C. After stirring at 38° C. for additional 2 hours, the mixture is cooled down to 25° C. within 6 hours. Cooling is continued from 25° C. to 10° C. within 5 hours, from 10° C. to 5° C. within 4 hours and from 5° C. to 2° C. within 1 hour. The suspension is stirred for additional 2 hours at 2° C., and the product is isolated by filtration. The wet filter cake is washed with water (2×30 g), and the product is dried in vacuo at 45° C. to obtain crystalline anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A).
EXAMPLE 28 Formation of Lactate Monohydrate Salt
3.67 g (10 mmol) of the free base monohydrate (N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide) and 75 ml of acetone were charged in a 250 ml 3-neck flask equipped with a magnetic stirrer and an addition funnel. To the stirred suspension were added dropwise 10 ml of 1 M lactic acid in water (10 mmol) dissolved in 20 ml acetone, affording a clear solution. Stirring continued at ambient and a white solid precipitated out after approximately 1 hour. The mixture was cooled in an ice bath and stirred for an additional hour. The white solid was recovered by filtration and washed once with cold acetone (15 ml). It was subsequently dried under vacuum to yield 3.94 g of the lactate monohydrate salt of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (86.2%).
References
- Revill, P; Mealy, N; Serradell, N; Bolos, J; Rosa, E (2007). “Panobinostat”. Drugs of the Future 32 (4): 315. doi:10.1358/dof.2007.032.04.1094476. ISSN 0377-8282.
- Table 3: Select epigenetic inhibitors in various stages of development from Mack, G. S. (2010). “To selectivity and beyond”. Nature Biotechnology 28 (12): 1259–1266.doi:10.1038/nbt.1724. PMID 21139608. edit
- ClinicalTrials.gov NCT00425555 Study of Oral LBH589 in Adult Patients With Refractory Cutaneous T-Cell Lymphoma
- ClinicalTrials.gov: LBH-589
- Prince, HM; M Bishton (2009). “Panobinostat (LBH589): a novel pan-deacetylase inhibitor with activity in T cell lymphoma”. Hematology Meeting Reports (Parkville, Australia: Peter MacCallum Cancer Centre and University of Melbourne) 3 (1): 33–38.
- Simons, J (27 April 2013). “Scientists on brink of HIV cure”. The Telegraph.
- ClinicalTrials.gov NCT01680094 Safety and Effect of The HDAC Inhibitor Panobinostat on HIV-1 Expression in Patients on Suppressive HAART (CLEAR)
- Mayo Clinic Researchers Formulate Treatment Combination Lethal To Pancreatic Cancer Cells
- Garbes, L; Riessland, M; Hölker, I; Heller, R; Hauke, J; Tränkle, Ch; Coras, R; Blümcke, I; Hahnen, E; Wirth, B (2009). “LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate”. Human Molecular Genetics 18 (19): 3645–3658. doi:10.1093/hmg/ddp313.PMID 19584083.
- Tate, CR; Rhodes, LV; Segar, HC; Driver, JL; Pounder, FN; Burow, ME; and Collins-Burow, BM (2012). “Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat”. Breast Cancer Research 14 (3).
- TA Rasmussen, et al. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Human Vaccines & Immunotherapeutics 9:5, 1-9, May 2013.
- Drugs of the Future 32(4): 315-322 (2007)
- WO 2002022577…
- WO 2007146718
- WO 2013110280
- WO 2010009285
- WO 2010009280
- WO 2005013958
- WO 2004103358
- WO 2003048774…
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
-
23009203 11-26-2012 Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. Journal of medicinal chemistry -
21634430 7-14-2011 Discovery of (2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an orally active histone deacetylase inhibitor with a superior preclinical profile. Journal of medicinal chemistry -
21417419 4-28-2011 Discovery, synthesis, and pharmacological evaluation of spiropiperidine hydroxamic acid based derivatives as structurally novel histone deacetylase (HDAC) inhibitors. Journal of medicinal chemistry -
19317450 4-23-2009 Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. Journal of medicinal chemistry -
15650931 1-1-2005 The American Society of Hematology–46th Annual Meeting and Exposition. HDAC, Flt and farnesyl transferase inhibitors. IDrugs : the investigational drugs journal -
US7989639 8-3-2011 PROCESS FOR MAKING SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2010286409 11-12-2010 SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2010179208 7-16-2010 Use of HDAC Inhibitors for the Treatment of Bone Destruction US2010160257 6-25-2010 USE OF HDAC INHIBITORS FOR THE TREATMENT OF MYELOMA US2010137398 6-4-2010 USE OF HDAC INHIBITORS FOR THE TREATMENT OF GASTROINTESTINAL CANCERS US2009306405 12-11-2009 PROCESS FOR MAKING N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE AND STARTING MATERIALS THEREFOR US2009281159 11-13-2009 USE OF HDAC INHIBITORS FOR THE TREATMENT OF LYMPHOMAS US2009264439 10-23-2009 Combination of a) N–4-(3-pyridyl)-2-pyrimidine-amine and b) a histone deacetylase inhibitor for the treatment of leukemia US2009197936 8-7-2009 SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2009012066 1-9-2009 Method of Use of Deacetylase Inhibitors
| US2008319045 | 12-26-2008 | Combination of Histone Deacetylase Inhibitors and Radiation |
| US2008221126 | 9-12-2008 | Use of Hdac Inhibitors for the Treatment of Myeloma |
| US2008176849 | 7-25-2008 | DEACETYLASE INHIBITORS |
| US2006189674 | 8-25-2006 | Deacetylase inhibitors |
| US7067551 | 6-28-2006 | Deacetylase inhibitors |
| US2006100140 | 5-12-2006 | Combination of a) n-{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]2-methylphenyl}-4- (3-pyridyl)-2-pyrimidine-amine and b) a histone deacetylase inhibitor for the treatment of leukemia |
| US6833384 | 12-22-2004 | Deacetylase inhibitors |
| US6552065 | 4-23-2003 | Deacetylase inhibitors |
| GB776693A | Title not available | |||
| GB891413A | Title not available | |||
| GB2185020A | Title not available | |||
| WO2002022577A2 | Aug 30, 2001 | Mar 21, 2002 | Kenneth Walter Bair | Hydroxamate derivatives useful as deacetylase inhibitors |
| WO2003016307A1 | Aug 6, 2002 | Aug 19, 1993 | Jolie Anne Bastian | β3 ADRENERGIC AGONISTS |
| WO2003039599A1 | Nov 5, 2002 | May 15, 2003 | Ying-Nan Pan Chen | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| WO2005105740A2 | Apr 26, 2005 | Nov 10, 2005 | Serguei Fine | Preparation of tegaserod and tegaserod maleate |
| WO2006021397A1 | Aug 22, 2005 | Mar 2, 2006 | Recordati Ireland Ltd | Lercanidipine salts |
…………………………………..
extras
5. Mocetinostat (MGCD0103), including pharmaceutically acceptable salts thereof. Balasubramanian et al., Cancer Letters 280: 211-221 (2009).
Mocetinostat, has the following chemical structure and name:

Vorinostat, including pharmaceutically acceptable salts thereof. Marks et al., Nature Biotechnology 25, 84 to 90 (2007); Stenger, Community Oncology 4, 384-386 (2007).
Vorinostat has the following chemical structure and name:

Belinostat (PXD-101 , PX-105684)
(2E)-3-[3-(anilinosulfonyl)phenyl]-N-hydroxyacrylamide

……………………………………………….
Dacinostat (LAQ-824, NVP-LAQ824,)
((E)-N-hydroxy-3-[4-[[2-hydroxyethyl-[2-(1 H-indol-3-yl)ethyl]amino]methyl]phenyl]prop-2-enamide

Entinostat (MS-275, SNDX-275, MS-27-275)
4-(2-aminophenylcarbamoyl)benzylcarbamate

(a) The HDAC inhibitor Vorinostat™ or a salt, hydrate, or solvate thereof.

Vorinostat………………..
(b) The HDAC inhibitor Givinostat or a salt, hydrate, or solvate thereof.

Givinostat or a salt, hydrate, or solvate thereof.
BELINOSTAT, FAST TRACK, ORPHAN DRUG, A hydroxamate-type inhibitor of histone deacetylase.

Belinostat (PXD101)
PHASE 2, FAST TRACK FDA , ORPHAN STATUS
- PDX101
- PX 105684
- PXD-101
- PXD101
- UNII-F4H96P17NZ
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
A hydroxamate-type inhibitor of histone deacetylase.
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
………………………………………………………………………..
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


…………………………………..
GENERAL SYNTHESIS
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A

Scheme 3B

Scheme 4


Scheme 8

Scheme 9

……………………………………………………………………..
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)

3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)

To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
……………………………………………………………………….
SYNTHESIS
US20100286279

…………………………………………………….
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,
J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1,
140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
………………………………………………..
SYNTHESIS
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
……………….
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.
………………………..

- Plumb, Jane A.; Finn, Paul W.; Williams, Robert J.; Bandara, Morwenna J.; Romero, M. Rosario; Watkins, Claire J.; La Thangue, Nicholas B.; Brown, Robert (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101”. Molecular Cancer Therapeutics 2 (8): 721–728. PMID 12939461.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
- Helvetica Chimica Acta, 2005 , vol. 88, 7 PG. 1630 – 1657, MP 172
- WO2009/40517 A2, ….
- WO2006/120456 A1, …..
- Synthetic Communications, 2010 , vol. 40, 17 PG. 2520 – 2524, MP 172
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 PG. 4694 – 4720, NMR IN SUP INFO
| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
………………………..
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
……………………………..


LEUCODERMA CASE ; PROGRESSING FOR CURE ; सफेद दाग , लियूकोडर्मा का एक केस जिसे ई०टी०जी० आयुर्वेदास्कैन तकनीक आधारित आयुर्वेदिक इलाज से फायदा
दिनान्क २८ अगस्त २०१३ को एक २७ साल के लड़्के ने सफेद दाग के इलाज के लिये मेरे OUT DOOR HOSPITAL मे consultation के लिये समपर्क किया था / इस लड़्के के सारे शरीर पर छोटे बड़े सैकड़ों की सन्ख्या मे LUECODERMA यानी सफेद दाग के चकत्ते पड़े हुये थे, जो उसको पिछले १५ साल पहले हुये थे / इसके पिता एक होम्योपैथी के डाक्टर है जो प्रैक्टिस करते है / वे ही इसे लेकर इलाज के लिये मेरे OUT-DOOR HOSPITAL मे लेकर आये थे /
मैने उनको बताया कि बिना ई०टी०जी० आयुर्वेदास्कैन और आयुर्वेद के रकत और पेशाब के परीक्शन के इलाज कराना बेकार है / LEUCODERMA के ईलाज के लिये परीक्षण कराना सबसे पहली आवश्यकता है /
दिनाक २८ अगस्त २०१३ को इस लड़्के का शरीर के दो हिस्सो का PHOTOGRAPH लिया गया था / नीचे दिया गया photograph इसी दिन का है /
यह PHOTOGRAPH मरीज के…
View original post 1,012 more words
CLAZOSENTAN

Clazosentan
READ ALL AT
http://www.allfordrugs.com/2014/01/22/clazosentan/

READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
Tezosentan Disodium for pulmonary hypertension

TEZOSENTAN
180384-57-0 CAS OF FREE ACID
N-[6-(2-Hydroxyethoxy)-5-(2-methoxyphenoxy)-2-[2-(2H-tetrazol-5-yl)pyridin-4-yl]pyrimidin-4-yl]-5-propan-2-ylpyridine-2-sulfonamide
5-isopropyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5- (2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide
| Formula | C27H27N9O6S |
|---|---|
| Mol. mass | 605.624 |
…………………………………………………………………………….

Tezosentan disodium, Ro-61-0612, Veletri
5-isopropyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide sodium salt (1:2)
180384-58-1 of disodium salt, 180384-57-0 (free acid)

Tezosentan is a non-selective ETA and ETB receptor antagonist.[1] It acts as a vasodilator and was designed as a therapy for patients with acuteheart failure. Recent studies have shown however, that tezosentan does not improve dyspnea or reduce the risk of fatal or nonfatal cardiovascular events.[2]
Pulmonary disease (COPD), which may possibly be associated with pulmonary hypertension, as well as allergic and non-allergic rhinitis, provided that treatment with endothelin from a therapeutic standpoint is not contraindicated.
Tezosentan disodium is an endothelin ETB receptor antagonist in phase II clinical development for the treatment of stable, chronic pulmonary arterial hypertension. The drug was previously being evaluated for heart failure, but trials in that indication have been discontinued. The compound is being developed by Actelion.
………………………………..
SYNTHESIS

………………………………..
SYNTHESIS

Reaction of 4-cyano-pyridine (I) with Na in methanol followed by treatment with ammonium chloride provides 4-amidino-pyridine hydrochloride (II), which is then converted into 5-(2-methoxyphenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (IV) by condensation with diethyl malonate derivative (III) by means of Na in MeOH. By heating compound (IV) with phosphorus oxychloride (POCl3), 4,6-dichloro-5-(2-methoxyphenoxy)-2-pyridin-4-yl)pyrimidine (V) is obtained, which in turn is oxidized with peracetic acid in refluxing acetonitrile to afford N-oxide derivative (VI). Condensation of (VI) with 5-isopropylpyridine-2-sulfonamide potassium (VII) furnishes 5-isopropylpyridine-2-sulfonic acid 6-chloro-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl amide (VIII), which is then dissolved in dimethoxyethane and subjected to reaction with Na in hot ethylene glycol (IX) to provide N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl]-5-isopropylpyridine-2-sulfonamide (X). Refluxing of (X) with trimethylsilylcyanide and Et3N in acetonitrile yields cyano derivative (XI), which is then converted into the tetrazole derivative (XII) by reaction with sodium azide and NH4Cl in DMF at 70 C. Finally, the disodium salt of tezosentan is obtained by treatment of (XII) with Na/MeOH in THF. refEP 0799209; JP 1998509182; WO 9619459
…………………………………..
SYNTHESIS PROCEDURE as in EP0979822A1
Examples
- 1360 ml of formamide were added to 136 g (437 mmol) of 5-(2-methoxy-phenoxy)-2-pyridine-4-yl-pyrimidine-4,6-diole. Then, at a temperature of 0°C, 11.7 ml (219 mmol) of concentrated sulfuric acid and thereafter 36.5 g (130 mmol) of iron(II)sulfate heptahydrate were added to the suspension. After that, 89 ml (874 mmol) of 30% hydrogen peroxide were added dropwise within 1 hr at a temperature of 0°C to 5°C. The viscous yellow-brownish suspension was stirred at 0°C for 1.5 hr. Subsequently, a solution of 83 g (437 mmol) of sodium pyrosulfite in 680 ml of de-ionized water was added dropwise to the reaction mixture within 30 min. at 0°C to 5°C and the reaction mixture was stirred at 0°C to 5°C for 30 min. The suspension was then filtered under reduced pressure. The filtrate was first washed with 1750 ml of de-ionized water and thereafter with 700 ml of ethanol. Then the solid was dried at 80°C, 2000 Pa for 16 hr. There were obtained 132.4 g (91% of theory) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide with a HPLC purity of 91.4% (w/w).
-
Preparation of starting material:
- a) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hr 29.5 g of NH4Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydrochloride (decomposition point 245-247°C).
- b) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 min. to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in a) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridine-4-yl)-pyrimidine-4,6-diole (or tautomer), melting point above 250°C.
- Example 1
Example 2
- Within 20 min. 61 ml (633 mmol) of POCl3 were added dropwise to 34 ml (200 mmol) of diisopropyl ethylamine at 5°C to 10°C followed by stirring at 5°C to 10°C for 15 min. Then 23.5 g (66 mmol) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide were added in four portions under cooling followed by stirring at 90°C for 25 hr. The reaction mixture was cooled down to 20°C and transferred to a new flask together with 50 ml of dichloromethane. Volatile components (i.e. excess of POCl3) was removed by evaporation from 20°C to 70°C followed by re-distillation with 100 ml of toluene. After adding 250 ml of dichloromethane to the residue (88 g of a black oil) the solution was heated to 35°C to 40°C and 80 ml of de-ionized water were added dropwise within 30 min. whereby the pH was kept constant by the subsequent addition of 28% NaOH solution (60 ml) within 5 to 6 hr. The mixture was stirred at 35°C to 40°C for 30 min. followed by removal of dichloromethane by distillation. The resulting suspension was allowed to cool down to 20°C and was stirred for additional 2 hr. The solid was filtered off under suction, washed with 500 ml of water and dried at 70°C, 2000 Pa for 16 hr. There were obtained 21.3 g (86% of theory) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile with a HPLC purity of 94.3% (w/w).
- 8.95 g (24 mmol) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile were suspended in 100 ml of acetone. At a temperature of 20°C, 5.04 g (25 mmol) of 5-isopropyl-pyridine-2-sulfonamide, 1 ml of de-ionized water, 10.6 g (77 mmol) of potassium carbonate and 135 mg (1.2 mmol) 1,4-diazobicyclo[2.2.2]octane were added. The mixture was stirred at 40°C for 20 hr. Thereafter, another 240 mg (1.2 mmol) of 5-isopropyl-pyridine-2-sulfonamide and 80 mg (0.7 mmol) of 1,4-diazobicyclo[2.2.2]octane were added. The reaction mixture was stirred for 24 hr at 40°C followed by cooling to 20°C. Then 50 ml of de-ionized water and 45 ml of 3 N aqueous hydrochloric acid were added slowly until pH = 1. The acetone was removed by distillation and the resulting suspension was stirred at 20°C for 1.5 hr. The solid was filtered off under suction, washed first with 100 ml of de-ionized water and thereafter with 50 ml of t-butylmethylether. Then the solid was dried at 70°C, 2000 Pa for 20 hr. There were obtained 13.2 g (102% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 87.8% (w/w).
- Example 4
- 122 g (233 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide was suspended in 450 ml of N,N-dimethyl formamide and the mixture was cooled down to 15°C. At this temperature, 35 ml of hydrazine hydrate were added dropwise within 1 hr. The resulting solution was stirred at 15°C to 20°C for 16 hr and thereafter diluted with 600 ml of de-ionized water. Then 50 ml of glacial acetic acid were added dropwise at 0°C to 5°C until pH = 5.5. 600 g of ice were added and the suspension was stirred for 1 hr. The solid was filtered off under suction, washed with 3000 ml of water and dried at 40°C, 2000 Pa for 24 hr. There were obtained 126 g (97% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 91.8% (w/w).
- Example 6
- 20 g (35 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide were added to 160 ml of N,N-dimethyl formamide. The solution was kept at 15°C to 20°C and 23 ml of 6 N aqueous hydrochloric acid were added, followed by addition of a solution containing 4.8 g (7 mmol) of sodium nitrite in 20 ml de-ionized water within 10 min. The mixture was stirred at 20°C for 1 hr, then 140 ml of de-ionized water were added and the suspension was stirred at 0°C for 1 hr. The solid was filtered, firstly washed with 80 ml of de-ionized water and thereafter with 80 ml of t-butylmethylether. Then the solid was dried at 70°C and 2000 Pa for 16 hr. The crude product (23.4 g) was taken up with 117 ml of tetrahydrofuran for 1 hr. After filtration at 0°C the crystallized product was washed with 25 ml of t-butylmethylether and was then dried at 70°C, 2000 Pa for 16 hr. There were obtained 17.3 g (84% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 91.1% (w/w).
- Example 8
- Example 10
- 6.2 g of sodium hydroxide were added to 15 g (26 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amid and 75 ml of ethylene glycol. The mixture was heated to 85°C for 5 hr. Then 55 ml of de-ionized water were added and thereafter 55 ml of 3 N hydrochloric acid were added dropwise. The mixture was allowed to cool down to 20°C and was stirred for 1 hr. The solid was filtered off and dried at 70°C, 2000 Pa for 18 hr. There were obtained 16.2 g (103%) of 5-isopropyl-pyridine-2-sulfonic acid 16-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 92% (w/w). 80 ml of dioxane and 80 ml of ethanol were added to this solid. At a temperature of 60°C, gaseous ammonia was introduced into the liquid until pH = 9 to 10. The resulting suspension was allowed to cool down to 20°C and was stirred at 20°C for 20 hr and thereafter at 0°C for 2.5 hr. Then the solid was filtered off and dried at 70°C, 2000 Pa for 18 hr. There were obtained 14.2 g of mono ammonium salt with a HPLC purity of 96.2% (w/w). The solid was heated (reflux) in 70 ml of methanol, cooled down slowly to 20°C and stirred at 20°C for 19 hr and thereafter at 0°C for 2 hr. Then the solid was filtered off and dried at 70°C, 2000 Pa for 19 hr. There were obtained 11.5 g (66% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide sodium salt (1:2) with a HPLC purity of 98.6% (w/w).

Reaction of 2-chloro-5-ispropylpyridine (VII) with thiourea (A) in aqueous HCl gives 5-isopropyl- pyridine-2-thiol (VIII), which is chlorinated with chlorine in acetic acid to yield 5-isopropylpyridine-2-sulfochloride (IX). This compound is converted into 5-isopropylpyridine-2-sulfonamide potassium salt (X).
…………………………
synthesis
. Example 1
a) 200 ml of dimethoxyethane and 1 10.9 g of 4-[4-(4-tert- butyl-phenyl-sulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide are added all at once to a solution of 23.80 g of sodium in 660 ml of ethylene glycol. The solution is heated at 90°C for 20 hours while stirring, thereafter cooled, poured into 2500 ml of H2O and thereafter treated with CH3COOH to pH 5. The mixture is extracted three times with EtOAc, the organic phase is washed with H2O, dried with Na2Sθ4 and evaporated under reduced pressure. The residue is recrystall- ized from CH3CN and thereafter twice from a mixture of acetone and CH3CN. There is thus obtained 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide.
Preparation of the starting material:
b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After
6 hours 29.5 g of NH4CI are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydro- chloride (decomposition point 245-247°C).
c) 1 12.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydro- chloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)- pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
d) A suspension of 1 54.6 g of 5-(2-methoxy-phenoxy)-2- (pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCI3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2CI2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2CI2. The combined CH2CI2 extracts are dried with MgSθ4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2CI2 remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)- pyrimidine, melting point 1 78-1 80°C.
e) A solution of 1 7.4 g of 4,6-dichloro-5-(2-methoxy- phenoxy)-2-pyridin-4-yl)-pyrimidine in 100 ml of CH3CN is boiled at reflux for 3 hours with 1 5 ml of a 32% peracetic acid solution, thereafter cooled and stored in a refrigerator overnight. The crystals are filtered off under suction and dried at 50°C under reduced pressure. There is thus obtained 4-[4,6-dichloro- 5-(2-methoxy-phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide, melting point 189-1 90°C.
f) A solution of 36.4 g of 4-[4,6-dichloro-5-(2-methoxy- phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide and 52.8 g of p-tert- butylphenyl-sulphonamide potassium in 1 50 ml of abs. DMF is stirred at room temperature for 24 hours. Thereafter, it is poured into a mixture of 1 500 ml of H2O and 1000 ml of ether while stirring mechanically, whereby a precipitate forms. The suspension is adjusted to pH 5 with CH3COOH, suction filtered, the crystals are washed with cold water and thereafter with ether and dried at 50°C. There is thus obtained 4-[4-(4-tert- butyl-phenylsulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide as a colourless material of melting point 247-249°C.
Example 2
A solution of 78.45 g of 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide, 122.5 g of trimethylsilyl cyanide, 127.8 g of triethylamine and 1200 ml of CH3CN is boiled at reflux for 20 hours and thereafter evaporated under reduced pressure. The oily residue is taken up in 1000 ml of EtOAc and the solution is washed with CH3COOH:H2θ 9:1 and then with H2O. The EtOAc extracts are dried with Na2SO4. After evaporation of the solvent the residue is taken up in a mixture of CH3CN and CF3COOH (20:1 ), whereby a crystalline precipitate separates. There is thus obtained 4-tert-butyl-N-[2-(2-cyano-pyridin-4- yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4- yl]-benzenesulphonamide of melting point 176-1 79°C.
Example 3 for analogy only compd is different
A suspension of 50.0 g of 4-tert-butyl-N-[2-(2-cyano- pyridin-4-yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-4-yl]-benzenesulphonamide, 46.33 g of NH4CI and 56.47 g of NaN3 in 1600 ml of DMF is heated to 70°C for 24 hours while stirring vigorously. The majority of the solvent is distilled off under reduced pressure, the residue is dissolved in H2O, the solution is extracted four times at pH 6.5 with ether, thereafter treated with CH3COOH to pH = 4.5 and extracted with EtOAc. After working up there is obtained a residue which is treated with ether and filtered off under suction therefrom. There is thus obtained 4-tert-butyl-N-[6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-yl]-benzenesulphonamide, melting point 225-227°C.
Example 30 final product
In analogy to Example 3, from 5-isopropyl-pyridine-2- sulphonic acid 2-(2-cyano-pyridin-4-yl)-6-(2-hydroxy-ethoxy)- 5-(2-methoxy-phenoxy)-pyrimidin-4-ylamide there is obtained 5-isopropyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5- (2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide (tezosantan free base) as a white substance of melting point 1 98- 200°C from acetonitrile.
The corresponding disodium salt (tezosantan di sodium salt) is obtained as a white powder from this product using sodium methylate in analogy to Example 5
Example 5 for analogy only, compd is different
A solution of 47.8 g of 2-[6-(4-tert-butyl-phenylsulphonyl- amino)-5-(2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin- 4-yl)-pyrimidin-4-yloxy]-ethyl pyridin-2-ylcarbamate in 500 ml of abs. THF is treated dropwise with a cold solution of 2.8 g of sodium in 50 ml of methanol, whereby there forms gradually a solid precipitate which, after stirring at room temperature for 1 hour, is filtered off under suction, dried under greatly reduced pressure at 35°C for 3 days and thereafter at 50°C for 2 days. There is thus obtained the bis-sodium salt, decomposition point above 250°C.
References
- Urbanowicz, W; Sogni, P, Moreau, R, Tazi, K A, Barriere, E, Poirel, O, Martin, A, Guimont, M C, Cazals-Hatem, D, Lebrec, D (2004). “Tezosentan, an endothelin receptor antagonist, limits liver injury in endotoxin challenged cirrhotic rats”. Gut (BMJ Publishing Group Ltd & British Society of Gastroenterology) 53 (12): 1844–1849. doi:10.1136/gut.2003.036517. PMC 1774327. PMID 15542526.
- “Tezosentan does not appear to improve symptoms for patients with acute heart failure”. Medical Studies/Trials. news-medical.net. 7 Nov 2007. Retrieved 2007-11-24.
4 US2003/100507 A1
5 Drugs Fut 2003,28(8),754
6 WO 1996019459……
7 EP 0897914
8 WO 2011163085
9 WO 2004082637
| 10 WO 2002074034 |
11…
| 15055997 | 4-8-2004 | Discovery, modeling, and human pharmacokinetics of N-(2-acetyl-4,6-dimethylphenyl)-3-(3,4-dimethylisoxazol-5-ylsulfamoyl)thiophene-2-carboxamide (TBC3711), a second generation, ETA selective, and orally bioavailable endothelin antagonist. | Journal of medicinal chemistry |
12 ..
| 10610277 | 7-1-1999 | RO 610612 . | Drugs in R&D |
13….
| 3-27-2003 | Aqueous pharmaceutical composition comprising Tezosentan | |
| US6103902 | 8-16-2000 | Carbamoylation process |
| WO0036918 | 6-30-2000 | METHODS AND COMPOSITIONS FOR TREATMENT OF CELL PROLIFERATIVE DISORDERS METHODS AND COMPOSITIONS FOR TREATMENT OF CELL PROLIFERATIVE DISORDERS |
| US6063911 | 5-17-2000 | Methods and compositions for treatment of cell proliferative disorders |
READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
DR ANTHONY MELVIN CRASTO
ORGANIC SPECTROSCOPY

