
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Triphala : A Digestive Miracle


| Terminalia bellirica | |
|---|---|

|
| Terminalia chebula | |
|---|---|
_leafless_tree_at_23_Mile,_Duars,_WB_W_IMG_5905.jpg) |

Triphala (/triːˈfɑːlə/ or /triːˈfælə/; Hindi/Sanskrit: त्रिफला, triphalā [trɪˈpʰɐlaː], “three fruits”)[1] is an Ayurvedic[2] herbal rasayana formula consisting of equal parts of three myrobalans, taken without seed: Amalaki (Emblica officinalis), Bibhitaki (Terminalia bellirica), and Haritaki (Terminalia chebula).[1]
Medicinal use
In traditional Ayurvedic medicine, Triphala is used for:
- immune system stimulation[3]
- improvement of digestion[4][1]
- relief of constipation[4][1]
- gastrointestinal tract cleansing[4]
- relief of gas[1]
- treatment of diabetes[1]
- treatment of eye disease[1]
These health claims have not been yet tested in clinical trials. Even within the practice of Ayurvedic medicine, there are controversies about the composition (amlaki, haritaki and bibhitaki), preparation, and medicinal uses of Triphala.[5]
The active constituents are unknown. Triphala contains several compounds that have been proposed to be responsible for its claimed health benefits, including gallic acid, chebulagic acid, and chebulinic acid. [6][7]
Contemporary research on triphala
There is preliminary evidence that Triphala contains compounds with antioxidant properties in isolated cells and rats, however this has not yet been demonstrated in people.[6][8][9][10]
Triphala, widely used by natural Ayurvedic healers in India for thousands of years, contains 3 different fruits: Harada, Amla and Bihara. The word “Triphala”literally means “three fruits”. The combination of these three fruits cleanses the gastro-intestinal tract in a natural and gentle way. Basically our “bathroom experience” becomes a better one That is the best way I can put it!!
Why should we cleanse?
It’s always a good idea to cleanse! Get rid of toxins that build up in our bodies so that our bodies can function most efficiently and have that bright glowing skin we all crave and want! More energy and feel less bloated!
And I’m not talking about cleansing with juicing or not eating. No no, that’s a whole other conversation. I absolutely believe in still eating a healthy diet while “cleansing”/taking Triphala.
I have suggested Triphala to many clients, students and friends and all of them have seen results. You can call it a form of laxative if you’d like but this is totally safe and gentle on the body. Yes, we are all different but seems like this one might be a miracle worker and work for everyone!
Suggested use: Take one pill before bedtime. *Take on and off for a period of time OR once in a while when you feel you need it. I usually take it when I feel I need a cleanse- about one or two times a week (usually when I have consumed a bigger meal or more food than usual).
Benefits of Triphala:
- detoxify and cleanses the colon of toxins
- removes excess fats
- purifies the blood
- removes toxins from the liver
- reduces some forms of cholesterol (serum cholesterol)
- reduces high blood pressure
- high nutritional value: including high levels of vitamin C
- high in antioxidants
- strengthens hair roots and enriches hair color


The three fruits contained in Triphala are
Amalaki (Indian Gooseberry),
Haritaki (Indian Gallnut or Terminalia chebula),
and Bibhitaki (Beleric Myrobalan or Terminalia bellerica).
The prokinetic cleanser
An immensely popular Ayurvedic herbal formula,Triphala(Terminalia chebula,Terminalia bellirica and Emblica officinalis) is an effective bowel cleanser. It combines the goodness of Indian Gooseberry, Belleric Myrobalan and Chebulic Myrobalan, which work together to produce effective bowel movements.
The herbal compound provides overall support for digestion and helps ensure that the digestive tract works at optimal levels. Triphala relieves constipation and regularizes the digestive system, without disrupting the fluid-electrolyte balance in the body.
The herbs that make up Triphala are found in abundance in India.
Triphala, the well-known traditional Ayurvedic formulation, makes an excellent skin tonic. It is one of the most popular Ayurvedic medicinal herbs, prescribed by a number of Ayurvedic practitioners. Triphala literally means ‘three fruits’. The three fruits contained in Triphala are Amalaki (Indian Gooseberry), Haritaki (Indian Gallnut or Terminalia chebula), and Bibhitaki (Beleric Myrobalan or Terminalia bellerica). Since Triphala is tridoshic – equally balancing for Vata, Pitta and Kapha – it is beneficial for all skin types. Triphala nourishes the skin, both directly and indirectly. Amla (Indian gooseberry), one of the three ingredients in Triphala, is the richest known natural source of Vitamin C. Apart from the rich source of Vitamin C, Triphala also contains calcium – an important nutrient that helps enhance skin clarity and brings dull, tired skin to life.
Preparation Of Triphala Rasayana
Triphala Rasayana is usually prepared by mixing triphala with equal quantity of madhuka (mahua tree), tavakshir (East Indian arrowroot) pippali (long pepper), saindhava (long salt), and each one of the loha (iron), suvarna (gold), vacha (Acorus calamus) with either honey, ghee or sugar, in equal quantity.
Benefits Of Triphala
Triphala Rasayana is beneficial is promoting ojas, the finest product of digestion that prevents the occurrence of many diseases, creates luster and make the skin exude its natural glow and radiance.
It nourishes both the body and the mind, thereby promoting longevity of life. Therefore, Triphala Rasayana is very much beneficial for adults and children alike.
The Rasayana is especially beneficial for eyes. In case one has problems in eye sight, opting for Triphala Rasayana would be the best bet.
The Rasayana creates a balance in the cholesterol level, by removing ama from the fat tissue.
It helps in the purification of urinary tract, thereby helping the prevention of urinary tract diseases.
It also strengthens and cleanses the liver, which is one of its main functions. This ensures that the liver, one of the important parts of the body, stays healthy. It can also be said that the consumption of Rasayana prevents diseases related to the functioning of liver.
The medicine also helps the management of weight. Thus, it is beneficial for people, who want to loose weight.
It enhances the thirteen agnis (digestive fires), especially the main digestive fire in the stomach.
Triphala Rasayana is helpful in pacifying Kapha and Pitta. If taken on a regular basis, the Rasayana can be a powerful anti-aging medicine.
People suffering from skin inflammation, heat, infection, obesity will find the consumption of Triphala Rasayana as beneficial.
Diseases such as fatigue and anemia can be effectively cured by the regular consumption of Triphala Rasayana, if taken according to the prescribed doses.
- Ayurvedic pharmacopoeia committee. The Ayurvedic Formulary of India, Part I, 2nd English ed. New Delhi: Controller of Publications; 2003
- Anne McIntyre (7 September 2005). Herbal treatment of children: Western and Ayurvedic perspectives. Elsevier Health Sciences. pp. 278–. ISBN 9780750651745. Retrieved 24 July 2010.
- Juss SS. Triphala – the wonder drug. Indian Med Gaz 1997;131:94-6.
- Nadkarni AK. Indian Materia Medica. 3rd ed. Mumbai: Popular Press; 1976. p. 1308-15.
- Harbans Singh Puri (2003). Rasayana: ayurvedic herbs for longevity and rejuvenation. CRC Press. pp. 30–. ISBN 9780415284899. Retrieved 24 July 2010.
- Reddy TC, Aparoy P, Babu NK, Kalangi SK, Reddanna P (May 2010). “Kinetics and Docking Studies of a COX-2 Inhibitor Isolated from Terminalia bellerica Fruits”. Protein Pept Lett. PMID 20441561.
- Pawar V, Lahorkar P, Anantha Narayana DB. Development of a RP-HPLC method for analysis of Triphala curna and its applicability to test variations in Triphala curna preparations. Indian J Pharm Sci [serial online] 2009 [cited 2010 Aug 1];71:382-6. Available from:http://www.ijpsonline.com/text.asp?2009/71/4/382/57286
- Mahesh R, Bhuvana S, Begum VM (August 2009). “Effect of Terminalia chebula aqueous extract on oxidative stress and antioxidant status in the liver and kidney of young and aged rats”. Cell Biochem. Funct. 27 (6): 358–63. doi:10.1002/cbf.1581. PMID 19548245.
- Sandhya T, Lathika KM, Pandey BN, et al. (October 2006). “Protection against radiation oxidative damage in mice by Triphala”. Mutat. Res. 609 (1): 17–25.doi:10.1016/j.mrgentox.2006.05.006. PMID 16860592.
- Srikumar R, Parthasarathy NJ, Manikandan S, Narayanan GS, Sheeladevi R (February 2006). “Effect of Triphala on oxidative stress and on cell-mediated immune response against noise stress in rats”. Mol. Cell. Biochem. 283 (1-2): 67–74. doi:10.1007/s11010-006-2271-0.PMID 16444587.

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link




Idrabiotaparinux for anticoagulant therapy.
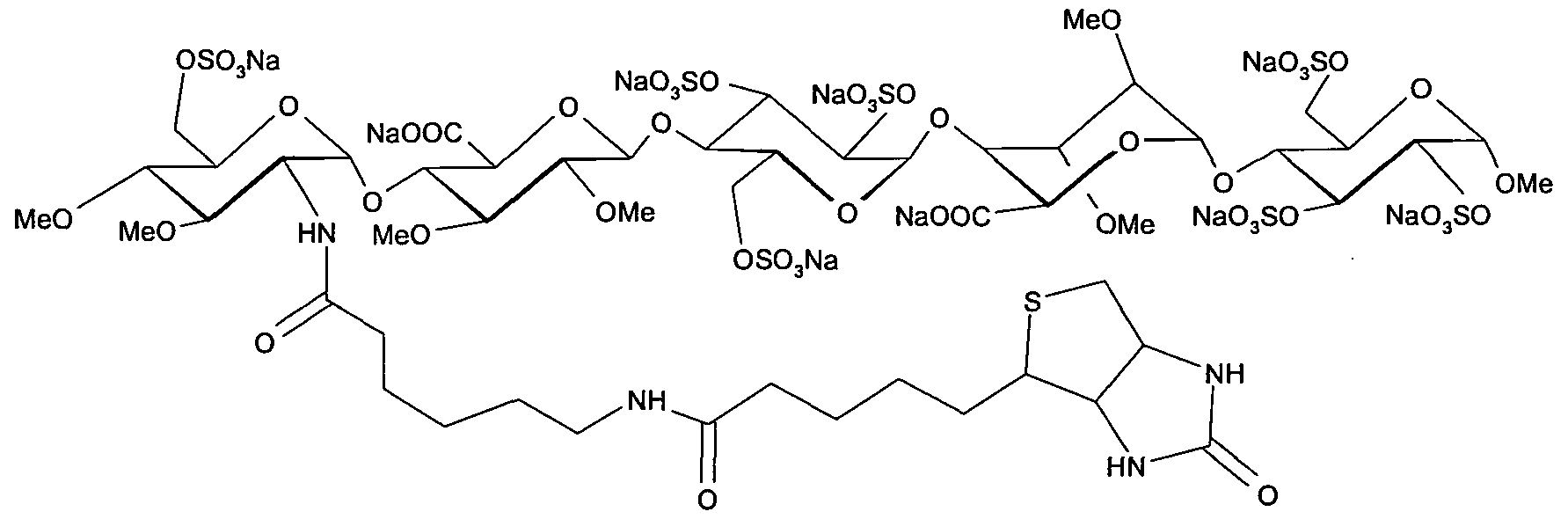
Idrabiotaparinux
(biotinylated idraparinux, SSR-126517, SSR-126517E)
405159-59-3 9 x Na salt
774531-07-6 (free acid)
Idrabiotaparinux has an attached biotin moiety at the non-reducing end unit, which allows its neutralisation with avidin, an egg-derived protein with low antigenicity. This compound is currently investigated in clinical trials for prevention of recurrent VTE in patients with acute pulmonary embolism. The future of idrabiotaparinux depends also on the safety and efficacy of avidin.
Symptomatic deep vein thrombosis (DVT) and/or pulmonary embolism (PE) – treatment and secondary prevention of recurrent venous thromboembolism (VTE).
SSR-126517, a biotinylated idraparinux, had been in phase III clinical trials at Sanofi (formerly known as sanofi-aventis) for the treatment of pulmonary embolism, deep venous thrombosis (DVT) and atrial fibrillation. However, in 2009, development of the compound was discontinued.

Idrabiotaparinux (biotinylated idraparinux, SSR-126517, SSR-126517E) is a long-acting selective pentasaccharide indirect factor Xa coagulation inhibitor, administered by once weekly subcutaneous (SC) injection at a dose of 3mg in patients without severe renal insufficiency and, after an initial dose of 3mg, at 1.8mg in those with renal insufficiency.
Warfarin, heparin and their derivatives have been the traditional anticoagulants used for prophylaxis and treatment of venous thromboembolism. While the modern clinician is familiar with the efficacy and pharmacokinetics of these agents, their adverse effects have provided the impetus for the development of newer anticoagulants with improved safety, ease of administration, more predictable pharmacodynamics and comparable efficacy. Research into haemostasis and the coagulation cascade has made the development of these newer anticoagulants possible.
These drugs include the factor Xa inhibitors and IIa (thrombin) inhibitors. Direct and indirect factor Xa inhibitors are being developed with a relative rapid onset of action and stable pharmacokinetic profiles negating the need for close monitoring; this potentially makes them a more attractive option than heparin or warfarin. Examples of direct factor Xa inhibitors include apixaban, rivaroxaban, otamixaban, betrixaban and edoxaban. Examples of indirect factor Xa inhibitors include fondaparinux, idraparinux and idrabiotaparinux.
Direct thrombin inhibitors (factor IIa inhibitors) were developed with the limitations of standard heparin and warfarin in mind. Examples include recombinant hirudin (lepirudin), bivalirudin, ximelagatran, argatroban, and dabigatran etexilate. This review will discuss emerging novel anticoagulants and their use for the prophylaxis and management of venous thromboembolism, for stroke prevention in nonvalvular atrial fibrillation and for coronary artery disease.
Idrabiotaparinux is intended as a substitute for current long-term oral anticoagulation (e.g. with warfarin) and has no known food or drug interactions, no need for overlapping with other anticoagulants or for laboratory blood monitoring.
Idrabiotaparinux has superseded the development and marketing of the non-biotinylated idraparinux. Idrabiotaparinux is also in phase III clinical trials for the prevention of stroke in patients with atrial fibrillation (AF).
Idrabiotaparinux will be the first once a week anticoagulant for the treatment of patients with VTE. It is intended to provide a predictable response with fixed dosing, no interactions with food, no requirement for overlapping with other therapy and no routine laboratory monitoring.
Developer Sanofi-aventis.
Standard treatment of venous thromboembolism,including deep vein thrombosis and pulmonary
embolism, is started with a rapidly acting parenteral anticoagulant such as heparin or low-molecular-weight
heparin for at least 5 days and is overlapped with a Vitamin K antagonist such as warfarin.
Warfarin is then continued for at least 3 months. Although eff ective, this drug has important limitations. Lifestyle changes are necessary because of interactions with food, alcohol,and other drugs, and the unpredictable anticoagulant eff ect of warfarin necessitates frequent coagulation monitoring and dose adjustments to optimise the balance between effi cacy and safety. Warfarin reduces the risk of recurrent venous thromboembolism by up to 90%, but there is a catch-up eff ect if warfarin is stopped in patients with unprovoked venous thromboembolism. This eff ect means that, by 2 years,the risk of recurrence in patients treated for 3 months is akin to that in patients treated for 12 months.

Consequently, some experts recommend life-long warfarin therapy for patients with unprovoked venousthromboembolism. The complexity of such treatment has prompted the development of new oral and parenteral anticoagulants that are more convenient to administer than is warfarin Idraparinux is a synthetic pentasaccharide that accel erates antithrombin-dependent inhibition of factor Xa and has a half-life of about 80 h. When compared with conventional anticoagulation therapy,
idraparinux given once-weekly by subcutaneous injection was non-inferior for treatment of deep vein thrombosis,but was inferior for treatment of pulmonary embolism. In patients with venous thromboembolism who received a 6 month course of anticoagulant treatment, idraparinux was better than was placebo for prevention of recurrent venous thromboembolism.6However, when compared with warfarin for stroke prevention in patients with atrial fi brillation, there was an excess of major bleeding with idraparinux (including intracranial haemorrhage).7Prompted by these safety concerns, idrabiotaparinux was developed as a replacement for idraparinux.

Idrabiotaparinux (International Non-proprietary Name), or SSR126517 (laboratory code), is developed by sanofi-aventis as the first long-acting anticoagulant administered once-weekly by subcutaneous route, with the unique property to be almost instantly and specifically neutralizable by intravenous administration of avidin. It is developed as an alternative to vitamin K antagonists (VKA). Idrabiotaparinux is the biotinylated pentasaccharide corresponding to the structure depicted below.

The pentasaccharide structure of idrabiotaparinux is the same as idraparinux, another antithrombotic agent developed by sanofi-aventis (see structure below). However in idrabiotaparinux, the presence of a biotin hook covalently linked to the first saccharidic unit enables the compound to be neutralized by avidin or streptavidin, as described in the international patent application WO 02/24754.

Idraparinux
In the EQUINOX trial, which enrolled 757 patients with DVT treated for 6 months with equimolar doses of either idrabiotaparinux or idraparinux, the administration of idrabiotaparinux was demonstrated to provide bioequipotent results to idraparinux in terms of pharmacokinetics and pharmacodynamics, in patients with symptomatic deep venous thrombosis (Journal of Thrombosis and Haemostasis, 2010, Vol. 9, p. 92-99). The results of this bioequipotency trial indicated that idrabiotaparinux could be a suitable treatment for patients with deep venous thrombosis. However, the apparent failure of idraparinux in patients with pulmonary embolism indicated the need for a formal evaluation of idrabiotaparinux in this patient group (N. Eng. J. Med., 2007, Vol. 357, p. 1094-104).
IDRABIOTAPARINUX
It has now been demonstrated, in a phase III study involving 3202 patients with pulmonary embolism, that idrabiotaparinux is a safe and effective drug in the treatment of pulmonary embolism in patients with or without deep venous thrombosis and in the secondary prevention of venous thromboembolic events in said patients. The invention therefore relates to idrabiotaparinux for use in the treatment of pulmonary embolism in patients with or without deep venous thrombosis and the secondary prevention of venous thromboembolic events in said patients, wherein the efficacy and safety of said uses are clinically proven by a phase III clinical trial. According to the instant invention, the terms below have the following meanings:
“idrabiotaparinux” designates the sodium salt of this compound, as defined above, or any other pharmaceutically acceptable salt thereof;
-a “phase III clinical trial” refers to an international, multicenter, randomized, double-blind, double-dummy, parallel group study involving a large patients group (3202 patients in the instant invention), aiming at being the definitive assessment of how effective and safe the drug is, in comparison with current standard treatment; – “deep venous thrombosis” refers to a blood clot in a deep vein of the lower limbs;
new polysaccharides of the invention, are comparable to the oligosaccharides of the prior art antithrombotic activity. But they also have the advantage of being quickly neutralized by a specific antidote in an emergency. This specific antidote avidin (The Merck Index, Twelfth Edition, 1996, MN 920, pages 151-152) or streptavidin, two tetrameric protein with respective masses equal to approximately 66 000 and 60 000 Da, which have a very high affinity for biotin. In general, the invention relates to synthetic polysaccharides antithrombotic activity has at least one covalent bond with biotin or a biotin derivative. As a derivative of biotin include the biotin derivatives listed in the catalog Pierce 1999-2000 pages 62-81, for example 6-biotinamido hexanoate,
you,

or 2-biotinamido éthanethiole

Patent application WO 02/24754 describes synthetic polysaccharides which have a covalent bond with biotin (hexahydro-2-oxo-1H-thieno[3,4-d]imidazole-4-pentanoic acid) or with a biotin derivative. Such polysaccharides have an antithrombotic activity which means that they can be used as anticoagulants, and also have the advantage of being able to be rapidly neutralized with a specific antidote, in an emergency situation. This specific antidote is avidin (The Merck Index, Twelfth edition, 1996, M.N. 920, pages 151-152) or streptavidin, two tetrameric proteins of respective weights equal to approximately 66 000 and 60 000 Da, which have a very strong affinity for biotin.
Patent application WO 02/24754 describes in particular the following compound, known as idrabiotaparinux:

In the mammalian body, idrabiotaparinux is partly metabolized at the level of the amide bond adjacent to the biotin group, thus producing a pentasaccharide compound bearing an amine chain —NH—CO—(CH2)5—NH2 on the first glucosamine unit, as described in patent application WO 2010/023374.
It may be desirable, in particular in the context of clinical developments of molecules of pharmaceutical interest, to limit or even prevent the metabolization of compounds of this type.
Novel polysaccharides with structures analogous to some of those described in patent application WO 02/24754 have now been identified, which polysaccharides have antithrombotic properties and a neutralization capacity, for example via avidin, which are comparable to those described in that patent application, but which also have improved metabolic stability.
Generally, the invention therefore relates to synthetic polysaccharides with antithrombotic activity having at least one covalent bond with biotin or a biotin derivative, characterized in that said covalent bond is resistant to metabolic cleavage and comprises a linkage X chosen from —O—, —N(R)—, —N(R)—CO— and —N(R′)—CO—N(R″)—, in which R is an alkyl group and R′ and R″, which may be identical or different, are, independently of one another, hydrogen atoms or alkyl groups.
For the purposes of the present invention, and unless otherwise mentioned in the text, the term “alkyl” is intended to mean a linear or branched, saturated aliphatic group comprising from 1 to 6 carbon atoms, and advantageously a methyl group.
Biotin, or hexahydro-2-oxo-1H-thieno[3,4-d]imidazole-4-pentanoic acid, is the compound having the following formula:

By way of biotin derivatives, mention may be made of those indicated in the Pierce catalog 1999-2000, pages 62 to 81, or in patent application WO 02/24754.
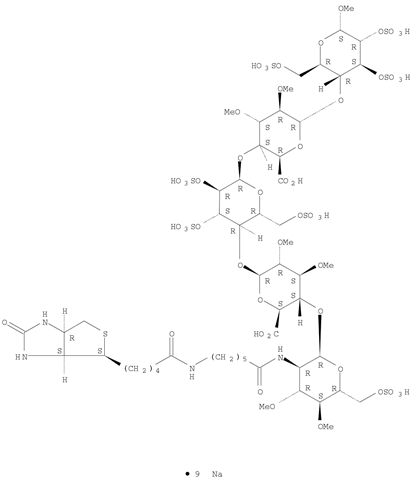
Idrabiotaparinux sodium;
Molecular Formula:C53H88N4O51S8.9NaCAS
Registry Number:405159-59-3
nonasodium methyl (2-deoxy-3 ,4-di-O-methyl-2-{6 – [5 – (2-oxohexahydro-1H-thieno [3,4-d] imidazol-4-yl) pentanamido] hexanamido} -6-O-sulfo-α-D-glucopyranosyl) – (1 → 4) – (2,3-di-O-methyl-β-D-glucopyranosyluronate) – (1 → 4) – (2,3,6 -tri-O-sulfo-α-D-glucopyranoside) – (1 → 4) – (2,3-di-O-methyl-α-L-idopyranosyluronate) – (1 → 4) -2,3,6 – tri-O-sulfo-α-D-glucopyranoside

………………..
SYNTHESIS
FIGURE 9
Synthesis of the pentasaccharide 39

39
PREPARATION 34
Methyl (6-O-acetyl-2-azido-2-deoxy-3) 4-di-0-methyl-O-glucopyranosyl) – (1 → 4) – (benzyl 2,3-di-O-methyl- β-D-glucopyranosyluronate) – (1 – → 4) – (3,6-di-O-acetyl-2-0-benzyl–D-glucopyranosyl) – (1 -> 4) – (benzyl 2,3 -di-O-methyl-a-idopyranosyl-uronate) – (1 → 4) -2,3,6-tri-0-benzyLa-D-glucopyranoside (39)
Compound 6-0-acetyl-2 was dissolved -azido-2-deoxy-3 ,4-di-0-methyl-, β-D-glucopyranose trichloroacetimidate 38 (265 mg, 0.631 mmol) (obtained by J. Basten, and Chem. Lett Bioorg. Med. al.. (1992), 2 (9), 901)
and
compound 32 (584 mg, 0.420 mmol) (obtained by P. Westerduin and Med. Bioorg Chem. al., 1994, 2, 1267) in a dichloromethane / diethyl ether 1/2 (v / v) (28.5 mL).
After addition of 4 Å molecular sieves powder, the mixture is cooled to -20 ° C. and a 0.1 M solution of trimethylsilyl trifluoromethanesulfonate in dichloromethane (94.6 uL). After 10 minutes, again added the imidate (53.8 mg) and a 0.1 M solution of trimethylsilyl trifluoromethanesulfonate in dichloromethane (19.2 uL). After 10 minutes, the mixture was neutralized by addition of solid sodium hydrogen carbonate. After filtration and concentration, the residue was purified by column chromatography on silica gel (toluene / ethyl acetate 3/1 (v / v)) to give 499 mg of compound 39. [Α] = +66 (c = 1, 07, dichloromethane).
FIGURE 10 -Summary of the pentasaccharide 44 (Method I)
COMPOUND 39

COMPD 40

COMPD 44
……………………
In scheme 1, the starting, intermediate and final compounds are the following:
-
- compound (I): N-succinimidyl N-biotinyl-6-aminocaproate,
- compound (II): N-biotinyl-6-aminocaproic acid,
- compound (II′): N-biotinyl-6-aminocaproate carboxylate,
- compound (III): biotin,
- compound (III′): cyanomethyl biotinate.

EXAMPLE 1 Preparation of the Compound (I)

The reactions are monitored by LC with the following conditions: Symmetry C18 150×4.6 mm 5μ column (Waters); eluent A: 0.01 M KH2PO4 buffer adjusted to pH=3; eluent B: acetonitrile; flow rate 1 ml/min; gradient: t=0 min A/B 85/15, t=9 min A/B 65/35, t=10 min A/B 85/15, t=15 min A/B 85/15. This method makes it possible to visualize the biotin (compound (III), tR=4.5 min), the intermediate activated ester (III′) (tR=8.4 min), the N-biotinyl-6-aminocaproic acid (compound (II), tR=5.5 to 5.6 min), the intermediate mixed anhydride (II′) (tR=11.2 min) and the N-succinimidyl N-biotinyl-6-aminocaproate (compound (I), tR=7.9 to 8.2 min).
1.1: Preparation of the Compound (II)


7.5 kg of biotin (III), triethylamine (15 l, 2 V, 3.5 eq), NMP (15 l, 2 V) and, finally, chloroacetonitrile (3.5 kg, 0.47 OU, 1.5 eq) are charged to a reactor. The medium is heated to 60° C. After this temperature has been maintained for 2 h, an LC analysis shows that all the biotin has been converted into compound (III′) (<2%). The medium is cooled to 50° C. and then transferred into another reactor, containing aminocaproic acid (9.05 kg, 1.206 OU, 2.2 eq). Rinsing is carried out with NMP (0.1 V). The medium is heated to 100° C. and maintained at this temperature for 2 h. An LC analysis shows that less than 2% of activated biotin (III′) remains. The medium is cooled to 60° C. Acetonitrile (60 l, 8 V) preheated to 55° C. is run in. The mixture is stirred for 30 minutes at 60° C., and then cooled to 20° C. Stirring is carried out for 1 h. The suspension is filtered, then rinsing is carried out with 3 times acetonitrile (5 V) and then with THF (5 V). Drying is carried out under vacuum at a maximum of 60° C. until there is no change in weight. 12.0 kg of the compound (II) are thus obtained, with a yield of 109% and an organic purity, measured by LC, of 98.6%.
10.0 kg of the compound (II) are recharged to a reactor. Hydrochloric acid (90 l, 9 V of water+10 l, 1 V of 36% HCl) is then added. The suspension is stirred at 20° C. for 30 min. The suspension is filtered and rinsing is carried out 3 times with water (4 V, 40 l), then twice with THF (3.5 V). Drying is carried out under vacuum at a maximum of 45° C. until there is no change in weight. 6.1 kg of the compound (II) are thus obtained, with a yield of 66%.
1.2: Preparation of the Compound (I)
In a reactor, 3 kg of the compound (II) are suspended in DMF (25 l, 8.3 V) and the temperature is brought to −5° C. Triethylamine (1.02 kg, 0.34 OU, 1.2 eq) is then added. After stirring for 15 minutes, ethyl chloroformate (1.1 kg, 0.365 OU, 1.2 eq) is added gently (over the course of at least 1 h). Rinsing is carried out with DMF (0.9 l, 0.3 V). The medium is stirred at −5° C. for at least 2 h. The suspension becomes finer and yellow. An LC analysis shows that all the compound (II) (<3%) has reacted.
N-Hydroxysuccinimide (1.04 kg, 0.386 OU, 1.2 eq) in solution in DMF (3 l, 1 V) is then introduced in 1 step (over the course of at least 20 min). Rinsing is carried out with DMF (1.5 l, 0.5 V). The medium is stirred for 1 h 30 at −5° C. An LC analysis shows that the presence of residual compound (II) is less than 3%. The temperature is brought to 22° C., the suspension is taken up in DCM (12 V, 36 l) and the resulting organic phase is washed with water (15 l, 5 V). The organic phase is drawn off and the aqueous phase is extracted twice with DCM (30 l, 3 V). The organic phases are mixed and are washed with water (1.5 l, 0.5 V). The organic phase is concentrated to 6 V, i.e. 181. Heating is carried out at 40° C. and MTBE (6.25 V, 19 l) is added over the course of a minimum of 1 h. The mixture is maintained at 40° C. for 1 h, and then MTBE (8.75 V, 26 l) is added over the course of a minimum of 2 h. The mixture is maintained at 40° C. for at least 30 minutes, and then cooled to 20° C. over the course of a minimum of 2 h, and maintained at this temperature for 30 minutes. The suspension is filtered by suction and the cake is washed with acetone (5 V, 15 l) and then twice more with acetone (2 V, 6 l). The resulting product is filtered by suction and dried in an oven under vacuum at a maximum of 40° C. until there is no change in weight.
3 kg of the compound (I) are thus obtained in the form of a cream powder, with a yield of 80% and an organic purity, measured by LC, of 96.0%. Except for the compound (II), the presence of which is not problematic for a subsequent coupling reaction with a polysaccharide since it will be inert during this coupling, the purity of the compound (I) is 98%.
The biotinylated polysaccharides, the preparation of which is described above, are for example such as those described in patent applications WO 02/24754 and WO 2006/030104. They may in particular be the biotinylated pentasaccharide known under the International Nonproprietary Name “idrabiotaparinux” and described in patent application WO 02/24754, or the biotinylated hexadecasaccharides described in examples 1 and 2 of patent application WO 2006/030104.
In order to prepare these biotinylated polysaccharides, the compound (I) is coupled, respectively, with the pentasaccharide 44 described in patent application WO 02/24754
pentasaccharide 44: methyl (2-amino-2-deoxy-3,4-di-O-methyl-6-O-sulfonato-α-D-glucopyranosyl)-(1→4)-(2,3-di-O-methyl-β-D-glucopyranosyluronic acid)-(1→4)-(2,3,6-tri-O-sulfonato-α-D-glucopyranosyl)-(1→4)-(2,3-di-O-methyl-α-L-idopyranosyluronic acid)-(1→4)-2,3,6-tri-O-sulfonato-α-D-glucopyranoside
EXAMPLE 2
Preparation of a biotinylated polysaccharide, idrabiotaparinux

A solution of 1.22 kg of the crude pentasaccharide 44 (containing salts), as described in patent application WO 02/24754, is prepared in 8.51 of water (7 V). 0.5 kg (1.6 eq) of the compound (I), 0.12 kg (2.0 eq) of NaHCO3 and 0.37 kg of NaCl are added thereto. The solution is in the form of a white suspension. 3.7 l of acetone are added thereto and the reaction medium is stirred at approximately 25° C. for at least 22 h. This suspension is then slowly run into a mixture of ethanol (120 l) and MTBE (60 l) cooled beforehand to approximately 4° C., which makes it possible to precipitate the biotinylated pentasaccharide. The resulting suspension is then filtered and rinsed successively with absolute ethanol and acetone. The precipitate is oven-dried under a vacuum until there is no change in weight. 1.60 kg of crude idrabiotaparinux (containing salts) are thus obtained in the form of a cream powder, with an organic purity of 99%, and with a yield of 109% with respect to the pentasaccharide 44 and a chemical yield of 70% over the last 3 stages.
………………………….
Compound of Preparation Example 1:
Methyl (2 – [N-(6-aminohexanoyl)]-2-deoxy-3 ,4-di-O-methyl-6-O-sulfonato-UD-glucopyranosyl) – (1 → 4) – (acid 2, 3 -di-O-methyl-β-D-glucopyranosyluronic) – (1-rf) – (2,3,6-tri-O-sulphonate-D-glucopyranosyl) – (1 → 4) – (2,3 – di-O-methyl-alpha-L-idopyranosyluronique) – (1 → 4) -2,3,6-tri-Osulfonato-D-glucopyranoside, sodium salt

Compound 1
1) Preparation of 6 – (benzyloxycarbonylamino) hexanoate succinimidyl
To a solution of 6 – (benzyloxycarbonyl amino) hexanoic acid (1.00 g, 3.77 mmol) in dimethylformamide (20 mL) was added triethylamine (0.63 mL, 4.52 mmol) and stirring the mixture at room temperature under argon for 30 minutes. The solution was cooled to 0 ° C and added dropwise ethyl chloroformate (0.43 mL, 4.52 mmol). After two hours at room temperature, N-hydroxysuccinimide (0.52 g, 4.52 mmol) and stirring the mixture overnight at room temperature. Evaporated to dryness before the residue in water to which is added with ethyl acetate. The phases were separated and the aqueous phase is extracted with ethyl acetate. The organic phases are combined, dried over sodium sulfate, filtered and evaporated to dryness before purification on a column of silica gel with pentane mixture of ethyl acetate / (75/25 v / v) as eluent. Once the fractions evaporated to give 1.13 g 6 – (benzyloxycarbonylamino) succinimidyl hexanoate as an oil. TLC: R f = 0.22 on silica gel plate with a mixture of n-heptane/ethyl acetate (30/70 v / v) as eluent.
2) Preparation of compound the
Grafting the amine is carried out on the chain 44 pentasaccharide, or methyl (2 – amino-2-deoxy-3 ,4-di-0-methyl-6-0-sulfonato-α-D-glucopyranosyl) – (1 → 4) – (2,3 – di-0-methyl-β-D-glucopyranosyluronic) – (1 -> 4) – (2,3,6-tri-0-sulphonato-α-D-gluco-pyranosyl) – (1 → 4) – (2,3-di-O-methyl-α-L-idopyranosyl-uronic acid) – (1 → 4) – 2,3,6-tri-O-sulfo-α-nato- D-glucopyranoside, sodium salt, the preparation of which is described in patent application WO 02/24754:

44
To a solution of 6 – (benzyloxycarbonylamino) succinimidyl hexanoate (783 mg, 2.16 mmol) in N, N-dimethylformamide (10 mL) was added the pentasaccharide 44 (505 mg, 0.29 mmol). After stirring for 24 hours in an inert atmosphere and at room temperature, the solvent was evaporated under reduced pressure and the residue (40 mL) before washing the solution with chloroform (2 x 30 mL) dissolved in water. The chloroform phase is washed with water (10 mL) and aqueous phases were combined and evaporated to dryness under reduced pressure. The solid residue was triturated with 2-propanol (10 mL) and the suspension centrifuged for 5 minutes at 2500 rpm. The alcoholic phase is removed and replaced with 2-propanol (10 mL) and centrifugation was repeated. After having extracted the solvent, the crude product was dried under vacuum.
-2-deoxy-3 ,4-di-0-methyl-6-0-sulfonato-α-D [N-(benzyloxycarbonyl-6-aminohexanoyl)] – thus obtained 399 mg of the compound “, or methyl (2 -glucopyranosyl) – (1 → 4) – (the acid 2 3-di-0-methyl-β-D-glucopyranosyluronic) – (1 → 4) – (2,3,6-tri-0-sulfonato-α- D-glucopyranosyl) – (1 – »4) – (2,3-di-0-methyl-α-L-idopyranosyluronique) – (1 – → 4) -2,3,6 – tri-O-sulphonate- α-D-glucopyranoside, wherein Pg is benzyloxycarbonyl:

January 1 compound
Proton NMR at 200 MHz in deuterated water: The structure of the expected product is confirmed that the spectrum is identical to that performed on a product synthesized according to Example 5 of WO 02/24754 without the signals due to the biotin portion atoms but with signals of 7.4 to 7.5 ppm due to the benzyloxy group.
3) Preparation of compound 1
The product ‘s obtained at the end of the previous step (399 mg) is dissolved in deuterated water (10 mL). The solution of palladium on charcoal are treated with 10% (25 mg) and the solution was allowed to stir 20 hours in the presence of hydrogen at atmospheric pressure. Mixed with water (15 mL) was diluted, the catalyst was filtered and the solution was washed with chloroform (2 x 15 mL) before evaporating to dryness under reduced pressure. An aliquot (98 mg to 320 mg) of this product was purified on a column of Sephadex G-25 (2.5 x 50 cm) with water as eluent to give 25 mg of compound 1.
HPLC: Tr = 15.4 min column X-Terra RP-18 15W x 4.6 mm, 5μ particles of Waters in SA. With detection at 211 nm UV lamp.Eluent 1: water containing 0.02 M ammonium acetate and 0.05 M di-n-butylamine, adjusted to pH 7 with acetic acid. 2 Eluant: acetonitrile / water (90/10 v / v) containing 0.05 M di-n-butylamine and 0.08M acetic acid. The proportions of eluents are programmed so that the eluent composition is 10% 2 0 min. , 20% at 25 min. , 50% at 40 min. , 50% at 43 min. and 5% to 50 minutes. Proton NMR at 600 MHz in deuterated water: The structure of the expected product is confirmed that the spectrum is identical to that performed on a product synthesized according to Example 5 of WO 02/24754 without the signals due to the biotin portion atoms.
…………………………
 IDRABIOTAPARINUX
IDRABIOTAPARINUX
REFERENCES
JOURNAL OF THROMBOSIS AND HAEMOSTASIS vol. 9, 2010, pages 92 – 99
BULLER HARRY R ET AL: “Idraparinux versus standard therapy for venous thromboembolic disease“, NEW ENGLAND JOURNAL OF MEDICINE, vol. 357, no. 11, September 2007 (2007-09) , pages 1094-1104,
EQUINOX INVESTIGATORS: “Efficacy and safety of once weekly subcutaneous idrabiotaparinux in the treatment of patients with symptomatic deep venous thrombosis.“, JOURNAL OF THROMBOSIS AND HAEMOSTASIS : JTH JAN 2011 LNKD- DOI:10.1111/J.1538-7836.2010.04100.X PUBMED:20946157, vol. 9, no. 1, January 2011 (2011-01), pages 92-99,
N. ENG. J. MED. vol. 357, 2007, pages 1094 – 104
PRANDONI P ET AL: “Idraparinux: review of its clinical efficacy and safety for prevention and treatment of thromboembolic disorders“, EXPERT OPINION ON INVESTIGATIONAL DRUGS, ASHLEY PUBLICATIONS LTD., LONDON, GB, vol. 17, no. 5, 1 May 2008 (2008-05-01), pages 773-777,
SAVI P ET AL: “Reversible biotinylated oligosaccharides: A new approach for a better management of anticoagulant therapy“, JOURNAL OF THROMBOSIS AND HAEMOSTASIS, BLACKWELL PUBLISHING, OXFORD, GB, vol. 6, no. 10, 1 January 2008 (2008-01-01), pages 1697-1706,
Emerging anticoagulants.Kennedy B, Gargoum FS, Kennedy L, Khan F, Curran DR, O’Connor TM.Curr Med Chem. 2012;19(20):3388-416. Review.
| 1 | * | ANONYMOUS: “Bioequipotency Study of SSR126517E and Idraparinux in Patients With Deep Venous Thrombosis of the Lower Limbs (EQUINOX)” INTERNET CITATION, [Online] 10 April 2008 (2008-04-10), pages 1-4, XP002503606 Retrieved from the Internet: URL:http://www.clinicaltrials.gov/ct2/show/NCT00311090?term=equinox&rank=1> [retrieved on 2008-11-11] |
| 2 | * | BULLER HARRY ROGER ET AL: “Idrabiotaparinux, a Biotinylated Long-Acting Anticoagulant, in the Treatment of Deep Venous Thrombosis (EQUINOX Study): Safety, Efficacy, and Reversibility by Avidin” BLOOD, vol. 112, no. 11, November 2008 (2008-11), page 18, XP009118800 & 50TH ANNUAL MEETING OF THE AMERICAN- SOCIETY-OF-HEMATOLOGY; SAN FRANCISCO, CA, USA; DECEMBER 06 -09, 2008 ISSN: 0006-4971 |
| 3 | * | HIRSH J ET AL: “Beyond unfractionated heparin and warfarin: Current and future advances” CIRCULATION, LIPPINCOTT WILLIAMS & WILKINS, US, vol. 116, no. 5, 1 July 2007 (2007-07-01), pages 552-560, XP002503605 ISSN: 0009-7322 |
| 4 | * | PRANDONI P ET AL: “Idraparinux: review of its clinical efficacy and safety for prevention and treatment of thromboembolic disorders” EXPERT OPINION ON INVESTIGATIONAL DRUGS, ASHLEY PUBLICATIONS LTD., LONDON, GB, vol. 17, no. 5, 1 May 2008 (2008-05-01), pages 773-777, XP008098574 ISSN: 1354-3784 |
| 5 | * | SAVI P ET AL: “Reversible biotinylated oligosaccharides: A new approach for a better management of anticoagulant therapy” JOURNAL OF THROMBOSIS AND HAEMOSTASIS, BLACKWELL PUBLISHING, OXFORD, GB, vol. 6, no. 10, 19 July 2008 (2008-07-19), pages 1697-1706, XP002503607 ISSN: 1538-7933 |
PATENTS
| WO2002024754A1 | Sep 20, 2001 | Mar 28, 2002 | Akzo Nobel Nv | Polysaccharides with antithrombotic activity comprising at least a covalent bond with biotin or a biotin derivative |
| EP2145624A1 * | Jul 18, 2008 | Jan 20, 2010 | Sanofi-Aventis | Use of idrabiotaparinux for decreasing the incidence of bleedings during an antithrombotic treatment |
| WO2008113918A1 * | Feb 12, 2008 | Sep 25, 2008 | Sanofi Aventis | Heparins including at least one covalent bond with biotin or a biotin derivative, method for preparing same and use thereof |
| WO2008113919A1 * | Feb 12, 2008 | Sep 25, 2008 | Sanofi Aventis | Low molecular weight heparins including at least one covalent bond with biotin or a biotin derivative, method for making same and use thereof |
| WO2010007530A1 * | Jul 17, 2009 | Jan 21, 2010 | Sanofi-Aventis | Use of idrabiotaparinux for decreasing the incidence of bleedings during an antithrombotic treatment |
| WO2010023375A1 * | Aug 24, 2009 | Mar 4, 2010 | Sanofi-Aventis | Hexadecasaccharides with antithrombotic activity, including a covalent bond and an amino chain |
| WO2011061449A1 | Nov 19, 2010 | May 26, 2011 | Sanofi-Aventis | Method for preparing n-succinimidyl n-biotinyl-6-aminocaproate |
| EP2145624A1 * | Jul 18, 2008 | Jan 20, 2010 | Sanofi-Aventis | Use of idrabiotaparinux for decreasing the incidence of bleedings during an antithrombotic treatment |
| EP2233143A1 * | Mar 24, 2009 | Sep 29, 2010 | Sanofi-Aventis | Use of idrabiotaparinux for decreasing the incidence of bleedings during an antithrombotic treatment |


HEPARIN SODIUM



HEPARIN SODIUM
LAUNCHED 1937
9041-08-1 NA SALT
9005-49-6 (heparin)
Thromboliquine, Calciparine, Certoparin, Dalteparin, Fraxiparin, Heparinate, Multiparin, Novoheparin, Parnaparin
Unfractionated heparin (UH) is a heterogenous preparation of anionic, sulfated glycosaminoglycan polymers with weights ranging from 3000 to 30,000 Da. It is a naturally occurring anticoagulant released from mast cells. It binds reversibly to antithrombin III (ATIII) and greatly accelerates the rate at which ATIII inactivates coagulation enzymes thrombin (factor IIa) and factor Xa. UH is different from low molecular weight heparin (LMWH) in the following ways: the average molecular weight of LMWH is about 4.5 kDa whereas it is 15 kDa for UH; UH requires continuous infusions; activated partial prothrombin time (aPTT) monitoring is required when using UH; and UH has a higher risk of bleeding and higher risk of osteoporosis in long term use. Unfractionated heparin is more specific than LMWH for thrombin. Furthermore, the effects of UH can typically be reversed by using protamine sulfate.
Unfractionated heparin is indicated for prophylaxis and treatment of venous thrombosis and its extension, prevention of post-operative deep venous thrombosis and pulmonary embolism and prevention of clotting in arterial and cardiac surgery. In cardiology, it is used to prevent embolisms in patients with atrial fibrillation and as an adjunct antithrombin therapy in patients with unstable angina and/or non-Q wave myocardial infarctions (i.e. non-ST elevated acute coronary artery syndrome) who are on platelet glycoprotein (IIb/IIIa) receptor inhibitors. Additionally, it is used to prevent clotting during dialysis and surgical procedures, maintain the patency of intravenous injection devices and prevent in vitro coagulation of blood transfusions and in blood samples drawn for laboratory values.
Indication: For anticoagulant therapy in prophylaxis and treatment of venous thrombosis and its extension, for prevention of post-operative deep venous thrombosis and pulmonary embolism and for the prevention of clotting in arterial and cardiac surgery.
Mechanism of action: The mechanism of action of heparin is antithrombin-dependent. It acts mainly by accelerating the rate of the neutralization of certain activated coagulation factors by antithrombin, but other mechanisms may also be involved. The antithrombotic effect of heparin is well correlated to the inhibition of factor Xa. Heparin interacts with antithrombin III, prothrombin and factor X.
Heparin (from Ancient Greek ηπαρ (hepar), liver), also known as unfractionated heparin, a highly sulfated glycosaminoglycan, is widely used as an injectable anticoagulant, and has the highest negative charge density of any known biological molecule.[3] It can also be used to form an inner anticoagulant surface on various experimental and medical devices such as test tubes and renal dialysis machines.
Although it is used principally in medicine for anticoagulation, its true physiological role in the body remains unclear, because blood anticoagulation is achieved mostly by heparan sulfate proteoglycans derived from endothelial cells.[4] Heparin is usually stored within the secretory granules of mast cells and released only into the vasculature at sites of tissue injury. It has been proposed that, rather than anticoagulation, the main purpose of heparin is defense at such sites against invading bacteria and other foreign materials.[5] In addition, it is conserved across a number of widely different species, including some invertebrates that do not have a similar blood coagulation system.
 HEPARIN
HEPARIN
Heparin structure

Native heparin is a polymer with a molecular weight ranging from 3 to 30 kDa, although the average molecular weight of most commercial heparin preparations is in the range of 12 to 15 kDa.[6] Heparin is a member of the glycosaminoglycan family of carbohydrates (which includes the closely related molecule heparan sulfate) and consists of a variably sulfated repeating disaccharide unit.[7] The main disaccharide units that occur in heparin are shown below. The most common disaccharide unit is composed of a 2-O-sulfated iduronic acid and 6-O-sulfated, N-sulfated glucosamine, IdoA(2S)-GlcNS(6S). For example, this makes up 85% of heparins from beef lung and about 75% of those from porcine intestinal mucosa.[8] Not shown below are the rare disaccharides containing a 3-O-sulfated glucosamine (GlcNS(3S,6S)) or a free amine group (GlcNH3+). Under physiological conditions, the ester and amide sulfate groups are deprotonated and attract positively charged counterions to form a heparin salt. Heparin is usually administered in this form as an anticoagulant.
One unit of heparin (the “Howell unit”) is an amount approximately equivalent to 0.002 mg of pure heparin, which is the quantity required to keep 1 ml of cat’s blood fluid for 24 hours at 0°C.[9]
-
 GlcA-GlcNAc
GlcA-GlcNAc -
 GlcA-GlcNS
GlcA-GlcNS -
 IdoA-GlcNS
IdoA-GlcNS -
 IdoA(2S)-GlcNS
IdoA(2S)-GlcNS -
 IdoA-GlcNS(6S)
IdoA-GlcNS(6S) -
 IdoA(2S)-GlcNS(6S)
IdoA(2S)-GlcNS(6S)



-GlcNS.png)
.png)
-GlcNS(6S).png)
Abbreviations
- GlcA = β-D–glucuronic acid
- IdoA = α-L–iduronic acid
- IdoA(2S) = 2-O-sulfo-α-L-iduronic acid
- GlcNAc = 2-deoxy-2-acetamido-α-D-glucopyranosyl
- GlcNS = 2-deoxy-2-sulfamido-α-D-glucopyranosyl
- GlcNS(6S) = 2-deoxy-2-sulfamido-α-D-glucopyranosyl-6-O-sulfate
Three-dimensional structure
The three-dimensional structure of heparin is complicated because iduronic acid may be present in either of two low-energy conformations when internally positioned within an oligosaccharide. The conformational equilibrium is influenced by sulfation state of adjacent glucosamine sugars.[10] Nevertheless, the solution structure of a heparin dodecasaccharide composed solely of six GlcNS(6S)-IdoA(2S) repeat units has been determined using a combination of NMR spectroscopy and molecular modeling techniques.[11] Two models were constructed, one in which all IdoA(2S) were in the 2S0 conformation (A and B below), and one in which they are in the 1C4 conformation (C and D below). However, no evidence suggests that changes between these conformations occur in a concerted fashion. These models correspond to the protein data bank code 1HPN.

In the image above:
- A = 1HPN (all IdoA(2S) residues in 2S0 conformation) Jmol viewer
- B = van der Waals radius space filling model of A
- C = 1HPN (all IdoA(2S) residues in 1C4 conformation) Jmol viewer
- D = van der Waals radius space filling model of C
In these models, heparin adopts a helical conformation, the rotation of which places clusters of sulfate groups at regular intervals of about 17 angstroms (1.7 nm) on either side of the helical axis.
Medical use

A sample of Heparin Sodium for injection
Heparin is a naturally occurring anticoagulant produced by basophils and mast cells.[12] Heparin acts as an anticoagulant, preventing the formation of clots and extension of existing clots within the blood. While heparin does not break down clots that have already formed (unlike tissue plasminogen activator), it allows the body’s natural clot lysis mechanisms to work normally to break down clots that have formed. Heparin is generally used for anticoagulation for the following conditions:
- Acute coronary syndrome, e.g., NSTEMI
- Atrial fibrillation
- Deep-vein thrombosis and pulmonary embolism
- Cardiopulmonary bypass for heart surgery
- ECMO circuit for extracorporeal life support
- Hemofiltration
- Indwelling central or peripheral venous catheters
Mechanism of action
Heparin and its low-molecular-weight derivatives (e.g., enoxaparin, dalteparin, tinzaparin) are effective at preventing deep vein thromboses and pulmonary emboli in patients at risk,[13][14] but no evidence indicates any one is more effective than the other in preventing mortality.[15] Heparin binds to the enzyme inhibitor antithrombin III (AT), causing a conformational change that results in its activation through an increase in the flexibility of its reactive site loop.[16] The activated AT then inactivates thrombin and other proteases involved in blood clotting, most notably factor Xa. The rate of inactivation of these proteases by AT can increase by up to 1000-fold due to the binding of heparin.[17]
AT binds to a specific pentasaccharide sulfation sequence contained within the heparin polymer:
GlcNAc/NS(6S)-GlcA-GlcNS(3S,6S)-IdoA(2S)-GlcNS(6S)
The conformational change in AT on heparin-binding mediates its inhibition of factor Xa. For thrombin inhibition, however, thrombin must also bind to the heparin polymer at a site proximal to the pentasaccharide. The highly negative charge density of heparin contributes to its very strong electrostatic interaction with thrombin.[3] The formation of a ternary complex between AT, thrombin, and heparin results in the inactivation of thrombin. For this reason, heparin’s activity against thrombin is size-dependent, with the ternary complex requiring at least 18 saccharide units for efficient formation.[18] In contrast, antifactor Xa activity requires only the pentasaccharide binding site.

Chemical structure of fondaparinux
This size difference has led to the development of low-molecular-weight heparins (LMWHs) and, more recently, to fondaparinux as pharmaceutical anticoagulants. LMWHs and fondaparinux target antifactor Xa activity rather than antithrombin activity, with the aim of facilitating a more subtle regulation of coagulation and an improved therapeutic index. The chemical structure of fondaparinux is shown above. It is a synthetic pentasaccharide, whose chemical structure is almost identical to the AT binding pentasaccharide sequence that can be found within polymeric heparin and heparan sulfate.
With LMWH and fondaparinux, the risk of osteoporosis and heparin-induced thrombocytopenia (HIT) is reduced. Monitoring of the activated partial thromboplastin time is also not required and does not reflect the anticoagulant effect, as APTT is insensitive to alterations in factor Xa.
Danaparoid, a mixture of heparan sulfate, dermatan sulfate, and chondroitin sulfate can be used as an anticoagulant in patients having developed HIT. Because danaparoid does not contain heparin or heparin fragments, cross-reactivity of danaparoid with heparin-induced antibodies is reported as less than 10%.[19]
The effects of heparin are measured in the lab by the partial thromboplastin time (aPTT), one of the measures of the time it takes the blood plasma to clot. Partial thromboplastin time should not be confused with prothrombin time, or PT, which measures blood clotting time through a different pathway of the coagulation cascade.
Administration
Heparin is given parenterally because it is not absorbed from the gut, due to its high negative charge and large size. It can be injected intravenously or subcutaneously (under the skin); intramuscular injections (into muscle) are avoided because of the potential for forming hematomas. Because of its short biologic half-life of about one hour, heparin must be given frequently or as a continuous infusion. Unfractionated heparin has a half-life of about one to two hours after infusion, [20] whereas LMWH has a half-life of four to five hours.[21] The use of LMWH has allowed once-daily dosing, thus not requiring a continuous infusion of the drug. If long-term anticoagulation is required, heparin is often used only to commence anticoagulation therapy until an oral anticoagulant e.g. warfarin takes effect.
Details of administration are available in clinical practice guidelines by the American College of Chest Physicians:[22]
Production
Pharmaceutical-grade heparin is derived from mucosal tissues of slaughtered meat animals such as porcine (pig) intestines or bovine (cattle) lungs.[23] Advances to produce heparin synthetically have been made in 2003 and 2008.[24]
Protamine sulfate (1 mg per 100 units of heparin that had been given over the past four hours) has been given to counteract the anticoagulant effect of heparin.[26]
Heparin is one of the oldest drugs currently in widespread clinical use. Its discovery in 1916 predates the establishment of the Food and Drug Administration of the United States, although it did not enter clinical trials until 1935.[27] It was originally isolated from canine liver cells, hence its name (hepar or “ήπαρ” is Greek for “liver”). Heparin’s discovery can be attributed to the research activities of Jay McLean and William Henry Howell.
In 1916, McLean, a second-year medical student at Johns Hopkins University, was working under the guidance of Howell investigating procoagulant preparations, when he isolated a fat-soluble phosphatide anticoagulant in canine liver tissue. In 1918, Howell coined the term ‘heparin’ for this type of fat-soluble anticoagulant. In the early 1920s, Howell isolated a water-solublepolysaccharide anticoagulant, which was also termed ‘heparin’, although it was distinct from the phosphatide preparations previously isolated. McLean’s work as a surgeon probably changed the focus of the Howell group to look for anticoagulants, which eventually led to the polysaccharide discovery.
In the 1930s, several researchers were investigating heparin. Erik Jorpes at Karolinska Institutet published his research on the structure of heparin in 1935,[28] which made it possible for the Swedish company Vitrum AB to launch the first heparin product for intravenous use in 1936. Between 1933 and 1936, Connaught Medical Research Laboratories, then a part of the University of Toronto, perfected a technique for producing safe, nontoxic heparin that could be administered to patients in a salt solution. The first human trials of heparin began in May 1935, and, by 1937, it was clear that Connaught’s heparin was a safe, easily available, and effective blood anticoagulant. Prior to 1933, heparin was available, but in small amounts, and was extremely expensive, toxic, and, as a consequence, of no medical value.[29]
A posthumous attempt to nominate McLean for a Nobel Prize failed
Heparin Sodium Injection, USP is a sterile, nonpyrogenic solution of heparin sodium (derived from porcine intestinal mucosa) in water for injection. Each container contains 10000, 12500, 20000 or 25,000 USP Heparin Units; 40 or 80 mg sodium chloride added to render isotonic (see HOW SUPPLIEDsection for various sizes and strength). May contain sodium hydroxide and/or hydrochloric acid for pH adjustment. pH 6.0 (5.0 to 7.5).
The solution contains no bacteriostat, antimicrobial agent or added buffer and is intended for use only as a single-dose injection. When smaller doses are required, the unused portion should be discarded.
Heparin sodium in the ADD-Vantage™ system is intended for intravenous administration only after dilution.
Heparin Sodium, USP is a heterogenous group of straight-chain anionic mucopolysaccharides, called glycosamino-glycans having anticoagulantproperties. Although others may be present, the main sugars occurring in heparin are: (1) α- L-iduronic acid 2-sulfate, (2) 2-deoxy-2-sulfamino-α-D-glucose-6-sulfate, (3) β-D-glucuronic acid, (4) 2-acetamido-2-deoxy-α-D-glucose, and (5) α-L-iduronic acid. These sugars are present in decreasing amounts, usually in the order (2) > (1) > (4) > (3) > (5), and are joined by glycosidic linkages, forming polymers of varying sizes. Heparin is strongly acidic because of its content of covalently linked sulfate and carboxylic acid groups. In heparin sodium, the acidic protons of the sulfate units are partially replaced by sodium ions. The potency is determined by a biological assay using a USP reference standard based on units of heparin activity per milligram.
Structure of Heparin Sodium (representative subunits):

………………..

http://www.medgadget.com/2008/08/on_the_road_to_a_fully_synthetic_heparin.html
…………

The chemoenzymatic synthesis of heparin from E. coli’s carbohydrate coat
Now, Linhardt’s team – who were also the first to identify the contaminant in the tainted batches as oversulfated chondroitin sulfate – have come up with a potentially safer way to produce heparin. The researchers grew flasks of the gut bacteria E. coli, then converted its naturally produced carbohydrate coat to heparin in just a few steps using enzymes and chemical treatment.
Linhardt says the key to the procedure was starting with the carbohydrate capsule that E coli produces to hide itself from the human immune system. The capsule is made from heparosan – a polysachharide that is already quite similar to heparin.
The team first chemically removed acetyl groups from the heparosan with sodium hydroxide and added a sulfate group using sulfur trioxide trimethylamine. Then, using four enzymes found in all mammals that produce heparin, they introduced further modifications, including the addition of three more sulfates at different positions on the molecule to get to heparin.
The checked the structure of the compound using NMR and showed that the synthetic compound could stop blood clotting as well as heparin derived from animals. To date, however, the team have only made a total of around 100mg of pure heparin – barely enough for a single dose. That is still a million times more than produced by a 2003 total synthesis of heparin, from researchers at the Massachusetts Institute of Technology, US.
- Heparin Sodium injection
- heparin. In: Lexi-Drugs Online [database on the Internet]. Hudson (OH): Lexi-Comp, Inc.; 2007 [cited 2/10/12]. Available from: http://online.lexi.com. subscription required to view.
- Cox, M.; Nelson D. (2004). Lehninger, Principles of Biochemistry (4). Freeman. p. 1100. ISBN 0-7167-4339-6.
- Marcum JA, McKenney JB. et al. (1986). “Anticoagulantly active heparin-like molecules from mast cell-deficient mice”.Am. J. Physiol. 250 (5 Pt 2): H879–888. PMID 3706560.
- Nader, HB et al.; Chavante, S.F.; Dos-Santos, E.A.; Oliveira, F.W.; De-Paiva, J.F.; Jerônimo, S.M.B.; Medeiros, G.F.; De-Abreu, L.R.D. et al. (1999). “Heparan sulfates and heparins: similar compounds performing the same functions in vertebrates and invertebrates?”. Braz. J. Med. Biol. Res. 32 (5): 529–538. doi:10.1590/S0100-879X1999000500005. PMID 10412563.
- Francis CW, Kaplan KL (2006). “Chapter 21. Principles of Antithrombotic Therapy”. In Lichtman MA, Beutler E, Kipps TJ, et al. Williams Hematology (7th ed.). ISBN 978-0-07-143591-8.
- Bentolila, A. et al.. “Synthesis and heparin-like biological activity of amino acid-based polymers” (Subscription required). Wiley InterScience. Retrieved 2008-03-10.
- Gatti, G., Casu, B. et al. (1979). “Studies on the Conformation of Heparin by lH and 13C NMR Spectroscopy” (PDF). Macromolecules 12 (5): 1001–1007. Bibcode:1979MaMol..12.1001G.doi:10.1021/ma60071a044.
- “Online Medical Dictionary”. Centre for Cancer Education. 2000. Retrieved 2008-07-11.
- Ferro D, Provasoli A, et al. (1990). “Conformer populations of L-iduronic acid residues in glycosaminoglycan sequences”. Carbohydr. Res. 195 (2): 157–167.doi:10.1016/0008-6215(90)84164-P. PMID 2331699.
- Mulloy B, Forster MJ, Jones C, Davies DB. (1 January 1993). “N.m.r. and molecular-modelling studies of the solution conformation of heparin”. Biochem. J. 293 (Pt 3): 849–858. PMC 1134446. PMID 8352752.
- Guyton, A. C.; Hall, J. E. (2006). Textbook of Medical Physiology (11). Elsevier Saunders. p. 464. ISBN 0-7216-0240-1.
- Agnelli G, Piovella F, Buoncristiani P et al. (1998). “Enoxaparin plus compression stockings compared with compression stockings alone in the prevention of venous thromboembolism after elective neurosurgery”. N Engl J Med 339 (2): 80–5. doi:10.1056/NEJM199807093390204.PMID 9654538.
- Bergqvist D, Agnelli G, Cohen AT et al. (2002). “Duration of prophylaxis against venous thromboembolism with enoxaparin after surgery for cancer”. N Engl J Med 346(13): 975–980. doi:10.1056/NEJMoa012385.PMID 11919306.
- Handoll HHG, Farrar MJ, McBirnie J, Tytherleigh-Strong G, Milne AA, Gillespie WJ (2002). “Heparin, low molecular weight heparin and physical methods for preventing deep vein thrombosis and pulmonary embolism following surgery for hip fractures”. In Handoll, Helen HG. Cochrane Database Syst Rev 4 (4): CD000305.doi:10.1002/14651858.CD000305. PMID 12519540.
- Chuang YJ, Swanson R. et al. (2001). “Heparin enhances the specificity of antithrombin for thrombin and factor Xa independent of the reactive center loop sequence. Evidence for an exosite determinant of factor Xa specificity in heparin-activated antithrombin”. J. Biol. Chem. 276 (18): 14961–14971. doi:10.1074/jbc.M011550200. PMID 11278930.
- Bjork I, Lindahl U. (1982). “Mechanism of the anticoagulant action of heparin”. Mol. Cell. Biochem. 48 (3): 161–182.doi:10.1007/BF00421226. PMID 6757715.
- Petitou M, Herault JP, Bernat A, Driguez PA et al. (1999). “Synthesis of Thrombin inhibiting Heparin mimetics without side effects”. Nature 398 (6726): 417–422.Bibcode:1999Natur.398..417P. doi:10.1038/18877.PMID 10201371.
- Shalansky, Karen. DANAPAROID (Orgaran) for Heparin-Induced Thrombocytopenia. Vancouver Hospital & Health Sciences Centre, February 1998 Drug & Therapeutics Newsletter. Retrieved on 8 January 2007.
- Eikelboom JW, Hankey GJ (2002). “Low molecular weight heparins and heparinoids”. The Medical Journal of Australia 177 (7): 379–383. PMID 12358583.
- Weitz JI (2004). “New anticoagulants for treatment of venous thromboembolism”. Circulation 110 (9 Suppl 1): I19–26. doi:10.1161/01.CIR.0000140901.04538.ae.PMID 15339877.
- Hirsh J, Raschke R (2004). “Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy”. Chest 126 (3 Suppl): 188S–203S.doi:10.1378/chest.126.3_suppl.188S. PMID 15383472.
- Linhardt RJ, Gunay NS. (1999). “Production and Chemical Processing of Low Molecular Weight Heparins”. Sem. Thromb. Hem. 3: 5–16. PMID 10549711.
- Bhattacharya, Ananyo (August 2008). “Flask synthesis promises untainted heparin”. Chemistry World. Royal Society of Chemistry. Retrieved 6 February 2011.
- Kusmer, Ken (20 September 2006). “3rd Ind. preemie infant dies of overdose”. Fox News (Associated Press). Retrieved 2007-01-08.
- Internal medicine, Jay H. Stein, page 635
- Linhardt RJ. (1991). “Heparin: An important drug enters its seventh decade”. Chem. Indust. 2: 45–50.
- Jorpes E (August 1935). “The chemistry of heparin”. The Biochemical Journal 29 (8): 1817–30. PMC 1266692.PMID 16745848.
- Rutty, CJ. “Miracle Blood Lubricant: Connaught and the Story of Heparin, 1928–1937”. Health Heritage Research Services. Retrieved 2007-05-21.
|
9-1-2004
|
Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy.
|
Chest
|
|
|
4-1-1999
|
Synthesis of thrombin-inhibiting heparin mimetics without side effects.
|
Nature
|
|
|
1-1-1999
|
Production and chemical processing of low molecular weight heparins.
|
Seminars in thrombosis and hemostasis
|
|
|
8-1-1993
|
N.m.r. and molecular-modelling studies of the solution conformation of heparin.
|
The Biochemical journal
|
|
|
1-15-1990
|
Conformer populations of L-iduronic acid residues in glycosaminoglycan sequences.
|
Carbohydrate research
|
Heparin, a highly sulfated glycosaminoglycan (GAG), is used extensively as an anticoagulant. It consists of repeating disaccharide units, containing iduronic acid (or glucuronic acid) and glucosamine, exhibiting variable degrees of sulfation. Heparin, and its analogues, are used during surgery and dialysis, and are often used to coat indwelling catheters and other devices where the vascular system is exposed. Administered parenterally, often continuously due to its short half-life, over 0.5 billion doses are required per year. Currently obtained from mucosal tissue of meat animals, mainly porcine intestine, and to a lesser extent bovine lung, its early stage production is poorly controlled, due to the source of the material (Figure 1). This problem came into sharp focus in 2008 when the presence of contaminating over-sulfated chondroitin sulfate in heparin, sourced from pigs, resulted in almost 100 deaths in the USA. This, coupled with the fact that only two doses are obtained per animal means that the demand for alternative and more controlled sources of heparin is high.




Figure.3. (A) The structure of heparosan disaccharide unit. (B) the structures of the major and minor variable repeating disaccharides comprising heparin where X = SO3- or H and Y = SO3- or COCH3.1

Figure.4. Time course of dry cell weight (g/L) and heparosan concentration in the fermentation supernatant (g/L) during the fermentation in a 20 L fermentor1
3. Z.Wang, M.Ly, F.Zhang, W. Zhong, A.Suen, A.M.Hickey, J.S.Dordick, R.J.Linhardt,”E. coli K5 fermentation and the preparation of heparosan, a bioengineered heparin precursor“, Biotechnol. Bioeng. 107, 964-973 (2010).
- M.Ly, Z.Wang, T.N.Laremore, F.Zhang, W.Zhong, D.Pu, D.V.Zagorevski, J.S.Dordick, R.J.Linhardt, “Analysis of E. coli K5 capsular polysaccharide heparosan.” Analytical and Bioanalytical Chemistry 399, 737-745 (2011).
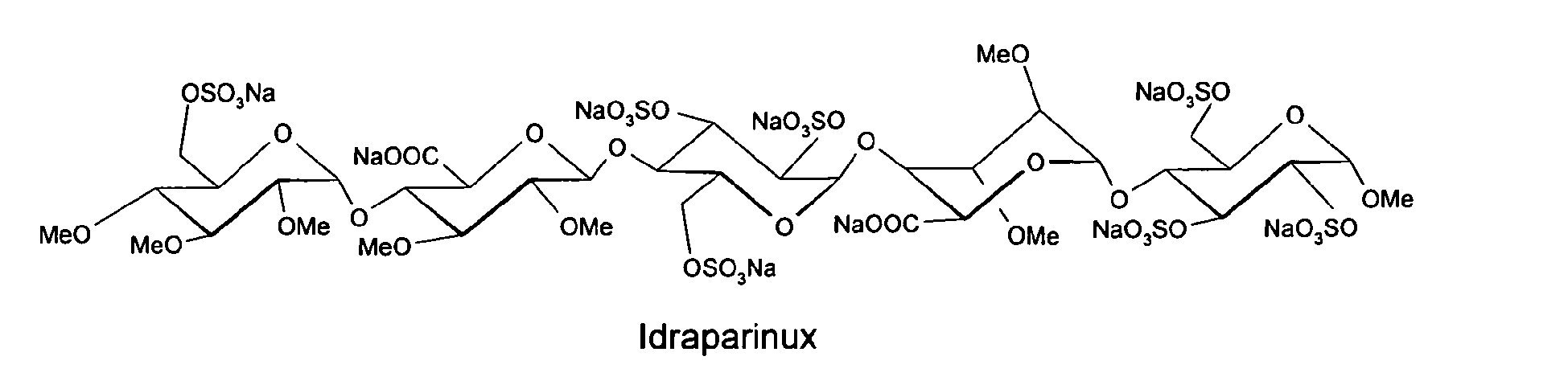
IDRAPARINUX… Sanofi (PHASE III)

IDRAPARINUX
Nonasodium (2S,3S,4S,5R,6R)-6-[(2R,3R,4S,5R,6R)-6-[(2R,3S,4S,5R,6R)-2-carboxy-4,5-dimethoxy-6-[(2R,3R,4S,5R,6S)-6-methoxy-4,5-disulfooxy-2-(sulfooxymethyl)oxan-3-yl]oxyoxan-3-yl]oxy-4,5-disulfooxy-2-(sulfooxymethyl)oxan-3-yl]oxy-4,5-dimethoxy-3-[(2R,3R,4S,5R,6R)-3,4,5-trimethoxy-6-(sulfooxymethyl)oxan-2-yl]oxyoxane-2-carboxylic acid |
| CAS number | 149920-56-9 |
|---|
| Formula | C38H55Na9O49S7 |
|---|---|
| Mol. mass | 1727.17683 g/mol |
CAS 162610-17-5 (free acid)
SANORG34006, SR-34006, SanOrg 34006, SanOrg-34006, UNII-H84IXP29FN, AC1MJ0N4, Org-34006
Methyl O-2,3,4-tri-O-methyl-6-O-sulfo-alpha-D-glucopyranosyl-(1–4)-O-2,3-di-O-methyl-beta-D-glucopyranuronosyl-(1–4)-O-2,3,6-tri-O-sulfo-alpha-D-glucopyranosyl-(1–4)-O-2,3-di-O-methyl-alpha-L-idopyranuronosyl-(1–4)-2,3,6-tri-O-sulfo-alpha-D-glucopyran
Sanofi-Syn(Originator), Organon (Codevelopment) , PHASE 3
, PHASE 3
Methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose
methyl O-2,3,4-tri-O-methyl-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-O-di-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt. [α]D²⁰ = +46.2° (c=1; water). Anomeric protons chemical shifts: 5.43; 5.37; 5.16; 5.09; and 5.09 ppm.
Idraparinux sodium, or methyl O-2,3,4-tri-O-methyl-6-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranose, is a pentasaccharide with antithrombotic activity.
The preparation of idraparinux by sulfatation of a deprotected pentasaccharide is described in Bioorganic & Medicinal Chemistry, 1994, Vol. 2, No. 11, pp. 1267-1280, and also in patent EP 0 529 715 B1.
Idraparinux sodium is an anticoagulant medication in development by Sanofi-Aventis.[1]
It has a similar chemical structure and the same method of action as fondaparinux, but with an elimination half-life about five to six times longer (an increase from fondaparinux’s 17 hours to approximately 80 hours), which means that the drug should only need to be injected once a week.
As of July 2007, it has completed the Phase III clinical trial AMADEUS.
Idraparinux selectively blocks coagulation factor Xa.[2]
See Heparin: Mechanism of anticoagulant action for a comparison of the mechanism of heparin, low-molecular-weight heparins, fondaparinux and idraparinux.
Idraparinux sodium is a synthetic pentasaccharide with indirect coagulation factor Xa inhibitor activity. The drug candidate had been in phase III clinical development at Sanofi (formerly known as sanofi-aventis) for the once-weekly long-term treatment and secondary prevention of venous thromboembolic events in patients with pulmonary embolism (PE) and deep vein thrombosis (DVT), as well as for the prevention of thromboembolic complications related to atrial fibrillation (AF).
However, no recent development has been reported for this research. The oligosaccharide is delivered by subcutaneous injection. Unlike other products, idraparinux is administered once weekly rather than daily, thereby increasing patient convenience.
Originally developed under a collaboration between sanofi-sventis and Akzo Nobel’s human healthcare business Organon, all rights to idraparinux were transferred to Sanofi in January 2004 in exchange for revenues based on future sales.
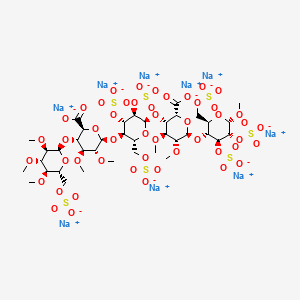
Several synthetic pentasaccharides have been developed, such as Idraparinux, where all hydroxyl groups are methylated or sulphated, as illustrated below:

Initially, the firm Organon developed a way of synthesis for the preparation of the “active pentasaccharide”. This synthesis, using the 3-0-benzyl-1 ,2-0-isopropylidene-a-D- glucofuranose as substrate (Van Boeckel et al., J. Carbohydr. Chem. 1985, 4, p.293-321 ), comprises more than 50 steps, and the inversion of configuration of the C5 carbon is carried out by the opening of an epoxide. After a step of protection followed by a bromination, the G unit is thus obtained. It is well known that the synthesis of said G unit is very tedious, due to the number of steps for obtaining such unit and the known tendency of L-idose derivatives to exist as furanoses. After being coupled to the H unit, successive steps of protection-deprotection then an oxidation reaction carried out on C6 carbon, lead to the GH disaccharide.
In the preparation of Idraparinux, the synthesis of the disaccharide GH is nearly similar to the above synthesis of early synthetic pentasaccharides. The major innovation lies in the obtaining of disaccharide EF by epimerization of disaccharide GH. The coupling of both disaccharides leads to the tetrasaccharide EFGH, which is further coupled to the D unit for obtaining said pentasaccharide. The preparation of the disaccharide EF from GH allows notably the decrease of the total number of the steps to approximatively 25 (Petitou, M.; Van Boeckel, C.A. Angew. Chem., Int.Ed. 2004, 43, p.31 18-3133).
Hence, all current syntheses of the “active pentasaccharide” comprise a large number of steps and more particularly involves the complex synthesis of key L-iduronic acid derivative (G unit). Indeed, the preparation of the G unit of the “active pentasaccharide” of heparin has always been a limiting step in the synthesis of antithrombotic heparin derivatives.
Thus, there is still a need for a new efficient process of preparation of L-iduronic acid derivative, which would not possess the drawbacks established above and would be compatible with industrial scales. Besides, there is a need for such process which would in addition lead to an improved process of preparation of the “active pentasaccharide” constituting the heparin derivatives.
-
Idrabiotaparinux, developed by sanofi-aventis, is the biotinylated pentasaccharide corresponding to the structure depicted below. The pentasaccharide structure of idrabiotaparinux is the same as idraparinux, another antithrombotic agent developed by sanofi-aventis (see structure below). However in idrabiotaparinux, the presence of a biotin hook covalently linked to the first saccharidic unit enables the compound to be neutralized by avidin or streptavidin, as described in the international patent application WO 02/24754 .


-
In the EQUINOX trial, which enrolled patients with DVT treated for 6 months with equimolar doses of either idrabiotaparinux or idraparinux, idrabiotaparinux, with the same anti-activated factor X pharmacological activity (hereafter “anti-Xa activity”) as idraparinux, was shown to have a similar efficacy, but, surprisingly, a better safety with less observed bleedings, in particular major bleedings.
-
Therefore, the subject-matter of the invention is the use of idrabiotaparinux for the manufacture of a medicament useful for the treatment and secondary prevention of thrombotic pathologies, wherein the use of idrabiotaparinux involves a decrease in the incidence of bleedings during said treatment.
-
In other words, the invention relates to the use of idrabiotaparinux as an antithrombotic treatment, wherein said use minimizes the risk of bleedings during the antithrombotic treatment. Indeed, idrabiotaparinux enables to increase the benefit-risk ratio during the antithrombotic treatment.

The L-ioduronic acid methyl ester derivative (XII) is then converted into its D-glucuronic acid methyl ester counterpart (XIII) by epimerization with NaOMe in refluxing MeOH, followed by esterification with MeI and KHCO3 in DMF.
Protection of the ester (XIII) with levulinic acid (IX) by means of DCC and DMAP in dioxane, followed by acetolysis of the anomeric center with sulfuric acid in acetic anhydride furnishes the disaccharide (XIV), which is then saponified with piperidine and subjected to reaction with trichloroacetonitrile and Cs2CO3 in THF to yield the imidate (XV).
Glycosylation of the disaccharide (XII) with the imidate (XV) by means of trimethylsilyl triflate in CH2Cl2, followed by removal of the levulinoyl group by means of hydrazine acetate, furnishes the tetrasaccharide (XVI), which is coupled with the glucosyl trichloroacetimidate (XVIII) by means of trimethylsilyl trifluoromethanesulfonate in CH2Cl2 providing the pentasaccharide (XVII).
Glucosyl imidate (XVIII) is prepared by methylation of 1,6-anhydroglucose (XIX) with MeI and NaH in DMF, followed by acetolysis with Ac2O/TFA to give compound (XX), which is treated with piperidine in THF and finally with trichloroacetonitrile in dichloromethane in the presence of Cs2CO3.
The pentasaccharide (XVII) is deprotected by saponification with LiOH in THF/H2O2, and then hydrogenated over Pd/C in tert-butanol/water to provide a fully deprotected pentamer, which is finally subjected to sulfation with triethylamine sulfur trioxide complex in DMF and converted into the corresponding sodium salt by elution in a Dowex 50 XW4-Na+ or a Mono-Q anion-exchange column.

……………..
Glycosylation of sugar (I) with the idopyranosyl fluoride (II) by means of BF3.Et2O and molecular sieves in dichloromethane gives the disaccharide fragment (III), which is then converted into acetonide (V) by saponification of the ester functions with t-BuOK, followed by reaction with 2,2-dimethoxypropane (IV) in DMF and acidification with p-toluensulfonic acid. Methylation of acetonide (V) with MeI and NaH in DMF/MeOH provides the disaccharide (VI), which is then treated with HOAc to yield the 4′,6′-diol (VII). Selective silylation of the diol (VII) with tert-butyldimethylsilyl chloride (TBDMSCl) in pyridine leads to the 6′-O-TBDMS derivative (VIII), which is condensed with levulinic acid (IX) by means of dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) in dioxane to give the ester (X). Compound (X) is then submitted to simultaneous Jones oxidation and TBDMS removal with CrO3 and H2SO4/H2O in acetone to provide the iduronic acid derivative (XI), which is converted into the key intermediate (XII), first by esterification with MeI and KHCO3 in DMF and then by removal of the 4′-O-levulinoyl protecting group with HOAc and hydrazine hydrate in pyridine.

………………………
Idraparinux sodium, or methyl O-2,3,4-tri-O-methyl-6-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranose, is a pentasaccharide with antithrombotic activity.
The preparation of idraparinux by sulfatation of a deprotected pentasaccharide is described in Bioorganic & Medicinal Chemistry, 1994, Vol. 2, No. 11, pp. 1267-1280, and also in patent EP 0 529 715 B1.
A crystalline form of the pentasaccharide methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose has now been isolated. This compound in its crystalline form has proven to be very useful for the preparation of idraparinux, since it makes it possible to obtain this product in a particularly interesting chemical yield and with a significant gain in quality, the purity being improved as regards the crude product obtained, as will be detailed hereinbelow. These gains in reaction yield and in purity for the production of idraparinux are considerable advantages from an industrial viewpoint, since improving the robustness of a process is a constant cause for concern, especially in the case of large-scale syntheses.
One subject of the invention is thus the compound methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic glucopyranose in crystalline form.
Methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose, referred to hereinbelow as the compound of formula (I), corresponds to the following formula:

The compound of formula (I) in crystalline form according to the invention has a powder X-ray diffractogram whose characteristic lines are approximately at 12.009; 7.703; 7.300; 7.129; 5.838; 4.665; 4.476 and 3.785 angströms (interplanar distances). It also has a melting point of about 203° C. (203° C.±1° C.).
EXAMPLE 1 Preparation of the Compound of Formula (I) in Crystalline Form (Scheme 1)

Methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose, referred to hereinbelow as the compound of formula (I)
1.1: Preparation of the Compound of Formula (I′)
The compound of formula (I″) is obtained, for example, according to the teaching of patent EP 0 529 715 B1 or of the articles “Bioorg. Med. Chem.” (1994, Vol. 2, No. 11, pp. 1267-1280), “Bioorg. Med. Chem. Letters” (1992, Vol. 2, No. 9, pp. 905-910) or “Magnetic Resonance in Chemistry” (2001, Vol. 39, pp. 288-293). The compound of formula (I″) (5 g, 3.06 mmol) is dissolved in acetonitrile (10 mL). Deionized water (12.2 mL) and aqueous 30% sodium hydroxide solution (4.1 g) are then added. The mixture is heated to 40° C. and maintained at this temperature for 5 hours. The reaction medium is then cooled to 20° C. and acidified to pH 6.25 with aqueous 1N hydrochloric acid solution (about 17.7 g) before extraction with MTBE of certain impurities, the saponified product remaining in the aqueous phase. The residual acetonitrile, contained in the aqueous phase, is then removed by concentration, followed by diluting with deionized water (125 mL). The saponified product is finally precipitated at pH 1.5 by adding aqueous 1N hydrochloric acid solution (about 17.6 g) at 20° C. The suspension is maintained for 4 hours at 20° C. before filtration. The wet solid is finally dried in a vacuum oven at 30° C. to give 2.93 g (93.6%) of compound of formula (I).
NMR (anomeric protons of the saccharide units D, E, F, G, H): 5.79, 5.14, 5.55, 5.92, 4.94 ppm.
1.2 Preparation of the Crude Compound of Formula (I)
The compound of formula (I′) obtained after the preceding step is dissolved in tetrahydrofuran (18 mL). Palladium-on-charcoal (0.3 g) is added. The reaction medium is hydrogenated at 0.3 bar of hydrogen (relative pressure) for 4 hours. After filtering and evaporating, 2.12 g (99%) of the crude compound of formula (I) are obtained.
1.3: Preparation of the Compound of Formula (I) in Crystalline Form Using an Isopropanol/MTBE Mixture
The crude hydrogenated product obtained after the preceding step is dissolved in isopropanol (13 mL) at 65° C., and then crystallized at room temperature. The suspension is then cooled to 40° C., followed by addition of MTBE (13 mL), and is then cooled slowly to 10° C. After maintenance at 10° C. for 2 hours, the crystalline hydrogenated product is filtered off, washed and dried. 1.66 g of the compound of formula (I) in crystalline form are thus obtained, in the form of a cream-white powder. The reaction yield for the production of the compound of formula (I) in crystalline form, from the compound of formula (I′), is 92.5%. When expressed relative to the starting compound (I″), the reaction yield for the production of the compound of formula (I) in crystalline form is 86.6%.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (I) in crystalline form: 5.77, 5.11, 5.51, 5.84, 5.01 ppm.
1.4: Preparation of the Compound of Formula (I) in Crystalline Form Using Isopropanol
The crude hydrogenated product obtained after step 1.2 is dissolved in isopropanol (5 volumes) at 75° C. The medium is then cooled slowly until crystals appear, according to the known standard techniques for crystallization. The process is performed, for example, by a first step of cooling at 65° C. for 1 hour, and than a second step of cooling to a final temperature of 25° C. over 4 hours or of 5° C. over 6 hours, and finally maintenance at this final temperature for 30 minutes. The suspension is then filtered and rinsed with isopropanol (2×0.1 V) and compound (I) is isolated in the form of white crystals, which appear under a microscope in the form of needles. The 1H NMR analysis of these crystals is identical to that described after step 1.3 above.
EXAMPLE 4 Preparation of Idraparinux from the Compound of Formula (I) in Crystalline Form (Scheme 2)
The preparation of idraparinux (II) from the compound of formula (I) is summarized in Scheme 2.

The compound of formula (I) in crystalline form, as obtained according to Example 1.3, is dissolved in N,N′-dimethylformamide (6.6 mL) and then heated to 30° C. Under an inert atmosphere, 3.8 g of pyridine-sulfur trioxide complex are added slowly, followed by maintenance at 30° C. for 4 hours. The reaction medium is then poured into aqueous 23.8% sodium hydrogen carbonate solution (16.3 g) maintained at a maximum of 25° C., to obtain the compound of formula (II). The reaction medium is kept stirring for hours. The solution of sulfated product is then poured onto an MTBE/isopropanol/ethanol mixture (171 mL/70 mL/70 mL). Precipitation of the product is observed, and, after filtering off, washing and drying the cake, 4.99 g (96.8%) of compound of formula (II) are obtained, and are then purified by anion-exchange chromatography according to the usual techniques.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (II): 5.48, 4.68, 5.44, 5.08, 5.18 ppm.
It thus appears that the process according to the invention makes it possible to obtain idraparinux (compound of formula (II)) in a chemical yield of about 84% (precisely 83.8% according to the protocols described above) starting from the compound of formula (I″), i.e. a gain in yield of about 30% relative to the process described in patent EP 0 529 715 B1.
………………..
methyl O-2,3,4-tri-O-methyl-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-O-di-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt. [α]D²⁰ = +46.2° (c=1; water). Anomeric protons chemical shifts: 5.43; 5.37; 5.16; 5.09; and 5.09 ppm.
WAS PREPARED AS PER
- Example 3
methyl O-4-O-(4-sulfoaminophenyl)-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt.
NOTE THIS IS ANALOGOUS PROCEDURE AND NOT SIMILAR
-
Methyl O-4-O-(4-nitrophenyl)-6-O-acetyl-2,3-O-di-phenylmethyl-α-D-glucopyranosyl-(1→4)-O-(methyl 3-O-methyl-2-O-acetyl-α-L-idopyranosyluronate)-(1→4)-O-2,3,6-tri-O-acetyl-α-D-glucopyranoside (100 mg, 0.09 mmol), obtained by the known imidate coupling of the trichloroacetimidate of O-4-O-(4-nitrophenyl)-6-O-acetyl-2,3-O-di-phenylmethyl-α-D-glucopyranoside and methyl O-(methyl 3-O-methyl-2-O-acetyl-α-L-idopyranosyluronate)-(1→4)-O- 2,3,6-tri-O-acetyl-α-D-glucopyranoside, was dissolved in tetrahydrofuran (9 ml) and cooled to -5 °C. At this temperature a 30% aq. solution of hydrogen peroxide (4.5 ml) was added to the reaction mixture, and after 10 min a 1.25 M lithium hydroxide solution (4.7 ml) was added. The mixture was stirred for 1 h at -5 °C, after which time the temperature was raised to 0 °C and the mixture was stirred overnight. The reaction mixture was acidified with 6N hydrogen chloride at 0 °C to pH 1.5, after which the saponified compound was extracted with ethyl acetate. The organic layers were pooled, dried over magnesium sulfate, and evaporated to give 63 mg (84%) of methyl O-4-O-(4-nitrophenyl)-2,3-O-di-phenylmethy1-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-α-L-idopyranuronosyl-(1→4)-O-α-D-glucopyranoside, which was dissolved in methanol (8 ml). 10% Pd on charcoal (63 mg) was added and the mixture hydrogenolyzed overnight. After filtration and evaporation 27 mg (50%) of methyl O-4-O-(4-aminophenyl)-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-α-L-idopyranuronosyl-(1→4)-O-α-D-glucopyranoside were obtained.
13 mg of methyl O-4-O-(4-aminophenyl)-O-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-α-L-idopyranuronosyl-(1→4)-O-α-D-glucopyranoside were dissolved in 2 ml of dry N,N-dimethylformamide, and under an atmosphere of nitrogen 148 mg of triethylamine sulfurtrioxide complex were added. The mixture was stirred overnight at 50 °C, after which an aq. solution of sodium hydrogen carbonate was added under ice cooling. The mixture was stirred for 1 h at room temperature, concentrated to a small volume and desalted on a Sephadex G-10 column with water. The crude product obtained was purified by HPLC using a Mono-Q anion exchange column to give 11 mg (37%) of methyl O-4-O-(4-sulfoaminophenyl)-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt. [α]D²⁰ = +52.2° (c=0.67; water). Anomeric protons chemical shifts: 5.5; 5.17; and 5.15 ppm.
………………………………..
BMCL Volume 19, Issue 14, 15 July 2009, Pages 3875–3879
http://www.sciencedirect.com/science/article/pii/S0960894X0900482X


Final elaboration of the pentasaccharide 1. Reagents and conditions: (a) TMSOTf, Et2O, 4 Å MS, rt, 66% (28α), 15% (28β); (b) CAN, CH3CN, toluene, H2O, rt, 72%; (c) CCl3CN, DBU, CH2Cl2, rt, 98%; (d) TMSOTf, 4 Å MS, CH2Cl2, rt, 51% (73% based on recovery of 4); (e) Pd/C (10%), H2, t-BuOH, H2O, rt; (f) SO3·Et3N, DMF, 50 °C, 93% (2 steps).
The final elaboration of the pentasaccharide 1 was illustrated in IN ABOVE SCHEME Coupling of the glucopyranosyl trichloroacetimidate 6 with disaccharide acceptor 5 in the presence of trimethylsilyl trifluoromethylsulfonate and powdered 4 Å molecular sieves at room temperature in diethyl ether afforded the desired α-coupled trisaccharide 28α in a yield of 66%, together with 15% of the separable β-coupled product 28β. The anomeric 4-methoxyphenyl group in trisaccharide 28α was removed with CAN, and the resulting lactol was readily converted into the trisaccharide trichloroacetimidate 3. Coupling of donor 3 with the disaccharide acceptor 4 in the presence of trimethylsilyl trifluoromethylsulfonate and powdered 4 Å molecular sieves at room temperature in dichloromethane afforded the fully protected pentasaccharide 2 in 51% yield (73% based on recovery of 4). Finally, pentasaccharide 2 was subject to hydrogenolysis of the benzyl protecting groups. The highly polar product without purification was O-sulfated directly with triethylamine-sulfur trioxide complex to afford the sulfated pentasaccharide 1 in an excellent yield of 93% (for two steps).
Summarizing, the potent anti-thromboembolic pentasaccharide Idraparinux (1) was synthesized in total 51 steps and in 4% overall yield from d-glucose and methyl α-d-glucopyranoside.18 The synthetic route is convergent with a linear sequence of 27 steps, and the transformations are scalable. The 4-methoxyphenol glycoside intermediates are easy to be purified by crystallization.
Compound 1: ![]() 54.2 (c 1.0, H2O);
54.2 (c 1.0, H2O);
1H NMR (400 MHz, D2O) δ 3.27 (t, J = 8.4 Hz, 1H), 3.30–3.38 (m, 2H), 3.47 (s, 3H), 3.53 (s, 3H), 3.56 (s, 6H), 3.58 (s, 3H), 3.62 (s, 3H), 3.63 (s, 3H), 3.64 (s, 6H), 3.75 (d, J = 10.0 Hz, 1H), 3.83–3.97 (m, 4H), 3.98 (t, J = 8.8 Hz, 1H), 4.06–4.18 (m, 3H), 4.19–4.45 (m, 8H), 4.56 (br t, J = 9.6 Hz, 1H), 4.65 (t, J = 9.2 Hz, 1H), 4.66 (d, J = 7.6 Hz, 1H), 5.00 (br s, 1H), 5.11 (br s, 1H), 5.17 (d, J = 3.6 Hz, 1H), 5.43 (d,J = 3.2 Hz, 1H), 5.47 (d, J = 4.0 Hz, 1H);
ESI-MS m/z 774.1 [M−8Na+6H]2−, 763.0; [M−9Na+7H]2−, 508.5 [M−9Na+6H]3−.
………………….
The process of preparation of Idraparinux having the following formula:

may comprise the following steps :
1 ) preparation a compound of formula (IXB)

(IXB) wherein Ra is methyl, Rb is methyl, Rc is methyl, T-i is benzyl, T2 is benzyl, T3 is benzyl and T is methyl, by the process according to the invention;
2) epimerisation of the disaccharide (IXB) so as to form disaccharide D of formula :

3) protection of the 4′-OH of D with a levulinoyl ester;
4) acetolysis of the disaccharide resulting from step 3), followed by preparation of the corresponding imidate;
5) coupling the disaccharide imidate resulting from step 4) with (IXB) obtainable by the process of the invention, wherein Ra is methyl, Rb is methyl, Rc is methyl, T-i is benzyl, T2 is benzyl, T3 is benzyl and T is methyl, to obtain a tetrasaccharide;
6) coupling the fully protected tetrasaccharide with a monosaccharide glycosyl imidate;
7) deprotection of the protecting groups by the successive saponification and hydrogenolysis;
8) sulfation of the hydroxyl groups.
In one embodiment, the present invention concerns a process of preparation of Idraparinux:

said process comprising the following steps:
preparation of a compound of formula (VI) such as defined above, from a compound of formula (V) such as defined above; preparation of a compound of formula (VII) such as defined above, from a compound of formula (VI) such as defined above;
preparation of a compound of formula (VIII) such as defined above, from a compound of formula (VII) such as defined above;
– preparation of a compound of formula (IX) such as defined above, from a compound of formula (VIII) such as defined above;
wherein in compounds of formulae (V), (VI), (VII), (VIII) and (IX), R-i , R2, R3 and X are as defined above, Ra is methyl, Rb is methyl, Rc is methyl, Rd is methyl and R’ is the monosaccharide of formula :

wherein T-i is benzyl, T2 is benzyl, T3 is benzyl and T is methyl. The inventors advantageously found that the process of preparation of Idraparinux comprising the decarboxylation/intramolecular cyclisation tandem reaction, which allows the inversion of configuration of C5 carbon of the compound of formula (VI), is more efficient than the processes previously described in the literature. Indeed, the process according to the invention allows advantageously a significant decrease of the number of steps and thus an improvement of the overall yield. Thus, the process of preparation of Idraparinux may be carried out in industrial scales. The inventors found an efficient process of preparation of Idraparinux.
According to another object, the present invention concerns the use of compounds of formulae (V), (VI), (VI I), (VIII) and (IX), as intermediates for the preparation of Idraparinux. In particular, the present invention concerns the use of a compounds of formulae (VB), (VI B), (VI IB), (VI I IB) and (IXB), as intermediates for the preparation of Idraparinux The invention is further illustrated but not restricted by the description in the following examples. Example 1 :
Preparation of Methyl-4,6-0-benzylidene-a-D-glucopyranoside (la)
CHC13)

Tf = 166-167°C (litt. 165-166°C) To a solution of benzaldehyde (400 mL, 3.94 mol, 5.9 eq.) was added zinc chloride (100.3 g, 0.74 mol, 1 .1 eq.) under vigorous stirring. After homogenization of the solution methyl- a-D-glucopyranoside (129.6 g, 0.67 mol, 1.0 eq.) was added portionwise. After 16 hours stirring at room temperature the reaction mixture was diluted with diethyl ether (100 mL). The mixture was then poured dropwise and under vigorous stirring in a solution containing ice water (1 .5 L) and hexane (350 mL). The precipitate was filtered, washed with diethyl ether (3 x 300 mL) and dried under vacuum over KOH. The product was then recrystallised from CH2CI2 (720 mL) and washed with a Et20/CH2CI2 solution (75:25, 2 x 200 mL). The filtrate was repeatedly recrystallised five times from CH2CI2 to afford compound la as white crystals (136.97 g, 0.49 mol, 72%).
1H NMR (CDCI3, 250 MHz): δ 2.35 (d, JCH-OH = 9.2 Hz, 1 H, OH), 2.83 (d, JCH-OH = 2.2 Hz, 1 H, OH), 3.46 (s, 3H, -OCH3), 3.43-3.46 (m, 1 H, H-4), 3.63 (td, JCH–OH = ^2,3 = 9.2 Hz, J1 2 = 3.9 Hz, 1 H, H-2), 3.70-3.81 (m, 2H, H-5, H-6), 3.93 (td, J = 9.2 Hz, JCH–OH = 2.2 Hz, 1 H, H- 3), 4.29 (m, 1 H, H-6 ), 4.79 (d, J1i2 = 3.9 Hz, 1 H, H-1 ), 5.54 (s, 1 H, Ph-CH), 7.35-7.38 (m, 3H, HAr), 7.47-7.51 (m, 2H, HAr).
13C NMR (CDCI3, 62.9 MHz): δ 55.6 (-OCH3), 62.5 (C-5), 69.0 (C-6), 71.1 (C-3), 72.9 (C- 2), 81 .0 (C-4), 99.9 (C-1 ), 102.0 (Ph-CH), 126.4, 128.5, 129.4, 137.1 (6xCAr). IR (film) v (cm“1): 3369 (O-H). Preparation of Methyl-2,3-di-0-methyl-4,6-0-benzylidene-a-D-glucopyranoside (Ma)
13)

°C)
To a solution of compound la (47.60 g, 0.17 mol, 1.0 eq.) in anhydrous THF (750 mL) under an argon atmosphere and cooled to 0°C, was added portionwise sodium hydride (60%, 16.93 g, 0.42 mol, 2.5 eq.). After 20 minutes methyl iodide was added dropwise (30 mL, 0.48 mol, 2.8 eq.) and the reaction mixture was allowed to reach room temperature. After 16 hours, methanol was added portionwise (75 mL) and the solution was stirred for another 15 minutes before being concentrated. The resulting residue was dissolved in EtOAc (400 mL) and washed with water (2 x 250 mL). The organic layer was dried (MgS04), filtered and concentrated. The resulting solid was dissolved in diethyl ether (1000 mL), hexane was added (400 mL) and the solvent was partially evaporated at low temperature. The crystals obtained were washed with hexane and the filtrate once again partially evaporated, filtered and the precipitate washed with hexane. The combined precipitates afforded compound Ma as white crystals (46.10 g, 0.15 mol, 88%).
1H NMR (CDCI3, 250 MHz): δ 3.30 (dd, J2-3 = 9.1 Hz, J1-2 = 3.7 Hz, 1 H, H-2), 3.44 (s, 3H, – OCH3), 3.49-3.87 (m, 4H, H-4, H-5, H-6, H-3), 3.55 (s, 3H, -OCH3), 3.64 (s, 3H, -OCH3), 4.28 (dd, J6-6. = 9.1 Hz, J5-6 = 3.7 Hz, 1 H, H-6 ), 4.85 (d, J1-2 = 3.7 Hz, 1 H, H-1 ), 5.54 (s, 1 H, Ph-CH), 7.36-7.41 (m, 3H, HAr), 7.48-7.52 (m, 2H, HAr).
13C NMR (CDCI3, 62.9 MHz): δ 55.4, 59.5, 61.1 (3x-OCH3), 62.3 (C-5), 69.1 (C-6), 79.9 (C-3), 81 .5 (C-4), 82.2 (C-2), 98.5 (C-1 ), 101.4 (Ph-CH), 126.2, 128.3, 129.0, 137.4 (6xCAr).
Preparation of Methyl-2,3-di-0-methyl-a-D-glucopyranoside (Ilia)
13)

To a suspension of compound Ma (10.33 g, 33.29 mmol, 1.0 eq.) in methanol (150 mL) was added para-toluenesulfonic acid monohydrate (322 mg, 1 .69 mmol, 0.05 eq.). After 4 hours stirring at room temperature, sodium carbonate (300 mg) was added and the reaction mixture was stirred an additional 15 minutes before filtration through a pad of Celite®. Then the filtrate was concentrated and the residue obtained was dissolved in a mixture of distilled water/diethyl ether (3:1 , 150 mL). The organic layer was extracted with water (2 x 50 mL) then the combined aqueous phases were concentrated and dried one night under vacuum over KOH. The resulting residue was recrystallised from toluene using petroleum ether as a co-solvent. The crystals obtained were washed with hexane and dried under vacuum over KOH to obtain compound Ilia as white crystals (6.63 g, 29.83 mmol, 90%).
1H NMR (DMSO, 400 MHz): δ 3.03 (dd, J2-3 = 9.3 Hz, J1-2 = 3.5 Hz, 1 H, H-2), 3.12-3.20 (m, 2H, H-3, H-4), 3.27 (s, 3H, -OCH3), 3.32 (s, 3H, -OCH3), 3.30-3.33 (m, 1 H, H-5), 3.44 (s, 3H, -OCH3), 3.40-3.46 (m, 1 H, H-6), 3.62 (ddd, J6-6‘ = 1 1.6 Hz, JCH-OH = 5.7 Hz, J5-6 = 1 ,9 Hz, 1 H, H-6′), 4.52 (t, J = 5.7 Hz, 1 H, OH), 4.78 (d, Ji-2 = 3.5 Hz, 1 H, H-1 ), 5.09 (d,
13C NMR (DMSO, 100.6 MHz): δ 54.1 , 57.4, 60.0 (3x-OCH3), 60.6 (C-6), 69.5 (C-3), 72.5 (C-5), 80.9 (C-2), 82.8 (C-4), 96.4 (C-1 ).
IR (film) v (cm“1): 3419 (O-H).
Preparation of Methyl methyl-2,3-di-0-methyl-a-D-glucopyranosiduronate (IVa)
C13)

To a solution of compound Ilia (500 mg, 2.25 mmol, 1.0 eq.) in distilled water (15 mL) were successively added NaBr (50 mg, 0.49 mmol, 0.2 eq.) and TEMPO (7 mg, 0.05 mmol, 0.02 eq.). The reaction mixture was cooled with the aid of an ice bath then a solution of NaOCI (13% v/v, 5.2 mL, 9.1 mmol, 4.0 eq.) was added. After 5 hours stirring at 0°C ethanol was added (96% v/v, 8 mL), then the pH was reduced to 2-3 by addition of HCI (10 % v/v). The solvent was evaporated and the residue obtained was suspended in methanol, filtered in order to remove the remaining salts and washed several times with dichloromethane and methanol. The filtrate was concentrated then dissolved, under an argon atmosphere, in dry methanol (40 mL). para-toluenesulfonic acid (85 mg, 0.45 mmol, 0.2 eq.) was added then the reaction mixture was heated under reflux overnight. The solvent was evaporated and the residue obtained was dissolved in EtOAc (60 mL). The organic layer was washed with a 5% aqueous NaHC03 solution (2 χ 20 mL) and with brine (1 χ 20 mL). The aqueous phase was extracted with dichloromethane (3 χ 20 mL). The combined organics were dried (MgS04), filtered and evaporated. Column chromatography (hexane/ethyl acetate 50:50) gave compound IVa as a colourless oil (503 mg, 2.00 mmol, 89%).
1H NMR (CDCIs, 400 MHz): δ 3.10 (d, JCH-OH = 3.0 Hz, 1 H, OH), 3.26 (dd, J2-3 = 9.3 Hz, J1 -2 = 3.4 Hz, 1 H, H-2), 3.47 (s, 3H, -OCH3), 3.49-3.52 (m, 1 H, H-3), 3.50 (s, 3H, -OCH3), 3.62 (s, 3H, -OCH3), 3.74 (td, J = 9.5 Hz, JCH-OH = 3.0 Hz, 1 H, H-4), 3.82 (s, 3H, -OCH3), 4.14 (d, J4.5 = 9.6 Hz, 1 H, H-5), 4.91 (d, Ji-2 = 3.4 Hz, 1 H, H-1 ).
13C NMR (CDCI3, 100.6 MHz): δ 52.9, 56.0, 59.1 , 61 .3 (4x-OCH3), 70.6 (C-5), 71.7 (C-4), 80.9 (C-2), 81.8 (C-3), 98.1 (C-1 ), 170.9 (C=0).
IR (film) v (cm“1): 3475 (O-H), 1750 (C=0).
Preparation of Methyl methyl^-O-il’-ethoxy^’-propyn-l’-ylJ^.S-di-O-methyl-a-D- glucopyranosiduronate (Va)

To a solution of compound IVa (4.56 g, 18.21 mmol, 1.0 eq.) in chloroform (stabilised with amylene, 200 mL) were added, under an argon atmosphere, P205 (5.31 g, 36.29 mmol, 2.0 eq.) and propargylaldehyde diethylacetal (5.2 mL, 36.27 mmol, 2.0 eq.), then the reaction mixture was heated at 60°C. After 4 hours stirring, the reaction mixture was filtered through a pad of Celite® then the solvent was removed under vacuum. The crude mixture was suspended in EtOAc (300 mL), washed with a 5% NaHC03 aqueous solution (1 x 30 mL) and brine (1 x 30 ml_). The organic layer was dried (MgS04), filtered, and evaporated. Column chromatography (gradient hexane/ethyl acetate 80:20 – 20:80) afforded compound Va as a colourless oil (4.07 g, 12.24 mmol, 67%) in a diastereomeric mixture (64:36) (the relative composition of the mixture was determined by 1H NMR from integrations of protons EtO-CH), along with some unreacted compound IVa (1.17 g, 4.68 mmol, 26%).
1H NMR (CDCI3, 400 MHz): δ 1.18-1.25 (m, 3H, -OCH2CH3) (diastereomeric mixture), 2.56 (m, 1 H, H-C≡C-) (mixture), 3.26-3.31 (m, 1 H, H-2) (mixture), 3.43 (s, 3H, -OCH3) (major), 3.44 (s, 3H, -OCH3) (minor), 3.50 (s, 3H, -OCH3) (mixture), 3.59 (s, 3H, -OCH3) (minor), 3.62 (s, 3H, -OCH3) (major), 3.47-3.62 (m, 2H, H-3, -OCHaHbCH3) (mixture), 3.65-3.73 (m, 1 H, -OCHaHbCH3) (mixture), 3.78 (s, 3H, -OCH3) (major), 3.80 (s, 3H, -OCH3) (minor), 3.78-3.86 (m, 1 H, H-4) (mixture), 4.15 (d, J4-5 = 10.0 Hz, 1 H, H-5) (major), 4.18 (d, J4-5 = 10.0 Hz, 1 H, H-5) (minor), 4.86-4.88 (m, 1 H, H-1 ) (mixture), 5.35 (d, J = 1.7 Hz, 1 H, EtO- CH) (minor), 5.58 (d, J = 1.7 Hz, 1 H, EtO-CH) (major).
13C NMR (CDCI3, 100.6 MHz): δ 15.0 (-OCH2CH3) (mixture), 52.6 (-OCH3) (major), 52.7 (- OCH3) (minor), 55.8 (-OCH3) (mixture), 59.2 (-OCH3) (major), 59.3 (-OCH3) (minor), 60.4 (-OCH2CH3) (major), 61 .3 (-OCH2CH3) (minor), 61.4 (-OCH3) (mixture), 70.1 (C-5) (minor), 70.2 (C-5) (major), 74.0 (H-C≡C-) (major), 74.2 (H-C≡C-) (minor), 76.4 (C-4) (minor), 76.7 (C-4) (major), 78.6 (H-C≡C-) (minor), 78.9 (H-C≡C-) (major), 81 .5 (C-2) (major), 81 .8 (C-2) (minor), 81.9 (C-3) (minor), 82.9 (C-3) (major), 92.6 (EtO-CH) (mixture), 97.9 (C-1 ) (minor), 98.0 (C-1 ) (major), 169.6 (C=0) (major), 169.9 (C=0) (minor). Elemental analysis: Calculated: C: 54.21 ; H: 7.28. Found: C: 54.17 ; H: 7.13.
ESI-MS (pos. mode): m/z = 355 [M+Na]+.
IR (film) v (cm“1): 1752 (C=0), 3266 (≡C-H). Preparation of 4-0-(1′-ethoxy-2′-propyn-1,-yl)-1 ,2,3-tri-0-methyl-a-D-gluco- pyranosiduronic acid (Via)

To a solution of compound Va (1.12 g, 3.37 mmol, 1.0 eq.) in EtOH/H20 (3:1 , 100 mL) was added sodium hydroxide (156 mg, 3.90 mmol, 1 .3 eq.). After 5 hours stirring at room temperature the solvent was evaporated. The residue obtained was dissolved in water (50 mL). The pH of the aqueous layer was reduced to 2-3 with a 5% citric acid aqueous solution, then the layer was saturated with sodium chloride before extraction with dichloromethane (10 x 20 mL). If necessary the pH was adjusted by addition of more citric acid aqueous solution. The combined organics were dried (MgS04), filtered and removed under vacuum. Compound Via was obtained without further purification as a colourless oil (1.020 g, 3.20 mmol, 95%), in a mixture of diastereomers (75:25) (the relative composition of the mixture was determined by 1H NMR from integrations of protons EtO-CH).
1H NMR (CDCIs, 400 MHz): δ 1.16-1.24 (m, 3H, -OCH2CH3) (diastereomeric mixture), 2.59 (d, J = 1 .6 Hz, 1 H, H-C≡C-) (major), 2.62 (s I, 1 H, H-C≡C-) (minor), 3.25-3.33 (m, 1 H, H- 2), 3.44 (s, 3H, -OCH3) (mixture), 3.51 (s, 3H, -OCH3) (mixture), 3.62 (s, 3H, -OCH3) (mixture), 3.54-3.62 (m, 2H, H-3, -OCHaHbCH3) (mixture), 3.68-3.77 (m, 1 H, -OCHaHbCH3) (mixture), 3.81-3.87 (m, 1 H, H-4) (mixture), 4.13-4.18 (m, 1 H, H-5) (mixture), 4.88-4.90 (m, 1 H, H-1 ), 5.45 (s I, 1 H, EtO-CH) (minor), 5.63 (d, J = 1.6 Hz, 1 H, EtO-CH) (major).
13C NMR (CDCI3, 100.6 MHz): δ 14.9 (-OCH2CH3) (mixture), 55.9 (-OCH3) (mixture), 59.2 (-OCH3) (minor), 59.3 (-OCH3) (major), 60.7 (-OCH2CH3) (mixture), 61 .2 (-OCH3) (minor), 61.3 (-OCH3) (major), 70.1 (C-5) (mixture), 74.3 (H-OC-) (major), 74.8 (H-OC-) (minor), 75.7 (C-4) (minor), 76.4 (C-4) (major), 78.5 (H-C≡C-) (minor), 78.8 (H-C≡C-) (major), 81.4 (C-2) (major), 81 .7 (C-2) (minor), 81 .8 (C-3) (minor), 82.9 (C-3) (major), 92.5 (EtO-CH) (mixture), 97.8 (C-1 ) (minor), 98.0 (C-1 ) (major), 173.8 (C=0) (major), 174.0 (C=0) (minor).
ESI-MS (pos. mode): m/z = 341 [M+Na]+. IR (film) v (cm“1): 1751 (C=0), 3268 (≡C-H).
Preparation of Methyl-4,7-anhydro-6-deoxy-6-methylene-7-ethoxy-2,3-di-0-methyl- a-L-/‘d -heptopyranoside (Vila)

To a solution of compound Via (1.89 g, 5.92 mmol, 1 .0 eq.) in anhydrous THF (40 mL) and cooled to 0°C, were added IBCF (0.84 mL, 6.48 mmol, 1.1 eq.) and N- methylmorpholine (0.72 mL, 6.55 mmol, 1.1 eq.). After 20 minutes stirring the flask was covered with aluminium foil, 2-mercaptopyridine /V-oxide sodium salt (1 .77 g, 1 1.80 mmol, 2.0 eq.) was added and the reaction mixture was stirred at ambient temperature. After 2 hours, anhydrous THF (80 mL) then ie f-butylthiol (0.28 mL, 2.61 mmol, 1.6 eq.) were added. The aluminium foil was removed and the reaction mixture was irradiated and heated 30 minutes with a UV lamp (300W). The thiol excess was neutralized with a NaOCI aqueous solution (13% v/v, 10 mL). The reaction mixture was concentrated then dissolved in EtOAc (100 mL), washed successively with a 5% NaHC03 aqueous solution (2 x 15 mL), a 5 % citric acid aqueous solution (1 x 15 mL) and brine (1 x 25 mL), then the aqueous layer was extracted with dichloromethane (2 x 20 mL). The combined organics were dried (MgS04), filtered and concentrated. Column chromatography (gradient dichloromethane/ethyl acetate 95:5 – 75:25) afforded compound Vila as a colourless oil (218 mg, 0.79 mmol, 48%), in a mixture of diastereomers (67:33) (the relative composition of the mixture was determined by 1H NMR from integrations of protons H-2).
1H NMR (CDCIs, 400 MHz): δ 1.23 (t, J = 7.1 Hz, 3H, -OCH2CH3) (diastereomeric mixture), 3.13 (dd, J2-3 = 9.6 Hz, J1-2 = 3.0 Hz, 1 H, H-2) (minor), 3.30 (dd, J2-3 = 5.0 Hz, J1-2 = 1.6 Hz, 1 H, H-2) (major), 3.41 (s, 3H, -OCH3) (minor), 3.47 (s, 3H, -OCH3) (major), 3.50 (s, 3H, -OCH3) (mixture), 3.53 (s, 3H, -OCH3) (major), 3.55-3.61 (m, 1 H, -OCHaHbCH3) (mixture), 3.72 (dd, J2-3 = 5.0 Hz, J3-4 = 2.8 Hz, 1 H, H-3) (major), 3.78-3.90 (m, 1 H, – OCHaHbCH3) (mixture), 3.93-4.07 (m, 2H, H-3, H-4) (mixture), 4.59 (d I, J4-5 = 4.0 Hz, 1 H, H-5) (major), 4.62 (d, J1-2 = 1.7 Hz, 1 H, H-1 ) (major), (td, J4-5 = 7.9 Hz, J = 2.6 Hz, 1 H, H- 5), (minor), 4.79 (d, J1-2 = 3.0 Hz, 1 H, H-1 ) (minor), 5.35-5.57 (m, 3H, H-7, -C=CH2) (mixture). 13C NMR (CDCI3, 100.6 MHz): δ 15.3 (-OCH2CH3) (minor), 15.4 (-OCH2CH3) (major), 56.5 (-OCH3) (minor), 56.8 (-OCH3) (major), 58.6 (-OCH3) (major), 59.1 (-OCH3) (minor), 59.9 (- OCH3) (major), 60.3 (-OCH3) (minor), 63.3 (-OCH2CH3) (minor), 63.8 (-OCH2CH3) (major), 74.2 (C-5) (minor), 74.7 (C-5) (major), 76.4 (C-3) (major), 77.0 (C-4) (major), 77.7 (C-2) (major), 79.0 (C-3) (minor), 79.7 (C-4) (minor), 80.1 (C-2) (minor), 99.3 (C-1 ) (major), 99.5 (C-1 ) (minor), 102.2 (C-7) (major), 103.0 (C-7) (minor), 1 1 1.6 (-C=CH2) (minor), 1 15.2 (- C=CH2) (major), 147.4 (C-6) (minor), 148.1 (C-6) (major).
ESI-MS (pos. mode): m/z = 297 [M+Na]+.
Preparation of Methyl-4,7-anhydro-7-ethoxy-2,3-di-0-methyl-a-L-/ o-hepto- pyranosid-6-ulose (Villa)

Through a solution of compound Vila (449 mg, 1 .64 mmol, 1.0 eq.) in anhydrous dichloromethane (10 ml_), under an argon atmosphere and cooled to -78°C, was bubbled ozone (0.2 L/min, 1 10 V). When the solution had turned dark blue, oxygen was bubbled through in order to remove the excess ozone. When the solution became colorless dimethylsulfide (5 drops) was added and the solution was brought to room temperature. After 1 h15 the reaction mixture was concentrated. Column chromatography (gradient dichloromethane/ethyl acetate 95:5 – 80:20) afforded compound Villa as a white solid (364 mg, 1.32 mmol, 80%), in a mixture of diastereomers (79:21 ) (the relative composition of the mixture was determined by 1H NMR from integrations of protons H-2).
1H NMR (CDCI3, 400 MHz): δ 1.24-1 .28 (m, 3H, -OCH2CH3) (diastereomeric mixture), 3.10 (dd, J2-3 = 10.2 Hz, J1-2 = 2.9 Hz, 1 H, H-2) (minor), 3.17 (dd, J2-3 = 9.4 Hz, J1-2 = 2.8 Hz, 1 H, H-2) (major), 3.38 (s, 3H, -OCH3) (major), 3.42 (s, 3H, -OCH3) (minor), 3.50 (s, 3H, – OCH3) (mixture), 3.63 (s, 3H, -OCH3) (major), 3.66 (s, 3H, -OCH3) (minor), 3.48-3.73 (m, 3H, H-3 major, -OCHaHbCH3 major, -OCHaHbCH3 minor), 3.77-3.95 (m, 2H, -OCHaHbCH3 minor, -OCHaHbCH3 major), 4.07 (dd, J2-3 = 10.2 Hz, J3-4 = 7.7 Hz, 1 H, H-3) (minor), 4.34 (d, J4-5 = 9.1 Hz, 1 H, H-5) (minor), 4.39-4.44 (m, 2H, H-4 minor, H-5 major), 4.50 (dd, J3-4 = 9.5 Hz, J4-5 = 6.2 Hz, 1 H, H-4) (major), 4.76 (d, Ji-2 = 2.8 Hz, 1 H, H-1 ) (major), 4.79 (d, J1-2 = 2.9 Hz, 1 H, H-1 ) (minor), 4.89 (d, J = 1 ,1 Hz, 1 H, H-7) (minor), 4.93 (s I, 1 H, H-7) (major).
13C NMR (CDCI3, 100.6 MHz): δ 15.1 (-OCH2CH3) (minor), 15.2 (-OCH2CH3) (major), 56.7 (-OCH3) (minor), 57.2 (-OCH3) (major), 59.3 (-OCH3) (mixture), 59.8 (-OCH3) (major), 60.6 (-OCH3) (minor), 65.0 (-OCH2CH3) (minor), 65.5 (-OCH2CH3) (major), 70.2 (C-5) (major), 72.4 (C-5) (minor), 75.9 (C-4) (major), 79.2 (C-4) (minor), 79.4 (C-3) (major), 79.8 (C-2 major, C-3 minor), 80.2 (C-2) (minor), 96.1 (C-7) (major), 97.2 (C-7) (minor), 98.7 (C-1 ) (major), 99.0 (C-1 ) (minor), 205.3 (C-6) (minor), 205.6 (C-6) (major).
IR (film) v (cm“1): 1783 (C=0).
ESI-MS (pos. mode): m/z = 299 [M+Na]+, 331 [M+Na+MeOH]+.
Preparation of Methyl methyl-2,3-di-0-methyl-a-L-idopyranosiduronate (IXa)
; CHCI3)

To a solution of compound Villa (50 mg, 0.18 mmol, 1 .0 eq.) in dichloromethane (3 mL), under an argon atmosphere and cooled to 0°C, were added m-CPBA (77%, 120 mg, 0.54 mmol, 3.0 eq.) and NaHC03 (20 mg, 0.23 mmol, 1 .3 eq.). After 3 hours stirring the solvent was removed under vacuum. The resulting residue was dissolved in EtOAc (30 mL), extracted with distilled water (2 x 10 mL), and the aqueous phase was concentrated. The crude mixture was dissoved in methanol (10 mL), para-toluenesulfonic acid monohydrate was added (4 mg, 0.02 mmol, 0.1 eq.) then the reaction mixture was heated to reflux and the reaction monitored by 1H NMR in deuterated methanol. After 8 hours the solvent was evaporated. The residue obtained was dissolved in DMF (5 mL) then triethylamine (28 μί, 0.20 mmol, 1 .1 eq.) and methyl iodide (56 μί, 0.90 mmol, 5 eq.) were added. After 3h30 the reaction mixture was concentrated, dissolved in EtOAc (30 mL) and the organic phase was washed with a 5% NaHC03 aqueous solution (2 x 10 mL), a 5% citric acid aqueous solution (2 x 10 mL) and brine (1 x 10 mL). The aqueous phase was extracted with dichloromethane (5 x 10 mL) and the combined organics were dried (MgS04), filtered and concentrated. Column chromatography (dichloromethane/ethyl acetate 85:15) afforded compound Xla as a colourless oil (25 mg, 0.10 mmol, 56%). 1H NMR (CDCI3, 400 MHz): δ 3.41 (d I, J2-3 = 3.5 Hz, 1 H, H-2), 3.47 (s, 3H, -OCH3), 3.56 (s, 3H, -OCH3), 3.57 (s, 3H, -OCH3), 3.69 (t, J2-3 = J3-4 = 3.5 Hz, 1 H, H-3), 3.75-3.78 (m, 1 H, OH), 3.80 (s, 3H, -OCH3), 3.97 (m, 1 H, H-4), 4.42 (d, J4-5 = 1 .6 Hz, 1 H, H-5), 4.61 (d,
13C NMR (CDCI3, 100.6 MHz): δ 52.4, 57.5, 58.4, 60.8 (4x-OCH3), 67.7 (C-4), 74.8 (C-5), 77.2 (C-2), 77.5 (C-3), 100.9 (C-1 ), 169.6 (C=0).
Elemental analysis: Calculated: C: 48.00 ; H: 7.25. Found: C: 47.62 ; H: 7.15.
ESI-MS (pos. mode): m/z = 272 [M+Na]+.
IR (film) v (cm“1): 3491 (O-H), 1765 (C=0). Example 2 :
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(2′,3′-di-0-methyl-p-D-glucopyranosyl- uronate)-a-D-glucopyranoside (IVb)

To a solution of the co

Me
(3.279 g, 5.00 mmol, 1.0 eq.) in a water/acetonitrile mixture (1 :1 , 300 mL) were added NaBr (105 mg, 1 .02 mmol, 0.2 eq.) and TEMPO (33 mg, 0.21 mmol, 0.04 eq.). The reaction mixture was cooled with the aid of an ice bath then a solution of NaOCI (13% v/v, 1 1.5 mL, 20.08 mmol, 4.0 eq.) was added. After 3 hours stirring at 0°C, NaOCI was added anew (13% v/v, 1 1 .5 mL, 20.08 mmol, 4.0 eq.). After two more hours ethanol was added (96% v/v, 20 mL), then the pH was reduced to 2-3 by addition of HCI (10% v/v). The solvent was evaporated and the residue obtained was suspended in DMF (40 mL) then triethylamine (2.8 mL, 2.032 g, 20.0 mmol, 4.0 eq.) and methyl iodide (6.2 mL, 14.136 g, 99.6 mmol, 20.0 eq.) were added. After 4 hours stirring at room temperature the solvent was evaporated and the residue obtained was dissolved in EtOAc (200 mL). The organic layer was washed with a 5% citric acid aqueous solution (1 χ 20 mL) and brine (1 χ 20 mL). The aqueous layer was extracted with dichloromethane (2 χ 20 mL). The combined organics were dried (MgS04), filtered and evaporated. The residue obtained was dissolved in DMF (20 mL), then triethylamine (1.4 mL, 1 .016 g, 10.0 mmol, 2.0 eq.) and methyl iodide (3.1 mL, 7.068 g, 49.8 mmol, 10.0 eq) were added. After 60 hours stirring at room temperature the solvent was evaporated and the residue obtained was dissolved in EtOAc (200 mL). The organic layer was washed with a 5% citric acid aqueous solution (2 x 20 mL) and brine (1 χ 20 mL). The organic layer was dried (MgS04), filtered and evaporated. Column chromatography (gradient hexane/ethyl acetate 80:20 – 50:50) gave compound IVb as a colourless oil which was dissolved in a diethyl ether/hexane mixture and evaporated at room temperature to afford a white solid (2.500 g, 3.66 mmol, 73%).
1H NMR (CDCI3, 400 MHz): δ 2.92 (d, 1 H), 2.93-2.98 (m, 2H), 3.41 (s, 3H), 3.48-3.76 (m, 5H), 3.51 (s, 3H), 3.60 (s, 3H), 3.63 (s, 3H), 3.87-3.98 (m, 3H), 4.36-4.41 (m, 1 H), 4.49- 5.08 (m, 6H), 4.61 (d, 1 H), 7.24-7.42 (m, 15H).
Elemental analysis: Calculated: C: 65.09 ; H: 6.79. Found: C: 65.29 ; H: 6.96.
ESI-MS (pos. mode): m/z = 705 [M+Na]+.
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′-0-(1 “-ethoxy-2”-propyn-1 “-yl)-2′,3′- di-0-methyl-p-D-glucopyranosyluronate)-a-D-glucopyranoside (Vb)

E F To a solution of compound IVb (385 mg, 0.56 mmol, 1 .0 eq.) in chloroform (stabilised with amylene, 30 mL) were added, under an argon atmosphere, P205 (410 mg, 2.80 mmol, 5.0 eq.) and propargylaldehyde diethylacetal (0.4 mL, 2.79 mmol, 5.0 eq.), then the reaction mixture was heated at reflux. After 5 hours stirring, the reaction mixture was filtered through a pad of Celite® then the solvent was removed under vacuum. The crude mixture was suspended in EtOAc (60 mL), washed with a 5% NaHC03 aqueous solution (1 x 15 mL) and brine (1 x 15 mL). The organic layer was dried (MgS04), filtered, and evaporated. Column chromatography (gradient hexane/ethyl acetate 90:10 – 70:30) afforded compound Vb as a colourless oil (275 mg, 0.36 mmol, 64%) in a diastereomeric mixture (64:36).
1H NMR (CDCI3, 250 MHz): δ 1.17-1 .27 (m, 3H), 2.55 (d, 0.36H), 2.57 (d, 0.64H), 2.92- 3.08 (m, 2H), 3.38 (s, 3H), 3.49 (s, 1.92H), 3.50 (s, 1.08H), 3.57 (s, 1.08H), 3.59 (s, 1.92H), 3.60 (s, 1.92H), 3.62 (s, 1.08H), 3.44-3.97 (m, 10H), 4.35 (t, 1 H), 4.46-4.76 (m, 6H), 5.03 (d, 1 H), 5.32 (d, 0.36H), 5.56 (d, 0.64H), 7.21-7.42 (m, 15H).
Elemental analysis: Calculated: C: 65.95 ; H: 6.85. Found: C: 65.92 ; H: 6.75.
ESI-MS (pos. mode): m/z = 787 [M+Na]+.
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′-0-(1 “-ethoxy-2″-propyn-1 ” 1 ‘,2′,3’-tri-0-methyl-a-D-glucopyranosiduronic acid)-a-D-glucopyranoside (Vlb)

E F
To a solution of compound Vb (1.02 g, 1.33 mmol, 1.0 eq.) in EtOH/H20 (1 :1 , 100 mL) was added sodium hydroxide (82 mg, 2.05 mmol, 1.5 eq.). After 3 hours stirring at room temperature sodium hydroxide was added anew (27 mg, 0.68 mmol, 0.5 eq.). After an additional hour stirring the solvent was evaporated. The residue obtained was dissolved in water (40 mL). The pH of the aqueous layer was reduced to 2-3 with a 10% HCI aqueous solution then the layer was saturated with sodium chloride before extraction with dichloromethane (3 x 20 mL). The combined organics were dried (MgS04), filtered and removed under vacuum. Compound VIb was obtained without further purification as a white solid (930 mg, 1.24 mmol, 93%), in a diastereomeric mixture (63:37).
1H NMR (CDCIs, 400 MHz): δ 1.17-1.28 (m, 3H), 2.65 (d, 0.63H), 2.68 (d, 0.37H), 2.90- 3.09 (m, 2H), 3.37 (s, 3H), 3.46 (s, 1.1 1 H), 3.57 (s, 1.89H), 3.58 (s, 1.1 1 H), 3.60 (s, 1.89H), 3.42-3.90 (m, 10H), 4.29 (d, 1 H), 4.46-4.97 (m, 7H), 5.44 (d, 0.37H), 5.61 (d, 0.63H), 7.28-7.44 (m, 15H).
ESI-MS (pos. mode): m/z = 773 [M+Na]+ , 795 [M-H+2Na]+ . ESI-MS (neg. mode): m/z = 749 [M-H]\
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′,7′-anhydro-6′-deoxy-6′-methylene-7′- ethoxy-2 3′-di-0-methyl-α-L-/‘ o-heptopyranosyl)-α-D-glucopyranoside (Vllb)

To a solution of compound VIb (647 mg, 0.86 mmol, 1.0 eq.) in anhydrous THF (20 mL) and cooled to 0°C, were added IBCF (0.1 1 mL, 0.85 mmol, 1.0 eq.) and N- methylmorpholine (0.10 mL, 0.91 mmol, 1.1 eq.). After 10 minutes stirring, the flask was covered with aluminium foil, 2-mercaptopyridine /V-oxide sodium salt (512 mg, 3.43 mmol, 4.0 eq.) was added and the reaction mixture was stirred at ambient temperature. After 20 minutes anhydrous THF (100 mL) then ie f-butylthiol (0.18 mL, 1 .68 mmol, 2.0 eq.) were added. The aluminium foil was removed and the reaction mixture was irradiated and heated 15 minutes with a UV lamp (300W). The thiol excess was neutralized with a NaOCI aqueous solution (13%, 10 mL). The reaction mixture was concentrated then dissolved in EtOAc (100 mL), washed successively with a 5% NaHC03 aqueous solution (1 x 15 mL), a 5 % citric acid aqueous solution (1 x 15 mL) and brine (1 x 15 mL), then the aqueous layer was extracted with dichloromethane (2 x 20 mL). The combined organics were dried (MgS04), filtered and concentrated. Column chromatography (gradient hexane/ethyl acetate 90 :10 – 70:30) afforded compound Vllb as a colourless oil (251 mg, 0.36 mmol, 42%) in a mixture of diastereomers (61 :39).
1H NMR (CDCI3, 250 MHz): δ 1.18-1.29 (m, 3H), 2.90-3.07 (m, 1 H), 3.37 (s, 1.17H), 3.38 (s, 1 .83H), 3.46 (s, 3H), 3.54 (s, 1.83H), 3.60 (s, 1 .17H), 3.29-4.00 (m, 10H), 4.1 1 -4.26 (m, 1 H), 4.50-4.96 (m, 8H), 5.09-5.48 (m, 3H), 7.23-7.39 (m, 15H).
ESI-MS (pos. mode): m/z = 720 [M+Na]+ .
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′,7′-anhydro-7′-ethoxy-2′,3′-di-0- methyl-a-L-/‘ o-heptopyranosid-6′-ulosyl)-a-D-glucopyranoside (Vlllb)

Through a solution of compound Vllb (145 mg, 0.21 mmol, 1 .0 eq.) in anhydrous dichloromethane (10 mL), under an argon atmosphere and cooled to -78°C, was bubbled ozone (0.2 L/min, 1 10 V). When the solution had turned dark blue, oxygen was bubbled through in order to remove the excess ozone. When the solution became colorless dimethylsulfide (4 drops) was added and the solution was brought to room temperature. After 30 min stirring the reaction mixture was concentrated. Column chromatography (gradient hexane/ethyl acetate 90:10 – 60:40) afforded compound Vlllb as a white solid (100 mg, <67%) in a mixture of diastereomers (67:33).
1H NMR (CDCI3, 400 MHz): δ 1.21 -1 .28 (m, 3H), 2.93-3.07 (m, 1 H), 3.31-4.26 (m, 20H), 4.52-5.02 (m, 9H), 7.20-7.45 (m, 15H).
ESI-MS (pos. mode): m/z = 731 [M+Na]+ .
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(methyl 2′,3′-di-0-methyl-a-L- idopyranosiduronate)-a-D-glucopyranoside (IXb)

To a solution of compound Vlllb (62 mg, 87 μηηοΙ, 1 .0 eq.) in dichloromethane (5 mL), under an argon atmosphere and cooled to 0°C, were added m-CPBA (77%, 58 mg, 259 μηηοΙ, 3.0 eq.) and NaHC03 (1 1 mg, 130 μηηοΙ, 1 .5 eq.). After 5 hours stirring the solvent was removed under vacuum. The reaction mixture was then dissolved in EtOAc (50 mL) and washed successively with a 5% NaHC03 aqueous solution (1 x 10 mL), a 5 % citric acid aqueous solution (1 x 10 mL) and brine (1 x 10 mL). The organic layer was dried (MgS04), filtered and concentrated. The crude mixture was dissolved in anhydrous methanol (10 mL) and sodium methoxide was added to reach pH = 10. After 30 minutes stirring at room temperature the reaction mixture was neutralized with Dowex®, filtered through a pad of Celite®, and concentrated. The residue obtained was dissolved in DMF (10 mL) then triethylamine (13 μί, 93 μηηοΙ, 1.1 eq.) and methyl iodide (27 μί, 434 μηηοΙ, 5.0 eq.) were added. After 2h30 stirring the reaction mixture was concentrated, dissolved in EtOAc (40 mL) and washed with a 5% citric acid aqueous solution (2 x 10 mL), a 5% NaHC03 aqueous solution (2 x 10 mL), and brine (1 x 10 mL). The organic layer was dried (MgS04), filtered and concentrated. Column chromatography (gradient hexane/ethyl acetate 60:40-50:50) afforded compound IXb as a colourless oil (12 mg, 18 μηηοΙ, 20% over two steps).
1H NMR (CDCIs, 400 MHz): δ 3.23 (s, 3H), 3.20-3.25 (m, 1 H), 3.36 (s, 3H), 3.43 (s, 3H), 3.46 (s, 3H), 3.33-3.58 (m, 3H), 3.60-3.69 (m, 2H), 3.72-3.95 (m, 4H), 4.53-4.60 (m, 4H), 4.68-4.97 (m, 4H), 5.14 (s, 1 H), 7.24-7.37 (15H).
ESI-MS (pos. mode): m/z = 705 [M+Na]+ .
……………
Volume 69, Issue 15, 15 April 2013, Pages 3149–3158
http://www.sciencedirect.com/science/article/pii/S0040402013003025
Abstract
Idraparinux, the fully O-sulfated, O-methylated, heparin-related pentasaccharide possessing selective factor Xa inhibitory activity, was prepared by two novel synthetic pathways. Each route was based on a 2+3 block synthesis utilizing the same l-iduronic acid-containing trisaccharide acceptor, which was glycosylated with either a glucuronide disaccharide donor or its non-oxidized precursor. The latter route, involving the oxidation of the glucose unit into d-glucuronic acid at a pentasaccharide level proved to be much more efficient, providing the target pentasaccharide in a reasonable overall yield.
Graphical abstract

- ……………………………
- SYNTHESIS
- US 20120041189 A1,
- http://www.patexia.com/us-publications/20120041189
- EXAMPLE 1Preparation of the Compound of Formula (I) in Crystalline Form (Scheme 1)
1.1: Preparation of the Compound of Formula (I′)
The compound of formula (I″) is obtained, for example, according to the teaching of patent EP 0 529 715 B1 or of the articles “Bioorg. Med. Chem.” (1994, Vol. 2, No. 11, pp. 1267-1280), “Bioorg. Med. Chem. Letters” (1992, Vol. 2, No. 9, pp. 905-910) or “Magnetic Resonance in Chemistry” (2001, Vol. 39, pp. 288-293). The compound of formula (I″) (5 g, 3.06 mmol) is dissolved in acetonitrile (10 mL). Deionized water (12.2 mL) and aqueous 30% sodium hydroxide solution (4.1 g) are then added. The mixture is heated to 40° C. and maintained at this temperature for 5 hours. The reaction medium is then cooled to 20° C. and acidified to pH 6.25 with aqueous 1N hydrochloric acid solution (about 17.7 g) before extraction with MTBE of certain impurities, the saponified product remaining in the aqueous phase. The residual acetonitrile, contained in the aqueous phase, is then removed by concentration, followed by diluting with deionized water (125 mL). The saponified product is finally precipitated at pH 1.5 by adding aqueous 1N hydrochloric acid solution (about 17.6 g) at 20° C. The suspension is maintained for 4 hours at 20° C. before filtration. The wet solid is finally dried in a vacuum oven at 30° C. to give 2.93 g (93.6%) of compound of formula (I).
NMR (anomeric protons of the saccharide units D, E, F, G, H): 5.79, 5.14, 5.55, 5.92, 4.94 ppm.
1.2 Preparation of the Crude Compound of Formula (I)
The compound of formula (I′) obtained after the preceding step is dissolved in tetrahydrofuran (18 mL). Palladium-on-charcoal (0.3 g) is added. The reaction medium is hydrogenated at 0.3 bar of hydrogen (relative pressure) for 4 hours. After filtering and evaporating, 2.12 g (99%) of the crude compound of formula (I) are obtained.
1.3: Preparation of the Compound of Formula (I) in Crystalline Form Using an Isopropanol/MTBE Mixture
The crude hydrogenated product obtained after the preceding step is dissolved in isopropanol (13 mL) at 65° C., and then crystallized at room temperature. The suspension is then cooled to 40° C., followed by addition of MTBE (13 mL), and is then cooled slowly to 10° C. After maintenance at 10° C. for 2 hours, the crystalline hydrogenated product is filtered off, washed and dried. 1.66 g of the compound of formula (I) in crystalline form are thus obtained, in the form of a cream-white powder. The reaction yield for the production of the compound of formula (I) in crystalline form, from the compound of formula (I′), is 92.5%. When expressed relative to the starting compound (I″), the reaction yield for the production of the compound of formula (I) in crystalline form is 86.6%.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (I) in crystalline form: 5.77, 5.11, 5.51, 5.84, 5.01 ppm.
1.4: Preparation of the Compound of Formula (I) in Crystalline Form Using Isopropanol
The crude hydrogenated product obtained after step 1.2 is dissolved in isopropanol (5 volumes) at 75° C. The medium is then cooled slowly until crystals appear, according to the known standard techniques for crystallization. The process is performed, for example, by a first step of cooling at 65° C. for 1 hour, and than a second step of cooling to a final temperature of 25° C. over 4 hours or of 5° C. over 6 hours, and finally maintenance at this final temperature for 30 minutes. The suspension is then filtered and rinsed with isopropanol (2×0.1 V) and compound (I) is isolated in the form of white crystals, which appear under a microscope in the form of needles. The 1H NMR analysis of these crystals is identical to that described after step 1.3 above.
EXAMPLE 4
Preparation of Idraparinux from the Compound of Formula (I) in Crystalline Form (Scheme 2)
The preparation of idraparinux (II) from the compound of formula (I) is summarized in Scheme 2.
The compound of formula (I) in crystalline form, as obtained according to Example 1.3, is dissolved in N,N’-dimethylformamide (6.6 mL) and then heated to 30.degree. C. Under an inert atmosphere, 3.8 g of pyridine-sulfur trioxide complex are added slowly, followed by maintenance at 30.degree. C. for 4 hours. The reaction medium is then poured into aqueous 23.8% sodium hydrogen carbonate solution (16.3 g) maintained at a maximum of 25.degree. C., to obtain the compound of formula (II). The reaction medium is kept stirring for hours. The solution of sulfated product is then poured onto an MTBE/isopropanol/ethanol mixture (171 mL/70 mL/70 mL). Precipitation of the product is observed, and, after filtering off, washing and drying the cake, 4.99 g (96.8%) of compound of formula (II) are obtained, and are then purified by anion-exchange chromatography according to the usual techniques.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (II): 5.48, 4.68, 5.44, 5.08, 5.18 ppm.It thus appears that the process according to the invention makes it possible to obtain idraparinux (compound of formula (II)) in a chemical yield of about 84% (precisely 83.8% according to the protocols described above) starting from the compound of formula (I”), i.e. a gain in yield of about 30% relative to the process described in patent EP 0 529 715 B1.

IDRAPARINUX
References
- Bousser MG, Bouthier J, Büller HR, et al. (January 2008). “Comparison of idraparinux with vitamin K antagonists for prevention of thromboembolism in patients with atrial fibrillation: a randomised, open-label, non-inferiority trial”. Lancet 371 (9609): 315–21. doi:10.1016/S0140-6736(08)60168-3.PMID 18294998.
- Buller HR, Cohen AT, Davidson B, et al. (September 2007). “Idraparinux versus standard therapy for venous thromboembolic disease”. N. Engl. J. Med. 357 (11): 1094–104. doi:10.1056/NEJMoa064247. PMID 17855670.
- Bioorg Med Chem1994,2,(11):1267
- Drugs Fut2002,27,(7):639
- EP 0454220, JP 1992225994, US 5378829, US 5382570, US 5529985, US 5773605
- EP 0529715
-
Bioorg Med Chem Lett. 2009 Jul 15;19(14):3875-9. doi: 10.1016/j.bmcl.2009.03.155. Epub 2009 Apr 5.
- Chemistry – A European Journal, 2012 , vol. 18, 34 p. 10643 – 10652
- Magnetic Resonance in Chemistry, 2001 , vol. 39, 5 p. 288 – 293
- Tetrahedron, 2013 , vol. 69, 15 p. 3149 – 3158…….. MP 210-15 DEG CENT
-
- I. Capila, R.J. Linhardt
Angew. Chem., Int. Ed., 41 (2002), p. 390
- I. Capila, R.J. Linhardt
-
- L. Roden
D.A. Lane, U. Lindahl (Eds.), Chemical and Biological Properties, Clinical Applications, CRC Press, Boca Raton, FL (1989), p. 1
- L. Roden
-
- (a) C.A.A. van Boeckel, M. Petitou
Angew. Chem., Int. Ed. Engl., 32 (1993), p. 1671
- (b) M. Petitou, C.A.A. van Boeckel
Angew. Chem., Int. Ed., 43 (2004), p. 3118
- (a) C.A.A. van Boeckel, M. Petitou
-
- M. Petitou, B. Casu, U. Lindahl
Biochimie, 85 (2003), p. 83Article |
- M. Petitou, B. Casu, U. Lindahl
-
- (a) M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, J.C. Jacquinet, P. Sinay, G. Torri
Carbohydr. Res., 167 (1987), p. 67Article |
- (b) P.-A. Driguez, I. Lederman, J.-M. Strassel, J.-M. Herbert, M. Petitou
J. Org. Chem., 64 (1999), p. 9512
- (c) J. Choay, M. Petitou, J.C. Lormeau, P. Sinay, B. Casu, G. Gatti
Biochem. Biophys. Res. Commun., 116 (1983), p. 492Article |
- (d) P. Sinay, J.C. Jacquinet, M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, G. Torri
Carbohydr. Res., 132 (1984), p. C5
- (a) M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, J.C. Jacquinet, P. Sinay, G. Torri
-
- (a) P. Westerduin, C.A.A. van Boeckel, J.E.M. Basten, M.A. Broekhoven, H. Lucas, A. Rood, H. van der Heijden, R.G.M. van Amsterdam, T.G. van Dinther, D.G. Meuleman, A. Visser, G.M.T. Vogel, J.B.L. Damm, G.T. Overklift
- Bioorg. Med. Chem., 2 (1994), p. 1267Article |
 PDF (1579 K)
PDF (1579 K)
- (b) J.M. Herbert, J.P. Herault, A. Bernat, R.G.M. van Amsterdam, J.C. Lormeau, M. Petitou, C. van Boeckel, P. Hoffmann, D.G. Meuleman
Blood, 91 (1998), p. 4197
-
- (a) P. Prandoni, D. Tormene, M. Perlati, B. Brandolin, L. Spiezia
Expert Opin. Investig. Drugs, 17 (2008), p. 773
- (b) A.S. Go, D.E. Singer
Lancet, 371 (2008), p. 278Article|
- (a) P. Prandoni, D. Tormene, M. Perlati, B. Brandolin, L. Spiezia
-
- I.M. Pinilla, M.B. Martinez, J.A. Galbis
Carbohydr. Res., 338 (2003), p. 549Article |
- I.M. Pinilla, M.B. Martinez, J.A. Galbis
-
- (a) K. Yoza, N. Amanokura, Y. Ono, T. Akao, H. Shinmori, M. Takeuchi, S. Shinkai, D.N. Reinhoudt
Chem. Eur. J., 5 (1999), p. 2722
- (b) J. Elhalabi, K.G. RiceCarbohydr. Res., 335 (2001), p. 159Article| PDF (177 K)
- (a) K. Yoza, N. Amanokura, Y. Ono, T. Akao, H. Shinmori, M. Takeuchi, S. Shinkai, D.N. Reinhoudt
-
- S.D. Debenham, E.J. Toone
Tetrahedron: Asymmetry, 11 (2000), p. 385Article |
- S.D. Debenham, E.J. Toone
-
- A. Meijer, U. Ellervik
J. Org. Chem., 69 (2004), p. 6249 and references therein
- A. Meijer, U. Ellervik
-
- P. Duchaussoy, G. Jaurand, P.-A. Driguez, I. Lederman, F. Gourvenec, J.-M. Strassel, P. Sizun, M. Petitou, J.-M. Herbert
Carbohydr. Res., 317 (1999), p. 63Article |
- P. Duchaussoy, G. Jaurand, P.-A. Driguez, I. Lederman, F. Gourvenec, J.-M. Strassel, P. Sizun, M. Petitou, J.-M. Herbert
-
- J. Lee, X.-A. Lu, S.S. Kulkarni, Y. Wen, S.-C. Hung
J. Am. Chem. Soc., 126 (2004), p. 476
- J. Lee, X.-A. Lu, S.S. Kulkarni, Y. Wen, S.-C. Hung
-
- L.A.G.M. van den Broek, D.J. Vermaas, B.M. Heskamp, C.A.A. van Boeckel
Recl. Trav. Chim. Pays-Bas., 112 (1993), p. 82
- R.R. Schmidt, J. Michel
Angew. Chem., Int. Ed. Engl., 19 (1980), p. 731
- L.A.G.M. van den Broek, D.J. Vermaas, B.M. Heskamp, C.A.A. van Boeckel
-
- (a) F. Lin, W. Peng, W. Xu, X. Han, B. Yu
Carbohydr. Res., 339 (2004), p. 1219
- (b) P.L. Anelli, C. Biffi, F. Montanari, S. Quici
J. Org. Chem., 52 (1987), p. 2559
- (c) P.L. Anelli, S. Banfi, F. Montanari, S. Quici
J. Org. Chem., 54 (1989), p. 2970
- (a) F. Lin, W. Peng, W. Xu, X. Han, B. Yu
-
- (a) P. Bourhis, F. Machetto, P. Duchaussoy, J.-P. Herault, J.-M. Mallet, J.-M. Herbert, M. Petitou, P. Sinay
- For other examples of 4,6-locked idose glycosyl donors, see: Bioorg. Med. Chem. Lett., 7 (1997), p. 2843
- (b) N. Barroca, J.-C. Jacquinet
Carbohydr. Res., 337 (2002), p. 673
- (c) J. Kuszmann, G. Medgyes, S. Boros
Carbohydr. Res., 339 (2004), p. 1569
- (d) J. Tatai, P. Fugedi
Org. Lett., 9 (2007), p. 4647
| WO2002024754A1 | Sep 20, 2001 | Mar 28, 2002 | Akzo Nobel Nv | Polysaccharides with antithrombotic activity comprising at least a covalent bond with biotin or a biotin derivative |
| WO2006030104A1 * | Sep 7, 2005 | Mar 23, 2006 | Sanofi Aventis | Biotinylated hexadecasaccharides, preparation and use thereof |
| WO2007042469A2 * | Oct 6, 2006 | Apr 19, 2007 | Organon Nv | Anticoagulant antithrombotic dual inhibitors comprising a biotin label |
| EP0230023A2 * | Dec 19, 1986 | Jul 29, 1987 | Marion Merrell Dow Inc. | Pharmaceutical compositions for the enhancement of wound healing |
| EP0300099A1 * | Jul 20, 1987 | Jan 25, 1989 | Akzo N.V. | New pentasaccharides |
| EP0301618A2 * | Jul 4, 1988 | Feb 1, 1989 | Akzo N.V. | New pentasaccharides |
| EP0454220A1 * | Apr 16, 1991 | Oct 30, 1991 | Akzo Nobel N.V. | Carbohydrate derivatives comprising a trisaccharide unit |
| GB1110939A * | Title not available | |||
| US3017407 * | Aug 18, 1958 | Jan 16, 1962 | Riker Laboratories Inc | Process for producing polysulfuric acid esters of polysaccharides |


Why FDA Supports a Flexible Approach to Drug Development

We all know that just as every person is different, so too is every disease and every drug. And so we weren’t surprised by the results of a new study published in the Journal of … Continue reading →
FONDAPARINUX

FONDAPARINUX
Fondaparinux is a drug belonging to the group of the antithrombotic agents and are used to prevent deep vein thrombosis in patients undergoing orthopedic surgery. It is also used for the treatment of severe venous thrombosis and pulmonary
114870-03-0 ………..10x SODIUM SALT
| CAS number | 114870-03-0 FREE FORM |
|---|
| MF | C31H43N3Na10O49S8 10X SODIUM |
|---|---|
| MW | 1726.77 g/mol 10X SODIUM |
GSK-576428 Org-31540 SR-90107SR-90107A
launched 2002
Arixtra, Quixidar, Fondaparinux sodium, Fondaparin sodium, Arixtra (TN), Fondaparinux, Org-31540, AC1LCS4W, SR-90107A
Fondaparinux (Arixtra) is a synthetic pentasaccharide anticoagulant. Apart from the O-methyl group at the reducing end of the molecule, the identity and sequence of the five monomeric sugar units contained in fondaparinux is identical to a sequence of five monomeric sugar units that can be isolated after either chemical or enzymatic cleavage of the polymeric glycosaminoglycan heparin and heparan sulfate (HS). This monomeric sequence in heparin and HS is thought to form the high affinity binding site for the natural anti-coagulant factor, antithrombin III (ATIII).
Binding of heparin/HS to ATIII has been shown to increase the anti-coagulant activity of antithrombin III 1000-fold. Fondaparinux potentiates the neutralizing action ofATIII on activated Factor X 300-fold. Fondaparinux may be used: to prevent venous thromboembolism in patients who have undergone orthopedic surgery of the lower limbs (e.g. hip fracture, hip replacement and knee surgery); to prevent VTE in patients undergoing abdominal surgery who are are at high risk of thromboembolic complications; in the treatment of deep vein thrombosis (DVT) and pumonary embolism (PE); in the management of unstable angina (UA) and non-ST segment elevation myocardial infarction (NSTEMI); and in the management of ST segment elevation myocardial infarction (STEMI).

FONDAPARINUX
Fondaparinux (trade name Arixtra) is an anticoagulant medication chemically related to low molecular weight heparins. It is marketed byGlaxoSmithKline. A generic version developed by Alchemia is marketed within the US by Dr. Reddy’s Laboratories.
Fondaparinux is a synthetic pentasaccharide Factor Xa inhibitor. Apart from the O-methyl group at the reducing end of the molecule, the identity and sequence of the five monomeric sugar units contained in fondaparinux is identical to a sequence of five monomeric sugar units that can be isolated after either chemical or enzymatic cleavage of the polymeric glycosaminoglycans heparin and heparan sulfate (HS). Within heparin and heparan sulfate this monomeric sequence is thought to form the high affinity binding site for the anti-coagulant factor antithrombin III (ATIII). Binding of heparin/HS to ATIII has been shown to increase the anti-coagulant activity of antithrombin III 1000 fold. In contrast to heparin, fondaparinux does not inhibit thrombin.
Fondaparinux is given subcutaneously daily. Clinically, it is used for the prevention of deep vein thrombosis in patients who have had orthopedic surgery as well as for the treatment of deep vein thrombosis and pulmonary embolism.
One potential advantage of fondaparinux over LMWH or unfractionated heparin is that the risk for heparin-induced thrombocytopenia (HIT) is substantially lower. Furthermore, there have been case reports of fondaparinux being used to anticoagulate patients with established HIT as it has no affinity to PF-4. However, its renal excretion precludes its use in patients with renal dysfunction.
Unlike direct factor Xa inhibitors, it mediates its effects indirectly through antithrombin III, but unlike heparin, it is selective for factor Xa.[1]
Fondaparinux is similar to enoxaparin in reducing the risk of ischemic events at nine days, but it substantially reduces major bleeding and improves long term mortality and morbidity.[2]
It has been investigated for use in conjunction with streptokinase.[3]
Fondaparinux sodium, a selective coagulation factor Xa inhibitor, was first launched in the U.S. in 2002 by GlaxoSmithKline in a subcutaneous injection formulation for the prophylaxis of deep venous thrombosis (DVT) which may lead to pulmonary embolism in patients at risk for thromboembolic complications who are undergoing hip replacement, knee replacement, hip fracture surgery or abdominal surgery. The product is available in Japan for the treatment of acute deep venous thrombosis and acute pulmonary thromboembolism. In 2004, GlaxoSmithKline launched fondaparinux as an injection to be used in conjunction with warfarin sodium for the subcutaneous treatment of acute pulmonary embolism and DVT.
In 2007, GlaxoSmithKline received approval in the E.U. for the treatment of acute coronary syndrome (ACS), specifically unstable angina or non-ST segment elevation myocardial infarction (UA/NSTEMI) and ST-segment elevation myocardial infarction (STEMI), while in the U.S. an approvable letter was received for this indication. Currently, the drug is in clinical development at GlaxoSmithKline for the treatment of venous limb superficial thrombosis.

Fondaparinux Molecule
GlaxoSmithKline had filed a regulatory application in the E.U. seeking approval of fondaparinux sodium for the prevention of venous thromboembolic events (VTE), however; in 2008, the application was withdrawn for commercial reasons. Commercial launch in Japan for the product for the prevention of venous thromboembolism in high risk patients undergoing surgery in the abdomen took place in 2008.
In 2010, the EMA approved a regulatory application filed by GlaxoSmithKline seeking approval of a once-daily formulation of fondaparinux sodium for the treatment of adults with acute symptomatic spontaneous superficial-vein thrombosis (SVT) of the lower limbs without concomitant DVT. Product launch took place in the U.K. for this indication the same year.
The antithrombotic activity of fondaparinux is the result of antithrombin III (ATIII)-mediated selective inhibition of Factor Xa. By selectively binding to ATIII, the drug potentiates (about 300 times) the innate neutralization of Factor Xa by ATIII. Neutralization of Factor Xa, in turn, interrupts the blood coagulation cascade and thus inhibits thrombin formation and thrombus development. Fondaparinux does not inactivate thrombin (activated Factor II) and has no known effect on platelet function. At the recommended dose, no effects have been demonstrated on fibrinolytic activity or bleeding time.
Originally developed by Organon and Sanofi (formerly known as sanofi-aventis), fondaparinux sodium is currently available in approximately 30 countries. In 2004, Organon transferred its rights to the drug to Sanofi in exchange for revenues based on future sales from jointly developed antithrombotic products and in early 2005, GlaxoSmithKline also acquired the antithrombotic.
At the beginning of 2005, GlaxoSmithKline signed a two-year agreement with Adolor (acquired by Cubist in 2011) for the copromotion of fondaparinux sodium in the U.S. In Sepetember 2013, Aspen Pharmacare acquired Arixtra global rights (excluding China, India and Pakistan) from GlaxoSmithKline for the treatment of thrombosis with GlaxoSmithKline commercializing the product in Indonesia under licence from Aspen.
Chemical structure
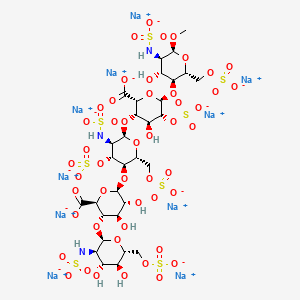
Abbreviations
- GlcNS6S = 2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside
- GlcA = β-D-glucopyranuronoside
- GlcNS3,6S = 2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl
- IdoA2S = 2-O-sulfo-α-L-idopyranuronoside
- GlcNS6SOMe = methyl-O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside
The sequence of monosaccharides is D-GlcNS6S-α-(1,4)-D-GlcA-β-(1,4)-D-GlcNS3,6S-α-(1,4)-L-IdoA2S-α-(1,4)-D-GlcNS6S-OMe, as shown in the following structure:
ARIXTRA (fondaparinux sodium) Injection is a sterile solution containing fondaparinux sodium. It is a synthetic and specific inhibitor of activatedFactor X (Xa). Fondaparinux sodium is methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-β-D-glucopyranuronosyl-( 1→4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-2-Osulfo-α-L-idopyranuronosyl-(1→4)-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside, decasodium salt.
The molecular formula of fondaparinux sodium is C31H43N3Na10O49S8 and its molecular weight is 1728. The structural formula is provided below:
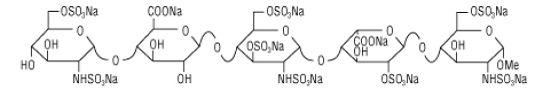
ARIXTRA is supplied as a sterile, preservative-free injectable solution for subcutaneous use.
Each single-dose, prefilled syringe of ARIXTRA, affixed with an automatic needle protection system, contains 2.5 mg of fondaparinux sodium in 0.5 mL, 5.0 mg of fondaparinux sodium in 0.4 mL, 7.5 mg of fondaparinux sodium in 0.6 mL, or 10.0 mg of fondaparinux sodium in 0.8 mL of an isotonic solutionof sodium chloride and water for injection. The final drug product is a clear and colorless to slightly yellow liquid with a pH between 5.0 and 8.0.
……………….
INTRODUCTION
In U.S. Patent No. 7,468,358, Fondaparinux sodium is described as the “only anticoagulant thought to be completely free of risk from HIT-2 induction.” The biochemical and pharmacologic rationale for the development of a heparin pentasaccharide in Thromb. Res., 86(1), 1-36, 1997 by Walenga et al. cited the recently approved synthetic pentasaccharide Factor Xa inhibitor Fondaparinux sodium. Fondaparinux has also been described in Walenga et al., Expert Opin. Investig. Drugs, Vol. 11, 397-407, 2002 and Bauer, Best Practice & Research Clinical Hematology, Vol. 17, No. 1, 89-104, 2004.
Fondaparinux sodium is a linear octasulfated pentasaccharide (oligosaccharide with five monosaccharide units ) molecule having five sulfate esters on oxygen (O-sulfated moieties) and three sulfates on a nitrogen (N- sulfated moieties). In addition, Fondaparinux contains five hydroxyl groups in the molecule that are not sulfated and two sodium carboxylates. Out of five saccharides, there are three glucosamine derivatives and one glucuronic and one L-iduronic acid. The five saccharides are connected to each other in alternate α and β glycosylated linkages (see Figure 1).
Figure 1 Fondaparinux Sodium

Monosaccharide E Monosaccharide D Monosaccharide C Monosaccharide B Monosaccharide A derived from derived from derived from derived from derived from
Monomer E Monomer D Monomer C Monomer B1 Monomer A2
Fondaparinux Sodium
Fondaparinux sodium is a chemically synthesized methoxy derivative of the natural pentasaccharide sequence, which is the active site of heparin that mediates the interaction with antithrombin (Casu et al., J. Biochem., 197, 59, 1981). It has a challenging pattern of O- and N- sulfates, specific glycosidic stereochemistry, and repeating units of glucosamines and uronic acids (Petitou et al, Progress in the Chemistry of Organic Natural Product, 60, 144-209, 1992).
The monosaccharide units comprising the Fondaparinux molecule are labeled as per the convention in Figure 1, with the glucosamine unit on the right referred to as monosaccharide A and the next, an uronic acid unit to its left as B and subsequent units, C, D and E respectively. The chemical synthesis of Fondaparinux starts with monosaccharides of defined structures that are themselves referred to as Monomers A2, Bl, C, D and E, for differentiation and convenience, and they become the corresponding monosaccharides in fondaparinux sodium.
Due to this complex mixture of free and sulfated hydroxyl groups, and the presence of N- sulfated moieties, the design of a synthetic route to Fondaparinux requires a careful strategy of protection and de-protection of reactive functional groups during synthesis of the molecule. Previously described syntheses of Fondaparinux all adopted a similar strategy to complete the synthesis of this molecule. This strategy can be envisioned as having four stages.
The strategy in the first stage requires selective de-protection of five out of ten hydroxyl groups. During the second stage these five hydroxyls are selectively sulfonated. The third stage of the process involves the de -protection of the remaining five hydroxyl groups. The fourth stage of the process is the selective sulfonation of the 3 amino groups, in the presence of five hydroxyl groups that are not sulfated in the final molecule. This strategy can be envisioned from the following fully protected pentasaccharide, also referred to as the late-stage intermediate.

In this strategy, all of the hydroxyl groups that are to be sulfated are protected with an acyl protective group, for example, as acetates (R = CH3) or benzoates (R = aryl) (Stages 1 and 2) All of the hydroxyl groups that are to remain as such are protected with benzyl group as benzyl ethers (Stage 3). The amino group, which is subsequently sulfonated, is masked as an azide (N3) moiety (Stage 4). R1 and R2 are typically sodium in the active pharmaceutical compound (e.g., Fondaparinux sodium).
This strategy allows the final product to be prepared by following the synthetic operations as outlined below: a) Treatment of the late- stage intermediate with base to hydrolyze (deprotect) the acyl ester groups to reveal the five hydroxyl groups. The two R1 and R2 ester groups are hydrolyzed in this step as well.

b) Sulfonation of the newly revealed hydroxyl groups.

c) Hydrogenation of the O-sulfated pentasaccharide to de-benzylate the five benzyl- protected hydroxyls, and at the same time, unmask the three azides to the corresponding amino groups.

d) On the last step of the operation, the amino groups are sulfated selectively at a high pH, in the presence of the five free hydroxyls to give Fondaparinux (Figure 1). While the above strategy has been shown to be viable, it is not without major drawbacks. One drawback lies in the procedure leading to the fully protected pentasaccharide (late stage intermediate), especially during the coupling of the D-glucuronic acid to the next adjacent glucose ring (the D-monomer to C-monomer in the EDCBA nomenclature shown in Figure 1). Sugar oligomers or oligosaccharides, such as Fondaparinux, are assembled using coupling reactions, also known as glycosylation reactions, to “link” sugar monomers together. The difficulty of this linking step arises because of the required stereochemical relationship between the D-sugar and the C-sugar, as shown below:

The stereochemical arrangement illustrated above in Figure 2 is described as having a β- configuration at the anomeric carbon of the D-sugar (denoted by the arrow). The linkage between the D and C units in Fondaparinux has this specific stereochemistry. There are, however, competing β- and α-glycosylation reactions.
The difficulties of the glycosylation reaction in the synthesis of Fondaparinux is well known. In 1991 Sanofi reported a preparation of a disaccharide intermediate in 51% yield having a 12/1 ratio of β/α stereochemistry at the anomeric position (Duchaussoy et al., Bioorg. & Med. Chem. Lett., 1(2), 99-102, 1991).
In another publication (Sinay et al, Carbohydrate Research, 132, C5-C9, 1984) yields on the order of 50% with coupling times on the order of 6- days are reported. U.S. Patent No. 4,818,816 {see e.g., column 31, lines 50-56) discloses a 50% yield for the β-glycosylation.
Alchemia’s U.S. Patent No. 7,541,445 is even less specific as to the details of the synthesis of this late-stage Fondaparinux synthetic intermediate. The ‘445 Patent discloses several strategies for the assembly of the pentasaccharide (1+4, 3+2 or 2+3) using a 2-acylated D-sugar (specifically 2-allyloxycarbonyl) for the glycosylation coupling reactions. However, Alchemia’s strategy involves late-stage pentasaccharides that all incorporate a 2-benzylated D- sugar.
The transformation of acyl to benzyl is performed either under acidic or basic conditions. Furthermore, these transformations, using benzyl bromide or benzyl trichloroacetimidate, typically result in extensive decomposition and the procedure suffers from poor yields. Thus, such transformations (at a disaccharide, trisaccharide, and pentasaccharide level) are typically not acceptable for industrial scale production.
Examples of fully protected pentasaccharides are described in Duchaussoy et al, Bioorg. Med. Chem. Lett., 1 (2), 99-102, 1991; Petitou et al, Carbohydr. Res., 167, 67-75, 1987; Sinay et al, Carbohydr. Res., 132, C5-C9, 1984; Petitou et al., Carbohydr. Res., 1147, 221-236, 1986; Lei et al., Bioorg. Med. Chem., 6, 1337-1346, 1998; Ichikawa et al., Tet. Lett., 27(5), 611-614, 1986; Kovensky et al, Bioorg. Med. Chem., 1999, 7, 1567-1580, 1999.
These fully protected pentasaccharides may be converted to the O- and N-sulfated pentasaccharides using the four steps (described earlier) of: a) saponification with LiOHZH2CVNaOH, b) O-sulfation by an Et3N- SO3 complex; c) de-benzylation and azide reduction via H2/Pd hydrogenation; and d) N-sulfation with a pyridine-SO3 complex.
Even though many diverse analogs of the fully protected pentasaccharide have been prepared, none use any protective group at the 2-position of the D unit other than a benzyl group. Furthermore, none of the fully protected pentasaccharide analogs offer a practical, scaleable and economical method for re-introduction of the benzyl moiety at the 2-position of the D unit after removal of any participating group that promotes β-glycosylation.
Furthermore, the coupling of benzyl protected sugars proves to be a sluggish, low yielding and problematic process, typically resulting in substantial decomposition of the pentasaccharide (prepared over 50 synthetic steps), thus making it unsuitable for a large [kilogram] scale production process.

Ref. 1. Sinay et al, Carbohydr. Res., 132, C5-C9, 1984.
Ref. 2. Petitou et al., Carbohydr. Res., 147, 221-236. 1986
It has been a general strategy for carbohydrate chemists to use base-labile ester-protecting group at 2-position of the D unit to build an efficient and stereoselective β-glycosidic linkage. To construct the β-linkage carbohydrate chemists have previously acetate and benzoate ester groups, as described, for example, in the review by Poletti et al., Eur. J. Chem., 2999-3024, 2003.
The ester group at the 2-position of D needs to be differentiated from the acetate and benzoates at other positions in the pentasaccharide. These ester groups are hydrolyzed and sulfated later in the process and, unlike these ester groups, the 2-hydroxyl group of the D unit needs to remain as the hydroxyl group in the final product, Fondaparinux sodium.
Some of the current ester choices for the synthetic chemists in the field include methyl chloro acetyl and chloro methyl acetate [MCA or CMA] . The mild procedures for the selective removal of theses groups in the presence of acetates and benzoates makes them ideal candidates. However, MCA/CMA groups have been shown to produce unwanted and serious side products during the glycosylation and therefore have not been favored in the synthesis of Fondaparinux sodium and its analogs. For by-product formation observed in acetate derivatives see Seeberger et al., J. Org. Chem., 2004, 69, 4081-93.
Similar by-product formation is also observed using chloroacetate derivatives. See Orgueira et al., Eur. J. Chem., 9(1), 140-169, 2003.
The levulinyl group can be rapidly and almost quantitatively removed by treatment with hydrazine hydrate as the deprotection reagent as illustrated in the example below. Under the same reaction conditions primary and secondary acetate and benzoate esters are hardly affected by hydrazine hydrate. See, e.g., Seeberger et al, J. Org. Chem., 69, 4081-4093, 2004.

The syntheses of Fondaparinux sodium described herein takes advantage of the levulinyl group in efficient construction of the trisaccharide EDC with improved β- selectivity for the coupling under milder conditions and increased yields.

Substitution of the benzyl protecting group with a THP moiety provides its enhanced ability to be incorporated quantitatively in position-2 of the unit D of the pentasaccharide. Also, the THP group behaves in a similar manner to a benzyl ether in terms of function and stability. In the processes described herein, the THP group is incorporated at the 2-position of the D unit at this late stage of the synthesis (i.e., after the D and C units have been coupled through a 1,2-trans glycosidic (β-) linkage). The THP protective group typically does not promote an efficient β- glycosylation and therefore is preferably incorporated in the molecule after the construction of the β-linkage.
Fondaparinux and sodium salt thereof can be prepared from pure compound of Formula II by following the teachings from Bioorganic and Medicinal Chemistry Letters, 1(2), p. 95-98 (1991). A second aspect of the present invention provides a process for the preparation of 4-0- -D-glucopyranosyl-l,6-anhydro- -D-glucopyranose, represented by STR BELOW

……………………………..
SYNTHESIS
EP2464668A2 AND US8288515
The scheme below exemplifies some of the processes of the present invention disclosed herein.

The tetrahydropyranyl (THP) protective group and the benzyl ether protective group are suitable hydroxyl protective groups and can survive the last four synthetic steps (described above) in the synthesis of Fondaparinux sodium, even under harsh reaction conditions. Certain other protecting groups do not survive the last four synthetic steps in high yield.
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:


Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; ACN is acetonitrile; MS is molecular sieves; DMF is dimethyl formamide; PMB is p-methoxybenzyl; Bn is benzyl; DCM is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; CSA is camphor sulfonic acid; TEA is triethylamine; MeOH is methanol; DMAP is dimethylaminopyridine; RT is room temperature; CAN is ceric ammonium nitrate; Ac2O is acetic anhydride; HBr is hydrogen bromide; TEMPO is tetramethylpiperidine-N-oxide; TBACl is tetrabutyl ammonium chloride; EtOAc is ethyl acetate; HOBT is hydroxybenzotriazole; DCC is dicyclohexylcarbodiimide; Lev is levunlinyl; TBDPS is tertiary-butyl diphenylsilyl; TCA is trichloroacetonitrile; O-TCA is O-trichloroacetimidate; Lev2O is levulinic anhydride; DIPEA is diisopropylethylamine; Bz is benzoyl; TBAF is tetrabutylammonium fluoride; DBU is diazabicycloundecane; BF3.Et2O is boron trifluoride etherate; TMSI is trimethylsilyl iodide; TBAI is tetrabutylammonium iodide; TES-Tf is triethylsilyl trifluoromethanesulfonate (triethylsilyl triflate); DHP is dihydropyran; PTS is p-toluenesulfonic acid.
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:


The ester moieties in EDCBA Pentamer were hydrolyzed with sodium and lithium hydroxide in the presence of hydrogen peroxide in dioxane mixing at room temperature for 16 hours to give the pentasaccharide intermediate API1. The five hydroxyl moieties in API1 were sulfated using a pyridine-sulfur trioxide complex in dimethylformamide, mixing at 60° C. for 2 hours and then purified using column chromatography (CG-161), to give the pentasulfated pentasaccharide API2. The intermediate API2 was then hydrogenated to reduce the three azides on sugars E, C and A to amines and the reductive deprotection of the five benzyl ethers to their corresponding hydroxyl groups to form the intermediate API3. This transformation occurs by reacting API2 with 10% palladium/carbon catalyst with hydrogen gas for 72 hours. The three amines on API3 were then sulfated using the pyridine-sulfur trioxide complex in sodium hydroxide and ammonium acetate, allowing the reaction to proceed for 12 hours. The acidic work-up procedure of the reaction removes the THP group to provide crude fondaparinux which is purified and is subsequently converted to its salt form. The crude mixture was purified using an ion-exchange chromatographic column (HiQ resin) followed by desalting using a size exclusion resin or gel filtration (Biorad Sephadex G25) to give the final API, fondaparinux sodium
Experimental Procedures Preparation of IntD1 Bromination of Glucose Pentaacetate
To a 500 ml flask was added 50 g of glucose pentaacetate (C6H22O11) and 80 ml of methylene chloride. The mixture was stirred at ice-water bath for 20 min HBr in HOAc (33%, 50 ml) was added to the reaction mixture. After stirring for 2.5 hr another 5 ml of HBr was added to the mixture. After another 30 min, the mixture was added 600 ml of methylene chloride. The organic mixture was washed with cold water (200 ml×2), Saturated NaHCO3(200 ml×2), water (200 ml) and brine (200 ml×2). The organic layer was dried over Na2SO4 and the mixture was evaporated at RT to give white solid as final product, bromide derivative, IntD1 (˜95% yield). C14H19BrO9, TLC Rf=0.49, SiO2, 40% ethyl acetate/60% hexanes; Exact Mass 410.02.
Preparation of IntD2 by Reductive Cyclization
To a stirring mixture of bromide IntD1 (105 g), tetrabutylammonium iodide (60 g, 162 mmol) and activated 3 Å molecular sieves in anhydrous acetonitrile (2 L), solid NaBH4 (30 g, 793 mmol) was added. The reaction was heated at 40° C. overnight. The mixture was then diluted with dichloromethane (2 L) and filtered through Celite®. After evaporation, the residue was dissolved in 500 ml ethyl acetate. The white solid (Bu4NI or Bu4NBr) was filtered. The ethyl acetate solution was evaporated and purified by chromatography on silica gel using ethyl acetate and hexane as eluent to give the acetal-triacetate IntD2 (˜60-70% yield). TLC Rf=0.36, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD3 by De-Acetylation
To a 1000 ml flask was added triacetate IntD2 (55 g) and 500 ml of methanol. After stirring 30 min, the reagent NaOMe (2.7 g, 0.3 eq) was added and the reaction was stirred overnight. Additional NaOMe (0.9 g) was added to the reaction mixture and heated to 50° C. for 3 hr. The mixture was neutralized with Dowex 50Wx8 cation resin, filtered and evaporated. The residue was purified by silica gel column to give 24 g of trihydroxy-acetal IntD3. TLC Rf=0.36 in SiO2, 10% methanol/90% ethyl acetate.
Preparation of IntD4 by Benzylidene Formation
To a 1000 ml flask was added trihydroxy compound IntD3 (76 g) and benzaldehyde dimethyl acetate (73 g, 1.3 eq). The mixture was stirred for 10 min, after which D(+)-camphorsulfonic acid (8.5 g, CSA) was added. The mixture was heated at 50° C. for two hours. The reaction mixture was then transferred to separatory funnel containing ethyl acetate (1.8 L) and sodium bicarbonate solution (600 ml). After separation, the organic layer was washed with a second sodium bicarbonate solution (300 ml) and brine (800 ml). The two sodium carbonate solutions were combined and extracted with ethyl acetate (600 ml×2). The organic mixture was evaporated and purified by silica gel column to give the benzylidene product IntD4 (77 g, 71% yield). TLC Rf=0.47, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD5 by Benzylation
To a 500 ml flask was added benzylidene acetal compound IntD4 (21 g,) in 70 ml THF. To another flask (1000 ml) was added NaH (2 eq). The solution of IntD4 was then transferred to the NaH solution at 0° C. The reaction mixture was stirred for 30 min, then benzyl bromide (16.1 ml, 1.9 eq) in 30 ml THF was added. After stirring for 30 min, DMF (90 ml) was added to the reaction mixture. Excess NaH was neutralized by careful addition of acetic acid (8 ml). The mixture was evaporated and purified by silica gel column to give the benzyl derivative IntD5. (23 g) TLC Rf=0.69, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD6 by Deprotection of Benzylidene
To a 500 ml flask was added the benzylidene-acetal compound IntD5 (20 g) and 250 ml of dichloromethane, the reaction mixture was cooled to 0° C. using an ice-water-salt bath. Aqueous TFA (80%, 34 ml) was added to the mixture and stirred over night. The mixture was evaporated and purified by silica gel column to give the dihydroxy derivative IntD6. (8 g, 52%). TLC Rf=0.79, SiO2 in 10% methanol/90% ethyl acetate.
Preparation of IntD7 by Oxidation of 6-Hydroxyl
To a 5 L flask was added dihydroxy compound IntD6 (60 g), TEMPO (1.08 g), sodium bromide (4.2 g), tetrabutylammonium chloride (5.35 g), saturated NaHCO3 (794 ml) and EtOAc (1338 ml). The mixture was stirred over an ice-water bath for 30 min To another flask was added a solution of NaOCl (677 ml), saturated NaHCO3 (485 ml) and brine (794 ml). The second mixture was added slowly to the first mixture (over about two hrs). The resulting mixture was then stirred overnight. The mixture was separated, and the inorganic layer was extracted with EtOAc (800 ml×2). The combined organic layers were washed with brine (800 ml). Evaporation of the organic layer gave 64 g crude carboxylic acid product IntD7 which was used in the next step use without purification. TLC Rf=0.04, SiO2 in 10% methanol/90% ethyl acetate.
Preparation of Monomer D by Benzylation of the Carboxylic Acid
To a solution of carboxylic acid derivative IntD7 (64 g) in 600 ml of dichloromethane, was added benzyl alcohol (30 g) and N-hydroxybenzotriazole (80 g, HOBt). After stirring for 10 min triethylamine (60.2 g) was added slowly. After stirring another 10 min, dicyclohexylcarbodiimide, (60.8 g, DCC) was added slowly and the mixture was stirred overnight. The reaction mixture was filtered and the solvent was removed under reduced pressure followed by chromatography on silica gel to provide 60.8 g (75%, over two steps) of product, Monomer D. TLC Rf=0.51, SiO2 in 40% ethyl acetate/60% hexanes.
Synthesis of the BA Dimer
Step 1. Preparation of BMod1, Levulination of Monomer B1
A 100 L reactor was charged with 7.207 Kg of Monomer B1 (21.3 moles, 1 equiv), 20 L of dry tetrahydrofuran (THF) and agitated to dissolve. When clear, it was purged with nitrogen and 260 g of dimethylamino pyridine (DMAP, 2.13 moles, 0.1 equiv) and 11.05 L of diisopropylethylamine (DIPEA, 8.275 kg, 63.9 moles, 3 equiv) was charged into the reactor. The reactor was chilled to 10-15° C. and 13.7 kg levulinic anhydride (63.9 mol, 3 equiv) was transferred into the reactor. When the addition was complete, the reaction was warmed to ambient temperature and stirred overnight or 12-16 hours. Completeness of the reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. When the reaction was complete, 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. The mixture was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L of water, 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight. The yield of the isolated syrup of BMod1 was 100%.
Synthesis of the BA Dimer
Step 2. Preparation of BMod2, TFA Hydrolysis of BMod1
A 100 L reactor was charged with 9296 Kg of 4-Lev Monomer B1 (BMod1) (21.3 mol, 1 equiv). The reactor chiller was turned to <5° C. and stirring was begun, after which 17.6 L of 90% TFA solution (TFA, 213 mole, 10 equiv) was transferred into the reactor. When the addition was complete, the reaction was monitored by TLC and HPLC. The reaction took approximately 2-3 hours to reach completion. When the reaction was complete, the reactor was chilled and 26.72 L of triethylamine (TEA, 19.4 Kg, 191.7 mole, 0.9 equiv) was transferred into the reactor. An additional 20 L of water and 20 L ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer was extracted (EtOAc layer) with 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 50:50, 80:20 (EtOAc/heptane), 100% EtOAc, 5:95, 10:90 (MeOH/EtOAc). The pure fractions were pooled and evaporated to a syrup. The yield of the isolated syrup, BMod2 was 90%.
Synthesis of the BA Dimer
Step 3. Preparation of BMod3, Silylation of BMod2
A 100 L reactor was charged with 6.755 Kg 4-Lev-1,2-DiOH Monomer B1 (BMod2) (17.04 mol, 1 equiv), 2328 g of imidazole (34.2 mol, 2 equiv) and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., then 5.22 L tert-butyldiphenylchloro-silane (TBDPS-Cl, 5.607 Kg, 20.4 mol, 1.2 equiv) was transferred into the reactor. When addition was complete, the chiller was turned off and the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (40% ethyl acetate/hexane) and HPLC. The reaction took approximately 3 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. Dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The yield of BMod3 was about 80%.
Synthesis of the BA Dimer
Step 4. Preparation of BMod4, Benzoylation
A 100 L reactor was charged with 8.113 Kg of 4-Lev-1-Si-2-OH Monomer B1 (BMod3) (12.78 mol, 1 equiv), 9 L of pyridine and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., after which 1.78 L of benzoyl chloride (2155 g, 15.34 mol, 1.2 equiv) was transferred into the reactor. When addition was complete, the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (40% ethyl acetate/heptane) and HPLC. The reaction took approximately 3 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The DCM solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). Isolated syrup BMod4 was obtained in 91% yield.
Synthesis of the BA Dimer
Step 5. Preparation of BMod5, Desilylation
A 100 L reactor was charged with 8.601 Kg of 4-Lev-1-Si-2-Bz Monomer B1 (BMod4) (11.64 mol, 1 equiv) in 30 L terahydrofuran. The reactor was purged with nitrogen and chilled to 0° C., after which 5.49 Kg of tetrabutylammonium fluoride (TBAF, 17.4 mol, 1.5 equiv) and 996 mL (1045 g, 17.4 mol, 1.5 equiv) of glacial acetic acid were transferred into the reactor. When the addition was complete, the reaction was stirred at ambient temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. The reaction took approximately 6 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20 (EtOAc/heptane) and 200 L 100% EtOAc. Pure fractions were pooled and evaporated to a syrup. The intermediate BMod5 was isolated as a syrup in 91% yield.
Synthesis of the BA Dimer
Step 6: Preparation of BMod6, TCA Formation
A 100 L reactor was charged with 5.238 Kg of 4-Lev-1-OH-2-Bz Monomer B1 (BMod5) (10.44 mol, 1 equiv) in 30 L of DCM. The reactor was purged with nitrogen and chilled to 10-15° C., after which 780 mL of diazabicyclo undecene (DBU, 795 g, 5.22 mol, 0.5 equiv) and 10.47 L of trichloroacetonitrile (TCA, 15.08 Kg, 104.4 mol, 10 equiv) were transferred into the reactor. Stirring was continued and the reaction was kept under a nitrogen atmosphere. After reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by HPLC and TLC (40:60 ethyl acetate/heptane). The reaction took approximately 2 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of dichloromethane were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was separated with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/Heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of BMod6 was 73%.
Synthesis of the BA Dimer
Step 7. Preparation of AMod1, Acetylation of Monomer A2
A 100 L reactor was charged with 6.772 Kg of Monomer A2 (17.04 mole, 1 eq.), 32.2 L (34.8 Kg, 340.8 moles, 20 eq.) of acetic anhydride and 32 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C. When the temperature reached −20° C., 3.24 L (3.63 Kg, 25.68 mol, 1.5 equiv) of boron trifluoride etherate (BF3.Et2O) was transferred into the reactor. After complete addition of boron trifluoride etherate, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 3-5 hours for completion. When the reaction was complete, extraction was performed with 3×15 L of 10% sodium bicarbonate and 20 L of water. The organic phase (DCM) was evaporated to a syrup (bath temp. 40° C.) and allowed to dry overnight. The syrup was purified in a 200 L silica column using 140 L each of the following gradient profiles: 5:95, 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of AMod1 was 83%.
Synthesis of the BA Dimer
Step 8. Preparation of AMod3,1-Methylation of AMod1
A 100 L reactor was charged with 5891 g of acetyl Monomer A2 (AMod1) (13.98 mole, 1 eq.) in 32 L of dichloromethane. The reactor was purged with nitrogen and was chilled to 0° C., after which 2598 mL of trimethylsilyl iodide (TMSI, 3636 g, 18 mol, 1.3 equiv) was transferred into the reactor. When addition was complete, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 2-4 hours to reach completion. When the reaction was complete, the mixture was diluted with 20 L of toluene. The solution was evaporated to a syrup and was co-evaporated with 3×6 L of toluene. The reactor was charged with 36 L of dichloromethane (DCM), 3.2 Kg of dry 4 Å Molecular Sieves, 15505 g (42 mol, 3 equiv) of tetrabutyl ammonium iodide (TBAI) and 9 L of dry methanol. This was stirred until the TBAI was completely dissolved, after which 3630 mL of diisopropyl-ethylamine (DIPEA, 2712 g, 21 moles, 1.5 equiv) was transferred into the reactor in one portion. The completion of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 16 hours for completion. When the reaction was complete, the molecular sieves were removed by filtration. Added were 20 L EtOAc and extracted with 4×20 L of 25% sodium thiosulfate and 20 L 10% NaCl solutions. The organic layer was separated and dried with 8-12 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 5:95, 10:90, 20:80, 30:70 and 40:60 (EtOAc/heptane). The pure fractions were pooled and evaporated to give intermediate AMod3 as a syrup. The isolated yield was 75%.
Synthesis of the BA Dimer
Step 9. Preparation of AMod4, DeAcetylation of AMod3
A 100 L reactor was charged with 4128 g of 1-Methyl 4,6-Diacetyl Monomer A2 (AMod3) (10.5 mol, 1 equiv) and 18 L of dry methanol and dissolved, after which 113.4 g (2.1 mol, 0.2 equiv) of sodium methoxide was transferred into the reactor. The reaction was stirred at room temperature and monitored by TLC (40% ethyl acetate/hexane) and HPLC. The reaction took approximately 2-4 hours for completion. When the reaction was complete, Dowex 50Wx8 cation resin was added in small portions until the pH reached 6-8. The Dowex 50Wx8 resin was filtered and the solution was evaporated to a syrup (bath temp. 40° C.). The syrup was diluted with 10 L of ethyl acetate and extracted with 20 L brine and 20 L water. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight at the same temperature. The isolated yield of the syrup AMod4 was about 88%.
Synthesis of the BA Dimer
Step 10. Preparation of AMod5,6-Benzoylation
A 100 L reactor was charged with 2858 g of Methyl 4,6-diOH Monomer A2 (AMod4) (9.24 mol, 1 equiv) and co-evaporated with 3×10 L of pyridine. When evaporation was complete, 15 L of dichloromethane, 6 L of pyridine were transferred into the reactor and dissolved. The reactor was purged with nitrogen and chilled to −40° C. The reactor was charged with 1044 mL (1299 g, 9.24 mol, 1 equiv) of benzoyl chloride. When the addition was complete, the reaction was allowed to warm to −10° C. over a period of 2 hours. The reaction was monitored by TLC (60% ethyl acetate/hexane). When the reaction was completed, the solution was evaporated to a syrup (bath temp. 40° C.). This was co-evaporated with 3×15 L of toluene. The syrup was diluted with 40 L ethyl acetate. Extraction was carried out with 20 L of water and 20 L of brine solution. The Ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 5:95, 10:90, 20:80, 25:70 and 30:60 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup. The isolated yield of the intermediate AMod5 was 84%.
Synthesis of the BA Dimer
Step 11. Preparation of BA1, Coupling of Amod5 with BMod6
A 100 L reactor was charged with 3054 g of methyl 4-Hydroxy-Monomer A2 (AMod5) from Step 10 (7.38 mol, 1 equiv) and 4764 g of 4-Lev-1-TCA-Monomer B1 (BMod6) from Step 6 (7.38 mol, 1 equiv). The combined monomers were dissolved in 20 L of toluene and co-evaporated at 40° C. Co evaporation was repeated with an additional 2×20 L of toluene, after which 30 L of dichloromethane (DCM) was transferred into the reactor and dissolved. The reactor was purged with nitrogen and was chilled to below −20° C. When the temperature was between −20° C. and −40° C., 1572 g (1404 mL, 11.12 moles, 1.5 equiv) of boron trifluoride etherate (BF3.Et2O) were transferred into the reactor. After complete addition of boron trifluoride etherate, the reaction was allowed to warm to 0° C. and stirring was continued. The completeness of the reaction was monitored by HPLC and TLC (40:70 ethyl acetate/heptane). The reaction required 3-4 hours to reach completion. When the reaction was complete, 926 mL (672 g, 6.64 mol, 0.9 equiv) of triethylamine (TEA) was transferred into the mixture and stirred for an additional 30 minutes, after which 20 L of water and 10 L of dichloromethane were transferred into the reactor. The solution was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was separated with 2×20 L water and 20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and used in the next step. The isolated yield of the disaccharide BA1 was about 72%.
Synthesis of the BA Dimer
Step 12, Removal of Levulinate Methyl [(methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate)-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl]-2-deoxy-α-D-glucopyranoside
A 100 L reactor was charged with 4.104 Kg of 4-Lev BA Dimer (BA1) (4.56 mol, 1 equiv) in 20 L of THF. The reactor was purged with nitrogen and chilled to −20 to −25° C., after which 896 mL of hydrazine hydrate (923 g, 18.24 mol, 4 equiv) was transferred into the reactor. Stirring was continued and the reaction was monitored by TLC (40% ethyl acetate/heptane) and HPLC. The reaction took approximately 2-3 hour for the completion, after which 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (ETOAc layer) was extracted with 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). The pure fractions were pooled and evaporated to dryness. The isolated yield of the BA Dimer was 82%. Formula: C42H43N3O13; Mol. Wt. 797.80.
Synthesis of the EDC Trimer
Step 1. Preparation of EMod1, Acetylation
A 100 L reactor was charged with 16533 g of Monomer E (45 mole, 1 eq.), 21.25 L acetic anhydride (225 mole, 5 eq.) and 60 L of dichloromethane. The reactor was purged with nitrogen and was chilled to −10° C. When the temperature was at −10° C., 1.14 L (1277 g) of boron trifluoride etherate (BF3.Et2O, 9.0 moles, 0.2 eq) were transferred into the reactor. After the complete addition of boron trifluoride etherate, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by TLC (30:70 ethyl acetate/heptane) and HPLC. The reaction took approximately 3-6 hours to reach completion. When the reaction was completed, the mixture was extracted with 3×50 L of 10% sodium bicarbonate and SOL of water. The organic phase (DCM) was evaporated to a syrup (bath temp. 40° C.) and allowed to dry overnight. The isolated yield of EMod1 was 97%.
Synthesis of the EDC Trimer
Step 2. Preparation of EMod2, De-Acetylation of Azidoglucose
A 100 L reactor was charged with 21016 g of 1,6-Diacetyl Monomer E (EMod1) (45 mole, 1 eq.), 5434 g of hydrazine acetate (NH2NH2.HOAc, 24.75 mole, 0.55 eq.) and 50 L of DMF (dimethyl formamide). The solution was stirred at room temperature and the reaction was monitored by TLC (30% ethyl acetate/hexane) and HPLC. The reaction took approximately 2-4 hours for completion. When the reaction was completed, 50 L of dichloromethane and 40 L of water were transferred into the reactor. This was stirred for 30 minutes and the layers were separated. This was extracted with an additional 40 L of water and the organic phase was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight at the same temperature. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of intermediate EMod2 was 100%.
Synthesis of the EDC Trimer
Step 3. Preparation of EMod3, Formation of 1-TCA
A 100 L reactor was charged with 12752 g of 1-Hydroxy Monomer E (EMod2) (30 mole, 1 eq.) in 40 L of dichloromethane. The reactor was purged with nitrogen and stirring was started, after which 2.25 L of DBU (15 moles, 0.5 eq.) and 15.13 L of trichloroacetonitrile (150.9 moles, 5.03 eq) were transferred into the reactor. Stirring was continued and the reaction was kept under nitrogen. After the reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (30:70 ethyl acetate/Heptane) and HPLC. The reaction took approximately 2-3 hours to reach completion. When the reaction was complete, 40 L of water and 20 L of DCM were charged into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 40 L water and the DCM solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90 (DCM/EtOAc/heptane), 20:5:75 (DCM/EtOAc/heptane) and 20:10:70 DCM/EtOAc/heptane). Pure fractions were pooled and evaporated to give Intermediate EMod3 as a syrup. Isolated yield was 53%.
Synthesis of the EDC Trimer
Step 4. Preparation of ED Dimer, Coupling of E-TCA with Monomer D
A 100 L reactor was charged with 10471 g of 6-Acetyl-1-TCA Monomer E (EMod3) (18.3 mole, 1 eq., FW: 571.8) and 6594 g of Monomer D (16.47 mole, 0.9 eq, FW: 400.4). The combined monomers were dissolved in 20 L toluene and co-evaporated at 40° C. This was repeated with co-evaporation with an additional 2×20 L of toluene, after which 60 L of dichloromethane (DCM) were transferred into the reactor and dissolved. The reactor was purged with nitrogen and was chilled to −40° C. When the temperature was between −30° C. and −40° C., 2423 g (2071 mL, 9.17 moles, 0.5 eq) of TES Triflate were transferred into the reactor. After complete addition of TES Triflate the reaction was allowed to warm and stirring was continued. The completeness of the reaction was monitored by HPLC and TLC (35:65 ethyl acetate/Heptane). The reaction required 2-3 hours to reach completion. When the reaction was completed, 2040 mL of triethylamine (TEA, 1481 g, 0.8 eq.) were transferred into the reactor and stirred for an additional 30 minutes. The organic layer (DCM layer) was extracted with 2×20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 15:85, 20:80, 25:75, 30:70 and 35:65 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The ED Dimer was obtained in 81% isolated yield.
Synthesis of the EDC Trimer
Step 5. Preparation of ED1 TFA, Hydrolysis of ED Dimer
A 100 L reactor was charged with 7.5 Kg of ED Dimer (9.26 mol, 1 equiv). The reactor was chilled to <5° C. and 30.66 L of 90% TFA solution (TFA, 370.4 mol, 40 equiv) were transferred into the reactor. When the addition was completed the reaction was allowed to warm to room temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexanes) and HPLC. The reaction took approximately 3-4 hours to reach completion. When the reaction was completed, was chilled and 51.6 L of triethylamine (TEA, 37.5 Kg, 370.4 mole) were transferred into the reactor, after which 20 L of water & 20 L ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. Ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 20:80, 30:70, 40:60, 50:50, 60:40 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup. Isolated yield of ED1 was about 70%.
Synthesis of the EDC Trimer
Step 6. Preparation of ED2, Silylation of ED1
A 100 L reactor was charged with 11000 g of 1,2-diOH ED Dimer (ED1) (14.03 mol, 1 equiv), 1910.5 g of imidazole (28.06 mol, 2 equiv) and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., after which 3.53 L butyldiphenylchloro-silane (TBDPS-Cl, 4.628 Kg, 16.835 mol, 1.2 equiv) was charged into the reactor. When the addition was complete, the chiller was turned off and the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (50% ethyl acetate/hexane) and HPLC. The reaction required 4-6 hours to reach completion. When the reaction was completed, 20 L of water and 10 L of dichloromethane were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. Dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). Intermediate ED2 was obtained in 75% isolated yield.
Synthesis of the EDC Trimer
Step 7. Preparation of ED3, D-Levulination
A 100 L reactor was charged with 19800 g of 1-Silyl ED Dimer (ED2) (19.37 moles, 1 equiv) and 40 L of dry tetrahydrofuran (THF) and agitated to dissolve. The reactor was purged with nitrogen and 237 g of dimethylaminopyridine (DMAP, 1.937 moles, 0.1 equiv) and 10.05 L of diisopropylethylamine (DIPEA, 63.9 moles, 3 equiv) were transferred into the reactor. The reactor was chilled to 10-15° C. and kept under a nitrogen atmosphere, after which 12.46 Kg of levulinic anhydride (58.11 moles, 3 eq) was charged into the reactor. When the addition was complete, the reaction was warmed to ambient temperature and stirred overnight or 12-16 hours. The completeness of the reaction was monitored by TLC (40:60 ethyl acetate/hexane) and by HPLC. 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L of water, 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The ED3 yield was 95%.
Synthesis of the EDC Trimer
Step 8. Preparation of ED4, Desilylation of ED3
A 100 L reactor was charged with 19720 g of 1-Silyl-2-Lev ED Dimer (ED3) (17.6 mol, 1 equiv) in 40 L of THF. The reactor was chilled to 0° C., after which 6903 g of tetrabutylammonium fluoride trihydrate (TBAF, 26.4 mol, 1.5 equiv) and 1511 mL (26.4 mol, 1.5 equiv) of glacial acetic acid were transferred into the reactor. When the addition was complete, the reaction was stirred and allowed to warm to ambient temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. The reaction required 3 hours to reach completion. When the reaction was completed, 20 L of water and 10 L of dichloromethane were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified using a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20 (EtOAc/heptane) and 200 L 100% EtOAc. The pure fractions were pooled and evaporated to a syrup and used in the next step. The isolated yield of ED4 was about 92%.
Synthesis of the EDC Trimer
Step 9. Preparation of ED5, TCA Formation
A 100 L reactor was charged with 14420 g of 1-OH-2-Lev ED Dimer (ED4) (16.35 mol, 1 equiv) in 30 L of dichloromethane. The reactor was purged with nitrogen and stirring was begun, after which 1222 mL of diazabicycloundecene (DBU, 8.175 mol, 0.5 equiv) and 23.61 Kg of trichloroacetonitrile (TCA, 163.5 mol, 10 equiv) were transferred into the reactor. Stirring was continued and the reaction was kept under nitrogen. After reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by HPLC and TLC (40:60 ethyl acetate/heptane). The reaction took approximately 2 hours for reaction completion. When the reaction was completed, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified using a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup and used in the next step. The isolated yield of intermediate ED5 was about 70%.
Synthesis of the EDC Trimer
Step 10.
Preparation of EDC Trimer, Coupling of ED5 with Monomer C 6-O-acetyl-2-azido-3,4-di-O-benzyl-2-deoxy-α-D-glucopyranosyl-(1→4)-benzyl (3-O-benzyl-2-O-levulinoyl)-β-D-glucopyranosyluronate-(1→4)-(3-O-acetyl-1,6-anhydro-2-azido)-2-deoxy-β-D-glucopyranose
A 100 L reactor was charged with 12780 g of 2-Lev 1-TCA ED Dimer (ED5) (7.38 mole, 1 eq., FW) and 4764 g of Monomer C (7.38 mole, 1 eq). The combined monomers were dissolved in 20 L toluene and co-evaporated at 40° C. Repeated was co-evaporation with an additional 2×20 L of toluene, after which 60 L of dichloromethane (DCM) was transferred into the reactor and dissolved. The reactor was purged with nitrogen and chilled to −20° C. When the temperature was between −20 and −10° C., 2962 g (11.2 moles, 0.9 eq) of TES Triflate were transferred into the reactor. After complete addition of TES Triflate the reaction was allowed to warm to 5° C. and stirring was continued. Completeness of the reaction was monitored by HPLC and TLC (35:65 ethyl acetate/Heptane). The reaction required 2-3 hours to reach completion. When the reaction was completed, 1133 g of triethylamine (TEA, 0.9 eq.) were transferred into the reactor and stirred for an additional 30 minutes. The organic layer (DCM layer) was extracted with 2×20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. Dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 15:85, 20:80, 25:75, 30:70 and 35:65 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of EDC Trimer was 48%. Formula: C55H60N6O18; Mol. Wt. 1093.09. The 1H NMR spectrum (d6-acetone) of the EDC trimer is shown in FIG. 3.

Preparation of EDC1
Step 1:
Anhydro Ring Opening & Acetylation 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-1,3,6-tri-O-acetyl-β-D-glucopyranose
7.0 Kg (6.44 mol) of EDC Trimer was dissolved in 18 L anhydrous Dichloromethane. 6.57 Kg (64.4 mol, 10 eq) of Acetic anhydride was added. The solution was cooled to −45 to −35° C. and 1.82 Kg (12.9 mol, 2 eq) of Boron Trifluoride etherate was added slowly. Upon completion of addition, the mixture was warmed to 0-10° C. and kept at this temperature for 3 hours until reaction was complete by TLC and HPLC. The reaction was cooled to −20° C. and cautiously quenched and extracted with saturated solution of sodium bicarbonate (3×20 L) while maintaining the mixture temperature below 5° C. The organic layer was extracted with brine (1×20 L), dried over anhydrous sodium sulfate, and concentrated under vacuum to a syrup. The resulting syrup of EDC1 (6.74 Kg) was used for step 2 without further purification. The 1H NMR spectrum (d6-acetone) of the EDC-1 trimer is shown in FIG. 4.

Preparation of EDC2
Step 2:
Deacetylation 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-β-D-glucopyranose
The crude EDC1 product obtained from step 1 was dissolved in 27 L of Tetrahydrofuran and chilled to 15-20° C., after which 6 Kg (55.8 mol) of benzylamine was added slowly while maintaining the reaction temperature below 25° C. The reaction mixture was stirred for 5-6 hours at 10-15° C. Upon completion, the mixture was diluted with ethyl acetate and extracted and quenched with 10% citric acid solution (2×20 L) while maintaining the temperature below 25° C. The organic layer was extracted with 10% NaCl/1% sodium bicarbonate (1×20 L). The extraction was repeated with water (1×10 L), dried over anhydrous sodium sulfate and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel yielded 4.21 Kg (57% yield over 2 steps) of EDC2[ also referred to as 1-Hydroxy-6-Acetyl EDC Trimer]. The 1H NMR spectrum (d6-acetone) of the EDC-2 trimer is shown in FIG. 5.

Preparation of EDC3
Step 3:
Formation of TCA Derivative 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-1-O-trichloroacetimidoyl-β-D-glucopyranose
4.54 Kg (3.94 mol) of EDC2 was dissolved in 20 L of Dichloromethane. 11.4 Kg (78.8 mol, 20 eq) of Trichloroacetonitrile was added. The solution was cooled to −15 to −20° C. and 300 g (1.97 mol, 0.5 eq) of Diazabicycloundecene was added. The reaction was allowed to warm to 0-10° C. and stirred for 2 hours or until reaction was complete. Upon completion, water (20 L) was added and the reaction was extracted with an additional 10 L of DCM. The organic layer was extracted with brine (1×20 L), dried over anhydrous sodium sulfate, and concentrated under vacuum to a syrup. Column chromatographic separation using silica gel and 20-60% ethyl acetate/heptane gradient yielded 3.67 Kg (72% yield) of 1-TCA derivative, EDC3. The 1H NMR spectrum (d6-acetone) of the EDC-3 trimer is shown in FIG. 6.

Preparation of EDCBA1
Step 4:
Coupling of EDC3 with BA Dimer Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl)-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
3.67 Kg (2.83 mol) of EDC3 and 3.16 Kg (3.96 mol, 1.4 eq) of BA Dimer was dissolved in 7-10 L of Toluene and evaporated to dryness. The resulting syrup was coevaporated with Toluene (2×15 L) to remove water. The dried syrup was dissolved in 20 L of anhydrous Dichloromethane, transferred to the reaction flask, and cooled to −15 to −20° C. 898 g (3.4 mol, 1.2 eq) of triethylsilyl triflate was added while maintaining the temperature below −5° C. When the addition was complete, the reaction was immediately warmed to −5 to 0° C. and stirred for 3 hours. The reaction was quenched by slowly adding 344 g (3.4 mol, 1.2 eq) of Triethylamine. Water (15 L) was added and the reaction was extracted with an additional 10 L of DCM. The organic layer was extracted with a 25% 4:1 Sodium Chloride/Sodium Bicarbonate solution (2×20 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. The resulting syrup of the pentasaccharide, EDCBA1 was used for step 5 without further purification. The 1H NMR spectrum (d6-acetone) of the EDCBA-1 pentamer is shown in FIG. 7.

Preparation of EDCBA2
Step 5:
Hydrolysis of Levulinyl moiety Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl)-(1→4)—O-[benzyl 3-O-benzyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl)-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
The crude EDCBA1 from step 4 was dissolved in 15 L of Tetrahydrofuran and chilled to −20 to −25° C. A solution containing 679 g (13.6 mol) of Hydrazine monohydrate and 171 g (1.94 mol) of Hydrazine Acetate in 7 L of Methanol was added slowly while maintaining the temperature below −20° C. When the addition was complete, the reaction mixture was allowed to warm to 0-10° C. and stirred for several hours until the reaction is complete, after which 20 L of Ethyl acetate was added and the reaction was extracted with 10% citric acid (2×12 L). The organic layer was washed with water (1×12 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel and 10-45% ethyl acetate/heptane gradient yielded 2.47 Kg (47.5% yield over 2 steps) of EDCBA2. The 1H NMR spectrum (d6-acetone) of the EDCBA-2 pentamer is shown in FIG. 8.

Preparation of EDCBA Pentamer
Step 6:
THP Formation Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
2.47 Kg (1.35 mol) of EDCBA2 was dissolved in 23 L Dichloroethane and chilled to 10-15° C., after which 1.13 Kg (13.5 mol, 10 eq) of Dihydropyran and 31.3 g (0.135 mol, 0.1 eq) of Camphorsulfonic acid were added. The reaction was allowed warm to 20-25° C. and stirred for 4-6 hours until reaction was complete. Water (15 L) was added and the reaction was extracted with an additional 10 L of DCE. The organic layer was extracted with a 25% 4:1 Sodium Chloride/Sodium Bicarbonate solution (2×20 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel and 10-35% ethyl acetate/heptane gradient yielded 2.28 Kg (88.5% yield) of fully protected EDCBA Pentamer. The 1H NMR spectrum (d6-acetone) of the EDCBA pentamer is shown in FIG. 9.

Preparation of API1
Step 1:
Saponification Methyl O-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-azido-2-deoxy-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-α-L-Idopyranosyluronosyl-(1→4)-2-azido-3-O-benzyl-2-deoxy-α-D-glucopyranoside disodium salt
To a solution of 2.28 Kg (1.19 mol) of EDCBA Pentamer in 27 L of Dioxane and 41 L of Tetrahydrofuran was added 45.5 L of 0.7 M (31.88 mol, 27 eq) Lithium hydroxide solution followed by 5.33 L of 30% Hydrogen peroxide. The reaction mixture was stirred for 10-20 hours to remove the acetyl groups. Then, 10 L of 4 N (40 mol, 34 eq) sodium hydroxide solution was added. The reaction was allowed to stir for an additional 24-48 hours to hydrolyze the benzyl and methyl esters completely. The reaction was then extracted with ethyl acetate. The organic layer was extracted with brine solution and dried with anhydrous sodium sulfate. Evaporation of the solvent under vacuum gave a syrup of API1 [also referred to as EDCBA(OH)5] which was used for the next step without further purification.
Preparation of API2
Step 2:
O-Sulfonation Methyl O-2-azido-2-deoxy-3,4-di-O-benzyl-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-azido-2-deoxy-3,6-di-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-azido-2-deoxy-6-O-sulfo-α-D-glucopyranoside, heptasodium salt
The crude product of API1 [aka EDCBA(OH)5] obtained in step 1 was dissolved in 10 L Dimethylformamide. To this was added a previously prepared solution containing 10.5 Kg (66 moles) of sulfur trioxide-pyridine complex in 10 L of Pyridine and 25 L of Dimethylformamide. The reaction mixture was heated to 50° C. over a period of 45 min. After stiffing at 1.5 hours at 50° C., the reaction was cooled to 20° C. and was quenched into 60 L of 8% sodium bicarbonate solution that was kept at 10° C. The pH of the quench mixture was maintained at pH 7-9 by addition of sodium bicarbonate solution. When all the reaction mixture has been transferred, the quench mixture was stirred for an additional 2 hours and pH was maintained at pH 7 or greater. When the pH of quench has stabilized, it was diluted with water and the resulting mixture was purified using a preparative HPLC column packed with Amberchrom CG161-M and eluted with 90%-10% Sodium Bicarbonate (5%) solution/Methanol over 180 min. The pure fractions were concentrated under vacuum and was then desalted using a size exclusion resin or gel filtration (Biorad) G25 to give 1581 g (65.5% yield over 2 steps) of API2 [also referred to as EDCBA(OSO3)5]. The 1H NMR spectrum (d6-acetone) of API-2 pentamer is shown in FIG. 10.

Preparation of API3
Step 3:
Hydrogenation Methyl O-2-amino-2-deoxy-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-amino-2-deoxy-3,6-di-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-amino-2-deoxy-6-O-sulfo-α-D-glucopyranoside, heptasodium salt
A solution of 1581 g (0.78 mol) of O-Sulfated pentasaccharide API2 in 38 L of Methanol and 32 L of water was treated with 30 wt % of Palladium in Activated carbon under 100 psi of Hydrogen pressure at 60-65° C. for 60 hours or until completion of reaction. The mixture was then filtered through 1.0μ and 0.2μ filter cartridges and the solvent evaporated under vacuum to give 942 g (80% yield) of API3 [also referred to as EDCBA(OSO3)5(NH2)3]. The 1H NMR spectrum (d6-acetone) of API-3 pentamer is shown in FIG. 11.

Preparation of Fondaparinux Sodium
Step 4:
N-Sulfation & Removal of THP Methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)—O-β-D-glucopyranuronosyl-(1→4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside, decasodium salt
To a solution of 942 g (0.63 mol) of API3 in 46 L of water was slowly added 3.25 Kg (20.4 mol, 32 eq) of Sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2 N sodium hydroxide solution. The reaction was allowed to stir for 4-6 hours at pH 9.0-9.5. When reaction was complete, the pH was adjusted to pH 7.0 using 50 mM solution of Ammonium acetate at pH 3.5. The resulting N-sulfated EDCBA(OSO3)5(NHSO3)3 mixture was purified using Ion-Exchange Chromatographic Column (Varian Preparative 15 cm HiQ Column) followed by desalting using a size exclusion resin or gel filtration (Biorad G25). The resulting mixture was then treated with activated charcoal and the purification by ion-exchange and desalting were repeated to give 516 g (47.6% yield) of the purified Fondaparinux Sodium form.
Analysis of the Fondaparinux sodium identified the presence of P1, P2, P3, and P4 in the fondaparinux. P1, P2, P3, and P4 were identified by standard analytical methods.
INTERMEDIATES
The monomers used in the processes described herein may be prepared as described in the art, or can be prepared using the methods described herein.

The synthesis of Monomer A-2 (CAS Registry Number 134221-42-4) has been described in the following references: Arndt et al., Organic Letters, 5(22), 4179-4182, 2003; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; and Sakairi et al., Journal of the Chemical Society, Chemical Communications, (5), 289-90, 1991, and the references cited therein, which are hereby incorporated by reference in their entireties.

Monomer C(CAS Registry Number 87326-68-9) can be synthesized using the methods described in the following references: Ganguli et al., Tetrahedron: Asymmetry, 16(2), 411-424, 2005; Izumi et al., Journal of Organic Chemistry, 62(4), 992-998, 1997; Van Boeckel et al., Recueil: Journal of the Royal Netherlands Chemical Society, 102(9), 415-16, 1983; Wessel et al.,Helvetica Chimica Acta, 72(6), 1268-77, 1989; Petitou et al., U.S. Pat. No. 4,818,816 and references cited therein, which are hereby incorporated by reference in their entireties.

Monomer E (CAS Registry Number 55682-48-9) can be synthesized using the methods described in the following literature references: Hawley et al., European Journal of Organic Chemistry, (12), 1925-1936, 2002; Dondoni et al., Journal of Organic Chemistry, 67(13), 4475-4486, 2002; Van der Klein et al., Tetrahedron, 48(22), 4649-58, 1992; Hori et al., Journal of Organic Chemistry, 54(6), 1346-53, 1989; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; Tailler et al.,Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio–Organic Chemistry, (23), 3163-4, (1972-1999) (1992); Paulsen et al., Chemische Berichte, 111(6), 2334-47, 1978; Dasgupta et al., Synthesis, (8), 626-8, 1988; Paulsen et al., Angewandte Chemie, 87(15), 547-8, 1975; and references cited therein, which are hereby incorporated by reference in their entireties.

Monomer B-1 (CAS Registry Number 444118-44-9) can be synthesized using the methods described in the following literature references: Lohman et al., Journal of Organic Chemistry, 68(19), 7559-7561, 2003; Orgueira et al., Chemistry—A European Journal, 9(1), 140-169, 2003; Manabe et al., Journal of the American Chemical Society, 128(33), 10666-10667, 2006; Orgueira et al., Angewandte Chemie, International Edition, 41(12), 2128-2131, 2002; and references cited therein, which are hereby incorporated by reference in their entireties.
Synthesis of Monomer D
Monomer D was prepared in 8 synthetic steps from glucose pentaacetate using the following procedure:

Pentaacetate SM-B was brominated at the anomeric carbon using HBr in acetic acid to give bromide derivative IntD1. This step was carried out using the reactants SM-B, 33% hydrogen bromide, acetic acid and dichloromethane, stirring in an ice water bath for about 3 hours and evaporating at room temperature. IntD1 was reductively cyclized with sodium borohydride and tetrabutylammonium iodide in acetonitrile using 3 Å molecular sieves as dehydrating agent and stirring at 40° C. for 16 hours to give the acetal derivative, IntD2. The three acetyl groups in IntD2 were hydrolyzed by heating with sodium methoxide in methanol at 50° C. for 3 hours and the reaction mixture was neutralized using Dowex 50WX8-100 resin (Aldrich) in the acid form to give the trihydroxy acetal derivative IntD3.
The C4 and C6 hydroxyls of IntD3 were protected by mixing with benzaldehyde dimethyl acetate and camphor sulphonic acid at 50° C. for 2 hours to give the benzylidene-acetal derivative IntD4. The free hydroxyl at the C3 position of IntD4 was deprotonated with sodium hydride in THF as solvent at 0° C. and alkylated with benzyl bromide in THF, and allowing the reaction mixture to warm to room temperature with stirring to give the benzyl ether IntD5. The benzylidene moiety of IntD5 was deprotected by adding trifluoroacetic acid in dichloromethane at 0° C. and allowing it to warm to room temperature for 16 hours to give IntD6 with a primary hydroxyl group. IntD6 was then oxidized with TEMPO (2,2,6,6-tetramethyl-1-piperidine-N-oxide) in the presence of tetrabutylammonium chloride, sodium bromide, ethyl acetate, sodium chlorate and sodium bicarbonate, with stirring at room temperature for 16 hours to form the carboxylic acid derivative IntD7. The acid IntD7 was esterified with benzyl alcohol and dicyclohexylcarbodiimide (other reactants being hydroxybenzotriazole and triethylamine) with stirring at room temperature for 16 hours to give Monomer D.
Synthesis of the BA Dimer
The BA Dimer was prepared in 12 synthetic steps from Monomer B1 and Monomer A2 using the following procedure:


The C4-hydroxyl of Monomer B-1 was levulinated using levulinic anhydride and diisopropylethylamine (DIPEA) with mixing at room temperature for 16 hours to give the levulinate ester BMod1, which was followed by hydrolysis of the acetonide with 90% trifluoroacetic acid and mixing at room temperature for 4 hours to give the diol BMod2. The C1 hydroxyl of the diol BMod2 was silylated with tert-butyldiphenylsilylchloride by mixing at room temperature for 3 hours to give silyl derivative BMod3. The C2-hydroxyl was then benzoylated with benzoyl chloride in pyridine, and mixed at room temperature for 3 hours to give compound BMod4. The silyl group on BMod4 was then deprotected with tert-butyl ammonium fluoride and mixing at room temperature for 3 hours to give the C1-hydroyl BMod5. The C1-hydroxyl is then allowed to react with trichloroacetonitrile in the presence of diazobicycloundecane (DBU) and mixing at room temperature for 2 hours to give the trichloroacetamidate (TCA) derivative BMod6, which suitable for coupling, for example with Monomer A-2.
Monomer A-2 was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1.
Monomer A2 was prepared for the coupling reaction by opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1. This transformation occurs using boron trifluoride etherate, acetic anhydride and dichloromethane, between −20° C. and room temperature for 3 hours. The C1-Acetate of AMod1 was then hydrolyzed and methylated in two steps to give the diacetate AMod3. That is, first AMod1 was reacted with trimethylsilyl iodide and mixed at room temperature for 2 hours, then reacted with and tetrabutyl ammonium iodide. This mixture was reacted with diisoproylethylamine and methanol and stirred for 16 hours at room temperature, thus forming AMod3. The C4 and C6 acetates of AMod3 are hydrolyzed with sodium methoxide to give the diol Amod4. The AMod3 mixture was also subjected to mixing at room temperature for 3 hours with Dowex 50 Wx4x8-100 resin in the acid form for neutralization. This formed Amod4. The C6-hydroxyl of AMod4 is then benzoylated by treating with benzoyl chloride in pyridine at −40° C. and then allowing it to warm up to −10° C. over 2 hours to give AMod5.
Coupling of monomer AMod5 with the free C4-hydroxyl group of BMod6 was performed in the presence of BF3.Et2O and dichloromethane with mixing between −20° C. and room temperature for 3 hours to provide disaccharide BA1. The C4-levulinyl moiety of the disaccharide was then hydrolyzed with hydrazine to give the BA Dimer, which is suitable for subsequent coupling reactions.
Synthesis of EDC Trimer
The EDC Trimer was prepared in 10 synthetic steps from Monomer E, Monomer D and Monomer C using the following procedure:


Monomer E was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give diacetate EMod1. This occurs by the addition of Monomer E with boron trifluoride etherate, acetic anhydride and dichloromethane at −10° C., and allowing the reaction to warm to room temperature with stirring for 3 hours. The C1-Acetate of EMod1 is then hydrolyzed to give the alcohol, EMod2. This occurs by reacting Emod1 with hydrazine acetate and dimethylformamide and mixing at room temperature for 3 hours. The C1-hydroxyl of Emod2 is then reacted with trichloroacetonitrile to give the trichloro acetamidate (TCA) derivative EMod3 suitable for coupling, which reaction also employs diazabicycloundecane and dichloromethane and mixing at room temperature for 2 hours.
Monomer D, having a free C4-hydroxyl group, was coupled with monomer EMod3 in the presence of triethylsilyl triflate with mixing at −40° C. for 2 hours to give the disaccharide ED Dimer. The acetal on ring sugar D of the ED Dimer is hydrolyzed to give the C1,C2-diol ED1. This occurs by reacting the ED Dimer with 90% trifluoro acetic acid and mixing at room temperature for 4 hours. The C1-hydroxyl moiety of ED1 was then silylated with tert-butyldiphenylsilyl chloride to give the silyl derivative ED2. The C2-hydroxyl of ED2 was then allowed to react with levulinic anhydride in the presence of dimethylaminopyridine (DMAP) and diethylisopropylamine for approximately 16 hours to give the levulinate ester ED3. The TBDPS moiety is then deprotected by removal with tert-butylammonium fluoride in acetic acid with mixing at room temperature for 3 hours to give ED4 having a C1-hydroxyl. The C1-hydroxyl moiety of ED4 was then allowed to react with trichloroacetonitrile to give the TCA derivative ED5, which is suitable for coupling.
The C1-hydroxyl moiety of ED4 is then allowed to react with trichloroacetonitrile to give the TCA derivative ED5 suitable for coupling using diazabicycloundecane and dichloromethane, and mixing at room temperature for 2 hours. Monomer C, havinga free C4-hydroxyl group, was then coupled with the disaccharide ED5 in the presence of triethylsilyl triflate and mixed at −20° C. for 2 hours to give the trisaccharide EDC Trimer.
Synthesis of the EDCBA Pentamer
The EDCBA Pentamer was prepared using the following procedure:

The preparation of EDCBA Pentamer is accomplished in two parts as follows. In part 1, the EDC Trimer, a diacetate intermediate, is prepared for the coupling reaction with Dimer BA by initially opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the tetraacetate derivative EDC1. This occurs by reacting the EDC Trimer with boron trifluoride etherate, acetic anhydride and dichlormethane and stirring between −10° C. and room temperature for 3 hours. The C1-Acetate of EDC1 is then hydrolyzed to give the alcohol, EDC2, by reacting EDC1 with benzylamine [BnNH2] and tetrahydrofuran and mixing at −10° C. for 3 hours. The C1-hydroxyl of EDC2 is then reacted with trichloroacetonitrile and diazabicycloundecane, with mixing at room temperature for 2 hours, to give the trichloro acetamidate (TCA) derivative EDC3 suitable for coupling.


In Part 2 of the EDCBA Pentameter synthesis, the Dimer BA, having a free C4-hydroxyl group, is coupled with trisaccharide EDC3 in the presence of triethylsilyltriflate at −30° C. mixing for 2 hours to give the pentasaccharide EDCBA1. The levulinyl ester on C2 of sugar D in EDCBA1 is hydrolyzed with a mixture of deprotecting agents, hydrazine hydrate and hydrazine acetate and stiffing at room temperature for 3 hours to give the C2-hydroxyl containing intermediate EDCBA2. The C2-hydroxyl moiety on sugar D of EDCBA2 is then alkylated with dihydropyran (DHP) in the presence of camphor sulfonic acid (CSA) and tetrahydrofuran with mixing at room temperature for 3 hours to give the tetrahydropyranyl ether (THP) derivative, EDCBA Pentamer.
…………………………
A fast and effective hydrogenation process of protected pentasaccharide: A key step in the synthesis of fondaparinux sodium, Org Process Res Dev 2013, 17: 869, http://pubs.acs.org/doi/full/10.1021/op300367c

An improved method for the simultaneous removal of O-benzyl and N-carboxybenzyl groups as well as reducing azide groups to amines in protected heparin-like pentasaccharides, a key process in fondaparinux sodium synthesis, is reported. Under catalytic transfer hydrogenation conditions, using readily available and inexpensive ammonium formate, the hydrogenolysis is done in less than an hour in good yield and purity. This procedure represents a major advantage over the previously published procedures, the latter of which involve several hours/days of hydrogenation reaction under catalytic reduction using gaseous hydrogen.

Synthesis of Compound 1 (FONDAPARINUX)
………………
SYNTHESIS
In the synthesis of Fondaparinux sodium, the monomers XII, XVIII, XXVII, XXXVIII, XXXXI and dimers XIX, XX, XL described herein may be made either by processes described in the art or, by a process as described herein. The XII and XVIII monomers may then linked to form a disaccharide XX, XXXIX and XXVII monomers may then linked to form a disaccharide XL, XLIII and XX dimers may then linked to form a tetrasaccharide, XLVII tetramer and XLV monomer may be linked to form a pentasaccharide (XLVIII) pentamer. The XLVIII pentamer is an intermediate that may be converted through a series of reactions to fondaparinux sodium. This strategy described herein provides an efficient method for multi-kilogram preparation of fondaparinux in high yields and highly stereoselective purity.
Fondaparinux sodium (LIII) was prepared in 3 synthetic steps from O – S pentasaccharide (L) using the following procedure:

Fondaparinux Sodium (LIII)
Preparation of Fondaparinux sodium (LIII)—
N- sulfonation of Deprotected Pentasaccharide (LI) methyl 0-2-deoxy-3,6-di-0- sulfo-2-(sulfoamino)-a-D-glucopyranosyl-(l— >4)-0-2-0-sulfo-a-L- idopyranurosyl-( 1— >4)-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D-glucopyranoside,decasodium salt
A solution of deprotected pentasaccharide (LI) (145 gm) in water (2.54 V) was adjusted to a pH of 9.5 – 10.5 with 1 N NaOH solution. S03-pyridine complex (156 gm) was added into 3 lots every 15 min, the pH being maintained at 9.5-10.5 by automatic addition of 1 N NaOH. The mixture was stirred for 2 hrs at RT, during this aqueous NaOH (IN solution) was added to maintain pH at 9.5 – 10.5. After neutralization to pH 7 – 7.5 by addition of HC1 solution, the mixture was evaporated using vacuum. The residue was dissolved in water (1.6 L) at RT, to this solution was added acetone (1.6 L) at RT. The reaction mass was cooled to 5°C – 1 0 °C and stirred for 1 hr. The solid was filtered and washed with cold acetone: water (1 :1). The clear filtrate was distilled off completely under vacuum below 55°C. The residue was dissolved in water (1.6 L) at RT, and to this solution was added acetone(1.6 L) at RT. The mixture was cooled to 5 to 10°C and stirred for 1 hr. The solid was filtered and washed with cold acetone/water (1 :1). The clear filtrate was distilled off completely under vacuum below 55°C. The residue was dissolved in water (0.7 L) and charcoal (40 gm) was added at RT. The mixture was stirred for 30 min at RT then filtered. To the filtrate was added charcoal (40 gm) at RT. The mixture was stirred for 30 min at RT then filtered. To the filtrate was added charcoal (40 gm) at RT. The mixture was stirred for 30 min at RT then filtered. The pH of the clear filtrate was adjusted to 8.0 – 8.5 with IN NaOH solution and distilled off completely under vacuum below 55 °C. The residue was dissolved in 0.5 M NaCl solution and layered onto a column of Dowex® 1×2 -400 resins using a gradient of NaCl solution (0.5 to 10M). The product fractions were combined and distilled off under vacuum below 55 °C up to 1 – 2 L volume. The solid was filtered off and the clear filtrate was distilled off under vacuum below 55 °C up to slurry stage and subjected to azeotropic distillation with methanol two times. The solid residue was stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The wet solid was again stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The wet solid was again stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The above solid was dissolved in water and the pH adjusted to 4 – 4.5 with IN HC1 solution and charcoalized three times with 26 gm of charcoal at RT for 15-30 minutes and filtered off. To the clear filtrate was added 0.39 kg of NaCl, then methanol was added (35 volume) at RT and the mixture was stirred for 15-30 minutes. The solution was decanted and the sticky mass was stirred with methanol (0.65 L) at RT for 15-30 minutes. The solid was filtered off and dissolved in water, and the pH adjusted to 8 – 8.5 with IN NaOH solution. The solution was filtered through 0.45 micron paper & distilled off completely under vacuum below 55°C. The solution was subjected to azeotropic distillation with methanol to give highly pure fondaparinux sodium (97.17 gm) (HPLC purity 99.7%).
SOR Results
Three batches of product made in accordance with the present processes provided the following stereoisomeric optical rotation results:
Specification: Between +50.0° and +60.0°.
Batch- 1 = +55.1°
Batch-2 = +55.7° Batch-3 = +55.4°.
INTERMEDIATES
Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; MS is molecular sieve; DMF is dimethyl formamide; Bn is benzyl; MDC is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; MeOH is methanol; RT is room temperature; Ac2O is acetic anhydride; HBr is hydrogen bromide; EtOAc is ethyl acetate; Cbz is benzyloxycarbonyl; CADS is chloro acetyl disaccharide; HDS is hydroxy disaccharide; NMP is N-methylpyrrolidone.
Methyl 3-O-benzyl-4-O-monochloro acetyl-β-L-idopyranuronate

Route of Synthesis for α-Methyl-6-o-acetyl-3-o-benzyl-2-(benzyloxy carbonyl)amino-2-deoxy-α-D-glucopyranoside

Methyl 6-O-acetyl-3-O-benzyl-2-(benzyloxy carbonyl)amino-2-deoxy-4-O-(methyl-2-O acetyl-3-O-benzyl-α-L-idopyranosyluronate)-glucopyranoside

Route of Synthesis for 1,6-Anhydro-2-azido-3-O-acetyl-2-deoxy-beta-D-glucopyranose

Route of synthesis for Methyl 2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranuronate

Route of synthesis for 3-O-Acetyl-1,6-anhydro-2-azido-4-O-2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranosyl methyluronate-beta-D-glucopyranose
(or)
3-O-Acetyl-1,6-anhydro-2-azido-2-deoxy-4-O-(methyl 2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranosyluronate)-beta-D-glucopyranose

Route of Synthesis for 1,6-Anhydro-2-azido-3,4-di-O-benzyl-2-deoxy-beta-D-glucopyranose

Synthesis of Disaccharide XLIII
Disaccharide XLIII was prepared in 2 synthetic steps from CADS sugar (XL) using the following procedure:

CADS sugar XL was acetylated at the anomeric carbon using AC2O and TFA to give acetyl derivative XLII. This step was carried out using the reactants CADS, AC2O and TFA, stirring in an ice water bath for about 5-24 hours, preferably 20 hours, and evaporating to residue under vacuum. Residue was recrystallized in ether. Acetyl CADS (XLII) was brominated at the anomeric carbon using titanium tetra bromide in MDC andethylacetate and stirring at 20° C.-50° C. for 6-16 hours, preferably 6 hours, to give the bromo derivative, (XLIII) after work-up and recrystallization from solvent/alcohol.
Synthesis of the Monosaccharide (XLV)
The monosaccharide (XLV) was prepared in 2 synthetic steps from monomer (XLI) using the following procedure:

Mono sugar (XLI) was acetylated at the anomeric carbon using AC2O and TFA to give acetyl derivative (XLIV). This step was carried out using the reactants Mono sugar (XLI), AC2O and TFA, stirring in an ice water bath for about 5-24 hours, preferably 24 hours, and evaporating to residue under vacuum. Residue was recrystallized in ether. Acetyl Mono sugar (XLIV) was brominated at the anomeric carbon using titanium tetra bromide in MDC and ethyl acetate and stirring at 20° C.-50° C. for 6-20 hours, preferably 16 hours, to give the bromo derivative, (XLV) after work-up and recrystallization from ether.
Synthesis of the Hydroxy Tetrasaccharide (XLVII)
The hydroxy tetrasaccharide (XLVII) was prepared in 2 synthetic steps from disaccharide (XLIII) and HDS (XX) using the following procedure:

Disaccharide (XLIII), was coupled with disaccharide (XX) in the presence of silver carbonate, silver per chlorate and 4 A° MS in MDC and stirred at ambient temperature for 5-12 hrs, preferably 4-6 hours, in the dark followed by work-up and purification in water/methanol to give the tetrasaccharide (XLVI). The d echloroacetylation of tetrasaccharide (XLVI) was carried out in THF, ethanol and pyridine in the presence of thiourea at reflux for 6 to 20 hrs, preferably 12 hours, to give the hydroxy tetrasaccharide (XLVIII).
Synthesis of the Pentasaccharide (XLVIII)
The pentasaccharide (XLVIII) was prepared in 2 synthetic steps from monosaccharide (XLV) and tetrasaccharide (XLVII) using the following procedure:

Monosaccharide (XLV), was coupled with tetrasaccharide (XLVII) in the presence of 2,4,6-collidine, silver triflate and 4 A° MS in MDC and stirred at −10° C. to −20° C. for 1 hr in the dark followed by work-up and purification by column chromatography to give the pentasaccharide (XLVIII).
Synthesis of OS Pentasaccharide (L)
The OS pentasaccharide (L) was prepared in 2 synthetic steps from pentasaccharide (XLVIII) using the following procedure:

Pentasaccharide (XLVIII) was deacetylated in the presence of NaOH in mixture of solvents of MDC, methanol and water at 0° C. to 35° C., for 1-2 hrs followed by work-up and distillation to obtain deacetylated pentasaccharide (XLIX) which was subjected to O-sulfonation in DMF in the presence of SO3-trimethylamine (TMA) at 50° C. to 100° C., preferably 50° C.-55° C., for 6-24 hrs, preferably 12 hours, followed by salt removal through Sephadex® resin and column chromatography purification, then pH adjustment by dilute NaOH to give OS pentasaccharide (L).
…………………………
INTERMEDIATE
highly pure 4-Ο-β-ϋ- glucopyranosyl- 1 ,6-anhydro- -D-glucopyranose

Example 1 : Preparation and purification of 4-0- -D-grucopyranosyl-L6-anhvdro- -D- glucopyranose
A solution of pentachlorophenyl 2,3,6,2′,3′,4′,6′-hepta-(9-acetyl- -D-ceilobioside represented by Formula I;

(400 g) in isopropyl alcohol (4 L) at ambient temperature was cooled to 2°C to 5°C and pulverized potassium hydroxide (355 g) was added to it. This reaction mixture was stirred and the temperature was allowed to rise to ambient temperature. At ambient temperature, the mixture was stirred until the reaction was complete (about 2 hours). The mixture was then heated to 50°C to 55°C and stirred for 30 minutes. The solid obtained was filtered and washed with isopropyl alcohol (400 mL). The solid was stirred with isopropyl alcohol (2.8 L) at 50°C for 30 minutes followed by filtering and washing with isopropyl alcohol (400 mL). The resultant solid was suspended into methanol (800 mL to 1600 mL) followed by cooling to 2°C to 5°C. The pH of the suspension was adjusted to 2 to 3 using 15% methanolic hydrochloride. The solid so obtained was filtered and washed with methanol (400 mL). Solvent was recovered from the filtrate to dryness under vacuum to obtain the pure compound of Formula II as foamy solid.
Yield: 142 g
Example 2: Preparation and purification of 4-Q- -D-grucopyranosyl-l,6-anhvdro- -D- glucopyranose
A solution of pentachlorophenyl 2,3,6,2 ,3 ^ ^‘-hepta-O-acetyl- -D-cellobioside of Formula I (100 g) in methanol (300 mL) at ambient temperature was cooled to 2°C to 5°C and pulverized potassium hydroxide (88.6 g) was added to it. This reaction mixture was stirred and the temperature was allowed to rise to ambient temperature. At ambient temperature, the mixture was stirred until the reaction was complete (about 2 hours). The mixture was cooled to 2°C to 5°C and 15% methanolic hydrogen chloride was added to it until the pH of the mixture reached 2 to 3. At this pH, the reaction mixture was filtered and the residual solid was washed with methanol (100 mL). The solvent was recovered from the filtrate under vacuum. The solid material so obtained was stirred with dichloromethane (500 mL) followed by removal of solvent through decantation/filtration. The resultant solid was stirred with isopropyl alcohol (500 mL), filtered and dried to obtain the pure compound of Formula II.
Yield: 29 g
………………………
SYNTHESIS
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:
Conversion of FPP (also referred to a Fully Protected Pentamer) to FondaparinuxSodium:

Reagents: 1. NaOH, H202, LiOH, Dioxane, RT, 24-48 h; 2. Py.S03, DMF, 60°C, 2h, CG-161 purification; 3. 10% Pd/C, H2, 72h; 4. (a) Py.S03, NaOH, NH4OAc, 12h, (b) HiQ NH4OAc/ NaCl ion-exchange, Sephadex Desalt and (c) HiQ NaCl ion-exchange, Sephadex Desalt. The ester moieties in EDCBA Pentamer-CB were hydrolyzed with sodium and lithium hydroxide in the presence of hydrogen peroxide in dioxane mixing at room temperature for 24- 48 hours to give the pentasaccharide intermediate API1-CB. The five hydroxyl moieties in API1-CB were sulfated using a pyridine-sulfur trioxide complex in dimethylformamide, mixing at 60°C for 2 hours and then purified using column chromatography (CG-161), to give the pentasulfated pentasaccharide API2-CB. The intermediate API2-CB was then hydrogenated to reduce the three azides on sugars E, C and A to amines and the reductive deprotection of the six benzyl ethers to their corresponding hydroxyl groups to form the intermediate API3-CB. This transformation occurs by reacting API2-CB with 10% palladium/carbon catalyst with hydrogen gas for 72 hours. The three amines on API3-CB were then sulfated using the pyridine-sulfur trioxide complex in sodium hydroxide and ammonium acetate, allowing the reaction to proceed for 12 hours . The crude fondaparinux is purified and is subsequently converted to its salt form. The crude mixture was purified using an ion-exchange chromatographic column (HiQ resin) followed by desalting using a size exclusion resin or gel filtration (Biorad Sephadex G25) to give the final product, fondaparinux sodium.
Preparation of Fondaparinux Sodium – Step 4: N-Sulfation of API-3-CB:
Methyl 0-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D-glucopyranosyl-(l→4)-0^-D- glucopyranuronosyl-(l→4)-0-2-deoxy-3,6-di-0-sulfo-2-(sulfoamino)-a-D-glucopyranosyl- (l→4)-0-2-0-sulfo-a-L-idopyranuronosyl-(l→4)-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D- glucopyranoside, decasodium salt
To a solution of 25.4 gram (16.80 mmol, leq) of API-3-CB in 847 mL of water was slowly added 66.85 gram (446.88 mmol, 25eq) of sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2N sodium hydroxide solution. The reaction was allowed to stir for 4 hours at pH 9.0 – 9.5. When reaction was completed, the pH was adjusted 7.0 by using 70 mL of 50 mmol Ammonium acetate solution pH -3.5. The resulting N-Sulfated Cellobiose mixture was purified using Ion-Exchange
Chromatographic Column followed by desalting using size exclusion resin to gave gram ( %) of the purified Fondaparinux Sodium form.
To a solution of 942 g (0.63 mol) of API3 in 46 L of water was slowly added 3.25 Kg (20.4 mol, 32 eq) of Sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2 N sodium hydroxide solution. The reaction was allowed to stir for 4-6 hours at pH 9.0-9.5. When reaction was complete, the pH was adjusted to pH 7.0 using 50 mM solution of Ammonium acetate at pH 3.5. The resulting N- sulfated EDCBA(OS03)5(NHS03)3 mixture was purified using Ion-Exchange Chromatographic Column (Varian Preparative 15 cm HiQ Column) followed by desalting using a size exclusion resin or gel filtration (Biorad G25). The resulting mixture was then treated with activated charcoal and the purification by ion-exchange and desalting were repeated to give 516 g (47.6% yield) of the purified Fondaparinux sodium form.
INT
SCHEME 1 – Synthesis of Monomer A-2 & AMod5 fBuildinq Block Al

Reagents: 1. NaOMe, MeOH, RT, 2hr, 50wx resin; 2. (Bu3Sn)20 (0.8equiv), ACN, MS, reflux, 3h; 3.l2 (1.5 equiv), 5°C to RT, 2h; 4. NaH (2 equiv), DMF, p-MeOC6H4CH2Br (PMB-Br, 2.5 equiv), -20°C to RT, 2h; 5. NaN3, DMF, 120°C, 12h; 6. NaH, DMF, BnBr, 0°C to RT, 3h.; 7. BF3.Et20, Ac20, DCM, -20°C to RT, 3h; 8. (a) TMS-I, TBAI, RT, 2h; (b) DIPEA, MeOH, 16h, RT; 9. NaOMe, Dowex 50WX8-100 resin H+ form, RT, 3h; 10. Pyridine, Bz-CI, -40°C to -10°C, 2h;
Scheme 2 – Synthesis of Monomer B-1 and BMod6 fBuildinq Block B1

Reagents: 1. NaH, BnBr, THF, DMF, 0° to 65°C, 3h; 2. 66% Acetic Acid/H20, 40 °C, 16h; 3. Nal04, (Bu)4NBr, DCM, H20, Dark, 3h; 4. (PhS)3CH, n-BuLi, THF, -78 °C, 3h; 5. CuCI2/CuO, MeOH, H20, 3h; 6. 90% TFA/H20, DCM, RT, 2h; 7. DMF, CSA 2-methoxypropene, 0° to RT, 16hrs; MeOH, TEA. 8. Lev20, DIPEA, RT, 16h; 9. 90% TFA, RT, 4h; 10. Imidazole, TBDPSi-CI, RT, 3h; 11. Pyridine, BzCI, RT, 3h; 12. TBAF, RT, 3h; 13. TCA, DBU, RT, 2h; Also see, e.g., Bonnaffe et al., Tetrahedron Lett., 41, 307-311, 2000; Bonnaffe et al., Carbohydr. Res., 2003, 338, 681-686, 2003; and Seeberger et al., J. Org. Chem., 2003, 68, 7559- 7561, 2003.
……………………..
Carbohydrate Research, 2012 , vol. 361, p. 155 – 161
1H NMR (D2O) δ: 5.68 (d, J = 3.8 Hz, 1H, H-1D), 5.56 (d, J = 3.4 Hz, 1H, H-1F), 5.24 (d, J = 3.8 Hz, 1H, H-1G), 5.07 (d, J = 3.5 Hz, 1H, H-1H), 4.68 (d, J = 7.9 Hz, 1H, H-5G), 4.54 (dd, J = 11.4, 2.2 Hz, 1H, H-1E), 4.48-4.34 (m, 6H, H-6F, 6H, 6’H, 6D, 3E, 2G), 4.33-4.30 (m, 1H, H-6’F), 4.25-4.17 (m, 4H, H-4G, 3G, 6’D, 5F), 4.06-3.98 (m, 2H, H-4F, 5H), 3.94 (dd, J = 9.7, 2.2 Hz, 1H, H-5D), 3.92-3.86 (m, 2H,H-3E, H-4E), 3.85-3.80 (m, 2H, H-5E, 4H), 3.73-3.60 (m, 3H, H-3H, 3D, 4D), 3.53-3.44 (m, 2H, H-2F, H-2E), 3.47 (s, 3H, OMe), 3.34 (dd, J = 10.2, 3.7 Hz, 1H, H-2H), 3.31(dd, J = 10.2, 3.7 Hz, 1H, H-2D)

FONDAPARINUX
……………………………………..
Synthesis of intermediates
Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; ACN is acetonitrile; MS is molecular sieves; DMF is dimethyl formamide; PMB is p-methoxybenzyl; Bn is benzyl; DCM is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; CSA is camphor sulfonic acid; TEA is triethylamine; MeOH is methanol; DMAP is dimethylaminopyridine; RT is room temperature; CAN is ceric ammonium nitrate; Ac2O is acetic anhydride; HBr is hydrogen bromide; TEMPO is tetramethylpiperidine-N-oxide; TBACl is tetrabutyl ammonium chloride; EtOAc is ethyl acetate; HOBT is hydroxybenzotriazole; DCC is dicyclohexylcarbodiimide; Lev is levunlinyl; TBDPS is tertiary-butyl diphenylsilyl; TCA is trichloroacetonitrile; O-TCA is O-trichloroacetimidate; Lev2O is levulinic anhydride; DIPEA is diisopropylethylamine; Bz is benzoyl; TBAF is tetrabutylammonium fluoride; DBU is diazabicycloundecane; BF3.Et2O is boron trifluoride etherate; TMSI is trimethylsilyl iodide; TBAI is tetrabutylammonium iodide; TES-Tf is triethylsilyl trifluoromethanesulfonate (triethylsilyl triflate); DHP is dihydropyran; PTS is p-toluenesulfonic acid.
The monomers used in the processes described herein may be prepared as described in the art, or can be prepared using the methods described herein.
The synthesis of Monomer A-2 (CAS Registry Number 134221-42-4) has been described in the following references: Arndt et al., Organic Letters, 5(22), 4179-4182, 2003; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; and Sakairi et al., Journal of the Chemical Society, Chemical Communications, (5), 289-90, 1991, and the references cited therein, which are hereby incorporated by reference in their entireties.
Monomer C(CAS Registry Number 87326-68-9) can be synthesized using the methods described in the following references: Ganguli et al., Tetrahedron: Asymmetry, 16(2), 411-424, 2005; Izumi et al., Journal of Organic Chemistry, 62(4), 992-998, 1997; Van Boeckel et al., Recueil: Journal of the Royal Netherlands Chemical Society, 102(9), 415-16, 1983; Wessel et al.,Helvetica Chimica Acta, 72(6), 1268-77, 1989; Petitou et al., U.S. Pat. No. 4,818,816 and references cited therein, which are hereby incorporated by reference in their entireties.
Monomer E (CAS Registry Number 55682-48-9) can be synthesized using the methods described in the following literature references: Hawley et al., European Journal of Organic Chemistry, (12), 1925-1936, 2002; Dondoni et al., Journal of Organic Chemistry, 67(13), 4475-4486, 2002; Van der Klein et al., Tetrahedron, 48(22), 4649-58, 1992; Hori et al., Journal of Organic Chemistry, 54(6), 1346-53, 1989; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; Tailler et al.,Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio–Organic Chemistry, (23), 3163-4, (1972-1999) (1992); Paulsen et al., Chemische Berichte, 111(6), 2334-47, 1978; Dasgupta et al., Synthesis, (8), 626-8, 1988; Paulsen et al., Angewandte Chemie, 87(15), 547-8, 1975; and references cited therein, which are hereby incorporated by reference in their entireties.
Monomer B-1 (CAS Registry Number 444118-44-9) can be synthesized using the methods described in the following literature references: Lohman et al., Journal of Organic Chemistry, 68(19), 7559-7561, 2003; Orgueira et al., Chemistry—A European Journal, 9(1), 140-169, 2003; Manabe et al., Journal of the American Chemical Society, 128(33), 10666-10667, 2006; Orgueira et al., Angewandte Chemie, International Edition, 41(12), 2128-2131, 2002; and references cited therein, which are hereby incorporated by reference in their entireties.
Synthesis of Monomer D
Monomer D was prepared in 8 synthetic steps from glucose pentaacetate using the following procedure:
Pentaacetate SM-B was brominated at the anomeric carbon using HBr in acetic acid to give bromide derivative IntD1. This step was carried out using the reactants SM-B, 33% hydrogen bromide, acetic acid and dichloromethane, stirring in an ice water bath for about 3 hours and evaporating at room temperature. IntD1 was reductively cyclized with sodium borohydride and tetrabutylammonium iodide in acetonitrile using 3 Å molecular sieves as dehydrating agent and stirring at 40° C. for 16 hours to give the acetal derivative, IntD2. The three acetyl groups in IntD2 were hydrolyzed by heating with sodium methoxide in methanol at 50° C. for 3 hours and the reaction mixture was neutralized using Dowex 50WX8-100 resin (Aldrich) in the acid form to give the trihydroxy acetal derivative IntD3.
The C4 and C6 hydroxyls of IntD3 were protected by mixing with benzaldehyde dimethyl acetate and camphor sulphonic acid at 50° C. for 2 hours to give the benzylidene-acetal derivative IntD4. The free hydroxyl at the C3 position of IntD4 was deprotonated with sodium hydride in THF as solvent at 0° C. and alkylated with benzyl bromide in THF, and allowing the reaction mixture to warm to room temperature with stirring to give the benzyl ether IntD5. The benzylidene moiety of IntD5 was deprotected by adding trifluoroacetic acid in dichloromethane at 0° C. and allowing it to warm to room temperature for 16 hours to give IntD6 with a primary hydroxyl group. IntD6 was then oxidized with TEMPO (2,2,6,6-tetramethyl-1-piperidine-N-oxide) in the presence of tetrabutylammonium chloride, sodium bromide, ethyl acetate, sodium chlorate and sodium bicarbonate, with stirring at room temperature for 16 hours to form the carboxylic acid derivative IntD7. The acid IntD7 was esterified with benzyl alcohol and dicyclohexylcarbodiimide (other reactants being hydroxybenzotriazole and triethylamine) with stirring at room temperature for 16 hours to give Monomer D.
Synthesis of the BA Dimer
The BA Dimer was prepared in 12 synthetic steps from Monomer B1 and Monomer A2 using the following procedure:
The C4-hydroxyl of Monomer B-1 was levulinated using levulinic anhydride and diisopropylethylamine (DIPEA) with mixing at room temperature for 16 hours to give the levulinate ester BMod1, which was followed by hydrolysis of the acetonide with 90% trifluoroacetic acid and mixing at room temperature for 4 hours to give the diol BMod2. The C1 hydroxyl of the diol BMod2 was silylated with tert-butyldiphenylsilylchloride by mixing at room temperature for 3 hours to give silyl derivative BMod3. The C2-hydroxyl was then benzoylated with benzoyl chloride in pyridine, and mixed at room temperature for 3 hours to give compound BMod4. The silyl group on BMod4 was then deprotected with tert-butyl ammonium fluoride and mixing at room temperature for 3 hours to give the C1-hydroyl BMod5. The C1-hydroxyl is then allowed to react with trichloroacetonitrile in the presence of diazobicycloundecane (DBU) and mixing at room temperature for 2 hours to give the trichloroacetamidate (TCA) derivative BMod6, which suitable for coupling, for example with Monomer A-2.
Monomer A-2 was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1.
Monomer A2 was prepared for the coupling reaction by opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1. This transformation occurs using boron trifluoride etherate, acetic anhydride and dichloromethane, between −20° C. and room temperature for 3 hours. The C1-Acetate of AMod1 was then hydrolyzed and methylated in two steps to give the diacetate AMod3. That is, first AMod1 was reacted with trimethylsilyl iodide and mixed at room temperature for 2 hours, then reacted with and tetrabutyl ammonium iodide. This mixture was reacted with diisoproylethylamine and methanol and stirred for 16 hours at room temperature, thus forming AMod3. The C4 and C6 acetates of AMod3 are hydrolyzed with sodium methoxide to give the diol Amod4. The AMod3 mixture was also subjected to mixing at room temperature for 3 hours with Dowex 50 Wx4x8-100 resin in the acid form for neutralization. This formed Amod4. The C6-hydroxyl of AMod4 is then benzoylated by treating with benzoyl chloride in pyridine at −40° C. and then allowing it to warm up to −10° C. over 2 hours to give AMod5.
Coupling of monomer AMod5 with the free C4-hydroxyl group of BMod6 was performed in the presence of BF3.Et2O and dichloromethane with mixing between −20° C. and room temperature for 3 hours to provide disaccharide BA1. The C4-levulinyl moiety of the disaccharide was then hydrolyzed with hydrazine to give the BA Dimer, which is suitable for subsequent coupling reactions.
Synthesis of EDC Trimer
The EDC Trimer was prepared in 10 synthetic steps from Monomer E, Monomer D and Monomer C using the following procedure:
Monomer E was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give diacetate EMod1. This occurs by the addition of Monomer E with boron trifluoride etherate, acetic anhydride and dichloromethane at −10° C., and allowing the reaction to warm to room temperature with stirring for 3 hours. The C1-Acetate of EMod1 is then hydrolyzed to give the alcohol, EMod2. This occurs by reacting Emod1 with hydrazine acetate and dimethylformamide and mixing at room temperature for 3 hours. The C1-hydroxyl of Emod2 is then reacted with trichloroacetonitrile to give the trichloro acetamidate (TCA) derivative EMod3 suitable for coupling, which reaction also employs diazabicycloundecane and dichloromethane and mixing at room temperature for 2 hours.
Monomer D, having a free C4-hydroxyl group, was coupled with monomer EMod3 in the presence of triethylsilyl triflate with mixing at −40° C. for 2 hours to give the disaccharide ED Dimer. The acetal on ring sugar D of the ED Dimer is hydrolyzed to give the C1,C2-diol ED1. This occurs by reacting the ED Dimer with 90% trifluoro acetic acid and mixing at room temperature for 4 hours. The C1-hydroxyl moiety of ED1 was then silylated with tert-butyldiphenylsilyl chloride to give the silyl derivative ED2. The C2-hydroxyl of ED2 was then allowed to react with levulinic anhydride in the presence of dimethylaminopyridine (DMAP) and diethylisopropylamine for approximately 16 hours to give the levulinate ester ED3. The TBDPS moiety is then deprotected by removal with tert-butylammonium fluoride in acetic acid with mixing at room temperature for 3 hours to give ED4 having a C1-hydroxyl. The C1-hydroxyl moiety of ED4 was then allowed to react with trichloroacetonitrile to give the TCA derivative ED5, which is suitable for coupling.
The C1-hydroxyl moiety of ED4 is then allowed to react with trichloroacetonitrile to give the TCA derivative ED5 suitable for coupling using diazabicycloundecane and dichloromethane, and mixing at room temperature for 2 hours. Monomer C, having a free C4-hydroxyl group, was then coupled with the disaccharide ED5 in the presence of triethylsilyl triflate and mixed at −20° C. for 2 hours to give the trisaccharide EDC Trimer.
Synthesis of the EDCBA Pentamer
The EDCBA Pentamer was prepared using the following procedure:
The preparation of EDCBA Pentamer is accomplished in two parts as follows. In part 1, the EDC Trimer, a diacetate intermediate, is prepared for the coupling reaction with Dimer BA by initially opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the tetraacetate derivative EDC1. This occurs by reacting the EDC Trimer with boron trifluoride etherate, acetic anhydride and dichlormethane and stirring between −10° C. and room temperature for 3 hours. The C1-Acetate of EDC1 is then hydrolyzed to give the alcohol, EDC2, by reacting EDC1 with benzylamine [BnNH2] and tetrahydrofuran and mixing at −10° C. for 3 hours. The C1-hydroxyl of EDC2 is then reacted with trichloroacetonitrile and diazabicycloundecane, with mixing at room temperature for 2 hours, to give the trichloro acetamidate (TCA) derivative EDC3 suitable for coupling.
In Part 2 of the EDCBA Pentameter synthesis, the Dimer BA, having a free C4-hydroxyl group, is coupled with trisaccharide EDC3 in the presence of triethylsilyltriflate at −30° C. mixing for 2 hours to give the pentasaccharide EDCBA1. The levulinyl ester on C2 of sugar D in EDCBA1 is hydrolyzed with a mixture of deprotecting agents, hydrazine hydrate and hydrazine acetate and stiffing at room temperature for 3 hours to give the C2-hydroxyl containing intermediate EDCBA2. The C2-hydroxyl moiety on sugar D of EDCBA2 is then alkylated with dihydropyran (DHP) in the presence of camphor sulfonic acid (CSA) and tetrahydrofuran with mixing at room temperature for 3 hours to give the tetrahydropyranyl ether (THP) derivative, EDCBA Pentamer.
………………………………
Intermediates listed on the internet
Fondaparinux sodium Intermediates
Fondaparinux sodium N-4

……………………………….
Fondaparinux sodium N-3
114903-05-8

a-D-Glucopyranoside, Methyl O-2-azido-2-deoxy-3,4-bis-O-(phenylMethyl)-a-D-glucopyranosyl-(14) -O-2,3-bis-O-(phenylMethyl)-b-D-glucopyranuronosyl-(14)-O-2-azido- 2-deoxy-a-D-glucopyranosyl-(14)-O-3-O-(phenylMethyl)-a-L-idopyranu ronosyl-(14)-2-deoxy-2
FSC

114903-05-8

|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||
|
|||||||
| Description | |||||||
|
|||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
References
- “Medscape.com”. Retrieved 2009-01-23.
- “NEJM — Comparison of Fondaparinux and Enoxaparin in Acute Coronary Syndromes”. Retrieved 2009-01-23.
- Peters RJ, Joyner C, Bassand JP, et al. (February 2008). “The role of fondaparinux as an adjunct to thrombolytic therapy in acute myocardial infarction: a subgroup analysis of the OASIS-6 trial”.Eur. Heart J. 29 (3): 324–31. doi:10.1093/eurheartj/ehm616. PMID 18245119.
- WO 2013003001
- Synthesis of heparin fragments: A methyl alpha-pentaoside with high affinity for antithrombin III
Carbohydr Res 1987, 167: 67 - A fast and effective hydrogenation process of protected pentasaccharide: A key step in the synthesis of fondaparinux sodiumOrg Process Res Dev 2013, 17: 869, http://pubs.acs.org/doi/full/10.1021/op300367c
- WO 2012047174
- US 2012116066
- WO 2013011460 RANBAXY
- WO 2013115817
- The unique antithrombin III binding domain of heparin: A lead to new synthetic antithrombotics
Angew Chem Int Ed Engl 1993, 32(12): 1671 - Bioorganic and Medicinal Chemistry Letters, 1(2), p. 95-98 (1991).
- Carbohydrate Research, 101, p. 148-151 (1982),
- Chemistry – A European Journal, 2012 , vol. 18, 34 pg. 10643 – 10652
- Carbohydrate Research, 2012 , vol. 361, p. 155 – 161
- Analytical Chemistry, 2006 , vol. 78, 6 pg. 1774 – 1779
PATENTS
| US4818816 * | Oct 26, 1987 | Apr 4, 1989 | Choay, S.A. | Process for the organic synthesis of oligosaccharides and derivatives thereof |
| US6376663 * | Nov 29, 1996 | Apr 23, 2002 | Macquarie Research Ltd. | Desalting and purification of oligosaccharides and their derivatives |
| US7541445 * | Sep 6, 2002 | Jun 2, 2009 | Alchemia Limited | Synthetic heparin pentasaccharides |
| US20040048785 * | Jun 18, 2003 | Mar 11, 2004 | Societe L’oreal S.A. | C-glycoside compounds for stimulating the synthesis of glycosaminoglycans |
| US20040149200 * | Jun 11, 2002 | Aug 5, 2004 | Tsuyoshi Shimose | Crystals of an oligosaccharides and process for preparation thereof |
| US20110105418 * | Jul 30, 2010 | May 5, 2011 | Reliable Biopharmaceutical Corporation | Process for preparing fondaparinux sodium and intermediates useful in the synthesis thereof |
| WO2011014793A2 * | Jul 30, 2010 | Feb 3, 2011 | Reliable Biopharmaceutical Corporation | Process for preparing fondaparinux sodium and intermediates useful in the synthesis thereof | |
| AU2008200616A1 | Title not available | ||||
| JPS63218691A * | Title not available | ||||
| US4818816 | Oct 26, 1987 | Apr 4, 1989 | Choay, S.A. | Process for the organic synthesis of oligosaccharides and derivatives thereof | |
| US7468358 | Oct 27, 2004 | Dec 23, 2008 | Paringenix, Inc. | Method and medicament for sulfated polysaccharide treatment of heparin-induced thrombocytopenia (HIT) syndrome | |
| US84771910 | Title not available | ||||
| USPP23055709 | Title not available |
FONDAPARINUX


The three specialties available in the United States – dalteparin (Fragmin, Pfizer), enoxaparin (Lovenox, Sanofi-Aventis) and tinzaparin (Innohep, Bristol-Myers Squibb) – the first two are found in Brazil, enoxaparin under the names Lovenox, Cutenox and Dripanina.

Eluxadoline …Diarrhea-predominant irritable bowel syndrome
Eluxadoline
5 JAN 2014
Furiex Pharmaceuticals Inc. more than doubled in its best day of trading after its experimental drug alleviated diarrhea and abdominal pain caused by irritable bowel syndrome in two studies.
The drug eluxadoline met targets for improvements in stool consistency and abdominal pain that were developed in conjunction with U.S. and European regulators, the company said today. Furiex will apply for approval in June, Chairman Fred Eshelman said in an investor call today. He estimated annual sales of $750 million to $1 billion.
“By our math, it looks like a pretty doggone good market,” Eshelman said on the call, noting that there is only one currently approved drug available in the U.S. for the condition.
Diarrhea-predominant irritable bowel syndrome is a chronic disorder that affects about 28 million patients in the U.S. and Europe, Furiex said in the statement.Furiex said it would apply by mid-year for U.S. approval of the drug, eluxadoline, to treat diarrhea-predominant irritable bowel syndrome (IBS-d), a debilitating bowel disorder that affects about 28 million people in the United States and major European markets.
Furiex said it expected to seek European approval in early 2015.
“We believe that there are a lot of patients out there who need this drug. There is a huge unmet need,” Furiex Chief Medical Officer June Almenoff said in a telephone interview.
Currently approved drugs for IBS address constipation associated with the disorder, but there are few options for diarrhea predominant IBS.
Furiex founder and chairman Fred Eshelman said he believes the drug has the potential for blockbuster sales, which he defined as annual sales of between $750 million and $1 billion.
Eluxadoline was tested at two doses against a placebo over the course of 12 weeks to meet requirements by the U.S. Food and Drug Administration, and for 26 weeks for European health regulators, in Phase III studies involving 2,428 patients, Furiex said.
For the combined goal of improvement in abdominal pain and stool consistency for at least half the days in the study, eluxadoline achieved a statistically significant improvement at the 100 milligram and 75 mg doses through 12 weeks in both studies.
On the 26-week measure, the higher dose succeeded in both studies but the lower dose missed statistical significance in one of the two trials, according to initial results released by the company.
The success appeared to be driven by the percentage of patients reporting improvements in diarrhea, which ranged from 30 percent to 37 percent versus 22 percent and 20.9 percent for the placebo groups.
When the composite goal was broken into its two components, researchers found a numerical improvement in pain response rates that did not achieve statistical significance.
The drug appeared to be safe and well-tolerated in both studies, Furiex said. The most commonly reported side effects were constipation and nausea.
The company plans to present a far more detailed analysis of the late stage studies at an upcoming medical meeting.
“We’re very excited about the path ahead and about how this can transform patients’ lives,” Almenoff said.
Eluxadoline
5-({[(2S)-2-amino-3-(4-carbamoyl-2,6-dimethylphenyl)propanoyl][(1S)-1-(4-phenyl-1H-imidazol-2-yl)ethyl]amino}methyl)-2-methoxybenzoic acid
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
864821-90-9 CAS
JNJ-27018966
Molecular Formula: C32H35N5O5
Molecular Weight: 569.6508
Agents for Irritable Bowel Syndrome, mu-Opioid Agonists, delta-Opioid Antagonists
Mu Delta is a locally active mu opioid receptor agonist and delta opioid receptor antagonist in phase III clinical evaluation at Furiex Pharmaceuticals for the oral treatment of diarrheal predominant irritable bowel syndrome (d-IBS).
The product candidate holds an advantage over currently marketed products for this indication because it acts locally on the enteric nervous system, possibly decreasing adverse effects on the central nervous system. In 2011, fast track designation was assigned in the U.S. for the treatment of d-IBS. In 2011, Mu Delta was licensed to Furiex Pharmaceuticals by Janssen for the treatment of d-IBS, granting an option to Furiex to continue development and commercialization following phase II proof of concept studies.
The opioid receptors were identified in the mid-1970’s, and were quickly categorized into three sub-sets of receptors (mu, delta and kappa). More recently the original three types of receptors have been further divided into sub-types. Also known is that the family of opioid receptors are members of the G-protein coupled receptor (GPCR) super-family. More physiologically pertinent are the well established facts that opioid receptors are found throughout the central and peripheral nervous system of many mammalian species, including humans, and that modulation of the respective receptors can elicit numerous, albeit different, biological effects, both desirable and undesirable (D. S. Fries, “Analgesics”, inPrinciples of Medicinal Chemistry, 4th ed.; W. O. Foye, T. L. Lemke, and D. A. Williams, Eds.; Williams and Wilkins: Baltimore, Md., 1995; pp. 247-269; J. V. Aldrich, “Analgesics”, Burger’s Medicinal Chemistry and Drug Discovery, 5thEdition, Volume 3: Therapeutic Agents, John Wiley & Sons, Inc., 1996, pp. 321-441). In the most current literature, the likelihood of heterodimerization of the sub-classes of opioid receptors has been reported, with respective physiological responses yet undetermined (Pierre J. M. Riviere and Jean-Louis Junien, “Opioid receptors: Targets for new gastrointestinal drug development”, Drug Development 2000, pp. 203-238).
A couple biological effects identified for opioid modulators have led to many useful medicinal agents. Most significant are the many centrally acting mu opioid agonist modulators marketed as analgesic agents to attenuate pain (e.g., morphine), as well as peripherally acting mu agonists to regulate motility (e.g., loperamide). Currently, clinical studies are continuing to evaluate medicinal utility of selective delta, mu, and kappa modulators, as well as compounds possessing combined sub-type modulation. It is envisioned such explorations may lead to agents with new utilities, or agents with minimized adverse side effects relative to currently available agents (examples of side effects for morphine includes constipation, respiratory depression, and addiction potential). Some new GI areas where selective or mixed opioid modulators are currently being evaluated includes potential treatment for various diarrheic syndromes, motility disorders (post-operative ileus, constipation), and visceral pain (post operative pain, irritable bowel syndrome, and inflammatory bowel disorders) (Pierre J. M. Riviere and Jean-Louis Junien, “Opioid receptors: Targets for new gastrointestinal drug development” Drug Development, 2000, pp. 203-238).
Around the same time the opioid receptors were identified, the enkephalins were identified as a set of endogenous opioid ligands (D. S. Fries, “Analgesics”, inPrinciples of Medicinal Chemistry, 4th ed.; W. O. Foye; T. L. Lemke, and D. A. Williams, Eds.; Williams and Wilkins: Baltimore, Md., 1995; pp. 247-269). Schiller discovered that truncating the original pentapeptide enkephalins to simplified dipeptides yielded a series of compounds that maintained opioid activity (Schiller, P. WO 96/06855). However one potential drawback cited for such compounds is the likelihood of their inherent instability (P. W. Schiller et al., Int. J. Pept. Protein Res. 1993, 41 (3), pp. 313-316).
More recently, a series of opioid pseudopeptides containing heteroaromatic or heteroaliphatic nuclei were disclosed, however this series is reported showing a different functional profile than that described in the Schiller works. (L. H. Lazarus et al., Peptides 2000, 21, pp. 1663-1671).
Most recently, works around morphine related structures were reported by Wentland, et al, where carboxamido morphine derivatives and it’s analogs were prepared (M. P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, pp. 1717-1721; M. P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, pp. 623-626). Wentland found that substitution for the phenol moiety of the morphine related structures with a primary carboxamide led anywhere from equal activities up to 40 fold reduced activities, depending on the opioid receptor and the carboxamide. It was also revealed that any additional N-substitutions on the carboxamide significantly diminished the desired binding activity.
Compounds of the present invention have not been previously disclosed and are believed to provide advantages over related compounds by providing improved pharmacological profiles.
Opioid receptor modulators, agonists or antagonists are useful in the treatment and prevention of various mammalian disease states, for example pain and gastrointestinal disorders such as diarrheic syndromes, motility disorders including post-operative ileus and constipation, and visceral pain including post-operative pain, irritable bowel syndrome and inflammatory bowel disorders.
It is an object of the present invention to provide opioid receptor modulators. It is a further object of the invention to provide opioid receptor agonists and opioid receptor antagonists. It is an object of the present invention to provide opioid receptor ligands that are selective for each type of opioid receptor, mu, delta and kappa. It is a further object of the present invention to provide opioid receptor ligands that modulate two or three opioid receptor types, mu, delta and kappa, simultaneously.
It is an object of the invention to provide certain instant compounds that are also useful as intermediates in preparing new opioid receptor modulators. It is also an object of the invention to provide a method of treating or ameliorating a condition mediated by an opioid receptor. And, it is an object of the invention to provide a useful pharmaceutical composition comprising a compound of the present invention useful as an opioid receptor modulator.
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1 h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid is an opoid receptor modulator (mu receptor agonist and delta receptor antagonist) and may be useful for treating irritable bowel syndrome, pain or other opioid receptor disorders.
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid and methods of making this molecule are disclosed in
US application 2005/02033143. Example 9 of US application 2005/02033143 makes the hydrochloride salt of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
Applicants have discovered a process of making the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid and two novel crystals of this zwitterion. In Applicant’s hands, these novel crystals provide improved properties and can be purified at higher purity. Applicant’s new process results in improved and less costly process manufacturing conditions than the procedure disclosed in US application 2005/02033143.
………………..
FIG. 6 is the molecular structure of the zwitterion 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.

…………………..
SYNTHESIS OF 5-formyl-2- methoxy-benzoic acid methyl ester
Example 8: 2-Methoxy-5-formylbenzoic acid

Lithium hydroxide (1.04g, 0.043mol, 3eq) in water (lOmL) was added to a stirred solution of methyl 2-methoxy-5-formylbenzoate (2.8g, 0.014mol, leq) in a mixture of tetrahydrofuran (30mL) and methanol (20mL). The solution was stirred overnight, acidified to pH 1 with 10% HCl and the organic solvents removed in vacuo. The aqueous solution was extracted with ethyl acetate (lOOmL) and the organic solution washed with brine (lOOmL), then extracted with saturated aqueous sodium bicarbonate (3 x lOOmL). The basic solution was washed with ethyl acetate (lOOmL), then acidified to pH 1 with 10% HCl and back extracted with dichloromethane (3 x lOOmL). The organic solution was dried over sodium sulfate and evaporated in vacuo to give a cream coloured powder (2.01g, 77%). 1H NMR (CDC13) δ 9.99 (s, IH, O=C- H), 4.14 (s, 3H, CH3).
………………
ANALOGOUS METHOD TO PREPARE..2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester

USE 5-formyl-2- methoxy-benzoic acid methyl ester for 3,4- dimethoxybenzaldehyde, TO GET 2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester
Example 4
(3,4-Dimethoxy-benzyl)-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amine

A solution of 1-(4-phenyl-1 W-imidazol-2-yl)-ethylamine (0.061 g, 0.33 mmol) of Example 3, and 0.55 g (0.33 mmol) of 3,4-dimethoxybenzaldehyde in 5 ml_ of anhydrous methanol was stirred at room temperature for 1 h and then cooled to about 0-100C in an ice bath for 1 h. The reaction was treated carefully with 0.019 g (0.49 mmol) of sodium borohydride in one portion and maintained at about 0-100C for 21 h. Cold 2M aqueous HCI was added dropwise (30 drops), the mixture was stirred for 5 min, and then partially concentrated in vacuo unheated. The residual material was taken up in EtOAc to yield a suspension that was treated with 5 ml_ of cold 3M aqueous NaOH and stirred vigorously until clear. The phases were separated and the aqueous layer was extracted three times additional with EtOAc. The combined extracts were dried over MgSO4, filtered, and concentrated to yield (3,4-dimethoxy- benzyl)-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amine as a light yellow oil (HPLC: 87% @ 254nm and 66% @ 214 nm).
MS (ES+) (relative intensity): 338.1 (100) (M+1)
This sample was of sufficient quality to use in the next reaction without further purification.
…………………..
SYNTHESIS
In an embodiment, the present invention is directed to processes for the preparation of the compound of formula (IV)

also known as, 5-({[2-amino-3-(4-carbamoyl-2,5-dimethyl-phenyl)- propionyl]-[1 -(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy- benzoic acid
Example 1
(S)-2-ferf-Butoxycarbonylamino-3-(4-carbamoyl-2.6-dimethyl-phenyl)- propionic acid


STEP A: Trifluoromethanesulfonic acid 4-bromo-3,5-dimethyl-phenyl ester
To a cooled (0°C) solution of 4-bromo-3,5-dimethylphenol (3.05 g, 15.2 mmol) in pyridine (8 ml_) was added trifluoromethanesulfonic anhydride (5.0 g, 17.7 mmol) dropwise. After completion of addition, the resulting mixture was stirred at 0°C for 15 min, and then at room temperature overnight. The reaction was quenched by addition of water, and then extracted with EtOAc. The organic extracts were washed sequentially with water, 2N HCI (2x ), brine, and then dried over MgSO4. Filtration and evaporation to dryness yielded compound 1 b as a colorless oil.
1H NMR (300 MHz, CDCI3): δ 2.45 (6H, s), 7.00 (2H, s).
Step B: 4-Bromo-3,5-dimethylbenzoic acid
Into a solution of compound 1 b (6.57 g, 19.7 mmol) in DMF (65 ml_) were added K2CO3 (13.1 g, 94.7 mmol), Pd(OAc)2 (0.44 g, 1.97 mmol) and 1 ,1′-bis(diphenylphosphino)ferrocene (2.29 g, 4.14 mmol). The resulting mixture was bubbled in gaseous CO for 10 min and was heated to 60°C for 7.5h with a CO(9) balloon. The cooled mixture was partitioned between aqueous NaHCO3 and EtOAc, and filtered. The aqueous phase was separated, acidified with aqueous 6N HCI, extracted with EtOAc, and then dried over Na2SO4. Filtration and concentration of the filtrate yielded crude compound 1c as a brown residue, which was used in the next step without further purification. STEP C: Method A: 4-Bromo-3,5-dimethyl-benzamide
Into a suspension of compound 1c in DCM (40 ml_) was added SOCI2 (3.1 rnL, 42 mmol) and the mixture was heated at reflux for 2 h. Upon removal of the solvent by evaporation, the residue was dissolved in DCM (40 ml_) and then ammonium hydroxide (28% NH3 in water, 2.8 ml_) was added. The reaction mixture was heated at 5O0C for 2 h and concentrated. The residue was diluted with H2O, extracted with EtOAc, and the organic portion was dried over Na2SO4. After filtration and evaporation, the residue was purified by flash column chramotagraphy (eluent: EtOAc) to yield compound 1 d as an off-white solid.
1H NMR (300 MHz, CD3CN): δ 2.45 (6H, s), 5.94 (1 H, br s), 6.71 (1 H, br s), 7.57 (2H, s)
MS(ES+)(relative intensity): 228.0 (100%) (M+1).
Step C: Method B: 4-Bromo-3,5-dimethyl-benzamide
A mixture of compound 1 b (3.33 g, 10 mmol), PdCI2 (0.053 g, 0.3 mmol), hexamethyldisilazane (HMDS, 8.4 ml_, 40 mmol), and DPPP (0.12 g, 0.3 mmol) was bubbled with a gaseous CO for 5 min and then stirred in a CO balloon at 80°C for 4 h. To the reaction mixture was added MeOH (5 ml_). The reaction mixture was stirred for 10 min, diluted with 2N H2SO4 (200 ml_), and then extracted with EtOAc. The EtOAc extract was washed with saturated aqueous NaHCO3, brine, and then dried over Na2SO4. Filtration and evaporation of the resultant filtrate yielded a residue, which was purified by flash column chromatography (eluent: EtOAc) to yield compound 1d as a white solid.
Step D: 2-terf-Butoxycarbonylaminoacrylic acid methyl ester
To a suspension of /V-Boc-serine methyl ester (Compound 1e, 2.19 g, 10 mmol) and EDCI (2.01 g, 10.5 mmol) in DCM (70 ml_) was added CuCI (1.04 g, 10.5 mmol). The reaction mixture was stirred at room temperature for 72 h. Upon removal of the solvent, the residue was diluted with EtOAc, washed sequentially with water and brine and then dried over MgSO4. The crude product was purified by flash column chromatography (eluent: EtOAc:hexane ~1 :4) to yield compound 1f as a colorless oil.
1H NMR (300 MHz, CDCI3): δ 1.49 (9H, s), 3.83 (3H, s), 5.73 (1 H, d, J = 1.5 Hz), 6.16 (1 H1 S), 7.02 (1 H, s).
STEP E: (2)-2-fert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)acrylic acid methyl ester
A flask charged with compound 1d (0.46 g, 2.0 mmol), compound 1f (0.80 g, 4.0 mmol), tri-o-tolylphosphine (0.098 g, 0.32 mmol) and DMF (8 ml_) was purged with N2(g) 3 times. After the addition of tris(dibenzylideneacetone)dipalladium (0) (0.074 g, 0.08 mmol) and TEA (0.31 ml_, 2.2 mol), the reaction mixture was heated at 110°C for 24 h. At that time, the reaction was quenched by addition of water, and then extracted with EtOAc. The organic phase was washed with 1 N HCI, saturated aqueous NaHCO3, brine, and dried over MgSO4. The mixture was concentrated to a residue, which was purified by flash column chromatography (eluent: EtOAc:hexane~1 :1 to EtOAc only) to yield compound 1g as a white solid.
1H NMR (300 MHz, CD3OD): δ 1.36 (9H, s), 2.26 (6H, s), 3.83 (3H, s), 7.10 (1 H, s), 7.56 (2H, s); 13C NMR (75 MHz, DMSO-d6): δ 17.6, 25.7, 50.2, 78.7, 124.9, 126.4,
128.3, 131.2, 135.2, 135.5, 152.8, 164.3, 169.6;
MS (ES+) (relative intensity): 349.1 (38%)(M+1).
STEP F: (S)-2-ferf-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid methyl ester
Into a reactor charged with a solution of compound 1g (0.56 g, 1.6 mmol) in degassed MeOH (80 mL) was added [Rh(COd)(H1R-DIPAMP)J+BF4 “ under a stream of argon. The reactor was sealed and flushed with H2, stirred at 6O0C under 1000 psi of H2 for 14 days. The crude product was purified by flash column chromatography (eluent: EtOAc:hexane ~1 :1) to yield compound 1 h as a white solid. ee: >99%; 1H NMR (300 MHz, CDCI3): δ 1.36 (9H, s), 2.39 (6H, s), 3.11 (2H, J = 7.2 Hz), 3.65 (3H, s), 4.53-4.56 (1 H, m), 5.12 (1 H, d, J = 8.7 Hz), 5.65 (1 H, br s), 6.09 (1 H, br s), 7.46 (2H, s);
MS(ES+) (relative intensity): 250.9 (100) (M-BoC)+.
STEP G: (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid
Into an ice-cooled solution of compound “I h (0.22 g, 0.63 mmol) in THF (3.5 ml_) was added an aqueous LiOH solution (1 N, 3.5 ml_) and the reaction mixture stirred at 0°C. Upon completion of the reaction, the reaction mixture was concentrated and the aqueous phase was neutralized with cooled aqueous 1 N HCI at 0°C, and then extracted with EtOAc. The combined extracts were dried over Na2SO4 overnight. Filtration and evaporation of the filtrate to dryness yielded compound 1j as a white solid. 1H NMR (300 MHz, DMSO-cfe): δ 1.30 (9H, s), 2.32 (6H, s), 2.95(1 H, dd,
J= 8.8, 13.9 Hz), 3.10 (1 H, dd, J= 6.2, 14.0 Hz), 4.02-4.12 (1 H, m), 7.18-7.23 (2H, m), 7.48 (2H1 s), 7.80 (1 H, s);
MS(ES+) (relative intensity): 236.9 (6) (M-BoC)+.
Example 5
5-((r2-Amino-3-(4-carbamoyl-2.6-dimethyl-phenyl)-propionvn-n-(4-phenyl- 1 H-imidazol-2-yl)-ethvπ-aminol-methyl)-2-methoxy-benzoic acid


STEP A. 2-Methoxy-5-{[1-(4-phenyl-1 W-imidazol-2-yl)-ethylamino]-methyl}- benzoic acid methyl ester
Using the procedures described for Example 4, substituting 5-formyl-2- methoxy-benzoic acid methyl ester (WO 02/22612) for 3,4- dimethoxybenzaldehyde, 2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester was prepared.
STEP B. 5-({[2-ferf-ButoxycarbonylmethyI-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl]-[1 -(4-phenyl-1 H-imidazoI-2-yl)-ethyl]-amino}-methyl)-2- methoxy-benzoic acid methyl ester
Using the procedure of Example 3 for the conversion of Cpd 3d to Cpd 3e, substituting 2-methoxy-5-{[1-(4-phenyl-1 /-/-imidazol-2-yl)-ethylamino]- methylj-benzoic acid methyl ester for Cpd 3d and substituting 2-tert- Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionic acid for 2- tø/t-Butoxycarbonylamino-3-(4-hydroxy-2,6-dimethyl-phenyl)-propionic acid, Cpd 5a was prepared.
STEP C. 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl]-[1 -(4-phenyl-1 W-imidazol-2-yl)-ethyl]-amino}-methyl)-2- methoxy-benzoic acid
5-({[2-tørf-Butoxycarbonylmethyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)- propionyl]-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy- benzoic acid methyl ester was dissolved in an ice-chilled (0-10°C), mixed solvent system of THF (10 ml_) and MeOH (5 ml_). A LiOH H2O/water suspension (2.48 M; 3.77 ml_) was added dropwise, then the reaction was allowed to warm to room temperature and stirred overnight. The resulting mixture was cooled in an ice bath and the basic solution was neutralized with 2N citric acid until slightly acidic. The mixture was concentrated under reduced pressure to remove the volatile materials, after which time the remaining aqueous phase was extracted with EtOAc (3 x 26 ml_). These combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure to yield a pale yellowish white solid. This crude material was dissolved in a 10% MeOH/CH2CI2 solution and adsorbed onto 30 g of silica. The adsorbed material was divided and chromatographed on an ISCO normal phase column over two runs, using a 40 g Redi-Sep column for both runs. The solvent system was a gradient MeOHZCH2CI2 system as follows: Initial 100% CH2CI2, 98%-92% over 40 min; 90% over 12 min, and then 88% over 13 min. The desired product eluted cleanly between 44-61 min. The desired fractions were combined and concentrated under reduced pressure to yield 5-({[2-terf- butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4- phenyl-1 /-/-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid, Cpd 5b, as a white solid.
STEP D. 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1 – (4-phenyl-1 W-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A portion of Cpd 5b (0.27g, 0.41 mmol) was dissolved in EtOAc (39 ml_)/THF (5 ml_), filtered, and subsequently treated with gaseous HCI for 15 min. After completion of the HCI addition, the reaction was slowly warmed to room temperature and a solid precipitate formed. After 5 h the reaction appeared >97% complete by LC (@214nm; 2.56 min.). The stirring was continued over 3 d, then the solid was collected and rinsed with a small amount of EtOAc. The resulting solid was dried under high vacuum under refluxing toluene for 2.5 h to yield Cpd 5c as a white solid di-HCI salt.
Example 2
Racemic 2-terf-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethvl- phenvD-propionic acid

STEP A: Racemic 2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)propionic acid methyl ester
To a reactor charged with a solution of compound 1g (0.68 g, 1.95 mmol) in MeOH (80 mL) was added 10% Pd-C (0.5 g). The reactor was connected to a hydrogenator and shaken under 51 psi of H2 overnight. The mixture was filtered through a pad of Celite and the filtrate was concentrated to dryness to yield compound 2a as a white solid.
The 1H NMR spectrum was identical to that of (S)-2-tert- butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)propionic acid methyl ester, compound 1 h.
STEP B: Racemic 2-terf-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)propionic acid
Following the procedure described for Example 1 , STEP G (preparation of (S)-2-teAt-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid), compound 2b – racemic 2-te/?-butoxycarbonylamino-3- (4-carbamoyl-2,6-dimethyl-phenyl)propionic acid – was prepared.
…………….
POLYMORPHS
Example 1 Preparation of the zwitterion of 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A 1 L three-necked round-bottomed flask equipped with a mechanical stirrer, addition funnel and a thermocouple was charged without agitation. 34.2 g of 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid (see Example 9 of US 2005/0203143), 340 ml of acetone, and 17 ml of 204 mmolar concentrated HCl were combined in the flask. The stirring was started and the resulting slurry formed a clear solution. This solution was heated to 45° C. under vigorous stirring and aged at this temperature for a period of two hours. After the completion, the reaction mass was cooled to ambient temperature and the supernatant was removed by suction. The vessel along with the residue was rinsed with 20 ml of acetone and then removed as previously. 170 ml of water was added and the reaction mass and was aged under stirring until a homogeneus solution resulted. This solution was then added over a period of ˜½ hr to a solution of 90 ml of 1N NaOH and water. The pH was adjusted to 6.5-7.0 accordingly. The resulting slurry was aged for about 2 hrs at ambient temperature, cooled to 10-15° C., aged at that temperature for about 1 hr, and then filtered. The solid was washed with 10 ml water, air-dried for a period of 4 to 5 hrs, and then placed in a vacuum oven at 50-55° C. until the water content was less than 3%.
Example 2 Preparation of the Form α Crystal
The Form α crystal can be prepared by storing the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid at 0-25% relative humidity for 3 days. Representative PXRD, TGA, and DSC data are shown in FIGS. 1-3 respectively.
Example 3 Preparation of the Form β crystal
The Form β crystal can be prepared by storing the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid at greater than 60% relative humidity for 3 days. Representative PXRD, TGA, and DSC data are shown in FIGS. 1, 4, and 5 respectively.
…………….
SYNTHESIS
Example 9 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid

A. 2-Methoxy-5{[1-(4-phenyl-1 H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester.
Using the procedures described for Example 3, substituting 5-formyl-2-methoxy-benzoic acid methyl ester (WO 02/22612) for 3,4-dimethoxybenzaldehyde, 2-methoxy-5-{[1-(4-phenyl-1H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester was prepared.
B. 5-({[2-tert-Butoxycarbonyl methyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid methyl ester.
Using the procedure of Example 1 for the conversion of Cpd 1d to Cpd 1e, substituting 2-methoxy-5-{[1-(4-phenyl-1H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester for Cpd 1 d and substituting 2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl-propionic acid of Example 8 for 2-tert-Butoxycarbonylamino-3-(4-hydroxy-2,6-dimethyl-phenyl)-propionic acid, Cpd 9a was prepared.
C. 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[11-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
5-({[2-tert-Butoxycarbonyl methyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid methyl ester was dissolved in an ice-chilled (0-10° C.), mixed solvent system of THF (10 mL) and MeOH (5 mL). A LiOH.H2O/water suspension (2.48 M; 3.77 mL) was added dropwise, then the reaction was allowed to warm to room temperature and stirred overnight. The resulting mixture was cooled in an ice bath and the basic solution was neutralized with 2N citric acid until slightly acidic. The mixture was concentrated under reduced pressure to remove the volatile materials, after which time the remaining aqueous phase was extracted with EtOAc (3×26 mL). These combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure to give 2.26 g (146% of theory) of pale yellowish white solid. This crude material was dissolved in a 10% MeOH/CH2Cl2 solution and adsorbed onto 30 g of silica. The adsorbed material was divided and chromatographed on an ISCO normal phase column over two runs, using a 40 g Redi-Sep column for both runs. The solvent system was a gradient MeOH/CH2Cl2 system as follows: Initial 100% CH2Cl2, 98%-92% over 40 min; 90% over 12 min, and then 88% over 13 min. The desired product eluted cleanly between 44-61 min. The desired fractions were combined and concentrated under reduced pressure to yield 1.74 g (113% of theory) of 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid, Cpd 9b, as a white solid.
D. 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
A portion of Cpd 9b (0.27g, 0.41 mmol) was dissolved in EtOAc (39 mL)/THF (5 mL), filtered, and subsequently treated with gaseous HCl for 15 min. After completion of the HCl addition, the reaction was slowly warmed to room temperature and a solid precipitate formed. After 5 h the reaction appeared >97% complete by LC (@214 nm; 2.56 min.). The stirring was continued over 3 d, then the solid was collected and rinsed with a small amount of EtOAc. The resulting solid was dried under high vacuum under refluxing toluene for 2.5 h to yield 0.19 g (71%) of desired Cpd 9c as a white solid di-HCl salt.
Example 8 (S)-2-tert-Butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester

A. (S)-2-tert-Butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester. Into a cool solution of Boc-L-(2,6-diMe)Tyr-OMe (7.0 g, 21.6 mmol; Sources: Chiramer or RSP AminoAcidAnalogues) and N-phenyltrifluoromethanesulfonimide (7.9 g, 22.0 mmol) in dichloromethane (60 mL) was added triethylamine (3.25 mL, 23.3 mmol). The resulting solution was stirred at 0° C. for 1 h and slowly warmed to rt. Upon completion, the reaction was quenched by addition of water. The separated organic phase was washed with 1 N NaOH aqueous solution, water and dried over Na2SO4 overnight. After filtration and evaporation, the residue was purified by flash column chromatography (eluent: EtOAc-hexane: 3:7) to give the desired product (9.74 g, 99%) as a clear oil; 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.39 (6H, s), 3.06 (2H, d, J=7.7 Hz), 3.64 (3H, s), 4.51-4.59 (1H, m), 5.12 (1H, d, J=8.5 Hz), 6.92 (2H, s); MS (ES+) (relative intensity): 355.8 (100) (M−Boc)+.
B. (S)4-(2-tert-Butoxycarbonylamino-2-methoxycarbonylethyl)-3,5-dimethylbenzoic acid. To a suspension of (S)-2-tert-butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester (9.68 g, 21.3 mmol), K2CO3 (14.1 g, 0.102 mol), Pd(OAc)2 (0.48 g, 2.13 mmol) and 1,1′-bis(diphenylphosphino)ferrocene (2.56 g, 4.47 mmol) in DMF (48 mL) was bubbled in gaseous CO for 15 min. The mixture was heated to 60° C. for 8 h with a CO balloon. The cool mixture was partitioned between NaHCO3 and EtOAc, and filtered. The aqueous layer was separated, acidified with 10% citric acid aqueous solution, extracted with EtOAc, and finally dried over Na2SO4. Filtration and concentration of the filtrate resulted in a residue. The residue was recrystallized from EtOAc-hexanes to afford the desired product (7.05 g, 94%); 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.42 (6H, s), 3.14 (2H, J=7.4 Hz), 3.65 (3H, s), 4.57-4.59 (1H, m), 5.14 (1H, d, J=8.6 Hz), 7.75 (2H, s); MS(ES+) (relative intensity): 251.9 (100) (M−Boc)+.
C. (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid methyl ester. Into a stirring solution of (S)-4-(2-tert-butoxycarbonylamino-2-methoxycarbonylethyl)-3,5-dimethyl benzoic acid (3.00 g, 8.54 mmol), PyBOP (6.68 g, 12.8 mmol) and HOBt (1.74 g, 12.8 mmol) in DMF (36 mL) was added DIPEA (5.96 mL, 34.2 mmol) and NH4Cl (0.92 g, 17.1 mmol). The resulting mixture was stirred at rt for 40 min before being partitioned between aqueous NH4Cl solution and EtOAc. The separated organic phase was washed sequentially with 2N citric acid aqueous solution, saturated aqueous NaHCO3 solution, and brine, then dried over Na2SO4 overnight. After filtration and concentration, the residue was purified by flash column chromatography (eluent: EtOAc) to give the product. (3.00 g, 100%); 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.39 (6H, s), 3.11 (2H, J=7.2 Hz), 3.65 (3H, s), 4.53-4.56 (1H, m), 5.12 (1H, d, J=8.7 Hz), 5.65 (1H, brs), 6.09 (1H, br s), 7.46 (2H, s); MS(ES+) (relative intensity): 250.9 (100) (M−Boc)+.
D. (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid. Into an ice-cooled solution of methyl ester from Step C (2.99 g, 8.54 mmol) in THF (50 mL) was added an aqueous LiOH solution (1N, 50 mL) and stirred at 0° C. Upon consumption of the starting materials, the organic solvents were removed and the aqueous phase was neutralized with cooled 1N HCl at 0° C., and extracted with EtOAc, and dried over Na2SO4 overnight. Filtration and evaporation to dryness led to the title acid (S)-2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid (2.51 g, 87%); 1H NMR (300 MHz, DMSO-d6): δ 1.30 (9H, s), 2.32 (6H, s), 2.95 (1H, dd, J=8.8, 13.9 Hz), 3.10 (1H, dd, J=6.2, 14.0 Hz), 4.02-4.12 (1H, m), 7.18-7.23 (2H, m), 7.48 (2H, s), 7.80 (1H, s); MS(ES+) (relative intensity): 236.9 (6) (M−Boc)+.
…………………..
PATENTS
1.WO 2005090315
2..WO 2006099060
3.WO 2009009480
4. WO 2010062590
5.US 2011263868 *
|
12-24-2010
|
NOVEL COMPOUNDS AS OPIOID RECEPTOR MODULATORS
|
|
|
8-32-2010
|
Compounds as opioid receptor modulators
|
|
|
6-23-2010
|
Compounds as opioid receptor modulators
|
|
|
2-12-2010
|
PROCESS FOR THE PREPARATION OF OPIOD MODULATORS
|
|
|
12-9-2009
|
Process for the preparation of opioid modulators
|
| US7629488 * | Mar 6, 2006 | Dec 8, 2009 | Janssen Pharmaceutica N.V. | Process for the preparation of opioid modulators |
| US7741356 * | Mar 14, 2005 | Jun 22, 2010 | Janssen Pharmaceutica N.V. | Compounds as opioid receptor modulators |
| US7786158 * | Oct 24, 2007 | Aug 31, 2010 | Janssen Pharmaceutica N.V. | Compounds as opioid receptor modulators |
| US7994206 | Jul 7, 2008 | Aug 9, 2011 | Janssen Pharmaceutica, N.V. | Crystals and process of making 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid |
| CN1950342A | Mar 14, 2005 | Apr 18, 2007 | 詹森药业有限公司 | Novel compounds as opioid receptor modulators |
Update july 2015
Eluxadoline
Trade Name: Viberzi®
Research Code: JNJ-27018966, JNJ27018966, JNJ 27018966
Chemical Name: 5 – [[[(2S) -2-amino-3- [4- (aminocarbonyl) -2,6-dimethylphenyl ] -1- oxopropyl] [(1S) -1- (4-phenyl-1H-imidazol-2-yl) ethyl] amino] methyl] -2-methoxybenzoic acid
CAS No: 864821-90-9
MOA: mu opioid receptor agonist
Indication: Irritable bowel syndrome with diarrhea (IBS-D)
Approval Date: May 27, 2015 (US)
Originator: Furiex Pharmaceuticals Inc ( Furiex acquired Eluxadoline from Janssen in 2011 )
Developer: Forest Laboratories Inc. (acquired by Actavis PLC in 2014 )

FDA grants breakthrough therapy designation to Promacta (EU trade name: Revolade)
![]()
Earlier this week, Ligand Pharmaceuticals Inc. ( LGND ) announced that the U.S. Food and Drug Administration (FDA) granted breakthrough therapy designation to Promacta (EU trade name: Revolade). Ligand and its partner GlaxoSmithKline ( GSK ) are looking to get Promacta approved for the treatment of cytopenias in patients suffering from severe aplastic anemia (SAA), who are unresponsive to immunosuppressive therapy.
The FDA granted breakthrough therapy designation to Promacta based on data from an open-label phase II National Institute of Health (NIH) study (n = 43) evaluating Promacta in treatment experienced SAA patients, who showed insufficient response to immunosuppressive therapy.
The designation, which was enacted as part of the 2012 Food and Drug Administration Safety and Innovation Act, is granted to potential new treatments for serious or life-threatening diseases or conditions where the initial clinical data shows that the treatment has the potential to demonstrate substantial improvement on one or more clinically significant endpoints compared to existing treatments. The designation should help fasten the development and review process for the candidate.
We note that Promacta is already approved for the treatment of thrombocytopenia (reduced platelet count) in patients with chronic hepatitis C virus (HCV) infection to enable the initiation and maintenance of interferon-based therapy. Promacta is also approved for thrombocytopenia in patients with chronic idiopathic thrombocytopenia (ITP).
PROMACTA (eltrombopag) Tablets contain eltrombopag olamine, a small molecule thrombopoietin (TPO) receptor agonist for oral administration. Eltrombopag interacts with the transmembrane domain of the TPO receptor (also known as cMpl) leading to increased platelet production. Each tablet contains eltrombopag olamine in the amount equivalent to 12.5 mg, 25 mg, 50 mg, 75 mg, or 100 mg of eltrombopag free acid.
Eltrombopag olamine is a biphenyl hydrazone. The chemical name for eltrombopag olamine is 3′-{ (2Z)-2-[1 -(3,4-dimethylphenyl)-3-methyl-5-oxo- 1,5-dihydro-4H-pyrazol-4- ylidene]hydrazino}-2′-hydroxy-3-biphenylcarboxylic acid – 2-aminoethanol (1:2). It has the molecular formula C25H22N4O4•2(C2H7NO). The molecular weight is 564.65 for eltrombopag olamine and 442.5 for eltrombopag free acid. Eltrombopag olamine has the following structural formula:
 |
Eltrombopag olamine is practically insoluble in aqueous buffer across a pH range of 1 to 7.4, and is sparingly soluble in water.
The inactive ingredients of PROMACTA are: Tablet Core: magnesium stearate, mannitol, microcrystalline cellulose, povidone, and sodium starch glycolate. Coating: hypromellose, polyethylene glycol 400, titanium dioxide, polysorbate 80 (12.5 mg tablet), FD&C Yellow No. 6 aluminum lake (25 mg tablet), FD&C Blue No. 2 aluminum lake (50 mg tablet), Iron Oxide Red and Iron Oxide Black (75 mg tablet), or Iron Oxide Yellow and Iron Oxide Black (100 mg tablet).
Biogen’s multiple sclerosis drug Tecfidera obtains EU approval
US-based Biogen Idec has received approval from the European Commission (EC) for its Tecfidera (dimethyl fumarate) as a first-line oral treatment for people with relapsing-remitting multiple sclerosis (RRMS), the most common form of multiple sclerosis (MS).
Biogen’s multiple sclerosis drug Tecfidera obtains EU approval click here
Lysosomal Storage Disorders: Advocacy Group Receives FDA Orphan Designations
This is the second Blog Post in a series over the next week that will examine Lysosomal Storage Disorders (LSDs) in the rare disease and orphan drug space. This Blog Post presents an advocacy group that receives two FDA Orphan Drug Designations (ODDs) in 2013 for treatment of rare diseases. The chart below identifies the gene therapy that receives FDA ODD where the sponsor is a rare disease advocacy group.
Row Num | Generic Name | Designation Date | Orphan Designation |
1 | recombinant adeno- associated virus vector AAV2/rh8 expressing human B-hexosaminidase A and B subunits | 03-25-2013 | Sandhoff Disease |
2 | recombinant adenovirus vector AAV2/rh8 expressing human B-hexosaminidase A & B subunits | 03-25-2013 | Tay-Sachs Disease |
.
** “Generic Name” Column Link = Is the FDA Orphan Drug Product Designation Database Record.
The National Tay-Sachs and Allied Diseases Association (NTSAD) announces in June 2013 that the FDA…
View original post 211 more words
DR ANTHONY MELVIN CRASTO
ORGANIC SPECTROSCOPY

