
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64



Benefits of ginger in prostate cancer
Regular consumption of fruits, vegetables and Herbs & Spices are linked with some anti-cancer benefits.

Ginger (Zingiber officinale) is also coming under increasing scrutiny and is known to be an source of several bioactive phenolics (gingerols, paradols, shogaols and gingerones).
On these pages recently we have seen how Ginger has been studied and can display anti-inflammatory & antioxidant activities.
In this study, Karna et al investigated ginger extract and its growth-inhibitory and death-inductory effects in a spectrum of prostate cancer cells.
Comprehensive studies have confirmed that ginger extracts disturbed cell-cycle progression, impaired reproductive capacity, modulated cell-cycle and apoptosis regulatory molecules and induced apoptosis in human prostate cancer cells.
Tumour tissue from an animal model showed reduced proliferation index and widespread apoptosis compared with controls and did not exert any detectable toxicity in normal, rapidly dividing tissues such as gut and bone marrow.
The authors add that to the best of their knowledge…
View original post 23 more words
Researchers reverse bone loss in immune disorder

The absence of recruited neutrophils to the periodontal tissue in LAD patients leads to unrestrained local production of IL-23 and hence IL-17 and G-CSF. Increased IL-17 leads to inflammatory bone loss and dysbiosis, whereas increased G-CSF leads to excessive release of mature neutrophils from the bone marrow. In contrast, normal recruitment of neutrophils regulates the expression of the same cytokines maintaining homeostasis in terms of periodontal health and release of mature neutrophils from the bone marrow. Credit: Copyright Niki Moutsopoulos and George Hajishengallis
Patients with leukocyte adhesion deficiency, or LAD, suffer from frequent bacterial infections, including the severe gum disease known as periodontitis. These patients often lose their teeth early in life. New research by University of Pennsylvania School of Dental Medicine researchers, teaming with investigators from the National Institutes of Health, has demonstrated a method of reversing this bone loss and inflammation.
The work was led by Penn Dental Medicine’s…
View original post 671 more words
CHINESE HERBS……Atractylodes ( Bai Zhu ) can adjust gastrointestinal motility, fight ulcer, protect liver, improve immune system
Bai Zhu (Atractylodes Macrocephala)
 Awarded the title as The First Herb of Invigorating Qi and Strengthening Spleen, no doubt Bai Zhu (Atractylodes Macrocephala) lives up to that reputation thanks for its consistent performance. As one of eight well-known medicinal specialties in Zhejiang province, this Chinese herb is produced mainly in Shao Xing.
Awarded the title as The First Herb of Invigorating Qi and Strengthening Spleen, no doubt Bai Zhu (Atractylodes Macrocephala) lives up to that reputation thanks for its consistent performance. As one of eight well-known medicinal specialties in Zhejiang province, this Chinese herb is produced mainly in Shao Xing.
Given its special effect in Traditional Chinese Medicine (TCM), it is treated as an equal to Ren Shen (Ginseng). Thus an old saying goes: “Ren Shen in the north and Bai Zhu in the south.” Through the famous classic formula of Si Jun Zi Tang, Four Gentleman Decoction, a quick glance will be given to their significance. Just a quick footnote here, it is the fundamental formula for deficiency of spleen and stomach Qi, which is the inspiration source of numerous subsequent formulas aiming to tonify spleen and benefit vital energy.
What is Bai Zhu?
Also known as White Atractylodes Rhizome or Atractylodes Macrocephala Rhizome, it refers to the root of Atractylodes macrocephala Koidz., which is a perennial herb, 30 to 60 in height. Rhizome is fleshy and clenched like a fist. Stem is erect and branching in upper part. Leaves grow alternatively, 3-parted or undivided in upper stem, elliptic lobes, and margined with spinescents. This perennial flowering plant is with terminal capitulum, bell-shaped involucre, purple-red corolla, and slightly flattened ellipsoid achene. Flowering period is July to September and fruit-bearing stage is August to October.
The medicinal part is the root, which is collected in winter, dirt removed, dried over a fire or in the sun, and fibril removed. It clenches like a fist, 3 to 13cm long, and 1.5 to 7cm in diameter. The surface is grayish yellow or grayish brown in color, with tubercule and intermittent lengthwise wrinkles and fibril scars, and remnant stem base and bud scars on top. The texture is hard and difficult to break. Traverse cross section is uneven, yellowish white to light brown, and scattered with brownish yellow oil spots. It has a delicate fragrance and sweet but pungent taste. But it is sticky when chewing.
What is it used for?
Now modern researches show that it can adjust gastrointestinal motility, fight ulcer, protect liver, improve immune system, relieve stress, enhance hematopoietic function, induce diuresis, fight oxidation, slow down aging, regulate blood sugar level, and fight cancer. Compared with the traditional applications, above-mentioned findings is perfectly in line with them, which to some extent gives more scientific proof to this amazing herb.
Property and indications
From the TCM’s perspective, it is bitter, sweet, and warm in nature and goes to meridians of spleen and stomach. Main functions are to invigorate Qi and strengthen the spleen, eliminate dampness and promote diuresis, stop sweat, and prevent miscarriage. Main clinical usage and indications are lack of appetite due to spleen deficiency, abdominal distension and diarrhea, dizziness and palpitation caused by phlegm and retained fluid, edema, spontaneous sweating, and fetal irritability. Regular dosage is 6 to 12 grams.
Atractylodes ( Bai Zhu )
Atractylodes ( Bai Zhu ) 白朮 Chinese Herbs Articles, also known as dong zhu 冬朮、xia zhu 夏朮,yun zhu 云朮,tai bai zhu 台白朮,wa zhu 蛙朮,ji yabn zhu 雞眼朮. It belong to the “” family.
Atractylodes ( Bai Zhu ) 白朮 has a aromatic, slightly acrid, non toxic and sweet and it is a little sticky when chewed. It is use for treating the spleen and stomach.

Atractylodes ( Bai Zhu ) 白朮 Medical Function:
1. Digestive System
• Protects Liver: Extraction of bai zhu (by boiling with water) was given to lab mice that had liver damage caused by carbon chloride. It lessened the necrosis and mutation of liver cells, and improved the new growth of the liver cells. It lowered the glutamate-pyruvate transaminase (GPT) that was increased.
• Improves gall secretion
• Prevents ulcer of stomach
• Improves movements of intestines and bowels
2. Diuretics
3. Improves immune system
4. Anti Cancer
Laboratory tests showed that neutral oil of the vaporizing oil bai zhu could inhibit esophagus cancer cells. 10mg/ml/hour could detach all the cancer cells. 5mg/ml/hour could detach most of the cancer cells and damaged the remaining cells. The nucleus became hazy and the cells became empty bubbles. 5. Affects heart and blood vessels
6. Lowers blood sugar
7. Anti coagulation of blood
8. Anti bacteria
Atractylodes ( Bai Zhu ) 白朮 Use Cautions:
Atractylodes ( Bai Zhu ) 白朮 should be use cautiously in cases of giddiness[ yinxu ] (yin deficient).
ACTIVE COMPONENTS
The investigation of the aromatic oils is a key to understanding the atractylodes herbal materials, particularly cangzhu. Atractylodes lancea is rich in a volatile oil, making up 3.5-7% of the dried rhizome, with atractylodin, β-eudesmol, hinesol, elemol, atractylone, and β-selinene; A. chinensis and other substitute species have less essential oil. The main constituents in the essential oils from the rhizome of A. chinensis are β-eudesmol and atractylone; A. lancea also has hinesol as a major constituent. β-eudesmol is a major component of the essential oil of magnolia bark, an herb in the same Materia Medica category as cangzhu. The fraction comprising the combination of hinesol and eudesmol in A. lancea is called atractylol, and this is the reddish substance appearing on the surface of the sliced rhizome, giving the name red atractylodes.
 β-eudesmol |
 Atractylodin |
Atractylodes macrocephala (baizhu) has less essential oil than the cangzhu varieties, with only 0.35-1.35% and with atractylone as the main component, along with smaller amounts of other lactones having similar structure. The differences in chemical composition help confirm that the two herbs (cangzhu and baizhu) may have differing properties, further justifying their separation in the Materia Medica.
Since white atractylodes has little essential oil, and even less of it after being fried (the heat drives off or destroys volatile components), other active ingredients may be present to explain its functions. A component called atractylenolide (a group of sesquiterpene lactones; three noted thus far) is found in baizhu; this component increases with frying of the herb (highest in lightly fried herb, which has turned yellowish, not brown). In terms of the atractylodes effects, it is thought that these components may serve as antispasmodic agents, thus reducing intestinal contractions associated with diarrhea. Diuretic action, measured in laboratory animal experiments, has been attributed to both volatile and non-volatile compounds of atractylodes, including ß-eudesmol, sesquiterpene lactones, and polyacetylenes
Atractylodes refers mainly to Atractylodes macrocephala (macro = big; cephala = head; so, big-headed atractylodes) known in Chinese as baizhu. Less frequently used is Atractylodes lancea (lancea = lance-like, so lance-leaved atractylodes) or its less-desirable (somewhat weaker) substitutes, such as A. chinensis, A. japonicum, and A. ovata, known in Chinese as cangzhu (see plant photos below). The basic term zhu was the only one used when atractylodes was first recorded in the ancient Shennong Bencao Jing (ca. 100 A.D.); the division between these two related herb materials first occurred in the Mingyi Bielu (ca. 500 A.D.). At that time, the tuber-like rhizomes of these plants were specified as either baizhu (bai = white) and chizhu (chi = red), referring to the color observed in the sliced rhizomes, the red being due to spots of accumulated oils. Later,chizhu was renamed cangzhu (cang = gray or black), which refers to the appearance of the outer skin of the rhizome, a dark gray-black color.
A. macrocephela |
A. japonica |
A. ovata |
A. lancea |
A. chinensis |
 Rhizomes with rootlets and stems, freshly pulled Atractylodes lancea. |
 Dried, whole rhizomes of Atractylodes macrocephala with rootlets and stems removed. |
Related Chinese herbal formulas
This herb is widely used in TCM practice. Only in Shang Han Lun (Treatise on Febrile Diseases) and Jin Gui Yao Lue (Synopsis of Golden Chamber), it has been enlisted in 35 formulas. The list can be much longer if taking all later formulas into consideration. However, just a few of them are shared here just for your reference.
(1). Li Zhong Tang or Wan, from Shang Han Lun, has four ingredient herbs. The other three are Ren Shen (Ginseng), Gan Cao (Licorice), and Gan Jiang (Dried Ginger). It is mainly used for epigastric distention and pain and difficulty in urination.
(2). Si Jun Zi Tang, from Tai Ping Hui Min He Ji Ju Fang (Formulas of the Bureau of People’s Welfare Pharmacy), exchanges Fu Ling (Poria) for Gan Jiang on the basis of Li Zhong Tang or Wan. Its indications are pale complexion, loss of appetite, shortness of breath, fatigue, light-colored tongue with white coating, and weak pulse. This formula is derived from the famous Li Zhong Wan. It is well known that Gan Jiang rescues devastated yang for its warm nature while Fu Ling is much mild. Thus the whole formula has changed its nature and they turn into four gentleman.
(3). Shen Ling Bai Zhu San, from Tai Ping Hui Min He Ji Ju Fang (Formulas of the Bureau of People’s Welfare Pharmacy), is the formula that add Shan Yao (Chinese Yam), Lian Zi (Lotus Seed), Bai Bian Dou (Hyacinth Bean), Yi Yi Ren (Seeds of Job’s Tears), Sha Ren (Cardamon), and Jie Geng (Balloon Flower Rhizome) on the basis of Si Jun Zi Tang. It is typically used for excessive damp due to spleen deficiency. According to interpromotion of Five Elements, it is a typical application of reinforcing earth to generate metal. By the way, its other forms like Pian (tablet) and Wan (teapills) are popular over-the-counter drugs in China up to this day.
(4). Ban Xia Bai Zhu Tian Ma Tang, from Yi Xue Xin Wu (Medical Revelations), is mainly used for abnormal ascending of phlegm and retained fluid, palpitation caused by excessive phlegm, dizziness and headache. Besides the mentioned three herbs, others are Chen Pi (Tangerine Peel), Fu Ling (Poria), Gan Cao, Sheng Jiang (Fresh Ginger Rhizome), Da Zao (Chinese Date, Jujube), and Man Jing Zi (Vitex Fruit Seed).
REFERENCES
- Yang Shouzhong (translator), The Divine Farmer’s Materia Medica, 1998 Blue Poppy Press, Boulder, CO.
- Hsu HY and Peacher WG (editors), Shang Han Lun: The Great Classic of Chinese Medicine, 1981 Oriental Healing Arts Institute, Long Beach, CA.
- Hsu HY and Wang SY (translators), Chin Kuei You Lueh, 1983 Oriental Healing Arts Institute, Long Beach, CA.
- Mitchell C, et al. (translators), Ten Lectures on the Use of Medicinals from their Personal Experience of Jiao Shude, 2003 Paradigm Publications, Brookline, Mass.
- State Administration of Traditional Chinese Medicine, Advanced Textbook on Traditional Chinese Medicine and Pharmacology, (vol. 2) 1995-6 New World Press, Beijing.
- Sionneau P, Pao Zhi: An Introduction to the Use of Processed Chinese Medicinals, 1995 Blue Poppy Press, Boulder, CO.
- Yang Yifang, Chinese Herbal Medicines Comparisons and Characteristics, 2002 Churchill Livingstone, London.
- Ding HY, Wu, YC, and Liu HC, Phytochemical and pharmacological studies on Chinese cangzhu, Journal of the Chinese Chemical Society 2000; 47: 561-566.
- Huang Bingshan and Wang Yuxia, Thousand Formulas and Thousand Herbs of Traditional Chinese Medicine, vol. 2, 1993 Heilongjiang Education Press, Harbin.
- Sionneau P, Dui Yao: The Art of Combining Chinese Medicinals, 1997 Blue Poppy Press, Boulder, CO.
- He Shanan and Sheng Ning, Utilization and conservation of medicinal plants in China with special reference to Atractylodes lancea, 1995 Food and Agriculture Organization of the United Nations.
CHINESE HERBS…….ASTRAGALUS HUANG QI, an immune system booster, known to stimulate body´s natural production of interferon

ASTRAGALUS HUANG QI
A Chinese herb; an immune system booster, known to stimulate body´s natural production of interferon. It also helps the immune system identify rogue cells. Work with the herb in both cancer and AIDS cases has been encouraging. The MD Anderson Cancer Centre in Texas conducted research showing that taking Astragalus when having Radiotherapy doubled survival times.
Astragalus is a large genus of about 3,000 species[1] of herbs and small shrubs, belonging to the legume family Fabaceae and the subfamily Faboideae. The genus is native to temperate regions of the Northern Hemisphere. Common names include milkvetch (most species), locoweed (in North America, some species)[2] and goat’s-thorn (A. gummifer, A. tragacanthus). Some pale-flowered vetches are similar in appearance, but vetches are more vine-like.

Astragalus species are used as food plants by the larvae of some Lepidoptera species including many case-bearing moths of the genus Coleophora: C. cartilaginella, C. colutella, C. euryaula, and C. onobrychiella feed exclusively on Astragalus, C. astragalella and C. gallipennella feed exclusively on the species Astragalus glycyphyllos, and C. hippodromica is limited to Astragalus gombo.
Traditional uses
The natural gum tragacanth is made from several species of Astragalus occurring in the Middle East, including A. adscendens, A. gummifer, A. brachycalyx,[3][4] and A. tragacanthus. Also Astragalus propinquus (syn. A. membranaceus) has a history of use as a herbal medicine used in systems of traditional Chinese medicine.[5] and Persian medicine [6]
popular qi tonic (especially the wei qi), these large roots of Astragalus are sweet and slightly warm in energy. Our roots are cut from long robust plants with a nice yellow colored pith, that possess a nice sweetness when chewed.

Research
Biotechnology firms are working on deriving a telomerase activator from Astragalus. The chemical constituent cycloastragenol (also called TAT2) is being studied to help combat HIV, as well as infections associated with chronic diseases or aging.[7] However, the National Institutes of Health states: “The evidence for using astragalus for any health condition is limited. High-quality clinical trials (studies in people) are generally lacking. There is some preliminary evidence to suggest that astragalus, either alone or in combination with other herbs, may have potential benefits for the immune system, heart, and liver, and as an adjunctive therapy for cancer”.[8]
Research at the UCLA AIDS Institute focused on the function of cycloastragenol in the aging process of immune cells, and its effects on the cells’ response to viral infections. It appears to increase the production of telomerase, an enzyme that mediates the replacement of short bits of DNA known as telomeres, which play a key role in cell replication, including in cancer processes.[9]
Supplement use
Extracts of Astragalus propinquus ( syn. A. membranaceus) are marketed as life-prolonging extracts for human use. A proprietary extract of the dried root of A. membranaceus, called TA-65, “was associated with a significant age-reversal effect in the immune system, in that it led to declines in the percentage of senescent cytotoxic T cells and natural killer cells after six to twelve months of use”.[10] There are mixed data regarding Astragalus, its effects on telomerase, and cancer. For example while 80% of cancer cells utilize telomerase for their proliferation – a factor which might theoretically be exacerbated by Astragalus – the shortening of telomeres (resulting from such factors as stress and aging and possible contributors to malignancy), might also be mitigated by Astragalus. Thus, short telomeres result in chromosome instability, and the potential for telomere lengthening as a protection against cancer is possible.[11] Additionally, scientists recently reported in Molecular and Cellular Biology that cancer cells may proliferate precisely because of the lack of differentiation occurring via damaged or shortened telomere length. They propose that “forced” elongation of telomeres promotes the differentiation of cancer cells, probably reducing malignancy, which is strongly associated with a loss of cell differentiation.
Side effects and toxicology
Astragalus may interact with medications that suppress the immune system, such as cyclophosphamide.[8] It may also affect blood sugar levels andblood pressure.[8] Some Astragalus species can be toxic. For example, several species native to North America contain the neurotoxin swainsonine.[8]The toxicity of Astragalus taxa varies.[12]
Ornamental use
Several species, including A. alpinus (bluish-purple flowers), A. hypoglottis (purple flowers), and A. lotoides, are grown as ornamental plants in gardens.
Astragalus ( Huang Qi ) 黃耆 Chinese Herbs Articles
Astragalus ( Huang Qi ) 黃耆 Chinese Herbs Articles also written as 黃芪 also known as: Astragali, Beg Kei, Bei Qi, Buck Qi, Huang Qi, Hwanggi, Membranous Milk Vetch, Milk Vetch, Mongolian Milk, Ogi. Astragalus membranaceus; Astragalus mongholicus. It belong to the Leguminosae or Fabaceae family.
Astragalas ( Huang Qi ) 黃耆 has a sweet taste and a warm properties and it is use for treating the spleen and lung.

Astragalus ( Huang Qi ) 黃耆 Usage:
- tonify spleen & lung Qi – raises Spleen & Stomach Qi (prolapse)
- tonify Wei Qi – stabilize exterior
- tonify Qi and blood due to loss of blood – postpartum fever
- promotes urination – Edema – discharge of pus – generates flesh
Astragalus ( Huang Qi ) 黃耆 Other Use:
1. Orally, Astragalus ( Huang Qi ) 黃耆 is used for treating the common cold and upper respiratory infections; to strengthen and regulate the immune system; and to increase the production of blood cells particularly in individuals with chronic degenerative disease or in individuals with cancer undergoing chemotherapy or radiation therapy. It is also used orally for chronic nephritis and diabetes. Astragalus is also used orally as an antibacterial and antiviral; a tonic; liver protectant; anti-inflammatory; antioxidant; and as a diuretic, vasodilator, or hypotensive agent.
2. Topically, Astragalus ( Huang Qi ) 黃耆 is used as a vasodilator and to speed healing.
3. In combination with Ligustrum lucidum (glossy privet), astragalus is used orally for treating breast, cervical, and lung cancers.
Astragalus ( Huang Qi ) 黃耆 Use Cautions:
There are many varieties of astragalus ( Huang Qi ) 黃耆. Some are toxic. The varieties used in Chinese herbal medicine is relatively safe but in rare cases it might cause rash.
Huang Qi (Astragalus membranaceus) Root
Astragalus membranaceus

Our freshly harvested root are completely chemical free and extremely high quality to preserve all of its benefits.
Traditional & Modern Use:
Huang Qi root is harvested white but becomes a pale yellow. The roots are a staple in Chinese medicine praised for its energizing effects. The root pieces can be simmered for long periods of time and served as a tea or soup but the root pieces are too tough to chew so they are not consumed unless powdered. The roots are a very powerful herb for aiding the kidneys as well as a preventative medicine for senility. Chinese holistic healers also believe strongly that the regular use of Astralagus rejuvinates debilitated patients and fights off serious disease. News studies in the West have now begun to show some amazing results in the treatment of cancer and that the root can restore normal immune function in cancer patients. Impressively, patients undergoing chemotherapy or radiotherapy recover much quicker and can live longer when using the root simultaneously to their treatments.
References
- Frodin, D. G. (2004). “History and concepts of big plant genera”. Taxon 53 (3): 753–776. doi:10.2307/4135449.
- “Astragalus (Locoweed) flowers”. Rootcellar.us. Retrieved 2013-07-05.
- [1]
- “Astragalus brachycalyx Fisch.”. Germplasm Resources Information Network (GRIN) online database. Retrieved 24 December 2010.
- “Astragalus | University of Maryland Medical Center”. Umm.edu. 2013-05-07. Retrieved 2013-07-05.
- Zargary, A. Medicinal plants. 5th Edition.Tehran: Tehran University Publications 1990; pp. 312-314
- “Herbal chemical helps combat HIV”. United Press International. January 1, 2009. Retrieved January 28, 2011.
- Astragalus, NCCAM
- Fauce, S. R., et al. (2008). “Telomerase-Based Pharmacologic Enhancement of Antiviral Function of Human CD8+ T Lymphocytes”. Journal of Immunology 181 (10): 7400–7406. PMC 2682219. PMID 18981163. Retrieved 2012-08-18.
- Harley, C. B., et al. (2011). “A natural product telomerase activator as part of a health maintenance program”.Rejuvenation Research 14 (1): 45–56.doi:10.1089/rej.2010.1085. PMC 3045570.PMID 20822369.
- Hiyama, K., et al. (2009). “Role of telomeres and telomerase in cancer”. In K. Hiyama. Telomeres and Telomerase in Cancer. Cancer Drug Discovery and Development II. Humana Press. pp. 171–180.doi:10.1007/978-1-60327-879-9_7. ISBN 978-1-60327-879-9.
- Rios, J. L.; P. G. Waterman (1998). “A review of the pharmacology and toxicology of Astragalus“. Phytotherapy Research 11 (6): 411–418. doi:10.1002/(SICI)1099-1573(199709)11:6<411::AID-PTR132>3.0.CO;2-6.
Astragalus- Large list of species
- Very large list of species (with synonyms).
- Astragalus at a Glance This fact sheet from the U.S. National Institutes of Health provides basic information about Astragalus – common names, uses, potential side effects, and resources for more information.
- Astragalus alpinus This Rare Species Guide profile from the Minnesota Department of Natural Resources provides information about the basis for the species’ listing, habitat, biology and life history, conservation and management, and conservation efforts.
- Chinese Milkvetch, Astragalus membranaceus, Kansas State University
- Astragalus Root
CHINESE MEDICINE..Scutellaria baicalensis fights cancer

Scutellaria baicalensis (or Baikal Skullcap, as opposed to Scutellaria lateriflora, a Skullcap native to North America) is a species of flowering plant in the Lamiaceae family.
Traditional Chinese medicine
It is one of the 50 fundamental herbs used in traditional Chinese medicine, where it has the name huáng qín (Chinese: 黄芩).[2] As a Chinese traditional medicine, Huang Qin usually refers to the dried root of Scutellaria baicalensis Georgi, S. viscidula Bge., S. amoena C.H. Wright, and S. ikoninkovii Ju.
Chemistry
Several chemical compounds have been isolated from the root; among them, baicalein, baicalin, wogonin, norwogonin, oroxylin A[3] and β-sitosterol are the major ones.
Etymology confusion
It is important to note the Latin name of the Skullcap being used as there are over 200 varieties, some used for various ailments, each with varying degrees of effectiveness. Sometimes Scutellaria lateriflora (North American Skullcap) is mistaken for Scutellaria baicalensis (Baikal Skullcap). This confusion can result in the intake of the lateriflora variety which is often processed and contaminated with other plants with high enough levels of toxicity to be of concern.
Baikal skullcap (scientific name Scutellaria baicalensis) is a plant. The root is used to make medicine. Common substitutions for Baikal skullcap in Chinese medicine include related plants whose scientific names are Scutellaria viscidula, Scutellaria amonea, and Scutellaria ikoninikovii.
Baikal skullcap is used to treat respiratory infections, hay fever, and fever. It is also used for gastrointestinal (GI) infections, as well as liver problems including viralhepatitis and jaundice.
Some people use Baikal skullcap for HIV/AIDS, kidney infections, pelvic inflammation, and sores or swelling. It is also used for scarlet fever, headache, irritability, red eyes, flushed face, seizures, epilepsy, hysteria, nervous tension, and to relieve a bitter taste in the mouth.

The active ingredient in Baikal skullcap, baicalin, is used in combination with shung hua (ephedra) to treat upper respiratory tract infections. In combination with other herbs, Baikal skullcap is used to treat attention deficit-hyperactivity disorder (ADHD),prostate cancer, a lung condition called bronchiolitis, arthritis, and hemorrhoids.
Baikal skullcap is also sometimes applied to the skin for psoriasis.
How does it work?
It is thought that the active chemicals in Baikal skullcap might be able to decrease inflammation, stop tumor growth, and prevent tumor cell reproduction.

Scutellaria baicalensis , also called Chinese skullcap, is a member of the mint family and has long been used in traditional Chinese herbal medicine . Chinese skullcap has been incorporated in herbal formulas designed to treat such widely varying conditions as cancer, liver disease, allergies, skin conditions, and epilepsy. The root is the part used medicinally.
Note: Chinese skullcap is substantially different from American skullcap ( Scutellaria lateriflora ).
Skullcap (Scutellaria baicalensis) has been widely used as a dietary ingredient and traditional herbal medicine owing to its anti-inflammatory and anticancer properties. In this study, we investigated the anti-allergic effects of skullcap and its active compounds, focusing on T cell-mediated responses ex vivoand in vivo. Splenocytes from mice sensitized with ovalbumin (OVA) were isolated for analyses of cytokine production and cell viability. Mice sensitized with OVA were orally administered skullcap or wogonin for 16 days, and then immunoglobulin (Ig) and cytokine levels were measured by enzyme-linked immunosorbent assays. Treatment with skullcap significantly inhibited interleukin (IL)-4 production without reduction of cell viability. Moreover, wogonin, but not baicalin and baicalein, suppressed IL-4 and interferon-gamma production. In vivo, skullcap and wogonin downregulated OVA-induced Th2 immune responses, especially IgE and IL-5 prediction. Wogonin as an active component of skullcap may be applied as a therapeutic agent for IgE- and IL-5-mediated allergic disorders…….http://www.mdpi.com/1420-3049/19/2/2536

References
- “Scutellaria baicalensis information from NPGS/GRIN”. USDA. Retrieved 2008-02-19.
- Zhang XW, Li WF, Li WW, Ren KH, Fan CM, Chen YY, Shen YL (2011). “Protective effects of the aqueous extract of Scutellaria baicalensis against acrolein-induced oxidative stress in cultured human umbilical vein endothelial cells”. Pharm Biol 49 (3): 256–261. doi:10.3109/13880209.2010.501803. PMID 20979538.
- Isolation and purification of baicalein, wogonin and oroxylin A from the medicinal plant Scutellaria baicalensis by high-speed counter-current chromatography. Hua-Bin Li and Feng Chen, Journal of Chromatography A, 13 May 2005, Volume 1074, Issues 1–2, pages 107–110, doi:10.1016/j.chroma.2005.03.088
- Scutellaria baicalensis List of Chemicals (Dr. Duke’s Databases)
- Scutellaria baicalensis (Plants for a Future)
- Sung Mun Jung et al., “Reduction of urate crystal-induced inflammation by root extracts from traditional oriental medicinal plants: elevation of prostaglandin D2 levels”, Arthritis Research & Therapy 2007, 9:R64 doi:10.1186/ar2222. Considers anti-inflammatory properties of dried roots from the species Angelica sinensis (Dong Quai), Acanthopanax senticosus (now known as Eleutherococcus senticosus, or Siberian Ginseng), andScutellaria baicalensis (Baikal Skullcap).
GREEN CHEMISTRY…Reduction of amides without hydride reagents

Clostridium sporogenes
Essentially all medicines and current drug candidates contain at least one basic nitrogen atom. A common approach to the synthesis of amines is to reduce the corresponding amide with a hydride reagent such as LiAlH4, DIBAL, RedAl, B2H6, Et3SiH, or polymethylhydroxysilane (PMHS).
The reaction survey reported that reduction of amides to amines was used in only 0.6% of chemical transformations; this number would surely be higher if safer methods for use on scale were available. The survey indicated that the number of amide reductions was equally split between diborane and hydride reagents.
Lithium aluminium hydride,

having a molecular weight of 38 and four hydrides per molecule, has the highest hydride density and is frequently used, even though it co-generates an inorganic by-product (lithium aluminum hydroxide) which is difficult to separate from the product. The workup procedure recommended by one bulk supplier (Chemetall) is to precipitate and filter the aluminum hydroxide salts. However, slow filtrations and product loss through occlusion or adsorption are typical problems that can be encountered.
Options for disposal of the cake include dissolving in water and sending to a waste water treatment plant or drying the cake and sending to a chemical waste dump that accepts solids.1 Both options have an environmental impact. Therefore, a generally applicable, safe, environmentally benign and economically viable method for the reduction of amides to amines would have an appreciable benefit to numerous processes.
Hydrogen gas is the ideal reductant because the only by-product is water. Thus, much research has been directed towards discovery of a transition metal catalyst selective for hydrogenation of amides. However, even with the best catalysts, both high temperature (![[similar]](https://i0.wp.com/www.rsc.org/images/entities/char_223c.gif) 150 °C) and pressure (>100 bar) are required. These conditions involve expensive high pressure hydrogenation equipment not typically available in a common pharmaceutical manufacturing plant.
150 °C) and pressure (>100 bar) are required. These conditions involve expensive high pressure hydrogenation equipment not typically available in a common pharmaceutical manufacturing plant.
The harsh conditions also preclude the use of these catalysts with substrates that contain other reducible or thermally labile functional groups. Recent research has led to the discovery of catalysts that are effective at lower temperature and pressure, giving encouragement that the goal of finding a selective, low pressure/temperature catalyst is realistic.2
Another approach would be to use a biotransformation to reduce the amide. It is notable that a number of bacteria and fungi reduce carboxylic acids to aldehydes or ketones.3 The usual fate of amides in biological pathways is hydrolysis. However, an anaerobic bacteria, Clostridium sporogenes, has been reported to reduce benzamide to benzylamine. 4

A key challenge in this technology area is gaining a detailed understanding of these complex enzyme-catalysed processes that require ATP/NADPH co-factor recycling, and getting the enzymes cloned and produced on a large scale in suitable expression systems.
The acylation/reduction strategy for N-alkylation avoids the need to handle alkylating agents and would be more widely used if a safer, more atom economical or preferably catalytic method for amide reduction were developed. The solution to this problem could be either chemical or biochemical.
- Chemetall brochures, Lithium Aluminum Hydride… strong, concentrated and economical, Oct. 2000, pp. 18–19 Search PubMed .
- A. A. Smith, P. Dani, P. D. Higginson and A. J. Pettman, World Pat., WO2005/066112 A1, 2005 Search PubMed .
- (a) A. Hage, H. E. Schoemaker and J. A. Field, Appl. Microbiol. Biotechnol., 1999, 52, 834–838 CrossRef CAS Search PubMed ; (b) A. He, T. Li, L. Daniels, I. Fotheringham and J. P. N. Rosazza, Appl. Environ. Microbiol., 2004, 70, 1874–1881 CrossRef CAS Search PubMed .
- O. Dipeolu, J. Gardiner and G. Stephens, Biotechnol. Lett., 2005, 27, 1803–1807 CrossRef CAS Search PubMed .
Delamanid……….an experimental drug for the treatment of multi-drug-resistant tuberculosis.

Delamanid
http://www.ama-assn.org/resources/doc/usan/delamanid.pdf
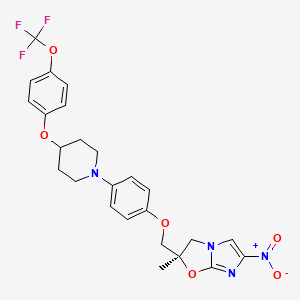
(2R)-2-Methyl-6-nitro-2-[(4-{4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl}phenoxy)methyl]-2,3-dihydroimidazo[2,1-b][1,3]oxazole
2(R)-Methyl-6-nitro-2-[4-[4-[4-(trifluoromethoxy)phenoxy]piperidin-1-yl]phenoxymethyl]-2,3-dihydroimidazo[2,1-b]oxazole
(R) -2-methyl-6-nitro-2- { 4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl } -2 , 3- dihydroimidazo [2 , 1-b] oxazole
Imidazo[2,1-b]oxazole, 2,3-dihydro-2-methyl-6-nitro-2-[[4-[4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl]phenoxy]methyl]-, (2R)-
(R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole
681492-22-8 cas no
Delamanid, 681492-22-8, Delamanid (JAN/USAN), Delamanid [USAN:INN],UNII-8OOT6M1PC7,
- OPC 67683
- OPC-67683
- UNII-8OOT6M1PC7
Molecular Formula: C25H25F3N4O6
Molecular Weight: 534.48441
Clinical trials……….http://clinicaltrials.gov/search/intervention=OPC+67683+OR+Delamanid
CLINICAL TRIALS
Primary Sponsor: Otsuka Pharmaceutical Development & Commercialization, Inc.
Trial ID / Reg # / URL: http://clinicaltrials.gov/ct2/show/NCT00685360

Delamanid (USAN, codenamed OPC-67683) is an experimental drug for the treatment of multi-drug-resistant tuberculosis. It works by blocking the synthesis of mycolic acids in Mycobacterium tuberculosis, the organism which causes tuberculosis, thus destabilising its cell wall.[1][2][3]
In phase II clinical trials, the drug was used in combination with standard treatments, such as four or five of the drugs ethambutol, isoniazid,pyrazinamide, rifampicin, aminoglycoside antibiotics, and quinolones. Healing rates (measured as sputum culture conversion) were significantly better in patients who additionally took delamanid.[3][4]
The European Medicines Agency (EMA) recommended conditional marketing authorization for delamanid in adults with multidrug-resistant pulmonary tuberculosis without other treatment options because of resistance or tolerability. The EMA considered the data show that the benefits of delamanid outweigh the risks, but that additional studies were needed on the long-term effectiveness.[5]
Delamanid, an antibiotic active against Mycobacterium tuberculosis strains, has been filed for approval in the E.U. and by Otsuka for the treatment of multidrug-resistant tuberculosis. In 2013, a positive opinion was received in the E.U. for this indication. Phase III trials for treatment of multidrug-resistant tuberculosis are under way in the U.S. Phase II study for the pediatric use is undergone in the Europe.
The drug candidate’s antimycobacterial mechanism of action is via specific inhibition of the synthesis pathway of mycolic acid, which is a cell wall component unique to M. tuberculosis.
In 2008, orphan drug designation was received in Japan for the treatment of pulmonary tuberculosis.
Tuberculosis (TB), an airborne lung infection, still remains a major public health problem worldwide. It is estimated that about 32% of the world population is infected with TB bacillus, and of those, approximately 8.9 million people develop active TB and 1.7 million die as a result annually according to 2004 figures. Human immunodeficiency virus (HIV) infection has been a major contributing factor in the current resurgence of TB. HIV-associated TB is widespread, especially in sub-Saharan Africa, and such an infectious process may further accelerate the resurgence of TB.
Moreover, the recent emergence of multidrug-resistant (MDR) strains ofMycobacterium tuberculosis that are resistant to two major effective drugs, isonicotinic acid hydrazide (INH) and rifampicin (RFP), has further complicated the world situation.
The World Health Organization (WHO) has estimated that if the present conditions remain unchanged, more than 30 million lives will be claimed by TB between 2000 and 2020. As for subsequent drug development, not a single new effective compound has been launched as an antituberculosis agent since the introduction of RFP in 1965, despite the great advances that have been made in drug development technologies.
Although many effective vaccine candidates have been developed, more potent vaccines will not become immediately available. The current therapy consists of an intensive phase with four drugs, INH, RFP, pyrazinamide (PZA), and streptomycin (SM) or ethambutol (EB), administered for 2 months followed by a continuous phase with INH and RFP for 4 months. Thus, there exists an urgent need for the development of potent new antituberculosis agents with low-toxicity profiles that are effective against both drug-susceptible and drug-resistant strains of M. tuberculosis and that are capable of shortening the current duration of therapy.
………………………
(R)-2-bromo-4-nitro-1-(2-methyl-2-oxiranylmethyl)imidazole
4-[4-(4-Trifluoromethoxyphenoxy)piperidin-1-yl]phenol
ARE THE INTERMEDIATES
Example 1884
Production of (R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole
4-[4-(4-Trifluoromethoxyphenoxy)piperidin-1-yl]phenol (693 mg, 1.96 mmol) was dissolved in N,N′-dimethylformamide (3 ml), and sodium hydride (86 mg, 2.16 mmol) was added while cooling on ice followed by stirring at 70-75° C. for 20 minutes. The mixture was cooled on ice. To the solution, a solution prepared by dissolving (R)-2-bromo-4-nitro-1-(2-methyl-2-oxiranylmethyl)imidazole (720 mg, 2.75 mmol) in N,N′-dimethylformamide (3 ml) was added followed by stirring at 70-75° C. for 20 minutes. The reaction mixture was allowed to return to room temperature, ice water (25 ml) was added, and the resultant solution was extracted with methylene chloride (50 ml) three times. The organic phases were combined, washed with water 3 times, and dried over magnesium sulfate. After filtration, the filtrate was concentrated, and the residue was purified by silica gel column chromatography (methylene chloride/ethyl acetate=3/1). Recrystallization from ethyl acetate/isopropyl ether gave (R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole (343 mg, 33%) as a light yellow powder.
…………………………
WO 2010021409 AND http://worldwide.espacenet.com/publicationDetails/biblio?CC=IN&NR=203704A1&KC=A1&FT=D
FOR 2, 4 DINITROIMIDAZOLE
…………………………………………
These patent literatures disclose Reaction Schemes A and B below as the processes for producing the aforementioned 2, 3-dihydroimidazo [2, 1-b] oxazole compound.
Reaction Scheme A:

wherein R1 is a hydrogen atom or lower-alkyl group; R2 is a substituted pxperidyl group or a substituted piperazinyl group; and X1 is a halogen atom or a nitro group.
Reaction Scheme B:


wherein X2 is a halogen or a group causing a substitution reaction similar to that of a halogen; n is an integer from 1 to 6; and R1, R2 and X1 are the same as in Reaction Scheme A.
An oxazole com ound represented by Formula (la) :

, i.e., 2-methyl-6-nitro-2-{4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl }-2, 3- dihydroimidazo [2, 1-b] oxazole (hereunder, this compound may be simply referred to as “Compound la”) is produced, for example, by the method shown in the Reaction Scheme C below (Patent
Literature 3) . In this specification, the term “oxazole compound’ means an oxazole derivative that encompasses compounds that contain an oxazole ring or an oxazoline ring (dihydrooxazole ring) in the molecule.
Reaction Scheme C:


However, the aforementioned methods are unsatisfactory in terms of the yield of the objective compound. For example, the method of Reaction Scheme C allows the objective oxazole Compound (la) to be obtained from Compound (2a) at a yield as low as 35.9%. Therefore, alternative methods for producing the compound in an industrially advantageous manner are desired. Citation List
Patent Literature
PTL 1: WO2004/033463
PTL 2: WO2004/035547
PTL 3: WO2008/140090
Example 9
Production of (R) -2-methyl-6-nitro-2- { 4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl } -2 , 3- dihydroimidazo [2 , 1-b] oxazole
{R) -1- [ – {2 , 3-epoxy-2-methylpropoxy ) phenyl] -4- [4- ( trifluoromethoxy ) phenoxy ] piperidine (10.0 g, 23.6 mmol, optical purity of 94.3%ee), 2-chloro-4-nitroimidazole (4.0 g, 27.2 mmol), sodium acetate (0.4 g, 4.9 mmol), and t- butyl acetate (10 ml) were mixed and stirred at 100°C for 3.5 hours. Methanol (70 ml) was added to the reaction mixture, and then a 25% sodium hydroxide aqueous solution (6.3 g, 39.4 mmol) was added thereto dropwise while cooling with ice. The resulting mixture was stirred at 0°C for 1.5 hours, and further stirred at approximately room
temperature for 40 minutes. Water (15 ml) and ethyl acetate (5 ml) were added thereto, and the mixture was stirred at 45 to 55°C for 1 hour. The mixture was cooled to room temperature, and the precipitated crystals were collected by filtration. The precipitated crystals were subsequently washed with methanol (30 ml) and water (40 ml) . Methanol (100 ml) was added to the resulting
crystals, followed by stirring under reflux for 30 minutes. The mixture was cooled to room temperature. The crystals were then collected by filtration and washed with methanol (30 ml) . The resulting crystals were dried under reduced pressure, obtaining 9.3 g of the objective product (yield: 73%) .
Optical purity: 99.4%ee.
……………….
Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles
J Med Chem 2006, 49(26): 7854
http://pubs.acs.org/doi/abs/10.1021/jm060957y
(R)-2-Methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole (19, DELAMANID).
To a mixture of 27 (127.56 g, 586.56 mmol) and 4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenol (28g) (165.70 g, 468.95 mmol) in N,N-dimethylformamide (1600 mL) was added 60% sodium hydride (22.51 g, 562.74 mmol) at 0 °C portionwise. After the mixture was stirred at 50 °C for 2 h under a nitrogen atmosphere, the reaction mixture was cooled in an ice bath and carefully quenched with ethyl acetate (230 mL) and ice water (50 mL). The thus-obtained mixture was poured into water (3000 mL) and stirred for 30 min. The resulting precipitates were collected by filtration, washed with water, and dried at 60 °C overnight. This crude product was purified by silica gel column chromatography using a dichloromethane and ethyl acetate mixture (5/1) as solvent. The appropriate fractions were combined and evaporated under reduced pressure. The residue was recrystallized from ethyl acetate (1300 mL)−isopropyl alcohol (150 mL) to afford 19 (119.11 g, 48%) as a pale yellow crystalline powder.
Mp 195−196 °C.
1H NMR (CDCl3) δ 1.77 (3H, s), 1.87−2.16 (4H, m), 2.95−3.05 (2H, m), 3.32−3.41 (2H, m), 4.02 (1H, d, J = 10.2 Hz), 4.04 (1H, d, J = 10.2 Hz), 4.18 (1H, J = 10.2 Hz), 4.36−4.45 (1H, m), 4.49 (1H, d, J = 10.2 Hz), 6.76 (2H, d, J = 6.7 Hz), 6.87−6.94 (4H, m), 7.14 (2H, d, J = 8.6 Hz), 7.55 (1H, s).
[α  −9.9° (c 1.01, CHCl3).
−9.9° (c 1.01, CHCl3).
MS (DI) m/z 535 (M+ + 1). Anal. (C25H25F3N4O6) C, H, N.
http://pubs.acs.org/doi/suppl/10.1021/jm060957y/suppl_file/jm060957ysi20061113_095044.pdf
References
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. (2006). “OPC-67683, a Nitro-Dihydro-Imidazooxazole Derivative with Promising Action against Tuberculosis in Vitro and in Mice”. PLoS Medicine 3 (11): e466.doi:10.1371/journal.pmed.0030466. PMC 1664607. PMID 17132069.
- Skripconoka, V.; Danilovits, M.; Pehme, L.; Tomson, T.; Skenders, G.; Kummik, T.; Cirule, A.; Leimane, V.; Kurve, A.; Levina, K.; Geiter, L. J.; Manissero, D.; Wells, C. D. (2012). “Delamanid Improves Outcomes and Reduces Mortality for Multidrug-Resistant Tuberculosis”. European Respiratory Journal41 (6): 1393–1400. doi:10.1183/09031936.00125812. PMC 3669462. PMID 23018916.
- H. Spreitzer (18 February 2013). “Neue Wirkstoffe – Bedaquilin und Delamanid”. Österreichische Apothekerzeitung (in German) (4/2013): 22.
- Gler, M. T.; Skripconoka, V.; Sanchez-Garavito, E.; Xiao, H.; Cabrera-Rivero, J. L.; Vargas-Vasquez, D. E.; Gao, M.; Awad, M.; Park, S. K.; Shim, T. S.; Suh, G. Y.; Danilovits, M.; Ogata, H.; Kurve, A.; Chang, J.; Suzuki, K.; Tupasi, T.; Koh, W. J.; Seaworth, B.; Geiter, L. J.; Wells, C. D. (2012). “Delamanid for Multidrug-Resistant Pulmonary Tuberculosis”. New England Journal of Medicine 366 (23): 2151–2160. doi:10.1056/NEJMoa1112433.PMID 22670901.
- Drug Discovery & Development. EMA Recommends Two New Tuberculosis Treatments. November 22, 2013.
- Synthesis and antituberculous activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles
45th Intersci Conf Antimicrob Agents Chemother (ICAAC) (December 16-19, Washington DC) 2005, Abst F-1473
|
12-28-2006
|
Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles.
|
Journal of medicinal chemistry
|
|
|
11-1-2006
|
OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice.
|
PLoS medicine
|
|
1-1-2008
|
New anti-tuberculosis drugs with novel mechanisms of action.
|
Current medicinal chemistry
|
|
11-11-2010
|
Synthesis and Structure-Activity Relationships of Aza- and Diazabiphenyl Analogues of the Antitubercular Drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824).
|
Journal of medicinal chemistry
|
|
5-1-2012
|
Tuberculosis: the drug development pipeline at a glance.
|
European journal of medicinal chemistry
|
|
|
1-12-2012
|
Structure-activity relationships for amide-, carbamate-, and urea-linked analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824).
|
Journal of medicinal chemistry
|
|
9-11-2009
|
Pharmaceutical Composition Achieving Excellent Absorbency of Pharmacologically Active Substance
|
|
|
1-16-2009
|
Sulfonamide Derivatives for the Treatment of Bacterial Infections
|
| WO2004033463A1 | Oct 10, 2003 | Apr 22, 2004 | Otsuka Pharma Co Ltd | 2,3-DIHYDRO-6-NITROIMIDAZO[2,1-b]OXAZOLES |
| WO2004035547A1 | Oct 14, 2003 | Apr 29, 2004 | Otsuka Pharma Co Ltd | 1-substituted 4-nitroimidazole compound and process for producing the same |
| WO2008140090A1 | May 7, 2008 | Nov 20, 2008 | Otsuka Pharma Co Ltd | Epoxy compound and method for manufacturing the same |
| JP2009269859A * | Title not available |
TB

It is estimated that a third of the world’s population is currently infected with tuberculosis, leading to 1.6 million deaths annually. The current drug regimen is 40 years old and takes 6-9 months to administer. In addition, the emergence of drug resistant strains and HIV co-infection mean that there is an urgent need for new anti-tuberculosis drugs. The twenty-first century has seen a revival in research and development activity in this area, with several new drug candidates entering clinical trials. This review considers new potential first-line anti-tuberculosis drug candidates, in particular those with novel mechanisms of action, as these are most likely to prove effective against resistant strains.
From among acid-fast bacteria, human Mycobacterium tuberculosis has been widely known. It is said that the one-third of the human population is infected with this bacterium. In addition to the human Mycobacterium tuberculosis, Mycobacterium africanum and Mycobacterium bovis have also been known to belong to the Mycobacterium tuberoculosis group. These bacteria are known as Mycobacteria having a strong pathogenicity to humans.
Against these tuberculoses, treatment is carried out using three agents, rifampicin, isoniazid, and ethambutol (or streptomycin) that are regarded as first-line agents, or using four agents such as the above three agents and pyrazinamide.
However, since the treatment of tuberculosis requires extremely long-term administration of agents, it might result in poor compliance, and the treatment often ends in failure.
Moreover, in respect of the above agents, it has been reported that: rifampicin causes hepatopathy, flu syndrome, drug allergy, and its concomitant administration with other drugs is contraindicated due to P450-associated enzyme induction; that isoniazid causes peripheral nervous system disorder and induces serious hepatopathy when used in combination with rifampicin; that ethambutol brings on failure of eyesight due to optic nerve disorder; that streptomycin brings on diminution of the hearing faculty due to the 8th cranial nerve disorder; and that pyrazinamide causes adverse reactions such a hepatopathy, gouty attack associated with increase of uric acid level, vomiting (A Clinician’s Guide To Tuberculosis, Michael D. Iseman 2000 by Lippincott Williams & Wilkins, printed in the USA, ISBN 0-7817-1749-3, Tuberculosis, 2nd edition, Fumiyuki Kuze and Takahide Izumi, Igaku-Shoin Ltd., 1992).
Actually, it has been reported that cases where the standard chemotherapy could not be carried out due to the adverse reactions to these agents made up 70% (approximately 23%, 52 cases) of the total cases where administration of the agents was discontinued (the total 228 hospitalized patients who were subject to the research) (Kekkaku, Vol. 74, 77-82, 1999).
In particular, hepatotoxicity, which is induced by rifampicin, isoniazid, and ethambutol out of the 5 agents used in combination for the aforementioned first-line treatment, is known as an adverse reaction that is developed most frequently. At the same time, Mycobacterium tuberculosis resistant to antitubercular agents, multi-drug-resistant Mycobacterium tuberculosis, and the like have been increasing, and the presence of these types of Mycobacterium tuberculosismakes the treatment more difficult.
According to the investigation made by WHO (1996 to 1999), the proportion ofMycobacterium tuberculosis that is resistant to any of the existing antitubercular agents to the total types of Mycobacterium tuberculosis that have been isolated over the world reaches 19%, and it has been published that the proportion of multi-drug-resistant Mycobacterium tuberculosis is 5.1%. The number of carriers infected with such multi-drug-resistant Mycobacterium tuberculosis is estimated to be 60,000,000, and concerns are still rising that multi-drug-resistantMycobacterium tuberculosis will increase in the future (April 2001 as a supplement to the journal Tuberculosis, the “Scientific Blueprint for TB Drug Development.”)
In addition, the major cause of death of AIDS patients is tuberculosis. It has been reported that the number of humans suffering from both tuberculosis and HIV reaches 10,700,000 at the time of year 1997 (Global Alliance for TB drug development). Moreover, it is considered that the mixed infection of tuberculosisand HIV has an at least 30 times higher risk of developing tuberculosis than the ordinary circumstances.
Taking into consideration the aforementioned current situation, the profiles of the desired antitubercular agent is as follows: (1) an agent, which is effective even for multi-drug-resistant Mycobacterium tuberculosis, (2) an agent enabling a short-term chemotherapy, (3) an agent with fewer adverse reactions, (4) an agent showing an efficacy to latent infecting Mycobacterium tuberculosis (i.e., latentMycobacterium tuberculosis), and (5) an orally administrable agent.
Examples of bacteria known to have a pathogenicity to humans include offending bacteria of recently increasing MAC infection (Mycobacterium avium—intracellulare complex infection) such as Mycobacterium avium andMycobacterium intracellulare, and atypical acid-fast bacteria such asMycobacterium kansasii, Mycobacterium marinum, Mycobacterium simiae, Mycobacterium scrofulaceum, Mycobacterium szulgai, Mycobacterium xenopi, Mycobacterium malmoense, Mycobacterium haemophilum, Mycobacterium ulcerans, Mycobacterium shimoidei, Mycobacterium fortuitum, Mycobacterium chelonae, Mycobacterium smegmatis, and Mycobacterium aurum.
Nowadays, there are few therapeutic agents effective for these atypical acid-fast bacterial infections. Under the presence circumstances, antitubercular agents such as rifampicin, isoniazid, ethambutol, streptomycin and kanamycin, a newquinolone agent that is a therapeutic agent for common bacterial infections, macrolide antibiotics, aminoglycoside antibiotics, and tetracycline antibiotics are used in combination.
However, when compared with the treatment of common bacterial infections, the treatment of atypical acid-fast bacterial infections requires a long-term administration-of agents, and there have been reported cases where the infection is changed to an intractable one, finally leading to death. To break the afore-mentioned current situation, the development of an agent having a stronger efficacy is desired.
For example, National Publication of International Patent Application No. 11-508270 (WO97/01562) discloses that a 6-nitro-1,2,3,4-tetrahydro[2,1-b]-imidazopyran compound has a bactericidal action in vitro to Mycobacterium tuberculosis (H37Rv strain) and multi-drug-resistant Mycobacterium tuberculosis, and that the above compound has a therapeutic effect to a tuberculosis-infected animal model when it is orally administered and thus useful as antitubercular agent.
Balsam for the bones: Chemists develop a nanopaste for the repair of bone defects
Following accidents or cancer surgery surgeons often have to transplant healthy bone tissue or synthetic material to repair the resulting bone defects. Unfortunately, these procedures do not always have the desired effect.
Now a professor for inorganic chemistry, Matthias Epple was attracted to the interface between biology and medical science. “We have been investigating the impact of mineral tissue such as teeth, bone and sea shells for many years and are now using the knowledge we have gained to produce new biomaterials.” To achieve this he has collaborated closely with medical scientists and his current project – carried out with three of his doctoral students – was no exception.
“The repair of bone defects presents a real challenge for surgeons,” he relates. “When possible they collect the patient’s own bone from various locations, such as the iliac crest, and implant it where needed to fill defects.” The researcher explained…
View original post 267 more words
Glenmark Pharmaceuticals Ltd. through its Swiss Subsidiary receives USD 4 Mn. as research fee payment from Forest Laboratories Inc.

Total Payment received for the mpges-1 program from Forest Laboratories is USD 15 million
March 25, 2014: Glenmark Pharmaceuticals Ltd. has informed the Stock Exchange today that the company through its Swiss subsidiary has received
USD 4 million as research fee payment from Forest Laboratories Inc. on a collaboration for the development of novel mPGES-1 inhibitors to treatchronic inflammatory conditions, including pain.
Under the terms of the agreement signed in FY 2012-13, Forest made USD 6 million upfront payment and also provided an additional USD 3 million
to support the next phase of work. In September 2013, Glenmark received an additional amount of USD 2 million as research fee payment from Forest Laboratories Inc.
Hence, the total amount received by Glenmark from Forest Laboratories Inc towards its novel mPEGS-1 inhibitors program is USD15 million.
read at
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

