
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

AbbVie’S Investigational Oncology Compound ABT-199/GDC-0199, Venetoclax

ABT 199, RG 7601, GDC 0199
Venetoclax
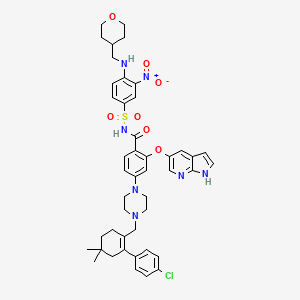
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
SYNTHESIS UPDATED BELOW …………..

CAS 1257044-40-8 [RN]
2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)-4-(4-((2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-enyl)methyl)piperazin-1-yl)-N-(3-nitro-4-((tetrahydro-2H-pyran-4-yl)methylamino)phenylsulfonyl)benzamide
4-[4-[[2-(4-chlorophenyl)-4,4-dimethylcyclohexen-1-yl]methyl]piperazin-1-yl]-N-[3-nitro-4-(oxan-4-ylmethylamino)phenyl]sulfonyl-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
ABT 199
- Molecular Formula: C45H50ClN7O7S
- Average mass: 868.439209 Da
- Monoisotopic mass: 867.318115 Da
-
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
NORTH CHICAGO, Ill., May 31, 2014/NEWS.GNOM.ES/ — AbbVie (NYSE: ABBV) released interim results from a Phase Ib clinical trial of ABT-199/GDC-0199, an investigational B-cell lymphoma 2 (BCL-2) selective inhibitor, in combination with rituximab (Abstract 7013). Results showed anoverall response rate (ORR) of 84 percent, in patients with relapsed/refractory chronic lymphocytic leukemia(CLL), the most common leukemia in the UnitedStates. These results were presented at the 50thAnnual Meeting of the American Society of ClinicalOncology (ASCO), May 30 – June 3 in Chicago.
ABT-199 is a so-called BH3-mimetic drug, which is designed to block the function of the protein Bcl 2. In 1988, it was discovered that Bcl-2 allowed leukaemia cells to become long-lived, a discovery made at the Walter and Eliza Hall Institute by Professors David Vaux, Suzanne Cory and Jerry Adams. Subsequent research led by them and other institute scientists, including Professors Andreas Strasser, David Huang, Peter Colman and Keith Watson, has explained much about how Bcl-2 and related molecules function to determine if a cell lives or dies. These discoveries have contributed to the development of a new class of drugs called BH3-mimetics that kill, and thereby rapidly remove, leukaemic cells by blocking Bcl-2. (source:http://www.wehi.edu.au).
|
Highlights of recent research using this agent |
GDC-0199 (RG7601) is a novel small molecule Bcl-2 selective inhibitor designed to restore apoptosis, also known as programmed cell death, by blocking the function of a pro-survival Bcl-2 family protein. The Bcl-2 family proteins, which are expressed at high levels in many tumors, play a central role in regulating apoptosis and, consequently, are thought to impact tumor formation, tumor growth and resistance.
Venetoclax (previously: GDC-0199, ABT-199, RG7601 )[1] is a small molecule oral drug being investigated to treat chronic lymphocytic leukemia (CLL).[2][3]
In 2015, the FDA granted Breakthrough Therapy Designation to venetoclax for CLL in previously treated (relapsed/refractory) patients with the 17p deletion genetic mutation.[3]
Mechanism of action
Venetoclax (a BH3-mimetic[4]) acts as a Bcl-2 inhibitor, ie. it blocks the anti-apoptotic B-cell lymphoma-2 (BCL2) protein, leading toprogrammed cell death in CLL cells.[2]
Clinical trials
A phase 1 trial established a dose of 400mg/day.[2]
A trial of venetoclax in combination with rituximab had an encouraging complete response rate.[5]
A phase 2 trial met its primary endpoint which was overall response rate.[3] Interim results from a Phase 2b trial are encouraging, especially in patients with the 17p deletion.[2]
A phase 3 trial (NCT02005471)[1] has started.[3]
NOW IN PHASE 3 UPDATED…………
4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (hereafter, “Compound 1”) is a potent and selective Bcl-2 inhibitor having, inter alia, antitumor activity as an apoptosis-inducing agent. Compound 1 has the formula:

Compound 1 is currently the subject of ongoing clinical trials for the treatment of chronic lymphocytic leukemia. U.S. Patent Publication No. 2010/0305122 describes Compound 1, and other compounds which exhibit potent binding to a Bcl-2 family protein, and pharmaceutically acceptable salts thereof. U.S. Patent Publication Nos. 2012/0108590 and 2012/0277210 describe pharmaceutical compositions comprising such compounds, and methods for the treatment of neoplastic, immune or autoimmune diseases comprising these compounds. U.S. Patent Publication No. 2012/0157470 describes pharmaceutically acceptable salts and crystalline forms of Compound 1. The disclosures of U.S. 2010/0305122; 2012/0108590; 2012/0157470 and 2012/0277210 are hereby incorporated by reference in their entireties.



PATENT
US 2015183783
http://www.google.com/patents/US20150183783
PATENT
CN 104370905
http://www.google.com/patents/CN104370905A?cl=en

ABT-199 is developed AbbVie Bel-2 inhibitors, I trial (NCT01328626) enrolled 84 patients with relapsed type / refractory CLL / SLL patients and 44 cases of relapsing / refractory non-Hodgkin lymphoma patients. ABT-199 treatment response CLL / SLL rate of 79% (complete response rate of 22%), median duration of response time was 20.5 months; ABT-199 treatment of non-Hodgkin’s lymphoma response rate of 48% (complete response rate was 7.5%). The efficacy of ABT-199 is capable of obinutuzumab, idelalisib, ibrutinib rival, is expected to become the first listed Bcl_2 inhibitors, ABT-199 is currently ongoing Phase III clinical study.
ABT-199 compound CAS number 1257044-40-8, the compound is structured as follows:

Patent W02012058392, W02012071336, W02010138588 et al. Discloses the preparation of ABT-199 in order to -IH- 5-bromo-pyrrolo [2, 3-b] pyridine as raw material to protect hydroxylation, after replacing the compound 5, and reaction of compound 6, hydrolysis to give compound 9, compound 10 and compound 9 obtained by condensation of ABT-199, a specific line as follows:

use of 2-fluoro-4-nitrobenzoate (A) as a raw material, and substituted 5-hydroxy-7-aza-indole (B), reduction to produce compound ( D), the compound (D) with the compound by cyclization after (H) substitution, hydrolysis to yield compound (J), and then with the compound (K) to afford ABT-199.

Preparation of a compound of Example (F) of the
Example

First step: Synthesis of Compound (C)
2-fluoro-4-nitrobenzoate in IL three-necked flask 50. 0g, dissolved with dimethylformamide N’N- 250ml, was added successively 5-hydroxy-7-aza-indole indole 33. 6g, potassium carbonate 34. 7g, the reaction was heated to 50 degrees under nitrogen gas protection for 2 hours, poured into 2L of ice water was added and extracted three times with ethyl acetate, the organic phase was dried with saturated sodium chloride spin dry to give Compound (C) crude 82. 0g, crude without purification in the next reaction direct investment.
Step two: Synthesis of Compound (D)
The compound of the previous step (C) of the crude product was dissolved in methanol 400ml, was added 10% palladium on carbon 4. 0g, through the reaction of hydrogen at atmospheric pressure, after the end of the reaction by TLC spin solvent to give compound (D) The crude product 73. 2g, crude without purification in the next reaction direct investment.
The third step: Synthesis of compound (F)
Take the previous step the compound (D) crude 20. 0g, t-butanol were added 150ml, compound (E) 10. g, potassium carbonate 9. 7g, completion of the addition the reaction was refluxed for 48 hours the reaction solution was cooled, added acetic acid ethyl ester was diluted, washed with water three times, the combined aqueous phases extracted once with ethyl acetate, the combined ethyl acetate phases twice, dried over anhydrous sodium sulfate and the solvent was spin, the crude product obtained was purified by silica gel column chromatography to give 13. 9g, three-step overall yield of 57.4%.
Preparation Example II Compound (H),

[0029] Take compound (G) (prepared according to W02012058392 method) 5. 0g, dissolved with 50ml of dichloromethane, was added triethylamine 5. 6ml, the reaction solution was cooled to 0-5 ° with stirring, was added dropwise methanesulfonyl chloride 2. 7g, the addition was complete the reaction was warmed to room temperature overnight, after the end of the reaction by TLC the reaction was quenched with water, the organic phase was dried over anhydrous sodium sulfate and the solvent was spin, purified by silica gel column chromatography to give compound (H) 6. 5g , a yield of 99%.
Three ABT-199 Preparation of Example

First step: Synthesis of Compound (I)
In IOOml three-necked flask were added the compound (F) 2. 5g, compound ⑶2. 3g, potassium carbonate I. 9g, Ν ‘was added and reacted at 50 degrees N- dimethylformamide 15ml, nitrogen atmosphere, TLC detection After the reaction, the reaction solution was poured into ice-water, extracted with ethyl acetate twice added ethyl acetate phase was dried over anhydrous sodium sulfate spin, and purified by silica gel column chromatography to give compound (I) 3. 6g, yield 88 %.
Step two: Synthesis of Compound (J)
In IOml single jar Compound (I) I. 0g, followed by adding water 5ml, ethanol 5ml, tetrahydrofuran 5ml, sodium hydroxide 136mg, the reaction was stirred at room temperature the reaction, ethyl acetate was added after dilution of the reaction by TLC, adjusted with IN hydrochloric acid PH4-5, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate and spin dried to give compound (J) 907mg, 93% yield.
Step two: Synthesis of ABT-199
In a 25ml single neck flask was added the compound (J) 100mg, EDCI67mg, dichloromethane 10ml, the reaction was stirred for 30 minutes, was added the compound (K) (prepared in accordance with W02012058392) 55mg, finally added a catalytic amount of DMAP, the force After opening the reaction was stirred overnight, after the end of the reaction by TLC the solvent was spin, HPLC purified preparation obtained by pure ABT-199 ^ 9811, 65% yield.
PATENT
WO 2014165044
http://www.google.com/patents/WO2014165044A1?cl=en
PATENT
US 2014275540
http://www.google.com/patents/US20140275540

-

-
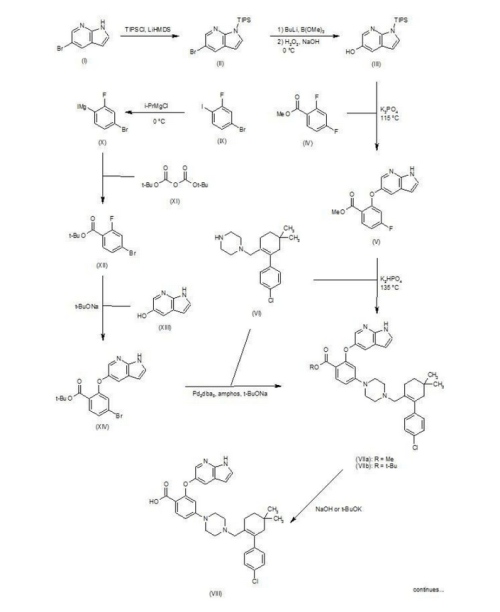
An exemplary reaction according to Scheme 2 is shown below.
 Scheme 3 below. Compound (E) is commercially available or may be prepared by techniques known in the art, e.g., as shown in U.S. Pat. No. 3,813,443 and Proceedings of the Chemical Society, London, 1907, 22, 302.
Scheme 3 below. Compound (E) is commercially available or may be prepared by techniques known in the art, e.g., as shown in U.S. Pat. No. 3,813,443 and Proceedings of the Chemical Society, London, 1907, 22, 302. -
 Scheme 4 below. Compound (M) is commercially available or may be prepared by techniques known in the art, e.g., as shown in GB 585940 and J. Am. Chem. Soc., 1950, 72, 1215-1218.
Scheme 4 below. Compound (M) is commercially available or may be prepared by techniques known in the art, e.g., as shown in GB 585940 and J. Am. Chem. Soc., 1950, 72, 1215-1218. -

-
In another embodiment, the compound of formula (1) is prepared from compound (D) and compound (I) as shown in Scheme 5 below. Compound (J) may be prepared by techniques known in the art, e.g., as shown in WO 2009/117626 and Organometallics, 2008, 27 (21), 5605-5611.
-

 Example 1 Synthesis of tert-butyl 4-bromo-2-fluorobenzoate (Compound (C))To a 100 ml jacketed reactor equipped with a mechanical stirrer was charged 4-bromo-2-fluoro1-iodobenzene, “Compound (A)” (5 g, 1.0 eq) and THF (25 ml). The solution was cooled to −5° C. 2 M isopropyl magnesium chloride in THF (10.8 ml, 1.3 eq) was slowly added maintaining the internal temperature below 0° C. The mixture was stirred at 0° C. for 1 h. Di-tert-butyl dicarbonate (5.44 g, 1.5 eq) in THF (10 ml) was added. After 1 h, the solution was quenched with 10% citric acid (10 ml), and then diluted with 25% NaCl (10 ml). The layers were separated and the organic layer was concentrated to near dryness and chased with THF (3×10 ml). The crude oil was diluted with THF (5 ml), filtered to remove inorganics, and concentrated to dryness. The crude oil (6.1 g, potency=67%, potency adjusted yield=88%) was taken to the next step without further purification. 1H NMR (DMSO-d6): δ 1.53 (s, 9H), 7.50-7.56 (m, 1H), 7.68 (dd, J=10.5, 1.9 Hz, 1H), 7.74 (t, J=8.2 Hz, 1H).
Example 1 Synthesis of tert-butyl 4-bromo-2-fluorobenzoate (Compound (C))To a 100 ml jacketed reactor equipped with a mechanical stirrer was charged 4-bromo-2-fluoro1-iodobenzene, “Compound (A)” (5 g, 1.0 eq) and THF (25 ml). The solution was cooled to −5° C. 2 M isopropyl magnesium chloride in THF (10.8 ml, 1.3 eq) was slowly added maintaining the internal temperature below 0° C. The mixture was stirred at 0° C. for 1 h. Di-tert-butyl dicarbonate (5.44 g, 1.5 eq) in THF (10 ml) was added. After 1 h, the solution was quenched with 10% citric acid (10 ml), and then diluted with 25% NaCl (10 ml). The layers were separated and the organic layer was concentrated to near dryness and chased with THF (3×10 ml). The crude oil was diluted with THF (5 ml), filtered to remove inorganics, and concentrated to dryness. The crude oil (6.1 g, potency=67%, potency adjusted yield=88%) was taken to the next step without further purification. 1H NMR (DMSO-d6): δ 1.53 (s, 9H), 7.50-7.56 (m, 1H), 7.68 (dd, J=10.5, 1.9 Hz, 1H), 7.74 (t, J=8.2 Hz, 1H). -
Example 2 Synthesis of tert-butyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-bromobenzoate (Compound (D))
-
To a 3 L three-neck Morton flask were charged 1H-pyrrolo[2,3-b]pyridin-5-ol (80.0 g, 1.00 eq.), tert-butyl 4-bromo-2-fluorobenzoate (193 g, 1.15 eq.), and anhydrous DMF (800 mL). The mixture was stirred at 20° C. for 15 min. The resulting solution was cooled to about zero to 5° C. A solution of sodium tert-butoxide (62.0 g) in DMF (420 mL) was added slowly over 30 min while maintaining the internal temperature at NMT 10° C., and rinsed with DMF (30 mL). The reaction mixture was stirred at 10° C. for 1 hour (an off-white slurry) and adjusted the internal temperature to ˜45° C. over 30 min. The reaction mixture was stirred at 45-50° C. for 7 hr and the reaction progress monitored by HPLC (IP samples: 92% conversion % by HPLC). The solution was cooled to ˜20° C. The solution was stirred at 20° C. overnight.
-
Water (1200 mL) was added slowly to the reaction mixture at <30° C. over 1 hour (slightly exothermic). The product slurry was adjusted to ˜20° C., and mixed for NLT 2 hours. The crude product was collected by filtration, and washed with water (400 mL). The wet-cake was washed with heptane (400 mL) and dried under vacuum at 50° C. overnight to give the crude product (236.7 g).
-
Re-crystallization or Re-slurry: 230.7 g of the crude product, (potency adjusted: 200.7 g) was charged back to a 3 L three-neck Morton flask. Ethyl acetate (700 mL) was added, and the slurry heated slowly to refluxing temperature over 1 hr (small amount of solids left). Heptane (1400 mL) was added slowly, and the mixture adjusted to refluxing temperature (78° C.). The slurry was mixed at refluxing temperature for 30 min., and cooled down slowly to down to ˜−10° C. at a rate of approximate 10° C./hour), and mixed for 2 hr. The product was collected by filtration, and rinsed with heptane (200 ml).
-
The solid was dried under vacuum at ˜50° C. overnight to give 194.8 g, 86% isolated yield of the product as an off-white solid. MS-ESI 389.0 (M+1); mp: 190-191° C. (uncorrected). 1H NMR (DMSO-d6): δ 1.40 (s, 9H), 6.41 (dd, J=3.4, 1.7 Hz, 1H), 7.06 (d, J=1.8 Hz, 1H), 7.40 (dd, J=8.3, 1.8 Hz, 1H), 7.51 (t, J=3.4 Hz, 1H), 7.58 (d, J=2.6 Hz, 1H), 7.66 (d, J=8.3 Hz, 1H), 8.03 (d, J=2.7 Hz, 1H), 11.72 (s, 1H, NH).
-
Example 3 Synthesis of 2-chloro-4,4-dimethylcyclohexanecarbaldehyde (Compound (F))
-
To a 500 mL RB flask were charged anhydrous DMF (33.4 g, 0.456 mol) and CH2Cl2 (80 mL). The solution was cooled down <−5° C., and POCl3 (64.7 g, 0.422 mol) added slowly over 20 min @<20° C. (exothermic), rinsed with CH2Cl2 (6 mL). The slightly brown solution was adjusted to 20° C. over 30 min, and mixed at 20° C. for 1 hour. The solution was cooled back to <5° C. 3,3-Dimethylcyclohexanone (41.0 g, 90%, ˜0.292 mol) was added, and rinsed with in CH2Cl2 (10 mL) (slightly exothermic) at <20° C. The solution was heated to refluxing temperature, and mixed overnight (21 hours).
-
To a 1000 mL three neck RB flask provided with a mechanical stirrer were charged 130 g of 13.6 wt % sodium acetate trihydrate aqueous solution, 130 g of 12% brine, and 130 mL of CH2Cl2. The mixture was stirred and cooled down to <5° C. The above reaction mixture (clear and brown) was transferred, quenched into it slowly while maintaining the internal temperature <10° C. The reaction vessel was rinsed with CH2Cl2 (10 mL). The quenched reaction mixture was stirred at <10° C. for 15 min. and allowed to rise to 20° C. The mixture was stirred 20° C. for 15 min and allowed to settle for 30 min. (some emulsion). The lower organic phase was separated. The upper aq. phase was back extracted with CH2Cl2 (50 mL). The combined organic was washed with a mixture of 12% brine (150 g)-20% K3PO4 aq. solution (40 g). The organic was dried over MgSO4, filtered and rinsed with CH2Cl2 (30 ml). The filtrate was concentrated to dryness under vacuum to give a brown oil (57.0 g, potency=90.9 wt % by qNMR, ˜100%). 1H NMR (CDCl3): δ 0.98 (s, 6H), 1.43 (t, J=6.4 Hz, 2H), 2.31 (tt, J=6.4, 2.2 Hz, 2H), 2.36 (t, J=2.2 Hz, 2H), 10.19 (s, 1H).
-
Example 4 Synthesis of 2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-enecarbaldehyde (Compound (G))
-
To a 250 mL pressure bottle were charged 2-chloro-4,4-dimethylcyclohex-1-enecarbaldehyde (10.00 g), tetrabutylammonium bromide (18.67 g), and acetonitrile (10 mL). The mixture was stirred at 20° C. for 5 min. 21.0 wt % K2CO3 aq. solution (76.0 g) was added. The mixture was stirred at room temperature (rt) for NLT 5 min. followed by addition of 4-chlorophenylboronic acid (9.53 g) all at once. The mixture was evacuated and purged with N2 for three times. Palladium acetate (66 mg, 0.5 mol %) was added all at once under N2. The reaction mixture was evacuated and purged with N2 for three times (an orange colored mixture). The bottle was back filled with N2 and heated to ˜35° C. in an oil bath (bath temp ˜35° C.). The mixture was stirred at 30° C. overnight (15 hours). The reaction mixture was cooled to RT, and pulled IP sample from the upper organic phase for reaction completion, typically starting material <2% (orange colored mixture). Toluene (100 mL) and 5% NaHCO3-2% L-Cysteine aq. solution (100 mL) were added. The mixture was stirred at 20° C. for 60 min. The mixture was filtered through a pad of Celite to remove black solid, rinsing the flask and pad with toluene (10 mL). The upper organic phase was washed with 5% NaHCO3 aq. solution-2% L-Cysteine (100 mL) once more. The upper organic phase was washed with 25% brine (100 mL). The organic layer (105.0 g) was assayed (118.8 mg/g, 12.47 g product assayed, 87% assayed yield), and concentrated to ˜1/3 volume (˜35 mL). The product solution was directly used in the next step without isolation. However, an analytical sample was obtained by removal of solvent to give a brown oil. 1HNMR (CDCl3): δ 1.00 (s, 6H), 1.49 (t, J=6.6 Hz, 2H), 2.28 (t, J=2.1 Hz, 2H), 2.38 (m, 2H), 7.13 (m, 2H), 7.34 (m, 2H), 9.47 (s, 1H).
-
Example 5 Synthesis of tert-butyl 4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazine-1-carboxylate (Compound (H))
-
To a 2 L three neck RB flask provided with a mechanical stirrer were charged a solution of 4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-carbaldehyde (50.0 g) in toluene (250 mL), BOC-piperazine (48.2 g) and anhydrous THF (250 mL). The yellow solution was stirred at 20° C. for 5 min. Sodium triacetoxyborohydride (52.7 g) was added in portion (note: the internal temperature rose to ˜29.5° C. in 15 min cooling may be needed). The yellow mixture was stirred at ˜25° C. for NLT 4 hrs. A conversion of starting material to product of 99.5% was observed by HPLC after a 3 hour reaction time.
-
12.5 wt % brine (500 g) was added slowly to quench the reaction. The mixture was stirred at 20° C. for NLT 30 min and allowed to settle for NLT 15 min. The lower aq. phase (˜560 mL) was separated (note: leave any emulsion in the upper organic phase). The organic phase was washed with 10% citric acid solution (500 g×2). 500 g of 5% NaHCO3 aq. solution was charged slowly into the flask. The mixture was stirred at 20° C. for NLT 30 min., and allowed to settle for NLT 15 min. The upper organic phase was separated. 500 g of 25% brine aq. solution was charged. The mixture was stirred at 20° C. for NLT 15 min., and allowed to settle for NLT 15 min. The upper organic phase was concentrated to ˜200 mL volume under vacuum. The solution was adjusted to −30° C., and filtered off the inorganic salt. Toluene (50 mL) was used as a rinse. The combined filtrate was concentrated to ˜100 mL volume. Acetonitrile (400 mL) was added, and the mixture heated to ˜80° C. to achieve a clear solution. The solution was cooled down slowly to 20° C. slowly at rate 10° C./hour, and mixed at 20° C. overnight (the product is crystallized out at ˜45-50° C., if needed, seed material may be added at 50° C.). The slurry was continued to cool down slowly to ˜−10° C. at rate of 10° C./hours. The slurry was mixed at ˜−10° C. for NLT 6 hours. The product was collected by filtration, and rinsed with pre-cooled acetonitrile (100 mL). The solid was dried under vacuum at 50° C. overnight (72.0 g, 85%). MS-ESI: 419 (M+1); mp: 109-110° C. (uncorrected); 1H NMR (CDCl3): δ 1.00 (s, 6H), 1.46 (s, 9H), 1.48 (t, J=6.5 Hz, 2H), 2.07 (s, br, 2H), 2.18 (m, 4H), 2.24 (t, J=6.4 Hz, 2H), 2.80 (s, 2H), 3.38 (m, 4H), 6.98 (m, 2H), 7.29 (m, 2H).
-
Example 6 Synthesis of 1-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazine dihydrochloride (Compound (I))
-
To a 2.0 L three-neck RB flask equipped with a mechanical stirrer were charged the Boc reductive amination product (Compound (H), 72.0 g) and IPA (720 mL). The mixture was stirred at rt for 5 min, and 59.3 g of concentrated hydrochloride aq. solution added to the slurry. The reaction mixture was adjusted to an internal temperature of ˜65° C. (a clear and colorless solution achieved). The reaction mixture was agitated at ˜65° C. for NLT 12 hours.
-
The product slurry was cooled down to −5° C. slowly (10° C./hour). The product slurry was mixed at ˜−5° C. for NLT 2 hours, collected by filtration. The wet cake was washed with IPA (72 mL) and dried at 50° C. under vacuum overnight to give 73.8 g (95%) of the desired product as a bis-hydrochloride IPA solvate (purity >99.5% peak area at 210 nm). MS-ESI: 319 (M+1); 1HNMR (CDCl3): δ 0.86 (s, 6H), 1.05 (d, J=6.0 Hz, 6H, IPA), 1.42 (t, J=6.1 Hz, 2H), 2.02 (s, br, 2H), 2.12 (m, 2H), 3.23 (m, 4H), 3.4 (s, br, 4H), 3.68 (s, 2H), 3.89 (septet, J=6.0 Hz, 1H, IPA), 7.11 (d, J=8.1 Hz, 2H), 7.41 (d, J=8.1 Hz, 2H).
-
Example 7 Synthesis of 3-nitro-4-(((tetrahydro-2H-pyran-4-yl)methyl)amino)-benzenesulfonamide (Compound (N))
-
To a 500 mL three-neck RB flask equipped with a mechanical stirrer were charged the 4-chloro-3-nitrobenzenesulfonamide, Compound M (10.0 g), diisopropylethylamine (17.5 g), (tetrahydro-2H-pyran-4-yl)methanamine (7.0 g) and acetonitrile (150 mL). The reaction mixture was adjusted to an internal temperature of 80° C. and agitated for no less than 12 hours.
-
The product solution was cooled down to 40° C. and agitated for no less than 1 hour until precipitation observed. The product slurry was further cooled to 20° C. Water (75 mL) was slowly charged over no less than 1 hour, and the mixture cooled to 10° C. and agitated for no less than 2 hours before collected by filtration. The wet cake was washed with 1:1 mix of acetonitrile:water (40 mL). The wet cake was then reslurried in water (80 mL) at 40° C. for no less than 1 hour before collected by filtration. The wet cake was rinsed with water (20 mL), and dried at 75° C. under vacuum to give 12.7 g of the desired product in 99.9% purity and in 91% weight-adjusted yield. 1H NMR (DMSO-d6): δ 1.25 (m, 2H), 1.60 (m, 2H), 1.89 (m, 1H), 3.25 (m, 2H), 3.33 (m, 2H), 3.83 (m, 2H), 7.27 (d, J=9.3 Hz, 1H), 7.32 (s, NH2, 2H), 7.81 (dd, J=9.1, 2.3 Hz, 1H), 8.45 (d, J=2.2 Hz, 1H), 8.54 (t, J=5.9 Hz, 1H, NH).
-
Example 8 Synthesis of tert-butyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoate (Compound (K))
-
General Considerations:
-
this chemistry is considered air and moisture sensitive. While the catalyst precursors in their solid, dry form can be handled and stored in air without special precautions, contact with even small amounts of solvent may render them susceptible to decomposition. As a result, traces of oxygen or other competent oxidants (e.g., solvent peroxides) must be removed prior to combination of the catalyst precursors with solvent and care must be used to prevent ingress of oxygen during the reaction. Also, care must be taken to use dry equipment, solvents, and reagents to prevent formation of undesirable byproducts. The sodium t-butoxide used in this reaction is hygroscopic and it should be properly handled and stored prior to or during use.
-
To a 2.0 L three-neck RB flask equipped with a mechanical stirrer were charged the bis-hydrochloride salt (Compound (I), 42.5 g) and toluene (285 ml). 20% K3PO4 (285 ml) was added and the biphasic mixture was stirred for 30 min. The layers were separated and the organic layer was washed with 25% NaCl (145 ml). The organic layer concentrated to 120 g and used in the coupling reaction without further purification.
-
NaOtBu (45.2 g) and Compound (I) in toluene solution (120 g solution −30 g potency adjusted) were combined in THF (180 ml) in a suitable reactor and sparged with nitrogen for NLT 45 min. Pd2dba3 (0.646 g), Compound (J) (0.399 g), and Compound (D) (40.3 g) were combined in a second suitable reactor and purged with nitrogen until oxygen level was NMT 40 ppm. Using nitrogen pressure, the solution containing Compound (I) and NaOtBu in toluene/THF was added through a 0.45 μm inline filter to the second reactor (catalyst, Compound (J) and Compound (D)) and rinsed with nitrogen sparged THF (30 ml).
-
The resulting mixture was heated to 55° C. with stirring for NLT 16 h, then cooled to 22° C. The mixture was diluted with 12% NaCl (300 g) followed by THF (300 ml). The layers were separated.
-
The organic layer was stirred with a freshly prepared solution of L-cysteine (15 g), NaHCO3 (23 g), and water (262 ml). After 1 h the layers were separated.
-
The organic layer was stirred with a second freshly prepared solution of L-cysteine (15 g), NaHCO3 (23 g), and water (262 ml). After 1 h the layers were separated. The organic layer was washed with 12% NaCl (300 g), then filtered through a 0.45 μm inline filter. The filtered solution was concentrated in vacuo to ˜300 mL, and chased three times with heptane (600 mL each) to remove THF.
-
The crude mixture was concentrated to 6 volumes and diluted with cyclohexane (720 ml). The mixture was heated to 75° C., held for 15 min, and then cooled to 65° C. over NLT 15 min. Seed material was charged and the mixture was held at 65° C. for 4 hours. The suspension was cooled to 25° C. over NLT 8 h, then held at 25° C. for 4 hours. The solids were filtered and washed with cyclohexane (90 ml) and dried at 50° C. under vacuum.
-
Isolated 52.5 g (88.9% yield) as a white solid. Melting point (uncorrected) 154-155° C. 1H NMR (DMSO-d6): δ 0.93 (s, 6H), 1.27 (s, 9H), 1.38 (t, J=6.4 Hz, 2H), 1.94 (s, 2H), 2.08-2.28 (m, 6H), 2.74 (s, 2H), 3.02-3.19 (m, 4H), 6.33 (dd, J=3.4, 1.9 Hz, 1H), 6.38 (d, J=2.4 Hz, 1H), 6.72 (dd, J=9.0, 2.4 Hz, 1H), 6.99-7.06 (m, 2H), 7.29 (d, J=2.7 Hz, 1H), 7.30-7.36 (m, 2H), 7.41-7.44 (m, 1H), 7.64 (t, J=6.7 Hz, 1H), 7.94 (d, J=2.7 Hz, 1H), 11.53 (s, 1H).
-
Example 9 Synthesis of 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoic acid (Compound (L))
-
Solution preparation: 10% KH2PO4 (aq): KH2PO4 (6 g) in water (56 g); 2:1 heptane/2-MeTHF:heptane (16 mL) in 2-MeTHF (8 mL).
-
Compound (K) (5.79 g), potassium tert-butoxide (4.89 g), 2-methyltetrahydrofuran (87 mL), and water (0.45 mL) were combined in a suitable reactor under nitrogen and heated to 55° C. until reaction completion. The reaction mixture was cooled to 22° C., washed with the 10% KH2PO4 solution (31 g) twice. The organic layer was then washed with water (30 g).
-
After removal of the aqueous layer, the organic layer was concentrated to 4 volumes (˜19 mL) and heated to no less than 50° C. Heptane (23 ml) was slowly added. The resulting suspension was cooled to 10° C. Solids were then collected by vacuum filtration with recirculation of the liquors and the filter cake washed with 2:1 heptane/2-MeTHF (24 ml). Drying of the solids at 80° C. under vacuum yielded 4.0 g of Compound (L) in approximately 85% weight-adjusted yield. 1H NMR (DMSO-d6): δ 0.91 (s, 6H), 1.37 (t, J=6.4 Hz, 2H), 1.94 (s, br, 2H), 2.15 (m, 6H), 2.71 (s, br, 2H), 3.09 (m, 4H), 6.31 (d, J=2.3 Hz, 1H), 6.34 (dd, J=3.4, 1.9 Hz, 1H), 6.7 (dd, J=9.0, 2.4 Hz, 1H), 7.02 (m, 2H), 7.32 (m, 2H), 7.37 (d, J=2.6 Hz, 1H), 7.44 (t, J=3.0 Hz, 1H), 7.72 (d, J=9.0 Hz, 1H), 7.96 (d, J=2.7 Hz, 1H) & 11.59 (m, 1H).
-
Example 10 Synthesis of 4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (Compound (I))
-
Solution preparation prior to reaction: 10% Acetic Acid:Acetic Acid (37 mL) in water (333 g); 5% NaHCO3:NaHCO3 (9 g) in water (176 g); 5% NaCl:NaCl (9 g) in water (176 g).
-
Compound (N) (13.5 g), DMAP (10.5 g), EDAC (10.7 g) and dichloromethane (300 mL) were combined in a suitable reactor and agitated at 25° C. In a second suitable reactor was charged the Acid (Compound (L), 25 g), Et3N (8.7 g) and dichloromethane (120 mL). The resulting Acid (Compound (L)) solution was slowly charged to the initial suspension of Compound (N) and agitated until reaction completion.








-
N,N-dimethylethylenediamine (9.4 g) was then charged to the reaction mixture with continued agitation. The reaction mixture was warmed to 35° C. and washed with 10% Acetic acid solution (185 mL) twice. The lower organic layer was diluted with more dichloromethane (75 mL) and methanol (12.5 mL). The organic, product layer was then washed with 5% NaHCO3 solution (185 mL) and then washed with 5% NaCl solution (185 mL) at 35° C. The lower, organic layer was separated and then concentrated to 8 vol (˜256 mL) diluted with methanol (26 mL) and warmed to 38° C. Ethyl Acetate (230 mL) was slowly charged. The resulting suspension was slowly cooled to 10° C. and then filtered. The wet cake was washed twice with a 1:1 mix of dichloromethane and ethyl acetate (˜2 vol, 64 mL). After drying the wet cake at 90° C., 32 g (84%) of Compound (I) was isolated.
-
1H NMR (DMSO-d6): δ 0.90 (s, 6H), 1.24 (m, 2H), 1.36 (t, J=6.4 Hz, 2H), 1.60 (m, 2H), 1.87 (m, 1H), 1.93 (s, br, 2H), 2.12 (m, 2H), 2.19 (m, 4H), 2.74 (s, br, 2H), 3.06 (m, 4H), 3.26 (m, 4H), 3.83 (m, 2H), 6.17 (d, J=2.1 Hz, 1H), 6.37 (dd, J=3.4, 1.9 Hz, 1H), 6.66 (dd, J=9.1, 2.2 Hz, 1H), 7.01 (m, 2H), 7.31 (m, 2H), 7.48 (m, 3H), 7.78 (dd, J=9.3, 2.3 Hz, 1H), 8.02 (d, J=2.61 Hz, 1H), 8.54 (d, J=2.33 Hz, 1H), 8.58 (t, J=5.9 Hz, 1H, NH), 11.65 (m, 1H).
PATENT

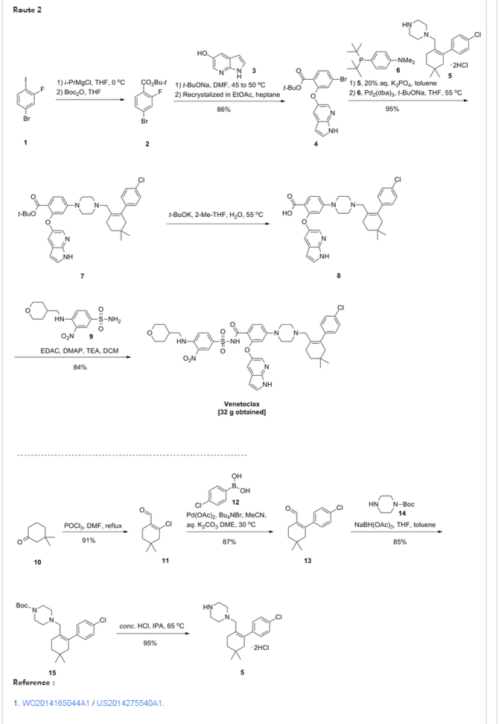
| Patent | Submitted | Granted |
|---|---|---|
| APOPTOSIS-INDUCING AGENTS FOR THE TREATMENT OF CANCER AND IMMUNE AND AUTOIMMUNE DISEASES [US2014275082] | 2014-02-10 | 2014-09-18 |
| Processes For The Preparation Of An Apoptosis-Inducing Agent [US2014275540] | 2014-03-12 | 2014-09-18 |
| APOPTOSIS INDUCING AGENTS FOR THE TREATMENT OF CANCER AND IMMUNE AND AUTOIMMUNE DISEASES [US2010305122] | 2010-12-02 | |
| Panel of micrornas that silence the MCL-1 gene and sensitize cancer cells to ABT-263 [US8742083] | 2010-12-23 | 2014-06-03 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
| METHODS OF TREATMENT USING SELECTIVE BCL-2 INHIBITORS [US2012129853] | 2011-11-22 | 2012-05-24 |
| INHIBITION OF MCL-1 AND/OR BFL-1/A1 [US2015051249] | 2013-03-14 | 2015-02-19 |
| COMBINATION THERAPY OF A TYPE II ANTI-CD20 ANTIBODY WITH A SELECTIVE BCL-2 INHIBITOR [US2014248262] | 2013-09-06 | 2014-09-04 |
References
- New Drugs Online Report for venetoclax
- Hard-to-Treat CLL Yields to Investigational Drug. ASH Dec 2015 refs: Roberts AW, et al “Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia” N Engl J Med 2015; DOI: 10.1056/NEJMoa1513257.
- Phase 2 Study of Venetoclax in Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia with 17p Deletion Meets Primary Endpoint
- ABT-199 BH-3 Mimetic Enters Phase Ia Trial For Chronic Lymphocytic Leukemia. 2011
- For Refractory CLL, Venetoclax’s Complete Response Rate Is Tops. 2015
External links
- ABT-199 inc formula and structure
|
References |
1: Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2
inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013 Jan 6. doi: 10.1038/nm.3048. [Epub ahead of print] PubMed PMID: 23291630.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
|
|
| Identifiers | |
| CAS Number | 1257044-40-8 |
| PubChem | CID: 49846579 |
| ChemSpider | 29315017 |
| Chemical data | |
| Formula | C45H50ClN7O7S |
| Molecular mass | 868.44 g/mol |
/////////
CC1(CCC(=C(C1)c2ccc(cc2)Cl)CN3CCN(CC3)c4ccc(c(c4)Oc5cc6cc[nH]c6nc5)C(=O)NS(=O)(=O)c7ccc(c(c7)[N+](=O)[O-])NCC8CCOCC8)C
OR
CC1(CCC(=C(C1)C2=CC=C(C=C2)Cl)CN3CCN(CC3)C4=CC(=C(C=C4)C(=O)NS(=O)(=O)C5=CC(=C(C=C5)NCC6CCOCC6)[N+](=O)[O-])OC7=CN=C8C(=C7)C=CN8)C
AMUVATINIB
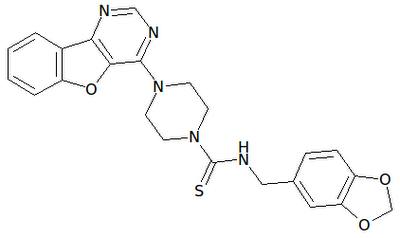
AMUVATINIB
| Name | N-(3,4-Methylenedioxiphenylmethyl) -4 – (benzofuro [3,2-d] pyrimidin-4-yl) piperazine-1-carbothioamide. |
| CAS | 850879-09-3 |
| Formula | C 23 H 21 N 5 O 3 S |
| MW | 447.51 |
| Synonim | MN-470, SGI-0470-03 |
Amuvatinib (MP-470) is an orally bioavailable synthetic carbothioamide with potential antineoplastic activity. Multitargeted receptor tyrosine kinase inhibitor MP470 binds to mutant forms of the stem cell factor receptor (c-Kit; SCFR), inhibiting clinically relevant mutants of this receptor tyrosine kinase that may be associated with resistance to therapy. In addition, MP470 inhibits activities of other receptor tyrosine kinases, such as c-Met, Ret oncoprotein, and mutant forms of Flt3 and PDGFR alpha, which are frequently dysregulated in variety of tumors. This agent also suppresses the induction of DNA repair protein Rad51, thereby potentiating the activities of DNA damage-inducing agents. Mutant forms of c-Kit are often associated with tumor chemoresistance.

http://www.google.co.in/patents/EP1678166A2?cl=en
Scheme 1

EXAMPLE 34 Synthesis and Analysis of Further Illustrative Compounds Compound (111-1-3), also referred to herein as HPK56/MP-470, is an illustrative compound of the present invention having the following structure:

Analogues of (111-1 -3) were designed and synthesized in order to evaluate and optimize kinase selectivity, aqueous solubility, and to improve pharmacokinetic and pharmacodynamic profiles. Illustrative synthesis approaches for generating (111-1 -3) analogues are depicted in the synthesis schemes below. Synthesis of R-i substituted benzofuranopyrimidines was undertaken. The methyl 3-guanidinobenzofuran-2-carboxylate is prepared from methyl 3-aminobenzofuran-2-carboxylate by reacting with cyanoacetamide in presence of dioxane and dry HCI gas. The obtained guanidine is cyclized in the presence of aqueous NaOH. Similar procedures were utilized for preparing 2- substituted (111-1-3) and its analogues as depicted in the Schemes 8-10 set forth below. Introduction of -NH2 at the 2 position was utilized for various sulfonic, inorganic and hydroxyacid salts. Illustrative compounds are shown in Table 4 below.Table 4


Scheme 1

Scheme 2 Scheme 3
,-NH2 /N Cl S

EXAMPLE 35 Analysis of Compound Binding and Inhibitory Activity against c-kit Mutants
The published crystal structure of c-kit kinase (pdb code:1 PKG) and its mutated structure were used to study the mode of binding of compound (111-1-3) (HPK56/MP-470), a benzofuranopyrimidine compound, its 2-substituted analogs, and quinazoline derivatives.

(Ill- 1-3)
|
Information about this agent |
According to news published on 15 Apr 2008; Research data build upon previous results showing that MP-470 exhibits anti-tumor activity in breast and prostate cancer cells. The fact that MP-470 in combination with erlotinib effectively suppressed the HER pathway suggests that concurrent administration of both compounds could represent a new treatment for prostate and breast cancers.
|
References |
1. Kreidberg, Jordan A.; Qin, Shan. Method using a cMET inhibitor for the treatment of polycystic kidney disease. PCT Int. Appl. (2009), 43pp. CODEN: PIXXD2 WO 2009111529 A2 20090911 CAN 151:350826 AN 2009:1107388
2. Fujiwara, Masahiro; Fujita, Masayuki. Curing agents, adhesive curable compositions, articles and coating materials therefrom, and optical materials formed by using the compositions. Jpn. Kokai Tokkyo Koho (2008), 39pp. CODEN: JKXXAF JP 2008260894 A 20081030 CAN 149:472741 AN 2008:1303932
3. Janne, Pasi A.; Engelman, Jeffrey; Cantley, Lewis C. Methods for treating cancer resistant to ErbB therapeutics. PCT Int. Appl. (2008), 96pp. CODEN: PIXXD2 WO 2008127710 A2 20081023 CAN 149:486836 AN 2008:1282443
4. Nakagawa, Kiyoshi; Kurushima, Yoshiaki; Mizuma, Masahiro. Fabric with highly-expanded layer and process for production thereof. PCT Int. Appl. (2007), 44pp. CODEN: PIXXD2 WO 2007083641 A1 20070726 CAN 147:213021 AN 2007:815020
5. Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; Nagle, R.; Bearss, D. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene (2007), 26(27), 3909-3919. CODEN: ONCNES ISSN:0950-9232. CAN 147:226484 AN 2007:613908
6. Gong, QingJie; Han, DongYu; Wang, YuRong. Experimental determination of scheelite solubility in 4.0% NaCl solution in critical region. Yanshi Xuebao (2006), 22(12), 3052-3058. CODEN: YANXEU ISSN:1000-0569. CAN 147:34728 AN 2007:301330
7. Tokunaga, Koji; Mukai, Takashi; Kishigami, Akira. Two-component weak solvent-based coating compositions with no curing interference by dewing. Jpn. Kokai Tokkyo Koho (2005), 15 pp. CODEN: JKXXAF JP 2005239815 A 20050908 CAN 143:249850 AN 2005:979155
8. Lauer, S. J.; Shuster, J. J.; Mahoney, D. H., Jr.; Winick, N.; Toledano, S.; Munoz, L.; Kiefer, G.; Pullen, J. D.; Steuber, C. P.; Camitta, B. M. A comparison of early intensive methotrexate/mercaptopurine with early intensive alternating combination chemotherapy for high-risk B-precursor acute lymphoblastic leukemia: A pediatric oncology group phase III randomized trial. Leukemia (2001), 15(7), 1038-1045. CODEN: LEUKED ISSN:0887-6924. CAN 135:338846 AN 2001:586045
9. Sen, Ayusman; Hennis, April. Palladium (II) catalyzed polymerization of norbornene and acrylates. PCT Int. Appl. (2001), 22 pp. CODEN: PIXXD2 WO 2001021670 A1 20010329 CAN 134:252774 AN 2001:228935
10. Patel, Raman; Mallin, Dan; Saunders, Keith; Tiberio, Patrick; Andries, John. Polymer compositions containing polyolefins, polar polymers and block or graft copolymer compatibilizers and their preparation. PCT Int. Appl. (1999), 25 pp. CODEN: PIXXD2 WO 9950350 A1 19991007 CAN 131:272658 AN 1999:640932
11. Koehler, Burkhard; Imai, Seisaku; Doering, Joachim; Ruesseler, Wolfgang; Dorf, Ernst Ullrich. Mixtures of polyarylene sulfides, glass fibers, and polymaleimides with good mechanical properties. Ger. Offen. (1992), 4 pp. CODEN: GWXXBX DE 4105913 A1 19920827 CAN 118:82122 AN 1993:82122
12. Itoh, Michiya; Fuke, Kiyokazu; Kobayashi, Sachiko. Direct observation of intramolecular anthracene excimer in 1,3-dianthrylpropane. Journal of Chemical Physics (1980), 72(2), 1417-18. CODEN: JCPSA6 ISSN:0021-9606. CAN 92:155340 AN 1980:155340
13. Tomillero A; Moral M A Gateways to clinical trials. Methods and findings in experimental and clinical pharmacology (2009), 31(9), 597-633. Journal code: 7909595. ISSN:0379-0355. PubMed ID 20094643 AN 2010048694
14. Tomillero A; Moral M A Gateways to clinical trials. Methods and findings in experimental and clinical pharmacology (2009), 31(8), 541-57. Journal code: 7909595. ISSN:0379-0355. PubMed ID 19967103 AN 2009808578
15. Gorrand Jean-Marie; Doly Michel; Bacin Franck Macular pigment density assessed by directional fundus reflectance. Journal of the Optical Society of America. A, Optics, image science, and vision (2009), 26(8), 1847-54. Journal code: 9800943. ISSN:1084-7529. PubMed ID 19649122 AN 2009529589
16. Tomillero A; Moral M A Gateways to clinical trials. Methods and findings in experimental and clinical pharmacology (2009), 31(4), 263-98. Journal code: 7909595. ISSN:0379-0355. PubMed ID 19557204 AN 2009445725

Italy’s Newron files Parkinson’s drug with FDA

SAFINAMIDE
cas 202825-46-5 (mesylate)
N2-{4-[(3-fluorobenzyl)oxy]benzyl}-L-alaninamide
Newron Pharmaceuticals and fellow Italy-headquartered partner Zambon have filed their investigational Parkinson’s disease treatment safinamide with regulators in the USA.
The submission to the US Food and Drug Administration is for safinamide as add-on therapy in early and mid-to late stage PD patients. Newron said the filing was based on “completion of activities agreed upon during meetings” with the FDA, noting that a marketing authorisation application was submitted to the European Medicines Agency in December.
Read more at: http://www.pharmatimes.com/Article/14-05-30/Italy_s_Newron_files_Parkinson_s_drug_with_FDA.aspx#ixzz33LlGLEt7


Safinamide (EMD 1195686) is a candidate drug against Parkinson’s disease with multiple methods of action.[1] In 2007, a Phase III clinical trial was started. It was scheduled to run until 2011.[2] The compound was originally discovered at Farmitalia-Carlo Erba and developed by Newron Pharmaceuticals, which sold the rights to Merck-Serono in 2006. In October 2011 Merck-Serono announced that they would give all rights to develop the compound back to Newron.[3]
Potential additional uses might be restless legs syndrome (RLS) and epilepsy.[4] They were being tested in Phase II trials in 2008, but no results are available.
Adverse effects
Common adverse events in clinical trials were nausea, dizziness, tiredness, headache and backache. There was no significant difference in the occurrence of these effects between safinamide and placebo treated patients.[5]
Methods of action
Parkinson and RLS relevant mechanisms
Safinamide is a reversible and selective monoamine oxidase B inhibitor, reducing degradation of dopamine, and a glutamate release inhibitor.[6][5] It also seems to inhibit dopamine reuptake.[7] Additionally, safinamide blocks sodium and calcium channels.[6]
References
- Fariello, RG (2007). “Safinamide”. Neurotherapeutics 4 (1): 110–116. doi:10.1016/j.nurt.2006.11.011. PMID 17199024.
- Study of Safinamide in Early Parkinson’s Disease as Add-on to Dopamine Agonist (MOTION)
- Merck Returns Rights for Safinamide to Newron, 21 October 2011.
- Chazot, PL (2007). “Drug evaluation: Safinamide for the treatment of Parkinson’s disease, epilepsy and restless legs syndrome”. Current Opinion in Investigational Drugs 8 (7): 570–579. PMID 17659477.
- H. Spreitzer (14 April 2014). “Neue Wirkstoffe – Safinamid”. Österreichische Apothekerzeitung (in German) (8/2014): 30.
- Caccia, C; Maj, R; Calabresi, M; Maestroni, S; Faravelli, L; Curatolo, L; Salvati, P; Fariello, RG (2006). “Safinamide: From molecular targets to a new anti-Parkinson drug”. Neurology 67 (7 Suppl 2): S18–23. PMID 17030736.
- Merck Serono: Vielversprechende Daten zur kognitiven Wirkung von Safinamid bei Parkinson im Frühstadium.(German) 8 June 2007.
|
1-13-2012
|
PHARMACEUTICAL COMPOSITION
|
|
|
10-12-2011
|
Pharmaceutical composition
|
|
|
10-22-2004
|
Methods of treating lower urinary tract disorders using sodium channell modulators
|
|
|
7-16-1999
|
ALPHA-AMINOAMIDE DERIVATIVES USEFUL AS ANALGESIC AGENTS
|

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link
blogs are

MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ
VIETNAM
ICELAND
RUSSIA


FDA Breakthrough Therapy Designation: Another For Genentech And Cancer
On May 31st, Genentech (member of the Roche Group) strategically announces that the FDA grants the Breakthrough Therapy Designation (BTD) to the company’s investigational cancer immunotherapy MPDL3280A (anti-PDL1) for the treatment of Bladder Cancer. At the 50th Annual Meeting of the American Society of Clinical Oncology (ASCO), now in progress in Chicago, Dr. Thomas Powles, M.D., clinical Professor of Genitourinary Oncology, Barts Cancer Institute at the Queen Mary University of London, is presenting on May 31st, Abstract #5011 (Results of the Phase I MPDL3280A Study).
The Phase I MPDL3280A Study, a single-arm, multi-center, open label trial, shows that MPDL3280A “shrank tumors (ORR – Overall Response Rate) in 43% (13/30) of people previously treated for metastatic Urothelial Bladder Cancer (UBC), whose tumors were characterized as PD-L1 (Programmed Death Ligand-1) positive by a test being developed by Roche.”
Per the Genentech Press Release, bladder cancer:
• Is the 9th most…
View original post 111 more words

FDA May 2014 Products Receiving Orphan Designation
.
.
.
.
The chart below identifies FDA May 2014 Products Receiving Orphan Designation as of 05/31/14 in ascending “Orphan Drug Designation Date” order.
FDA May 2014 Products Receiving Orphan Designation
| # | Generic Name/ODD Date | Sponsor Company | Indication |
| 1 | Filanesib/ 05.06 | Array BioPharma | Multiple Myeloma |
| 2 | Autologous dendritic cells pulsed with allogeneic tumor cell lysate/ 05.06 | Amphera BV (Netherlands) | Malignant Mesothelioma |
| 3 | Ex vivo cultured human mesenchymal stromal cells / 05.08 | iCell Science AB (Sweden) | Prevention of graft rejection following solid organ transplantation |
| 4 | Adalimumab/ 05.13 | AbbVie | Uveitis |
| 5 | Diazoxide choline / 05.13 | Essentialis | Prader-Willi Syndrome |
| 6 | Vasoactive intestinal peptide (VIP)-elastin-like peptide (ELP) fusion protein / 05.13 | PhaseBio Pharmaceuticals | Pulmonary arterial hypertension |
| 7 | (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1H-1,2,4-triazol-1-yl)-N-(pyrazin-2-yl)acrylohydrazide / 05.14 | Karyopharm Therapeutics | Diffuse large B-cell lymphoma |
| 8 | 177Lu-tetraxetan-tetulomab / 05.14 | Nordic Nanovector AS (Norway) | Follicular Lymphoma |
| 9 | Selinexor; (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1H-1,2,4-triazol-1-yl)-N-(pyrazin-2-yl)acrylohydrazide / 05.14 | Karyopharm Therapeutics | Acute myeloid leukemia |
| 10 | Menadione Sodium Bisulfite/ 05.14 |
View original post 198 more words
Cocrystals

Active pharmaceutical ingredients (APIs) are frequently delivered to the patient in the solid state as part of such dosage forms as tablets, capsules, etc.In this context the ability to deliver the drug to the patient in a safe, efficacious and cost-effective way depends largely on the physicochemical properties of the APIs in the solid state, and ……..read more
API SCALEUP (R AND D)
|
|
| The ultimate goal of drug synthesis is to scale up from producing milligram quantities in a laboratory to producing kilogram to ton quantities in a plant, all while maintaining
all at |
Polymer Nanoflower Encapsulates Two Cancer Drugs to Hit Tumors with More Punch
Many existing anti-cancer drugs can be disappointingly ineffective in clinical practice, but often it is the delivery method and not the medication itself that limits effectiveness. Being able to deliver multiple drugs together, each with a different mechanism of action, to their target can be considerably more powerful than separate administrations. Researchers at North Carolina State University and the University of North Carolina at Chapel Hill have developed a “nanoflower” made out of a hydrophilic polymer that carries camptothecin and doxorubicin directly into cancer cells.

The hydrophobic drugs are encapsulated within the polyethylene glycol structure similarly to how proteins fold in on themselves. At about 50 nanometers in diameter, the nanoflowers can be injected into the bloodstream to seek out cancer cells. In an animal study, the structures stayed together until they penetrated lung cancer cells by taking advantage of “lipid raft and clathrin-mediated endocytotic pathway without premature leakage,” according…
View original post 39 more words
BioCryst Pharmaceuticals Inc. ( BCRX ) will be reporting results from OPuS-1, a phase IIa trial of orally-administered BCX4161 in patients with hereditary angioedema

(RTTNews.com) – BioCryst Pharmaceuticals Inc. ( BCRX ) will be reporting results from OPuS-1, a phase IIa trial of orally-administered BCX4161 in patients with hereditary angioedema, on Tuesday, May 27, 2014 at 8:30 a.m. Eastern Time.
The OPuS-1 clinical trial is testing 400 mg of BCX4161 administered three times daily for 28 days in up to 25 hereditary angioedema patients who have a high frequency of attacks (≥ 1 per week), in a randomized, placebo-controlled, two-period cross-over design.
Read more: http://www.nasdaq.com/article/bcrx-to-watch-out-for-gtiv-adopts-poison-pill-teva-qgen-drtx-get-fda-nod-20140527-00005#ixzz335Khl0sk
BCX-4161 is a novel, selective inhibitor of plasma kallikrein in development for prevention of attacks in patients with hereditary angioedema (HAE). By inhibiting plasma kallikrein, BCX-4161 suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
……………………………….
old article
BCRX – BioCryst – Entering The HAE Market
BioCryst announced on Monday July 22 the successful completion of a Phase I study on the safety and PK of BCX4161, a candidate for the treatment of Hereditary angioedema (HAE). HAE is a genetic disorder resulting from the loss or dysfunction of complement C1 Inhibitor (C1INH).
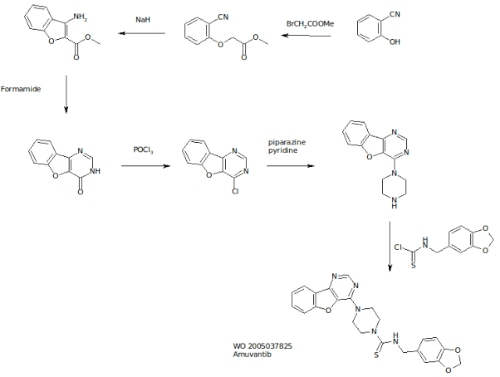
Among the functions performed by C1INH is regulation of the hormone bradykinin, which when activated, leads to the dilation of blood vessels. Left unchecked, excess bradykinin can cause painful attacks of swelling, or angioedemas, in any part of the body, including the face, abdomen, hands, and larynx. Death can occur from asphyxiation, particularly in children.
The mechanics involved in HAE are fairly well understood today. There are several approved drugs available today that work at three major points in the pathway. Ultimately, each prevents bradykinin from activating its receptor on endothelial cells.
C1 Inhibitors, of which four have been approved, prevent Factor XIIa activation of Plasma Kallikrein and inhibit Kallikrein itself. The single specific Kallikrein inhibitor is Kalbitor from Dyax. C1INHs and kallikrein inhibitors prevent the formation of bradykinin (labeled “BK” in this diagram). Then there is Firazyr from Shire, a B2 bradykinin receptor antagonist; while not preventing overproduction of the hormone, activation of downstream activity is suppressed.
Interestingly, of all the available therapies, only C1INH Cinryze from Viropharma is approved for prophylactic use- all others are designated strictly for treatment of acute attacks. A key reason for this is Cinryze’s long half-life, allowing sustained activity over longer intervals. As each of these drugs are given by injection, frequent treatment is not practical. Consider, for instance, Kalbitor has a half life of just two hours.
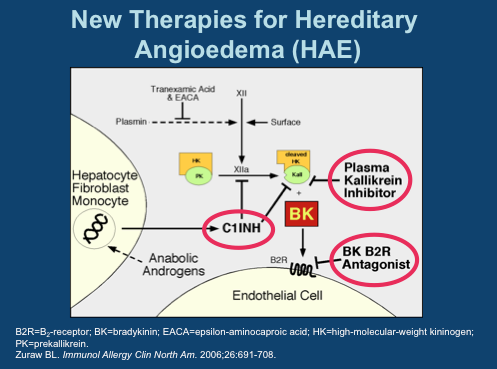
This is where BioCryst comes in. The company is pursuing the less crowded prophylaxis indication. It has the only orally available (although just barely) plasma kallikrein inhibitor. And while PK is not great, requiring three-times daily dosing to ensure adequate drug levels, pills make this a feasible option. As you can see, 800 mg appears optimal, however, 400 mg was selected as the Phase IIa dose due to 3 cases of moderate AEs seen at 800. This study was in healthy volunteers and the drug was otherwise well tolerated [ref].
(From Company Presentation)
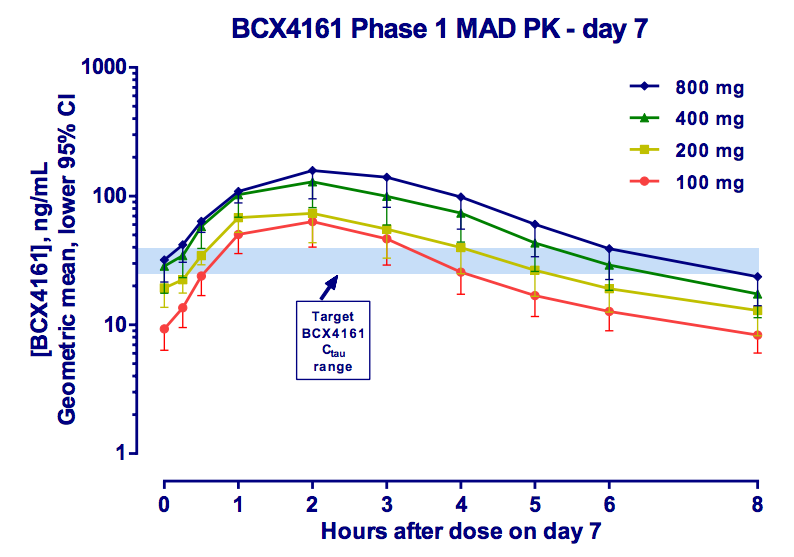
BCX4161 is an interesting compound. Based on patent literature, we believe the molecule has a similar structure to the one illustrated below:
BCX4161 is not a specific inhibitor of kallikrein, and in fact has near equal potency against Factor XIIa. This dual-activity is also seen with C1INH, setting the compound apart from Kalbitor and Firazyr.
The different profile may improve efficacy, but that is unknown at this point. Along with Factor XIIa, BCX4161 inhibits additional factors involved in coagulation. Bleeding issues has been something the company has been testing and will be certain to monitor. As a drug designed for chronic use, safety will be a major concern.
A 25 patient Phase IIa study set for Q4 will be placebo-controlled double-blind crossover of the following design:
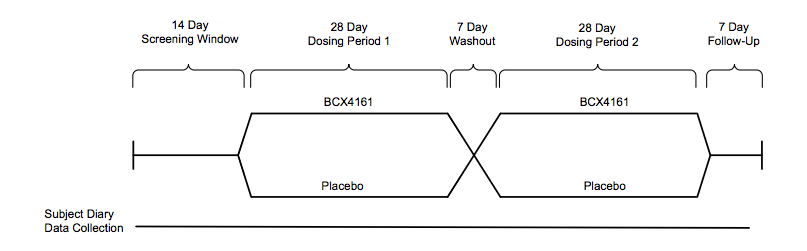
(From Company Presentation)
Individuals with a high frequency of attacks(~1/week) will be enrolled, the primary endpoint is attack frequency. Viropharma conducted a pivotal trial of similar design (but two twelve week dosing periods), reporting ~50% reduction in attacks vs. placebo. We imagine BioCryst would need to achieve results in this range for the drug to be competitive.
A major impedance toward these efficacy goals will likely be individual adherence to dosing every eight hours schedule. Missed doses will mean severe drops in drug levels, potentially putting the patient at risk for an attack. The company noted patients on Cinryze occasionally miss doses with no apparent adverse effect. We will see if this holds true for their own compound.
The Phase IIa is being run in Germany, ostensibly because of the country’s well organized HAE medical treatment system. The study is expected to initiate in 4Q 2013. BioCryst aims to market the drug in the U.S. on their own, likely partnering in the EU.
Handicapping this Phase II is rather difficult with the lack of any prior efficacy results. BioCryst has selected a well-validated target in a fairly well understood disease. The data suggests BCX4161 is an active drug. What we will soon find out is whether the compound is active enough and has a sufficiently clean profile. As attractive as oral dosing is- it has an achilles heel. Regardless of the medication, patients continue to have attacks, only of less frequency and severity. If a patient should suffer major laryngeal swelling, pills may not be an option as a rescue medicine. Cinryze on the hand can serve as both prophylaxis and acute treatment.
Commercially, we believe the compound will have a difficult time competing with Cinryze. True, Cinryze has its own issues, namely a requirement for infusions every 3 to 4 days, but it is difficult to see how a 3-times/day treatment is much of an improvement. In any case, by the time BCX4161 reaches the market, Viropharma should have a much simpler subcutaneous version of its C1INH available, allowing it to maintain a strong monopoly in prophylaxis HAE treatments. Additional competition may come in the form of a follow-up kallikrein inhibitor in development at Dyax; the long acting antibody is designed specifically for the prophylaxis market and is expected to enter the clinic 2H 2013.
आयुर्वेद न्यूट्रास्यूटीकल्स ; अधिक गर्मी के कारण होने वाली तकलीफो से बचने के लिये ………
आयुर्वेद की विशेषता यही है कि सभी मौसम के लिये सभी के लिये आयुर्वेद के मनीषियों ने मौसम से प्राप्त सभी वस्तुओ के सटीक उपयोग के लिये combinations दे दिये है /
गर्मी के मौसम मे गर्म हवाओ के चलने और लू लपट high degree temperature के चलते हुये बहुत सी तकलीफे शरीर मे अचानक पैदा होने की स्तिथि बन जाती है , इन सभी अवस्थाओ से बचने के लिये नीचे लिखे nueutraceutical को अपनाइये और फायदा उठाइये ;
साम्ग्री सब आप्के किचन मे मिल जायेगी /
१- एक छोटा प्याज , बड़ा हो तो आधा कर ले
२- एक कच्चा छोटा आम / अमिया
३-१५ -२० पुदीना की पत्ती / अधिक भी छोड़ सकते है
४- एक टुकड़ा अदरख
५- एक चम्मच जल जीरा मसाला पाउडर
५- आधा चम्मच काली मिर्च
६- स्वादानुसार काला नमक
७- एक या दो चम्मच शक्कर / चीनी / गुड़
८- आधा या…
View original post 277 more words
DR ANTHONY MELVIN CRASTO
ORGANIC SPECTROSCOPY

