
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64



GSK launches huge Phase III trial for heart drug losmapimod (GW856553)

losmapimod
Losmapimod is a p38 mitogen-activated protein kinase inhibitor.
Smithkline Beecham Corporation
6-[5-(cyclopropylcarbamoyl)-3-fluoro-2-methylphenyl]-N-(2,2-dimethylpropyl)pyridine-3-carboxamide
GW856553X, 585543-15-3, Losmapimod (USAN/INN), UNII-F2DQF16BXE, AGN-PC-00BFXU,
Molecular Formula: C22H26FN3O2
Molecular Weight: 383.459143
cas 585543-15-3
Synonym: Losmapimod; GW856553; GW-856553; GW 856553)
IUPAC/Chemical name:
6-[5-(cyclopropylcarbamoyl)-3-fluoro-2-methylphenyl]-N-(2,2-dimethylpropyl)pyridine-3-carboxamide
GlaxoSmithKline has begun a Phase III study cardiovascular outcomes study of its investigational compound losmapimod in patients with acute coronary syndrome.
The trial will assess whether losmapimod can reduce the risk of a subsequent cardiac event when administered orally twice a day for three months immediately after presentation with an ACS, such as heart attack. GSK says that some 25,500 patients will be enrolled over the study period across 39 countries.
Read more at: http://www.pharmatimes.com/Article/14-06-06/GSK_launches_huge_Phase_III_trial_for_heart_drug_losmapimod.aspx#ixzz33vFHAK14
Losmapimod, also know as GW856553 or GW856553X, is a drug developed by GlaxoSmithKline which acts as a selective inhibitor of the enzyme family known as p38 mitogen-activated protein kinases. p38 mitogen-activated protein kinases are mediators of inflammation. A Phase II human clinical trial for the treatment of COPD (chronic obstructive pulmonary disease) is underway. Inhibiting these enzymes has been shown to produce antidepressant and antipsychotic effects in animal studies, with the mechanism thought to involve increased neurogenesis probably related to BDNF release. Losmapimod has completed Phase II human clinical trials for the treatment of depression although its safety and efficacy have yet to be proven in further trials. Losmapimod is also being studied for cardiovascular disease. A Phase II trial to study its effects in myocardial infarction (heart attack) is ongoing.

Losmapimod (GW856553X) is a drug developed by GlaxoSmithKline which acts as a selective inhibitor of the enzyme family known as p38 mitogen-activated protein kinases.[1]
p38 mitogen-activated protein kinases are mediators of inflammation. A Phase II human clinical trial for the treatment of COPD(chronic obstructive pulmonary disease)[2] is underway. Inhibiting these enzymes has been shown to produce antidepressant andantipsychotic effects in animal studies, with the mechanism thought to involve increased neurogenesis[3] probably related to BDNFrelease. Losmapimod has completed Phase II human clinical trials for the treatment of depression although its safety and efficacy have yet to be proven in further trials.[4]
Losmapimod is also being studied for cardiovascular disease.[5] A Phase II trial to study its effects in myocardial infarction (heart attack) is ongoing.[6]
………………………
http://www.google.com/patents/US8252818

| Example 36 6-(5- Cyclopropylcarbamoyl- 3-fluoro-2-methyl- phenyl)-N-(2,2- dimethylpropyl)- nicotinamide |
 |
6-Chloro-N-(2,2- dimethylpropyl))nicotin- amide (Intermediate 24) | 384 | 3.01 |
……………….
https://www.google.com/patents/US7514456
General Method A
6-Bromonicotinic acid (100 mg, 0.5 mmol) was heated at 95° C. in thionyl chloride (0.63 ml) for 2 hours. The excess thionyl chloride was evaporated under vacuum and the residue dissolved in DCM (2 ml). To this solution, amine (0.5 mmol) and sodium carbonate (100 mg) were added and the reaction was stirred at room temperature for 2 hours. The reaction was filtered and the residue washed with DCM. The combined filtrate and washings were reduced to dryness to give the desired 6-chloronicotinamide.
| Retention time | |||
| Compound | Amine | MH+ | (minutes) |
| Intermediate 22: 6-Chloro-N-(3- | 3-methylbutylamine | 227 | 2.92 |
| methylbutyl)nicotinamide | |||
| Intermediate 23: 6-Chloro-N-(1- | 1-cyclopropylethylamine | 225 | 2.65 |
| cyclopropylethyl)nicotinamide | |||
| Intermediate 24: 6-Chloro-N-(2,2- | 2,2-dimethylpropylamine | 227 | 2.82 |
| dimethylpropyl))nicotinamide | |||
| Intermediate 25: 6-Chloro-N-(2,2- | 2,2- | 225 | 2.67 |
|
8-29-2012
|
Nicotinamide derivatives useful as P38 inhibitors
|
|
|
8-3-2011
|
Use of a p38 Kinase Inhibitor for Treating Psychiatric Disorders
|
|
|
11-24-2010
|
3-Aminocarbonyl, 6-phenyl substituted pyridine-1-oxides as p38 kinase inhibitors
|
|
|
8-27-2010
|
NICOTINAMIDE DERIVATES USEFUL AS P38 INHIBITORS
|
|
|
5-5-2010
|
Nicotinamide Derivatives Useful as p38 Inhibitors
|
|
|
4-8-2009
|
Nicotinamide Derivatives Useful as p38 Inhibitors
|
|
|
10-25-2006
|
Nicotinamide derivatives useful as p38 inhibitors.
|
References
- Aston N, Bamborough P, Buckton J, Edwards C, Holmes D, Jones K, Patel V, Smee P, Somers D, Vitulli G, Walker A. p38α Mitogen-Activated Protein Kinase Inhibitors: Optimization of a Series of Biphenylamides to Give a Molecule Suitable for Clinical Progression.Journal of Medicinal Chemistry 2009, 52(20), 6257. doi:10.1021/jm9004779
- Randomised, Double-Blind, Placebo-Controlled, Parallel-Group, Multi-centre, Dose Ranging Study to Evaluate the Efficacy and Safety of Losmapimod Tablets Administered Twice Daily Compared With Placebo for 24 Weeks in Adult Subjects With Chronic Obstructive Pulmonary Disease (COPD)
- Noh JS, Kang HJ, Kim YE, Sohn S, Chung YK, Kim SU, Gwag BJ. Haloperidol-Induced Neuronal Apoptosis: role of p38 and c-Jun-NH(2)-terminal protein kinase. Journal of Neurochemistry 2000, 75(6), 2327. PMID 11080184 doi:10.1046/j.1471-4159.2000.0752327.x
- A Study of GW856553X For the Treatment of Depression
- Cheriyan et al., Circulation 2011, 123(5), 515-523. Inhibition of p38 Mitogen-Activated Protein Kinase Improves Nitric Oxide–Mediated Vasodilatation and Reduces Inflammation in Hypercholesterolemia doi:10.1161/CIRCULATIONAHA.110.971986
- A Study to Evaluate the Safety of 12 Weeks of Dosing With GW856553 and Its Effects on Inflammatory Markers, Infarct Size, and Cardiac Function in Subjects With Myocardial Infarction Without ST-segment Elevation (Solstice)
|
more References |
1: Yang S, Beerahee M. Losmapimod concentration-QT relationship in healthy volunteers: meta-analysis of data from six clinical trials. Eur J Clin Pharmacol. 2013 Jun;69(6):1261-7. doi: 10.1007/s00228-012-1469-1. Epub 2013 Jan 17. PubMed PMID: 23325437.
2: Yang S, Lukey P, Beerahee M, Hoke F. Population pharmacokinetics of losmapimod in healthy subjects and patients with rheumatoid arthritis and chronic obstructive pulmonary diseases. Clin Pharmacokinet. 2013 Mar;52(3):187-98. doi: 10.1007/s40262-012-0025-6. PubMed PMID: 23254770.
3: Dewenter M, Vettel C, El-Armouche A. [Losmapimod: a novel drug against cardiovascular diseases?]. Dtsch Med Wochenschr. 2013 Jan;138(1-2):39-42. doi: 10.1055/s-0032-1327368. Epub 2012 Dec 18. Review. German. PubMed PMID: 23250695.
4: Ostenfeld T, Krishen A, Lai RY, Bullman J, Baines AJ, Green J, Anand P, Kelly M. Analgesic efficacy and safety of the novel p38 MAP kinase inhibitor, losmapimod, in patients with neuropathic pain following peripheral nerve injury: a double-blind, placebo-controlled study. Eur J Pain. 2013 Jul;17(6):844-57. doi: 10.1002/j.1532-2149.2012.00256.x. Epub 2012 Dec 14. PubMed PMID: 23239139.
5: Barbour AM, Sarov-Blat L, Cai G, Fossler MJ, Sprecher DL, Graggaber J, McGeoch AT, Maison J, Cheriyan J. Safety, tolerability, pharmacokinetics and pharmacodynamics of losmapimod following a single intravenous or oral dose in healthy volunteers. Br J Clin Pharmacol. 2013 Jul;76(1):99-106. doi: 10.1111/bcp.12063. PubMed PMID: 23215699; PubMed Central PMCID: PMC3703232.
6: Melloni C, Sprecher DL, Sarov-Blat L, Patel MR, Heitner JF, Hamm CW, Aylward P, Tanguay JF, DeWinter RJ, Marber MS, Lerman A, Hasselblad V, Granger CB, Newby LK. The study of LoSmapimod treatment on inflammation and InfarCtSizE (SOLSTICE): design and rationale. Am Heart J. 2012 Nov;164(5):646-653.e3. doi: 10.1016/j.ahj.2012.07.030. Epub 2012 Oct 16. PubMed PMID: 23137494.
7: Elkhawad M, Rudd JH, Sarov-Blat L, Cai G, Wells R, Davies LC, Collier DJ, Marber MS, Choudhury RP, Fayad ZA, Tawakol A, Gleeson FV, Lepore JJ, Davis B, Willette RN, Wilkinson IB, Sprecher DL, Cheriyan J. Effects of p38 mitogen-activated protein kinase inhibition on vascular and systemic inflammation in patients with atherosclerosis. JACC Cardiovasc Imaging. 2012 Sep;5(9):911-22. doi: 10.1016/j.jcmg.2012.02.016. PubMed PMID: 22974804.
8: Lomas DA, Lipson DA, Miller BE, Willits L, Keene O, Barnacle H, Barnes NC, Tal-Singer R; Losmapimod Study Investigators. An oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary disease. J Clin Pharmacol. 2012 Mar;52(3):416-24. doi: 10.1177/0091270010397050. Epub 2011 Nov 16. PubMed PMID: 22090363.
9: Cheriyan J, Webb AJ, Sarov-Blat L, Elkhawad M, Wallace SM, Mäki-Petäjä KM, Collier DJ, Morgan J, Fang Z, Willette RN, Lepore JJ, Cockcroft JR, Sprecher DL, Wilkinson IB. Inhibition of p38 mitogen-activated protein kinase improves nitric oxide-mediated vasodilatation and reduces inflammation in hypercholesterolemia. Circulation. 2011 Feb 8;123(5):515-23. doi: 10.1161/CIRCULATIONAHA.110.971986. Epub 2011 Jan 24. PubMed PMID: 21262998.
10: Welchman R. Advances and Progress in Drug Design – SMi’s ninth annual meeting. IDrugs. 2010 Apr;13(4):239-42. PubMed PMID: 20373252.
11: Willette RN, Eybye ME, Olzinski AR, Behm DJ, Aiyar N, Maniscalco K, Bentley RG, Coatney RW, Zhao S, Westfall TD, Doe CP. Differential effects of p38 mitogen-activated protein kinase and cyclooxygenase 2 inhibitors in a model of cardiovascular disease. J Pharmacol Exp Ther. 2009 Sep;330(3):964-70. doi: 10.1124/jpet.109.154443. Epub 2009 Jun 25. PubMed PMID: 19556450.
GSK Announces Phase III Cardiovascular Outcomes Study with Losmapimod in Patients with Acute Coronary Syndrome
GlaxoSmithKline plc Thursday 5 June 2014, London UK (LSE/NYSE: GSK) today announced the start of a pivotal phase III study, LATITUDE-TIMI 60, to evaluate the effects of losmapimod in patients presenting with acute coronary syndrome. The global, phase III study will assess whether losmapimod can reduce the risk of a subsequent cardiac event when administered…
Fasting for three days can regenerate entire immune system, study finds
Fasting for as little as three days can regenerate the entire immune system, even in the elderly, scientists have found in a breakthrough described as “remarkable”.
Although fasting diets have been criticised by nutritionists for being unhealthy, new research suggests starving the body kick-starts stem cells into producing new white blood cells, which fight off infection.
Scientists at the University of Southern California say the discovery could be particularly beneficial for people suffering from damaged immune systems, such as cancer patients on chemotherapy.
It could also help the elderly whose immune system becomes less effective as they age, making it harder for them to fight off even common diseases.
The researchers say fasting “flips a regenerative switch” which prompts stem cells to create brand new white blood cells, essentially regenerating the entire immune system.
“It gives the ‘OK’ for stem cells to go ahead and begin proliferating and rebuild the…
View original post 575 more words
Tie up with Emcure…..Roche to launch cheaper cancer drugs in India
Reuters | Updated On: June 06, 2012 12:36 (IST)
Mumbai:
Swiss drugmaker Roche Holding AG plans to offer cut-price versions of two blockbuster cancer drugs for the Indian market soon, a company spokesman said on Friday, days after New Delhi moved to slash the price of a rival cancer treatment.
India stripped German’s Bayer AG of its exclusive rights to Nexavar earlier this month and licensed a local drugs company to produce a cheap, generic version, on the grounds that poor Indians could not otherwise afford the life-saving drug.
Roche, the world’s biggest maker of cancer drugs, said it would offer “significantly” cheaper, locally branded versions of its two cancer drugs, Herceptin and MabThera, by early next year, under an alliance with India’s Emcure Pharmaceuticals Ltd.
http://profit.ndtv.com/news/corporates/article-roche-to-launch-cheaper-cancer-drugs-in-india-300344
Russian scientists create cancer cure that doubles life span

Scientists in the Russian city of Novosibirsk have created a vaccine that can double a cancer patient’s life expectancy. The drug has already been tested clinically and may soon appear in stores.
When given to patients suffering from the third and fourth stage of colorectal, breast and prostate cancer, the treatment can significantly prolong their life span, says Vladimir Kozlov, the head of the Clinical Immunology Institute that did the research.
The vaccine is extracted from the dendritic cells, which are immune cells producing antigen material. The patient’s dendritic cells are then altered with the tumor cell culture and injected into the organizm, triggering an immune response, which means they attack cancer.
The treatment is now available to people after a cancer surgery, although scientists envisage the drug would be able to cure cancer in the early stages.
New therapy wipes out cervical cancer in two women
Aricca Wallace knew she was nearly out of time. For more than three years, she had suffered cramping and irregular bleeding, which her doctor thought was a side effect of her birth control implant, known as an intrauterine device, or IUD. Her annual Pap smears were always normal, so no one suspected cancer.
Except it was cancer, and by the time the 34-year-old mother of two had the IUD removed and was finally diagnosed, her tumors had reached stage three and the disease was spreading through the lymph nodes in her abdomen and chest.
“I was told by a specialist that there wasn’t any chemo that could kill it,” Wallace told AFP. “And that I’d be gone in a year.”
That was in February 2012. A few months later, Wallace’s doctor told her about an immunotherapy trial at the National Institutes of Health Clinical Center, a research hospital just outside the…
View original post 646 more words
Hey, did you consider the impact of waste disposal as part of the cost of your API synthesis ?
Hi Everyone, I want to thank Chemjobber for the renewed interest in an article I posted a while back. The posting was about selecting solvents, my post was “Which solvent should I choose ?”, referring to an article that was published by some colleagues at GSK, Green Chem., 2011,13, 854-862, DOI: 10.1039/c0gc00918k. I have an article that is somewhat related to picking solvents in my current grab-bag of chemical literature.
I have to admit that it never crossed my mind to figure out how I was going to dispose of the chemical waste my reactions were generating. Health & Safety comes by and disposed of my chemical waste. I think that there is a movement to introduce some self-consciousness in the chemistry we are using and part of this responsibility is to consider what impact we have on the environment (did I dare say that ?) …
View original post 272 more words
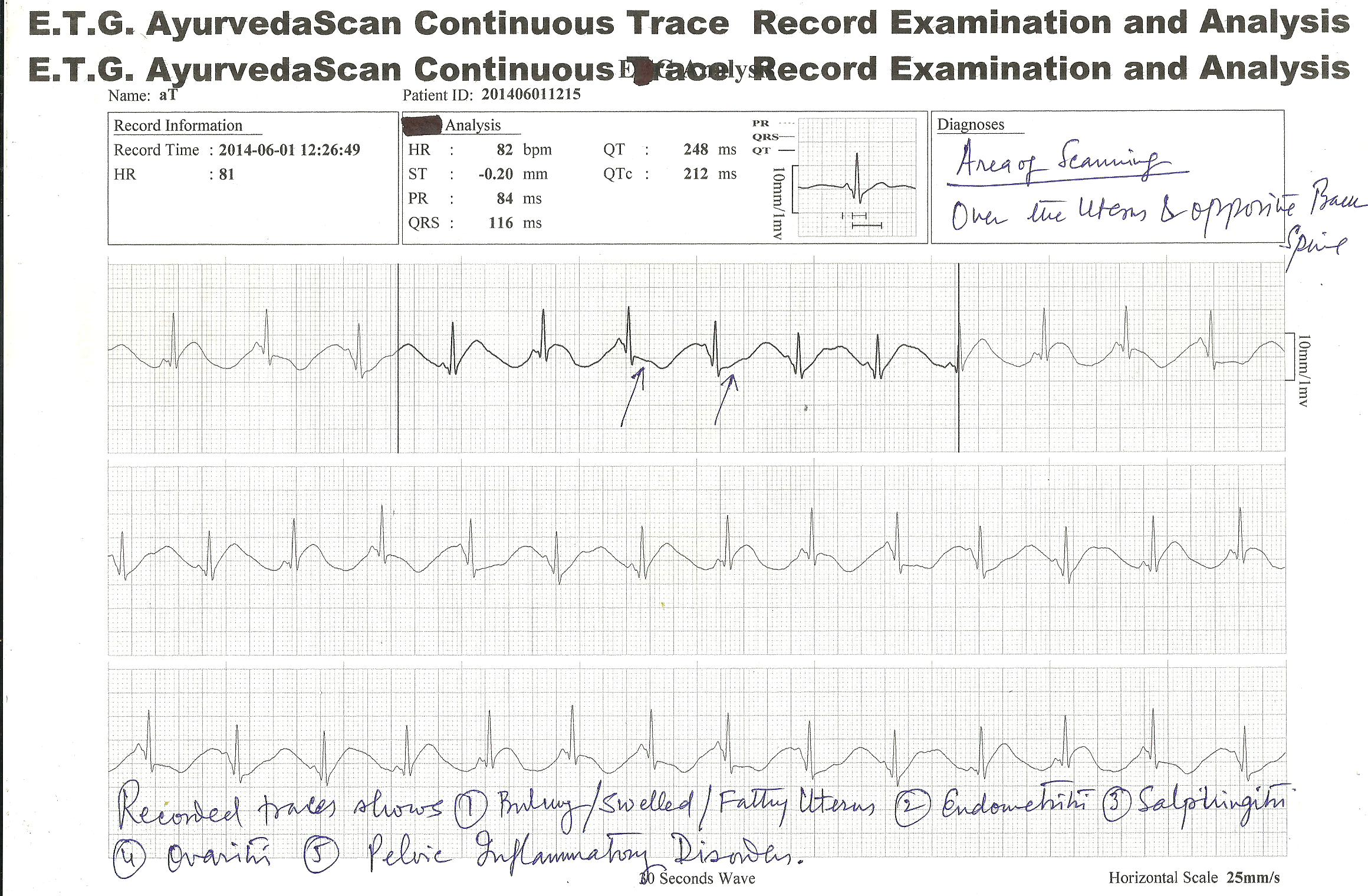
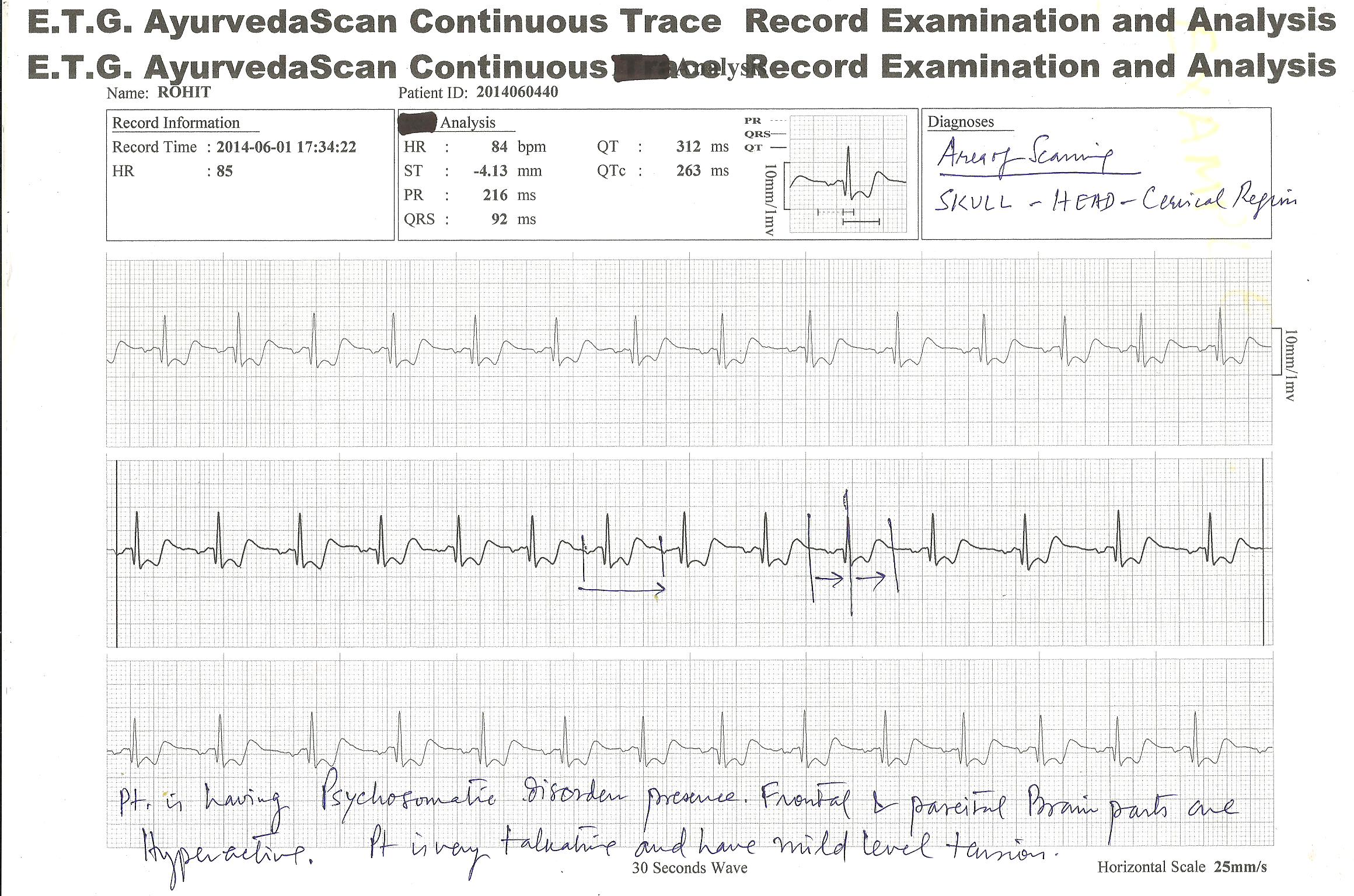
E.T.G. AyurvedaScan CONTINUOUS TRACE RECORD TEST RESULTS ; FEW ILLUSTRATION OF CASES
NEW INVENTION of ETG AyurvedaScan system have now introduced “continuous test” of the organs or viscera affected for their minute patho-physiological and pathological studies for diagnosis and other purposes.
Below is given three patient’s traces , which was recorded at least 04 hours continuously for best diagnosis and pin-point problem solutions.
………………………..
……………………………
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
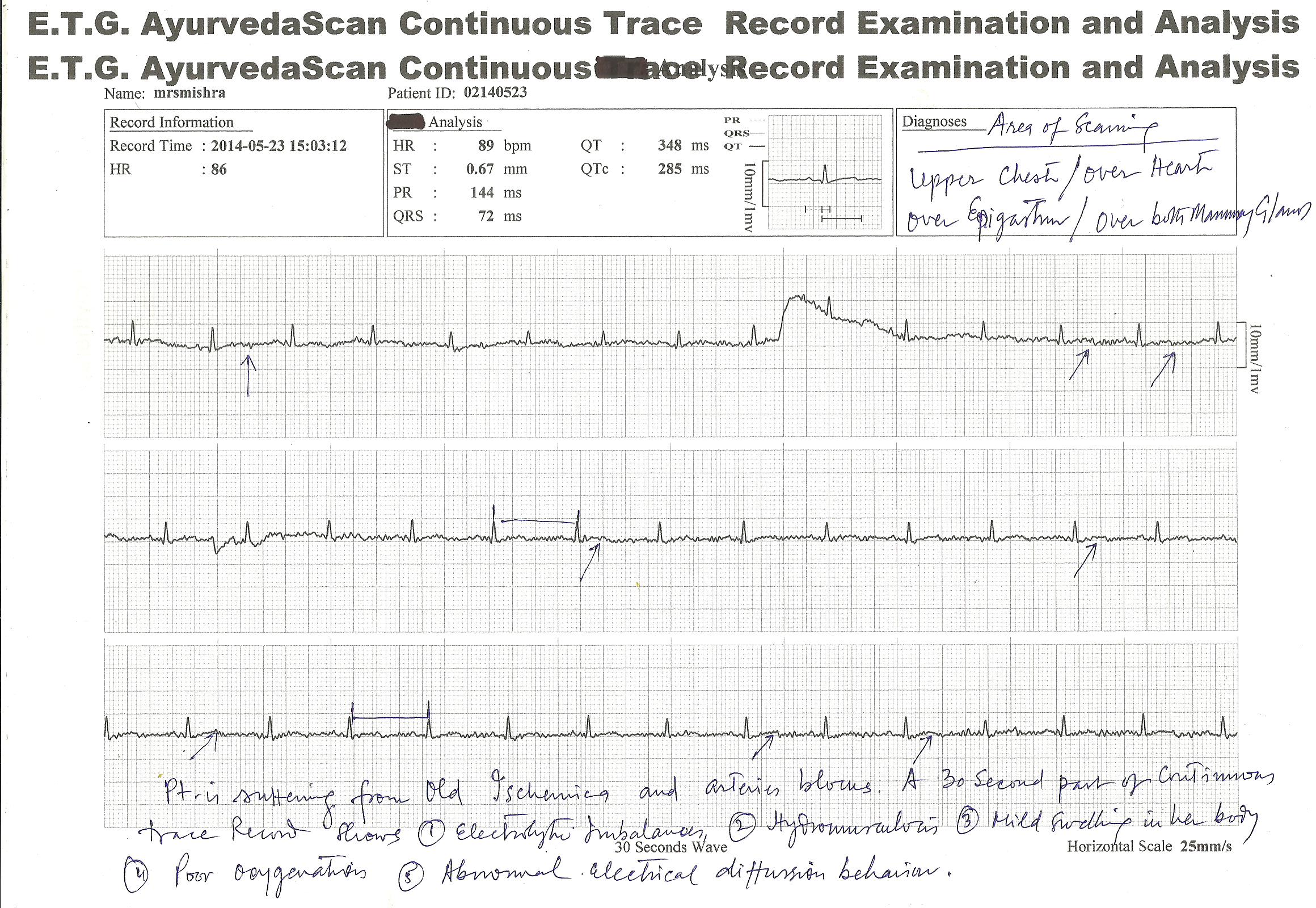
E.T.G. AyurvedaScan CONTINUOUS TRACE RECORDING SYSTEM provides regular monitoring of the affected area or patient problems diagnosis and to know , what is happening with the patient within 24 hours or within 48 hours oe more , may be monitorised by this system.
Many patient complaints that they feel problems at any hours of the day or night , for them this process is beneficial to trace the problems they have.
Few hours like 3 to 4 hours continuous monitoring is also beneficial for short term observations. These observations are beneficial for treatment and management of…
View original post 139 more words
Autism linked to ‘male hormones’
http://www.bbc.com/news/health-27662080
Exposure to high levels of “male” hormones in the womb increases the chance of a baby boy developing autism, according to researchers.
The University of Cambridge researchers say their findings from more than 300 boys help unravel the causes of autism – a condition that affects both sexes but is far more common in males.
But they say it does not mean a prenatal test for autism is near.
Nor will it necessarily be possible to stop autism by blocking the hormones.
“Because some of these hormones are produced in much higher quantities in males than in females, this may help us explain why autism is more common in males” – Prof Baron-Cohen Study author
The hormones in question – testosterone and three other steroid hormones – were important for foetal development, which meant it could be too risky to block them, they told the journal Molecular Psychiatry.
Autism…
View original post 435 more words
A New Approach for Heart Disease: G Strophantin
Coronary artery disease is currently the leading cause of death in the United States. Despite the increasing sophistication of surgical techniques, the introduction of new techniques such as balloon angioplasty, and a number of new drugs (e.g. beta blockers, calcium antagonists), it is estimated that over 1 million heart attacks will occur this year, resulting in 500,000 deaths. In short, we do not have an adequate therapeutic solution to the problem of myocardial infarction (heart attack).
The cornerstone of therapy for treatment and prevention of myocardial infarction is to remove blockages in coronary arteries that are thought to be the cause of the infarction. This adheres to the widely accepted coronary artery thrombosis theory of infarction; that is, arteries become clogged with plaque, damaged from such things as smoking or high cholesterol. A clot forms a fissure in the plaque. The clot may shut off the blood flow of the…
View original post 1,784 more words
Billions and billions…of molecules?
Billions and billions…of molecules?

READ
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

