
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64



Plerixafor…………..an immunostimulant used to mobilize hematopoietic stem cells in cancer patients.

Plerixafor
cas 110078-46-1
CXCR4 chemokine antagonist
Stem cell mobilization [CXCR4 receptor antagonist]
A bicyclam derivate, highly potent & selective inhibitor of HIV-1 & HIV-2.
Bone marrow transplantation; Chronic lymphocytic leukemia; Chronic myelocytic leukemia; Myelodysplastic syndrome; Neutropenia; Sickle cell anemia
Plerixafor; Mozobil; AMD3100; 110078-46-1; Amd 3100; bicyclam JM-2987; AMD-3100; UNII-S915P5499N; JM3100
- JKL 169
- Mozobil
- Plerixafor
- SDZ SID 791
- UNII-S915P5499N
Molecular Formula: C28H54N8
Molecular Weight: 502.78196
1,4-bis((1,4,8,11-tetraazacyclotetradecan-1-yl)methyl)benzene
1,4,8,11-Tetraazacyclotetradecane, 1,1′-(1,4-phenylenebis(methylene))bis-

1,1′-[1,4-phenylenebis(methylene)]bis [1,4,8,11-tetraazacyclotetradecane]
1,1′- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane
Johnson Matthey (Innovator)
Plerixafor is a hematopoietic stem cell mobilizer. It is used to stimulate the release of stem cells from the bone marrow into the blood in patients with non-Hodgkin lymphoma and multiple myeloma for the purpose of stimulating the immune system. These stem cells are then collected and used in autologous stem cell transplantation to replace blood-forming cells that were destroyed by chemotherapy. Plerixafor has orphan drug status in the United States and European Union; it was approved by the U.S. Food and Drug Administration on December 15, 2008.

Mozobil (plerixafor injection) is a sterile, preservative-free, clear, colorless to pale yellow, isotonic solution for subcutaneous injection. Each mL of the sterile solution contains 20 mg of plerixafor. Each single-use vial is filled to deliver 1.2 mL of the sterile solution that contains 24 mg of plerixafor and 5.9 mg of sodium chloride in Water for Injection adjusted to a pH of 6.0 to 7.5 with hydrochloric acid and with sodium hydroxide, if required.
Plerixafor is a hematopoietic stem cell mobilizer with a chemical name l, 1′-[1,4phenylenebis (methylene)]-bis-1,4,8,11-tetraazacyclotetradecane. It has the molecular formula C28H54N8. The molecular weight of plerixafor is 502.79 g/mol. The structural formula is provided in Figure 1.
Figure 1: Structural Formula
 |
Plerixafor is a white to off-white crystalline solid. It is hygroscopic. Plerixafor has a typical melting point of 131.5 °C. The partition coefficient of plerixafor between 1octanol and pH 7 aqueous buffer is < 0.1.

Plerixafor (hydrochloride hydrate) |
(CAS 155148-31-5)
| Formal Name | 1,4-bis((1,4,8,11-tetraazacyclotetradecan-1-yl)methyl)benzene, octahydrochloride |
|---|---|
| CAS Number | 155148-31-5 |
| Molecular Formula | C28H54N8 • 8HCl • [XH2O] |
| Formula Weight | 794.5 |
The α-chemokine receptor, CXCR4, on CD4+ T-cells is used by CXCR4-selective HIV forms as a gateway for T-cell infection. In mammalian cell signaling, CXCR4 activation promotes the homing of hematopoietic stem cells, chemotaxis and quiescence of lymphocytes, and growth and metastasis of certain cancer cell types. Plerixafor (hydrochloride) is a macrocyclic compound that acts as an irreversible antagonist against the binding of CXCR4 with its ligand, SDF-1 (CXCL12). It suppresses infection by HIV with an IC50 value of 1-10 ng/ml with selectivity toward CXCR4-tropic virus. Plerixafor mobilizes hematopoietic stem and progenitor cells for transplant better than the ‘gold standard’, G-CSF alone 4and synergizes with G-CSF. It also increases T-cell trafficking in the blood and spleen as well as the central nervous system. Plerixafor regulates the growth of primary and metastic breast cancer cells7 and inhibits dissemination of ovarian carcinoma cells.
Plerixafor hydrochloride (AMD-3100), a chemokine CXCR4 (SDF-1) antagonist, is launched in the U.S. for the following indications: to enhance mobilization of hematopoietic stem cells for autologous transplantation in patients with lymphoma and to enhance mobilization of hematopoietic stem cells for transplantation in patients with multiple myeloma.
In 2009, the product was approved in EU for these indications.AnorMED filed an orphan drug application for AMD-3100 with the FDA in January 2003 and received approval in July 2003 as immunostimulation for increasing the stem cells available in patients with multiple myeloma and non-Hodgkin’s lymphoma. Orphan drug status was also granted by the EMEA in October 2004 as a treatment to mobilize progenitor cells prior to stem cell transplantation.
In 2011, orphan drug designation was assigned by the FDA for the treatment of AML and by the EMA for the adjunctive treatment to cytotoxic therapy in acute myeloid leukemia.
Plerixafor (rINN and USAN, trade name Mozobil) is an immunostimulant used to mobilize hematopoietic stem cells in cancer patients. The stem cells are subsequently transplanted back to the patient. The drug was developed by AnorMED which was subsequently bought by Genzyme.
History
The molecule 1,1′-[1,4-phenylenebis(methylene)]bis [1,4,8,11-tetraazacyclotetradecane], consisting of two cyclam rings linked at the amine nitrogen atoms by a 1,4-xylyl spacer, was first synthesised by Fabbrizzi et al. in 1987 to carry out basic studies on the redox chemistry of dimetallic coordination compounds.[1] Then, it was serendipitously discovered by De Clercq that such a molecule, could have a potential use in the treatment of HIV[2] because of its role in the blocking of CXCR4, a chemokine receptor which acts as a co-receptor for certain strains of HIV (along with the virus’s main cellular receptor, CD4).[2]Development of this indication was terminated because of lacking oral availability and cardiac disturbances. Further studies led to the new indication for cancer patients.[3]
Indications
Peripheral blood stem cell mobilization, which is important as a source of hematopoietic stem cells for transplantation, is generally performed using granulocyte colony-stimulating factor (G-CSF), but is ineffective in around 15 to 20% of patients. Combination of G-CSF with plerixafor increases the percentage of persons that respond to the therapy and produce enough stem cells for transplantation.[4] The drug is approved for patients with lymphoma and multiple myeloma.[5]
Contraindications
Pregnancy and lactation
Studies in pregnant animals have shown teratogenic effects. Plerixafor is therefore contraindicated in pregnant women except in critical cases. Fertile women are required to use contraception. It is not known whether the drug is secreted into the breast milk. Breast feeding should be discontinued during therapy.[5]
Adverse effects
Nausea, diarrhea and local reactions were observed in over 10% of patients. Other problems with digestion and general symptoms like dizziness, headache, and muscular pain are also relatively common; they were found in more than 1% of patients. Allergies occur in less than 1% of cases. Most adverse effects in clinical trials were mild and transient.[5][6]
The European Medicines Agency has listed a number of safety concerns to be evaluated on a post-marketing basis, most notably the theoretical possibilities of spleen rupture and tumor cell mobilisation. The first concern has been raised because splenomegaly was observed in animal studies, and G-CSF can cause spleen rupture in rare cases. Mobilisation of tumor cells has occurred in patients with leukaemia treated with plerixafor.[7]
Phase III clinical development in combination with G-CSF (granulocyte colony-stimulating factor) is under way at Genzyme (which acquired the product through its acquisition of AnorMED in late 2006) in a stem cell mobilization regimen in non-Hodgkin’s lymphoma (NHL). The trials are designed to evaluate the potential of plerixafor in combination with G-CSF, to rapidly increase the number of peripheral blood stem cells capable of engraftment, thereby increasing the proportion of patients reaching a peripheral blood stem cell target and, as a result, reducing the number of apheresis sessions required for patients to collect a target number of peripheral blood stem cells. A phase I safety trial had been under way for the treatment of renal cancer, however, no recent development for this indication has been reported. An IND has been filed in the U.S. seeking approval to initiate clinical evaluation of the drug candidate to help repair damaged heart tissue in patients who have suffered heart attacks. Currently, an investigator-sponsored study is ongoing to evaluate plerixafor as a single agent in allogeneic transplant. AMD-3100, in combination with mitoxantrone, etoposide and cytarabine, is also in phase I/II clinical trials at the University of Washington for the treatment of acute myeloid leukemia (AML).
The University has also been conducting early clinical trials for increasing the stem cells available for transplantation in patients with advanced hematological malignancies, however, no recent developments on this trial have been reported. Genzyme has completed a phase I/II clinical study of plerixafor hydrochloride in combination with rituximab for the treatment of chronic lymphocytic leukemia. The former AnorMED had been developing plerixafor for the treatment of rheumatoid arthritis (RA), but no clinical development has been reported as of late. AnorMED was also developing plerixafor for the treatment of HIV, but discontinued the trials in 2001 due to abnormal cardiac activity and lack of efficacy.
By blocking CXCR4, a specific cellular receptor, plerixafor triggers the rapid movement of stem cells out of the bone marrow and into circulating blood. Once in the circulating blood, the stem cells can be collected for use in stem cell transplant. In terms of use for cardiac applications, there is clinical evidence that the presence of stem cells circulating in the bloodstream or directly injected into the hearts of patients who have suffered a heart attack may result in improved cardiac function.
Chemical properties
Plerixafor is a macrocyclic compound and a bicyclam derivative.[4] It is a strong base; all eight nitrogen atoms accept protons readily. The two macrocyclic rings form chelate complexes with bivalent metal ions, especially zinc, copper and nickel, as well as cobalt and rhodium. The biologically active form of plerixafor is its zinc complex.[8]
Synthesis
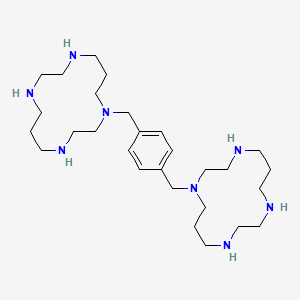
Three of the four nitrogen atoms of the macrocycle 1,4,8,11-tetraazacyclotetradecan are protected with tosyl groups. The product is treated with 1,4-dimethoxybenzene or 1,4-bis(brommethyl)benzene and potassium carbonate in acetonitrile. After cleaving of the tosyl groups with hydrobromic acid, plerixafor octahydrobromide is obtained.[9]
SEE CHINESE JOURNAL OF MEDICINAL CHEMISTRY 2010 20 (6): 511-513 ISSN: 1005-0108 CN: 21-1313/R
DOWNLOAD………http://download.bioon.com.cn/upload/201207/24113552_9395.pdf
http://www.zgyhzz.cn/qikan/epaper/zhaiyao.asp?bsid=14753
( 1 ) BASE FORM
0155g ( 8016% ), m p 129 ~ 131 e 。
1H-NM R
( CDC l3 ) D: 7.28( s, 4H, A r-H ), 3.55 ( br s, 4H,A r-CH2 ), 2.82 ~ 2.52( m, 32H, NCH2, NHCH2 ),
1.86 ~ 1.68 ( m, 8H, CCH2C )。 ESI-M S m /z:
503.55 [M + H]+ 。
………………………………………..
SEE
http://doc.sciencenet.cn/upload/file/2011531154034454.pdf
…………………………………..

………………………….
http://www.google.com/patents/US5756728
U.S. Pat. No. 5,021,409 is directed to a method of treating retroviral infections comprising administering to a mammal in need of such treatment a therapeutically effective amount of a bicyclic macrocyclic polyamine compound. Although the usefulness of certain alkylene and arylene bridged cyclam dimers is generically embraced by the teachings of the reference, no arylene bridged cyclam dimers are specifically disclosed.
WO 93/12096 discloses the usefulness of certain linked cyclic polyamines in combating HIV and pharmaceutical compositions useful therefor. Among the specifically disclosed compounds is 1,1′- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11 tetraazacyclotetradecane (and its acid addition salts), which compound is a highly potent inhibitor of several strains of human immune deficiency virus type 1 (HIV-1) and type 2 (HIV-2).
European Patent Appln. 374,929 discloses a process for preparing mono-N-alkylated polyazamacrocycles comprising reacting the unprotected macrocycle with an electrophile in a non-polar, relatively aprotic solvent in the absence of base. Although it is indicated that the monosubstituted macrocycle is formed preferentially, there is no specific disclosure which indicates that linked bicyclams can be synthesized by this process.
U.S. Pat. No. 5,047,527 is directed to a process for preparing a monofunctionalized (e.g., monoalkylated)cyclic tetramine comprising: 1) reacting the unprotected macrocycle with chrominum hexacarbonyl to obtain a triprotected tetraazacyloalkane compound; 2) reacting the free amine group of the triprotected compound prepared in 1) with an organic (e.g., alkyl) halide to obtain a triprotected monofunctionalized (e.g., monoalkylated) tetraazacycloalkane compound; and 3) de-protecting the compound prepared in 2) by simple air oxidation at acid pH to obtain the desired compound. In addition, the reference discloses alternative methods of triprotection employing boron and phosphorous derivatives and the preparation of linked compounds, including the cyclam dimer 1,1′- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane, by reacting triprotected cyclam prepared as set forth in 1) above with an organic dihalide in a molar ratio of 2:1, and deprotecting the resultant compound to obtain the desired cyclam dimer.
J. Med. Chem., Vol. 38, No. 2, pgs. 366-378 (1995) is directed to the synthesis and anti-HIV activity of a series of novel phenylenebis(methylene)-linked bis-tetraazamacrocyclic analogs, including the known cyclam dimer 1,1′- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane. The cyclam dimers disclosed in this reference, including the afore-mentioned cyclam dimer, are prepared by: 1) forming the tritosylate of the tetraazamacrocycle; 2) reacting the protected tetraazamacrocycle with an organic dihalide, e.g., dibromo-p-xylene, in acetonitrile in the presence of a base such as potassium carbonate; and 3) de-protecting the bis-tetraazamacrocycle prepared in 2) employing freshly prepared sodium amalgam, concentrated sulfuric acid or an acetic acid/hydrobromic acid mixture to obtain the desired cyclam dimer, or an acid addition salt thereof.
Although the processes disclosed in U.S. Pat. No. 5,047,527 and the J. Med. Chem. reference are suitable to prepare the cyclam dimer 1,1′- 1,4-phenylene bis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane, they involve the use of cyclam as a starting material, a compound which is expensive and not readily available. Accordingly, in view of its potent anti-HIV activity, a number of research endeavors have been undertaken in an attempt to develop a more practical process for preparing 1,1′- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane.
EXAMPLE 1
a) Preparation of the 1,4-phenylenebis-methylene bridged hexatosyl acylic precursor of formula III
To a 4-necked, round-bottom flask, equipped with a mechanical stirrer, heating mantle, internal thermometer and addition funnel, is added 43.5 g (0.25 mol) of N,N’-bis(3-aminopropyl) ethylenediamine and 250 ml of tetrahydrofuran. To the resultant solution is added, over a period of 30 minutes with external cooling to maintain the temperature at 20° C., 113.6 g (0.8 mol) of ethyl trifluoroacetate. The reaction mixture is then stirred at room temperature for 4 hours, after which time 52.25 ml. (0.3 mol) of diisopropylethylamine is added. The resultant reaction mixture is warmed to 60° C. and, over a period of 2 hours, is added a solution of 33.0 g (0.125 mol) of α,α’-dibromoxylene in 500 ml. of tetrahydrofuran. The reaction mixture is then maintained at a temperature of 60° C., with stirring, for an additional 2 hours after which time a solution of 62.0 g. (1.55 mol) of sodium hydroxide in 250 ml. of water is added. The resultant mixture is then stirred vigorously for 2 hours, while the temperature is maintained at 60° C. A solution of 152.5 g. (0.8 mol) of p-toluenesulfonyl-chloride in 250 ml. of tetrahydrofuran is then added, over a period of 30 minutes, while the temperature is maintained at between 20° C. and 30° C. The reaction is then allowed to proceed for another hour at room temperature. To the reaction mixture is then added 1 liter of isopropyl acetate, the layers are separated and the organic layer is concentrated to dryness under vacuum to yield the desired compound as a foamy material.
b) Preparation of the hexatosyl cyclam dimer of formula IV
To a 4-necked, round-bottom flask, equipped with a mechanical stirrer, heating mantle, internal thermometer and addition funnel, is added 114.6 g. (0.10 mol) of the compound prepared in a) above and 2.5 liters of dimethylformamide. After the system is degassed, 22.4 g. (0.56 mol) of NaOH beads, 27.6 g (0.2 mol) of anhydrous potassium carbonate and 5.43 g. (0.016 mol) of t-butylammonium sulfate are added to the solution, and the resultant mixture is heated to 100° C. and maintained at this temperature for 2.5 hours. A solution of 111.0 g (0.3 mol) of ethyleneglycol ditosylate in 1 liter of dimethylformamide is then added, over a period of 2 hours, while the temperature is maintained at 100° C. After cooling the reaction mixture to room temperature, it is poured into 4 liters of water with stirring. The suspension is then filtered and the filter cake is washed with 1 liter of water. The filter cake is then thoroughly mixed with 1 liter of water and 2 liters of ethyl acetate. The solvent is then removed from the ethyl acetate solution and the residue is re-dissolved in 500 ml. of warm acetonitrile. The precipitate that forms on standing is collected by filtration and then dried to yield the desired compound as a white solid.
c) Preparation of 1,1′- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane
In a 4-necked, round-bottom flask, equipped with a mechanical stirrer, heating mantle, internal thermometer and addition funnel, is added 26.7 g.(0.02 mol) of the compound prepared in b) above, 300 ml. of 48% hydrobromic acid and 1 liter of glacial acetic acid. The resultant mixture is then heated to reflux and maintained at reflux temperature, with stirring, for 42 hours. The reaction mixture is then cooled to between 22° C. and 23° C. over a period of 4 hours, after which time it is stirred for an additional 12 hours. The solids are then collected using suction filtration and added to 400 ml. of deionized water. The resultant solution is then stirred for 25 to 30 minutes at a temperature between 22° C. and 23° C. and filtered using suction filtration. After washing the filter pad with a small amount of deionized water, the solution is cooled to between 10° C. and 15° C. 250 g. of a 50% aqueous solution of sodium hydroxide is then added, over a period of 30 minutes, while the temperature is maintained at between 5° C. and 15° C. The resultant suspension is stirred for 10 to 15 minutes, while the temperature is maintained at between 10° C. and 15° C. The suspension is then warmed to between 22° C. and 23° C. and to the warmed suspension is added 1.5 liters of dichloromethane. The mixture is then stirred for 30 minutes, the layers are separated and the organic layer is slurried with 125 g. of sodium sulfate for 1 hour. The solution is then filtered using suction filtration, and the filtrate is concentrated under reduced pressure (40°-45° C. bath temperature, 70-75 mm Hg) until approximately 1.25 liters of solvent is collected. To the slurry is then added 1.25 liters of acetone, and the filtrate is concentrated under reduced pressure (40°-45° C. bath temperature, 70-75 mm Hg) until approximately 1.25 liters of solvent is collected. The slurry is then cooled to between 22° C. and 23° C. and the solids are collected using suction filtration. The solids are then washed with three 50 ml. portions of acetone and dried in a vacuum oven to obtain the desired compound as a white solid.
EXAMPLE 2
The following is an alternate procedure for the preparation of the 1,4-phenylenebis-methylene bridged hexatosyl acyclic precursor of formula III.
To a 3-necked, round-bottomed flask, equipped with a mechanical stirrer, heating mantle, internal thermometer and addition funnel, is added 3.48 g. (20 mmol) of N,N’-bis-(3-aminopropyl)ethylenediamine and 20 ml. of tetrahydrofuran. To the resultant solution is added, over a period of 20 minutes with external cooling to maintain the temperature at 20° C., 5.2 ml. (42 mmol) of ethyl trifluoroacetate. The reaction mixture is then stirred at room temperature for 1 hour, after which time a solution of 2.64 g. (10 mmol) of α,α’-dibromoxylene in 20 ml. of tetrahydrofuran is added. The resultant reaction mixture is then stirred at room temperature for 4 hours. A solution of 4.8 g. (120 mmol) of sodium hydroxide in 20 ml. of water is then added and the resultant mixture is warmed to 60° C. and maintained at this temperature, with vigorous stirring, for 2 hours. Over a period of 20 minutes, 13.9 g. (73 mmol) of p-toluenesulfonylchloride is then added portionwise, while the temperature is maintained at 20° C. The reaction is then allowed to proceed for another hour at room temperature. To the reaction mixture is then added 100 ml. of isopropyl acetate, the layers are separated and the organic layer is washed with saturated sodium bicarbonate aqueous solution. The solution is then condensed to 40 ml., cooled to 4° C. and kept at that temperature overnight. The resultant suspension is filtered and the solid is washed with 10 ml. of isopropyl acetate. The solvents are then removed from the filtrate to yield the desired compound as a brown gel.
…………………………
see
Synthesis and structure-activity relationships of phenylenebis(methylene)linked bis-tetraazamacrocycles that inhibit HIV replication. Effects of macrocyclic ring size and substituents on the aromatic linker
J Med Chem 1995, 38(2): 366
http://pubs.acs.org/doi/abs/10.1021/jm00002a019
…………………………………………………
see
New bicyclam-AZT conjugates: Design, synthesis, anti-HIV evaluation, and their interaction with CXCR-4 coreceptor
J Med Chem 1999, 42(2): 229
http://pubs.acs.org/doi/full/10.1021/jm980358u
……………………………………………………..
CN 102584732
http://www.google.com/patents/CN102584732B?cl=en
[0003]

[0004] plerixafor (trade name Mozobil ™) was developed by the U.S. company Genzyme chemokine receptor 4 (CXCR4) antagonist specificity. The drug is a hematopoietic stem (progenitor) cell activator, and can stimulate hematopoietic stem cell proliferation and differentiation into functional blood circulation.
[0005] As the non-Hodgkin’s lymphoma (NHL) and multiple myeloma (Korea) most of the cases and the progress of cases to alleviate the need for autologous peripheral blood stem cell transplantation, and plerixafor joint G-CSF can significantly improve the number of patients with ⑶ 34 + cells, about 60% of the patient’s peripheral blood can ⑶ 34 + cells increased to ensure that the NHL and MM patients with autologous hematopoietic stem cell transplantation success.
[0006] U.S. FDA approval on December 15, 2008 its listing, clinical studies showed that the drug can greatly increase the number of white blood cells of patients and to promote hematopoietic stem cells from bone marrow to the blood flow, and granulocyte colony-stimulating factor (G-CSF ) have a synergistic effect; has been used in multiple myeloma and Hodgkin’s lymphoma patients with stem cell transplantation in clinical trials.
[0007] About plerixafor or synthetic analogs have some at home and abroad reported in the literature, there are J.0rg.Chem.2003, 68,6435-6436; J.Med Chem.1995, 38 (2): 366-378; J.SynthCommun.1998 ,28:2903-2906; Tetrahedron, 1989,45 (1) :219-226; Chinese Journal of Pharmaceuticals 2007,38 (6); World Patent W09634860A1; W09312096A1; U.S. Patent US5047527, US5606053, US5801281, US5064956, Chinese patent CN1466579A.
[0008] J.Med Chem.1995, 38 (2) = 366-378 relates to a preparation method comprises the following steps: a) forming a salt of trimethoxy benzene tetraaza macrocycles; 2) reacting the protected tetrazole hetero macrocycle in acetonitrile under the presence of a base such as potassium carbonate as dibromo-p-xylene is reacted with an organic dihalide; 3) using freshly prepared sodium amalgam, concentrated sulfuric acid or acetic acid / hydrobromic acid mixture deprotected target product.
[0009] US 5047527 relates to preparation of the cyclic four monofunctional amine, the method comprising: a) reacting the unprotected macrocycle of reaction with chromium hexacarbonyl to obtain protection tetraazadecalin three compounds; 2) 3 Protection of the free amino compound with an organic halide to obtain three-protected monofunctional tetraaza naphthenic compounds; 3) simple air oxidation, deprotection to obtain the desired product. [0010] J.Synth Commun.1998 ,28:2903-2906 describes an improved method for synthesizing intermediates Plerixafor, the method using phosphor protection, deprotection to give a smooth 1,1 ‘- [1,4 – phenylene bis (methylene)] _ two _1, 4,8,11 – tetraazacyclododecane fourteen burn.
[0011] US 5606053 relates to a process for preparing dimers 1, I ‘- [1,4 – phenylene bis (methylene)] – two -1,4,8,11 – tetraazacyclododecane-tetradecane method. The preparation of compounds include: 1) the four-amine as the starting material, obtained by acylation of toluene Juan acyclic intermediates and three xylene sulfonate and toluene sulfonate and toluene intermediates; 2) and xylene sulfonate and intermediates trimethylbenzene toluenesulfonic acid intermediates after alkylation separation dibromo xylene, toluene sulfonate and then obtain a non-cyclic dimers of six toluenesulfonic acylated; 3) six isolated bridged acyclic toluenesulfonic acid dimer form is reacted with ethylene glycol ditosylate three equivalents of cyclization; 4) deprotection to obtain the objective product was purified by hydrobromic acid and acetic acid.
[0012] US 5801281 relates to preparation of dimer 1, I ‘- [1,4 _-phenylene bis (methylene)] – two _1, 4,8,11
[0013] – tetraazacyclo tetradecane, comprising: a) reacting the acyclic tetraamine with 3 equivalents of ethyl trifluoroacetate, the reaction; 2) with 0.5 equivalents of the tri-dibromo-p-xylene-protected acyclic alkylation of the amine obtained form four non-cyclic dimers; 3) hydrolysis to remove the six trifluoroacetyl compound group; 4) acylation of the compound toluenesulfonic bridged tetraamine dimer; 5) B Juan xylene glycol ester cyclization; 6) and glacial acetic acid mixed with hydrobromic acid deprotection was the target product.
Under the [0014] US 5064956 discloses a multi-alkylated single-ring nitrogen of the compound prepared, the method involves reacting the unprotected macrocycle in an aprotic, relatively non-polar solvent in presence of alkali electrophilic reagent. Not mentioned in this document similar to the embodiment Seclin dimer synthesis.
[0015] Through the open Plerixafor synthetic route research and meta-analysis of the literature, mainly in the following four synthetic routes:
[0016] Route One, is 1,4,8,11 – tetraazacyclododecane cyclotetradecane as raw material, NI, N4, N8 three protected with 1,4 – bis (halomethyl) benzene-bridged deprotection to obtain the finished product. The following reaction scheme, wherein R is p-toluenesulfonyl group, a methanesulfonyl group, a trifluoroacetyl group, a tert-butoxycarbonyl group and the like:
[0017]

[0018] Route II is di (2 – aminopropyl) ethylenediamine as raw material, the ring and the reaction with 1,4 – bis (halomethyl) benzene-bridged, and then deprotection Bullock Suffolk.
[0019] Route 3 to 1,4,8,11 – tetraazacyclododecane cyclotetradecane as raw material, under anhydrous, anaerobic conditions, after the ring protection with 1,4 – bis (halomethyl ) benzene bridging, and then deprotection plerixafor. Synthesis scheme below, wherein R is P, Ni, etc.;

[0021] line four, based on acrylate as starting material, first with ethylene diamine as raw material by Michael addition of the amine solution, then with malonate cyclization 1,4,8,11 – Tetraaza _5, 7,12 – three oxo cyclotetradecane by α, α ‘- dibromo-p-xylene bridging, the final deprotection plerixafor. Reaction Roadmap follows:
[0022]

[0023] The above synthesis route and the existing methods have the following disadvantages:
[0024] In an intermediate of the synthesis route, the existing technology, the need for column purification of the intermediates, low yield.
[0025] route to protect the stability of the two because of the strong, leading to the final deprotection step difficult, long production cycle, low yield, and finished organic residues can not be achieved within the standard limits.
Higher dry anaerobic demands [0026] Route 3 on, harsh reaction conditions, deprotection is not complete, intermediates need to repeatedly purified, low yield, after repeated recrystallization, finished monohetero difficult to control in 0.1% less.
[0027] Anhydrous ethylene diamine route and need four anhydrous THF, more stringent requirements on the process, and to use dangerous borane dimethyl sulfide, while the second step is only about 35% lower yield. Selectivity of the reaction is not high shortcomings, so do not be the most economical and reasonable synthetic route.
[0028] We prepared by Plerixafor prepared by methods disclosed above may Plerixafor single impurity of 0.1% or less is difficult to achieve, it is difficult to meet the quality requirements of the injection material, the same techniques can not reach the European Quality of ICH guidelines of the relevant technical requirements, low yield, high cost required for each step of the intermediate column to afford a large amount of solvent, time consuming, and the greater the elution solvent toxicity, is not suitable for industrial production.
(I) Preparation of 1,4,8 _ tris (p-toluenesulfonyl) -1,4,8,11 – tetraazacyclododecane-tetradecane: the raw 1,4,8,11 – tetraazacyclododecane cyclotetradecane suspended in methylene chloride, in the role of acid binding agent, at a temperature 10 ~ 30 ° C, p-toluenesulfonyl chloride and 3 ~ 8h, filtered, and the filtrate was collected and concentrated to dryness to obtain a residue; will have The residue of said C ^ C3 alkyl group in a mixed solvent of alcohol and an aprotic solvent, purification, crystallization segment greater than 95% purity of 1,4,8 – tris (p-toluenesulfonyl) _1, 4,8,11 – tetraaza cyclotetradecane;
[0032] (2) Preparation of 1,1 ‘- [1,4 – (phenylene methylene)] – two – [4,8,11 – tris (p-toluenesulfonyl)] -1,4, 8,11 – tetraazacyclododecane-tetradecane: A (I) the resulting 1,4,8 – tris (p-toluenesulfonyl) _1, 4,8,11 – tetraazacyclododecane-tetradecane, α, α two bromo-p-xylene in place of anhydrous acetonitrile, was added acid-binding agent, the reaction was refluxed under nitrogen for 5 to 24 hours; After the reaction was cooled to room temperature, the reaction mixture was then collected by filtration and the filter cake was purified to obtain a mixed solvent I , I, – [1,4 – (phenylene methylene)] – two – [4,8,11 – tris (p-toluenesulfonyl)] _1, 4,8,11 – tetraazacyclododecane ten four alkyl;
[0033] (3) Synthesis Plerixafor: A (2) the resultant I, 1′-[1,4 _ (phenylene methylene)] – two – [4,8,11 – tris (p-toluene sulfonyl)] -1,4,8,11 – tetraazacyclododecane myristic acid solution was added to the mixture, stirred and dissolved, the reaction was warmed to reflux for 10 to 24 hours, cooled, filtered, and filter cake was collected; the filter cake was dissolved in purified water, adjusted with sodium hydroxide solution or potassium hydroxide solution to the PH-12, filtered, and the filtrate was extracted with a halogenated solvent, and the organic layer was dried over anhydrous sodium sulfate and then filtered, the filtrate was concentrated under reduced pressure P Le Suffolk crude;
[0034] (4) Purification Plerixafor: Plerixafor the crude was dissolved into a solvent and heated to reflux to dissolve, filtered, and the crystallization solvent is added dropwise at 40 ~ 45 ° C crystallization 30min, filtered and the filtrate then cooled to 20 ~ 25 ° C crystallization I hour at O ~ 5 ° C crystallization three hours, filtered, and the filter cake was dried Plerixafor.
Plerixafor Preparation: 6 [0075] Implementation
[0076] The starting material 1,4,8,11 – tetraazacyclo tetradecane (5g, 25mmol) was suspended in dichloromethane (50g) was added N, N-diisopropylethylamine (7.5ml) , a solution of p-toluenesulfonyl chloride (10.8g, 56.5mmol) and methylene chloride (50g) in a solution of, at 25 ~ 30 ° C reaction temperature 3h, filtered, and the filtrate was collected and concentrated to dryness and to the residue in methanol (30g), toluene (IOg) was heated to reflux, filtered, and the filtrate was cooled to 40 ° C crystallization 30min, filtered to remove impurities little over protection, and the filtrate was added methyl tert-butyl ether (30g), stirring rapidly cooled to O ~ 5 ° C crystallization 3h, filtered, and dried to give 1,4,8 – tris (p-toluenesulfonyl) -1, 4,8,11 – tetraazacyclododecane-tetradecane (9.6g, 61.9%), purity of 97.2%.
[0077] The 4,8 _ tris (p-toluenesulfonyl) _1, 4,8,11 – tetraazacyclododecane-tetradecane (9g, 13.6mmol) α, α ‘- dibromo-p-xylene (1.81 g, 6.8mmol) in dry acetonitrile was placed (90ml) was added potassium carbonate (15.0g, 108.5mmol), the reaction was refluxed under nitrogen for 5 hours. Cooled to room temperature and filtered to collect the filter cake, was added anhydrous methanol (10ml), ethyl acetate (30ml), dichloromethane (IOml) hot melt, whereby the cooling crystallization, filtration, and dried under reduced pressure to obtain white solid (16. lg, 83%), purity 97.5%.
[0078] The intermediate obtained above (5g, 3.5mmol) was added to glacial acetic acid (25ml) and concentrated hydrochloric acid (25ml) was stirred until dissolved in the mixed solution was heated to reflux for 24 hours, cooled, collected by filtration cake. The filter cake was dissolved in purified water (20ml), adjusting the PH value of the solution with sodium hydroxide to 12, filtered, and the filtrate was extracted with dichloromethane (50mlX3), the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain sand Bullock Fu crude (1.4g, 79.5%), purity 98.6%.
[0079] The crude Plerixafor (1.4g) is placed in tetrahydrofuran (14g), heated to reflux to dissolve, filtered, and added dropwise n-hexane (42g), and 40 ~ 45 ° C crystallization 30min, filtered little solid, The filtrate was rapidly cooled to 20 ~ 25 ° C crystallization I hour and then at O ~ 5 ° C crystallization three hours, filtered, 45 ° C and dried under reduced pressure to obtain the finished Plerixafor (1.2g, 85.7%), purity 99.93 %, the largest single miscellaneous 0.04%.
………………………………….
http://www.google.com/patents/US8420626?cl=en

wherein, n is 0 or 1, Ts is tosyl radical, P is trifluoroacetyl or p-tosyl radical;
To the NaOH solution of the starting material 7 is dropwise added ether solution of tosyl chloride. The system is stirred over night. A white solid is formed and filtrated. The filter cake is washed with water and ethyl ether, respectively, recrystallized to give a white solid intermediate of formula 8. To the dried acetonitrile solution of the compound of formula 8 is slowly dropwise added dried acetonitrile solution of 1,2-di-p-tosyloxypropane under reflux state, refluxed for 2-4 days, stood until room temperature. A white solid is precipitated and filtrated. The filter cake is washed with water and ethyl acetate, respectively, recrystallized to give a white solid compound of formula 9. The compound of formula 9 is dissolved in 90% concentrated sulfuric acid, allowed to react at 100° C. for 24-48 hours, stood until room temperature. To the reaction solution are dropwise added successively ethanol and ethyl ether. A white solid is precipitated, filtrated, dried, and dissolved in NaOH solution. The aqueous phase is extracted with chloroform. The chloroform phase is combined, concentrated, recrystallized to give a white solid compound of formula 10. To the chloroform solution of the compound of formula 10 and triethylamine is dropwise added chloroform solution of tosyl chloride. The mixture is allowed to react at room temperature over night, concentrated and column separated (eluant: dichloromethane/methanol system) to give a white solid compound of formula 11 (protective group is tosyl); or to the methanol solution of the compound of formula 10 is dropwise added ethyl trifluoroacetate. The mixture is allowed to react at room temperature over night, concentrated and column separated (eluant: ethyl acetate) to give a white solid compound of formula 11 (protective group is trifluoroacetyl);
Pharmacokinetics
Following subcutaneous injection, plerixafor is absorbed quickly and peak concentrations are reached after 30 to 60 minutes. Up to 58% are bound to plasma proteins, the rest mostly resides in extravascular compartments. The drug is not metabolized in significant amounts; no interaction with the cytochrome P450 enzymes or P-glycoproteins has been found. Plasma half life is 3 to 5 hours. Plerixafor is excreted via the kidneys, with 70% of the drug being excreted within 24 hours.[5]
Pharmacodynamics
In the form of its zinc complex, plerixafor acts as an antagonist (or perhaps more accurately a partial agonist) of the alpha chemokine receptor CXCR4 and an allosteric agonist ofCXCR7.[10] The CXCR4 alpha-chemokine receptor and one of its ligands, SDF-1, are important in hematopoietic stem cell homing to the bone marrow and in hematopoietic stem cell quiescence. The in vivo effect of plerixafor with regard to ubiquitin, the alternative endogenous ligand of CXCR4, is unknown. Plerixafor has been found to be a strong inducer of mobilization of hematopoietic stem cells from the bone marrow to the bloodstream as peripheral blood stem cells.[11]
Interactions
No interaction studies have been conducted. The fact that plerixafor does not interact with the cytochrome system indicates a low potential for interactions with other drugs.[5]
Legal status
Plerixafor has orphan drug status in the United States and European Union for the mobilization of hematopoietic stem cells. It was approved by the U.S. Food and Drug Administration for this indication on December 15, 2008.[12] In Europe, the drug was approved after a positive Committee for Medicinal Products for Human Use assessment report on 29 May 2009.[7] The drug was approved for use in Canada by Health Canada on December 8, 2011.[13]
Research
Small molecule cancer therapy
Plerixafor was seen to reduce metastasis in mice in several studies.[14] It has also been shown to reduce recurrence of glioblastoma in a mouse model after radiotherapy. In this model, the cancer surviving radiation are critically depended on bone marrow derived cells for vasculogenesis whose recruitment mediated by SDF-1 CXCR4 interaction is blocked by plerixafor.[15]
Use in generation of other stem cells
Researchers at Imperial College have demonstrated that plerixafor in combination with vascular endothelial growth factor (VEGF) can produce mesenchymal stem cells andendothelial progenitor cells in mice.[16]
Other uses
Blockade of CXCR4 signalling by plerixafor (AMD3100) has also unexpectedly been found to be effective at counteracting opioid-induced hyperalgesia produced by chronic treatment with morphine, though only animal studies have been conducted as yet.[17]
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1,1′-[1,4-Phenylenebis(methylene)]bis [1,4,8,11-tetraazacyclotetradecane] | |
| Clinical data | |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a609018 |
| Pregnancy cat. | D (US) |
| Legal status | ℞-only (US) |
| Routes | Subcutaneous injection |
| Pharmacokinetic data | |
| Protein binding | Up to 58% |
| Metabolism | None |
| Half-life | 3–5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS number | 110078-46-1 |
| ATC code | L03AX16 |
| PubChem | CID 65015 |
| IUPHAR ligand | 844 |
| DrugBank | DB06809 |
| ChemSpider | 58531 |
| UNII | S915P5499N |
| Synonyms | JM 3100, AMD3100 |
| Chemical data | |
| Formula | C28H54N8 |
| Mol. mass | 502.782 g/mol |
http://www.google.com/patents/CN102653536A?cl=en
(Plerixafor), chemical name: 1, I ‘- [I, 4_ phenylene ni (methylene)] – ni -1,4,
8,11 – tetraazacyclo tetradecane, its molecular structure is as follows:
[0004]

Synthesis of domestic and foreign literature in general, all require 1,4,8,11 – tetraazacyclo-tetradecane for 3 protection (eg of formula I), of the three methods are used to protect the p-toluenesulfonamide chloride, trifluoroacetic acid ko ko cool, tert-butyl carbonate ni. Use of p-toluenesulfonamide-protected deprotection step into strict step because deprotecting reagent (such as hydrobromic acid / glacial acetic acid, concentrated sulfuric acid, etc.) side reactions often occur.The use of trifluoroacetic acid ko ko ester protecting, since the trifluoromethyl group strongly polar ko, resulting fourth-NH unprotected decrease in activity, usually not fully reflect the subsequent reaction, thereby further into ー is introduced after deprotection difficult to remove impurities 1,4,8,11 – tetraazacyclo-tetradecane.
[0006] tert-butyl carbonate ni selective protection of the amino group is widely used (polyamines, amino acids, p printed tidic chains, etc.), but to use it for 1,4,8,11 – tetraazacyclo tetradecane rarely reported, abroad it for 1,4,8,11 – tetraazacyclo tetradecane protection coverage, we use the t-butyl carbonate brother attempted 3 protection, he was surprised to find that in certain conditions, the three protection up to 90% (see Figure I), with high selectivity, significantly higher than the reported domestic Boc protected
Selectivity of the reaction (see table below).
[0007]

[0008] 2 by three protection product with quite different polarity protection products, flash column chromatography using silica gel column to separate the protector 3 of sufficient purity, and deprotection conditions milder (only hydrochloric acid solution), in a certain extent reduce the incidence of side effects, so capable of synthesizing high purity products.
[0009]

SUMMARY OF THE INVENTION


xample I: 3Boc protection 1,4,8,11 _ tetraazacyclo Preparation tetradecane
[0048] 1,4,8,11 taken tetraazacyclo tetradecane _ 10g (0.05mol), and acetone – water (2: l) 50ml, tris ko amine 10. 119g (0. Lmol), ni ko isopropyl amine 3. 225g (0. 025mol), at room temperature was added dropwise tert-butyl carbonate, brother 38. 194g (0. 175mol), dropwise at room temperature after stirring for 24 hours, HPLC monitoring of the reaction. After completion of the reaction 50 ° C under reduced pressure to dryness to give a pale yellow oil, 150g on a silica gel column, and eluted with ko acid esters ko collecting ko ko acid ester liquid evaporated to dryness under reduced pressure to give a white foam 23. 12g, yield of 92.36%. 1HNMR (400MHz, CDCl3, 6 ppm): 1. 74 (2H, q, 5. 5);
I. 96 (2H, q, 6. 5); 2. 66 (2H, t, 5. 5); 2. 82 (2H, t, 5. 5); 3. 33 (4H, m); 3. 34 (2H, m); 3. 37 (2H, m), 3. 43 (4H, m).
[0049] Implementation Example 2: 6Boc protection Bullock Suffolk Preparation
[0050] Take 3Boc protection 1,4,8,11 _ tetraazacyclo tetradecane 20. 03g (0. 04mol), dissolved in anhydrous ko nitrile 400ml, anhydrous potassium carbonate 20g, aa ‘ni chlorine ni toluene 3.5012g (0.02mol), sodium iodide 75mg, at reflux for 24 hours under nitrogen, TLC monitoring of the reaction. After completion of the reaction, cooled to room temperature, filtered, the filter cake was washed with 200ml of ko nitrile, nitrile ko combined solution was evaporated to dryness under reduced pressure to give the protected Bullock 6Boc Suffolk 21. 20g, yield of 96.06%. Alcohol with ko – a mixed solvent of water and recrystallized to give a white solid. [0051] Implementation Example 3: Bullock Suffolk • 8HC1 • 3H20 Preparation of compounds
[0052] Protection Bullock Suffolk take 6Boc 20g, add methanol 200ml, stirring to dissolve, concentrated hydrochloric acid was added dropwise at room temperature, 60ml, was stirred at room temperature after the addition was complete 48 inches, TLC monitoring of the reaction. After completion of the reaction, filtration, the filter cake was dried 50 ° C under reduced pressure to give a white solid 13. 54g, yield of 88.04%.

[0053] Implementation Example 4: Preparation of Suffolk Bullock…………Plerixafor BASE
[0054] Take Bullock Suffolk • 8HC1 • 3H20 compound 13. 54g, add water 40ml ultrasound to dissolve after stirring constantly with 50% sodium hydroxide solution to adjust the pH to 12 and filtered, the filter cake 50 ° C minus pressure and dried to give a white solid 7. 24g, yield 90.24 V0o
1H NMR (400MHz, CDCl3, 6 ppm): 1. 75 (4H, bs); 1. 87 (4H, bs); 2. 95-2. 51 (32H, m); 3. 54 (4H, s); 4. 23 (4H, bs); 7. 30 (4H, s).
IR (KBr) 3280,2927,2883,2805,1458,1264,1117 cm,
NEW PATENT…………….WO-2014125499
Improved and commercially viable process for the preparation of high pure plerixafor base
Process for the preparation of more than 99.8% pure plerixafor base by HPLC. Also claims solid forms of plerixafor base and composition comprising the same. Appears to be the first filing from the assignee on this API. FDA Orange book lists US6987102 and US7897590, expire in July 2023.
|
3-5-1997
|
Process for preparing 1,4,8,11-tetraazacyclotetradecane
|
|
|
2-26-1997
|
Process for preparing 1,1′-[1,4-phenylenebis-(methylene)]-bis-1,4,8,11-tetraazacyclotetradecane
|
|
|
12-11-1996
|
Aromatic-linked polyamine macrocyclic compounds with anti-HIV activity
|
|
|
11-8-1996
|
PROCESS FOR PREPARING 1,1′-[1,4-PHENYLENEBIS-(METHYLENE)]-BIS-1,4,8,11-TETRAAZACYCLOTETRADECANE
|
|
|
10-4-1996
|
PROCESS FOR PREPARING 1,1′-[1,4-PHENYLENEBIS-(METHYLENE)]-BIS-1,4,8,11-TETRAAZACYCLOTETRADECANE
|
|
|
7-14-1995
|
CYCLIC POLYAMINES
|
|
|
6-25-1993
|
LINKED CYCLIC POLYAMINES WITH ACTIVITY AGAINST HIV
|
|
9-2-2005
|
Substituted benzodiazepines as inhibitors of the chemokine receptor CXCR4
|
|
|
2-4-2005
|
Methods and compositions for the treatment or prevention of human immunodeficiency virus and related conditions using cyclooxygenase-2 selective inhibitors and antiviral agents
|
|
|
12-4-2002
|
Process for preparation of N-1 protected N ring nitrogen containing cyclic polyamines and products thereof
|
|
|
10-2-2002
|
Prodrugs
|
|
|
10-25-2001
|
PROCESS FOR PREPARING 1,1′- 1,4-PHENYLENEBIS-(METHYLENE)]-BIS-1,4,8,11-TETRAAZACYCLOTETRADECANE
|
|
|
9-29-2000
|
CHEMOKINE RECPETOR BINDING HETEROCYCLIC COMPOUNDS
|
|
|
8-11-2000
|
METHODS AND COMPOSITIONS TO ENHANCE WHITE BLOOD CELL COUNT
|
|
|
1-15-1998
|
PROCESS FOR PREPARING 1,1′- 1,4-PHENYLENEBIS-(METHYLENE) -BIS-1,4,8,11-TETRAAZACYCLOTETRADECANE
|
|
|
3-19-1997
|
Process for preparing 1,1′-[1,4-phenylenebis-(methylene)]-bis-1,4,8,11-tetraazacyclotetradecane
|
|
|
3-7-1997
|
PROCESS FOR PREPARING 1,4,8,11-TETRAAZACYCLOTETRADECANE PROCESS FOR PREPARING 1,4,8,11-TETRAAZACYCLOTETRADECANE
|
|
6-24-2011
|
BETULINIC ACID DERIVATIVES AS ANTI-HIV AGENTS
|
|
|
11-3-2010
|
Antiviral methods employing double esters of 2′, 3′-dideoxy-3′-fluoroguanosine
|
|
|
2-5-2010
|
Chemokine Receptor Modulators
|
|
|
1-29-2010
|
NOVEL POLYNITROGENATED SYSTEMS AS ANTI-HIV AGENTS
|
|
|
9-4-2009
|
Combination of CXCR4 Antagonist and Morphogen to Increase Angiogenesis
|
|
|
11-28-2008
|
Chemokine receptor modulators
|
|
|
10-24-2008
|
Chemokine receptor modulators
|
|
|
8-32-2006
|
Compositions and methods for treating tissue ischemia
|
|
|
7-5-2006
|
ANTIVIRAL METHODS EMPLOYING DOUBLE ESTERS OF 2′, 3′-DIDEOXY-3′-FLUOROGUANOSINE
|
|
|
12-14-2005
|
Treatment of viral infections using prodrugs of 2′,3-dideoxy,3′-fluoroguanosine
|
References
- Jump up^ Ciampolini, M.; Fabbrizzi, L.; Perotti, A.; Poggi, A.; Seghi, B.; Zanobini, F. (1987). “Dinickel and dicopper complexes with N,N-linked bis(cyclam) ligands. An ideal system for the investigation of electrostatic effects on the redox behavior of pairs of metal ions”.Inorganic Chemistry 26 (21): 3527. doi:10.1021/ic00268a022. edit
- Jump up^ Davies, S. L.; Serradell, N.; Bolós, J.; Bayés, M. (2007). “Plerixafor Hydrochloride”.Drugs of the Future 32 (2): 123. doi:10.1358/dof.2007.032.02.1071897. edit
- Jump up^ Davies, S. L.; Serradell, N.; Bolós, J.; Bayés, M. (2007). “Plerixafor Hydrochloride”.Drugs of the Future 32 (2): 123. doi:10.1358/dof.2007.032.02.1071897. edit
- ^ Jump up to:a b &Na; (2007). “Plerixafor”. Drugs in R & D 8 (2): 113–119. doi:10.2165/00126839-200708020-00006. PMID 17324009. edit
- ^ Jump up to:a b c d e Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. ISBN 3-85200-196-X.
- Jump up^ Wagstaff, A. J. (2009). “Plerixafor”. Drugs 69 (3): 319. doi:10.2165/00003495-200969030-00007. PMID 19275275. edit
- ^ Jump up to:a b “CHMP Assessment Report for Mozobil”. European Medicines Agency.
- Jump up^ Esté, J. A.; Cabrera, C.; De Clercq, E.; Struyf, S.; Van Damme, J.; Bridger, G.; Skerlj, R. T.; Abrams, M. J.; Henson, G.; Gutierrez, A.; Clotet, B.; Schols, D. (1999). “Activity of different bicyclam derivatives against human immunodeficiency virus depends on their interaction with the CXCR4 chemokine receptor”. Molecular Pharmacology 55 (1): 67–73.PMID 9882699. edit
- Jump up^ Bridger, G.; et al. (1993). “Linked cyclic polyamines with activity against HIV. WO/1993/012096”.
- Jump up^ Kalatskaya, I.; Berchiche, Y. A.; Gravel, S.; Limberg, B. J.; Rosenbaum, J. S.; Heveker, N. (2009). “AMD3100 is a CXCR7 Ligand with Allosteric Agonist Properties”.Molecular Pharmacology 75: 1240. doi:10.1124/mol.108.053389.PMID 19255243. edit
- Jump up^ Cashen, A. F.; Nervi, B.; Dipersio, J. (2007). “AMD3100: CXCR4 antagonist and rapid stem cell-mobilizing agent”. Future Oncology 3 (1): 19–27.doi:10.2217/14796694.3.1.19. PMID 17280498. edit
- Jump up^ “Mozobil approved for non-Hodgkin’s lymphoma and multiple myeloma” (Press release). Monthly Prescribing Reference. December 18, 2008. Retrieved January 3, 2009.
- Jump up^ Notice of Decision for MOZOBIL
- Jump up^ Smith, M. C. P.; Luker, K. E.; Garbow, J. R.; Prior, J. L.; Jackson, E.; Piwnica-Worms, D.; Luker, G. D. (2004). “CXCR4 Regulates Growth of Both Primary and Metastatic Breast Cancer”. Cancer Research 64 (23): 8604–8612. doi:10.1158/0008-5472.CAN-04-1844. PMID 15574767. edit
- Jump up^ Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R. M.; Harsh, G. R.; Brown, J. M. (2010).“Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice”. Journal of Clinical Investigation 120 (3): 694–705. doi:10.1172/JCI40283. PMC 2827954. PMID 20179352. edit
- Jump up^ Pitchford, S.; Furze, R.; Jones, C.; Wengner, A.; Rankin, S. (2009). “Differential Mobilization of Subsets of Progenitor Cells from the Bone Marrow”. Cell Stem Cell 4 (1): 62–72. doi:10.1016/j.stem.2008.10.017. PMID 19128793. edit
- Jump up^ Wilson NM, Jung H, Ripsch MS, Miller RJ, White FA (March 2011). “CXCR4 Signaling Mediates Morphine-induced Tactile Hyperalgesia”. Brain, Behavior, and Immunity 25(3): 565–73. doi:10.1016/j.bbi.2010.12.014. PMC 3039030. PMID 21193025.
- http://worlddrugtracker.blogspot.in/2013/11/plerixafor-new-treatment-approaches-for.html
External links

http://pubs.rsc.org/en/content/articlehtml/2012/dt/c2dt31137b


http://www.thno.org/v03p0047.htm
SEE ALSO……….http://www.scipharm.at/download.asp?id=1427
SEE…………..https://www.academia.edu/5549712/2011531154034454SCHEME 15 IS SYNTHESIS OF PLEXIXAFOR
read
ncur_powerpoint Courtney.ppt
faculty.swosu.edu/tim.hubin/share/ncur_powerpoint%20Courtney.ppt
… trials against cancer and for stem cell mobilization as “Mozobil” or “Plerixafor” …NMR studies of AMD-3100 suggest that complex configuration is important.
Eliquis, Apixaban for the Treatment of Deep Vein Thrombosis and Pulmonary Embolism
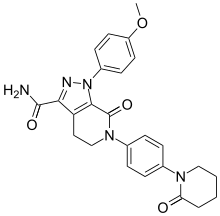
Apixaban
CAS 503612-47-3
APPROVALS
EMA————MAY 18, 2011
FDA…………………DEC28, 2012
PMDA………….. DEC25, 2012
CFDA………………JAN 22, 2013

Apixaban, sold under the tradename Eliquis, is a anticoagulant for the treatment of venous thromboembolic events. It is taken by mouth. It is a direct factor Xa inhibitor.
Apixaban was approved in Europe in 2012.[1] It was approved in the U.S. in 2014 for treatment and secondary prophylaxis of deep vein thrombosis (DVT) and pulmonary embolism (PE).[2] It is being developed in a joint venture by Pfizer and Bristol-Myers Squibb.[3][4]


INVENTIVE
Donald Pinto (left) and Michael Orwat (right) work on developing new products for BMS.
Credit: Bristol-Myers Squibb
Ruth R. Wexler, executive director of cardiovascular diseases chemistry at Bristol-Myers Squibb, who led the group that designed and synthesized Eliquis (apixaban) to reduce the risk of stroke in patients with an abnormal heart rhythm called atrial fibrillation, recalls hearing about the drug’s success in late-stage clinical trials for the first time.
“I was at the European Society of Cardiology meeting when the results of ARISTOTLE, our large Phase 3 trial, were announced,” she says. “I was sitting in the audience, and it was just amazing to see the data released for the first time. It blew my mind that the data was that spectacular.”
In the trial, which compared apixaban with the workhorse anticoagulant Coumadin (warfarin), apixaban reduced the risk of stroke in patients with atrial fibrillation by 21%, major bleeding by 31%, and mortality by 11%. Unlike Coumadin, apixaban doesn’t require regular monitoring of the blood.
Medical uses
Apixaban is indicated for the following:[5]
- To lower the risk of stroke and embolism in patients with nonvalvular atrial fibrillation.
- Deep vein thrombosis (DVT) prophylaxis. DVT’s may lead to pulmonary embolism (PE) in knee or hip replacement surgery patients.
- Treatment of both DVT and PE.
- To reduce the risk of recurring DVT and PE after initial therapy.
Atrial fibrillation
Apixaban is recommended by the National Institute for Health and Clinical Excellence for the prevention of stroke and systemic embolism in people with non-valvular atrial fibrillation and at least one of the following risk factors: prior stroke or transient ischemic attack, age 75 years or older, diabetes mellitus, or symptomatic heart failure.[6]
Apixaban and other newer anticoagulants (dabigatran and rivaroxaban) appear equally effective as warfarin in preventing non-hemorrhagic stroke in people with atrial fibrillation and are associated with lower risk of intracranial bleeding.[7]
Mechanism of action
Apixaban is a highly selective, orally bioavailable, and reversible direct inhibitor of free and clot-bound factor Xa. Factor Xa catalyzes the conversion of prothrombin to thrombin, the final enzyme in the coagulation cascade that is responsible for fibrin clot formation.[10] Apixaban has no direct effect on platelet aggregation, but by inhibiting factor Xa, it indirectly decreases clot formation induced by thrombin.[5]
FDA approval
A new drug application (NDA) for the approval of apixaban was submitted to the FDA by Bristol-Myers Squibb and Pfizer jointly after conclusion of the ARISTOTLE clinical trial in 2011.[11]
Apixaban was approved for the prevention of stroke in people with atrial fibrillation on December 28, 2012.[12] On March 14, 2014, it was approved for the additional use of preventing deep vein thrombosis and pulmonary embolism in people that had recently undergone knee or hip replacement.[13] On August 21, 2014, the FDA approved apixaban for the treatment of recurring deep vein thrombosis and pulmonary embolism.[2]
During development it was known as BMS-562247-01.




Thursday, August 21, 2014 – Bristol-Myers Squibb Company (NYSE: BMY) and Pfizer Inc. (NYSE: PFE) today announced the U.S. Food and Drug Administration (FDA) has approved a Supplemental New Drug Application (sNDA) for Eliquis for the treatment of DVT and PE, and for the reduction in the risk of recurrent DVT and PE following initial therapy. Combined, DVT and PE are known as VTE. It is estimated that every year, approximately 900,000 Americans are affected by DVT and PE.
See more at: http://worlddrugtracker.blogspot.in/2014/08/fda-approves-eliquis-apixaban-for.html


![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide](https://i0.wp.com/pic8.molbase.net/molpic/ae/b8/543077.png) APIXABAN
APIXABAN
PREDICTIONS
1H NMR

13C NMR

COSY
1H NMR PREDICT


13 C NMR PREDICT


|
|
![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-08/000/543/077/503612-47-3-1h.png)
![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-08/000/543/077/503612-47-3-13c.png)
C-NMR spectral analysis
|
CAS NO. 503612-47-3, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectral analysis
l-(4-Methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l -yl)phenyl]-4, 5,6,7- tetrahydro- lH-pyrazolo[3,4-c]pyridine-3-carboxyamide of formula (I), also known come apixaban, is a powerful inhibitor of coagulation factor Xa disclosed in US 6,967,208. Said compound is used in the prevention and treatment of thromboembolic disorders.
 (I)
US 7, 153,960 discloses a process for the preparation of apixaban wherein the key step is the formation of intermediate (A) by 1 ,3 dipolar cycloaddition reaction between the compounds of formula (B) and (C) and its subsequent conversion to the compound of formula (D) by treatment with an acid. The compound of formula (D), after simple manipulations of functional groups, is converted to apixaban
 B C A D
Said patent discloses the preparation of the compounds of formula (B) and (C). While the synthesis of the hydrazone of formula (B) has been known for some time, the preparation of the key intermediate of formula (C) is complex and uses reagents which are expensive and potentially hazardous, such as phosphorus pentachloride (PC15), and drastic reaction conditions.
US 7, 153,960, for example, discloses as preferred the preparation of an enamine intermediate of formula (C) wherein the amine residue NRbRc is a morpholine. The conditions used for the success of the reaction actually involve the use of morpholine as solvent at high temperatures, such as reflux temperature (about 130- 135°C).
The complexity of the known processes for the preparation of the intermediate of formula C, the expense and danger of the reagents and the drastic reaction conditions used make said processes difficult to apply and scale up industrially, especially for the purpose of preparing the intermediates of formula A and D and apixaban.
Example 6. Synthesis of compound of formula (I): l-(4- Methoxyphenyl)-6-[4-(2-oxo-piperidinyl)phenyl]-7-oxo-4,5,6,7-tetrahydro- l//-pyrazolo[3,4-c]pyridine-3-carboxyamide: Apixaban (I)
 The compound of formula II, prepared as in Example 5 (17.50 g, 35.82 mmol), is suspended in 100 ml of 33% NH3 and 200 ml of MeOH in a 1L 4-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere, and heated to 45°. MeOH (250 ml) is added until completely dissolved, and the solution is left under stirring for 2h. Another addition of 33% NH3 (50 ml) is performed, and the progress of the reaction is monitored by TLC (AcOEt/MeOH 9: 1) and HPLC. After 18h the solvent is evaporated under low pressure, and the solid residue obtained is suspended in 200 ml of H2O and left under stirring for 2h. The white solid is filtered through a Buchner funnel, and washed with H2O (50 ml). The product of formula (I) is stove-dried at 50°C to a constant weight (12.60 g, yield 76%). The HPLC purity of the product exceeds 99%
 .
1H NMR (300 MHz, CDC13): DELTA
7.47 (2H, dd, J0=8.7 Hz, Ar-H),
7.31(2H, dd, J0=8.7 Hz, Ar-H),
7.23 (2H, dd, J0=8.7 Hz, Ar-H),
6.93 (2H, dd, J0=8.7 Hz, Ar-H),
6.83 (1H, s, N-H),
5.53 (1H, s, N-H),
4.1 1 (2H, t, J=6.6 Hz, CH2CH2N),
3.81 (3H, s, Ar-OCH3),
3.59 (2H, m, NCH2CH2CH2CH2CO)
3.37 (2H, t, J=6.6 Hz, CH2CH2N),
2.55 (2H, m, NCH2CH2CH2CH2CO),
1.93 (4H, m, NCH2CH^CH2CH2CO).
SEE
NMR For apixaban (in CDCl3) 1  Zhou , J. C. ; Oh , L. M. ; Ma , P. ; Li , H. Y. Synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones. WO Patent 2003/0 49681, June 19 , 2003 .
  J. Med. Chem. 2007 , 50 , 5339 – 5356 .
1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (1)[ref ]J. Med. Chem. 2007 , 50 , 5339 – 5356 .To the advanced intermediate 2 (2.44 g, 5.0 mmol) was added 25% ammonia water (1.5 mL, 20 mmol) in methanol (20 mL), and the mixture was heated to 65 °C for 5 h in an autoclave of 50 mL. The resulting mixture was cooled to room temperature, poured into water (30 mL), and crystalized below 0°C. The precipitate was filtrated and dried in vacuo at 50°C to afford the desired product 1 as a pale white solid. Yield: 2.09 g, 91%; mp 171–173 °C; IR (KBr, cm−1): 3448 and 3298 (N-H stretching), 2940 (C-H aliphatic), 1669 (C˭N stretching), 1614 (C˭O stretching), 1544 (aliphatic C˭C), 1513, 1463 and 1441 (aromatic C˭C), 1334, 1300 and 1254 (C-N stretching), 1146, 1111, 1090 and 1024 (C-O stretching), 835, 816, 794 and 758 (Ar-H aromatic bending); 1H NMR (500 MHz, CDCl3, ppm), δ: 7.48 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 8.0 Hz, 2H), 5.66 (brs, 2H), 4.12 (t, J = 5.6 Hz, 2H), 3.84 (s, 3H), 3.55–3.65 (m, 2H), 3.39 (t, J = 5.6 Hz, 2H), 2.57 (t, J = 6.2 Hz, 2H), 1.91–2.01 (m, 4H); 13C NMR (125 MHz, CDCl3, ppm), δ: 170.9, 164.4, 160.5, 158.0, 142.1, 140.6 (2C), 134.0, 133.2, 127.4 (4C), 126.9 (2C), 126.5, 114.4 (2C), 56.2, 52.3, 51.8, 33.5, 24.2, 22.1, 21.9; MS/EI m/z = 459.2 (M+).
SEE
http://www.google.com/patents/WO2014056434A1?cl=en ………………….. http://www.google.com/patents/WO2014108919A2?cl=en HPLC method of Analysis:
Apixaban compound of formula- 1 of the present invention is analyzed by HPLC using the following conditions:
Apparatus: A liquid chromatographic system is to be equipped with variable wavelength UV- detector; Column: Zorbax Bonus RP, 250 x 4.6 mm, 5μιη or equivalent; Flow rate: 1.2 ml/min; wavelength: 270 nm; column temperature: 40°C; Injection volume; 5 uL; Run time: 35 minutes; Needle wash: diluent; Diluent: Acetonitrile: water (90: 10 v/v); Elution: Gradient; Mobile phase-A: Buffer; Mobile phase-B: acetonitrile:water (90:10 v/v); Buffer: Weigh accurately about 1.36 g of potassium dihydrogen ortho phosphate in 1000 10 ml of milli-Q water and adjust pH 6.0 with dil KOH solution, then filter through 0.22 μιη nylon membrane filter paper. The following impurities have been observed during the preparation of Apixaban.
 methyl esterImpurity Chloro Impurity Dehydro Impurity
Scheme-I:
Apixaban
 Scheme-II:
 Pure Apixaban Formula-1 [Apixaban]
Example-1: Preparation of 3-chloro-l-(4-iodophenyI)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (4.08 gm) followed by lithium chloride (2.28 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (30 gm) and dimethylformamide (60 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 25.0 gm; MR: 120-130°C.
Example-2: Preparation of 3-chIoro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (2.99 gm) followed by sodium chloride (2.76 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (50 gm) and dimethylformamide (150 ml) at 30-35°C and stirred for 10 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 6 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound.
Yield: 42.0 gm; M.R: 120-130°C.
Example-3: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (5.09 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydro pyridin-2(lH)-one compound of formula-6 (5 gm) and toluene (5 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 115-120°C and stirred for 3 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 15 hrs at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 3.8 gm.
Example-4: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (28.73 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6- dihydropyridin-2(lH)-one compound of formula-6 (50 gm) and toluene (50 ml) at 30-35°C. Heated the reaction mixture to 115-120°C and stirred for 8 hrs at 115-120°C. After completion of the reaction, cooled the reaction mixture to 25-30°C. Methyl tert-butyl ether (100 ml) followed by water were slowly added to the reaction mixture at 25-30°C. Cooled the reaction mixture to 5- 10°C and stirred for 2 hours at 5-10°C. Filtered the precipitated solid and then dried to get the title compound. Yield: 45 gm.
Example-5: Preparation of ethyl 6-(4-iodophenyl)-l-(4-methoxyphenyI)-7-oxo-4,5,6,7-tetra hydro-lH-pyrazoIo[3,4-c]pyridine-3-carboxyIate (FormuIa-13)
A mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one compound of formula-6 (79.2 gm), (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula-9 (65 gm) and toluene (450 ml) was heated to 90-100°C and stirred for 5 min at the same temperature. Triethyl amine (72 gm) was slowly added to the reaction mixture at 95-100°C and stirred for 2½ hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water (110 ml) was added to the reaction mixture at 25-30°C and stirred for 8 hrs at the same temperature. Filtered the solid, washed with water and then dried to get the title compound.
Yield: 78.5 gm.
Example-6: Preparation of 5-bromo-N-(4-iodophenyl)pentanamide (Formula-3)
A mixture of 5-bromopentanoic acid (54 g), thionyl chloride (41 g), dimethylformamide (2 ml) and toluene (100 ml) was heated to 40-45°C and stirred for 2 hours at the same temperature. Distilled off the reaction mixture to remove the un-reacted thionyl chloride under reduced pressure at a temperature below 40°C. Toluene (50 ml) was added to the reaction mixture and stirred for 15 minutes. The reaction mixture was cooled to 25-30°C under nitrogen atmosphere and it slowly added to a pre-cooled mixture of 4-iodoaniline compound of formula-2 (50 g) and toluene (350 ml) at 0-5°C. Triethyl amine (29 g) was added to it at 0-5°C. The above reaction mixture containing acid chloride was slowly added to the reaction mixture containing 4- iodoaniline under nitrogen atmosphere and stirred for 2 hours at 0-5°C. Water (250 ml) was added to the reaction mixture and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and then dried to get title compound. Yield: 83 gm; MR: 135-140°C; HPLC purity: 99%.
Example-7: Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Step-a) Preparation of l-(4-iodophenyl)piperidin-2-one (Formula-4)
Sodium tert-butoxide (18.86 g) was added to a mixture of 5-bromo-N-(4- iodophenyl)pentanamide compound of formula-3 (50 g) and toluene (250 ml) at 0-5°C and stirred for 2 hours at 0-5°C. Water (100 ml) followed by aqueous hydrochloric acid solution (50 ml) were added to the reaction mixture and stirred for 10 minutes at 5-10°C. Both the organic and aqueous layers were separated; the organic layer was washed with water. Distilled off the solvent from the organic layer under reduced pressure at a temperature below 60°C to get title compound as a solid.
Step-b) Preparation of 3,3-dichIoro-l-(4-iodophenyI)piperidin-2-one (Formula-5)
The compound obtained in step-a) was dissolved in dichloromethane (100 ml) and slowly added to a mixture of phosphorous pentachloride (95 g) and dichloromethane (150 ml) at 25- 30°C. The reaction mixture was heated to 35-40°C and stirred for 4 hours at the same temperature. Cooled the reaction mixture to 5-10°C. Chilled water (150 ml) was added to the reaction mixture and stirred for 1.5 hours at 10-15°C. Both the organic and aqueous layers were separated; the organic layer was washed with water followed by 10% aqueous sodium carbonate solution. Distilled off the solvent completely from the organic layer to get title compound as a solid.
Step-c) Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula- 6)
To the obtained compound in step-b), dimethylformamide (100 ml), followed by lithium carbonate (2.2 g) and sodium chloride (2.0 g) were added at 25-30°C. The reaction mixture was heated to 115-120°C and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 30-35°C, water (350 ml) was added to it and stirred for 2 hours at 25-30°C. Filtered the precipitated solid and washed with water. Methanol (360 ml) was added to the obtained solid and the reaction mixture was heated to 65-70°C. Stirred the reaction mixture for 20 minutes at the same temperature. Carbon (3.0 g) was added to the reaction mixture and stirred for 20 minutes at 65-70°C. Filtered the reaction mixture through hyflow bed and washed with methanol. Distilled off the solvent from the filtrate under reduced pressure and methanol (300 ml) was added to the residue and stirred for 20 minutes at 25-30°C. Cooled the reaction mixture to -5 to 0°C and stirred for 60 minutes at the same temperature. Filtered the precipitated solid, washed with methanol and then dried to get title compound.
Yield: 25 gm; MR: 115- 120°C: HPLC purity: 98%.
Example-8: Preparation of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydro pyridin-2(lH)-one (Formula-8)
A mixture of l-(4-iodophenyl)-3-mo holino-5,6-dihydropyridin-2(lH)-one compound of formula-7 (50 g), piperidin-2-one (32.25 g) and o-xylene (75 ml) was stirred for 10 minutes at 25-30°C. Potassium carbonate (27.0 g), followed by copper iodide (7.43 g) were added to the reaction mixture. The reaction mixture was heated to 140-145°C under azeotropic distillation condition and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 35- 40°C, water (175 ml) was slowly added to the reaction mixture at 35-40°C. Cooled the reaction mixture to 10-15°C and ammonia (125 ml) was added to the reaction mixture at 10-15°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 2 hours at the same temperature. Filtered the precipitated solid, washed with water and then dried to get title compound.
Yield: 35 gm; MR: 195-200°C; HPLC purity: 95%.
Example-9: Preparation of (Z)-ethyl 2-chloro-2-(2-(4-nlethoxyphenyl)hydrazono)acetate (FormuIa-9)
A mixture of 4-methoxyaniline compound of formula- 12 (50 g) and water (150 ml) was cooled to 5-10°C. Hydrochloric acid (100 ml), followed by a solution of sodium nitrite (30.81 g) in water (50 ml) were slowly added to the reaction mixture at 5-10°C and stirred for 2 hours at 5- 10°C to provide diazotized compound. Ethyl acetate (250 ml) was added to the reaction mixture. Ethyl 2-chloro acetoacetate (76.84 g) was slowly added to a mixture of sodium acetate (76.6 g), ethyl acetate (250 ml) and water (150 ml) at 25-30°C and the reaction mixture was stirred for 2 hours at 25-30°C. The reaction mixture was slowly added to the reaction mixture containing diazotized compound at a temperature below 10°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 16 hours at the same temperature. Both the organic and aqueous layers were separated and the organic layer was washed with 10% aqueous sodium bicarbonate solution followed by 10% aqueous sodium chloride solution. Distilled off the solvent completely from the organic layer under reduced pressure and then co-distilled with toluene. Toluene was added to the obtained compound and stirred for 15 minutes at 25-30°C. Silica-gel was added to the reaction mixture and stirred for 30 minutes at 25-30°C. Filtered the reaction mixture and the solvent from the filtrate was distilled off completely under reduced pressure. Cyclohexane (400 ml) was added to the obtained compound and the reaction mixture was stirred for 60 minutes at 25-30°C. Filtered the precipitated solid, washed with cyclohexane and then dried to get title compound. Yield: 60 gm; MR: 95-100°C; HPLC purity: 99%.
ExampIe-10: Preparation of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl) phenyl)-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate (Formula-11)
A mixture of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydropyridin-2(lH)- one compound of formula-8 (30 g), sodium carbonate (26.83 g) and acetone (150 ml) was heated to 45-50°C. (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula- 9 (32.5 g) was added to the reaction mixture at 45-50°C and stirred for 3 hours at the same temperature. Cooled the reaction mixture to 25-30°C and aqueous hydrochloric acid (50 ml) in 50 ml of water was added to it at 25-30°C. Stirred the reaction mixture for 2 hours at 25-30°C. Water was slowly added to the reaction mixture and stirred for 45 minutes at 25-30°C. Filtered the obtained solid and washed with water. The obtained solid was recrystallized from toluene (150 ml) to get the title compound. Yield: 35 gm; MR: 155-160°C; HPLC purity: 97%.
Example- 11: Preparation of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
A mixture of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl)phenyl)- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate compound of formula-11 (50 g), formamide (150 ml), sodium methoxide (30 ml) and isopropanol (300 ml) was heated to 65-70°C and stirred for 2 hours at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid and washed with isopropanol. Methanol (150 ml) was added to the obtained solid, the reaction mixture was heated to 65-70°C and stirred for 15 minutes at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid, washed with methanol and then dried to get title compound. Yield: 35 g. MR: 230-235°C; HPLC purity: 98%.
The PXRD of the crystalline solid obtained from the above example is matches with the PXRD of crystalline form-M of the present invention.
Example-12: Purification of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6, 7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (100 g) was dissolved in a mixture of dichloromethane (1200 ml) and methanol (200 ml) at 25-30°C. 10% aqueous sodium carbonate solution (200 ml) was added to the reaction mixture and stirred for 15 minutes at 25- 30°C. Both the organic and aqueous layers were separated, methanol (100 ml) was added to the organic layer and again 200 ml of 10% aqueous sodium carbonate solution was added to the reaction mixture. The reaction mixture was stirred for 15 minutes at 25-30°C and separated the organic and aqueous layers. To the organic layer methanol (100 ml) followed by water (200 ml) were added. Both the organic and aqueous layers were separated. The solvent from organic layer was distilled under reduced pressure at a temperature below 40°C. 3000 ml of a mixture of dichloromethane and methanol (in the ratio of 3:7) was added to the crude compound and the reaction mixture was heated to reflux temperature and stirred for 10 minutes. Carbon (10 g) was added to the reaction mixture and stirred for 15 minutes at the reflux temperature. Filtered the reaction mixture through hyflow bed, washed with a mixture of dichloromethane and methanol. The filtrate was cooled to 0-5°C and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and washed with a mixture of dichloromethane and methanol. Isopropanol (1000 ml) was added to the reaction mixture. Heated the reaction mixture to 80-85°C and stirred for 15 minutes. Cooled the reaction mixture to 25-30°C and stirred for 2 hours at 35-30°C. Filtered the precipitated solid, washed with isopropanol and then dried to get title compound.
Yield: 80 gm; MR: 235-240°C.
The PXRD pattern of crystalline solid obtained from the above example is matches with PXRD of crystalline form-M of the present invention.
Example-13: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to isopropanol (400 ml) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min the same temperature. Filtered the solid, washed with isopropanol and then dried to get the title compound. Yield: 4.5 gm; Water content: 0.30% w/w. HPLC purity: 99.8%; Acid impurity: 0.02%; Amino acid impurity: Not detected; Chloro impurity: 0.01%; Methyl ester impurity: 0.05%; Ethyl ester impurity: 0.01%; Dehydro impurity: 0.07%.
Particle size distribution: D(0.1): 9.183 μπι; D(0.5): 25.991 um; D(0.9): 60.749 μιη; D[4,3]: 31.066 μπι.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-14: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yI)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to 50% aqueous isopropanol (60 ml) at 25-30°C. Heated the reaction mixture to 50-60°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get the title compound.
Yield: 4.1 gm; Water content: 0.35% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-15: Preparation of crystalline form-S of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0-5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 24.0 gm; M.R: 235-245°C; Water content: 7.38% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-3 and figure-4 respectively.
Example-16: Preparation of crystalline form-N of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3- carboxamide(Formula-l)
A mixture of dichloromethane and ethyl acetate (625 ml, in 3:7 ratio) was added to l-(4- methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula- 1 (6.25 gm) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get title compound. Yield: 3.9 g; Water content: 5.21% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-5 and figure-6 respectively.
Example-17: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to isopropanol (510 ml). Heated the reaction mixture to reflux temperature and stirred for 15 Minutes at the same temperature. The reaction mixture was cooled to 0-5°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1. Yield: 23 g; Water content: 0.30%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-18: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to aq.isopropanol (340 ml). Heated the reaction mixture to 50-60°C and stirred for 15 minutes at the same temperature. The reaction mixture was cooled to 25-35°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1.
Yield: 23 g; Water content: 0.35%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively
/…………………
|
References
- “ELIQUIS® (apixaban) Approved In Europe For Preventing Venous Thromboembolism After Elective Hip Or Knee Replacement” (Press release). Pfizer. April 20, 2012. Retrieved 2012-05-29.
- http://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_eliquis_apixaban_for_the_treatment_of_deep_vein_thrombosis_dvt_and_pulmonary_embolism_pe_and_for_the_reduction_in_the_risk_of_recurrent_dvt_and_pe_following_initial_therapy
- “Bristol-Myers Squibb News Release 26 April 2007”. Archived from the original on 11 September 2007. Retrieved 2007-09-15.
- Nainggolan, Lisa. “Apixaban better than European enoxaparin regimen for preventing VTE”. Retrieved 2011-04-01.
- “Eliquis (apixaban) [prescribing information]” (PDF). Princeton, NJ: Bristol-Myers Squibb. March 2014. Retrieved 2014-10-29.
- “www.nice.org.uk”.
- Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). “Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.”. Thrombosis 2013: 640723. PMID 24455237.
- “ELIQUIS® (apixaban) tablets Factor Xa Inhibitor” (PDF). FDA. August 2014. Retrieved 2014-11-02.
- Enriquez A, Lip GY, Baranchuk A (2015). “Anticoagulation reversal in the era of the non-vitamin K oral anticoagulants”. Europace. doi:10.1093/europace/euv030. PMID 25816811.
- Frost C, Wang J, Nepal S; et al. (February 2013). “Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects”. Br J Clin Pharmacol 75 (2): 476–87. doi:10.1111/j.1365-2125.2012.04369.x. PMID 22759198.
- Granger, M.D. et. al., Christopher (September 15, 2011). “Apixaban versus Warfarin in Patients with Atrial Fibrillation”. New England Journal of Medicine (365): 981–992. doi:10.1056/NEJMoa1107039. Retrieved 17 September 2015.
- FDA approves Eliquis to reduce the risk of stroke, blood clots in patients with non-valvular atrial fibrillation
Neale, Todd (March 14, 2014). “FDA OKs Apixaban for DVT Prevention”. MedPage Today. Retrieved 17 September 2015.
//////////
SEE ABAN SERIES……http://organicsynthesisinternational.blogspot.in/p/aban-series.html
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
 COCK WILL TEACH YOU
COCK WILL TEACH YOU
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
 amcrasto@gmail.com
amcrasto@gmail.com
Etirinotecan pegol (NKTR-102) エチリノテカンペゴル: A Next-Generation Topoisomerase I Inhibitor


Etirinotecan pegol (NKTR-102)
848779-32-8
PEG-irinotecan
Also known as: NKTR-102; UNII-LJ16641SFT; 848779-32-8
Molecular Formula: C161H192N20O40 Molecular Weight: 3047.35718
Nektar Therapeutics innovator
http://www.acsmedchem.org/mediabstractf2013.pdf

CAS: 1193151-09-5
Synonym: NKTR102; NKTR 102; NKTR-102; pegylated irinotecan NKTR 102; Etirinotecan pegol.
IUPAC/Chemical name: (1). Tetrakis{(4S)-9-[([1,4′-bipiperidinyl]-1′-carbonyl)oxy]-4,11-diethyl-3,14-dioxo-3,4,12,14- tetrahydro-1H-pyrano[3′,4′:6,7]indolizino[1,2-b]quinolin-4-yl} N,N’,N”,N”’- {methanetetrayltetrakis[methylenepoly(oxyethylene)oxy(1-oxoethylene)]}tetraglycinate tetrahydrochloride
(2). Poly(oxy-1,2-ethanediyl), α-hydro-ω-[2-[[2-[[(4S)-9-[([1,4′-bipiperidin]-1′-ylcarbonyl)oxy]- 4,11-diethyl-3,4,12,14-tetrahydro-3,14-dioxo-1H-pyrano[3′,4′:6,7]indolizino[1,2- b]quinolin-4-yl]oxy]-2-oxoethyl]amino]-2-oxoethoxy]-, ether with 2,2-bis(hydroxymethyl)- 1,3-propanediol, hydrochloride (4:1:4)

Etirinotecan pegol tetratriflutate [USAN]
RN: 1193151-12-0
MF and MW
-
C153-H176-N20-O36 (C8-H16-O4)n.4(C2-H-F3-O2)
- 3503.4754
Tetrakis((4S)-9-(((1,4′-bipiperidinyl)-1′-carbonyl)oxy)-4,11-diethyl-3,14-dioxo-3,4,12,14- tetrahydro-1H-pyrano(3′,4′:6,7)indolizino(1,2-b)quinolin-4-yl) N,N’,N”,N”’- (methanetetrayltetrakis(methylenepoly(oxyethylene)oxy(1-oxoethylene)))tetraglycinate tetrakis(trifluoroacetate)
NKTR-102 is currently being developed by Nektar. According to the company’s news release, this agent exhibits a very high response rate and excellent clinical benefit rate in patients with metastatic breast cancer, and importantly, this anti-tumor activity is maintained in each of the poor prognosis subsets within the study. The data from the Phase 2 study also shows highly promising PFS of 5.3 months and OS of 13.1 months in the every three week dose schedule, which was also very well-tolerated. As a novel topoisomerase I inhibitor in breast cancer, NKTR-102 holds great therapeutic potential and allows us to address the challenge of resistance in this setting
NKTR-102 (PEG-irinotecan), a PEGylated form of irinotecan, is in clinical development by Nektar Therapeutics for the treatment of multiple solid tumors, including colorectal cancer, metastatic or locally advanced breast cancer, metastatic or locally advanced ovarian cancer and gastrointestinal cancer. No recent development has been reported for phase I clinical trials for the treatment of gastrointestinal cancer.
In preclinical studies, NKTR-102 resulted in significantly higher reduction in tumor growth than irinotecan in colon, lung and breast tumors. The company believes that following intravenous administration of NKTR-102, irinotecan will be released slowly, resulting in prolonged systemic exposure of irinotecan. Irinotecan is a cytotoxic anticancer agent used extensively to treat colorectal, lung, esophageal and other solid tumors. In 2011, orphan drug designation was assigned to the compound in the U.S. for the treatment of ovarian cancer.
In 2011, orphan drug designation was assigned in the E.U. for the treatment of ovarian cancer. In 2012, fast track designation was assigned by the FDA for the treatment of locally recurrent or metastatic breast cancer progressing after treatment with an anthracycline, a taxane and capecitabine.
| Therapeutic Area | Nektar Discovered |
Indication | Phase |
| Oncology | |||
| Etirinotecan pegol (NKTR-102) | |||
| Metastatic Breast Cancer |  Phase 3 |
||
| Platinum-Resistant Ovarian Cancer |  Phase 2 Completed |
||
| Second-Line Colorectal Cancer | Phase 2 Completed |
||
| Bevacizumab (Avastin)-refractory high-grade glioma | Phase 2 |
||
| Non-Small Cell Lung Cancer (NSCLC) | Phase 2 |
||
| Small Cell Lung Cancer (SCLC) | Phase 2 |
||
| GI and solid tumors In combination with 5-FU |
 Phase 1 Completed |
||
http://www.nektar.com/product_pipeline/all_phases.html#BAX855
Market Overview
Etirinotecan pegol is in Phase 3 clinical development for patients with metastatic or locally recurrent breast cancer and Phase 2 clinical development for patients with solid tumor malignancies, including ovarian, colorectal, glioma, small cell and non-small cell lung cancers. Each year, approximately 5.3 million patients worldwide are diagnosed with one of these types of cancer.1
Etirinotecan Pegol Clinical Data and Product Profile
Etirinotecan pegol (NKTR-102) is the first long-acting topoisomerase I-inhibitor (Topo I) designed to concentrate in tumor tissue, provide sustained tumor suppression throughout the entire chemotherapy cycle, and to reduce the peak exposures that are associated with toxicities of other cytotoxics. Etirinotecan pegol was invented by Nektar using its advanced polymer conjugate technology platform, and is the first oncology product candidate to leverage Nektar’s releasable polymer technology platform.
Topo I-inhibitors are important chemotherapeutic agents used to treat cancer. Immediately after dosing, however, standard topo I-inhibitors reach high peak concentrations and diffuse quickly throughout the body—penetrating and damaging healthy tissue, such as bone marrow, as well as tumor tissue. Subsequent rapid metabolism limits topo I exposure in tumor cells, reducing the duration of their effect and resulting in a much lower tumor exposure to the active metabolite that may limit their efficacy.
Etirinotecan pegol is a novel chemotherapeutic designed to enhance the anti-cancer effects of topo I-inhibition while minimizing its toxicities. Unlike first generation topo I-inhibitors that exhibit a high initial peak concentration and short half-life, etirinotecan pegol’s unique pro-drug design results in a lowered initial peak concentration of active topo I inhibitor in the blood. The large etirinotecan pegol molecule is inactive when administered. Over time, the body’s natural enzymatic processes slowly metabolize the linkers within the molecule, continuously freeing active drug that then works to stop tumor cell division through inhibition of topo I.
Clinical and preclinical studies have shown that the half-life of active drug generated from etirinotecan pegol is greatly extended to 50 days (compared to 48 hours for irinotecan) and that active drug remains in circulation throughout the entire chemotherapy cycle, providing sustained exposure to topo I inhibition. In preclinical models, etirinotecan pegol achieved a 300-fold increase in tumor concentration as compared to a first generation topo I-inhibitor. Because etirinotecan pegol is a large molecule, it is believed to penetrate the leaky vasculature within the tumor environment more readily than normal vasculature, concentrating and trapping etirinotecan pegol in tumor tissue.
Etirinotecan pegol is currently in development for the treatment of breast, ovarian, colorectal, glioma, small cell and non-small cell lung cancers.
Ongoing clinical development for etirinotecan pegol:
- In metastatic breast cancer, a Phase 3 randomized, head to head study (The BEACON Study) of etirinotecan pegol compared to Treatment of Physician’s Choice (TPC) completed enrollment of 864 patients in August 2013. Data from the study on the primary endpoint of overall survival is expected by the end of 2014 or early 2015.
- In ovarian cancer, an expanded Phase 2 study of single-agent etirinotecan pegol in platinum refractory/resistant ovarian cancer in 177 women who failed prior Doxil therapy was completed at the end of 2012.
- In colorectal cancer, a 174-patient Phase 2 randomized, head-to-head study of etirinotecan pegol compared to irinotecan in patients with second-line colorectal cancer with the KRAS gene mutation is in progress.
- Etirinotecan pegol is also being evaluated in glioma, small cell and non-small cell lung cancers.
Highlighted Data Presentations:
Data from a Phase 2 clinical study of etirinotecan pegol in metastatic breast cancer were published in the November 2013 issue of The Lancet Oncology (click here to view manuscript) These data were previously presented at the 2011 ASCO Annual meeting (click here to download this presentation).
Data from a Phase 2 clinical study of etirinotecan pegol in platinum-resistant/refractory ovarian cancer were published in the September 30, 2013 online edition of the Journal of Clinical Oncology (click here to view abstract). These data were previously presented at the 2010 ASCO Meeting (click here to download this presentation).
Data from a Phase 2 clinical study of etirinotecan pegol in metastatic breast cancer were presented in an oral abstract session at the 2011 ASCO Breast Cancer Symposium by Agustin Garcia, MD. View presentation slides.
Data from a Phase 2 clinical study of NKTR-102 in a subpopulation of patients with platinum-resistant/refractory ovarian cancer and prior Doxil® (pegylated liposomal doxorubicin or PLD) treatment were presented at the 2011 ASCO Annual Meeting by Agustin Garcia, MD. (click here to download this presentation).
Data from a Phase 2 clinical study of etirinotecan pegol in metastatic breast cancer were presented at the 2010 33rd Annual CTRC-AACR San Antonio Breast Cancer Symposium by Amad Awada, MD. (click here to download this presentation).
2014 events related to this product
| January 16-18, 2014 | 2014 Gastrointestinal Cancers SymposiumPoster C55: “A phase I study of etirinotecan pegol in combination with 5-fluorouracil and leucovorin in patients with advanced cancer.” January 18, 2014 | San Francisco, CA |
| February 22, 2014 | 26.2 with Donna Marathon sponsored by Mayo Clinic | Jacksonville, FL |
| March 5-7, 2014 | TAT 2013: International Congress on Targeted Anticancer Therapies | Washington, DC |
| April 5-9, 2014 | AACR Annual Meeting 2013 | San Diego, CA |
| May 19-21, 2014 | 10th International Symposium on Polymer Therapeutics | Valencia, Spain |
| May 30-June 3, 2014 | 2014 ASCO 50th Annual MeetingPoster Presentation: “Combination Immunotherapy: Synergy of a Long-Acting Engineered Cytokine (NKTR-214) and Checkpoint Inhibitors Anti-CTLA-2 or Anti-PD-1 in Murine Tumor Models,” Kantak et al. Abstract Number: 3082 Session Title/Track: Developmental Therapies – Immunotherapy Date: June 1, 2014, 8:00 a.m. – 11:45 a.m. Central Time |
Chicago, Illinois |
| September 4-6, 2014 | ASCO Breast Cancer Symposium | San Francisco, CA |
| September 26-30, 2014 | ESMO 2014 Congress | Madrid, Spain |
| December 9-13, 2014 | San Antonio Breast Cancer Symposium | San Antonio, TX |
……………………….
http://www.google.com.ar/patents/US7744861?cl=pt-PT
Example 1 SYNTHESIS OF PENTAERYTHRITOLYL-4-ARM-(PEG-1-METHYLENE-2 OXO-VINYLAMINO ACETATE LINKED-IRINOTECAN)-20K
A. Synthesis of t-Boc-Glycine-Irinotecan

In a flask, 0.1 g Irinotecan (0.1704 mmoles), 0.059 g t-Boc-Glycine (0.3408 mmoles), and 0.021 g DMAP (0.1704 mmoles) were dissolved in 13 mL of anhydrous dichloromethane (DCM). To the solution was added 0.070 g DCC (0.3408 mmoles) dissolved in 2 mL of anhydrous DCM. The solution was stirred overnight at room temperature. The solid was removed through a coarse frit, and the solution was washed with 10 mL of 0.1N HCL in a separatory funnel. The organic phase was further washed with 10 mL of deionized H2O in a separatory funnel and then dried with Na2SO4. The solvent was removed using rotary evaporation and the product was further dried under vacuum. 1H NMR (DMSO): δ 0.919 (t, CH2CH 3), 1.34 (s, C(CH3)3), 3.83 (m, CH2), 7.66 (d, aromatic H).
B. Deprotection of t-Boc-Glycine-Irinotecan

0.1 g t-Boc-Glycine-Irinotecan (0.137 mmoles) was dissolved in 7 mL of anhydrous DCM. To the solution was added 0.53 mL trifluoroacetic acid (6.85 mmoles). The solution was stirred at room temperature for 1 hour. The solvent was removed using rotary evaporation. The crude product was dissolved in 0.1 mL MeOH and then precipitated in 25 mL of ether. The suspension was stirred in an ice bath for 30 minutes. The product was collected by filtration and dried under vacuum. 1H NMR (DMSO): δ 0.92 (t, CH2CH 3), 1.29 (t, CH2CH 3), 5.55 (s, 2H), 7.25 (s, aromatic H).
C. Covalent Attachment of a Multi-Armed Activated Polymer to Glycine Irinotecan.

0.516 g Glycine-Irinotecan (0.976 mmoles), 3.904 g 4arm-PEG(20K)-CM (0.1952 mmoles), 0.0596 g 4-(dimethylamino)pyridine (DMAP, 0.488 mmoles), and 0.0658 g 2-hydroxybenzyltriazole (HOBT, 0.488 mmoles) were dissolved in 60 mL anhydrous methylene chloride. To the resulting solution was added 0.282 g 1,3-dicyclohexylcarbodiimide (DCC, 1.3664 mmoles). The reaction mixture was stirred overnight at room temperature. The mixture was filtered through a coarse frit and the solvent was removed using rotary evaporation. The syrup was precipitated in 200 mL of cold isopropanol over an ice bath. The solid was filtered and then dried under vacuum. Yield: 4.08 g. 1H NMR (DMSO): δ 0.909 (t, CH2CH 3), 1.28 (m, CH2CH 3), 3.5 (br m, PEG), 3.92 (s, CH2), 5.50 (s, 2H).
Example 2 ANTI-TUMOR ACTIVITY OF PENTAERYTHRITOLYL-4-ARM-(PEG -1-METHYLENE-2 OXO-VINYLAMINO ACETATE LINKED-IRINOTECAN)-20K, “4-ARM-PEG-GLY-IRINO-20K” IN A COLON CANCER MOUSE XENOGRAFT MODEL
Human HT29 colon tumor xenografts were subcutaneously implanted in athymic nude mice. After about two weeks of adequate tumor growth (100 to 250 mg), these animals were divided into different groups of ten mice each. One group was dosed with normal saline (control), a second group was dosed with 60 mg/kg of irinotecan, and the third group was dosed with 60 mg/kg of the 4-arm PEG-GLY-Irino-20K (dose calculated per irinotecan content). Doses were administered intraveneously, with one dose administered every 4 days for a total of 3 administered doses. The mice were observed daily and the tumors were measured with calipers twice a week. FIG.1 shows the effect of irinotecan and PEG-irinotecan treatment on HT29 colon tumors in athymic nude mice.
As can be seen from the results depicted in FIG. 1, mice treated with both irinotecan and 4-arm-PEG-GLY-Irino-20K exhibited a delay in tumor growth (anti-tumor activity) that was significantly improved when compared to the control. Moreover, the delay in tumor growth was significantly better for the 4-arm-PEG-GLY-Irino-20K group of mice when compared to the group of animals administered unconjugated irinotecan.
Example 3 SYNTHESIS OF PENTAERYTHRITOLYL-4-ARM-(PEG-1-METHYLENE-2 OXO-VINYLAMINO ACETATE LINKED-IRINOTECAN)-40K, “4-ARM-PEG-GLY-IRINO-40K”
4-arm-PEG-GLY-IRINO-40K was prepared in an identical fashion to that described for the 20K compound in Example 1, with the exception that in step C, the multi-armed activated PEG reagent employed was 4 arm-PEG(40K)-CM rather than the 20K material.
Example 4 SYNTHESIS OF PENTAERYTHRITOLYL-4-ARM-(PEG-1-METHYLENE-2 OXO-VINYLAMINO ACETATE LINKED-SN-38)-20K, “4-ARM-PEG-GLY-SN-38-20K”
4-arm PEG-GLY-SN-38-20K was prepared in a similar fashion to its irinotecan counterpart as described in Example 1, with the exception that the active agent employed was SN-38, an active metabolite of camptothecin, rather than irinotecan, where the phenolic-OH of SN-38 was protected with MEMCI (2-methoxyethoxymethyl chloride) during the chemical transformations, followed by deprotection with TEA to provide the desired multi-armed conjugate.
Example 5 SYNTHESIS OF PENTAERYTHRITOLYL-4-ARM-(PEG-1-METHYLENE-2 OXO-VINYLAMINO ACETATE LINKED-SN-38)-40K, “4-ARM-PEG-GLY-SN-38-40K”
4-arm PEG-GLY-SN-38-40K was prepared in a similar fashion to the 20K version described above, with the exception that the multi-armed activated PEG reagent employed was 4 arm-PEG(40K)-CM rather than the 20K material.
Example 8 SYNTHESIS OF PENTAERYTHRITOLYL-4-ARM-(PEG-2-{2-[2-1-HYDROXY-2-OXO-VINYLOXY)-ETHOXY]-ETHYLAMINO}-PROPEN-1-ONE LINKED-IRINOTECAN)-20K AND -40K


A. 2-(2-t-Boc-aminoethoxy)ethanol (1)
2-(2-Aminoethoxy)ethanol (10.5 g, 0.1 mol) and NaHCO3 (12.6 g, 0.15 mol) were added to 100 mL CH2Cl2 and 100 mL H2O. The solution was stirred at RT for 10 minutes, then di-tert-butyl dicarbonate (21.8 g, 0.1 mol) was added. The resulting solution was stirred at RT overnight, then extracted with CH2Cl2 (3×100 mL). The organic phases were combined and dried over anhydrous sodium sulfate and evaporated under vacuum. The residue was subjected to silica gel column chromatography (CH2Cl2:CH3OH=50:1˜10:1) to afford 2-(2-t-Boc-aminoethoxy)ethanol (1) (16.0 g, 78 mmol, yield 78%)
B. 2-(2-t-Boc-aminoethoxy)ethoxycarbonyl-Irinotecan (2)
2-(2-t-Boc-aminoethoxy)ethanol (1) (12.3 g, 60 mmol) and 4-dimethylaminopyridine (DMAP) (14.6 g, 120 mmol) were dissolved in 200 ml anhydrous CH2Cl2. Triphosgene (5.91 g, 20 mmol) was added to the solution while stirring at room temperature. After 20 minutes, the solution was added to a solution of irinotecan (6.0 g, 10.2 mmol) and DMAP (12.2 g, 100 mmol) in anhydrous CH2Cl2 (200 mL). The reaction was stirred at RT for 2 hrs, then washed with HCI solution (pH=3, 2L) to remove DMAP. The organic phases were combined and dried over anhydrous sodium sulfate. The dried solution was evaporated under vacuum and subjected to silica gel column chromatography (CH2Cl2:CH3OH=40:1˜10:1) to afford 2-(2-t-Boc-aminoethoxy)ethoxycarbonyl-irinotecan (2) (4.9 g, 6.0 mmol, yield 59%).
C. 2-(2-aminoethoxy)ethoxycarbonyl-irinotecan TFA salt (3)
2-(2-t-Boc-aminoethoxy)ethoxycarbonyl-irinotecan (2) (4.7 g, 5.75 mmol) was dissolved in 60 mL CH2Cl2, and trifluoroacetic acid (TFA) (20 mL) was added at RT. The reaction solution was stirred for 2 hours. The solvents were removed under vacuum and the residue was added to ethyl ether and filtered to give a yellow solid as product 3 (4.3 g, yield 90%).
D. 4-arm-PEG20k-carbonate-inotecan (4)
4-arm-PEG20k-SCM (16.0 g) was dissolved in 200 mL CH2Cl2. 2-(2-aminoethoxy)ethoxycarbonyl-irinotecan TFA salt (3) (2.85 g, 3.44 mmol) was dissolved in 12 mL DMF and treated with 0.6 mL TEA, then added to a solution of 4-arm-PEG20k-SCM. The reaction was stirred at RT for 12 hrs then precipitated in Et2O to yield a solid product, which was dissolved in 500 mL IPA at 50° C. The solution was cooled to RT and the resulting precipitate collected by filtration to give 4-arm-PEG20k-glycine -irinotecan (4) (16.2 g, drug content 7.5% based on HPLC analysis). Yield: 60%.
E. 4-arm-PEG40k-carbonate-irinotecan (5)
4-arm-PEG40k-SCM (32.0 g) was dissolved in 400 mL CH2Cl2. 2-(2-aminoethoxy)ethoxycarbonyl-irinotecan TFA salt (3) (2.85 g, 3.44 mmol) was dissolved in 12 mL DMF and treated with 0.6 mL TEA, then added to the solution of 4-arm -PEG40k-SCM. The reaction was stirred at RT for 12 hrs and then precipitated in Et2O to get solid product, which was dissolved in 1000 mL isopropyl alcohol (IPA) at 50° C. The solution was cooled to RT and the precipitate collected by filtration to gave 4-arm-PEG40k-glycine-irinotecan (4) (g, drug content 3.7% based on HPLC analysis). Yield: 59%.
References
1: Iwase Y, Maitani Y. Dual functional octreotide-modified liposomal irinotecan leads to high therapeutic efficacy for medullary thyroid carcinoma xenografts. Cancer Sci. 2011 Oct 24. doi: 10.1111/j.1349-7006.2011.02128.x. [Epub ahead of print] PubMed PMID: 22017398.
2: Matsuzaki T, Takagi A, Furuta T, Ueno S, Kurita A, Nohara G, Kodaira H, Sawada S, Hashimoto S. Antitumor activity of IHL-305, a novel pegylated liposome containing irinotecan, in human xenograft models. Oncol Rep. 2012 Jan;27(1):189-97. doi: 10.3892/or.2011.1465. Epub 2011 Sep 20. PubMed PMID: 21935577.
3: Cobleigh MA. Other options in the treatment of advanced breast cancer. Semin Oncol. 2011 Jun;38 Suppl 2:S11-6. Review. PubMed PMID: 21600380.
4: Li C, Cui J, Wang C, Li Y, Zhang L, Xiu X, Li Y, Wei N, Zhang L, Wang P. Novel sulfobutyl ether cyclodextrin gradient leads to highly active liposomal irinotecan formulation. J Pharm Pharmacol. 2011 Jun;63(6):765-73. doi: 10.1111/j.2042-7158.2011.01272.x. Epub 2011 Apr 7. PubMed PMID: 21585373.
5: Iwase Y, Maitani Y. Octreotide-targeted liposomes loaded with CPT-11 enhanced cytotoxicity for the treatment of medullary thyroid carcinoma. Mol Pharm. 2011 Apr 4;8(2):330-7. Epub 2011 Jan 18. PubMed PMID: 21166471.
6: Xenidis N, Vardakis N, Varthalitis I, Giassas S, Kontopodis E, Ziras N, Gioulbasanis I, Samonis G, Kalbakis K, Georgoulias V. Α multicenter phase II study of pegylated liposomal doxorubicin in combination with irinotecan as second-line treatment of patients with refractory small-cell lung cancer. Cancer Chemother Pharmacol. 2011 Jul;68(1):63-8. Epub 2010 Sep 10. PubMed PMID: 20830475.
7: Pastorino F, Loi M, Sapra P, Becherini P, Cilli M, Emionite L, Ribatti D, Greenberger LM, Horak ID, Ponzoni M. Tumor regression and curability of preclinical neuroblastoma models by PEGylated SN38 (EZN-2208), a novel topoisomerase I inhibitor. Clin Cancer Res. 2010 Oct 1;16(19):4809-21. Epub 2010 Aug 11. PubMed PMID: 20702613.
8: Morgensztern D, Baggstrom MQ, Pillot G, Tan B, Fracasso P, Suresh R, Wildi J, Govindan R. A phase I study of pegylated liposomal doxorubicin and irinotecan in patients with solid tumors. Chemotherapy. 2009;55(6):441-5. Epub 2009 Dec 8. PubMed PMID: 19996589.
9: Meckley LM, Neumann PJ. Personalized medicine: factors influencing reimbursement. Health Policy. 2010 Feb;94(2):91-100. Epub 2009 Oct 7. PubMed PMID: 19815307.
10: Skak K, Søndergaard H, Frederiksen KS, Ehrnrooth E. In vivo antitumor efficacy of interleukin-21 in combination with chemotherapeutics. Cytokine. 2009 Dec;48(3):231-8. Epub 2009 Aug 25. PubMed PMID: 19709902.
11: Murphy CG, Seidman AD. Evolving approaches to metastatic breast cancer previously treated with anthracyclines and taxanes. Clin Breast Cancer. 2009 Jun;9 Suppl 2:S58-65. Review. PubMed PMID: 19596644.
12: Fox ME, Guillaudeu S, Fréchet JM, Jerger K, Macaraeg N, Szoka FC. Synthesis and in vivo antitumor efficacy of PEGylated poly(l-lysine) dendrimer-camptothecin conjugates. Mol Pharm. 2009 Sep-Oct;6(5):1562-72. PubMed PMID: 19588994; PubMed Central PMCID: PMC2765109.
13: Atyabi F, Farkhondehfai A, Esmaeili F, Dinarvand R. Preparation of pegylated nano-liposomal formulation containing SN-38: In vitro characterization and in vivo biodistribution in mice. Acta Pharm. 2009 Jun;59(2):133-44. PubMed PMID: 19564139.
14: Liu Z, Robinson JT, Sun X, Dai H. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J Am Chem Soc. 2008 Aug 20;130(33):10876-7. Epub 2008 Jul 29. PubMed PMID: 18661992; PubMed Central PMCID: PMC2597374.
15: Scott LC, Yao JC, Benson AB 3rd, Thomas AL, Falk S, Mena RR, Picus J, Wright J, Mulcahy MF, Ajani JA, Evans TR. A phase II study of pegylated-camptothecin (pegamotecan) in the treatment of locally advanced and metastatic gastric and gastro-oesophageal junction adenocarcinoma. Cancer Chemother Pharmacol. 2009 Jan;63(2):363-70. Epub 2008 Apr 9. PubMed PMID: 18398613.
16: Almubarak M, Newton M, Altaha R. Reinduction of bevacizumab in combination with pegylated liposomal Doxorubicin in a patient with recurrent glioblastoma multiforme who progressed on bevacizumab/irinotecan. J Oncol. 2008;2008:942618. Epub 2008 Sep 2. PubMed PMID: 19259336; PubMed Central PMCID: PMC2648641.
17: Krauze MT, Noble CO, Kawaguchi T, Drummond D, Kirpotin DB, Yamashita Y, Kullberg E, Forsayeth J, Park JW, Bankiewicz KS. Convection-enhanced delivery of nanoliposomal CPT-11 (irinotecan) and PEGylated liposomal doxorubicin (Doxil) in rodent intracranial brain tumor xenografts. Neuro Oncol. 2007 Oct;9(4):393-403. Epub 2007 Jul 24. PubMed PMID: 17652269; PubMed Central PMCID: PMC1994096.
18: Li YF, Fu S, Hu W, Liu JH, Finkel KW, Gershenson DM, Kavanagh JJ. Systemic anticancer therapy in gynecological cancer patients with renal dysfunction. Int J Gynecol Cancer. 2007 Jul-Aug;17(4):739-63. Epub 2007 Feb 16. Review. PubMed PMID: 17309673.
19: Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2006 Dec;28(10):719-40. PubMed PMID: 17235418.
20: Lokich J. Same-day pegfilgrastim and chemotherapy. Cancer Invest. 2005;23(7):573-6. PubMed PMID: 16305982.
21: Honig A, Rieger L, Sutterlin A, Kapp M, Dietl J, Sutterlin MW, Kämmerer U. Brain metastases in breast cancer–an in vitro study to evaluate new systemic chemotherapeutic options. Anticancer Res. 2005 May-Jun;25(3A):1531-7. PubMed PMID: 16033055.
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy- 3,14-dioxo1H-pyrano[3’,4’:6,7]-indolizino[1,2-b]quinolin- 9-yl-[1,4’bipiperidine]-1’-carboxylate |
|
| Clinical data | |
| Trade names | Camptosar |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a608043 |
| Pregnancy cat. | D (Australia, United States) |
| Legal status | POM (UK), ℞-only (U.S.) |
| Routes | Intravenous |
| Pharmacokinetic data | |
| Bioavailability | NA |
| Metabolism | Hepatic glucuronidation |
| Half-life | 6 to 12 hours |
| Excretion | Biliary and renal |
| Identifiers | |
| CAS number | 100286-90-6 |
| ATC code | L01XX19 |
| PubChem | CID 60838 |
| DrugBank | DB00762 |
| ChemSpider | 54825 |
| UNII | 7673326042 |
| KEGG | D08086 |
| ChEMBL | CHEMBL481 |
| Chemical data | |
| Formula | C33H38N4O6 e |
| Mol. mass | 586.678 g/mol (Irinotecan) 623.139 g/mol (Irinotecan hydrochloride) 677.185 g/mol (Irinotecan hydrochloride trihydrate)) |
…………..
Irinotecan (Camptosar, Pfizer; Campto, Yakult Honsha) is a drug used for the treatment of cancer.
Irinotecan prevents DNA from unwinding by inhibition of topoisomerase 1.[1] In chemical terms, it is a semisynthetic analogue of the natural alkaloid camptothecin.
Its main use is in colon cancer, in particular, in combination with other chemotherapy agents. This includes the regimen FOLFIRI, which consists of infusional 5-fluorouracil, leucovorin, and irinotecan.
Irinotecan received accelerated approval by the U.S. Food and Drug Administration (FDA) in 1996[2] and full approval in 1998.[3] During development, it was known as CPT-11.
Mechanism
Irinotecan is activated by hydrolysis to SN-38, an inhibitor of topoisomerase I. This is then inactivated by glucuronidation by uridine diphosphate glucoronosyltransferase 1A1 (UGT1A1). The inhibition of topoisomerase I by the active metabolite SN-38 eventually leads to inhibition of both DNA replication and transcription.
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles. [§ 1]
[[File:

|{{{bSize}}}px]]
Irinotecan Pathway edit
- The interactive pathway map can be edited at WikiPathways: “IrinotecanPathway_WP46359”.
Side-effects
The most significant adverse effects of irinotecan are severe diarrhea and extreme suppression of the immune system.
Diarrhea
Irinotecan-associated diarrhea is severe and clinically significant, sometimes leading to severe dehydration requiring hospitalization or intensive care unit admission. This side-effect is managed with the aggressive use of antidiarrheals such as loperamide or Lomotil with the first loose bowel movement.
Immunosuppression
The immune system is adversely impacted by irinotecan. This is reflected in dramatically lowered white blood cell counts in the blood, in particular the neutrophils. The patient may experience a period of neutropenia (a clinically significant decrease of neutrophils in the blood) while the bone marrow increases white cell production to compensate.
Pharmacogenomics
Irinotecan is converted by an enzyme into its active metabolite SN-38, which is in turn inactivated by the enzyme UGT1A1 by glucuronidation.
*28 variant patients
People with variants of the UGT1A1 called TA7, also known as the “*28 variant”, express fewer UGT1A1 enzymes in their liver and often suffer from Gilbert’s syndrome. During chemotherapy, they effectively receive a larger than expected dose because their bodies are not able to clear irinotecan as fast as others. In studies this corresponds to higher incidences of severe neutropenia and diarrhea.[4]
In 2004, a clinical study was performed that both validated prospectively the association of the *28 variant with greater toxicity and the ability of genetic testing in predicting that toxicity before chemotherapy administration.[4]
In 2005, the FDA made changes to the labeling of irinotecan to add pharmacogenomics recommendations, such that irinotecan recipients with a homozygous (both of the two gene copies) polymorphism in UGT1A1 gene, to be specific, the *28 variant, should be considered for reduced drug doses.[5] Irinotecan is one of the first widely used chemotherapy agents that is dosed according to the recipient’s genotype.[6]
Research
Recently it was shown that antitumor activity of irinotecan against glioblastoma can be enhanced by co-treatment with statins.[7] Similarly, it was shown that berberine may enhance chemosensitivity to irinotecan in colon cancercells. [8]
References
- Pommier, Y., Leo, E., Zhang, H., Marchand, C. 2010. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17: 421-433.
- New York Times Article http://www.nytimes.com/1996/06/18/science/new-cancer-drug-approved.html
- FDA Review Letter http://www.accessdata.fda.gov/drugsatfda_docs/appletter/1998/20571s8ltr.pdf
- Innocenti F, Undevia SD, Iyer L, et al. (April 2004). “Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan”. J. Clin. Oncol. 22 (8): 1382–8. doi:10.1200/JCO.2004.07.173. PMID 15007088.
- Camptosar® irinotecan hydrochloride injection August 2010 http://labeling.pfizer.com/ShowLabeling.aspx?id=533
- O’Dwyer PJ, Catalano RB (October 2006). “Uridine diphosphate glucuronosyltransferase (UGT) 1A1 and irinotecan: practical pharmacogenomics arrives in cancer therapy”. J. Clin. Oncol. 24 (28): 4534–8. doi:10.1200/JCO.2006.07.3031. PMID 17008691.
- Jiang PF (Jan 2014). “Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs.”. J Transl Med. 12. doi:10.1186/1479-5876-12-13. PMC 3898565. PMID 24433351.
- Yu M (Jan 2014). “Berberine enhances chemosensitivity to irinotecan in colon cancer via inhibition of NF-κB”. J Mol Med Rep 9 (1): 249–54. doi:10.3892/mmr.2013.1762. PMID 24173769.
- DNA Topoisomerases and Cancer. Yves Pommier, Editor. Human Press. 2012
External links
With Persistence And Phase 3 Win, Amicus Nears First Drug Approval …….Migalastat

Migalastat hydrochloride
CAS Number: 75172-81-5 hydrochloride
CAS BASE….108147-54-2
ABS ROT = (+)
|
Conc: 1 g/100mL; Solv: water ; 589.3 nm; Temp: 24 °C |
IN Van den Nieuwendijk, Adrianus M. C. H.; Organic Letters 2010, 12(17), 3957-3959
3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride (1:1), (2R,3S,4R,5S)-
Molecular Structure:
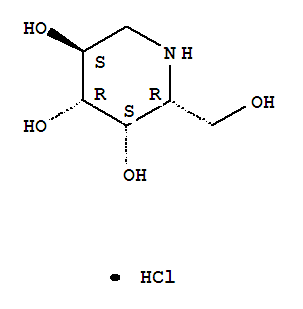
Formula: C6H14ClNO4
Molecular Weight:199.63
Synonyms: 3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, (2R,3S,4R,5S)- (9CI);
3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, [2R-(2a,3a,4a,5b)]-;
Migalastat hydrochloride;Galactostatin hydrochloride;
(2S,3R,4S,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol hydrochloride;
- 1-Deoxygalactonojirimycin
- 1-Deoxygalactostatin
- Amigal
- DDIG
- Migalastat
- UNII-C4XNY919FW
Melting Point:160 °C-162…….http://www.google.com/patents/DE3906463A1?cl=de
Boiling Point:382.7 °C at 760 mmHg
Flash Point:185.2 °C
Amicus Therapeutics, Inc. innovator
Aug 2014
Amicus Therapeutics was on the ropes in late 2012 when its pill for a rare condition called Fabry Disease108147-54-2 failed a late-stage trial. It had already put seven years of work into the drug, and the setback added even more development time and uncertainty to the mix. But the Cranbury, NJ-based company kept plugging away, and now it looks like all the effort could lead to its first approved drug.
Amicus (NASDAQ: FOLD) is reporting today that the Fabry drug, migalastat, succeeded in the second of two late-stage trials. It hit two main goals that essentially measured its ability to slow the decline of Fabry patients’ kidney function comparably to enzyme-replacement therapy (ERT)—the standard of care for the often-fatal disorder.
Amicus believes the results, along with those from an earlier Phase 3 trial comparing migalastat to a placebo, are good enough to ask regulators in the U.S. and Europe for market approval.
“These are the good days to be a CEO,” says Amicus CEO John Crowley (pictured above). “It’s great when a plan comes together and data cooperates.”
Crowley says Amicus will seek approval of migalastat first in Europe and is already in talks with regulators there. In the next few months, Amicus will begin talking with the FDA about a path for approval in the U.S. as well.
End feb 2013
About Amicus Therapeutics
Amicus Therapeutics is a biopharmaceutical company at the forefront of therapies for rare and orphan diseases. The Company is developing orally-administered, small molecule drugs called pharmacological chaperones, a novel, first-in-class approach to treating a broad range of human genetic diseases. Amicus’ late-stage programs for lysosomal storage disorders include migalastat HCl monotherapy in Phase 3 for Fabry disease; migalastat HCl co-administered with enzyme replacement therapy (ERT) in Phase 2 for Fabry disease; and AT2220 co-administered with ERT in Phase 2 for Pompe disease.
About Migalastat HCl
Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease. Amicus has commercial rights to all Fabry products in the United States and GSK has commercial rights to all of these products in the rest of world.
As a monotherapy, migalastat HCl is designed to bind to and stabilize, or “chaperone” a patient’s own alpha-galactosidase A (alpha-Gal A) enzyme in patients with genetic mutations that are amenable to this chaperone in a cell-based assay. Migalastat HCl monotherapy is in Phase 3 development (Study 011 and Study 012) for Fabry patients with genetic mutations that are amenable to this chaperone monotherapy in a cell-based assay. Study 011 is a placebo-controlled study intended primarily to support U.S. registration, and Study 012 compares migalastat HCl to ERT to primarily support global registration.
For patients currently receiving ERT for Fabry disease, migalastat HCl in combination with ERT may improve ERT outcomes by keeping the infused alpha-Gal A enzyme in its properly folded and active form thereby allowing more active enzyme to reach tissues.2Migalastat HCl co-administered with ERT is in Phase 2 (Study 013) and migalastat HCl co-formulated with JCR Pharmaceutical Co. Ltd’s proprietary investigational ERT (JR-051, recombinant human alpha-Gal A enzyme) is in preclinical development.
About Fabry Disease
Fabry disease is an inherited lysosomal storage disorder caused by deficiency of an enzyme called alpha-galactosidase A (alpha-Gal A). The role of alpha-Gal A within the body is to break down specific lipids in lysosomes, including globotriaosylceramide (GL-3, also known as Gb3). Lipids that can be degraded by the action of α-Gal are called “substrates” of the enzyme. Reduced or absent levels of alpha-Gal A activity leads to the accumulation of GL-3 in the affected tissues, including the kidneys, heart, central nervous system, and skin. This accumulation of GL-3 is believed to cause the various symptoms of Fabry disease, including pain, kidney failure, and increased risk of heart attack and stroke.
It is currently estimated that Fabry disease affects approximately 5,000 to 10,000 people worldwide. However, several literature reports suggest that Fabry disease may be significantly under diagnosed, and the prevalence of the disease may be much higher.
2. Benjamin, et al., Molecular Therapy: April 2012, Vol. 20, No. 4, pp. 717–726.
http://clinicaltrials.gov/show/NCT01458119
http://www.docstoc.com/docs/129812511/migalastat-hcl
Migalastat hydrochloride is a pharmacological chaperone in phase III development at Amicus Pharmaceuticals for the oral treatment of Fabry’s disease. Fabry’s disease occurs as the result of an inherited genetic mutation that results in the production of a misfolded alpha galactosidase A (alpha-GAL) enzyme, which is responsible for breaking down globotriaosylceramide (GL-3) in the lysosome. Migalastat acts by selectively binding to the misfolded alpha-GAL, increasing its stability and promoting proper folding, processing and trafficking of the enzyme from the endoplasmic reticulum to the lysosome.
In February 2004, migalastat hydrochloride was granted orphan drug designation by the FDA for the treatment of Fabry’s disease.
The EMEA assigned orphan drug designation for the compound in 2006 for the treatment of the same indication. In 2007, the compound was licensed to Shire Pharmaceuticals by Amicus Therapeutics worldwide, with the exception of the U.S., for the treatment of Fabry’s disease.
In 2009, this license agreement was terminated. In 2010, the compound was licensed by Amicus Therapeutics to GlaxoSmithKline on a worldwide basis to develop, manufacture and commercialize migalastat hydrochloride as a treatment for Fabry’s disease, but the license agreement terminated in 2013.
| Chemical Name: | DEOXYGALACTONOJIRIMYCIN, HYDROCHLORIDE |
| Synonyms: | DGJ;Amigal;Unii-cly7m0xd20;GALACTOSTATIN HCL;DGJ, HYDROCHLORIDE;Migalastat hydrochloride;Galactostatin hydrochloride;DEOXYGALACTONOJIRIMYCIN HCL;1-DEOXYGALACTONOJIRIMYCIN HCL;1,5-dideoxy-1,5-imino-d-galactitol |

………………………..

http://www.google.co.in/patents/WO1999062517A1?cl=en
Example 1
A series of plant alkaloids (Scheme 1, ref. 9) were used for both in vitro inhibition and intracellular enhancement studies of α-Gal A activity. The results of inhibition experiments are shown in Fig. 1 A.

f^

Among the tested compounds, 1-deoxy-galactonojirimycin (DGJ, 5) known as a powerful competitive inhibitor for α-Gal A, showed the highest inhibitory activity with IC50 at 4.7 nM. α-3,4-Di-epi-homonojirimycin (3) was an effective inhibitor with IC50 at 2.9 μM. Other compounds showed moderate inhibitory activity with IC50 ranging from 0.25 mM (6) to 2.6 mM (2). Surprisingly, these compounds also effectively enhanced α-Gal A activity in COS-1 cells transfected with a mutant α-Gal A gene (R301Q), identified from an atypical variant form of Fabry disease with a residual α- Gal A activity at 4% of normal. By culturing the transfected COS-1 cells with these compounds at concentrations cat 3 – 10-fold of IC50 of the inhibitors, α-Gal A activity was enhanced 1.5 – 4-fold (Fig. 1C). The effectiveness of intracellular enhancement paralleled with in vitro inhibitory activity while the compounds were added to the culture medium at lOμM
concentration (Fig. IB).
………………………
WO 2008045015
or http://www.google.com/patents/EP2027137A1?cl=en, http://www.google.com/patents/US7973157?cl=en
This invention relates to a process for purification of imino or amino sugars, such as D-1-deoxygalactonojirimycin hydrochloride (DGJ’HCl). This process can be used to produce multi-kilogram amounts of these nitrogen-containing sugars.
Sugars are useful in pharmacology since, in multiple biological processes, they have been found to play a major role in the selective inhibition of various enzymatic functions. One important type of sugars is the glycosidase inhibitors, which are useful in treatment of metabolic disorders. Galactosidases catalyze the hydrolysis of glycosidic linkages and are important in the metabolism of complex carbohydrates. Galactosidase inhibitors, such as D-I- deoxygalactonojirimycin (DGJ), can be used in the treatment of many diseases and conditions, including diabetes (e.g., U.S. Pat. 4,634,765), cancer (e.g., U.S. Pat. 5,250,545), herpes (e.g. , U.S. Pat. 4,957,926), HIV and Fabry Disease (Fan et al, Nat. Med. 1999 5:1, 112-5).
Commonly, sugars are purified through chromatographic separation. This can be done quickly and efficiently for laboratory scale synthesis, however, column chromatography and similar separation techniques become less useful as larger amounts of sugar are purified. The size of the column, amount of solvents and stationary phase (e.g. silica gel) required and time needed for separation each increase with the amount of product purified, making purification from multi-kilogram scale synthesis unrealistic using column chromatography.
Another common purification technique for sugars uses an ion- exchange resin. This technique can be tedious, requiring a tedious pre-treatment of the ion exchange resin. The available ion exchange resins are also not necessarily able to separate the sugars from salts (e.g., NaCl). Acidic resins tend to remove both metal ions found in the crude product and amino- or imino-sugars from the solution and are therefore not useful. Finding a resin that can selectively remove the metal cations and leave amino- or imino-sugars in solution is not trivial. In addition, after purification of a sugar using an ion exchange resin, an additional step of concentrating the diluted aqueous solution is required. This step can cause decomposition of the sugar, which produces contaminants, and reduces the yield.
U.S. Pats. 6,740,780, 6,683,185, 6,653,482, 6,653,480, 6,649,766, 6,605,724, 6,590,121, and 6,462,197 describe a process for the preparation of imino- sugars. These compounds are generally prepared from hydroxyl-protected oxime intermediates by formation of a lactam that is reduced to the hexitol. However, this process has disadvantages for the production on a multi-kg scale with regard to safety, upscaling, handling, and synthesis complexity. For example, several of the disclosed syntheses use flash chromatography for purification or ion-exchange resin treatment, a procedure that is not practicable on larger scale. One particularly useful imino sugar is DGJ. There are several DGJ preparations disclosed in publications, most of which are not suitable for an industrial laboratory on a preparative scale (e.g., >100 g). One such synthesis include a synthesis from D-galactose (Santoyo-Gonzalez, et al, Synlett 1999 593-595; Synthesis 1998 1787-1792), in which the use of chromatography is taught for the purification of the DGJ as well as for the purification of DGJ intermediates. The use of ion exchange resins for the purification of DGJ is also disclosed, but there is no indication of which, if any, resin would be a viable for the purification of DGJ on a preparative scale. The largest scale of DGJ prepared published is 13 g (see Fred-Robert Heiker, Alfred Matthias Schueller, Carbohydrate Research, 1986, 119-129). In this publication, DGJ was isolated by stirring with ion-exchange resin Lewatit MP 400 (OH“) and crystallized with ethanol. However, this process cannot be readily scaled to multi- kilogram quantities.
Similarly, other industrial and pharmaceutically useful sugars are commonly purified using chromatography and ion exchange resins that cannot easily be scaled up to the purification of multi-kilogram quantities.
Therefore, there is a need for a process for purifying nitrogen- containing sugars, preferably hexose amino- or imino-sugars that is simple and cost effective for large-scale synthesis
FIG. 1. HPLC of purified DGJ after crystallization. The DGJ is over 99.5% pure.

FIG. 2A. 1H NMR of DGJ (post HCl extraction and crystallization), from 0 – 15 ppm in DMSO.

FIG. 2B. 1H NMR of DGJ (post HCl extraction and crystallization), from 0 – 5 ppm, in DMSO.

FIG. 3 A. 1H NMR of purified DGJ (after recrystallization), from 0 – 15 ppm, in D2O. Note OH moiety has exchanged with OD.

FIG. 3B. 1H NMR of purified DGJ (after recrystallization), from 0 –
4 ppm, in D2O. Note OH moiety has exchanged with OD.

FIG. 4. 13C NMR of purified DGJ, (after recrystallization), 45 – 76 ppm.

One amino-sugar of particular interest for purification by the method of the current invention is DGJ. DGJ, or D-l-deoxygalactonojirimycin, also described as (2R,3S,4R,5S)-2-hydroxymethyl-3,4,5-trihydroxypiperidine and 1- deoxy-galactostatin, is a noj irimycin (5-amino-5-deoxy-D-galactopyranose) derivative of the form:

Example 1: Preparation and Purification of DGJ
A protected crystalline galactofuranoside obtained from the technique described by Santoyo-Gonzalez. 5-azido-5-deoxy-l,2,3,6-tetrapivaloyl-α-D- galactofuranoside (1250 g), was hydrogenated for 1-2 days using methanol (10 L) with palladium on carbon (10%, wet, 44 g) at 50 psi of H2. Sodium methoxide (25% in methanol, 1.25 L) was added and hydrogenation was continued for 1-2 days at 100 psi ofH2. Catalyst was removed by filtration and the reaction was acidified with methanolic hydrogen chloride solution (20%, 1.9 L) and concentrated to give crude mixture of DGJ • HCl and sodium chloride as a solid. The purity of the DGJ was about 70% (w/w assay), with the remaining 30% being mostly sodium chloride.
The solid was washed with tetrahydrofuran (2 x 0.5 L) and ether (I x 0.5 L), and then combined with concentrated hydrochloric acid (3 L). DGJ went into solution, leaving NaCl undissolved. The obtained suspension was filtered to remove sodium chloride; the solid sodium chloride was washed with additional portion of hydrochloric acid (2 x 0.3 L). All hydrochloric acid solution were combined and slowly poured into stirred solution of tetrahydrofuran (60 L) and ether (11.3 L). The precipitate formed while the stirring was continued for 2 hours. The solid crude DGJ* HCl, was filtered and washed with tetrahydrofuran (0.5 L) and ether (2 x 0.5 L). An NMR spectrum is shown in FIGS. 2A-2B.
The solid was dried and recrystallized from water (1.2 mL /g) and ethanol (10 ml/1 ml of water). This recrystallization step may be repeated. This procedure gave white crystalline DGJ* HCl, and was usually obtained in about 70- 75% yield (320 – 345 g). The product of the purification, DGJ-HCl is a white crystalline solid, HPLC >98% (w/w assay) as shown in FIG. 1. FIGS. 3A-3D and FIG. 4 show the NMR spectra of purified DGJ, showing the six sugar carbons.
Example 2: Purification of 1-deoxymannojirimycin 1 -deoxymannojirimycin is made by the method described by Mariano
(J. Org. Chem., 1998, 841-859, see pg. 859, herein incorporated by reference). However, instead of purification by ion-exchange resin as described by Mariano, the 1-deoxymannojirimycin is mixed with concentrated HCl. The suspension is then filtered to remove the salt and the 1-deoxymannojirimycin hydrochloride is precipitated crystallized using solvents known for recrystallization of 1- deoxymannojirimycin (THF for crystallization and then ethanol/water.
Example 3: Purification of (+)-l-deoxynojirimycin
(+)-l-deoxynojirimycin is made by the method Kibayashi et al. (J. Org. Chem., 1987, 3337-3342, see pg. 334I5 herein incorporated by reference). It is synthesized from a piperidine compound (#14) in HCl/MeOH. The reported yield of 90% indicates that the reaction is essentially clean and does not contain other sugar side products. Therefore, the column chromatography used by Kibayashi is for the isolation of the product from non-sugar related impurities. Therefore, instead of purification by silica gel chromatography, the (+)-l-deoxynojirimycin is mixed with concentrated HCl. The suspension is then filtered to remove the salt and the nojirimycin is crystallized using solvents known for recrystallization of nojirimycin.
Example 4: Purification of Nojirimycin
Nojirimycin is made by the method described by Kibayashi et al. (J.
Org. Chem., 1987, 3337-3342, see pg. 3342). However, after evaporating of the mixture at reduced pressure, instead of purification by silica gel chromatography with ammonia-methanol-chloroform as described by Kibayashi, the nojirimycin is mixed with concentrated HCl. The suspension is then filtered to remove the impurities not dissolved in HCl and the nojirimycin is crystallized using solvents known for recrystallization of nojirimycin.
……………………….
Synthesis of (+)-1-deoxygalactonojirimycin and a related indolizidine
Tetrahedron Lett 1995, 36(5): 653
-
Synthesis of (+)-1-deoxygalactonojirimycin and a related indolizidine
Original Research Article
- Pages 653-654
- Carl R. Johnson, Adam Golebiowski, Hari Sundram, Michael W. Miller, Renee L. Dwaihy
-
Amido-alcohol 1 is transformed via aminal 2 into 1-deoxygalactonojirimycin (3) and indolizidine 4.

Amido-alcohol 1 is transformed via aminal 2 into 1-deoxygalactonojirimycin (3) and the structurally related indolizidine 4.
………………………
Synthesis of D-galacto-1-deoxynojirimycin (1,5-dideoxy-1,5-imino-D-galactitol) starting from 1-deoxynojirimycin
Carbohydr Res 1990, 203(2): 314
-
Synthesis of d-galacto-1-deoxynojirimycin (1,5-dideoxy-1,5-imino-d-galactitol) starting from 1-deoxynojirimycin
- Pages 314-318
- Fred-Robert Heiker, Alfred Matthias Schueller
………………………………..
Synthesis of (+)-1,5-dideoxy-1,5-imino-D-galactitol, a potent alpha-D-galactosidase inhibitor
Carbohydr Res 1987, 167: 305
-
Synthesis of (+)-1,5-dideoxy-1,5-imino-d-galactitol, a potent α-d-galactosidase inhibitor
- Pages 305-311
- Ronald C. Bernotas, Michael A. Pezzone, Bruce Ganem
……………………………..
SEE
Monosaccharides containing nitrogen in the ring, XXXVII. Synthesis of 1,5-didexy-1,5-imino-D-galactitol
Chem Ber 1980, 113(8): 2601
…………………………
Org. Lett., 2010, 12 (17), pp 3957–3959
DOI: 10.1021/ol101556k
http://pubs.acs.org/doi/abs/10.1021/ol101556k
|
Conc: 1 g/100mL; Solv: water ; 589.3 nm; Temp: 24 °C |
IN
van den Nieuwendijk, Adrianus M. C. H.; Organic Letters 2010, 12(17), 3957-3959

The chemoenzymatic synthesis of three 1-deoxynojirimycin-type iminosugars is reported. Key steps in the synthetic scheme include a Dibal reduction−transimination−sodium borohydride reduction cascade of reactions on an enantiomerically pure cyanohydrin, itself prepared employing almond hydroxynitrile lyase (paHNL) as the common precursor. Ensuing ring-closing metathesis and Upjohn dihydroxylation afford the target compounds.
http://pubs.acs.org/doi/suppl/10.1021/ol101556k/suppl_file/ol101556k_si_002.pdf
COMPD 18
D-galacto-1-deoxynojirimicin.HCl (18).
D-N-Boc-6-OBn-galacto-1-deoxynojirimicin (159 mg, 0.450 mmol) was dissolved in a mixture of MeOH
(10 mL) and 6 M HCl (2 mL). The flask was purged with argon, Pd/C-10% (20 mg) was added and a balloon
with hydrogen gas was placed on top of the reaction. The mixture was stirred overnight at room temperature.
Pd/C was removed by filtration and the filtrate evaporated to yield the crude product (90 mg, 100%) as a
white foam that needed no further purification.
[α]24D = + 53.0 (c = 1, H2O);
[lit4a [α]24D = +44.6 (c = 0.9, H2O); lit4b [α]20D = +46.1 (c = 0.9, H2O)].
HRMS calculated for [C6H13NO4 + H]+164.09173; Found 164.09160.
1H NMR (400 MHz, D2O) δ 4.20 (dd, J = 2.7, 1.1 Hz, 1H), 4.11 (ddd, J = 11.4, 9.7, 5.4 Hz, 1H), 3.88 (ddd,
J = 20.9, 12.2, 6.8 Hz, 2H), 3.68 (dd, J = 9.7, 3.0 Hz, 1H), 3.55 (dd, J = 12.5, 5.4 Hz, 1H), 3.46 (ddd, J = 8.6,
4.8, 1.0 Hz, 1H), 2.97 – 2.86 (t, J = 12.0 Hz, 1H). [lit4c supporting information contains 1
H NMR-spectrumof an authentic sample].
13C NMR (101 MHz, D2O) δ 73.01, 66.97, 64.69, 60.16, 59.15, 46.15
4a) Ruiz, M.; Ruanova, T. M.; Blanco, O.; Núñez, F.; Pato, C.; Ojea, V. J. Org. Chem. 2008, 73, 2240
– 2255.
4b) Paulsen, H.; Hayauchi, Y.; Sinnwell, V. Chem. Ber. 1980, 113, 2601 – 2608. c)
McDonnell, C.; Cronin, L.; O’Brien, J. L.; Murphy, P. V. J. Org. Chem. 2004, 69, 3565 – 3568.
……………………………………….
(- ) FORM………… BE CAREFUL
Short and straightforward synthesis of (-)-1-deoxygalactonojirimycin
Org Lett 2010, 12(6): 1145
http://pubs.acs.org/doi/abs/10.1021/ol100037c

The mildness and low basicity of vinylzinc species functioning as a nucleophile in addition to α-chiral aldehydes is characterized by lack of epimerization of the vulnerable stereogenic center. This is demonstrated by a highly diastereoselective synthesis of 1-deoxygalactonojirimycin in eight steps from commercial starting materials with overall yield of 35%.

Figure 1. Structures of nojirimycin (1) and DGJ (2).
SEE SUPP INFO
http://pubs.acs.org/doi/suppl/10.1021/ol100037c/suppl_file/ol100037c_si_001.pdf
(-)-1-deoxygalactojirimycin hydrochloride as transparent colorless needles.
[α]D -51.4 (D2O, c 1.0)
1H-NMR (D2O) δ ppm 4.09 (dd, 1H, J 2.9 Hz, 1.3 Hz), 4.00 (ddd, 1H, J = 11.3 Hz, 9.7 Hz, 5.3 Hz),
3.80 (dd, 1H, J = 12,1 Hz, 8.8 Hz), 3.73 (dd, 1H, J = 12.1 Hz, 8.8 Hz), 3.56 (dd, 1H, J = 9.7 Hz, 2.9
Hz), 3.44 (dd, 1H, J = 12.4 Hz, 5.3 Hz), 3.34 (ddd, 1H, J = 8.7 Hz, 4.8 Hz, 1.0 Hz), 2.8 (app. t, 1H,
J = 12.0 Hz)
13C-NMR (D2O, MeOH iSTD) δ 73.6, 67.5, 65.3, 60.7, 59.7, 46.7
HRMS Measured 164.0923 (M + H – Cl) Calculated 164.0923 (C6H13NO4 + H – Cl)
…………………………………………………..
Concise and highly stereocontrolled synthesis of 1-deoxygalactonojirimycin and its congeners using dioxanylpiperidene, a promising chiral building block
Org Lett 2003, 5(14): 2527
http://pubs.acs.org/doi/abs/10.1021/ol034886y

A concise and stereoselective synthesis of the chiral building block, dioxanylpiperidene 4 as a precursor for deoxyazasugars, starting from the Garner aldehyde 5 using catalytic ring-closing metathesis (RCM) for the construction of the piperidine ring is described. The asymmetric synthesis of 1-deoxygalactonojirimycin and its congeners 1−3 was carried out via the use of 4in a highly stereocontrolled mode.
mp 135-135.5 °C [lit.3mp 137-139 °C];
[α]D25 +27.8° (c 0.67, H2O)
[lit.3[α]D23 +28° (c 0.5, H2O)];
1H NMR (300 MHz, D2O) δ 2.59–2.65 (m, 1H), 2.81–2.87 (m, 1H),
3.02–3.08 (m, 1H), 3.46–3.48 (m, 2H), 3.59–3.66 (m, 3H); 13C NMR (75 MHz, D2O) δ 44.7, 57.1,
58.4, 70.9, 71.4, 73.3 [lit4 13C NMR (125 MHz, D2O) δ 44.5, 56.8, 58.3, 70.1, 70.7, 72.3];
HRMScalcd for C6H13NO4 (M+) 163.0855, Found 163.0843. Anal. calcd for C6H13NO4: C, 44.16; N,
8.58; H, 8.03. Found: C, 44.31; N, 8.55; H, 7.71.
3. Schaller, C.; Vogel, P.; Jager, V. Carbohydrate Res. 1998, 314, 25-35.
4. Lee, B. W.; Jeong, Ill-Y.; Yang, M. S.; Choi, S. U.; Park, K. H. Synthesis 2000, 1305-1309.
…………………………………………..
Applications and limitations of the I2-mediated carbamate annulation for the synthesis of piperidines: Five- versus six-membered ring formation
J Org Chem 2013, 78(19): 9791
http://pubs.acs.org/doi/abs/10.1021/jo401512h

A protecting-group-free synthetic strategy for the synthesis of piperidines has been explored. Key in the synthesis is an I2-mediated carbamate annulation, which allows for the cyclization of hydroxy-substituted alkenylamines into piperidines, pyrrolidines, and furans. In this work, four chiral scaffolds were compared and contrasted, and it was observed that with both d-galactose and 2-deoxy-d-galactose as starting materials, the transformations into the piperidines 1-deoxygalactonorjirimycin (DGJ) and 4-epi-fagomine, respectively, could be achieved in few steps and good overall yields. When d-glucose was used as a starting material, only the furan product was formed, whereas the use of 2-deoxy-d-glucose resulted in reduced chemo- and stereoselectivity and the formation of four products. A mechanistic explanation for the formation of each annulation product could be provided, which has improved our understanding of the scope and limitations of the carbamate annulation for piperidine synthesis.
……………………………………………
Ruiz, Maria; Journal of Organic Chemistry 2008, 73(6), 2240-2255
http://pubs.acs.org/doi/abs/10.1021/jo702601z
ROT +44.6 ° Conc: 0.9 g/100mL; Solv: water ; 589.3 nm; Temp: 24 °C

A general strategy for the synthesis of 1-deoxy-azasugars from a chiral glycine equivalent and 4-carbon building blocks is described. Diastereoselective aldol additions of metalated bislactim ethers to matched and mismatched erythrose or threose acetonides and intramolecular N-alkylation (by reductive amination or nucleophilic substitution) were used as key steps. The dependence of the yield and the asymmetric induction of the aldol addition with the nature of the metallic counterion of the azaenolate and the γ-alkoxy protecting group for the erythrose or threose acetonides has been studied. The stereochemical outcome of the aldol additions with tin(II) azaenolates has been rationalized with the aid of density functional theory (DFT) calculations. In accordance with DFT calculations with model glyceraldehyde acetonides, hightrans,syn,anti-selectivitity for the matched pairs and moderate to low trans,anti,anti-selectivity for the mismatched ones may originate from (1) the intervention of solvated aggregates of tin(II) azaenolate and lithium chloride as the reactive species and (2) favored chair-like transition structures with a Cornforth-like conformation for the aldehyde moiety. DFT calculations indicate that aldol additions to erythrose acetonides proceed by an initial deprotonation, followed by coordination of the alkoxy-derivative to the tin(II) azaenolate and final reorganization of the intermediate complex through pericyclic transition structures in which the erythrose moiety is involved in a seven-membered chelate ring. The preparative utility of the aldol-based approach was demonstrated by application in concise routes for the synthesis of the glycosidase inhibitors 1-deoxy-d-allonojirimycin, 1-deoxy-l-altronojirimycin, 1-deoxy-d-gulonojirimycin, 1-deoxy-d-galactonojirimycin, 1-deoxy-l-idonojirimycin and 1-deoxy-d-talonojirimycin.
…………………..
J. Org. Chem., 1991, 56 (2), pp 815–819
DOI: 10.1021/jo00002a057
http://pubs.acs.org/doi/abs/10.1021/jo00002a057
………………
Hinsken, Werner; DE 3906463 A1 1990
http://www.google.com/patents/DE3906463A1?cl=de
Example 1 Preparation of 1,5-dideoxy-1,5-imino-D-glucitol hydrobromide
A suspension of 1,5-dideoxy-1,5-imino-D-glucitol (500 g) in isopropanol (2 l) with 48% hydrochloric acid, bromine (620 g). The suspension is stirred for 2 hours at 40 ° C, cooled to 0 ° C and the product isolated by filtration.
Yield: 700 g (93% of theory),
mp: 184 ° C.
Example 2 Preparation of 1,5-dideoxy-1,5-imino-D-mannitol hydrobromide
The prepared analogously to Example 1 from 1,5-dideoxy 1,5-imino-D-mannitol and 48% hydrobromic acid.
Yield: 89% of theory;
C₆H₁₄NO₄Br (244.1)
Ber .: C 29.5%; H 5.8%; N 5.7%; Br 32.7%;
vascular .: C 29.8%; H 5.8%; N 5.8%; Br 32.3%.
Example 3 Preparation of 1,5-dideoxy-1,5-imino-D-Galactitol- hydrochloride
The preparation was carried out analogously to Example 1 from 1,5-dideoxy-1,5-imino-D-galactitol and corresponding mole ratios of 37% hydrochloric acid.
yield: 91% of theory
, mp: 160-162 ° C.
| Amat et al., “Eantioselective Synthesis of 1-deoxy-D-gluonojirimycin From A Phenylglycinol Derived Lactam,” Tetrahedron Letters, pp. 5355-5358, 2004. | ||
| 2 | Chernois, “Semimicro Experimental Organic Chemistry,” J. de Graff (1958), pp. 31-48. | |
| 3 | Encyclopedia of Chemical Technology, 4th Ed., 1995, John Wiley & Sons, vol. 14: p. 737-741. | |
| 4 | Heiker et al., “Synthesis of D-galacto-1-deoxynojirimycin (1, 5-dideoxy-1, 5-imino-D-galactitol) starting from 1-deoxynojirimycin.” Carbohydrate Research, 203: 314-318, 1990. | |
| 5 | Heiker et al., 1990, “Synthesis of D-galacto-1-deoxynojirimycin (1,5-dideoxy-1, 5-imino-D-galactitol) starting from 1-deoxynojirimycin,” Carbohydrate Research, vol. 203: p. 314-318. | |
| 6 | * | Joseph, Carbohydrate Research 337 (2002) 1083-1087. |
| 7 | * | Kinast et al. Angew. Chem. Int. Ed. Engl. 20 (1998), No. 9, pp. 805-806. |
| 8 | * | Lamb, Laboratory Manual of General Chemistry, Harvard University Press, 1916, p. 108. |
| 9 | Linden et al., “1-Deoxynojirimycin Hydrochloride,” Acta ChrystallographicaC50, pp. 746-749, 1994. | |
| 10 | Mellor et al., Preparation, biochemical characterization and biological properties of radiolabelled N-alkylated deoxynojirimycins, Biochem. J. Aug. 15, 2002; 366(Pt 1):225-233. | |
| 11 | * | Mills, Encyclopedia of Reagents for Organic Synthesis, Hydrochloric Acid, 2001 John Wily & Sons. |
| 12 | Santoyo-Gonzalez et al., “Use of N-Pivaloyl Imidazole as Protective Reagent for Sugars.” Synthesis 1998 1787-1792. | |
| 13 | Schuller et al., “Synthesis of 2-acetamido-1, 2-dideoxy-D-galacto-nojirimycin (2-acetamido-1, 2, 5-trideoxy-1, 5-imino-D-galacitol) from 1-deoxynojirimycin.” Carbohydrate Res. 1990; 203: 308-313. | |
| 14 | Supplementary European Search Report dated Mar. 11, 2010 issued in corresponding European Patent Application No. EP 06 77 2888. | |
| 15 | Uriel et al., A Short and Efficient Synthesis of 1,5-dideoxy-1,5-imino-D-galactitol (1-deoxy-D-galactostatin) and 1,5-dideoxy-1,5-dideoxy-1,5-imino-L-altritol (1-deoxy-L-altrostatin) From D-galactose, Synlett (1999), vol. 5, pp. 593-595. |
1-Deoxygalactonojirimycin:
(a) Liguchi, T.; Tajiri, K.; Ninomiya, I.; Naito, T. Tetrahedron2000, 56, 5819−5833.
(b) Mehta, G.; Mohal, N. Tetrahedron Lett. 2000, 41, 5741−5745.
(c) Asano, K.; Hakogi, T.; Iwama, S.; Katsumura, S. Chem. Commun. 1999, 41−42.
(d) Johnson, C. R.; Golebiowsky, A.; Sundram, H.; Miller, M. W.; Dwaihy, R. L. TetraherdonLett. 1995, 36, 653−654.
(e) Uriel, C.; Santoyo-Gonzalez, F. Synlett 1999, 593−595.
(f) Ruiz, M.; Ruanova, T. M.; Ojea, V.; Quintela, J. M. Tetrahedron Lett. 1999, 40, 2021−2024.
(g) Shilvock, J. P.; Fleet, G. W. J. Synlett 1998, 554−556.
(h) Chida, N.; Tanikawa, T.; Tobe, T.; Ogawa, S. J. Chem. Soc., Chem. Commun. 1994, 1247−1248.
(i) Aoyagi, S.; Fujimaki, S.; Yamazaki, N.; Kibayashi, C. J. Org. Chem. 1991, 56, 815−819.
(j) Kajimoto, T.; Chen, L.; Liu, K. K. C.; Wong, C. H. J. Am. Chem. Soc. 1991, 113, 6678−6680.
(k) Bernotas, R. C.; Pezzone, M. A.; Ganem, B. Carbohydr. Res. 1987, 167, 305−311. 1-Deoxyidonojirimycin:
(l) Singh, O. V.; Han, H. Tetrahedron Lett. 2003, 44, 2387−2391.
(m) Schaller, C.; Vogel, P.; Jager, V. Carbohydr. Res. 1998, 314, 25−35.
(n) Fowler, P. A.; Haines, A. H.; Taylor, R. J. K.; Chrystal, E. J. T.; Gravestock, M. B. Carbohydr. Res. 1993,246 377−381.
(o) Liu, K. K. C.; Kajimoto, T.; Chen, L.; Zhong, Z.; Ichikawa, Y.; Wong, C. H.J. Org. Chem. 1991, 56, 6280−6289. 1-Deoxygulonojirimycin: ref 5l.
(p) Haukaas, M. H.; O’Doherty, G. A. Org. Lett. 2001, 3, 401−404.
(q) Ruiz, M.; Ojea, V.; Ruanova, T. M.; Quintela, J. M. Tetrahedron: Asymmetry 2002, 13, 795−799. (r) Liao, L.-X.; Wang, Z.-M.; Zhang, H.-X.; Zhou, W.-S. Tetrahedron: Asymmetry 1999, 10, 3649−3657.
Indian Sterile Manufacturer receives FDA Warning Letter and changes company name from Marck Biosience to Amanta Healthcare
DRUG REGULATORY AFFAIRS INTERNATIONAL

Marck Biosciences Ltd. to “Amanta Healthcare Ltd.”, effective from June 24, 2014. Click here to visit website at
|
| Marck Biosciences Limited is a producer of sterile products which has been producing sterile products for the US market. The FDA Warning Letter dated July 8, 2014 contains shocking details about the GMP situation at this facility. Read more here about the FDA Warning Letter to Marck Biosciences and about the name change to Amanta Healthcare. |
Which SOPs are required by GMP?
DRUG REGULATORY AFFAIRS INTERNATIONAL

ECA is receiving a lot of questions on SOPs (Standard Operating Procedures) needed in a GMP environment. The most interesting is the one on which SOPs are required by law. Here is an Overview.

GMP News: Which SOPs are required by GMP?
|
View original post 608 more words
Drug from Mediterranean weed kills tumor cells in mice – Thapsia garganica
Just a three-day course reduced the size of human prostate tumors grown in mice by an average of 50 percent within 30 days
‘Molecular grenade’ now in clinical trials for advanced cancer
12 JUL 2012
Scientists at the Johns Hopkins Kimmel Cancer Center, working with Danish researchers, have developed a novel anticancer drug designed to travel — undetected by normal cells — through the bloodstream until activated by specific cancer proteins. The drug, made from a weedlike plant, has been shown to destroy cancers and their direct blood supplies, acting like a “molecular grenade,” and sparing healthy blood vessels and tissues.
In laboratory studies, researchers said they found that a three-day course of the drug, called G202, reduced the size of human prostate tumors grown in mice by an average of 50 percent within 30 days. In a direct comparison, G202 outperformed the chemotherapy drug docetaxel, reducing seven of nine human prostate…
View original post 572 more words
FDA approves new drug Cerdelga (eliglustat) to treat a form of Gaucher disease

August 19, 2014
Release
The U.S. Food and Drug Administration today approved Cerdelga (eliglustat) for the long-term treatment of adult patients with the Type 1 form of Gaucher disease, a rare genetic disorder.
Gaucher disease occurs in people who do not produce enough of an enzyme called glucocerebrosidase. The enzyme deficiency causes fatty materials to collect in the spleen, liver and bone marrow. The major signs of Gaucher disease include liver and spleen enlargement, low red blood cell counts (anemia), low blood platelet counts and bone problems.
Cerdelga is a hard gelatin capsule containing eliglustat that is taken orally. In patients with Gaucher disease Type 1, the drug slows down the production of the fatty materials by inhibiting the metabolic process that forms them. Type 1 Gaucher disease is estimated to affect about 6,000 people in the United States.
“Today’s approval offers another important treatment option for patients with Type 1 Gaucher disease,” said Amy G. Egan, M.D., M.P.H., deputy director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research. “In addition, Cerdelga received orphan drug designation from the FDA, reflecting the agency’s focus and commitment to the development of treatments for rare diseases.”
The safety and effectiveness of Cerdelga were evaluated in two clinical trials with 199 participants with Type 1 Gaucher disease.
In one randomized, double-blind, placebo-controlled, multicenter clinical trial the safety and effectiveness of Cerdelga were evaluated in 40 participants with Type 1 Gaucher’s disease who had not previously received enzyme replacement therapy. Subjects received the drug at a starting dose of 42 mg two times a day, with most receiving a dose of 84 mg two times a day after four weeks. Study participants continued the drug for nine months.
Compared to placebo, treatment with Cerdelga resulted in a greater reduction in spleen volume from baseline to the end of the study (by the 39th week), the trial’s primary endpoint. Cerdelga also resulted in greater improvement in liver volume, blood platelet count, and red blood cell (hemoglobin) level, compared to placebo.
The other trial sought to determine the safety and effectiveness of Cerdelga compared to enzyme replacement therapy in 159 participants with Type 1 Gaucher disease previously treated and stabilized on enzyme replacement therapy. Subjects in the trial received either the enzyme replacement therapy drug imiglucerase or Cerdelga. The trial demonstrated that treatment with Cerdelga resulted in similar stabilization of hemoglobin level, platelet count and spleen and liver volume as imiglucerase.
The most commonly observed side effects in the Cerdelga clinical trials were fatigue, headache, nausea, diarrhea, back pain, pain in extremities, and upper abdominal pain.
Cerdelga is manufactured by Cambridge, Massachusetts-based Genzyme.
read synthesis at
https://newdrugapprovals.org/2013/12/14/the-us-food-and-drug-administration-fda-has-granted-a-six-month-priority-review-designation-to-genzymes-new-drug-application-nda-for-cerdelga-eliglustat/ – See more at: http://worlddrugtracker.blogspot.in/#sthash.tJzVgHVT.dpuf
Wednesday, 20 August 2014 Glenmark enters Oncology with the Discovery and the Initiation of IND enabling Studies of an innovative bispecific Antibody
![]()
August 20, 2014: Glenmark Pharmaceuticals S.A. (GPSA), a wholly owned subsidiary of Glenmark Pharmaceuticals Limited India (GPL), announces the discovery and initiation of IND enabling studies of a novel clinical development candidate, GBR 1302, a HER2xCD3 bispecific antibody. GBR 1302 was discovered and developed by the Glenmark Biologics Research Centre located in La Chaux-de-Fonds, Switzerland. GBR 1302 is based on Glenmark’s innovative BEAT antibody technology platform which facilitates the efficient development and manufacture of antibodies with dual specificities, so-called bispecific antibodies. GBR 1302 is the first clinical development candidate based on the BEAT technology. Glenmark expects to obtain approval for the initiation of clinical studies during this financial year.
·GBR 1302 is the first bispecific antibody based on Glenmark’s proprietary BEAT platform
- GBR 1302 is Glenmark’s first clinical candidate targeting oncology indications
Glenmark Pharmaceuticals announced the discovery and initiation of IND enabling studies of a novel clinical development candidate, GBR 1302, a HER2xCD3 bispecific antibody. GBR 1302 was discovered and developed by the Glenmark Biologics Research Centre located in La Chaux-de-Fonds, Switzerland. GBR 1302 is based on Glenmark’s innovative BEAT antibody technology platform which facilitates the efficiend development and manufacture of antibodies with dual specificities, so-called bispecific antibodies. GBR 1302 is the first clinical development candidate based on the BEAT technology.Glenmark expect to obtain approval for the initiation of clinical studies during this financial year.
read at
– See more at: http://worlddrugtracker.blogspot.in/#sthash.tJzVgHVT.dpuf
Antimicrobial resistance

Antimicrobials are medicines that kill or inactivate microbes, small disease-causing organisms. They include antibiotics, which are used against bacteria. After being exposed to an antimicrobial repeatedly, microbes can undergo changes that stop them being killed or inactivated by the treatments. This is known as antimicrobial resistance.
The European Medicines Agency is concerned about the development of antimicrobial resistance, particularly resistance to antibiotics. A well-known example of a bacterium that is resistant to a number of antibiotics is meticillin-resistant Staphylococcus aureus(MRSA), which has caused infections that are difficult to treat across the European Union (EU).

This problem is being made worse by the fact that few new antimicrobials have been authorised over the past few years. This may lead to infections becoming more difficult to treat in the future.
Antimicrobial resistance is a growing problem in humans and in animals. Resistance can also spread from animals to humans through the food chain or direct contact.
The role of the Agency
The Agency works in collaboration with its EU and international partners in a number of initiatives aiming to limit the development of resistance. It is also monitoring and evaluating the risks to human and animal health.
A major such initiative is the Transatlantic Task Force on Antimicrobial Resistance![]() (TATFAR), which was established following the EU-United States summit in November 2009. The Task Force aims to increase levels of communication, coordination and co-operation between the EU and the United States on human and veterinary antimicrobials.
(TATFAR), which was established following the EU-United States summit in November 2009. The Task Force aims to increase levels of communication, coordination and co-operation between the EU and the United States on human and veterinary antimicrobials.

Human health
In human medicine, the availability of medicines to treat infections with resistant organisms has become a major problem in recent years.
In September 2009, the Agency published a joint report together with the European Centre for Disease Prevention and Control![]() (ECDC) and the international network ReAct – Action on Antibiotic Resistance
(ECDC) and the international network ReAct – Action on Antibiotic Resistance![]() . This report highlights the gap between infections due to resistant bacteria and the development of new antibiotics.
. This report highlights the gap between infections due to resistant bacteria and the development of new antibiotics.

The report states that at least 25,000 patients in the EU die each year from infections due to bacteria that are resistant to many medicines, and that infections due to these bacteria in the EU result in extra healthcare costs and productivity losses of at least €1.5 billion each year. Although it identified 15 antibiotics under development, most of these were early in development and were targeted against bacteria for which treatment options were already available.
Authorisation of new antibiotics
The Agency plays a key role in the authorisation of new antibiotics, because medicines with a significant therapeutic innovation or that are in the interest of public or animal health are authorised centrally in the EU, on the recommendation of the Agency.
In January 2012, the Agency updated its guidance to companies developing antibiotics, covering how they should carry out studies to test these medicines’ benefits and risks:
This is accompanied by an addendum giving information on how to study medicines for specific indications. The final addendum was published in November 2013 following a public consultation:

Animal health
The Agency is focused on promoting the prudent use of antimicrobials in animals, to limit the development of resistance. This goal is addressed in this document:
In line with this strategy, the Agency published a revised version of its guideline onefficacy for public consultation in May 2013. This draft guideline provides detailed recommendations for the design and conduct of pre-clinical and clinical studies to support clinical efficacy for antimicrobial veterinary products:
Since early 2010, the Agency has been leading a project collecting information on the sale of veterinary antimicrobials across the EU:

The CVMP has also published a large number of documents on microbial resistance in animals and its risks for humans.
Reports published by the Agency together with other European bodies, including ECDC, EFSA and the European Commission’s Scientific Committee on Emerging and Newly Identified Health Risks![]() (SCENIHR) have emphasised the need for the prudent use of antibiotics in animals and the role of basic hygiene, and called for strengthened surveillance of resistance, the development of new antimicrobials and new strategies to combat the spread of resistance:
(SCENIHR) have emphasised the need for the prudent use of antibiotics in animals and the role of basic hygiene, and called for strengthened surveillance of resistance, the development of new antimicrobials and new strategies to combat the spread of resistance:
- Joint scientific report of ECDC, EFSA and the European Medicines Agency on MRSA in livestock, companion animals and food
- Joint opinion on antimicrobial resistance focused on zoonotic infections
In 2013 and 2014, the Agency carried out a large body of work to provide advice to the European Commission on the use of antibiotics in animals and the impact on public health and animal health.
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

