| FDA Orange Book Patents: 1 of 2 | |
|---|---|
| Patent | 7459561 |
| Expiration | Oct 31, 2020 |
| Applicant | ASTELLAS |
| Drug Application | N207500 (Prescription Drug: CRESEMBA. Ingredients: ISAVUCONAZONIUM SULFATE) |
from FDA Orange Book
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusDRUG REGULATORY AFFAIRS INTERNATIONAL

At the beginning of each year the FDA always publishes a list of the guidances it plans to publish during that year. It has done so again in 2015. The document is relatively comprehensive, containing five pages. Find out more about the Guidances the FDA plans on publishing in 2015.
At the beginning of each year the FDA always publishes a list of the guidances it plans to publish during that year. It has done so again in 2015. The document is relatively comprehensive, containing five pages. The list is subdivided into different categories. It contains for example also guidances planned in connection with the topics Clinical Pharmacology or Clinical/Statistical.
CGMP is a category of its own for which “only” three new guidances are planned for 2015:
View original post 190 more words

![]()
March 6, 2015
The U.S. Food and Drug Administration today approved Zarxio (filgrastim-sndz), the first biosimilar product approved in the United States.
Biological products are generally derived from a living organism. They can come from many sources, including humans, animals, microorganisms or yeast.
A biosimilar product is a biological product that is approved based on a showing that it is highly similar to an already-approved biological product, known as a reference product. The biosimilar also must show it has no clinically meaningful differences in terms of safety and effectiveness from the reference product. Only minor differences in clinically inactive components are allowable in biosimilar products.
Sandoz, Inc.’s Zarxio is biosimilar to Amgen Inc.’s Neupogen (filgrastim), which was originally licensed in 1991. Zarxio is approved for the same indications as Neupogen, and can be prescribed by a health care professional for:
“Biosimilars will provide access to important therapies for patients who need them,” said FDA Commissioner Margaret A. Hamburg, M.D. “Patients and the health care community can be confident that biosimilar products approved by the FDA meet the agency’s rigorous safety, efficacy and quality standards.”
The Biologics Price Competition and Innovation Act of 2009 (BPCI Act) was passed as part of the Affordable Care Act that President Obama signed into law in March 2010. The BPCI Act created an abbreviated licensure pathway for biological products shown to be “biosimilar” to or “interchangeable” with an FDA-licensed biological product, called the “reference product.” This abbreviated licensure pathway under section 351(k) of the Public Health Service Act permits reliance on certain existing scientific knowledge about the safety and effectiveness of the reference product, and enables a biosimilar biological product to be licensed based on less than a full complement of product-specific preclinical and clinical data.
A biosimilar product can only be approved by the FDA if it has the same mechanism(s) of action, route(s) of administration, dosage form(s) and strength(s) as the reference product, and only for the indication(s) and condition(s) of use that have been approved for the reference product. The facilities where biosimilars are manufactured must also meet the FDA’s standards.
The FDA’s approval of Zarxio is based on review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data and other clinical safety and effectiveness data that demonstrates Zarxio is biosimilar to Neupogen. Zarxio has been approved as biosimilar, not as an interchangeable product. Under the BPCI Act, a biological product that that has been approved as an “interchangeable” may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.
The most common expected side effects of Zarxio are aching in the bones or muscles and redness, swelling or itching at injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome, a lung disease that can cause shortness of breath, difficulty breathing or increase the rate of breathing.
For this approval, the FDA has designated a placeholder nonproprietary name for this product as “filgrastim-sndz.” The provision of a placeholder nonproprietary name for this product should not be viewed as reflective of the agency’s decision on a comprehensive naming policy for biosimilar and other biological products. While the FDA has not yet issued draft guidance on how current and future biological products marketed in the United States should be named, the agency intends to do so in the near future.
Sandoz, a Novartis company, is based in Princeton, New Jersey. Neupogen is marketed by Amgen, based in Thousand Oaks, California.

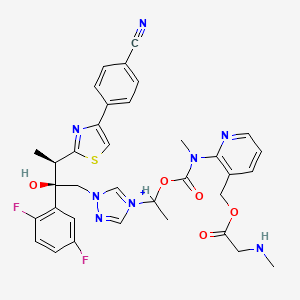
| Molecular Formula: | C35H36F2N8O9S2 |
|---|---|
| Molecular Weight: | 814.837 g/mol |


.syn……https://newdrugapprovals.org/2013/10/02/isavuconazole-basilea-reports-positive-results-from-study/
March 6, 2015
The U.S. Food and Drug Administration today approved Cresemba (isavuconazonium sulfate), a new antifungal drug product used to treat adults with invasive aspergillosis and invasive mucormycosis, rare but serious infections.
Aspergillosis is a fungal infection caused by Aspergillus species, and mucormycosis is caused by the Mucorales fungi. These infections occur most often in people with weakened immune systems.
Cresemba belongs to a class of drugs called azole antifungal agents, which target the cell wall of a fungus. Cresemba is available in oral and intravenous formulations.
“Today’s approval provides a new treatment option for patients with serious fungal infections and underscores the importance of having available safe and effective antifungal drugs,” said Edward Cox, M.D., M.P.H, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Cresemba is the sixth approved antibacterial or antifungal drug product designated as a Qualified Infectious Disease Product (QIDP). This designation is given to antibacterial or antifungal drug products that treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act.
As part of its QIDP designation, Cresemba was given priority review, which provides an expedited review of the drug’s application. The QIDP designation also qualifies Cresemba for an additional five years of marketing exclusivity to be added to certain exclusivity periods already provided by the Food, Drug, and Cosmetic Act. As these types of fungal infections are rare, the FDA also granted Cresemba orphan drug designations for invasive aspergillosis and invasive mucormycosis.
The approval of Cresemba to treat invasive aspergillosis was based on a clinical trial involving 516 participants randomly assigned to receive either Cresemba or voriconazole, another drug approved to treat invasive aspergillosis. Cresemba’s approval to treat invasive mucormycosis was based on a single-arm clinical trial involving 37 participants treated with Cresemba and compared with the natural disease progression associated with untreated mucormycosis. Both studies showed Cresemba was safe and effective in treating these serious fungal infections.
The most common side effects associated with Cresemba include nausea, vomiting, diarrhea, headache, abnormal liver blood tests, low potassium levels in the blood (hypokalemia), constipation, shortness of breath (dyspnea), coughing and tissue swelling (peripheral edema). Cresemba may also cause serious side effects including liver problems, infusion reactions and severe allergic and skin reactions.
Cresemba is marketed by Astellas Pharma US, Inc., based in Northbrook, Illinois.

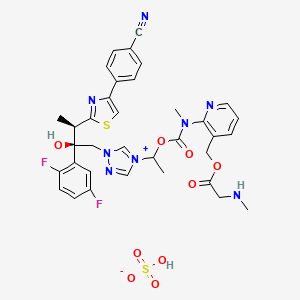
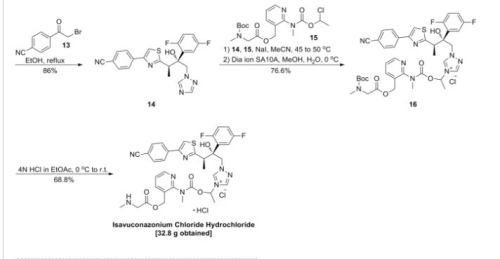
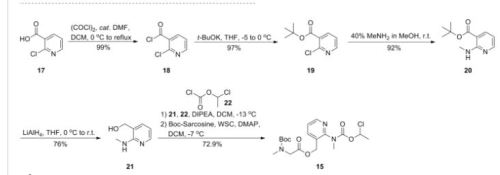
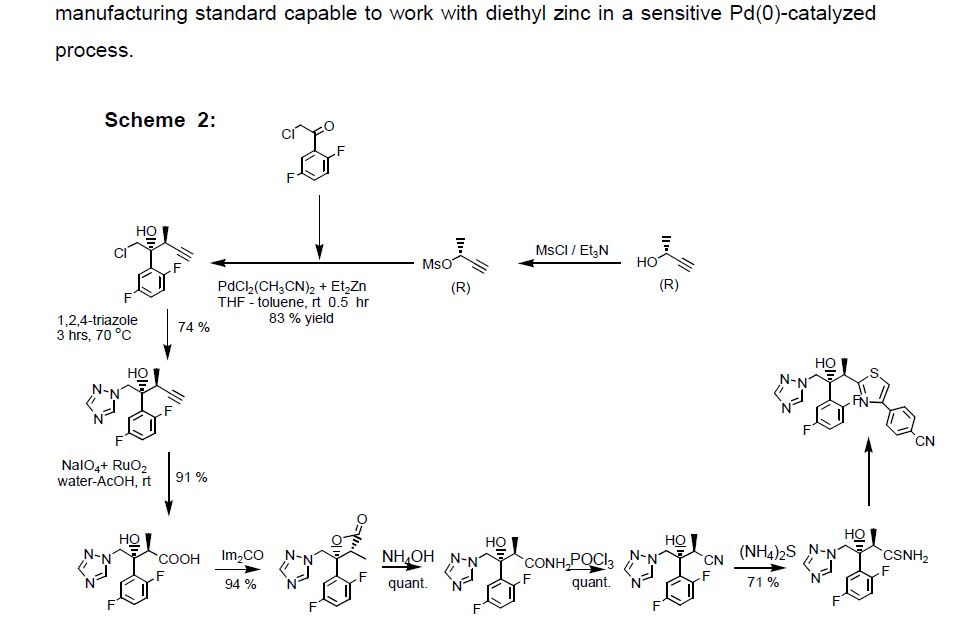
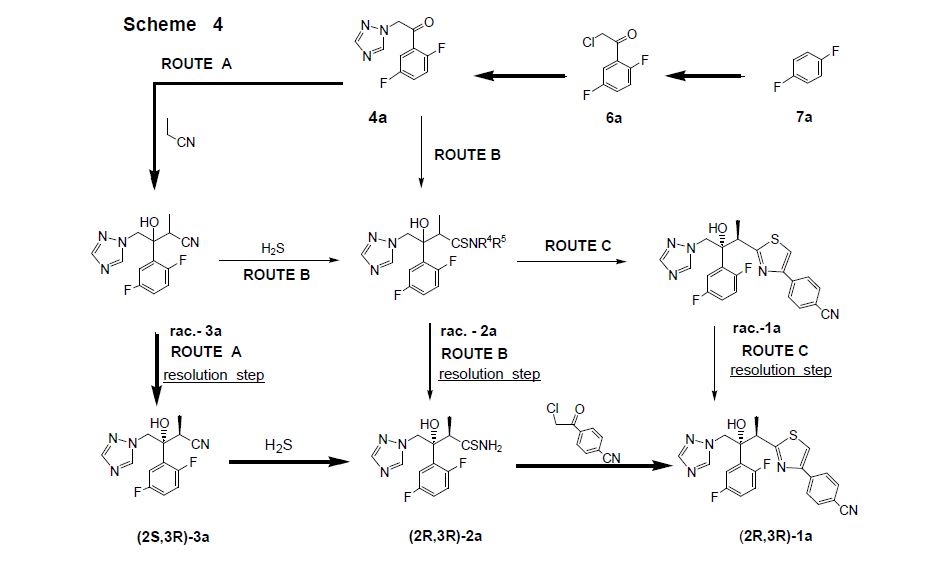
The active substance is isavuconazonium sulfate, a highly water soluble pro-drug of the active triazole isavuconazole. The chemical name of the active substance isavuconazonium sulfate is 1-{(2R,3R)-3-[4-(4-cyanophenyl)-1,3- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxybutyl}-4-[(1RS)-1-({methyl[3-({[(methylamino)acetyl] oxy}methyl) pyridin-2-yl]carbamoyl}oxy)ethyl]-1H-1,2,4-triazol-4-ium monosulfate (IUPAC), corresponding to the molecular formula C35H35F2N8O5S·HSO4 and has a relative molecular mass of 814.84 g/mol. The relative molecular mass of isavuconazole is 437.47. The active substance has the following structure:
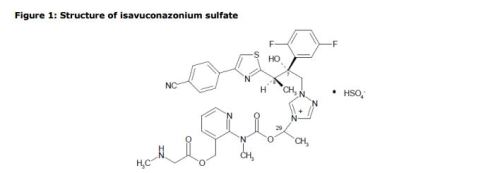
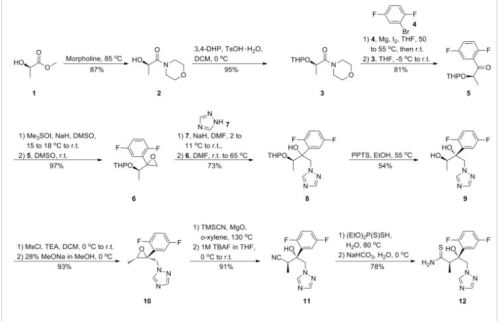
The structure of the active substance has been confirmed by elemental analysis, mass spectrometry, UV, IR, 1H-, 13C- and 19F-NMR spectrometry, and single crystal X-ray analysis, all of which support the chemical structure. It appears as a white, amorphous, hygroscopic powder. It is very soluble in water and over the pH range 1-7. It is also very soluble in methanol and sparingly soluble in ethanol. Two pKa values have been found and calculated to be 2.0 and 7.3. Its logPoct/wat calculated by software is 1.31. Isavuconazonium sulfate has three chiral centres. The stereochemistry of the active substance is introduced by one of the starting materials which is controlled by appropriate specification. The two centres, C7 and C8 in the isavuconazole moiety and in an intermediate of the active substance, have R configuration. The third chiral centre, C29, is not located on isavuconazole moiety and has both the R and S configurations. The nondefined stereo centre at C29 has been found in all batches produced so far to be racemic. Erosion of stereochemical purity has not been observed in the current process. The active substance is a mixture of two epimers of C29. An enantiomer of drug substance was identified as C7 (S), C8 (S) and C29 (R/S) structure. The control of the stereochemistry of isavuconazonium sulfate is performed by chiral HPLC on the active substance and its two precursors. Subsequent intermediates are also controlled by relevant specification in the corresponding steps. Two crystal forms have been observed by recrystallisation studies. However the manufacturing process as described yields amorphous form only.
Two different salt forms of isavuconazonuium (chloride and sulfate) were identified during development. The sulfate salt was selected for further development. A polymorph screening study was also performed. None of the investigated salts could be obtained in crystalline Form………http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002734/WC500196130.pdf

clip






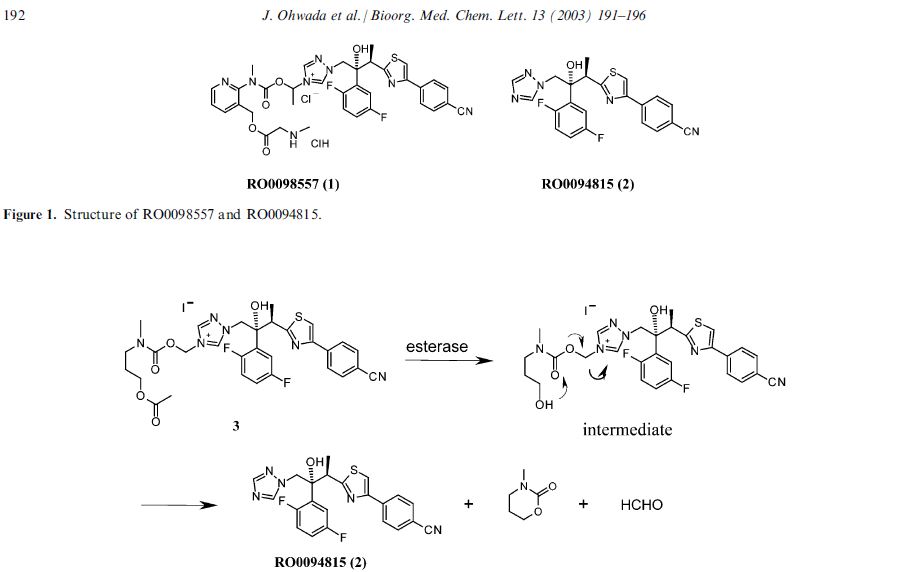


Isavuconazonium (Cresemba ) is a water-soluble prodrug of the triazole antifungal isavuconazole (BAL4815), a 14-a-demethylase inhibitor, under development byBasilea Pharmaceutica International Ltd and Astellas Pharma Inc. Isavuconazonium, in both its intravenous and oral formulations, was approved for the treatment of invasive aspergillosis and invasive mucormycosis (formerly termed zygomycosis) in the US in March 2015. Isavuconazonium is under regulatory review in the EU for invasive aspergillosis and mucormycosis. It is also under phase III development worldwide for the treatment of invasive candidiasis and candidaemia. This article summarizes the milestones in the development of isavuconazonium leading to the first approval for invasive spergillosis and mucormycosis.
Introduction
The availability of both an intravenous (IV) and an oral formulation of isavuconazonium (Cresemba ), as a result of its water solubility, rapid hydrolysis to the active entity isavuconazole and very high oral bioavailability, provides maximum flexibility to clinicians for treating seriously ill patients with invasive fungal infections [1]. Both the IV and oral formulations have been approved by the US Food and Drug Administration (FDA) to treat adults with invasive aspergillosis and invasive mucormycosis [2]. The recommended dosages of each formulation are identical, consisting of loading doses of 372 mg (equivalent to 200 mg of isavuconazole) every eight hours for six doses, followed by maintenance therapy with 372 mg administered once daily [3]. The Qualified Infectious Disease Product (QIDP) designation of the drug with priority review status by the FDA isavuconazonium in the US provided and a five year extension of market exclusivity from launch. Owing to the rarity of the approved infections,
isavuconazonium was also granted orphan drug designation by the FDA for these indications [2]. It has also been granted orphan drug and QIDP designation in the US for the treatment of invasive candidiasis [4]. In July 2014, Basilea Pharmaceutica International Ltd submitted a Marketing Authorization Application to the European Medicines Agency (EMA) for isavuconazonium in the treatment of invasive aspergillosis and invasive mucormycosis, indications for which the EMA has granted isavuconazonium orphan designation [5, 6]. Isavuconazonium is under phase III development in many countries worldwide for the treatment of invasive candidiasis and candidaemia.
1.1 Company agreements
In 2010, Basilea Pharmaceutica International Ltd (a spinoff from Roche, founded in 2000) entered into a licence agreement with Astellas Pharma Inc in which the latter would co-develop and co-promote isavuconazonium worldwide, including an option for Japan. In return for milestone payments, Astellas Pharma was granted an exclusive right to commercialize isavuconazonium, while Basilea Pharmaceutica retained an option to co-promote the drug in the US, Canada, major European countries and China [7]. The companies amended their agreement in 2014, making Astellas Pharma responsible for all regulatory filings, commercialization and manufacturing of isavuconazonium in the US and Canada. Basilea Pharmaceutica waived its right to co-promote the product in the US and Canada, in order to assume all rights in the rest of the world [8]. However, Astellas Pharma remains as sponsor of the multinational, phase III ACTIVE trial in patients with invasive candidiasis.
2 Scientific Summary
Isavuconazonium (as the sulphate; BAL 8557) is a prodrug that is rapidly hydrolyzed by esterases (mainly butylcholinesterase) in plasma into the active moiety isavuconazole
(BAL 4815) and an inactive cleavage product (BAL 8728).
References
1. Falci DR, Pasqualotto AC. Profile of isavuconazole and its potential in the treatment of severe invasive fungal infections. Infect Drug Resist. 2013;6:163–74.
2. US Food and Drug Administration. FDA approves new antifungal drug Cresemba. 2015. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm437106.htm. Accessed 12 Mar 2015.
3. US Food and Drug Administration. Cresemba (isavuconazonium sulfate): US prescribing information. 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207500Orig1s000lbl.pdf. Accessed 18 Mar 2015.
4. Astellas Pharma US Inc. FDA grants Astellas Qualified Infectious Disease Product designation for isavuconazole for the treatment of invasive candidiasis (media release). 2014. http://newsroom astellas.us/2014-07-16-FDA-Grants-Astellas-Qualified-Infectious-Disease-Product-Designation-for-Isavuconazole-for-the-Treatmentof-Invasive-Candidiasis.
5. European Medicines Agency. Public summary of opinion on orphan designation: isavuconazonium sulfate for the treatment of invasive aspergillosis. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2014/07/WC500169890.pdf. Accessed 18 Mar 2015.
European Medicines Agency. Public summary of opinion on orphan designation: isavuconazonium sulfate for the treatment of mucormycosis. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2014/07/WC500169714.pdf. Accessed 18 Mar 2015.
7. Basilea Pharmaceutica. Basilea announces global partnership with Astellas for its antifungal isavuconazole (media release).2010. http://www.basilea.com/News-and-Media/Basilea-announcesglobal-partnership-with-Astellas-for-its-antifungal-isavuconazole/343.
8. Basilea Pharmaceutica. Basilea swaps its isavuconazole North American co-promote rights for full isavuconazole rights outside of North America (media release). 2014. http://www.basilea.com/News-and-Media/Basilea-swaps-its-isavuconazole-North-Americanco-promote-rights-for-full-isavuconazole-rights-outside-


CLIP
http://www.jpharmsci.org/article/S0022-3549(15)00035-0/pdf
http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/207500Orig1207501Orig1s000ChemR.pdf








US 6812238
US 7459561
| FDA Orange Book Patents: 1 of 2 | |
|---|---|
| Patent | 7459561 |
| Expiration | Oct 31, 2020 |
| Applicant | ASTELLAS |
| Drug Application | N207500 (Prescription Drug: CRESEMBA. Ingredients: ISAVUCONAZONIUM SULFATE) |
FREE FORM
Isavuconazonium; Isavuconazonium ion; Cresemba; BAL-8557; 742049-41-8;
[2-[1-[1-[(2R,3R)-3-[4-(4-cyanophenyl)-1,3-thiazol-2-yl]-2-(2,5-difluorophenyl)-2-hydroxybutyl]-1,2,4-triazol-4-ium-4-yl]ethoxycarbonyl-methylamino]pyridin-3-yl]methyl 2-(methylamino)acetate
| Molecular Formula: | C35H35F2N8O5S+ |
|---|---|
| Molecular Weight: | 717.773 g/mol |
Patent IDDatePatent TitleUS20102494262010-09-30STABILIZED PHARMACEUTICAL COMPOSITIONUS74595612008-12-02N-substituted carbamoyloxyalkyl-azolium derivativesUS71898582007-03-13N-phenyl substituted carbamoyloxyalkyl-azolium derivativesUS71511822006-12-19Intermediates for N-substituted carbamoyloxyalkyl-azolium derivativesUS68122382004-11-02N-substituted carbamoyloxyalkyl-azolium derivatives
REF
http://www.drugbank.ca/drugs/DB06636
//////////
CC(C1=NC(=CS1)C2=CC=C(C=C2)C#N)C(CN3C=[N+](C=N3)C(C)OC(=O)N(C)C4=C(C=CC=N4)COC(=O)CNC)(C5=C(C=CC(=C5)F)F)O
CC(C1=NC(=CS1)C2=CC=C(C=C2)C#N)C(CN3C=[N+](C=N3)C(C)OC(=O)N(C)C4=C(C=CC=N4)COC(=O)CNC)(C5=C(C=CC(=C5)F)F)O.OS(=O)(=O)[O-]
March 4, 2015
The U.S. Food and Drug Administration today expanded the approved use of Opdivo (nivolumab) to treat patients with advanced (metastatic) squamous non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy.
Lung cancer is the leading cause of cancer death in the United States, with an estimated 224,210 new diagnoses and 159,260 deaths in 2014. The most common type of lung cancer, NSCLC affects seven out of eight lung cancer patients, occurring when cancer forms in the cells of the lung.
Opdivo works by inhibiting the cellular pathway known as PD-1 protein on cells that blocks the body’s immune system from attacking cancerous cells. Opdivo is intended for patients who have previously been treated with platinum-based chemotherapy.
“The FDA worked proactively with the company to facilitate the early submission and review of this important clinical trial when results first became available in late December 2014,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This approval will provide patients and health care providers knowledge of the survival advantage associated with Opdivo and will help guide patient care and future lung cancer trials.”
Opdivo’s efficacy to treat squamous NSCLC was established in a randomized trial of 272 participants, of whom 135 received Opdivo and 137 received docetaxel. The trial was designed to measure the amount of time participants lived after starting treatment (overall survival). On average, participants who received Opdivo lived 3.2 months longer than those participants who received docetaxel.
The safety and efficacy of Opdivo to treat squamous NSCLC was supported by a single-arm trial of 117 participants who had progressed after receiving a platinum-based therapy and at least one additional systemic regimen. The study was designed to measure objective response rate (ORR), or the percentage of participants who experienced partial shrinkage or complete disappearance of the tumor. Results showed 15 percent of participants experienced ORR, of whom 59 percent had response durations of six months or longer.
The most common side effects of Opdivo are fatigue, shortness of breath, musculoskeletal pain, decreased appetite, cough, nausea and constipation. The most serious side effects are severe immune-mediated side effects involving healthy organs, including the lung, colon, liver, kidneys and hormone-producing glands.
Opdivo for squamous NSCLC was reviewed under the FDA’s priority review program, which provides for an expedited review of drugs that treat serious conditions and, if approved, would provide significant improvement in safety or effectiveness in the treatment of a serious condition. Opdivo is being approved more than three months ahead of the prescription drug user fee goal date of June 22, 2015, the date when the agency was scheduled to complete its review of the application.
The FDA previously approved Opdivo to treat patients with unresectable (cannot be removed by surgery) or metastatic melanoma who no longer respond to other drugs.
Opdivo is marketed by Princeton, New Jersey-based Bristol-Myers Squibb.
pronunciation (help·info) (Marathi: शिर्डी) is a town and falls under the jurisdiction of municipal council popularly known as Shirdi Nagar Panchayat, located …
.




Shraddha Inn,Shirdi

SHIRDI PRASADALAYA BOJAN

Solar Kitchen Feeds Many at Shirdi, India Shrine


Rajdhani Restaurant: Rajdhani at Shirdi

The well equipped kitchen provides food two times a day, daily. Around 27, 000 of people are distributed food at cheap rate. The food comprises of dal,

/////////



Givinostat (INN[1]) or gavinostat (originally ITF2357) is a histone deacetylase inhibitor with potential anti-inflammatory, anti-angiogenic, and antineoplastic activities.[2] It is a hydroxamate used in the form of its hydrochloride.
Givinostat is in numerous phase II clinical trials (including for relapsed leukemias and myelomas),[3] and has been granted orphan drug designation in the European Union for the treatment of systemic juvenile idiopathic arthritis[4] and polycythaemia vera.[5]
In 2010, orphan drug designation was assigned in the E.U. for the treatment of systemic-onset juvenile idiopathic arthritis and for the treatment of polycythemia vera. In 2013, this designation was assigned by the FDA for the treatment of Duchenne’s muscular dystrophy and for the treatment of Becker’s muscular dystrophy.
ITF2357 was discovered at Italfarmaco of Milan, Italy. It was patented in 1997 and first described in the scientific literature in 2005.[6][7]
Givinostat hydrochloride, an orally active, synthetic inhibitor of histone deacetylase, is being evaluated in several early clinical studies at Italfarmaco, including studies for the treatment of myeloproliferative diseases, polycythemia vera, Duchenne’s muscular dystrophy and periodic fever syndrome. The company was also conducting clinical trials for the treatment of Crohn’s disease and chronic lymphocytic leukemia; however, the trials were terminated.
No recent development has been reported for research into the treatment of juvenile rheumatoid arthritis, for the treatment of multiple myeloma and for the treatment of Hodgkin’s lymphoma.
Muscular dystrophies (MDs) include a heterogeneous group of genetic diseases invariably leading to muscle degeneration and impaired function. Mutation of nearly 30 genes gives rise to various forms of muscular dystrophy, which differ in age of onset, severity, and muscle groups affected (Dalkilic I, Kunkel LM. (2003) Muscular dystrophies: genes to pathogenesis. Curr. Opin. Genet. Dev. 13:231-238). The most common MD is the Duchenne muscular dystrophy (DMD), a severe recessive X-linked disease which affects one in 3,500 males, characterized by rapid progression of muscle degeneration, eventually leading to loss of ambulation and death within the second decade of life.
Attempts to replace or correct the mutated gene, by means of gene or cell therapy, might result in a definitive solution for muscular dystrophy, but this is not easy to achieve. Alternative strategies that prevent or delay muscle degeneration, reduce inflammation or promote muscle metabolism or regeneration might all benefit patients and, in the. future, synergize with gene or cell therapy. Steroids that reduce inflammation are currently the only therapeutic tool used in the majority of DMD patients (Cossu G, Sampaolesi M . (2007) New therapies for Duchenne muscular dystrophy: challenges, prospects and clinical trials. TRENDS Mol . Med. 13:520-526).
Diethyl- [ 6- ( 4-hydroxycarbamoyl-phenyl-carbamoyloxy- methyl ) -naphthalen-2-yl-methyl ] -ammonium chloride , which is described in WO 97/43251 (anhydrous form) and in WO 2004/065355 (monohydrate crystal form), herein both incorporated by reference, is an anti-inflammatory agent which is able to inhibit the synthesis of the majority of pro-inflammatory cytokines whilst sparing anti-inflammatory ones. Diethyl- [ 6- ( 4-hydroxycarbamoyl-phenyl-carbamoyloxy- methyl ) -naphthalen-2-yl-methyl ] -ammonium chloride is also known as ITF2357.
The monohydrate crystal form of diethyl- [ 6- ( 4- hydroxycarbamoyl-phenyl-carbamoyloxy-methy1 ) – naphthalen-2-yl-methyl ] -ammonium chloride is known as Givinostat .
Givinostat is being evaluated in several clinical studies, including studies for the treatment of myeloproliferative diseases, polycythemia vera, periodic fever syndrome, Crohn’s disease and systemic- onset juvenile idiopathic arthritis. Orphan drug designation was assigned in the E.U. for the treatment of systemic-onset juvenile idiopathic arthritis and for the treatment of polycythemia vera.
Givinostat has been recently found to act also as a Histone Deacetylase inhibitor (WO 2011/048514).
Histone deacetylases ( HDAC ) are a family of enzymes capable of removing the acetyl group bound to the lysine residues in the N-terminal portion of histones or in other proteins.
HDACs can be subdivided into four classes, on the basis of structural homologies. Class I HDACs (HDAC 1, 2, 3 and 8) are similar to the RPD3 yeast protein and are located in the cell nucleus. Class II HDACs (HDAC 4, 5, 6, 7, 9 and 10) are similar to the HDA1 yeast protein and are located both in the nucleus and in the cytoplasm. Class III HDACs are a structurally distinct form of NAD-dependent enzymes correlated with the SIR2 yeast protein. Class IV (HDAC 11) consists at the moment of a single enzyme having particular structural characteristics. The HDACs of classes I, II and IV are zinc enzymes and can be inhibited by various classes of molecule: hydroxamic acid derivatives, cyclic tetrapeptides , short-chain fatty acids, aminobenzamides , derivatives of electrophilic ketones, and the like. Class III HDACs are not inhibited by hydroxamic acids, and their inhibitors have structural characteristics different from those of the other classes .
The expression “histone deacetylase inhibitor” in relation to the present invention is to be understood as meaning any molecule of natural, recombinant or synthetic origin capable of inhibiting the activity of at least one of the enzymes classified as histone deacetylases of class I, class II or class IV.
Although HDAC inhibitors, as a class, are considered to be potentially useful as anti-tumor agents, it is worth to note that, till now, only two of them (Vorinostat and Romidepsin) have been approved as drugs for the cure of a single tumor form (Cutaneous T-cell lymphoma ) .
It is evident that the pharmaceutical properties of each HDAC inhibitor may be different and depend on the specific profile of inhibitory potency, relative to the diverse iso-enzymes as well as on the particular pharmacokinetic behaviour and tissue distribution.
Some HDAC inhibitors have been claimed to be potentially useful, in combination with other agents, for the treatment of DMD (WO 2003/033678, WO 2004/050076, Consalvi S. et al. Histone Deacetylase Inhibitors in the Treatment of Muscular Dystrophies: Epigenetic Drugs for Genetic Diseases. (2011) Mol. Med. 17 : 457-465 ) .
The potential therapeutic use of HDAC inhibitors in DMD may however be hampered by the possible harmful effects of these relatively toxic agents, especially when used for long-term therapies in paediatric patients .
Givinostat, as anti-inflammatory agent, has been already used in a phase II study in children with Systemic Onset Juvenile Idiopathic Arthritis; Givinostat administered at 1.5 mg/kg/day for twelve weeks achieved ACR Pedi 30, 50 and 70 improvement of approximately 70% (Vojinovic J, Nemanja D. (2011) HDAC Inhibition in Rheumatoid Arthritis and Juvenile Idiopathic Arthritis. Mol. Med 17:397-403) showing only a limited number of mild or moderate but short lasting, adverse effects.
To date more than 500 patients (including 29 children) have been treated with Givinostat. Repeated dose toxicity studies were carried out in dogs, rats and monkeys. Oral daily doses of the drug were administered up to nine consecutive months. The drug was well tolerated with no overt toxicity at high doses. The “no adverse effect levels” (NOAEL) ranged from 10 to 25 mg/kg/day depending on the animal species and the duration of treatment.
In juvenile animals Givinostat at 60 mg/kg/day did not affect the behavioural and physical development and reproductive performance of pups.
No genotoxic effect was detected for Givinostat in the mouse lymphoma assay and the chromosomal aberration assay in vitro and in the micronucleus test and UDS test in vivo.
| Patent | Submitted | Granted |
|---|---|---|
| Monohydrate hydrochloride of the 4-hydroxycarbamoyl-phenyl)-carbamic acid (6-diethylaminomethyl-naphtalen-2-yl) ester [US7329689] | 2005-11-03 | 2008-02-12 |
In clinical trials of givinostat as a salvage therapy for advanced Hodgkin’s lymphoma, the most common adverse reactions were fatigue (seen in 50% of participants), mild diarrhea or abdominal pain (40% of participants), moderate thrombocytopenia (decreased platelet counts, seen in one third of patients), and mild leukopenia (a decrease in white blood cell levels, seen in 30% of patients). One-fifth of patients experienced prolongation of the QT interval, a measure of electrical conduction in the heart, severe enough to warrant temporary suspension of treatment.[8]
Givinostat inhibits class I and class II histone deacetylases (HDACs) and several pro-inflammatory cytokines. This reduces expression of tumour necrosis factor (TNF), interleukin 1α and β, and interleukin 6.[7]
It also has activity against cells expressing JAK2(V617F), a mutated form of the janus kinase 2 (JAK2) enzyme that is implicated in the pathophysiology of many myeloproliferative diseases, including polycythaemia vera.[9][10] In patients with polycythaemia, the reduction of mutant JAK2 concentrations by givinostat is believed to slow down the abnormal growth of erythrocytes and ameliorate the symptoms of the disease.[5]
………………….

PATENT
https://www.google.com/patents/WO2004065355A1?cl=en
Hydrochloride of (6-diethylaminomethyl-naphthalen-2-yl)- methyl ester of (4-hydroxycarbamoylphenyl)-carbamic acid (II)

has been described in US patent 6,034,096 as a derivative of hydroxamic acid having anti-inflammatory and immunosuppressive activity, probably owing to the ability thereof to inhibit the production of pro-inflammatory cyto ines. This compound is obtained according to
Example 12 of the above-mentioned patent as an anhydrous, amorphous, hygroscopic, deliquescent solid which is difficult to handle.
crystalline form of monohydrous hydrochloride of
(6-diethylaminomethyl-naphthalen-2-yl)-methyl ester of
(4~hydroxycarbamoylphenyl)-carbamic acid (I).

This form is particularly advantageous from the industrial perspective because it is stable and simpler to handle than the anhydrous and amorphous form described above.
………………
PATENT
http://www.google.co.in/patents/US7329689
Hydrochloride of (6-diethylaminomethyl-naphthalen-2-yl)-methyl ester of (4-hydroxycarbamoylphenyl)-carbamic acid (II)

has been described in U.S. Pat. No. 6,034,096 as a derivative of hydroxamic acid having anti-inflammatory and immunosuppressive activity, probably owing to the ability thereof to inhibit the production of pro-inflammatory cytokines. This compound is obtained according to Example 12 of the above-mentioned patent as an anhydrous, amorphous, hygroscopic, deliquescent solid which is difficult to handle.
The 4-(6-diethylaminomethyl-naphthalen-2-ylmethoxycarbonylamino)-benzoic acid can be prepared as described in Example 12, point C, of U.S. Pat. No. 6,034,096.
The acid (1.22 kg, 3 moles) was suspended in THF (19 l) and the mixture was agitated under nitrogen over night at ambient temperature. The mixture was then cooled to 0° C. and thionyl chloride (0.657 l, 9 moles) was added slowly, still under nitrogen, with the temperature being maintained below 10° C. The reaction mixture was heated under reflux for 60 minutes, DMF (26 ml) was added and the mixture was further heated under reflux for 60 minutes.
The solvent was evaporated under vacuum, toluene was added to the residue and was then evaporated. This operation was repeated twice, then the residue was suspended in THF (11.5 l) and the mixture was cooled to 0° C.
The mixture was then poured into a cold solution of hydroxylamine (50% aq., 1.6 l, 264 moles) in 5.7 l of water. The mixture was then cooled to ambient temperature and agitated for 30 minutes. 6M HCl was added until pH 2 was reached and the mixture was partially evaporated under vacuum in order to eliminate most of the THF. The solid was filtered, washed repeatedly with water and dissolved in a solution of sodium bicarbonate (2.5%, 12.2 l). The solution was extracted with 18.6 l of a mixture of THF and ethyl acetate (2:1 v/v). 37% HCl (130 ml) were added to the organic layer in order to precipitate the monohydrate of the (6-diethylaminomethyl-naphthalen-2-yl)-methyl ester hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid. If necessary, this operation can be repeated several times to remove any residues of the original acid.
Finally, the solid was dried under vacuum (approximately 30 mbar, 50° C.), producing 0.85 kg (60%) of compound (I).
HPLC purity: 99.5%; water content (Karl Fischer method): 3.8%; (argentometric) assay: 99.8%.
| Elemental analysis | |||||
| C % | H % | Cl % | N % | ||
| Calculated for | 60.56 | 6.35 | 7.45 | 8.83 | |
| C24H30ClN3O5 | |||||
| Found | 61.06 | 6.48 | 7.48 | 8.90 | |
PATENT
http://www.google.co.in/patents/US20120302633
The hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphtalenyl) ester, also known as ITF 2357 and having the International Non Proprietary Name (INN) of Givinostat® is an organic compound with immunosuppressive and anti-inflammatory activity,

…………………..
http://www.google.com/patents/US6034096
A. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) (22.2 g, 115 mmol) was added to a solution of 2,6-naphthalenedicarboxylic acid (25 g, 115 mmol) and hydroxybenzotriazole (15.6 g, 115 mmol) in dimethylformamide (1800 ml) and the mixture was stirred at room temperature for 2 hours. Diethyl amine (34.3 ml, 345 mmol) was added and the solution was stirred overnight at room temperature. The solvent was then evaporated under reduced pressure and the crude was treated with 1N HCl (500 ml) and ethyl acetate (500 ml), insoluble compounds were filtered off and the phases were separated. The organic phase was extracted with 5% sodium carbonate (3×200 ml) and the combined aqueous solutions were acidified with concentrated HCl and extracted with ethyl acetate (3×200 ml). The organic solution was then washed with 1N HCl (6×100 ml), dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure yielding 18.5 g (Yield 60%) of pure 6-(diethylaminocarbonyl)-2-naphthalenecarboxylic acid; m.p.=122-124° C.
1 H-NMR d 8.67 (s, 1H), 8.25-8.00 (m, 4H), 7.56 (d, 1H), 3.60-3.20 (m, 4H), 1.30-1.00 (m, 6H).
B. A solution of 6-(diethylaminocarbonyl)-2- naphthalenecarboxylic acid (18 g, 66 mmol) in THF (200 ml) was slowly added to a refluxing suspension of lithium aluminium hydride (7.5 g, 199 mmol) in THF (500 ml). The mixture was refluxed for an hour, then cooled at room temperature and treated with a mixture of THF (25 ml) and water (3.5 ml), with 20% sodium hydroxide (8.5 ml) and finally with water (33 ml). The white solid was filtered off and the solvent was removed under reduced pressure. Crude was dissolved in diethyl ether (200 ml) and extracted with 1N HCl (3×100 ml). The aqueous solution was treated with 32% sodium hydroxide and extracted with diethyl ether (3×100 ml). The organic solution was dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure yielding 12.7 g (79% yield) of pure 6-(diethylaminomethyl)-2-naphthalenemethanol as thick oil.
1 H-NMR d 7.90-7.74 (m, 4H), 7.49 (m, 2H), 5.32 (t, 1H, exchange with D2 O), 4.68 (d, 2H), 3.69 (s, 2H), 2.52 (q, 4H), 1.01 (t, 6H).
C. A solution of 6-(diethylaminomethyl)-2-naphthalene-methanol (12.5 g, 51 mmol) and N,N’-disuccinimidyl carbonate (13.2 g, 51 mmol) in acetonitrile (250 ml) was stirred at room temperature for 3 hours, then the solvent was removed and the crude was dissolved in THF (110 ml). This solution was added to a solution of 4-amino benzoic acid (7.1 g, 51 mmol) and sodium carbonate (5.5 g, 51 mmol) in water (200 ml) and THF (100 ml). The mixture was stirred overnight at room temperature, then THF was removed under reduced pressure and the solution was treated with 1N HCl (102 ml, 102 mmol). The precipitate was filtered, dried under reduced pressure, tritured in diethyl ether and filtered yielding 13.2 g (yield 64%) of pure 4-[6-(diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]-benzoic acid; m.p.=201-205° C. (dec.)
1 H-NMR d 10.26 (s, 1H), 8.13 (s, 1H), 8.05-7.75 (m, 6H), 7.63 (m, 3H), 5.40 (s, 2H), 4.32 (s, 2H), 2.98 (q, 4H), 1.24 (t, 6H).
D. A solution of 4-[6-(diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]benzoic acid (13.1 g, 32 mmol) and thionyl chloride (7 ml, 96 mmol) in chloroform (300 ml) was refluxed for 4 hours, then the solvent and thionyl chloride were evaporated. Crude was dissolved in chloroform (100 ml) and evaporated to dryness three times. Crude was added as solid to a solution of hydroxylamine hydrochloride (2.7 g, 39 mmol) and sodium bicarbonate (5.4 g, 64 mmol) and 1N sodium hydroxide (39 ml, 39 mmol) in water (150 ml) and THF (50 ml). The mixture was stirred overnight at room temperature, then THF was removed under reduced pressure and the aqueous phase was extracted with ethyl acetate (3×100 ml). The combined organic phases were dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. Crude was dissolved in THF and treated with a 1.5 N etheric solution of HCl. The solid product was filtered and dried yielding 6 g (yield 41%) of pure 4-[6-(diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]benzohydroxamic acid hydrochloride as white solid; m.p.=162-165° C., (dec.)
1 H-NMR d 11.24 (s, 1H, exchange with D2 O), 10.88 (s, 1H, exchange with D2 O), 10.16 (s, 1H), 8.98 (bs, 1H, exchange with D2 O), 8.21 (s, 1H), 8.10-7.97 (m, 3H), 7.89 (d, 1H), 7.80-7.55 (m, 5H), 5.39 (s, 2H), 4.48 (d, 2H), 3.09 (m, 4H), 1.30 (t, 6H).

Some nmr predictions
CAS NO. 497833-27-9, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate H-NMR spectral analysis
![[6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate NMR spectra analysis, Chemical CAS NO. 497833-27-9 NMR spectral analysis, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-20/001/566/1566912_1h.png)
13 C NMR PREDICTIONS
![[6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate NMR spectra analysis, Chemical CAS NO. 497833-27-9 NMR spectral analysis, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-20/001/566/1566912_13c.png)

COSY NMR…..http://www.nmrdb.org/
HMBC /HSQC
| US6034096 | 12 May 1997 | 7 Mar 2000 | Italfarmaco S.P.A. | Compounds with anti-inflammatory and immunosuppressive activities |
| WO1997043251A1 | May 12, 1997 | Nov 20, 1997 | Italfarmaco Spa | Compounds with anti-inflammatory and immunosuppressive activities |
| WO2004063146A1 | Jan 7, 2004 | Jul 29, 2004 | Italfarmaco Spa | Hydroxamic acid derivatives having anti-inflammatory action |
| WO2004065355A1 | Jan 8, 2004 | Aug 5, 2004 | Italfarmaco Spa | Monohydrate hydrochloride of the 4-hydroxycarbamoyl-phenyl)-carbamic acid (6-diethylaminomethyl-naphtalen-2-yl) ester |
| WO2006003068A2 | Jun 7, 2005 | Jan 12, 2006 | Italfarmaco Spa | Alpha-amino acid derivatives with antiinflammatory activity |
| WO2008097654A1 | Feb 8, 2008 | Aug 14, 2008 | Nancie M Archin | Methods of using saha for treating hiv infection |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8518988 * | 3 Dec 2010 | 27 Aug 2013 | Chemi Spa | Polymorph of the hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphthalenyl) ester |
| US20120302633 * | 3 Dec 2010 | 29 Nov 2012 | Chemi Spa | Novel polymorph of the hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphthalenyl) ester |
| WO2011092556A1 | 3 Dec 2010 | 4 Aug 2011 | Chemi Spa | Novel polymorph of the hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphtalenyl) ester |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| {6-[(diethylamino)methyl]naphthalen-2-yl}methyl [4-(hydroxycarbamoyl)phenyl]carbamate | |
| Clinical data | |
|
|
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 497833-27-9 |
| ATC code | None |
| PubChem | CID 9804992 |
| ChemSpider | 7980752 |
| UNII | 5P60F84FBH |
| Chemical data | |
| Formula | C24H27N3O4 |
| Molecular mass | 421.489 g/mol |

| Italfarmaco S.p.A. | |
|---|---|
|
|
|
| Stato | |
| Tipo | Società per azioni |
| Fondazione | 1938 a Milano |
| Fondata da | Gastone De Santis |
| Sede principale | Milano |
| Filiali | |
| Persone chiave | Francesco De Santis, [Presidente Holding] |
| Settore | sanità |
| Prodotti | Farmaci |
| Fatturato | >500 milioni di Euro (gruppo) (2011) |
| Dipendenti | >1900 (gruppo) (2011) |
| Sito web | www.italfarmaco.com |
MILAN ITALY



| Chemical Formula: | C5H4FN3O2 | |
| CAS #: | 259793-96-9 | |
| Molecular Weight: | 157.1 | |
ANTI-INFLUENZA COMPOUND |
||
| clinical trials | http://clinicaltrials.gov/search/intervention=Favipiravir | |
| Chemical Name: | 6-fluoro-3-hydroxy-2-pyrazinecarboxamide | |
| Synonyms: | T-705, T705, Favipiravir |
ファビピラビル
Favipiravir
6-Fluoro-3-hydroxypyrazine-2-carboxamide
C5H4FN3O2 : 157.1
[259793-96-9]
The drug substance is a white to light yellow powder. It is sparingly soluble in acetonitrile and in methanol, and slightly soluble in water and in ethanol (99.5). It is slightly soluble at pH 2.0 to 5.5 and sparingly soluble at pH 5.5 to 6.1. The drug substance is not hygroscopic at 25°C/51% to 93%RH. The melting point is 187°C to 193°C, and the dissociation constant (pKa) is 5.1 due to the hydroxyl group of favipiravir. Measurement results on the partition ratio of favipiravir in water/octanol at 25°C indicate that favipiravir tends to be distributed in the 1-octanol phase at pH 2 to 4 and in the water phase at pH 5 to 13.
Any batch manufactured by the current manufacturing process is in Form A. The stability study does not show any change in crystal form over time; and a change from Form A to Form B is unlikely.
Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 187℃ to 193℃ | https://www.pmda.go.jp/files/000210319.pdf |
| water solubility | slightly soluble in water | https://www.pmda.go.jp/files/000210319.pdf |
| pKa | 5.1 | https://www.pmda.go.jp/files/000210319.pdf |
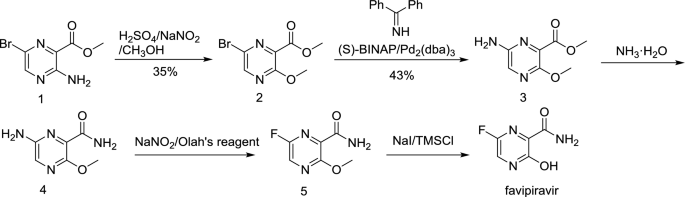
Favipiravir, also known as T-705, Avigan, or favilavir is an antiviral drug being developed by Toyama Chemical (Fujifilm group) of Japan with activity against many RNA viruses. Like certain other experimental antiviral drugs (T-1105 and T-1106), it is a pyrazinecarboxamide derivative. In experiments conducted in animals Favipiravir has shown activity against influenza viruses, West Nile virus, yellow fever virus, foot-and-mouth disease virus as well as other flaviviruses, arenaviruses, bunyaviruses and alphaviruses.[1]Activity against enteroviruses[2] and Rift Valley fever virus has also been demonstrated.[3] Favipiravir has showed limited efficacy against Zika virus in animal studies, but was less effective than other antivirals such as MK-608.[4] The agent has also shown some efficacy against rabies,[5] and has been used experimentally in some humans infected with the virus.[6]
In February 2020 Favipiravir was being studied in China for experimental treatment of the emergent COVID-19 (novel coronavirus)disease.[7][8] On March 17 Chinese officials suggested the drug had been effective in treating COVID in Wuhan and Shenzhen.[9][10]
Discovered by Toyama Chemical Co., Ltd. in Japan, favipiravir is a modified pyrazine analog that was initially approved for therapeutic use in resistant cases of influenza.7,9 The antiviral targets RNA-dependent RNA polymerase (RdRp) enzymes, which are necessary for the transcription and replication of viral genomes.7,12,13
Not only does favipiravir inhibit replication of influenza A and B, but the drug shows promise in the treatment of influenza strains that are resistant to neuramidase inhibitors, as well as avian influenza.9,19 Favipiravir has been investigated for the treatment of life-threatening pathogens such as Ebola virus, Lassa virus, and now COVID-19.10,14,15
The mechanism of its actions is thought to be related to the selective inhibition of viral RNA-dependent RNA polymerase.[11] Other research suggests that favipiravir induces lethal RNA transversion mutations, producing a nonviable viral phenotype.[12] Favipiravir is a prodrug that is metabolized to its active form, favipiravir-ribofuranosyl-5′-triphosphate (favipiravir-RTP), available in both oral and intravenous formulations.[13][14] Human hypoxanthine guanine phosphoribosyltransferase (HGPRT) is believed to play a key role in this activation process.[15] Favipiravir does not inhibit RNA or DNA synthesis in mammalian cells and is not toxic to them.[1] In 2014, favipiravir was approved in Japan for stockpiling against influenza pandemics.[16] However, favipiravir has not been shown to be effective in primary human airway cells, casting doubt on its efficacy in influenza treatment.[17]
In 2014, Japan approved Favipiravir for treating viral strains unresponsive to current antivirals.[18]
In March 2015, the US Food and Drug Administration completed a Phase III clinical trial studying the safety and efficacy of Favipiravir in the treatment of influenza.[19]
Some research has been done suggesting that in mouse models Favipiravir may have efficacy against Ebola. Its efficacy against Ebola in humans is unproven.[20][21][22] During the 2014 West Africa Ebola virus outbreak, it was reported that a French nurse who contracted Ebola while volunteering for MSF in Liberia recovered after receiving a course of favipiravir.[23] A clinical trial investigating the use of favipiravir against Ebola virus disease was started in Guéckédou, Guinea, during December 2014.[24] Preliminary results showed a decrease in mortality rate in patients with low-to-moderate levels of Ebola virus in the blood, but no effect on patients with high levels of the virus, a group at a higher risk of death.[25] The trial design has been criticised by Scott Hammer and others for using only historical controls.[26] The results of this clinical trial were presented in February 2016 at the annual Conference on Retroviruses and Opportunistic Infections (CROI) by Daouda Sissoko[27] and published on March 1, 2016 in PLOS Medicine.[28]
In March 2020, Chinese officials suggested Favipiravir may be effective in treating COVID-19.[29]
SYN
https://link.springer.com/article/10.1007/s11696-018-0654-9

https://pdfs.semanticscholar.org/be8e/cb882b99204983d2f60077c7ab8b53f4d62c.pdf
Drug Discoveries & Therapeutics. 2014; 8(3):117-120.
As a RNA polymerase inhibitor, 6-fluoro-3-hydroxypyrazine-2-carboxamide commercially named favipiravir has been proved to have potent inhibitory activity against RNA viruses in vitro and in vivo. A four-step synthesis of the compound is described in this article, amidation, nitrification, reduction and fluorination with an overall yield of about 8%. In addition, we reported the crystal structure of the title compound. The molecule is almost planar and the intramolecular O−H•••O hydrogen bond makes a 6-member ring. In the crystal, molecules are packing governed by both hydrogen bonds and stacking interactions.


2.2.1. Preparation of 3-hydroxypyrazine-2-carboxamide To a suspension of 3-hydroxypyrazine-2-carboxylic acid (1.4 g, 10 mmol) in 150 mL MeOH, SOCl2 was added dropwise at 40°C with magnetic stirring for 6 h resulting in a bright yellow solution. The reaction was then concentrated to dryness. The residue was dissolved in 50 mL 25% aqueous ammonia and stirred overnight to get a suspension. The precipitate was collected and dried. The solid yellow-brown crude product was recrystallization with 50 mL water to get the product as pale yellow crystals (1.1 g, 78%). mp = 263-265°C. 1 H-NMR (300 MHz, DMSO): δ 13.34 (brs, 1H, OH), 8.69 (s, 1H, pyrazine H), 7.93-8.11 (m, 3H, pyrazine H, CONH2). HRMS (ESI): m/z [M + H]+ calcd for C5H6N3O2 + : 140.0460; found: 140.0457.
2.2.2. Preparation of 3-hydroxy-6-nitropyrazine-2- carboxamide In the solution of 3-hydroxypyrazine-2-carboxamide (1.0 g, 7 mmol) in 6 mL concentrate sulfuric acid under ice-cooling, potassium nitrate (1.4 g, 14 mmol) was added. After stirring at 40°C for 4 h, the reaction mixture was poured into 60 mL water. The product was collected by fi ltration as yellow solid (0.62 g, 48%). mp = 250-252°C. 1 H-NMR (600 MHz, DMSO): δ 12.00- 15.00 (br, 1H, OH), 8.97 (s, 1H, pyrazine H), 8.32 (s, 1H, CONH2), 8.06 (s, 1H, CONH2). 13C-NMR (75 MHz, DMSO): δ 163.12, 156.49, 142.47, 138.20, 133.81. HRMS (ESI): m/z [M + H]+ calcd for C5H5N4O4 + : 185.0311; found: 185.0304.
2.2.3. Preparation of 6-amino-3-hydroxypyrazine-2- carboxamide 3-Hydroxy-6-nitropyrazine-2-carboxamide (0.6 g, 3.3 mmol) and a catalytic amount of raney nickel were suspended in MeOH, then hydrazine hydrate was added dropwise. The resulting solution was refl uxed 2 h, cooled, filtered with diatomite, and then MeOH is evaporated in vacuo to get the crude product as dark brown solid without further purification (0.4 g, 77%). HRMS (ESI): m/z [M + H]+ calcd for C5H7N4O2 + : 155.0569; found:155.0509.
2.2.4. Preparation of 6-fluoro-3-hydroxypyrazine-2- carboxamide To a solution of 6-amino-3-hydroxypyrazine-2- carboxamide (0.4 g, 2.6 mmol) in 3 mL 70% HFpyridine aqueous at -20°C under nitrogen atmosphere, sodium nitrate (0.35 g, 5.2 mmol) was added. After stirring 20 min, the solution was warmed to room temperature for another one hour. Then 20 mL ethyl acetate/water (1:1) were added, after separation of the upper layer, the aqueous phase is extracted with four 20 mL portions of ethyl acetate. The combined extracts are dried with anhydrous magnesium sulfate and concentrated to dryness to get crude product as oil. The crude product was purified by chromatography column as white solid (0.12 g, 30%). mp = 178-180°C. 1 H-NMR (600 MHz, DMSO): δ 12.34 (brs, 1H, OH), 8.31 (d, 1H, pyrazine H, J = 8.0 Hz), 7.44 (s, 1H, CONH2), 5.92 (s, 1H, CONH2). 13C-NMR (75 MHz, DMSO): δ 168.66, 159.69, 153.98, 150.76, 135.68. HRMS (ESI): m/z [M + H]+ calcd for C5H5FN3O2 + : 158.0366; found: 158.0360.
SEE
Chemical Papers (2019), 73(5), 1043-1051.
PAPER
Medicinal chemistry (Shariqah (United Arab Emirates)) (2018), 14(6), 595-603
http://www.eurekaselect.com/158990/article
PATENT
CN 107641106
PAPER
Chemical Papers (2017), 71(11), 2153-2158.
https://link.springer.com/article/10.1007%2Fs11696-017-0208-6

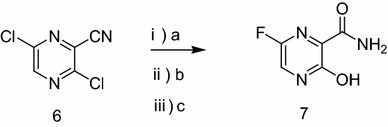
Below is the link to the electronic supplementary material.





CLIP
Influenza virus is a central virus of the cold syndrome, which has attacked human being periodically to cause many deaths amounting to tens millions. Although the number of deaths shows a tendency of decrease in the recent years owing to the improvement in hygienic and nutritive conditions, the prevalence of influenza is repeated every year, and it is apprehended that a new virus may appear to cause a wider prevalence.
For prevention of influenza virus, vaccine is used widely, in addition to which low molecular weight substances such as Amantadine and Ribavirin are also used
CLIP
Synthesis of Favipiravir
ZHANG Tao1, KONG Lingjin1, LI Zongtao1,YUAN Hongyu1, XU Wenfang2*
(1. Shandong Qidu PharmaceuticalCo., Ltd., Linzi 255400; 2. School of Pharmacy, Shandong University, Jinan250012)
ABSTRACT: Favipiravir was synthesized from3-amino-2-pyrazinecarboxylic acid by esterification, bromination with NBS,diazotization and amination to give 6-bromo-3-hydroxypyrazine-2-carboxamide,which was subjected to chlorination with POCl3, fluorination with KF, andhydrolysis with an overall yield of about 22%.
PATENT
US6787544

| subs G1 | G2 | G3 | G4 | R2 |
| compd 32 N | CH | C—CF3 | N | H |
…………………
EP2192117
Example 1-1

To a 17.5 ml N,N-dimethylformamide solution of 5.0 g of 3,6-difluoro-2-pyrazinecarbonitrile, a 3.8 ml water solution of 7.83 g of potassium acetate was added dropwise at 25 to 35° C., and the solution was stirred at the same temperature for 2 hours. 0.38 ml of ammonia water was added to the reaction mixture, and then 15 ml of water and 0.38 g of active carbon were added. The insolubles were filtered off and the filter cake was washed with 11 ml of water. The filtrate and the washing were joined, the pH of this solution was adjusted to 9.4 with ammonia water, and 15 ml of acetone and 7.5 ml of toluene were added. Then 7.71 g of dicyclohexylamine was added dropwise and the solution was stirred at 20 to 30° C. for 45 minutes. Then 15 ml of water was added dropwise, the solution was cooled to 10° C., and the precipitate was filtered and collected to give 9.44 g of dicyclohexylamine salt of 6-fluoro-3-hydroxy-2-pyradinecarbonitrile as a lightly yellowish white solid product.
1H-NMR (DMSO-d6) δ values: 1.00-1.36 (10H, m), 1.56-1.67 (2H, m), 1.67-1.81 (4H, m), 1.91-2.07 (4H, m), 3.01-3.18 (2H, m), 8.03-8.06 (1H, m), 8.18-8.89 (1H, broad)
Example 1-2
4.11 ml of acetic acid was added at 5 to 15° C. to a 17.5 ml N,N-dimethylformamide solution of 5.0 g of 3,6-difluoro-2-pyrazinecarbonitrile. Then 7.27 g of triethylamine was added dropwise and the solution was stirred for 2 hours. 3.8 ml of water and 0.38 ml of ammonia water were added to the reaction mixture, and then 15 ml of water and 0.38 g of active carbon were added. The insolubles were filtered off and the filter cake was washed with 11 ml of water. The filtrate and the washing were joined, the pH of the joined solution was adjusted to 9.2 with ammonia water, and 15 ml of acetone and 7.5 ml of toluene were added to the solution, followed by dropwise addition of 7.71 g of dicyclohexylamine. Then 15 ml of water was added dropwise, the solution was cooled to 5° C., and the precipitate was filtered and collected to give 9.68 g of dicyclohexylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile as a slightly yellowish white solid product.
Examples 2 to 5
The compounds shown in Table 1 were obtained in the same way as in Example 1-1.
| TABLE 1 | |||
 |
|||
| Example No. | Organic amine | Example No. | Organic amine |
| 2 | Dipropylamine | 4 | Dibenzylamine |
| 3 | Dibutylamine | 5 | N-benzylmethylamine |
Dipropylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) 6 values: 0.39 (6H, t, J=7.5 Hz), 1.10 (4H, sex, J=7.5 Hz), 2.30-2.38 (4H, m), 7.54 (1H, d, J=8.3 Hz)
Dibutylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) 6 values: 0.36 (6H, t, J=7.3 Hz), 0.81 (4H, sex, J=7.3 Hz), 0.99-1.10 (4H, m), 2.32-2.41 (4H, m), 7.53 (1H, d, J=8.3 Hz)
Dibenzylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) δ values: 4.17 (4H, s), 7.34-7.56 (10H, m), 8.07 (1H, d, J=8.3 Hz)
N-benzylmethylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) δ values: 2.57 (3H, s), 4.14 (2H, s), 7.37-7.53 (5H, m), 8.02-8.08 (1H, m)
Preparation Example 1

300 ml of toluene was added to a 600 ml water solution of 37.5 g of sodium hydroxide. Then 150 g of dicyclohexylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile was added at 15 to 25° C. and the solution was stirred at the same temperature for 30 minutes. The water layer was separated and washed with toluene, and then 150 ml of water was added, followed by dropwise addition of 106 g of a 30% hydrogen peroxide solution at 15 to 30° C. and one-hour stirring at 20 to 30° C. Then 39 ml of hydrochloric acid was added, the seed crystals were added at 40 to 50° C., and 39 ml of hydrochloric acid was further added dropwise at the same temperature. The solution was cooled to 10° C. the precipitate was filtered and collected to give 65.6 g of 6-fluoro-3-hydroxy-2-pyrazinecarboxamide as a slightly yellowish white solid.
1H-NMR (DMSO-d6) δ values: 8.50 (1H, s), 8.51 (1H, d, J=7.8 Hz), 8.75 (1H, s), 13.41 (1H, s)
CLIP
jan 2014
| US3631036 * | Nov 4, 1969 | Dec 28, 1971 | American Home Prod | 5-amino-2 6-substituted-7h-pyrrolo(2 3-d) pyrimidines and related compounds |
| US3745161 * | Apr 20, 1970 | Jul 10, 1973 | Merck & Co Inc | Phenyl-hydroxy-pyrazine carboxylic acids and derivatives |
| US4404203 * | May 14, 1981 | Sep 13, 1983 | Warner-Lambert Company | Substituted 6-phenyl-3(2H)-pyridazinones useful as cardiotonic agents |
| US4545810 * | Mar 25, 1983 | Oct 8, 1985 | Sds Biotech Corporation | Herbicidal and plant growth regulant diphenylpyridazinones |
| US4565814 * | Jan 18, 1984 | Jan 21, 1986 | Sanofi | Pyridazine derivatives having a psychotropic action and compositions |
| US4661145 * | Sep 20, 1984 | Apr 28, 1987 | Rohm And Haas Company | Plant growth regulating 1-aryl-1,4-dihydro-4-oxo(thio)-pyridazines |
| US5420130 | May 16, 1994 | May 30, 1995 | Synthelabo | 2-aminopyrazine-5-carboxamide derivatives, their preparation and their application in therapeutics |
| US5459142 * | Aug 23, 1993 | Oct 17, 1995 | Otsuka Pharmaceutical Co., Ltd. | Pyrazinyl and piperazinyl substituted pyrazine compounds |
| US5597823 | Jun 5, 1995 | Jan 28, 1997 | Abbott Laboratories | Tricyclic substituted hexahydrobenz [e]isoindole alpha-1 adrenergic antagonists |
| US6159980 * | Sep 15, 1997 | Dec 12, 2000 | Dupont Pharmaceuticals Company | Pyrazinones and triazinones and their derivatives thereof |
| EP0023358A1 * | Jul 28, 1980 | Feb 4, 1981 | Rohm And Haas Company | Process for the preparation of pyridazine derivatives |
| GB1198688A | Title not available | |||
| HU9401512A | Title not available | |||
| JPH09216883A * | Title not available | |||
| JPS5620576A | Title not available |
 |
|
| Names | |
|---|---|
| IUPAC name
5-Fluoro-2-hydroxypyrazine-3-carboxamide
|
|
| Other names
T-705; Avigan; favilavir
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEMBL | |
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C5H4FN3O2 | |
| Molar mass | 157.104 g·mol−1 |
| Pharmacology | |
| J05AX27 (WHO) | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
////////////

Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
WORLD DRUG TRACKER
one time
$10.00
PALINAVIR, BILA-2011-BS
| Patent | Submitted | Granted |
|---|---|---|
| Substituted pipecolinic acid derivatives as HIV protease inhibitors [US5614533] | 1997-03-25 | |
| Substituted pipecolinic acid derivatives as HIV protease inhibitors. [EP0560268] | 1993-09-15 | 1995-01-04 |

……………………….
PATENT
http://www.google.com/patents/WO2013105118A1?cl=en
Scheme 5: Synthesis of Palinavir (6):

The organic solvent mentioned according to the invention is selected from the group consisting of organic solvents, wherein the organic solvents are polar aprotic such as DCM, THF, Ethyl acetate, acetone, DMF, acetonitrile, DMSO ; polar protic solvents such as lower alcohol particularly (C1-C6) alkyl alcohol, water, acetic acid ; non-polar solvents such as hexane, benzene, toluene, chloroform, pet. ether, 1,4-dioxane, heptane either alone or mixtures thereof . Additionally the purification or separation of crude product can be accomplished by known techniques viz. extraction, column chromatography in a suitable organic solvent with the aid of instruments such as TLC, HPLC, GC, mass spectroscopy, or distillation, crystallization, derivatization.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
………………………….
J Org Chem 1997,62(11),3440

The reaction of tert-butoxycarbonyl-L-phenylalanine (I) with isobutyl chloroformate in THF gives the expected mixed anhydride which is treated with diazomethane and HCl yielding the corresponding chloromethyl ketone (II). The reduction of (II) with NaBH4 in THF affords the (S)-chlorohydrin (IV), which is treated with KOH in ethanol to obtain the chiral epoxide (V)(1,2). Ring opening of (V) with (?(cis)-N-tert-butyl-4-(4-pyridylmethoxy)piperidine-2-carboxamide (VI) by a treatment with LiCl in refluxing ethanol gives a mixture of diastereomers that is separated by chromatography giving the pure isomer (VII). The reaction of (VII) with tert-butoxycarbonyl-L-valine (VIII) by treatment first with trifluoroacetic acid (TFA), and condesation by means of BOP ((benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate) and NMM (N-methylmorpholine) affords the expected condensation product (IX). Finally, this compound is condensed with quinoline-2-carboxylic acid (X) by means of BOP and NMM as before. 2) The piperidine (VI) has been obtained by condensation of (?(cis)-N-(tert-butoxycarbonyl)-4-hydroxypiperidine-2-carboxamide (XI) with 4-(chloromethyl)pyridine (XII) by means of NaH in DMS, followed by hydrolysis with HCl.

Palinavir can also be obtained as follows: The controlled oxidation of 2(S)-(dibenzylamino)-3-phenyl-1-propanol (XIII) with pyridine-SO3 complex in DMSO gives the corresponding aldehyde (XIV), which is condensed with bromochloromethane (XV) by means of Li in THF followed by hydrolysis with HCl yielding regioselectively the 1-chloro-2-butanol (XVI). The debenzylation of (XVI) by hydrogenation over Pd/C affords the free amine (XVII), which is treated with tert-butoxycarbonyl anhydride/triethylamine and dehydrochlorinated with KOH in methanol to give the desired chiral epoxide (V).

The chiral piperidine (2S,4R)(VI) has been obtained as follows: The cyclization of 3-buten-1-ol (XXII) with (S)-1-phenylethylamine (XXIII) and glyoxylic acid (XXIV) by means of tosyl chloride in THF gives a mixture of the (2S,4R) and (2R,4S) lactones (XXV), which is resolved by fractional crystallyzation of their salts with the chiral camphorsulfonic acid (XXVI), followed by elimination of the acid with ammonia to afford (2S,4R)(XXVII). The reaction of lactone (XXVII) with isopropylmagnesium chloride and tert-butylamine in THF gives (2S,4R)-N-tert-butyl-4-hydroxy-1-(1(S)-phenylethyl)piperidine-2-carboxamide (XXVIII), which is debenzylated by hydrogenation and protected with tert-butoxycarbonyl anhydride yielding (2S,4R)-N-(tert-butoxycarbonyl)-4-hydroxypiperidine-2-carboxamide (2S,4R)(XI), which is finally condensed with 4-(chloromethyl)pyridine (XII) as before to obtain the chiral piperidine (2S,4R)(VI), already reported.

The condendsation of epoxide (V) with (2S,4R)(VI) by means of basic alumina in THF, followed by elimination of the protecting group with HCl and NaOH yields directly the condensation product (XVIII) as a pure diastereomer and with a free amino group. Finally, this compound is condensed with N-(2-quinolylcarbonyl)-L-valine (XIX) through its activation compound with isobutyl chloroformate (the 4(S)-isopropyl-2-(2-quinolyl)oxazol-5(4H)-one (XX)). The N-acyl-L-valine (XIX) has been obtained by acylation of L-valine (XXI) with quinoline-2-carboxylic acid (X) through its acyl chloride obtained with SOCl2.
………………………..

Palinavir is an inhibitor with five chiral centers. It contains the amino acid valine and pipecolinin acid. The previous way to create this drug faced three major obstacles. First, the reaction from 2 to 3 used diazomethane. Therefore, is is difficult, if not impossible, to produce large quantities. Secondly, the steps included in going from 4 to 5 gave way to racemers which is very inefficient. Finally, chromatography is needed at two separate times.

Four issues were addresses in route to product 1. First, because of the number of chiral centers, stereochemical control was a concern. high chemical yields were a second concern. Also, multi step procedures were advantageous to cut down on purification steps. Finally, the synthesis tried to restrict the use of hazardous reagents. The following retrosynthesis reaction was conceived and three target molecules were identified as seen in figure 1.

Molecule 3 uses a diaseteroselective addition of in situ (chloromethyl)lithium to N,N-dibenzylphenylalaninol and is derived from a four step process.

Recrystallization of 13 is required. Molecule 14 was not reached because it posed a problem later in the reaction. The N-benzyl protection group could not be removed to react with 9.

8 is a derivative of naturally occurring pipocolic acid, 16, named 3-buten-1-ol. Selective crystallization of diastereomeric salts can lead to 17a, but a more efficient way is by having a 60:40 mixture of lactones 17a,b. This leads to 18a,b using a Brodroux process. Crystallization of 18a,b lead to a poor overall yield. Instead, 18a,b undergoes salt crystallization with (-)-camphorsulfonic acid. Finally, 18a underwent hydrolysis and then addition of di-tert butyl dicarbonate leads to 8.
8 was then transformed to 5 in a three step process.
 8 was added to NaOH and alkylated with 4-picolyl chloride. The protecting group was lost with the addition of acid.
8 was added to NaOH and alkylated with 4-picolyl chloride. The protecting group was lost with the addition of acid.
 Derivation of 9 was started by a simple substitution of 19, quinoline-2-carboxylic acid, to 20, an acid chloride, with the help of thionyl chloride. Acylation of amino acid L-valine to 20 was accomplished by a biphasic system.
Derivation of 9 was started by a simple substitution of 19, quinoline-2-carboxylic acid, to 20, an acid chloride, with the help of thionyl chloride. Acylation of amino acid L-valine to 20 was accomplished by a biphasic system.
 In the original synthesis of palinavir, a 2:1 mixture of 3 to 5 was needed to produce only ~35% of 6 and flash chromatography was needed. On a large scale without chromatography, 6 was produced with a 85% yield, but 21 was also produced. To keep the production of 21 to a minimum, the reaction was performed in a solution that was degassed. This insured that the pyridine ring would not react in the presence of air. With this precaution, only 1-2% of the yield was 21. A washing of the solution with 1 M KH2PO4 removed and left over 5. Deprotection was achieved with the addition of concentrated HCl and followed by adding NaOH. The product of 10 was a “viscous syrup”. 22 was 1-1.5% of the product and was not removed before the addition of 9 to form 80-85% palinavir.
In the original synthesis of palinavir, a 2:1 mixture of 3 to 5 was needed to produce only ~35% of 6 and flash chromatography was needed. On a large scale without chromatography, 6 was produced with a 85% yield, but 21 was also produced. To keep the production of 21 to a minimum, the reaction was performed in a solution that was degassed. This insured that the pyridine ring would not react in the presence of air. With this precaution, only 1-2% of the yield was 21. A washing of the solution with 1 M KH2PO4 removed and left over 5. Deprotection was achieved with the addition of concentrated HCl and followed by adding NaOH. The product of 10 was a “viscous syrup”. 22 was 1-1.5% of the product and was not removed before the addition of 9 to form 80-85% palinavir.

Coupling of 10 and 9 is the final step in the synthesis , although there are still some purification steps left.

Two recrystallizations were required for the final 99.6% purity.

………………………..

Palinavir is a potent peptidomimetic-based HIV protease inhibitor. We have developed a highly convergent and stereoselective synthesis which is amenable to the preparation of multikilogram quantities of this compound. The synthetic sequence proceeds in 24 distinct chemical steps (with several integrated, multistep operations) from commercially available starting materials. No chromatographies are required throughout the process, and the final product is purified by crystallization of its dihydrochloride salt to >99% homogeneity.
crude palinavir (1) as a thick brown oil (yield not determined). HPLC analysis (Supelcosil LZ-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): 1, tR 17.80 min (84.1%); 24, tR 18.47 min (2.0%); 25, tR 19.97 min (1.45%).
palinavir dihydrochloride (1750 g, 51% yield) containing 0.25% w/w isopropanol (by 1H NMR):
mp 175−185 °C.
[α]25D −13.0° (c 1, MeOH). [α]25Hg365 +44.9° (c 1, MeOH).
IR (KBr) ν 3700−2300, 1660, 1555, 1520 cm-1.
1H NMR (DMSO-d6) δ 10.00 (broad s, 1H), 8.88 (d, J = 6.3 Hz, 2H), 8.61 (d, J = 8.4 Hz, 1H), 8.60 (s, 1H), 8.51 (d, J = 9.6 Hz, 1H), 8.35 (d, J = 8.7 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 7.94 (d, J = 6.0 Hz, 2H), 7.89 (t, J = 7.6 Hz, 1H), 7.74 (t, J = 7.5 Hz, 1H), 7.19 (d, J = 7.2 Hz, 2H), 7.08 (t, J = 7.5 Hz, 2H), 6.91 (t, J = 7.3 Hz, 1H), 4.86 (AB quartet, 2H), 4.37 (broad t, J = 7.8 Hz, 1H), 4.21 (d, J = 11.4 Hz, 1H), 4.11 (broad m, 1H), 3.96 (broad m, 1H), 3.80−3.65 (m, 2H), 3.26 (t, J = 7.4 Hz, 1H), 3.15−3.01 (m, 2H), 2.94 (broad d, J = 12.0 Hz, 1H), 2.62 (dd, J = 13.6, 10.6 Hz, 1H), 2.56 ((broad d, J = 12.0 Hz, 1H), 2.20−2.05 (m, 2H), 1.86 (m, 1H), 1.69 (q, J = 11.7 Hz, 1H), 1.31 (s, 9H), 0.81 (d, J = 6.3 Hz, 3H), 0.80 (d, J = 6.6 Hz, 3H).
13C NMR (DMSO-d6) δ 170.4, 166.4, 163.3, 158.3, 149.5, 145.9, 141.9, 138.6, 138.2, 130.7, 129.3, 129.1, 129.0, 128.3, 128.2, 128.0, 125.9, 124.1, 118.6, 72.3, 68.8, 67.2, 64.8, 58.0, 57.8, 54.4, 51.3, 51.1, 35.4, 34.1, 31.1, 28.2, 19.5, 17.9.
FAB-MS m/z 709 (MH+ of free base). Anal. Calcd for C41H54Cl2N6O5 (corrected for 8% water content as determined by Karl Fisher analysis and 0.25% w/w isopropanol as determined by 1H NMR): C, 58.31; H, 7.29; N, 9.93. Found: C, 57.76; H, 7.25; N, 9.89. Titration of HCl content using NaOH: 2.09 ± 0.03 mol HCl. HPLC homogeneity (Supelcosil LC-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): palinavir dihydrochloride, tR 18.24 min (99.51%); 25 tR 20.39 min (0.33%). HPLC homogeneity (Nova-Pak C8, 20−80% MeCN/50 mM NaH2PO4 in 25 min, 1 mL/min flow rate): palinavir dihydrochloride, tR 15.52 min (99.67%); 25 tR 13.52 min (0.33%).
PURE palinavir (1) as a white amorphous powder (1902 g, 84% yield):
mp 100−107 °C. [α]25D −11.5° (c 1, MeOH).
IR (KBr) ν 3700−3100, 1660, 1520, 1495 cm-1.
1H NMR (CDCl3) δ 8.54 (d, J = 5.7 Hz, 2H), 8.48 (d, J = 8.6 Hz, 1H), 8.31 (d, J = 8.6 Hz, 1H, part of AB), 8.22 (d, J = 8.3 Hz, 1H, part of AB), 8.13 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.80 (t, J = 7.6 Hz, 1H), 7.65 (t, J = 7.6 Hz, 1H), 7.25 (d, J = 5.4 Hz, 2H), 7.13 (d, J = 7.3 Hz, 2H), 7.07 (t, J = 7.5 Hz, 1H), 6.92 (t, J = 7.3 Hz, 1H), 6.59 (d, J = 8.3 Hz, 1H), 6.57 (s, 1H), 4.61 (d, J = 13.4 Hz, 1H, part of AB), 4.51 (d, J = 13.4 Hz, 1H, part of AB), 4.32 (dd, J = 8.6, 6.4 Hz, 1H), 4.22 (m, 1H), 3.97 (m, 1), 3.47−3.33 (m, 2H), 2.94 (dd, J = 14.3, 4.1 Hz, 1H), 2.89 (d, J= 8.6 Hz, 1H), 2.79−2.72 (m, 1H), 2.77 (dd, J = 14.3, 10.8 Hz, 1H), 2.43 (dd, J = 13.4, 8.3 Hz, 1H), 2.40−2.25 (m, 3H), 1.95 (broad d, J = 12.4 Hz, 1H), 1.65 (q J = 11.8 Hz, 2H), 1.32 (s, 9H), 0.95 (d, J = 7.0 Hz, 3H), 0.83 (d, J = 6.7 Hz, 3H).
13C NMR (CDCl3) δ 171.6, 171.2, 165.0, 149.8, 148.8, 147.9, 146.5, 137.6, 137.5, 130.3, 129.9, 129.5, 129.4, 129.0, 128.8, 128.5, 128.2, 127.7, 126.4, 121.7, 118.8, 75.0, 71.9, 68.1, 66.7, 59.4, 56.9, 54.6, 50.9, 50.2, 34.8, 33.3, 29.8, 29.7, 28.7, 19.6, 17.5.
FAB-MS m/z 709 (MH+). Anal. Calcd for C41H52N6O5(corrected for 0.7% water content as determined by Karl Fisher analysis): C, 68.98; H, 7.42; N, 11.77. Found: C, 68.71; H, 7.47; N, 11.71. HPLC homogeneity (Supelcosil LC-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): palinavir (1), tR 17.83 min (99.59%); 25 tR20.00 min (0.41%). HPLC homogeneity (Nova-Pak C8, 10−80% MeCN/50 mM NaH2PO4 in 25 min, 1 mL/min flow rate): palinavir (1), tR 17.37 min (99.51%); 25 tR 15.87 min (0.49%).
| Reference | ||
|---|---|---|
| 1 | * | ARUN K. GHOSH ET AL: “The Development of Cyclic Sulfolanes as Novel and High-Affinity P2 Ligands for HIV-1 Protease Inhibitors“, JOURNAL OF MEDICINAL CHEMISTRY, vol. 37, no. 8, 1 April 1994 (1994-04-01), pages 1177-1188, XP055057710, ISSN: 0022-2623, DOI: 10.1021/jm00034a016 |
| 2 | * | KAY BRICKMANN ET AL: “Synthesis of Conformationally Restricted Mimetics of [gamma]-Turns and Incorporation into Desmopressin, an Analogue of the Peptide Hormone Vasopressin“, CHEMISTRY – A EUROPEAN JOURNAL, vol. 5, no. 8, 2 August 1999 (1999-08-02), pages 2241-2253, XP055057517, ISSN: 0947-6539, DOI: 10.1002/(SICI)1521-3765(19990802)5:8<2241: :AID-CHEM2241>3.0.CO;2-L |
| 3 | * | KIRAN I N C ET AL: “A concise enantioselective synthesis of (+)-goniodiol and (+)-8-methoxygoniodiol via Co-catalyzed HKR of anti-(2SR, 3RS)-3-methoxy-3-phenyl-1, 2-epoxypropane“, TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 52, no. 3, 19 January 2011 (2011-01-19), pages 438-440, XP027558447, ISSN: 0040-4039 [retrieved on 2010-12-14] |
| 4 | * | M. TOKUNAGA: “Asymmetric Catalysis with Water: Efficient Kinetic Resolution of Terminal Epoxides by Means of Catalytic Hydrolysis“, SCIENCE, vol. 277, no. 5328, 15 August 1997 (1997-08-15), pages 936-938, XP055057541, ISSN: 0036-8075, DOI: 10.1126/science.277.5328.936 |
| 5 | * | PARKES K E B ET AL: “STUDIES TOWARD THE LARGE-SCALE SYNTHESIS OF THE HIV PROTEINASE INHIBITOR RO 31-8959“, JOURNAL OF ORGANIC CHEMISTRY, ACS, US, vol. 59, no. 13/16, 1 January 1994 (1994-01-01), pages 3656-3664, XP002011975, ISSN: 0022-3263, DOI: 10.1021/JO00092A026 |
| 6 | * | R. SANTHOSH REDDY ET AL: “Co(iii)(salen)-catalyzed HKR of two stereocentered alkoxy- and azido epoxides: a concise enantioselective synthesis of (S,S)-reboxetine and (+)-epi-cytoxazone“, CHEMICAL COMMUNICATIONS, vol. 46, no. 27, 1 January 2010 (2010-01-01), page 5012, XP055057537, ISSN: 1359-7345, DOI: 10.1039/c0cc00650e |
| 7 | * | SHINJI NAGUMO ET AL: “Intramolecular Friedel-Crafts type reaction of vinyloxiranes linked to an ester group“, TETRAHEDRON, vol. 65, no. 47, 1 November 2009 (2009-11-01), pages 9884-9896, XP055057655, ISSN: 0040-4020, DOI: 10.1016/j.tet.2009.09.037 |
| 8 | * | SUNITA K. GADAKH ET AL: “Enantioselective synthesis of HIV protease inhibitor amprenavir via Co-catalyzed HKR of 2-(1-azido-2-phenylethyl)oxirane“, TETRAHEDRON: ASYMMETRY, vol. 23, no. 11-12, 1 June 2012 (2012-06-01), pages 898-903, XP055057475, ISSN: 0957-4166, DOI: 10.1016/j.tetasy.2012.06.003
Want to know everything on vir series click http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html AND http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html |

Amprenavir
KVX-478, 141W94, VX-478,
(3S)-Tetrahydro-3-furanyl ((1S,2R)-3-(((4-aminophenyl)sulfonyl)(2-methylpropyl)amino)-2-hydroxy-1-(phenylmethyl)propyl)carbamate
(3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl] carbamate
CAS NO. 161814-49-9, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate
| 161814-49-9 | |
| Weight | 505.224656557 |
|---|---|
| Chemical Formula | C25H35N3O6S |
| Amprenavir is a protease inhibitor used to treat HIV infection. |
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available.
| Amprenavir is a protease inhibitor with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Protease inhibitors block the part of HIV called protease. HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Amprenavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. Protease inhibitors are almost always used in combination with at least two other anti-HIV drugs. |

| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| United States | 5585397 | 1993-12-17 | 2013-12-17 |
| United States | 6730679 | 1997-11-11 | 2017-11-11 |
Research aimed at development of renin inhibitors as potential antihypertensive agents had led to the discovery of compounds that blocked the action of this peptide cleaving enzyme. The amino acid sequence cleaved by renin was found to be fortuitously the same as that required to produce the HIV peptide coat. Structure–activity studies on renin inhibitors proved to be of great value for developing HIV protease inhibitors. Incorporation of an amino alcohol moiety proved crucial to inhibitory activity for many of these agents. This unit is closely related to the one found in the statine, an unusual amino acid that forms part of the pepstatin, a fermentation product that inhibits protease enzymes.
R.D. Tung, M.A. Murcko, G.R. Bhisetti, U.S. Patent 5,558,397 (1996). The scheme shown here is partly based on that used to prepare darunavir and fosamprenavir due to difficulty in deciphering the patent.
AGENERASE (amprenavir) is an inhibitor of the human immunodeficiency virus (HIV) protease. The chemical name of amprenavir is (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulfonamido)-1-benzyl-2-hydroxypropyl]carbamate. Amprenavir is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H35N3O6S and a molecular weight of 505.64. It has the following structural formula:
 |
Amprenavir is a white to cream-colored solid with a solubility of approximately 0.04 mg/mL in water at 25°C.
AGENERASE Capsules (amprenavir capsules) are available for oral administration. Each 50- mg capsule contains the inactive ingredients d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), polyethylene glycol 400 (PEG 400) 246.7 mg, and propylene glycol 19 mg. The capsule shell contains the inactive ingredients d-sorbitol and sorbitans solution, gelatin, glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink. Each 50- mg AGENERASE Capsule contains 36.3 IU vitamin E in the form of TPGS. The total amount of vitamin E in the recommended daily adult dose of AGENERASE is 1,744 IU.
………………………………
paper
DOI: 10.1039/B404071F
http://pubs.rsc.org/en/content/articlelanding/2004/ob/b404071f#!divAbstract

Efficient and industrially applicable synthetic processes for precursors of HIV protease inhibitors (Amprenavir, Fosamprenavir) are described. These involve a novel and economical method for the preparation of a key intermediate, (3S)-hydroxytetrahydrofuran, from L-malic acid. Three new approaches to the assembly of Amprenavir are also discussed. Of these, a synthetic route in which an (S)-tetrahydrofuranyloxy carbonyl is attached to L-phenylalanine appears to be the most promising manufacturing process, in that it offers satisfactory stereoselectivity in fewer steps.

Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html

The reaction of N,N-dibenzyl-L-alaninal (I) with nitromethane, catalyzed by the chiral ammonium salt (II) and KF in THF gives the chiral nitroalcohol (III), which is reduced with NiCl2 and NaBH4 to yield the aminoalcohol (IV). The condensation of (IV) with isobutyraldehyde (V) affords the Schiff base (VI), which is reduced with NaBH4 to provide the secondary amine (VII). The reaction of (VII) with 4-nitrobenzenesulfonyl chloride (VIII) and TEA in dichloromethane furnishes the sulfonamide (IX), which is deprotected by hydrogenation with H2 over Pd/C in methanol, giving the diamino compound (X). Finally, this compound is condensed with 3(S)-tetrahydrofuryl (N-oxysuccinimidyl) carbonate (XI) by means of TEA in dichloromethane to afford the target carbamate.
Angew Chem. Int Ed Engl1999,38,(13-14):1931
……………………………………………………….

The reaction of the chiral epoxide (I) with isobutylamine (II) in refluxing ethanol gives the secondary amine (III), which is protected with benzyl chloroformate (IV) and TEA, yielding the dicarbamate (V). Selective deprotection of (V) with dry HCl in ethyl acetate affords the primary amine (VI), which is treated with 3(S)-tetrahydrofuryl N-succinimidinyl carbonate (VII) (prepared by condensation of tetrahydrofuran-3(S)-ol (VIII) with phosgene and N-hydroxysuccinimide (IX)) and DIEA in acetonitrile to provide the corresponding carbamate (X). The deprotection of (X) by hydrogenation with H2 over Pd/C in ethanol gives the secondary amine (XI), which is condensed with 4-nitrophenylsulfonyl chloride (XII) by means of NaHCO3 in dichloromethane/water to yield the sulfonamide (XIII). Finally, the nitro group of (XIII) is reduced with H2 over Pd/C in ethyl acetate to afford the target compound.
https://www.google.com/patents/WO1999048885A1?cl=ensynthesis of (3S)-tetrahydro-3-furyl N-[(1S,2R)-3(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl]carbamate, hereinafter referred to as the compound of formula (I), and to novel intermediates thereto.The compound of formula (I) has the following structure

and was first described in PCT patent publication number WO94/05639 at Example 168. Currently there is considerable interest in the compound of formula (I) as a new chemotherapeutic compound in the treatment of human immunodeficiency virus (HIV) infection and the associated conditions such as acquired immune deficiency syndrome (AIDS) and AIDS dementia.
There exists at the present time a need to produce large quantities of the compound of formula (I) for clinical investigation into the efficacy and safety of the compound as a chemotherapeutic agent in the treatment of HIV infections.
An ideal route for the synthesis of the compound should produce the compound of formula (I) in high yields at a reasonable speed and at low cost with minimum waste materials and in a manner that is of minimum impact to the environment in terms of disposing of waste-materials and energy consumption.
We have found a new process for the synthesis of the compound of formula (I) with many advantages over previously known routes of synthesis. Such advantages include lower cost, less waste and more efficient use of materials. The new process enables advantageous preparation of the compound of formula (I) on a manufacturing scale.
The route of synthesis of the compound of formula (I) described in the specification of WO94/05639 is specifically described therein in examples 39A, 51A, 51B, 51C, 51D, 167 and 168. The overall yield from these examples is 33.2% of theory.
Generally the route described in WO94/05639 involves protecting the amino alcohol of formula (A) (Ex.39)

wherein P is a protecting group to form a compound of formula (B);

wherein P and P′ are each independently a protecting group;
deprotecting the compound of formula (B) to form a compound of formula (C) (Ex 51A);

wherein P′ is a protecting group;
forming a hydrochloride salt of compound (C) (Ex 51B) then reacting with N-imidazolyl-(S)-tetrahydrofuryl carbamate to form the compound of formula (D) (Ex 51C);

wherein P′ is a protecting group;
deprotecting the compound of formula (D) (Ex 51D) wherein P′ is a protecting group to form the compound of formula (D) wherein P′ is H (Ex 51E); and coupling the resultant secondary amine on the compound of formula (D) to a p-nitrophenylsulphonyl group to form a compound of formula (E) (Ex 167);

the resultant compound of formula (E) is then reduced to form the compound of formula (I) (Ex 168).
In summary, the process disclosed in WO94/05639 for producing the compound of formula (I) from the compound of formula (A) comprises 6 distinct stages:
1) protecting,
2) deprotecting,
3) reacting the resultant compound with an activated tetrahydrofuranol group,
4) deprotecting,
5) coupling with a p-nitrophenylsulfonyl group, and
6) reducing the resultant compound to form a compound of formula (I).
Applicants have now found a process by which the compound of formula (I) may be prepared on a manufacturing scale from the same starting intermediate, the compound of formula (A), in only 4 distinct stages instead of 6. In addition to the associated benefits of fewer stages, such as savings in time and cost, the improved process reduces the number of waste products formed. Furthermore, product may be obtained in a higher yield, of approximately 50% of theory
 .
.
EXAMPLESExample 1
(1S,2R)-tert-butyl N-[1-benzyl-2-hydroxy-3-(isobutylamino)propyl]carbamate (127.77 g, 379.7 mmol) was heated in toluene (888 ml) to 80° C. and triethylamine (42.6 g, 417.8 mmol) added. The mixture was heated to 90° C. and a solution of p-nitrobenzene sulphonyl chloride (94.3 g, 425.4 mmol) in toluene (250 ml) was added over 30 minutes then stirred for a further 2 hours. The resultant solution of the nosylated intermediate {(1S,2R)-tert-butyl N-[1-benzyl-2-hydroxy-3-(N-isobutyl- 4-nitrobenzenesulphonamido)propyl]carbamate } was then cooled to 80° C. The solution was maintained at approximately 80° C., and concentrated hydrochloric acid (31.4 ml, 376.8 mmol) was added over 20 minutes. The mixture was heated to reflux (approx 86° C.) and maintained at this temperature for an hour then a further quantity of concentrated hydrochloric acid (26.4 ml, 316.8 mmol) was added. Solvent (water and toluene mixture) was removed from the reaction mixture by azeotropic distillation (total volume of solvent removed approx 600 ml), and the resultant suspension was cooled to 70-75° C. Denatured ethanol (600 ml) was added, and the solution was cooled to 20° C. The mixture was further cooled to approximately −10° C. and the precipitate formed was isolated by filtration, washed with denatured ethanol (50 ml) and dried at approximately 50° C., under vacuum, for approximately 12 hours, to give (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride (160 g; 73% of theory yield corrected for assay). NMR: 1H NMR (300Mhz, dmso-d6): 8.37(2H, d, J=9 Hz), 8.16(NH3 +s), 8.06(2H, d, J=9 Hz), 7.31(5H, m), 5.65(1H, d, J=5 Hz), 3.95(1H, m), 3.39(2H, m), 2.95(5H, m), 1.90(1H, m), 0.77(6H, dd, J=21 Hz and 6 Hz).
1,1′-carbonyidiimidazole (27.66 kg, 170.58 mol) was added to ethyl acetate (314.3 kg) with stirring to give 3-(S)-tetrahydrofuryl imidazole-1-carboxylate. (S)-3-hydroxytetrahydrofuran (157 kg, 178.19 mol) was added over 30 minutes, washed in with ethyl acetate (9.95 kg), then the mixture was stirred for a further hour. (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride (65.08 kg, 142.10 mol) was added and the mixture heated to reflux for approximately 22 hours. The solution was cooled slightly, and denatured ethanol (98 l) was added. The solution was stirred at 60° C. for 10 minutes then cooled and the product allowed to crystallise. The mixture was cooled to <10° C. and stirred for 2 hours. The product was isolated by filtration, washed with denatured ethanol (33 l) and dried at approximately 50° C., under vacuum to give (3S)-tetrahydro-3-furyl N-[(1S,2R)-1-benzyl-2-hydroxy-3-(N-isobutyl-4-nitrobenzene sulphonamido)propyl]carbamate in a yield of 82% of theory.
NMR: 1H NMR (500 Mhz, dmso-d6): 8.38(2H, d, J=9Hz), 8.06(2H, d, J=9 Hz), 7.20(6H, m), 5.02(1H, d, J=5 Hz), 4.94(1H, m), 4.35(EtOH, broad s), 3.71(EtOH, q), 3.65(1H, m), 3.60(1H, m), 3.51(2H, broad m), 3.40(2H, m), 3.15(1H, dd, J=8 Hz and 14 Hz), 3.07(1H, dd, J=8 Hz and 15 Hz), 2.94(2H, m), 2.48(1H, m), 2.06(1H, m), 1.97(1H, m), 1.78(1H, m), 1.05(EtOH, t), 0.83(6H, dd, J=7 Hz and 16 Hz).
Product from the above stage (80.0 g, 149.4 mmol) was hydrogenated in isopropanol (880 ml) with 5% palladium on carbon (16 g, of a wet paste) and hydrogen pressure (approx 0.5 to 1.5 bar) at 25-50° C. for approximately 5 hours. The mixture was cooled and the catalyst removed by filtration. The solution was distilled to a volume of approximately 320 ml and water (80 ml) was added. This solution was divided into two for the crystallisation step.
To half of the above solution, decolourising charcoal (2 g) was added, the mixture stirred at approximately 32° C. for 4 hours, then filtered. The filtercake was washed with isopropanol (20 ml) then further water (40 ml) was added to the filtrate. The solution was seeded to induce crystallisation and stirred for 5 hours. Water (130 ml) was added slowly over 1 hour then the mixture was stirred for 4 hours. The resultant slurry was cooled to approximately 20° C. and the product was isolated by filtration and washed with a 1:4 mixture of isopropano/water (120 ml). The product was dried at approximately 50° C., under vacuum, for approximately 12 hours to give (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl] carbamate (30.3 g; 80% of theory yield).
NMR: 1H NMR (300 Mhz, dmso-d6): 7.39(2H, d, J=9 Hz), 7.18(6H, m), 6.60(2H, d, J=9 Hz), 6.00(2H, s), 4.99(1H, d, J=6 Hz), 4.93(1H, ddt), 3.64(5H, m), 3.34(1H, m), 3.28(1H, dd, J=14 Hz and 3 Hz), 3.01(1H, m, J=14 Hz and 3 Hz), 2.91(1H, m), 2.66(2H, m), 2.50(1H, m), 2.05(1H, m), 1.94(1H, m), 1.78(1H, m), 0.81(6H, dd, J=16 Hz and 7 Hz). m/z: 506.2(M+H+)
Example 11Synthesis of Amprenavir (1)To a solution of carbamate nitro derivative 15 (0.05 g, 0.09 mmol) in 2 mL of EtOAc was added SnCl2.2H2O (0.1 g, 0.5 mmol) at 70° C. The reaction mixture was heated for 1 h until starting material disappeared and the solution cooled to room temperature. It was then poured into saturated aq. NaHCO3 solution and extracted with EtOAc. The organic extract was dried over anhyd. Na2SO4 and concentrated under reduced pressure. It was purified over chromatography using petroleum ether:EtOAc (3:2) to give amprenavir 1 (0.04 g, 90%).IR: (CHCl3, cm−1): υmax 757, 1090, 1149, 1316, 1504, 1597, 1633, 1705, 2960, 3371; 1H NMR (200 MHz, CDC3): δ 0.86 (d, J=5.7 Hz, 3H), 0.90 (d, J=6.6 Hz, 3H), 1.78-2.21 (m, 3H), 235-3.11 (m, 6H), 3.58-4.11 (m, 7H), 4.25 (s, 2H), 5.01 (br s, 1H), 5.07 (br s, 1H), 6.65 (d, J=8.4 Hz, 2H), 7.20-7.28 (m, 5H), 7.51 (d, J=8.4 Hz, 2H); 13C NMR (50 MHz, CDC3): δ 19.9, 20.2, 27.3, 32.8, 35.4, 35.7, 53.8, 55.0, 58.6, 66.8, 72.6, 73.2, 75.3, 114.0, 125.9, 126.5, 1280.4, 129.5, 137.7, 150.9, 155.9;
Anal. Calcd for C25H35N3O6S: C, 59.39; H, 6.98; N, 8.31; S, 6.34. Found: C, 59.36; H, 6.81; N, 8.25; S, 6.29%.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-23/000/115/669/161814-49-9-1h.png)
![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-23/000/115/669/161814-49-9-13c.png)
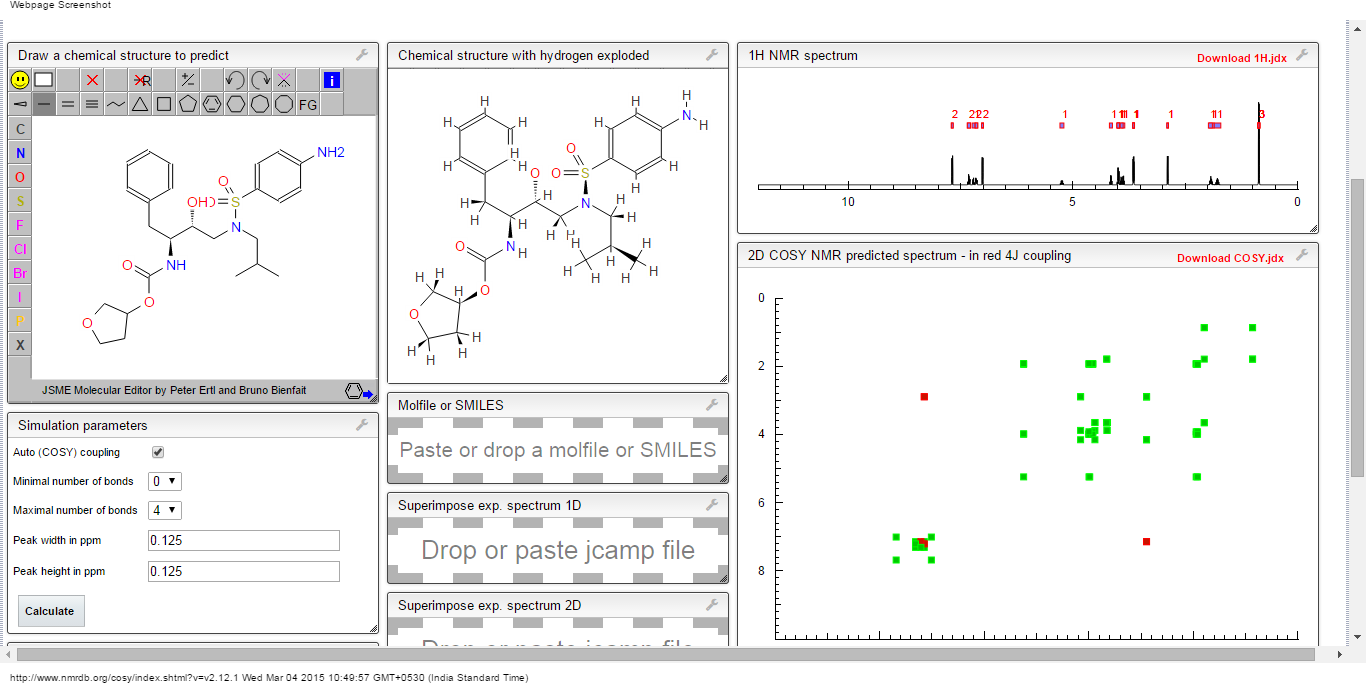
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3S)-oxolan-3-yl N-[(2S,3R)-3-hydroxy-4-[N-(2-methylpropyl)(4-aminobenzene)sulfonamido]-1-phenylbutan-2-yl]carbamate | |
| Clinical data | |
| Trade names | Agenerase |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699051 |
| Licence data | EMA:Link, US FDA:link |
|
|
| Legal status |
?
|
| Routes | oral |
| Pharmacokinetic data | |
| Protein binding | 90% |
| Metabolism | hepatic |
| Half-life | 7.1-10.6 hours |
| Excretion | <3% renal |
| Identifiers | |
| CAS number | 161814-49-9 |
| ATC code | J05AE05 |
| PubChem | CID 65016 |
| DrugBank | DB00701 |
| ChemSpider | 58532 |
| UNII | 5S0W860XNR |
| KEGG | D00894 |
| ChEBI | CHEBI:40050 |
| ChEMBL | CHEMBL116 |
| NIAID ChemDB | 006080 |
| Chemical data | |
| Formula | C25H35N3O6S |
| Molecular mass | 505.628 g/mol |
![]()
India-based Sun Pharmaceutical Industries has agreed to acquire GlaxoSmithKline’s (GSK) opiates business in Australia.
The agreement has been signed by wholly-owned subsidiaries of both firms, with the financial terms not disclosed.
Sun Pharma API business executive vice-president Iftach Seri said: “The global opiates market holds good potential and the addition of GSK’s Opiates business will strengthen our positioning further.”
Sun Pharmaceutical Industries Ltd(SUN.NS), India’s largest drugmaker by sales, said on Tuesday it has agreed to buy GlaxoSmithKline’s(GSK.L) opiates business in Australia to strengthen its pain management portfolio.
The business consists of analgesics made from raw materials found in opium poppy plants, and includes two manufacturing sites in the states of Tasmania and Victoria.
Financial details of the deal were not disclosed. A Sun Pharma spokesman declined to comment. Glaxo did not immediately respond to a request seeking comment.
Glaxo supplies a quarter of the world’s medicinal opiate needs from poppies grown by farmers in Tasmania, according to the company website. The company’s Australian opiates business brought in revenue of A$89 million ($69.63 million) in 2013.
Australia’s poppy industry is the world’s largest legal supplier of pharmaceutical grade opiates for painkillers, and Glaxo is one of three firms that control the crop and production in Tasmania.
The other two are Johnson & Johnson’s (JNJ.N) unit Tasmanian Alkaloids, and privately-held TPI Enterprises.
Glaxo’s decision to part with the opiates business comes as Tasmania’s poppy industry is facing a tough crop and the United Nations is expected to cut the state’s poppy crop area this week.
Glaxo said the deal would allow it to “focus on delivering its innovative product portfolio” in Australia.
“The opiates business has been an important part of our Australian business for many years, but as our portfolio transitions, we believe now is the right time to hand this business over to someone else,” Steve Morris, general manager of GSK Opiates, said in a statement.
The business employs 185 staff, including 155 in Victoria state and 30 in Tasmania state. Sun Pharma said it would hire all employees from both sites.
“The acquisition is a part of our strategy towards building our portfolio of opiates and accessing strong capabilities in this segment,” said Iftach Seri, executive vice president of the active pharmaceuticals ingredients business at Sun Pharma.
Both companies said they expect to close the deal by August.
Sun Pharma shares closed 1.93 percent higher on Tuesday, while the broader Nifty rose 0.44 percent.
($1 = 1.2781 Australian dollars)
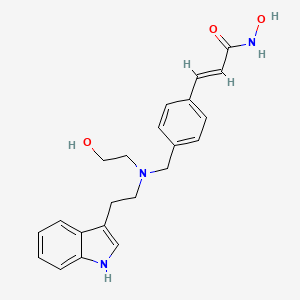
C22H25N3O3
Exact Mass: 379.18959
Molecular Weight: 379.45

Reversible acetylation of histones is a major regulator of gene expression that acts by altering accessibility of transcription factors to DNA. In normal cells, histone deacetylase (HDA) and histone acetyltrasferase together control the level of acetylation of histones to maintain a balance. Inhibition of HDA results in the accumulation of hyperacetylated histones, which results in a variety of cellular responses.
Inhibitors of HDA have been studied for their therapeutic effects on cancer cells. For example, butyric acid and its derivatives, including sodium phenylbutyrate, have been reported to induce apoptosis in vitro in human colon carcinoma, leukemia and retinoblastoma cell lines. However, butyric acid and its derivatives are not useful pharmacological agents because they tend to be metabolized rapidly and have a very short half-life in vivo. Other inhibitors of HDA that have been widely studied for their anti-cancer activities are trichostatin A and trapoxin. Trichostatin A is an antifungal and antibiotic and is a reversible inhibitor of mammalian HDA. Trapoxin is a cyclic tetrapeptide, which is an irreversible inhibitor of mammalian HDA.
Although trichostatin and trapoxin have been studied for their anti-cancer activities, the in vivo instability of the compounds makes them less suitable as anti-cancer drugs. There remains a need for an active compound that is suitable for treating tumors, including cancerous tumors, that is highly efficacious and stable

|
References |
1: Wang H, Cheng F, Woan K, Sahakian E, Merino O, Rock-Klotz J, Vicente-Suarez I, Pinilla-Ibarz J, Wright KL, Seto E, Bhalla K, Villagra A, Sotomayor EM. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol. 2011 Apr 1;186(7):3986-96. doi: 10.4049/jimmunol.1001101. Epub 2011 Mar 2. PubMed PMID: 21368229.
2: Schwarz K, Romanski A, Puccetti E, Wietbrauk S, Vogel A, Keller M, Scott JW, Serve H, Bug G. The deacetylase inhibitor LAQ824 induces notch signalling in haematopoietic progenitor cells. Leuk Res. 2011 Jan;35(1):119-25. doi: 10.1016/j.leukres.2010.06.024. Epub 2010 Jul 31. PubMed PMID: 20674020.
3: Cho YS, Whitehead L, Li J, Chen CH, Jiang L, Vögtle M, Francotte E, Richert P, Wagner T, Traebert M, Lu Q, Cao X, Dumotier B, Fejzo J, Rajan S, Wang P, Yan-Neale Y, Shao W, Atadja P, Shultz M. Conformational refinement of hydroxamate-based histone deacetylase inhibitors and exploration of 3-piperidin-3-ylindole analogues of dacinostat (LAQ824). J Med Chem. 2010 Apr 8;53(7):2952-63. doi: 10.1021/jm100007m. PubMed PMID: 20205394.
4: Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, de la Rocha P, Yang MY, Mok S, Garban HJ, Craft N, Economou JS, Marincola FM, Wang E, Ribas A. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009 Nov 15;69(22):8693-9. doi: 10.1158/0008-5472.CAN-09-1456. Epub 2009 Oct 27. PubMed PMID: 19861533; PubMed Central PMCID: PMC2779578.
5: Ellis L, Bots M, Lindemann RK, Bolden JE, Newbold A, Cluse LA, Scott CL, Strasser A, Atadja P, Lowe SW, Johnstone RW. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood. 2009 Jul 9;114(2):380-93. doi: 10.1182/blood-2008-10-182758. Epub 2009 Apr 21. PubMed PMID: 19383971.
6: de Bono JS, Kristeleit R, Tolcher A, Fong P, Pacey S, Karavasilis V, Mita M, Shaw H, Workman P, Kaye S, Rowinsky EK, Aherne W, Atadja P, Scott JW, Patnaik A. Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin Cancer Res. 2008 Oct 15;14(20):6663-73. doi: 10.1158/1078-0432.CCR-08-0376. PubMed PMID: 18927309.
7: Chung YL, Troy H, Kristeleit R, Aherne W, Jackson LE, Atadja P, Griffiths JR, Judson IR, Workman P, Leach MO, Beloueche-Babari M. Noninvasive magnetic resonance spectroscopic pharmacodynamic markers of a novel histone deacetylase inhibitor, LAQ824, in human colon carcinoma cells and xenografts. Neoplasia. 2008 Apr;10(4):303-13. PubMed PMID: 18392140; PubMed Central PMCID: PMC2288545.
8: Cuneo KC, Fu A, Osusky K, Huamani J, Hallahan DE, Geng L. Histone deacetylase inhibitor NVP-LAQ824 sensitizes human nonsmall cell lung cancer to the cytotoxic effects of ionizing radiation. Anticancer Drugs. 2007 Aug;18(7):793-800. PubMed PMID: 17581301.
9: Kato Y, Salumbides BC, Wang XF, Qian DZ, Williams S, Wei Y, Sanni TB, Atadja P, Pili R. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol Cancer Ther. 2007 Jan;6(1):70-81. PubMed PMID: 17237267.
10: Leyton J, Alao JP, Da Costa M, Stavropoulou AV, Latigo JR, Perumal M, Pillai R, He Q, Atadja P, Lam EW, Workman P, Vigushin DM, Aboagye EO. In vivo biological activity of the histone deacetylase inhibitor LAQ824 is detectable with 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography. Cancer Res. 2006 Aug 1;66(15):7621-9. PubMed PMID: 16885362.
SEE MORE AT……….http://drugsynthesisint.blogspot.in/p/nostat-series.html
SEE MORE AT……….http://drugsynthesisint.blogspot.in/p/nostat-series.html