

TIRUPATI, INDIA
 .
.
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64


β-Sitosterol, 후박(厚朴)

β-Sitosterol
http://www.herbdb.co.kr/herb/dbsearch3/separation_view.asp?key=302
| C29H50O, 414.00 | |||
| White needles | |||
| m.p(℃) | 283-285 | ||
| IR(cm-¹) | νmax (KBr): 3400, 1680 | ||
| UV(nm) | λmax (MeOH): 216 | ||
| MS | EIMS m/z: 414 [M]+ | ||


β-Sitosterol (β-谷甾醇); CAS: 83-46-5

(300 MHz, CDCl3) δ: 5.36 (1H, d, J = 5.2 Hz, H-6), 3.53 (1H, m, H-3),1.01 (3H, s, CH3-19), 0.94 (3H, d, J = 6.5 Hz, CH3-21), 0.92 (3H, d, J = 6.5 Hz,CH3-26), 0.83 (3H, t, J = 6.6 Hz, CH3-29), 0.69 (3H, s, CH3-18)
13c nmr

(75 MHz, CDCl3) δ: 37.2 (C-1), 32.1 (C-2), 72.0 (C-3), 42.5 (C-4), 141.0 (C-5), 121.9 (C-6), 32.1 (C-7), 31.9 (C-8), 50.4 (C-9), 36.7 (C-10), 21.3 (C-11), 40.0 (C-12), 42.5 (C-13), 57.0 (C-14), 24.5 (C-15), 28.4 (C-16), 56.3 (C-17), 12.2 (C-18), 19.2 (C-19), 36.3 (C-20), 19.0 (C-21), 34.2 (C-22), 26.4 (C-23),46.1 (C-24), 29.4 (C-25), 19.6 (C-26), 20.0 (C-27), 23.3 (C-28),12.0 (C-29)

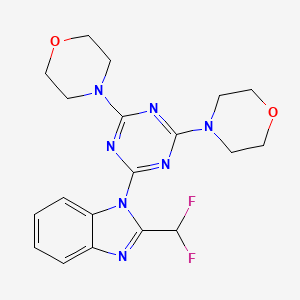
ZSTK 474

4-[4-[2-(difluoromethyl)benzimidazol-1-yl]-6-morpholin-4-yl-1,3,5-triazin-2-yl]morpholine
ZSTK474; 475110-96-4; 4,4′-(6-(2-(Difluoromethyl)-1H-benzo[d]imidazol-1-yl)-1,3,5-triazine-2,4-diyl)dimorpholine; ZSTK-474; ZSTK 474; TCMDC-137004;
2-(2-Difluoromethylbenzimidazol-1-yl)-4,6-bis(morpholino)-1,3,5-triazine
2-(2-difluoromethylbenzimidazol-1-yl)-4,6-dimorpholino-1,3,5-triazine
Zenyaku Kogyo (Innovator)
phase2………Treatment of Solid Tumors Therapy
ZSTK474 is a cell permeable and reversible P13K inhibitor with an IC₅₀ at 6nm. It was identified as part of a screening library, selected for its ability to block tumor cell growth. ZSTK474 has shown strong antitumor activities against human cancer xenographs when administered orally to mice without a significant toxic effect.
Phosphatidylinositol 3-kinase (PI3K) has been implicated in a variety of diseases including cancer. A number of PI3K inhibitors have recently been developed for use in cancer therapy. ZSTK474 is a highly promising antitumor agent targeting PI3K. We previously reported that ZSTK474 showed potent inhibition against four class I PI3K isoforms but not against 140 protein kinases.
However, whether ZSTK474 inhibits DNA-dependent protein kinase (DNA-PK), which is structurally similar to PI3K, remains unknown. To investigate the inhibition of DNA-PK, we developed a new DNA-PK assay method using Kinase-Glo. The inhibition activity of ZSTK474 against DNA-PK was determined, and shown to be far weaker compared with that observed against PI3K. The inhibition selectivity of ZSTK474 for PI3K over DNA-PK was significantly higher than other PI3K inhibitors, namely NVP-BEZ235, PI-103 and LY294002.
Other Names: ZSTK-474
Chemical Formula: C19H21F2N7O2
CAS Number: 475110-96-4
Molecular Weight: 417.41
WO 2002088112
http://www.google.co.in/patents/EP1389617A1?cl=en
The condensation of 2,4-dichloro-6-(4-morpholinyl)-1,3,5-triazine

with 2-(difluoromethyl)-1H-benzimidazole by means of K2CO3 in DMF gives

2-chloro-4-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-6-(4-morpholinyl)-1,3,5-triazine ,

which is then condensed with morpholine by means of K2CO3 in DMF to afford the target trisubstituted triazine.
aReagents and conditions: (i) K2CO3, DMF, room temp; (ii) morpholine, DMF or THF, room temp; (iii) NaH or K2CO3, DMF or DMSO, 120 °C.

- 2-(2-difluoromethylbenzimidazol-1-yl)-4,6-dimorpholino-1,3,5-triazine(compound 19)
Melting point: 211-214°C
NMR(CDCl3) δ : 3.79(8H, t, J=4Hz), 3.88(8H, t, J=4Hz), 7.3-7.4(2H, m), 7.56(1H, t, J=53Hz), 7.88(1H, d, J=7Hz), 8.32(1H, d, J=7Hz)
MS m/z: 417(M+
……………………
J. Med. Chem., 2011, 54 (20), pp 7105–7126
DOI: 10.1021/jm200688y
1 (0.35 g, 84% yield): mp (EtOH) 217–219 °C (lit. 211–214 °C);
1H NMR (CDCl3) δ 8.33 (dd, J = 7.3, 1.4 Hz, 1H), 7.89 (dd, J = 7.2, 1.5 Hz, 1H), 7.56 (t, JHF= 53.6 Hz, 1H), 7.46–7.37 (m, 2H), 3.91–3.86 (m, 8H), 3.81–3.76 (m, 8H).
Kawashima, S.; Matsuno, T.; Yaguchi, S.; Sasahara, H.; Watanabe, T.Preparation of Heterocyclic Compounds as Antitumor Agents. PCT Int. Appl. WO 02088112, 2002;
Chem. Abstr. 2002, 137, 370113.
………………………………….
2-(difluoromethyl)-1H-benzimidazole
A mixture of o-phenylenediamine (5.41 g, 50 mmol) and difluoroacetic acid (9.6 g, 100 mmol) in 4 M HCl (20 mL) was heated under reflux for 1 h and diluted with hot water (50 mL). The solution was treated with charcoal and filtered through Celite before being neutralized with aqueous NH3. The resulting white precipitate was collected, washed with water, and dried to give 2-(difluoromethyl)-1H-benzimidazole (6.07 g, 72% yield): mp 156–158 °C; 1H NMR (DMSO-d6) δ 13.28 (br, 1H), 7.76–7.68 (m, 1H), 7.61–7.54 (m, 1H), 7.36–7.26 (m, 2H), 7.26 (t,JHF= 53.3 Hz, 1H).
Ge, F.; Wang, Z.; Wan, W.; Lu, W.; Hao, J.One-pot synthesis of 2-trifluoromethyl and 2-difluoromethyl substituted benzo-1,3-diazoles Tetrahedron Lett. 2007, 48, 3251–3254
TRIAZINE, PYRIMIDINE AND PYRIDINE ANALOGS AND THEIR USE AS THERAPEUTIC AGENTS AND DIAGNOSTIC PROBES [US2011275762]2011-11-10
| Patent | Submitted | Granted |
|---|---|---|
| Heterocyclic compound and antitumor agent containing the same as active ingredient [US7071189] | 2004-06-17 | 2006-07-04 |
| Treatment of prostate cancer, melanoma or hepatic cancer [US2007244110] | 2007-10-18 | |
| Heterocyclic compound and antitumor agent containing the same as effective ingredient [US7307077] | 2006-11-02 | 2007-12-11 |
| IMMUNOSUPPRESSIVE AGENT AND ANTI-TUMOR AGENT COMPRISING HETEROCYCLIC COMPOUND AS ACTIVE INGREDIENT [US7750001] | 2008-05-15 | 2010-07-06 |
| PYRIMIDINYL AND 1,3,5-TRIAZINYL BENZIMIDAZOLES AND THEIR USE IN CANCER THERAPY [US2011009405] | 2011-01-13 | |
| SUBSTITUTED PYRIMIDINES AND TRIAZINES AND THEIR USE IN CANCER THERAPY [US2011053907] | 2011-03-03 | |
| IMMUNOSUPPRESSIVE AGENT AND ANTI-TUMOR AGENT COMPRISING HETEROCYCLIC COMPOUND AS ACTIVE INGREDIENT [US2010267700] | 2010-10-21 | |
| AMORPHOUS BODY COMPOSED OF HETEROCYCLIC COMPOUND, SOLID DISPERSION AND PHARMACEUTICAL PREPARATION EACH COMPRISING THE SAME, AND PROCESS FOR PRODUCTION OF THE SAME [US8227463] | 2010-09-30 | 2012-07-24 |
| PYRAZOLO[1,5-a]PYRIDINES AND THEIR USE IN CANCER THERAPY [US2010226881] | 2010-09-09 | |
| PYRIMIDINYL AND 1,3,5-TRIAZINYL BENZIMIDAZOLE SULFONAMIDES AND THEIR USE IN CANCER THERAPY [US2010249099] | 2010-09-30 |
…………..
Zenyaku Kogyo


Sector: Health Care
Industry: Biotech & Pharma
Sub-Industry: Specialty Pharma
Zenyaku Kogyo Co. Ltd. produces pharmaceuticals. The Company manufactures and sells over-the-counter drugs, health foods, and prescription medicines, as well as skin care products.
Address:
5-6-15 Otsuka
Bunkyo, 112-8650
Japan
Phone: 81-3-3946-1111
Fax: 81-3-3946-1130
Web url: www.zenyaku.co.jp
Otsuka
Bunkyo




……
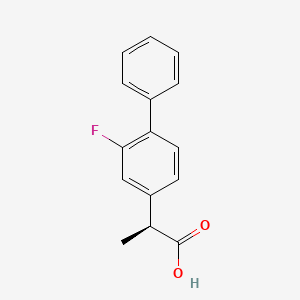
S-flurbiprofen (TT-063)
Cas 51543-39-6,
MW 244.26,
MF C15 H13 F O2
[1,1′-Biphenyl]-4-acetic acid, 2-fluoro-α-methyl-, (αS)-
- [1,1′-Biphenyl]-4-acetic acid, 2-fluoro-α-methyl-, (S)-
- (+)-(S)-Flurbiprofen
- (+)-Flurbiprofen
- (2S)-2-(2-Fluoro-1,1′-biphenyl-4-yl)propanoic acid
- (2S)-2-(2-Fluoro-4-biphenyl)propanoic acid
- (S)-Flurbiprofen
- Dexflurbiprofen
- Esflurbiprofen
- S-(+)-Flurbiprofen
- d-Flurbiprofen
On October 20, 2014, Taisho filed for manufacturing and marketing approval for TT-063 from the Ministry of Health, Labour and Welfare as a new drug candidate that will follow the Type 2 diabetes treatment Lusefi®, which was launched in May 2014. TT-063 is a patch formulation that has been co-developed by Taisho and TOKUHON Corporation with the aim of obtaining an indication for osteoarthritis. In Phase 3 clinical trials comparing TT-063 with therapeutic drugs already on the market, TT-063 has been found to be more effective than the control drugs in patients with osteoarthritis of the knee joint (January 16, 2014 announcement ).
Furthermore, Taisho is also preparing to file for approval from the Ministry of Health, Labour and Welfare for CT-064, an oral formulation of the osteoporosis treatment agent Bonviva launched in August 2013. Taisho has confirmed the effectiveness of CT-064 for osteoporosis patients through Phase 3 clinical trials (September 22, 2014 announcement).

In the central nervous system field, TS-091 transitioned from Phase 1 to Phase 2 in Japan in May 2014. Clinical trials of TS-091 have commenced to confirm the effectiveness of this drug in patients with central disorders of hypersomnolence. In addition, Phase 1 clinical trials of TS-091 have commenced overseas. TS-111 and TS-121 are undergoing Phase 1 clinical trials overseas with the aim of obtaining an indication for depression.
Faced with intensifying competition in new drug discovery, we will jointly implement R&D activities with research institutions outside the Taisho Group, and with companies in Japan and overseas, as we work to enhance our drug development pipeline (lineup of drugs in development). Our goal is to discover many more new drugs, primarily in our priority fields.
| Company | Taisho Pharmaceutical Holdings Co. Ltd. |
| Description | Topical anti-inflammatory analgesic patch containing S-flurbiprofen |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase III |
| Standard Indication | Osteoarthritis |
| Indication Details | Treat osteoarthritis (OA) and scapulohumeral periarthritis |
| Regulatory Designation | |

Scheme 2.
Reagents and conditions: (a) THF, EDC, Et3N; (b) TFA; (c) 0.5 equiv 2,5-dimethoxybenzoquinone, EtOH, 50–80 °C for 3–5 h; (d) 1 equiv naphthoquinone, MeOH, rt, overnight.
http://www.sciencedirect.com/science/article/pii/S0960894X13011773
……………………………………………
2-(6-methoxynaphthalen-2-yl) propanoic acid By way of illustration, chemically, flurbiprofen is 2-(2-fluoro-4-biphenylyl) propionic acid and is described in US Patent No. 3,755,427. NSAIDs, such as flurbiprofen, are usually supplied as a racemate. However, recently there has been renewed interest in the separate enantiomers of flurbiprofen, i.e. S-flurbiprofen and R-flurbiprofen.

R-Flurbιprofen

S-Flurtιprofen
Flurbiprofen is a potent inhibitor of cyclooxygenase (both COX-I and COX-2) in humans and it is understood that the inhibitory effect lies predominantly in the S- enantiomer.
Flurbiprofen is generally produced in the form of a racemic compound. It is known that from the racemic compound, flurbiprofen having a high optical purity can be produced by an optical resolution method using, for example, an optically active amine compound, such as α-phenylethylamine, as an optical resolution agent, as is described in US Patent No. 5,599,969. In addition, whether dealing with racemic, S- or R- 2-aryl propionic acid, there is also a need to make the synthetic process as efficient as possible.
Example 2 – Ibuprofen
Example 2.1 Resolution procedure
Racemic ibuprofen (530g) is dissolved in toluene (1335ml) and methanol (900ml).
The mixture is heated to dissolve the solid. S-1-Phenylethylamine (247g) is dissolved in toluene (200ml) and the solution is added with stirring at 600C over about 3 hours while the temperature is maintained at about 65-700C. The mixture is cooled gradually to 0 to 50C to induce crystallisation and stirred at this temperature for 1 hour. The crystals are filtered off, washed with toluene (600ml) and dried in a Vacuum oven at 550C to form crude S-ibuprofen / S-1-phenylethylamine salt (635g).
Crude S-ibuprofen / S-1-phenylethylamine salt (635g) is stirred with toluene (1930ml) and methanol (800ml) and the mixture is heated to 6O0C to dissolve the solid. The solution is cooled gradually to 0 to 5°C to induce crystallisation. The crystals are filtered off and dried in a vacuum oven at 55°C to form pure S-ibuprofen / S-I- phenylethylamine salt (510g). This recrystallisation of the S-ibuprofen / S-I- phenylethylamine salt may be repeated if necessary to upgrade the enantiomeric purity if required.
Pure S-ibuprofen / S-1-phenylethylamine salt (485g) is mixed with toluene (1700ml) with stirring. Water (300ml) and concentrated hydrochloric acid (17Og) are added and
÷ibe mixture is stirred at 600C. The lower aqueous layer is separated off and the upper organic layer is retained. The hydrochloric acid wash is repeated, then the toluene solution is washed with water. Water (370ml) and 47% sodium hydroxide
(118g) are added and the solution is heated to 600C and allowed to settle. The lower aqueous layer is separated and the upper toluene layer is washed with water. The aqueous phases are combined and heptane (420ml) is added. Hydrochloric acid
(130g) is added and the mixture is heated to 600C, stirred and settled. The organic layer is separated off and washed with water. The solution is cooled to -100C to induce crystallisation and the crystals are separated off by filtration, washed with heptane and dried under vacuum to yield (S)-ibuprofen (28Og) at an enantiomeric purity of over 99%.
Example 2.2 Racemisation procedure
Toluene/methanol mother liquors from the filtration of crude S-ibuprofen / S-I- phenylethylamine salt in the resolution procedure (2400ml, containing an estimated 130g of ibuprofen) is charged into a 3 L 3 necked round bottomed flask and methanol and toluene are distilled out at atmospheric pressure (volume removed approximately 1400 ml). The batch is then cooled to around 60°C and washed twice with hydrochloric acid (20 ml concentrated hydrochloric acid in 200 ml of water), and then twice with water (200 ml). Toluene is charged (80 ml) followed by methanol (200 ml) and caustic soda solution (45Og of 28% w/w solution, 5 molar equivalents). The mixture is heated to reflux for about 6 hours. Solvent is then removed at atmospheric pressure until the vapour temperature reaches approximately 85°C. The mixture is cooled to around 60°C and concentrated hydrochloric acid is charged at about 60 to 70°C until the pH of the mixture is 1 or less. The layers are allowed to separate and the bottom aqueous layer removed. The organic layer is washed with water (200 ml) and then azeotroped to dryness using a Dean and Stark trap. A solution of racemic ibuprofen in toluene remains.
…………………………………………
PATENT
http://www.google.com/patents/CN104478703A?cl=en
Preparation of R – (+) _ flurbiprofen:
The racemic flurbiprofen as a starting material, to obtain an intermediate product of formula I as shown and then the ester prepared as shown in Formula II with 5-isosorbide monobenzyl ether, ester hydrolysis after obtained R – (+) – flurbiprofen;

wherein, in formula I, X is Cl or Br;
(2) by the R – (+) _ flurbiprofen obtained (RS) – flurbiprofen:
The R _ (+) _ flurbiprofen 200mg, potassium hydroxide 150mg, 0. 5mL water into IOmL reaction flask and heated to 120 ° C and held for 2h, then water was added 15mL, cooled to room temperature, the resulting stirring the mixed solution with 10% hydrochloric acid to pH = 0. 5, extracted with ethyl acetate, combined several layers, washed with water until neutral, the organic solvent is recovered, the resulting residue was added at 60~90 ° C under an appropriate amount of petroleum ether by recrystallization, obtained (RS) – flurbiprofen 100mg, 50% yield.
(3) Preparation of (S) -⑴- flurbiprofen:
In 25mL single-necked flask, followed by adding (RS) – flurbiprofen 123mg, Portugal TOA 29. 8mg, isopropanol lmL, the mixture was stirred at reflux until clear, half the amount of the solvent evaporated under reduced pressure except , set the refrigerator overnight. The precipitate was collected by suction filtration as white crystals, after washing a small amount of isopropanol, which was dissolved in water, washed with 10% aqueous sodium hydroxide (10% NaOH mean mass fraction) adjusted pH = 13, the sheet-like precipitate was filtered off Portuguese octylamine white crystals. The resulting filtrate was added dropwise with stirring 10% hydrochloric acid to pH = 1, extracted with ethyl acetate, the organic layer was washed with water to recover the solvent, the resulting residue was purified by an appropriate amount of petroleum ether and recrystallized at 60~90 ° C. The product was collected by filtration, and dried in vacuo to give a white (S) – (+) _ flurbiprofen needle crystal 45. 3mg, 65% yield, mp 102~103 ° C, [α] = + 44 ° (C = 1, methanol), ee value of 92.6% (ee value measurement method: (S) – (+) – flurbiprofen after chiral amine derivatization reagents, by HPLC analysis).
wherein in step (3) is a byproduct eleven R _ (+) _ flurbiprofen, its follow step (1) of racemic reused.
Step (1) of the specific operation is as follows:
(la) 1:. Synthesis of 2,6-sorbitol dehydration -D- -5- benzyl ether: 4: 3
250ml volumetric flask isosorbide 18. 25g (125mmol), lithium hydroxide monohydrate 5. 25g (125mmol) and 60ml of dimethyl sulfoxide (DMSO), heated to 90 ° C, stirred for 30min, constant pressure equalizing dropping funnel was added dropwise benzyl chloride 14. 4ml (125mmol), 90 ° C the reaction 19-20h, reaction mixture was adjusted to pH 1 with 2M hydrochloric acid, extracted with ethyl acetate (50ml * 3), the organic layers combined, washed with water ( 30ml * 2), dried over anhydrous sodium sulfate overnight, filtered and concentrated residue Cheng baby gel column chromatography (petroleum ether: ethyl acetate = 5: 1) to give a cream solid, that is 1: 4: 3: 2,6 Dehydration -D- sorbitol -5- benzyl ether 24. 5g, m.p. 59 ~61 ° C.
(Ib) · 2- (2- fluoro-4-biphenylyl) propionyl chloride Synthesis
50ml vial before racemic flurbiprofen was added 2. 44g (IOmmol), anhydrous toluene 20ml, freshly distilled thionyl chloride was added dropwise 0. 8ml (Ilmmol), N, N- dimethylformamide amide (DMF) 2 dropwise, stirred at room temperature 2h, the solvent was distilled off under reduced pressure to give a pale yellow gum, i.e., 2- (2-fluoro-4-biphenylyl) propionyl chloride, it was used directly in the reaction without isolation.
(lc). R-2- (2- fluoro-4-biphenylyl) propionic acid 5- isosorbide monobenzyl ether ester synthesis
The (Ib) resulting acid chloride was dissolved in 20ml of dry toluene was added dropwise at room temperature, dimethyl amine 3. 5ml, solid precipitation, stirred for about Ih, ice salt bath, a bath temperature of minus 10-15Ό, stirred at this temperature IOmin so, and then the constant pressure dropping funnel (Ia) 5 isosorbide monobenzyl ether (2. 83g, 12mmol) in toluene, keeping the reaction temperature, stirring 8h. The ice bath was removed and the reaction mixture under reduced pressure to remove the solvent, the residue was extracted with ethyl acetate. The extract was washed with water, dried over anhydrous sodium sulfate overnight, ethyl acetate was removed under reduced pressure, the residue was a white gel, recrystallized from petroleum ether to give a white solid that R-2- (2- fluoro-4-biphenylyl) propionic acid 5- isosorbide monobenzyl ether ester 3. 65g (7. 88mmol), in order to put the racemic flurbiprofen yield based on 78.8%.
(ld) R – Synthesis of flurbiprofen – (+)
Under ice bath (Ic) obtained R-2- (2- fluoro-4-biphenylyl) propionic acid monobenzyl ether isosorbide 5- ester 2. 3Ig (5mmol) was dissolved in 20ml of acetone / water (1/1) was added Iml hydrochloric acid to adjust pH to 3, stirred for 3-4h, the reaction solution was extracted with ethyl acetate (20ml * 2), sash organic layer was washed with ice (10ml * 2), dried over anhydrous sodium sulfate overnight , filtration, and the filtrate was concentrated, the residue was recrystallized from ether to give white crystals, i.e. L-flurbiprofen 1.02g (4 18mmol.), yield 83.5%, optical purity 93% (HPLC method); input-racemic flurbiprofen dollars, the total yield of 78.8% * 83.5% = 65.8%.
Step (1) reaction of the formula:

FLURBIPROFEN RACEMIC
3-Fluoro-4-phenyl-α-methylphenylacetic acid 1
M.p. 110-113°C (lit.3d 111-113.5°C).
1 H NMR (CDCl3, δ ppm) 7.51-7.55 (m, 2H), 7.49-7.37 (m, 4H), 7.21-7.16 (m, 2H), 3.85-3.78 (q, 1H, J = 7.1 Hz, CH), 1.60-1.57 (d, 3H, J = 7.1 Hz, CH3);
13C NMR (CDCl3 δ ppm) 180.4 (COOH), 161.3 & 158.0 (3-Ar-C), 140.9 & 140.8, 135.4, 130.9 & 130.8 (5-Ar-C), 128.9, 128.4, 128.2 & 128.0 (4-Ar-C), 127.7 (4′-Ar-C), 123.7 & 123.7 (6-Ar-C), 115.5 & 115.2 (2-Ar-C), 44.8 (CH), 18.0 (CH3).
(d) Sagami Chemical Research Center. Jpn. Kokai Tokkyo Koho JP 8216840, 1982 (Chem. Abstr. 1982, 97: 5996s).

 |
| RACEMIC |
|
Flurbiprofen
CAS : 5104-49-4
: 2-Fluoro-a-methyl[1,1¢-biphenyl]-4-acetic acid
Additional Names: 2-(2-fluoro-4-biphenylyl)propionic acid; 3-fluoro-4-phenylhydratropic acid
Manufacturers’ Codes: BTS-18322; U-27182
Trademarks: Adfeed (Lead Chem.); Ansaid (Pfizer); Antadys (Thamex); Cebutid (Boots); Froben (Boots); Flurofen (Boots); Ocufen (Allergan); Stayban (Boots); Zepolas (Mikasa)
Molecular Formula: C15H13FO2
Molecular Weight: 244.26
Percent Composition: C 73.76%, H 5.36%, F 7.78%, O 13.10%
Literature References: Prepn: FR M5737; Adams et al., US 3755427 (1968, 1973 both to Boots Co., Ltd.). Pharmacology: Chalmers et al., Ann. Rheum. Dis. 31, 319 (1972); ibid. 32, 58 (1973); Glenn et al., Agents Actions 3, 210 (1973); Nishizawa et al.,Thromb. Res. 3, 577 (1973). HPLC determn in urine and plasma: J. M. Hutzler et al., J. Chromatogr. B 749, 119 (2000). Symposium on pharmacokinetics and clinical efficacy in pain management: Am. J. Med. 80, Suppl. 3A, 1-157 (1986).
Properties: Crystals from petr ether, mp 110-111°. Slightly sol in water (pH 7.0); readily sol in most polar solvents.
Melting point: mp 110-111°
Therap-Cat: Anti-inflammatory; analgesic.
|
racemic


s form


| Patent | Submitted | Granted |
|---|---|---|
| Methods to accelerate the isolation of novel cell strains from pluripotent stem cells and cells obtained thereby [US2008070303] | 2006-11-21 | 2008-03-20 |
| Herpes Virus-Based Compositions and Methods of Use in the Prenatal and Perinatal Periods [US2008226601] | 2006-06-05 | 2008-09-18 |
| METHOD OF REDUCING ABETA42 AND TREATING DISEASES [US2008021085] | 2007-06-21 | 2008-01-24 |
| METHODS TO ACCELERATE THE ISOLATION OF NOVEL CELL STRAINS FROM PLURIPOTENT STEM CELLS AND CELLS OBTAINED THEREBY [US2010184033] | 2009-07-16 | 2010-07-22 |
| Pyridyl Amide T-Type Calcium Channel Antagonists [US2011112064] | 2011-05-12 | |
| PROCESS FOR THE MANUFACTURE OF RACEMIC 2-ARYL-PROPIONIC ACID [US2011172460] |
| Patent | Submitted | Granted |
|---|---|---|
| Nitroxyderivatives having antinflammatory, analgesic and antithrombotic activity [US6613784] | 2003-09-02 | |
| Global method for mapping property spaces [US6675136] | 2004-01-06 | |
| Method of reducing Abeta42 and treating diseases [US2006004086] | 2006-01-05 | |
| 11-Beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia [US7179802] | 2004-06-03 | 2007-02-20 |
| 11-BETA-HYDROXYSTEROID DEHYDROGENASE 1 INHIBITORS USEFUL FOR THE TREATMENT OF DIABETES, OBESITY AND DYSLIPIDEMIA [US6730690] | 2004-03-11 | 2004-05-04 |
| Process for producing optically active flurbiprofen [US7214820] | 2006-06-22 | 2007-05-08 |
| Pyridyl Amide T-Type Calcium Channel Antagonists [US7875636] | 2009-11-05 | 2011-01-25 |
| METHOD FOR PRODUCING OPTICALLY ACTIVE ESTER AND METHOD FOR PRODUCING OPTICALLY ACTIVE CARBOXYLIC ACID [US8115008] | 2010-09-16 | 2012-02-14 |
| DRUG SUBSTANCE PREPARATIONS, PHARMACEUTICAL COMPOSITIONS AND DOSAGE FORMS [US2010087538] | 2010-04-08 | |
| (R)-2-(3-Benzoylphenyl)propionic acid salts and pharmaceutical preparations containing them [EP0935961] | 1999-08-18 | 2008-04-02 |
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

Taisho Pharmaceutical Co., Ltd. (大正製薬株式会社 Taishō Seiyaku Kabushiki-gaisha?) (TYO: 4535) is a Japanese pharmaceutical company based in Tokyo.

.
////////////
| Tirupati తిరుపతి |
|
|---|---|
| City | |

Clockwise from top: Tirumala Venkateswara Temple, Tirumala ghat road, City skyline and Chandragiri fort
|
|
|
Tirupati
Location in Andhra Pradesh, India |
|
| Coordinates: 13.65°N 79.42°ECoordinates: 13.65°N 79.42°E | |
| Country | India |
| State | Andhra Pradesh |
| Region | Rayalaseema |
| District | Chittoor |
| Government | |
| • Member of Parliament | Varaprasad Rao Velagapalli |
| Area | |
| • City | 24 km2 (9 sq mi) |
| Elevation | 161 m (528 ft) |
| Population (2011)[1] | |
| • City | 287,035 |
| • Density | 12,000/km2 (31,000/sq mi) |
| • Metro[2] | 459,985 |
| Languages | |
| • Official | Telugu |
| Time zone | IST (UTC+5:30) |
| PIN | 517501 |
| Telephone code | +91–877 |
| Vehicle registration | AP 03 |
| Website | Tirupati Mucnicipal Corporation |


 .
.
 .
.
Kapila Theertham in Tirupati








Food Service During Tirumala Tirupati Devastanam’s ‘Srinivasa Kalyanam Utsavam’ at MARG Swarnabhoomi



Harbin Gloria to Commercialize Constipation Drug in China
![]()
Harbin Gloria to Commercialize Constipation Drug in China

Harbin Gloria Pharma in-licensed China rights to Amitiza, a novel anti-constipation drug from Sucampo Pharma of the US. Amitiza is a chloride channel activator, approved for US use in 2006, which acts in the small intestine. Gloria will be responsible for obtaining CFDA approval of the drug and then commercializing it in China. Gloria paid $1 million upfront and will be liable for additional milestone payments. More details….
– See more at: http://www.chinabiotoday.com/articles/20150512_1#sthash.YbauZ6qM.dpuf
http://www.chinabiotoday.com/articles/20150512_1
 AMITIZA (lubiprostone)
AMITIZA (lubiprostone)
Harbin Gloria Pharmaceuticals Co., Ltd. engages in the research, development, production, and sale of pharmaceutical products primarily in the People’s Republic of China. The company offers orthopedic medicines, antineoplastic products, medical-nutrition products, rheumatology drugs, digestive and respiratory system medicines, cardiovascular medicines, liver disease medications, gynecology medications, and antibiotics. It also provides circulatory system, pediatrics, uropoiesis and reproduction, immune regulation, and other products. Harbin Gloria Pharmaceuticals Co., Ltd. was founded in 2000 and is based in Harbin, the People’s Republic of China.
No. 29, Beijing Road
Limin Economic & Technological Development Zone
Harbin, 150025
China
Founded in 2000
Phone:
86 451 5735 1368
Fax:
86 451 5735 1992

Harbin




Lascufloxacin, KRP-AM1977, by Kyorin

Lascufloxacin
CAS 848416-07-9
Kyorin Pharmaceutical Co., Ltd., 杏林製薬株式会社
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-
7-((3S,4S)-3-((Cyclopropylamino)methyl)-4-fluoropyrrolidin-1-yl)-6-fluoro-1-(2-fluoroethyl)-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
{(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
(KRP-AM1977X)
-
C21-H24-F3-N3-O4
- 439.4316
- SMILES……COc1c2c(cc(c1N3C[C@H](C(C3)CNC4CC4)F)F)c(=O)c(cn2CCF)C(=O)O
![]()
…………………………
Lascufloxacin hydrochloride
-
C21-H24-F3-N3-O4.Cl-H
- 475.8925
- CAS 1433857-09-0
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, hydrochloride (1:1)
……………….
Lascufloxacin mesylate
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, methanesulfonate (1:1)
-
C21-H24-F3-N3-O4.C-H4-O3-S
- 535.5372
- CAS 1433857-41-0
The other non-fluorinated quinolone under clinical development is KRP-AM1977, by Kyorin, which is in Phase I of clinical trials. The oral formulation of the compound (KRP-AM1977X) is being tested for treatment of respiratory infections and the I.V. formulation is under development for treatment of MRSA infections [1,2].
………………………………..
PATENT
WO 2013069297
http://www.google.co.in/patents/WO2013069297A1?cl=en
The present invention is represented by Formula (1) – {(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (hereinafter, compound (1) crystals of a salt also referred to), and a method for their preparation.
Typically, the pharmaceutical, in addition to the therapeutic effects on diseases, such as safety and quality are required. Therefore, the compound is the active ingredient of drugs, a variety of conditions and that is excellent in storage stability in the (light, temperature, humidity etc. influence the compound) are determined. Also, if the medicament is a dosage form such as oral preparations and injections, it is preferred that higher solubility in active ingredients of the water contained.
Compound (1) is safe, not only exhibit a strong antimicrobial action, conventional hard Gram-positive bacteria antimicrobial agents shown efficacy, particularly MRSA, PRSP, to VRE such resistant strains, to exhibit strong antibacterial activity It is known (for example, Patent Document 1).
WO 2005/026147
Patent Document 1, as the physicochemical characteristics of the compound (1) only has been shown to be a light brown free crystals. Also, Patent Document 1, the solubility in water of Compound (1), stability, no disclosure whatsoever information including characteristics of the crystal.
The present invention aims to provide a technique capable of improving the solubility and storage stability in water of the compound (1).
(Reference Example 4)
Bis (acetato -O) – [6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylate -O 3, O 4] boron Under a nitrogen atmosphere, boric acid (catalyst preparation) 86.4 g (1.40mol) was added acetic anhydride 17.9 L (190mol), and was heated and stirred for 30 minutes at 70.0 ~ 77.7 ℃. It was then cooling the mixture to an internal temperature of 24.7 ℃ (hot water set temperature 23.0 ℃). Subsequently, it was added portionwise boric acid to 4 times to the mixture. Specifically, the addition of boric acid (1 time) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.7 ~ 27.4 ℃. The addition of boric acid (second) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.3 ~ 26.3 ℃. In addition boric acid (third time) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 24.3 ~ 26.8 ℃. In addition boric acid (4 th) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 25.1 ~ 28.3 ℃. The mixture was stirred for 30 minutes at 50.0 ~ 54.9 ℃, was with boric acid triacetate adjusted solution.
In the boric acid triacetate adjusted solution, 6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylic acid ethyl ester 4.60kg (14. In a reaction preparation solution are added 0mol), and stirred for 3 hours at 53.7 ~ 56.9 ℃. The reaction preparation was cooled to 30.0 ℃, and allowed to stand overnight at room temperature. The reaction preparation was allowed to dissolve with heating to precipitate up to 55.0 ℃, acetone 13.8L was added and the reaction solution (1).
Separately, under nitrogen atmosphere, it is mixed Tsunemizu 161L and aqueous ammonia (28%) 28.2L (464mol), and cooled the mixture to 1.6 ℃. To the mixture, it was added the reaction solution of the above (1), to obtain a crude crystal acquisition solution crowded washed with acetone 9.20L. After cooling the crude crystal acquisition solution to 15.0 ℃, it was stirred for 1 hour at 6.2 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with Tsunemizu 46.0L, to give 9.07kg of wet crude crystals. Set temperature 65.0 to about 16 hours and dried under reduced pressure at ℃, the crude crystals were obtained 5.89kg.
Under a nitrogen atmosphere, it is mixed acetone and 29.5L crude crystal, the resulting mixture was heated and dissolved (melting temperature 52.6 ℃). When heated, it was dropped until the crystallization of diisopropyl ether 58.9L in a mixture (dropping amount 10.0L; 52.8 → 48.7 ℃; crystallization temperature 49.0 ℃). After crystallization confirmation, stirred for 15 minutes the mixture at 49.0 ~ 50.1 ℃, it was dropped the rest of diisopropyl ether to the mixture (50.1 → 46.4 ℃), 46.7 ~ 51.7 It was stirred for 15 minutes mixture at ℃. After cooling the mixture to 15 ℃, it was stirred for 30 minutes at 8.1 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with acetone and diisopropyl ether 5.89L 11.8L, to obtain 6.19kg of wet crystals. For about 20 hours drying under reduced pressure at warm water set temperature 65.0 ℃, bis (acetato -O) – [6,7-difluoro-1- (2-fluoroethyl) -8-methoxy-4-oxo-1,4- dihydro-3-carboxylate -O 3, O 4] was obtained 5.42kg boron (90.4% yield).
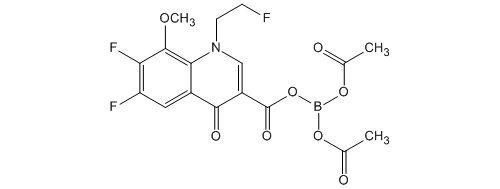
Melting point: 183 ~ 185 ℃ (dec).
Elemental analysis (%): calculated as C 17 H 15 BF 3 NO 8: C, 47.58; H, 3.52; N, 3.26.
Measured value: C, 47.91; H, 3.44; N, 3.04.
1 H-NMR (CDCl 3, 400 MHz) δ: 2.04 (6H, s), 4.22 (3H, d, J = 2.4Hz), 4.88 (2H, dt, J = 47.0 , 4.4Hz), 5.21 (2H, dt, J = 24.9,4.4Hz), 8.17 (1H, t, J = 8.8Hz), 9.11 (1H, s).
ESI MS (positive) m / z: 430 (M + H) +.
IR (KBr) cm -1: 3080,1703.
………………………………………….
WO 2005026147
http://www.google.com/patents/EP1666477A1?cl=en
KEY INTERMEDIATE

604798-54-1
3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-
| Chemical Name:3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-CAS: 604798-54-1Molecular Formula: C8H15FN2Molecular Weight: 158.2165032 |
………………………….
KEY INTERMEDIATE
CAS 848498-67-9
Boron, bis(acetato-κO)[6,7-difluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-(oxo-κO)-3-quinolinecarboxylato-κO]-, (T-4)-
Coordination Compound
ビス(アセチルオキシ)[6,7-ジフルオロ-1-(2-フルオロエチル)
-8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ
キシ]ボラン
-8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ
キシ]ボラン
| 化学物質名 | ビス(アセチルオキシ)[6,7-ジフルオロ-1-(2-フルオロエチル) -8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ キシ]ボラン |
|---|---|
| 構造別分類コード番号 | F60622212422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
|
| 安衛法官報通し番号 | 21534 |
| 安衛法官報公示整理番号 | 8-(1)-3764 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 848498-67-9 |
| 出典 | 厚生労働省 |
……………………………….
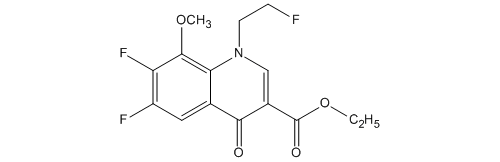
KEY INTERMEDIATE
3-Quinolinecarboxylic acid, 6,7-difluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, ethyl ester
114214-60-7
C15H14F3NO4
6,7-ジフルオロ-1-(2-フルオロエチル)-8-メトキシ-4-オキ
ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル
ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル
| 化学物質名 | 6,7-ジフルオロ-1-(2-フルオロエチル)-8-メトキシ-4-オキ ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル |
|---|---|
| 構造別分類コード番号 | F60622322422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
 |
| 安衛法官報通し番号 | 21467 |
| 安衛法官報公示整理番号 | 8-(1)-3758 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 114214-60-7 |
| 出典 | 厚生労働省 |
| WO2003076428A1 * | 8 Mar 2002 | 18 Sep 2003 | Toshifumi Akiba | Quinolonecarboxylic acid derivative |
| WO2005026147A1 | 8 Sep 2004 | 24 Mar 2005 | Yoshikazu Asahina | 7-(4-substituted 3- cyclopropylaminomethyl-1 pyrrolidinyl) quinolonecarboxylic acid derivative |
| WO2007082471A1 * | 18 Jan 2007 | 26 Jul 2007 | Guangzhou Baiyunshan Pharmaceu | Anti-infective compound, preparation method thereof and use thereof |
| CN1158846A * | 9 May 1995 | 10 Sep 1997 | 昆山市康壮达兽药厂 | Synthesis technology of norfluxacini hydrochloride |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014174846A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174847A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174848A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Tablet |
- Kyorin. Kyorin—Main R&D Activities-1 (4 February 2013 Release). Available online: http://www.kyorin-pharm.co.jp/en/business/pdf/main_rd_activities_20130204_en.pdf (accessed on 4 February 2013).
- Kyorin. Drug discovery, development, and lcm with medical professionals and patients in mind. Available online: http://www.kyorin-gr.co.jp/en/business/gensen/r_and_d.shtml (accessed on 11 April 2013).
-
……….
![]()




Ochyanomizu Sola City 16F,
Kanda Surugadai 4-6, Chiyoda-ku,
Tokyo 101-8311 Japan
TEL: 03-3525-4711
Access
One-minute walk from the Hijiribashi exit of Ochanomizu station on JR Chuo and Sobu lines
One-minute walk from the B2 exit of Shin-Ochanomizu station on Tokyo Metro Chiyoda line
Four-minutes walk from the No.1 exit of Ochanomizu station on Tokyo Metro Marunouchi line
Six-minutes walk from the B3 exit of Ogawamachi station on Toei Subway Shinjuku line



| Trade Name | KYORIN Pharmaceutical Co.,Ltd. |
|---|---|
| Business | Manufacture and sales of prescription medicines |
| Head Office | Ochyanomizu Sola City 16F, Kanda Surugadai 4-6, Chiyoda-ku, Tokyo 101-8311 Japan (Access Map) |
| Telephone | 03-3525-4711 |
| Foundation | 1923 |
| Establishment | 1940 |
Shimotsuga-gun, Tochigi

 Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park –
Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park – .
.MARKET
Ochanomizu station


Lupin buys Brazilian pharmaceutical firm Medquímica

Lupin buys Brazilian pharmaceutical firm Medquímica
India-based drugmaker Lupin has acquired 100% equity interest in Brazilian pharmaceutical firm Medquímica Indústria Farmacêutica.

read at

Group president and CEO, Lupin Pharmaceuticals, and director on the board of Lupin Limited, Vinita Gupta
Medquímica
 .
.



A Medquímica Indústria Farmacêutica é uma empresa genuinamente brasileira que atua na produção de medicamentos para o uso humano. Com sua linha de produção instalada em Juiz de Fora, a Medquimica está presente em todo o Brasil através de seus representantes.
Working in syntony with the market. There are 30 years that it is the north that takes the Medquímica Pharmaceutical Industry Ltda. to dedicate to the production of pharmaceutical products of proven effectiveness and certain return for all its net of customers. For such, the Medquímica invests in physical and human resources, forming a qualified and highly experienced technical team, who detaches in the marketplace for producing medicines of uncontestable quality and trustworthiness.
With the quality certified through the Good Practical of Manufacture, the Medquímica detaches for having a laboratory of reference inside its installations. Moreover, respecting the resolution 134 from Anvisa, gradually, all its medicines are being evaluated for the tests of pharmaceutical equivalence, proving, in this way, the quality of Medquímica’s product. Such actions had resulted in a supported growth of credibility and sales.
For giving support to all this development, the Medquímica will start the construction of the first projected national plant in the most rigorous and recent requirements of the ANVISA. Thirty years of history, qualified technician team, tests of equivalence and bioequivalência and a model plant makes from Medquímica the certain alternative for the success of its businesses
Fábrica:
(32) 3224-4087 (Telefax)
Administração:
(32) 2101-4000 (Telefax)
The Medquímica Pharmaceutical Industry is a genuinely Brazilian company that acts in the medicine production for the human use. With its line of production installed in Juiz de Fora, the Medquimica is present in all over Brazil through its representatives.
Industry
Rua Otacílio Esteves da Silva, 40
Bairro Granjas Betânia
CEP: 36047-400
Juiz de Fora – MG
Administration
Rua Fernando Lamarca, 255
Bairro Distrito Industrial
CEP: 36092-030
Juiz de Fora – MG
Juiz de Fora
 Paço Municipal de Juiz de Fora
Paço Municipal de Juiz de Fora
 No todo parece bem interessante. Dá pra tirar muitas fotos boas. Já teve outro thread bem legal sobre a cidade.
No todo parece bem interessante. Dá pra tirar muitas fotos boas. Já teve outro thread bem legal sobre a cidade.

CARMEN DRAHL….Tribute to a Great Writer

CARMEN DRAHL
Award-winning science communicator and social media power user based in Washington, DC.
Carmen Drahl is a multimedia science journalist and chemistry communicator based in Washington, DC.
A social media evangelist, Carmen started her first chemistry blog in 2006. Today, she regularly leverages Twitter, Facebook, and Google Plus Hangouts in her reporting.
Carmen has written about how life may have originated on Earth, explained how new medications get their names, and covered the ongoing issues plaguing the forensic science community. Her video on the food science behind 3D printed cocktail garnishes won the 2014 Folio Eddie Award for Best Association Video.
Until December 2014, Carmen worked at Chemical & Engineering News magazine. Her work has also been featured at Scientific American’s blog network, SiriusXM’s Doctor Radio, and elsewhere.
Carmen holds a Ph.D. in chemistry from Princeton University.
Specialties:
interviewing, science writing, social media, Twitter, Storify, YouTube,
public speaking, hosting, video production, iPhone videography,
non-linear video editing, blogging (WordPress and Blogger), HTML website
coding
We have been reading her for the past several years and a inspiration for many

Links
https://www.facebook.com/carmenwrites
Carmen Drahl (@carmendrahl) | Twitter
www.linkedin.com/in/carmendrahl/en
http://cenblog.org/the-safety-zone/
Carmen Drahl – Google+
Carmen Drahl – YouTube
Carmen Drahl’s profile – Vine
Carmen Drahl, Introductory Remarks for Forensics on Vimeo
Phone: 202-872-4502
Fax: 202-872-8727 or -6381

Education


Princeton University
Ph.D., Chemistry
2002 – 2007
Ph.D. with Erik J. SorensenShe was on a team that completed the first total synthesis of
abyssomicin C, a molecule found in small quantities in nature that
showed hints of promise as a potential antibiotic. I constructed
molecular probes from abyssomicin for proteomics studies of its
biological activity.
abyssomicin C, a molecule found in small quantities in nature that
showed hints of promise as a potential antibiotic. I constructed
molecular probes from abyssomicin for proteomics studies of its
biological activity.
M.A. with George L. McLendon
worked
toward developing a drug conjugate as a potential treatment for cancer. I
synthesized a photosensitizer dye-peptide conjugate for targeting the
cell death pathway called apoptosis.

At a reception before the Alumni Day luncheon, President Tilghman (third
from left) congratulated the winners of the University’s highest awards
for students: (from left) Pyne Prize winners Lester Mackey and Alisha
Holland; and Jacobus Fellowship recipients Sarah Pourciau, Egemen
Kolemen and Carmen Drahl. Unable to attend the event was Jacobus Fellowship winner William Slauter. (photo: Denise Applewhite

Jennifer Maclachlan, Carmen Drahl, Antony Williams


Drew University
B.A., Chemistry
1998 – 2002
Graduated
summa cum laude with specialized honors in chemistry. Honors thesis
entitled “Structural, kinetic, and mechanistic studies: the protein
tyrosine phosphatases CD45 and PTP1B”
summa cum laude with specialized honors in chemistry. Honors thesis
entitled “Structural, kinetic, and mechanistic studies: the protein
tyrosine phosphatases CD45 and PTP1B”
Activities and Societies: Phi Beta Kappa

Carmen Drahl, Class of 2002,
Experience
Science Journalist
Freelance
January 2014 – Present Washington D.C. Metro Area
Multimedia
science journalist – I deliver clean products on time. Experience in
reporting on chemistry, food science, history of science, drug
development, science education.
science journalist – I deliver clean products on time. Experience in
reporting on chemistry, food science, history of science, drug
development, science education.
Senior Editor, Chemical & Engineering News
American Chemical Society

August 2007 – December 2014 (7 years 5 months)Washington D.C. Metro Area
Reporting:Cover the science of chemistry for C&EN, the American Chemical
Society’s weekly magazine, circulation 160,000. Track new research
findings daily, particularly in forensic science, drug discovery,
organic chemistry, and food science.
Society’s weekly magazine, circulation 160,000. Track new research
findings daily, particularly in forensic science, drug discovery,
organic chemistry, and food science.
Video:
Doubled circulation to C&EN’s YouTube channel in 2013. Scripted, narrated, edited footage.
Managed a core team of 4 and collaborated with other reporters to
produce 30 videos, some reproduced in The Atlantic, Scientific American,
Eater National, The Daily Mail.
Incepted, scripted, and co-hosted
“Speaking of Chemistry”, a monthly web show that summarizes top
chemistry news for the busy scientist.
Social Media:
Developed magazine-wide best practices for YouTube videos and Twitter. Ran staff workshops about Storify, Slashdot, and Reddit.
Hosting/Public Speaking:
Topics include communicating chemistry simply, transitioning from a
Ph.D. to careers in science communication. Moderated discussions on
chemophobia, social media usage in the chemical sciences. On-camera
co-host for web newscasts produced by ACS.
Innovation:
With
C&EN art and web teams, developed first-for-the-magazine features,
including a 90th anniversary commemorative timeline poster, a pullout
guide to top conference speakers, interactive quizzes and database
searches.
Carmen Drahl, senior editor of Chemical and Engineering News,
used her Ph.D. in chemistry as a springboard into the career she
envisioned for herself. Here she shares some advice that helped her make
the decision.
Carmen Drahl made the transition to a writing
career while earning a Ph.D. in chemistry at Princeton University. Born
and raised in New Jersey, she now lives in Washington, D.C., and reports
for Chemical and Engineering News (C&EN). At C&EN
she has written about how new medications get their names, explained
the science behind a controversial hair-straightening product, and
covered the scientific firestorm sparked by an alleged arsenic life
form. Her work has been featured on SiriusXM’s Doctor Radio, Radio New Zealand’s This Way Up, and elsewhere. Her coverage has also been recognized by MIT’s Knight Science Journalism Tracker.
- (Open)1 honor or award

Scientific Cocktails: Award-winning video

Speaking of Chemistry: All About Tinsel

Carmen Drahl
Twitter Maven
World Central Kitchen
March 2013 – August 2014 (1 year 6 months)Washington D.C. Metro Area
she was the “voice of Twitter” for World Central Kitchen, the humanitarian
organization founded by renowned Chef José Andrés. Doubled followers to
Twitter account in 2013, developed Twitter strategy for projects and
events. Edited Annual Report, press releases and other communication
materials. Volunteered in person at outreach events.
Twitter account in 2013, developed Twitter strategy for projects and
events. Edited Annual Report, press releases and other communication
materials. Volunteered in person at outreach events.

Contributing Editor, AWIS Magazine
Association of Women in Science
December 2005 – August 2007 (1 year 9 months)
sHE
reported and wrote profiles of prominent women scientists in a range of
fields (molecular biology, physics, geoscience) for the Research
Advances column in AWIS Magazine.
reported and wrote profiles of prominent women scientists in a range of
fields (molecular biology, physics, geoscience) for the Research
Advances column in AWIS Magazine.

Writer, various publications
Princeton University
April 2005 – May 2007 (2 years 2 months)
She
reported and wrote news for the Princeton University News Office’s
Research Notes, and wrote news and features for the Princeton University
Chemistry Department’s Industrial Affiliates Program Newsletter and
Chemistry Alumni Newsletter.
reported and wrote news for the Princeton University News Office’s
Research Notes, and wrote news and features for the Princeton University
Chemistry Department’s Industrial Affiliates Program Newsletter and
Chemistry Alumni Newsletter.
Honors & Awards
Eddie Digital Award- Best Video (B-to-B)
FOLIO Magazine
December 2014
Porter Ogden Jacobus Fellowship
Princeton University
February 2007
NSF Graduate Research Fellowship
National Science Foundation
2002
Volunteer Experience & Causes
Board Member
Princeton Alumni Weekly Magazine
October 2013
Advisory Committee
American Institute of Physics News and Media Services
October 2013
Member, Graduate Alumni Leadership Council
Princeton University
2009 – 2012 (3 years)
INTERVIEW
Continuing with the tradition from last two years, I will occasionally post interviews with some of the participants of the ScienceOnline2010 conference that was held in the Research Triangle Park, NC back in January. See all the interviews in this series here. You can check out previous years’ interviews as well: 2008 and 2009.Today, I asked Carmen Drahl, Associate Editor for Science/Technology/Education at Chemical & Engineering News (find her as @carmendrahl on Twitter) to answer a few questions.Welcome
to A Blog Around The Clock. Would you, please, tell my readers a little
bit more about yourself? Where are you coming from (both geographically
and philosophically)? What is your (scientific) background? It’s a pleasure and a privilege to be interviewed, Bora.Good
It’s a pleasure and a privilege to be interviewed, Bora.Good
conversations make me happy. School was fun for me (well, maybe not
grad school) and that’s evolved into a desire to always be learning
something new. I enjoy doing nothing as much as I enjoy doing things. On
Mondays, if I’m not too busy, I take hip-hop dance classes.her hometown is Hackettstown, New Jersey. M&M’s are made there. I got a
bachelor’s in chemistry from Drew University and a Ph.D. in chemistry at
Princeton. Scientifically my expertise hovers somewhere around the
interface between organic chemistry and biochemistry. A short while
after defending my dissertation, I moved to Washington DC to write for Chemical & Engineering News, and that’s where I’ve been for almost three years now.When and how did you first discover science blogs?Scandal
led me to science blogs. Seriously. In March 2006 I was still an
organic chemistry grad student. Everyone in my lab was buzzing about a
set of retractions in the Journal of the American Chemical Society
(disclosure: today I work for the American Chemical Society, which
publishes JACS). A rising young organic chemistry star retracted the
papers because work by one of his graduate students couldn’t be
reproduced. It was a big deal and became an even bigger deal as the
inevitable rumors (salacious and otherwise) surfaced. The blogosphere
had the details first. So that’s where Google pointed me and the other
members of my lab when we searched for more information. I learned about
the awesome (but sadly now defunct) blogs Tenderbutton and The Endless
Frontier, by Dylan Stiles and Paul Bracher, both chemistry grad students
like me. I also discovered the solid mix of chemistry and pharma at
Derek Lowe’s In the Pipeline, which is still the first blog I visit every day.Tell us a little more about your career trajectory so far: interesting projects past and present?
to A Blog Around The Clock. Would you, please, tell my readers a little
bit more about yourself? Where are you coming from (both geographically
and philosophically)? What is your (scientific) background?
It’s a pleasure and a privilege to be interviewed, Bora.Goodconversations make me happy. School was fun for me (well, maybe not
grad school) and that’s evolved into a desire to always be learning
something new. I enjoy doing nothing as much as I enjoy doing things. On
Mondays, if I’m not too busy, I take hip-hop dance classes.her hometown is Hackettstown, New Jersey. M&M’s are made there. I got a
bachelor’s in chemistry from Drew University and a Ph.D. in chemistry at
Princeton. Scientifically my expertise hovers somewhere around the
interface between organic chemistry and biochemistry. A short while
after defending my dissertation, I moved to Washington DC to write for Chemical & Engineering News, and that’s where I’ve been for almost three years now.When and how did you first discover science blogs?Scandal
led me to science blogs. Seriously. In March 2006 I was still an
organic chemistry grad student. Everyone in my lab was buzzing about a
set of retractions in the Journal of the American Chemical Society
(disclosure: today I work for the American Chemical Society, which
publishes JACS). A rising young organic chemistry star retracted the
papers because work by one of his graduate students couldn’t be
reproduced. It was a big deal and became an even bigger deal as the
inevitable rumors (salacious and otherwise) surfaced. The blogosphere
had the details first. So that’s where Google pointed me and the other
members of my lab when we searched for more information. I learned about
the awesome (but sadly now defunct) blogs Tenderbutton and The Endless
Frontier, by Dylan Stiles and Paul Bracher, both chemistry grad students
like me. I also discovered the solid mix of chemistry and pharma at
Derek Lowe’s In the Pipeline, which is still the first blog I visit every day.Tell us a little more about your career trajectory so far: interesting projects past and present?
 By
By
the time I discovered science blogs I knew my career goals were
changing. I’d already been lucky enough to audit a science writing
course at Princeton taught by Mike Lemonick from TIME, and thought that
maybe science writing was a good choice for me. After reading chemistry
blogs for a while I realized “Hey, I can do this!” and started my own
blog, She Blinded Me with Science, in July 2006. It was the typical grad student blog, a mix of posts about papers I liked and life in the lab.
At C&E News I’ve contributed to its C&ENtral Science
blog, which premiered in spring 2008. I’ve experimented with a few
different kinds of posts- observations and on-the-street interviews when
I run into something chemistry-related in DC, in-depth posts from
meetings, and video demos of iPod apps. One of my favorite things to do
is toy with new audio/video/etc technology for the blog.
What is taking up the most of your time and passion these days? What are your goals?
In March I just started a new era in my web existence- I’m becoming a pharma blogger. I’m the science voice at The Haystack,
C&E News’s new pharma blog and one of seven new blogs the magazine
launched last month. My co-blogger is the talented Lisa Jarvis, who’s
written about the business side of pharma for ten years and who brings a
solid science background to the table as well. I kicked us off by
liveblogging/livetweeting a popular session at the American Chemical
Society’s meeting in San Francisco where drug companies reveal for the
first time the chemical structures of potential new drugs being tested
in clinical trials. The whole thing synced to FriendFeed as well. Folks
followed the talks from all three venues, which was great. I hope I can
continue doing that sort of thing in the future.
For
this August, I’m co-organizing a mini-symposium at the American
Chemical Society meeting in Boston about the chem/pharma blogosphere and
its impact on research and communication. I’m in the process of
inviting speakers right now. It’s my first time doing anything like this
and part of me is petrified that no one will show up. Tips on
organizing a conference session and how not to stress when doing so are
welcome!
More broadly, I’d love to get more chemistry bloggers to
connect with the community that attends ScienceOnline. I don’t ever want
to become that old (or not-so-old) person who is clueless about
them-thar newfangled whosiwhatsits that the kids are using nowadays.
What aspect of science communication and/or particular use of the Web in science interests you the most?
A
few things come to mind, actually. I’d like to think that the web has
made grad school a helluva lot less isolating for science grad students.
You have the virtual journal clubs like Totally Synthetic, posts like SciCurious’s letter to a grad student, etc.
As
a journalist the web’s capacity to equalize fascinates me. I’m
extremely lucky to have a staff gig as a science writer without having
gone to journalism school or landed a media fellowhip and it’s weird to
think that my old blog might’ve helped my visibility. I didn’t know Ed
Yong’s story until Scio10 but I think he’s a highly talented example of
how the web can open doors.
The web’s equalizing power goes to
readers of science content as well as writers, of course. In the ideal
situation a reader can give a writer instant feedback and you can get a
real conversation going, something that was much harder with the
snail-paced system of letters to the editor and reader surveys. Not that
the conversation is always civil. Most of C&EN’s readers have a
decent amount of scientific training, but the debate that rages whenever
we run an editorial about climate change is as intense as any I’ve
seen.
In cases like that I don’t know that the web gives people a
good representation of what the consensus is. For folks who don’t have
scientific training, how do you ensure that people don’t just go to the
content that already confirms their pre-existing beliefs about autism or
global warming? John Timmer touched on this more eloquently in his interview with you,
and I agree with him that I don’t think we have an answer yet. Though
on a slightly different note, I will mention that I’ve been enjoying the
New York Times’s recent attempts to recapture the spontaneity of
flipping through the newspaper in online browsing, like the Times Skimmer for Google Chrome.
What are some of your favourite science blogs? Have you discovered any cool science blogs by the participants at the Conference?
In addition to the blogs I’ve already mentioned I enjoy Carbon-Based Curiosities, Wired Science, Chemistry Blog, and Terra Sigillata, to name a few of the 50 or so blogs on my feed reader.
I discovered scads of new blogs at Scio10 but I’ll focus on the one that’s become required reading for me these days: Obesity Panacea.
I’d covered obesity drug development for C&EN but I’d never met
Travis Saunders and Peter Janiszewski or heard of their blog until the
conference.
What was the best aspect of ScienceOnline2010 for
you? Is there anything that happened at this Conference – a session,
something someone said or did or wrote – that will change the way you
think about science communication, or something that you will take with
you to your job, blog-reading and blog-writing?
Dave Mungeris
my hero – his blogging 102 session was packed with practical tips that I
brought back to C&EN for incorporating into our blogs, such as the
use of the Disqus plugin for catching conversations on social networks,
getting smart about using stats and surveys, etc. Some of that’s already
happened, and some of the ideas are still in the works.
I came
for the nuts-and-bolts blogging tips but I stayed for the conversations,
especially the ones at the bar after the official program was done for
the night. And the icing on the cake was seeing folks I’d worked with
but never met, like Cameron Neylon and you, Bora, and catching up with
people I hadn’t seen in months, like Jean-Claude Bradley, Aaron Rowe,
Jennifer Ouellette and Nancy Shute.
It was so nice to meet you in person and thank you for the interview. I hope to see you again next January.


Fall 2006
Back row: Junjia Liu, Carmen Drahl, Joel Freundlich, Adam Charnley, Jess Frie, Doug McLeod, Jay Schneekloth
Front row: Qian Qin, Jimin Kim, Hao Xie, Jillian Spangler, Julia Clay, Rachel MacCoss, Annie Nalbandian, Erik Sorensen

Spring 2005
Back row: Erik Alexanian, Jason Rohde, Brian Goess, Christoph Zapf, Steve Miller, David Richard, Jamie McBean
Middle row: Glenn Sammis, Hao Xie, Eric Flamme, Rachel MacCoss, Amanda Menco, Sy-Yeu Chern, Amanda Chi, Ryan Foss
Front row: Jeff Celaje, Qihui Jin, Bob Moreau, Jeewoo Lim, Carmen Drahl, Jimin Kim, Annie Nalbandian, Erik Sorensen

Spring 2004
Back row: Jeewoo Lim, William Shipe, Eric Flamme, Steve Miller, Bryce Harrison, Christoph Zapf
Front row: Erik Sorensen, Jeff Celaje, Bob Moreau, Erik Alexanian, Carmen Drahl, Jason Rohde

Company: GlaxoSmithKline
Meant to treat: tumors with loss-of-function in the tumor suppressor
protein PTEN (phosphatase and tensin homolog)- 2nd most inactivated
tumor suppressor after p53- cancers where this is often the case include
prostate and endometrial
Mode of action: inhibitor of
phosphoinositide 3-kinase-beta (PI3K-beta). Several lines of evidence
suggest that proliferation in certain PTEN-deficient tumor cell lines is
driven primarily by PI3K-beta.
Medicinal chemistry tidbits: The GSK
team seemed boxed in because in 3 out of 4 animals used in preclinical
testing, promising drug candidates had high clearance. It turned out
that a carbonyl group that they thought was critical for interacting
with the back pocket of the PI3K-beta enzyme wasn’t so critical after
all. When they realized they could replace the carbonyl with a variety
of functional groups, GSK2636771 eventually emerged. GSK2636771B (shown)
is the tris salt of GSK2636771.
Status in the pipeline: Phase I clinical trials……….http://cenblog.org/the-haystack/2012/03/liveblogging-first-time-disclosures-from-acssandiego/
 CARMEN
CARMEN
Posted By Carmen Drahl on Mar 24, 2012
Phone: 202-872-4502
Fax: 202-872-8727 or -6381
Company: GlaxoSmithKline
Meant to treat: tumors with loss-of-function in the tumor suppressor
protein PTEN (phosphatase and tensin homolog)- 2nd most inactivated
tumor suppressor after p53- cancers where this is often the case include
prostate and endometrial
Mode of action: inhibitor of
phosphoinositide 3-kinase-beta (PI3K-beta). Several lines of evidence
suggest that proliferation in certain PTEN-deficient tumor cell lines is
driven primarily by PI3K-beta.
Medicinal chemistry tidbits: The GSK
team seemed boxed in because in 3 out of 4 animals used in preclinical
testing, promising drug candidates had high clearance. It turned out
that a carbonyl group that they thought was critical for interacting
with the back pocket of the PI3K-beta enzyme wasn’t so critical after
all. When they realized they could replace the carbonyl with a variety
of functional groups, GSK2636771 eventually emerged. GSK2636771B (shown)
is the tris salt of GSK2636771.
Status in the pipeline: Phase I clinical trials……….http://cenblog.org/the-haystack/2012/03/liveblogging-first-time-disclosures-from-acssandiego/
CARMEN
Posted By Carmen Drahl on Mar 24, 2012
Phone: 202-872-4502
Fax: 202-872-8727 or -6381

Washington, D.C.
.
 .
.



 .
.
 .
.
Motesanib (AMG-706)

Motesanib (AMG-706)
Amgen Inc.

Motesanib (AMG 706) is an experimental drug candidate originally developed by Amgen[1] but is now being investigated by theTakeda Pharmaceutical Company. It is an orally administered small molecule belonging to angiokinase inhibitor class which acts as an antagonist of VEGF receptors, platelet-derived growth factor receptors, and stem cell factor receptors.[2] It is used as thephosphatesalt motesanib diphosphate.
Motesanib, also known as AMG-706, is an orally administered multikinase inhibitor that selectively targets VEGF receptors, platelet-derived growth factor receptors, and Kit receptors.
Clinical trials
Motesanib was originally investigated for effectiveness against advanced nonsquamous non-small-cell lung cancer (NSCLC), withPhase II trials indicating an effectiveness comparable to bevacizumab when they were both used in combination withpaclitaxel/carboplatin.[3] However a later and more detailed Phase III trial failed to show any benefit for the treatment of NSCLC.[2][4]A second Phase III trial was started in 2012,[5] which focused on patients from Asian backgrounds (performed on the bases ofsubgroup analysis)[6] however this also failed to meet its primary endpoint.[7]
The drug has undergone a Phase II evaluation as first-line therapy for breast cancer[2] however this study found no evidence to support further investigation.[8] Phase II testing against persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas was also unsuccessful.[9]
There have also been 2 separate Phase II clinical trials for thyroid cancer which have both shown promising results.[10][11][12]
Developed at Amgen, the compound is also being evaluated as both monotherapy and in combination with other agents in the treatment of breast, colorectal, lung, thyroid and ovarian cancers. Clinical trials for the treatment of bladder cancer have been terminated.
The National Cancer Institute had been evaluating the potential of the drug in patients with low-grade neuroendocrine tumors; however, no recent development has been reported for this research. The FDA awarded fast track status to motesanib in 2004. In 2008, the compound was licensed to Takeda in Japan.

AMG-706 is synthesized as follows: 1-Acetyl-3,3-dimethyl-6-nitroindoline (I) is reduced by catalytic hydrogenation over Pd/C, giving the aminoindoline (II), which is then coupled with 2-chloronicotinoyl chloride (III) in the presence of DIEA to yield the corresponding nicotinamide (IV). Subsequent condensation of (IV) with neat 4-(aminomethyl)pyridine (V) at 120 °C affords the 2-aminonicotinamide derivative (VI). The N-acetyl group of (VI) is finally removed by acidic hydrolysis to furnish the title compound (1,2).
,………………………………………
US 2003125339
http://www.google.com/patents/US20030125339
………………………………………………….
US 2003225106
https://www.google.com/patents/US20030225106
EXAMPLE 133
[2295]

N-(3,3-Dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
Step A—Preparation of 1-acetyl-6-amino-3,3-dimethylindoline
1-Acetyl-3,3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL), the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc:CH2Cl2 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O—204.27.
Step B—Preparation of N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from 1-acetyl-6-amino-3,3-dimethylindoline (Step A) by the method described in Example 82.
Step C—Preparation of N-(3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N5O—373.45.
…………………….
http://www.google.com/patents/WO2012063085A3?cl=en
Example 133

N- (3, 3-Dimethy1indolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) }carboxamide Step A – Preparation of l-acetyl-6-amino-3 , 3- dimethylindoline l-Acetyl-3 , 3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL) , the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc :CH2C12 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O-204.27.
Step B – Preparation of N-(l-acetyl- 3 , 3-dimethylindolin-6- yl) (2-[ (4-pyridylmethyl) amino] (3-pyridyl) } carboxamide The titled compound was prepared from l-acetyl-6- amino-3 , 3-dimethylindoline (Step A) by the method described in Example 82.
Step C – Preparation of N- (3 , 3-dimethylindolin-6-yl) {2- [ (4- pyridylmethyl) amino] (3-pyridyl) }carboxamide
The titled compound was prepared from N-(l-acetyl- 3 , 3-dimethylindolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) } carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N50-373.45.
References
- Stafford, edited by Rongshi Li, Jeffrey A. (2009). “Chapter 5. Discovery of Motesanib”. Kinase inhibitor drugs. Hoboken, N.J.: Wiley. pp. 113–130. ISBN 978-0-470-27829-1.
- “Amgen and Takeda’s NSCLC Drug Fails in Phase III Study”. 30 Mar 2011.
- Blumenschein Jr, G. R.; Kabbinavar, F.; Menon, H.; Mok, T. S. K.; Stephenson, J.; Beck, J. T.; Lakshmaiah, K.; Reckamp, K.; Hei, Y.- J.; Kracht, K.; Sun, Y.- N.; Sikorski, R.; Schwartzberg, L. (14 February 2011). “A phase II, multicenter, open-label randomized study of motesanib or bevacizumab in combination with paclitaxel and carboplatin for advanced nonsquamous non-small-cell lung cancer”. Annals of Oncology 22 (9): 2057–2067. doi:10.1093/annonc/mdq731.
- Jump up^ Scagliotti, G. V.; Vynnychenko, I.; Park, K.; Ichinose, Y.; Kubota, K.; Blackhall, F.; Pirker, R.; Galiulin, R.; Ciuleanu, T.-E.; Sydorenko, O.; Dediu, M.; Papai-Szekely, Z.; Banaclocha, N. M.; McCoy, S.; Yao, B.; Hei, Y.-j.; Galimi, F.; Spigel, D. R. (2 July 2012). “International, Randomized, Placebo-Controlled, Double-Blind Phase III Study of Motesanib Plus Carboplatin/Paclitaxel in Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer: MONET1”. Journal of Clinical Oncology 30 (23): 2829–2836. doi:10.1200/JCO.2011.41.4987. PMID 22753922.
- “Takeda Initiates Phase 3 Trial of Motesanib in Japan and Additional Asian Countries”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Kubota, K.; Ichinose, Y.; Scagliotti, G.; Spigel, D.; Kim, J. H.; Shinkai, T.; Takeda, K.; Kim, S.- W.; Hsia, T.- C.; Li, R. K.; Tiangco, B. J.; Yau, S.; Lim, W.- T.; Yao, B.; Hei, Y.- J.; Park, K. (13 January 2014). “Phase III study (MONET1) of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous nonsmall-cell lung cancer (NSCLC): Asian subgroup analysis”.Annals of Oncology 25 (2): 529–536. doi:10.1093/annonc/mdt552.
- Jump up^ “Takeda Announces Phase 3 MONET-A Study Evaluating Motesanib (AMG 706) in Patients with Advanced Non-Squamous Non-Small Cell Lung Cancer Does Not Meet Primary Endpoint”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Martin, Miguel; Roche, Henri; Pinter, Tamas; Crown, John; Kennedy, M John; Provencher, Louise; Priou, Frank; Eiermann, Wolfgang; Adrover, Encarna; Lang, Istvan; Ramos, Manuel; Latreille, Jean; Jagiełło-Gruszfeld, Agnieszka; Pienkowski, Tadeusz; Alba, Emilio; Snyder, Raymond; Almel, Sachin; Rolski, Janusz; Munoz, Montserrat; Moroose, Rebecca; Hurvitz, Sara; Baños, Ana; Adewoye, Henry; Hei, Yong-Jiang; Lindsay, Mary-Ann; Rupin, Matthieu; Cabaribere, David; Lemmerick, Yasmin; Mackey, John R (April 2011). “Motesanib, or open-label bevacizumab, in combination with paclitaxel, as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: a phase 2, randomised, double-blind, placebo-controlled study”. The Lancet Oncology 12 (4): 369–376. doi:10.1016/S1470-2045(11)70037-7. PMID 21429799.
- Schilder, R.J.; Sill, M.W.; Lankes, H.A.; Gold, M.A.; Mannel, R.S.; Modesitt, S.C.; Hanjani, P.; Bonebrake, A.J.; Sood, A.K.; Godwin, A.K.; Hu, W.; Alpaugh, R.K. (April 2013). “A phase II evaluation of motesanib (AMG 706) in the treatment of persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas: A Gynecologic Oncology Group study”. Gynecologic Oncology 129 (1): 86–91. doi:10.1016/j.ygyno.2013.01.006. PMID 23321064.
- Motesanib Diphosphate Provides Anticancer Activity Among Patients with Progressive Thyroid Cancer, CancerConnect.com
- Jump up^ Schlumberger, M. J.; Elisei, R.; Bastholt, L.; Wirth, L. J.; Martins, R. G.; Locati, L. D.; Jarzab, B.; Pacini, F.; Daumerie, C.; Droz, J.-P.; Eschenberg, M. J.; Sun, Y.-N.; Juan, T.; Stepan, D. E.; Sherman, S. I. (29 June 2009). “Phase II Study of Safety and Efficacy of Motesanib in Patients With Progressive or Symptomatic, Advanced or Metastatic Medullary Thyroid Cancer”.Journal of Clinical Oncology 27 (23): 3794–3801. doi:10.1200/JCO.2008.18.7815. PMID 19564535.
- Sherman, Steven I.; Wirth, Lori J.; Droz, Jean-Pierre; Hofmann, Michael; Bastholt, Lars; Martins, Renato G.; Licitra, Lisa; Eschenberg, Michael J.; Sun, Yu-Nien; Juan, Todd; Stepan, Daniel E.; Schlumberger, Martin J. (3 July 2008). “Motesanib Diphosphate in Progressive Differentiated Thyroid Cancer”. New England Journal of Medicine 359 (1): 31–42.doi:10.1056/NEJMoa075853. PMID 18596272.
External links

Motesanib Diphosphate (AMG-706)
857876-30-3 diphosphate
453562-69-1 (free base)
N-(2,3-Dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-3-pyridinecarboxamide diphosphate
3-Pyridinecarboxamide, N-(2,3-dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-, phosphate (1:2)
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-(pyridin-4-ylmethylamino)pyridine-3-carboxamide diphosphate
| 569.4 | |
| Formula | C22H23N5O.2H3PO4 |
|---|
 |
|
| Names | |
|---|---|
| IUPAC name
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-[(pyridin-4-ylmethyl)amino]pyridine-3-carboxamide
|
|
| Other names
AMG 706
|
|
| Identifiers | |
| 453562-69-1 |
|
| ChEMBL | ChEMBL572881 |
| ChemSpider | 9842625 |
| Jmol-3D images | Image |
| PubChem | 11667893 |
| Properties | |
| C22H23N5O | |
| Molar mass | 373.45 g·mol−1 |
Stats today
6.8 lakh views on this blog
…………………..
TAKEDA, JAPAN
![]()

TOKYO HO

Takeda Pharmaceutical CEO Yasuchika Hasegawa
 Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.
Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.

Christophe Weber (L), the new president of Takeda Pharmaceutical Co., and CEO Yasuchika Hasegawa pose

 Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
 OSAKA
OSAKA

Dotonbori, Osaka, Japan
 OSAKA
OSAKA

AZD 3264 an IKK2 Inhibitor from Astra Zeneca

AZD 3264
MW 441.50
CAS 1609281-86-8
MF C21 H23 N5 O4 S
3-Thiophenecarboxamide, 2-[(aminocarbonyl)amino]-5-[4-(3,5-dimethyl-4-isoxazolyl)-2-[(3S)-3-pyrrolidinyloxy]phenyl]-
2-(Carbamoylamino)-5-[4-(3,5-dimethyl-1,2-oxazol-4-yl)-2-[(3S)-pyrrolidin-3-yloxy]phenyl]thiophene-3-carboxamide
Inhibition of IkB-kinase IKK2 has been identified as one of the novel pathways to treat inflammatory conditions such as asthma, chronic pulmonary obstructive disorder (COPD) and rheumatoid arthritis
……………………..
PATENT
WO 2003010158
https://www.google.com/patents/WO2003010158A1?cl=en

The synthesis began with the aromatic nucleophilic substitution reaction of 2-fluorobromobenzene (2) with (S)-N-Boc-3-pyrrolidinol 3 to give the bromo intermediate 4, which was borylated via halogen metal exchange using n-hexLi in THF followed by treatment with triisopropyl borate and acidic work-up to give the boronic acid intermediate 5. Suzuki coupling of the boronic acid 5 with bromothiophene 6(2)afforded the intermediate 7. Intermediate 7 was subjected to regioselective bromination using bromine in acetic acid. This reaction was nonregioselective and yielded 17% of the required isomer 8. The bromo compound 8 was coupled with isoxazole boronate ester 9 by another Suzuki reaction to get the title compound. The overall yield of the synthesis was <6%.


………………………..
PAPER
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/op500105n
http://pubs.acs.org/doi/full/10.1021/op500105n

An efficient and scalable synthesis of AZD3264 is described in which the differential reactivities of various halogen atoms have been employed. The process involves five linear chemical steps with three isolated stages starting from commercially available fragments.
AZD3264 (1)
A stirred solution of tert-butyl (3S)-3-[2-(4-carbamoyl-5-methyl-2-thienyl)-5-(3,5-dimethylisoxazol-4-yl)phenoxy]pyrrolidine-1-carboxylate (16) (2.65 kg, 4.63 mol) in tetrahydrofuran (25 L) w……………………………………………………title compound in 91% yield.
Purification

To a stirred suspension of crude AZD3264 (1) (1.75 kg, 3.98 mol) in methanol (23.75 L) and water (2.64 L) was added formic acid (0.24 kg, 5.18 mol), and the mixture was heated to 40 °C for 1.5 h, cooled to 25 °C, and basified with aqueous ammonia (12.29 M in water, 1.62 L, 19.92 mol). The product was isolated by filtration.
1H NMR (DMSO-d6, 400 MHz): δ 1.92–2.10 (m, 2H), 2.28 (s, 3H), 2.46 (s, 3H), 2.75–2.82 (m, 1H), 3.00–3.12 (m, 3H), 5.11–5.12 (m, 1H), 6.90 (br, 2H), 7.00–7.03 (m, 2H), 7.30 (br, 1H), 7.70–7.72 (m, 2H), 7.83 (s, 1H), 10.93 (s, 1H).
13C NMR (DMSO-d6, 100.6 MHz): δ 10.54, 11.42, 32.94, 45.51, 53.00, 79.37, 111.76, 114.17, 115.66, 120.70, 121.20, 122.77, 125.39, 126.92, 128.84, 150.12, 152.54, 154.50, 158.13, 165.14, 167.06.
DEPT NMR (DMSO-d6, 100.6 MHz): δ 10.54, 11.43, 32.94, 45.51, 53.01, 79.35, 114.17, 120.70, 121.20, 126.92.
HRMS calcd for C21H24N5O4S (M + H)+: 442.1543, found 442.1554.
[α]25D −13.80 (c 0.5, DMSO)
Journal of Medicinal Chemistry (2013), 56(18), 7232-7242 reports similar analogues
Elemental impurities – A database to facilitate the risk assessment of active ingredients and excipients
DRUG REGULATORY AFFAIRS INTERNATIONAL

One of the main demands of the Guideline ICH Q3D is to carry out risk assessments on metallic impurities. A database with analytical data provides a valuable support. Learn more about the data sharing using the new elemental impurities database.
Released in December 2014, the ICH Q3D Guideline on Elemental Impurities contains extensive specifications for the control of a total of 24 elements (21 metals, 3 metalloids) that can be present as impurities in pharmaceutical products. Main sources can be
- Active ingredients
- Excipients (including water)
- Processing auxiliaries and catalysts
- Production equipment
- Container and closure systems
The Guideline ICH Q3D calls for a risk assessment with regard to the presence of metallic impurities in various dosage forms, taking into account the respective limit values. The main factors of influence are to be included (see fishbone diagram on p. 6 of the Guideline). The risks identified in a comprehensive analysis…
View original post 343 more words
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

