
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

How to document a Product Transfer? Example templates!
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
All participants of the GMP training course “GMP-compliant Product Transfer” will receive a special version of the Guideline Manager CD including documents and templates useable for site change projects. Read more.
According to the European GMP-Rules, written procedures for tranfser activities and their documentation are required. For example, a Transfer SOP, a transfer plan and a report are now mandatory and will be checked during inspections.
As a participant of the GMP education course “GMP-compliant Product Transfer” in Prague, from 20-22 October 2015 you will receive a special version of the Guideline Manager CD with a special section concerning product transfers. This section contains, amongst others, a Transfer SOP and a template for a Transfer Plan. Both documents are in Word format and can immediately be used after adoption to your own situation.

Regulatory Guidance Documents like the WHO guideline on transfer of technology in pharmaceutical manufacturing and the…
View original post 36 more words
How to become a QP for Europe
DRUG REGULATORY AFFAIRS INTERNATIONAL

Both the ECA and the European QP Association are often contacted by people who would like to become a Qualified Person in a Member State of the European Union or outside the EU to release products for the EU market.
Both the ECA Academy and the European Qualified Person Association (EQPA) are often contacted by people who would like to become a Qualified Person (QP according the EU Directives) in a Member State of the European Union or outside the EU to release products for the EU market. Questions are for example:
- “Can I become a QP and live and work outside the EU?”
- “I work for an American company that would like to export medicinal product to the EU. How can we hire a QP here in the U.S.?”
- “I am an Irish Citizen living and working in Australia. I am thinking of studying a course by distance learning…
View original post 411 more words
FDA approves new drug treatment for nausea and vomiting from chemotherapy

September 2, 2015
Release
The U.S. Food and Drug Administration approved Varubi (rolapitant) to prevent delayed phase chemotherapy-induced nausea and vomiting (emesis). Varubi is approved in adults in combination with other drugs (antiemetic agents) that prevent nausea and vomiting associated with initial and repeat courses of vomit-inducing (emetogenic and highly emetogenic) cancer chemotherapy.
Nausea and vomiting are common side effects experienced by cancer patients undergoing chemotherapy. Symptoms can persist for days after the chemotherapy drugs are administered. Nausea and vomiting that occurs from 24 hours to up to 120 hours after the start of chemotherapy is referred to as delayed phase nausea and vomiting, and it can result in serious health complications. Prolonged nausea and vomiting can lead to weight loss, dehydration and malnutrition in cancer patients leading to hospitalization.
“Chemotherapy-induced nausea and vomiting remains a major issue that can disrupt patients’ lives and sometimes their therapy,” said Amy Egan, M.D., M.P.H., deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “Today’s approval provides cancer patients with another treatment option for the prevention of the delayed phase of nausea and vomiting caused by chemotherapy.”
Varubi is a substance P/neurokinin-1 (NK-1) receptor antagonist. Activation of NK-1 receptors plays a central role in nausea and vomiting induced by certain cancer chemotherapies, particularly in the delayed phase. Varubi is provided to patients in tablet form.
The safety and efficacy of Varubi were established in three randomized, double-blind, controlled clinical trials where Varubi in combination with granisetron and dexamethasone was compared with a control therapy (placebo, granisetron and dexamethasone) in 2,800 patients receiving a chemotherapy regimen that included highly emetogenic (such as cisplatin and the combination of anthracycline and cyclophosphamide) and moderately emetogenic chemotherapy drugs. Those patients treated with Varubi had a greater reduction in vomiting and use of rescue medication for nausea and vomiting during the delayed phase compared to those receiving the control therapy.
Varubi inhibits the CYP2D6 enzyme, which is responsible for metabolizing certain drugs. Varubi is contraindicated with the use of thioridazine, a drug metabolized by the CYP2D6 enzyme, because use of the two drugs together may increase the amount of thioridazine in the blood and cause an abnormal heart rhythm that can be serious.
The most common side effects in patients treated with Varubi include a low white blood cell count (neutropenia), hiccups, decreased appetite and dizziness.
Varubi is marketed by Tesaro Inc., based in Waltham, Massachusetts.
Ximelagatran

Ximelagatran
192939-46-1, EXANTA
N-[(1R)-1-cyclohexyl-2-[(2S)-2-[[[[4-[(hydroxyamino)iminomethyl]phenyl]methyl]amino]carbonyl]-1-azetidinyl]-2-oxoethyl]-glycine, ethyl ester
| C24H35N5O5 | |
| MW | 473.6 |
CAS 260790-58-7 (Monohydrate)
CAS 260790-59-8 (MonoHBr)
CAS 260790-60-1 (Monomethanesulfonate)
ASTRAZENECA INNOVATOR
Ximelagatran (Exanta or Exarta, H 376/95) is an anticoagulant that has been investigated extensively as a replacement forwarfarin[1] that would overcome the problematic dietary, drug interaction, and monitoring issues associated with warfarin therapy. In 2006, its manufacturer AstraZeneca announced that it would withdraw pending applications for marketing approval after reports ofhepatotoxicity (liver damage) during trials, and discontinue its distribution in countries where the drug had been approved (Germany, Portugal, Sweden, Finland, Norway, Iceland, Austria, Denmark, France, Switzerland, Argentina and Brazil).[2]
Ximelagatran is an ester prodrug of melagatran, a potent, direct, and reversible thrombin inhibitor (Ki = 1.2 nM). While melagatran has poor oral bioavailability, ximelagatran displays good bioavailability resulting, in part, from rapid absorption at the gastrointestinal tract, as well as rapid onset of action.Ximelagatran is converted to melagatran by reduction and hydrolysis at the liver and other tissues. It is used as an anticoagulant in a variety of situations, including thromboembolic disorders, stroke prevention in atrial fibrillation, and therapy in vein thrombosis
Method of action
Ximelagatran, a direct thrombin inhibitor,[3] was the first member of this class that can be taken orally. It acts solely by inhibiting the actions of thrombin. It is taken orally twice daily, and rapidly absorbed by the small intestine. Ximelagatran is a prodrug, being converted in vivo to the active agent melagatran. This conversion takes place in the liver and many other tissues throughdealkylation and dehydroxylation (replacing the ethyl and hydroxyl groups with hydrogen).
Uses
Ximelagatran was expected to replace warfarin and sometimes aspirin and heparin in many therapeutic settings, including deep venous thrombosis, prevention of secondary venous thromboembolism and complications of atrial fibrillation such as stroke. The efficacy of ximelagatran for these indications had been well documented,[4][5][6] except for non valvular atrial fibrillation.
An advantage, according to early reports by its manufacturer, was that it could be taken orally without any monitoring of its anticoagulant properties. This would have set it apart from warfarin and heparin, which require monitoring of the international normalized ratio (INR) and the partial thromboplastin time (PTT), respectively. A disadvantage recognised early was the absence of an antidote in case acute bleeding develops, while warfarin can be antagonised by vitamin K and heparin by protamine sulfate.
Side-effects
Ximelagatran was generally well tolerated in the trial populations, but a small proportion (5-6%) developed elevated liver enzymelevels, which prompted the FDA to reject an initial application for approval in 2004. The further development was discontinued in 2006 after it turned out hepatic damage could develop in the period subsequent to withdrawal of the drug. According to AstraZeneca, a chemically different but pharmacologically similar substance, AZD0837, is undergoing testing for similar indications.[2]
Melagatran synthesis

Sobrera, L. A.; Castaner, J.; Drugs Future, 2002, 27, 201.
SYNTHESIS

SYNTHESIS

SYNTHESIS



……
WO 1997023499/http://www.google.com/patents/EP0869966A1?cl=en
…………
References
- Hirsh J, O’Donnell M, Eikelboom JW (July 2007). “Beyond unfractionated heparin and warfarin: current and future advances”. Circulation 116 (5): 552–560.doi:10.1161/CIRCULATIONAHA.106.685974. PMID 17664384.
- “AstraZeneca Decides to Withdraw Exanta” (Press release). AstraZeneca. February 14, 2006. Retrieved 2012-07-16.
- Ho SJ, Brighton TA (2006). “Ximelagatran: direct thrombin inhibitor”. Vasc Health Risk Manag 2 (1): 49–58. doi:10.2147/vhrm.2006.2.1.49. PMC 1993972.PMID 17319469.
- Eriksson, H; Wahlander K; Gustafsson D; Welin LT; Frison L; Schulman S; THRIVE Investigators (January 2003). “A randomized, controlled, dose-guiding study of the oral direct thrombin inhibitor ximelagatran compared with standard therapy for the treatment of acute deep vein thrombosis: THRIVE I”. Journal of Thrombosis and Haemostasis 1 (1): 41–47. doi:10.1046/j.1538-7836.2003.00034.x. PMID 12871538.
- Francis, CW; Berkowitz SD, Comp PC, Lieberman JR, Ginsberg JS, Paiement G, Peters GR, Roth AW, McElhattan J, Colwell CW Jr; EXULT A Study Group (October 2003). “Comparison of ximelagatran with warfarin for the prevention of venous thromboembolism after total knee replacement”. New England Journal of Medicine 349 (18): 1703–1712.doi:10.1056/NEJMoa035162. PMID 14585938.
- Schulman, S; Wåhlander K; Lundström T; Clason SB; Eriksson H; THRIVE III investigators (October 2003). “Secondary prevention of venous thromboembolism with the oral direct thrombin inhibitor ximelagatran”. New England Journal of Medicine 349 (18): 1713–1721. doi:10.1056/NEJMoa030104. PMID 14585939.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
ethyl 2-[[(1R)-1-cyclohexyl-2-
[(2S)-2-[[4-(N’-hydroxycarbamimidoyl) phenyl]methylcarbamoyl]azetidin-1-yl]- 2-oxo-ethyl]amino]acetate |
|
| Clinical data | |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 20% |
| Metabolism | None |
| Biological half-life | 3-5h |
| Excretion | Renal (80%) |
| Identifiers | |
| CAS Registry Number | 192939-46-1 |
| ATC code | B01AE05 |
| PubChem | CID: 9574101 |
| IUPHAR/BPS | 6381 |
| DrugBank | DB04898 |
| ChemSpider | 7848559 |
| UNII | 49HFB70472 |
| KEGG | D01981 |
| ChEMBL | CHEMBL522038 |
| Chemical data | |
| Formula | C24H35N5O5 |
| Molecular mass | 473.57 g·mol−1 (429 g/mol after conversion) |

See full gatran series at………………http://apisynthesisint.blogspot.in/p/argatroban.html
///////
Neovacs Receives First Regulatory Approvals for a Phase IIb Trial of IFNa-Kinoid in Lupus
NEOVACS, a leader in active immunotherapies for the treatment of autoimmune diseases, today announced that it has been granted first approvals by regulatory agencies and ethics committees in several European countries for a Phase IIb clinical trial of IFNα-Kinoid in Systemic Lupus Erythematosus (SLE) or lupus.
The upcoming trial was notably assessed favorably using the Voluntary Harmonization Procedure (VHP) of Europe’s Heads of Medicine Agencies, which allows for a harmonized assessment of clinical trials by relevant national health authorities.
Acceptance by competent authorities enables Neovacs to initiate IFN-K-002, a Phase IIb clinical study to assess the biological and clinical efficacy of Neovacs’ lead active immunotherapy product candidate IFNα-Kinoid in patients suffering from lupus. Inclusion of first patients is expected to begin in the coming weeks. Approvals from other European, Asian and Latin American countries are expected in the second half of 2015.
Phase IIB trial design for IFN-K-002 in SLE
IFN-K-002 is a double-blind, randomized, placebo-controlled multicentric Phase IIb clinical trial designed to assess the efficacy and safety of IFNα-Kinoid in moderate to severe lupus patients. The study will recruit 166 patients across 19 countries in Europe, Asia and Latin America.
The co-primary endpoints for the trial are biological efficacy and clinical efficacy nine months after first immunization with IFNα-Kinoid. Biological efficacy is defined as IFNα-signature neutralization, while clinical efficacy will be measured by the BILAG-based1 Composite Lupus Assessment (BICLA) response.
Timelines for the study
Regulatory and ethics committee approvals pave the way for a rapid initiation of the study IFN-K-002. These centers will begin screening and immunizing patients in the coming weeks. Results of the clinical trial are expected in the first quarter of 2017.
About Neovacs
Created in 1993, Neovacs is today a leading biotechnology company focused on an active immunotherapy technology platform (Kinoids) with applications in autoimmune and/or inflammatory diseases. On the basis of the company’s proprietary technology for inducing a polyclonal immune response (covered by five patent families that potentially run until 2032) Neovacs is focusing its clinical development efforts on IFNα-Kinoid, an immunotherapy being developed for the indication of lupus and dermatomyositis. Neovacs is also conducting preclinical development works on other therapeutic vaccines in the fields of auto-immune diseases, oncology and allergies. The goal of the Kinoid approach is to enable patients to have access to safe treatments with efficacy that is sustained in these life-long diseases.
1 The British Isles Lupus Assessment Group (BILAG) is a validated index to measure lupus disease activity listed in FDA guidance on lupus. See FDA Systemic Lupus Erythematosus working group report at: www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072063.pdf#sthash.qR2f2REj.dpuf
For more information on Neovacs, please visit www.neovacs.fr
CONTACT:
NEOVACS - Investor Relations Nathalie Trepo +33 (0)1
53 10 93 00 ntrepo@neovacs.com Investor
Relations / Financial Communications - NewCap Valentine
Brouchot / Pierre Laurent +33 (0)1 44 71 94 94
neovacs@newcap.fr Investor Relations /
Financial Communications Germany - MC Services Raimund Gabriel
+49-89-210228-30 raimund.gabriel@mc-services.eu
Press / U.S. Inquiries - The Ruth Group Melanie
Sollid-Penton 1.646.536.7023 msollid@theruthgroup.com

DABIGATRAN PART 3/3
WO2015124764
ERREGIERRE S.P.A. [IT/IT]; Via Francesco Baracca, 19 I-24060 San Paolo D’argon (IT)
Erregierre SpA
DABIGATRAN ETEXILATE MESYLATE, INTERMEDIATES OF THE PROCESS AND NOVEL POLYMORPH OF DABIGATRAN ETEXILATE”
Abstract
A novel process is described for the production of Dabigatran etexilate mesylate, a 5 compound having the following structural formula: and two novel intermediates of said process.
(WO2015124764) SYNTHESIS PROCESS OF DABIGATRAN ETEXILATE MESYLATE, INTERMEDIATES OF THE PROCESS AND NOVEL POLYMORPH OF DABIGATRAN ETEXILATE click herefor patent
Dabigatran etexilate mesylate is an active substance developed by Boehringer
Ingelheim and marketed under the name Pradaxa® in the form of tablets for oral administration; Dabigatran etexilate mesylate acts as direct inhibitor of thrombin (Factor I la) and is used as an anticoagulant, for example, for preventing strokes in patients with atrial fibrillation or blood clots in the veins (deep vein thrombosis) that could form following surgery.
Dabigatran etexilate mesylate is the INN name of the compound 3-({2-[(4-{Amino-[(E)-hexyloxycarbonylimino]-methyl}-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate methanesulphonate, having the following structural formula:

The family of compounds to which Dabigatran etexilate belongs was described for the first time in patent US 6,087,380, which also reports possible synthesis pathways.
The preparation of polymorphs of Dabigatran etexilate or Dabigatran etexilate mesylate is described in patent applications US 2006/0276513 A1 , WO 2012/027543 A1 , WO 2008/059029 A2, WO 2013/124385 A2, WO 2013/124749 A1 , WO 2013/1 1 1 163 A2 and WO 2013/144903 A1 , while patent applications WO 2012/044595 A1 , US 2006/0247278 A1 , US 2009/0042948 A2, US 2010/0087488 A1 and WO 2012/077136 A2 describe salts of these compounds.
One of the objects of the invention is to provide an alternative process for the preparation of Dabigatran etexilate mesylate and two novel intermediates of the process.
These objects are achieved with the present invention, which, in a first aspect thereof, relates to a process for the production of Dabigatran etexilate mesylate, comprising the following steps:
a) reacting 4-methylamino-3-nitrobenzoic acid (I) with thionyl chloride to give 4- methylamino-3-nitrobenzoyl chloride hydrochloride (II):

(I) (ID
b) reacting compound (II) with 3-(2-pyridylamino) ethyl propanoate (III) to give the compound 3-[(4-methylamino-3-nitro-benzoyl)-pyridyn-2-yl-amino]-ethyl propanoate (IV):

(II) (IV)
reducing compound (IV) with hydrogen to 3-[(3-amino-4-methyl benzoyl)-pyridin-2-yl-amino]ethyl propanoate (V):

(IV) (V)
d) reacting N-(4-cyanophenyl)glycine (VI) with 1 ,1 -carbonyldiimidazole (CDI) to give 4-(2-imidazol-1 -yl-2-oxo-ethylamino)-benzonitrile (VII):

(VI) (VII)
e) reacting compound (VII) with compound (V) obtained in step c) to give one of compounds 3-({3-[2-(4-cyano-phenylamino)-acetylamino]-4-methylamino- benzoyl}-pyridin-2-yl-amino)-ethyl propanoate (VIII) and 3-[(3-amino-4-{[(2- (4-cyano-phenylamino)-acetyl]-methylamino}-benzoyl)-pyridin-2-yl- amino]ethyl propanoate (IX), or a mixture of the two compounds (VIII) and (IX):

f) transforming, through treatment with acetic acid, compounds (VIII) or (IX) or the mixture thereof into the compound 3-({2-[(4-cyano-phenylamino)-methyl]- 1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate (X), and then treating compound (X) with hydrochloric or nitric acid to form the corresponding salt (XI):
CHsCOOH
[(VIII) ; (IX)]


wherein A is a chlorine or nitrate anion;
liberating in solution compound (X) from salt (XI), and reacting compound (X) in solution with ethyl alcohol in the presence of hydrochloric acid and 2,2,2-trifluoroethanol to give the compound 3-({2-[(4-ethoxycarbonimidoyl-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate hydrochloride (XII):

reacting compound (XII) with ammonium carbonate to form compound Dabigatran ethyl ester (XIII):

reacting compound (XIII) with maleic acid to produce the maleate salt thereof (XI 11 ‘) and isolating the latter:

j) reacting maleate salt (XI 11 ‘) with hexyl chloroformate to give compound Dabigatran etexilate (XIV :
hexyl chloroformate

k) reacting compound (XIV) with methanesulfonic acid to give the salt Dabigatran etexilate mesylate:

a gatran etex ate mesy ate
EXAMPLE 12
Preparation of Dabigatran etexilate mesylate (step k).
All the Dabigatran etexilate obtained in Example 1 1 (4.7 kg; 7.49 moles) is loaded into a reactor along with 28.2 kg of acetone and the mass is heated at 50-60 °C until a complete solution is obtained; it is then filtered to remove suspended impurities. The filtered solution is brought to 28-32 °C. Separately, a second solution is prepared by dissolving 0.705 kg (7.34 moles) of methanesulfonic acid in 4.7 kg of acetone; the second solution is cooled down to 0-10 °C. The second solution is poured into the Dabigatran etexilate solution during 30 minutes, while maintaining the temperature of the resulting solution at 28-32 °C with cooling. The salt of the title is formed. The mass is maintained at 28-32 °C for 2 hours, then cooled to 18-23 °C to complete precipitation and the system is maintained at this temperature for 2 hours; lastly, centrifugation takes place, washing the precipitate with 5 kg of acetone. The precipitate is dried at 60 °C.
4.88 kg of Dabigatran etexilate mesylate, equal to 6.74 moles of compound, are obtained, with a yield in this step of 90%.
EXAMPLE 13
0.5 g of the crystalline compound (XIV) obtained in Example 1 1 are ground thoroughly and loaded into the sample holder of a Rigaku Miniflex diffractometer with copper anode.
The diffractogram shown in Figure 1 is obtained; a comparison with the XRPD data of the known Dabigatran etexilate polymorphs allows to verify that the polymorph of Example 1 1 is novel.
EXAMPLE 14
0.7 g of the crystalline compound (XIV) obtained in Example 1 1 are loaded into
the sample holder of a Perkin-Elmer DSC 6 calorimeter, performing a scan from ambient T to 350 °C at a rate of 10 °C/min in nitrogen atmosphere. The graph of the test is shown in Figure 2, and shows three endothermic phenomena with peaks at 83.0-85.0 °C, 104.0-104.2 °C and 129.9 °C; events linked to the thermal decomposition of the compound are evident at about 200 °C.
Figure 1 is an XRPD spectrum of the novel polymorph of Dabigatran etexilate of the invention;
Figure 2 is the graph of a DSC test on the novel polymorph of Dabigatran etexilate of the invention.
1H NMR OF
Dabigatran etexilate mesylate 872728-81-9
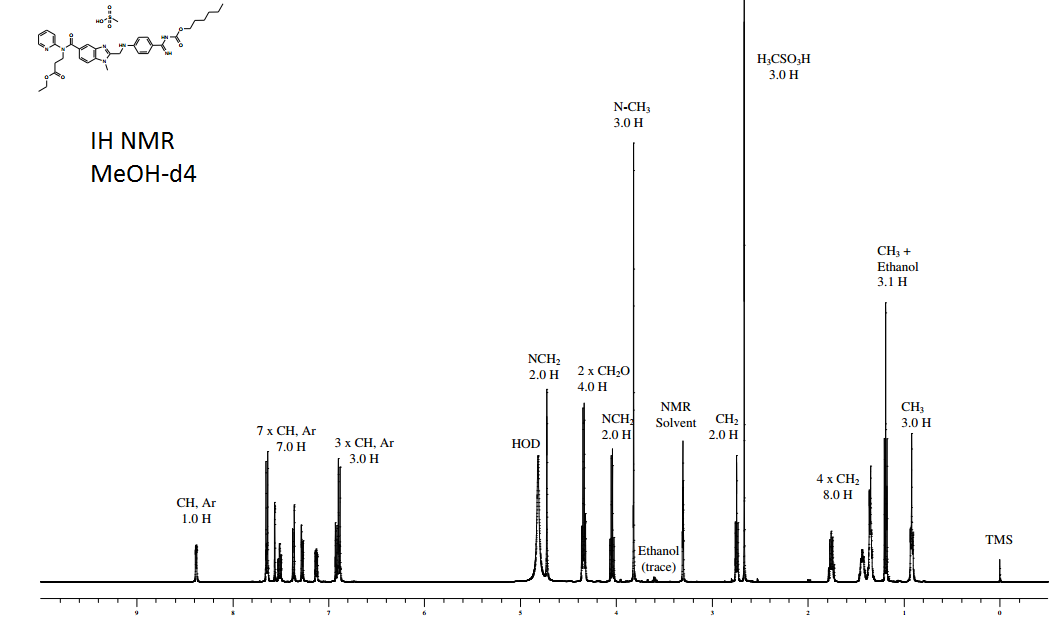 .
.
…
PATENT
http://www.google.com/patents/WO2012044595A1?cl=en
Examples
Reference examples:
Preparation of starting material: Dabigatran etexilate mesylate form I according to US 2005/0234104 example 1:
Ethyl 3 – [(2- { [4-(hexyloxycarbonylarninoimmomemyl)phenylammo]methyl } –
1 – methyl- lH-benzimidazole-5-carbonyl)pyridm-2-ylamino]propionate base (52.6 kg) (which has preferably been purified beforehand by recrystallization from ethyl acetate) is placed in an agitator apparatus which has been rendered inert and then 293 kg of acetone is added. The contents of the apparatus are heated to 40° C to 46° C with stirring. After a clear solution has formed, the contents of the apparatus is filtered into a second agitator apparatus through a lens filter and then cooled to 30° C to 36° C. 33 kg of acetone precooled to 0° C to 5° C, 7.9 kg of 99.5% methanesulfonic acid, and for rinsing another 9 kg of acetone are placed in the suspended container of the second apparatus. The contents of the suspended container are added in metered amounts to the solution of ethyl 3-[(2-{[4-(hexyloxycarbonylamino- iminomethyl)phenylamino]methyl} – 1 -methyl- 1 H-benzimidazole-5-carbonyl)pyridin-
2- ylamino]propionate base at 26° C to 36° C within 15 to 40 minutes. Then the mixture is stirred for 40 to 60 minutes at 26° C to 33° C. It is then cooled to 17° C to 23° C and stirred for a further 40 to 80 minutes. The crystal suspension is filtered through a filter dryer and washed with a total of 270 L of acetone. The product is dried in vacuum at a maximum of 50° C for at least 4 hours. Yield: 54.5-59.4 kg;
90%-98% of theory based on ethyl 3-[(2-{[4-(hexyloxycarbonyl- ammoiminomethyl)phenylamino]methyl} – 1 -methyl- 1 H-benzimidazole-5-carbonyl)- pyridm-2-ylamino]propionate base.
Preparation of starting material: Dabigatran Etexilate free base
Dabigatran Etexilate free base can be prepared according to the procedures disclosed in US 6087380 – example 113 or US 7202368 – example 5 Example 1
2.08 g of dabigatran etexilate free base was dissolved in 14.7 ml of acetone at 30 – 36 °C. 0.210 ml of methanesulfonic acid diluted in 2.20 ml of acetone was added within 15 – 40 min. at 26 – 36 °C. The resulting mixture was first steered for 40 – 60 min. at 26 – 36 °C and then for 40 – 80 min at 17 – 23 °C.
The resulting crystalline product was filtered off, washed with 17.87 ml of acetone and dried at 50 °C for 18 hours at 540 mbar.
………………..
PAPER
| Chinese Journal of Applied Chemistry |
|
Synthesis of Dabigatran Etexilate
|
| LIU Xiaojun, CHEN Guohua* |
| (Department of Medicinal Chemistry,China Pharmaceutical University,Nanjing 210009,China) |
4-Methylamino-3-nitrobenzoic acid(3) was prepared from 3-nitro-4-chlorobenzoic acid by methylamination. 3-[(Pyridin-2-yl)amino]propinoic acid ethyl ester(5) was prepared from 2-aminopyridine and ethyl acrylate by Michael addition. Dabigatran etexilate was synthesized from compounds 3 and 5 via condensation, catalytic hydrogenation, acylation with N-(4-cyanophenyl)glycine(9), cyclization, Pinner reaction, followed by reaction with n-hexyl chlorofomate. The overall yield is about 40% and the structure of the product was determined by IR, 1H NMR and MS.
ESIMS(m/z):628[M+H]+;1 HNMR(500MHz,DMSOd6), δ:091(t,3H,J=90Hz,CH3),116(t,3H,J=85Hz,CH3),125~169(m,8H,4×CH2),274(t, 2H,J=145Hz,CH2CO),379(s,3H,CH3N),395~403(m,4H,2CH2O),428(t,2H,J=140Hz, CH2),451(d,2H,J=55Hz,CH2N),676(d,J=85Hz,2H,Ar—H),688(d,J=75Hz,1H,Ar— H), 702(s,1H,N—H),713~721(m,2H,Py—H),740(d,J=85Hz,1H,Ar—H),747(d,J=15Hz, 1H,Ar—H),755~759(m,1H,Py—H),786(d,J=85Hz,2H,Ar—H),836~843(m,1H,Py—H), 902(brs,2H,NH2);IR(KBr),σ/cm-1 :3374,2953,2929,1730,1640,1610,1470,1389,1326,1256,1192, 1145,1127,1021,945,835,811,768,747。
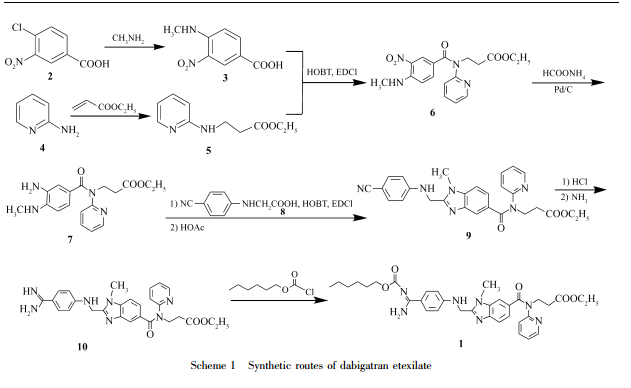


…………
PAPER
Identification, Synthesis, and Strategy for the Reduction of Potential Impurities Observed in Dabigatran Etexilate Mesylate Processes

Synthetic impurities that are present in dabigatran etexilate mesylate were studied, and possible pathways by which these impurities are formed during the manufacturing process were examined. The impurities were monitored by high-performance liquid chromatography, and their structures were determined by mass spectrometry and 1H and 13C NMR. Potential causes for the formation of these impurities are discussed, and strategies to minimize their formation are also described.

…………….
1H NMR PREDICT
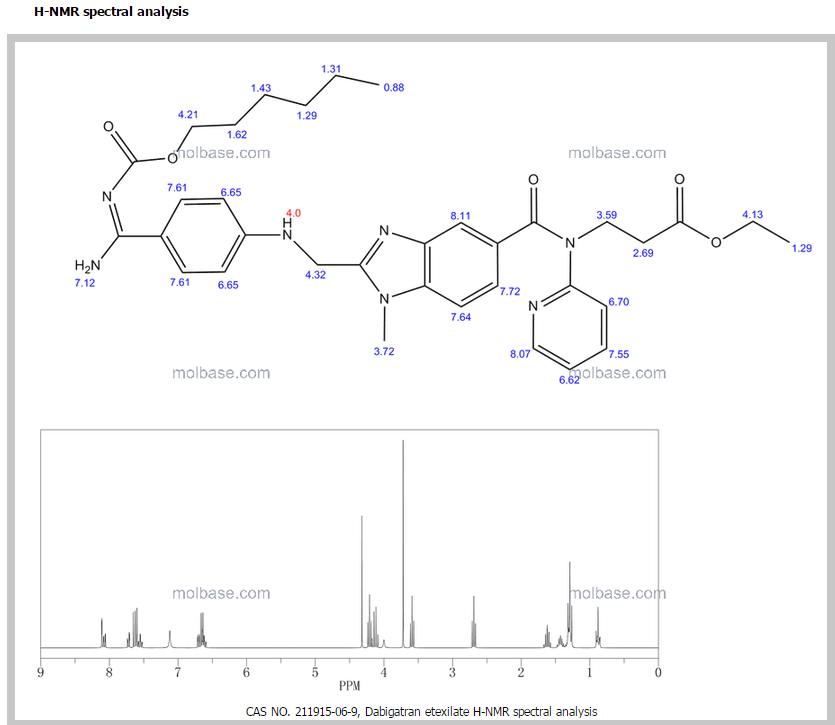
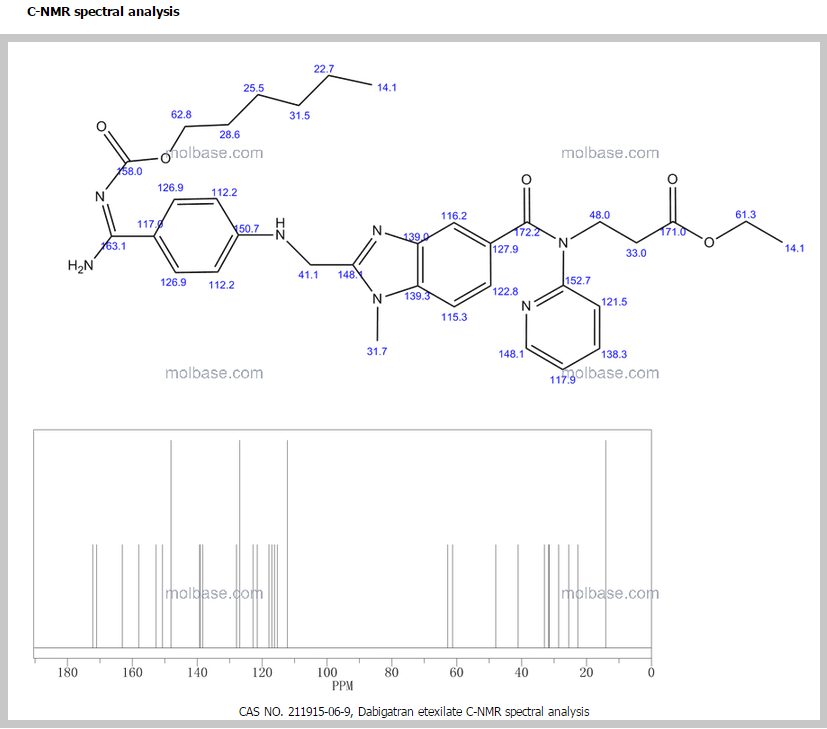
13 C NMR PREDICT ABOVE
| WO2015044375A1 * | Sep 26, 2014 | Apr 2, 2015 | Ratiopharm Gmbh | Pharmaceutical preparation comprising dabigatran etexilate bismesylate |
See full gatran series at………………http://apisynthesisint.blogspot.in/p/argatroban.html
///////////
DABIGATRAN PART 1/3

Dabigatran (Pradaxa in Australia, Canada, Europe and USA, Prazaxa in Japan) is an oral anticoagulant from the class of the direct thrombin inhibitors. It is being studied for various clinical indications and in some cases it offers an alternative towarfarin as the preferred orally administered anticoagulant (“blood thinner”) since it cannot be monitored by blood tests forinternational normalized ratio (INR) monitoring while offering similar results in terms of efficacy. There is no specific way to reverse the anticoagulant effect of dabigatran in the event of a major bleeding event,[2][3] unlike warfarin,[4] although a potential dabigatran antidote (pINN: idarucizumab) is undergoing clinical studies.[5] It was developed by the pharmaceutical company Boehringer Ingelheim.

Medical uses
Dabigatran is used to prevent strokes in those with atrial fibrillation (afib) due to non heart valve causes, as well as deep venous thrombosis (DVT) and pulmonary embolism (PE) in persons who have been treated for 5–10 days with parenteral anticoagulant (usually low molecular weight heparin), and to prevent DVT and PE in some circumstances.[6]
It appears to be as effective as warfarin in preventing nonhemorrhagic strokes and embolic events in those with afib not due to valve problems.[7]

Contraindications
Dabigatran is contraindicated in patients who have active pathological bleeding since dabigatran can increase bleeding risk and can also cause serious and potentially life-threatening bleeds.[8] Dabigatran is also contraindicated in patients who have a history of serious hypersensitivity reaction to dabigatran (e.g. anaphylaxis or anaphylactic shock).[8] The use of dabigatran should also be avoided in patients with mechanical prosthetic heart valve due to the increased risk of thromboembolic events (e.g. valve thrombosis, stroke, and myocardial infarction) and major bleeding associated with dabigatran in this population.[8][9][10]
Adverse effects
The most commonly reported side effect of dabigatran is GI upset. When compared to people anticoagulated with warfarin, patients taking dabigatran had fewer life-threatening bleeds, fewer minor and major bleeds, including intracranial bleeds, but the rate of GI bleeding was significantly higher. Dabigatran capsules contain tartaric acid, which lowers the gastric pH and is required for adequate absorption. The lower pH has previously been associated with dyspepsia; some hypothesize that this plays a role in the increased risk of gastrointestinal bleeding.[11]
A small but significantly increased risk of myocardial infarctions (heart attacks) has been noted when combining the safety outcome data from multiple trials.[12]
Reduced doses should be used in those with poor kidney function.[13]
Pharmacokinetics
Dabigatran has a half-life of approximately 12-14 h and exert a maximum anticoagulation effect within 2-3 h after ingestion.[14] Fatty foods delay the absorption of dabigatran, although the bio-availability of the drug is unaffected.[1] One study showed that absorption may be moderately decreased if taken with a proton pump inhibitor.[15] Drug excretion through P-glycoprotein pumps is slowed in patients taking strong p-glycoprotein pump inhibitors such as quinidine, verapamil, and amiodarone, thus raising plasma levels of dabigatran.[16]
History
Dabigatran (then compound BIBR 953) was discovered from a panel of chemicals with similar structure to benzamidine-based thrombin inhibitor α-NAPAP (N-alpha-(2-naphthylsulfonylglycyl)-4-amidinophenylalanine piperidide), which had been known since the 1980s as a powerful inhibitor of various serine proteases, specifically thrombin, but also trypsin. Addition of ethyl ester and hexyloxycarbonyl carbamide hydrophobic side chains led to the orally absorbed prodrug, BIBR 1048 (dabigatran etexilate).[17]
On March 18, 2008, the European Medicines Agency granted marketing authorisation for Pradaxa for the prevention of thromboembolic disease following hip or knee replacement surgery and for non-valvular atrial fibrillation.[18]
The National Health Service in Britain authorised the use of dabigatran for use in preventing blood clots in hip and knee surgery patients. According to a BBC article in 2008, Dabigatran was expected to cost the NHS £4.20 per day, which was similar to several other anticoagulants.[19]
Pradax received a Notice of Compliance (NOC) from Health Canada on June 10, 2008,[20] for the prevention of blood clots in patients who have undergone total hip or total knee replacement surgery. Approval for atrial fibrillation patients at risk of stroke came in October 2010.[21][22]
The U.S. Food and Drug Administration (FDA) approved Pradaxa on October 19, 2010, for prevention of stroke in patients with non-valvular atrial fibrillation.[23][24][25][26] The approval came after an advisory committee recommended the drug for approval on September 20, 2010[27] although caution is still urged by some outside experts.[28]
On February 14, 2011, the American College of Cardiology Foundation and American Heart Association added dabigatran to their guidelines for management of non-valvular atrial fibrillation with a class I recommendation.[29]
In May 2014 the FDA reported the results of a large study comparing dabigatran to warfarin in 134,000 Medicare patients. The Agency concluded that dabigatran is associated with a lower risk of overall mortality, ischemic stroke, and bleeding in the brain than warfarin. Gastrointestinal bleeding was more common in those treated with dabigatran than in those treated with warfarin. The risk of heart attack was similar between the two drugs. The Agency reiterated its opinion that dabigatran’s overall risk/benefit ratio is favorable.[30]
On July 26, 2014, the British Medical Journal (BMJ) published a series of investigations that accused Boehringer of withholding critical information about the need for monitoring to protect patients from severe bleeding, particularly in the elderly. Review of internal communications between Boehringer researchers and employees, the FDA and the EMA revealed that Boehringer researchers found evidence that serum levels of dabigatran vary widely. The BMJ investigation suggested that Boehringer had a financial motive to withhold this concern from regulatory health agencies because the data conflicted with their extensive marketing of dabigatran as an anticoagulant that does not require monitoring.[31][32]
Research
In August 2015, an article found that idarucizumab was able to reverse the anticoagulation effects of dabigatran within minutes.[33]
References
- Pradaxa Full Prescribing Information. Boehringer Ingelheim. October 2010.
- Eerenberg, ES; Kamphuisen, PW; Sijpkens, MK; Meijers, JC; Buller, HR; Levi, M (2011-10-04). “Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects”. Circulation 124(14): 1573–9. doi:10.1161/CIRCULATIONAHA.111.029017. PMID 21900088. Retrieved 2012-03-15.
- van Ryn J, Stangier J, Haertter S, Liesenfeld KH, Wienen W, Feuring M, Clemens A (Department of Drug Discovery Support, Boehringer Ingelheim Pharma) (Jun 2010).“Dabigatran etexilate–a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity”. Thrombosis and Haemostasis103 (6): 1116–27. doi:10.1160/TH09-11-0758. PMID 20352166. Retrieved2012-03-15.
Although there is no specific antidote to antagonise the anticoagulant effect of dabigatran, due to its short duration of effect drug discontinuation is usually sufficient to reverse any excessive anticoagulant activity.
- Hanley JP, J P (Nov 2004). “Warfarin reversal”. Journal of Clinical Pathology 57 (11): 1132–9. doi:10.1136/jcp.2003.008904. PMC 1770479. PMID 15509671.
- “Boehringer Ingelheim’s Investigational Antidote for Pradaxa® (dabigatran etexilate mesylate) Receives FDA Breakthrough Therapy Designation” (Press release). Ridgefield, CT: Boehringer Ingelheim’. 2014-06-26. Retrieved 2014-07-26.
- http://www.drugs.com/pro/pradaxa.html Pradaxa
- Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). “Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.”. Thrombosis 2013: 640723. doi:10.1155/2013/640723. PMC 3885278.PMID 24455237.
- Pradaxa (dabigatran etexilate mesylate) Prescribing Information:http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=ba74e3cd-b06f-4145-b284-5fd6b84ff3c9#Section_5.4, accessed October 29, 2014.
- “FDA Drug Safety Communication: Pradaxa (dabigatran etexilate mesylate) should not be used in patients with mechanical prosthetic heart valves”. U.S. Food and Drug Administration (FDA). Retrieved October 29, 2014.
- Eikelboom, JW; Connolly, SJ; Brueckmann, M et al. (September 2013). “Dabigatran versus Warfarin in Patients with Mechanical Heart Valves”. N Engl J Med 369: 1206–1214.doi:10.1056/NEJMoa1300615. PMID 23991661.
- ML Blommel et al. (2011). “Dabigatran etexilate: A novel oral direct thrombin inhibitor”.Am J Health Syst Pharm 68 (16): 1506–19. doi:10.2146/ajhp100348. PMID 21817082.
- Uchino K, Hernandez AV; Hernandez (2012). “Dabigatran associated with higher risk of acute coronary events – meta-analysis of noninferiority randomized controlled trials”.Arch. Intern. Med. Online first (5): 397–402. doi:10.1001/archinternmed.2011.1666.PMID 22231617.
- 18/12/2014 Pradaxa -EMEA/H/C/000829 -II/0073
- Chongnarungsin D; Ratanapo S; Srivali N; Ungprasert P; Suksaranjit P; Ahmed S; Cheungpasitporn W (2012). “In-Depth Review of Stroke Prevention in Patients with Non-Valvular Atrial Fibrillation”. Am. Med. J. 3 (2): 100. doi:10.3844/amjsp.2012.100.103.
- Stangier J, Eriksson BI, Dahl OE et al. (May 2005). “Pharmacokinetic profile of the oral direct thrombin inhibitor dabigatran etexilate in healthy volunteers and patients undergoing total hip replacement”. J Clin Pharmacol 45 (5): 555–63.doi:10.1177/0091270005274550. PMID 15831779.
- “Pradaxa Summary of Product Characteristics”. European Medicines Agency.
- Hauel NH, Nar H, Priepke H, Ries U, Stassen JM, Wienen W; Nar; Priepke; Ries; Stassen; Wienen (April 2002). “Structure-based design of novel potent nonpeptide thrombin inhibitors”. J Med Chem 45 (9): 1757–66. doi:10.1021/jm0109513.PMID 11960487. Lay summary.
- “Pradaxa EPAR”. European Medicines Agency. Retrieved 2011-01-30.
- “Clot drug ‘could save thousands'”. BBC News Online. 2008-04-20. Retrieved2008-04-21.
- “Summary Basis of Decision (SBD): Pradax” Health Canada. 2008-11-06.
- Kirkey, Sharon (29 October 2010). “Approval of new drug heralds ‘momentous’ advance in stroke prevention”. Montreal Gazette. Retrieved 29 October 2010.
- “Pradax (Dabigatran Etexilate) Gains Approval In Canada For Stroke Prevention In Atrial Fibrillation” Medical News Today. 28 October 2010.
- Connolly, SJ; Ezekowitz, MD; Yusuf, S et al. (September 2009). “Dabigatran versus warfarin in patients with atrial fibrillation” (PDF). N Engl J Med 361 (12): 1139–51.doi:10.1056/NEJMoa0905561. PMID 19717844.
- Turpie AG (January 2008). “New oral anticoagulants in atrial fibrillation”. Eur Heart J 29(2): 155–65. doi:10.1093/eurheartj/ehm575. PMID 18096568.
- “Boehringer wins first US OK in blood-thinner race”. Thomson Reuters. 2010-10-19. Retrieved 2010-10-20.
- “FDA approves Pradaxa to prevent stroke in people with atrial fibrillation”. U.S. Food and Drug Administration (FDA). 2010-10-19.
- Shirley S. Wang (2010-09-20). “New Blood-Thinner Recommended by FDA Panel”. The Wall Street Journal. Retrieved 2010-10-20.
- Merli G, Spyropoulos AC, Caprini JA; Spyropoulos; Caprini (August 2009). “Use of emerging oral anticoagulants in clinical practice: translating results from clinical trials to orthopedic and general surgical patient populations”. Ann Surg 250 (2): 219–28.doi:10.1097/SLA.0b013e3181ae6dbe. PMID 19638915.
- Wann LS, Curtis AB, Ellenbogen KA, Estes NA, Ezekowitz MD, Jackman WM, January CT, Lowe JE, Page RL, Slotwiner DJ, Stevenson WG, Tracy CM, Jacobs AK; Curtis; Ellenbogen; Estes Na; Ezekowitz; Jackman; January; Lowe; Page; Slotwiner; Stevenson; Tracy; Fuster; Rydén; Cannom; Crijns; Curtis; Ellenbogen; Halperin; Kay; Le Heuzey; Lowe; Olsson; Prystowsky; Tamargo; Wann; Jacobs; Anderson; Albert et al. (March 2011). “2011 ACCF/AHA/HRS Focused Update on the Management of Patients With Atrial Fibrillation (Update on Dabigatran): A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines”. Circulation123 (10): 1144–50. doi:10.1161/CIR.0b013e31820f14c0. PMID 21321155.
- “FDA Drug Safety Communication: FDA study of Medicare patients finds risks lower for stroke and death but higher for gastrointestinal bleeding with Pradaxa (dabigatran) compared to warfarin”.
- Cohen, D (July 2014). “Dabigatran: how the drug company withheld important analyses”.BMJ 349: g4670. doi:10.1136/bmj.g4670. PMID 25055829.
- Moore TJ, Cohen MR, Mattison DR; Cohen; Mattison (July 2014). “Dabigatran, bleeding, and the regulators”. BMJ 349: g4517. doi:10.1136/bmj.g4517. PMID 25056265.
- Pollack, Charles V.; Reilly, Paul A.; Eikelboom, John; Glund, Stephan; Verhamme, Peter; Bernstein, Richard A.; Dubiel, Robert; Huisman, Menno V.; Hylek, Elaine M. (2015-01-01).“Idarucizumab for Dabigatran Reversal”. New England Journal of Medicine 373 (6).doi:10.1056/nejmoa1502000.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Ethyl N-[(2-{[(4-{N ’-[(hexyloxy)carbonyl]carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl]-N-2-pyridinyl-β-alaninate
|
|
| Clinical data | |
| Trade names | Pradaxa, Pradax, Prazaxa |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
oral |
| Pharmacokinetic data | |
| Bioavailability | 3–7%[1] |
| Protein binding | 35%[1] |
| Biological half-life | 12–17 hours[1] |
| Identifiers | |
| CAS Registry Number | 211915-06-9 |
| ATC code | B01AE07 |
| PubChem | CID: 6445226 |
| DrugBank | DB06695 |
| ChemSpider | 4948999 |
| ChEMBL | CHEMBL539697 |
| Chemical data | |
| Formula | C34H41N7O5 |
| Molecular mass | 627.734 g/mol |
External links
- Pradaxa.com. Boehringer Ingelheim.
- dabigatran.com. Boehringer Ingelheim.
- Pradaxa For U.S. Health Care Professionals. Boehringer Ingelheim.
- Pradaxa Prescribing Information. Boehringer Ingelheim.
- Pradaxa Medication Guide. Boehringer Ingelheim.
- Dabigatran. MedlinePlus. United States National Library of Medicine (NLM).
- Dabigatran. Drug Information Portal. United States National Library of Medicine (NLM).
The chemical name for dabigatran etexilate mesylate, a direct thrombininhibitor, is β-Alanine, N-[[2-[[[4-[[[(hexyloxy)carbonyl]amino]iminomethyl] phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl]-N-2-pyridinyl-,ethyl ester, methanesulfonate. The empirical formula is C34H41N7O5 • CH4O3S and the molecular weight is 723.86 (mesylate salt), 627.75 (free base). The structural formula is:
 |
Dabigatran etexilate mesylate is a yellow-white to yellow powder. A saturated solution in pure water has a solubility of 1.8 mg/mL. It is freely soluble in methanol, slightly soluble in ethanol, and sparingly soluble in isopropanol.
The 150 mg capsule for oral administration contains 172.95 mg dabigatran etexilate mesylate, which is equivalent to 150 mg of dabigatran etexilate, and the following inactive ingredients: acacia, dimethicone, hypromellose, hydroxypropyl cellulose, talc, and tartaric acid. The capsule shell is composed of carrageenan, FD&C Blue No. 2 (150 mg only), FD&C Yellow No. 6, hypromellose, potassium chloride, titanium dioxide, and black edible ink. The 75 mg capsule contains 86.48 mg dabigatran etexilate mesylate, equivalent to 75 mg dabigatran etexilate, and is otherwise similar to the 150 mg capsule.
See full gatran series at………………http://apisynthesisint.blogspot.in/p/argatroban.html
/////////Dabigatran, Pradaxa
Polymorph case study……….Duvelisib


Duvelisib
Infinity and AbbVie partner to develop and commercialise duvelisib for cancer
INK 1197; IPI 145; 8-Chloro-2-phenyl-3-[(1S)-1-(9H-purin-6-ylamino)ethyl]-1(2H)-isoquinolinone
1(2H)-Isoquinolinone, 8-chloro-2-phenyl-3-((1S)-1-(9H-purin-6-ylamino)ethyl)-
8-Chloro-2-phenyl-3-((1S)-1-(7H-purin-6-ylamino)ethyl)isoquinolin-1(2H)-one
(S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
UNII-610V23S0JI; IPI-145; INK-1197;
Originator…….. Millennium Pharmaceuticals
| Molecular Formula | C22H17ClN6O | |
| Molecular Weight | 416.86 | |
| CAS Registry Number | 1201438-56-3 |

Infinity Pharmaceuticals has partnered with AbbVie to develop and commercialise its duvelisib (IPI-145), an oral inhibitor of phosphoinositide-3-kinase (PI3K)-delta and PI3K-gamma, to treat patients with cancer.

Duvelisib
The filing of patents claiming new crystalline forms, usually 4−6 years after the original product patent, is a typical strategy applied by such companies to extend patent protection. This patent protection approach by big pharma forces generic bulk producers to discover and file patents on new polymorphs if they want to market the drug after expiry of the product patents.
Polymorphism is of paramount importance due to its effect on some physical characteristics of powders such as melting point, flowability, vapour pressure, bulk density, chemical reactivity, apparent solubility and dissolution rate, and optical and electrical properties. In other words, polymorphism can affect drug stability, manipulation, and bioavailability
the principal aim of generic bulk producers was to generate a competitive market advantage by protecting their new crystal form.
Polymorphic forms of a compound of Formula (I):.US8809349

herein referred to as Form A, Form B, Form C, Form D, Form E, Form F, Form G, Form H, Form I, Form J, or an amorphous form of a compound of Formula (I), or a salt, solvate, or hydrate thereof; or a mixture of two or more thereof. In one embodiment, the polymorphic form of a compound of Formula (I) can be a crystalline form, a partially crystalline form, an amorphous form, or a mixture of crystalline form(s) and/or amorphous form(s).
(XRPD) peaks
“Enantiomerically pure”
As used herein, and unless otherwise specified, the term “enantiomerically pure” means a stereomerically pure composition of a compound having one or more chiral center(s).
As used herein, and unless otherwise specified, the terms “enantiomeric excess” and “diastereomeric excess” are used interchangeably herein. In some embodiments, compounds with a single stereocenter can be referred to as being present in “enantiomeric excess,” and those with at least two stereocenters can be referred to as being present in “diastereomeric excess.” For example, the term “enantiomeric excess” is well known in the art and is defined as:
eea=(conc.ofa-conc.ofbconc.ofa+conc.ofb)×100
Thus, the term “enantiomeric excess” is related to the term “optical purity” in that both are measures of the same phenomenon. The value of ee will be a number from 0 to 100, zero being racemic and 100 being enantiomerically pure. A compound which in the past might have been called 98% optically pure is now more precisely characterized by 96% ee. A 90% ee reflects the presence of 95% of one enantiomer and 5% of the other(s) in the material in question.
Some compositions described herein contain an enantiomeric excess of at least about 50%, 75%, 90%, 95%, or 99% of the S enantiomer. In other words, the compositions contain an enantiomeric excess of the S enantiomer over the R enantiomer. In other embodiments, some compositions described herein contain an enantiomeric excess of at least about 50%, 75%, 90%, 95%, or 99% of the R enantiomer. In other words, the compositions contain an enantiomeric excess of the R enantiomer over the S enantiomer.
GRAPHS
FIG. 1 shows an X-ray powder diffraction (XRPD) for Polymorph Form A.
FIG. 2 shows an XRPD for Polymorph Form B.
FIG. 3 shows an XRPD for Polymorph Form C.
FIG. 4 shows an XRPD for Polymorph Form D.
FIG. 5 shows an XRPD for Polymorph Form E.
FIG. 6 shows an XRPD for Polymorph Form F.
FIG. 7 shows an XRPD for Polymorph Form G.
FIG. 8 shows an XRPD for Polymorph Form H.
FIG. 9 shows an XRPD for Polymorph Form I.
FIG. 10 shows an XRPD for Polymorph Form J.
FIG. 11 shows an XRPD for amorphous compound of Formula (I).
FIG. 12 shows a differential scanning calorimetry (DSC) thermogram for Polymorph Form A.
FIG. 13 shows a DSC for Polymorph Form B.
FIG. 14 shows a DSC for Polymorph Form C.
FIG. 15 shows a DSC for Polymorph Form D.
FIG. 16 shows a DSC for Polymorph Form E.
FIG. 17 shows a DSC for Polymorph Form F.
FIG. 18 shows a DSC for Polymorph Form G.
FIG. 19 shows a DSC for Polymorph Form H.
FIG. 20 shows a DSC for Polymorph Form I.
FIG. 21 shows a DSC for Polymorph Form J.
FIG. 22 shows a DSC thermogram and a thermogravimetric analysis (TGA) for Polymorph Form A.
FIG. 23 shows two DSC thermograms for Polymorph Form C.
FIG. 24 shows a DSC and a TGA for Polymorph Form F.
FIG. 25 shows a panel of salts tested for formation of crystalline solids in various solvents.
FIG. 26 shows a single crystal X-ray structure of Polymorph Form G MTBE (t-butyl methyl ether) solvate of a compound of Formula (I).
FIG. 27 shows an FT-IR spectra of Polymorph Form C.
FIG. 28 shows a 1H-NMR spectra of Polymorph Form C.
FIG. 29 shows a 13C-NMR spectra of Polymorph Form C.
FIG. 30 shows a dynamic vapor sorption (DVS) analysis of Polymorph Form C.
FIG. 31 shows representative dissolution profiles of capsules containing Polymorph Form C.
DRAWINGS
FIG. 1 shows an X-ray powder diffraction (XRPD) for Polymorph Form A.

FIG. 1 shows a representative X-ray powder diffraction (XRPD) for polymorph Form A.
In one embodiment, polymorph Form A can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 1. In one embodiment, polymorph Form A can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 12.2° (±0.2°), and 18.3° (±0.2°). In one embodiment, polymorph Form A can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 12.2° (±0.2°), and 18.3° (±0.2°) in combination with at least one XRPD peak selected from 2θ=15.6° (±0.2°) and 19.2° (±0.2°). In another embodiment, polymorph Form A can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 12.2° (±0.2°), 15.6° (±0.2°), 18.3° (±0.2°), and 19.2° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.1° (±0.2°), 9.4° (±0.2°), 12.4° (±0.2°), 14.8° (±0.2°), 16.3° (±0.2°), 17.7° (±0.2°), 21.1° (±0.2°), 21.9° (±0.2°), 24.0° (±0.2°), and 26.9° (±0.2°). In one embodiment, polymorph Form A can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 1.
FIG. 2 shows an XRPD for Polymorph Form B.

FIG. 2 shows a representative XRPD for polymorph Form B.
In one embodiment, polymorph Form B can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 2. In one embodiment, polymorph Form B can be characterized as having at least one XRPD peak selected from 2θ=7.9° (±0.2°), 13.4° (±0.2°), and 23.4° (±0.2°). In one embodiment, polymorph Form B can be characterized as having at least one XRPD peak selected from 2θ=7.9° (±0.2°), 13.4° (±0.2°), and 23.4° (±0.2°) in combination with at least one XRPD peak selected from 2θ=14.0° (±0.2°) and 15.0° (±0.2°). In another embodiment, polymorph Form B can be characterized as having at least one XRPD peak selected from 2θ=7.9° (±0.2°), 13.4° (±0.2°), 14.0° (±0.2°), 15.0° (±0.2°), and 23.4° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.5° (±0.2°), 12.7° (±0.2°), 13.6° (±0.2°), 14.2° (±0.2°), 15.7° (±0.2°), 19.0° (±0.2°), 22.3° (±0.2°), 24.2° (±0.2°), 24.8° (±0.2°), and 26.9° (±0.2°). In one embodiment, polymorph Form B can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 2.
FIG. 3 shows an XRPD for Polymorph Form C.
In one embodiment, polymorph Form C can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 3. In one embodiment, Form C can be characterized by having at least one XRPD peak selected from 2θ=10.5° (±0.2°), 13.7° (±0.2°), and 24.5° (±0.2°). In another embodiment, Form C can be characterized by having at least one XRPD peak selected from 2θ=10.4° (±0.2°), 13.3° (±0.2°), and 24.3° (±0.2°). In one embodiment, polymorph Form C can be characterized as having at least one XRPD peak selected from 2θ=10.4° (±0.2°), 13.3° (±0.2°), and 24.3° (±0.2°) in combination with at least one XRPD peak selected from 2θ=6.6° (±0.2°) and 12.5° (±0.2°). In another embodiment, polymorph Form C can be characterized as having at least one XRPD peak selected from 2θ=6.6° (±0.2°), 10.4° (±0.2°), 12.5° (±0.2°), 13.3° (±0.2°), and 24.3° (±0.2°) in combination with at least one XRPD peak selected from 2θ=8.8° (±0.2°), 9.9° (±0.2°), 13.4° (±0.2°), 15.5° (±0.2°), 16.9° (±0.2°), 19.8° (±0.2°), 21.3° (±0.2°), 23.6° (±0.2°), 25.3° (±0.2°), and 27.9° (±0.2°). In one embodiment, polymorph Form C can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 3.

FIG. 4 shows an XRPD for Polymorph Form D.

In one embodiment, polymorph Form D can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 4. In one embodiment, polymorph Form D can be characterized as having at least one XRPD peak selected from 2θ=11.4° (±0.2°), 17.4° (±0.2°), and 22.9° (±0.2°). In one embodiment, polymorph Form D can be characterized as having at least one XRPD peak selected from 2θ=11.4° (±0.2°), 17.4° (±0.2°), and 22.9° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.2° (±0.2°) and 18.3° (±0.2°). In another embodiment, polymorph Form D can be characterized as having at least one XRPD peak selected from 2θ=9.2° (±0.2°), 11.4° (±0.2°), 17.4° (±0.2°), 18.3° (±0.2°), and 22.9° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.8° (±0.2°), 12.2° (±0.2°), 15.8° (±0.2°), 16.2° (±0.2°), 16.8° (±0.2°), 18.9° (±0.2°), 19.9° (±0.2°), 20.0° (±0.2°), 24.9° (±0.2°), and 29.3° (±0.2°). In one embodiment, polymorph Form D can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 4.
FIG. 5 shows an XRPD for Polymorph Form E. US8809349
In one embodiment, polymorph Form E can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 5. In one embodiment, polymorph Form E can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.3° (±0.2°), and 24.4° (±0.2°). In one embodiment, polymorph Form E can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.3° (±0.2°), and 24.4° (±0.2°) in combination with at least one XRPD peak selected from 2θ=12.7° (±0.2°) and 13.9° (±0.2°). In another embodiment, polymorph Form E can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.3° (±0.2°), 12.7° (±0.2°), 13.9° (±0.2°), and 24.4° (±0.2°) in combination with at least one XRPD peak selected from 2θ=12.4° (±0.2°), 13.3° (±0.2°), 14.3° (±0.2°), 15.5° (±0.2°), 17.4° (±0.2°), 18.5° (±0.2°), 22.0° (±0.2°), 23.9° (±0.2°), 24.1° (±0.2°), and 26.4° (±0.2°). In one embodiment, polymorph Form E can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 5.
FIG. 6 shows an XRPD for Polymorph Form F. US8809349
In one embodiment, polymorph Form F can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 6. In one embodiment, polymorph Form F can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 17.3° (±0.2°), and 24.6° (±0.2°). In one embodiment, polymorph Form F can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 17.3° (±0.2°), and 24.6° (±0.2°) in combination with at least one XRPD peak selected from 2θ=14.0° (±0.2°) and 19.2° (±0.2°). In another embodiment, polymorph Form F can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 14.0° (±0.2°), 17.3° (±0.2°), 19.2° (±0.2°), and 24.6° (±0.2°) in combination with at least one XRPD peak selected from 2θ=12.4° (±0.2°), 16.1° (±0.2°), 16.6° (±0.2°), 17.1° (±0.2°), 20.8° (±0.2°), 21.5° (±0.2°), 22.0° (±0.2°), 24.3° (±0.2°), 25.2° (±0.2°), and 25.4° (±0.2°). In one embodiment, polymorph Form F can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 6.
FIG. 7 shows an XRPD for Polymorph Form G. US8809349
In one embodiment, polymorph Form G can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 7. In one embodiment, polymorph Form G can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.5° (±0.2°), and 19.0° (±0.2°). In one embodiment, polymorph Form G can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.5° (±0.2°), and 19.0° (±0.2°) in combination with at least one XRPD peak selected from 2θ=10.6° (±0.2°) and 19.6° (±0.2°). In another embodiment, polymorph Form G can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.5° (±0.2°), 10.6° (±0.2°), 19.0° (±0.2°), and 19.6° (±0.2°) in combination with at least one XRPD peak selected from 2θ=13.4° (±0.2°), 15.0° (±0.2°), 15.8° (±0.2°), 17.8° (±0.2°), 20.7° (±0.2°), 21.2° (±0.2°), 22.8° (±0.2°), 23.8° (±0.2°), 24.3° (±0.2°), and 25.6° (±0.2°). In one embodiment, polymorph Form G can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 7.
FIG. 8 shows an XRPD for Polymorph Form H. US8809349
In one embodiment, polymorph Form H can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 8. In one embodiment, polymorph Form H can be characterized as having at least one XRPD peak selected from 2θ=8.9° (±0.2°), 9.2° (±0.2°), and 14.1° (±0.2°). In one embodiment, polymorph Form H can be characterized as having at least one XRPD peak selected from 2θ=8.9° (±0.2°), 9.2° (±0.2°), and 14.1° (±0.2°) in combination with at least one XRPD peak selected from 2θ=17.3° (±0.2°) and 18.5° (±0.2°). In another embodiment, polymorph Form H can be characterized as having at least one XRPD peak selected from 2θ=8.9° (±0.2°), 9.2° (±0.2°), 14.1° (±0.2°), 17.3° (±0.2°), and 18.5° (±0.2°) in combination with at least one XRPD peak selected from 2θ=7.1° (±0.2°), 10.6° (±0.2°), 11.3° (±0.2°), 11.6° (±0.2°), 16.2° (±0.2°), 18.3° (±0.2°), 18.8° (±0.2°), 20.3° (±0.2°), 21.7° (±0.2°), and 24.7° (±0.2°). In one embodiment, polymorph Form H can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 8.
FIG. 9 shows an XRPD for Polymorph Form I.
In one embodiment, polymorph Form I can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 9. In one embodiment, polymorph Form I can be characterized as having at least one XRPD peak selected from 2θ=9.7° (±0.2°), 19.3° (±0.2°), and 24.5° (±0.2°). In one embodiment, polymorph Form I can be characterized as having at least one XRPD peak selected from 2θ=9.7° (±0.2°), 19.3° (±0.2°), and 24.5° (±0.2°) in combination with at least one XRPD peak selected from 2θ=11.4° (±0.2°) and 14.2° (±0.2°). In another embodiment, polymorph Form I can be characterized as having at least one XRPD peak selected from 2θ=9.7° (±0.2°), 11.4° (±0.2°), 14.2° (±0.2°), 19.3° (±0.2°), and 24.5° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.2° (±0.2°), 14.7° (±0.2°), 15.5° (±0.2°), 16.7° (±0.2°), 17.3° (±0.2°), 18.4° (±0.2°), 21.4° (±0.2°), 22.9° (±0.2°), 29.1° (±0.2°), and 34.1° (±0.2°). In one embodiment, polymorph Form I can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 9.
FIG. 10 shows an XRPD for Polymorph Form J.
In one embodiment, polymorph Form J can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 10. In one embodiment, polymorph Form J can be characterized as having at least one XRPD peak selected from 2θ=9.1° (±0.2°), 17.3° (±0.2°), and 18.3° (±0.2°). In one embodiment, polymorph Form J can be characterized as having at least one XRPD peak selected from 2θ=9.1° (±0.2°), 17.3° (±0.2°), and 18.3° (±0.2°) in combination with at least one XRPD peak selected from 2θ=16.4° (±0.2°) and 17.9° (±0.2°). In another embodiment, polymorph Form J can be characterized as having at least one XRPD peak selected from 2θ=9.1° (±0.2°), 16.4° (±0.2°), 17.3° (±0.2°), 17.9° (±0.2°), and 18.3° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.4° (±0.2°), 10.1° (±0.2°), 10.7° (±0.2°), 14.0° (±0.2°), 14.3° (±0.2°), 15.5° (±0.2°), 16.9° (±0.2°), 19.9° (±0.2°), 24.0° (±0.2°), and 24.7° (±0.2°). In one embodiment, polymorph Form J can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 10.
FIG. 11 shows an XRPD for amorphous compound of Formula (I).
FIG. 12 shows a differential scanning calorimetry (DSC) thermogram for Polymorph Form A.
FIG. 13 shows a DSC for Polymorph Form B.
FIG. 14 shows a DSC for Polymorph Form C.
FIG. 15 shows a DSC for Polymorph Form D.
FIG. 16 shows a DSC for Polymorph Form E.
FIG. 17 shows a DSC for Polymorph Form F.
FIG. 18 shows a DSC for Polymorph Form G.
FIG. 19 shows a DSC for Polymorph Form H.
FIG. 20 shows a DSC for Polymorph Form I.
FIG. 21 shows a DSC for Polymorph Form J.
FIG. 22 shows a DSC thermogram and a thermogravimetric analysis (TGA) for Polymorph Form A.
FIG. 23 shows two DSC thermograms for Polymorph Form C.
FIG. 24 shows a DSC and a TGA for Polymorph Form F.
FIG. 25 shows a panel of salts tested for formation of crystalline solids in various solvents.
FIG. 26 shows a single crystal X-ray structure of Polymorph Form G MTBE (t-butyl methyl ether) solvate of a compound of Formula (I).
FIG. 27 shows an FT-IR spectra of Polymorph Form C.

FIG. 28 shows a 1H-NMR spectra of Polymorph Form C.

FIG. 29 shows a 13C-NMR spectra of Polymorph Form C.

FIG. 30 shows a dynamic vapor sorption (DVS) analysis of Polymorph Form C.
FIG. 31 shows representative dissolution profiles of capsules containing Polymorph Form C.
Enantiomers
Enantiomers can be isolated from racemic mixtures by any method known to those skilled in the art, including chiral high pressure liquid chromatography (HPLC), the formation and crystallization of chiral salts, or prepared by asymmetric syntheses. See, for example, Enantiomers, Racemates and Resolutions (Jacques, Ed., Wiley Interscience, New York, 1981); Wilen et al., Tetrahedron 33:2725 (1977); Stereochemistry of Carbon Compounds (E. L. Eliel, Ed., McGraw-Hill, NY, 1962); and Tables of Resolving Agents and Optical Resolutions p. 268 (E. L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, Ind. 1972).
“Tautomer”
The term “tautomer” is a type of isomer that includes two or more interconvertable compounds resulting from at least one formal migration of a hydrogen atom and at least one change in valency (e.g., a single bond to a double bond, a triple bond to a single bond, or vice versa). “Tautomerization” includes prototropic or proton-shift tautomerization, which is considered a subset of acid-base chemistry. “Prototropic tautomerization” or “proton-shift tautomerization” involves the migration of a proton accompanied by changes in bond order. The exact ratio of the tautomers depends on several factors, including temperature, solvent, and pH. Where tautomerization is possible (e.g., in solution), a chemical equilibrium of tautomers can be reached. Tautomerizations (i.e., the reaction providing a tautomeric pair) can be catalyzed by acid or base, or can occur without the action or presence of an external agent. Exemplary tautomerizations include, but are not limited to, keto-to-enol; amide-to-imide; lactam-to-lactim; enamine-to-imine; and enamine-to-(a different) enamine tautomerizations. An example of keto-enol tautomerization is the interconversion of pentane-2,4-dione and 4-hydroxypent-3-en-2-one tautomers. Another example of tautomerization is phenol-keto tautomerization. Another example of phenol-keto tautomerization is the interconversion of pyridin-4-ol and pyridin-4(1H)-one tautomers.
As defined herein, the term “Formula (I)” includes (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one in its imide tautomer shown below as (1-1) and in its lactim tautomer shown below as (1-2):

“polymorph” can be used herein to describe a crystalline material, e.g., a crystalline form. In certain embodiments, “polymorph” as used herein are also meant to include all crystalline and amorphous forms of a compound or a salt thereof, including, for example, crystalline forms, polymorphs, pseudopolymorphs, solvates, hydrates, co-crystals, unsolvated polymorphs (including anhydrates), conformational polymorphs, tautomeric forms, disordered crystalline forms, and amorphous forms, as well as mixtures thereof, unless a particular crystalline or amorphous form is referred to. Compounds of the present disclosure include crystalline and amorphous forms of those compounds, including, for example, crystalline forms, polymorphs, pseudopolymorphs, solvates, hydrates, co-crystals, unsolvated polymorphs (including anhydrates), conformational polymorphs, tautomeric forms, disordered crystalline forms, and amorphous forms of the compounds or a salt thereof, as well as mixtures thereof.
As used herein, and unless otherwise specified, a particular form of a compound of Formula (I) described herein (e.g., Form A, B, C, D, E, F, G, H, I, J, or amorphous form of a compound of Formula (I), or mixtures thereof) is meant to encompass a solid form of a compound of Formula (I), or a salt, solvate, or hydrate thereof, among others.
The polymorphs made according to the methods provided herein can be characterized by any methodology known in the art. For example, the polymorphs made according to the methods provided herein can be characterized by X-ray powder diffraction (XRPD), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), dynamic vapor sorption (DVS), hot-stage microscopy, optical microscopy, Karl Fischer analysis, melting point, spectroscopy (e.g., Raman, solid state nuclear magnetic resonance (ssNMR), liquid state nuclear magnetic resonance (1H- and 13C-NMR), and FT-IR), thermal stability, grinding stability, and solubility, among others.
“Solid form”
The terms “solid form” and related terms herein refer to a physical form comprising a compound provided herein or a salt or solvate or hydrate thereof, which is not in a liquid or a gaseous state. Solid forms can be crystalline, amorphous, disordered crystalline, partially crystalline, and/or partially amorphous.
“Crystalline,”
The term “crystalline,” when used to describe a substance, component, or product, means that the substance, component, or product is substantially crystalline as determined, for example, by X-ray diffraction. See, e.g., Remington: The Science and Practice of Pharmacy, Lippincott Williams & Wilkins, 21st ed. (2005).
As used herein, and unless otherwise specified, the term “crystalline form,” “crystal form,” and related terms herein refer to the various crystalline material comprising a given substance, including single-component crystal forms and multiple-component crystal forms, and including, but not limited to, polymorphs, solvates, hydrates, co-crystals and other molecular complexes, as well as salts, solvates of salts, hydrates of salts, other molecular complexes of salts, and polymorphs thereof. In certain embodiments, a crystal form of a substance can be substantially free of amorphous forms and/or other crystal forms. In other embodiments, a crystal form of a substance can contain about 1%, about 2%, about 3%, about 4%, about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 35%, about 40%, about 45% or about 50% of one or more amorphous form(s) and/or other crystal form(s) on a weight and/or molar basis.
Certain crystal forms of a substance can be obtained by a number of methods, such as, without limitation, melt recrystallization, melt cooling, solvent recrystallization, recrystallization in confined spaces, such as, e.g., in nanopores or capillaries, recrystallization on surfaces or templates, such as, e.g., on polymers, recrystallization in the presence of additives, such as, e.g., co-crystal counter-molecules, desolvation, dehydration, rapid evaporation, rapid cooling, slow cooling, vapor diffusion, sublimation, grinding, solvent-drop grinding, microwave-induced precipitation, sonication-induced precipitation, laser-induced precipitation, and/or precipitation from a supercritical fluid. As used herein, and unless otherwise specified, the term “isolating” also encompasses purifying.
Characterizing crystal forms and amorphous forms
Techniques for characterizing crystal forms and amorphous forms can include, but are not limited to, thermal gravimetric analysis (TGA), differential scanning calorimetry (DSC), X-ray powder diffractometry (XRPD), single crystal X-ray diffractometry, vibrational spectroscopy, e.g., infrared (IR) and Raman spectroscopy, solid-state nuclear magnetic resonance (NMR) spectroscopy, optical microscopy, hot stage optical microscopy, scanning electron microscopy (SEM), electron crystallography and quantitative analysis, particle size analysis (PSA), surface area analysis, solubility studies, and dissolution studies.
PEAK
As used herein, and unless otherwise specified, the term “peak,” when used in connection with the spectra or data presented in graphical form (e.g., XRPD, IR, Raman, and NMR spectra), refers to a peak or other special feature that one skilled in the art would recognize as not attributable to background noise. The term “significant peak” refers to peaks at least the median size (e.g., height) of other peaks in the spectrum or data, or at least 1.5, 2, or 2.5 times the background level in the spectrum or data.
“Pharmaceutically acceptable carrier”
“pharmaceutically acceptable carrier” or “pharmaceutically acceptable excipient” includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like. The use of such media and agents for pharmaceutically active substances is known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, its use in the therapeutic compositions of the present disclosure is contemplated. Supplementary active ingredients can also be incorporated into the compositions.
“Substantially pure”
the term “substantially pure” when used to describe a polymorph, a crystal form, or a solid form of a compound or complex described herein means a solid form of the compound or complex that comprises a particular polymorph and is substantially free of other polymorphic and/or amorphous forms of the compound. A representative substantially pure polymorph comprises greater than about 80% by weight of one polymorphic form of the compound and less than about 20% by weight of other polymorphic and/or amorphous forms of the compound; greater than about 90% by weight of one polymorphic form of the compound and less than about 10% by weight of other polymorphic and/or amorphous forms of the compound; greater than about 95% by weight of one polymorphic form of the compound and less than about 5% by weight of other polymorphic and/or amorphous forms of the compound; greater than about 97% by weight of one polymorphic form of the compound and less than about 3% by weight of other polymorphic and/or amorphous forms of the compound; or greater than about 99% by weight of one polymorphic form of the compound and less than about 1% by weight of other polymorphic and/or amorphous forms of the compound.
“Stable”
The term “stable” refers to a compound or composition that does not readily decompose or change in chemical makeup or physical state. A stable composition or formulation provided herein does not significantly decompose under normal manufacturing or storage conditions. In some embodiments, the term “stable,” when used in connection with a formulation or a dosage form, means that the active ingredient of the formulation or dosage form remains unchanged in chemical makeup or physical state for a specified amount of time and does not significantly degrade or aggregate or become otherwise modified (e.g., as determined, for example, by HPLC, FTIR, or XRPD). In some embodiments, about 70 percent or greater, about 80 percent or greater, about 90 percent or greater, about 95 percent or greater, about 98 percent or greater, or about 99 percent or greater of the compound remains unchanged after the specified period. In one embodiment, a polymorph provided herein is stable upon long-term storage (e.g., no significant change in polymorph form after about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 18, 24, 30, 36, 42, 48, 54, 60, or greater than about 60 months).
Amorphous form
In one embodiment, an amorphous form of a compound of Formula (I), or a pharmaceutically acceptable salt, solvate, or hydrate thereof, can be made by dissolution of a crystalline form followed by removal of solvent under conditions in which stable crystals are not formed. For example, solidification can occur by rapid removal of solvent, by rapid addition of an anti-solvent (causing the amorphous form to precipitate out of solution), or by physical interruption of the crystallization process. Grinding processes can also be used. In other embodiments, an amorphous form of a compound of Formula (I), or a pharmaceutically acceptable salt, solvate, or hydrate thereof, can be made using a process or procedure described herein elsewhere.
In certain embodiments, an amorphous form can be obtained by fast cooling from a single solvent system, such as, e.g., ethanol, isopropyl alcohol, t-amyl alcohol, n-butanol, methanol, acetone, ethyl acetate, or acetic acid. In certain embodiments, an amorphous form can be obtained by slow cooling from a single solvent system, such as, e.g., ethanol, isopropyl alcohol, t-amyl alcohol, or ethyl acetate.
In certain embodiments, an amorphous form can be obtained by fast cooling from a binary solvent system, for example, with acetone or DME as the primary solvent. In certain embodiments, an amorphous form can be obtained by slow cooling from a binary solvent system, for example, with ethanol, isopropyl alcohol, THF, acetone, or methanol as the primary solvent. In some embodiments, an amorphous form can be obtained by dissolution of a compound of Formula (I) in t-butanol and water at elevated temperature, followed by cooling procedures to afford an amorphous solid form.
Salt Forms
In certain embodiments, a compound of Formula (I) provided herein is a pharmaceutically acceptable salt, or a solvate or hydrate thereof. In one embodiment, pharmaceutically acceptable acid addition salts of a compound provided herein can be formed with inorganic acids and organic acids. Inorganic acids from which salts can be derived include, but are not limited to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like. Organic acids from which salts can be derived include, but are not limited to, acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid, and the like. In other embodiments, if applicable, pharmaceutically acceptable base addition salts of a compound provided herein can be formed with inorganic and organic bases. Inorganic bases from which salts can be derived include, but are not limited to, sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum, and the like. Organic bases from which salts can be derived include, but are not limited to, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like. Exemplary bases include, but are not limited to, isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, and ethanolamine. In some embodiments, a pharmaceutically acceptable base addition salt is ammonium, potassium, sodium, calcium, or magnesium salt. In one embodiment, bis salts (i.e., two counterions) and higher salts (e.g., three or more counterions) are encompassed within the meaning of pharmaceutically acceptable salts.
In certain embodiments, salts of a compound of Formula (I) can be formed with, e.g., L-tartaric acid, p-toluenesulfonic acid, D-glucaronic acid, ethane-1,2-disulfonic acid (EDSA), 2-naphthalenesulfonic acid (NSA), hydrochloric acid (HCl) (mono and bis), hydrobromic acid (HBr), citric acid, naphthalene-1,5-disulfonic acid (NDSA), DL-mandelic acid, fumaric acid, sulfuric acid, maleic acid, methanesulfonic acid (MSA), benzenesulfonic acid (BSA), ethanesulfonic acid (ESA), L-malic acid, phosphoric acid, and aminoethanesulfonic acid (taurine).
(R)- and (S)-isomers
In some embodiments, the (R)- and (S)-isomers of the non-limiting exemplary compounds, if present, can be resolved by methods known to those skilled in the art, for example by formation of diastereoisomeric salts or complexes which can be separated, for example, by crystallization; via formation of diastereoisomeric derivatives which can be separated, for example, by crystallization, gas-liquid or liquid chromatography; selective reaction of one enantiomer with an enantiomer-specific reagent, for example enzymatic oxidation or reduction, followed by separation of the modified and unmodified enantiomers; or gas-liquid or liquid chromatography in a chiral environment, for example on a chiral support, such as silica with a bound chiral ligand or in the presence of a chiral solvent. Alternatively, a specific enantiomer can be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
XRPD
Compounds and polymorphs provided herein can be characterized by X-ray powder diffraction patterns (XRPD). The relative intensities of XRPD peaks can vary depending upon the sample preparation technique, the sample mounting procedure and the particular instrument employed, among other parameters. Moreover, instrument variation and other factors can affect the 2θ peak values. Therefore, in certain embodiments, the XRPD peak assignments can vary by plus or minus about 0.2 degrees theta or more, herein referred to as “(±0.2°)”.
XRPD patterns for each of Forms A-J and amorphous form of the compound of Formula (I) were collected with a PANalytical CubiX XPert PRO MPD diffractometer using an incident beam of CU radiation produced using an Optix long, fine-focus source. An elliptically graded multilayer mirror was used to focus Cu Kα X-rays through the specimen and onto the detector. Samples were placed on Si zero-return ultra-micro sample holders. Analysis was performed using a 10 mm irradiated width and the following parameters were set within the hardware/software:
| X-ray tube: | Cu Kα, 45 kV, 40 mA | ||
| Detector: | X′Celerator | ||
| Slits: | ASS Primary Slit: Fixed 1° | ||
| Divergence Slit (Prog): | Automatic – 5 mm irradiated length | ||
| Soller Slits: | 0.02 radian | ||
| Scatter Slit (PASS): | Automatic – 5 mm observed length | ||
| Scanning | |||
| Scan Range: | 3.0-45.0° | ||
| Scan Mode: | Continuous | ||
| Step Size: | 0.03° | ||
| Time per Step: | 10 s | ||
| Active Length: | 2.54° | ||
DSC
Compounds and polymorphs provided herein can be characterized by a characteristic differential scanning calorimeter (DSC) thermogram. For DSC, it is known in the art that the peak temperatures observed will depend upon the rate of temperature change, the sample preparation technique, and the particular instrument employed, among other parameters. Thus, the peak values in the DSC thermograms reported herein can vary by plus or minus about 2° C., plus or minus about 3° C., plus or minus about 4° C., plus or minus about 5° C., plus or minus about 6° C., to plus or minus about 7° C., or more. For some polymorph Forms, DSC analysis was performed on more than one sample which illustrates the known variability in peak position, for example, due to the factors mentioned above. The observed peak positional differences are in keeping with expectation by those skilled in the art as indicative of different samples of a single polymorph Form of a compound of Formula (I).
Impurities in a sample can also affect the peaks observed in any given DSC thermogram. In some embodiments, one or more chemical entities that are not the polymorph of a compound of Formula (I) in a sample being analyzed by DSC can result in one or more peaks at lower temperature than peak(s) associated with the transition temperature of a given polymorph as disclosed herein.
DSC analyses were performed using a Mettler 822e differential scanning calorimeter. Samples were weighed in an aluminum pan, covered with a pierced lid, and then crimped. General analysis conditions were about 30° C. to about 300° C.-about 350° C. ramped at about 10° C./min. Several additional ramp rates were utilized as part of the investigation into the high melt Form B, including about 2° C./min, about 5° C./min, and about 20° C./min. Samples were analyzed at multiple ramp rates to measure thermal and kinetic transitions observed.
Isothermal holding experiments were also performed utilizing the DSC. Samples were ramped at about 10° C./min to temperature (about 100° C. to about 250° C.) and held for about five minutes at temperature before rapid cooling to room temperature. In these cases, samples were then analyzed by XRPD or reanalyzed by DSC analysis.
TGA
A polymorphic form provided herein can give rise to thermal behavior different from that of an amorphous material or another polymorphic form. Thermal behavior can be measured in the laboratory by thermogravimetric analysis (TGA) which can be used to distinguish some polymorphic forms from others. In one embodiment, a polymorph as disclosed herein can be characterized by thermogravimetric analysis.
TGA analyses were performed using a Mettler 851e SDTA/TGA thermal gravimetric analyzer. Samples were weighed in an alumina crucible and analyzed from about 30° C. to about 230° C. and at a ramp rate of about 10° C./min.
DVS
Compounds and polymorphs provided herein can be characterized by moisture sorption analysis. This analysis was performed using a Hiden IGAsorp Moisture Sorption instrument. Moisture sorption experiments were carried out at about 25° C. by performing an adsorption scan from about 40% to about 90% RH in steps of about 10% RH and a desorption scan from about 85% to about 0% RH in steps of about −10% RH. A second adsorption scan from about 10% to about 40% RH was performed to determine the moisture uptake from a drying state to the starting humidity. Samples were allowed to equilibrate for about four hours at each point or until an asymptotic weight was reached. After the isothermal sorption scan, samples were dried for about one hour at elevated temperature (about 60° C.) to obtain the dry weight. XRPD analysis on the material following moisture sorption was performed to determine the solid form.
Optical Microscopy
Compounds and polymorphs provided herein can be characterized by microscopy, such as optical microscopy. Optical microscopy analysis was performed using a Leica DMRB Polarized Microscope. Samples were examined with a polarized light microscope combined with a digital camera (1600×1200 resolution). Small amounts of samples were dispersed in mineral oil on a glass slide with cover slips and viewed with 100× magnification.
Karl Fischer Analysis
Compounds and polymorphs provided herein can be characterized by Karl Fischer analysis to determine water content. Karl Fischer analysis was performed using a Metrohm 756 KF Coulometer. Karl Fisher titration was performed by adding sufficient material to obtain 50 μg of water, about 10 to about 50 mg of sample, to AD coulomat.
Raman Spectroscopy
Compounds and polymorphs provided herein can be characterized by Raman spectroscopy. Raman spectroscopy analysis was performed using a Kaiser RamanRXN1 instrument with the samples in a glass well. Raman spectra were collected using a PhAT macroscope at about 785 nm irradiation frequency and about 1.2 mm spot size. Samples were analyzed using 12 to 16 accumulations with about 0.5 to about 12 second exposure time and utilized cosmic ray filtering. The data was processed by background subtraction of an empty well collected with the same conditions. A baseline correction and smoothing was performed to obtain interpretable data when necessary.
FT-IR
Compounds and polymorphs provided herein can be characterized by FT-IR spectroscopy. FT-IR spectroscopy was performed using either a Nicolet Nexus 470 or Avatar 370 Infrared Spectrometer and the OMNIC software. Samples were analyzed using a diamond Attenuated Total Reflection (ATR) accessory. A compound sample was applied to the diamond crystal surface and the ATR knob was turned to apply the appropriate pressure. The spectrum was then acquired and analyzed using the OMNIC software. Alternative sample preparations include solution cells, mulls, thin films, and pressed discs, such as those made of KBr, as known in the art.
NMR
Compounds and polymorphs provided herein can be characterized by nuclear magnetic resonance (NMR). NMR spectra were obtained using a 500 MHz Bruker AVANCE with 5-mm BBO probe instrument. Samples (approximately 2 to approximately 10 mg) were dissolved in DMSO-d6 with 0.05% tetramethylsilane (TMS) for internal reference. 1H-NMR spectra were acquired at 500 MHz using 5 mm broadband observe (1H-X) Z gradient probe. A 30 degree pulse with 20 ppm spectral width, 1.0 s repetition rate, and 32-64 transients were utilized in acquiring the spectra.
High-Performance Liquid Chromatography
Compounds and polymorphs provided herein can be analyzed by high-performance liquid chromatography using an Agilent 1100 instrument. The instrument parameters for achiral HPLC are as follows:
| Column: | Sunfire C18 4.6 × 150 mm | ||
| Column Temperature: | Ambient | ||
| Auto-sampler Temperature: | Ambient | ||
| Detection: | UV at 250 nm | ||
| Mobile Phase A: | 0.05% trifluoroacetic acid in water | ||
| Mobile Phase B: | 0.05% trifluoroacetic acid in MeCN | ||
| Flow Rate: | 1.0 mL/minute | ||
| Injection Volume: | 10 μL | ||
| Data Collection time: | 20 minutes | ||
| Re-equilibration Time: | 5 minutes | ||
| Diluent & Needle Wash: | MeOH | ||
Gradient Conditions:
| Time (minutes) | % A | % B |
| 0.0 | 90 | 10 |
| 3.5 | 90 | 10 |
| 10.0 | 10 | 90 |
| 15.0 | 10 | 90 |
| 18.0 | 90 | 10 |
| 20.0 | 90 | 10 |
Compounds and polymorphs provided herein can be analyzed by high-performance liquid chromatography using a chiral HPLC column to determine % ee values:
| Column: | Chiralpak IC, 4.6 mm × 250 mm, 5 μm. |
| Column Temperature: | Room Temperature |
| Sample Temperature: | Room Temperature |
| Detection: | UV at 254 nm |
| Mobile Phase A: | 60% Hexane 40% (IPA: EtOH = 2:3) with 0.2% |
| Acetic Acid and 0.1% DEA | |
| Isocratic: | 100% A |
| Flow Rate: | 1 mL/min |
| Diluent: | Methanol |
| Injection Volume: | 10 μL |
| Analysis Time: | 25 min |
Example 8
Analytical Data of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one
Provided herein are analytical data of various purified samples of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one, the compound of Formula (I). Confirmation of the structure of the compound of Formula (I) was obtained via single crystal X-ray diffraction, FT-IR, 1H-NMR and 13C-NMR spectra.
A single crystal structure of a tert-butyl methyl ether solvate of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one (e.g., polymorph Form G) was generated and single crystal X-ray data was collected. The structure is shown in FIG. 26, which further confirmed the absolute stereochemistry as the S-enantiomer.
FT-IR spectra of Form C of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one was obtained, and shown in FIG. 27.
1H-NMR and 13C-NMR spectra of a sample of Form C of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one were obtained, and are provided in FIG. 28 and FIG. 29, respectively.
Example 9
General Methods for the Preparation of Polymorphs Form A, B, C, D, E, F, G, H, I, J of the Compound of Formula (I)
General Method A: Single Solvent Crystallization with Fast Cooling or Slow Cooling
A sample of a compound of Formula (I) (e.g., Form A or Form C) is placed into a vial equipped with stir bar and dissolved with a minimal amount of solvent (such as about 0.2 mL to about 0.3 mL) at an elevated temperature. The resulting solution is polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, the vial is placed in a refrigerator (e.g., about 4° C.) overnight in a fast cooling procedure, or cooled to ambient temperature at a rate of about 20° C./h and allowed to equilibrate without stiffing at ambient temperature overnight in a slow cooling procedure. Optionally, a sample without solids can be scratched with an implement known in the art (e.g., a spatula) to initiate crystallization. The solution can be allowed to equilibrate for a period of time, such as approximately 8 hours. For a slow cooling sample, if scratching does not provide solids after about 8 hours, then a stir bar can be added and the sample then stirred overnight. A sample without precipitation can be evaporated to dryness under a gentle gas stream, such as argon, nitrogen, ambient air, etc. The precipitated solids can be recovered by vacuum filtration, centrifuge filtration, or decanted as appropriate to afford the Form as indicated below.
General Method B: Multi-Solvent Crystallization with Fast Cooling or Slow Cooling
Multi-solvent (e.g., binary) solvent crystallizations can be performed. Primary solvents include, but are not limited to, ethanol, isopropyl alcohol, methanol, tetrahydrofuran, acetone, methyl ethyl ketone, dioxane, NMP, DME, and DMF. Anti-solvents include, but are not limited to, MTBE, DCM, toluene, heptane, and water.
A sample of a compound of Formula (I) (e.g., Form A or Form C) is placed into a vial equipped with stir bar and dissolved with a minimal amount of solvent (such as about 0.2 mL to about 0.3 mL) at an elevated temperature. The resulting solution is polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, the anti-solvent is added until turbidity is observed. After hot filtration, the vial is placed in a refrigerator (e.g., about 4° C.) overnight in a fast cooling procedure, or cooled to ambient temperature at a rate of about 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight in a slow cooling procedure. Optionally, a sample without solids can be scratched with an implement known in the art (e.g., a spatula) to initiate crystallization. The solution can be allowed to equilibrate for a period of time, such as approximately 8 hours. For a slow cooling sample, if scratching does not provide solids after about 8 hours, then a stir bar can be added and the sample then stirred overnight. A sample without precipitation can be evaporated to dryness under a gentle gas stream, such as argon, nitrogen, ambient air, etc. The precipitated solids can be recovered by vacuum filtration, centrifuge filtration, or decanted as appropriate to afford the Form as indicated below.
General Method C: Slurry Procedures to Afford Formula (I) Polymorph Forms
A mixture of one or more Forms (e.g., Form A or Form C) of the compound of Formula (I) are placed in a vial equipped with a stir bar. A minimal amount of solvent (e.g., a single solvent or a mixture/solution of two or more solvents) is added to the vial to form a heterogeneous slurry. Optionally, the vial can be sealed to prevent evaporation. The slurry is stirred for a period of time ranging from less than about an hour, to about 6 hours, to about 12 hours, to about 24 hours, to about 2 days, to about 4 days, to about 1 week, to about 1.5 weeks, to about 2 weeks or longer. Aliquots can be taken during the stirring period to assess the Form of the solids using, for example, XRPD analysis. Optionally, additional solvent(s) can be added during the stirring period. Optionally, seeds of a given polymorph Form of the compound of Formula (I) can be added. In some cases, the slurry is then stirred for a further period of time, ranging as recited above. The recovered solids can be recovered by vacuum filtration, centrifuge filtration, or decanted as appropriate to afford the Form as indicated below.
Example 10
Preparation of Polymorphs Form A, B, C, D, E, F, G, H, I, J of the Compound of Formula (I)
Form A
Single Solvent Crystallizations to Afford Formula (I) Form A
1. Fast Cooling Procedure From MeCN: Approximately 23 mg of Formula (I) Form A was placed into a 20-mL glass vial equipped with a stir bar. To the vial was added a minimal amount of acetonitrile (7.4 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, the vial was placed in a refrigerator (4° C.) overnight. Once at 4° C., the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by decanting off the liquid and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
2. Slow Cooling Procedure From MeCN: Approximately 24 mg of Formula (I) Form A was placed into a 20-mL glass vial equipped with a stir bar. To the vial was added a minimal amount of acetonitrile (8 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, the vial was cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by decanting off the liquids and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
3. Slow Cooling Procedure From n-Butanol: Approximately 23 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of n-butanol (0.6 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, the vials were cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. To further induce crystallization, a stir bar was added to the vial and the contents stirred overnight. The resulting crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
Binary Solvent Crystallizations to Afford Formula (I) Form A
1. Fast Cooling Procedure From Acetone/DCM: Approximately 23.5 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of acetone (2.6 ml) to just dissolve the solids at 50° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (5.0 ml) was added portion-wise. After the anti-solvent addition, the vials were placed in a refrigerator (4° C.) overnight. Once at 4° C., the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
2. Fast Cooling Procedure From MEK/DCM: Approximately 23 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of MEK (2.2 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (5.0 ml) was added portion-wise. After the anti-solvent addition, the vial was placed in a refrigerator (4° C.) overnight. Once at 4° C., the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
3. Fast Cooling Procedure From DMF/DCM: Approximately 24 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of DCM (0.2 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (7.0 ml) was added portion-wise. After the anti-solvent addition, the vial was placed in a refrigerator (4° C.) overnight. Once at 4° C., the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
4. Fast Cooling Procedure From Dioxane/DCM: Approximately 24.4 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of dioxane (0.8 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (7.0 ml) was added portion-wise. After the anti-solvent addition, the vial was placed in a refrigerator (4° C.) overnight. Once at 4° C., the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
5. Slow Cooling Procedure From Acetone/DCM: Approximately 22 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of acetone (2.5 ml) to just dissolve the solids at 50° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (5.0 ml) was added portion-wise. After the anti-solvent addition, the vial was cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. To further induce crystallization, a stir bar was added to the vial and the contents stirred overnight. The resulting crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
6. Slow Cooling Procedure From MEK/DCM: Approximately 23.4 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of MEK (2.2 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (5.0 ml) was added portion-wise. After the anti-solvent addition, the vial was cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The resulting crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
7. Slow Cooling Procedure From Dioxane/DCM: Approximately 24 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of dioxane (0.8 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (7.0 ml) was added portion-wise. After the anti-solvent addition, the vial was cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. To further induce crystallization, a stir bar was added to the vial and the contents stirred overnight. The resulting crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
8. Slow Cooling Procedure From DMF/DCM: Approximately 23.5 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of DMF (0.2 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (7.0 ml) was added portion-wise. After the anti-solvent addition, the vial was cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. To further induce crystallization, a stir bar was added to the vial and the contents stirred overnight. To further induce crystallization, the contents of the vial were concentrated under a gentle stream of nitrogen to near dryness. The resulting crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
Slurry Procedure to Afford Formula (I) Form A
1. Procedure from CH2Cl2 and from IPA: Form C (1 g) was slurried in five volumes of dichloromethane. After holding for 15 hours, filtration, and drying, Form A was isolated in 82% yield. Scale-up was performed on a 20 g scale with a water-wet cake of Form C to yield Form A in 92% yield. Drying at 70° C. for six days indicated no degradation in chemical or chiral purity. Slurrying dry Form C in isopropyl alcohol using a similar method also yielded Form A.
2. Procedure for Competitive Slurry Experiment (using forms A, B and C): Competitive slurries were performed by charging approximately a 50/50 mixture of Forms A and C (11.2 mg of Form A and 11.7 mg Form C) to a 1-dram glass vial equipped with a glass stir bar. To the vial was added 600 μL of MeCN. The vial cap was wrapped with parafilm to prevent evaporation. The slurry was stirred for 1 day and an aliquot was taken. The contents of the vial were allowed to stir for an additional week and another aliquot was taken. Both aliquots were centrifuge filtered for five minutes at 8000 RPM. XRPD analysis was performed on the solids from each aliquot to show that the Formula (I) had converted to Form A at both time points. After the one week aliquot was taken, an additional 300 μL of acetonitrile was added to the remaining slurry and allowed to equilibrate for one day. The slurry was then seeded with approximately 3.2 mg of Form B and allowed to equilibrate for an additional three days. The solids were isolated by centrifuge filtration (5 minutes at 8000 RPM) and dried over night under vacuum. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
3. Procedure for Competitive Slurry Experiment (using forms A, C, D, and E): Competitive slurries were performed by charging an approximately equal mixture of each form (7.8 mg of Form A, 7.7 mg Form C, 7.7 mg of Form D, and 8.2 mg of Form E) to a 1-dram glass vial equipped with a glass stir bar. To the vial was added 1 ml of 2-propanol. The vial cap was wrapped with parafilm to prevent evaporation. The slurry was mixed for 1 day and an aliquot was taken. The contents of the vial were allowed to stir for an additional week and another aliquot was taken. Both aliquots were centrifuge filtered for five minutes at 8000 RPM. XRPD analysis was performed on the solids from each aliquot to show that the Formula (I) had converted to Form A at both time points. After the one week aliquot was taken, the remaining solids were isolated by centrifuge filtration (5 minutes at 8000 RPM) and dried over night under vacuum. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
Using the General Method B of Example 9, the following experiments detailed in Tables 4 and 5 were performed to afford Formula (I) Form C. Table 4 experiments were conducted using the fast cooling procedure, while Table 5 experiments were conducted using the slow cooling procedure.
Table 4. Fast Cooling Procedure

Table 5. Slow Cooling Procedure

Using General Method C of Example 9, the following experiments detailed in Table 6 were performed to afford the polymorph Form of the compound of Formula (I) as indicated.
Table 6:

Example 12
XRPD Studies
[00653] Using the XRPD instrument and parameters described above, the following XRPD peaks were observed for Formula (I) Polymorph Forms A, B, C, D. E, F, G, H, I, and J. The XRPD traces for these ten polymorph forms are given in Figures 1-10, respectively. In Table 7, peak position units are °2Θ. In one embodiment, a given polymorph Form can be characterized as having at least one of the five XRPD peaks given in Set 1 in Table 7. In another embodiment, the given Form can be characterized as having at least one of the five XRPD peaks given in Set 1 in combination with at least one of the XRPD peaks given in Set 2 in Table 7. In some embodiments, one or more peak position values can be defined as being modified by the term “about” as described herein. In other embodiments, any given peak position is with ±0.2 2Θ (e.g., 9.6+0.2 2Θ).
Table 7.

Example 13
Differential Scanning Calorimetry (DSC) Studies
[00654] Using the DSC instrument and parameters described above, the following DSC peaks were observed for the compound of Formula (I) polymorph Forms A, B, C, D. E, F, G, H, I, and J. The DSC thermograms for these nine polymorph forms are given in FIGS. 12-24, respectively, and peak positions are given in Table 8. Further DSC data for Polymorph Forms A, B, C, D. E, F, G, H, I, and J is given in Table 9 below. Unless marked with a Λ that indicates an exothermic peak, all peaks are endothermic.
Table 8.

Table 9 summarizes non-limiting exemplary preparation techniques for Formula (I) Polymorph Forms A-J and representative analytical data as described below and elsewhere.
Table 9.


http://www.google.com/patents/US8809349?cl=en
Extras…….
SEE NMR ……….http://www.medkoo.com/Product-Data/IPI-145/IPI-145-QC-SSC20130422Web.pdf
http://www.chemietek.com/Files/Line2/CHEMIETEK,%20IPI-145%20(01),%20NMR.pdf

New Parathyroid Disease Drug Etelcalcetide Seeks FDA Approval
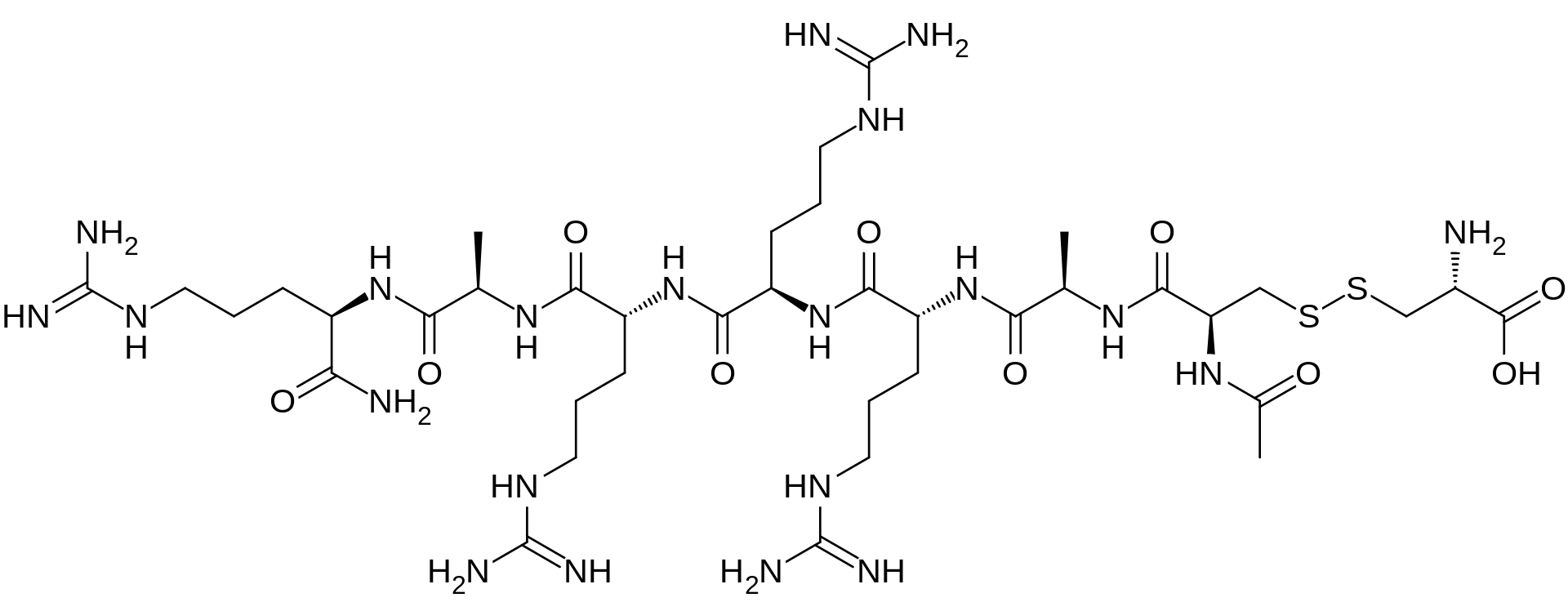
Etelcalcetide, AMG 416
AMG-416; Etelcalcetide hydrochloride; KAI-4169; KAI-4169-HCl; ONO-5163; Telcalcetide; Velcalcetide; Velcalcetide hydrochloride
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine,
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine
Secondary hyperparathyroidism
- Originator KAI Pharmaceuticals…Kai Pharmaceuticals, Inc.
- Developer Amgen; KAI Pharmaceuticals; Ono Pharmaceutical
- ClassDisulfides; Peptides
- Mechanism of ActionCalcium-sensing receptor agonists
New Parathyroid Disease Drug Seeks FDA Approval
Amgen is seeking FDA approval for etelcalcetide (AMG 461), the first calcimimetic agent administered intravenously after dialysis to treat secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease (CKD).
SHPT is a common and serious condition that is often progressive among CKD patients. It usually manifests as high amounts of parathyroid hormone (PTH) associated with abnormal calcium and phosphorus levels in the body.
– See more at: http://www.pharmacytimes.com/product-news/new-parathyroid-disease-drug-seeks-fda-approval
Etelcalcetide is a D-amino peptide calcimimetic undergoing clinical evaluation for the treatment of secondary hyperparathyroidismfor patients with chronic kidney disease (CKD) on hemodialysis. Etelcalcetide is administered intravenously at the end of each dialysis session.[1][2] It exerts a pharmacological effect by binding to and activating the calcium-sensing receptor (CaSR) in theparathyroid gland, resulting in parathyroid hormone (PTH) reduction and suppression.[1] Elevated PTH is often observe in patients with CKD.[3]
On August 25, 2015 Amgen Inc. announced its submission of a New Drug Application to the Food and Drug Administration for etelcalcetide.[1]
| CAS Registry Number | 1262780-97-1 |
|---|---|
| Synonyms | Velcalcetide |
| Chemical data | |
| Formula | C38H73N21O10S2 |
| Molecular mass | 1,048.26 g·mol−1 |
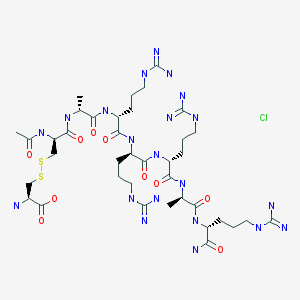
Etelcalcetide hydrochloride
RN: 1334237-71-6
UNII: 72PT5993DU
The term “AMG 416” refers to the compound having the chemical name: JV-acetyl-D- cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-arginamide disulfide with L- cysteine, which may be represented as:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The terms “AMG 416 hydrochloride” or “AMG 416 HQ” are interchangeable and refer to the compound having the chemical name: N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl- D-arginyl-D-alanyl-D-arginamide disulfide with L-cysteine hydrochloride, which may be represented as:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · xHCl
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine, hydrochloride (1:?)
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
Amgen today announced the submission of a New Drug Application (NDA) with the United States Food and Drug Administration (FDA) for etelcalcetide (formerly AMG 416) for the treatment of secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease (CKD) on hemodialysis. If approved, etelcalcetide will be the first calcimimetic agent that can be administered intravenously at the end of the dialysis session.
“Secondary hyperparathyroidism is a serious, progressive disease that can lead to significant clinical consequences and is also associated with a high pill burden for patients,” said Sean E. Harper, M.D., executive vice president of Research and Development at Amgen. “We look forward to working with regulatory authorities during the review process to bring this important treatment to market, helping to fill an unmet need for the many patients impacted by this disease.”
Etelcalcetide is a novel calcimimetic agent that suppresses the secretion of parathyroid hormone and is in clinical development for the treatment of SHPT in patients with CKD on hemodialysis. Etelcalcetide is administered intravenously three times per week at the end of each dialysis session. It acts by binding to and activating the calcium-sensing receptor on the parathyroid gland, thereby causing decreases in parathyroid hormone (PTH). Sustained elevations in PTH are known to be associated with significant clinical consequences for patients with CKD.
The submission includes data from three Phase 3 studies, all of which met the primary endpoints, including two pooled placebo-controlled trials in more than 1,000 patients and a head-to-head study evaluating etelcalcetide compared with cinacalcet.
About Secondary Hyperparathyroidism
SHPT is a common and serious condition that is often progressive among patients with CKD, and it affects many of the approximately two million people throughout the world who are receiving dialysis, including 450,000 people in the U.S. The disorder develops early in the course of CKD and usually manifests as increased levels of PTH as a result of increased production from the parathyroid glands (four small glands in the neck). Patients with end stage renal disease who require maintenance dialysis often have substantial elevations of PTH that are commonly associated with abnormal calcium and phosphorus levels and an increased risk of significant clinical consequences.
About Etelcalcetide (AMG 416)
Etelcalcetide is a novel calcimimetic agent in clinical development for the treatment of SHPT in CKD patients on hemodialysis that is administered intravenously at the end of the dialysis session. Etelcalcetide binds to and activates the calcium-sensing receptor on the parathyroid gland, thereby decreasing PTH levels.
About Sensipar® (cinacalcet)
Sensipar® (cinacalcet) is the first oral calcimimetic agent approved by the FDA for the treatment of SHPT in adult patients with CKD on dialysis. Sensipar is not indicated for use in adult patients with CKD who are not on dialysis because of an increased risk of hypocalcemia. The therapy is also approved in the U.S. for treatment of hypercalcemia in adult patients with parathyroid carcinoma and hypercalcemia in adult patients with primary HPT for whom parathyroidectomy would be indicated on the basis of serum calcium levels, but who are unable to undergo parathyroidectomy. Sensipar binds to the calcium-sensing receptor, resulting in a drop in PTH levels by inhibiting PTH synthesis and secretion. In addition, the reductions in PTH lower serum calcium and phosphorus levels.
…………………
WO 2011014707
http://www.google.com/patents/WO2011014707A2?cl=en
……………………..
WO 2014210489
http://www.google.com/patents/WO2014210489A1?cl=en
A variety of compounds having activity for lowering parathyroid hormone levels have been described. See International Publication No. WO 2011/014707. In one embodiment, the compound may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The main chain has 7 amino acids, all in the D-configuration and the side-chain cysteine residue is in the L-configuration. The amino terminal is acetylated and the carboxyl-terminal is amidated. This compound (“AMG-416”) has utility for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis patients. A liquid formulation comprising AMG-416 may be administered to a subject intravenously. The hydrochloride salt of AMG-416 may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
Therapeutic peptides pose a number of challenges with respect to their formulation. Peptides in general, and particularly those that contain a disulfide bond, typically have only moderate or poor stability in aqueous solution. Peptides are prone to amide bond hydrolysis at both high and low pH. Disulfide bonds can be unstable even under quite mild conditions (close to neutral pH). In addition, disulfide containing peptides that are not cyclic are particularly prone to dimer formation. Accordingly, therapeutic peptides are often provided in lyophilized form, as a dry powder or cake, for later reconstitution. A lyophilized formulation of a therapeutic peptide has the advantage of providing stability for long periods of time, but is less convenient to use as it requires the addition of one or more diluents and there is the potential risk for errors due to the use of an improper type or amount of diluent, as well as risk of contamination. In addition, the lyophilization process is time consuming and costly.
Accordingly, there is a need for an aqueous liquid formulation comprising a peptide agonist of the calcium sensing receptor, such as AMG 416. It would be desirable for the liquid formulation to remain stable over a relevant period of time under suitable storage conditions and to be suitable for administration by intravenous or other parenteral routes.
…………………………………
Milestones
- 25 Aug 2015Preregistration for Secondary hyperparathyroidism in USA (IV)
- 29 May 2015Pooled analysis efficacy and adverse events data from two phase III trials in secondary hyperparathyroidism released by Amgen
- 21 Apr 2015Amgen plans to submit Biological License Application to USFDA and Marketing Authorisation Application to EMA for Secondary hyperparathyroidism
References
- “Amgen Submits New Drug Application For Novel Intravenous Calcimimetic Etelcalcetide (AMG 416)”
- “Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects
- “Evidence for Chronic Kidney Disease-Mineral and Bone Disorder Associated With Metabolic Pathway Changes”
KAI-4169, a novel calcium sensing receptor agonist, decreases serum iPTH, FGF-23 and improves serum bone markers in a phase 2 study in hemodialysis subjects with chronic kidney disease-mineral and bone disorder
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO054
KAI-4169, a novel peptide agonist of the calcium sensing receptor, attenuates PTH and soft tissue calcification and restores parathyroid gland VDR levels in uremic rats
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO014
Long term safety and efficacy of velcalcetide (AMG 416), a calcium-sensing receptor (CaSR) agonist, for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis (HD) patients
Kidney Week (November 5-10, Atlanta, GA) 2013, Abst SA-PO575
Preclinical PK and PD relationship for KAI-4169, a novel calcimimetic
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P1-198
KAI-4169, a novel calcimimetic for the treatment of secondary hyperparathyroidism
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P2-98
Characterization of KAI-4169, a novel peptide for the treatment of chronic kidney disease – Mineral and bone disorder, in a phase I study in healthy males
44th Annu Meet Am Soc Nephrol (ASN) (November 8-13, Philadelphia) 2011, Abst FR-PO1238
| WO2011014707A2 | Jul 29, 2010 | Feb 3, 2011 | Kai Pharmaceuticals, Inc. | Therapeutic agents for reducing parathyroid hormone levels |
////Etelcalcetide, Parathyroid Disease, Amgen Inc, AMG 416, KAI-4169; KAI-4169-HCl, ONO-5163, Telcalcetide, Velcalcetide, Velcalcetide hydrochloride
FDA approves Repatha to treat certain patients with high cholesterol
August 27, 2015
Release
The U.S. Food and Drug Administration today approved Repatha (evolocumab) injection for some patients who are unable to get their low-density lipoprotein (LDL) cholesterol under control with current treatment options.
Repatha, the second drug approved in a new class of drugs known as PCSK9 inhibitors, is approved for use in addition to diet and maximally-tolerated statin therapy in adult patients with heterozygous familial hypercholesterolemia (HeFH), homozygous familial hypercholesterolemia (HoFH), or clinical atherosclerotic cardiovascular disease, such as heart attacks or strokes, who require additional lowering of LDL cholesterol.
Familial hypercholesterolemia (encompassing both HeFH and HoFH) is an inherited condition that causes high levels of LDL cholesterol. A high level of LDL cholesterol in the blood is linked to cardiovascular or heart disease. Heart disease is the number one cause of death for Americans, both men and women. According to the Centers for Disease Control and Prevention, about 610,000 people die of heart disease in the United States every year– that equals one in every four deaths.
“Repatha provides another treatment option in this new class of drugs for patients with familial hypercholesterolemia or with known cardiovascular disease who have not been able to lower their LDL cholesterol enough with statins,” said John Jenkins, M.D., director of the Office of New Drugs, Center for Drug Evaluation and Research. “Cardiovascular disease is a serious threat to the health of Americans, and the FDA is committed to facilitating the development and approval of effective and safe drugs to address this important public health problem.”
Repatha is an antibody that targets a specific protein, called PCSK9. PCSK9 reduces the number of receptors on the liver that remove LDL cholesterol from the blood. By blocking PCSK9’s ability to work, more receptors are available to get rid of LDL cholesterol from the blood and, as a result, lower LDL cholesterol levels.
The efficacy and safety of Repatha were evaluated in one 52-week placebo-controlled trial and eight 12-week placebo-controlled trials in participants with primary hyperlipidemia, including two that specifically enrolled participants with HeFH and one that enrolled participants with HoFH. In one of the 12-week studies, 329 participants with HeFH, who required additional lowering of LDL cholesterol despite statins with or without other lipid-lowering therapies, were randomized to receive Repatha or placebo for 12 weeks. Participants taking Repatha had an average reduction in LDL cholesterol of approximately 60 percent, compared to placebo.
The most common side effects of Repatha include nasopharyngitis, upper respiratory tract infection, flu, back pain, and reactions such as redness, pain, or bruising where the injection is given. Allergic reactions, such as rash and hives, have been reported with the use of Repatha. Patients should stop using Repatha and get medical help if they experience symptoms of a serious allergic reaction.
Multiple clinical trials have demonstrated that statins lower the risk of having a heart attack or stroke. A trial evaluating the effect of adding Repatha to statins for reducing cardiovascular risk is ongoing.
Repatha is marketed by Amgen Inc., of Thousand Oaks, Calif.
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

