
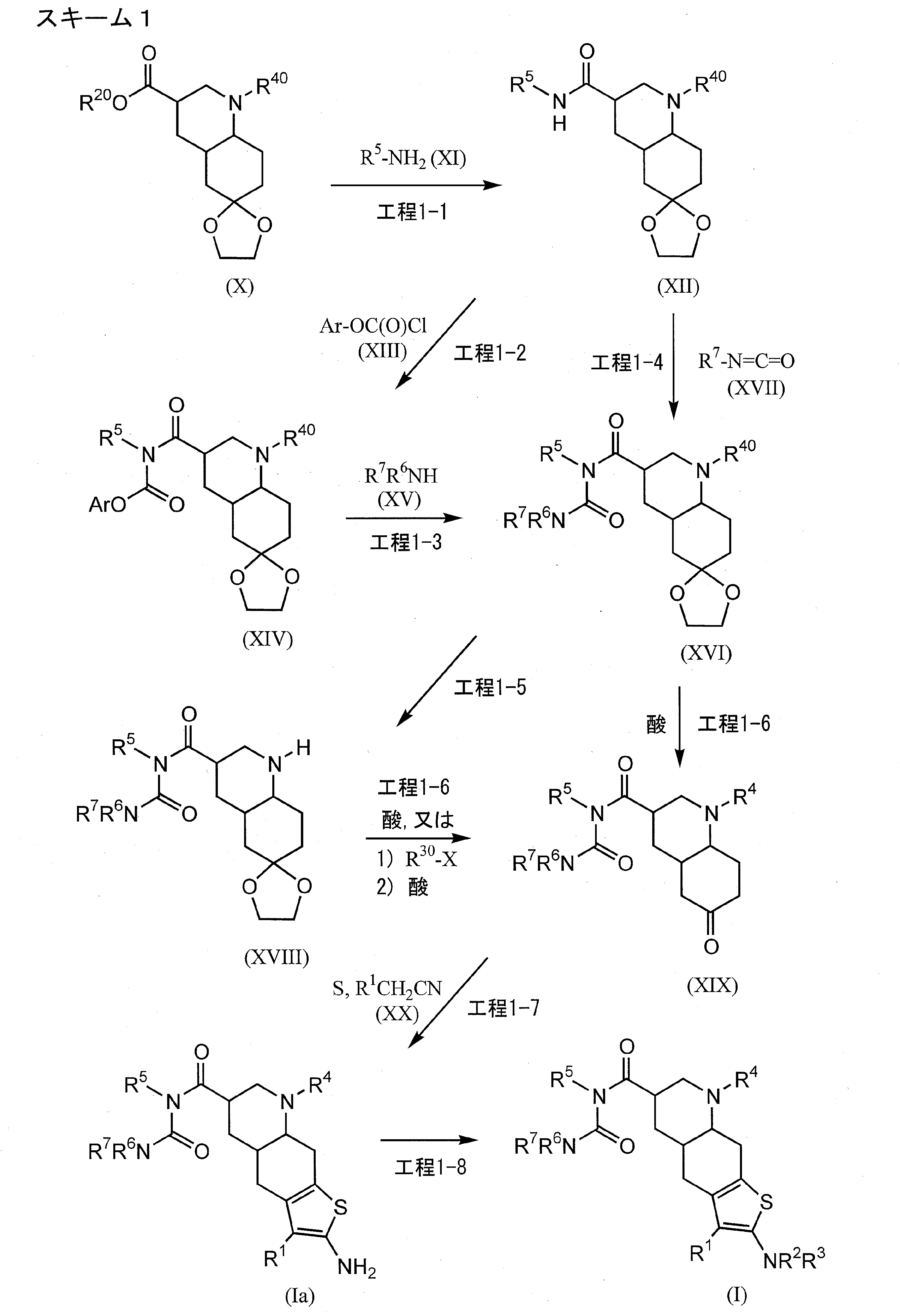
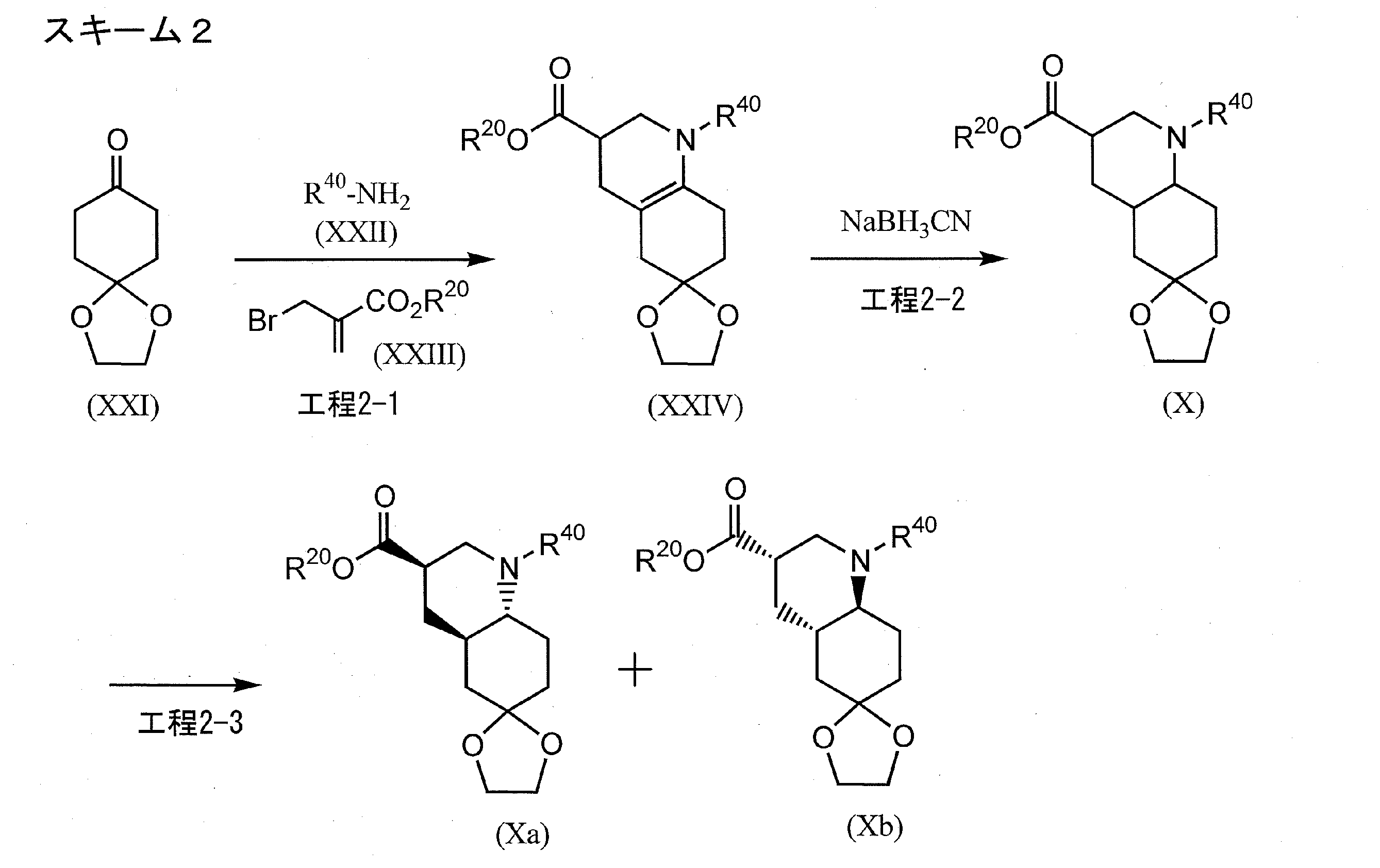
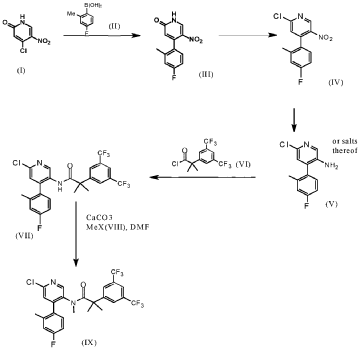
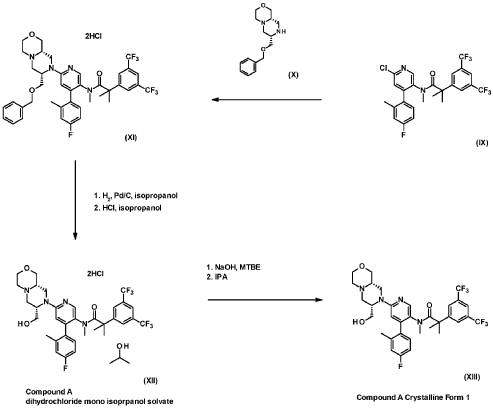
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

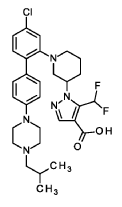
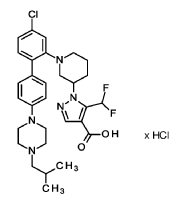
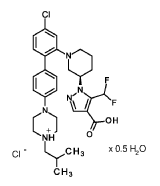
Nurandociguat
Nurandociguat
CAS 2781965-75-9
MF C30H36ClF2N5O2 MW 572.1 g/mol
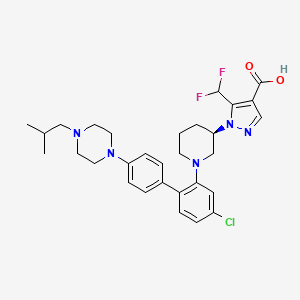
1-[(3R)-1-{4-chloro-4′-[4-(2-methylpropyl)piperazin-1-yl][1,1′-biphenyl]-2-yl} piperidin-3-yl]-5-(difluoromethyl)-1H-pyrazole-4-carboxylic acid
1-[(3R)-1-[5-chloro-2-[4-[4-(2-methylpropyl)piperazin-1-yl]phenyl]phenyl]piperidin-3-yl]-5-(difluoromethyl)pyrazole-4-carboxylic acid
guanylate cyclase activator, BAY 3283142, LPU8429UK5
Nurandociguat is a small molecule drug candidate, previously known as BAY 3283142, that is a guanylate cyclase activator being developed by Bayer for cardiovascular conditions. The “ciguat” stem in its name indicates its function as a guanylate cyclase activator, a mechanism that is also being investigated for related drugs like runcaciguat. It is currently in clinical trials, including a Phase 2 program for chronic kidney disease (CKD).
- Drug class: Guanylate cyclase activator
- Developer: Bayer
- Previous name: BAY 3283142
- Indication: Investigated for cardiovascular conditions
- Current status: In clinical development, including a Phase 2 study for chronic kidney disease (CKD)
- OriginatorBayer
- ClassAntihypertensives; Cardiovascular therapies; Hepatoprotectants; Urologics
- Mechanism of ActionGuanylate cyclase stimulants
- Phase IIRenal failure
- Phase ICardiovascular disorders; Diabetic retinopathy; Hypertension; Liver disorders
- 28 Sep 2025No recent reports of development identified for phase-I development in Renal-failure in Germany (PO, Immediate release)
- 16 Sep 2025(CTIS2024-510856-11-00) (EudraCT2024-510856-11-00): Trial initiation and completion info added; updated DevT; Corrected intro to match DevT as most of the info about indication and countries missing
- 28 May 2025No recent reports of development identified for phase-I development in Renal-failure(In volunteers, In adults) in Japan (PO, Immediate release)
Nurandociguat is a small molecule drug. The usage of the INN stem ‘-ciguat’ in the name indicates that Nurandociguat is a guanylate cyclase activator and stimulator. Nurandociguat has a monoisotopic molecular weight of 571.25 Da.
PAT
- Soluble guanylate cyclase activators for use in the treatment of heart failure with preserved ejection fraction in womenPublication Number: WO-2023237577-A1Priority Date: 2022-06-09
- Substituted pyrazolo piperidine carboxylic acidsPublication Number: WO-2022122910-A1Priority Date: 2020-12-10
- The use of sgc activators for the treatment of ophthalmologic diseasesPublication Number: WO-2022122917-A1Priority Date: 2020-12-10
- Use of sGC activators for the treatment of ophthalmic diseasesPublication Number: CN-115175681-APriority Date: 2020-12-10
- Use of sgc activators for the treatment of ophthalmologic diseasesPublication Number: US-2022241273-A1Priority Date: 2020-12-10
- Use of sGC activators for the treatment of ophthalmic diseasesPublication Number: JP-2023514928-APriority Date: 2020-12-10
- The use of sgc activators for the treatment of ophthalmologic diseasesPublication Number: EP-4259140-A1Priority Date: 2020-12-10
- Use of sGC activators for the treatment of ophthalmic diseasesPublication Number: KR-20230118143-APriority Date: 2020-12-10
- Substituted pyrazolo piperidine carboxylic acidsPublication Number: US-2023265072-A1Priority Date: 2020-12-10
- Use of sGC activators for the treatment of ophthalmic diseasesPublication Number: JP-2024073585-APriority Date: 2020-12-10
- Use of sGC activators for the treatment of ophthalmological diseasesPublication Number: JP-7458683-B2Priority Date: 2020-12-10Grant Date: 2024-04-01
- Use of sgc activators for the treatment of ophthalmologic diseasesPublication Number: US-2023346777-A1Priority Date: 2020-12-10
- Use of sGC activators for treating ophthalmic diseasesPublication Number: CN-115175681-BPriority Date: 2020-12-10Grant Date: 2024-10-25
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022122917&_cid=P20-MHVQYD-96133-1
soluble guanylate cyclase (sGC) activators for use in the treatment and/or prophylaxis of ophthalmologic diseases, including non-proliferative diabetic retinopathy (NPDR), diabetic macular edema (DME), retinal ganglion cell/photoreceptor neurodegeneration and cataract, especially wherein the soluble guanylate cyclase (sGC) activators are compounds selected from the group consisting of
Example 1
1 – [ 1 – { 4-Chloro-4′- [4-(2-methylpropyl)piperazin- 1 -yl] [1,1 ’-biphenyl] -2-yl }piperidin-3-yl] -5- (difluoromethyl)-lH-pyrazole-4-carboxylic acid hydrochloride (Enantiomer 1)

Ethyl 1 – [ 1 – { 5-chloro-2- [(trifluoromethanesulfonyl)oxy]phenyl }piperidin-3-yl] -5-(difluoromethyl)- 1 H-pyrazole-4-carboxylate (prepared in analogy to Example 11A, Enantiomer 1, 80.0 mg, 147 pmol) and l-(2-methylpropyl)-4- [4-(4,4,5 ,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)phenyl]piperazine (Example 18 A 62.8 mg, 97 % purity, 177 pmol) were placed under argon in toluene/ethanol (820/820 pl). 2 M sodium carbonate solution (220 pl, 2.0 M, 440 pmol) and tetrakis(triphenylphosphine)palladium(0) (8.52 mg, 7.37 pmol) were added and the mixture was stirred at 100°C. overnight. The reaction mixture was diluted with ethyl acetate and 1 M hydrochloric acid was added. The aqueous phase was extracted three times with ethyl acetate. The organic phase was dried with sodium sulfate, filtered off and evaporated. The crude mixture was dissolved with THF/ethanol (2.0/0.2 ml), 1 M lithium hydroxide solution (1.5 ml, 1.5 mmol) was added and the mixture was stirred at room temperature overnight. A I M lithium hydroxide solution (740 pl, 740 pmol) was added again. After about 6 h the reaction mixture was evaporated at 50°C. The residue was dissolved in
SUBSTITUTE SHEET (RULE 26)
acetonitrile/water/0.25 ml trifluoroacetic acid and purified by preparative HPLC (RP18 column, acetonitrile/water gradient with the addition of 0.1% trifluoroacetic acid). The crude product was purified by means of thick layer chromatography (dichloromethane/methanol/formic acid: 10/1/0.1). The silica gel mixture was stirred with dichloromethane/1 M hydrochloric acid in dioxane (10/1) in ethanol, filtered off and carefully evaporated at 30°C and lyophilized. 34 mg of the target compound (36% of theory, purity 95%) were obtained.
LC-MS (Method 6): Rt = 1.23 min; MS (ESIpos): m/z = 572 [M-HC1+H]+
‘H-NMR (600 MHz, DMSO-d6) 5 [ppm]: 1.004 (15.87), 1.015 (16.00), 1.500 (0.51), 1.521 (0.57), 1.728 (0.73), 1.750 (0.61), 1.897 (0.57), 1.917 (0.62), 1.975 (0.79), 2.122 (0.42), 2.133 (0.84), 2.144 (1.02), 2.156
(0.79), 2.571 (0.47), 2.587 (0.91), 2.610 (0.52), 3.004 (0.84), 3.022 (2.01), 3.026 (2.20), 3.038 (3.72), 3.048
(2.50), 3.065 (0.75), 3.154 (2.66), 3.161 (2.75), 3.169 (2.36), 3.177 (1.88), 3.224 (0.84), 3.237 (0.70), 3.589
(1.41), 3.602 (1.80), 3.825 (1.02), 3.841 (0.78), 3.866 (1.05), 3.882 (0.75), 4.223 (2.57), 4.445 (0.68), 4.463
(0.97), 4.481 (0.57), 7.045 (0.55), 7.055 (3.63), 7.070 (3.72), 7.084 (2.72), 7.087 (3.09), 7.110 (1.47), 7.113
(1.11), 7.123 (2.19), 7.127 (2.02), 7.163 (3.67), 7.177 (2.19), 7.215 (0.46), 7.428 (0.83), 7.495 (4.24), 7.510
(4.02), 7.515 (2.07), 7.602 (0.82), 7.959 (4.79), 9.484 (0.54).
Example 2
1 – [ 1 – { 4-Chloro-4′- [4-(2-methylpropyl)piperazin- 1 -yl] [1,1 ’-biphenyl] -2-yl }piperidin-3-yl] -5-(difluoromethyl)-lH-pyrazole-4-carboxylic acid (Enantiomer 2)
Method A
A solution of ethyl l-[l-{4-chloro-4′-[4-(2-methylpropyl)piperazin-l-yl][l,T-biphenyl]-2-yl}piperidin-3-yl]-5-(difluoromethyl)-lH-pyrazole-4-carboxylate (prepared in analogy to Example 17A, Enantiomer 2, 50.8 g, 84.6 mmol) in a THF/methanol mixture 9:1 (1.0 1) was treated with an aqueous solution of lithium hydroxide (850 ml, 1.0 M, 850 mmol) and stirred overnight at room temperature. The reaction mixture was
SUBSTITUTE SHEET (RULE 26)
concentrated, diluted with dichloromethane (1.5 1) and adjusted to pH = 2 with an aqueous solution of hydrogen chloride (2N). The resulting suspension was stirred 45 minutes at room temperature. The solid was filtered, washed with water and dried under vacuum affording 43 g (90 % yield) of the title compound.
LC-MS (Method 7): Rt = 1.27 min; MS (ESIpos): m/z = 572 [M+H]+
‘H-NMR (600 MHz, DMSO-d6) 5 [ppm]: 1.002 (15.68), 1.013 (16.00), 1.080 (0.57), 1.092 (1.18), 1.103 (0.63), 1.498 (0.74), 1.519 (0.83), 1.719 (1.03), 1.741 (0.88), 1.902 (0.78), 1.908 (0.74), 1.922 (0.88), 1.928
(0.83), 1.943 (0.45), 1.978 (1.13), 1.994 (0.74), 2.102 (0.71), 2.112 (0.85), 2.123 (0.70), 2.571 (1.40), 2.591
(0.77), 2.882 (1.10), 3.018 (1.27), 3.035 (3.01), 3.053 (2.14), 3.239 (2.40), 3.254 (2.32), 3.368 (1.13), 3.379
(1.40), 3.391 (1.33), 3.403 (0.92), 3.493 (0.76), 4.463 (0.65), 4.482 (1.12), 4.500 (0.62), 7.033 (4.22), 7.048
(4.45), 7.074 (3.47), 7.077 (4.04), 7.100 (1.85), 7.103 (1.52), 7.113 (2.53), 7.117 (2.34), 7.162 (4.18), 7.175
(2.71), 7.439 (1.03), 7.481 (4.88), 7.495 (4.57), 7.526 (2.04), 7.613 (0.91), 7.952 (5.28).
Method B
1 – { 1 – [4-Chloro-4′-(4-isobutylpiperazin- 1 -yl) [biphenyl] -2-yl]piperidin-3-yl } -5-(difluoromethyl)- 1 H-pyrazole-4-carboxylic acid hydrochloride (prepared in analogy to Example 3, Enantiomer 2, 31.2 mg, 51.3 pmol) were dissolved in 17 ml of dichloromethane and 1 ml of methanol. The solution was shaken once with 1.5 ml of saturated, aqueous sodium bicarbonate solution. The phases were separated. 5 ml of dichloromethane and 3 ml of methanol were added to the organic phase. The organic phase was then dried over sodium sulfate, filtered, evaporated and purified by preparative HPLC (RP18 column, acetonitrile/water gradient, neutral without acid addition). Product fractions were combined and lyophilized. 22 mg of the target compound (74% of theory) were obtained.
LC-MS (Method 3): Rt = 1.73 min; MS (ESIpos): m/z = 572 [M+H]+
‘H-NMR (600 MHz, DMSO-d6) 5 [ppm]: 0.887 (15.60), 0.898 (16.00), 1.493 (0.64), 1.514 (0.70), 1.695 (0.89), 1.718 (0.74), 1.799 (0.48), 1.811 (0.88), 1.822 (1.12), 1.833 (0.92), 1.844 (0.48), 1.890 (0.68), 1.910
(0.74), 1.977 (0.93), 1.995 (0.62), 2.118 (3.91), 2.130 (3.66), 2.516 (5.14), 3.017 (1.09), 3.035 (2.76), 3.053
(1.94), 3.181 (5.03), 3.185 (5.02), 3.267 (1.53), 4.473 (0.55), 4.491 (0.96), 4.509 (0.54), 6.963 (3.96), 6.977
(4.06), 7.048 (3.13), 7.051 (3.31), 7.081 (1.60), 7.084 (1.26), 7.095 (2.21), 7.098 (1.89), 7.152 (3.52), 7.165
(2.42), 7.434 (4.45), 7.448 (4.50), 7.533 (1.51), 7.621 (0.67), 7.930 (4.14).
Example 3
1 – { 1 – [4-Chloro-4′-(4-isobutylpiperazin- 1 -yl) [biphenyl] -2-yl]piperidin-3-yl } -5-(difluoromethyl)- 1 H-pyrazole-4-carboxylic acid hydrochloride (Enantiomer 2)
SUBSTITUTE SHEET (RULE 26)
Method A
A suspension of 1 – [ 1 – { 4-chloro-4′- [4-(2-methylpropyl)piperazin- 1 -yl] [1,1 ’-biphenyl] -2-yl }piperidin-3-yl] -5-(difluoromethyl)-lH-pyrazole-4-carboxylic acid (prepared in analogy to Example 2, Enantiomer 2, 43.5 g, 76.0 mmol) in diethyl ether (870 ml) was treated with a solution of hydrogen chloride in diethyl ether (84 ml, 1.0 M, 84 mmol). The resulting mixture was stirred overnight at room temperature and evaporated affording 46.1 g (quant.) of the title compound.
LC-MS (Method 3): Rt = 1.72 min; MS (ESIpos): m/z = 572 [M+H]+
‘H-NMR (600 MHz, DMSO-d6) 5 [ppm]: 1.026 (15.64), 1.037 (16.00), 1.497 (0.56), 1.519 (0.61), 1.722 (0.78), 1.743 (0.65), 1.903 (0.59), 1.910 (0.53), 1.924 (0.66), 1.930 (0.61), 1.978 (0.82), 1.994 (0.50), 2.142
(0.45), 2.154 (0.91), 2.165 (1.11), 2.176 (0.89), 2.187 (0.45), 2.557 (0.64), 2.577 (1.02), 2.594 (0.55), 2.992
(1.81), 3.002 (2.77), 3.012 (1.87), 3.018 (1.15), 3.036 (2.40), 3.054 (1.60), 3.133 (1.12), 3.148 (1.19), 3.168
(0.53), 3.237 (0.88), 3.250 (0.76), 3.338 (0.81), 3.360 (1.42), 3.379 (0.88), 3.580 (1.61), 3.791 (0.89), 3.819
(1.25), 3.844 (0.81), 4.463 (0.89), 4.474 (0.97), 4.481 (1.26), 4.488 (0.99), 4.499 (0.88), 7.051 (3.56), 7.065
(3.77), 7.077 (2.72), 7.080 (3.14), 7.103 (1.42), 7.106 (1.13), 7.116 (2.00), 7.120 (1.84), 7.165 (3.40), 7.178
(2.22), 7.443 (0.84), 7.489 (4.04), 7.504 (3.79), 7.531 (1.66), 7.618 (0.72), 7.954 (4.33), 10.519 (0.49).
Method B
Ethyl 1 – [ 1 – { 5-chloro-2- [(trifluoromethanesulfonyl)oxy]phenyl }piperidin-3-yl] -5-(difluoromethyl)- 1 H-pyrazole-4-carboxylate (prepared in analogy to Example 14A, Enantiomer 2, 80.0 mg, 150 pmol) and l-(2-methylpropyl)-4- [4-(4,4,5 ,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)phenyl]piperazine (Example 18 A 64.1 mg, 97 % purity, 180 pmol) were dissolved under argon in toluene/ethanol (0.83/0.83 ml). Tetrakis(triphenylphosphine)palladium(0) (8.69 mg, 7.52 pmol) and 2 M sodium carbonate solution (226 pl, 452 pmol) were added and the mixture was stirred at 100°C overnight. The reaction mixture was diluted with ethyl acetate and water. The aqueous phase was acidified with 1 M hydrochloric acid. The phases were
SUBSTITUTE SHEET (RULE 26)
separated and the aqueous phase was extracted twice with ethyl acetate. The combined organic phases were dried over sodium sulfate, filtered and evaporated. The crude product was dissolved in THF/ethanol (3.9/0.39 ml), 1 M aqueous lithium hydroxide solution (1.5 ml, 1.5 mmol) was added and the mixture was stirred overnight at room temperature. The mixture was evaporated, the residue was dissolved in acetonitrile/TFA/water and purified using preparative HPLC (RP18 column, acetonitrile/water gradient with the addition of 0.1% TFA). The product fractions were combined and evaporated. The residue was mixed with 0.1 M hydrochloric acid in dioxane, carefully evaporated at 30°C (twice) and then lyophilized. 53 mg of the target compound (55% of theory, purity 95%) were obtained.
LC-MS (Method 4): Rt = 0.91 min; MS (ESIpos): m/z = 572 [M-HC1+H]+
‘H-NMR (400 MHz, DMSO-d6) 5 [ppm]: 1.004 (15.46), 1.020 (16.00), 1.491 (0.44), 1.522 (0.50), 1.722 (0.68), 1.753 (0.55), 1.890 (0.47), 1.920 (0.55), 1.967 (0.84), 2.129 (0.76), 2.146 (0.96), 2.163 (0.76), 2.582
(0.91), 2.613 (0.48), 2.999 (0.86), 3.010 (1.71), 3.025 (3.88), 3.041 (2.30), 3.131 (0.88), 3.161 (1.25), 3.177
(2.08), 3.213 (1.75), 3.242 (1.16), 3.467 (1.06), 3.496 (0.84), 3.503 (0.60), 3.519 (0.54), 3.525 (0.50), 3.549
(0.75), 3.555 (0.84), 3.572 (1.57), 3.582 (1.48), 3.589 (1.38), 3.601 (2.78), 3.608 (1.89), 3.633 (0.44), 3.640
(0.41), 3.811 (0.94), 3.847 (1.32), 3.878 (0.71), 4.329 (0.49), 4.439 (0.46), 4.466 (0.73), 4.477 (0.52), 4.839
(0.49), 7.047 (3.30), 7.070 (3.64), 7.082 (2.61), 7.087 (3.29), 7.104 (1.46), 7.109 (0.86), 7.124 (2.34), 7.129
(2.03), 7.160 (3.99), 7.181 (1.96), 7.388 (0.88), 7.490 (4.02), 7.512 (3.81), 7.519 (2.20), 7.650 (0.72), 7.959
(3.78), 9.708 (0.41).
[OC]D20 = -73.05°, c = 0.465g/100cm3, trichloromethane.
Enantiomer 2 has an absolute configuration of R as shown in example 3 A below.
1 – { 3(2?)- 1 – [4-Chloro-4′-(4-isobutylpiperazin- 1 -yl) [biphenyl] -2-yl]piperidin-3-yl } -5-(difluoromethyl)- 1H-pyrazole-4-carboxylic acid hydrochloride
Example 3A
1 – { 3(7?)- 1 – [4-Chloro-4′-(4-isobutylpiperazin- 1 -yl) [biphenyl] -2-yl]piperidin-3-yl } -5-(difluoromethyl)- 1H-pyrazole-4-carboxylic acid hydrochloride hemihydrate
SUBSTITUTE SHEET (RULE 26)
100 mg 1 – { 1 – [4-Chloro-4′-(4-isobutylpiperazin- 1 -yl) [biphenyl] -2-yl]piperidin-3-yl } -5-(difluoromethyl)-lH-pyrazole-4-carboxylic acid hydrochloride (Enantiomer 2) (example 3) were solved at 60°C in 3,5 ml 2 -propanol, wherein the 2-propanol was dosed portion wise in lOOpl -portions at 60°C until a clear solution was obtained. Afterwards the vessel was closed with a septum and placed into a slowly cooling sand bath from 60°C to roomtemperature over the weekend -> small amounts of solids were detected. Thereafter the septum was provided with a canula, in order to slowly let the solvent evaporate. After 4 weeks crystals were collected and inspected under a microscope.



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
/////////Nurandociguat, guanylate cyclase activator, BAY 3283142, LPU8429UK5
Nibrozetone
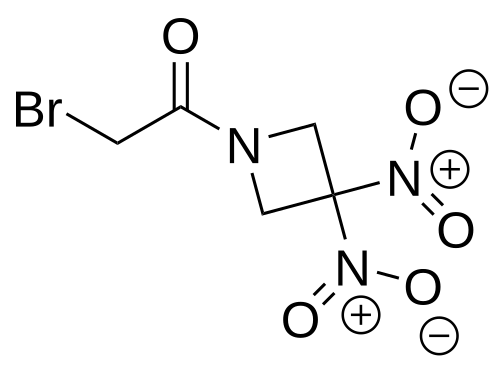
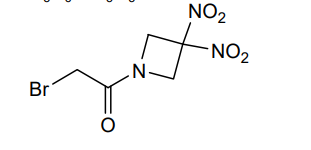
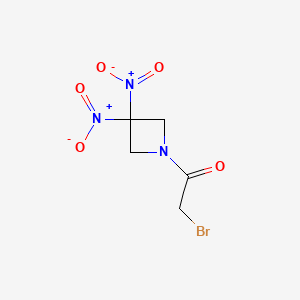
Nibrozetone
CAS 925206-65-1
MF C5H6BrN3O5 MW268.02 g/mol
2-bromo-1-(3,3-dinitroazetidin-1-yl)ethan-1-one
2-Bromo-1-(3,3-dinitroazetidin-1-yl)ethanone
2-BROMO-1-(3,3-DINITROAZETIDIN-1-YL)ETHAN-1-ONE
anti-inflammatory, RRx-001, RRx 001, ABDNAZ
Nibrozetone is an investigational new drug that is being evaluated by EpicentRx for the treatment of oral mucositis in head and neck cancer patients. It is a small molecule that combines direct inhibition of the NLRP3 inflammasome, induction of NRF2, and release of nitric oxide under hypoxic conditions.[1][2] It has received Fast Track designation from the FDA for severe oral mucositis in head and neck cancer patients.[3]
Nibrozetone (RRx-001) is an investigational, multi-action small molecule drug that is being developed by EpicentRx for a range of conditions, including head and neck cancers, small cell lung cancer, and neurodegenerative diseases like Parkinson’s and ALS. Its mechanism involves inhibiting the NLRP3 inflammasome, activating the Nrf2 pathway, and releasing nitric oxide in hypoxic tumor environments, while also protecting healthy tissues. It is being evaluated for its potential to reduce the side effects of cancer treatments and as a disease-modifying therapy itself.
How it works
- Anti-inflammatory: Nibrozetone inhibits the NLRP3 inflammasome, which is a key driver of inflammation in several diseases.
- Antioxidant: It activates the Nrf2 pathway, a cellular defense mechanism that protects against oxidative stress.
- Tumor-specific delivery: It acts as a “hypoxia-activated” drug, releasing a nitric oxide-releasing radical only in the low-oxygen environment of tumors, which can be toxic to cancer cells.
- Protective to normal tissue: The drug’s protective mechanisms are thought to keep it from causing harm to healthy tissues outside of the tumor environment.
Current and potential uses
- Oral mucositis: It is being studied to prevent and treat severe mouth sores that can be a side effect of head and neck cancer radiation therapy.
- Small cell lung cancer (SCLC): It is being investigated in a Phase 3 trial for the treatment of SCLC.
- Neurodegenerative diseases: Animal studies have shown promising neuroprotective effects in models of Parkinson’s and ALS.
- Other potential applications: Research is ongoing for its use as a treatment for other conditions, including endometriosis, toxic exposures, and various types of cancers.
- RRx-001 in Lung Cancer, Ovarian Cancer and Neuroendocrine Tumors Prior to Re-administration of Platinum Based Doublet Regimens (QUADRUPLE THREAT)CTID: NCT02489903Phase: Phase 2Status: CompletedDate: 2025-03-17
- RRx-001 for Reducing Oral Mucositis in Patients Receiving Chemotherapy and Radiation for Head and Neck CancerCTID: NCT05966194Phase: Phase 2Status: RecruitingDate: 2024-11-15
- Safety and Efficacy of RRx-001 in the Attenuation of Oral Mucositis in Patients Receiving Chemoradiation for the Treatment of Oral CancersCTID: NCT03515538Phase: Phase 2Status: CompletedDate: 2024-11-04
- Safety and Pharmacokinetic Study of RRx-001 in Cancer SubjectsCTID: NCT01359982Phase: Phase 1Status: CompletedDate: 2024-11-01
- RRx-001 Given With Irinotecan and Temozolomide for Pediatric Patients With Recurrent or Progressive Malignant Solid and Central Nervous System TumorsCTID: NCT04525014Phase: Phase 1Status: TerminatedDate: 2024-10-31
REF
- Dinitroazetidines Are a Novel Class of Anticancer Agents and Hypoxia-Activated Radiation Sensitizers Developed from Highly Energetic MaterialsPublication Name: Cancer ResearchPublication Date: 2012-05-14PMID: 22589277DOI: 10.1158/0008-5472.can-11-2303
- Properties of delta5-3beta-hydroxysteroid oxidoreductase isolated from Streptomyces griseocarneusPublication Name: Acta microbiologica Academiae Scientiarum HungaricaePublication Date: 1975PMID: 5856
PAT
- Dinitroazetidines Are a Novel Class of Anticancer Agents and Hypoxia-Activated Radiation Sensitizers Developed from Highly Energetic MaterialsPublication Name: Cancer ResearchPublication Date: 2012-05-14PMID: 22589277DOI: 10.1158/0008-5472.can-11-2303
- Properties of delta5-3beta-hydroxysteroid oxidoreductase isolated from Streptomyces griseocarneusPublication Name: Acta microbiologica Academiae Scientiarum HungaricaePublication Date: 1975PMID: 5856
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-8927527-B2Priority Date: 2005-08-12Grant Date: 2015-01-06
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-9226915-B2Priority Date: 2005-08-12Grant Date: 2016-01-05
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: WO-2007022225-A2Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-2022016077-A1Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-11925617-B2Priority Date: 2005-08-12Grant Date: 2024-03-12
- Methods of synthesizing and isolating N-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: US-8471041-B2Priority Date: 2010-02-09Grant Date: 2013-06-25
- Methods of synthesizing and isolating n-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: WO-2011100090-A1Priority Date: 2010-02-09
- Methods of synthesizing and isolating n-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: IL-221141-A0Priority Date: 2010-02-09
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: EP-1924253-A2Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: EP-1924253-B1Priority Date: 2005-08-12Grant Date: 2014-12-10
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011100090&_cid=P11-MHTYGA-61308-1
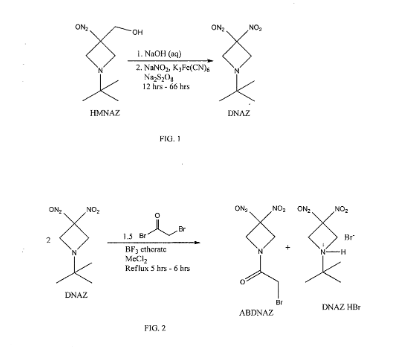
Cyclic nitro compounds, such as ABDNAZ, are being investigated for their potential use in treating cancer. Methods of synthesizing ABDNAZ have been described, such as in United States Patent No. 7,507,842 to Bednarski et al.
(“Bednarski”). In Bednarski, ABDNAZ is synthesized by reacting
l-½rt-butyl-3,3-dinitroazetidine (DNAZ) with bromoacetyl bromide and boron trifluoride etherate. For every mole of ABDNAZ produced, a mole of a hydrogen bromide salt of DNAZ (DNAZ HBr) is also produced as a coproduct. The ABDNAZ is isolated from the DNAZ HBr by cooling the reaction mixture, adding
dichloromethane, and filtering the DNAZ HBr. Solid DNAZ HBr is sensitive to impact, friction, and other external stimuli and, therefore, must be handled carefully. The dichloromethane filtrate is washed with water, dried, and then the dichloromethane is evaporated, producing a crude ABDNAZ mixture. The product is washed sequentially with diethyl ether and dried under vacuum, yielding ABDNAZ that is approximately 98% pure and at a yield of approximately 75% (based on bromoacetyl bromide). The 2% of impurities remaining in the ABDNAZ are believed to include
bromoacetic acid, unreacted DNAZ, and DNAZ HBr. This method of producing ABDNAZ is referred to herein as the Bednarski process. While the Bednarski process provides ABDNAZ at a reasonable purity and yield, the purity is not sufficient for pharmaceutical uses. In addition, solid DNAZ HBr produced during the Bednarski process is an explosive compound, which adds to the complexity of producing
Example 2
Synthesis of ABDNAZ from DNAZ
A three neck round bottom flask (3 L) equipped with a magnetic stir bar and a water jacketed reflux condenser was charged with the dichloromethane solution of DNAZ (produced as described in Example 1). A nitrogen gas purge of the apparatus was initiated and, after ten minutes, boron trifluoride diethyletherate (6.37 mL, 52 mmol) was added, followed by bromoacetyl bromide (33.77 mL, 388 mmol). The flask was sealed, except for a small vent at the top of the condenser, and the solution was heated to a mild reflux. After six hours (± 0.5 hour), heating was stopped and dichloromethane (1000 mL) and distilled water (800 mL) were added, in that order, to the heterogeneous mixture. The two-phase system was stirred vigorously for sixteen hours, until all solids (DNAZ HBr) were dissolved. The two-phase system was then transferred to a separatory funnel. The aqueous phase was removed and the organic phase was washed with additional distilled water (4 x 500 mL). The organic phase was dried with sodium sulfate (100 g – 150 g) and then transferred to a single neck, round bottom flask. The solution was concentrated on a rotary evaporator to approximately half of its initial volume and then ethanol (250 mL) was added. The remaining dichloromethane was removed by a rotary evaporator, causing precipitation of clear, colorless crystals. The flask was chilled in an ice bath for thirty minutes. The precipitate was isolated by vacuum filtration, rinsed with additional cold ethanol (5 x 150 mL), and dried to afford pure ABDNAZ (56.04 g, 81% yield): Ή NMR
(d6-acetone) δ 4.02 (s, 2H, -CH2Br ), 4.96 (br s, 2H, ring -CH2), 5.36 (br s, 2H, ring -CH2); 13C NMR (d6-acetone) δ 25.58, 58.58, 60.53, 107.69, 167.48.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007022225&_cid=P11-MHTYDP-59218-1
Example 5: Synthesis of ABDNAZ
[00139] A 25 ml, three-neck, round bottom flask was charged with 7 ml of methylene chloride and 2.50 g (12.3 mmol) of t-BuDNAZ prepared as described in Archibald et at, Journal of Organic Chemistry, 1990, 2920. Under nitrogen, 0.16 ml (1.23 mmol) of boron trifluoride etherate was added. After stirring 5 min. at ambient temperature, 0.54 ml (6.15 mol) of bromoacetyl bromide was added. The solution was heated between 50-600C for 2 h. The darkened reaction mixture was cooled to ambient temperature, diluted with 50 ml methylene chloride, and filtered. The solid was identified as the HBr salt of t-BuDNAZ. The methylene chloride filtrate was washed with two 20 ml portions of water, dried over sodium sulfate, filtered, and evaporated under reduced pressure. The resultant solid was washed with three 20 ml portions of ethyl ether and dried under vacuum to yield 1.24 g (75.2% based on bromoacetyl bromide) of BrADNAZ as a white solid (mp = 124-1250C). 1H NMR (CDCl3): δ 3.76 (s, 2H), 4.88 (br s, 2H), 5.14 (br s, 2H); 13C NMR (CDCl3): δ 165.2, 105.0, 59.72, 57.79, 23.90. CaIc. for C5H6BrN3O5: %C 22.41, %H 2.26, %N 15.68; Found: %C 22.61, %H 2.36, %N 15.58.
HPLC/MS C-8 reverse phase column with acetonitrile/water mobile phase – m/e 266.95 (100%), 268.95 (98.3%). FT-IR 3014.24 (weak), 1677.66, 1586.30, 1567.65, 1445.55 (NO2), 1367.80, 1338.00, 1251.27 cm‘1.
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Oronsky B, Takahashi L, Gordon R, Cabrales P, Caroen S, Reid T (2023). “RRx-001: a chimeric triple action NLRP3 inhibitor, Nrf2 inducer, and nitric oxide superagonist”. Frontiers in Oncology. 13 1204143. doi:10.3389/fonc.2023.1204143. PMC 10258348. PMID 37313460.
- Jayabalan N, Oronsky B, Cabrales P, Reid T, Caroen S, Johnson AM, et al. (April 2023). “A Review of RRx-001: A Late-Stage Multi-Indication Inhibitor of NLRP3 Activation and Chronic Inflammation”. Drugs. 83 (5): 389–402. doi:10.1007/s40265-023-01838-z. PMC 10015535. PMID 36920652.
- Ryan C (30 March 2023). “FDA Grants Fast Track Designation to RRx-001 for Severe Oral Mucositis in Head and Neck Cancer”. OncLive.
| Clinical data | |
|---|---|
| Other names | Rrx-001 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 925206-65-1 |
| PubChem CID | 15950826 |
| DrugBank | DB12060 |
| ChemSpider | 13092644 |
| UNII | 7RPW6SU9SC |
| KEGG | D12720 |
| ChEMBL | ChEMBL3526802 |
| Chemical and physical data | |
| Formula | C5H6BrN3O5 |
| Molar mass | 268.023 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Nibrozetone, anti-inflammatory, RRx-001, RRx 001, ABDNAZ
Neladalkib
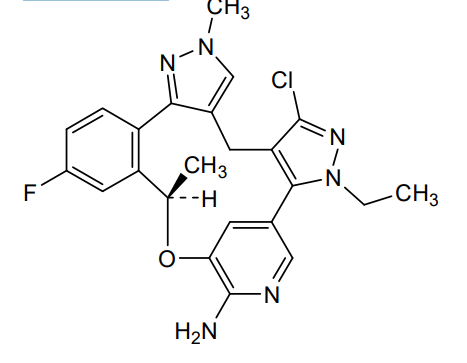
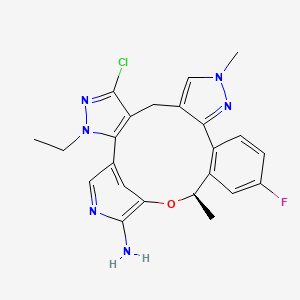
Neladalkib
CAS 2739866-40-9
MF C23H22ClFN6O MW 452.9 g/mol
(19R)-5-chloro-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,10,11,23-pentazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(25),2(6),4,8,11,13(18),14,16,21,23-decaen-22-amine

anaplastic lymphoma kinase (ALK) inhibitor, antineoplastic, NVL-655, NVL 655, J32P26A6BC, ALK-IN-27
Neladalkib is a small molecule drug. The usage of the INN stem ‘-alkib’ in the name indicates that Neladalkib is a ALK (anaplastic lymphoma kinase) inhibitor. Neladalkib is under investigation in clinical trial NCT06765109 (Neladalkib (NVL-655) for TKI-naive Patients With Advanced ALK-Positive NSCLC). Neladalkib has a monoisotopic molecular weight of 452.15 Da.
ALK Inhibitor NVL-655 is an orally bioavailable, brain-penetrant, selective small molecule inhibitor of the receptor tyrosine kinase (RTK) anaplastic lymphoma kinase (ALK), with potential antineoplastic activity. Upon oral administration, ALK inhibitor NVL-655 specifically targets, binds to and inhibits ALK fusion proteins and activating mutations, including the acquired resistance mutations solvent front mutation (SFM) G1202R and the compound mutations G1202R/L1196M and G1202R/G1269A. The inhibition of ALK leads to the disruption of ALK-mediated signaling and the inhibition of cell growth in ALK-expressing tumor cells. ALK belongs to the insulin receptor superfamily and plays an important role in nervous system development. ALK is not expressed in healthy adult human tissue but ALK dysregulation and gene rearrangements are associated with a variety of tumor cell types. NVL-655 is able to penetrate the blood-brain-barrier (BBB) and may therefore exert its activity against EGFR-driven central nervous system (CNS) primary tumors and CNS metastases.
- Expanded Access Program of Neladalkib (NVL-655) for Patients With Advanced ALK+ NSCLC or Other ALK+ Solid TumorsCTID: NCT06834074Status: AvailableDate: 2025-09-22
- Neladalkib (NVL-655) for TKI-naive Patients With Advanced ALK-Positive NSCLCCTID: NCT06765109Phase: Phase 3Status: RecruitingDate: 2025-08-29
- A Study of Neladalkib (NVL-655) in Patients With Advanced NSCLC and Other Solid Tumors Harboring ALK Rearrangement or Activating ALK Mutation (ALKOVE-1)CTID: NCT05384626Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-07-24
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023196910&_cid=P20-MHSIQF-58684-1
SYN
PAT
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-2022098212-A1Priority Date: 2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-2022340586-A9Priority Date: 2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-2023076627-A1Priority Date: 2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-11667649-B2Priority Date: 2020-05-05Grant Date: 2023-06-06
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number: US-2023322797-A1Priority Date: 2022-04-07
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number: WO-2023196900-A1Priority Date: 2022-04-07
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number: WO-2023196900-A9Priority Date: 2022-04-07
- Methods of treating solid tumor using (19r)-5-chloro-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,10,11,23-pentaazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(24),2(6),4,8,11,13,15,17,21(25),22-decaen-22-aminePublication Number: WO-2023196910-A1Priority Date: 2022-04-07
- Methods of treating solid tumor using (19r)-5-chloro-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,10,11,23-pentaazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(24),2(6),4,8,11,13,15,17,21(25),22-decaen-22-aminePublication Number: EP-4504189-A1Priority Date: 2022-04-07
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////neladalkib, antineoplastic, NVL-655, NVL 655, J32P26A6BC, ALK-IN-27
Nefextinib
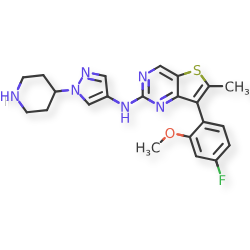
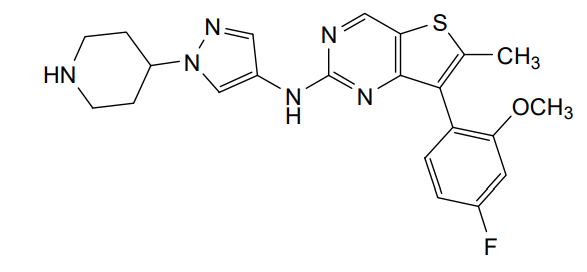
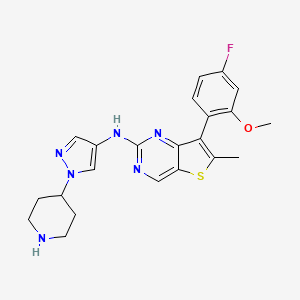
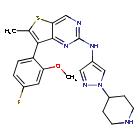
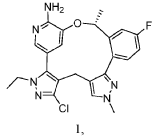
Nefextinib
CAS 2070931-57-4
MF C22H23FN6OS MW 438.52
7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-[1-(piperidin4-yl)-1H-pyrazol-4-yl]thieno[3,2-d]pyrimidin-2-amine
7-(4-FLUORO-2-METHOXYPHENYL)-6-METHYL-N-(1-(PIPERIDIN-4-YL)-1H-PYRAZOL-4-YL) THIENO (3,2-D)PYRIMIDIN-2-AMINE
tyrosine kinase inhibitor, antineoplastic, DL772G3NN7, MAX-40279, MAX 40279
Nefextinib is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) and FMS-like tyrosine kinase 3 (FLT3; CD135; STK1; FLK2), with potential antineoplastic activity. Upon oral administration, nefextinib binds to and inhibits both FGFR and FLT3, including FLT3 mutant forms, which results in the inhibition of FGFR/FLT3-mediated signal transduction pathways. This inhibits proliferation in FGFR/FLT3-overexpressing tumor cells. FGFR, a family of receptor tyrosine kinases, is upregulated in many tumor cell types. FLT3, a class III receptor tyrosine kinase (RTK), is overexpressed or mutated in most B-lineage neoplasms and in acute myeloid leukemias. They both play key roles in cellular proliferation and survival.
- A Phase 2 Study to Evaluate the Safety and Efficacy of Max-40279-01 in Patients With Advanced Gastric Cancer or Gastroesophageal Junction CancerCTID: NCT05395780Phase: Phase 2Status: Unknown statusDate: 2022-06-02
- MAX-40279 in Subjects With Acute Myelogenous Leukemia (AML)CTID: NCT03412292Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- MAX-40279-01 in Patients With Advanced Solid TumorsCTID: NCT04183764Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- Study of MAX-40279 in Patients With Relapsed or Refractory Acute Myelogenous Leukemia (AML)CTID: NCT04187495Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- A Clincal Study of Max-40279-01 in Patients With Advanced Colorectal CancerCTID: NCT05130021Phase: Phase 2Status: Unknown statusDate: 2021-12-06
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017012559&_cid=P22-MHRG1L-67142-1
[0488]N-[7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidin-4-yl)-1H-pyrazol-4-amine (compound 31)
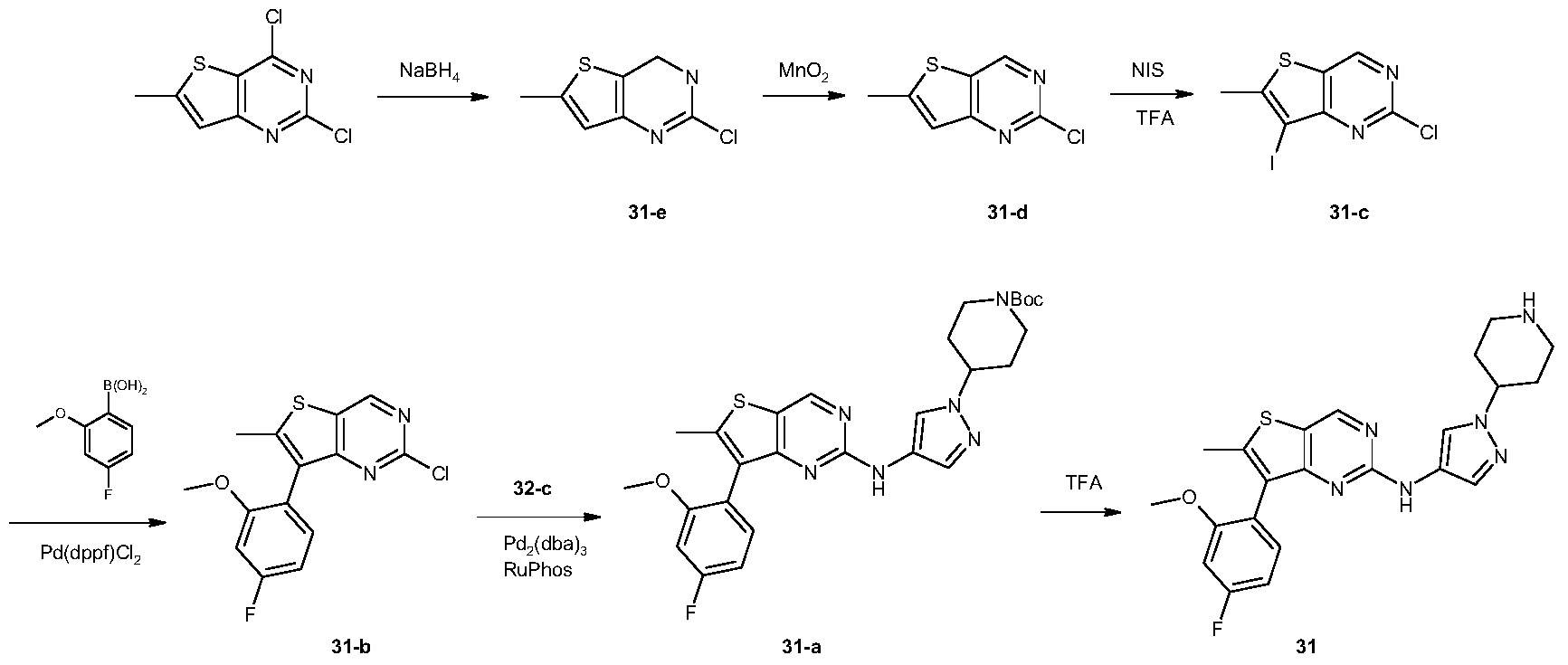
[0491]2,4-Dichloro-6-methylthiophene[3,2-d]pyrimidine (10 g, 45.6 mmol) was dissolved in tetrahydrofuran (100 mL) and ethanol (100 mL). The reaction mixture was cooled to 0 °C, and sodium borohydride (12.5 g, 198 mmol) was added in portions. The reaction mixture was brought to room temperature and stirred for 16 hours. It was then diluted with water (500 mL) and adjusted to pH 7 with 1 N hydrochloric acid solution. The aqueous phase was extracted with ethyl acetate (150 mL × 3). The organic phase was washed successively with water (100 mL × 3) and saturated brine (100 mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give a white solid 31-e (7.5 g, yield: 88%). This product required no further purification. LC-MS (ESI): m/z = 187 [M+H] + .
[0492]Synthesis of compound 31-d
[0493]Compound 31-e (7.5 g, 40 mmol) was dissolved in chloroform (300 mL) at 0 °C, and activated manganese dioxide (35 g, 400 mmol) was added. The reaction mixture was brought to room temperature and stirred for 16 hours. The reaction mixture was filtered through diatomaceous earth, and the filter cake was washed with chloroform (100 mL × 3). The combined filtrates were concentrated under reduced pressure to give a white solid 31-d (6.6 g, yield: 89%), which did not require further purification. LC-MS (ESI): m/z = 185 [M + H]+.
[0494]Synthesis of compound 31-c
[0495]Compound 31-d (3.1 g, 16.8 mmol) was dissolved in trifluoroacetic acid (30 mL) at 0 °C. N-iodosuccinimide (5.7 g, 25.3 mmol) was added in portions. The reaction mixture was brought to room temperature and stirred for 1 hour. The reaction was quenched with water (50 mL) and extracted with dichloromethane (50 mL × 3). The organic phase was washed successively with water (50 mL × 3) and saturated brine (50 mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give a white solid 31-c (4.9 g, yield: 94%). This product required no further purification. LC-MS (ESI): m/z = 311 [M + H] + .
[0496]Synthesis of compound 31-b
[0497]Compound 31-c (615 mg, 1.98 mmol), 2-methoxy-4-fluorophenylboronic acid (405 mg, 2.38 mmol), and sodium carbonate (630 mg, 5.94 mmol) were suspended in dioxane (5 mL) and water (5 mL). A [1,1′-bis(diphenylphosphine)ferrocene]palladium dichloride dichloromethane complex (163 mg, 0.2 mmol) was added. The mixture was purged three times with nitrogen and heated to 80 °C for 16 hours. After cooling to room temperature, the reaction solution was concentrated under reduced pressure. The residue was separated into layers by dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated and purified by silica gel column chromatography (petroleum ether:dichloromethane = 1:1) to give a white solid 31-b (240 mg, yield: 39%). LC-MS (ESI): m/z = 309 [M+H] + .
[0498]Synthesis of compound 31-a
[0499]Compound 31-b (240 mg, 0.78 mmol) and compound 32-c (208 mg, 0.78 mmol) were dissolved in N,N-dimethylformamide (3 mL), and potassium carbonate (323 mg, 2.34 mmol), 2-dicyclohexylphosphine-2′,6′-diisopropoxy-1,1′-biphenyl (112 mg, 0.24 mmol), and tris(dibenzylacetone)palladium (134 mg, 0.24 mmol) were added. The reaction was carried out under nitrogen protection at 110 °C for 16 hours. After cooling to room temperature, the reaction mixture was separated into layers by dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel thin-layer chromatography (petroleum ether: ethyl acetate = 1:1) to give a yellow viscous oil 31-a (190 mg, yield: 45%). LC-MS(ESI): m/z = 539[M+H] + .
[0500]Synthesis of Compound 31
[0501]31-a (190 mg, 0.35 mmol) was dissolved in dichloromethane (3 mL), and trifluoroacetic acid (3 mL) was added. The mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure, and the residue was separated into layers by ethyl acetate (50 mL) and 1N hydrochloric acid aqueous solution (50 mL). The aqueous phase was adjusted to pH = 10 with saturated potassium carbonate aqueous solution, and a solid precipitated. The solid was filtered, and the filter cake was washed with water (20 mL × 3). The solid was dried under vacuum to give a light yellow solid 31 (22 mg, yield: 14%). LC-MS (ESI): m/z = 439 [M+H] + .
[0502]
1H-NMR(400MHz,MeOD)δ:8.78(d,J=5Hz,1H),7.87(s,1H),7.48(s,1H),7.35(m,1H),7.05(dd,J=11Hz,J=2Hz,1H),6.91(m,1H),4.10(m,1H),3.79(s,3H),3.22(m,2H),2.77(m,2H),2.47(s,3H),2.03(m,2H),1.73(m,2H)ppm
PAT
- Condensed ring pyrimidine compound, intermediate, its preparation method, composition and applicationPublication Number: CN-106366093-BPriority Date: 2015-07-21Grant Date: 2020-08-18
- Condensation ring pyrimidine compounds, intermediates, methods for producing them, compositions and applicationsPublication Number: JP-6875372-B2Priority Date: 2015-07-21Grant Date: 2021-05-26
- Condensed ring pyrimidine compounds, intermediates, preparation methods, compositions and applications thereofPublication Number: KR-102591886-B1Priority Date: 2015-07-21Grant Date: 2023-10-20
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: EP-3354653-B1Priority Date: 2015-07-21Grant Date: 2019-09-04
- Fused ring pyrimidine compounds, intermediates, production methods, compositions and applications thereofPublication Number: JP-2018520202-APriority Date: 2015-07-21
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: US-10494378-B2Priority Date: 2015-07-21Grant Date: 2019-12-03
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: US-2018208604-A1Priority Date: 2015-07-21
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: WO-2017012559-A1Priority Date: 2015-07-21
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////nefextinib, tyrosine kinase inhibitor, antineoplastic, DL772G3NN7, MAX-40279, MAX 40279
Muvadenant
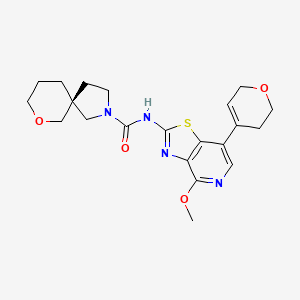
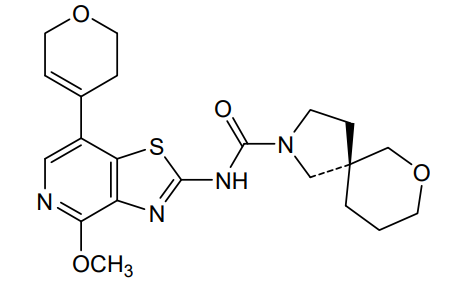
Muvadenant
CAS 2459881-03-7
MF C21H26N4O4S , 430.5 g/mol
(5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy[1,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5] decane-2-carboxamide
(5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide
adenosine receptor antagonist, antineoplastic, 6LSF69F6A8, M1069 , M 1069
Muvadenant is a small molecule drug. The usage of the INN stem ‘-adenant’ in the name indicates that Muvadenant is a adenosin receptor antagonist. Muvadenant has a monoisotopic molecular weight of 430.17 Da.
Adenosine is an ubiguitous modulator of numerous physiological activities, particularly within the cardiovascular, nervous and immune systems. Adenosine is related both structurally and metabolically to the bioactive nucleotides adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP) and cyclic adenosine monophosphate (cAMP), to the biochemical methylating agent S-adenosyl-L-methione (SAM) and structurally to the coenzymes NAD, FAD and coenzym A and to RNA.
Via cell surface receptors, adenosine modulates diverse physiological functions including induction of sedation, vasodilatation, suppression of cardiac rate and contractility, inhibition of platelet aggregability, stimulation of gluconeogenesis and inhibition of lipolysis. Studies show that adenosine is able to activate adenylate cyclases, open potassium channels, reduce flux through calcium channels, and inhibit or stimulate phosphoinositide turnover through receptor-mediated
mechanisms (Muller C. E. and Stein B., Current Pharmaceutical Design, 2: 501 , 1996; Muller C. E., Exp. Opin. Ther. Patents, 7(5): 419, 1997).
Adenosine receptors belong to the superfamily of G-protein-coupled receptors (GPCRs). Four major subtypes of adenosine receptors have been
pharmacologically, structurally and functionally characterized (Fredholm et al., Pharm. Rev., 46: 143-156, 1994) and referred to as A1, A2A, A2B and A3. Though the same adenosine receptor can couple to different G-proteins, adenosine A1 and A3 receptors usually couple to inhibitory G-proteins referred to as G, and Go which inhibit adenylate cyclase and down-regulate cellular cAMP levels. In contrast, the adenosine A2A and A2B receptors couple to stimulatory G-proteins referred to as Gs that activate adenylate cyclase and increase intracellular levels of cAMP (Linden J., Annu. Rev. Pharmacol. Toxicol., 41 : 775-87 2001).
PAT
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: US-2022119412-A1Priority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: EP-3914600-B1Priority Date: 2019-01-22Grant Date: 2024-08-07
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: CN-113939520-BPriority Date: 2019-01-22Grant Date: 2024-11-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: ES-2992081-T3Priority Date: 2019-01-22Grant Date: 2024-12-09
- Thiazolopyridine derivatives as adenosine receptor antagonists.Publication Number: JP-7600119-B2Priority Date: 2019-01-22Grant Date: 2024-12-16
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: AU-2020211697-A1Priority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: KR-20210116572-APriority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: CN-113939520-APriority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: EP-3914600-A1Priority Date: 2019-01-22
- Thiazolopyridine derivative as an adenosine receptor antagonistPublication Number: JP-2022524914-APriority Date: 2019-01-22
- Combination therapy for cancerPublication Number: CN-117858723-APriority Date: 2021-06-07
- Combination treatment of cancerPublication Number: EP-4351640-A1Priority Date: 2021-06-07
- Combination Treatment of CancerPublication Number: JP-2024520764-APriority Date: 2021-06-07
- Combination Treatment of CancerPublication Number: US-2024279338-A1Priority Date: 2021-06-07
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: WO-2020152132-A1Priority Date: 2019-01-22
- Novel crystalline forms of (s)-7-oxa-2-aza-spiro[4.5]decane-2-carboxylic acid [7-(3,6-dihydro-2h-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin-2-yl]-amide and co-crystal forms thereofPublication Number: TW-202415667-APriority Date: 2022-08-02
- Novel crystalline forms of (s)-7-oxa-2-aza-spiro[4.5]decane-2-carboxylic acid [7-(3,6-dihydro-2h-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin-2-yl]-amide and co-crystal forms thereofPublication Number: WO-2024028273-A1Priority Date: 2022-08-02
- Combination treatment of cancerPublication Number: WO-2022258622-A1Priority Date: 2021-06-07
- Combination treatment of cancerPublication Number: AU-2022288571-A1Priority Date: 2021-06-07
- Combination treatment of cancerPublication Number: CA-3220380-A1Priority Date: 2021-06-07
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020152132&_cid=P10-MHPOEV-06540-1
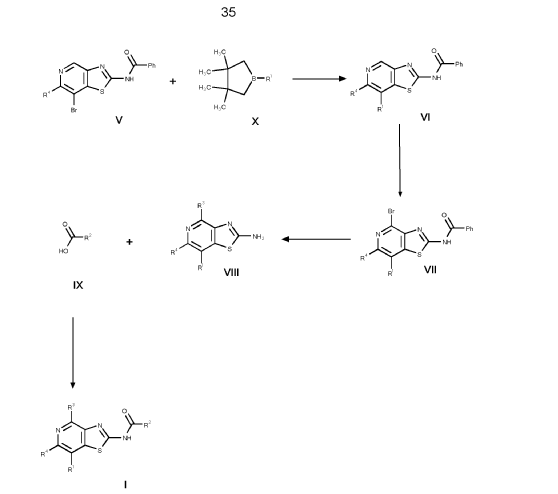
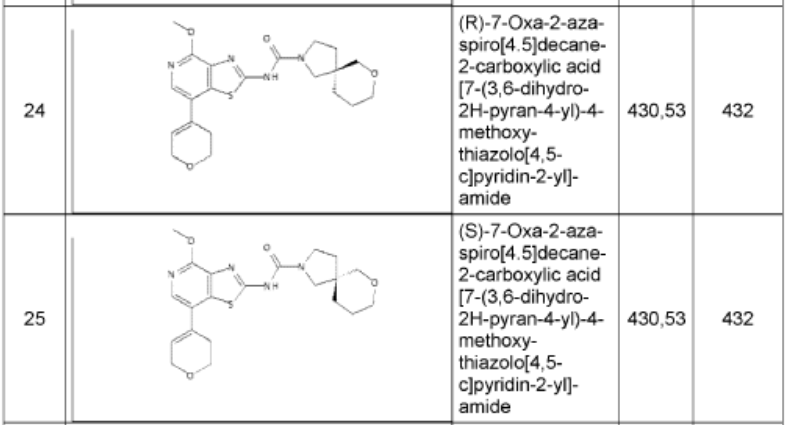
1. (5R)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin- 2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 24
and (5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5- c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 25
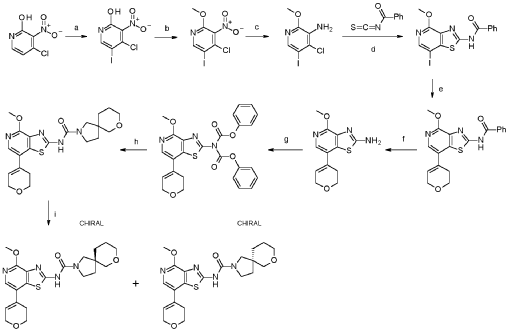
a. 4-chloro-5-iodo-3-nitropyridin-2-ol
Into a 250-mL round-bottom flask was placed 4-chloro-3-nitropyridin-2-ol (10.0 g, 54.4 mmol, 95%), N-lod-succinimid (NIS, 14.2 g, 59.9 mmol, 95%) in acetonitrile (115 mL). The solution was stirred for 1 h overnight at 80°C in an oil bath. The mixture was concentrated and the precipitate formed collected by filtration. The residue was washed with twice with petrol ether (500 mL) dried under vacuum at 60°C overnight. This resulted in 4-chloro-5-iodo-3-nitropyridin-2-ol (16.5 g, 97.9%, 97% purity) as a yellow solid. MS: m/z = 300.9 [M+H]+.
b. 4-chloro-5-iodo-2-methoxy-3-nitropyridine
Into a 500-mL round-bottom flask was placed 4-chloro-5-iodo-3-nitropyridin-2-ol (16.5 g, 53.3 mmol, 97%), Ag2CO3 (15.5 g, 53.3 mmol, 95%) in toluene (310 mL). To this suspension CH3I (15.9 g, 107 mmol, 95%) was added at 50°C and the mixture was stirred at 80°C for 4 h. The precipitate was collected by filtration and discarded. The filtrate was evaporated to dryness under vacuum and the residue purified by silica gel chromatography with ethyl acetate/petroleum ether (15:85).
This resulted in 4-chloro-5-iodo-2-methoxy-3-nitropyridine (9.90 g, 52.6%, 89% purity) as a light yellow solid. MS: m/z = 315.5 [M+H]+.
c. 4-chloro-5-iodo-2-methoxypyridin-3-amine
Into a 250-mL 3-necked round-bottom flask was placed 4-chloro-5-iodo-2-methoxy-3-nitropyridine (9.90 g, 28.0 mmol, 89%), iron (16.5 g, 281 mmol, 95%) and NH 4C (9.40 g, 174 mmol, 99%) in ethanol (152 mL) and water (30 mL). The mixture was stirred for 2 h at 80°C in an oil bath. The reaction mixture was filtered over Celite, washed with ethanol and the mother liquor was concentrated to dryness. The residue was stirred for 30 min. with 100 ml water at 60°dried in vacuo. This resulted in 4-chloro-5-iodo-2-methoxypyridin-3-amine (7.20 g, 75%, 83% purity) as an off-white solid. It was used without further purification in the next step. MS: m/z = 285.9 [M+H]+.
d. N-[7-iodo-4-methoxy-[1,3]thiazolo[4,5-c]pyridin-2-yl]benzamide
Into a 500-mL round-bottom flask was placed 4-chloro-5-iodo-2-methoxypyridin-3-amine (7.20 g, 21.0 mmol, 83%) in acetone (150 mL) and benzoyl isothiocyanate (5.21 g, 31.5 mmol, 99%) was added dropwise at room temperature. The solution was stirred for 1 h at 50 °C in an oil bath. The solids were collected by filtration, washed with acetone and dried in vacuo to give N-[7-iodo-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide (8.73 g, 91 %, 90% purity) as a white solid. MS: m/z = 412.2 [M+H]+.
e. N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin- 2-yl]benzamide
To a solution of N-[7-iodo-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide (6.00 g, 13.1 mmol, 90%) and 2-(3,6-dihydro-2H-pyran-4-yl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane (6.13 g, 27.7 mmol, 95%) in dioxane (200 mL) and water (40.00 mL) were added NaOH (2.90 g, 68.9 mmol, 95%) and Pd(dppf)Cl2* dichloromethane (1.20 g, 1.40 mmol, 95%). After stirring for 1 h at 100°C under a nitrogen atmosphere, the mixture was concentrated to dryness under vacuo. The residue was purified by silica gel chromatography with ethyl acetate/hexane (95:5). This resulted in 3.32 g (62%, 90% purity) of N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide as colorless solid. MS: m/z = 368.1 [M+H]+.
f. 7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin-2- amine
To a stirred mixture of N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide (3.27 g, 8.00 mmol, 90%) in water/methanol (1 :1 , 300 mL) was added NaOH (3.36 g, 80.0 mmol, 95%) at room temperature under nitrogen atmosphere. The mixture was stirred for overnight at 90°C under nitrogen atmosphere and evaporated to dryness. The residue was taken up in water and extracted 3 times with dichloromethane (100 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel column chromatography, eluted with petrol ether/ethyl acetate (1 :1) to afford 7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-amine (1.50 g, 68%, 96% purity) as a light brownish solid. MS: m/z = 264.1 [M+H]+.
g. phenyl N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy- [1,3]thiazolo[4,5c]pyridin-2-yl]-N-(phenoxycarbonyl)carbamate
To a stirred solution of 7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-amine (600 mg, 2.19 mmol, 96%) and phenyl chloroformate (1.81 g,
11.0 mmol, 95%) in THF (50 mL) was added K2CO3 (1.59 g, 11.0 mmol, 95%) and pyridine (913 mg, 11.0 mmol, 95%) at room temperature under nitrogen
atmosphere. The mixture was stirred for 6 h at 50° and then after re-cooling to room temperature quenched by the addition of water (300 mL). The mixture was extracted 3 times with dichloromethane (200 mL), the combined organic layers were washed once with brine (200 mL), dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. This resulted in phenyl N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-N-(phenoxycarbonyl)carbamate (1.00 g, 69%, 76% purity) as a light yellow solid. The crude product was used in the next step directly without further purification. MS: m/z = 504.1 [M+H]+.
h. N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin- 2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide
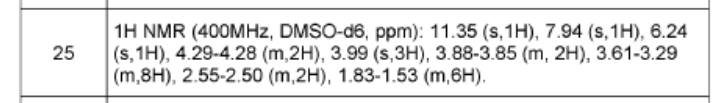
To a mixture of phenyl N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-N-(phenoxycarbonyl)carbamate (1.00 g, 1.52 mmol, 76.) and bis(7-oxa-2-azaspiro[4.5]decane), oxalic acid (1.19 g, 3.03 mmol, 95%) in THF (50 mL) was added diisopropylethyl amine (1.24 g, 9.09 mmol, 95%) at room temperature under nitrogen atmosphere. The mixture was stirred for 1 h at 60°. After re-cooling to room temperature, the mixture was extracted twice with dichloromethane (100 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel column chromatography, eluted with petrol ether/ethyl acetate (1 :1) to afford N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (600 mg, 92%) as a white solid. HPLC: 99.9 % purity, RT = 1.17 min. MS: m/z = 431.1 [M+H]+. 1 H NMR (300 MHz, DMSO-d6) d 1 1.37 (s, 1 H), 7.95 (s, 1 H), 6.25 (s, 1 H), 4.30-4.29 (m, 2H), 3.99 (s, 3H), 3.89 (t, J=5.4Hz, 2H), 3.61-3.29 (m, 8H), 2.55-2.51 (m, 2H), 1.82-1.54 (m, 6H).
i. (5R)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin- 2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 24
and (5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5- c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 25
N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (450 mg, 1.044 mmol, 1 equiv, 99.9%) was purified by chiral-preparative HPLC (Preparative HPLC-032, column: ChiralPak IA, 2*25cm, 5 mm; mobile phase, dichloromethane:ethanol (20:80); detector, UV). This resulted in (5R)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (178 mg, 39%) as a white solid. HPLC: 99.7 % purity, RT (chiral) = 3.86 min, 100% ee. MS: m/z = 431.2 [M+H]+. 1 H NMR (400 MHz, DMSO-d6) d 1 1.36 (s, 1 H), 7.94 (s, 1 H), 6.24 (s, 1 H), 4.29-4.27 (m, 2H), 3.97 (s,3H), 3.88 (t, J=5.2 Hz, 2H), 3.51-3.19 (m, 8H), 2.55-2.50 (m, 2H), 1.83-1.53 (m, 6H) and (5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (171 mg, 38%) as a white solid. HPLC: 99.8 % purity, RT (chiral) = 5.23 min, 99.9% ee. MS: m/z = 431.2 [M+H]+. 1 H NMR (400 MHz, DMSO-d6) d 1 1.35 (s, 1 H), 7.94 (s, 1 H), 6.24 (s, 1 H), 4.29-4.28 (m, 2H), 3.99 (s, 3H), 3.88-3.85 (m, 2H), 3.61-3.29 (m, 8H), 2.55-2.50 (m,2H), 1.83-1.53 (m,6H).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024028273&_cid=P10-MHPOFP-06905-1
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////muvadenant, adenosine receptor antagonist, antineoplastic, 6LSF69F6A8, M1069 , M 1069
Matsupexole
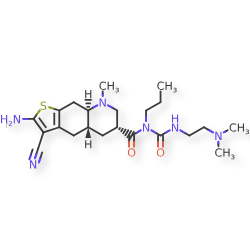
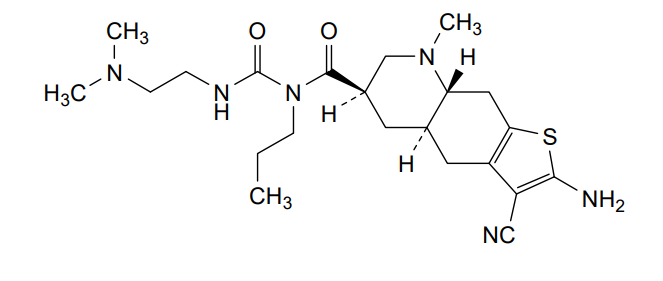
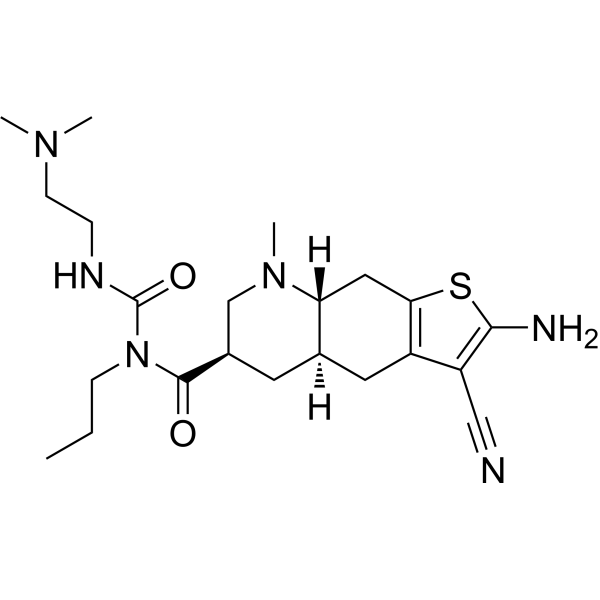

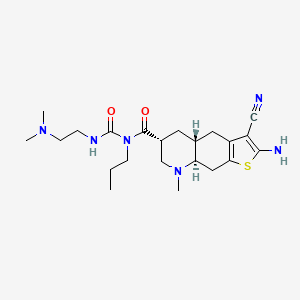
Matsupexole
CAS 1399442-97-7
MF C22H34N6O2S, Molecular Weight, 446.61
(4aR,6R,8aR)-2-amino-3-cyano-N-{[2-(dimethylamino)ethyl]carbamoyl}-8-methyl-N-propyl 4,4a,5,6,7,8,8a,9-octahydrothieno[3,2-g]quinoline-6-carboxamide
(4aR,6R,8aR)-2-amino-3-cyano-N-[2-(dimethylamino)ethylcarbamoyl]-8-methyl-N-propyl-4a,5,6,7,8a,9-hexahydro-4H-thieno[3,2-g]quinoline-6-carboxamide
dopamine receptor agonist, Phase 2, Parkinson’s disease, K4UEG65HTX
- OriginatorKissei Pharmaceutical
- DeveloperAffaMed Therapeutics; Kissei Pharmaceutical
- ClassAmides; Amines; Antiparkinsonians; Dimethylamines; Ethylenediamines; Nitriles; Quinolines; Small molecules; Thiophenes; Urea compounds
- Mechanism of ActionDopamine receptor agonists
- Phase IIParkinson’s disease
- 28 Aug 2025Chemical structure information added.
- 06 Sep 2021Kissei Pharmaceutical completes a phase II trial in Parkinson’s disease (In adults, In elderly) in Japan (PO) (NCT04867551)
- 04 Aug 2021Phase-II clinical trials in Parkinson’s disease in China (PO) (Kissei Pharmaceutical pipeline, August 2021)
PAT
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: WO-2012124649-A1Priority Date: 2011-03-14
- COMPOUNDS DERIVED FROM OCTAIDROTHIENOQUINOLINE, PHARMACEUTICAL COMPOSITION AND PHARMACEUTICAL AGENT COMPRISING SUCH COMPOUNDSPublication Number: BR-112013023575-B1Priority Date: 2011-03-14
- New octahydrothienoquinoline derivative, pharmaceutical composition containing derivative, and using themPublication Number: RU-2573399-C2Priority Date: 2011-03-14Grant Date: 2016-01-20
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: SG-193400-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing the same, and their usesPublication Number: TW-I537274-BPriority Date: 2011-03-14Grant Date: 2016-06-11
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: US-2014243311-A1Priority Date: 2011-03-14
- Octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: US-9138434-B2Priority Date: 2011-03-14Grant Date: 2015-09-22
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: HU-E033449-T2Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing them and their usePublication Number: JP-5563716-B2Priority Date: 2011-03-14Grant Date: 2014-07-30
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing them and their usePublication Number: JP-WO2012124649-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing them and uses thereofPublication Number: KR-20140010137-APriority Date: 2011-03-14
- NEW OCTAHYDROTHYENOCHINOLINE DERIVATIVE, PHARMACEUTICAL COMPOSITION CONTAINING A DERIVATIVE AND THEIR APPLICATIONPublication Number: RU-2013145799-APriority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions comprising said derivatives and their usesPublication Number: CN-103443106-BPriority Date: 2011-03-14Grant Date: 2015-09-30
- New octahydrothienoquinoline derivative, pharmaceutical composition comprising the derivative, and use thereofPublication Number: DK-2687532-T3Priority Date: 2011-03-14Grant Date: 2017-02-20
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: EP-2687532-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: EP-2687532-B1Priority Date: 2011-03-14Grant Date: 2016-12-14
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising the derivative, and use thereofPublication Number: ES-2613658-T3Priority Date: 2011-03-14Grant Date: 2017-05-25
- Novel dopamine D2 receptor agonistPublication Number: JP-2014074013-APriority Date: 2012-09-12
- Novel dopamine D2 receptor agonistPublication Number: JP-6177061-B2Priority Date: 2012-09-12Grant Date: 2017-08-09
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: AU-2012227428-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: AU-2012227428-B2Priority Date: 2011-03-14Grant Date: 2016-05-05
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions comprising said derivatives and their usesPublication Number: CN-103443106-APriority Date: 2011-03-14
- Succinate of octahydrothienoquinoline compound, and crystals thereofPublication Number: WO-2022009815-A1Priority Date: 2020-07-06
- Succinate of octahydrothienoquinoline compound and its crystalPublication Number: CN-115803329-APriority Date: 2020-07-06
- Succinate salts of octahydrothienoquinoline compounds and crystals thereofPublication Number: KR-20230035050-APriority Date: 2020-07-06
- Succinate of octahydrothienoquinoline compound, and crystals thereofPublication Number: EP-4177257-A1Priority Date: 2020-07-06
- Succinate salts of octahydrothienoquinoline compound and crystals thereofPublication Number: US-2023286998-A1Priority Date: 2020-07-06
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022009815&_cid=P22-MHO952-66657-1
[0018]Example 11-{[(4aR,6R,8aR)-2-amino-3-cyano-8-methyl-4,4a,5,6,7,8,8a,9-octahydrothieno[3,2-g]quinolin-6-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea sesquisuccinate monohydrate (Form I crystals of salt (A-1)) 102.8 g of acetone was added to 1-{[(4aR,6R,8aR)-2-amino-3-cyano-8-methyl-4,4a,5,6,7,8,8a,9-octahydrothieno[3,2-g]quinolin-6-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (22.00 g), the mixture was suspended, and the suspension was heated and stirred at an external temperature of 52°C to dissolve the suspension. Activated carbon (2.2 g) was added to this solution and stirred for 10 minutes. This suspension was hot filtered and washed with 35.2 g of acetone. 220.0 g of acetone was then added, and the reaction solution was heated to an external temperature of 52°C and stirred. Next, 44.0 g of water was added to the reaction solution. Separately, 8.73 g of succinic acid was dissolved in a mixed solution of 156.1 g of acetone and 19.8 g of water. This succinic acid solution was added dropwise to the reaction solution over approximately 10 minutes. The dropping funnel was washed with a mixed solution of 17.4 g of acetone and 2.2 g of water and then added dropwise to the reaction solution. The reaction solution was stirred at an internal temperature of 50°C for 1 hour and cooled to 15°C over 30 minutes. The reaction solution was stirred at an external temperature of 10°C for 2 hours, and the crystals were collected by filtration. The crystals were washed twice with 52.8 g of acetone. The obtained wet crystals were dried under reduced pressure at 50°C for 37 hours and then returned to room temperature under reduced pressure over 3 hours. The crystals were stored under air for 24 hours to obtain crystals (27.75 g) of the title compound.
1 H-NMR (DMSO-d6) (δ (ppm)): 0.85 (3H, t, J = 7.4Hz), 1.32 (1H, ddd, J=12.2Hz, 12.2Hz, 12.2Hz), 1.42-1.57 (2H, m), 1.57-1.70 (1H, m ), 1.89-2.00 (2H, m), 2.20-2.13 (1H, m), 2.13-2.28 (2H, m), 2.21 (3H, s), 2.24 (6H, s ), 2.35-2.48 (1H, m), 2.40 (6H, s), 2.46 (2H, t, J = 6.4Hz), 2.81-2.96 (2H, m), 3.00-3 .12 (1H, m), 3.21-3.33 (2H, m), 3.47-3.66 (2H, m), 6.99 (2H, s), 8.50-8.90 (1H, br).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012124649&_cid=P22-MHO8UB-55660-1
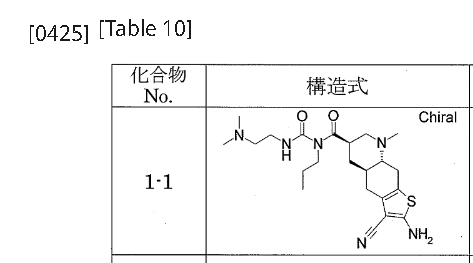
[0422]Example 1-11-{[(4aR,6R,8aR)-2-amino-3-cyano-8-methyl-4H,4aH,5H,6H,7H,8H,8aH,9H-thieno[3,2-g]quinolin-6-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (Compound 1-1) To a mixture of 1-{[(3R,4aR,8aR)-1-methyl-6-oxodecahydroquinolin-3-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (Reference Example 10-1) (1.602 g) and ethanol (44 mL) were added malononitrile (435 mg), morpholine (0.572 mL), and then elemental sulfur (282 mg) with stirring at room temperature, and the mixture was heated to 55°C and stirred for 1.5 hours. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure, and the residue was purified by column chromatography on aminopropyl silica gel (eluent: 0%-5% methanol/ethyl acetate, gradient elution) to give the title compound (1.479 g) as a solid.
1 H-NMR (CDCl
3 ) δ ppm: 0.94(3H, t, J=7.4Hz), 1.45-1.85(4H, m), 1.95-2.15(2H, m), 2.15-2.30(7H, m), 2.30-2.55(7H, m), 2.60-2.75(1H, m), 2.90-3.00(2H, m), 3.00-3.10(1H, m), 3.35-3.45(2H, m), 3.60-3.85(2H, m), 4.65(2H, s), 9.27(1H, br)[α]
D 29 =-105.54°(c=0.30, MeOH)
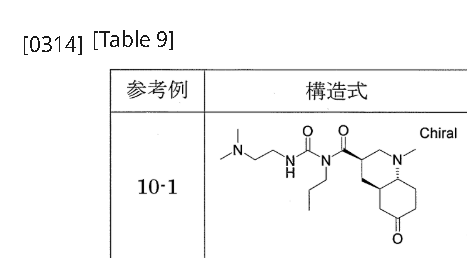
[0311]Reference Example 10-11-{[(3R,4aR,8aR)-1-methyl-6-oxodecahydroquinolin-3-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea 1-{[(3’R,4’aR,8’aR)-1′-methyloctahydro-1’H-spiro[1,3-dioxolane-2,6′-quinoline]-3′-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (Reference Example 8-1) (2.366 g) was added to 2 mol/L hydrochloric acid (30 mL), and the mixture was stirred at room temperature for 2 hours. The reaction mixture was washed with diethyl ether, and then potassium carbonate was added to the aqueous layer to make it alkaline. The mixture was extracted with a methylene chloride/methanol mixed solvent (methylene chloride:methanol = 9:1). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to give the title compound (1.605 g).
1 H-NMR (CDCl
3 ) δ ppm: 0.94 (3H, t, J=7.4 Hz), 1.45-1.90 (6H, m), 1.95-2.05 (1H, m), 2.10-2.55 (17H, m), 2.90-3.10 (2H, m), 3.30-3.45 (2H, m), 3.60-3.80 (2H, m), 9.22 (1H, brs).[α]
D 28 =-37.56° (c=0.38, MeOH).
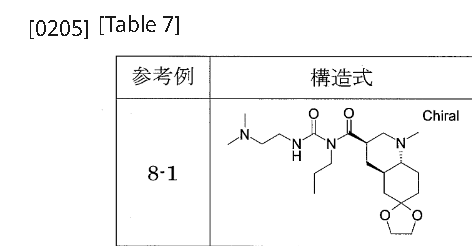
[0198]Reference Example 8-1To a mixture of phenyl 1-{[(3’R,4’aR,8’aR)-1′-methyloctahydro-1’H-spiro[1,3-dioxolane-2,6′-quinoline]-3′-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea N-{[(3’R,4’aR,8’aR)-1′-methyloctahydro-1’H-spiro[1,3-dioxolane-2,6′-quinoline]-3′-yl]carbonyl}-N-propylcarbamate (Reference Example 6-1) (2.401 g) and 2-propanol (30 mL), N,N-dimethylethylenediamine (1.26 mL) was added with stirring at room temperature, and the mixture was heated to 53°C and stirred for 13 hours. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was purified by aminopropyl silica gel column chromatography (eluent: 0%-100% ethyl acetate/hexane, gradient elution) to give the title compound (2.383 g).
1 H-NMR (CDCl
3 ) δ ppm: 0.92(3H, t, J=7.4Hz), 1.35-1.50(3H, m), 1.50-1.90(8H, m), 2.00-2.15(1H, m), 2.26(6H, s), 2.31(3H, s), 2.37(1H, t, J=11.2Hz), 2.46(2H, t, J=6.4Hz), 2.85-3.10(2H, m), 3.35-3.45(2H, m), 3.60-3.70(1H, m), 3.70-3.80(1H, m), 3.90-4.00(4H, m), 9.33(1H, br)[α]
D 28 =-6.62°(c=0.31, MeOH)
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////matsupexole, dopamine receptor agonist, Phase 2, Parkinson’s disease, K4UEG65HTX
Mangaciclanol
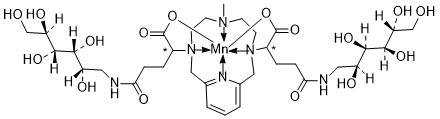
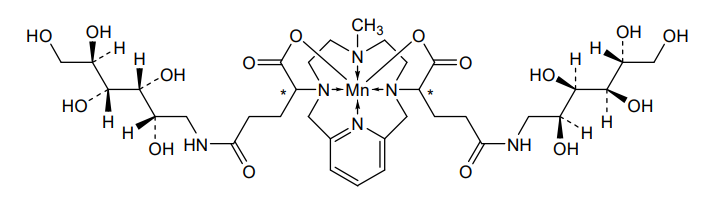
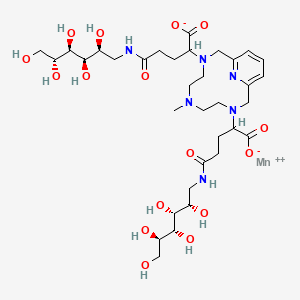
Mangaciclanol
Cas 2169771-05-3
MF C34H56MnN6O16 MW859.8 g/mol
- ANU6AE7NAP
- [[1,1′-[(6-Methyl-3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-1(15),11,13-triene-3,9-diyl-kappaN3,kappaN6,kappaN9,kappaN15)bis[[4-(carboxy-kappaO)-1-oxo-4,1-butanediyl]imino]]bis[1-deoxy-D-glucitolato]](2-)]manganese
2-[9-[1-carboxylato-4-oxo-4-[[(2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl]amino]butyl]-6-methyl-3,6,9,15-tetrazabicyclo[9.3.1]pentadeca-1(15),11,13-trien-3-yl]-5-oxo-5-[[(2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl]amino]pentanoate;manganese(2+)

diagnostic imaging agent, ANU6AE7NAP
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////mangaciclanol, diagnostic imaging agent, ANU6AE7NAP
Elinzanetant
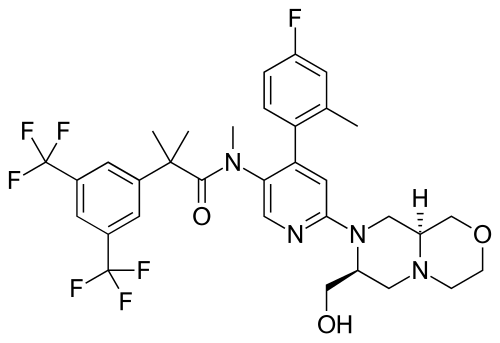
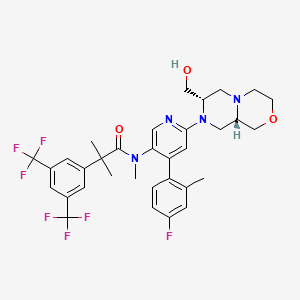
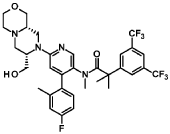
Elinzanetant
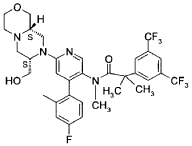
CAS 929046-33-3
MW 668.6 g/mol MF C33H35F7N4O3
N-[6-[(7S,9aS)-7-(hydroxymethyl)-3,4,6,7,9,9a-hexahydro-1H-pyrazino[2,1-c][1,4]oxazin-8-yl]-4-(4-fluoro-2-methylphenyl)-3-pyridinyl]-2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethylpropanamide
FDA 10/24/2025, Lynkuet, To treat moderate-to-severe vasomotor symptoms due to menopause
BAY-3427080; GSK-1144814; NT-814, UNII-NZW2BOW35N
Elinzanetant, sold under the brand name Lynkuet, is a medication used for the treatment of moderate to severe vasomotor symptoms due to menopause.[4] It is an neurokinin 1 and neurokinin 3 receptor antagonist.[4] It was developed by Bayer Healthcare.[4] It is taken by mouth.[4]
Elinzanetant is a non-hormonal, selective, neurokinin 1 (NK-1) and neurokinin 3 (NK-3) receptor antagonist.[5] By blocking NK-1 and NK-3 receptors signaling, elinzanetant is postulated to normalize neuronal activity involved in thermo- and sleep regulation in the hypothalamus.[5]
Elinzanetant is an orally bioavailable neurokinin/tachykinin 1 receptor (NK1-receptor; NK1R; NK-1R) and NK3 receptor (NK-3R; NK3R) antagonist, that may be used to treat vasomotor symptoms in menopausal woman. Upon oral administration, elinzanetant targets, competitively binds to and blocks the activity of the NK1R and NK3R in the central nervous system (CNS), thereby inhibiting the binding of the endogenous ligands and neuropeptides substance P (SP; neurokinin-1; NK1) and neurokinin B (NKB). This inhibits NK1R/NK3R-mediated signal transduction and may prevent certain menopausal symptoms such as hot flashes. Neurokinin-mediated signaling may increase during hormone deficiency and may cause hot flashes.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021094247&_cid=P20-MHLSZY-53200-1
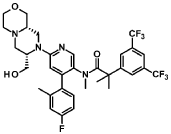
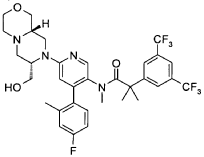
“Compound A” refers to 2-[3,5-Bis(trifluoromethyl)phenyl]-N-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hydroxymethyl)hexahydropyrazino[2,l-c][l,4]oxazin-8(lH)-yl]-3-pyridi-nyl}-N,2-dimethylpropanamide, and has the chemical structure depicted below.
(Compound A).
Example 8
2-[3.5-Bis(trifluoromethyl¾phenyl1-N-{4-(4-fluoro-2-methylphenyl¾-6-[(7S.9aS¾-7-(hvdroxymethyl¾hexa-hvdropyrazino[2,l-c1[l,41oxazin-8(lH)-yl1-3-pyridinyl}-N, 2-dimethyl propanamide as anhydrous crys talline form (Compound A)
Example 7 (2-[3,5-Bis(trifluoromethyl)phenyl]-N-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hy-droxymethyl)hexahydropyrazino[2,l-c][l,4]oxazin-8(lH)-yl]-3-pyridinyl}-N,2-dimethylpropanamide dihydrochloride salt mono-isopropanol solvate (Compound XII)) (3.4 kg), methyl-f-butyl ether (from now on, MTBE) (15.0 L/kg of Example 7) and NaOH 2.5N (4.9 L/kg of Example 7) were loaded, heated to 40QC and stirred for 10 to 30 min. The layers were settled for not less than 30 min at 40QC and the bottom aqueous layer discarded.
An aqueous solution of L-cysteine 9 wt% (5.0 L water per kg of Example 7+ 0.5 w/w L-cysteine per Ex ample 7) was added over the organic layer and stirred at 40QC for not less than 60 min. The layers were settled for not less than 30 min at 40QC and the bottom aqueous layer discarded.
Water (5.0 L/kg of Example 7) was added over the organic layer and stirred at 40QC for not less than 15 min. The layers were settled for not less than 60 min at 40QC and the bottom aqueous layer dis carded.
Water (5.0 L/kg of Example 7) was added over the organic layer and stirred at 40QC for not less than 15 min. The layers were settled for not less than 60 min at 40QC and the bottom aqueous layer dis carded.
The organic layer was concentrated at atmospheric pressure to 2.5 L/kg of Example 7. Iso-octane (8.3 L/kg of Example 7) was added at 50/55QC in not less than lh and the solution distilled under light vac uum to 4.0 L/kg of Example 7. A sample was taken for controlling the water and MTBE removal.
Isopropanol (0.8 L/kg of Example 7) was added and stirred at 65/75QC until total dissolution. The solu tion was cooled down to 45/55QC and filtered to remove any foreign matters. Iso-octane (4.5 L/kg of Example 7) was added and the batch heated to 70QC for not less than 30 min. The solution was cooled down to 50QC and seeded with a slurry of 2-[3,5-Bis(trifluoromethyl)phenyl]-N-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hydroxymethyl)hexahydropyrazino[2,l-c][l,4]oxazin-8(lH)-yl]-3-pyridi-nyl}-N,2-dimethylpropanamide(0.008% w/w of Example 7) in iso-octane (0.07 L/kg of Example 7) and isopropanol (0.01 L/kg of Example 7). The seeds were aged at 50QC for not less than 3h and additional iso-octane (4.2 L/kg of Example 7) was added in not less than 3h keeping the temperature at 50/55QC. The slurry was held at 50QC for not less than 8h, cooled down to 0QC in not less than 5h and aged for not less than 3h before proceeding with the centrifugation step.
The slurry was centrifuged and the cake washed with iso-octane (2 x 3.3 L/kg of Example 7).
The wet product was dried under vacuum at 50QC to obtain 2.34 kg of the title compound (yield = 82.7%). This product was sieved for delumping to obtain 2.26kg of the title compound with a 99.8% purity as a white powder.
NMR spectrometer: Varian Agilent Mercury Vx 400 (16 scans, sw 6400 Hz, 25 °C).
*H NMR (400 MHz, DMSO-ds): d 8.02 (s, 1 H), 7.85 (s, 1 H), 7.74 (bd, 2 H), 7.22-6.92 (m, 3 H), 6.61 (s, 1 H), 4.70 (m, 1 H), 4.21 (bd, 1 H), 4.09 (bd, 1 H), 3.75 (m, 3 H), 3.55 (td, 11.3 Hz, 2.2 Hz, 1 H), 3.40 (bd, 1 H), 3.15 (t, 10.5 Hz, 1 H), 3.02 (d, 11.3 Hz, 1 H), 2.63 (d, 11.3 Hz, 1 H), ca. 2.5 (bd, 2 H), 2.31-2.00 (m, 7 H), 1.58-1.10 (m, 6 H).
SYN
- WO2007028654
- https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007028654&_cid=P20-MHLT4M-58180-1
Example 34
2-[3,5-Bis(trifluoromethyl)phenyl]-yV-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hydroxymethyl)hexahydropyrazino[2,1 -c][1 ,4]oxazin-8(1 H)-yl]-3-pyridinyl}-A/,2-dimethylpropanamide (E34)
2-[3,5-bis(trifluoromethyl)phenyl]-Λ/-[6-[(7S,9aS)-7-({[(1 , 1 -dimethylethyl)(dimethyl)silyl]oxy}methyl)hexahydropyrazino[2,1-c][1 ,4]oxazin-8(1H)-yl]-4-(4-fluoro-2-methylphenyl)-3-pyridinyl]-Λ/,2-dimethylpropanamide (D24) (390 mg, 0.498 mmol) was dissolved 17 ml. of methanol. To this solution was added concentrated HCI (0.9 mL) at 00C, and stirring was continued at room temperature for 3h (complete conversion). The reaction mixture was loaded on a SCX cartridge and washed with MeOH. The product was eluted with 0.5 M methanolic ammonia. The product-containing fractions were evaporated, leaving the target compound as a white solid: 310 mg, 0.464 mmol, 93%.
UPLC/MS: m/z= 669 (M+1 ).
1H-NMR (DMSO-d6): δ (ppm) 8.07-7.97 (s, 1 H), 7.88-7.81 (s, 1 H), 7.79-7.69 (br. s, 2H), 7.19-7.11 (d, 1 H), 7.14-7.06 (br. s, 2H) 6.64-6.56 (s, 1 H), 4.75-4.65 (m, 1 H), 4.31-4.13 (br. S, 1 H), 4.15-4.01 (br. s, 1 H), 3.80-3.68 (m, 3H), 3.58-3.49 (t, 1 H); 3.43-3.34 (m, 1 H); 3.18-3.09 (t, 1 H); 3.04-2.98 (d, 1 H); 2.68-2.58 (d, 1 H); 2.51-2.45 (s, 3H); 2.20-2.13 (s, 3H); 2.29-2.00 (m, 4H); 1.54-1.39 (s, 3H); 1.39-1.28 (s, 3H).
SYN
Crystalline forms of a pyridine derivative
Publication Number: WO-2010015626-A1
Priority Date: 2008-08-05
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010015626&_cid=P20-MHLT79-60873-1
ntermediate 7
2-r3.5-bis(trifluoromethyl)phenvn-Λ/-f4-(4-fluoro-2-methylphenyl)-6-r(7S,9aS)-7-(hvdroxymethyl)hexahvdro-pyrazinoF2,1-c1f1 ,41oxazin-8(1/-/)-vn-3-pyridinyl}-Λ/.2-dimethylpropanamide
16.90 g of bis(trifluoromethyl)phenyl]-Λ/-[6-chloro-4-(4-fluoro-2-methylphenyl)-3-pyridinyl]- Λ/,2-dimethylpropanamide (WO 2005/002577) 4.58 g sodium tert-butoxide and 2.1O g Bis-(tri-terf-butylphposphine-palladium(O) catalyst was loaded into the vessel under nitrogen.
10.00 g intermediate 6 dissolved in 338 mL toluene was charged to afford a dark brown solution. The solution was heated to 800C and stirred for at least 16 h (thin suspension obtained).
The reaction mixture was cooled down to 20-25°C and 16.90 g celite was added to give a brown suspension. The suspension was filtered over 16.90 g celite and washed with 33.8 mL toluene. 338 mL sat. sodium bicarbonate solution was added and the biphasic system was stirred for 5 min. at 20-250C. After phase separation, the aqueous layer was extracted twice with 118 mL toluene. The combined organic layers were treated with 90 mL of a 10% aqueous cysteine solution and stirred for 1 h at 25°C. After phase separation the organic layer was treated again with 90 mL of a 10% cysteine-solution and stirred for a further 1 h at 25°C. After phase separation , the organic layer was washed with 85 mL half saturated sodium bicarbonate solution then solvent exchanged to dioxane. The dioxane solution was cooled down to 10-150C. 63.5 mL of 4M hydrogen chloride in dioxane was added at 10-150C over at least 10 min. The solution was warmed to 20-25°C and stirred for 2 h.
Dioxane was concentrated down to 85 mL at 45°C under reduced pressure. 85 mL water and 254 mL dichloromethane were added to the residue to give a thin suspension. The biphasic system was stirred for 5 min. at 20-250C. The layers were separated and the organic phase was washed with 33.8 mL saturated sodium bicarbonate solution at 20-25°C (pH adjusted to 7-8). The biphasic system was stirred for 5 min. at 20-250C and the organic layer separated and concentrated under reduced pressure at 500C to afford crude title compound as a pale brown solid. 8.00 g of the title compound (78.8% a/a HPLC) was dissolved in 16 mL ethyl acetate. The filter was loaded with 80 to 104 g silica gel and conditioned with ethyl acetate. The product solution was loaded on top of the column and chromatography was started using ethyl acetate as solvent. The product fractions were combined and partially concentrated at 45-50°C under reduced pressure. To the mixture was added 2.64 g to 4.00 g silicycle (Si-Thiol, 1.2 mmol/g) at rt and stirred for 2 h. Filtration over 8.00 g celite and washing with 32 mL ethyl acetate gave the filtrate which was concentrated to dryness at 45°C afford the title compound as a light brown solid. ( Weight yield 72%) 1H-NMR [ppm, CDCI3]: 8.04-7.91 , (m, 1 H); 7.77, (s, 1 H); 7.72-7.60, (m, 2H); 7.59-7.16, (m, 1H); 7.06-6.74, (m, 2H); 6.44, (s, 1 H); 4.64-4.43, (m, 1 H); 4.38-4.18, (m, 1 H); 4.07-3.96, (m, 2H); 3.95-3.76, (m, 3H); 3.76-3.61 , (m, 1 H); 3.37-3.27, (m, 1 H); 3.16-2.98, (m, 2H); 2.84-2.70, (m, 1 H); 2.67-2.51 , (m, 2H); 2.49-2.22, (m, 5H); 2.19-2.06, (m, 2H); 1.64-1.31 , (m, 5H), OH broad and not observed
LIT
- Elinzanetant: a phase III therapy for postmenopausal patients with vasomotor symptomsPublication Name: Expert Opinion on Investigational DrugsPublication Date: 2024-01-02PMID: 38224099DOI: 10.1080/13543784.2024.2305122
- Elinzanetant (NT-814), a Neurokinin 1,3 Receptor Antagonist, Reduces Estradiol and Progesterone in Healthy WomenPublication Name: The Journal of clinical endocrinology and metabolismPublication Date: 2021-02-24PMCID: PMC8277204PMID: 33624806DOI: 10.1210/clinem/dgab108
- Pleiotypic responses of regenerating liverPublication Name: Advances in Enzyme RegulationPublication Date: 1976PMID: 9791DOI: 10.1016/0065-2571(76)90023-6
PAT
Pyridine Derivatives and Their Use in the Treatment of Psychotic Disorders
Publication Number: US-2008269208-A1
Priority Date: 2005-06-06
- Pyridine Derivatives And Their Use In The Treatment Of Psychotic DisordersPublication Number: US-2011190276-A1Priority Date: 2005-09-09
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: US-7683056-B2Priority Date: 2005-09-09Grant Date: 2010-03-23
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: US-7919491-B2Priority Date: 2005-09-09Grant Date: 2011-04-05
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: US-8097618-B2Priority Date: 2005-09-09Grant Date: 2012-01-17
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: WO-2007028654-A1Priority Date: 2005-09-09
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Elinzanetant is indicated for the treatment of moderate to severe vasomotor symptoms associated with menopause.[4]
Society and culture
Legal status
In September 2025, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Lynkuet, intended for the treatment of moderate to severe vasomotor symptoms (hot flushes).[5] The applicant for this medicinal product is Bayer AG.[5]
Lynkuet was approved for medical use in the United States in October 2025.[6]
Names
Elinzanetant is the international nonproprietary name.[7]
Elinzanetant is sold under the brand name Lynkuet.[8]
References
- “Details for: Lynkuet”. Drug and Health Products Portal. 23 July 2025. Retrieved 28 September 2025.
- “Lynkuet product information”. Lynkuet. 23 July 2025. Retrieved 28 September 2025.
- “MHRA approves elinzanetant to treat moderate to severe vasomotor symptoms (hot flushes) caused by menopause”. Medicines and Healthcare products Regulatory Agency (Press release). 8 July 2025. Retrieved 28 September 2025.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219469s000lbl.pdf
- “Lynkuet EPAR”. European Medicines Agency (EMA). 19 September 2025. Retrieved 27 September 2025. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 24 October 2025. Retrieved 29 October 2025.
- World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 84”. WHO Drug Information. 34 (3). hdl:10665/340680.
- “Bayer’s Lynkuet (elinzanetant), the First and Only Neurokinin 1 and Neurokinin 3 Receptor Antagonist, Receives FDA Approval for Moderate to Severe Hot Flashes Due to Menopause” (Press release). Bayer. 24 October 2025. Retrieved 29 October 2025 – via Business Wire.
External links
- Clinical trial number NCT05042362 for “A Study to Learn More About How Well Elinzanetant Works and How Safe it is for the Treatment of Vasomotor Symptoms (Hot Flashes) That Are Caused by Hormonal Changes Over 26 Weeks in Women Who Have Been Through the Menopause (OASIS-1)” at ClinicalTrials.gov
- Clinical trial number NCT05099159 for “A Study to Learn More About How Well Elinzanetant Works and How Safe it is for the Treatment of Vasomotor Symptoms (Hot Flashes) That Are Caused by Hormonal Changes Over 26 Weeks in Women Who Have Been Through the Menopause (OASIS-2)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lynkuet |
| Other names | BAY-3427080; GSK-1144814; NT-814 |
| License data | US DailyMed: Elinzanetant |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | CA: ℞-only[1][2]UK: POM (Prescription only)[3]US: ℞-only[4] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 929046-33-3 |
| PubChem CID | 16063568 |
| ChemSpider | 17223178 |
| UNII | NZW2BOW35N |
| KEGG | D12123 |
| CompTox Dashboard (EPA) | DTXSID101337049 |
| Chemical and physical data | |
| Formula | C33H35F7N4O3 |
| Molar mass | 668.657 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////elinzanetant, Lynkuet, FDA 2025, APPROVALS 2025, BAY 3427080, GSK 1144814, NT 814
Lunresertib
Lunresertib
CAS 2719793-90-3
MF C18H20N4O2 MW 324.4 g/mol
(1P)-2-amino-1-(3-hydroxy-2,6-dimethylphenyl)-5,6-dimethyl1H-pyrrolo[2,3-b]pyridine-3-carboxamide
serine/ threonine kinase inhibitor, antineoplastic, N95U3A7N57, RP-6306, RP 6306
2-Amino-1-(3-hydroxy-2,6-dimethylphenyl)-5,6-dimethylpyrrolo[2,3-b]pyridine-3-carboxamide
Lunresertib is an investigational new drug that is being evaluated for the treatment of cancer. It is an oral small molecule inhibitor of PKMYT1, developed by Repare Therapeutics.[1] This drug targets cell cycle regulation in tumors with specific genetic alterations, including CCNE1 amplifications or FBXW7 and PPP2R1A loss of function mutations. It is currently in phase 1/2 clinical trials, both as monotherapy or in combination with camonsertib, an ATR inhibitor.[2]
Lunresertib is an orally bioavailable inhibitor of the human membrane-associated tyrosine– and threonine-specific cdc2-inhibitory kinase (PKMYT1), with potential antineoplastic activity. Upon oral administration, lunresertib targets, binds to and inhibits the activity of PKMYT1. This results in the inhibition of CDK1 phosphorylation, which may promote both premature mitosis and a prolonged mitotic arrest, and lead to the accumulation of unrepaired DNA damage and apoptosis in susceptible tumor cells, such as CCNE1-overexpressing tumor cells. PKMYT1 phosphorylates CDK1 specifically when CDK1 is complexed to cyclins, which blocks progression from G2 into mitosis.NCI Thesaurus (NCIt)
- Study of RP-6306 With FOLFIRI in Advanced Solid TumorsCTID: NCT05147350Phase: Phase 1Status: TerminatedDate: 2025-08-20
- Study of RP-6306 Alone or in Combination With RP-3500 or Debio 0123 in Patients With Advanced Solid TumorsCTID: NCT04855656Phase: Phase 1Status: RecruitingDate: 2025-08-06
- RP-6306 in Patients With Advanced CancerCTID: NCT05605509Phase: Phase 2Status: Active, not recruitingDate: 2025-07-14
- Study of RP-6306 With Gemcitabine in Advanced Solid TumorsCTID: NCT05147272Phase: Phase 1Status: TerminatedDate: 2025-06-17
- Liquid-biopsy Informed Platform Trial to Evaluate CDK4/6-inhibitor Resistant ER+/HER2- Metastatic Breast CancerCTID: NCT05601440Phase: Phase 2Status: RecruitingDate: 2025-01-14
- Phase 1 Study of RP-6306 With Carboplatin and Paclitaxel in TP53 Ovarian and Uterine Cancer
- CTID: NCT06107868
- Phase: Phase 1
- Status: Active, not recruiting
- Date: 2024-03-22
PAT
- Compounds, Pharmaceutical Compositions, and Methods of Preparing and Using CompoundsPublication Number: JP-2023521633-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: US-2023151014-A1Priority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: US-2023158022-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: EP-4126879-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: IL-296934-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of making the compounds and methods of using themPublication Number: KR-20230011279-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions and methods of making compounds and methods of their usePublication Number: CN-115916783-APriority Date: 2020-04-01
- Methods of using MYT1 inhibitorsPublication Number: JP-2023519430-APriority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: WO-2021195782-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: AU-2021250744-A1Priority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: CA-3173955-A1Priority Date: 2020-04-01
- Methods of using MYT1 inhibitorsPublication Number: CN-115811976-APriority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: EP-4125907-A1Priority Date: 2020-04-01
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021195781&_cid=P20-MHLE6P-37080-1
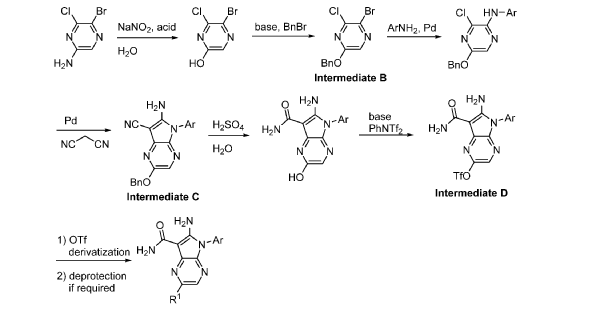
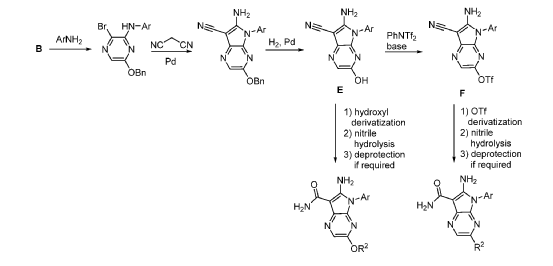
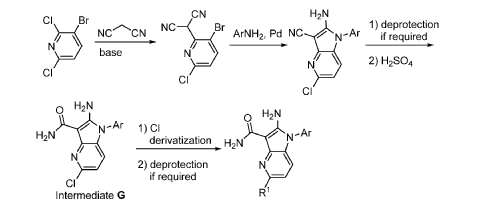
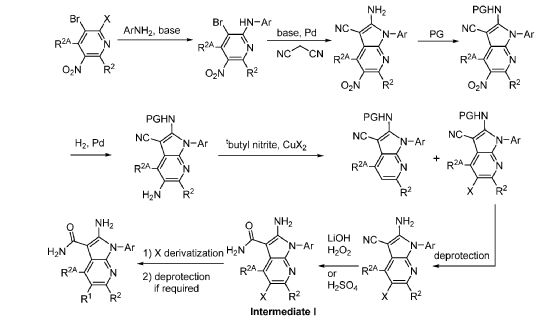
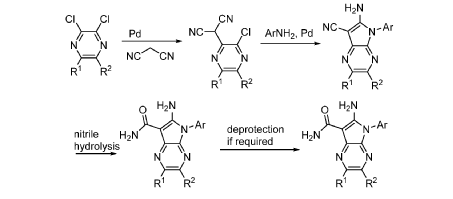
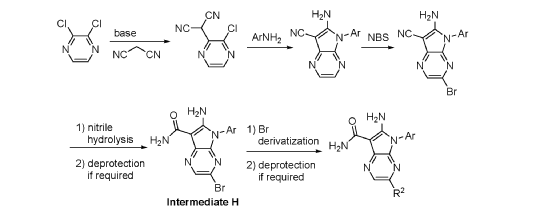
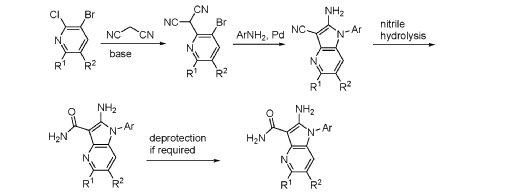
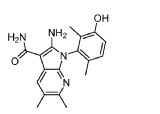
Step 9. To a suspension of 2-amino-1-(3-methoxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (2.22 g, 6.56 mmol, 77% purity) in DCM (25 mL) was added tribromoborane in DCM (1 M, 26 mmol, 26 mL) dropwise. The reaction mixture was stirred at RT for 45 min, then concentrated to dryness. The crude product was taken in DCM and placed in an ice bath and MeOH was added carefully (exotherm). The mixture was concentrated to dryness then co-evaporated twice with MeOH. The residue was triturated with saturated aqueous NaHCO3. The solids were collected by filtration on a Buchner funnel, washed with H2O and air-dried. The still wet solid was dissolved in DCM/MeOH, concentrated to dryness and triturated in 20% MeOH/DCM (50 mL). The solid was collected by filtration, washed with 20% MeOH/DCM, air-dried then dried in vacuo to afford 2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (1.60g, 75% yield) as a light beige solid. MS: [M+1]: 325.1. A different batch was purified by preparative HPLC to yield 2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (63% yield) as an off-white fluffy solid.
1H NMR (400 MHz, DMSO-d6) δ 9.51 (s, 1H), 7.82 (s, 1H), 7.05 (d, J = 8.3 Hz, 1H), 6.90 (d, J =
8.2 Hz, 1H), 6.71 (br s, 2H), 6.64 (br s, 2H), 2.26 (s, 3H), 2.23 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
Chiral SFC separation of Compound 181 (1.60g, 4.93 mmol) (Instrument: Waters Prep 100 SFC-MS; Column: Phenomenex Lux Cellulose-2, 30 x 250 mm, 5 μm; Conditions: isocratic at 55% IPA + 10mM Ammonium Formate with 45% CO2 ; Flow Rate: 70 mL/min) provided
Compound 182 and Compound 183.
Compound 182 from SFC separation of 181. Peak 1 (retention time 3.94 min, 99.86%): (S)-2- amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (381 mg) was obtained as an off white fluffy solid. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 7.83 (s, 1 H), 7.05 (d, J = 8.3 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H), 6.72 (s, 2H), 6.65 (s, 2H), 2.26 (s, 3H), 2.24 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
Compound 183 from SFC separation of 181. Peak 2 (retention time 4.35 min, 98.09%): (R)-2- amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (495 mg) was obtained as an off white fluffy solid. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 7.83 (s, 1 H), 7.05 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.72 (s, 2H), 6.66 (s, 2H), 2.26 (s, 3H), 2.24 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
SYN
https://pubs.acs.org/doi/full/10.1021/acs.oprd.4c00493
REF
- The Science and Art of Structure-Based Virtual ScreeningPublication Name: ACS Medicinal Chemistry LettersPublication Date: 2024-03-25PMCID: PMC11017385PMID: 38628791DOI: 10.1021/acsmedchemlett.4c00093
- Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306Publication Name: Journal of Medicinal ChemistryPublication Date: 2022-07-26PMCID: PMC9837800PMID: 35880755DOI: 10.1021/acs.jmedchem.2c00552
- CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibitionPublication Name: NaturePublication Date: 2022-04-20PMCID: PMC9046089PMID: 35444283DOI: 10.1038/s41586-022-04638-9
- Contributions in the domain of cancer research: Review¶Negative regulators of cyclin-dependent kinases and their roles in cancersPublication Name: Cellular and molecular life sciences : CMLSPublication Date: 2001-11PMCID: PMC11337304PMID: 11766887DOI: 10.1007/pl00000826
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | RP-6306 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2719793-90-3 |
| PubChem CID | 156869388 |
| ChemSpider | 115008046 |
| UNII | N95U3A7N57 |
| KEGG | D12736 |
| ChEMBL | ChEMBL5199076 |
| Chemical and physical data | |
| Formula | C18H20N4O2 |
| Molar mass | 324.384 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Szychowski J, Papp R, Dietrich E, Liu B, Vallée F, Leclaire ME, et al. (August 2022). “Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306”. Journal of Medicinal Chemistry. 65 (15): 10251–10284. doi:10.1021/acs.jmedchem.2c00552. PMC 9837800. PMID 35880755.
- Previtali V, Bagnolini G, Ciamarone A, Ferrandi G, Rinaldi F, Myers SH, et al. (July 2024). “New Horizons of Synthetic Lethality in Cancer: Current Development and Future Perspectives”. Journal of Medicinal Chemistry. 67 (14): 11488–11521. doi:10.1021/acs.jmedchem.4c00113. PMC 11284803. PMID 38955347.
///////lunresertib, Serine/ threonine kinase inhibitor, antineoplastic, N95U3A7N57, RP-6306, RP 6306
Lunbotinib
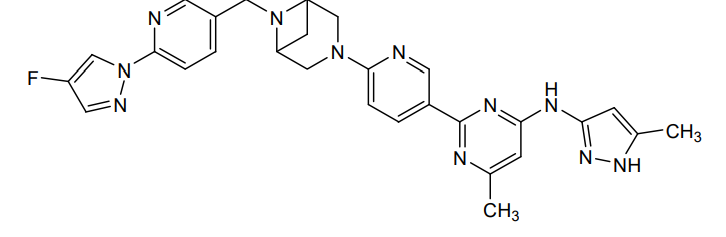
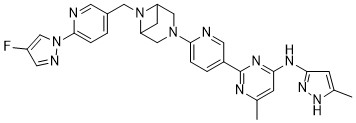
Lunbotinib
CAS 2479961-46-9
MF C28H28FN11 MW537.6 g/mol
2-[6-(6-{[6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl]methyl}-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl]-6-methyl-N-(5-methyl1H-pyrazol-3-yl)pyrimidin-4-amine
tyrosine kinase inhibitor, antineoplastic, KL3T9ZU6HQ
- 2-(6-(6-((6-(4-fluoropyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo(3.1.1)heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine
- 2-[6-[6-[[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine
Lunbotinib is an orally bioavailable selective inhibitor of the proto-oncogene receptor tyrosine kinase rearranged during transfection (RET), with potential antineoplastic activity. Upon oral administration, lunbotinib selectively binds to various RET fusions and mutations, including solvent front resistance mutations, and inhibits the activity of RET. This results in an inhibition of cell growth of tumors that exhibit increased RET activity due to these fusions and mutations. RET overexpression, activating mutations, and fusions result in the upregulation and/or overactivation of RET tyrosine kinase activity in various cancer cell types. Dysregulated RET activity plays a key role in the development and progression of certain cancers. Lunbotinib is able to penetrate the blood-brain barrier (BBB) and may also be able to overcome resistance mechanisms to first generation selective RET inhibitors (SRIs).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020168939&_cid=P12-MHKH7H-14851-1
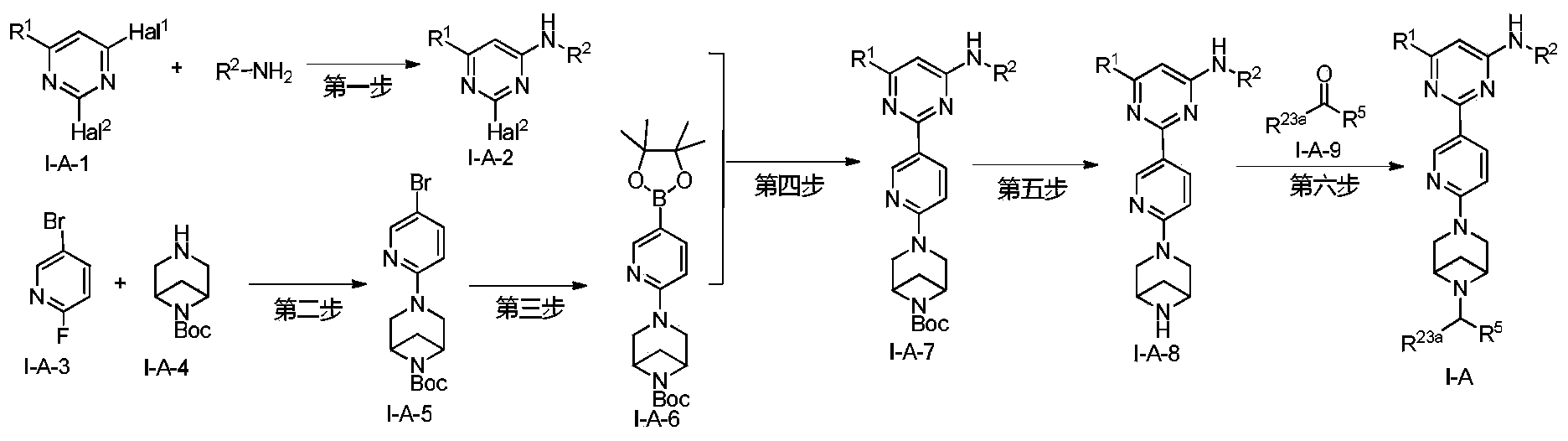
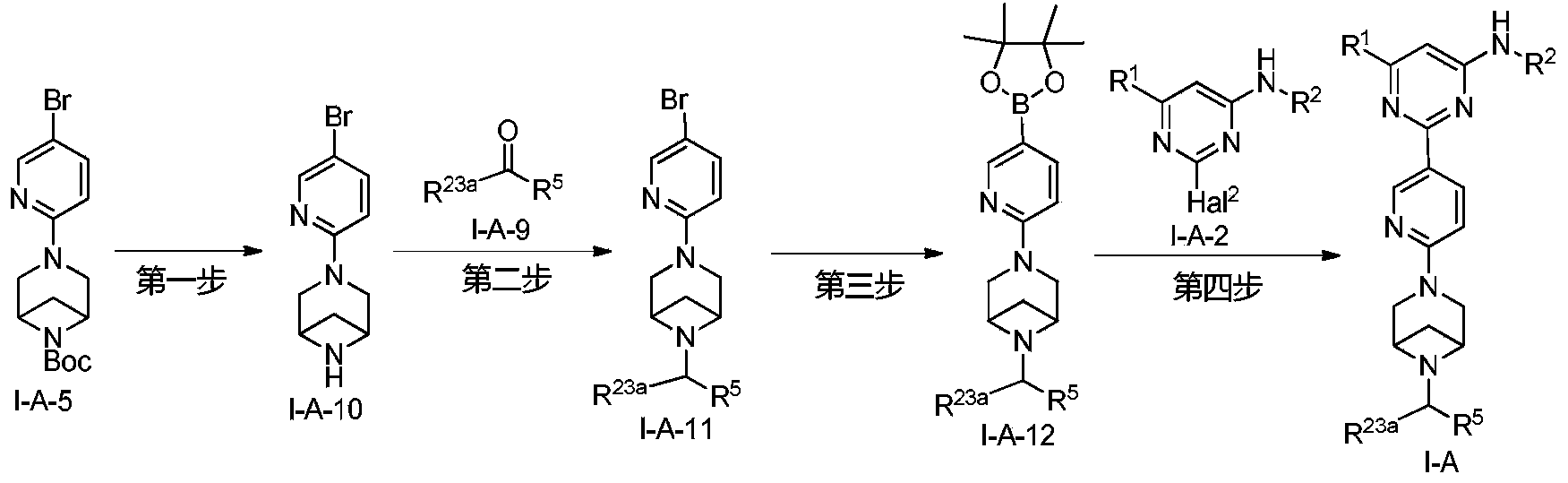
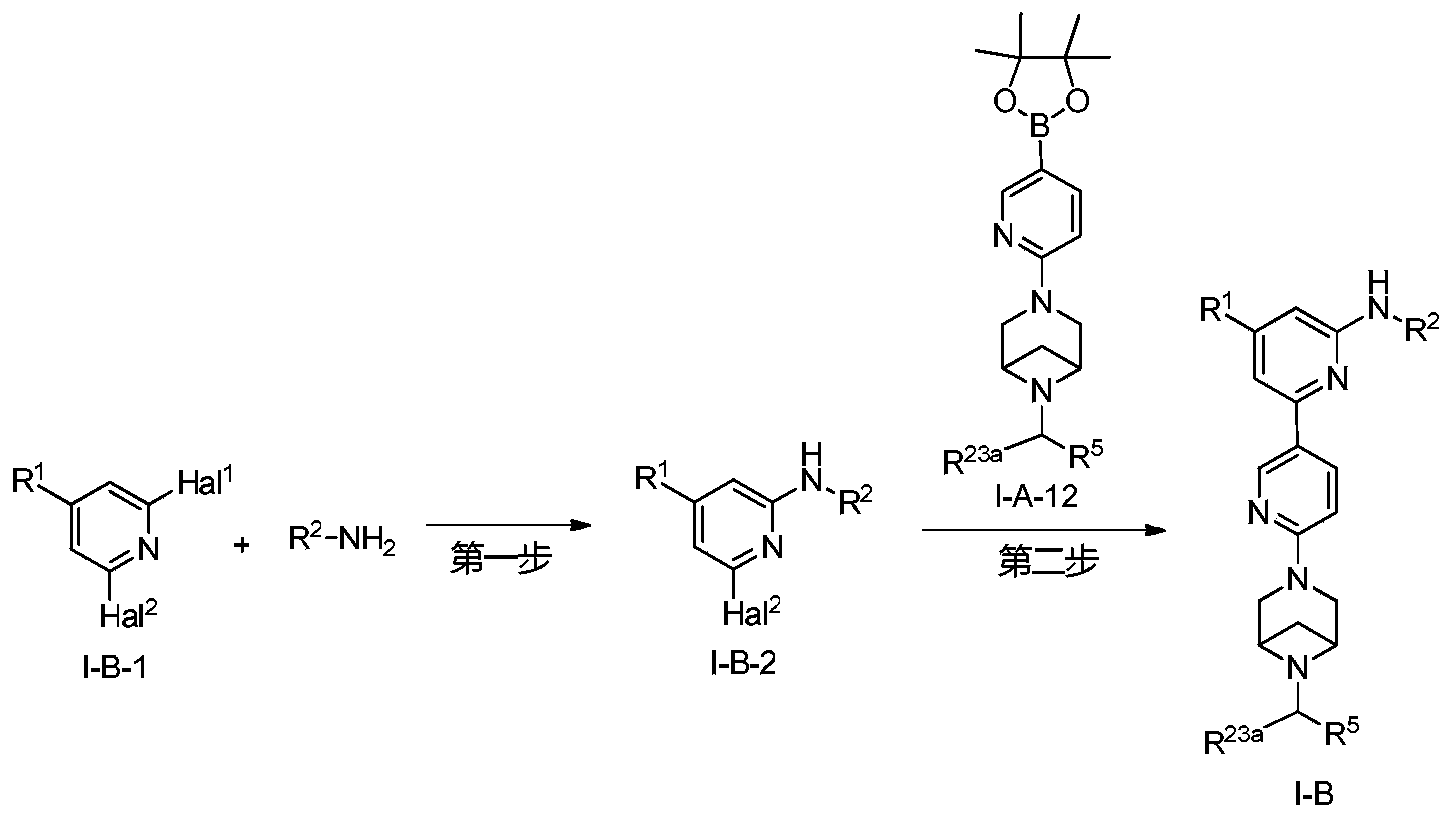
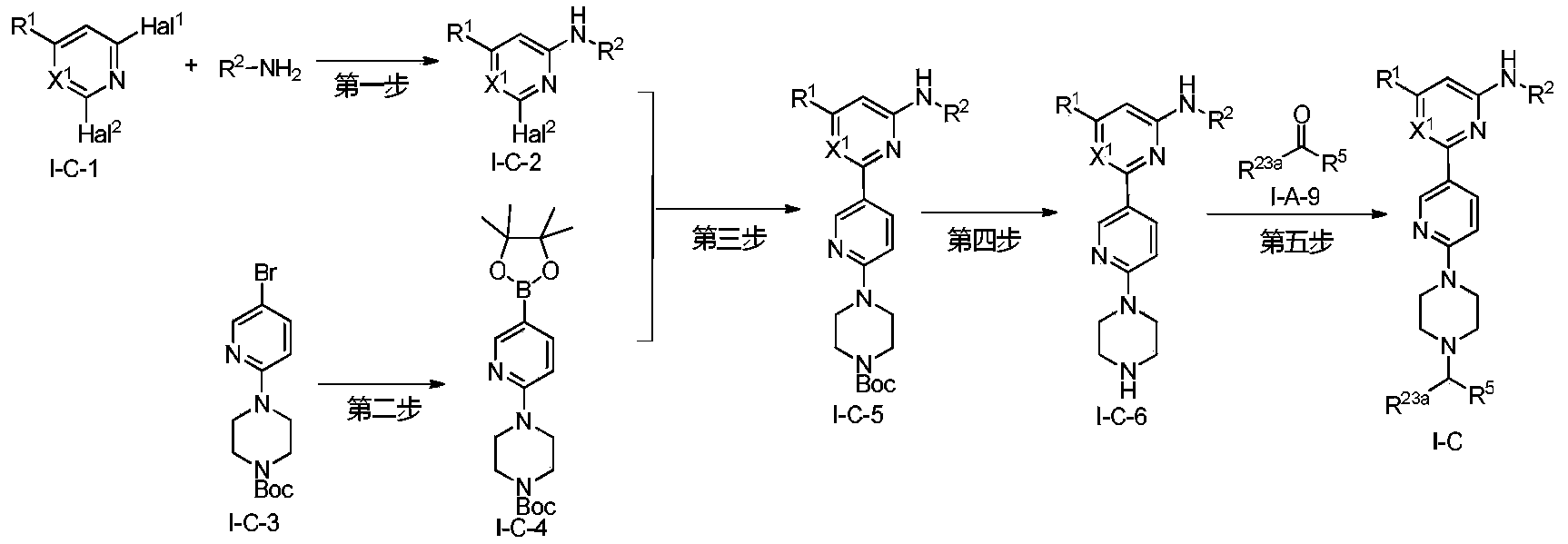
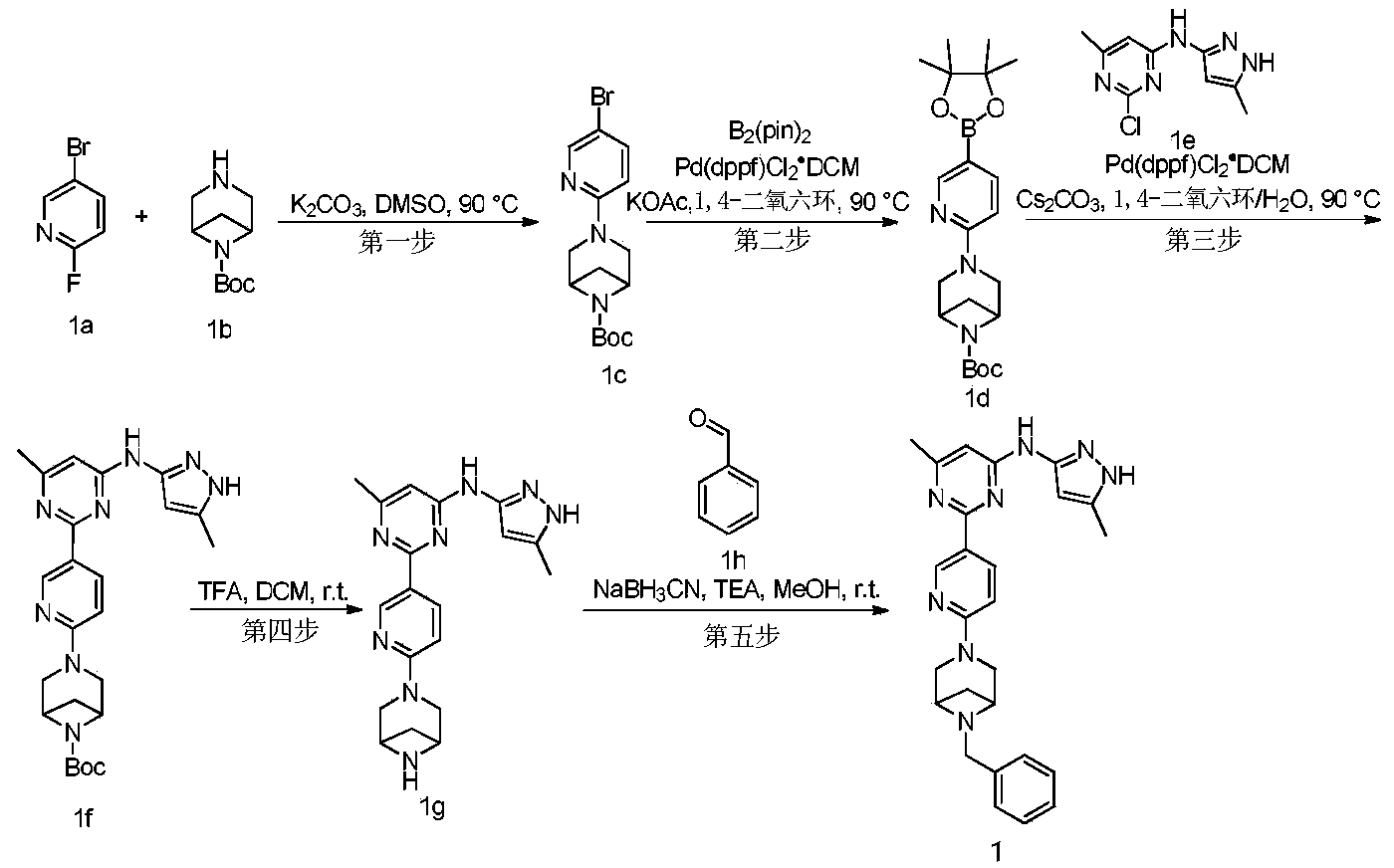
Example 6: 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 17)
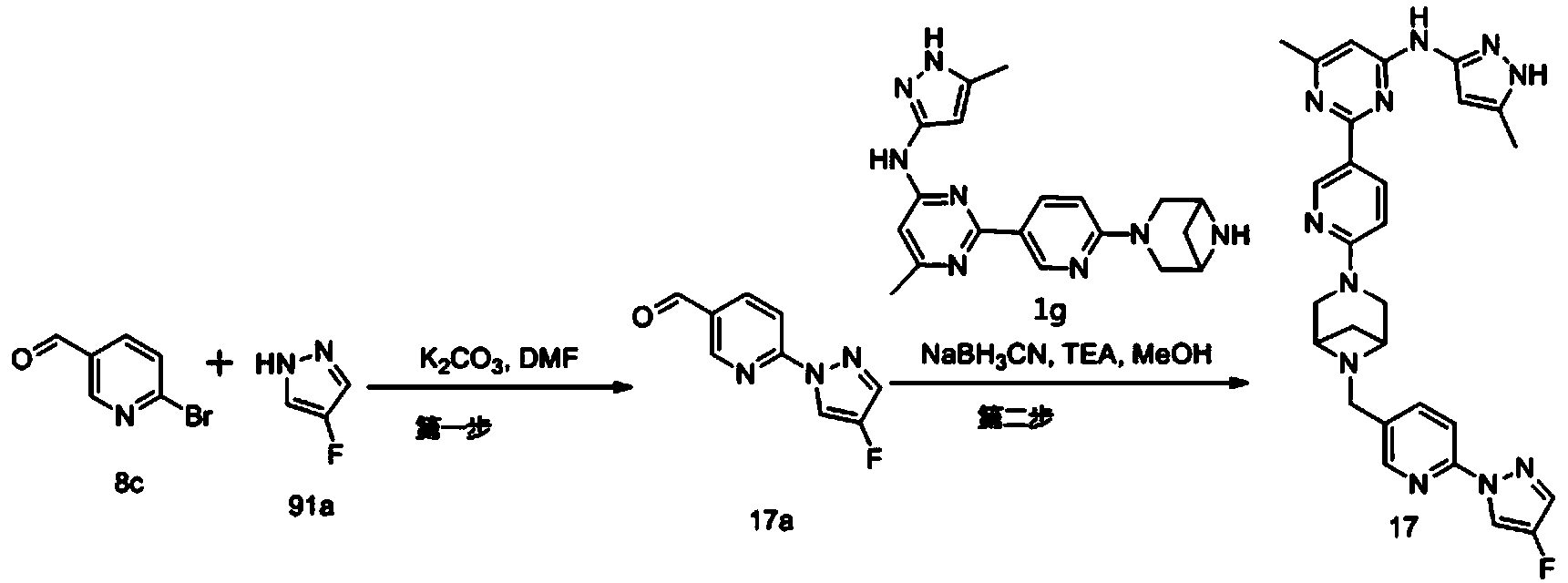
Step 1: Preparation of 6-(4-fluoro-1H-pyrazol-1-yl)nicotinaldehyde (compound 17a)
[0396]Compound 8c (2.0 g), 91a hydrochloride (1.58 g), and potassium carbonate (4.45 g) were sequentially added to DMF (15 mL), and the mixture was heated to 80 °C and stirred for 14 h. The reaction mixture was cooled to room temperature, diluted with water (100 mL), and extracted with DCM (50 mL x 2). The organic phases were combined, washed with water and saturated brine, dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography (PE:EA = 10:1) to give compound 17a (0.81 g). MS m/z (ESI): 192.1 [M+H]
[0397]Step 2: Preparation of 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 17)
[0398]1 g of trifluoroacetate (22.82 mg) and compound 17a (27.47 mg) were added to methanol (1.0 mL), followed by the sequential addition of triethylamine (4.45 mg) and sodium cyanoborohydride (13.86 mg), and the reaction was carried out at room temperature for 14 h. After the reaction was completed, the reaction solution was concentrated to dryness under reduced pressure and purified by Prep-HPLC to obtain compound 17 (7.0 mg). MS m/z (ESI): 538.3 [M+H]
[0399]
1H NMR(400MHz,DMSO-d 6)δ11.98(s,1H),9.66(s,1H),9.12(d,J=2.16Hz,1H),8.67(dd,J=4.54,0.64Hz,1H),8.43(dd,J=8.94,2.28Hz,1H),8.41(d,J=1.68,1H),7.98(dd,J=8.48Hz,2.12 1H),7.92(d,J=4.28,1H),7.87(d,J=8.4,1H),6.78(d,J=9.0Hz,2H),6.31(br,1H),3.78-3.71(m,4H),3.68-3.52(m,4H),2.59-2.52(m,1H),2.33(s,3H),2.25(s,3H),1.60(d,J=8.36Hz,1H).
PAT
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: US-2022144847-A1Priority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-113316578-BPriority Date: 2019-02-19Grant Date: 2023-10-31
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-117263945-APriority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-117327078-APriority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing same, methods for their preparation and usePublication Number: JP-7615056-B2Priority Date: 2019-02-19Grant Date: 2025-01-16
- Salt and crystal form of pyrimidine compound, and preparation methods thereforPublication Number: US-2023295174-A1Priority Date: 2020-07-28
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: WO-2020168939-A1Priority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same, and preparation methods and uses thereofPublication Number: CN-113316578-APriority Date: 2019-02-19
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: EP-3929198-A1Priority Date: 2019-02-19
- Heterocyclic compounds, drug compositions containing them, methods of their manufacture and usePublication Number: JP-2022521859-APriority Date: 2019-02-19
- Use of heterocyclic compound for treating diseases related to ret genetic change and method thereforPublication Number: WO-2024240017-A1Priority Date: 2023-05-19
- Uses and methods of heterocyclic compounds for treating diseases associated with kinase resistance mutationsPublication Number: CN-116801882-APriority Date: 2021-03-24
- Use of heterocyclic compound in treating diseases related to kinase drug-resistant mutation and method thereforPublication Number: EP-4316490-A1Priority Date: 2021-03-24
- Salt and crystal form of pyrimidine compound, and preparation methods thereforPublication Number: EP-4190781-A1Priority Date: 2020-07-28
- Salts, crystal forms of pyrimidine compounds and methods for their preparationPublication Number: JP-2023535361-APriority Date: 2020-07-28
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Lunbotinib, tyrosine kinase inhibitor, antineoplastic, KL3T9ZU6HQ
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}