
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Ribociclib, рибоциклиб , ريبوسيكليب , 瑞波西利
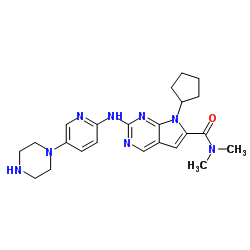
Ribociclib
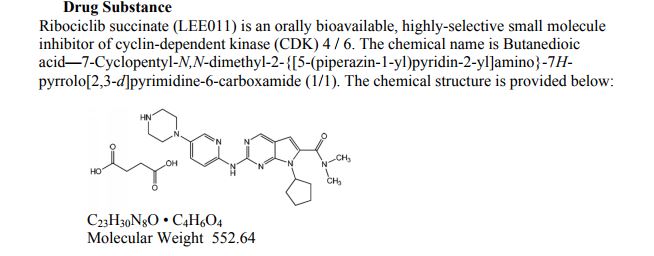
Ribociclib (LEE 011)
CAS: 1211441-98-3
Chemical Formula: C23H30N8O
Exact Mass: 434.25426
7-Cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide
FDA UNII
-
TK8ERE8P56
Current developer: Novartis /Astex Pharmaceuticals.
Novartis Ag, Astex Therapeutics Ltd.
NMR.http://file.selleckchem.com/downloads/nmr/S744002-LEE011-2-HNMR-Selleck%20.pdf
http://file.selleckchem.com/downloads/hplc/S744002-LEE011-2-HPLC-Selleck.pdf
Ribociclib (LEE011) is an orally available, and highly specific CDK4/6 inhibitor. Phase 3.
CDK4 AND 6
(Cell-free assay)Product Ingredients
NOW FDA APPROVED 2017 since the blog post was written
| Kisqali | FDA 3/13/2017 | To treat postmenopausal women with a type of advanced breast cancer Drug Trials Snapshot |

| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Ribociclib hydrochloride | 63YF7YKW7E | 1211443-80-9 | JZRSIQPIKASMEV-UHFFFAOYSA-N |
| Ribociclib succinate | BG7HLX2919 | 1374639-75-4 | NHANOMFABJQAAH-UHFFFAOYSA-N |
RIBOCICLIB SUCCINATE
STRUCTURE ….LINK
Ribociclib is in phase III clinical trials by Novatis for the treatment of postmenopausal women with advanced breast cancer.
Phase II clinical trials are also in development for the treatment of liposarcoma, ovarian cancer, fallopian tube cancer, peritoneum cancer, endometrial cancer, and gastrointestinal cancer.
Ribociclib, also known as LEE011, is an orally available cyclin-dependent kinase (CDK) inhibitor targeting cyclin D1/CDK4 and cyclin D3/CDK6 cell cycle pathway, with potential antineoplastic activity. CDK4/6 inhibitor LEE011 specifically inhibits CDK4 and 6, thereby inhibiting retinoblastoma (Rb) protein phosphorylation. Inhibition of Rb phosphorylation prevents CDK-mediated G1-S phase transition, thereby arresting the cell cycle in the G1 phase, suppressing DNA synthesis and inhibiting cancer cell growth. Overexpression of CDK4/6, as seen in certain types of cancer, causes cell cycle deregulation
Orally bioavailable CDK4/6-selective inhibitor that has been tested in Phase III clinical trials for treatment of advanced breast cancer.
CDK full name of cyclin-dependent kinases, there are many other subtypes CDK1-11, capable of binding to cell cycle proteins regulate the cell cycle. Pfizer Palbociclib been submitted for FDA review under phase II clinical data, Novartis Ribociclib (LEE011), Lilly Abemaciclib (LY2835219) the three CDK4 / 6 inhibitors have entered late stage development for the treatment of breast cancer
SYNTHESIS

WO2010020675
US20120115878
WO2010020675
http://www.google.co.in/patents/WO2010020675A1?cl=en
Example 74
7-Cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide

Following Buchwald Method B, then General Procedure A, 2-chloro-7-cyclopentyl-7H- pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide (300 mg, 1.02 mmol) and 5-piperazin-1- yl-pyridin-2-ylamine (314 mg, 1.13 mmol) gave 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2- ylamino)-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide (142 mg, 36%). MS(ESI) m/z 435.3 (M+H)+
POSTER

SYNTHESIS
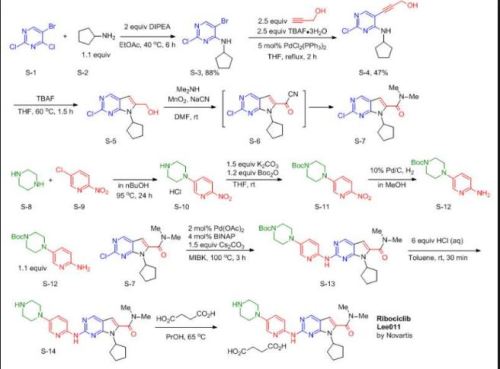


TAKEN FROM ….http://www.joygooo.com/news_71.htm?pageNum=21
PCT Int Appl, WO2012061156.
US Pat Appl Publ, US20120115878
PCT Int Appl, WO2011130232 5) Brain, Christopher Thomas et al; Preparation of pyrrolopyrimidine Derivatives for Use as CDK4 / 6 inhibitors;. PCT Int Appl, WO2011101409.
PCT Int Appl, WO2011101417. 7) Besong, Gilbert et al;.
PCT Int Appl, WO2010020675.
PCT Int Appl, WO2007140222.

Reference:1. WO2012064805A1 / US20120115878A1.
2. WO2010020675A1 / US8415355B2.
3. WO2011130232A1 / US20130035336A1.
Clinical Trial Information( data from http://clinicaltrials.gov, updated on 2015-10-17)
| NCT Number | Recruitment | Conditions | Sponsor /Collaborators |
Start Date | Phases |
|---|---|---|---|---|---|
| NCT02571829 | Not yet recruiting | Liposarcoma|Soft Tissue Sarcoma | Hadassah Medical Organization | December 2015 | Phase 2 |
| NCT02524119 | Not yet recruiting | Hepatocellular Carcinoma | University of Texas Southwestern Medical Center|Novartis …more | November 2015 | Phase 2 |
| NCT02494921 | Recruiting | Prostate Cancer | Rahul Aggarwal|University of California, San Francisco | September 2015 | Phase 1|Phase 2 |
| NCT02420691 | Recruiting | Gastrointestinal Cancer | M.D. Anderson Cancer Center|Novartis | August 2015 | Phase 2 |
| NCT02431481 | Not yet recruiting | Normal Renal Function|Impaired Renal Function | Novartis Pharmaceuticals|Novartis | August 2015 | Phase 1 |
Protocols from literature
|
In vitro protocol:: |
Pharmacologic growth inhibition: Clin Cancer Res. 2013 Nov 15;19(22):6173-82. Cell-cycle analysis: Clin Cancer Res. 2013 Nov 15;19(22):6173-82. Senescence and apoptosis assays: Clin Cancer Res. 2013 Nov 15;19(22):6173-82. |
|
In vivo protocol: |
Xenograft therapeutic trials: Clin Cancer Res. 2013 Nov 15;19(22):6173-82 Immunohistochemistry of xenografted neuroblastomas.Clin Cancer Res. 2013 Nov 15;19(22):6173-82 |
Ribociclib (LEE011) is a Me-Too version of palbociclib. Their structures are compared side-by-side as the following:
 |
Ribociclib (LEE011) is currently being developed by Novartis and Astex. According its Novartis’s website, LEE011 is a novel, orally available, selective inhibitor of CDK4/6 kinases, which induces complete dephosphorylation of Rb and G1 arrest in cancer cells. In preclinical in vitro and in vivo tumor models, LEE011 has been shown active in cancers harboring aberrations that increase CDK4/6 activity, including those directly linked to the kinases as well as activating alterations in the upstream regulators. First-in-human study of LEE011 in patients with solid tumors and lymphoma is currently ongoing. (source: http://www.novartisoncology.us/research/pipeline/lee011.jsp).
Treatment with LEE011 significantly reduced proliferation in 12 of 17 human neuroblastoma-derived cell lines by inducing cytostasis at nanomolar concentrations (mean IC50 = 307 ± 68 nmol/L in sensitive lines). LEE011 caused cell-cycle arrest and cellular senescence that was attributed to dose-dependent decreases in phosphorylated RB and FOXM1, respectively. In addition, responsiveness of neuroblastoma xenografts to LEE011 translated to the in vivo setting in that there was a direct correlation of in vitro IC50 values with degree of subcutaneous xenograft growth delay. Although our data indicate that neuroblastomas sensitive to LEE011 were more likely to contain genomic amplification of MYCN (P = 0.01), the identification of additional clinically accessible biomarkers is of high importance. LEE011 is active in a large subset of neuroblastoma cell line and xenograft models, and supports the clinical development of this CDK4/6 inhibitor as a therapy for patients with this disease. (Clin Cancer Res. 2013 Nov 15;19(22):6173-82)
|
References |
1. Rader J, Russell MR, Hart LS, Nakazawa MS, Belcastro LT, Martinez D, Li Y, Carpenter EL, Attiyeh EF, Diskin SJ, Kim S, Parasuraman S, Caponigro G, Schnepp RW, Wood AC, Pawel B, Cole KA, Maris JM. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013 Nov 15;19(22):6173-82. doi: 10.1158/1078-0432.CCR-13-1675. Epub 2013 Sep 17. PubMed PMID: 24045179; PubMed Central PMCID: PMC3844928.
2. Caponigro, Giordano; Stuart, Darrin; Kim, Sunkyu; Loo, Alice; Delach, Scott. Pharmaceutical combinations of a CDK4/6 inhibitor and a B-RAF inhibitor for treatment of proliferative diseases such as cancer. PCT Int. Appl. (2014), WO 2014018725 A1 20140130.
3. Kim, Sunkyu; Doshi, Shivang; Haas, Kristy; Kovats, Steven; Huang, Alan Xizhong; Chen, Yan. Combination therapy comprising a cyclin dependent kinase 4/6 (CDK4/6) inhibitor and a phosphatidylinositol 3-kinase (PI3K) inhibitor for use in the treatment of cancer. PCT Int. Appl. (2013), WO 2013006532 A1 20130110
4. Kim, Sunkyu; Doshi, Shivang; Haas, Kristy; Kovats, Steven. Combination of cyclin dependent kinase 4/6 (CDK4/6) inhibitor and fibroblast growth factor receptor (FGFR) kinase inhibitor for the treatment of cancer. PCT Int. Appl. (2013), WO 2013006368 A1 20130110
5. Calienni, John Vincent; Chen, Guang-Pei; Gong, Baoqing; Kapa, Prasad Koteswara; Saxena, Vishal. Salt(s) of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide and processes of making thereof. U.S. Pat. Appl. Publ. (2012), US 20120115878 A1 20120510.
6. Borland, Maria; Brain, Christopher Thomas; Doshi, Shivang; Kim, Sunkyu; Ma, Jianguo; Murtie, Josh; Zhang, Hong. Combination comprising a cyclin dependent kinase 4 or cyclin dependent kinase (cdk4/6) inhibitor and an Mtor inhibitor for treating cancer. PCT Int. Appl. (2011), WO 2011130232 A1 20111020
7. Besong, Gilbert; Brain, Christopher Thomas; Brooks, Clinton A.; Congreve, Miles Stuart; Dagostin, Claudio; He, Guo; Hou, Ying; Howard, Steven; Li, Yue; Lu, Yipin; et al. Preparation of pyrrolopyrimidine compounds as CDK inhibitors. PCT Int. Appl. (2010), WO 2010020675 A1 20100225.
CLIP
Cyclin-dependent kinase inhibitors (14 compounds) under clinical evaluation.
LEE-011 is one of the most selective inhibitors for CDK4 and CDK6 [59] and is being developed by Astex Pharmaceuticals™ and Novartis. In January 2014 this inhibitor entered phase III clinical trials for the treatment of breast cancer [60]. Due to encouraging results LEE-011 has now become the main competing drug-candidate with Pfizer’s PD0332991 (palbociclib), see Figure 3 [59].

Upon comparison of the chemical structure of Novartis’ LEE-011 and Pfizer’s PD0332991, the similarity is evident. The major difference lies in the bicyclic core since LEE-011 possesses a pyrrolo-pyrimidine and PD0332991 a pyridopyrimidine. The “east” part of the structure is also modified. The structural similarities make their analogous CDKs inhibition profiles (high selectivity for CDK4 and CDK6) quite obvious Moreover, both derivatives are orally administered which is pretty advantageous compared with dinaciclib, which is also in phase III clinical trials but is administered intravenously.
http://www.mdpi.com/1420-3049/19/9/14366/htm
- Kurt, S. LEE011 CDK Inhibitor Showing Early Promise in Drug-Resistant Cancers. Oncol. Times 2014, 36, 39–40. [Google Scholar]
- Macmillan Publishers Limited. CDK inhibitors speed ahead. Nat. Rev. Drug Discov. 2014, 13, 323. [Google Scholar] [CrossRef]
Sources:
1)Rader, JulieAnn et al.;Dual CDK4/CDK6 Inhibition Induces Cell-Cycle Arrest and Senescence in Neuroblastoma;Clinical Cancer Research (2013), 19(22), 6173-6182
2)Tavares, Francis X. and Strum, Jay C.;Preparation of pyrazinopyrrolopyrimidine derivatives and analogs for use as CDK inhibitors;PCT Int. Appl., WO2012061156
3)Calienni, John Vincent et al.;Salt(s) of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide and processes of making thereof;U.S. Pat. Appl. Publ., US20120115878
4)Borland, Maria et al;Combination comprising a cyclin dependent kinase 4 or cyclin dependent kinase (cdk4/6) inhibitor and an Mtor inhibitor for treating cancer;PCT Int. Appl., WO2011130232
5)Brain, Christopher Thomas et al;Preparation of pyrrolopyrimidine derivatives for use as CDK4/6 inhibitors;PCT Int. Appl., WO2011101409
6)Brain, Christopher Thomas and Perez, Lawrence Blas; Preparation of deuterated pyrrolopyrimidine compounds as inhibitors of CDK4/6 for treating cancer; PCT Int. Appl., WO2011101417
7)Besong, Gilbert et al.;Preparation of pyrrolopyrimidine compounds as CDK inhibitors;PCT Int. Appl., WO2010020675
8)Brain, Christopher Thomas et al.;Preparation of pyrrolopyrimidine compounds as protein kinase inhibitors; PCT Int. Appl., WO2007140222
9)A Randomized Double-blind, Placebo-controlled Study of LEE011 in Combination With Letrozole for the Treatment of Postmenopausal Women With Hormone Receptor Positive, HER2 Negative, Advanced Breast Cancer Who Received no Prior Therapy for Advanced Disease;ClinicalTrials.gov Identifier: NCT01958021
/////////Ribociclib, novartis, LEE011, astex, phase 3, CDK inhibitors
CN(C)C(=O)c1cc2cnc(nc2n1C3CCCC3)Nc4ccc(cn4)N5CCNCC5
VARDENAFIL

VARDENAFIL
224785-90-4 CAS NO
Vardenafil hydrochloride (CAS NO.224785-91-5)
| Formula | C23H32N6O4S |
|---|---|
| Mol. mass | 488.604 g/mol |
4-[2-Ethoxy-5-(4-ethylpiperazin-1-yl)sulfonyl-phenyl]-9-methyl-7-propyl-3,5,6,8-tetrazabicyclo[4.3.0]nona-3,7,9-trien-2-one
Vivanza, Vardenafil (INN), Levitra (TN), STK642629, , LEVITRA
Vardenafil (INN) is a PDE5 inhibitor used for treating erectile dysfunction that is sold under the trade names Levitra (Bayer AG, GSK, and SP) andStaxyn.
Vardenafil was co-marketed by Bayer Pharmaceuticals, GlaxoSmithKline, and Schering-Plough under the trade name Levitra. As of 2005, the co-promotion rights of GSK on Levitra have been returned to Bayer in many markets outside the U.S. In Italy, Bayer sells vardenafil as Levitra and GSK sells it as Vivanza. Thus, because of European Union trade rules, parallel imports might result in Vivanza sold next to Levitra in the EU.
Vardenafil (Levitra) is an oral therapy for the treatment of erectile dysfunction. It is a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). Penile erection is a hemodynamic process initiated by the relaxation of smooth muscle in the corpus cavernosum and its associated arterioles. During sexual stimulation, nitric oxide is released from nerve endings and endothelial cells in the corpus cavernosum. Nitric oxide activates the enzyme guanylate cyclase resulting in increased synthesis of cyclic guanosine monophosphate (cGMP) in the smooth muscle cells of the corpus cavernosum. The cGMP in turn triggers smooth muscle relaxation, allowing increased blood flow into the penis, resulting in erection. The tissue concentration of cGMP is regulated by both the rates of synthesis and degradation via phosphodiesterases (PDEs). The most abundant PDE in the human corpus cavernosum is the cGMPspecific phosphodiesterase type 5 (PDE5); therefore, the inhibition of PDE5 enhances erectile function by increasing the amount of cGMP.
An orally disintegrating form, marketed as Staxyn, has been gaining approvals in countries such as the United States[1] and Canada.[2]
Vardenafil’s indications and contra-indications are the same as with other PDE5 inhibitors; it is closely related in function to sildenafil citrate (Viagra) and tadalafil (Cialis). The difference between the vardenafil molecule and sildenafil citrate is a nitrogen atom’s position and the change of sildenafil’spiperazine ring methyl group to an ethyl group. Tadalafil is structurally different from both sildenafil and vardenafil. Vardenafil’s relatively short effective time is comparable to but somewhat longer than sildenafil’s.
Beyond its indications for erectile dysfunction, vardenafil may be effective in the treatment of premature ejaculation, where it may significantly increase the time from vaginal penetration to ejaculation.[3]
The common, adverse drug reactions (side-effects) are the same as with other PDE5 inhibitors. The frequent vardenafil-specific side-effect is nausea; the infrequent side-effects are abdominal pain, back pain, photosensitivity, abnormal vision, eye pain, facial edema, hypotension, palpitation,tachycardia, arthralgia, myalgia, rash, itch, and priapism.

One possibly serious, but rare, side-effect with vardenafil is heart attack. Also, in rare cases, vardenafil use may cause priapism, a very painful emergency condition that can cause impotence if left untreated.[4]
On 18 October 2007, the U.S. Food and Drug Administration (FDA) announced that a warning about possible deafness (sudden hearing loss) would be added to the drug labels of Vardenafil, and other PDE5 inhibitors.[5]
Vardenafil, as with all PDE5 inhibitors, should not be used by men taking nitrate medications, because combining them with vardenafil might provoke potentially life-threatening hypotension (low blood pressure).
Further, Vardenafil causing lengthening of the QT interval. Therefore it should not be taken by men taking other medications that affect the QT interval (such as amiodarone).

Levitra 20mg Oral Tablet
It is available in 2.5 mg, 5 mg, 10 mg, and 20 mg doses in round orange tablets. The normal starting dose is 10 mg (roughly equivalent to 50 mg of sildenafil). Vardenafil should be taken 1 to 2 hours prior to sexual activity, with a maximum dose frequency of once per day. In some territories, such as the UK, only certain doses may be available.
Vardenafil is also available under the name Staxyn as a tablet which dissolves on the tongue rather than being swallowed in the form of a pill.
STAXYN is an oral therapy for the treatment of erectile dysfunction. This monohydrochloride salt of vardenafil is a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific PDE5.
Vardenafil HCl is designated chemically as piperazine, 1-[[3-(1,4-dihydro-5-methyl-4-oxo-7-propylimidazo[5,1f][1,2,4]triazin-2-yl)-4-ethoxyphenyl]sulfonyl]-4-ethyl-, monohydrochloride and has the following structural formula:
 |
Vardenafil HCl is a nearly colorless, solid substance with a molecular weight of 579.1 g/mol and a solubility of 0.11 mg/mL in water.
TRIHYDRATE, HCL SALT


US2002137930A

vardenafil hydrochloride is piperazine, 1-[[3-(1,4-dihydro-5-methyl-4-oxo-7-propylimidazo[5,1-f][1,2,4]triazin-2-yl)-4-ethoxyphenyl]sulfonyl]-4-ethyl-, mono -hydrochloride and can be structurally represented by Formula I.

The monohydrochloride salt of vardenafil is a selective inhibitor of cyclic guaosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). It is commercially available in products sold under the brand name LEVITRA formulated as 2.5 mg, 5 mg, 10 mg, 20 mg film-coated tablets.
U.S. Pat. No. 6,362,178 B1 discloses vardenafil, its related compounds and processes for their preparation. The patent describes a process in which vardenafil is obtained by recrystallization in ether in Example 19. Vardenafil produced as per Example 19 is hereinafter referred as “crystalline Form I” of vardenafil. The patent also describes processes for the preparation of its monohydrochloride and dihydrochloride salts, which are formed in a combination of ether and dichloromethane. The patent also describes a process for the preparation of vardenafil monohydrochloride trihydrate.
U.S. Patent Application Publication No. 2005/0203298 also describes a process for the preparation of vardenafil, and its monohydrochloride trihydrate.
Chemical synthesis of vardenafil has mostly been directed to the preparation of the trihydrate of monohydrochloride of vardenafil.
In WO 99/24433, sulphonamide-substituted imidazotriazinones are described as potent inhibitors of either one or more of the cyclic guanosine 3′,5′-monophosphate-metabolizing phosphodiesterases (cGMP PDEs). According to the nomenclature of Beavo and Reifsnyder (Trends in Pharmacol. Sci. 11, 150-155, 1990), these cGMP PDEs are the phosphodiesterase isoenzymes PDE-I, PDE-II and PDE-V.
According to WO 99/24433, the sulphonamide-substituted imidazotriazinones described therein are prepared from corresponding 2-ethoxyphenyl-substituted imidazotriazinones by reaction with chlorosulphonic acid and subsequent reaction with an appropriate amine, as is illustrated by the following scheme (R1 to R6 here have the meanings indicated in WO 99/24433):
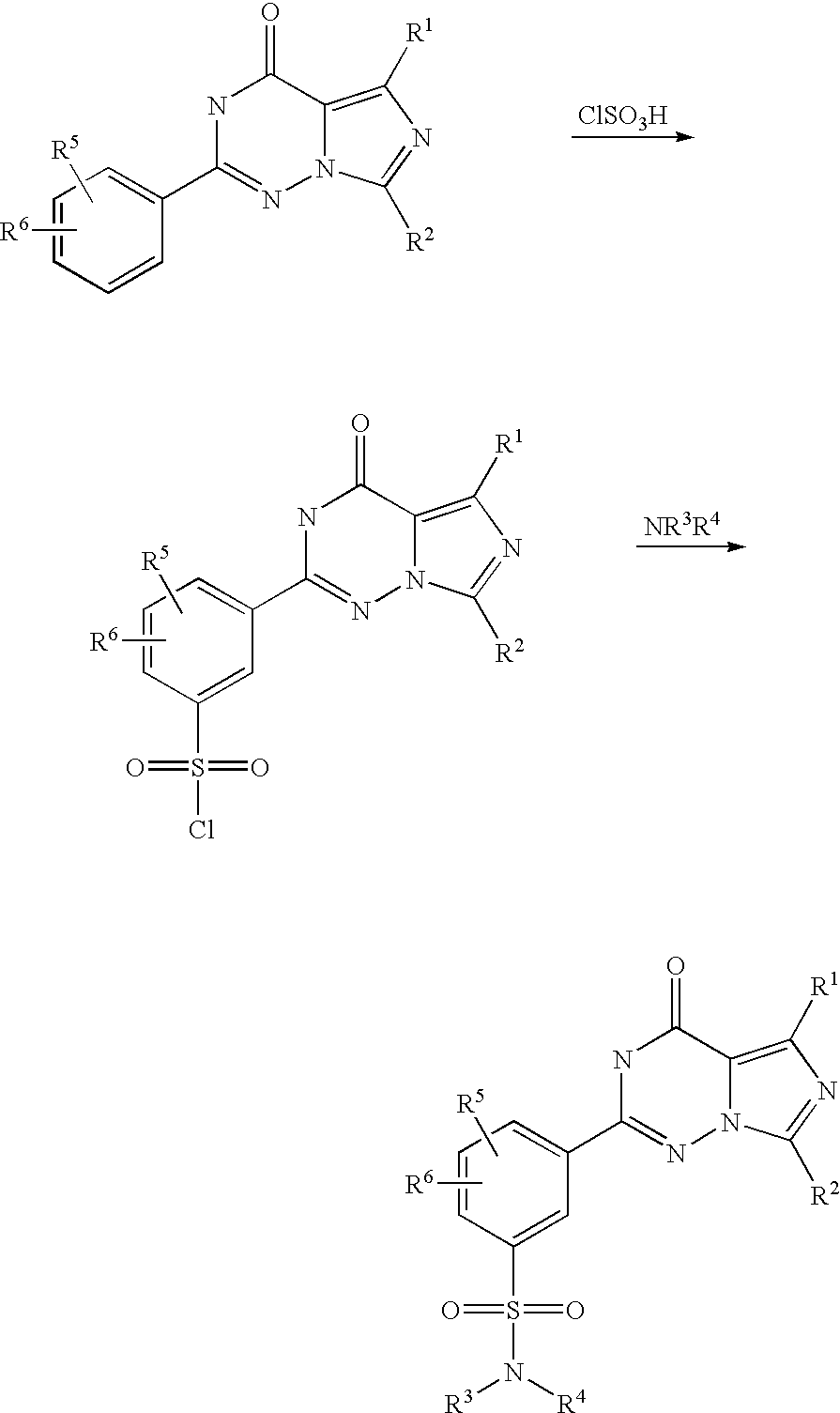
In this process, highly reactive chlorosulphonic acid has to be used as a reagent. Moreover, the imidazotriazinonesulphonyl chlorides formed as intermediates are sensitive to hydrolysis, which, in particular in the conversion of this preparation process to the industrial scale, can lead to not inconsiderable yield variations.
It was therefore the object of the present invention to make available a process for the preparation of sulphonamide-substituted imidazotriazinones in which the disadvantages of the above process known from the prior art are avoided.
This object is achieved according to the present invention by a process as in claim 1. In particular, in the process according to the invention as in claim 1 the use of chlorosulphonic acid is avoided by introduction of the sulphonic acid via a reaction with sulphuric acid and subsequent reaction with thionyl chloride. Moreover, the reaction with thionyl chloride and the subsequent reaction with an amine is carried out in a one-pot process, so that the imidazotriazinonesulphonyl chloride intermediate, which is sensitive to hydrolysis, does not need to be isolated. By means of this, yield variations on account of partial hydrolysis of this intermediate can be excluded. As a result of these advantages, the process according to the invention is much simpler to carry out on the industrial scale than the process described in WO 99/24433.
………………….
SYNTHESIS
2-butyrylamino-propionic acid
EXAMPLE 1A 2-Butyrylaminopropionic acid

22.27 g (250 mmol) of D,L-alanine and 55.66 g (550 mmol) of triethylamine are dissolved in 250 ml of dichloromethane, and the solution is cooled to 0° C. 59.75 g (550 mmol) of trimethylsilyl chloride are added dropwise, and the solution is stirred for 1 hour at room temperature and for 1 hour at 40° C. After cooling to −10° C., 26.64 g (250 mmol) of butyryl chloride are added dropwise, and the resulting mixture is stirred for 2 hours at −10° C. and for one hour at room temperature.
With ice-cooling, 125 ml of water are added dropwise and the reaction mixture is stirred at room temperature for 15 minutes. The aqueous phase is evaporated to dryness, the residue is titrated with acetone and the mother liquor is filtered off with suction. The solvent is removed and the residue is chromatographed. The resulting product is dissolved in 3N aqueous sodium hydroxide solution and the resulting solution is evaporated to dryness. The residue is taken up in conc. HCl and once more evaporated to dryness. The residue is stirred with acetone, precipitated solid is filtered off with suction and the solvent is removed under reduced pressure. This gives 28.2 g (71%) of a viscous oil which crystallizes after some time.
200 MHz 1H-NMR (DMSO-d6): 0.84, t, 3H; 1.22, d, 3H; 1.50, hex, 2H; 2.07, t, 2H; 4.20, quin., 1H; 8.09, d, 1H.
EXAMPLE 3A 2-Ethoxybenzonitrile

25 g (210 mmol) of 2-hydroxybenzonitrile are refluxed with 87 g of potassium carbonate and 34.3 g (314.8 mmol) of ethyl bromide in 500 ml of acetone overnight. The solid is filtered off, the solvent is removed under reduced pressure and the residue is distilled under reduced pressure. This gives 30.0 g (97%) of a colourless liquid.
200 MHz 1H-NMR (DMSO-d6): 1.48, t, 3H; 4.15, quart., 2H; 6.99, dt, 2H; 7.51, dt, 2H.
EXAMPLE 4A 2-Ethoxybenzamidine hydrochloride

21.4 g (400 mmol) of ammonium chloride are suspended in 375 ml of toluene, and the suspension is cooled to 0° C. 200 ml of a 2M solution of trimethylaluminium in hexane are added dropwise, and the mixture is stirred at room temperature until the evolution of gas has ceased. After addition of 29.44 g (200 mmol) of 2-ethoxybenzonitrile, the reaction mixture is stirred at 80° C. (bath) overnight.
With ice-cooling, the cooled reaction mixture is added to a suspension of 100 g of silica gel and 950 ml of chloroform, and the mixture is stirred at room temperature for 30 minutes. The mixture is filtered off with suction, and the filter residue is washed with the same amount of methanol. The mother liquor is concentrated, the resulting residue is stirred with a mixture of dichloromethane and methanol (9:1), the solid is filtered off with suction and the mother liquor is concentrated. This gives 30.4 g (76%) of a colourless solid.
200 MHz 1H-NMR (DMSO-d6): 1.36, t, 3H; 4.12, quart., 2H; 7.10, t, 1H; 7.21, d, 1H; 7.52, m, 2H; 9.30, s, broad, 4H.
EXAMPLE 10A 2-(2-Ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

7.16 g (45 mmol) of 2-butyrylamino-propionic acid and 10.67 g of pyridine are dissolved in 45 ml of THF and, after addition of a spatula tip of DMAP, heated to reflux. 12.29 g (90 mmol) of ethyl oxalyl chloride are slowly added dropwise, and the reaction mixture is refluxed for 3 hours. The mixture is poured into ice-water and extracted three times with ethyl acetate and the organic phase is dried over sodium sulphate and concentrated using a rotary evaporator. The residue is taken up in 15 ml of ethanol and refluxed with 2.15 g of sodium bicarbonate for 2.5 hours. The cooled solution is filtered.
With ice-cooling, 2.25 g (45 mmol) of hydrazine hydrate are added dropwise to a solution of 9.03 g (45 mmol) of 2-ethoxybenzamidine hydrochloride in 45 ml of ethanol, and the resulting suspension is stirred at room temperature for another 10 minutes. The ethanolic solution described above is added to this reaction mixture, and the mixture is stirred at a bath temperature of 70° C. for 4 hours. After filtration, the mixture is concentrated, the residue is partitioned between dichloromethane and water, the organic phase is dried over sodium sulphate and the solvent is removed under reduced pressure.
This residue is dissolved in 60 ml of 1,2-dichloroethane and, after addition of 7.5 ml of phosphorus oxychloride, refluxed for 2 hours. The mixture is diluted with dichloromethane and neutralized by addition of sodium bicarbonate solution and solid sodium bicarbonate. The organic phase is dried and the solvent is removed under reduced pressure. Chromatography using ethyl acetate and crystallization afford 4.00 g (28%) of a colourless solid, Rf=0.42 (dichloromethane/methanol=95:5)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.56, t, 3H; 1.89, hex, 2H; 2.67, s, 3H; 3.00, t, 2H; 4.26, quart., 2H; 7.05, m, 2H; 7.50, dt, 1H; 8.17, dd, 1H; 10.00, s, 1H.
EXAMPLE 15A 4-Ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride

At 0° C., 2.00 g (6.4 mmol) of 2-(2-ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are slowly added to 3.83 ml of chlorosulphonic acid. At room temperature, the reaction mixture is stirred ovemight, and then poured into ice-water and extracted with dichloromethane. This gives 2.40 g (91%) of a colourless foam.
200 MHz 1H-NMR (CDCl3): 1.03, t, 3H; 1.61, t, 2H; 1.92, hex, 2H; 2.67, s, 3H; 3.10, t, 2H; 4.42, quart., 2H; 7.27, t, 1H; 8.20, dd, 1H; 8.67, d, 1H; 10.18, s, 1H.
Example 19 2-[2-Ethoxy-5-(4-ethyl-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

470 mg (1.14 mmol) of 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride are dissolved in 20 ml of dichloromethane and cooled to 0° C. 390 mg (3.42 mmol) of N-ethylpiperazine are added, and the reaction mixture is stirred at room temperature overnight. The mixture is diluted with dichloromethane, the organic phase is washed twice with water and dried over sodium sulphate and the solvent is removed under reduced pressure. Crystallization from ether gives 370 mg (66%) of a colourless solid.
400 MHz 1H-NMR (CDCl3): 1.01, t, 3H; 1.59, t, 3H; 1.88, hex, 2H; 2.42, quart., 2H; 2.56, m, 4H; 2.63, s, 3H; 3.00, t, 2H; 3.10, m, 4H; 4.33, quart., 2H, 7.17, d, 1H; 7.88, dd, 1H; 8.44, d, 1H; 9.75, s, 1H.
…………………….
EXAMPLE 7 Preparation of the Trihydrate of Vardenafil Monohydrochloride
14 g of vardenafil hydrochloride was taken into a round bottom flask followed by the addition of 70 ml water and the pH of the reaction mass was adjusted using sodium hydroxide to 11 at 30° C. 280 ml of dichloromethane was added to the above reaction mass and the layers were separated. The organic layer was dried over sodium sulfate and the organic layer was transferred into a round bottom flask and subjected to heating for distillation at 40° C. for 1.5 hours. The solid material was transferred into a round bottom flask and 36 ml of a mixture of acetone and water in 12:1 ratio was added with stirring, then 2.2 ml of 36% aqueous hydrochloric acid was added with stirring. The reaction mass was heated to a temperature of about 45° C. and the undissolved particles were removed by filtration. The filtrate was taken into a round bottom flask and cooled to 5° C., maintained for 45 minutes at 3 to 5° C. followed by the filtration of the solid which was then subjected to suction drying and finally dried at 40° C. to yield 9.0 g of the trihydrate of vardenafil monohydrochloride.
……………………..
STARTING COMPOUNDS
Example I Preparation of 2-(2-ethoxyphenyl)-5-methyl-7-propyl-3H-imidazo-[5,1-f][2,4]triazin-4-oneIa) Preparation of 2-butyrylaminopropionic acid
A solution of 100 kg of D,L-alanine in aqueous sodium hydroxide solution is reacted in the cold with 119 kg of butyryl chloride. After addition of butyl acetate, the mixture is acidified with hydrochloric acid, the organic phase is separated off and the aqueous phase is re-extracted. The organic phase is dried by azeotropic distillation. The crystallizate is isolated, washed with butyl acetate and dried.
Yield: 132.6 kg (68%)
1H-NMR: δ=0.8 (t, 3H), 1.25 (d, 3H), 1.5 (m, 2H), 2.1 (t, 2H), 4.2 (q, 1H), 8.1 (d, NH), 12.0-12.7 (s, COOH)
MS: 336 (2M+NH4, 40), 319 (2M+H, 15), 177 (M+NH4, 100), 160 (M+H, 20)
Ib) Preparation of 2-ethoxybenzonitrile

260 kg of thionyl chloride are added at 85-95° C. to a suspension of 250 kg of 2-ethoxybenzamide in toluene under metering control. The reaction mixture is stirred in the presence of heat. Thionyl chloride and toluene are then distilled off in vacuo. The product is employed in the subsequent stage as a crude product.
Yield: 228.5 kg (crude product)
1H-NMR: δ=1.45 (t, 3H), 4.15 (q, 2H), 7.0 (m, 2H, phenyl), 7.5 (m, 2H, phenyl)
MS: 312 (2M+N4, 35), 165 (M+NH4, 100), 147 (5)
Ic) Preparation of 2-ethoxy-N-hydroxybenzamidine

111 kg of 2-ethoxybenzonitrile (crude product) from Example Ib are heated under reflux with 164 1 of triethylamine and 73 kg of hydroxylamine hydrochloride in isopropanol. The reaction mixture is treated with water and cooled. The crystallizate is isolated, washed and employed in the subsequent stage as a moist product.
Yield: 92.6 kg (moist product)
1H-NMR: δ=1.35 (t, 3H), 4.1 (q, 2H), 5.6 (s, 2H), 6.9-7.4 (4H, phenyl), 9.4 (s, 1H, OH)
MS: 361 (2M+H, 30), 198 (M+N, 30), 181 (M+H, 100)
Id) Preparation of 2-ethoxybenzamidine hydrochloride

135 kg of 2-ethoxy-N-hydroxybenzamidine (moist product) from Example Ic are hydrogenated at 50-60° C. in acetic acid using palladium on carbon as a catalyst. For the work-up, the hydrogenation reaction is freed from the catalyst, treated with hydrochloric acid and concentrated. Residual acetic acid and water are removed by azeotropic distillation with toluene. The crystallizate is isolated and dried in vacuo.
Yield: 136.4 kg
H-NMR: 1.35 (t, 3H), 4.15 (q, 2H), 7.1-7.7 (m, 4H, phenyl), 9.1-9.4 (2×s, 3H), 10.5-10.7 (s, 1H)
MS: 329 (2M+H, 10), 165 (M+H, 100)
Ie) Preparation of 2-(2-ethoxyphenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]-triazin-4-one

231 kg of 2-butyrylaminopropionic acid from Example Ia are treated in tetrahydrofuran with 341 kg of pyridine, catalytic amounts of 4-N,N-dimethylaminopyridine and 392 kg of ethyl chloroxalate and stirred with heating under reflux. The reaction mixture is taken up in ethyl acetate, washed with water and the ethyl acetate phase is concentrated. The distillation residue is taken up in methanol and reacted with the following solution.
192 kg of 2-ethoxybenzamidine hydrochloride from Example Id are treated in methanol with 47.5 kg of hydrazine hydrate and the mixture is stirred at room temperature. The solution is combined with the solution of 2-butyrylamino-1-ethoxycarbonylpropenyl ethyl oxalate prepared above. The reaction mixture thus obtained is stirred with heating under reflux. Methanol is removed by distillation and replaced by acetic acid.
Option A:
138.6 kg of phosphorus oxychloride are added and stirred in the presence of heat.
Acetic acid is distilled off in vacuo. The residue is treated with water and dichloromethane or optionally methyl isobutyl ketone and rendered neutral using sodium hydroxide solution. The organic phase is concentrated, and the residue is dissolved in acetone and crystallized with cooling. The crystallizate is isolated, washed and dried.
Option B:
At least 190 kg of acetyl chloride are added and stirred in the presence of heat. Acetic acid is distilled off in vacuo. The distillation residue is treated with acetone and water, and the product is crystallized by rendering neutral with sodium hydroxide solution. The product is isolated, washed and dried.
Yield: 90-160 kg
1H-NMR: δ=1.0 (t, 3H), 1.6 (t, 3H), 1.9 (m, 2H), 2.8 (s, 3H), 3.3 (t, 2H), 4.3 (q, 2H), 7.0-8.2 (Ar, 4H), 10.3 (CONH, 1H)
MS: 313 (M+H, 100), 149 (25), 151 (40), 121 (15)
HPLC: Kromasil C-18 phase, neutral phosphate buffer, acetonitrile, 233 nm, linear gradient of 30% acetonitrile ->80% acetonitrile (30 min.): 99 area % (Rt 19.1)
PREPARATION EXAMPLES Example 1a 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydroimidazo[5,1-fl-][1,2,4]triazin-2-yl)benzenesulphonic acid

194 kg of 2-(2-ethoxyphenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one from Example Ie are reacted with 504 kg of concentrated sulphuric acid. The reaction mixture is added to water, cooled, and the crystallizate is isolated and dried in vacuo.
Yield: 195.2 kg
1H-NMR: δ=0.95 (t, 3H), 1.3 (t, 3H), 1.8 (m, 2H), 2.6 (s, 3H), 3.05 (t, 2H), 4.1 (q, 2H), 7.15 (Ar, 1H), 7.75 (m, 2H), 12.3 (SO2OH)
MS: 393 (M+H, 100), 365 (25), 151 (40)
HPLC: X-Terra C-18 phase, aqueous phosphoric acid, acetonitrile, 242 nm, linear gradient of 10% acetonitrile ->90% acetonitrile (20 min.):
98 area % (R, 9.2)
Example 1b) 2-[2-ethoxy-5-(4-ethlylpiperazin-1-sulphonyl)phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

22.5 kg of 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]-triazin-2-yl)benzenesulphonic acid from Example 1a are reacted with 74 kg of thionyl chloride and catalytic amounts of dimethylformamide until the evolution of gas has ended. Xylene is repeatedly added to the reaction mixture and thionyl chloride is distilled off. 15.1 kg of N-ethylpiperazine are added to the suspension and it is stirred. After the addition of water, it is adjusted to pH 1 using hydrochloric acid, and the phases are separated. The aqueous phase is treated with acetone and rendered neutral by addition of sodium hydroxide solution. The mixture is cooled, and the crystallizate is isolated, washed and dried in vacuo.
Yield: 26.1 kg
1H-NMR: δ=1.0 (2×t, 6H), 1.6 (t, 3H), 1.9 (m, 2H), 2.45 (q, 2H), 2.55 (m, 4H), 2.65 (s, 3H), 3.0 (t, 2H), 3.1 (m, 4H), 4.35 (q, 2H), 7.15 (Ar, 1H), 7.9 (Ar, 1H), 8.4 (Ar, 1H), 9.8 (CONH)
MS: 489 (M+H, 100), 345 (10), 313, (10), 285 (10), 113 (20)
HPLC: X-Terra C-18 phase, neutral phosphate buffer, acetonitrile, 242 nm, linear gradient of 20% acetonitrile ->75% acetonitrile (20 min.): 98 area % (Rt 16.3)
1 c) 2-[2-ethoxy-5-(4-ethylpiperazin-1-sulphonyl)phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-fl][1,2,4]triazin-4-one hydrochloride trihydrate

22.5 kg of 2-[2-ethoxy-5-(4-ethylpiperazin-1-sulphonyl)phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one from Example 1b are dissolved in 5.1 kg of concentrated hydrochloric acid and acetone/water (12:1 v/v) in the presence of heat. The clear solution is filtered hot and crystallized by cooling and seeding. The crystallizate is isolated, washed and dried in vacuo at about 30° C. and about 300 mbar.
Yield: 25.4 kg
M.p. (DSC): 192° C.
HPLC: X-Terra C-18 phase, neutral phosphate buffer, acetonitrile, 242 nm, linear gradient of 20% acetonitrile ->75% acetonitrile (20 min.): 99 area % (Rt 16.3)
- http://www.pharmpro.com/News/Feeds/2010/06/pharmaceutical-companies-bayer-new-erectile-dysfunction-treatment-staxyn-approve/
- http://www.newswire.ca/en/story/832217/staxyn-new-innovation-in-erectile-dysfunction-helps-younger-men-rise-to-the-occasion
- A Aversa et al. “Effects of vardenafil administration on intravaginal ejaculatory latency time in men with lifelong premature ejaculation”. Retrieved 2010-12-14.
- Schools of Pharmacy (Glen L. Stimmel, Pharm.D., and Mary A. Gutierrez, Pharm.D.) and Medicine (Glen L. Stimmel, Pharm.D.), University of Southern California, Los Angeles, California. “Counseling Patients About Sexual Issues: Drug-Induced Priapism”. Medscape. Retrieved 2010-12-06.
- “FDA Announces Revisions to Labels for Cialis, Levitra and Viagra”. Food and Drug Administration. 2007-10-18. Retrieved 2009-08-06.
- Official Levitra website
- PubChem Information
-
2-1-2013Synthesis of quinoline derivatives: discovery of a potent and selective phosphodiesterase 5 inhibitor for the treatment of Alzheimer’s disease.European journal of medicinal chemistry
PATENTS
| US6362178 * | Oct 31, 1998 | Mar 26, 2002 | Bayer Aktiengesellschaft | 2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors |
| US20050203298 * | May 5, 2005 | Sep 15, 2005 | Bayer Healthcare Aktiengesellschaft | Process for the preparation of sulphonamide-substituted imidazotriazinones |
| US20060111354 * | Jul 3, 2003 | May 25, 2006 | Peter Serno | Medicaments containing vardenafil hydrochloride trihydrate |
| WO2004006894A1 * | Jul 3, 2003 | Jan 22, 2004 | Bayer Healthcare Ag | Medicaments containing vardenafil hydrochloride trihydrate |
|
11-4-2011
|
ROFLUMILAST FOR THE TREATMENT OF DIABETES MELLITUS
|
|
|
9-14-2011
|
Roflumilast for the Treatment of Diabetes Mellitus
|
|
|
8-5-2011
|
N-BUTYRAMIDE, THE PREPARATION METHOD AND USE THEREOF
|
|
|
3-4-2011
|
Fatty Acid Oxidation Inhibitors Treating Hyperglycemia and Related Disorders
|
|
|
1-14-2011
|
2-PHENYL SUBSTITUTED IMIDAZOTRIAZINONES AS PHOSPHODIESTERASE INHIBITORS
|
|
|
9-17-2010
|
SUBSTITUTED PDE5 INHIBITORS
|
|
|
7-16-2010
|
Combination treatment for diabetes mellitus
|
|
|
4-28-2010
|
2-Phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
|
|
|
4-14-2010
|
2-PHENYL SUBSTITUTED IMIDAZOTRIAZINONES AS PHOSPHODIESTERASE INHIBITORS
|
|
|
2-5-2010
|
Heterocyclic Compounds And Uses Thereof In The Treatment Of Sexual Disorders
|
|
12-25-2009
|
Therapeutic Compositions Comprising a Specific Endothelin Receptor Antagonist and a PDE5 Inhibitor
|
|
|
11-27-2009
|
Substituted PDE5 inhibitors
|
|
|
9-4-2009
|
Uses of 2-Phenyl-Substituted Imidazotriazinone Derivatives for Treating Pulmonary Hypertension
|
|
|
8-28-2009
|
Roflumilast for the Treatment of Pulmonary Hypertension
|
|
|
8-7-2009
|
Use of Phosphodiesterase Inhibitor as a Component of Implantable Medical Devices
|
|
|
6-26-2009
|
Method for healing a wound using a phosphodiesterase type five inhibitor
|
|
|
3-20-2009
|
Pde5 inhibitor compositions and methods for immunotherapy
|
|
|
3-6-2009
|
Pde5 inhibitor compositions and methods for treating cardiac indications
|
|
|
10-31-2008
|
Formulations with Controlled Release of Active Ingredient
|
|
|
8-15-2008
|
HIGHLY SELECTIVE and LONG-ACTING PDE5 MODULATORS
|
|
8-8-2008
|
Formulations With Controlled Release Of Active Ingredient
|
|
|
4-11-2008
|
Use of 2-alkoxyphenyl-substituted imidazotriazinones
|
|
|
1-2-2008
|
2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors, for treatment of hypertension
|
|
|
12-28-2007
|
Novel Uses of 2-Phenyl-Substituted Imidazotriazinone Derivatives
|
|
|
10-3-2007
|
Use of 2-alkoxyphenyl-substituted imidazotriazinones
|
|
|
11-24-2006
|
Methods for synthesizing imidazotriazinones
|
|
|
10-18-2006
|
2-Phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
|
|
|
2-15-2006
|
Process for the preparation of sulphonamide-substituted imidazotriazinones
|
|
|
8-17-2005
|
Use of 2-alkoxyphenol-substituted imidazotriazinones
|
|
|
5-11-2005
|
2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
|
|
1-21-2005
|
Process for the preparation of sulphonamide-substituted imidazotriazinones
|
|
|
8-18-2004
|
Process for the preparation of sulphonamide-substituted imidazotriazinones
|
|
|
8-6-2004
|
Novel use of 2-phenyl-substituted imidazotriazinones
|
|
|
7-32-2003
|
Daily treatment for erectile dysfunction using a PDE5 inhibitor
|
|
|
5-21-2003
|
2-phenyl substituted imidatriazinones as phosphodiesterase inhibitors
|
|
|
3-27-2002
|
2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
|
|
|
12-21-2001
|
Daily treatment for erectile dysfunction using a PDE5 inhibitor
|
|
|
5-21-1999
|
2-PHENYL SUBSTITUTED IMIDAZOTRIAZINONES AS PHOSPHODIESTERASE INHIBITORS
|
FDA approves Praxbind, Idarucizumab the first reversal agent for the anticoagulant Pradaxa
October 16, 2015
Release
The U.S. Food and Drug Administration today granted accelerated approval to Praxbind (idarucizumab) for use in patients who are taking the anticoagulant Pradaxa (dabigatran) during emergency situations when there is a need to reverse Pradaxa’s blood-thinning effects.
“The anticoagulant effects of Pradaxa are important and life-saving for some patients, but there are situations where reversal of the drug’s effects is medically necessary,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval offers the medical community an important tool for managing patients taking Pradaxa in emergency or life-threatening situations when bleeding can’t be controlled.”
The FDA approved Pradaxa in 2010 to prevent stroke and systemic blood clots in patients with atrial fibrillation, as well as for the treatment and prevention of deep venous thrombosis and pulmonary embolism. Praxbind is the first reversal agent approved specifically for Pradaxa and works by binding to the drug compound to neutralize its effect. Praxbind solution is for intravenous injection.
The safety and effectiveness of Praxbind were studied in three trials involving a total of 283 healthy volunteers taking Pradaxa (i.e., people who did not require an anticoagulant). In the healthy volunteers who were given Praxbind, there was an immediate reduction in the amount of Pradaxa in participants’ blood (measured as unbound dabigatran plasma concentration) that lasted for a period of at least 24 hours. In this study, the most common side effect from use of Praxbind was headache.
Another trial included 123 patients taking Pradaxa who received Praxbind due to uncontrolled bleeding or because they required emergency surgery. In this ongoing trial, based on laboratory testing, the anticoagulant effect of Pradaxa was fully reversed in 89 percent of patients within four hours of receiving Praxbind. In this patient trial, the most common side effects were low potassium (hypokalemia), confusion, constipation, fever and pneumonia.
Reversing the effect of Pradaxa exposes patients to the risk of blood clots and stroke from their underlying disease (such as atrial fibrillation). The Praxbind labeling recommends patients resume their anticoagulant therapy as soon as medically appropriate, as determined by their health care provider.
Praxbind is approved under the FDA’s accelerated approval program, which allows the agency to approve drugs for serious conditions that fill an unmet medical need based on an effect on a surrogate or an intermediate clinical endpoint that is reasonably likely to predict a clinical benefit to patients. The program is designed to provide patients with earlier access to promising new drugs, but the company will be required to submit additional clinical information after approval to confirm the drug’s clinical benefit.
Praxbind and Pradaxa are both marketed by Boehringer Ingelheim of Ridgefield, Connecticut.
Etelcalcetide, AMG 416, KAI-4169, velcalcetide
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2

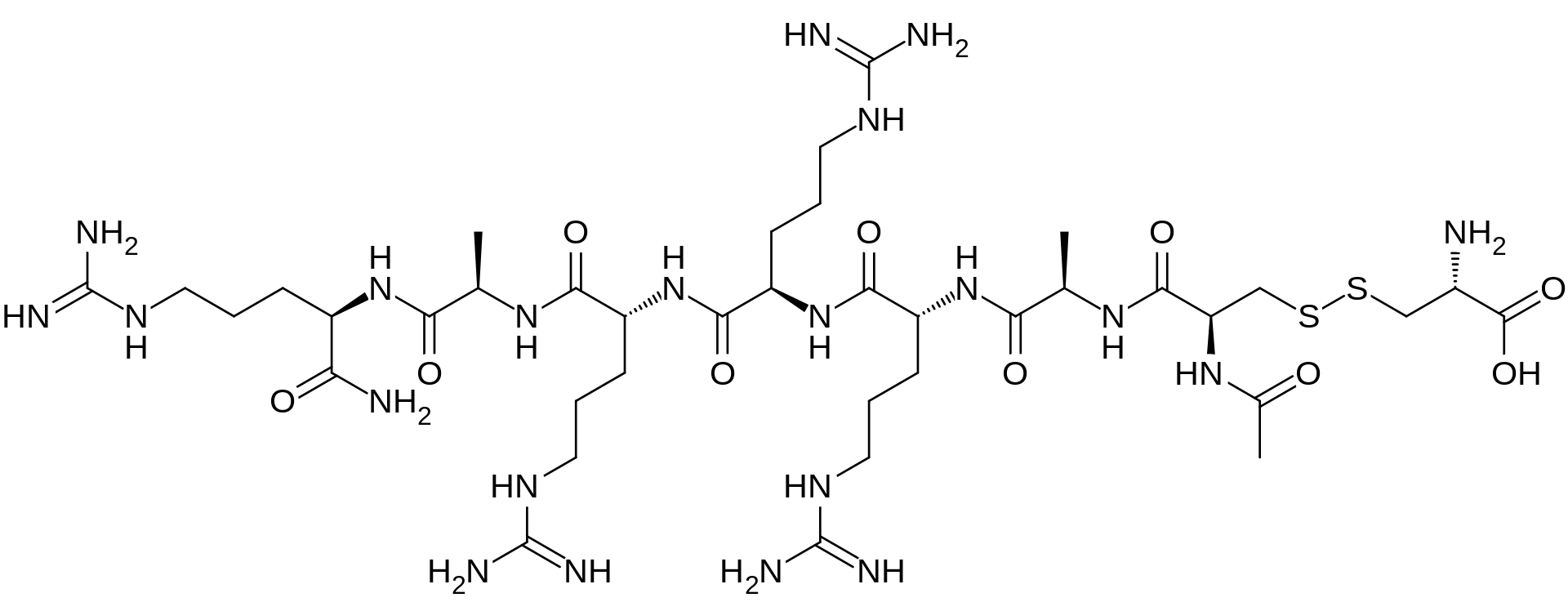
AMG 416 IS (Ac-D-Cys(L-Cys-OH)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2)
Etelcalcetide (AMG 416, KAI-4169, velcalcetide)
The main chain has 7 amino acids, all in the D-configuration. The side-chain cysteine residue is in the L-configuration. The molecular formula of AMG 416 (free base) is C38H73N21O10S2, and has a calculated average molecular mass of 1048.3 Da.
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine, hydrochloride (1:?)
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
http://www.amgenpipeline.com/pipeline/
WO 2011/014707. , the compound may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The main chain has 7 amino acids, all in the D-configuration and the side-chain cysteine residue is in the L-configuration. The amino terminal is acetylated and the carboxyl-terminal is amidated. This compound (“AMG-416”) has utility for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis patients. A liquid formulation comprising AMG-416 may be administered to a subject intravenously. The hydrochloride salt of AMG-416 may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
Therapeutic peptides pose a number of challenges with respect to their formulation. Peptides in general, and particularly those that contain a disulfide bond, typically have only moderate or poor stability in aqueous solution. Peptides are prone to amide bond hydrolysis at both high and low pH.
Disulfide bonds can be unstable even under quite mild conditions (close to neutral pH). In addition, disulfide containing peptides that are not cyclic are particularly prone to dimer formation. Accordingly, therapeutic peptides are often provided in lyophilized form, as a dry powder or cake, for later reconstitution.
A lyophilized formulation of a therapeutic peptide has the advantage of providing stability for long periods of time, but is less convenient to use as it requires the addition of one or more diluents and there is the potential risk for errors due to the use of an improper type or amount of diluent, as well as risk of contamination. In addition, the lyophilization process is time consuming and costly.
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
Generic Name:Etelcalcetide
Synonym:KAI-4169
CAS Number:1262780-97-1
N-acetyl-D-cysteinyl-S-(L-cysteine disulfide)-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide
Mechanism of Action:Activates calcium sensing receptor on parathyroid glands reducing PTH synthesis and secretion
Indication: secondary hyperparathyroidism associated with chronic kidney disease
Development Stage: Phase III
Developer:KAI Pharmaceuticals/Amgen Inc.
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
HYDROCHLORIDE
Generic Name:Etelcalcetide Hydrochloride
AMG 416, KAI-4169, previously also known as velcalcetide hydrochloride
CAS :1334237-71-6
Chemical Name:N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
Mechanism of Action:Activates calcium sensing receptor on parathyroid glands reducing PTH synthesis and secretion
Indication: secondary hyperparathyroidism associated with chronic kidney disease
Development Stage: Phase III
Developer:KAI Pharmaceuticals/Amgen Inc.
Method for preparing etelcalcetide and its salts, particularly hydrochloride. See WO2014210489, for a prior filing claiming stable liquid formulation of etelcalcetide. Amgen, following its acquisition of KAI Pharmaceuticals, and Japanese licensee Ono Pharmaceuticals are developing etelcalcetide, a long-acting iv isozyme-selective peptide-based protein kinase C epsilon inhibitor and agonist of the calcium-sensing receptor, for treating secondary hyperparathyroidism (SHPT) in patients with end-stage renal disease receiving dialysis.
In August 2015, an NDA was submitted seeking approval of the drug for SHPT in patients with chronic kidney disease (CKD) on hemodialysis (HD) in the US.
In September 2015, Amgen filed an MAA under the centralized procedure in the EU for the approval of etelcalcetide for treating SHPT in patients with CKD on HD therapy.
KAI is also investigating a transdermal patch formulation of the drug for treating primary HPT.
- 25 Aug 2015 Preregistration for Secondary hyperparathyroidism in USA (IV)
- 29 May 2015 Pooled analysis efficacy and adverse events data from two phase III trials in secondary hyperparathyroidism released by Amgen
- 21 Apr 2015 Amgen plans to submit Biological License Application to USFDA and Marketing Authorisation Application to EMA for Secondary hyperparathyroidism
PATENT
WO2011014707
http://www.google.com/patents/WO2011014707A2?cl=en
PATENT
WO 2015154031
The hydrochloride salt of AMG 416 has the chemical structure:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
(SEQ ID NO:l)
The main chain has 7 amino acids, all in the D-configuration. The side-chain cysteine residue is in the L-configuration. The molecular formula of AMG 416 (free base) is C38H73N21O10S2, and has a calculated average molecular mass of 1048.3 Da.
AMG 416 and a method for its preparation are described in International Pat. Publication No. WO 2011/014707, which is incorporated herein by reference for any purpose. As described in International Pat. Publication No. WO 2011/014707, AMG 416 may be assembled by solid-phase synthesis from the corresponding Fmoc-protected D-amino acids. After cleavage from the resin, the material may be treated with Boc-L-Cys(NPyS)-OH to form the disulfide bond. The Boc group may then be removed with trifluoroacetate (TFA) and the resulting product purified by reverse-phase high pressure liquid chromatography (HPLC) and isolated as the TFA salt form by lyophilization. The TFA salt can be converted to a pharmaceutically acceptable salt by carrying out a subsequent salt exchange procedure. Such procedures are well known in the art and include, e.g., an ion exchange technique, optionally followed by purification of the resultant product (for example by reverse phase liquid chromatography or reverse osmosis).
There is a need for an efficient method of producing AMG 416, or a pharmaceutically acceptable salt thereof (e.g., AMG 416 HC1), and particularly one appropriate for commercial scale manufacturing.
In a first aspect, provided is a method for preparing AMG 416, the method comprising: providing a resin-bound peptide having a structure selected from the group consisting of Fmoc-D-Cys(Trt)-D-Ala-D- Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:2) and Ac-D-Cys(Trt)-D-Ala-D- Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:3); cleaving the peptide from the solid support; and activating the side chain of the D-Cys residue of the cleaved peptide.
In a second aspect, provided is a method for preparing AMG 416, the method comprising: providing a peptide having a structure of Ac-D-Cys(SPy)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 (SEQ ID NO:4); and contacting the peptide with L-Cys to produce a conjugated product.
In yet a third aspect provided is a method for preparing AMG 416, the method comprising: providing a resin-bound peptide having a structure selected from the group consisting of Fmoc-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:2) and Ac-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:3); cleaving the peptide from the solid support, i.e., to provide an unsupported peptide, and activating the side chain of the D-Cys residue of the unsupported peptide to generate an AMG 416 SPy intermediate (where SPy is 2-pyridinesulfenyl or S-Pyr), dissolving the AMG 416 SPy intermediate in an aqueous 0.1% TFA (trifluoroacetic acid solution), and purifying the AMG 416 SPy derivative by HPLC.
The term “AMG 416”, also known as etelcalcetide, formerly known as velcalcetide or KAI-4169, refers to a compound having the chemical name: N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-arginamide disulfide with L-cysteine, which has the following structural formula:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
Reference to AMG 416, or to any compound or AMG 416 fragment, intermediate, or precursor as described herein, is intended to encompass neutral, uncharged forms thereof, as well as pharmaceutically acceptable salts, hydrates and solvates thereof.
The terms “AMG 416 hydrochloride” and “AMG 416 HC1” are interchangeable and refer to a hydrochloride salt form of AMG 416 having the following structural formula:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · xHCl
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1 shows the chemical structure of AMG 416 (Ac-D-Cys(L-Cys-OH)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2) (SEQ ID NO: l).
FIG. 2 shows the chemical structure of Rink Amide AM resin and Ac-D-Cys(Trt)- D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-Resin (SEQ ID NO:3).
FIG. 3 shows a reaction scheme in which the SPy intermediate product (Ac-D-Cys(SPy)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2) (SEQ ID NO:4) is formed from the peptidyl-resin (Ac-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-NH-Resin) (SEQ ID NO:3).
FIG. 4 shows a reaction scheme in which a TFA salt of AMG 416 is formed from the SPy intermediate (AA1_7(SPy)).
FIG. 5 shows a reaction scheme in which the HC1 salt of AMG 416 is formed from the TFA salt of AMG 416.
FIG. 6 shows a reaction scheme in which Boc-D-Arg(Pbf)-OH is formed from Boc-D-Arg-OH.
FIG. 7 shows a reaction scheme in which D-Arg(Pbf)-OH is formed from Boc-D-Arg(Pbf)-OH.
EXAMPLE 5
Purification of the SPy Intermediate and Production of AMG 416 HC1
An alternative method for preparation of AMG 416 HC1 salt is described here. As described in Example 2 above, the SPy intermediate product was dried at 20°C under full vacuum after cleavage from the resin, precipitation and filtration. The precipitate was then dissolved in a 0.1% TFA aqueous solution and loaded onto a C-18 column for HPLC purification. The column was run at <60 bar and the solution temperature was 15-25 °C throughout. The eluents were 0.1% TFA in acetonitrile and 0.1% TFA in water. The fractions were stored at 5°C, they were sampled and then fractions were pooled. The combined pools from two runs were diluted and a concentration/purification run was performed using the same HPLC column to decrease the total volume and remove additional impurities. The fractions were stored at 5°C.
The fractions containing the AMG 416 SPy intermediate were subjected to azeotropic distillation to change the solvent from the 0.1% TFA to a 15% water in IPA solution, charging with IPA as needed. To the resultant AMG 416 SPy intermediate in IPA solution was then added L-Cysteine 1.15 eq and the reaction was allowed to proceed at room temperature for conjugation to occur and to form the AMG 416 TFA salt as described above in Example 4. The AMG 416 TFA solution was added to a solution of 12M aqueous HC1, 0.27 L/kg and IPA 49.4 L/kg over 3 hours via subsurface addition, resulting in direct precipitation of the AMG 416 4.5 HC1 salt. The batch was aged for 3 hours and sampled for analysis.
The material was filtered and slurry washed with 96 wt% IPA, 10 L/kg. The cake was then re-slurried for 4 hours in 10 L/kg of 96% wt% IPA. The material was filtered and further slurry washed with 96% IPA, 10 L/kg and then IPA 10 L/kg. The material was dried under full vacuum at 25°C. The dry cake was dissolved in water 8 L/kg and the batch was concentrated via distillation to remove residual IPA and achieve the desired concentration. The solution temperature was kept below 25 °C throughout the distillation.
PATENT
WO2014210489
SEE
EXAMPLE 1
Solubility of AMG 416 in Succinate Buffered Saline
In this study, the solubility of AMG 416 in succinate buffered-saline was investigated. AMG 416 HC1 (103 mg powder, 80 mg peptide) was dissolved in 200 iL of sodium succinate buffered saline (25 mM succinate, 0.9% saline, pH 4.5). After briefly vortexing, a clear solution was obtained with a nominal concentration of 400 mg/mL. Because expansion of the solution volume was not determined, the solubility of AMG 416 can be conservatively stated as at least 200 mg/mL. Although the maximal solubility was not determined in this experiment, AMG 416 is soluble in pH 4.5 succinate buffered saline to concentrations of at least 200 mg/mL.
REFERENCES
- “Amgen Submits New Drug Application For Novel Intravenous Calcimimetic Etelcalcetide (AMG 416)”
- “Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects
- “Evidence for Chronic Kidney Disease-Mineral and Bone Disorder Associated With Metabolic Pathway Changes”
KAI-4169, a novel calcium sensing receptor agonist, decreases serum iPTH, FGF-23 and improves serum bone markers in a phase 2 study in hemodialysis subjects with chronic kidney disease-mineral and bone disorder
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO054
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO014
Kidney Week (November 5-10, Atlanta, GA) 2013, Abst SA-PO575
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P1-198
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P2-98
44th Annu Meet Am Soc Nephrol (ASN) (November 8-13, Philadelphia) 2011, Abst FR-PO1238
| WO2011014707A2 | Jul 29, 2010 | Feb 3, 2011 | Kai Pharmaceuticals, Inc. | Therapeutic agents for reducing parathyroid hormone levels |
//////////////Etelcalcetide, AMG 416, KAI-4169, velcalcetide, peptide drugs
Multistep Flow Synthesis of 5-Amino-2-aryl-2H-[1,2,3]-triazole-4- carbonitrilesultistep Flow Synthesis of 5-Amino-2-aryl-2H-[1,2,3]-triazole-4- carbonitriles

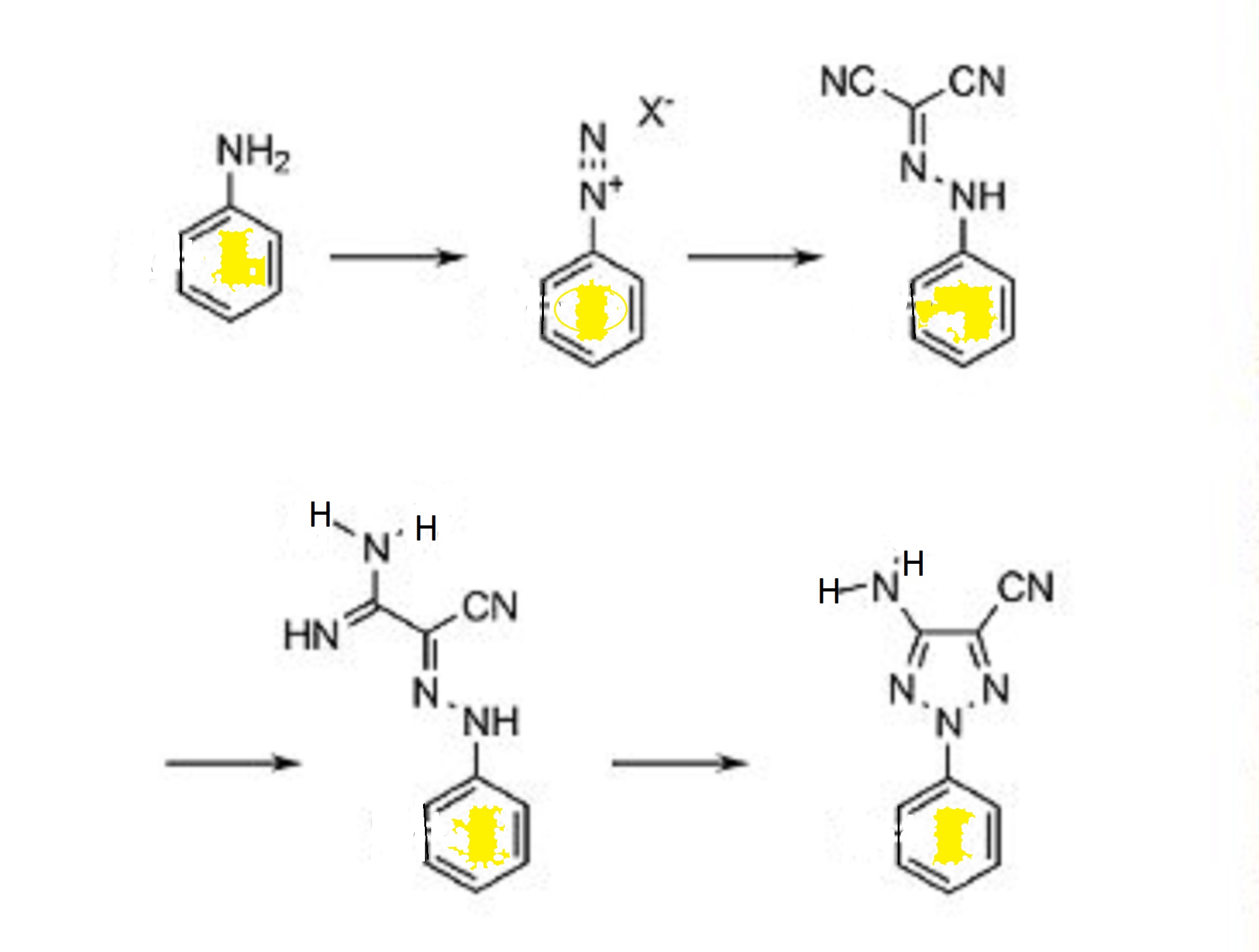
Using the Uniqsis FlowSyn flow chemistry system researchers from the UCB Biopharma. Belgium have developed a flow synthesis of 2-substituted 1,2,3-triazoles that demonstrates improvements over the conventional batch route.
The route involves the diazotisation of anilines and condensation with malononitrile followed by the nucleophilic addition of ammonia or an alkylamine and finally a novel copper catalysed cyclisation. The intermediate azide was generated and consumed in situ which enabled safe scale up under the flow-through conditions employed.
Multistep Flow Synthesis of 5-Amino-2-aryl-2H-[1,2,3]-triazole-4-carbonitriles
Corresponding author
1,2,3-Triazole has become one of the most important heterocycles in contemporary medicinal chemistry. The development of the copper-catalyzed Huisgen cycloaddition has allowed the efficient synthesis of 1-substituted 1,2,3-triazoles. However, only a few methods are available for the selective preparation of 2-substituted 1,2,3-triazole isomers. In this context, we decided to develop an efficient flow synthesis for the preparation of various 2-aryl-1,2,3-triazoles. Our strategy involves a three-step synthesis under continuous-flow conditions that starts from the diazotization of anilines and subsequent reaction with malononitrile, followed by nucleophilic addition of amines, and finally employs a catalytic copper(II) cyclization. Potential safety hazards associated with the formation of reactive diazonium species have been addressed by inline quenching. The use of flow equipment allows reliable scale up processes with precise control of the reaction conditions. Synthesis of 2-substituted 1,2,3-triazoles has been achieved in good yields with excellent selectivities, thus providing a wide range of 1,2,3-triazoles.http://onlinelibrary.wiley.com/wol1/doi/10.1002/chem.201402074/full
1H/13c NMR OF 1a
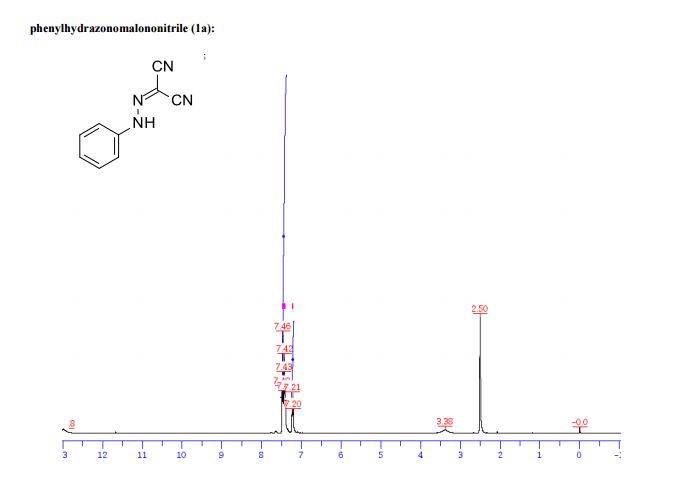

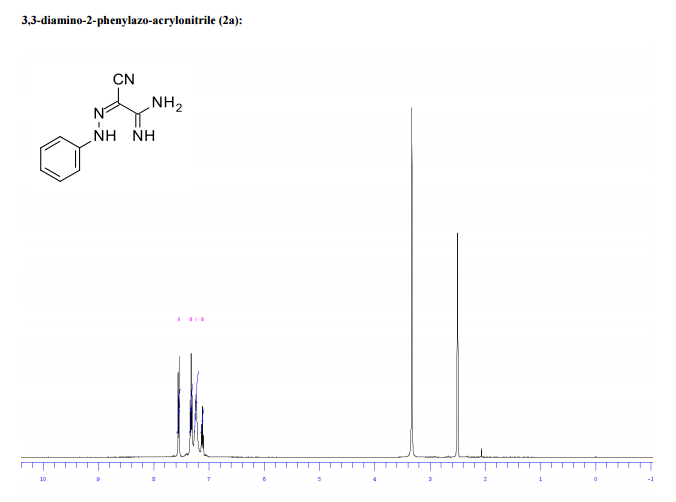
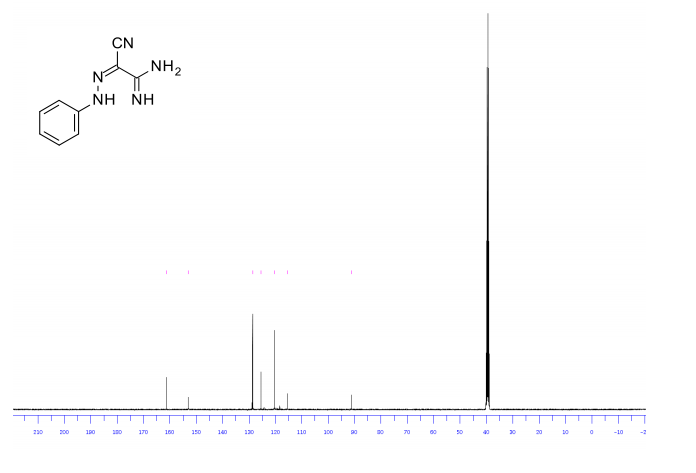
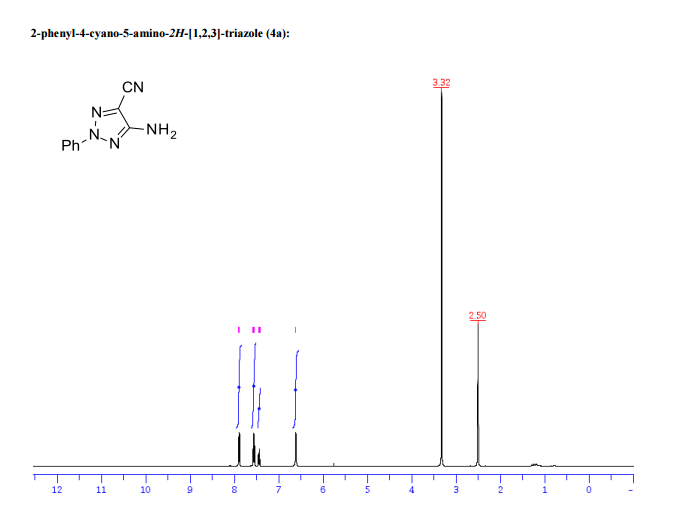
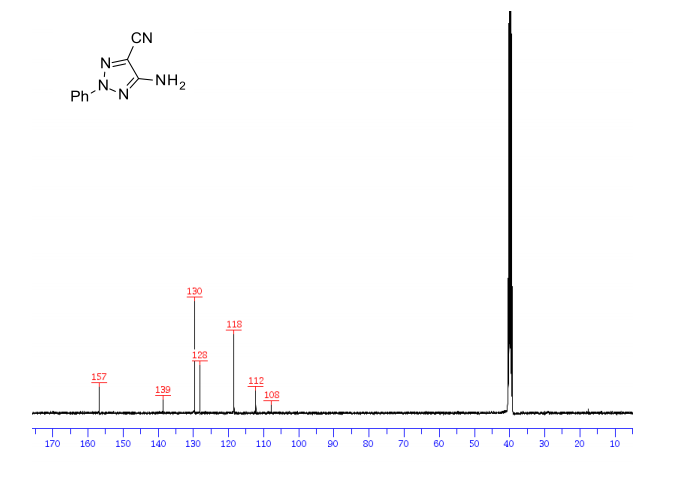
UCB Biopharma, Belgium



Uniqsis FlowSyn
![]()

| Uniqsis Ltd |
| 29 Station Road |
| Shepreth |
| Cambridgeshire |
| SG8 6GB |
| UK |
| Telephone |
| +44 (0)845 864 7747 |
| info@uniqsis.com |




 Halifax survey names South Cambridgeshire as best place to live in rural Britain
Halifax survey names South Cambridgeshire as best place to live in rural Britain
///////////FLOW SYNTHESIS, UCB Biopharma, Belgium, Uniqsis FlowSyn
Monoclonal Antibody Therapy: What is in the name or clear description?
Leaders in Pharmaceutical Business Intelligence Group, LLC, Doing Business As LPBI Group, Newton, MA


Monoclonal Antibody Therapy: What is in the name or clear description?
Curator: Demet Sag, PhD, CRA, GCP
What is in the name?
Nomenclature is important part of the scientific community so we can stay on the same page in all kinds of communications for clarity. Therefore, a defined nomenclature scheme for assigning generic, or nonproprietary, names to monoclonal antibody drugs is used by the World Health Organization’s International Nonproprietary Names (INN) and the United States Adopted Names (USAN). In general, word stems are used to identify classes of drugs, in most cases placed at the end of the word.
Knowing what Antibody relies on understanding of immune response system so that one can modify the cells, choose correct biomarkers from the primary pathways (like Notch, WNT etc), know signaling from outside to inside (like GPCRs, MAPKs, nuclear transcription receptors), personalized gene make up (genomics) and key gene regulation mechanisms. Thus…
View original post 2,927 more words
Finally published: new Annex 16 on QP Certification and Batch Release


Finally published: new Annex 16 on QP Certification and Batch Release
The European Commission finally has published the new EU-GMP Guideline Annex 16 “Certification by a Qualified Person and Batch Release“.
The European Commission has published the final version of the revised EU-GMP Guideline Annex 16 “Certification by a Qualified Person and Batch Release”. Deadline for coming into operation is 15 April 2016.
As one important topic, it has been pointed out that the major task of a Qualified Person (QP) is the certification of a batch for its release. In this context, the QP must personally ensure the responsibilities listed in chapter 1.6 are fulfilled. In chapter 1.7 a lot of additional responsibilities are listed which need to be secured by the QP. The work can be delegated and the QP can rely on the respective Quality Management Systems. However “the QP should have on-going assurance that this reliance is well founded” (1.7). Amongst these twenty-one tasks are for example:
- Starting materials comply and the supply chain is secured, including GMP assessments by third parties
- The necessary audits have been performed and the audit reports are available
- Manufacturing and testing performance are compliant with the MA
- Manufacturing and testing processes are validated
- Changes have been evaluated and investigations completed

It is important to mention in this context that “the ultimate responsibility for the performance of an authorised medicinal product over its lifetime; its safety, quality and efficacy lies with the marketing authorisation holder (MAH). However “the QP is responsible for ensuring that each individual batch has been manufactured and checked in compliance with laws in force (…), in accordance with the requirements of the marketing authorisation (MA) and with Good Manufacturing Practice (GMP)” (see General Principles).
In the case that the QP has to rely on the correct functioning of the quality management system of other sites, the QP “should ensure that a written final assessment and approval of third party audit reports has been made”. The QP should also “be aware of the outcome of an audit with critical impact on the product quality before certifying the relevant batches.”
Another important section clarifies the role of the QP when it comes to deviations, implementing main features of the EMA Position Paper on QP Discretion (which was issued in February 2006 and updated January 2008). Chapter 3 of the draft describes the “handling of unexpected deviations”. A batch with an unexpected deviation from details contained within the Marketing Authorisation and/or GMP may be certified if a risk assessment is performed, evaluating a “potential impact of the deviation on quality, safety or efficacy of the batch(es) concerned and conclusion that the impact is negligible.” Depending on the outcome of the investigation and the root cause, the submission of a variation to the MA for the continued manufacture of the product might be required.
During the consultation phase, stakeholders expressed their concerns regarding the sampling of imported products. Now the new annex is clear on this: “Samples may either be taken after arrival in the EU, or be taken at the manufacturing site in the third country in accordance with a technically justified approach which is documented within the company’s quality system. (…) Any samples taken outside the EU should be shipped under equivalent transport conditions as the batch that they represent.”
The new annex is rather short on other importation requirements. These requirements will probably be defined in the new Annex 21

.////////////published, new Annex 16, QP Certification and Batch Release
The new APIC Guidance on Handling of Insoluble Matter and Foreign Particles in the Manufacture of Active Pharmaceutical Ingredients

The new APIC Guidance on Handling of Insoluble Matter and Foreign Particles in the Manufacture of Active Pharmaceutical Ingredients
The occurrence of foreign particles in the manufacture of active pharmaceutical ingredients is always undesirable. For the responsible QA departments it involves an increased effort as concerns the search for the root causes and for CAPA measures. A new APIC Guidance offers concrete recommendations for the GMP compliant handling of foreign particles in APIs, intermediates and raw materials.

Foreign particles in APIs or medicinal preparations are undesirable and sometimes lead to a recall of the batches concerned. Depending on the type of particles their presence in active pharmaceutical ingredients may be harmless; in many cases they are inevitable. In any case the manufacturer must find an adequate way how to handle those impurities visible to the human eye. The search for a guideline or another official document in the relevant regulations is in vain. Visible particles or fibres are only mentioned in the USP chapter <790>, in chapter 2.9.20 of the European Pharmacopoeia as well as in the United States Food, Drug and Cosmetic Act (FD&C Act).
In order to remedy this lack of guidance or recommendations a group of experts within APIC has drawn up a guidance on the handling of foreign particles. This “Guidance on Handling of insoluble Matter and Foreign Particles in APIs” describes in detail
- the types of particles which can often occur during the manufacture of APIs, API intermediates and raw materials (including packaging materials),
- suitable measures to minimize the presence of particles or to remove them,
- how to determine them analytically
- how to identify the source and to carry out subsequent CAPA measures and an adequate risk management.
This APIC guidance offers valuable assistance for all API manufacturers that are confronted with the problem of the occurrence of foreign particles in their products, intermediates or raw materials. The implementation of the very concrete and practicable recommendations in this guidance offers also valuable supporting arguments for GMP inspections or audits and can help to avoid unpleasant surprises.
///////APIC Guidance, Handling of Insoluble Matter and Foreign Particles, Manufacture, Active Pharmaceutical Ingredients
Israeli scientists turn Nano science fiction into fact

Find out how Israeli scientists are manipulating the tiniest parts of matter to make life better for millions.
Think of a tiny robot transporting drugs to a cancer cell in your body. An artificial retina to restore lost sight. Self-cleaning windows and bullet-proof fabrics.
It’s all possible today with nanotechnology from Israel.
Tune into ISRAEL21c’s TLV1 radio show for a fascinating discussion of how Israeli scientists are turning science fiction into fact. Guests include Nava Swersky Sofer, founder and co-chair of NanoIsrael; Prof. Uriel Levy, head of the Nanotechnology Institute at the Hebrew University of Jerusalem; and Prof. Uri Sivan, one of the Technion’s leading nanotechnology experts……….http://www.israel21c.org/israeli-scientists-turn-science-fiction-into-fact-audio/
![]()

About the INNI mission

The mission of INNI — the Israel National Nanotechnology Initiative is to make nanotechnology the next wave of successful industry in Israel by creating an engine for global leadership.
- Establishing a national policy of resources for nanotechnology, with the aim of faster commercialization.
- Long-range nanotechnology programs for scientific research and technology development in academia and industry, and promoting development of world-class infrastructure in Israel to support them.
- Leading in the creation of projects that promote agreed national priorities; allocate their budgets and review development progress.
- Actively seeking funding resources from public and private sources in order to implement the selected projects.
- Promoting development of innovative local nanotechnology industries which will strongly impact Israeli economic growth and benefit investors.
- Encouraging Academia and Industry cooperation with public access to a national database of Israel’s nanotechnology researchers and industry. Effective access to information about Israel’s researchers and companies accelerates cooperation on R&D projects and on innovative new products. Israel’s nanotechnology National Database may be accessed here or from the link in the INNI website upper navigation menu.

Sivan Uri .
Room 611, Lidow Building
Physics

![]()
Nano Area: Nano Electronics, Nano Materials & Nano Particles, Nanobiotechnology & Nanomedicine
Phone: +972-4-8293452
Fax: +972-4-8292418
Email: phsivan@tx.technion.ac.il
Main
Ph.D.: Tel Aviv University 1988
M.Sc.: Physics, Tel Aviv University 1984
B.Sc.: Physics and Mathematics, Tel Aviv University 1982
Main Nano Field:
Selection of antibodies and peptides against electronic materials, electrical control over bioreactions, bioassembly of electronic devices.
Bertoldo Badler Chair in Physics
Former director of the Russell Berrie Nanotechnology Institute
Head of Ben and Esther Rosenbloom Center of Excellence in Nanoelectronics by Biotechnology

Prof. Uriel Levy of the Hebrew University of Jerusalem has received the Hebrew University President’s Prize as the Outstanding Young Researcher for 2010-11. The prize is awarded in memory of Prof. Yoram Ben-Porath, former president and rector of the Hebrew University.Hebrew University President Prof. Menahem Ben-Sasson said that the prize was being awarded to Prof. Levy “for his impressive list of scientific articles, for his creativity, and for his groundbreaking innovations.”
Prof. Levy is a member of the applied physics department at the Benin School of Computer Science and Engineering and is a renowned researcher in nanophotonics He is a member of the Harvey M. Kruger Family Center for Nanoscience and Nanotechnology at the Hebrew University.
A graduate of the Technion in physics and materials engineering, he subsequently earned a Ph.D. in electro-optics at Tel Aviv University in 2002. He then was awarded a Rothschild Fellowship for post-doctoral work at the University of California, San Diego, which he completed in 2006.
Prof. Levy has published until now 55 scientific articles and has had a number of his research discoveries patented.
Downloadable File: PresidentsPrize2010.doc
The NanoOpto group is affiliated with the Applied Physics Department at the Hebrew University of Jerusalem, Israel. Our research is mainly focused on Silicon Photonics, Polarization Optics, Plasmonics and Opto-Fluidics.
Our group host SPP7 in Jerusalem from 31 of may till the 5 of June 2015:


 Research highlights:
Research highlights:
|
Silicon Photonics
|
| In this work we study the optimization of interleaved Mach-Zehnder silicon carrier depletion electro-optic modulator. Following the simulation results we demonstrate a phase shifter with the lowest figure of merit (modulation efficiency multiplied by the loss per unit length) 6.7V-dB. This result was achieved by reducing the junction width to 200 nm along the phase-shifter and optimizing the doping levels of the PN junction for operation in nearly fully depleted mode. The demonstrated low FOM is the result of both low VπL of ~0.78 Vcm (at reverse bias of 1V), and low free carrier loss (~6.6 dB/cm for zero bias). Our simulation results indicate that additional improvement in performance may be achieved by further reducing the junction width followed by increasing the doping levels. (read more) |
|
Light vapor interactions on a chip
|
| Alkali vapours, such as rubidium, are being used extensively in many important fields of research. Recently, there is a growing effort towards miniaturizing traditional centimetre-size vapour cells. Owing to the significant reduction in device dimensions, light– matter interactions are greatly enhanced, enabling new functionalities due to the low power threshold needed for nonlinear interactions. Here, we construct an efficient and flexible platform for tailored light–vapour interactions on a chip, and demonstrate efficient interaction of the electromagnetic guided mode with absorption saturation at powers in the nanowatt regime. (read more) |

|
Active Silicon Plasmonics
|
| In this work, we experimentally demonstrate an on-chip nanoscale silicon surface-plasmon Schottky photodetector based on internal photoemission process and operating at telecom wavelengths. The responsivity of the nanodetector to be 0.25 and 13.3mA/W for incident optical wavelengths of 1.55 and 1.31 μm, respectively. The presented device can be integrated with other nanophotonic and nanoplasmonic structures for the realization of monolithic opto-electronic circuitry on-chip. (read more) |

|
Plasmonics
|
| Planar plasmonic devices are becoming attractive for myriad applications. Mitigating the challenges of using plasmonics in on-chip configurations requires precise control over the properties of plasmonic modes, in particular their shape and size. Here we achieve this goal by demonstrating a planar plasmonic graded index lens focusing surface plasmons propagating along the device. Focusing and divergence of surface plasmons is demonstrated experimentally. The demonstrated approach can be used for manipulating the propagation of surface plasmons, e.g. for beam steering, splitting, cloaking, mode matching and beam shaping applications (read more) |

|
Metamaterials
|
| The interaction of an incident plane wave with a metamaterial periodic structure consisting of alternating layers of positive and negative refractive index with average zero refractive index is studied. We show that the existence of very narrow resonance peaks for which giant absorption – 50% at layer thickness of 1% of the incident wavelength – is exhibited. Maximum absorption is obtained at a specific layer thickness satisfying the critical coupling condition. This phenomenon is explained by the Rayleigh anomaly and excitation of Fabry Perot modes. (read more) |

|
Plasmonics
|
| Great hopes rest on surface plasmon polaritons’ (SPPs) potential to bring new functionalities and applications into various branches of optics. In this work, we demonstrate a pin cushion structure capable of coupling light from free space into SPPs, split them based on the polarization content of the illuminating beam of light, and focus them into small spots. We also show that for a circularly or randomly polarized light, four focal spots will be generated at the center of each quarter circle comprising the pin cushion device. Furthermore, following the relation between the relative intensity of the obtained four focal spots and the relative position of the illuminating beam with respect to the structure, we propose and demonstrate the potential use of our structure as a miniaturized plasmonic version of the well-known four quadrant detector. (read more) |

|
Silicon Photonics
|
| We demonstrate a nanoscale mode selector supporting the propagation of the first antisymmetric mode of a silicon waveguide. The mode selector is based on embedding a short section of PhC into the waveguide. On the basis of the difference in k-vector distribution between orthogonal waveguide modes, the PhC can be designed to have a band gap for the fundamental mode, while allowing the transmission of the first antisymmetric mode. The device was tested by directly measuring the modal content before and after the PhC section using a near field scanning optical microscope. Extinction ratio was estimated to be ~23 dB. Finally, we provide numerical simulations demonstrating strong coupling of the antisymmetric mode to metallic nanotips. On the basis of the results, we believe that the mode selector may become an important building block in the realization of on chip nanofocusing devices. (read more) |

|
Plasmonics
|
 |
We experimentally demonstrate the focusing of surface plasmon polaritons by a plasmonic lens illuminated with radially polarized light . The field distribution is characterized by near-field scanning optical microscope. A sharp focal spot corresponding to a zero-order Bessel function is observed. For comparison, the plasmonic lens is also measured with linearly polarized light illumination, resulting in two separated lobes. Finally, we verify that the focal spot maintains its width along the optical axis of the plasmonic lens. The results demonstrate the advantage of using radially polarized light for nanofocusing applications involving surface plasmon polaritons. (read more) |






Cutting Edge of Pharmaceutical Nanotechnology

Nanoscience is the engineering of functional systems at the molecular scale. This covers both current work and concepts that are more advanced. In its original sense, nanotechnology refers to the projected ability to construct items from the bottom up, using techniques and tools being developed today to make complete, high performance products. Some researches and findings in the field of Nanoscience are selected and expended here: “Fabrication of Novel Poly (ethylene terephthalate)/TiO2 Nanofibers by Electrospinning and their Photocatalytic Activity” reports on functional nanocomposites PET/TiO2 nanofibers membranes prepared via simple electrospinning and hydrothermal processing, involving preparation of titania precursor sol solution, electrospinning the homogeneous mixture of PET solution and sol solution, and in-situ growth of nanoscale TiO2 within PET nanofibers in hot water.
“Oxidation of glyoxal to glyoxalic acid by Prepared Nano-Au/C catalysts” describes that Nano-Au/C catalysts were obtained by loading the gold nanoparticles which were prepared by photochemical reduction method to the activated carbon, and were used for the catalytic oxidation reaction of glyoxal into glyoxylic acid.
“Preparation of the Al-CNT (Carbon Nanotubes) Compound Material by High Energy Milling” using high energy ball milling (HEM), researched the technology of preparation of Al-CNT compound material.
“Theoretical Prediction of Tensile Behavior of Single-Walled Carbon Nanotubes” establishes a link between molecular and continuum mechanics based on the Morse potential function.
In the paper “Research on the stress-relaxation characteristics of cancer cells based on Atomic Force Microscope”, the AFM indentation experiments are carried out on two different transferring characteristic cancer cells (Anip-937 and AGZY-83a) under physiological conditions using the expansion of atomic force microscope (AFM) indentation and the improvement of Hertz model.
“Application of Nanoscale Zero-valent Iron (nZVI) to Enhance Microbial Reductive Dechlorination of TCE: A Feasibility Study” evaluates the feasibility of nanoscale zero-valent iron (nZVI) application to enhance microbial reductive dechlorination of trichloroethylene (TCE).
“Hydrothermal Processing-Assisted Synthesis of Nanocrystalline YFeO3 and its Visible-Light Photocatalytic Activity” finds that the single phase YFeO3 can be obtained through the calcination of hydrothermally processed YFeO3 precursors at 800°C, and the resulting product has a spherical shape and uniform size distribution.
“Preparation and exothermic characterization of HTPB-coated aluminum nano-powders prepared by laser-induction hybrid heating” calculates the temperature distribution of aluminum with the heating time and the distance from the crucible centre based on the ANSYS software.
“Application Thinking of Nanotechnology in Acupuncture” discusses the application of nanotechnology methods for the researches on meridians of Chinese medicine, acupoint catgut embedding therapy (ACET) and therapeutic mechanism in acupuncture field.
“The Research of Conjunction Calculated Relationships between Proteins with Gold Nanoparticles” researches the conjunction calculated relationship between proteins and gold nanoparticles.
“Engineered nanoparticles as precise drug delivery systems”- Nanoparticles, an evolvement of nanotechnology, are increasingly considered as a potential candidate to carry therapeutic agents safely into a targeted compartment in an organ, particular tissue or cell.

“Dendrimers: emerging polymers for drug-delivery systems”, the unique properties associated with these dendrimers such as uniform size, high degree of branching, water solubility, multivalency, welldefined molecular weight and available internal cavities make them attractive for biological and drug-delivery applications.
“Strategies for in vivo siRNA delivery in cancer”- As a research tool, siRNA has proven to be highly effective in silencing specific genes and modulating intracellular signaling pathways.
“Rapid delivery of drug carriers propelled and navigated by catalytic nanoshuttles”- nanoshuttles’ navigation ability is illustrated by the transport of the drug carriers through a microchannel from the pick-up to the release microwell. Such ability of nanomotors to rapidly deliver drug-loaded polymeric particles and liposomes to their target destination represents a novel approach towards transporting drug carriers in a target-specific manner.
“Multigram-scale fabrication of monodisperse conducting polymer and magnetic carbon nanoparticles” is an emerging tool for cutting edge nanotechnology approach.
Cutting Edge of Pharmaceutical Nanotechnology
Suryakanta Swain*
Suryakanta Swain
Roland Institute of Pharmaceutical Sciences
Department of Pharmaceutics
Khodasinghi, Berhampur-760 010 (Ganjam)
Odisha, India
Email: swain_suryakant@yahoo.co.in
Roland Institute of Pharmaceutical Sciences, Department of Pharmaceutics, IndiaCitation: Swain S (2012) Cutting Edge of Pharmaceutical Nanotechnology. Pharmaceut Reg Affairs 1:e110. doi: 10.4172/2167-7689.1000e110
/////////////Cutting Edge, Pharmaceutical Nanotechnology
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

