
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Fresolimumab

Fresolimumab
GC 1008, GC1008
UNII-375142VBIA
cas 948564-73-6
Structure
- immunoglobulin G4, anti-(human transforming growth factors beta-1, beta-2 (G-TSF or cetermin) and beta-3), human monoclonal GC-1008 γ4 heavy chain (134-215′)-disulfide with human monoclonal GC-1008 κ light chain, dimer (226-226”:229-229”)-bisdisulfide
- immunoglobulin G4, anti-(transforming growth factor β) (human monoclonal GC-1008 heavy chain), disulfide with human monoclonal GC-1008 light chain, dimer
For Idiopathic Pulmonary Fibrosis, Focal Segmental Glomerulosclerosis,and Cancer
An anti-TGF-beta antibody in phase I clinical trials (2011) for treatment-resistant primary focal segmental glomerulosclerosis.
A pan-specific, recombinant, fully human monoclonal antibody directed against human transforming growth factor (TGF) -beta 1, 2 and 3 with potential antineoplastic activity. Fresolimumab binds to and inhibits the activity of all isoforms of TGF-beta, which may result in the inhibition of tumor cell growth, angiogenesis, and migration. TGF-beta, a cytokine often over-expressed in various malignancies, may play an important role in promoting the growth, progression, and migration of tumor cells.

Fresolimumab (GC1008) is a human monoclonal antibody[1] and an immunomodulator. It is intended for the treatment of idiopathic pulmonary fibrosis (IPF), focal segmental glomerulosclerosis, and cancer[2][3] (kidney cancer and melanoma).
It binds to and inhibits all isoforms of the protein transforming growth factor beta (TGF-β).[2]
History
Fresolimumab was discovered by Cambridge Antibody Technology (CAT) scientists[4] and was one of a pair of candidate drugs that were identified for the treatment of the fatal condition scleroderma. CAT chose to co-develop the two drugs metelimumab (CAT-192) and fresolimumab with Genzyme. During early development, around 2004, CAT decided to drop development of metelimumab in favour of fresolimumab.[5]
In February 2011 Sanofi-Aventis agreed to buy Genzyme for US$ 20.1 billion.[6]
As of June 2011 the drug was being tested in humans (clinical trials) against IPF, renal disease, and cancer.[7][8] On 13 August 2012, Genzyme applied to begin a Phase 2 clinical trial in primary focal segmental glomerulosclerosis[9] comparing fresolimumab versus placebo.
As of July 2014, Sanofi-Aventis continue to list fresolimumab in their research and development portfolio under Phase II development.[10]

References
2 National Cancer Institute: Fresolimumab
- 3 Statement On A Nonproprietary Name Adopted By The USAN Council – Fresolimumab
- 4 Grütter, Christian; Wilkinson, Trevor; Turner, Richard; Podichetty, Sadhana; Finch, Donna; McCourt, Matthew; Loning, Scott; Jermutus, Lutz; Grütter, Markus G. (2008-12-23). “A cytokine-neutralizing antibody as a structural mimetic of 2 receptor interactions”. Proceedings of the National Academy of Sciences 105 (51): 20251–20256. doi:10.1073/pnas.0807200106. ISSN 0027-8424. PMC 2600578. PMID 19073914.
- 5 http://www.independent.co.uk/news/business/news/cat-may-abandon-skin-drug-after-trial-results-disappoint-569445.html
- 6 http://www.bbc.co.uk/news/business-12477750
- 7 http://www.genengnews.com/gen-news-highlights/scientists-trigger-white-fat-to-become-brown-fat-like-to-treat-obesty-and-type-2-diabetes/81245389/
- 8 Clinicaltrials.gov for Fresolimumab
- 9 http://clinicaltrials.gov/show/NCT01665391
- 10 http://en.sanofi.com/rd/rd_portfolio/rd_portfolio.aspx
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | TGF beta 1, 2 and 3 |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Number | 948564-73-6 |
| ATC code | None |
| ChemSpider | none |
| KEGG | D09620 |
| Chemical data | |
| Formula | C6392H9926N1698O2026S44 |
| Molar mass | 144.4 kDa |
////////////
GCC 4401C , GC 2107 , Nokxaban for treating thrombosis

GCC-4401C ( GC-2107), Nokxaban
In phase 1 for treating thrombosis
5-chloro-N-({(5S)-2-oxo-3-[4-(5,6-dihydro-4H-[1,2,4]triazin-1-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide methanesulfonate
5-chloro-N-[[3-[4-(5,6-dihydro-2H-1,2,4-triazin-1-yl)phenyl]-2-oxo-1,3-oxazolidin-5-yl]methyl]thiophene-2-carboxamide
CB02-0133; GC-2107; GC4401; GCC-2107; GCC-4401; GCC-4401C; I Fxa – LegoChem Biosciences; LCB02-0133; Nokxaban
WO2010002115; LegoChem Bioscience INNOVATOR
| Green Cross Corporation, Legochem Bioscience Ltd. |
![]()
DEVELOPER
CAS NO FREE FORM

CAS 1159610-29-3, 159610-29-3, C18 H18 Cl N5 O3 S
2-Thiophenecarboxamide, 5-chloro-N-[[(5S)-3-[4-(5,6-dihydro-1,2,4-triazin-1(2H)-yl)phenyl]-2-oxo-5-oxazolidinyl]methyl]-
Molecular Formula: C18H18ClN5O3S Molecular Weight: 419.88522 g/mol
METHANE SULFONATE

CAS 1261138-12-8, C18 H18 Cl N5 O3 S . C H4 O3 S,
2-Thiophenecarboxamide, 5-chloro-N-[[(5S)-3-[4-(5,6-dihydro-1,2,4-triazin-1(2H)-yl)phenyl]-2-oxo-5-oxazolidinyl]methyl]-, methanesulfonate (1:1)
HYDROCHLORIDE
CAS 1261138-08-2., C18 H18 Cl N5 O3 S . Cl H, 2-Thiophenecarboxamide, 5-chloro-N-[[(5S)-3-[4-(5,6-dihydro-1,2,4-triazin-1(2H)-yl)phenyl]-2-oxo-5-oxazolidinyl]methyl]-, hydrochloride (1:1)

SUMMARY
- 09 Jan 2015GC 2107 is available for licensing as of 09 Jan 2015. http://www.greencross.com
- 01 May 2014Green Cross Corporation completes a phase I trial in Healthy volunteers in USA (NCT01954238)
- 26 Sep 2013Green Cross initiates enrolment in a phase I trial in Healthy volunteers in USA (NCT01954238)
Used as factor Xa antagonist for treating coronary artery disease, inflammatory disease, myocardial infarction and thrombosis.
Green Cross Corp in collaboration with LegoChem Bioscience, is developing GCC-4401C ( phase I), for treating thrombosis including venous thromboembolism
Development and Market Objectives
Green Cross Corporation is developing an orally available direct Factor Xa inhibitor, GCC-4401C, which has shown an excellent safety profile during Phase I clinical study. After completion of Phase II and III studies for the prevention of venous thromboembolism (VTE) on hip or knee replacement surgery patients, we will explore additional indications for the treatment of acute coronary syndromes and the prevention of stroke in patients with atrial fibrillation.
Unmet Medical Need & Target Patients

GCC-4401C may prove its greatest impact in providing a much-needed and attractive alternative to warfarin in various indications. Prophylaxis of deep vein thrombosis (DVT), which may lead to pulmonary embolism in patients undergoing hip or knee arthroplasty, is considered to be a primary unmet medical need. It is the most common cause for rehospitalisation in this patient group. Each year in the United States, between 350,000 and 600,000 people experience a blood clot in the legs or in the lungs. The US and European hip and knee implant markets are the two largest, accounting for nearly 80 percent of total procedures conducted worldwide. The 2005 revenues for hip and knee implants in the US and Europe were $6.5 billion. Demand driven by an aging population and an increasing number of younger patients are contributing to the continuous growth of hip and knee replacement procedures.
Thromboembolism involving arterial or venous circulation is a common cause of morbidity and mortality. As an anticoagulation therapy, heparin and Vitamin K antagonists (VKAs) such as warfarin have been used in clinical settings for more than 50 years, but both are associated with several limitations requiring frequent coagulation monitoring due to unpredictable effects of anticoagulant . Therefore, there is an urgent need for novel, oral agents with a predictable anticoagulant action. The greatest unmet medical need in anticoagulation therapy is to find a replacement for VKAs for long-term therapy, particularly stroke prevention in patients with atrial fibrillation (a heart rhythm disorder). Recently, Factor Xa has emerged as an attractive target for novel anticoagulants and a number of Factor Xa inhibitors are currently under development as oral anticoagulants for long-term use.
A major unmet medical need is for direct FXa inhibitors that are simpler to administer than VKAs, with fewer strokes and less intracranial bleeding compared with warfarin and less bleeding yet similar or better efficacy with a lower-dose regimen. In addition, the availability of simple, fixed-dose, unmonitored therapies should increase the use of direct FXa inhibitor therapy in patients with atrial fibrillation at risk for stroke.
Status
Phase I Clinical Study
To investigate the safety and tolerability of single doses of GCC-4401C in healthy male subjects, a Phase Ia study (GCC-4401C-101) was recently conducted at Quintiles in the United States under the conditions of randomized, double-blind, placebo-controlled, and single ascending dose. Forty eight healthy male subjects were enrolled in 6 cohorts and administered at 6 dose-escalation levels up to 80 mg/subject. GCC-4401C was well-tolerated without any significant adverse events, and was detected in blood plasma dose-proportionally across the dose range of 2.5 mg to 80 mg per patient. The pharmacodynamic variables were also statistically correlated with GCC-4401C plasma concentrations.
We plan to characterize the safety, tolerability, pharmacokinetics and pharmacodynamics of multiple doses of GCC-4401C in healthy male subjects based on the safety margins of the SAD study. An appropriate dose and dosing regimen of oral GCC-4401C from subsequent clinical trials on VTE patients are expected to be identified. The Phase 1b study will be completed with Global CRO in the US in 3Q, 2014.
Intellectual Property
Material patent for GCC-4401C, covering a wide range of chemical structures, was awarded in early 2008 within S. Korea, followed by its production method patent in early 2011. Moreover, patent applications for both material and production method, are in progress in 21 and 5 overseas countries including the US, respectively.
– KR811865 : Pyrimidinone derivatives or pyridazinone derivatives for inhibition of factor VIIa activity
– KR109594 : FXa inhibitors with cyclic amidines as P4 subunit, processes for their preparations, and pharmaceutical compositions and derivatives thereof
– KR898361 : FXa inhibitors with cyclic amidoxime or cyclic amidrazone as P4 subunit, processes for their preparations, and pharmaceutical compositions and derivatives thereof
– KR1037051 : Method for preparing of (S)-5-chloro-N-((3-(4-(5,6-dihydro-4H-1,2,4-oxadiazin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methyl)thiophene-2-carboxamide derivatives
– KR1037052 : Method for preparing 5-chloro-N-(((5S)-2-oxo-3-(4-(5,6-dihydro-1,2,4-triazin-1(4H)-yl)phenyl)-1,3-oxazolidin-5-yl)methyl)thiophen-2-carboxamide derivatives, and their intermediates
– PCT/KR2010/004420 : Method for preparing (S)-5-chloro-N-((3-(4-(5,6-dihydro-4H-1,2,4-oxadiazin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methyl)thiophene-2-carboxamide derivatives
– PCT/KR2010/004421 : Method for preparing 5-chloro-N-({(5S)-2-oxo-3-[4-(5,6-dihydro-4H-[1,2,4]triazin-1-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide derivative and intermediate used therein
Competitive Advantages

GCC-4401C has been specifically designed for chronic, once-a-day treatment. It has a half-life that supports true, once-daily dosing and a low peak-to-trough drug concentration ratio that minimizes anticoagulant variability. Since GCC-4401C has an excellent aqueous solubility, there has been potential for the development of both po and iv formulations. Data from comparative efficacy studies in animals have also demonstrated the superiority of GCC-4401C against other direct FXa inhibitors with less bleeding effects. From the recent Phase Ia clinical study, GCC-4401C did not show any significant sign of adverse events. PK parameters and PD markers were predictable dose-proportionally across the all dose ranges. GCC-4401C is expected to show excellent safety profiles, less bleeding and less liver toxicity through human clinical studies.
Contact & Company Overview
![]()
-
 Company Name :Green Cross Corporation
Company Name :Green Cross Corporation -
 Homepage :
Homepage : -
 Contact Person :Hyoung Geun Beak, Deputy General Manager, Business
Contact Person :Hyoung Geun Beak, Deputy General Manager, Business -
 E-mail :
E-mail : -
 Contact :+82-31-260-9337
Contact :+82-31-260-9337
PATENT
GREEN CROSS CORPORATION [KR/KR]; 107, Ihyeon-ro 30beon-gil, Giheung-gu, Yongin-si, Gyeonggi-do 446-770 (KR).
LEGOCHEM BIOSCIENCES, INC. [KR/KR]; 8-26, Munpyeongseo-ro, Daedeok-gu, Daejeon 306-220 (KR)
The present invention relates to a novel crystalline form of 5-chloro-N-({(5S)-2-oxo-3-[4-(5,6-dihydro-4H-[1,2,4]triazin-1-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide methanesulfonate and a pharmaceutical composition containing the same. The novel crystalline form of a compound according to the present invention exhibits excellent stability even in high-temperature and humidity environments, and thus can be favorably used to prevent or treat diseases, such as thrombosis, myocardial infarction, atherosclerosis, inflammation, stroke, angina pectoris, restenosis after angioplasty, and thromboembolism.
According to the present invention 5-chloro -N – ({(5 S) -2- oxo-3- [4- (5,6-dihydro the -4H- [1, 2, 4] triazine-1-yl) phenyl] -1, 3-oxazolidin-5-yl} methyl) thiophene-2-mid copy methane sulfonic acid salt (hereinafter referred to as a new crystal form has excellent solubility referred to) in “GCO4401C”, Ko Un and wet environments It is excellent in stability.
Novel crystalline forms of GCC-4401C of the present invention, the organic solvent under reduced pressure crystallization method, a cooling crystallization method or solvent-can be easily obtained by the anti-solvent crystallization process.
Ateumyeo GCC-4401C is used as a reaction raw material can be prepared according to the procedure described in PCT Publication No. W02011 / 005029 No., dissolving the starting compound in an organic solvent the semi-adding a solvent after filtration to determine the resulting mixture was cooled and then dried to give the novel crystalline form can be a compound according to the invention.
PATENT
http://www.google.com/patents/WO2011005029A2?cl=en
5-Chloro-N-( {(5S)-2-oxo-3-[4-(5,6-dihydro-4H-[ 1 ,2,4]triazin- 1-yl)phenyl]-l,3-oxazolidin-5-yl}-methyl)thiophene-2-carboxamide of formula (A) has been known as an inhibitor of blood coagulation factor Xa and used for treating and preventing thrombosis, myocardial infarction, arteriosclerosis, inflammation, stroke, angina pectoris, recurrent stricture after angioplasty, and thromboembolism such as intermittent claudication.
Korea Patent No. 2008-64178, whose application has been filed by the present invetors, discloses a use of the compound as an inhibitor of blood coagulation factor Xa and a preparation method thereof. The preparation method comprises the step of preparing a cyclic amidrazone starting from 4-nitroaniline, as shown in reaction scheme 1 :
Reaction Scheme 1

Specifically, the cyclic amidrazone (A) is prepared by the steps of: preparing the compound (B) using 4-nitroaniline; treating the compound (B) with a t-butoxycarbonyl amine protecting group to prepare the compound (C); introducing a nitroso group into the compound (C) using NaNO2, followed by reduction using zinc to prepare the compound (D); and treating the compound (D) successively with hydrochloric acid and an ortho-formate.
However, the above preparation method is complicated and gives a low yield of the compound (A) (e.g., a total yield of 9 %), and it also requires the use of a column chromatography purification step, which limits mass production of the cyclic amidrazone. In particular, the step for preparing the compound (D) from the compound (C) is required to use a harmful heavy metal-containg materal such as zinc amalgam which gives an unsatisfactorily low yield, and the isolation step of the compound (D) does not proceed easily.
Reaction Scheme 2

Reaction Scheme 3

Example 1: Preparation of Ethyl formimidate hydrochloride

To a solution of benzoyl chloride (1212 g, 8.62 mol, 1 eq) in anhydrous ether (5.8 L) was added dropwise a solution of formamide (388 g, 8.62 mol, 1 eq) in EtOH (396 g, 8.60 mol, 0.998 eq) at 0 °C for lhr. The mixture thus obtained was stirred at 0 °C for 30min. The solid was filtered off, washed with ether (3 L) and EA (3 L). The solid was dried under high vacuum.
Yield : 625 g (66%)
Example 1: 5-chloro-N-({(5S)-2-oxo-3-[(5,6-dihydro-lH-[l,2,4]triazin-4-yl)phenyl]-l,3-oxazolidin-5-yl}-methyl)-2-thiophene carboxamide hydrochloride
Step 1: Preparation of 2- [N-(4-nitro-phenyl)-hydrazino]-ethanol

l-Fluoro-4-nitrobenzene (7.1 g, 50 mmol) was dissolved in CH3CN (70 ml), 2-hydroxyethylhyrazine (purity: 90 %, Aldrich, 5.0 g, 66 mmol) and K2CO3 (7.6 g, 55 mmol) were added thereto. The suspension thus obtained was stirred for 4 hrs with reflux. The resulting orange-colored suspension was concentrated under reduced pressure (reflux condenser, 10 torr, 40 °C) and ethylacetate (EA, 90 ml) and water (18 ml) were added thereto. The resulting mixture was stirred strongly at r.t. for 10 min. The organic layer was extracted and washed with the saturated brine (10 ml). The resulting solution was cooled to 10 °C and 48 % HBr solution (3.7 ml) was added thereto dropwise with stirring. The pale yellow colored solid thus obtained was filtered off and dried under high vacuum (1 torr, 40 “C) to obtain the title compound as an intermediate.
Yield: 7.1 g (51 %).
TLC : Rf= 0.62 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (600 MHz, DMSO-J6) δ 8.17 (d, J = 9.0 Hz, 2H), 7.12 (d, J = 9.0 Hz, 2H), 3.82 (t, J= 5.4 Hz, 2H), 3.69 (t, J= 5.4 Hz, 2H)
LCMS: 198 (M+H+) (C8H11N3O3)
Step 2: Preparation of l-bromo-2-[N-(4-nitro-phenyl)-hydrazino] -ethane

The compound obtained in Step 1 (38.9 g, 0.140 mol) was suspended in anhydrous 1 ,2-dimethoxyethane (585 ml). The resultant suspension was cooled to 0 °C and PBr3 (15.9 ml, 0.168 mol) was added thereto dropwise for 30 min. The mixture thus obtained was stirred at 60 °C for 4 hrs. The pale yellow colored solution thus obtained was concentrated under reduced pressure (reflux condenser, 10 torr, 45 °C). The resultant residue (oil) was suspended with water (150 ml) and stirred. Aq. sat’d NaHCO3 solution (150 m) was added to the resultant suspension to be pH 4. The resulting mixture was stirred for 30 min to precipitate the pale yellow colored precipitates. The precipitates were filtered off and washed with water (100 ml). The resulting solid was mixed with water (100 ml), aq. sat’d NaHCO3 solution (70 ml) and CH2Cl2 (500 ml). The resulting mixture was stirred for 10 min and stood to separate organic and aqueous layers. The organic layer was dried over 20 g of MgSO4 and filtered off. The resulting filterate was concentrated under reduced pressure (reflux condenser, 10 torr, 40 °C) to obtain the title compound as a pale yellow solid.
Yield : 31.3 g (86 %)
TLC : Rf= 0.91 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (600 MHz, CDCl3) δ 8.14 (d, J = 10.2 Hz, 2H), 6.92 (d, J= 10.2 Hz, 2H), 4.00 (t, J= 7.2 Hz, 2H), 3.65 (t, J= 7.2 Hz, 2H)
LCMS: 261 (M+H+) (C8H10BrN3O2)
Step 3: Preparation of 4-(5,6-dihydro-4H-[l,2,4]triazin-l-yl)-l-nitrobenzene

The compound obtained in Step 2 (13.0 g, 50.0 mmol) was completely dissolved in anhydrous 1,2-dimethoxyethane (200 ml) which is prepared by mixing 1,2-dimethoxyethane (purity: 99 %, Junsei Co. Ltd) with an desired amount of molecular sieve 4A and standing for 5 hrs or more with stirring at times. Ethyl formimidate HCl salt (5.8 g, 52.5 mmol) was added thereto. The suspension thus obtained was stirred at 25 °C for 10 min. Anhydrous sodium acetate (NaOAc, 8.6 g, 105 mmol) was added thereto and stirred for 15 hrs with reflux. The orange colored suspension thus obtained was concentrated under reduced pressure (10 torr, 50 “C). The orange colored residue thus obtained was mixed with IN HCl (140 ml), EA (50 ml) and hexane (100 ml), and stirred at r.t for 10 min. A small amount of insoluble suspended solids was remained in aqueous layer and filtered off. The resulting aqueous layer was washed with a mixture of EA (30 ml) and hexane (60 ml). 12 g of sodium carbonate was added to the resulting solution to be pH 8.5. The orange colored solid thus obtained was filtered off under reduced pressure, washed with water (15 ml) and dried under vacuum to obtain the title compound .
Yield : 7.7 g (75 %).
TLC : R/= 0.45 (EA/MeOH/AcOH = 20/1/0.5)
HPLC : R, = 8.65 (Gradient A), purity 91.1%
1H NMR (400 MHz, DMSO-^6) δ 8.03 (d, J= 9.6 Hz, 2H), 7.16 (d, J = 9.6 Hz, 2H), 7.12 (br s, IH), 7.01 (d, J= 4.0 Hz, 2H), 3.77 (t, J= 5.2 Hz, 2H), 3.43-3.40 (m, 2H)
LCMS: 207 (M+H+) (C9H10N4O2)
Step 4: Preparation of 4-(5,6-dihydro-4-t-butoxycarbonyl-[l,2,4]triazin-l-yl)-1-nitrobenzene
![]()
To the orange colored suspension prepared by suspending the compound obtained in Step 3 (12.4 g, 60 mmol) in tetrahydrofurane (THF, 200 ml), 4-dimethylaminopyridine (DMAP, 0.367 g, 3 mmol) and di-tert-butyl dicarbonate
(BoC2O, 19.6 g, 90 mmol) were added and stirred with reflux for 1.5 hrs. The yellow colored suspension thus obtained was concentrated under reduced pressure
(reflux condenser, 10 torr, 40 °C) to remove the solvent. The resulting yellow colored residue was completely dissolved in CH2Cl2 (700 ml) and washed with IN HCl (700 ml). The organic layer was extracted, dried over 25 g of MgSO4, and concentrated under reduced pressure (condenser, 10 torr, 40 °C). The resultant yellow colored residue was dissolved in cyclohexane (250 ml) and stirred strongly at r.t. for 30 min. The resulting mixture was concentrated under reduced pressure to obtain yellow colored solids. The solids were dried (1 torr, 50 °C ) to obtain a disried compound.
Yield: 15.6 g (85 %)
TLC : R/= 0.93 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (600 MHz, DMSO-J6) δ 8.14 (d, J= 9.6 Hz, 2H), 7.62 (br s, IH), 7.30 (d, J = 9.6 Hz, 2H), 3.89 (br s, 2H), 3.79 (br s, 2H), 1.50 (s, 9H)
LCMS: 307 (M+H+) (C14H18N4O4)
Step 5: Preparation of 4-(5,6-dihydro-4-t-butoxycarbonyl-[l,2,4]triazin-l-yl)aniline
![]()
To the yellow colored suspension prepared by suspending the compound obtained in Step 4 (19.9 g; 65 mmol) in methanol (200 ml), 10 % palladium on carbon (4.0 g) was added. The resulting mixture was subjected to vacuum outgassing and stirred at r.t., for 2 hrs in the flask connected with hydrogen bollum. The resulting mixture was filtered through celite 545 under redued pressure to remove the palladium on carbon. The fϊlterate was concentrated under reduced pressure (reflux condenser, 10 torr, 40 °C). The resulting pale brown colored residue was dissolved in isopropylalcohol (140 ml) and refluxed to dissolve completely. The resulting solution was stood at 0 °C for 2 hrs to cool, stirred for 30 min and filtered off under redued pressure. The resulting ivory crystalline solid was dried in vacuo to obtain the title compound (15.8 g, 88 %).
TLC : Rf= 0.38 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (400 MHz, DMSO-(I6) δ 7.34 (br s, IH), 6.91 (d, J = 12.0 Hz, 2H), 6.51 (d, J = 12.0 Hz, 2H), 6.64 (br s, 2H), 3.74 (br s, 2H), 3.41 (br s, 2H), 1.48 (s, 9H)
LCMS: 277 (M+H+) (C14H20N4O2)
Step 6: Preparation of N-(3-(5,6-dihydro-4-t-butoxycarbonyl-[l,2,4]triazin-l-yI)anilino-(2R)-2-hydroxypropyI)-5-chloro-2-thiophene carboxamide

The compound obtained in Step 5 (19.3 g, 70 mmol) and 5-chloro-N-(((S)-oxiran-2-yl)methyl)thiophene-2-carboxamide (19.1 g, 88 mmol) were suspended in isobutyl alcohol (350 ml) and stirred for 18 hrs with reflux. The dark blue colored solution thus obtained was concentrated under reduced pressure (reflux condenser, 10 torr, 50 °C). To the yellow solid residue thus obrained, ethylacetate (200 ml) was added and the resulting mixture was stirred at r.t. for 30 min and further stirred strongly at 0 °C for 30 min. The suspended solid thus obtained was filtered off under reduced pressure and dried in vaccum (1 torr, 50 °C ) to obtain the title compound as ivory crude.
Yield : 25.9 g (75 %)
TLC : R/= 0.34 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR of a crude sample (600 MHz, DMSO-</6) δ 8.62 (t, J = 5.4 Hz, IH), 7.69 (d, J = 3.6 Hz, IH), 7.36 (br s, IH), 7.18 (d, J = 4.2 Hz, IH), 6.95 (d, J = 9.0 Hz, 2H), 6.54 (d, J = 9.0 Hz, 2H), 5.10 (t, J = 6.6 Hz, IH), 5.05 (d, J = 5.4 Hz, IH), 3.81-3.75 (m, 3H), 3.44 (br s, 2H), 3.37-3.34 (m, IH), 3.25-3.21 (m, IH), 3.08-3.04 (m, IH), 2.94-2.89 (m, IH), 1.48 (s, 9H)
LCMS: 494 (M+H+) (C22H28ClN5O4S)
Step 7: Preparation of 5-chloro-N-({(5S)-2-oxo-3-[(5,6-dihydro-4-t-butoxycarbonyl-[l,2,4]triazin-l-yl)phenyl]-l,3-oxazolidin-5-yI}-methyl)-2-thiophene carboxamide

The compound obtained in Step 6 (25.2 g, 51 mmol) was completely dissolved in THF (325 ml), and Ll’-carbonyldiimidazole (10.8 g, 66 mmol) and DMAP (0.31 mg, 2.6 mmol) were added thereto. The resulting mixture was stirred with reflux for 18 hrs. The resulting pale yellow colored suspension was cooled to r.t, concentrated under reduced pressure and dried in vacuo (1 torr, 50 °C) to obtain the title compound as an ivory solid.
Yield : 23.3 g (88 %)
TLC : R/= 0.75 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (400 MHz, DMSO-J6) δ 8.97 (t, J = 5.4 Hz, IH), 7.69 (d, J= 4.2 Hz, IH), 7.43 (br s, IH), 7.41 (d, J = 9.0 Hz, 2H), 7.20 (d, J = 4.2 Hz, IH), 7.19 (d, J= 9.0 Hz, 2H), 4.82-4.77 (m, IH), 4.12 (t, J= 9.0 Hz, IH), 3.80-3.78 (m, 3H), 3.62 (br s, 2H), 3.59 (t, J= 6.0 Hz, 2H), 1.49 (s, 9H)
LCMS: 520 (M+H+) (C23H26ClN5O5S)
Step 8: Preparation of 5-chloro-N-({(5S)-2-oxo-3-[(5,6-dihydro-4H-[l,2,4]triazin-l-yl)phenyl]-l,3-oxazolidin-5-yl}-methyl)-2-thiophene
carboxamide hydrochloride

The compound obtained in Step 7 (16.1 g, 31 mmol) was completely
dissolved in THF (193 ml), 3N HCl (193 ml) was added thereto. The resulting solution was stirred with reflux for 1 hr. The white suspension thus obtained was cooled tq r.t, concentrated under reduced pressure and dried in vacuo (1 torr, 40 °C ) to obtain the title compound as a white solid.
Yield : 13.4 g (95 %)
TLC : R/= 0.82 (MC/MeOH/AcOH = 10/1/0.5)
HPLC : R, = 12.39 (Gradient A), purity 99.5%
1H NMR (600 MHz, OMSO-d6) δ 12.12 (br s, IH), 10.20 (br s, IH), 9.08
(t, J = 6.0 Hz, IH), 8.60 (d, J = 5.2 Hz, IH), 7.74 (d, J= 4.2 Hz, IH), 7.53 (d, J = 9.0 Hz, 2H), 7.20 (d, J= 4.2 Hz, IH), 7.13 (d, J= 9.0 Hz, 2H), 4.85-4.81 (m, IH),
4.15 (t, J = 8.8 Hz, IH), 3.85 (dd, J = 6.0, 9.2 Hz, IH), 3.66 (t, J = 4.8 Hz, 2H),
3.63-3.56 (m, 2H), 3.19 (br s, 2H)
LCMS: 420 (M+H+) (C18H18ClN5O3S)
Example 2: Preparation of 5-chloro-N-({(5S)-2-oxo-3-[(5,6-dihydro-4H-[l,2,4]triazin-l-yI)phenyl]-l,3-oxazolidin-5-yl}-methyI)-2-thiophene
carboxamide
The HCl salt obtained in Example 1 (6.9 g, 15 mmol) was completely dissolved in 33 % methanol aqueous solution (1.1 L) and heated to 50 °C while stirring. To the resulting colorlessness solution, 0.6M aq. Na2CO3 solution (25 ml) was added and the white suspension thus obtained was stood at 0 °C for 0.5 hr to cool. The white solid thus obtained was concentrated under reduced pressure, wished with H2O (150 ml) and dried in vacuo (1 torr, 40 “C) to obtain the title compound (yield: 5.5 g, 87 %). The title compound was dissolved in methanol (330 ml) and stirred with reflux. The pale yellow colored solution thus obtained was stood at 0 °C for 2 hrs to cool. The resulting white solid was concentrated under reduced pressure, washed with methanol (10 ml), and dried in vacuo (1 torr, 40 “C) to obtain a crystal of the title compound (yield: 5.0 g, 80 %).
HPLC : R, = 12.37 (Gradient A), purity 99.7 %
1H NMR (400 MHz, DMSO-^6) δ 8.97 (t, J = 6.0 Hz, IH), 7.69 (d, J = 4.0 Hz, IH), 7.32 (d, J = 9.2 Hz, 2H), 7.20 (d, J = 4.0 Hz, IH), 7.12 (d, J = 9.2 Hz, 2H), 6.79 (d, J = 4.0 Hz, IH), 6.52 (br s, IH), 4.80-4.75 (m, IH), 4.10 (t, J = 8.8 Hz, IH), 3.77 (dd, J= 6.0, 9.2 Hz, IH), 3.58 (t, J= 5.6 Hz, 2H), 3.33 (s, 4H)
LCMS: 420 (M+H+) (C18H18ClN5O3S)
Example 3: Preparation of 5-chloro-N-({(5S)-2-oxo-3-[(5,6-dihydro-4H-[l,2,4]triazin-l-yl)phenyl]-l,3-oxazolidin-5-yI}-methyI)-2-thiophene carboxamide methane sulfonate

To the compound obtained in Example 2 (3.3 g, 7.9 mmol), a mixture solution of MeOH/CH2Cl2 (1/4 v/v, 70 ml) was added and stirred with reflux. The pale yellow colored solution thus obtained was cooled to 0 °C and methylsulfonic acid (0.56 ml, 8.6 mmol) was added thereto. The resulting mixture was concentrated under reduced pressure (reflux condenser, 10 torr, 40 °C) to obtain pale yellow foamy solid. To the resultant solid, absolute ethanol (20 ml) was added and the resulting mixture was stirred with reflux to dissolve solid clearly. The resulting solution was cooled to 0 °C to 2 hrs. The resulting white solid was concentrated under reduced pressure, washed with absolute EtOH (5 ml), and dried in vacuo (1 torr, 40 “C) to obtain a crystalline methane sulfonate.
Yield : 3.8 g (93 %)
HPLC : R, – 12.35 (Gradient A), purity 99.8%
1H NMR (400 MHz, DMSO-CZ6) δ 11.97 (br s, IH), 10.07 (br s, IH), 8.99
(t, J= 6.0 Hz, IH), 8.59 (U1 J= 6.0 Hz, IH), 7.70 (d, J= 4.0 Hz, IH), 7.53 (d, J =
9.2 Hz, 2H), 7.20 (d, J= 4.0 Hz, IH), 7.13 (d, J= 9.2 Hz, 2H), 4.86-4.80 (m, IH),
4.16 (t, J = 9.2 Hz, IH), 3.82 (dd, J = 6.0, 9.2 Hz, IH), 3.67 (m, 2H), 3.60 (t, J = 5.6 Hz, 2H), 3.20 (br s, 2H), 2.31 (s, 3H)
LCMS: 420 (M+H+)(C18H18ClN5O3S)
Example 4: (S)-5-chloro-N-((3-(4-(5,6-dihydro-l,2,4-triazin-l(4H)-yl)phenyI)-2-oxooxazolidin-5-yl)methyl)thiophene-2-carboxamide methane sulfonate
Step 1: Preparation of (2-[N-(4-nitro-phenyl)-hydrazinyl]-ethanol) hydrobromide

l-Flouro-4-nitrobenzene (428 g, 3.03 mol, Aldrich Fl 1204) was dissolved in CH3CN (4.3 L), and 2 -hydroxy ethylhyrazine (300 g, 3.94 mol, 1.3 eq, imported from China, >98 %) and K2CO3 (461 g, 3.34 mol, 1.1 eq, Aldrich
347825) were added thereto. The mixture thus obtained was stirred at 80 °C for
19 hrs. The mixture was cooled to r.t. and evaporated to remove solvent. The residue was dissolved with EA (1.5 L) and H2O (1 L). The organic layer was extracted and washed with H2O (500 mL) and brine (200 mL). The extracted
EA layer was cooled to 0 °C and 48 % HBr solution (360 mL, Aldrich 244260) was added thereto dropwise at 0 °C with stirring. The resultant mixture was stirred at 0 °C for 1 hr. The solid thus obtained was filtered off and washed with
EA (5 L). The obtained solid was dried under high vacuum to obtain the title compound.
Yield : 531 g (63 %)
TLC : Rf= 0.62 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (400 MHz, OMSO-d6) δ 7.94 (d, J = 9.6 Hz, 2H), 7.12 (br s, 2H), 6.63 ![]()
5.8 Hz, 2H) LCMS: 198 (M+H+) (C8H11N3O3)
Step 2: Preparation of l-bromo-2-[N-(4-nitro-phenyl)-hydrazino]-ethane

The compound obtained in Step 1 (531 g, 1.90 mol) was suspended in
anhydrous 1,2-dimethoxyethane (4.5 L). The resultant suspension was cooled to 0 °C and PBr3 (220 niL, 2.29 mol, 1.2 eq, Aldrich 256536) was added thereto dropwise at 0 °C . The mixture thus obtained was warmed up to r.t. and stirred at 6O 0C for l5 hrs.
The mixture was cooled to r.t., and filtered off to remove remained insoluble solid. The filter cake thus obtained was washed with 1,2- dimethoxyethane (700 mL) and the filtrate was concentrated in vacuo. The resultant residue was suspended with H2O (2.5 L), stirred and cooled to 0 °C . Aq. 2N NaOH solution (1.7 L) was added thereto at 0°C to neutralize the suspension mixture (pH 6-7). The solid was filtered off and washed with H2O (5 L). The filtered solid was air-dried for 5 hrs.
The air-dried solid was dissolved with CH2Cl2 (3 L), and aq. sat’d
NaHCO3 solution (1.5 L) and H2O (700 mL) were added thereto. The resultant
– mixture was stirred for 15 min and stood to separate organic and aqueous layers. Insoluble solid which was not dissolved in organic layer and H2O was remained in the mixture. The mixture was filtered off to remove insoluble solid and the filter cake was washed with CH2Cl2 (700 mL). The organic layer was extracted, dried over MgSO4, filtered off, and concentrated in vacuo. The resultant solid was dried under high vacuum to obtain the title compound.
Yield : 383 g (77% : When product was dissolved in CDCl3 to check the
1H NMR spectroscopy, insoluble solid was stilled remained in CDCl3)
TLC : Rf= 0.91 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 9.6 Hz, 2H), 6.92 (d, J = 9.2 Hz, 2H), 4.00 (t, J = 6.6 Hz, 2H), 3.65 (t, J = 6.6 Hz, 2H)
LCMS: 261 (M+H+) (C8H10BrN3O2)
Step 3: Preparation of 4-(5,6-dihydro-4H-[l,2,4]triazin-l-yl)-l-nitrobenzene
Ethyl formimidate HCI, NaOAc
1 ,2-dimethoxyethane ![]()

The compound obtained in Step 2 (384 g, 1.48 mol) was dissolved in anhydrous 1,2-dimethoxyethane (4 L) and ethyl formimidate HCl salt (322 g, 2.94 mol, 2 eq) was added thereto at r.t. The resultant mixture was stirred at r.t. for 30 min. NaOAc (364 g, 4.44 mol, 3.0 eq, Aldrich 110191) was added to the mixture and the mixture was stirred at 75 °C for 15 hrs.
The mixture was cooled to r.t. and evaporated to remove solvent. The resultant residue was suspended in EA (2 L) and 1,2-dimethoxyethane (I L). Aq.
3N HCl solution (2.5 L) was added to the suspension. Insoluble solid was remained in resultant mixture. The solid was filtered off two times to remove insoluble solid. Ether (3 L) was added to the filtrate to separate organic and aqueous layers effectively. Aqueous layer was separated and washed with mixed organic solution (EA (1 L) + Hexane (500 mL)). The combined organic layer should be kept to recover the product.
(The treatment of aqueous layer)
The aqueous layer was cooled to 0 °C and aq. 6N NaOH solution (2.2 L) was added thereto slowly to basify the H2O layer (pH ~ 9). The resultant suspension was stirred at r.t. for 12 hrs. The solid was filtered off and washed with H2O (3 L) and dried under high vacuum.
(The treatment of combined organic layer)
The combined organic layer was concentrated in vacuo. The resultant residue was acidified with aq. 3N HCl solution (500 mL). Filtration was carried out to remove insoluble solid. The filtrate (H2O layer) thus obtained was washed with ether (700 mL X 2). The aqueous layer was stirred and cooled to 0 °C . Aq. 5N NaOH solution (1 L) was added to the cooled aqueous layer to basify (pH ~9). The mixture thus obtained was stirred at r.t. for 12 hrs. The solid thus obtained was filtered off and washed with H2O (1.5 L). The solid was dried under high vacuum to obtain the title compound.
Yield : 187 g (62 %)
TLC : Rf= 0.45 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (400 MHz, DMSO-</6) δ 7.99 (d, J = 9.6 Hz, 2H), 7.16 (d, J =
9.6 Hz, 2H), 7.09 (br s, IH), 6.97 (d, J = 3.6 Hz, 2H), 3.73 (t, J = 5.0 Hz, 2H), 3.45-3.46 (m, 2H)
LCMS: 207 (M+H+) (C9H10N4O2)
Step 4: Preparation of 4-(5,6-dihydro-4-t-butoxycarbonyl-[l,2,4]triazin-l-yl)- 1-nitrobenzene
![]()
The compound obtained in Step 3 (187g, 0.907 mol) was suspended in anhydrous THF (2.2 L), and BoC2O (30Og, 1.36 mol, 1.5 eq, Aldrich 205249) and DMAP (6g, 0.045 mol, 0.05 eq, Aldrich 107700) were added thereto. The mixture thus obtained was stirred at 65 °C for 5 hrs.
The mixture was cooled to 0 °C . MeOH (1.5 L) was added to the mixture at 0 °C and stirred at 0 °C for 1 hr. The solid thus obtained was filtered off, washed with MeOH (750 niL) and dried under high vacuum.
Filtrate thus obtained was concentrated in vacuo. MeOH (1 L) was added to the resultant residue with stirring. The mixture thus obtained was stirred at r.t for 12 hrs. Solid thus obtained was filtered off, washed with MeOH (500 mL), and dried under high vacuum to obtain the title compound.
Yield : 182 g (65 %)
TLC : Rf= 0.93 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (400 MHz, DMSO-J6) δ 8.17 (d, J= 9.6 Hz, 2H), 7.57 (br s, IH), 7.19 (d, J= 9.6 Hz, 2H), 3.93-3.86 (m, 2H), 3.83-3.745 (m, 2H), 1.56 (s, 9H)
LCMS: 307 (M+H+) (C14H18N4O4)
Step 5: Preparation of 4-(5,6-dihydro-4-t-butoxycarbonyl-[l,2,4]triazin-l-yl)aniline
![]()
The compound obtained in Step 4 (134 g, 438 mmol) was suspended in
MeOH (1.3 L) at r.t., and NH4Cl (12 g, 0.5 eq, Aldrich A4514) and Zn (15 g, 0.5 eq, Aldrich 209988) were added 6 times at intervals of 15 min at r.t. (total amounts Of NH4Cl = 73 g (1356 mmol, 3.1 eq) and total amounts of Zn = 88 g
(1356 mmol, 3.1 eq))
Temperature of the resultant mixture was risen gradually to 65 °C and the mixture was stirred at 65 °C for 12 hrs. The mixture was cooled to 40 °C and NH4Cl (12 g, 0.5 eq, Aldrich A4514) and Zn (15 g, 0.5 eq, Aldrich 209988) were added thereto. Temperature of the resultant mixture was risen gradually to 65 °C and the mixture was stirred at 65 “C for 1 hr.
The mixture was cooled to r.t. and filtered off through celite pad. The filter cake was washed with MeOH (700 mL) and THF (700 mL) and the filtrate was concentrated. The crude product thus obtained was dried under high vacuum and used without further purification.
Yield : 124 g (quantitative)
TLC : Rf= 0.38 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (400 MHz, OMSO-d6) δ 7.31 (br s, IH), 6.86 (d, J = 12.0 Hz, 2H), 6.48 (d, J = 12.0 Hz, 2H), 4.60 (s, 2H), 3.71 (br s, 2H), 3.38 (br s, 2H), 1.44 (s, 9H)
LCMS: 277 (M+H+) (C14H20N4O2)
Step 6: Preparation of N-(3-(5,6-dihydro-4-t-butoxycarbonyl-[l,2,4]triazin-l-yl)anilino-(2R)-2-hydroxypropyl)-5-chloro-2-thiophene carboxamide

The compound obtained in Step 5 (120 g, 435 mmol) and 5-chloro-N-(((S)-oxiran-2-yl)methyl)thiophene-2-carboxamide (123 g, 566 mmol, 1.3 eq, purchased from RStech (Daejeon, Korea) was suspended in absolute EtOH (1450 mL). The mixture thus obtained was stirred at 85 °C for 16 hrs. The mixture was cooled to r.t. and evaporated in vacuo to remove solvent. The resultant residue was dried under high vacuum for 18 hrs. The dried solid was suspended in EA (2 L). The suspension thus obtained was stirred at r.t. for 1 hr. The solid thus obtained was filtered off and washed with EA (500 mL) and ether (500 mL). The filtered solid was dried under high vacuum to obtain the title compound.
Aniline (starting material), epoxide, over-reacted by product were contained in crude product.
Yield : 158 g (74 %)
TLC : Rf= 0.34 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR of a crude sample (400 MHz, DMSO-^6) δ 8.57 (t, J = 5.4 Hz,
IH), 7.65 (d, J = 3.6 Hz, IH), 7.32 (br s, IH), 7.14 (d, J = 4.2 Hz, IH), 6.90 (d, J
= 9.0 Hz, 2H), 6.51 (d, J = 9.0 Hz, 2H), 5.04 (t, J = 6.6 Hz, IH), 5.00 (d, J = 5.4 Hz, IH), 3.87-3.65 (m, 3H), 3.40 (br s, 2H), 3.37-3.34 (m, IH), 3.25-3.21 (m, IH),
3.17-2.96 (m, IH), 2.94-2.84 (m, IH), 1.44 (s, 9H)
LCMS: 494 (M+H+) (C22H28ClN5O4S)
Step 7: Preparation of 5-chloro-N-({(5S)-2-oxo-3-[(5,6-dihydro-4-t-butoxycarbonyl-[l,2,4]triazin-l-yl)phenyl]-l,3-oxazolidin-5-yl}-methyl)-2-thiophene carboxamide

The compound obtained in Step 6 (158 g, 320 mmol) was suspended in
THF (1000 niL), and 1,1-carbonyldiimidazole (68 g, 416 mmol, 1.3 eq, Aldrich 115533) and DMAP (2 g, 16 mmol, 0.05 eq, Aldrich 107700) were added thereto. The mixture thus obtained was stirred at 75 °C for 3 hrs, cooled to r.t, and evaporated in vacuo to remove solvent. The resultant residue was suspended in EtOH (1300 mL). The suspension thus obtained was stirred at 0 °C for 1 hr. The solid thus produced was filtered off and washed with cold EtOH (800 mL) and cold MeOH (300 mL). The filtered solid was dried under high vacuum to obtain the title compound.
Yield : 101 g (61 %)
TLC : R/= 0.75 (EA/MeOH/AcOH = 20/1/0.5)
1H NMR (400 MHz, DMSO-^6) δ 8.93 (t, J= 5.4 Hz, IH), 7.66 (d, J= 4.2 Hz, IH), 7.43-7.33 (m, 3H),7.29-7.12 (m, 3H), 4.82-4.73 (m, IH), 4.09 (t, J = 9.0 Hz, IH), 3.82-3.70 (m, 3H), 3.65-3.52 (m, 4H), 1.45 (s, 9H)
LCMS: 520 (M+H+) (C23H26ClN5O5S)
Step 8: Preparation of 5-chloro-N-({(5S)-2-oxo-3-[(5,6-dihydro-4H-[l,2,4]triazin-l-yl)phenyl]-l,3-oxazolidin-5-yl}-methyl)-2-thiophene
carboxamide hydrochloride

The compound obtained in Step 7 (101 g, 194 mmol) was suspended in aq.
3N HCl solution (1.1 L) and THF (1.1 L), and stirred at 80 “C for 3 hrs. The mixture thus obtained was cooled to r.t. The solid thus produced was filtered off, washed with THF (700 mL) and dried under high vacuum to obtain the title compound.
Yield : 75 g (85 %)
TLC : Rf= 0.82 (MC/MeOH/AcOH = 10/1/0.5)
1H NMR (400 MHz, DMSO-J6) δ 12.12 (br s, IH), 10.32 (br s, IH), 9.13
(t, J = 6.0 Hz, IH), 8.57 (d, J= 5.2 Hz, IH), 7.75 (d, J = 4.2 Hz, IH), 7.49 (d, J =
9.0 Hz, 2H), 7.15 (d, J= 4.2 Hz, IH), 7.09 (d, J= 9.0 Hz, 2H), 4.85-4.74 (m, IH), 4.11 (t, J = 8.8 Hz, IH), 3.85 (dd, J = 6.0, 9.2 Hz, IH), 3.62 (t, J = 4.8 Hz, 2H),
3.59-3.49 (m, 2H), 3.15 (br s,2H)
LCMS: 420 (M+H+) (C18H18ClN5O3)
Example 5: Preparation of 5-chloro-N-({(5S)-2-oxo-3-[(5,6-dihydro-4H-[l,2,4]triazin-l^yl)phenyl]-l,3-oxazolidin-5-yl}-methyl)-2-thiophene
carboxamide

The compound obtained in Example 4 (20 g, 43.8 mmol) was suspended in MeOH/H2O (1/2 wt/wt, 3.2 L) and stirred at 100 °C until the compound obtained in Example 4 was dissolved clearly. 0.6M aq. Na2CO3 solution (75 mL) was added thereto. The mixture thus obtained was stood at 0 °C for 2 hrs. The solid thus produced was filtered off, washed with H2O (400 mL) and dried
under high vacuum to obtain the title compound.
Yield : 17 g (93 %)
1H NMR (400 MHz, DMSO-J6) δ 8.93 (t, J = 6.0 Hz, IH), 7.66 (d, J = 4.0 Hz, IH), 7.29 (d, J = 9.2 Hz, 2H), 7.16 (d, J = 4.0 Hz, IH), 7.08 (d, J = 9.2 Hz, 2H), 6.76 (d, J = 4.0 Hz, IH), 6.48 (br s, IH), 4.78-4.69 (m, IH), 4.07 (t, J = 8.8 Hz, IH), 3.74 (dd, J = 6.0, 9.2 Hz, IH), 3.54 (t, J = 5.6 Hz, 2H), 3.38 (s, 4H)
LCMS: 420 (M+H+) (C18H18ClN5O3)
Example 6: Preparation of 5-chIoro-N-({(5S)-2-oxo-3-[(5,6-dihydro-4H-[l,2,4]triazin-l-yl)phenyI]-l,3-oxazolidin-5-yl}-methyl)-2-thiophene
carboxamide methane sulfonate

The compound obtained in Example 5 (16.7 g, 39.8 mmol) was suspended in MeOH/CH2Cl2 (1/4 v/v, 350 mL) and stirred at 50 °C until the compound obtained in Example 5 was dissolved clearly. The mixture thus obtained was cooled to 0 °C and methylsulfonic acid (2.9 mL, 43.8 mmol, 1.3 eq, Aldrich 471356) was added thereto at 0 °C . The resulting mixture was evaporated in vacuo to remove solvent. The resultant solid was suspended in absolute EtOH (100 mL) and the suspension was stirred at 90 °C to dissolve solid clearly. The resulting mixture was cooled to 0 °C and stirred at 0 °C for 2 hrs. The solid thus produced was filtered off, washed with absolute EtOH (100 mL), and dried under high vacuum to obtain the title compound.
Yield : 18.4 g (89.7 %)
1H NMR (400 MHz, DMSO-J6) δ 11.93 (br s, IH), 10.03 (br s, IH), 8.94 (t, J = 6.0 Hz, IH), 8.55 (d, J = 6.0 Hz, IH), 7.66 (d, J = 4.0 Hz, IH), 7.49 (d, J = 9.2 Hz, 2H), 7.16 (d, J = 4.0 Hz, IH), 7.08 (d, J = 9.2 Hz, 2H), 4.93-4.87 (m, IH), 4.10 (t, J = 9.2 Hz, IH), 3.77 (dd, J = 6.0, 9.2 Hz, IH), 3.63 (m, 2H), 3.57 (t, J = 5.6 Hz, 2H), 3.16 (br s, 2H), 2.28 (s, 3H)
LCMS: 420 (M+H+) (C18H18ClN5O3)
PATENT
WO2010002115
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010002115
[Reaction Scheme 1] [96] A., O
NCONH2 + &J\ – NC NC- boc IPA, reflux O*B£.H. .κ> boc DMAP boc 2

Example 10: Preparation of compound 109

Compound 15a (450 mg, 0.88 mmol) obtained in Manufacturing Example 3 was dissolved in dichloromethane (10 mL), to which HCl (4 M 1,4-dioxane solution) (10 mL) was added, followed by stirring at room temperature for 1 hour. The reactant was concentrated under reduced pressure and dried to give light yellow solid compound (425 mg, 0.88 mmol, 100%). This compound (392 mg, 0.81 mmol) was dissolved in acetic acid (4 mL), to which trimethylorthoformate (2 mL) was added, followed by reflux with stirring. 10 hours later, after solvent was evaporated all, column chromatography (dichlorome thane/me thanol(v/v) 20/1 → 12/1) was performed to give the title compound 109 as a light yellow solid (215 mg, 5.12 mmol, 63 %).
1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 9.2 Hz, 2H), 7.33 (d, J = 4.4 Hz, IH), 7.14 (d, J = 9.2 Hz, 2H), 7.01 (t, J = 6.4 Hz, IH), 6.88 (s, IH), 6.85 (d, J = 4.4 Hz, IH), 4.87-4.79 (m, IH), 4.06 (t, J = 9 Hz, IH), 3.86 (ddd, J = 14.4 ,6, 3 Hz, IH), 3.81 (dd, J = 9, 6.4 Hz, IH), 3.69 (dt, J = 14.4, 6 Hz, IH), 3.62-3.58 (m, 2H), 3.55-3.51 (m, 2H); LCMS: 420 (M+H+) to Ci8H18ClN5O3S

REFERENCES
https://clinicaltrials.gov/ct2/show/NCT01954238
SEE EARLIER MOLECULE LCB01-0371…..https://newdrugapprovals.org/2014/03/31/lcb01-0371-new-oxazolidinone-has-improved-activity-against-gram-positive-pathogens/
////////////////phase 1, Green Cross Corp, LegoChem Bioscience, GCC 4401C, thrombosis, venous thromboembolism, GC 2107, CB02-0133, GC-2107, GC4401, GCC-2107, GCC-4401, GCC-4401C, I Fxa – LegoChem Biosciences, LCB02-0133, Nokxaban
O=C(NC[C@H]3CN(c1ccc(cc1)N2CCNC=N2)C(=O)O3)c4ccc(Cl)s4.CS(=O)(=O)O METHANE SULFONATE
O=C(NC[C@H]3CN(c1ccc(cc1)N2CCNC=N2)C(=O)O3)c4ccc(Cl)s4 FREE FORM
C1CN(NC=N1)C2=CC=C(C=C2)N3CC(OC3=O)CNC(=O)C4=CC=C(S4)Cl
What was the drug in Clinical Trial Tragedy In France Jan 2016
BIA 10-2474
cas 1233855-46-3
3-(1-(cyclohexyl(methyl)carbamoyl)-1H-imidazol-4-yl)pyridine 1-oxide
1H-Imidazole-1-carboxamide, N-cyclohexyl-N-methyl-4-(1-oxido-3-pyridinyl)-
C16 H20 N4 O2, 300.36
| Bial-Portela & Ca. S.A. |
BIA 10-2474 is an experimental fatty acid amide hydrolase inhibitor[1] developed by the Portuguese pharmaceutical company Bial-Portela & Ca. SA. The drug was developed to relieve pain,[2][3] to ease mood and anxiety problems, and to improve movement coordination linked to neurodegenerative illnesses.[4] It interacts with the human endocannabinoid system.[5][6] It has been linked to severe adverse events affecting 5 patients in a drug trial in Rennes, France, and at least one death, in January 2016.[7]

Synthesis
WO 2014017938
Example 5. 3-(l-(cyclohexyl(methyl)carbamoyl-lfl-imidazol-4-yl)pyridine l-oxide (compound A)

C16H20N4O C16H20N4O2
MW 284,36 MW 300,36
To a solution of N-cyclohexyl-N-methyl-4-(pyridm-3-yl)-lH-imidazole-l-carboxamide in dichioromethane at 25°C was added peracetic acid (38%; the concentration is not critical, and may be varied) in a single portion. The reaction mixture was then maintained at 25°C for at least 20 h, whereupon the reaction was washed four times with water (in some embodiments, the water for the extraction step may be supplemented with a small amount (e.g. 1%) of acetic acid, which helps to promote product solubility in the DCM). The dichioromethane solution was then filtered prior to diluting with 2-propanol. Dichioromethane (50%) was then distilled off under atmospheric pressure, whereupon, 2-propanol was charged at the same rate as the distillate was collected. The distillation was continued until >90% of the dichioromethane was collected. The resulting suspension was then cooled to 20°C and aged for at least 30 min. prior to cooling to 0°C and aging for a further 60 min. The reaction mixture was then filtered and the product washed with additional 2-propanol, before drying at 50°C under vacuum to afford the title compound as an off-white crystalline solid.
The purity of the product was ascertained by HPLC, with identity confirmable by NMR. The yield was consistently >80% in several production runs.
PATENT
Example 1. Preparation of N-cyclohexyl-N-methyl-4-(pyridin-3yl)-lH-imidazole-l-carboxamide

C8H7N3 C15H1 1N302 C16H20N4O
MW 145,16 MW 265,27 MW 284,36
To a suspension of 3-(l/ -imidazol-4-yl)pyridine in tetrahydrofuran (THF) containing pyridine at 25°C was slowly added a solution of phenyl chloroformate in THF over 60 to 90 min. The resulting fine white suspension was then maintained at 25°C for at least 60 min. before the addition of N-methyl- -cyclohexylamine in a single portion, causing the suspension to thin and become yellow in colour. The reaction mixture was then stirred for 90 min. before filtering and washing the filter cake with additional THF. The mother liquors were then maintained at 25°C for at least 18 h, whereupon 65% of the volume of THF was distilled off under atmospheric pressure. The resulting solution was then diluted with 2-propanol and maintained at > 50°C for 10 min. prior to cooling down to 20°C. The resulting suspension was aged at 20°C for 15 min. prior to cooling to 0°C and aging for a further 60 min. The reaction mixture was then filtered and the product was washed with additional 2-propanol, before drying at 50°C under vacuum to afford the title compound as an off-white crystalline solid.
The purity of the product was ascertained by HPLC, with identity confirmable by NMR. The yield was consistently around 50% in several production runs.
Example 2. 3-(l-(cyclohexyl(methyl)carbamoyl-l//-imidazol-4-yl)pyridine 1 -oxide (compound A)

C16H20N4O Ci6H2oN402
MW 284,36 MW 300,36
To a solution of N-cyclohexyl-N-methyl-4-(pyridin-3-yl)-lH-imidazole-l-carboxamide in dichloromethane at 25°C was added peracetic acid (38%; the concentration is not critical, and may be varied) in a single portion. The reaction mixture was then maintained at 25°C for at least 20 h, whereupon the reaction was washed four times with water. The dichloromethane solution was then filtered prior to diluting with 2-propanol. Dichloromethane (50%) was then distilled off under atmospheric pressure, whereupon, 2-propanol was charged at the same rate as the distillate was collected. The distillation was continued until >90% of the dichloromethane was collected. The resulting suspension was then cooled to 20°C and aged for at least 30 min. prior to cooling to 0°C and aging for a further 60 min. The reaction mixture was then filtered and the product washed with additional 2-propanol, before drying at 50°C under vacuum to afford the title compound as an off-white crystalline solid.
The purity of the product was ascertained by HPLC, with identity confirmable by NMR. The yield was consistently >80% in several production runs. It will be appreciated that this gives an overall yield of compound A many times greater than that achieved in the prior art.
In a further run of this synthesis, in a 2L reactor to a mixture of N-cyclohexyl-N-methyl-4-(pyridin-3-yl)-l H- imidazole-l-carboxamide (90 g, 317 mmol) and dichloromethane (1350 ml) was added peracetic acid (84 ml, 475 mmol). The reaction mixture was stirred at 25°C. Completion of the reaction was monitored by HPLC for the disappearance of N-cyclohexyl-N-methyl-4-(pyridin-3-yl)-lH- imidazole- 1-carboxamide. After reaction completion a solution of sodium metabisulfite (60.2 g, 317 mmol) in water (270ml) was added to the reaction mixture maintaining the temperature below 30°C. After phase separation the organic phase was washed with water. After phase separation the organic phase was concentrated at atmospheric pressure until 5 vol. Then solvent was swapped to isopropanol (1350 ml) and the suspension was cooled to 0°C during 4 hours and stirred at that temperature for 1 hour. The resulting solid was collected by filtration and was rinsed with water (270 ml) and isopropanol (270 ml) to afford a white crystalline solid in 84.8g (89%).
PATENT
Preparation of compound 362 a) N-cyclohexyl-N-methyl-4-(pyridin-3-yl)- 1 H-imidazole- 1 -carboxamide

To a stirred suspension of 3-( 1 H-imidazol-4-yl)pyridine dihydrochloride (1.745 g, 8 mmol) in a mixture of tetrahydrofuran (29 mL) and DMF (2.90 mL) was added potassium 2-methylpropan-2-olate (1.795 g, 16.0 mmol) and the mixture was refluxed for 30 minutes. The resulting brown suspension was cooled to room temperature and treated with pyridine (0.979 mL, 12 mmol) and N,N-dimethylpyridin-4-amine (0.098 g, 0.8 mmol), followed by the addition of cyclohexyl(methyl)carbamic chloride (1.476 g, 8.4 mmol). The reaction was heated to 90 0C overnight, whereupon the mixture was diluted with water and extracted with ethyl acetate. The organic phase was dried (MgSO^) and filtered. After evaporation, the crude product was chromatographed over silica gel using a dichloromethane/methanol (9:1) mixture. Homogenous fractions were pooled and evaporated to leave a white powder, (160 mg, 7 %).
b) 3-( 1 -(cyclohexyl(methyl)carbamoyl)- 1 H-imidazol-4-yl)pyridine 1 -oxide

To a stirred solution of N-cyclohexyl-N-methyl-4-(pyridin-3-yl)-l H-imidazole- 1 -carboxamide (90 mg, 0.317 mmol) in chloroform (5 mL) was added 3-chlorobenzoρeroxoic acid (149 mg, 0.475 mmol) in one portion. The reaction was allowed to stir at room temperature for 20 h. TLC showed the reaction to be complete and the mixture was evaporated to dryness. The residue was triturated with ether and the resulting white crystals were filtered off and dried in air. Recrystallisation from hot isopropanol gave a white powder (46 mg, 46 %).
Structure and action
French newspaper Le Figaro has obtained Bial study protocol documents listing the the chemical name of BIA-10-2474 as 3-(1-(cyclohexyl(methyl)carbamoyl)-1H-imidazol-4-yl)pyridine 1-oxide.[8] A Bial news release described BIA-10-2474 as “a long-acting inhibitor of FAAH”.[9]
Fatty acid amide hydrolase (FAAH) is an enzyme which degrades endocannabinoid neurotransmitters like anandamide,[10] which relieves pain and can affect eating and sleep patterns.[11][12] FAAH inhibitors have been proposed for a range of nervous-system disorders including anxiety, alcoholism, pain and nausea.
The Portuguese pharmaceutical company Bial holds several patents on FAAH enzyme inhibitors.[12][13][14][15]
No details of the preclinical testing of this molecule have been made public by the manufacturer Bial. However, the French newspaper Le Figaro has obtained and published an apparently legitimate copy of the full clinical trial protocol (BIA-102474-101).[8] The protocol presents a summary of what appears to be a full package of pharmacodynamic, pharmacokinetic and toxicological studies that might be expected to support a first-in-man study, including safety pharmacology studies in two species (rat, dog) and repeated dose toxicity studies in four species (13 week sub-chronic studies in mouse, rat, dog and monkey). The summary presented however includes no assessment of the relevance of the animal species selected for study (that is, in terms of physiological and genetic similarities with humans and the mechanism of action of the study drug).
Of note, few adverse events were observed in any of the studies, with the 13-week oral No Observed Adverse Effect Level (NOAEL) varying between 10 mg/kg/day in mice to 75 mg/kg/day in monkeys. The authors suggest that these were the maximum doses tested in these studies, though it is not clear. The authors also report no effects of significance in the animal models used for the CNS safety pharmacology studies, which studied a dose of up to 300 mg/kg/day.[8]
Notably absent from the protocol are calculations of receptor occupancy; predictions of in vivo ligand binding saturation levels; measures of target affinity; or assessment of the molecule’s activity in non-target tissues or non-target binding interactions as suggested by the European guidance for Phase I studies,[16] assuming BIA 10-2474 could be considered ‘high risk’).[8]
The trial protocol makes no reference to chimpanzee studies (only monkeys) which contradicts a previous statement to the media in which the French Health Minister stated that the drug had been tested on animals including chimpanzees.[4][17] [18] Some experts had remarked that drug testing in chimpanzees was unlikely.[19]
These findings provide no explanation for the type and severity of events observed in Rennes. In describing the rationale for the starting dose, the authors conclude that:
No target organ was identified during toxicology studies and few adverse clinical findings were observed at the highest dose tested. For the single ascending dose part [of the clinical trial], a starting dose of 0.25 mg was judged to be safe for a first-in-human administration. [8]
The protocol defines no starting dose for the multi-dose treatment groups, noting that this will be based on the outcome of the single dose portion of the trial (an approach known as adaptive trial design). The authors note that nonetheless, the starting dose will not exceed 33% of the maximum tolerated dose (MTD) identified in the single dose groups (or 33% of the maximum administered dose if the MTD is not reached).[8]
Death and serious adverse events during phase I clinical trial
In July 2015 Biotrial, a contract research organization, began testing the drug in a human phase one clinical trial for the manufacturer. The study was approved by French regulatory authority, the Agence Nationale de Sécurité du Médicament (ANSM), on June 26, 2015, and by the Brest regional ethics committee on July 3, 2015.[20] The trial commenced on July 9, 2015,[21] in the city of Rennes, and recruited 128 healthy volunteers, both men and women aged 18 to 55. According to French authorities, the study employed a three-stage design with 90 of the volunteers having received the drug during the first two stages of the trial, with no serious adverse events being reported .[17][20] Participants of the study were to receive €1,900 and, in turn, asked to stay at Biotrial’s facility for two weeks during which time they would take the drug for ten days and undergo tests.[22]
In the third stage of the trial evaluating multiple doses, six male volunteers received doses by mouth, starting on 7 January 2016. The first volunteer was hospitalized at the Rennes University Hospital on January 10, became brain dead,[17][23][24][25] and died on January 17.[26] The other five men in the same dosage group were also hospitalized, in the period of January 10 through January 13[27] four of them suffering injuries including deep hemorrhagic and necrotic lesions seen on brain MRI.[7] The six men who were hospitalised were the group which received the highest dose.[26] A neurologist at the University of Rennes Hospital Center, Professor Pierre-Gilles Edan, stated in a press conference with the French Minister for Health, that 3 of the 4 men who were displaying neurological symptoms “already have a severe enough clinical picture to fear that even in the best situation there will be an irreversible handicap” and were being given corticosteroids to control the inflammation.[27] The sixth man from the group was not showing adverse effects but had been hospitalized for observation.[25][28][29] Biotrial stopped the experiment on January 11, 2016.[4]
No details of the trial have been made public by the manufacturer Bial. The study does not appear in searches of any of the key clinical trial registries, including EudraCT and ClinicalTrials.gov which would normally contain details of approved clinical studies.[30][31][32][33] The trial protocol published by Le Figaro provides extensive detail on what was planned for the study, but many details of the key multi-dose part are not included and were to have been finalised at the conclusion of the single-dose part of the trial.[8]
The French health minister Marisol Touraine called the event “an accident of exceptional gravity” and promised to investigate the matter.[4] On January 18 it was reported authorities were investigating if a manufacturing or transport error might be involved.[34]
Le Figaro posted a 96-page clinical study protocol for BIA 10-2474 that the French newspaper procured from an unnamed source.
According to the document, BIA 10-2474 is 3-(1-(cyclohexyl(methyl)carbamoyl)-1H-imidazol-4-yl)pyridine 1-oxide.
BIA 10-2474 “is designed to act as a long-active and reversible inhibitor of brain and peripheral FAAH,” notes the protocol. The compound “increases anandamide levels in the central nervous system and in peripheral tissues.”
The clinical trial protocol also notes that the company tested BIA 10-2474 on mice, rats, dogs, and monkeys for effects on the heart, kidneys, and gastrointestinal tract, among other pharmacological and toxicological evaluations.

One man is dead and five men were hospitalized after participating in a Phase I clinical trial in Rennes, France
The clinical trial, conducted by the company Biotrial on behalf of the Portuguese pharmaceutical firm Bial, was evaluating a pain relief drug candidate called BIA 10-2474 that inhibits fatty acid amide hydrolase (FAAH) enzymes. Blocking these enzymes prevents them from breaking down cannabinoids in the brain, a family of compounds that includes the euphoria-inducing neurotransmitter anandamide and Δ9-tetrahydrocannabinol, the major psychoactive component of marijuana.
Phase I clinical trials are conducted to check a drug candidate’s safety profile in healthy, paid volunteers. In this case, the drug caused hemorrhagic and necrotic brain lesions in five out of six men in a group who received the highest doses of the drug, said Gilles Edan, a neurologist at the University Hospital Center of Rennes.

The most severely affected man was pronounced brain-dead after hospitalization and then died on Jan. 17. Four men remain in the hospital in stable condition. The only man in the high-dose group who had no adverse symptoms has been released from the hospital.
Clinical trials are an essential part of the drug development process. In order to get life-improving and life-saving medicines to patients, they first have to go through an extensive series of tests. Even before a drug makes it to Phase 1 testing, where its safety, dosage amount, and side effects are tested in a small group of humans, it will undergo testing in animals. As a result, it is not common for a medicine undergoing clinical tests to have a very serious adverse effect on a human. This makes you wonder what happened to a group of patients involved in a clinical study in Rennes, France.
According to news reports, a drug undergoing testing in a French clinic has left one person dead, two others with what may be permanent brain damage, and and two others critically ill. The drug has thus far been unnamed, but it appears to have been produced by the Portuguese company Bial. The French health minister has stated the drug acted on natural receptors found in the body known as endocannibinoids, which regulate mood and appetite. It did not contain cannabis or anything derived from it, as was originally reported. All six trial participants were administered the doses simultaneously.
The trial was being performed at Biotrial, a French-based firm that was formed in 1989 and has conducted thousands of trials. A message on the company’s website stated that they are working with health authorities to understand the cause of the accident, while extending thoughts to the patients and their families. Bial has disclosed the drug was a FAAH (fatty acid amide hydrolase) inhibitor, which is an enzyme produced in the brain and elsewhere that breaks down neurotransmitters called endocannabinoids. Two scientists from the Nottingham Medical School who have worked with FAAH tried over the weekend to try and identify the drug by examining a list of drugs Bial currently has in its pipeline. They believe the culprit is one identified by the codename BIA 10-2474. That same codename appeared on a recruitment form that was given to a volunteer, which was published in a French newspaper. Little more is known about it, and there does not appear to be any entry for it in clinical trial registries.
The French health ministry is reporting the six patients were all in good health prior to taking the oral medicine, which was administered to 90 volunteers. The trial recruited 128 individuals, and the remaining participants received a placebo. Health minister Marisol Touraine, describing the situation as a very serious accident, noted the patients were taking part in a trial in Brittany, Rennes involving a medicine developed by a “European laboratory”, refusing to comment further until additional information became available. She has also asked the Inspector General of Social Affairs to lead an investigation into the circumstances around the trial, which has obviously been suspended. She notes the drug had been tested on animals, including chimpanzees. France’s National Agency for Medicine and Health Products Safety approved the trial on in June 2015.
One thing we do know is that the trial was a Phase 1 clinical study that included 90 healthy volunteers. Regulations that oversee all clinical trials in Europe do attempt to minimize the risk associated with trials, but there is always a risk involved with administering an unapproved medicine to humans. At this time the chief neuroscientist at the hospital where the patients are being treated has said there is no known antidote for the drug.
The drug, administered to men between the ages of 28 and 49, was intended to treat mood disorders such as anxiety. While the men were administered varying doses, the patients who are hospitalized were taking the drug “regularly”.

Old 2006 case
While safety issues like this are rare, they are not unheard of. In 2006, a clinical trial in London left six men ill. All were taking part in a study testing a drug designed to fight auto-immune disease and leukemia. Within hours of taking the drug TGN1412, all experienced a serious reaction, were admitted to intensive care, and had to be treated for organ failure. Two became critically ill, with one eventually losing all of his fingers and toes. All were told they would have a higher risk of developing cancers or auto-immune diseases.
This of course led many to wonder about the future of trials, and whether the situation could happen again. The Duff Report, written in response to the TGN1412 trial, noted the medicine should have been tested in one person at a time. It also helped to put additional safety measures in place. The Medicines and Health Products Regulatory Agency (MHRA) now requires committees to look at pre-clinical data to determine the proper initial dose, and rules are in place to stop the trial if unintended reactions occur.
However, since patients can fall ill immediately after being administered a medication, certain risks will still exist.
The company that manufactured TGN1412, TeGenero Immuno Therapeutics, later went bankrupt. However the drug was later purchased by a Russian investor and renamed TABO8. TheraMAB, a Russian biotech company, then conducted a new trial of the drug in a much lower dose. A later Phase 2 study was started in patients with Rheumatoid Arthritis.
Other pharmaceutical companies, including Merck, Pfizer, Johnson & Johnson, Sanofi and Vernalis, have previously taken other FAAH inhibitors into clinical trials without experiencing such adverse events (e.g. respectively, MK-4409,[35][36] PF-04457845, JNJ-42165279,[37] SSR411298 and V158866.[38][39] Related enzyme inhibitor compounds such as URB-597 and LY-2183240 have been sold illicitly as designer drugs,[40][41] all without reports of this type of toxicity emerging, so the mechanism of the toxicity observed with BIA 10-2474 remains poorly understood.
Following the events in Rennes, Janssen announced that it was temporarily suspending dosing in two Phase II clinical trials with its own FAAH inhibitor JNJ-42165279, headlining the decision as “precautionary measure follows safety issue with different drug in class”. Janssen was emphatic that no serious adverse events had been reported in any of the clinical trials with JNJ-42165279 to date. The suspension is to remain in effect until more information is available about the BIA 10-2474 study.[42]
References
- 1 “BRIEF-Bial says firmly committed to ensure wellbeing of test participants”. Reuters. 15 January 2016.
- 2 “BIA 10-2474”. Biocentury.com. Retrieved 2016-01-17.
- 3 “BIA 102474”. Adisinsight/springer.com. Retrieved 2016-01-17.
- 4 Adamson B (January 15, 2016). “Botched Drug Trial Leaves 1 Brain Dead, 5 in Hospital”. ABC, AP. Retrieved January 16, 2016.
- 5 Angeline Benoit,Makiko Kitamura (15 January 2016). “France Ties Brain-Dead Person to Tests of Bial-Portela Drug”. Bloomberg.com.
- 6 “France/Monde – Essai thérapeutique : 90 personnes ont pris la molécule”. Ledauphine.com. Retrieved 2016-01-17.
- 7 Enserink, Martin (2016). “More Details Emerge on Fateful French Drug Trial” (online). Science (January 16). Retrieved 16 January 2016.
- 8 “Drame de Rennes : le protocole de l’essai clinique en accusation”. sante.lefigaro.fr. Retrieved 2016-01-21.
- 9 “News Release – Phase I Clinical Trial Rennes”. http://www.bial.com. Retrieved 2016-01-21.
- 10 Debora Mackenzie. “Six in hospital after French pain relief drug trial goes wrong”. New Scientist.
- 11 “The Discovery and Development of Inhibitors of Fatty Acid Amide Hydrolase (FAAH)”. PubMed Central.
- 12 “Patent WO2015012708A1 – Imidazolecarboxamides and their use as faah inhibitors – Google Patents”. Google.com. Retrieved 2016-01-17.
- 13 “Patent WO2015016729A1 – Urea compounds and their use as faah enzyme inhibitors”. Google.com. Retrieved 2016-01-17.
- 14 “PT2014000052 UREA COMPOUNDS AND THEIR USE AS FAAH ENZYME INHIBITORS”. Patentscope.wipo.int. Retrieved 2016-01-17.
- 15 WO application 2015016729, Laszlo Erno KISS, Rita GUSMÃO DE NORONHA, Carla Patrícia ROSA DA COSTA PEREIRA, Rui PINTO, “Urea compounds and their use as faah enzyme inhibitors”, published Feb 5, 2015, assigned to BIAL
- 16″Strategies to identify and mitigate risks for first-in-human clinical trials with investigational medicinal products (CHMP/SWP/28367/07)” (PDF). European Medicines Agency. 1 September 2007. Retrieved 22 January 2016.
- 17 “Accident grave dans le cadre d’un essai clinique – Intervention de Marisol Touraine à Rennes”. Ministère des Affaires Sociales, de la Santé et des Droits des Femmes, France. 15 January 2016.
- 18Barbara Casassus (23 January 2016). “France investigates drug trial disaster” (PDF). The Lancet 387 (10016): 326. doi:10.1016/S0140-6736(16)00154-9.
- 19
- “Expert reaction to French drug trial – reports of one patient dying and five others in hospital and of the Paris prosecutor’s office having opened an investigation into what happened”. Science Media Centre, London. 16 January 2016. Retrieved 21 January 2016.
- 20
- “La survenue d’effets graves ayant entraîné l’hospitalisation de 6 patients, dont un en état de mort cérébrale, a conduit à l’arrêt prématuré d’un essai clinique du laboratoire BIAL – Point d’information”. Agence Nationale de Sécurité du Médicament, France (ANSM). 15 January 2016.
- 21
- “Six hospitalized in Bial clinical trial in France”. BioWorld.com. Retrieved 2016-01-17.
- 22
- Enserink M (January 16, 2016). “More details emerge on fateful French drug trial”. Science Magazine. Retrieved January 18, 2016.
- 23
- “France clinical trial: 90 given drug, one man brain-dead”. BBC. January 15, 2016. Retrieved January 16, 2016.
- 24
- “Ce que l’on sait de l’accident survenu lors d’un essai clinique à Rennes”, Le Monde, 15 January 2016
- 25
- Blamont M (January 15, 2016). “French drug trial disaster leaves one brain dead, five injured”. Reuters. Retrieved January 16, 2016.
- 26
- Clarisse Lucas, AFP (January 17, 2016). “Man dies after being left brain-dead in French drug trial”. Yahoo. Retrieved January 17, 2016.
- 27
- “Accident “inédit” lors d’un essai clinique: un homme en état de mort cérébrale, cinq hospitalisés”. La Depeche. January 15, 2016. Retrieved January 18, 2016.
- 28
- “France clinical trial: ‘No known antidote’ to drug”. BBC News. 15 January 2016. Retrieved 18 January 2016.
- 29
- Enserink, Martin (2016). “What We Know So Far About the Clinical Trial Disaster in France” (online). Science (January 15). Retrieved 16 January 2016.
- 30
- “EU Clinical Trials Register”. European Medicines Agency. Retrieved 20 January 2016.
- 31
- “WHO International Clinical Trials Registry Platform”. World Health Organization. Retrieved 20 January 2016.
- 32
- “ANSM – Répertoire public des essais cliniques de médicaments”. Agence Nationale de Sécurité du Médicament et des Produits de Santé, France. Retrieved 20 January 2016.
- 33
- “ClinicalTrials.gov Registry”. U.S. National Institutes of Health. Retrieved 20 January 2016.
- 34
- “Drug trial volunteer dies as ‘manufacturing error’ theory investigated”. The Independent. January 18, 2016. Retrieved January 18, 2016.
- 35
- Chobanian; et al. (10 April 2014). “Discovery of MK-4409, a Novel Oxazole FAAH Inhibitor for the Treatment of Inflammatory and Neuropathic Pain”. ACS Med. Chem. Lett.
- 36
- Merck (15 October 2009). “Merck Pipeline, Oct 2009” (PDF). Merck.
- 37
- “Seven studies found for: jnj-42165279”. Clinicaltrials.gov. Retrieved 2016-01-19.
- 38
- “2 studies found for: V158866”. Clinicaltrials.gov. Retrieved 2016-01-19.
- 39
- Bisogno T, Maccarrone M. Latest advances in the discovery of fatty acid amide hydrolase inhibitors. Expert Opin Drug Discov. 2013 May;8(5):509-22. PMID 23488865 doi: 10.1517/17460441.2013.780021.
- 40
- Shanks KG, Behonick GS, Dahn T, Terrell A. Identification of novel third-generation synthetic cannabinoids in products by ultra-performance liquid chromatography and time-of-flight mass spectrometry. J Anal Toxicol. 2013 Oct;37(8):517-25. PMID 23946450 doi: 10.1093/jat/bkt062.
- 41
- Uchiyama N, Matsuda S, Kawamura M, Shimokawa Y, Kikura-Hanajiri R, Aritake K, Urade Y, Goda Y. Characterization of four new designer drugs, 5-chloro-NNEI, NNEI indazole analog, α-PHPP and α-POP, with 11 newly distributed designer drugs in illegal products. Forensic Sci Int. 2014 Oct;243:1-13. PMID 24769262 doi: 10.1016/j.forsciint.2014.03.013.
- “Janssen Research & Development, LLC Voluntarily Suspends Dosing in Phase 2 Clinical Trials of Experimental Treatment for Mood Disorders”. Janssen.com. 17 January 2016. Retrieved 21 January 2016.
External links
| WO2005073199A1 * | Jan 15, 2005 | Aug 11, 2005 | Aventis Pharma Gmbh | Indazole derivatives as inhibitors of hormone-sensitive lipases |
| WO2010074588A2 | Dec 23, 2009 | Jul 1, 2010 | BIAL – PORTELA & Cª, S.A. | Pharmaceutical compounds |
| WO2012015324A1 | Jul 28, 2011 | Feb 2, 2012 | Bial – Portela & Ca, S.A. | Process for the synthesis of substituted urea compounds |
| US4051252 * | Nov 24, 1975 | Sep 27, 1977 | Bayer Aktiengesellschaft | 3-aminoindazole-1 and 2-carboxylic acid derivatives |
| US4331678 * | Jan 14, 1980 | May 25, 1982 | Fbc Limited | Carbamoyl pyrazole compounds and their pesticidal application |
| US4973588 * | Feb 10, 1989 | Nov 27, 1990 | Mitsui Petrochemical Industries, Ltd. | Imidazole derivatives having anti-hypoxia properties |
| US5578627 * | Oct 27, 1993 | Nov 26, 1996 | Toyama Chemical Co., Ltd. | 1,2-benzoisoxazole derivative or its salt and brain-protecting agent comprising the same |
 |
|||||
| Systematic (IUPAC) name | |||||
|---|---|---|---|---|---|
|
3-(1-(cyclohexyl(methyl)carbamoyl)-1H-imidazol-4-yl)pyridine 1-oxide
|
|||||
| Clinical data | |||||
| Legal status |
|
||||
| Routes of administration |
Oral | ||||
| Identifiers | |||||
| PubChem | CID: 46831476 | ||||
| Chemical data | |||||
| Formula | C16H20N4O2
|
||||
/////////
C1C(CCCC1)N(C)C(=O)n2cc(nc2)c3ccc[n+](c3)O
Elotuzumab
Elotuzumab
Approved nov 30 2012
A SLAMF7-directed immunostimulatory antibody used to treat multiple myeloma.
(Empliciti®)
HuLuc-63;BMS-901608
cas 915296-00-3
![]()


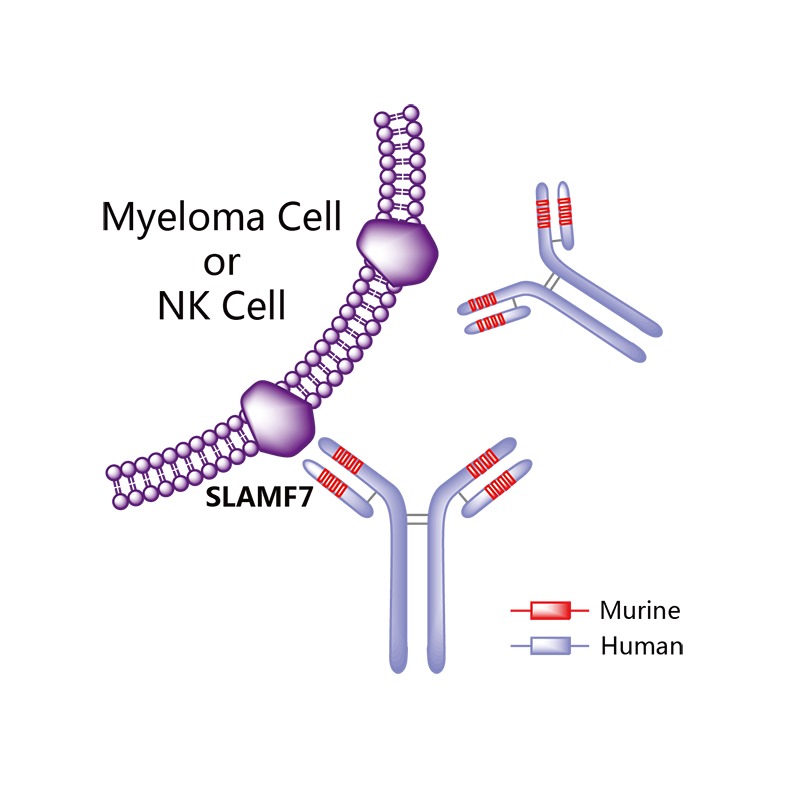
Elotuzumab (brand name Empliciti, previously known as HuLuc63) is a humanized monoclonal antibody used in relapsed multiple myeloma.[1] The package insert denotes its mechanism as a SLAMF7-directed (also known as CD 319) immunostimulatory antibody.[2]
Approvals and indications
In May 2014, it was granted “Breakthrough Therapy” designation by the FDA. [3] On November 30, 2015, FDA approved elotuzumab as a treatment for patients with multiple myeloma who have received one to three prior medications.[1] Elotuzumab was labeled for use with lenalidomide and dexamethasone. Each intravenous injection of elotuzumab should be premedicated with dexamethasone, diphenhydramine, ranitidine and acetaminophen.[2]
Elotuzumab is APPROVED for safety and efficacy in combination with lenalidomide and dexamethasone.
Monoclonal antibody therapy for multiple myeloma, a malignancy of plasma cells, was not very clinically efficacious until the development of cell surface glycoprotein CS1 targeting humanized immunoglobulin G1 monoclonal antibody – Elotuzumab. Elotuzumab is currently APPROVED in relapsed multiple myeloma.
Elotuzumab (HuLuc63) binds to CS1 antigens, highly expressed by multiple myeloma cells but minimally present on normal cells. The binding of elotuzumab to CS1 triggers antibody dependent cellular cytotoxicity in tumor cells expressing CS1. CS1 is a cell surface glycoprotein that belongs to the CD2 subset of immunoglobulin superfamily (IgSF). Preclinical studies showed that elotuzumab initiates cell lysis at high rates. The action of elotuzumab was found to be enhanced when multiple myeloma cells were pretreated with sub-therapeutic doses of lenalidomide and bortezomib. The impressive preclinical findings prompted investigation and analysis of elotuzumab in phase I and phase II studies in combination with lenalidomide and bortezomib.
Elotuzumab As Part of Combination Therapy: Clinical Trial Results
Elotuzumab showed manageable side effect profile and was well tolerated in a population of relapsed/refractory multiple myeloma patients, when treated with intravenous elotuzumab as single agent therapy. Lets’ take a look at how elotuzumab fared in combination therapy trials,
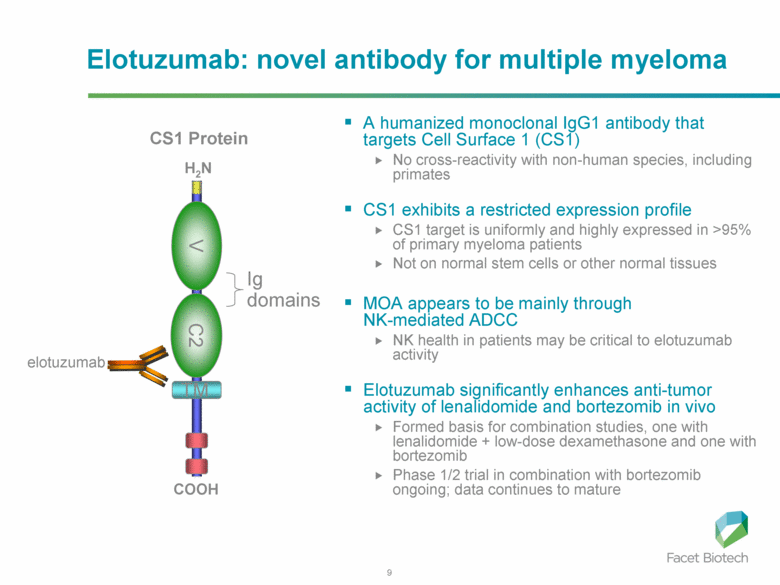
In phase I trial of elotuzumab in combination with Velcade/bortezomib in patients with relapsed/refractory myeloma, the overall response rate was 48% and activity was observed in patients whose disease had stopped responding to Velcade previously. The trial results found that elotuzumab enhanced Velcade activity.
A phase I/II trial in combination with lenalidomide and dexamethasone in refractory/relapsed multiple myeloma patients showed that 82% of patients responded to treatment with a partial response or better and 12% of patients showed complete response. Patients who had received only one prior therapy showed 91% response rate with elotuzumab in combination with lenalidomide and dexamethasone.
Phase I/II trials of the antibody drug has been very impressive and the drug is currently into Phase III trials. Two phase III trials are investigating whether addition of elotuzumab with Revlimid and low dose dexamethasone would increase the time to disease progression. Another phase III trial (ELOQUENT 2) is investigating and comparing safety and efficacy of lenalidomide plus low dose dexamethasone with or without 10mg/kg of elotuzumab in patients with relapsed/refractory multiple myeloma.
Elotuzumab is being investigated in many other trials too. It is being evaluated in combination with Revlimid and low-dose dexamethasone in multiple myeloma patients with various levels of kidney functions, while another phase II study is investigating elotuzumab’s efficacy in patients with high-risk smoldering myeloma.
The main target of multiple myeloma drug development is to satisfy the unmet need for drugs that would improve survival rates. Elotuzumab is an example that mandates much interest in this area and should be followed with diligence.

Empliciti’s Cost
Empliciti will be sold in the U.S. in two vials sizes: A smaller vial that contains 300 mg of the drug, and a larger vial that contains 400 mg.
Bristol-Myers Squibb has informed The Beacon that the wholesale price per vial of Empliciti will be $1,776 for the 300 mg vial and $2,368 for the 400 mg vial.
Using these prices and an assumed patient weight of between 154 and 176 pounds, Empliciti will cost $18,944 per four-week cycle for each of the first two cycles of treatment, and $9,472 per cycle thereafter. This means, in turn, that Empliciti’s cost per year will be $142,080 in the first year and $123,136 in subsequent years.
In comparison, Velcade costs between $4,800 and $8,500 per four-week cycle, depending on how often it is dosed. Ninlaro costs $8,670 per four-week cycle. And Kyprolis costs $10,500 per four-week cycle at the standard (20 – 27 mg/m2) dose.
Additional details about the FDA approval of Empliciti can be found in this press release from the FDA, a related press release from Bristol-Myers Squibb and AbbVie, and the full Empliciti prescribing information.
The results of the ELOQUENT-2 trial were published in Lonial, S. et al., “Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma,” The New England Journal of Medicine, June 2, 2015 (abstract). Slides from the ASCO presentation summarizing the ELOQUENT-2 results can be viewed here (PDF, courtesy of Dr. Lonial). This Beacon news article provides an in-depth look at the trial results.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | SLAMF7 (CD319) |
| Clinical data | |
| Trade names | Empliciti |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
IV |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Identifiers | |
| CAS Number | 915296-00-3 |
| ATC code | None |
| IUPHAR/BPS | 8361 |
| UNII | 1351PE5UGS |
| Chemical data | |
| Formula | C6476H9982N1714O2016S42 |
| Molecular mass | 145.5 kDa |
References
1 “Press Announcement—FDA approves Empliciti, a new immune-stimulating therapy to treat multiple myeloma”. U.S. Food and Drug Administration. Retrieved 3 December 2015.
2“Empliciti (elotuzumab) for Injection, for Intravenous Use. Full Prescribing Information” (PDF). Empliciti (elotuzumab) for US Healthcare Professionals. Bristol-Myers Squibb Company, Princeton, NJ 08543 USA.
3 “Bristol-Myers Squibb and AbbVie Receive U.S. FDA Breakthrough Therapy Designation for Elotuzumab, an Investigational Humanized Monoclonal Antibody for Multiple Myeloma” (Press release). Princeton, NJ & North Chicago, IL: Bristol-Myers Squibb. 2014-05-19. Retrieved 2015-02-05.
///////
BMS 911543

BMS 911543
N,N-dicyclopropyl-4-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-6-ethyl-1-methyl-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide
cas 1271022-90-2
Chemical Formula: C23H28N8O
Exact Mass: 432.23861
UNII-7N03P021J8;
N,N-dicyclopropyl-4-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-6-ethyl-1-methyl-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide
Bristol-Myers Squibb Company innovator
BMS-911543 is an orally available small molecule targeting a subset of Janus-associated kinase (JAK) with potential antineoplastic activity. JAK2 inhibitor BMS-911543 selectively inhibits JAK2, thereby preventing the JAK/STAT (signal transducer and activator of transcription) signaling cascade, including activation of STAT3. This may lead to an induction of tumor cell apoptosis and a decrease in cellular proliferation. JAK2, often upregulated or mutated in a variety of cancer cells, mediates STAT3 activation and plays a key role in tumor cell proliferation and survival.

The JAK2 selective compound BMS911543 (WO2011028864) is in phase II clinical trials for the treatment of m elofibrosis. BMS91 1543 is shown below.

PAPER
ACS Medicinal Chemistry Letters (2015), 6(8), 850-855
Discovery of a Highly Selective JAK2 Inhibitor, BMS-911543, for the Treatment of Myeloproliferative Neoplasms

JAK2 kinase inhibitors are a promising new class of agents for the treatment of myeloproliferative neoplasms and have potential for the treatment of other diseases possessing a deregulated JAK2-STAT pathway. X-ray structure and ADME guided refinement of C-4 heterocycles to address metabolic liability present in dialkylthiazole 1 led to the discovery of a clinical candidate, BMS-911543 (11), with excellent kinome selectivity, in vivo PD activity, and safety profile

MS (ESI) m/z 434.3 (M+H). 1H NMR (CDCl3) δ: 7.96 (s, 1H), 7.65 (s, 1H), 6.83 (s, 1H), 4.67 (q, J = 7.1 Hz, 2H), 4.01 (s, 3H), 3.82 (s, 3H), 2.77 – 2.84 (m, 2H), 2.43 (s, 3H), 1.48 (t, J = 7.2 Hz, 3H), 0.79 – 0.86 (m, 4H), 0.71 – 0.77 (m, 4H).
PAPER
Journal of Organic Chemistry (2015), 80(12), 6001-601
Click to access jo5b00572_si_001.pdf
Ni-Catalyzed C–H Functionalization in the Formation of a Complex Heterocycle: Synthesis of the Potent JAK2 Inhibitor BMS-911543

BMS-911543 is a complex pyrrolopyridine investigated as a potential treatment for myeloproliferative disorders. The development of a short and efficient synthesis of this molecule is described. During the course of our studies, a Ni-mediated C–N bond formation was invented, which enabled the rapid construction of the highly substituted 2-aminopyridine core. The synthesis of this complex, nitrogen-rich heterocycle was accomplished in only eight steps starting from readily available materials.
N,N-Dicyclopropyl-4-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-6-ethyl-1-methyl-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide, 1
PATENT
WO 2015031562
These Schemes are illustrative and are not meant to limit the possible techniques one skilled in the art may use to manufacture compounds disclosed herein.
As shown below in Scheme 1, the general preparation of compound 7 is described. Trichloroacetyl pyrrole (Compound 1) is reacted with a halogenating agent to give the C4-bromo pyrrole (Compound 2). Alcoho lysis occurs in the presence of an alcohol and base to generate ester (Compound 3), which can be selectively nitrated through contact with an appropriate nitrating agent (defined as a species that generates N02 ), yielding C5-nitro pyrrole (Compound 4). Compound 4 can be isolated as its free form, or optionally as a salt with an appropriate base. Ethylation with an appropriate alkylating agent generates the N-ethyl pyrrole (Compound 5), which in the presence of an imidazole, base, palladium and an appropriate phosphine ligand, will undergo a coupling process to form Compound 6. Reduction of the nitro-group of Compound 6 in the presence of hydrogen, a metal catalyst and optionally a base will produce Compound 7.
Scheme 1

As shown below in Scheme 2, the preparation of Compound 13 is described. Trichloroacetyl pyrrole is treated with NBS in acetonitrile to produce Compound 8. Treatment with sodium ethoxide in EtOH yields the ethyl ester Compound 9. This may be treated with a range of nitrating systems, in this example, NaNC /SCVPy, to generate nitro-pyrrole Compound 10, which can be isolated directly or as a salt form with an appropriate base, preferably dibenzylamine. Ethylation with ethyl iodide generates Compound 11 which may be isolated, or optionally telescoped directly into the arylation with Compound 32. Arylation proceeds in the presence of palladium, Xantphos, potassium pivylate and Hunig’s base to generate Compound 12. Hydrogenation presence of Pt/C followed by cyclization with NaOEt yields Compound 13.
Scheme 2

Another process of the invention is disclosed in Scheme 3 shown below. Compound 14 is prepared from Compound 3 in the presence of an alkylating agent. Treatment with a suitable diboron reagent produces Compound 15, which can then be coupled with a suitably functionalized imidazole derivative to yield Compound 16. Amino lysis with a suitable nitrogen donor produces Compound 17, which can cyclize under appropriate conditions to produce Compound 7.
Scheme 3
Step 3 Step 4 Step 5

As shown below in Scheme 4, ethylation of Compound 9 with ethyl iodide produces Compound 18. This may be directly reacted with dipinacol-diboron in the presence of Pd(OAc)2 and tricyclohexylphosphin hexafluorophosphate and
tetramethylammonium acetate to yield Compound 19. Subsequent coupling with 5-Br-imidazole derivative yields Compound 20. Treatment with hydroxylamine hydrochloride in the presence of triethylamine yields the Compound 21. Subsequent cyclization with Piv20 in the presence of PRICAT™ and hydrogen yields Compound 13.
Scheme 4
77% isolated over 2-steps%

18
Step 5 Pd(OAc)2
PPh3
78%

As shown below in Scheme 5, Compound 23 may be converted to Compound 26 by two pathways. In one option, Compound 23 can be treated with palladium, ligand and a mild base to prepare Compound 25. Reaction of Compound 25 with a metal hydroxide produces Compound 26.
Alternately, Compound 23 can be treated with palladium and ligand in the presence of a soluble hydroxide base, followed by treatment with the metal counter-ion to prepare Compound 26 directly. Once Compound 26 is formed, it can be coupled to Compound 27 to form compound I.


A solution of Compound 1 in acetonitrile (1238.0 kg, 264.9 kg after correction) was charged into a 5000 L glass-lined reactor at a temperature of 20-30 °C. The mixture was added with stirring over about 2 h and then cooled to 0 °C. NBS (221.8 kg) was charged into the mixture at intervals of 20-30 min at 0-20 °C. The mixture was cooled to 0-5 °C and reacted until the content of Compound 8was < 1.0%. Additional NBS (4.0 kg) was charged into the mixture at 0-20 °C. The mixture was reacted over 3 h until the content of Compound 8 was < 1.0%. Purified water (2650.0 kg) was added over about 1.5 – 2.5 h at 0-20 °C. The mixture was cooled to 0-5 °C and then stirred for about 1 h for crystallization. The mixture was filtered and the filter cake was rinsed with water.
Example 2

While maintaining the temperature at 20-30 °C, anhydrous ethanol (950.0 kg) was charged into a 3000 L glass-lined reactor followed by Compound 8 (342.7 kg). The mixture was cooled to 0-5 °C over about 2 h. Sodium alcoholate solution in ethanol (21%, 36.4 kg) was added dropwise over about 1-1.5 h at 0-5 °C. The reaction mixture was then heated to about 25-30 °C and tested until the content of Compounds 8/9 was < 1.0%. The reaction mixture was concentrated at a temperature < 50 °C until about 1.3-1.4 volume of Compound 8 was left. The concentrated mixture was cooled at 25-30 °C. The mixture was quenched into cooled water (3427.0 kg) over about 2 h. After addition, the mixture was stirred at 0-5 °C over about 2 h for crystallization. The mixture was filtered and the filter cake was rinsed. The solid was dried at 30-40 °C over 40-45 h to afford 234.3 kg of Compound 9 , 99.9% purity and 91.3% yield.
Example 3

9 10
A mixture of NaN03, NaHS04, and Na2S04 in CH3CN is wet-milled to constant particle size of -50 micron. To the slurry of inorganic salts is added S03 -pyridine and Compound 9. The reaction mixture is agitated at 25 °C until 90-95% conversion is achieved. The reaction is quenched with aqueous sodium hydroxide and the spent inorganic salts are removed by filtration. The filtrate is passed through a carbon pad and distilled under constant volume distillation and diluted with water to a target 15
volumes/kg of Compound 9 and a target ratio 1.0:2.0 vol/vol MeCN to water. The resulting solids are deliquored, washed, and dried to afford Compound 10.
Example 4

Toluene (10 L/Kg)
65 °C
Compound 10 (1.0 eq) and TBABr (1.0 eq) were added to a biphasic mixture of toluene (8 L/kg 10) and potassium carbonate (1.5 eq) in water (5 L/kg 10). The batch temperature was held at 25 °C. The resulting triphasic slurry was heated to 60-65 °C and diethylsulfate (1.5 eq, in a solution of toluene 2 L/kg 10) was slowly added over ~ 1 h. The reaction was aged until less than 1 RAP of Compound 10 (10:11) remained. The resulting homogeneous biphasic mixture was cooled to 20 °C and the lean aq. phase was removed. The rich organic phase was washed with water (2×7 L/kg 10) and concentrated to 6 mL/g 10. The concentrated stream was dried via azeotropic, constant volume distillation with toluene until the water content of the stream was <0.1 wt %. The resulting stream was telescoped into the subsequent direct arylation reaction.
Example 5

11 28 12
To the toluene stream of Compound 11, with potassium pivalate (1.5 equiv.) was charged, followed by DIPEA (3 eq.), Compound 28 (3 eq.) and Pd(Xantphos)Cl2 (0.04 eq.). The vessel was evacuated to < 200 torr and backfilled with nitrogen (3 X) followed by heating to 95 °C until residual Compound 11 was less than 1 RAP (11: 12). The reaction mixture was cooled to 25 °C and diluted with ethyl acetate (15 mL/g vs input pyrrole) and aq. N-acetylcysteine (0.2 eq., 5 wt % solution, 1.8 mL/g vs. input pyrrole) and heated to 50 °C for 1 h. The biphasic mixture was cooled to 25 °C. The lower aqueous layer was removed. The ethyl acetate stream was washed with water (2×7 mL/g vs. input pyrrole). The rich organic phase was polish filtered followed by a vessel/polish filter rinse with ethyl acetate (2 mL/g vs. input pyrrole). The rich organic stream was concentrated to 4 mL/g vs. input pyrrole via vacuum distillation, while maintaining the batch temperature above 50 °C. If spontaneous nucleation did not occur, Compound 12 seeds (1 wt %) were charged, followed by aging for 30 min at temperature. MTBE (5 mL/g vs. 11) was charged to the slurry over 1 hour while maintaining the batch temperature above 40 °C, followed by aging at 40 °C for 1 h. The slurry was cooled to 0 °C over 6 h and aged at 0°C for 6 h. The slurry was filtered and washed with
EtO Ac : Toluene : MTBE (1.5: 1.0: 1.5, 2 mL/g vs. input 11 ). The wet cake was dried (50 °C, 100 torr) until LOD was < 1 wt %.
Example 6

Compound 12 (1 eq., limiting reagent (LR)) is dissolved in THF/NMP (20 Vol wrt LR, 9/1 ratio) and submitted to hydrogenation using 10 wt% (wrt LR) Pt/C (5 wt%) at 25 to 40° C for 5-10 h. The reaction containing the corresponding amine is filtered. The rich organic stream is concentrated to Compound 12 Vol (wrt LR) and subjected to 0.1 eq of 21 wt% NaOEt/EtOH for 5 h at 20-25 °C, upon which Compound 13 forms. The stream is cooled to 0-10 °C, and water (5L/Kg, wrt to LR) is added and then filtered to isolate Compound 13. The product is dried at 50 °C under vacuum.
Example 7

in toluene solution
9
18
Compound 18 was prepared by treating the pyrrole with ethyl iodide and pulverized potassium carbonate in DMF at 25-30°C under inert atmosphere. After the reaction was completed, the batch mass was cooled to 15°C to 20°C and quenched by slow addition of water then MTBE. The MTBE layer was separated and washed with water. The MTBE layer was distilled to 4 Vol and solvent swapped with toluene. The toluene stream was then taken into the next step.
Example 8

18 19
Tetra-methyl ammonium acetate in toluene slurry was heated to 75-80°C to get a clear solution. The mass was cooled to below 30°C and pyrrole in toluene and bis (pinacolato) diborane were added. The reactor was inerted by nitrogen purging then the reaction was heated to 75-80°C. A freshly prepared catalyst/ligand complex (0.0 leq of palladium acetate, 0.025eq of tricyclohexyl phosphino hexafluoroborate and 0.2eq of tetra methyl ammonium acetate in toluene) was charged under nitrogen atmosphere at RT and stirred for 2h. The mass was then stirred at 75-80°C under nitrogen atmosphere. After the reaction was completed, the mixture was cooled below 30°C and quenched with aq. sodium bisulphate solution. The organic layer was polish filtered through a Celite bed and the filtrate was washed with water. The solvent swapped to ethanol until the toluene content became less than 0.5 %. The solution was cooled to 0-5°C and water was added for crystallization. The product was then isolated by filtration.
Example 9

Compound 20 was prepared by treating Compound 19 with Compound 34 in the presence of palladium acetate, triphenyl phosphine and potassium carbonate in dimethyl acetamide with the water mixture as the solvent. Dimethyl acetamide, water, potassium carbonate and the two starting materials were charged into the reactor. The mixture was made inert with nitrogen for 30 min and then charged with freshly prepared catalyst mixture (palladium acetate, triphenyl phosphine and potassium carbonate in dimethyl acetamide). The temperature was raised to 78-83 °C then the mass was stirred at this temperature. After the reaction was completed, the reaction mass was cooled to ambient temperature and purified water was added slowly into the mass for product
crystallization. The mass was stirred for a period of 3 h and filtered. The wet cake was washed with purified water and dried in VTD at 50-55 °C under vacuum.
Example 10

Compound 21 was prepared by treating Compound 20 with hydroxylamine hydrochloride and triethyl amine using ethanol as the solvent. Compound 20 was added into ethanol (15 Vol) and the reaction mass was heated to 38-40 °C. Hydroxylamine hydrochloride was charged and stirred for 10 min, then triethyl amine was added slowly at 38-40 °C over a period of lh. The above mass was stirred at 38-40 °C until Compound 20 becomes less than 5.0%, typically in about 15 h. After the reaction was completed, the above reaction mass was cooled to ambient temperature (below 30 °C) and filtered. The wet cake was washed with purified water (4 Vol) and dried under vacuum in VTD at 55-60 °C.
Example 11

Initially Compound 21 was treated with pivalic anhydride using toluene and acetic acid mixture as solvent under inert atmosphere until Compound 21 becomes less than 3.0% with respect to Compound 21, typically in about 30 min. PRICAT Nickel was then added under nitrogen atmosphere. The reaction mass was inerted with nitrogen for three cycle times and then degassed with hydrogen gas for three cycle times. Following this, 3.0 kg/cm2 hydrogen pressure was applied to the reaction mass which was stirred for about 12h. After the reaction was completed, the reaction mixture was filtered through a sparkler filter. The filtrate was distilled and the solvent exchanged with toluene until the ratio of acetic acid & toluene reaches 1 :20. At this time, n-Heptane was charged and cooled to 15°C. Then the product was filtered and the wet cake was dried in VTD at 50-55°C under vacuum.

Compound 30 was prepared by the coupling of Compound 22 with Compound 29, 3 -bromo- 1,5 -dimethyl- lH-pyrazole in the presence of
Tris(dibenzylideneacetone)dipalladium chloroform adduct, t-Brettphos and potassium phosphate in tert-amyl alcohol at 98-103 °C under inert atmosphere. After completion of the reaction (typical level of Int.9 -5% & typical reaction hrs 20 h), the mass was cooled to ambient temperature and t-amyl alcohol (4 Vol) and 20 Vol of water were charged into the reaction mass. The reaction mass was stirred for 15 min. and then phase split. The organic layer was diluted with 10 Vol of MTBE and product was extracted with 20 Vol of 1M methane sulphonic acid. The MSA stream was treated with 15 wt % charcoal to reduce the residual palladium numbers. The filtrate was cooled to below 20 °C and the pH was adjusted to 1.7-1.9 using IN NaOH for product crystallization and then iltered. The wet cake was washed with purified water (3 x 5 Vol), followed by methanol (5 Vol). The cake was vacuum dried for 3 h. then the wet cake and dimethyl sulfoxide (20 Vol) were charged into a reactor. The mass was heated to 120-125 °C to get clear solution then the mass was cooled to ambient temperature and stirred for 2 h, then filtered. The wet cake was washed with methanol (3x 4.0 Vol) and vacuum dried for 2 h. The wet cake was dried in VTD at below 55°C under vacuum.
Example 13

Compound 30 , ethanol (16.5 Vol), water and aq sodium hydroxide solution were charged into a reactor then the mass was heated to 70-75 °C and stirred until Compound 30 becomes less than 1.0%. After the reaction was completed, the mass was diluted with ethanol for complete product precipitation at 65-75 °C. Then the mass was cooled to 50 °C for a period of lh and stirred for lh at 50 °C. The mass was further cooled to 20 °C and stirred for lh at 20 °C and then filtered. The wet cake was washed with 5 Vol of 15% aqueous ethanolic solution followed by THF. The wet cake was dried under vacuum at 70-75 °C till LOD comes to less than 5.0 %, typically in about 40 h.
Example 14

In a vessel 36.5 mmol (-42.6 mL) of Compound 29 solution in 2-methyl-2-butanol was combined with 30.7g (65.1 mmol) tetrabutylammonium hydroxide (55 wt% in water), 8.01g (27.0 mmol) Compound 13 , and 10 mL 2-methyl-2-butanol. The mixture was heated at 70 °C until hydrolysis of Compound 13 was complete (full dissolution, <15 min). The solution was cooled to 60 °C and 1.12g (2.22 mmol) of tBuBippyPhos followed by 384 mg (1.028 mmol) allylpalladium chloride dimer (L:Pd = 1 :1) was added. The mixture was heated to 80 °C and was aged at this temperature for 20h before cooling to 22 °C.
Water was added and the mixture concentrated, a constant volume distillation was then performed to swap to ethanol (40-55 °C, 150 mbar). The resulting solution was passed through a 5 micron filter to remove any particulates. The solution was heated to 55 °C and 8.10 mL (40.52 mmol, 1.5 equiv) 5N NaOH (aq) was added dropwise over a 3 h period. Crystals of Compound 31 began to form, and after aging for an additional lh, the mixture was cooled to 20 °C over 3 h. After an additional 6h of aging, crystals were collected on a frit and the cake was washed with 40 mL of 90: 10 ethanol: water, followed by 48 mL acetone. After drying at 80 °C in a vacu-oven for 16 h, Compound 31 was collected as an off-white solid (8.89g, 85%).
Example 15

Compound 31 was added into dichloromethane (20 Vol) and cooled to 15-20 °C. The reaction mass was charged with DMC in DCM solution (1.4 eq of DMC in 5.0 Vol of DCM). The mixture was stirred until Compound 31 becomes less than 2.0% with respect to the corresponding acid chloride, typically in about lh. After completion of the reaction, Compound 27 (1.4 eq) and N,N-diisopropylethyleneamine (3.0 eq) were charged and the mixture was stirred. After completion of the reaction, the mass was quenched with 12 Vol of water then the layers were separated. The organic layer was washed with water and filtered through a celite bed. The filtrate was concentrated to ~6.0 vol and then the mass was cooled to 35 °C. To the resulting solution was added THF, followed by seeds of product, then stirred for 3 h. The solvent was swapped with THF until
dichloromethane becomes less than 2 wt% (wrt THF). The mass was cooled to -5 to 0 °C over a period of 2 h and stirred for 2 h. The reaction mass was then filtered under a nitrogen atmosphere. The material was slurried with pre-cooled THF (2*2 Vol) and filtered. The wet cake was dried in VTD at 60 °C under vacuum till LOD becomes < 1%, typically in about 20 h.
Example 16

DC , RT
I
To a slurry of Compound 31 (15.00 g, 40.0 mmol) in dichloromethane (300 ml) was added diphenylphosphinic chloride (12.29 g, 51.9 mmol). The mixture was stirred at room temperature for 2 h and Ν,Ν-diisopropylethylamine ( 16.53 g, 127.9 mmol) was then added and stirred for another 30 min. Compound 27 (6.94 g, 51.9 mmol) and 4-dimethylaminopyridine (0.49 g, 4.0 mmol) were subsequently added and stirred for 16 h until the reaction was completed. The reaction mixture was treated with N-acetyl-L-cysteine (3.26 g, 20.0 mmol) and citric acid (10.10 g, 48.0 mmol) in deionized water (180 ml) for 2 h. After phase split, the dichloromethane phase was washed once with 0.42 N NaOH solution (180 ml) and washed twice with deionized water (180 ml each). The final dichloromethane phase was concentrated (to 90 ml) and acetone (30 ml) was added. The solution was cooled to 35 °C and N-2 form seed of Compound 1 ( 150 mg ) was added and aged for 1 h. The resulting slurry was solvent-swapped to acetone (DCM < 10% v/v), and cooled to 0 °C. The solid was filtered and washed with cold acetone and dried to afford 14.69 g (85%) of Compound I (HPLC AP 99.8) as off-white crystals.
Patent
WO 2011028864
http://www.google.com/patents/WO2011028864A1?cl=en
Compounds of general formula I in which the R group is thiazole (as in Ial) and R1 and R2 groups are CF3 or alkyl or cycloalkyl or combine to form a saturated carbocyclic or heterocyclic ring or where R2 group is COORb could be prepared using the general method depicted in Scheme 1. Dichloro intermediate II (prepared using procedure reported in WO200612237) could be combined with a 2,4-dimethoxybenzyl and the resulting secondary amine is capped with suitable protective group (Boc) (III). The second chlorine atom could be converted into the
corresponding amine (IV) through the benzophenone imine intermediate. The amino compound could be halogenated to intermediate V. V could be subjected to transition metal mediated indole ring formation and the resulting indole nitrogen is capped with ethyl iodide to afford VI. Ester hydrolysis followed by amide bond formation and cleavage of protective groups with acid treatment would yield amine VII. Amine VII could be converted into thiourea VIII by first coupling with benzoyl isothiocyanate followed treatment with aqueous base. Formation of thiazole could be achieved by condensation with an a-bromoketone derivative (R^HBrCOR2).


a) 2,4-dimethoxybenzylamine, heat; b) NaHMDS, Boc20; c) (Ph)2=NH; d) HCl; e) NIS; f) Pd2(dba)3, ethyl pyruvate; g) Etl, Cs2C03; h) NaOH (aq); i) dicyclopropylamine HCl, HATU, DIPEA; j) TFA; k) Benzoyl isothiocyanate;
1) NaOH (aq); m) I^CHBrCOR1
Scheme 1
Compounds of general formula Ia2 in which the R1 group is CONRaRa could be made using Scheme 2. Thiourea intermediate (VIII) could be combined with Et02CCHBrCOR1 to afford the thiazole ester (IX). The ester could be hydrolyzed and the acid could be coupled with amine to afford thiazole amide derivative (la)

a) Et02CCHBrCOR1; b) NaOH (aq); c) HNRaRa, HATU, DIPEA
Scheme 2
Similarly, compounds of general formula Ia3 in which the R1 group is CONRaRa could be prepared using the general protocol depicted in Scheme 3.

a) R2CHBrCOC02Me; b) NaOH (aq); c) HNRaRa, HATU, DIPEA
Scheme 3
Compounds of general formula la in which R1 is halogen (CI, Br or I) could be prepared by condensing an a,a’-dihaloketone as depicted in Scheme 4.
a) R2COCH(Hal)2
Scheme 4
Alternatively, thiourea derivative VIII could be converted to room temperature into C-5 un-substituted thiazole XI and then directly halogenated using electrophilic halogen source or through metallation followed by quenching with an electrophilic halogenating agent (Scheme 5).

a) BrCH2COR2; b) Selectfluor or NCS or NBS or NIS or tBuLi followed Selectfluor or NBS or NCS
Scheme 5
Compounds of general formula Ia5 in which R1 is S02Rb could be synthesized using the general synthetic approach shown in Scheme 6


a) Br2-acetic acid; b) EtOH, heat
Scheme 6
Compounds with general formula la in which R1 and R2 combine to form an aromatic or heteroaromatic ring could be prepared using Scheme 7.

X = hal, -S02Me
a) Pd(0) catalyst, NaOtBu, phosphine ligand, heat
Scheme 7
Alternatively, these compounds could be made by first coupling aniline or heteroaniline (XVI) with the isothiocyanate (XV) followed by oxidative cyclization (Scheme 8).

a) 1, 1 ‘-Thiocarbonyldi-2( 1 H)-pyridone; b) NaH; c) NIS
Scheme 8
Compounds of general formula Ibl could be prepared using the general synthetic approach depicted in Scheme 9. Aniline VII could be combined with γ-dithiomethylketone compound XVII, (prepared using the procedure reported at room temperature in Synlett, p 2331 (2008)) under basic condition to afford XVIII.
Stepwise condensation of the Boc-protected hydrazine derivative would give the required pyrazole Ibl.

a) NaH, THF; b) R1N(Boc)NH2, AcOH, 35-40°C; c) HCO2H or TFA, 60°C
Scheme 9
Compounds of general formula Ibl or Ifl and If could also be prepared by coupling C-4 halo derivative (XIX) with an appropriately substituted 2-aminopyrazole derivative (XX) using a transition metal catalyzed reaction (Scheme 10).

a) isoamyl nitrite, CH2I2 or isoamyl nitrite, CH2Br2; b) Pd2(dba)3, Xanphos, Cs2C03
Scheme 10
Compounds of general formula Ib2 in which R2 group is CONRaRa could be synthesized using Scheme 11. Aniline VII could be combined with γ-dithiomethylketone derivative XXII, (prepared using the procedure from
Tetrahedron, p 2631 (2003)) to afford intermediate XXIII. Stepwise condensation of Boc-protected hydrazine derivative would give the required pyrazole aldehyde XXIV. Aldehyde could be oxidized using oxone or sodium hypochlorite to furnish carboxylic acid XXV. Coupling of acid XXV with amine would give pyrazole amide Ib2.

a) NaH, THF, heat; b) R1N(Boc)NH2, AcOH; c) TFA; d) oxone or sodium hypochlorite; e) HNRaRa, HATU, DIPEA
Scheme 11
Compounds of general formula Icl could be prepared using the general protocol as shown in Scheme 12. Aniline VII could be coupled with chloroacetyl chloride and the resulting amide could be treated with thioamide (R2CS H2) to furnish thiazole Icl .

a) chloroacetyl chloride, base; b) R2CSNH2
Scheme 12
00120] Compounds of general formula ldl could be made as per Scheme 13. Previously described isothiocyanate derivative XV could be combined with amidine XXV under dehydrating reaction conditions to give 1,2,4-thiadiazole (ldl).

Scheme 13
Compounds of general formula lei could be prepared using a synthetic approach as shown in Scheme 14. Isothiocyanate XV could be combined with azide XXVI in the presence of phosphine to yield 1,3-oxazole Iel .

Scheme 14
Compounds of general formula lgl could be prepared using a synthetic approach as shown in Scheme 15. Amine VII could be combined with acyl isothiocyanate XXVII. The acylthioureaido could be condensed with hydrazine derivative to yield the 1,2,4-triazol derivative lgl.

igi
Scheme 15
without a methyl

Preparation of 7V,7V-dicyclopropyl-6-ethyl-l-methyl-4-(5-m ethyl- lH-pyrazol-3- ylamino)-l,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide
[00437] Prepared using similar protocol as for example 72 from hydrazine.
[00438] MS (ESI) m/z 419.3 (M+H)
[00439] 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 8.70 (br s, 1 H), 7.91 (br s, 1 H), 6.87 (s, 1 H), 6.09 (br s, 1 H), 4.64 (q, 2 H, J= 7.03 Hz), 4.08 (s, 3 H), 2.74 -2.95 (m, 2 H), 2.41 (s, 3 H), 1.51 (t, 3 H, J= 7.15 Hz), 0.81 – 0.95 (m, 4 H), 0.70 -0.81 (m, 4 H)
with an ethyl

7V,iV-dicyclopropyl-6-ethyl-4-(l-ethyl-5-methyl-lH-pyrazol-3-ylamino)-l-methyl- 1,6-dihydroimidazo [4,5-d] pyrrolo [2,3-b] pyridine-7-carboxamide
74A Preparation of fe/t-butyl l,3-dioxoisoindolin-2-yl(ethyl)carbamate

Diisopropyl azodicarboxylate (2.92 mL, 15.00 mmol) was added in one portion to a solution of tert-butyl l,3-dioxoisoindolin-2-ylcarbamate (2.62 g, 10 mmol, prepared following the procedure described by Nicolas Brosse et al. in Eur. J. Org. Chem. 4757-4764, 2003), triphenylphosphine (3.93 g, 15.00 mmol) and ethanol (0.691 g, 15.00 mmol) in THF (20 mL) at 0 °C and the reaction solution was stirred at room temperature for lh (monitored by TLC until completion). Solvent was evaporated and the residue was purified by flash chromatography on silica gel using an automated ISCO system (80 g column, eluting with 5-35% ethyl acetate / hexanes) to provide tert-butyl l,3-dioxoisoindolin-2-yl(ethyl)carbamate (2.6 g, 90 % yield) as a white solid which was used as it in the next step
74B Preparation of fe/t-butyl l-ethylhydrazinecarboxylate
Boc
H2N-N
\
Methylhydrazine (1.415 niL, 26.9 mmol) was added to a solution oi tert-butyl l,3-dioxoisoindolin-2-yl(ethyl)carbamate (example 74A, 5.2 g, 17.91 mmol) in THF (40 mL) at 0 °C and the reaction mixture was stirred at room temperature overnight. A white precipitate formed and was filtered off through a pad of Celite, The filtrate was concentrated in vacuo. The residue was dissolved in ethyl acetate (50 ml) and extracted with IN HC1 (3×30 ml), the acid layer was washed with ethyl acetate (50 ml) and basified to pH 10 by addition of 20% NaOH. The basic solution was then extracted with ethyl acetate (3×50 ml) and the combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated in vacuo to give tert-butyl 1 -ethylhydrazinecarboxylate (2.5 g, 87 % yield) as colorless oil.
XH NMR (400 MHz, CDC13) δ: 3.90 (br. s., 2H), 3.35 (q, J = 7.0 Hz, 2H), 1.42 (s, 9H), 1.07 (t, J = 7.0 Hz, 3H)
74 Preparation of N.N-dicyclopropyl-6-ethyl-4-(l-ethyl-5-methyl-lH-pyrazol-3-ylamino)-l-methyl-l ,6-dihydroimidazor4,5-d1pyrrolor2,3-b1pyridine-7-carboxamide
A mixture of (Z)-N,N-dicyclopropyl-6-ethyl- 1 -methyl-4-( 1 -(methylthio)-3-oxobut-l-enylamino)-l,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide (example 74B, 70 mg, 0.155 mmol) and tert-butyl 1-ethylhydrazinecarboxylate (49.6 mg, 0.309 mmol) in acetic acid (1 mL) wan stirred at 35 °C for 4 h (monitored by LC/MS until no starting material left). Formic acid (1 mL) was added and the reaction mixture stirred at 60 °C for 6 h. The solvent was evaporated and the crude product was purified by flash chromatography on silica gel using an automated ISCO system (12 g column, eluting with 2-10% methanol / dichloromethane). The material was further purified by preparative HPLC to afford N,N-dicyclopropyl-6-ethyl-4-( 1 -ethyl-5-methyl- lH-pyrazol-3-ylamino)- 1 -methyl- 1 ,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide (38 mg, 53.4 % yield) as an off-white solid.
MS (ESI) m/z 447.3 (Μ+Η).
XH NMR (500 MHz, CDC13) δ: 8.08 (s, 1H), 7.61 (s, 1H), 6.93 (s, 1H),
6.84 (s, 1H), 4.66 (q, J = 7.1 Hz, 2H), 4.02 (q, J = 7.2 Hz, 2H), 3.98 (s, 3H), 2.79 – 2.85 (m, 2H), 2.34 (s, 3H), 1.49 (t, J = 7.1 Hz, 3H), 1.41 (t, J = 7.2 Hz, 3H), 0.82 -0.87 (m, 4H), 0.72 – 0.78 (m, 4H).
Patent
JAK2 INHIBITORS AND THEIR USE FOR THE TREATMENT OF MYELOPROLIFERATIVE DISEASES AND CANCER [US8202881]2011-03-102012-06-19
JAK2 inhibitors and their use for the treatment of myeloproliferative diseases and cancer [US8673933]2012-04-302014-03-18
: Purandare AV, McDevitt TM, Wan H, You D, Penhallow B, Han X, Vuppugalla R, Zhang Y, Ruepp SU, Trainor GL, Lombardo L, Pedicord D, Gottardis MM, Ross-Macdonald P, de Silva H, Hosbach J, Emanuel SL, Blat Y, Fitzpatrick E, Taylor TL, McIntyre KW, Michaud E, Mulligan C, Lee FY, Woolfson A, Lasho TL, Pardanani A, Tefferi A, Lorenzi MV. Characterization of BMS-911543, a functionally selective small-molecule inhibitor of JAK2. Leukemia. 2012 Feb;26(2):280-8. doi: 10.1038/leu.2011.292. Epub 2011 Oct 21. PubMed PMID: 22015772.
Characterization of BMS-911543, a functionally selective small-molecule inhibitor of JAK2http://www.nature.com/leu/journal/vaop/ncurrent/full/leu2011292a.html
GRAPHS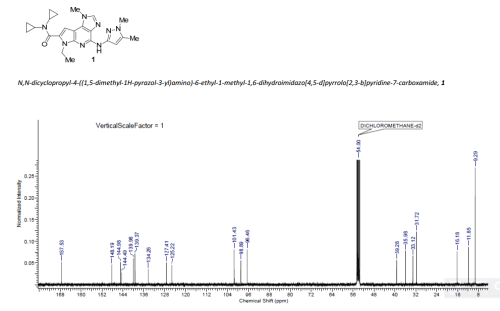
Click to access jo5b00572_si_001.pdf

//////BMS 911543, phase 2, bms,
Regorafenib, SHILPA MEDICARE LIMITED, New patent, WO 2016005874
![]()
WO2016005874, PROCESS FOR THE PREPARATION OF REGORAFENIB AND ITS CRYSTALLINE FORMS
SHILPA MEDICARE LIMITED [IN/IN]; 10/80,Second Floor,Rajendra Gunj, Raichur, ರಾಯಚೂರು , karnataka 584102 (IN)
RAMPALLI, Sriram; (IN).
UPALLA, Lav Kumar; (IN).
RAMACHANDRULA, Krishna Kumar; (IN).
PUROHIT, Prashant; (IN).
AKSHAY KANT, Chaturvedi; (IN)
The present invention relates to a process for the preparation of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methylpyridine-2- carboxamide or Regorafenib (I): Formula (I). The present invention further relates to a process for the purification of 4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl} amino)-3-fluorophenoxy]-N-methylpyridine-2- carboxamide or Regorafenib (I) to provide highly pure material. The present invention further relates to a process for the preparation stable crystalline material of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]- N-methyl pyridine-2-carboxamide or Regorafenib (I) useful in the preparation of pharmaceutical compositions for the treatment of cancer.
4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide or Regorafenib is low molecular weight, orally available, inhibitor of multiple protein kinases, including kinases involved in tumour angiogenesis (VEGFR1, -2, -3, TIE2), oncogenesis (KIT, RET, RAF-1, BRAF, BRAFV600E), and the tumour microenvironment (PDGFR, FGFR). In preclinical studies regorafenib has demonstrated antitumour activity in a broad spectrum of tumour models including colorectal tumour models which is mediated both by its antiangiogenic and antiproliferative effects. Major human metabolites (M-2 and M-5) exhibited similar efficacies compared to Regorafenib both in vitro and in vivo models.
Regorafenib was approved by USFDA in 2012 and is marketed under the brand name Stivarga®, is an important chemotherapeutic agent useful for the treatment of adult patients with metastatic colorectal cancer (CRC) who have been previously treated with, or are not considered candidates for, available therapies. These include fluoropyrimidine-based chemotherapy, an anti-VEGF therapy and an anti-EGFR therapy.
Regorafenib is chemically known as 4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl} amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide (I). Regorafenib is a white to slightly pink or slightly brownish solid substance with the empirical formula C2iHi5ClF4N403 and a molecular weight of 482.82. Regorafenib is practically insoluble in water, dilute alkaline solution, dilute acid solution, n-heptane, glycerine and toluene. It is slightly soluble in acetonitrile, dichloromethane, propylene glycol, methanol, 2-propanol, ethanol and ethyl acetate. It is sparingly soluble in acetone and soluble in PEG 400 (macrogol). Regorafenib is not hygroscopic.
Regorafenib is generically disclosed in US 7351834, and specifically disclosed in US 8637553. US ‘553 disclose a process for the preparation of Regorafenib starting from 3-fluoro-4-nitrophenol. The process is as demonstrated below:

The present inventors has repeated the above process and found the following disadvantages:
Unwanted reactions are observed during the formation of Regorafenib, due to the involvement of prolonged time in process.
> Incomplete reactions were observed with excessive impurity formations due to incomplete conversion.
Removal of impurities from final product
US 2010173953 disclose Regorafenib monohydrate and crystalline Form I of Regorafenib. This patent application further discloses that crystalline Form I of Regorafenib stated in this application is obtained as per the process disclosed in WO 2005009961 A2 (Equivalent to US ‘553). The compound obtained was having a melting point of 186-206° C.
This patent publication discloses a process for the preparation of Regorafenib monohydrate comprises dissolving Regorafenib Form I obtained as per WO ‘961 in acetone
and the solution is filtered, followed by addition of water until precipitation, which was filtered and dried at room temperature
US 2010/0113533 discloses crystalline Form II of Regorafenib, comprises dissolving Regorafenib Form I obtained as per WO ‘961 in ethyl acetate, the suspension was heated to 40-45°C, addition of isocyanate solution (isocyanate in ethyl acetate) and is cooled to room temperature to yield the crystals, which was filtered, washed with ethyl acetate and dried at room temperature.
US 2010/0063112 discloses Form III of Regorafenib, process comprises of heating
Regorafenib monohydrate at 100°C or 60 min, and further 15 min at 110°C, followed by cooling to room temperature.
As polymorphism has been given importance in the recent literatures owing to its relevance to the drugs having oral dosage forms due to its apparent relation to dose preparation/suitability in composition steps/ bioavailability and other pharmaceutical profiles, stable polymorphic form of a drug has often remained the clear choice in compositions due to various reasons of handling, mixing and further processing including bioavailability and stability.
Exploring new process for these stable polymorphic forms which are amenable to scale up for pharmaceutically active / useful compounds such as 4-[4-({[4-chloro-3-(trifluoro methyl)phenyl]carbamoyl } amino)-3 -fluorophenoxy] -N-methylpyridine-2 -carboxamide or Regorafenib may thus provide an opportunity to improve the drug performance characteristics of such products.
Hence, inventors of the present application report a process for the preparation of a stable and usable form of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluoi phenoxy]-N-methylpyridine-2-carboxamide or Regorafenib, which may be industrially amenable and usable for preparing the corresponding pharmaceutical compositions. The present invention provides an improved process for the preparation of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fiuorophenoxy]-N-methylpyridine-2-carboxamide or Regorafenib crystalline forms specifically for crystalline polymorphic forms Form I and Form III. Crystalline polymorphic forms of 4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl } amino)-3 -fluorophenoxy] -N-methylpyridine-2 -carboxamide or Regorafenib obtained by the process of the present invention is non-hygroscopic and chemically stable and has good dissolution properties.
The process related impurities that appear in the impurity profile of the Regorafenib may be substantially removed by the process of the present invention resulting in the formation of highly pure material. The process of the present invention is as summarized below:

Example 1
Preparation of 4-(4-amino-3-fluorophenoxy) pyridine-2-carboxylic acid methyl amide
4-Amino-3-fiuorophenol (l lg, 0.08 moles) and of 4-Chloro-N-methyl-2-pyridinecarboxamide (8.85 g, 0.05 moles) was added to a reaction flask containing N, N-dimethylacetamide (55 ml) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 110-115°C and then potassium tert-butoxide in tetrahydrofuran (60 ml, 0.06 moles) was added slowly over a period of 3 to 4hours. Distill off solvent at same temperature, cooled the reaction mass to 25-30°Cand water(110 ml) was added slowly over a period of 15min. and cooled the reaction mass to 0-5°C . Adjust the pH of the reaction mass in between 7 and 7.5 by using 10% aqueous hydrochloric acid (~7 ml). Stir the reaction mass for 30min at the same temperature. Filter the product, washed with water (22 mL) and Dried at 50-55 °C for 12hrs. The obtained crude material was added to the flask containing Ethyl acetate (55 mL).The reaction mass was heated to reflux to get a clear solution and stirred for 15min at reflux. Cooled to 0-5°C, stir for 2hrs at the same temperature. Filter the product, washed with Toluene (9 mL) and dried at 50-55°C for 3-5hrs.
Above recrystallized material was added to the reaction flask containing methylene dichloride (270 mL) at 25-30°C and stirred for 10-15 min. Activated carbon (1 g) and silica gel (4.4 g) was added to the reaction mass and stir for lh at the same temperature. Filter the reaction mass through hyflow bed and wash with methylene dichloride (18 mL).Distill off solvent still~l-2 volumes of methylene dichloride remains in the flask and then cooled to 25-30°C. Toluene (20 mL) was added and stirred for 30min at the same temperature. Filtered the product, washed with Toluene (9 mL) and dried at 50-55°C for 12h.
Yield: 9 gm
Chromatographic Purity (By HPLC): 98%
Example 2
Preparation of Regorafenib
4-(4-amino-3-fluorophenoxy) pyridine-2-carboxylic acid methyl amide (4g, 0.01 moles) was added in to a reaction flask containing acetone (20 ml) at 25-30°C and stirred for 15 minutes. 4-chloro-3-trifluoromethylisocyanate (6.1g, 0.02 moles) was added slowly over a period of 5 to 10 minutes and stirred the reaction mixture 3 to 4 hours. Toluene (20 n L) was added to the reaction mass and stirred for 30 min at 25-30°C.The obtained reaction mass was filtered and washed with toluene (8 mL). Dried the material still constant weight appears to yield title product a crystalline material.
Yield: 5.5 gm
Chromatographic Purity (By HPLC): 97%
Example 3
Purification of Regorafenib using acetone and toluene mixture
4- [4-( { [4-chloro-3 -(trifluoromethyl)phenyl] carbamoyl } amino)-3 -fluorophenoxy] -N-methylpyridine-2-carboxamide (I) or Regorafenib (1 g) was added slowly in to the reaction flask containing acetone (2 mL) and toluene (3 mL) at 25-30°C and stirred for 15 minutes.
The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes.
Cooled the reaction mass to 25-30°C and stirred for 1 hour. Filter the material, washed with toluene (2 mL) and suck dried for 15 min, followed by drying at 50-55°C for 10-12h to yield
Pure 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methyl pyridine-2-carboxamide (I) or Regorafenib.
Yield: 0.88gm
Chromatographic Purity (By HPLC): 99.3 %
Example 4
Purification of Regorafenib using acetone
4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl} amino)-3 -fluorophenoxy] -N-methylpyridine-2-carboxamide (I) or Regorafenib (1 g) was added slowly in to the reaction flask containing acetone (5 mL) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 0-5°C and stirred for 1 hour. Filter the material, washed with acetone (1 mL) and suck dried for 15 min. The obtained wet cake was added in to the reaction flask containing acetone (5 mL) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50- 55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 0-5°C and stirred for 1 hour. Filter the material, washed with acetone (1 mL) and dried at 60-65°C for 12 h to yield Pure 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methyl pyridine -2-carboxamide (I) or Regorafenib.
Yield: 0.7 gm
Chromatographic Purity (By HPLC): 99.77%
Example 5
Double – Purification of Regorafenib using acetone and toluene mixture
4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] Carbamoyl} amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide (I) or Regorafenib (1 g) was added slowly in to the reaction flask containing acetone (2 mL) and toluene (3 mL) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 25-30°C and stirred for 1 hour. Filter the material, washed with toluene (2 mL) and suck dried for 15 min. The obtained wet cake was added in to the reaction flask containing acetone (2 mL) and toluene (3 mL) mixture at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 25-30°C and stirred for 1 hour. Filter the material, washed with toluene (2 mL) and dry at 60-65°C for 12h.
Yield: 0.80gm
Chromatographic Purity (By HPLC): 99.79 %
Moisture content: 0.09%
Impurity-A: 0.03%
Impurity-B: Not detected
Impurity-C: 0.02%
Example 6
Preparation of Regorafenib Form I
4-(4-amino-3-fluorophenoxy) pyridine-2-carboxylic acid methyl amide (1.3 g, 0.004 moles) was added in to a reaction flask containing acetone (13 mL) at 25-30°C and stirred for 15 minutes.4-chloro-3-trifluoromethylisocyanate (6.6 g, 0.006 moles) wasadded slowly over a period of 15 to 20 minutes and stirred the reaction mixture 3 to 4 hours. The obtained reaction mass was filtered and washed with acetone. Dried the material still constant weight appears to yield title product a crystalline material.
Yield: 1.9 g
Chromatographic Purity (By HPLC): 98.4 %
XRPD was found to resemble similar to Fig-1.
![]()
Omprakash Inani – Chairman, Vishnukant C Bhutada – Managing Director, Namrata Bhutada – Non Executive Director, Ajeet Singh Karan – Independent Director, Carlton Felix Pereira – Independent Director, Pramod Kasat – Independent Director, Rajender Sunki Reddy – Independent Director, N P S Shinh – Independent Director,
 Mr. Omprakash Inani |
Mr. Omprakash Inani – CHAIRMAN
Mr. Omprakash Inani has more than 30 years of Business experience. He monitors business and functional aspects of the Company along with the operations of all the plants. Additionally, he is member of Audit and Remuneration committee of Shilpa Medicare Group of Companies. Currently he is also a council Member in “Academy of Medical Education, Dental College & V.L. College of Pharmacy”, “Taranath Shikshana Samsthe, Raichur” and a trustee in “Akhil Bhartiya Maheshwari Education Trust, Pune”. Mr. Omprakash Inani is also Managing Committee Member of “Karnataka State Cotton Assn., Hubli”. |
|
|
|
||
 Mr. Vishnukant C. Bhutada Mr. Vishnukant C. Bhutada |
Mr. Vishnukant C. Bhutada – MANAGING DIRECTOR
Mr. Vishnukant has vast and diverse Business experience of API and Intermediates and presently leads the core Business and functional teams which accelerate growth and performance by Innovating for Affordable solutions at Shilpa Medicare Group of Companies. He is the key decision maker with the teams for Shilpa Group for successful API and Generics formulation strategies. His untiring efforts have led the company to a leadership position in the Indian pharmaceutical domain and helped create a prominent presence for Oncology APIs globally. For his efforts on APIs Business, Mr. Vishnukant was awarded “Best Entrepreneur Award” by Late Dr Shankar Dayal Sharma – President of India in 1995. Subsequently, various state honours were conferred upon him -like -“Best Entrepreneur” from Karnataka State Govt. in 1996; “Excellence in Exports” from Vishweshwarayya Industrial Trade Centre, Bangalore 1996; and Export Excellence Award-2006” by FKCCI, Bangalore. Success has never stopped coming his way- as he was awarded “First runner up” at the Emerging India Awards London 2008 by CNBC TV18. Recently, his efforts in the Shilpa Group for environment sustainability, has led to “Best National Energy Conservation Award in Drugs & Pharmaceutical Sector for the year 2012” by Hon’ble President of India, Dr. Pranab Mukherjee. |
|
|
|
||
 Dr. Vimal Kumar Shrawat Dr. Vimal Kumar Shrawat |
Dr. Vimal Kumar Shrawat – CHIEF OPERATING OFFICER
Dr. Shrawat by qualification holds degrees of M.Sc (Organic Chemistry), Ph.D. (from Delhi University) and joined Shilpa Medicare in 2009. He has vast experience of more than 25 years of working in large pharma industries like Ranbaxy/ Dabur Pharma- presently known as Fresenius Kabi Oncology Ltd., spanning across activities of R&D, Pilot and Plant Productions, QA/QC, Administration, CRAMS, Project management etc. Presently, Dr. Shrawat is spearheading the entire Operations/ Control of Shilpa Medicare. His vision of team work and time bound approach always guides and motivates teams at all operational sites. His keen interest and consistent efforts for R&D has led him to become one of key contributor in large number of Patent/applications of Shilpa Medicare. |
|
|
|
||
 Dr. Pramod Kumar |
Dr. Pramod Kumar – MANAGING DIRECTOR(LOBA FEINCHEMIE GMBH AUSTRIA), SENIOR VICE-PRESIDENT (SHILPA MEDICARE LTD)
Dr. Pramod Kumar, who by qualification holds degrees of M.Pharm, Ph.D (Pharmaceutical chemistry) and a PGDBA, joined Shilpa Medicare in 1989. Since 2009 he is Managing Director of Loba FeinchemieGmBH, Austria and driving all R&D driven commercial processes. Dr. Pramod Kumar has more than 25 years of experience in Pharmaceutical industry, spanning across activities of production, QA/QC, administration, import/export, CRAMS etc. His efforts in CRAMS have led to the formation of Joint venture company RAICHEM MEDICARE Pvt LTD with Italian companies ICE SPA / P.C.A SPA. |
|
|
|
||
 Mr. Prashant Purohit |
Mr. Prashant Purohit – VICE-PRESIDENT-CRD
Mr. Prashant Purohit by qualification holds degrees of, M.Sc.(Organic Chemistry) and Diploma in Business Management and joined Shilpa Medicare in 1996. He is presently heading Chemical R&D wings of Shilpa Medicare Group. He has vast experience of handling CRAMS and Generics APIs R&D. His vast experience of nearly 35 years in R & D and production in Pharmaceutical Industry has consistently enriched the portfolio of Shilpa Medicare Group of Companies. He is one of key contributor in large number of Patent/applications of Shilpa Medicare. |
|
|
|
||
 Dr. Akshay Kant Chaturvedi |
Dr. Akshay Kant Chaturvedi – HEAD- CORPORATE IPM & LEGAL AFFAIRS
Dr. Akshay Kant by qualification holds degrees of M.Sc, Organic Chemistry (Univ. Gold Medalist), Ph.D. (Medicinal Chem), LL.B., M.B.A. and joined Shilpa Medicare in Jun 2012. Presently, Dr. Akshay is spearheading the entire IP portfolio management/ Legal Affairs of Contractual Business of Shilpa Medicare Group. His vision of innovative and creative thinking, team work and time bound approach always guide and motivate teams at all locations.His keen interest and consistent efforts for R&D has led him to become one of key contributor in large number of Patent/applications of Shilpa Medicare. |
|
|
|
||
 Dr. Seshachalam U. |
Dr. Seshachalam U. -ASSOCIATE VICEPRESIDENT- QUALITY AND RA
Dr. Seshachalam by qualification holds M.Sc (Chemistry) and Ph.D. (Chemistry) and joined Shilpa Medicare in 2008. He is presently heading Regulatory Affairs wings of Shilpa Medicare Group of Companies. He has vast experience of handling regulatory affairs related to Generics APIs. Being instrumental in Shilpa Medicare’s efforts to achieve recognition of different authorities, his key contribution in successful inspection and audit by various regulatory authorities is one of the core strength to the organization’s aims and objectives. |
|
|
|
||
 Mr. Sharath Reddy |
Mr. Sharath Reddy – VICE-PRESIDENT PROJECTS & OPERATIONS
Mr. Sharath Reddy by qualification holds M.Pharm from BITS Pilani and has overall experience of about 22 years predominately in the field of pharmaceuticals new projects and operations. His expertise of Oncology specialized equipment and Utilities designing has boosted organizations confidence to takeover new endeavors of upcoming projects with faster pace of time. His efforts have led to successfully executing Energy Saving projects of Shilpa Medicare Group of Companies and registration of the project under Clean Development Mechanism with UNFCC (Under Kyoto Protocol). |
|
|
|
||
 Mr. R K Somani |
Mr. R K Somani – VICE-PRESIDENT FORMULATION -BUSINESS DEVELOPMENT
Mr. R. K. Somani is a professional Chartered Accountant and holds a Diploma in Central Excise.He has overall business experience of more than 21 years predominately in the field of pharmaceuticals. Mr. Somani is one of the key drivers of Formulation business besides handling various key Contract Businesses of advanced oncology/ Non-Oncology APIs. He is known for successfully building formulations portfolio and spearheading the Generic sales operation. |
|
Shilpa Medicare Limited
1st Floor, 10/80,
Rajendra Gunj,
RAICHUR ರಾಯಚೂರು – 584 102.
Karnataka, India.
Telephone: +91-8532-236494
Fax: +91-8532-235876
Email: info@vbshilpa.com
RAICHUR, ರಾಯಚೂರು Karnataka, India








Historical Stone Elephants in Malayabad, Raichur Taluk …

View of Raichur city and lake Aam Talab
///Regorafenib, SHILPA MEDICARE LIMITED, new patent, WO 2016005874, raichur, ರಾಯಚೂರು , karnataka, india
FDA´s Emerging Technology Applications Program – Draft Guidance
![]()
FDA´s Emerging Technology Applications Program – Draft Guidance
The FDA recently published a draft guidance for industry on the “Advancement of Emerging Technology Applications”. The draft guidance provides recommendations to pharmaceutical companies interested in participating in a program involving the submission of CMC information containing emerging manufacturing (including testing, packaging and labeling, and quality control) technology to FDA. Find out more about the draft guidance for industry “Advancement of Emerging Technology Applications to Modernize the Pharmaceutical Manufacturing Base“..
On December 23, 2015, the FDA published a draft guidance for industry “Advancement of Emerging Technology Applications to Modernize the Pharmaceutical Manufacturing Base“. Comments and suggestions regarding this draft document should be submitted within 60 days of publication.
The draft guidance provides recommendations to pharmaceutical companies interested in participating in a program involving the submission of CMC (chemistry, manufacturing, and controls) information containing emerging manufacturing (including testing, packaging and labeling operations, and quality control) technology to FDA.
The program is open for new drug applications (INDs), original or supplemental new drug application (NDA), abbreviated new drug application (ANDA), or biologic license application (BLA). It only affects the quality section of a submission (CMC and facility-related information).
The development of emerging manufacturing technology, like, for example, aseptic manufacturing facilities with highly automated systems and isolators, may lead to improved manufacturing, and therefore improved product quality and availability throughout a product´s lifecycle.
Pharmaceutical companies can submit questions and proposals about the use of these technologies to a group within CDER (Emerging Technology Team – ETT).
The draft guidance is a follow-on to the FDA guidance for industry “PAT – A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance” which describes the concept that quality cannot be tested into products. It should be built-in or should be present by design. Through the ETT, FDA intends to encourage the adoption of innovative approaches by leveraging existing resources of FDA to facilitate regulatory reviews of submissions.
Examples of emerging technology elements include an innovative or novel:
- Product manufacturing technology, such as the dosage form;
- Manufacturing process (e.g., design, scale-up, and/or commercial scale);
- Testing technology.
Interested parties should submit a written meeting request to participate in the ETT program at least three months prior to the planned application (IND, ANDA, BLA, NDA) submission date. In addition to the items outlined in the FDA guidance “Formal Meetings Between the FDA and Sponsors or Applicants” the request should also include the following items:
- A brief description of the proposed testing, process, and/or proposed technology;
- A brief explanation why the proposed testing, process, and/or technology are substantially novel and unique;
- A description of how the proposed testing and/or technology could modernize pharmaceutical manufacturing and thus improve product safety, identity, strength, quality, or purity;
- A summary of the development plan and any perceived roadblocks to technical or regulatory implementation;
- A timeline for submission.
The request should generally not exceed five pages and FDA expects to notify companies of its decision regarding acceptance into the program within 60 days of receipt of the request. Once accepted into the program, the participant can engage with ETT and CMC in accordance with existing meeting procedures and guidances (e.g. above mentioned FDA guidance on Formal Meetings).
For further information, please find all the details in the draft guidance “Advancement of Emerging Technology Applications to Modernize the Pharmaceutical Manufacturing Base“.
Lupin Ltd, Patent, Pitavastatin, WO2014203045
![]()
Lupin Ltd, Patent, Pitavastatin, WO2014203045
A NOVEL, GREEN AND COST EFFECTIVE PROCESS FOR SYNTHESIS OF TERT-BUTYL (3R,5S)-6-OXO-3,5-DIHYDROXY-3,5-O-ISOPROPYLIDENE-HEXANOATE
ROY, Bhairabnath; (IN).
SINGH, Girij, Pal; (IN).
LATHI, Piyush, Suresh; (IN).
AGRAWAL, Manoj, Kunjabihari; (IN).
MITRA, Rangan; (IN).
TRIVEDI, Anurag; (IN).
PISE, Vijay, Sadashiv; (IN).
RUPANWAR, Manoj; (IN)
The present invention describes an eco-friendly and cost effective process for the synthesis of teri-butyl (3R,5S)-6-oxo-3,5-dihydroxy-3,5-0-isopropylidene-hexanoate [I]
 PITAVASTATIN
PITAVASTATIN
![]()
TEXT
tert-b tyl (3R,5S)-6-oxo-3,5-dihydroxy-3,5-0-isopropylidene-hexanoate [I] [CAS No. 124752-23-4] is key intermediate for the preparation of statins such as Atorvastatin (Tetrahedron 63, 2007, 8124 -8134), Cerivastatin (Journal of Labeled Compounds and Radiopharmaceuticals, 49, 2006 311-319), Fluvastatin [WO2007125547; US 4739073], Pitavastatin [WO2007/132482; US2012/22102 Al, WO2010/77062 A2; WO2012/63254 Al ; EP 304063; Tetrahedron Letters, 1993, 34, 513 – 516; Bulletin of the Chemical Society of Japan, 1995, 68, 364 – 372] and Rosuvastatin [WO2007/125547 A2; WO2011/132172 Al ; EP 521471]. Statins are used for treatment of hypercholesterolemia, which reduces the LDL cholesterol levels by inhibiting activity of HMG-CoA reductase enzyme, which is involved in the synthesis of cholesterol in liver.

[I]
Compound [I] is generally obtained by various methods of oxidation of teri-butyl 2- ((4R,65)-6-(hydroxymethyl)-2,2-dimethyl-l,3-dioxan-4-yl)acetate [compound II] and are discussed in details hereinafter. In addition, various methods for synthesis of compound [II] are also elaborated below.

[II]
[II]
A) tert-butyl2-((4«,6.S)-6-(hydroxymethyl)-2,2-dimethyl-l,3-dioxan-4-yl)acetate
[compound II]
US patent Number 5278313 describes a process for synthesis of compound [II]
(Schemel). In the said process, (5)-methyl 4-chloro-3-hydroxybutanoate has been obtained in only 70% yield through whole cell enzymatic reduction of methyl 4-chloro-3- oxobutanoate, which has a necessity of special equipment such as fermenters as well as other microbial facilities such as sterile area, autoclaves, incubator for growing seed culture, etc.
(S)-mefhyl 4-chloro-3-hydroxybutanoate upon reaction with teri-butyl acetate in presence of LiHMDS or LDA at -78°C, yielded (S)-ieri-butyl 6-chloro-5-hydroxy-3- oxohexanoate, which was further transformed to corresponding diol through syn selective reduction in presence of methoxydiethyl borane/sodium borohydride at -78°C. The diol thus obtained was converted to compound [II] .
The overall yield for this process is low and required special equipment such as fermenters, etc and in addition to that, this process is not cost effective due to use of costly reagent such as methoxydiethyl borane.
Moreover, methoxydiethylborane is highly pyrophoric (Encyclopedia for organic synthesis, editor in chief L. Paquette; 2, 5304; Published by John and Wiley Sons;
Organic Process Research & Development 2006, 10, 1292-1295) and hence safety is a major concern.

Scheme 1
EP 1282719 B l (PCT application WO 01/85975 Al ) discloses a process for synthesis of compound ( R, 5S)-tert-bv y\ 3,5,6-trihydroxyhexanoate from (S)-tert-b tyl-5,6-dihydroxy-3-oxohexanoate through a) asymmetric hydrogenation in presence of a chiral catalyst e.g. di-mu-chlorobis-[(p-cymene)chlororuthenium(II)] along with an auxiliary such as (IS, 2S)-(+)-N- (4-toluenesulfonyl)-l ,2-diphenylethylenediamine as ligand, which gave desired product only in 70% diastereomeric excess (de); b) Whole cell enzymatic reduction of (S)-tert- butyl 5,6-dihydroxy-3-oxohexanoate to obtain compound (3R, 5S)-tert-bv y\ 3,5,6-trihydroxyhexanoate in 99% de (80% yield).
It is needless to mention that it has necessity of fermenter and other microbiological equipment (Scheme 2).
Moreover, conversion of (2>R,5S)-tert-bv y\ 6-acetoxy-3,5-dihydroxyhexanoate to tert-bv yl 2-((4R,65)-6-(acetoxymethyl)-2,2-dimethyl-l ,3-dioxan-4-yl)acetate was accomplished in only 25% yield and also required the flash chromatography for isolation of desired product.
Thus, overall yield for this process is poor and process is not operation friendly especially at large scale hence cannot be considered feasible for commercial manufacturing.

Scheme 2
EP1317440 Bl (PCT Application WO 02/06266 Al) has disclosed the process for synthesis of compound [II] from 6-chloro-2,4,6-trideoxy-D-erythro-hexose (Scheme 3) .
In the said patent application 6-chloro-2,4,6-trideoxy-D-erythro-hexose was converted to (4R, 65)-4-hydroxy-6-chloromethyl-tetrahydropyran-2one with excess of bromine in presence of potassium bicarbonate, which liberates environmentally undesired gas i.e. carbon dioxide.
Moreover, starting material i.e. 6-chloro-2,4,6-trideoxy-D-erythro-hexose is not commercially available and conversion efficiency of starting material at large scale towards (4R, 65)-4-hydroxy-6-chloromethyl-tetrahydropyran-2-one is only 67%.

Scheme 3
US Patent No. 6689591 B2 has demonstrated the whole cell enzymatic reduction of teri-butyl 6-chloro-3,5-dioxohexanoate to compound [II] (Scheme 4).
In the said process, whole cell enzymatic reduction is not specific; yield for desired product is only 34% and other partially reduced products are also obtained.
Hence, further purification is required for obtaining the desired compound. Thus, this process is not suitable for commercial scale.

Scheme 4
Tatsuya et al (Tetrahedron Letters; 34, 1993,513 – 516) has reported synthesis of compound [I] from derivative of L-tartatric acid (Scheme 5).
In the said process, tartaric acid di-i‘sopropyl ester is doubly protected by tert-butyldimethylsilyl group, which was reacted with dianion of teri-butyl acetoacetate to give β, δ-diketo ester compound.
β,δ-diketo ester was reacted with 2 equivalent of diisobutylaluminium hydride (which is a pyrophoric reagent) to afford -hydroxy,8-keto ester in only 60% yield.
This process is not industrially viable as overall yield is very low and also because of use of costly and pyrophoric reagents/chemicals.

Scheme 5
US7205418 (PCT application WO03/053950A1) has described the process for synthesis of compound [II] from (S)-ieri-butyl-3,4-epoxybutanoate (Scheme 6).
The overall yield for this process is very low and moreover, it required the diastereomeric separation of teri-butyl 2-(6-(iodomethyl)-2-oxo-l,3-dioxan-4-yl)acetate by flash chromatography.
Since overall requirement of title compound is very high, any operation involving flash chromatography will tend to render the process commercially unviable.

Scheme 6
Fengali et al (Tetrahedron: Asymmetry 17; 2006; 2907-2913) has reported the process for synthesis of compound [II] from racemic epichlorohydrin (Scheme 7).
In this process, racemic epichlorohydrin was converted to corresponding nitrile intermediate through reaction with sodium cyanide; nitrile intermediate thus obtained was further resolved through lipase catalyzed stereo-selective esterification to obtain (5)-4-(benzyloxy)-3-hydroxybutanenitrile and (R)-l-(benzyloxy)-3-cyanopropan-2-yl acetate;
separation of desired product i.e. (S)-4-(benzyloxy)-3-hydroxybutanenitrile having 98% de (40% yield) was done by column chromatography.
Needless to mention a commodity chemical like compound [I] cannot be manufactured by such a laboratory method, which involved number of steps.

Scheme 7
Bode et al (Organic letters, 2002, 4, 619-621) has reported diastereomer- specific hydrolysis of 1,3-diol-acetonides (Scheme 8).
In this publication, duration of the reaction for diastereomer- specific hydrolysis of 1,3, diol-acetonides is reported to be 4 h, however, in our hand it was observed that hardly any reaction took place in 4 h, which made it non-reproducible.
In addition to that, separation of desired product is achieved by flash chromatography and it is needless to mention that any process which involved flash chromatography would render the process to be commercially unviable.
Hence, additional innovation needs to be put in for making the process industrially viable.

Scheme 8
CN 101613341A has reported the process for synthesis of compound [II] (Scheme
9).
In the same patent application tert-b tyl (S)-6-chloro-5-hydroxy-3-oxohexanoate was synthesized through Blaise condensation of (5)-4-chloro-3-hydorxy-butanenitrile with zinc enolate of tert butyl bromo acetate.
In the literature, synthesis of tert-bv yl (S)-6-chloro-5-hydroxy-3-oxohexanoate was reported through Blaise condensation of silyl protected (5)-4-chloro-3-(trimethylsilyl)oxy-butanenitrile with zinc enolate of tert butyl bromo acetate, in good yield (Synthesis 2004, 16, 2629-2632). Thus, protection of hydroxy group in (5)-4-chloro-3-hydorxy-butanenitrile is imperative.
In the said Chinese patent application, in claim 7, it was mentioned that solvent used for conversion of tert-bv yl (5)-6-chloro-5-hydroxy-3-oxohexanoate to ( R,5S)-tert-butyl 6-chloro-3,5-dihydroxyhexanoate is anyone or mixture of more than one from tetrahydrofuran, ether, methanol, ethanol, n-propanol, /so-propanol and ethylene glycol.
However, in enablement the only example using mixture of solvent was that of THF-methanol (Experimental section, Example 4: The preparation of (R,5)-6-chloro-3,5- dihydroxyhexanoate) and same outcome was expected in other individual or mixture of solvents.
Claim 8 of CN 101613341A mentioned that reduction was carried out by any one or mixture of more than one reducing agents such as sodium borohydride, potassium borohydride, lithium aluminum hydride, diethylmethoxy borane, triethyl borane and tributyl borane.
It implies that either any one of the reducing agents or a mixture of the same can be employed. From reaction mechanism it is very much clear that diethylmethoxy borane, triethyl borane and tributyl borane form the six membered complex between optically active hydroxyl and carbonyl group, which gets reduced by sodium borohydride, signifying that individually diethylmethoxy borane, triethyl borane and tributyl borane are not reducing agents
Moreover, in claims 12 and 13 (Experimental section, Example 4: The preparation of (R,S)-6-chloro-3,5-dihydroxyhexanoate), it is mentioned that reduction should be carried out in temperature range -80 °C to -60 °C, implying that reaction would not work beyond this temperature range i.e. it would work in the temperature window of -80 °C to -60 °C only.
Summarizing, the teachings of the application are not workable.

Scheme 9
Wolberg et al (Angewandte Chemie International Edition, 2000, 4306) has reported that diastereomeric excess for syn selective reduction using mixture of diethyl methoxy borane/sodium borohydride of compound [VI] gave 93% de for compound [VIII], which required further re-crystallization to obtain compound [VIII] in 99% de and 70% yield.
Thus, all the reported methods for stereo-selective hydride reduction of compound [VI] were achieved through mixture of trialkyl borane or diethyl methoxy borane & sodium borohydride in THF, at -78°C. As mentioned earlier, trialkyl borane or diethyl methoxy borane are pyrophoric in nature; in addition to that anhydrous THF is costly and moreover, reaction required large dilution.
Hence, there is need for developing efficient, environment friendly, cost effective and green process for stereo-selective reduction compound [VI].
B) The process of Oxidation of compound [II] to compound [I] has been discussed in following literature processes.
1) Swern oxidation (US4970313; Tetrahedron Letters, 1990, 2545
Synthetic Communications, 2003, 2275 – 2284).
2) Parrkh-Doering oxidation (J. Am. Chem. Soc, 1967, 89, 5505-5507)
3) TEMPO/NaOCl oxidization (EP2351762)
4) Trichloroisocyanuric acid/ TEMPO (CN 101747313A)
5) Oxidation of compound [II] to compound [I] through IBX [CN101475558A].
It would be evident that most of the reported methods are not “green” and
environmentally benign; none of the reported methods use molecular oxygen as oxidizing agent in presence of metal catalyst/co-catalyst.
Example 18: Process for synthesis of tert-butyl 2-((4R,6S)-6-formyl-2,2-dimethyl-l,3-dioxan-4-yl)acetate [I]

A reactor was charged with 1.1 g of copper (I) chloride and 10 mL of acetonitrile. 2-2′ Bipyridyl (156 mg) and TEMPO (156 mg) were added to the reactor under oxygen environment at 25°C. A solution of (6-Hydroxymethyl-2,2-dimethyl-[l,3]dioxan-4-yl)-acetic acid tert-butyl ester 2.6 g in 26 mL DCM was added dropwise over a period of 10 min into it. The reaction mass was stirred at 40°C and progress of reaction was monitored on GLC, which shows that 90% conversion for desired product.
Example 19: Process for synthesis of tert-butyl 2-((4R,6S)-6-formyl-2,2-dimethyl-l,3-dioxan-4-yl)acetate [I]
A reactor was charged with 1.1 g of copper (I) chloride and 10 mL of dichlorome thane. 2-2′ Bipyridyl (156 mg) and TEMPO (156 mg) were added to the reactor under oxygen environment at 25°C. A solution of (6-Hydroxymethyl-2,2-dimethyl-[l,3]dioxan-4-yl)-acetic acid tert-butyl ester 2.6 g in 26 mL DCM was added dropwise over a period of 10 min into it. The reaction mass was stirred at 40°C and progress of reaction was monitored on GLC, which shows that 90% conversion for desired product.
AUTHORS

///////
Lupin Ltd, New patent, Pitavastatin, WO 2016005919
Formula (1)
Lupin Ltd, New patent, Pitavastatin, WO 2016005919
MANE, Narendra, Dattatray; (IN).
NEHATE, Sagar, Purushottam; (IN).
GODBOLE, Himanshu, Madhav; (IN).
SINGH, Girij, Pal; (IN)
![]()
The present invention is directed to polymorphic forms of Pitavastatin sodium and processes for preparation of the same
Novel crystalline polymorphic forms (I and II) and an amorphous form of pitavastatin, useful for treating hyperlipidemia and mixed dyslipidemia.
Also claims a method for preparing the crystalline and amorphous forms of pitavastatin. In January 2016, Newport Premium™ reported that Lupin holds an active US DMF for pitavastatin calcium since July 2013.
Nissan Chemical Industries and licensee Kowa, with sub-licensees Sankyo, Eli Lilly, Esteve, JW Pharmaceutical, Recordati, Laboratorios Delta and Zydus-Cadila, have developed and launched pitavastatin.
WO2014203045, claiming a process for preparing an intermediate useful in the synthesis of statins (eg pitavastatin).
Pitavastatin is a cholesterol lowering agent of the class of HMG-CoA reductase inhibitor. The HMG-CoA reductase enzyme catalyzes the conversions of HMG- CoA to mevalonate. Inhibitors of HMG-CoA reductase are commonly referred to as “statins.” Statins are therapeutically effective drugs used for reducing low density lipoprotein (LDL) particle concentration in the blood stream of patients at risk for cardiovascular disease.
Pitavastatin is one of the synthetic statins which is chemically known as (3R, 5S, 6E)-7-[2-cyclopropyl-4-(4-fluorophenyl) quinoline-3-yl]-3, 5-dihydroxy-6- heptenoic acid represented by structural formula (1):
Formula (1)
Pitavastatin and its pharmaceutically acceptable salts are described in US 5,753,675 patent and US 5,856,336 patent, respectively.
Processes for the preparation of Pitavastatin are well documented in the literature. European patents, EP 0304063 and EP 1099694 and reports by Miyachi et al (Tetrahedron Letters
(1993) vol. 34, pages 8267-8270) and Takahashi et al (Bull. Chem. Soc. Japan (1995) Vol. 68, 2649-2656) describe processes for preparation of Pitavastatin.
US 5,872,130 patent discloses sodium salt of Pitavastatin. This patent, however, is silent about the solid state form of Pitavastatin Sodium.
It is generally known in the art that active pharmaceutical ingredients frequently do not exhibit the range of physical properties that makes them directly suitable for development. One of the approaches that is used to modify the characteristics of drug substances is to employ a salt form of the substance, since salts enable one to modify aqueous solubility, dissolution rate, solution pH, crystal form, hygroscopicity, chemical stability, melting point and even mechanical properties. The beneficial aspects of using salt forms of active pharmaceutical ingredients are well known and represent one of the means to increase the degree of solubility of otherwise intractable substances and to increase bioavailability.
Although the known salts of Pitavastatin like sodium, potassium, magnesium, calcium etc. and their polymorphic forms may address some of the deficiencies in terms of formulated product and its manufacturability. There remains a need for yet further improvement in these properties as well as improvements in other properties such as flowability, and solubility.
Polymorphism is a known phenomenon among pharmaceutical substances. It is commonly defined as the ability of any substance to exist in two or more crystalline phases that have a different arrangement and/or conformation of the molecules in the crystal lattice. Different polymorphic forms of the same pharmaceutically active moiety also differ in their physical properties such as melting point, solubility, chemical reactivity, etc. These properties may also appreciably influence pharmaceutical properties such as dissolution rate and bioavailability.
Further, the discovery of new polymorphic forms and solvates of an active pharmaceutical ingredient provides broader scope to a formulation scientist for formulation optimization, for example by providing a product with different properties, e.g., better processing or handling characteristics, improved dissolution profile, or improved shelf-life. For at least these reasons, there is a need for polymorphs of Pitavastatin salts such as Pitavastatin sodium.
New polymorphic forms and hydrates and/or solvates of a pharmaceutically acceptable salt of Pitavastatin can also provide an opportunity to improve the performance characteristics of a pharmaceutical product.
Therefore, there is a scope to prepare novel polymorphic forms of Pitavastatin sodium and hydrates and/or solvates.
Example-1: Preparation of Pitavastatin Sodium (Form-I)
A mixture of 40.0 gm Pitavastatin acid and 120 ml water was cooled to 15-20 °C temperature. Thereafter aqueous solution of sodium hydroxide (4.0 gm) in water (20 ml) was added to the reaction mixture. The reaction mixture was stirred for 30-45 min at 15-20 °C temperature. Ethyl acetate (80ml) was added into the reaction mixture at 15-20 °C temperature, stirred for 15-20 min and the layers were separated. The aqueous layer was filtered and acetonitrile (1200 ml) was gradually added to the aqueous layer under stirring till the precipitation was completed. The reaction mixture was cooled to 5-8 °C temperature and stirred for 2-3 hours at 5-8 °C temperature. The precipitated solid was filtered, washed with acetonitrile (40ml) and dried at 45-50 °C temperature under vacuum for 10-12 hours to afford the title compound (28.0 gm).
Yield (w/w): 0.70 (66.0%)
HPLC purity: 99.70 %
Example-2: Preparation of Pitavastatin Sodium (Form-II)
A mixture of 40.0 gm of Pitavastatin acid and 120 ml of water was cooled to 15-20°C temperature under stirring. Thereafter aqueous solution of sodium hydroxide (4.0 gm) in water (20 ml) was added to the reaction mixture. The reaction mixture was stirred for 30-45 min at 15-20 °C temperature. Ethyl acetate (80ml) was added to the reaction mixture at 15-20 °C temperature, stirred for 15-20 min and the layers were separated. The aqueous layer was filtered and acetonitrile (1200 ml) was gradually added to the aqueous layer under stirring till the precipitation was completed. The reaction mixture was cooled to 5-8 °C temperature and stirred for 2-3 hours at 5-8 °C temperature. The precipitated solid was filtered, washed with acetonitrile (40ml) and dried at 45-50 °C temperature under vacuum for 10-12 hours and kept in a petri dish at 25-30 °C and 60 ± 5 RH (relative humidity) for 18-24 hours to afford the title compound (31.6 gm).
Yield (w/w): 0.79 (65.8%)
HPLC purity: 99.70 %
Example-3: Preparation of Pitavastatin Sodium Amorphous
Pitavastatin sodium (3.0 gm) and ethanol (60 ml) were taken in a round bottomed flask at 25-30 °C temperature. The reaction mixture was filtered and the solvent was distilled off on rotatory evaporator under vacuum maintaining bath temperature at 45-50 °C temperature. Thereafter the reaction mixture was degassed under vacuum for 2-3 hours to afford the title compound (2.8gm).
HPLC purity: 99.70 %.
SEE……..https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016005919&redirectedID=true
/////////Lupin Ltd, New patent, Pitavastatin, WO 2016005919, statins, POLYMORPH
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel | |
| VÍDEOS – A Pfizer ac… on Temanogrel | |
| Pfizer Bought Arena… on Temanogrel | |
| Pfizer va profita de… on Temanogrel | |
| Pfizer to Cash In on… on Temanogrel | |
| Pfizer tocmai a achi… on Temanogrel | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer just purchase… on Temanogrel | |
| Tanya A on Dasotraline, ダソトラリン | |
| Julia Chernus on RIDINILAZOLE | |
| Adv.Siji Antony Kera… on Myself Anthony Crasto up for g… |

{kind=link}