
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

FDA Guideline on Dissolution Testing
DRUG REGULATORY AFFAIRS INTERNATIONAL

The FDA has presented the draft of a revised guideline on dissolution testing for immediate release. Under certain conditions, the tests can now be standardised. Read on to get more information about FDA’s Guideline on Dissolution Testing.
http://www.gmp-compliance.org/enews_05230_FDA-Guideline-on-Dissolution-Testing_15398,Z-QCM_n.html
In August 2015, the FDA published the draft of a guideline on dissolution testing for immediate release solid oral dosage forms. It is planned that after its finalisation, a part of this guideline will replace the current guideline from August 1997.
The Biopharmaceutics Classification System (BCS) distinguishes 4 different classes of APIs depending on their solubility and permeability.
On the basis of this classification, a decision can be taken for determining when bioavailability or bioequivalence studies are required, or when a successful in vitro-in vivo correlation (IVIVC) is likely.
The BCS proposes that, for certain medicinal products which contain a high soluble API, dissolution testing can be standardised. Due to their high solubility…
View original post 121 more words
What are the current Rules for Supplier Qualification?

Supplier Qualification is more than auditing. Supplier qualification can be seen as a risk assessment tool. But what are the exact requirements for qualifying suppliers?
Supplier Qualification is more than auditing. Supplier qualification can be seen as a risk assessment tool. It should provide an appropriate level of confidence that suppliers, vendors and contractors are able to supply consistent quality of materials, components and services in compliance with regulatory requirements. An integrated supplier qualification process should also identify and mitigate the associated risks of materials, components and services. But what are the exact requirements?
They are wide-ranging and complex. There are different directives and regulations for medicinal drug products for human or veterinary use and for investigational medicinal drug products. Certain requirements in different directives and the EU-GMP Guidelines define expectations. Here are some examples:
Article 8 of EU-Directive 2001/83/EC
“The application [of a marketing authorization] shall be accompanied […] by […] a written confirmation that the manufacturer of the medicinal product has verified compliance of the manufacturer of active substance with principles and guidelines of good manufacturing practice by conducting audits.”
Article 46 of EU-Directive 2001/83/EC
“The holder of a manufacturing and/or import authorisation shall at least be obliged […] to use only active substances, which have been manufactured in accordance with GMP for active substances and distributed in accordance with GDP for active substances and … to ensure that the excipients are suitable for use in medicinal products by ascertaining what the appropriate GMP is.”
Article 46b of EU-Directive 2001/83/EC
“Active substances shall only be imported if they have been manufactured in accordance with standards of good manufacturing practice at least equivalent to those laid down by the European Union”. This can be shown by a written confirmation, or the exporting country is included in the so called white list, or a waiver has been granted.
EU-GMP Guidelines Chapter 5:
5.25 “The purchase of starting materials is an important operation which should involve staff who have a particular and thorough knowledge of the supplier.”
5.26 “Starting materials should only be purchased from approved suppliers …”
5.40 “…printed packaging materials shall be accorded attention similar to that given to starting materials.”
The revised Chapter 7 of the EU-GMP Guidelines describe the responsibilities of the Contract Giver when it comes to contract manufacturing and testing. He needs to assure the control of the outsourced activities, incorporating quality risk management principles and including continuous reviews of the quality of the Contract Acceptor’s performance. Audits are a helpful tool to asses the “legality, suitability and the competence of the Contract Acceptor“. The new Chapter 7 was obviously designed to intensify the control of Contract Acceptors by the Contract Giver and extend those controls to subcontractors.
The holder of the manufacturing authorisation is responsible for the supplier qualification by law but in fact the supplier qualification is one of the duties of the Qualified Person (which can be delegated) as defined in Annex 16 of the EU-GMP Guidelines. The QP of the marketing authorisation holder is responsible for certifying the drug product for the market place and is now being held accountable to ensure that all aspects of the supply chain have been made under the appropriate GMPs. However, according to Chapter 2 of the EU-GMP Guidelines, the heads of Production, Quality Control and Quality Assurance share the responsibility of approving and monitoring suppliers of materials (2.9).
So how to proceed? At the beginning of a supplier qualification process, the regulatory requirements regarding the type of material, component or service and the type of product (human/veterinary drug product or IMP) should be identified and specified. Audits, if required, should be planned and executed. The compliance of the selected supplier(s) with the requirements and user requirement specification should be demonstrated. The scope of an audit should cover this. But a successful audit is not the end of the qualification process. After finalising the contract, the compliance of the selected supplier(s) with the applicable requirements should be evaluated periodically. Changes at the supplier´s site (for example manufacturing process etc.) that pose a particular risk to the compliance with the requirements should be assessed. There needs to be a mechanism in place so that any change made by the supplier which could have an impact on the GMP status or the production or testing parameters have to be agreed to before any such changes are implemented. A supplier must also notify the contract giver immediately upon discovery of any deviation/non-conformance/complaint that may have an impact on the services provided. Those need to be assessed and respective actions need to be defined.
The use of Brokers:
Some raw materials are only available at reasonable costs if purchased through an intermediary, i.e. a Broker. If the material is critical to the process, e.g. an API or a key excipient this can give an added complexity to the process and this must be fully investigated with the Quality and Regulatory units being involved, before any orders are placed.







////////
The new Elemental Impurities Database for Excipients – ECA offers a meeting at no costs

A step-wise integrated risk-based approach to determine a control strategy for according to ICH Q3D has to consider data from all kinds of potential sources for elemental impurities in particular from excipients. Read more about the newly created Elemental Impurities Database as a valuable support for performing risk assessments for drug products.
http://www.gmp-compliance.org/eca_mitt_05257_15499_n.html
The new ICH Q3D Guideline on Elemental Impurities strongly advocates the use of risk assessments in order to define a final control strategy. Specific challenges appear when risks associated with production equipment, packaging material and excipients have to be determined, and quantified. In particular the contribution of elemental impurities from excipients is not easy to assess due to their big variety and the lack of information from excipient vendors.
Quite recently a pharma consortium started an initiative which aims to collect and share data from pharmaceutical excipients by establishing a database. This Elemental Impurities (EI) Database provides information required for performing a comprehensive risk assessment of a drug product with respect to elemental impurities. Interested companies can contribute to this database by providing information about excipients and may also benefit from this database by taking out information needed for their risk assessments.
The “Impurities Workshop” from 14-16 June 2016 in Heidelberg, Germany provides a comprehensive and practical oriented review of impurities analysis and characterisation in drug substances and drug products. Part III of the workshop on 16 June 2016 is specifically dedicated to Elemental Impurites. In the subsequent post-Conference Workshop on 17 June 2016 the above mentioned EI Database will be explained. The following questions will be discussed:
- What is the procedure of providing data for the Database?
- How can information be obtained from the Database?
- What has to be considered in terms of confidentiality when data will be received or submitted to the Database?
This post-Conference Workshop is free of charge. It ideally complements the previous parts of the “Impurities Workshop” and can be booked in combination with either Part III or all Parts of the “Impurities Workshop”. As we expect a high interest in this post-Conference Workshop participants joining the “Impurities Workshop” (one day or all three days) will be registered first

////////////////
New Drug Approvals Blog has 2 lakh plus viewers in USA alone

New Drug Approvals Blog has 2 lakh plus viewers in USA alone
that is 200 thousand viewers
A record 1170135 views (11 lakh plus)all over the world in 211 countries
that is 1100 thousand plus views on this blog
I suffered a paralytic stroke in dec 2007 and bound to a wheelchair, this seems to have injected feul in me to help chemists around the world, I am more active than before and pushing boundaries, I have 2,5 lakh connections on all networking sites, I am available to all, contact me on +91 9323115463, amcrasto@gmail.com, Twitter @amcrasto

My son lionel was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, He cried bitterly and we had never seen him so depressed
Now I keep Lionel as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son and family happy.


////////////
Mehta Api Pvt Ltd, Cinacalcet hydrochloride, New patent, WO 2016027211
![]()

Mehta Api Pvt Ltd, Cinacalcet hydrochloride, New patent, WO 2016027211
Mehta Api had cinacalcet hydrochloride under development and holds US DMF and European DMF as listed on the company’s website. Amgen and Kyowa Hakko Kirin, under license from NPS Pharmaceuticals, have developed and launched cinacalcet.
The present filing represents the first PCT filing from the assignee, which focuses on developing (using green chemistry) manufacturing and marketing of API’s- multi step, highly complex, potent, chiral and semi-synthetic, advance intermediates, specialty chemicals and building blocks.
PROCESS FOR THE PREPARATION OF CINACALCET AND ITS PHARMACEUTICALLY ACCEPTABLE SALTS
MEHTA API PVT. LTD. [IN/IN]; 203, Centre Point, 2nd Floor, Near Hotel Kohinoor, J.B. Nagar, Andheri-Kurla Road, Andheri (East), Maharashtra, Mumbai 400 059 (IN)
KHAN, Rao, Uwais, Ahmad; (IN).
PATHAK, Rajesh, Harshnath; (IN).
PATIL, Chetan, Vinesh; (IN).
GAIKWAD, Sanjay, Ramrao; (IN).
APAR, Shrikrishna, Motiram; (IN).
LINGE, Govind, Udhavrao; (IN).
SHAIKH, Mohammad, Umar; (IN)
Cinacalcet (N-[l-(R)-(-)-(l-naphthyl) ethyl]-3-[3-(trifluoromethyl) phenyl]-l-aminopropane) of Formula II, belongs to a category of calcimimetics class of compounds. It is useful for the treatment of hyperparathyroidism and the preservation of bone density in patients with kidney failure or hypercalcemia due to cancer. It is marketed under the trade name of Senipar in United States and under the trade name of Mimpara in Europe.
US6211244 and Drugs of the future (2002) 27 (9): 831, discloses a synthesis of Cinacalcet by reductive amination which implies the reaction of (R)-(l-naphthyl) ethylamine of formula (IV) with 3 -[3- (trifluoromethyl) phenyl] propionaldehyde of formula (V) in the presence of titaniumisopropoxide to afford the corresponding cinacalcet imine of formula (III), which is reduced to cinacalcet of formula (II) with NaBH4CN in ethanol.
WO2012007954 A 1 discloses process for Cinacalcet by reductive amination in presence of titanium Isopropoxide using NaBH4CN, wherein an ether solvent is used instead of ethanol. Indian patent applications 2268/DEL/2008 and 87/MUM/2011 disclose preparation of Cinacalcet wherein reaction of (R)-(I-naphthyl)ethylamine of formula (IV) with 3-[3-(trifluoromethyl)phenyl] propionaldehyde of formula (V) is carried out in the presence of titaniumisopropoxide to afford the corresponding cinacalcet imine, which is further reduced to cinacalcet with NaBH4.
The above disclosed processes require the use of reagents such as NaBH4CN, titanium isopropoxide, which are extremely toxic and flammable as well as not being environmentally sound. These reagents therefore make the industrial application of the process difficult.
US20110124917A1 and WO2008068625A2 both disclose preparation of Cinacalcet by reductive amination wherein reduction is performed by using sodiumtriacetoxyborohydride as a selective reducing agent for imines.
Sodiumtriacetoxyborohydride is hygroscopic in nature hence demands anhydrous conditions to be maintained rendering it not suitable for use on industrial scale.
WO2012007954 A 1 discloses reaction and work-up in THF followed by salt formation in Di-isopropyl ether and further purification in two solvent system consisting of Water and Methanol or Water and Acetonitrile. US20110124917 discloses reaction in Methanol, Workup in toluene, Salt formation in Ethyl Acetate and purification in Isopropanol. WO2008068625A2 discloses reaction, salt formation and Purification in two solvent system consisting of isobutyl Acetate and n-Heptane. 2268/DEL/2008 discloses reaction in MDC, Salt formation in Ethyl Acetate and Purification in Ethyl Acetate and Di-isopropyl ether. 87/MUM/2011 discloses reaction in THF, work-up in toluene. Salt formation in two solvent system consisting of cyclohexane and MTBE.
All the above prior-art process employs use of different solvents for each unit operation or a two-solvent system for purification, thereby rendering the processes not easily scalable on industrial scale.
1367/MUM/2009 discloses reductive amination using sodium borohydride with 67.6% yield reported. 3068/MUM/2012 discloses reductive amination using sodium borohydride with 86% yield but with less purity. Further 3068/MUM/2012 requires the usage of sulphuric acid for the reaction of (R)-(I-naphthyl)ethylamine of formula (II) with 3-[3-(trifluoromethyl)phenyl] propionaldehyde of formula (III).
Thus the processes disclosed above have one or other drawbacks, ranging from poor yield, purity, use of difficult to handle and toxic reagents or use of different solvents for each unit operation.
In view of the problems occurred in above methods, there remains a need for more economical and efficient industrially scalable process for the preparation of Cinacalcet and its pharmaceutically acceptable salts, which overcomes the drawbacks as disclosed in the prior art.
The present inventors have surprisingly found that when the condensation of [3-(trifluoromethyl)phenyl]propionaldehyde of formula – (V) with (R)-(l- naphthyl)ethylamine formula – (IV) is carried out in the absence of any reagent and water is removed under vacuum by azeotropic distillation at low temperatures in the optional presence of water scavengers, than Cinacalcet.hydrochloride with high purity and yield is obtained. Further the process is also industrially feasible due to the non-usage of hazardous reagents as also due to the reduction in isolation and purification steps.

Example I:
Preparation of Cinacalcet Hydrochloride, Formula (la)
To (1000 ml) toluene in a 4Neck Round Bottom flask along with dean-stark apparatus coupled to a condenser, charge (80gms) (R)-(l- naphthyl) ethylamine of formula – (IV). Cool to 10-15°C. Charge (lOOgms) 3-[(3-Trifluoromethyl)phenyl] propionaldehyde of formula (V). Apply vacuum to the reaction mass through condenser and maintain for 8 hrs simultaneously azeotroping out water generated in the reaction till the reaction complies by thin layer chromatography to give Cinacalcet imine of formula (III) in-situ. Release vacuum after the reaction complies. Water collected after Azeotropic distillation: 7-7.5 ml. Cool the reaction mass to 5-10°C. Charge (35 gms) sodium borohydride in two lots to the reaction mass and raise the temperature to 25-30°C. Maintain the reaction mass for 8 hrs to give Cinacalcet of formula (II) in-situ. After the reaction complies by thin layer chromatography adjust the pH of the reaction mass to about pH 6 using acetic acid. Charge (200 ml) water to the reaction mass and stir for 30 mins. Separate Layers the organic layer and treat with 15% HC1 (150 ml). Stirr the Reaction mass at 40 – 50°C for one hour and separate the layer. Heat the toluene at same temperature. Adjust pH of toluene layer to below pH-2 by treating with 15% HC1 (150 ml) at 40-45 °C. Distill out 500 ml toluene under vacuum below 45 °C. Gradually charge 500 ml water to the reaction mass along with simultaneously distilling out 500 ml toluene approximately. Filter the reaction mass to give crude Cinacalcet Hydrochloride. Dry at 45-50°C for 8 hrs.
Weight: 182 gms
% Yield on theoretical basis: 98.9%
Purity: 97.54%
To (182 gms) of Crude cinacalcet Hydrochloride charge (800 ml) Methyl tert butyl ether and stirr for 60°C for 3 hrs. Cool gradually at 25-30°C and further chill the reaction mass to 0°C -5°C. Maintain the reaction mass at 0-5°C for 2 hrs and filter under vacuum followed by washing to the wet-cake with (100 ml) chilled Methyl tert butyl ether.
Wet cake is dried under vacuum at 40°C.
Weight: 163 gms
Yield on theoretical basis: 88.58%
Purity: 99.54%
To (163 gms) of MTBE pure Cinacalcet Hydrochloride is charged (400 ml) Isopropanol and heated to 70-75°C to get a clear solution which is then gradually cooled to 25-30°C and further chilled to 0-5 °C. The reaction mass is maintained for 2 hrs at same temperature and filtered under vacuum followed by washing with chilled isopropanol. Wet cake is dried under vacuum at 40°C.
Weight: 157 gms
Yield on theoretical basis: 85.32%
Purity: 99.91%
Example II:
Preparation of Crude Cinacalcet Hydrochloride, Formula (la)
To (1000 ml) toluene in a 4Neck Round Bottom flask, is charged (80gms) (R)-(l-naphthyl)ethylamine of formula (IV). Cooled to 10-15°C. Charged (lOOgms) 3-[(3-Trifluoromethyl)phenyl] propionaldehyde of formula (V) slowly. Charged (1 gm) Calcium Chloride and maintained for 8 hrs till the reaction complies by thin layer chromatography to give Cinacalcet imine of formula (III) in-situ. After the reaction complies, the reaction mass is cooled to 5-10°C. Charged (35 gms) sodium borohydride in two lots to the reaction mass and raised the temperature to 25-30°C.The reaction mass is maintained for 8 hrs to give Cinacalcet Free base of formula (II) in-situ. After the reaction complies by thin layer chromatography pH of the reaction mass is adjusted to about pH 6 using acetic acid. Charged (200 ml) water to the reaction mass and stirred for 30 mins. Layers separated and the organic layer is treated with 15% HC1 (150 ml). Reaction mass is stirred at 40 – 50°C for one hour and layer separated. Toluene layer is water washed at same temperature. pH of toluene layer adjusted to below pH-2 by treating with 15% HC1 (150 ml) at 40-45°C. Distill out 500 ml toluene under vacuum below 45 °C. Gradually charge 500 ml water to the reaction mass along with simultaneously distilling out 500 ml toluene approximately. Filter the reaction mass to give crude Cinacalcet Hydrochloride. Dry at 45-50°C for 8 hrs
Weight: 178 gms
Yield on theoretical basis: 96.73%
Purity: 94.88%
To (178 gms) of Crude cinacalcet Hydrochloride charged (800 ml) Methyl tert butyl ether and stirr for 60°C for 3 hrs. Allowed to cool gradually at 25-30°C and further chilled the reaction mass to 0-5°C. Maintained the reaction mass at 0-5°C for 2 hrs and filtered under vacuum followed by washing to the wet-cake with (100 ml) chilled Methyl tert butyl ether. Wet cake is dried under vacuum at 40°C.
Weight: 159 gms,
% Yield on theoretical basis: 86.40%
Purity: 99.77%
To (159 gms) of MTBE pure Cinacalcet Hydrochloride is charged (400 ml) Isopropanol and heated to 70-75°C to get a clear solution. Gradually cool to 25-30°C and further chill to 0-5 °C. Maintain the reaction mass is for 2 hrs at same temperature and filte under vacuum followed by washing with chilled isopropanol. Wet cake is dried under vacuum at 40°C. Weight: 150 gms
% Yield on theoretical basis: 81.51 %
Purity: 99.91 %
Example III:
Preparation of Cinacalcet Hydrochloride, Formula (la)
To (1000 ml) toluene in a 4Neck Round Bottom flask, charge (80gms) (R)-(l-naphthyl)ethylamine of formula (IV). Cool to 10-15°C. Charge (lOOgms) 3-[(3-Trifluoromethyl)phenyl] propionaldehyde of formula (V). Charge ( 1 gm) Molecular Sieves and maintain the reaction mass for 8 hrs till the reaction complies by thin layer chromatography to give Cinacalcet imine of formula (III) in-situ. After the reaction complies, cool the reaction mass to 5-10°C. Charge (35 gms) sodium borohydride in two lots to the reaction mass and raise the temperature to 25-30°C. Maintain the reaction mass for 8 hrs to give Cinacalcet of formula (II) in-situ. After the reaction complies by thin layer chromatography adjust the pH of the reaction mass to about pH 6 using acetic acid. Charge (200 ml) water to the reaction mass and stir for 30 mins. Separate the layers and treat organic layer with 15% HC1 (150 ml).Stirr Reaction mass is at 40 – 50°C for one hour and separate layers. Water wash toluene layer at same temperature. Adjust pH of toluene layer pH-2 by treating with 15% HC1 (150 ml) at 40-45 °C. Distill and degasse under vacuum below 70°C to give Cinacalcet Hydrochloride
Weight: 172 gms
Yield on theoretical basis: 93.47%
Purity: 97.29%
To (172 gms) of Crude cinacalcet Hydrochloride charge (800 ml) Methyl tert butyl ether and stirr for 60°C for 3 hrs. Cool gradually at 25-30°C and further chill the reaction mass to 0-5 °C. Maintain the reaction mass at 0-5 °C for 2 hrs and filter under vacuum followed by washing to the wet-cake with (100 ml) chilled Methyl tert butyl ether.
Wet cake is dried under vacuum at 40°C.
Weight: 155 gms
% Yield on theoretical basis: 84.23%
Purity: 99.57%
To (155 gms) of MTBE pure Cinacalcet Hydrochloride charge (400 ml) Isopropanol and heat to 70-75°C to get a clear solution which is then gradually cooled to 25-30°C and further chill to 0-5 °C. Maintain the reaction mass i for 2 hrs at same temperature and filter under vacuum followed by washing with chilled isopropanol. Wet cake is dried under vacuum at 40°C.
Weight: 146 gms
% Yield on theoretical basis: 79.34%
Purity: 99.83%
Mehta API Pvt. Ltd.

MR HARSHADRAI P MEHTA
Chairman & Managing Director

Devendra Mehta
Chief Executive Officer at MEHTA API PVT LTD
////////Mehta Api Pvt Ltd, Cinacalcet hydrochloride, New patent, WO-2016027211, WO 2016027211
Afatinib dimaleate, Dr Reddy’s, New patent, WO 2016027243
![]()
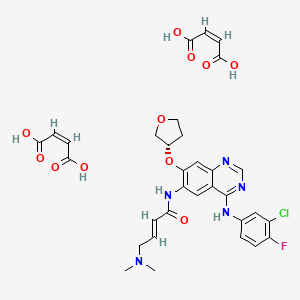
Afatinib dimaleate, Dr Reddy’s, New patent, WO-2016027243,
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara Hills, Hyderabad, Telangana, India – 500034. Hyderabad 500034 (IN)
RAMAKRISHNAN, Srividya; (IN).
PEDDY, Vishweshwar; (IN).
MAHAPATRA, Sudarshan; (IN).
KANNIAH, Sundara Lakshmi; (IN).
CHENNURU, Ramanaiah; (IN).
JOSE, Jithin; (IN).
DHAGE, Yogesh Mohanrao; (IN).
PEDDIREDDY, Subba Reddy; (IN).
YARRAGUNTLA, Sesha Reddy; (IN).
RAGHUVEER, Sherial; (IN).
KOLLA, Srinivasa Rao; (IN).
ANIL KSHIRSAGAR, Shivani; (IN).
JAFAR SHAIKH, Latif; (IN).
BANDARU, Srinivasulu; (IN)
The drug compound having the adopted name afatinib dimaleate, has a chemical name N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-,(2E)-, (2Z)-2-butenedioate (1 :2), and is represented by structure of formula I

Formula I
Afatinib dimaleate is an anticancer protein kinase inhibitor indicated for treatment of non-small-cell lung cancer. Process for preparation of afatinib, afatinib dimaleate and intermediates useful in preparation of afatinib dimaleate are described in US Patent Nos. 7,019,012; 8,426,586 and 7,960,546.
US Patent No. 8,426,586 discloses crystalline Form A of afatinib dimaleate salt and processes for preparation thereof. US Patent Application Publication No. 20140051713 discloses crystalline Form B of afatinib dimaleate salt and processes for preparation thereof. PCT Application Publication No. 2013052157 discloses crystalline Form C, Form D and Form E of afatinib dimaleate salt and processes for preparation thereof. The PCT publication also discloses crystalline Form A, B, C and Form D of afatinib base.
Polymorphism, the occurrence of different crystal forms, is a phenomenon of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties. Polymorphs in general will have different melting points, thermal behaviors (e.g. measured by thermogravimetric analysis – “TGA”, or differential scanning calorimetry – “DSC”), X-ray powder diffraction (XRPD or powder XRD) pattern, infrared absorption fingerprint, and solid state nuclear magnetic resonance (NMR) spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Discovering new polymorphic forms, hydrates and solvates of a pharmaceutical product can provide materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification or as desirable intermediate crystal forms that facilitate conversion to other polymorphic forms. New polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof can also provide an opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, e.g., better processing or handling characteristics, improved dissolution profile, or improved shelf-life. For at least these reasons, there is a need for additional solid state forms of Afatinib di-maleate.
SUMMARY
The present application provides novel solid state forms of Afatinib di-maleate, processes for preparing them, and pharmaceutical compositions containing them.
The present application also encompasses the use of novel solid state forms of Afatinib di-maleate provided herein, for the preparation of other afatinib salts, other solid state forms of afatinib dimaleate, and formulations thereof.
The present application also encompasses the use of any one of the novel solid state forms of Afatinib di-maleate disclosed herein for the preparation of a medicament, preferably for the treatment of cancer, particularly for the treatment of cancers mediated by epidermal growth factor receptor (EGFR) and human epidermal receptor 2 (HER2) tyrosine kinases, e.g., solid tumors including NSCLC, breast, head and neck cancer, and a variety of other cancers mediated by EGFR or HER2 tyrosine kinases. The present invention further provides a pharmaceutical composition comprising any one of the Afatinib di-maleate crystalline forms of the present invention and at least one pharmaceutically acceptable excipient.
The present application also provides a method of treating cancer, comprising administering a therapeutically effective amount of at least one of the Afatinib di-
maleate novel solid state forms of the present application, or at least one of the above pharmaceutical compositions to a person suffering from cancer, particularly a person suffering from a cancer mediated by epidermal growth factor receptor (EGFR) and human epidermal receptor 2 (HER2) tyrosine kinases, e.g., solid tumors including but not limited to NSCLC, breast, head and neck cancer, and a variety of other cancers mediated by EGFR or HER2 tyrosine kinases.
Example 1 : Preparation of amorphous form of afatinib dimaleate.
2.0 g of afatinib dimaleate was dissolved in 80 mL of a mixture of methanol and acetone (3:1 ) at 26°C and stirred for 15 min. The solution was filtered to remove the undissolved particles and the filtrate was distilled under reduced pressure at 50°C. After distillation the solid was dried under vacuum at 45°C to get 1 .29 g of amorphous afatinib dimaleate. PXRD pattern: Fig. 1 .
///////Afatinib dimaleate, Dr Reddy’s, New patent, WO-2016027243, WO 2016027243
Avoralstat

Avoralstat, BCX4161,
CAS 918407-35-9
UNII: UX17773O15
513.5513, C28-H27-N5-O5
2-Pyridinecarboxylic acid, 3-(2-(((4-(aminoiminomethyl)phenyl)amino)carbonyl)-4-ethenyl-5-methoxyphenyl)-6-(((cyclopropylmethyl)amino)carbonyl)-
3-(2-((4-Carbamimidoylphenyl)carbamoyl)-4-ethenyl-5-methoxyphenyl)-6-((cyclopropylmethyl)carbamoyl)pyridine-2-carboxylic acid
Hereditary angioedema (HAE)
Kallikrein inhibitor
BioCryst Pharmaceuticals
![]()
BioCryst is also investigating second-generation plasma kallikrein inhibitors to avoralstat, for treating HAE (in February 2016, this program was listed as being in preclinical development).
Prevent acute attacks in patients with hereditary angioedema (HAE); Treat hereditary angioedema (HAE)
U.S. – Fast Track (Treat hereditary angioedema (HAE));
U.S. – Orphan Drug (Prevent acute attacks in patients with hereditary angioedema (HAE))
26 Feb 2016Clinical trials in Hereditary angioedema (Prevention) in USA (PO, Hard-gelatin capsule) before February 2016
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in France (PO, Soft-gelatin capsule)
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in Germany (PO, Soft-gelatin capsule)

| Conditions | Interventions | Phases | Recruitment | Sponsor/Collaborators |
|---|---|---|---|---|
| Hereditary Angioedema|HAE | Drug: BCX4161|Drug: Placebo | Phase 2|Phase 3 | Recruiting | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161|Drug: Placebo | Phase 2 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
Avoralstat, also known as BCX-4161, is a potent and orally active Kallikrein inhibitor and Bradykinin inhibitor. Avoralstat may be potentially useful for treatment for Hereditary angioedema. Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.

Selective inhibitor of plasma kallikrein that subsequently suppresses bradykinin production
Hereditary angioedema (HAE) is a serious and potentially life-threatening rare genetic illness, caused by mutations in the C1-esterase inhibitor (C1 INH) gene, located on chromosome 11q. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by C1 INH gene mutations which produce low levels of C1 -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective C1 protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; 100(Suppl 2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herin by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that C1 INH is the most important inhibitor of kallikrein in human plasma and that C1 INH deficiency leads to unopposed activation of the kallikrein-bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack.
Approved therapies include purified plasma-derived C1 INH (Cinryze®, Berinert), the recombinant peptide kallikrein inhibitor ecallantide (Kalbitor®), and the bradykinin receptor B2 inhibitor iticabant (Firazyr®). All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
There are many delivery routes for active pharmaceutical ingredients (APIs). Generally, the oral route of administration is favored. Oral administration provides a number of advantages, such as, but not limited to, patient convenience, flexibility of timing of administration, location of administration and non-invasiveness. Oral administration also provides more prolonged drug exposure compared with intermittent intravenous infusion, which may be important for drugs with schedule-dependent efficacy. For example, a drug with a short half-life can achieve a greater exposure time by either continuous infusion or by continuous oral dosing. The use of oral therapy further has the potential to reduce the cost of healthcare resources for inpatient and ambulatory patient care services.
In the pharmaceutical arts, it is known that a number of APIs cannot be administered effectively by the oral route. The main reasons why these compounds cannot be administered by the oral route are: a) rapid enzymatic and metabolic degradation; b) chemical and/or biological instability; c) low solubility in aqueous medium; and/or d) limited permeability in the gastrointestinal tract. For such compounds, non-oral routes of delivery, such as parenteral administration, mainly via intramuscular or subcutaneous injections, may be developed. However, non-oral administration poses a disadvantage for the patient as well as healthcare providers, and for this reason, it is important to develop alternative routes of administration for such compounds, such as oral routes of administration.
While the oral route of administration is the most convenient for the patient and the most economical, designing formulations for administration by the oral route involves many complications. Several methods are available to predict the ease by which an API may be formulated into a formulation suitable for administration by the oral route. Such methods include, but are not limited to, and Lipinski rule (also referred to as the Rule of Five) and the Biopharmaceutical Drug Disposition Classification System (BDDCS).
The BDDCS divides APIs into four classifications, depending on their solubility and permeability. Class I APIs have high solubility and high permeability; Class II APIs have low solubility and high permeability; Class III APIs have high solubility and low permeability; and Class IV APIs have low solubility and low permeability. APIs in higher classes in the BDDCS face greater challenges in formulating into an effective, pharmaceutically acceptable product than those in lower classes. Of the four classes, APIs falling into Class IV are the most difficult to formulate into a formulation for administration by the oral route that is capable of delivering an effective amount of the API as problems of both solubility and permeability must be addressed (note the BDDCS does not inherently address chemical stability). The role of BDDCS in drug development is described generally in L.Z. Benet J Pharm Sci. 2013, 102(1), 34-42.
Lipinski’s rule (described in Lipinski et al. Adv. Drug Deliv. Rev. 46 (1-3): 3-26) states, in general, that in order to develop a successful formulation for administration by the oral route, an API can have no more than one violation of the following criteria:
i) not more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms)
ii) not more than 10 hydrogen bond acceptors (nitrogen or oxygen atoms) iii) a molecular mass less than 500 daltons
iv) an octanol-water partition coefficient log P not greater than 5.
J. Zhang et al. Medicinal Chemistry, 2006, 2, 545-553, describes a number of small molecule amidine compounds which have activity as inhibitors of kallikrein. The molecules described in this document fall into Class IV of the BDDCS as described above. The compounds are poorly soluble in aqueous and physiological fluids, and are poorly permeable as demonstrated by oral dosing in rats and in vitro experiments with Caco-2 cells.
Furthermore, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, one of the compounds described in Zhang et al., is a Class IV API and violates criteria iii) and iv) as set forth in the Lipinski Rule.
Furthermore, the compounds described in Zhang et al., including 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, exhibit poor stability with respect to oxidation in air, to light
(photodegradation) and in aqueous and physiological fluids, as well as to elevated temperatures.
Therefore, the compounds described by Zhang et al. including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, not only exhibit poor solubility and permeability characteristics, but also poor stability characteristics. As a result, such compounds are predicted to be especially difficult to formulate into an effective, orally deliverable
pharmaceutical composition that is capable of delivering an effective amount of the compound to a subject.
Polymorphism, the occurrence of different crystal forms, is a property of some molecules. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties, such as, but not limited to, melting point, thermal behaviors (e.g. measured by thermogravimetric analysis (TGA), or differential scanning calorimetry (DSC), x-ray diffraction pattern, infrared absorption fingerprint, and solid state NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Discovering new polymorphic forms and solvates of a pharmaceutical product can provide alternate forms of the compound that display a number of desirable and advantageous properties, such as, but not limited to, ease of handling, ease of processing, ease of formulation, storage stability, and/or ease of purification. Further, new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof may further provide for improved pharmaceutical products, by providing compounds that are more soluble in a set of pharmaceutical excipients. Still further, the provision of new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof enlarges the repertoire of compounds that a formulation scientist has available for formulation optimization, for example by providing a pharmaceutical product with different properties, such as, but not limited to, improved processing characteristics, improved handling characteristics, improved solubility profiles, improved dissolution profile and/or improved shelf-life. Therefore, there is a need for additional polymorphs of pharmaceutically useful compounds, such as, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid and the compounds disclosed herein.
In one aspect, the present invention provides an oral formulation that is capable of delivering an effective amount of the amidine compounds described by Zhang et al. to a subject. In particular, the present invention provides an oral formulation that is capable of delivering an effective amount of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid to a subject. In one specific aspect, the 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid is present in a particular crystal form designated Form A. In light of the art suggesting the difficulties in formulating such an oral formulation, this result was unexpected.
As described herein, the amidine compounds described in Zhang et al., including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (specifically including particular crystal Form A), may now be conveniently used in oral administration and further used in oral administration for the treatment of a number of diseases and conditions in a subject, such as, but not limited to, HAE as described herein.
Avoralstat & next generation kallikrein inhibitors for HAE
Avoralstat
![]()
May 16 is HAE awareness day
See BioCryst’s video regarding HAE to learn more
Avoralstat is being developed as an oral prophylactic treatment for patients suffering from Hereditary Angioedema (HAE). Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
In May 2014 BioCryst, announced that the OPuS-1 (OralProphylaxiS-1) Phase 2a proof of concept clinical trial met its primary efficacy endpoint, several secondary endpoints and all other objectives established for the trial. OpuS-1 enrolled 24 HAE patients with a history of HAE attack frequency of at least 1 per week. Treatment with avoralstat demonstrated a statistically significant mean attack rate reduction of 0.45 attacks per week versus placebo, p<0.001. The mean attack rate per week was 0.82 on BCX4161 treatment, compared to 1.27 on placebo.
In December 2014, BioCryst initiated enrollment in OPuS-2 (Oral ProphylaxiS-2). OPuS-2 is a blinded, randomized, 12-week, three-arm, parallel cohort design trial evaluating the efficacy and safety of two different dose regimens of avoralstat administered three-times daily, 300 mg and 500 mg, compared with placebo. The primary efficacy endpoint for the trial will be the mean angioedema attack rate, which will be reported for each avoralstat dose group compared to placebo. The trial is being conducted in the U.S., Canada and Europe. On October 8, 2015, announced that it has completed enrollment of approximately 100 HAE patients with a history of moderately frequent to very frequent attacks in OPuS-2. BioCryst expects to report the OPuS-2 trial results in early 2016.
PATENT
WO200234711
http://www.google.com/patents/WO2002034711A1?cl=en
PATENT
PATENT
Examples
Example 1 – Synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl- phenyll-6-(cvclopropylmethyl-carbarnoyl)-pyridine-2-carboxylic acid
The synthesis of the above compound and intermediates is described below. In this section, the following abbreviations are used:

The synthesis of starting material, (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) is described in Scheme 1.
f 0HCY ° ΒΓΥΥ°

Preparation of 6-bromobenzofdl[1,3ldioxole-5-carbaldehvde (1b)

1a 1b
To a mixture of piperonal (1a) (498 g, 3.32 mol) in glacial acetic acid (1000 mL) was added a solution of bromine (200 mL, 3.89 mol) in glacial acetic acid (500 mL) over a period of 30 min and stirred at room temperature for 24h. The reaction mixture was poured into water (2000 mL) and the solid that separated was collected by filtration. The solid was dissolved in boiling ethanol (4000 mL) and cooled to room temperature. The solid obtained on cooling was collected by filtration to furnish 6-bromobenzo[d][1 ,3]dioxole-5-carbaldehyde (lb) (365 g, 48 %) as a white solid, MP 126 °C; HNMR (300 MHz, DMSO-d6): δ 10.06 (s, 1 H), 7.42 (s,1 H), 7.29 (s, 1 H), 6.20 (d, J=12.3, 2H); IR (KBr) 3434, 2866, 1673,1489, 1413, 259, 1112, 1031 , 925 cm“1; Analysis calculated for CeH5BrO3.O 25H C, 41.15; H, 2.37; Found: C, 41.07; H, 2.11.
Preparation of 2-bromo-5-hvdroxy-4-methoxybenzaldehyde (1c)

1c
A solution of potassium tert-butoxide (397 g, 3.36 mol) in DMSO (1.5 L) was heated at 50 °C for 30 min. Methanol (1.5 L) was added to it and continued heating at 50 °C for additional 30 min. To the hot reaction mixture was added 6-bromo-benzo[d][1,3]dioxole-5-carbaldehyde (1 b) (350g, 1.53 mol) and continued heating at 50 °C for 30 min. The reaction mixture was cooled to room temperature and quenched with water (2.3 L) and sodium hydroxide (61.2 g, 1.53 mol). The reaction mixture was washed with ether (2 x 1.5 L), acidified to pH 2 using cone. HCI and extracted with ethyl acetate ( 1 L). The ethyl acetate layers were combined and concentrated under vacuum to dryness. The residue obtained was treated with water (1.5 L) and ethyl acetate (1 L). The solid obtained was collected by filtration to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (97 g, 27.5% as a first crop). The layers from the filtrate were separated and aqueous layer was extracted with ethyl acetate (200 ml_). The ethyl acetate layers were combined dried over MgS04 and concentrated under vacuum to dryness to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (192 g, 54.4%, second crop) as an orange solid, MP 108 °C; ‘HNMR (300MHz, DMSO-cfe): S 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H); IR (KBr) 3477, 2967, 2917,
2837, 2767, 2740, 1657, 1595, 1428, 1270, 1210, 1164, 1022 cm“‘; Analysis calculated for C8H7Br03.H20: C, 38.58; H, 3.64: Found: C, 38.60; H, 3.60.
Preparation of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehvde ( d)

To a solution 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (120 g, 520 mmol) in DMF (1000 mL) was added potassium carbonate (79 g, 572 mmol) and benzyl bromide (68 mL, 572 mmol). The reaction mixture was stirred at room temperature overnight and quenched with water (3000 mL). The solid obtained was collected by filtration, washed with ether and dried under vacuum to furnish 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (113.19 g, 67.9%) as a white solid, MP 144 °C;1HNMR (300 MHz, DMSO-c/6): δ 10.06 (s, 1H), 7.47-7.34 (m, 7H), 5.17 (s, 2H), 3.92 (s, 3H); IR (KBr) 2898, 2851 , 1673, 1592, 1502, 1437, 1402, 1264, 1210, 1158, 1017, 754 cm“1; Analysis calculated for C 5H13Br03: C, 56.10; H, 4.08; Found: C, 55.44; H, 4.08.
Preparation of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e)
15 046578
146

1d 1e
To a solution of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (100 g, 311 mmol) in
ethanol (1500 mL) was added triethyl orthoformate (103 mL, 622 mmol), ammonium nitrate
(7.5 g, 93.3 mmol) and stirred at room temperature overnight. The reaction mixture was
treated with ether (1200 mL) and stirred for 15 min before filtration. The filtrate was
concentrated under vacuum to dryness to give 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (134 g) as a brown syrup; The product was used in the next step
without further purification; 1H N R (300 MHz, DMSO-cf6) δ 7.45 – 7.37 (m, 4H), 7.36 – 7.33
(m, 1 H), 7.17 – 7.14 (m, 1 H), 7.10 (s, 1 H), 5.10 (s, 2H), 3.80 (s, 3H), 3.58 – 3.33 (m, 5H),
1.13 – 1.07 (m, 6H); IR (KBr) 2974, 2879, 1601 , 1503, 1377, 1260, 1163, 1060 cm“1;
Analysis calculated for C19H23Br04: C, 57.73; H, 5.86; Found: C, 57.21 ; H, 5.94.
acid (1fi

To a solution of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (120 g,
300 mmol) in dry ether (1000 mL) at -78 °C was added n-butyllithium (1.6 M solution in
hexanes, 244 mL, 390 mmol) over a period of 30 min and further stirred at -78 °C for 30 min.
A solution of tri-n-butylborate (110 mL, 405 mmol) in dry ether (300 mL) was added to this
solution at -78 °C over a period of 30 min. The reaction mixture was further stirred for 2 h at -78 °C and warmed to 0 °C. The reaction mixture was quenched with 3N HCI (300 mL) at 0
°C and heated at reflux for 1 h. After cooling to room temperature, the solid obtained was
collected by filtration washed with water (250 mL) dried in vaccum to afford (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (30.85 gm, 37.6% as a white solid. The organic
layer from above filtrate was extracted with 1.5 N NaOH (3 x 200 mL). The combined basic
extracts were acidified with cone. HCI (pH about 4). The solid obtained was collected by
filtration, washed with water and dried under vacuum to furnish a second crop of (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (22.3 g, 26%) as a light orange solid
MP 158 °C; 1H NMR (300 MHz, DMSO-cfe) δ 10.08 (s, 1 H), 7.52 (s, 1 H), 7.48 – 7.33 (m, 5H),
7.24 (s, 1H), 5.18 (s, 2H), 3.89 (s, 3H); 1H NMR (300 MHz, DMSO-d6/D20) δ 10.06 (s, 1H),
7.52 (s, 1H), 7.49 – 7.32 (m, 5H), 7.23 (s, 1 H), 5.18 (s, 2H), 3.89 (s, 3H); MS (ES+) 309.1 (M+Na); IR (KBr) 3335, 2937, 1647, 1545, 1388, 1348, 1268, 1146, 1095 cm-1; Analysis calculated for C15H15BO5.0.25H2O: C, 62.00; H, 5.38; Found: C, 61.77; H, 5.19.
Synthesis of methyl-6-(cvclopropylmethylcarbamoyl¾-3-ftrifluoromethylsulfonyloxyVpicolinate
The synthesis of the intermediate methyl 6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethyl sulfonyloxy)picolinate (2h) is described in Scheme 2.

Preparation of 2-bromo-3-hvdroxy-6-methylpyridine (2b)
![]()
H3C N Br
2a 2b
To a solution of 3-hydroxy-6-methylpyridine (2a) (3000 g, 27.5 mol) in pyridine (24 L) cooled to 15 °C was added a solution of bromine (4.83 kg, 1.55 L, 30.2 mol) in pyridine (3 L) over a period of 50 min maintaining the internal temperature between 20 to 25 DC. After stirring for 19 h at room temperature the solvent was removed under vacuum and the residue was triturated with water. The solid separated was collected by filtration, washed with water and dried under vacuum to give 2-bromo-3-hydroxy-6-methylpyridine (2b) (3502 g, 67.7 %) as a light brown solid which was used as such without further purification; 1H NMR (300 MHz, DMSO-d6) δ 10.43 (s, 1H), 7.18 (d, J = 8.0 Hz, 1 H), 7.08 (d, J
MS (ES+) 188.35, 186.36 (M+1).
(2c)
![]()
2b 2c
A mixture of 2-bromo-3-hydroxy-6-methylpyridine (2b) (3000 g, 15.96 mol), anhydrous potassium carbonate (3308 g, 23.94 mol), and iodomethane (2.491 kg, 1.09 L, 17.556 mol) in 30 L of acetone was heated at 40 °C overnight. The reaction mixture was cooled to room temperature and filtered through Celite. Evaporation of the solvent followed by silica gel chromatography (Hexane: ethyl acetate = 7:3) afforded the desired compound, 2-bromo-3-methoxy-6-methylpyridine (2c) which was used as such for the next step; 1H NMR (300 MHz, DMSO-cfe) δ 7.42 (dd, J = 8.3, 1.5 Hz, 1H), 7.29 – 7.19 (m, 1H), 3.84 (d, J = 1.6 Hz, 3H), 2.37 (d, J = 1.7 Hz, 3H).

2c
2d
To a solution of 2-bromo-3-methoxy-6-methylpyridine (2c) (310 g, 1.53 mol) in 6000 mL of water at 60 °C was added KMnO, (725 g, 4.59 mol) in small portions over a 90 min period with vigorous mechanical stirring. A dark purple solution resulted. This solution was kept at 90 °C for a further 3 h and filtered through Celite while still hot to give a colourless filtrate.
After cooling, the aqueous solution was acidified to pH 1-2 by adding 6 N HCI. The white solid obtained was collected by filtration to give on drying 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (302g, 85%) of product, which was used as such in the next reaction without further purification. An analytical sample was obtained by recrystallization from methanol to give 6-bromo-5-methoxy-2-pyridinecarboxylic acid; 1H NMR (300 MHz, DMSO-tfe) δ 7.40 – 7.28 (m, 1H), 7.17 (d, J = 8.3 Hz, 1 H), 3.83 (d, J = 1.7 Hz, 3H).
Preparation of 6-bromo-N-(cvclopropylmethyl)-5-methoxypicolinamide (2e)

To a solution of 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (12 g, 52 mol) in pyridine (70 mL) was added EDCI (11.5 g, 59 mmol) and cyclopropylmethylamine (3.6 g, 52 mmol). The reaction mixture was stirred at room temperature overnight and then concentrated under vacuum. The reaction mixture was diluted with water (100 mL) and ethyl acetate (100 mL). The organic layer was separated and the water layer was extracted with ethyl acetate (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), brine (500 mL), dried over magnesium sulphate, filtered and concentrated under vacuum to furnish 10.43g of crude product. The crude product was converted into a slurry (silica gel 20 g) and purified by flash column chromatography (silica gel 230 g, eluting with 0-100% ethyl acetate in hexane) to yield compound 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (8.02 g, 54%) as off white solid, mp 67-70 °C; 1HNMR (300 MHz, DMSO-d6) δ 8.51 (t, J = 5.8, 1 H), 8.02 (d, J = 8.4, 1 H), 7.65 (d, J = 8.5, 1 H), 3.96 (s, 3H), 3.14 (t, J = 6.5, 2H), 1.11 -0.99 (m, 1 H), 0.47 – 0.36 (m, 2H), 0.27 – 0.20 (m, 2H); MS (ES+) 307.0, 309.0 (100%
M+Na)
Preparation of methyl 6-(cvclopropylmethylcarbamoyl)-3-methoxypicolinate (2f)

To a solution of 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (7.5 g, 27.6 mol) in methanol (300 mL) in a 2-L stainless steel bomb was added Pd(OAc)2(750 mg), 1 ,1-bis(diphenylphosphino)-ferrocene (750 mg), and triethylamine (3.9 mL, 27.6 mmol). The reaction mixture was vacuum flushed and charged with CO gas to 150 psi. The reaction mixture was and heated with stirring at 150°C overnight and cooled to room temperature. The catalyst was filtered through a pad of celite, and concentrated to dryness to furnish crude product. The crude was purified by flash column chromatography (silica gel 150 g,
eluting with, 0%, 5%, 10%, 20%, 30%, 50% ethyl acetate/hexanes (250 mL each) as eluents to give methyl 6-(cyclopropylmethyl-carbamoyl)-3-methoxypicolinate (2f) (6.29 g, 86.1 %) as a salmon coloured solid, MP 107 °C; 1HNMR (300 MHz, DMSO-cfe) δ 8.28 (t, J = 6.0, 1H), 7.91 (d, J = 8.8, 1H), 7.55 (d, J = 8.8, 1 H), 3.68 (s, 3H), 3.64 (s, 3H), 2.90 (t, J = 6.5, 2H), 0.89 – 0.68 (m, 1 H), 0.26 – 0.09 (m, 2H), 0.08 – 0.00 (m, 2H); MS (ES+) 287.1 (M+Na); IR (KBr) 3316, 2921 , 1730, 1659, 1534, 1472, 1432, 1315, 1272, 1228, 1189, 1099, 1003, 929, 846, 680 cm“1; Analysis calculated for C13H16 204: C, 59.08; H, 6.10; N, 10.60; Found: C, 58.70; H, 5.97; N, 10.23.
Preparation of 6-(cvclopropylmethylcarbamoyl 3-hvdroxypicolinic acid (2q)

2f 2g
Aluminium chloride method:
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-methoxypicolinate (2f) (0.16 mmol) in dichloromethane (840 mL) was added AICI3 (193 g, 1.5 mol). The reaction mixture was heated at reflux for 12 h under nitrogen. After slowly adding ~2L of 1 N HCI, the organic layer was separated. The aqueous layer was re-extracted several times with ethyl acetate/DME. The combined organic layer was washed with brine, dried (MgSO.4), and evaporated in vacuo to furnish crude 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid. To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid was added a solution of acetyl chloride (1 10 mL) in methanol (1.1 L). The reaction mixture was stirred for 12 h at room temperature and then concentrated to dryness in vacuo. After co-evaporating once with methanol, the compound was purified by flash-column chromatography (silica gel, 500 g, eluted with chloroform and 3% methanol in chloroform) to furnish 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g).
Boron tribromide method:
To a stirring solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-ethoxypicolinate (2f) (58.0 g, 208 mmol) was added BBr3 (79 mL, 834 mmol) in CH2CI2 (1.3 L) at 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 18h. The reaction mixture was evaporated to dryness and anhydrous methanol (1 L) was added to the light yellowish solid residue. Insoluble solid was collected by filtration (36 g). Mother liquor was evaporated and co-evaporated with MeOH (2 x 200 mL). The insoluble solid (36 g) was treated with MeOH (500 mL) and acetyl chloride (50 mL) and stirred at room temperature for 18 h (at this point reaction mixture was clear). The mixture was evaporated to dryness and diluted with water and extracted with EtOAc. White solid that separated out from EtOAc layer was collected by filtration, washed with water (2 x 20 mL), dried in vacuo at 50 °C to afford 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (5.36 g, 10 %) as a white solid, MP 92-95 °C. 1HNMR (DMSO-cfe) δ 11.04 (s, 1 H, exchangeable with D20), 8.37 (t, J = 6.0, 1 H, exchangeable with D20), 8.12 (d, J = 8.7 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 3.90 (m, 3 H), 3.15 (m, 2 H), 1.04 ( m, 1 H), 0.41 (m, 2 H), 0.24 (m, 2 H). IR (KBr): 3346, 3205, 1684 cm“1; MS (ES+): 251.1 (M+1); Analysis calculated for C12H14N2O4.0.1 H2O: C, 57.18; H, 5.67; N, 11.14; Found: C, 57.11 ; H, 5.61; N, 11.09.
Preparation of methyl-6-(cvclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy) picolinate (2h

To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (28 mmol) in DMF (200 mL) were added triethylamine (12 mL, 84 mmol) and N-phenyl-bis(trifluoromethanesulfonimide) (12 g, 34 mmol). The reaction mixture was stirred for 1.5 h at room temperature and then poured into ice. After diluting with water and extracting with ethyl acetate, the aqueous phase was re-extracted, and then the combined organic layer was washed with water and concentrated under vacuum to give methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)picolinate (2h), which was used in the next step without purification.
1H NMR (300 MHz, CDCI3) δ 8.50 (d, J = 8.6, 1 H), 8.07 (s, 1 H), 7.88 (d, J = 8.6, 1 H), 4.09 (d, J = 12.6, 3H), 3.48 – 3.24 (m, 2H), 1.18 – 1.01 (m, 1 H), 0.69 – 0.44 (m, 2H), 0.42 – 0.20 (m, 2H). MS (ES*): 405.17, 100%, M+Na.
Synthesis of 3-f2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid:
The synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i) is described as shown in Scheme 3.

3-f4-Benzyloxy-2-formyl-5-methoxy-phenylV6-(cvcloDroDvlmethvl-carbarnovn-pyridine-2-carboxylic acid methyl ester (3a)
5 046578
153

3a
To a solution of methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)
picolinate (2h) (24.3g, 63 mmol) in DME (225 mL) were added water (25 mL), (4- (benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (27.3 g, 95 mmol), NaHC03(15.9 g,
5 189 mmol), and bis(triphenylphosphine)palladium(ll) chloride (0.885 g). The reaction
mixture was stirred at 70°C overnight under nitrogen. After extracting with ethyl acetate, the organic layer was washed with water and brine and dried (MgSO^), and then concentrated
under vacuum. The compound was purified by flash-column chromatography (silica gel, 300 g, eluting with 10%, 20%, 30% and 40% ethyl acetate in hexane) to furnish 3-(4-benzyloxy- 10 2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
methyl ester (3a) (25 g, 83%) as off white solid, MP 48-50°C: 1H NMR (300 MHz, DMSO-cfe) δ 9.61(s, 1 H), 8.40 (d, J= 7.9 Hz, 1H), 8.14 (t, J= 5.0 Hz, 1H), 7.87 (d, J= 8.1 Hz, 1 H), 7.58
(s, 1H), 7.54-7.30 (m, 5H), 6.71 (s, 1 H), 5.24 (s, 2H), 3.93 (s, 3H), 3.70 (s, 3H), 3.45-3.34 (m,
2H), 1.19-1.05 (m, 1 H), 0.64-0.54 (m, 2H), 0.37-0.30 (m, 2H); IR ( Br) 1735, 1678, 1594,
15 1513, 1437, 1283, 1217, 1141, 1092 cm“1; MS (ES+) 497.29 (M+Na); Analysis calculated for
C27H2eN206: C, 68.34; H, 5.52; N, 5.90; Found; C, 68.16; H, 5.62; N, 5.80.
2-(6-(Cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-vn-4-methoxy-5- vinylbenzoic acid (3b)

To a solution of 3-(4-benzyloxy-2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl- carbamoyl)-pyridine-2-carboxylic acid methyl ester (3a) (24g, 50.6 mmol) in acetonitrile (50
mL), 2-methyl-2-propanol (350 mL), and water (125 mL) were added sodium dihydrogen
phosphate (12.5 g) and 2-methyl-2-butene (55 mL, 519 mmol). The reaction mixture was cooled in an ice bath and then sodium chlorite (28 g) was added. After stirring for 1 h, the reaction mixture was extracted with ethyl acetate and washed with water. The aqueous layer was re-extracted and then the combined organic layers were dried (MgS04). The solvent was evaporated in vacuo to furnish 5-(benzyloxy)-2-(6- ((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (29 g) which was used for the next step. MS (ES+): 513.24, (M+Na(; (ES ): 489.26, M-1.
Methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxytoarbonyltohenyl)-6-(cvclopropylmethylcarbamovnpicolinate (3c)

To a mixture of 5-(benzyloxy)-2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxy-carbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (31 g, 63.2 mmol), and triethylamine (17.7 mL, 126.4 mmol) in dichloromethane (300 mL), was added MEM-chloride (9.03 mL, 79 mmol), and stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and dried over MgS04, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 40 g) to furnish methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 89%) as a thick gum; H NMR (300 MHz, CDCI3) δ 8.35 (d, J = 8.0 Hz, 1 H), 8.15 (t, J = 5.7 Hz, 1 H), 7.78 (d, J = 8.0 Hz, 1H), 7.71 (s, 1H), 7.49 (d, J = 6.8 Hz, 2H), 7.36 (ddd, J = 7.5, 14.8, 22.4 Hz, 3H), 6.66 (s, 1 H), 5.37-5.13 (m, 4H), 3.90 (s, 3H), 3.69 (s, 3H), 3.60-3.49 (m, 2H), 3.49 (s, 2H), 3.39 (dd, J = 4.4, 8.4 Hz, 2H), 3.34 (s, 3H), 1.19-1.00 (m, 1H), 0.57 (q, J = 5.8 Hz, 2H), 0.38-0.25 (m, 2H). MS (ES+): 601.24 (M+Na); (ES“): 577.27 (M-1);1H NMR (300 MHz, DMSO-cfe) δ 8.69 (t, 7 = 6.1 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1 H), 7.63 (s, 1H), 7.41 (m, 5H), 6.92 (s, 1 H), 5.20 (m, 4H), 3.83 (s, 3H), 3.57 (s, 3H), 3.44 (m, 2H), 3:33 (m, 2H), 3.21 (m, 5H), 1.14 (m, 1H), 0.44 (m, 2H), 0.27 (m, 2H). IR (KBr):
1732, 1671 cm“1. MS (ES+): 601.1(M+Na); Analysis calculated for C31H 2Oe: C, 64.35; H, 5.92; N, 4.84; Found: C, 64.27; H, 6.04; N, 4.79.
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(4-hvdroxy-5-methoxy-2-(((2-methoxyethoxy¾methoxy)carbonyl)phenyl)picolinate (3d)

3c 3d
To a solution of methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)-carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 56.68 mmol) in ethanol (650 mL) was added 10% Pd/C (4 g) and hydrogenated at 45 psi for 5 h. The catalyst was removed by filtration through Celite and the filtrate was concentrated under vacuum to yield methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)picolinate (3d) (31.87 g, 86%), which was pure enough to be used as such for the next step. An analytical sample of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) was obtained by purification of 350 mg of above crude using flash column chromatography (silica gel, eluting with ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethyl-carbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-phenyl)picolinate (3d) as a clear gum; 1HNMR (300 MHz, DMSO-d6) δ 9.74 (s, 1 H), 8.68 (t, J = 6.1 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1 H), 7.95 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 6.83 (s, 1H), 5.19 (s, 2H), 3.77 (m, 3H), 3.58 (s, 3H), 3.44 (m, 2H), 3.34 (m, 2H), 3.21 (m, 5H), 1.04 (m, 1 H), 0.44 (m, 2H), 0.27 (m, 2H); IR (KBr): 1731 , 1664 cm‘1. MS (ES*): 489.0 (M+1); Analysis calculated for C^e^O,,: C, 59.01; H, 5.78; N, 5.73; Found: C, 58.92; H, 6.15; N, 5.29.
6-(Cvclopropylmethylcarbamovn-3-(5-methoxy-2-(((2-methoxyethoxy^methoxy)-carbonyl)-4- (trifluoromethylsulfonyloxy)phenyl)picolinate (3e)

To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2- methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) (14.3 g, 29.3 mmol) in dichloromethane (150 mL) were added pyridine (12 mL, 146 mmol) and triflic anhydride (7.5 mL g, 44 mmol). After stirring overnight at room temperature under N2. the reaction mixture was poured into ice water and then extracted twice with dichloromethane. After washing the combined organic extracts with water and drying (MgS0 ), the solvent was evaporated in vacuo. The compound was purified by flash chromatography over silica gel column using ethyl acetate: hexane to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)-carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (1 g, 93%); H NMR (300 MHz, CDCy a 8.41 (d, J = 8.0, 1H), 8.17 (s, 1H), 8.03 (s, 1H), 7.79 (d, J = 8.0, 1 H), 6.82 (s, 1H), 5.32 (q, J = 6.1, 2H), 3.97 (s, 3H), 3.74 (s, 3H), 3.67 – 3.57 (m, 2H), 3.55 – 3.45 (m, 2H), 3.41 (dd, J = 8.2, 14.5, 2H), 3.34 (s, 3H), 1.36 – 1.17 (m, 1H), 0.58 (d, J = 7.1 , 2H), 0.33 (d, J = 5.1 , 2H).
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(5-methoxy-2-f((2-methoxyethoxy)- methoxy)carbonvn-4-vinylphenyl)picolinate (3f)

To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (37.4
g, 60.30 mmol) and potassium vinyltrifluoroborate (16.87 g, 120.6 mmol) in DMF (450 mL) and water (45 mL) was bubbled N2 for 5 min. To this mixture was added NaHC03 (20.26 g, 241.2 mmol) and dichloro-bis(triphenylphosphine)palladium (II) (6.34 g, 9.0 mmol). The reaction mixture was stirred at 70 °C for 20 h under N2(reaction progress was checked by 1H N R because product and starting material had same Rf in TLC). The reaction mixture was cooled down to room temperature and diluted with ethyl acetate. The organic layer was separated, washed with water, brine, dried ( gS04) and filtered. The filtrate was concentrated under vacuum to yield crude methyl 6-(cyclopropylmethyl-carbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-4-vinylphenyl)-picolinate (3f). The crude product was purified by flash column chromatography (silica gel, 1 kg, eluting with 0-100% ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate [31) (26.54 g, 88%) as an amber gum; H NMR (300 MHz, DMSO-c¾ δ 8.70 (t, J = 6.1 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1 H), 8.12 (s, 1 H), 8.00 (d, J = 8.0 Hz, 1 H), 6.98 (m, 2H), 5.94 (dd, J = 1.2, 17.8 Hz, 1H), 5.43 (d, J = 12.5 Hz, 1 H), 5.21 (d, J = 6.5 Hz, 2H), 3.88 (s, 3H), 3.64 (s, 3H), 3.48 (d, J = 3.1 Hz, 2H), 3.35 (m, 5H), 3.22 (m, 2H), 1.11 (s, 1H), 0.44 (dt, J = 4.9, 5.5 Hz, 2H), 0.28 (q, J = 4.8 Hz, 2H). IR (KBr); 1732, 1670 cm“1. MS (ES+) 499.1 (M+1).
2-(6-(cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzolc acid (3g)

A mixture of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate (3f) (27.4 mmol) in DME (160 mL) and 6N HCI (40 mL) was stirred at room temperature for 6 h or till TLC showed complete conversion. The solvent was removed under vacuum. The residue obtained was suspended in water, the solid separated out was collected by filtration, washed with water and dried under vacuum to give 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (7.0 g, 63%) as a white
solid MP 40 – 42 °C; H NMR (300 MHz, DMSO-de) δ 8.69 (t, J= 6.0 Hz, 1H, NH), 8.20 (d, J= 7.9 Hz, 1H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1H), 6.97 (dd, J= 18.0, 11.3 Hz, 1H), 6.88 (s, 1H), 5.92 (d, J= 7.9 Hz, 1H), 5.38 (d, J= 11.1 Hz, 1H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); IR (KBr): 3084, 1728, 1650, 1533, 1212, 1143 cm-1; MS (ES+) 433.26 (M+Na); (ES-): 409.28 (M-1); Analysis calculated for θ22Η22Ν2Ο6.0.25Η2Ο; C, 63.68; H, 5.47; N, 6.75; Found C, 63.75; H, 5.56; N, 6.65
Methyl-3-(2-(4-carbamimidoylprienylcarbamoyl)-5-metrioxy-4-vinylphenyl)-6- (cvclopropylmethylcarbamoyl)picolinate (3h)

To a solution of 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (2.35 g, 5.7 mmol) and 4-aminobenzimidamide dihydrochloride (3j) (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 mL) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 in CMA 50) yielding methyl-3-(2-(4-carbamimidoylphenyl-carbamoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (2.2 g, 65%) as a white solid MP 266 °C; 1H NMR (300 MHz, DMSO-c/6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 – 7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for ![]()
C, 58.93; H, 5.63; N,11.85; Found: C, 58.75; H, 5.65; N, 11.92.
46578
159
3-r2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy -vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i)

3h 3i
To a solution of methyl-3-(2-(4-carbamirriidoylphenylcarbarnoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (1 g, 1.9 mmol) in methanol (10 mL) and THF
(10 mL) was added 2 N NaOH (10 mL). The reaction mixture was stirred at room
temperature for 3 h, and concentrated in vacuo to remove methanol and THF. The aqueous layer was acidified with 6N HCI to pH 6-7 and the solid obtained was collected by filtration
washed with water and ether to furnish on drying 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
(3i)(0.775 g, 80%) as the hydrochloride salt as an off white solid.
1H NMR (300 MHz, DMSO-d6) δ 12.67 (s, 1 H), 9.11 (s, 2H), 8.97 (s, 2H), 8.74 (s, 1 H), 7.90
(d, J = 7.8, 1 H), 7.80 (s, 1 H), 7.72 – 7.58 (m, 4H), 6.99 (dd, J = 11.3, 17.7, 1 H), 6.78 (s, 1H),
5.95 (d, J = 17.2, 1H), 5.38 (d, J = 11.9, 1H), 3.82 (s, 3H), 3.18 (s, 2H), 1.06 (s, 1 H), 0.43 (d,
J = 7.9, 2H), 0.25 (d, J = 4.7, 2H); MS (ES+) 514.0 (M+1 ); Analysis calculated for
C2eH27N5O5.HCI.H2O: C, 59.21; H, 5.32; N, 12.33; Found: C, 59.43; H, 5.21; N, 12.06.
Example 1A- Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride in Form
C

The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 to -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1.6 kg) and water (0.57 kg). The mixture was heated to 46 °C. Smopex-234 (21 g) and Acticarbone 2SW (10 g) were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 “C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The
suspension was stirred at 0-3.0 “for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjackel= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 11 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C.
Example-1 B: Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbartiovQpyridine-2-carboxylic acid hydrochloride in Form A
The procedure was carried out in an identical manner to Example 1 A, with the exception that after the final filtration the filter cake was rinsed with 2.87 kg methyl ierf-butyl ether instead of 2.87 kg water, and pulled dry. The product was dried at 40-43 °C and 50 mbar to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) as Form A.
PATENT
Methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (compound 6a) is (I) (pages 85 and 86). Avoralstat hydrochloride (compound of formula XVIII) is (II) (claim 40, page 109). A Markush structures is presented (claim 1, page 99).
The synthesis of (II) via intermediate (I) is described (example 1, pages 80-93).
A synthesis of the compound 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (Compound 3i) is described in Schemes A-C.
O y OHCk n Br^ ^OCH3
B Brr22,, AAccOOHH Y^ V”“ \ \ tt–BBuuOOKK
OHC^^^O ” Br^\^0 MeOH ” OHC
1a 1b 66%

1d 95% 1 e
![]()
1f
Scheme A


3h 31
Scheme C
Examples. In this section, the following abbreviations are used:



Example-1 : Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b)

7b
Step (1): Preparation of 6-Bromobenzo 1 ,3]dioxole-5-carbaldehyde (1 b):

1b
A solution of bromine (33.0 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to a solution of piperonal (1a) (29.9 kg, 199.16 mol) in acetic acid (105 L) at room
temperature over a period of 50 min and the reaction mixture was stirred at room temperature for 14.2 h. Additional solution of bromine (33 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to the reaction mixture over a period of 2 h and the reaction mixture was stirred for 22 h. The reaction mixture was quenched by addition of ice water (500 L) with stirring over a period of 6 h and continued stirring for additional 1.25 h. The mixture was allowed to settle and most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (600 L) was added to the solid, stirred, mixture was allowed to settle and then most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (100 L) was added to the decanted mixture, stirred for 15 min and the solid obtained was collected by filtration using a centrifuge. The solid was washed with water (2 x 100 L) and air-dried in a tray drier for 3.75 h to afford the crude product 1 b (52 kg). The crude product (51.2 kg) was stirred in n-hexane (178 L) for 3 h, collected by filtration, washed with n-hexane (25 L) and dried to afford 6-bromobenzo[1 ,3]dioxole-5-carbaldehyde (1b) (40.1 1 kg, 87.9%) as a light brown solid. MP: 109-112°C. 1H NMR (300 MHz, CDCI3) δ 10.21 (s, 1 H), 7.37 (s, 1 H), 7.07 (s, 1 H), 6.10 (s, 2H); HNMR (DMSO-cf6): δ 10.06 (s, 1 H), 7.42 (s, 1 H), 7.29 (s, 1 H), 6.20 (d, J =12.3 Hz, 2H)
The process is also illustrated in Fig. 1.
Average yield of isolated 1 b from step-1 is 78 – 88%.
Step (2): Preparation of 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c)

A solution of potassium terf-butoxide (10.7 kg, 95.36 mol) in DMSO (49 L) was stirred at 50 °C for 30 min. Methanol (49 L) was added slowly over a period of 4.25 h and stirred at 50 °C for 30 min. 6-Bromobenzo[1 ,3]dioxole-5-carbaldehyde (1 b) (9.91 kg, 43.27 mol) was added to the reaction mixture in small portions over a period of 45 min and stirred at 50 °C for 1 h. The reaction mixture was cooled to room temperature and split into two equal portions. Each portion was quenched with water (50.9 L) and basified with 50% aqueous NaOH solution (2.4 L). Each portion was extracted with MTBE (4 x 36 L) to remove impurities. The aqueous layer was acidified with cone. HCI to pH ~ 3 to obtain
product as a yellow solid. The solid was collected by filtration using a centrifuge, washed with water (2 x 35 L) and air-dried to afford 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) (4.37 kg, 40.7%, contains 7 % water); Mp: 100-102°C; 1HNMR (300MHz, DMSO-d6): δ 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H).
The process is also illustrated in Fig. 2.
Average yield of isolated product 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) from step-2 is 40-50%.
Step (3): 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-y benzaldehyde (4a)

2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) [1.3 kg (93%, 7% water content), 5.25 mol] was dissolved in toluene (13 L) in a reaction flask equipped with a Dean Stark apparatus. The solution was heated at reflux with stirring to distil off about 25% of the toluene along with water (90 ml_). The solution was cooled to 90 °C then
bis(pinacolato)diboron (1.5 kg, 5.82 mol), KOAc (772.6 g, 7.87 mol) and Pd(PPh3) (24.3 g, 0.02 mol) were added and the reaction mixture was heated at reflux for 10h. After confirming the completion of reaction by TLC (mobile phase: 100% DCM), the reaction mixture was cooled to room temperature and was kept standing overnight. The reaction mixture was filtered through celite and the celite cake was washed with toluene (4 L). The filtrate of this batch was mixed with the filtrate of another batch (batch size 1.3 kg obtained from an identical reaction). The mixed filtrate was washed with water (17.5 L), brine (17.5 L), dried over Na2S04, filtered and the solution was passed through a pad of silica gel (2 kg, mesh size 230-400). The silica gel pad was washed with toluene. The combined filtrate and washing was concentrated under reduced pressure and the residual crude product was stirred with n-hexane (23 L) for 1 h to obtain a solid product. The solid was collected by filtration, washed with n-hexane (5 L) and dried to afford 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)benzaldehyde (4a) (2.47 kg, 84.6%). H NMR (300 MHz, CDCI3) δ 10.54 (s, 1 H), 7.57 (s, 1 H), 7.33 (s, 1 H), 5.89 (s, 1 H), 4.01 (s, 3H), 1.37 (s, 12H); 1H NMR (300 MHz, DMSO-d6) δ 10.35 (s, 1 H), 9.95 (s, 1 H), 7.33 (s, 1 H), 7.23 (s, 1 H), 3.87 (s, 3H), 1.33 (s, 12H); MS (ES+) 301.1 (M+Na); 579.1 (2M+Na); Analysis calculated for C14H19B05: C, 60.46; H, 6.89; Found: C, 60.60; H, 6.87
The average yield of 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) from step (3) is 78 – 90%.
The process is also illustrated in Fig. 3.
Step (4): Preparation of 3-Bromo-2,6-dimethylpyridine (5b)

2,6-lutidine (5a) (115 kg, 1073.3 mol) was added into pre-chilled oleum (20-23%, 1015 kg, 2276.7 mol) at 0 °C over a period of 4.5 h (temperature r6ached 14 °C during the addition). Bromine (88.18 kg, 1103.6 mol) was then added at 5-10 °C over a period of 1 h. The reaction mixture was slowly heated to 150 °C over a period of 12h. TLC analysis indicated about 40-50% conversion to product and the formation of a dimer by-product (5%). The reaction mixture was cooled to room temperature and then additional bromine (88.18 kg, 1103.6 mol) was added slowly. The reaction mixture was slowly heated to maintain a temperature of 65-75 °C over a period of 15h. TLC analysis indicated a 65-70 % conversion to product and the formation of 5% dimer by product. The reaction mixture was quenched by addition of water (500L) while maintaining the reaction temperature below 20 °C. The mixture was basified with 6.6 M NaOH (3800 L) while maintain the temperature at < 40 °C. EtOAc (220 L) was added and the mixture was stirred for 1 h then allowed to settle over a period of 2 h. The layers were separated and the aqueous layer was treated with NaOH (10 kg) in water (10 L) and extracted with EtOAc (160 L). The organic extracts were combined washed with brine (100 L), dried over Na2S04 (50.0 kg), filtered and the solvent was evaporated under atmospheric pressure. The residue was vacuum distilled and the desired product 3-bromo-2,6-dimethylpyridine (5b) was collected at 58-60 °C, 2 mmHg (98.45 kg, 49.2 %) as a colorless liquid.
The process is also illustrated in Fig. 4.
Step (5): Preparation of 3-Bromopyridine-2,6-dicarboxylic acid (5c)
![]()
5b 5c
To a stirred solution of 3-bromo-2,6-dimethylpyridine (5b) (98 kg, 5326 mol) in water (1310 L) was added KMn0 (225 kg, 1423.6 mol) in 5 equal portions in 1 h intervals at 70 °C. After stirring for 1 h at 70 °C, additional KMn04 (225 Kg, 1423.6 mol) was added in 5 equal portion in 1 h intervals at 90 °C. The reaction mixture was stirred for 12 h at 90 °C. The suspension was filtered hot through celite to obtain a clear solution. The solvent was distilled off to remove about 30% of the total volume. The remaining concentrated solution was chilled to 0 °C and made acidic (to pH 3-4) by the addition of cone. HCI (120 L). The white precipitate obtained was collected by filtration and dried at 70 °C to afford 3-bromopyridine-2,6-dicarboxylic acid (5c) as a white solid (109 kg, 84%).
The process is also illustrated in Fig. 5.
Step (6): Preparation of Dimethyl 3-Bromopyridine-2,6-dicarboxylate (5d)

To a stirred solution of 3-bromopyridine-2,6-dicarboxylic acid (5c) (20.0 kg, 81.29 mol) in methanol (100 L) was added cone. H2S04 (4.4 L) over a period of 30 min. The reaction mixture was heated to 65 °C and maintained at that temperature for 5 h (the reaction was monitored by TLC analysis to determine completion of reaction). The reaction mixture was cooled to room temperature basified by careful addition of aqueous NaHC03 solution (prepared from 10 kg NaHC03 in 120 L of water) and further diluted with water (120 L). The white solid obtained was collected by filtration, washed with plenty of water and then oven-dried at 40 °C to obtain dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (9.2 kg, 41.3%) as a white solid; 1HNMR (300 MHz, DMSO-cf6) δ 8.47 (d, J = 8.4, 1 H), 8.08 (dd, J = 4.5, 8.4, 1 H), 3.95 (s, 3H), 3.91 (s, 3H); MS (ES+) 570.6 (2M+Na); Analysis calculated for C9H8BrN04: C, 39.44; H, 2.94; Br, 29.15 N, 5. 1 ;
Found: C, 39.52; H, 2.92; Br, 29.28; N, 5.03.
The process is also illustrated in Fig. 6.
6582
Step (7): Preparation of Methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (

To a stirred solution of dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (27 kg, 98.52 mol) in ierf-butanol (135 L) was added at room temperature cyclopropylmethanamine (7.83 kg, 110.1 mol). The reaction mixture was heated at 65 °C for 17 h. The progress of reaction was monitored by TLC and HPLC (HPLC analysis showed the formation of 74% of the product 5e after 17 h. The reaction mixture was cooled to room temperature and then cone. HCI (2.7 L) was added slowly and the mixture was stirred for 15 min. The reaction mixture was concentrated under reduced pressure to obtain the crude product. The crude product was dissolved in hot /-PrOH (54 L) filtered through a celite pad. The filtrate was cooled with stirring to 10 °C to obtain a white precipitate. The solid obtained was collected by filtration, washed with cold
i-PrOH (13 kg), n-hexane (15 L) and dried to provide pure methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e) (15.7 kg, 50.9%). The filtrate was concentrated under reduced pressure and the crude product can be purified by silica gel column chromatography eluting with tert-butanol in hexanes to furnish additional 10% methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e). HNMR (300 MHz, DMSO-cf6) δ 8.83 (t, J = 5.9, 1 H), 8.47 – 8.41 (m, 1 H), 8.06 (d, J = 8.4, 1 H), 3.96 (s, 3H), 3.16 (t, J = 6.5, 2H), 1.14 – 0.99 (m, 1 H), 0.42 (m, 2H), 0.30 -0.19 (m, 2H); MS (ES+) 337.0 (M+23), 650.8 (2M+23); Analysis calculated for
C12H13BrN203: C, 46.03; H, 4.18; N, 8.95; Br, 25.52; Found: C, 46.15; H, 4.17; N, 8.72; Br, 25.26.
The average isolated yield for step (7) is 50% to 60%.
The process is also illustrated in Fig. 7.
Step (8): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a)
2

6a
THF (37.5 L) was charged to a 100 L reactor followed by ethyl 3-bromo-6- (cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate (5e) (2.5 kg, 7.98 mol) under a nitrogen atmosphere. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) (2.88 kg, 10.36 mol) was added, followed by the addition of PPh3 (53.13 g, 0.20 mol), PdCI2(PPh3)2 (120.4 g, 0.17 mol) and a solution of Na2C03(2.12 kg, 20.00 mol) in demineralized water (10.0 L) under nitrogen atmosphere. The reaction mixture was degassed again two times by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 6.5 h, cooled to room temperature and filtered through a Celite bed. Water (75 L) was added to the filtrate and the product was extracted with ethyl acetate (75 L). The aqueous layer was back extracted with ethyl acetate (2 χ 60 L). The combined ethyl acetate extract was divided into two equal portions and each portion was washed with brine (37 L), dried over Na2S04, filtered and concentrated under reduced pressure to give crude methyl 6- ((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) as a reddish viscous material (-4.5 Kg) which was used as such for the next step without further purification. An analytical sample was prepared by purification of a small sample by flash column chromatography (silica gel, eluting with 0-100% ethyl acetate in hexane) to furnish methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)-picolinate (6a) as an off-white solid; HNMR (300 MHz, DMSO-d6) δ 9.89 (s, 1 H), 9.52 (s, 1 H), 8.79 (t, J = 6.1 Hz, 1 H), 8.23 (d, J = 8.0 Hz, 1 H), 8.09 (d, J = 8.0 Hz, 1 H), 7.34 (s, 1 H), 6.90 (s, 1 H), 3.85 (s, 3H), 3.62 (s, 3H), 3.22 (m, 2H), 1.16 -1.02 (m, 1 H), 0.49 – 0.38 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 791.0 (2M+Na), (ES-) 382.7 (M-1), 767.3 (2M-1); Analysis calculated for C20H20N2O6.0.25 H20: C, 61.77; H, 5.31 ; N, 7.20; Found: C, 61.54; H, 5.13; N, 7.05.
The process is also illustrated in Fig. 8.
46582
Step (9): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b)

6a 6b
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) (2.11 kg, estimated about 3.83 mol from step-8) in dichloromethane (16.0 L) and pyridine (1.4 L, 17.4 mol) cooled to -10°C and maintained at that temperature for 1 h was added a solution of triflic anhydride (980.0 ml_, 5.8 mol) in dichloromethane (6.0 L) drop wise over a period of 3 h at -10 °C. The reaction mixture was stirred at -5°C for 1.3 h, quenched with saturated aqueous NaHCO3(10.4 L) and stirred for 30 mins. The organic layer was separated, washed successively with saturated aqueous NaHC03 (10.4 L), 1 HCI (2 x 16.6 L), water (13.2 L), brine (13.2 L), dried over MgS04, filtered and concentrated under reduced pressure to give the crude product. The crude product was stirred with 15% ethyl acetate in n-hexane (7.0 L) for 1 h. The solid obtained was collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (7.0 L) for 1 h, was collected by filtration and washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (8.0 L) for 1 h, collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was dried to afford methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)-oxy)phenyl)picolinate (6b) as a light brown solid (1.7 kg, 86% yield, for combined steps 8 & 9). Average isolated yield for combined steps 8 and 9 was 70% to 86%; Ή NMR (300 MHz, DMSO-cf6): δ 9.64 (s, 1 H), 8.78 (t, J = 6.1 , 1 H), 8.29 (d, J = 8.0, 1 H), 8.16 (d, J = 8.0, 1 H), 8.03 (s, 1H), 7.39 (s, 1 H), 4.00 (s, 3H), 3.63 (s, 3H), 3.22 (m, 2H), 1.11 (m, 1 H), 0.52 – 0.39 (m, 2H), 0.28 (m, 2H); MS (ES+) 538.9 (M+Na). The process is also illustrated in Fig. 9.
Step (10): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c)

A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4- (((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b) (12 kg, 23.24 mol) in DME (106 L) was charged into reactor under nitrogen. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. Potassium trifluoro(vinyl)borate (3.9 kg, 29.1 1 mol), PdCI2(PPh3)2 (815 g, 1.13 mol), KHC03 (4.65 g, 46.44 mol) and demineralized water (12 L) was then added under a N2 atmosphere. The reaction mixture was degassed by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 5 h. The reaction mixture was cooled to room temperature and then filtered through a Celite bed. Demineralized water (118 L) was added to the filtrate followed by ethyl acetate (124 L). The mixture was stirred for 20 min and then the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 95 L). The combined organic extract was washed with brine (95 L), dried over Na2S04, and filtered. The solvent was evaporated under reduced pressure to give the crude product. The crude product was purified by column chromatography (silica gel, 120 kg, 230-400 mesh size, eluting with ethyl acetate in n-hexane) to obtain methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (6 kg, 72%). 1H NMR (300 MHz, CDCI3): δ (ppm) 9.64 (s, 1 H), 8.35 (d, J = 7.8 Hz, 1 H), 8.06-8.03 (m, 2H), 7.78(d, J = 7.8 Hz, 1 H), 7.02-6.92 (m, 1 H), 6.61 (s, 1 H), 5.86 (d, J = 17.7 Hz, 1 H), 5.38 (d, J = 1 1.4 Hz, 1 H), 3.84 (s, 3H), 3.67 (s, 3H), 3.35-3.29 (m, 2H),1.08-1.03 (m, 1H), 0.55-0.49 (m, 2H), 0.29-0.2 4(m, 2H). 1HNMR (300 MHz, DMSO-d6) 6 9.68 (s, 1 H), 8.77 (t, J = 6.1 , 1 H), 8.35 – 8.21 (m, 1 H), 8.16 – 8.01 (m, 2H), 7.14 -6.87 (m, 2H), 6.01 (dd, J = 1.2, 17.8, 1 H), 5.45 (dd, J = 1.1 , 1 1.3, 1 H), 3.91 (s, 3H), 3.64 (s, 3H), 3.23 (m, 2H), 1.21 – 1.01 (m, 1H), 0.51 – 0.40 (m, 2H), 0.34 – 0.20 (m, 2H). MS
(ES+) 417.0 (M+Na); Analysis calculated for C22H22N205: C, 66.99; H, 5.62; N, 7.10;
Found: C, 66.75; H, 5.52; N, 7.06.
The process is also illustrated in Fig. 10.
Step (1 1): Preparation of 2-(6-((cyclopropylmethyl)carbamoyl)-2- (methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d)

To a stirred solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (1.57 kg, 3.80 mol) in acetonitrile (15.4 L) was added ferf-butyl alcohol (22.2 L), demineralized water (3.2 L) and sodium dihydrogen phosphate monohydrate (323.74 g, 2.346 mol). The reaction mixture was cooled to 0 °C and added 2-methyl-2-butene (5.3 L, 50.0 mol) and stirred at 0 °C for 30 min. A solution of 80% sodium chlorite (1.36 kg, 12.0 mol) in demineralized water (5.2 L) was added to the reaction mixture over a period of 2.5 h at 0 °C [temperature rises to 7 °C during the addition]. The reaction mixture was stirred at 0 °C for 2 h, diluted with water (40 L) and ethyl acetate (24 L). After stirring the mixture, it was allowed to settle and the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 20 L) then acidified with 5.9 % aqueous acetic acid (2 L) and extracted once with ethyl acetate (10 L). The organic extracts were combined washed with water (2 x 20 L), a solution of acetic acid (125 mL) in water (20.0 L), brine (2 χ 20 L), dried over Na2S04, filtered and concentrated under reduced pressure (vapor temperature below 40 °C). The residue obtained was dissolved in acetone (7 L) (residue didn’t dissolve completely). The solution was poured slowly into a reactor containing stirred n-hexane (70.0 L) to precipitate the solid product and the mixture was stirred for 2 h. The solid obtained was collected by filtration, washed with 10% acetone in n-hexane (6.3 L), AJ-hexane (6.3 L), dried to afford 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4- methoxy-5-vinylbenzoic acid (6d) as an off-white solid (1.29 Kg, yield: 79.0%). Average isolated yield for step 1 1 is 74% to 84%. 1H NMR (300 MHz, DMSO-d6): δ (ppm) 12.50 (brs, 1 H), 8.69(t, J= 6.0 Hz, 1 H, NH), 8.20 (d, J= 7.9 Hz, 1 H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1 H), 6.97 (dd, J= 18.0, 1 1.3 Hz, 1 H), 6.88 (s, 1 H), 5.92 (d, J= 7.9 Hz, 1 H), 5.38 (d, J= 1 1.1 Hz, 1 H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); MS (ES+) 433.26, (M+Na); (ES-) 409.28 (M-1). The process is also illustrated in Fig. 1 1.
Step (12): Preparation of Methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate methanesulfonate (7a

Pyridine (3.8 L, 47.17 mol) and EDCI (5.31 kg, 27.66 mol) were sequentially added to a cooled solution (0 °C) of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (9 kg, 21.92 mol) and 4-aminobenzamidine dihydrochloride (5.13 kg, 24.65 mol) in /-PrOH (90 L). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. TLC analysis indicated incomplete reaction. Additional EDCI (1.08 kg, 5.6 mol) was added and the reaction mixture was stirred for 8 h. The reaction was still incomplete as indicated by TLC analysis, additional EDCI (0.54 kg, 2.8 mol) was added and the reaction mixture was stirred for 5 h. TLC analysis indicated there was trace amount of unreacted starting material remaining. The reaction mixture was cooled to 0 °C and a solution of
methanesulfonic acid (MSA) (9.13 kg, 95 mol) in MeOH (38.7 L) was added to the cooled mixture over a period of 4 h. The reaction mixture was allowed to warm to room temperature and stirred for 15 h. The product was collected by filtration, washed with a mixture of /‘-PrOH and MeOH (4:1 , 45 L). The wet cake was slurried in a mixture of /-PrOH and MeOH (2:1 , 135 L) stirred for 1 h and the product was collected by filtration and washed with a mixture of /‘-PrOH and MeOH (4:1 , 46.8 L). The product was dried in
2015/046582
a vacuum oven at 45 °C to afford methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) as a pink-colored solid (12.71 kg, 93%). Average isolated yield for this step: >90%.
1H NMR (300 MHz, DMSO-c/6) δ 10.71 (s, 1 H), 9.16 (s, 2H), 8.80 (s, 2H), 8.68 (t, J = 6.1 Hz, 1 H), 8.22 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.1 Hz, 1 H), 7.93 (s, 1H), 7.84 – 7.72 (m, 4H), 7.12 – 6.97 (m, 2H), 6.04 (dd, J = 17.8, 1.3 Hz, 1 H), 5.45 (d, J = 12.6 Hz, 1H), 3.91 (s, 3H), 3.60 (s, 3H), 3.25 – 3.16 (m, 2H), 2.32 (s, 3H), 1.10 – 1.01 (m, 1 H), 0.48 – 0.37 (m, 2H), 0.30 – 0.22 (m, 2H); MS (ES+) 528.0 (M+1); Analysis calculated for
C29H29N5O5.CH3SO3H.2H2O. C, 54.62; H, 5.65; N, 10.62; S, 4.86; Found: C, 54.95; H, 5.55; N, 10.61 ; S, 4.87.
The process is also illustrated in Fig. 12.
Step (13): Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-rnethoxy-4- vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate

(3i) ,a 3i
A pre-cooled (0-5 °C) aq. NaOH solution [prepared from solid NaOH (4 kg, 100 mol) in water (86 L)] was added to a suspension of methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) (28.7 kg, 46 mol) in acetonitrile (86 L) cooled to 0 to 5 °C over a period of 25 mins. The reaction mixture was stirred at 0 to 5 °C for 2.5 h (TLC analysis showed the reaction was complete). The reaction mixture was filtered through a sparkler filter, washed with a mixture of 1 :1 CH3CN / H20 ( 57.4 L). Acetic acid (3.2 L, 55.9 mol) in water (56 L) was added to the filtrate at room temperature over a period of 25 mins and the resulting mixture was stirred at room temperature for 2.5 h. The solid product obtained was collected by filtration, washed with a 1 :4 mixture of CH3CN / H20 (57.5 L). The solid was dried at 45°C in a vacuum oven to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate (3i) as an off-white solid (12,77 kg, 54.1%). Average yield for this step is 50% to 75%. Mp: >200°C; H NMR (300 MHz, DMSO-d6): δ 13.49 (s, 1 H), 8.94 (bs, 4H), 8.56 (t, 1 H), 7.82 – 7.71 (m, 2H), 7.67 -7.56 (m, 4H), 7.51 (d, J = 7.8, 1 H), 6.98 (dd, J = 11.3, 17.8, 1 H), 6.68 (s, 1 H), 5.92 (d, J = 16.6, 1 H), 5.36 (d, J = 12.4, 1 H), 3.80 (s, 3H), 3.16 (m, 2H), 1.05 (m, 1 H), 0.43 (m, 2H), 0.24 (m, 2H); MS (ES+) 514.1 (M+1), 536.1 (M+Na), (ES-) 512.1 ; Analysis calculated for C28H27N5O5.3H2O: C, 59.25; H, 5.86; N, 12.34; Found C, 59.50; H,
5.75; N, 12.05. If needed this material can be crystallized from a mixture of acetone and water.
The process is also illustrated in Fig. 13.
Step 14: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b

A pre-cooled (5-8 °C) aqueous NaOH solution (prepared from solid NaOH (1.97 kg, 49.25 mol) in demineralized water (41 L) was added to a pre-cooled (0-5 °C) suspension of (3i) (13.8 kg, 26.9 mol) in acetonitrile (41 L). The reaction mixture was stirred at 0-5 °C for 30 min (until the reaction mixture becomes homogeneous). The reaction mixture was filtered through a sparkler filter washed with 50% acetonitrile in demineralized water (4.4 L). The filtrate was charged into a reactor and cooled to 0-5 °C. Aqueous HCI [prepared from cone. HCI (9.3 L) in demineralized water (36 L)] was added slowly with stirring to keep the reaction temperature at or below 15 °C, the resulting mixture was stirred at 10-15 °C for 13 h. The reaction mixture was cooled to 0-5 °C and stirred for 1 h. The solid obtained was collected by filtration and washed with demineralized water (36 L). The solid product was suspended in water (69 L) stirred for 30 mins and collected by filtration washed twice with water (20 L each). The solid product was dried in a vacuum oven at 45°C to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethyl carbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (1 1.21 Kg, 75.77%). Mp: >200°C; 1H NMR (300 MHz, DMSO-ci6): δ 12.98 (br s, 1 H), 10.86 (s, 1 H), 9.24 (s, 3H), 9.04 (s, 2H), 8.22 (d, J = 7.8 Hz, 1 H), 7.96 (d, J = 5.7 Hz, 2H), 7.78 (s, 4H), 7.09-6.99 (m, 2H), 6.07 (d, J = 17.7 Hz, 1 H), 5.45(d, J = 11.4 Hz, 1 H), 3.88 (s, 3H), 3.26-3.24 (m, 2H), 1.09 (m, 1 H), 0.47 (m, 2H), 0.28 (m, 2H).
Average isolated yield for this step varies from 63% to 80%.
The process is also illustrated in Fig. 14.
Example-2: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)

6d 8a
To a solution of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (2.35 g, 5.7 mmol) and 4-aminobenzamidine dihydrochloride (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 ml_) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The
reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and concentrated in vacuum. The residue obtained was purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 and 0-100% chloroform in CMA 50) to furnish methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)-carbamoyl)picolinate hydrochloride (8a) (2.2 g, 65%) as a white solid; MP 266 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 -7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1 H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1 H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for C29H29N505 (H20)1 5 (HCI): C, 58.93; H, 5.63; N, 1 1.85; Found: C, 58.75; H, 5.65; N, 1 1.92.
Step-2: preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)

8a 8b j0 a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml), was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with a solution of sulfuric acid (0.483 ml, 9.00 mmol) in water (5 mL) and stirred for 10 min at room temperature. To this cold water (5 ml) was added and stirred at room temperature until product crystallized out. Cold water (5 mL) was added to the slurry and stir for additional 20 min, additional cold water (5 mL) was added prior to filtration of solid. The solid obtained was collected by filtration washed with water (5 mL and 2.5 mL), dried under vacuum overnight to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b) (1.103 g, 90 % yield) as a white solid; MP 221.7 °C; H NMR (300 MHz, DMSO-d6) δ 12.30 – 10.91 (bs, 1 H, D20 exchangeable), 10.69 (bs, 1 H, D20 exchangeable), 9.24 (t, J = 6.0 Hz, 1 H), 9.16 (s, 2H, D2O exchangeable), 8.78 (s, 2H, D2O exchangeable), 8.24 (d, J = 8.0 Hz, 1 H), 8.04 – 7.91 (m, 2H), 7.84 – 7.67 (m, 4H), 7.13 – 6.94 (m, 2H), 6.03 (dd, J = 17.8, 1 .4 Hz, 1 H), 5.51 – 5.37 (m, 1 H), 3.88 (s, 3H), 3.24 (t, J = 6.4 Hz, 2H), 1.16 – 1.01 (m, 1 H), 0.52 – 0.41 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for: C28H27N605 1.0H2SO4 1.5H20: C, 52.66; H, 5.05; N, 10.97; S, 5.02; Found: C, 52.81 ; H, 4.95; N, 10.94; S, 4.64.
Example-3: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane s

To a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml) was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with methanesulfonic acid (0.584 ml, 9.00 mmol) and stirred for 1 h at room temperature. Cold water (5.00 ml) was added to the reaction mixture and stirred at room temperature until product crystallized out. To the slurry was added water (5 ml) of water stirred for additional 20 min, followed by the addition of water (5 ml) prior to filtration. The solid obtained was collected by filtration washed with water (5 ml and 2.5 ml), dried under vacuum to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane sulfonate salt (8c)
(1 .138 g, 1.867 mmol, 93 % yield) as a white solid; MP 221.2 °C; 1 H NMR (300 MHz,
DMSO-d6) δ 12.89 (s, 1 H, D2O exchangeable), 10.69 (s, 1 H, D2O exchangeable), 9.24
(t, J = 6.0 Hz, 1 H), 9.16 (s, 2H,), 8.85 (s, 2H), 8.24 (d, J = 8.0 Hz, 1 H), 8.06 – 7.91 (m, 2H), 7.86 – 7.70 (m, 4H), 7.15 – 6.96 (m, 2H), 6.03 (dd, J = 17.8, 1.4 Hz, 1 H), 5.52 – 5.35 (m, 1 H), 3.88 (s, 3H), 3.25 (t, J = 6.3 Hz, 2H), 2.34 (s, 3H), 1.17 – 1.01 (m, 1 H), 0.53 -0.43 (m, 2H), 0.32 – 0.23 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for:
CzeH^NsOsCHsSOsH 1.5H20: C, 54.71 ; H, 5.38; N, 11.00; S, 5.04; Found: C, 54.80; H, 5.14; N, 10.94; S, 4.90.
Example-4: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) in Form C (Compound XX)

The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled
46582
to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 – -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1 .6 kg) and 0.57 kg water. The mixture was heated to 46 °C. 21 g of Smopex-234 and 10 g Acticarbone 2SW were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 °C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The suspension was stirred at 0-3.0 °for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjacke,= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 1 17 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C (Compound XX).
/////avoralstat, BCX4161, Fast Track, Treat hereditary angioedema (HAE), Orphan Drug, PRECLINICAL
COc1cc(c(cc1C=C)C(=O)Nc2ccc(cc2)C(=N)N)c3cc(ncc3C(=O)O)C(=O)NCC4CC4
Fraud and Major GMP Violations at API Manufacturers in India and China
DRUG REGULATORY AFFAIRS INTERNATIONAL

Two Non-Compliance reports to API manufacturers from the Far East published in the EudraGMDP database reveal once more that basic requirements laid down in the ICH Q7 Guideline are not implemented. Read more details about those Non-Compliance Reports.
![]()
The Non-Compliance reports in the Eudra-GMDP database of the European Medicines Agency (EMA) are – to a certain extent – the European counterpart of FDA’s Warning Letters. These reports are first drawn up then put in the database after a GMP inspection performed by a representative of the European national competent authorities at an API or medicinal product manufacturer showed serious GMP deficiencies. Similar to Warning Letters, the consequences of Non-Compliance reports are for the companies concerned critical, e.g. withdrawal of the GMP certificate or product recalls.
Two Non-Compliance reports issued at the end of last year concerned API production sites in China and India.
Regarding the Chinese manufacturer “Minsheng…
View original post 346 more words
Which External GMP Audit Reports may be used?
We are often asked about the acceptance of third GMP audits at API manufacturers. The background for this is that more and more organisations offer such audits. Now, the question is what do you have to pay attention to?

We are often asked about the acceptance of third GMP audits at API manufacturers. The background for this is that more and more organisations offer such audits. Now, the question is what do you have to pay attention to?
It is essential to clarify who gave the order: has the audit been initiated from another pharmaceutical company? From the auditor himself/ herself or the organisation behind? Or from the API manufacturer?
Audits (and their reports) which have been initiated by the API manufacturers or their traders have to be viewed in a critical light. Also audits performed by the auditor – i.e. the audit organisation requires closer examination and analysis. Especially possible conflicts of interest have to be clarified.
At best, a contract audit is requested by one or several medicinal product manufacturers who buy themselves a product from the API manufacturer. If the API manufacturer is the customer, then the independence of the auditor has to be clearly demonstrated. In such a case, it is absolutely necessary to obtain a confirmation from the auditor in writing.
Acceptance, Accreditation and “Conflict of Interest”: what should you keep in mind?
A medicinal product manufacturer can basically perform an audit himself or let it perform by a so-called Third Party. Commonly, the medicinal product manufacturer assigns a consultant who has experience in the performance of audits. Yet, – here again – a few elements should be considered because the external auditor will be acting for the pharmaceutical company as if he were an own employee. The important thing here is to choose an auditor who knows the processes which have to be audited. If for example a biopharmaceutical API i.e. the manufacturer has to be audited, the auditor should have relevant experiences in biopharmaceutical processes. The auditor should also confirm that he/ she hasn’t been acting as a consultant in the area to be audited for at least the last 2 years. This should help to avoid eventual conflicts of interest. For this, a documented confirmation is helpful but often forgotten. The qualification of the GMP auditor is an essential aspect. You should require a CV of the auditor (education, work experience, audit history and audit trainings) and qualify him/ her. The execution by an accredited body doesn’t play any role. Accreditation is of no significance in pharmaceutical law.
Purchasing audit reports later:
More and more audit reports are available for purchase. In principle there is no objection to the purchase of an audit report. However, the same rules apply as those concerning the initiation of an audit. In any case, the auditors must be independent. This must also be confirmed in writing. You should check whether the audited products are the products which are also relevant for your supplier qualification. The audit of another product is quite unhelpful. The audit report should contain concrete information about the product-specific processes and procedures. This is the only way for the customer to decide on the basis of the available information whether the supplier can be suitably qualified.
Important: an audit report is only a part of the supplier qualification!
Audit reports are the main focus of interest. However, it is often forgotten that audit reports are only a part of the supplier qualification but a central one. Audit reports contain a description of the GMP situation on the audit day(s). The real assessment of the results is done by the customer – for example by a quality unit or the Qualified Person. Beside the audit report, further data should be consulted like the experiences with the supplier and the assessment of the products delivered. How valuable is an audit report with a good GMP rating when repeated deviations from the specifications are observed in the course of withdrawal of samples? The assessment of further information like for example inspection reports of the FDA which are generally accessible through the Freedom of Information Act, or EDQM’s database with the list of CEPs of API manufacturers which have been withdrawn because of GMP deficiencies. All these data should flow into a risk analysis to be used to qualify (or not) a supplier.
///////External GMP, Audit Reports, API manufacturers
Indacaterol

Indacaterol
QAB-149
CAS 753498-25-8 MALEATE
CAS 312753-06-3 (free base)
QAB-149 maleate
QAB-149-AFA
5-[2-(5,6-Diethylindan-2-ylamino)-1(R)-hydroxyethyl]-8-hydroxyquinolin-2(1H)-one maleate
R)-5-[2-[(5, 6-Diethyl-2, 3-dihydro-lH- inden-2-yl) amino]- 1 -hydroxy ethyl]-8-hydroxyquinolin-2(lH)-one, is an ultra long acting beta-adrenoceptor agonist developed by Novartis
Indacaterol (C 24 H 28 N 2 O 3 , M r = 392.49 g / mol) is chiral and is in the drug as R enantiomer and indacaterol ago. It is a derivative of 8-hydroxyquinoline and 2-aminoindan and has a certain structural similarity with other beta2-agonists , for example salbutamol . Indacaterol is lipophilic, which is a prerequisite for its long duration of action.

Indacaterol (INN) is an ultra-long-acting beta-adrenoceptor agonist[1] developed by Novartis. It was approved by the European Medicines Agency (EMA) under the trade name Onbrez Breezhaler on November 30, 2009,[2] and by the United States Food and Drug Administration (FDA), under the trade name Arcapta Neohaler, on July 1, 2011.[3] It needs to be taken only once a day,[4]unlike the related drugs formoterol and salmeterol. It is licensed only for the treatment of chronic obstructive pulmonary disease(COPD) (long-term data in patients with asthma are thus far lacking). It is delivered as an aerosol formulation through a dry powder inhaler.
Indacaterol maleate (QAB-149) is a long-acting inhaled beta2-adrenoceptor agonist. In 2008, it was filed for approval in the U.S. and the E.U. by Novartis for the treatment of chronic obstructive pulmonary disease (COPD).
In 2009, approval was granted by the EMEA and a complete response letter was assigned by the FDA.
In 2010, Novartis resubmitted an NDA seeking approval for the long-term maintenance bronchodilator treatment of airflow obstruction in adult patients with COPD, including bronchitis and/or emphysema.
In 2011, the FDA approved this indication and in 2012 the product was launched in the U.S.
The product was approved and launched in Japan in 2011 for the treatment of COPD.
In 2010, indacaterol was first launched by Novartis in Denmark and Ireland.
Clinical trials
A Phase III trial published in March 2010 examined the efficacy and safety of indacaterol in COPD patients.[5] This study, conducted in the U.S., New Zealand, and Belgium, compared indacaterol dry-powder inhaler to placebo in 416 COPD patients, mostly moderate to severe (mean FEV1 of 1.5 L). Indacaterol produced statistically improved FEV1 (both trough and AUC) and decreased use of rescue medication compared to placebo, but with safety and tolerability similar to those of placebo.
A year-long, placebo-controlled trial published in July 2010 suggests indacaterol may be significantly more effective than twice-daily formoterol in improving FEV1. There were some reductions in the need for rescue medication, but these were not significantly different; nor was there any difference in the rate of exacerbation between the 2 active treatments.[6]
A study published in October, 2011 in the European Respiratory Journal compared indacaterol with tiotropium over the study period of 12 weeks. The study found no statistical difference between the effects of the two drugs on FEV1. Indacaterol yielded greater improvements in transition dyspnoea index (TDI) total score and St. George’s Respiratory Questionnaire (SGRQ) total score.[7]
A recent Cochrane Library meta-analysis indicates that the clinical benefit in lung function from indacaterol is at least as good as that seen with twice-daily long-acting beta2-agonists. [8]
SYNTHESIS

Its synthesis is divided into two parts, a primary amine and a chiral epoxide.
Primary amine starting at 1,2 – diethyl benzene (JMC2010, 3676), two FC reaction into the ring post and then converted into oxime reduction, get four . Compound 5 obtained by Fries rearrangement 6 , phenolic hydroxyl group protected, chlorinated 7 , CBS asymmetric reduction to give the chiral secondary alcohols 8 , ring closure under alkaline conditions to obtain an epoxy compound 9 , a primary amine 4 on epoxy, to the benzyl protecting, salt to be Indacaterol Maleate.

PATENT
http://www.google.com/patents/WO2013132514A2?cl=en
Indacaterol chemically known as (R)-5-[2-[(5, 6-Diethyl-2, 3-dihydro-lH- inden-2-yl) amino]- 1 -hydroxy ethyl]-8-hydroxyquinolin-2(lH)-one, is an ultra long acting beta-adrenoceptor agonist developed by Novartis and has the following structural formula:

Indacaterol maleate is a long acting inhaled β2- agonist. Indacaterol maleate is marketed under the trade name Arcapta Neohaler in US and Onbrez in Europe.
Indacaterol maleate was disclosed in US6878721 by Novartis. The process for Indacaterol is depicted below.

Indacaterol Maleate
VII
In the above process for preparing Indacaterol maleate involves the step of reacting 8 substituted oxy-5-(R)-oxiranyl-(lH)-quinolin-2-one (III) with 2-amino- (5,6-diethyl)-indan (IV) to form a intermediate 5-[(R)-2-(5,6-diethyl-indan-2- ylamino)-l-hydroxy-ethyl]-8-substituted oxy-(lH)-quinolin-2-one (V). This epoxide ring opening is not region specific thereby along with 5-[(R)-2-(5,6- diethyl-indan-2-ylamino)- 1 -hydroxy-ethyl]-8-substituted oxy-( 1 H)-quinol intone, below mentioned products are being produced as impurities.

The above reaction mixture contains only about 60% of desired compound i.e. 5-[(R)-2-(5, 6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-substituted oxy- (lH)-quinolin-2-one. The purification of this intermediate is done using silica gel chromatography which is tedious and requires large amounts of solvents, not suitable for industrial synthesis.
To overcome the above draw backs of the process for preparing Indacaterol, the patent US7534890 discloses a process that avoids the column purification by the formation of acid addition salts of intermediate (formula – IV).
Therefore, there exists a need to develop a novel process for the preparation of indacaterol maleate.
Examples
Example -1 Preparation of compound of IIIA, wherein R is Benzyl

The compound of formula IA (25 gm) was dissolved in DMSO (75 ml) and stirred for 15 min, then compound of formula IIA (0.09 mol) was added to the reaction mixture at 25 – 30°C. The triethylamine (0. 1 mol) was added to above contents slowly, following by added sodium iodide (0.03 mol) at same temperature and stirred the reaction mixture for 3 hours at same temperature. The purified water (250 ml) was added to the reaction mixture and stirred for 1.0 hour. The contents were filtered and washed with water. The wet material was dissolved in methanol (250 ml) and stirred for 30 minutes, and then water was added. The contents were stirred for lhour at 25 – 30°C and filtered to obtain the title compound. Yield: 76%
Example -2 Preparation of compound of IIIA, wherein R is Benzyl

The compound of formula IA (25 gm) was dissolved in DMSO (75 ml) and stirred for 15 min, then compound of formula IIA (0.09 mol) was added to the reaction mixture at 25 – 30°C. Potassium carbonate (0. 1 mol) was added to above contents slowly, following by added sodium iodide (0.03 mol) at same temperature and stirred the reaction mixture for 3 hours at same temperature. The purified water (250 ml) was added to the reaction mixture and stirred for 1.0 hour. The contents were filtered and washed with water. The wet material was dissolved in methanol (250 ml) and stirred for 30 minutes, and then water was added. The contents were stirred for lhour at 25 – 30 °C and filtered to obtain the title compound. Yield: 82%
Exam le -3 Preparation of compound of IIIA, wherein R is Benzyl

The compound of formula IA (25 gm) was dissolved in DMSO (75 ml) and stirred for 15 min, then compound of formula IIA (0.09 mol) was added to the reaction mixture at 25 – 30°C, then Sodium iodide (0.03 mol) was added to the reaction mixture at same temperature and stirred the reaction mixture for 3 hours at same temperature. The purified water (250 ml) was added to the reaction mixture and stirred for 1.0 hour. The contents were filtered and washed with water. The wet material was dissolved in methanol (250 ml) and stirred for 30 minutes, and then water was added. The contents were stirred for lho‘ur at 25 – 30 °C and filtered to obtain the title compound. Yield: 84%
Exam le -4 Preparation of compound of IVA, wherein R is Benzyl

The Borane-dimethyl sulfide (0.11 mol) was added at 0-5°C, followed by addition of R – (2)-Methyl CBS (0.01 mol) and stirred the contents for 10 minutes at same temperature. The compound of example-1 (20 gm) was dissolved in methylene chloride (200 ml) at same temperature and stirred the reaction mixture for 1.0 hour. The methanol was added to the reaction mixture followed by addition of 5% hydrogen peroxide (0.01 mol) at 0-5 °C and stirred the contents for 15 minutes at same temperature, gradually increased the temperature to 20- 30°C. The 6. ON sulfuric acid (10 ml) solution was added to the reaction mixture and stirred for 15 minutes.The layers were separated. The separated organic layer was washed with 2. ON sulfuric acid solution followed by washings with water, then distilled and dissolved in ethyl acetate. The contents were stirred for 1.0 hour, filtered and dried at 60°C. Yield: 85%; E.e: > 95%.
Example -5 Preparation of compound of formula VA (Indacaterol)
The compound of example-4 (10 gm) was dissolved in methanol (100 ml), followed by addition of acetic acid (50 ml) to the reaction mixture. The 5% Pd/C was added to the reaction mixture and applied hydrogen pressure 3-4 Kg/cm3‘ and then the contents were stirred for 4.0 hours at 25-30°C, filtered and distilled. The residue was dissolved in ethyl acetate, stirred for 10 min and distilled to obtain the compound. Yield: 79%
Example -6 Preparation of Indacaterol Maleate
To a methanolic solution of Indacaterol, maleic acid (0.9 mol) in methanol was slowly added at 25 -30°C and stirred the isolated compound for 2.0 hours at same temperature. The reaction mass was cooled to 0 -10°C and maintained for 2.0 hrs at same temperature. The contents were filtered, washed with methanol and dried at 60 -65 °C. Yield: 93%; E.e: >99%.
Example -7 Preparation of compound of formula IXA, wherein R and Rl is benzyl

The (Bromo compound) of formula I (25 gm) was dissolved in DMF (150 ml) and stirred the contents for 15 min. The 5,6-Diethyl indane N-benzyl amine (0.9 mol) was added to the above mixture at 25 -30°C, followed by the slow addition of triethylamine, then the reaction mixture was stirred for 5.0 min. The sodium iodide (0.01 mol) was added to the reaction mixture at same temperature and stirred for 3 hours at same temperature. The purified water was added to the reaction mixture, and then the contents were filtered and washed with water. The wet compound was dissolved in methanol then water was added to the contents and stirred for lhour at 25 -30 °C. The contents were filtered and dried the compound at 60°C. Yield: 70%.
Example -8 Preparation of compound of formula XA, wherein R and Rl is benzyl
A mixture of Borane-dimethyl sulfide (0.11 mol), R-(2)-Methyl CBS (0.01 mol) and methylene chloride was stirred for 10 minutes at 0-5 C. The compound of example-7 (20 gm) was dissolved in methylene chloride (200 ml) and was added to the reaction mixture at same temperature. The reaction mixture was stirred for 1.0 hour. The methanol was added to the reaction mixture followed by addition of 5% hydrogen peroxide (0.01 mol) at 0-5 C. Stirred the contents for 15 minutes at same temperature, gradually increased the temperature to 20-30°C. The 6. ON sulfuric acid (10 ml) solution was added to the reaction mixture and stirred for 5minutes.The layers were separated. The organic layer was washed with 2. ON sulfuric acid solution followed by washing with water. The organic layer was distilled and dissolved in ethyl acetate. Stirred the contents for 1.0 hour and filtered the compound. The compound was dried at 60°C. Yield: 80%; Purity E.e: > 95%.
Example -9 Preparation of compound of formula VA (Indacaterol)
The compound of example-8 (10 gm) was dissolved in methanol (100 ml), followed by addition of acetic acid (50 ml) to the reaction mixture. Then 5% Pd/C was added to the reaction mixture and applied hydrogen pressure 3-4 Kg/cm3 The content was stirred for 4.0 hours at 25-30°C, filtered and the filtrate was distilled. The residue was dissolved in ethyl acetate (50 ml), stirred the contents for 10 min and distilled to obtain the compound. Yield: 80%
PATENT
http://www.google.com/patents/WO2014139485A1?cl=en
WO 0075114 Al is the first to describe preparation of indacaterol ((i?)-2) (Scheme 1).

Scheme 1 The synthesis is a follow-up of the previously published method for the preparation of 8- benzyloxy-5-(i?)-oxiranyl-(lH)-quinolin-2-one, published in WO 9525104 Al.This synthesis of indacaterol ((i?)-2) was further modified un WO 04076422 Al, WO 04087668 Al and WO 05123684 A2 to be better applicable for the industrial production. A weak point of the above mentioned synthesis is the use of the expensive benzyl trichloromethyl dichloroiodate as the chlorination agent in the first step. A considerable weak point of the above mentioned synthesis is the formation of undesired side products during the reaction of 8-benzyloxy-5-(R)- oxiranyl-(lH)-quinolin-2-one with 2-amino-5,6-diethylindane (Scheme 2).

Scheme 2
Crude 5-[(i?)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8-benzyloxy-(lH)-quinolin-2- one ((i?)-l) can be purified from these undesired side products by conversion to the benzoate, which is then re-crystallized, reduced with hydrogen, converted to indacaterol maleinate, which is finally re-crystallized. According to WO 04076422 Al, WO 04087668 Al and WO 05123684 A2, the yield of 5-[(i?)-2-(5,6-diethyl-indan-2-ylamino)- 1 -hydroxyethyl]-8- benzyloxy-(lH)-quinolin-2-one ((i?)-l) benzoate from 8-benzyloxy-5-(i?)-oxiranyl-(lH)- quinolin-2-one is only 67%.
Scheme 3.

The starting 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one and 2-amino-5,6- diethylindane were prepared according to US 2004167167 Al and F. Baur et al. J. Med.
Chem. 2010, 53, 3675-3684. Example 1. Preparation of 5-[2-(5,6-diethyI ndan-2-yIamino)-l-hydroxyethyl]-8- benzyloxy-(lH)-quinoIin-2-one (1)
A mixture of 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (1,15 g), 2-amino-5,6- diethylindane (0.83 g) and dimethyl sulfoxide (5 ml) was stirred at 20°C for 1 h. The resulting suspension was cooled down to 0°C and methanol (5 ml) was added at this temperature. Finely triturated NaB¾ (0.39 g) was added at 0°C and the resulting clear solution was stirred at 20°C for 16 hours. Water (20 ml) was added to the mixture and the mixture was stirred at 20°C for 6 h. The product was filtered off, washed with water and air-dried. The yield was 1.68 g (98%) of beige powder.
Example 2. Preparation of 5- [2-(5,6-diethyl-indan-2-yIamino)-l -hydrox ethyl] -8- benzyloxy-(lH)-quinolin-2-one (1) A mixture of 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (1.95 g), 2-amino-5,6- diethylindane (1.25 g), dimethyl sulfoxide (8 ml) and acetic acid (0.05 ml) was stirred at 20°C for 2 h. The resulting suspension was cooled down to 0°C and methanol (8 ml) was added at this temperature. Finely triturated NaBH (1.13 g) was added at 0°C and the produced clear solution was stirred at 20°C for 3 h. Water (32 ml) was added to the mixture and the mixture was stirred at 20°C for 16 h. The product was filtered off, washed with water and air-dried. The yield was 2.75 g (95%) of beige powder.
Example 3. Preparation of 5-[2-(5,6-diethyI-indan-2-ylamino)-l-hydroxyethyI]-8- benzyloxy-(lH)-quinolin-2-one (1)
A mixture of 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (115 mg), 2-amino-5,6- diethylindane (83 mg) and dimethyl acetamide (0.5 ml) was stirred at 20°C for 1 h. The resulting suspension was cooled down to 0°C and methanol (0.5 ml) was added at this temperature. Finely triturated NaBHU (39 mg) was added at 0°C and the obtained clear solution was stirred at 20°C for 16 h. Water (2 ml) was added to the mixture and the mixture was stirred at 20°C for 6 h. The product was filtered off, washed with water and air-dried. The yield was 160 mg (94%) of beige powder. Example 4. Preparation of 5-[2-(5,6-diethyI-indan-2-ylamino)-l-hydroxyethyI]-8- benzyloxy-(lH)-quinoLm-2-one (1)
A mixture of 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (115 mg), 2-amino-5,6- diethylindane (83 mg) and dichloromethane (2 ml) was stirred at 20°C for 2 h. Finely triturated NaBH(OAc)3 (250 mg) was added at 20°C. The resulting mixture was stirred at 20°C for 16 h and then evaporated until dry. Water (2 ml) was added to the evaporation product and the mixture was stirred at 20°C for 6 h. The product was filtered off, washed with water and air-dried. The yield was 164 mg (96%) of beige powder.
Example 5. Preparation of 5-[2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8- benzyloxy-(li?)-quinolin-2-one (1)
A mixture of 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (33 mg), 2-amino-5,6- diethylindane (21 mg) and tetrahydrofuran (1 ml) was stirred at 20°C for 1 h. The resulting suspension was cooled down to 0°C and 1 M BH3 in tetrahydrofuran (0.5 ml) was added at this temperature. The produced clear solution was stirred at 20°C for 16 h and then evaporated until dry. Water (1 ml) was added to the evaporation product and the mixture was stirred at 20°C for 6 h. The product was filtered off, washed with water and air-dried. The yield was 48 mg (99%) of beige powder.
Example 6. Preparation of 5-[2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8- hydroxy-(l/Z)-quinolin-2-one (2) A mixture of 5-[2-(5,6-diethyl-indan-2-ylamino)- 1 -hydroxyethyl]-8-benzyloxy-(lH)-quinolin- 2-one (1) (1.21 g), ethanol (100 ml) and 5 % Pd / C (80 mg) was stirred in a hydrogen atmosphere at 20°C at the pressure of 101 kPa for 2 h. A TLC analysis of the mixture showed the pure reactant, therefore the mixture was filtered and fresh 5% Pd / C (80 mg) was added to the filtrate. The mixture was stirred in a hydrogen atmosphere at 20°C at the pressure of 101 kPa for 2 h. A TLC analysis of the mixture showed the reactant accompanied by a small amount of the product, therefore the mixture was filtered and fresh 5 % Pd / C (80 mg) was again added to the filtrate. The mixture was stirred under a hydrogen atmosphere at 40°C at the pressure of 101 kPa for 4 h. A TLC analysis of the mixture showed the pure product, therefore the mixture was hot filtered and the residue on the filter was extensively washed with hot ethanol. The filtrate was evaporated in an evaporator at a reduced pressure. The yield was 0.97 g (99%) of yellow powder. Example 7. Preparation 5-[2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8-hydroxy- (lfl)-quinoIin-2-one (2)
A mixture of 5-[2-(5,6-diemyl-indan-2-ylammo)-l-hydroxyethyl]-8-benzyloxy-(lH)-quinolin- 2-one (1) (1,21 g), ethanol (100 ml) and Raney nickel (1 g) was stirred at 20°C for 2 h. The mixture was filtered and 5% Pd / C (0.1 g) was added to the filtrate. The mixture was stirred under a hydrogen atmosphere at 40°C at the pressure of 101 kPa at 40°C. A TLC analysis of the mixture showed the pure product, therefore the mixture was hot filtered and the residue on the filter was extensively washed with hot ethanol. The filtrate was evaporated in an evaporator at a reduced pressure. The yield was 0.96 g (98%) of yellow powder.
Example 8. Preparation of indacaterol ((R)-2)
Indacaterol ((i?)-2) was resolved from Z 5-[2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]- 8-hydroxy-(lH)-quinolin-2-one (2) (0.90 g) by means of preparative HPLC. Conditions of the resolution: UV detection at 260 nm, column length 500 mm, column internal diameter 50 mm, stationary phase Chiralcel OJ (20 μηι), temperature 25°C, flow rate 120 ml/min, mobile phase A: 500 ml of hexane + 1 ml triethylamine, mobile phase B: ethanol, isocratic elution 82% A + 18% B. The fractions containing indacaterol ((R)-2) were evaporated in an evaporator at a reduced pressure. The yield was 0.44 g (49%) of white powder. HPLC enantiomeric purity 99.0% ee.
Example 9. Preparation of 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8- benzyloxy-(lH)-quinolin-2-one ((R)-l) 5-[(i?)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8-benzyloxy-(lH)-quinolin-2-one ((R)-l) was resolved from 5-[2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8-benzyloxy- (lH)-quinolin-2-one (1) (1.00 g) by means of preparative HPLC. Conditions of the resolution: UV detection at 260 nm, column length 500 mm, column internal diameter 50 mm, stationary phase Chiralcel AS-V (20 μηι), temperature 25°C, flow rate 120 ml/min, mobile phase A: phosphate buffer (1.15 g of NH4H2P04, dissolved in 1000 ml of water, adjusted to pH 6.0 with 25% aqueous NH3), mobile phase B: acetonitrile, isocratic elution 20% A + 80% B. The fractions containing 5-[(i?)-2-(5,6-diethyl-indan-2-ylamino)-l -hydroxyethyl]-8-benzyloxy- (lH)-quinolin-2-one ((R)-l) were evaporated in an evaporator at a reduced pressure to the volume of about 50 ml. 25% aqueous NH3 was added dropwise to the resulting suspension up to pH 8-9 and the product was extracted with ethyl acetate. The combined extracts were dried with Na2S04 and evaporated in an evaporator at a reduced pressure. The yield was 0.48 g (48%) of white powder. HPLC enantiomeric purity 99.2% ee.
Example 10. Preparation of indacaterol ((R)-2)
A mixture of 5-[(i-)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8-benzyloxy-(lH)- quinolin-2-one ((R)-l) (0.42 g, HPLC enantiomeric purity of 99.2% ee), ethanol (50 ml) and Raney nickel (0.5 g) was stirred at 20°C for 2 h. The mixture was filtered and 5% Pd / C (0.05 g) was added to the filtrate. The mixture was stirred under a hydrogen atmosphere at 40°C at the pressure of 101 kPa for 4 h. A TLC analysis of the mixture showed the pure product, therefore the mixture was hot filtered and the residue on the filter was extensively washed with hot ethanol. The filtrate was evaporated in an evaporator at a reduced pressure. The yield was 0.33 g (97%) of white powder. HPLC enantiomeric purity 99.0% ee.
PATENT
The compound 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8- hydroxy-(lH)-quinolin-2-one, which is known as Indacaterol (INN), and its corresponding salts are beta-selective adrenoceptor agonists with a potent bronchodilating activity. Indacaterol is especially useful for the treatment of asthma and chronic obstructive pulmonary disease (COPD) and is sold commercially as the maleate salt. WO 00/75114 and WO 2004/076422 describe the preparation of Indacaterol for the first time through the process:

regioisomer impurity
Puri
Dep
Overall

The condensation between the indanolamine and the quinolone epoxide leads to the desired product but always with the presence of a significant amount of impurities, the most significant being the dimer impurity, which is the
consequence of a second addition of the product initially obtained with another quinolone epoxide, as well as the formation of another isomer which is the result of the addition of the indanolamine to the secondary carbon of the epoxide.
In addition, the reaction conditions to achieve the opening of the epoxide require high energies (ex. 21 of WO 00/75114) with temperatures of 110 °C or more for several hours, which favours the appearance of impurities.
WO 2004/076422 discloses the purification of the reaction mixture by the initial formation of a salt with an acid, such as tartaric acid or benzoic acid,
hydrogenation and final formation of the maleate salt. However, the yield achieved by the end of the process is only 49% overall.
It has been found that impurities of tartrate and benzoate salts can exist in the final product as a result of displacing the tartrate or benzoate with maleate without prior neutralization to Indacaterol base. In addition, WO 2004/076422 discloses that proceeding via the free base of Indacaterol is not viable due to its instability in organic solvents. WO 00/75114 does disclose a method proceeding via the Indacaterol free base, but it is not isolated in solid form.
WO 2004/076422 furthermore discloses the method for obtaining the quinolone epoxide from the corresponding a-haloacetyl compound by reduction in the presence of a chiral catalyst, such as an oxazaborolidine compound, by proceeding via the a-halohydroxy compound.
Documents WO 2007/124898 and WO 2004/013578 disclose 8-(benzyloxy)-5- [(lR)-2-bromo-l-{[tert-butyl(dimethyl)silyl]oxy}ethyl]quinolin-2(lH)-one and 8- (benzyloxy)-5-[(lR)-2-bromo-l-{tetrahydro-2H-pyran-2-yl-oxy}ethyl]quinolin- 2(lH)-one, respectively. Said documents are however not concerned with the preparation of Indacaterol. There exists, therefore, the need to develop an improved process for obtaining Indacaterol and salts thereof, which overcomes some or all of the problems associated with known methods from the state of the art. More particularly, there exists the need for a process for obtaining Indacaterol and pharmaceutically acceptable salts thereof, which results in a higher yield and/or having fewer impurities in the form of the dimer and regioisomers impurities and/or salts other than the desired pharmaceutically acceptable salt.
Examples
Example 1 – protecting the ot-halohydroxy compound of formula VI

A flask is charged with 5 ml of tetrahydrofuran (THF) and 5 ml of toluene, p- toluene sulfonic acid (0,15 mmol) and molecular sieves are added with stirring for 30 minutes. 6 mmol of butyl-vinylether and 3 mmol of 8-(phenylmethoxy)-5-((R)- 2-bromo-l-hydroxy-ethyl)-(lH)-quinolin-2-one are added. The mixture is agitated at 20/25° C until completion of the reaction, followed by filtration and distillation of the filtrate to remove the solvent. The product is obtained in quantitative yield as an oil consisting of 50% of each of the diastereomers.
^-NMR (DMSO-c/6, δ), mixture 50/50 of diastereomers: 0.61 and 0.82 (3H, t, J=7.2 Hz, CHs-Pr-O), 1.12 and 1.22 (3H, d, J=5.6 Hz, acetalic CH3), 0.90-1.40 (4H, m, CH2 + CH2), 3.20-3.80 (4H, m, CH2-OAr + CH2-Br), 4.51 and 4.82 (1H, q, J = 5.6 Hz, acetalic CH), 5.18 and 5.24 (1H, dd, J=4.0, 8.0 Hz, CH-O-acetal), 6.56 and 6.58 (1H, d, J = 10.0 Hz, H4), 7.00-7.57 (7H, m), 8.17 and 8.23 (1H, d, J = 10.0 Hz, H3), 10.71 (1H, s, NH)
13C-NMR (DMSO-c/6, δ), mixture 50/50 of diastereoisomers: 13.5 and 13.7 CH3), 18.5 and 18.8 (CH2), 19.9 and 20.0 (acetalic CH3), 30.9 and 31.4 (CH2), 36.8 and 37.3 (CH2), 63.7 and 64.2 (CH2-Br), 69.8 and 69.9 (CH2-OAr), 73.8 and 75.1 (CH- O), 97.5 and 100.4 (acetalic CH), 111.8 (CH), 116.9 and 117.2 (C), 121.2 and 122.4 (CH), 122.3 and 122.6 (CH), 127.7 and 127.8 (C), 127.8 and 127.9 (CH), 128.2 and 128.3 (CH), 128.8 and 129.1 (C), 129.4 and 129.6 (C), 136.1 and 136.5 (CH), 136.5 and 136.6 (C), 144.0 and 144.2 (C), 160.7 and 160.8 (C=0). Example 2 – protecting the ot-halohydroxy compound of formula VI

Pivaloyl chloride (0.72 g) is added to a stirred mixture of 8-(phenylmethoxy)-5- 5 ((R)-2-chloro-l-hydroxy-ethyl)-(lH)-quinolin-2-one (0.74 g), dichloromethane (15 ml) and 4-dimethylaminopyridine (0.89 g) at 20/25° C, and the reaction is stirred until all the starting material disappeared . Water (22 ml) is added and the phases are separated.
10 The organic phase is washed with 1 M HCI (22 ml) and then with water (22 ml).
The solvent is removed and the residue is crystallized from acetone to obtain 0.82 g of the product.
^-NMR (DMSO-c/6, δ) : 1.13 (9H, s, CH3), 3.92 (1H, dd, J= 4.0, 12.0 Hz, CH2-Br), 15 4.00 (1H, dd, J= 8.4, 12.0 Hz, CH2-CI), 5.28 (2H, s, Ph-CH2-0), 6.25 (1H, dd, J = 4.0, 8.4 Hz, CH-OPiv), 6.59 (1H, d, J= 10.0 Hz, H4), 7.15 (1H, d, J= 8.4 Hz, H6), 7.20 (1H, d, J= 8.4 Hz, H7), 7.27-7.30 (1H, m, Ph), 7.33-7.37 (2H, m, Ph), 7.54- 7.56 (2H, m, Ph), 8.18 (1H, d, J= 10.0 Hz, H3), 10.77 (1H, s, NH).
20 13C-NMR (DMSO-c/6, δ) : 26.7 (3 x CH3), 38.3 (C), 46.4 (CH2-CI), 69.8 (CH2-Ph), 71.3 (CH-OPiv), 111.9 (CH), 116.8 (C), 120.5 (CH), 122.9(CH), 126.0 (C), 127.8 (2 x CH), 127.9 (CH), 128.3 (2 x CH), 129.5 (C), 136.0 (C), 136.5 (CH), 144.5 (C), 160.7 (CON), 176.2 (COO). Example 3 – preparation of the compound of formula IV

A flask is charged with 2.5 ml of THF and 2.5 ml of toluene, p-toluene sulfonic 5 acid (5 mg) and molecular sieves (0.2 g) are added with stirring for 30 minutes.
1.5 ml of butyl-vinylether and 2 g of 8-(phenylmethoxy)-5-((R)-2-bromo-l- hydroxy-ethyl)-(lH)-quinolin-2-one are added . The mixture is agitated at 20/25° C until completion of the reaction. 0.015 ml of diisopropylethyl amine is added, the mixture is filtered, and the solvent is distilled off.
10
The residue is dissolved in 6 ml of dimethylformamide (DMF), 1.9 ml of
diisoproypylethyl amine, 1.2 g sodium iodide, and 1.5 g of 2-amino-5,6- diethylindane are added and the mixture is heated to 100° C. After completion of the reaction the mixture is cooled to 20/25° C, 0.4 ml of concentrated hydrochloric 15 acid and 0.4 ml of water are added, and the mixture is stirred for 30 minutes.
HPLC analysis shows the expected product with a purity of 75% and being free from the dimer and regioisomer impurities.
20 20 ml of water, 20 ml of methylene chloride, and 3 ml of 6N NaOH are added with stirring. The organic phase is separated and washed with 20 ml of water. The organic phase is distilled and the solvent is changed to ethyl acetate with a final volume of 100 ml. The mixture is heated to 70° C, 0.8 g of L-tartaric acid is added, and stirring continues for 30 minutes at 70° C. The mixture is cooled
25 slowly to 20/25° C, filtered, and washed with 8 ml of ethyl acetate to obtain 8- (phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-(lH)- quinolin-2-one tartrate in 68% yield. The purity of the product is >95% by HPLC analysis. Example 4 – preparation of the compound of formula IV

A flask is charged with 19 ml of THF and 19 ml of toluene, p-toluene sulfonic acid (75 mg) and molecular sieves (1.5 g) are added and the mixture is stirred for 30 minutes. 11.2 ml of butyl-vinylether and 15 g of 8-(phenylmethoxy)-5-((R)-2- bromo-l-hydroxy-ethyl)-(lH)-quinolin-2-one are added. The mixture is agitated at 20/25° C until completion of the reaction. 0.1 ml of diisopropylethyl amine are added, the mixture is filtered, and the solvent is distilled off.
The residue is dissolved in 40 ml of butanone, 14.5 ml of diisoproypylethyl amine, 9 g sodium iodide, and 11.3 g of 2-amino-5,6-diethylindane are added and the mixture is heated to 90-100° C. After completion of the reaction the mixture is cooled to 20/25° C, 3 ml of concentrated hydrochloric acid and 3 ml of water are added, and the mixture is stirred for 30 minutes.
HPLC analysis shows the expected product with a purity of 84% and being free from the dimer and regioisomer impurities. 150 ml of water, 150 ml of methylene chloride, and 22.5 ml of 6N NaOH are added with stirring. The organic phase is separated and washed with 10 ml of water. The organic phase is distilled and the solvent is changed to isopropyl alcohol with a final volume of 300 ml. The mixture is heated to 70° C, 4.9 g of benzoic acid is added, and stirring continues for 30 minutes at 70° C. The mixture is cooled slowly to 20/25° C, filtered, and washed with 30 ml of isopropanol to obtain 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy- ethyl]-(lH)-quinolin-2-one benzoate in 59 % yield. The purity of the product is > 99 % by HPLC analysis. Example 5 – preparation of the compound of formula IV

A flask is charged with 7.5 ml of THF and 7.5 ml of toluene, p-toluene sulfonic acid (30 mg) and molecular sieves (0.6 g) are added and the mixture is stirred for 30 minutes. 4.5 ml of butyl-vinylether and 6 g of 8-(phenylmethoxy)-5-((R)-2- bromo-l-hydroxy-ethyl)-(lH)-quinolin-2-one are added. The mixture is agitated at 20/25° C until completion of the reaction. 0.040 ml of diisopropylethyl amine are added, the mixture is filtered, and the solvent is distilled off.
The residue is dissolved in 18 ml of acetonitrile (ACN), 5,8 ml of diisoproypylethyl amine, 3.6 g sodium iodide, and 4.5 g of 2-amino-5,6-diethylindane are added and the mixture is heated to 80-90° C. After completion of the reaction the mixture is cooled to 20/25° C, 1.2 ml of concentrated hydrochloric acid and 1.2 ml of water are added, and the mixture is stirred for 30 minutes. HPLC analysis shows the expected product with a purity of 89% and being free from the dimer and regioisomer impurities.
60 ml of water, 60 ml of methylene chloride, and 9 ml of 6N NaOH are added with stirring. The organic phase is separated and washed with 60 ml of water. The organic phase is distilled and the solvent is changed to isopropyl alcohol with a final volume of 120 ml. The mixture is heated to 70° C, 1.9 g of succinic acid is added, and stirring continues for 30 minutes at 70° C. The mixture is cooled slowly to 20/25° C, filtered, and washed with 12 ml of isopropanol to obtain 8- (phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-(lH)- quinolin-2-one succinate in 56 % yield . The purity of the product is > 99 % by HPLC analysis. Example 6 : purification with EtOH/water

To 2.0 g of 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l- hydroxy-ethyl]-(lH)-quinolin-2-one, a mixture of 35 ml/g of EtOH and 5 ml/g of water are added and heated to reflux. Once this temperature is reached, benzoic acid is added (1.2 eq.) as a solution in 5 ml/g of the mixture of EtOH/water. The temperature is maintained for 30 minutes. The mixture is then cooled slowly overnight to 20-25°C. The resulting suspension is filtered and a white solid is obtained and dried in vacuum. The white solid is analyzed by HPLC to determine the chromatographic purity and by chiral HPLC to determine the enantiomeric purity, obtaining a white solid product with a proportion of enantiomeric impurity below 0.05%. No other impurities are detected.
Example 7 : purification with Acetone/water

To 2.0 g of 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l- hydroxy-ethyl]-(lH)-quinolin-2-one, a mixture of 35 ml/g of Acetone and 1 ml/g of water are added and heated to reflux. Once this temperature is reached, Dibenzoyl-L-tartaric monohydrate acid is added (1.2 eq.) as a solution in 5 ml/g of the mixture of Acetone /water. The temperature is maintained for 30 minutes. The mixture is then cooled slowly overnight to 20-25°C. The resulting suspension is filtered and a white solid is obtained and dried in vacuum. The white solid is analyzed by HPLC to determine the chromatographic purity and by chiral HPLC to determine the enantiomeric purity, obtaining a white solid product with a proportion of enantiomeric impurity below 0.05%. No other impurities are detected.
Example 8 : purification with EtOH/water

To 2.0 g of of 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l- hydroxy-ethyl]-(lH)-quinolin-2-one, a mixture of 35 ml/g of EtOH and 5 ml/g of water are added and heated to reflux. Once this temperature is reached, L Tartaric acid is added (1.2 eq.) as a solution in 5 ml/g of the mixture of
EtOH/water. The temperature is maintained for 30 minutes. The mixture is then cooled slowly overnight to 20-25°C. The resulting suspension is filtered and a white solid is obtained and dried in vacuum. The white solid is analyzed by HPLC to determine the chromatographic purity and by chiral HPLC to determine the enantiomeric purity, obtaining a white solid product with a proportion of enantiomeric impurity below 0.06%. No other impurities are detected.
Example 9 : synthesis of protected benzyl Indacaterol

A solution of sodium carbonate (0.57 kg/kg, 2 equivalents) in water (13 l/kg) is prepared in another reactor. This carbonate solution is added to the product solution from example 1, diethyl indanolamine HCI (0.72 kg/kg, 1.2 equivalents) is added and the mixture is heated and distilled at atmospheric pressure until a volume of 13 l/kg . Water (3 l/kg) is added and the mixture is distilled at atmospheric pressure until a volume of 13 l/kg . The system is placed in reflux position and reflux is maintained for 20 hours. When the reaction is complete, the mixture is cooled to 20-25°C and methylene chloride (15 l/kg) is added. The mixture is agitated, decanted, and the aqueous phase is extracted with methylene chloride (5 l/kg). The organic phases are washed with water (5 l/kg).
Example 10 – preparation of Indacaterol maleate

28 g of 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy- ethyl]-(lH)-quinolin-2-one tartrate is dissolved in a mixture of 560 ml of dichloromethane, 560 ml of water, and 30 ml of an aqueous solution of 6N sodium hydroxide under stirring . The phases are separated and the organic phase is washed with 280 ml of water. The organic phase is distilled to a final volume of 140 ml and 420 ml of methanol and 4.2 g of Pd/C (5% – 50% water) are added . The system is purged with nitrogen and subsequently with hydrogen at an overpressure of 0.3 bar and stirring until completion of the reaction. The catalyst is filtered off and the solvent is changed to isopropanol adjusting the final volume to 950 ml. The solution is heated to 70/80° C and a solution of 5.4 g maleic acid in 140 ml of isopropanol is added, maintaining the temperature between 70 and 80° C. The mixture is stirred at 70/80° C for 30 minutes and then slowly cooled to 20/25° C. The resulting suspension is filtered, the solid residue is washed with 90 ml of isopropanol and dried to obtain 18g of Indacaterol maleate (Yield : 79%). The product shows 99.6% purity by HPLC analysis.
Example 11 – Isolation of Indacaterol free base in solid form

lg of 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy- ethyl]-(lH)-quinolin-2-one tartrate is dissolved in a mixture of 20 ml of dichloromethane,20 ml of water, andl ml of an aqueous solution of 6N sodium hydroxide under stirring. The phases are separated and the organic phase is washed with 10 ml of water.
The organic phase is distilled to a final volume of 5 ml and 15 ml of methanol and 0.15 g of Pd/C (5% – 50% water) are added . The system is purged with nitrogen and subsequently with hydrogen at an overpressure of 0.3 bar and stirring until completion of the reaction.
The catalyst is filtered off and the solvent is changed to isopropanol adjusting the final volume to 8 ml. The resulting suspension is cooled to 0-5°C, filtered and the solid residue is washed with isopropanol and dried to obtain 0.47 g of Indacaterol free base (77%) showing 99.6% purity by HPLC analysis.
A sample of Indacaterol free base stored at 20-25°C is analysed one month later without showing any loss of purity. Example 12 – obtaining the maleate salt from Indacaterol free base

0.47 g of solid Indacaterol are suspended in 20 ml of isopropanol, heated to 70/80° C, and a solution of 0.15 g of maleic acid in 5 ml of isopropanol are added, maintaining the temperature between 70 and 80° C. The mixture is cooled to 0/5°C and filtration of the resulting solid affords 0.52 g of Indacaterol maleate with a purity of 99.7%.
Comparative example 13 – direct conversion to Indacaterol maleate
8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]- (lH)-quinolin-2-one benzoate (4 g) is dissolved in acetic acid (40 ml). Pd/C (5 %, 50% wet, 0.6 g) is added and the product is hydrogenated under a hydrogen atmosphere. When the reaction is complete the catalyst is filtered off and the filtrate is vacuum distilled until a volume of 8 ml is reached.
Ethanol (40 ml) is added and the mixture is heated to 50° C. A solution of 1.2 g of maleic acid in 2.4 ml of ethanol is added and the mixture is seeded with
indacaterol maleate and then slowly cooled to 0/5° C. The solid is filtered and washed with 5 ml of ethanol and 3 ml of isopropanol to obtain 6.0 g of indacaterol maleate.
1H-NMR analysis of the solid shows the presence of acetic acid in 2-4 % by integration of the peak at δ 1.88 (400 MHz, DMSO-c/6) corresponding to acetic acid.
References
- Cazzola M, Matera MG, Lötvall J (July 2005). “Ultra long-acting beta 2-agonists in development for asthma and chronic obstructive pulmonary disease”. Expert Opin Investig Drugs 14(7): 775–83. doi:10.1517/13543784.14.7.775.PMID 16022567.
- European Public Assessment Report for Onbrez Breezhaler
- “FDA approves Arcapta Neohaler to treat chronic obstructive pulmonary disease” (Press release). U.S. Food and Drug Administration. 2011-07-01. Retrieved 2011-07-02.[1]
- Beeh KM, Derom E, Kanniess F, Cameron R, Higgins M, van As A (May 2007). “Indacaterol, a novel inhaled beta2-agonist, provides sustained 24-h bronchodilation in asthma”. Eur. Respir. J. 29 (5): 871–8. doi:10.1183/09031936.00060006.PMID 17251236.
- Feldman, G; Siler, T; Prasad, N; Jack, D; Piggott, S; Owen, R; Higgins, M; Kramer, B; Study Group, I (2010). “Efficacy and safety of indacaterol 150 mcg once-daily in COPD: a double-blind, randomised, 12-week study”. BMC pulmonary medicine10: 11. doi:10.1186/1471-2466-10-11. PMC 2848004.PMID 20211002.
- Dahl R; Chung KF; Buhl R; et al. (June 2010). “Efficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol versus twice-daily formoterol in COPD”. Thorax 65 (6): 473–9.doi:10.1136/thx.2009.125435. PMID 20522841.
- R. Buhl; L.J. Dunn; C. Disdier; et al. (October 2011). “Blinded 12-week comparison of once-daily indacaterol and tiotropium in COPD”. European Respiratory Journal 38 (4): 797–803.doi:10.1183/09031936.00191810. PMID 21622587.
- http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD010139.pub2/abstract;jsessionid=2E0FA3EB220BD4ADED29D7B5707FC667.f01t04
| A. BORGHESE ET AL.: “Efficient Fast Screening Methodology for Optical Resolution Agents: Solvent Effects Are Used To Affect tge Efficiency of the Resolution Process“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 8, no. 3, 2004, pages 532-534, XP002725198, | ||
| 2 | * | D. BEATTIE ET AL.: “An investigation into the structure-activity relationships associated with the systematic modification of the beta2-adrenoreceptor agonist indacaterol“, BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 22, 2012, pages 6280-6285, XP002724553, |
| 3 | F. BAUR ET AL. J. MED. CHEM. vol. 53, 2010, pages 3675 – 3684 | |
| 4 | * | F. BAUR ET AL.: “The Identification of Indacaterol as an Ultralong-Acting Inhaled beta2-Adrenoceptor Agonist“, JOURNAL OF MEDICINAL CHEMISTRY, vol. 53, no. 9, 2010, pages 3675-3684, XP002724552, |
| 5 | * | KRAUSE M ET AL: “Optical resolution of flavanones by high-performance liquid chromatography on various chiral stationary phases“, JOURNAL OF CHROMATOGRAPHY, ELSEVIER SCIENCE PUBLISHERS B.V, NL, vol. 514, 1990, pages 147-159, XP026539395, ISSN: 0021-9673, DOI: 10.1016/S0021-9673(01)89386-9 [retrieved on 1990-01-01] |
| 6 | * | M. NISHIKATA ET AL.: “Method for Optical Resolution of Racemic Homochlorcyclizine and Comparison of Optical Isomers in Antihistamine Activity and Pharmacokinetics“, CHEMICAL AND PHARMACEUTICAL BULLETIN, vol. 40, no. 5, 1992, pages 1341-1342, XP002725199, |
| WO1995025104A1 | Mar 3, 1995 | Sep 21, 1995 | Lee James Beeley | Novel heterocyclic ethanolamine derivatives with beta-adrenoreceptor agonistic activity |
| WO2000075114A1 | Jun 2, 2000 | Dec 14, 2000 | Novartis Ag | Beta2-adrenoceptor agonists |
| WO2004074276A1 * | Feb 13, 2004 | Sep 2, 2004 | Theravance Inc | BIPHENYL DERIVATIVES HAVING β2 ADRENERGIC RECEPTOR AGONIST AND MUSCARINIC RECEPTOR ANTAGONIST ACTIVITY |
| WO2004076422A1 | Feb 27, 2004 | Sep 10, 2004 | Olivier Lohse | Process for preparing 5-‘(r)-2-(5,6-diethyl-indian-2-ylamino)-1-hydroxy-ethyl!-8-hydroxy-(1h)-quinolin-2-one salt, useful as an adrenoceptor agonist |
| WO2004087668A1 | Apr 1, 2004 | Oct 14, 2004 | Novartis Ag | A process for the preparation of 5-(haloacetyl)-8-(substituted oxy)-(1h)-quinolin-2-ones |
| WO2005123684A2 | Jun 21, 2005 | Dec 29, 2005 | Stephan Abel | Enantioselektive preparation of quinoline derivative |
| WO2007124898A1 * | Apr 24, 2007 | Nov 8, 2007 | Almirall Lab | DERIVATIVES OF 4-(2-AMINO-1-HYDROXIETHYL)PHENOL AS AGONISTS OF THE β2 ADRENERGIC RECEPTOR |
| WO2008046598A1 * | Oct 17, 2007 | Apr 24, 2008 | Almirall Lab | DERIVATIVES OF 4-(2-AMINO-1-HYDROXYETHYL)PHENOL AS AGONISTS OF THE β2 ADRENERGIC RECEPTOR |
| WO2009106351A1 * | Feb 27, 2009 | Sep 3, 2009 | Almirall, S.A. | Derivatives of 4-(2-amino-1-hydroxyethyl) phenol as agonists of the b2 adrenergic receptor |
| EP0147719A2 * | Dec 11, 1984 | Jul 10, 1985 | Tanabe Seiyaku Co., Ltd. | Novel carbostyril derivative and process for preparing same |
| EP1405844A1 * | Jun 27, 2002 | Apr 7, 2004 | Nikken Chemicals Company, Limited | Cycloalkenone derivative |
| US20040167167 | Feb 13, 2004 | Aug 26, 2004 | Mathai Mammen | Biphenyl derivatives |
| WO2000075114A1 * | Jun 2, 2000 | Dec 14, 2000 | Novartis Ag | Beta2-adrenoceptor agonists |
| WO2002045703A2 * | Dec 3, 2001 | Jun 13, 2002 | Bernard Cuenoud | Mixtures or organic compounds for the treatmentof airway diseases |
| WO2004076422A1 * | Feb 27, 2004 | Sep 10, 2004 | Olivier Lohse | Process for preparing 5-‘(r)-2-(5,6-diethyl-indian-2-ylamino)-1-hydroxy-ethyl!-8-hydroxy-(1h)-quinolin-2-one salt, useful as an adrenoceptor agonist |
| WO2004087668A1 * | Apr 1, 2004 | Oct 14, 2004 | Novartis Ag | A process for the preparation of 5-(haloacetyl)-8-(substituted oxy)-(1h)-quinolin-2-ones |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014154841A1 * | Mar 27, 2014 | Oct 2, 2014 | Laboratorios Lesvi, S.L. | Process for the manufacture of (r)-5-[2-(5,6-diethylindan-2-ylamino)-1-hydroxyethyl]-8-hydroxy-(1h)-quinolin-2-one |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-[2-[(5,6-Diethyl-2,3-dihydro-1H-inden-2-yl)amino]-1-hydroxyethyl]-8-hydroxyquinolin-2(1H)-one
|
|
| Clinical data | |
| Trade names | Onbrez, Arcapta |
| AHFS/Drugs.com | International Drug Names |
| Licence data |
|
| Pregnancy category |
|
| Routes of administration |
Inhalation |
| Legal status | |
| Identifiers | |
| CAS Number | 312753-06-3 |
| ATC code | R03AC18 |
| PubChem | CID 6433117 |
| IUPHAR/BPS | 7455 |
| ChemSpider | 5293751 |
| UNII | 8OR09251MQ |
| KEGG | D09318 |
| ChEBI | CHEBI:68575 |
| ChEMBL | CHEMBL1095777 |
| Chemical data | |
| Formula | C24H28N2O3 |
| Molar mass | 392.490 g/mol |

//////
O=C4/C=C\c1c(c(O)ccc1[C@@H](O)CNC3Cc2cc(c(cc2C3)CC)CC)N4
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

