
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

[18F]AMG 580
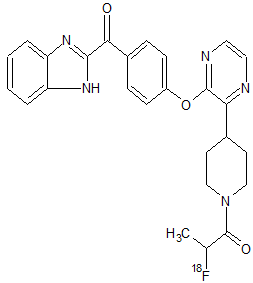
[18F]AMG 580
NOTE………CAS OF AMG 580 IS 1227067-71-1, WITHOUT 18F
AMG 580 [1-(4-(3-(4-(1H-benzo[d]imidazole-2-carbonyl)phenoxy)pyrazin-2-yl)piperidin-1-yl)-2-fluoropropan-1-one],
Phosphodiesterase 10A (PDE10A) inhibitors have therapeutic potential for the treatment of psychiatric and neurologic disorders, such as schizophrenia and Huntington’s disease. One of the key requirements for successful central nervous system drug development is to demonstrate target coverage of therapeutic candidates in brain for lead optimization in the drug discovery phase and for assisting dose selection in clinical development. Therefore, we identified AMG 580 [1-(4-(3-(4-(1H-benzo[d]imidazole-2-carbonyl)phenoxy)pyrazin-2-yl)piperidin-1-yl)-2-fluoropropan-1-one], a novel, selective small-molecule antagonist with subnanomolar affinity for rat, primate, and human PDE10A. We showed that AMG 580 is suitable as a tracer for lead optimization to determine target coverage by novel PDE10A inhibitors using triple-stage quadrupole liquid chromatography–tandem mass spectrometry technology. [3H]AMG 580 bound with high affinity in a specific and saturable manner to both striatal homogenates and brain slices from rats, baboons, and human in vitro. Moreover, [18F]AMG 580 demonstrated prominent uptake by positron emission tomography in rats, suggesting that radiolabeled AMG 580 may be suitable for further development as a noninvasive radiotracer for target coverage measurements in clinical studies. These results indicate that AMG 580 is a potential imaging biomarker for mapping PDE10A distribution and ensuring target coverage by therapeutic PDE10A inhibitors in clinical studies.
PAPER

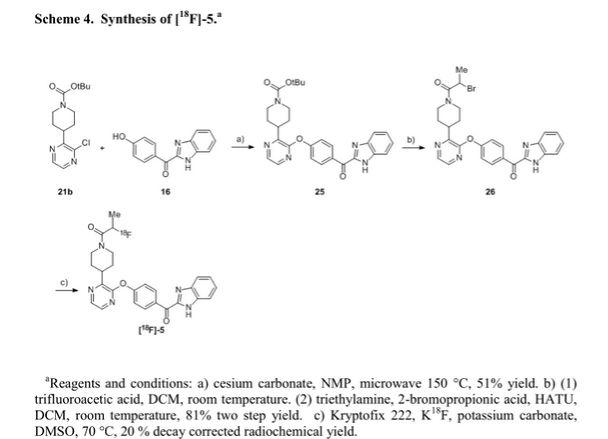
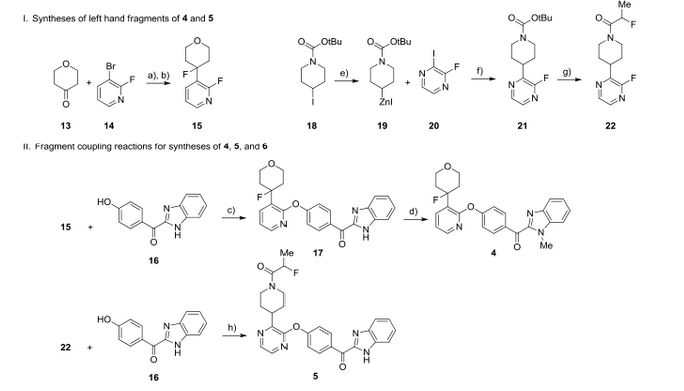
We report the discovery of PDE10A PET tracer AMG 580 developed to support proof of concept studies with PDE10A inhibitors in the clinic. To find a tracer with higher binding potential (BPND) in NHP than our previously reported tracer 1, we implemented a surface plasmon resonance assay to measure the binding off-rate to identify candidates with slower washout rate in vivo. Five candidates (2–6) from two structurally distinct scaffolds were identified that possessed both the in vitro characteristics that would favor central penetration and the structural features necessary for PET isotope radiolabeling. Two cinnolines (2, 3) and one keto-benzimidazole (5) exhibited PDE10A target specificity and brain uptake comparable to or better than 1 in the in vivo LC–MS/MS kinetics distribution study in SD rats. In NHP PET imaging study, [18F]-5 produced a significantly improved BPND of 3.1 and was nominated as PDE10A PET tracer clinical candidate for further studies.
Discovery of Phosphodiesterase 10A (PDE10A) PET Tracer AMG 580 to Support Clinical Studies
PATENT FOR AMG 580
WO 2010057121
https://www.google.com/patents/WO2010057121A1?cl=en
PAPER
Nuclear Medicine and Biology (2015), 42(8), 654-663.
http://www.sciencedirect.com/science/article/pii/S0969805115000724
Phosphodiesterase 10A (PDE10A) is an intracellular enzyme responsible for the breakdown of cyclic nucleotides which are important second messengers for neurotransmission. Inhibition of PDE10A has been identified as a potential target for treatment of various neuropsychiatric disorders. To assist drug development, we have identified a selective PDE10A positron emission tomography (PET) tracer, AMG 580. We describe here the radiosynthesis of [18 F]AMG 580 and in vitro and in vivo characterization results.
AMG 580 has an in vitro KD of 71.9 pM. Autoradiography showed specific uptake in striatum. Mean activity of 121 ± 18 MBq was used in PET studies. In Rhesus, the baseline BPND for putamen and caudate was 3.38 and 2.34, respectively, via 2TC, and 3.16, 2.34 via Logan, and 2.92, and 2.01 via SRTM. A dose dependent decrease of BPNDwas observed by the pre-treatment with a PDE10A inhibitor. In baboons, 0.24 mg/kg dose of AMG 580 resulted in about 70% decrease of BPND. The in vivo KD of [18 F]AMG 580 was estimated to be around 0.44 nM in baboons.
Conclusion
[18 F]AMG 580 is a selective and potent PDE10A PET tracer with excellent specific striatal binding in non-human primates. It warrants further evaluation in humans.
REFERNCES
http://jpet.aspetjournals.org/content/352/2/327.full
///Phosphodiesterase, tracer, receptor occupancy, positron emission tomography, radiotracer, brain penetration, AMG 580, Phosphodiesterase 10A, PDE10A, PET Tracer, [18F]AMG 580
FDA approves new diagnostic imaging agent FLUCICLOVINE F-18 to detect recurrent prostate cancer

FLUCICLOVINE F-18
Cyclobutanecarboxylic acid, 1-amino-3-(fluoro-18F)-, trans- [
- Molecular FormulaC5H818FNO2
- Average mass132.124 Da
May 27, 2016
Release
The U.S. Food and Drug Administration today approved Axumin, a radioactive diagnostic agent for injection. Axumin is indicated for positron emission tomography (PET) imaging in men with suspected prostate cancer recurrence based on elevated prostate specific antigen (PSA) levels following prior treatment.
Prostate cancer is the second leading cause of death from cancer in U.S. men. In patients with suspected cancer recurrence after primary treatment, accurate staging is an important objective in improving management and outcomes.
“Imaging tests are not able to determine the location of the recurrent prostate cancer when the PSA is at very low levels,” said Libero Marzella, M.D., Ph.D., director of the Division of Medical Imaging Products in the FDA’s Center for Drug Evaluation and Research. “Axumin is shown to provide another accurate imaging approach for these patients.”
Two studies evaluated the safety and efficacy of Axumin for imaging prostate cancer in patients with recurrent disease. The first compared 105 Axumin scans in men with suspected recurrence of prostate cancer to the histopathology (the study of tissue changes caused by disease) obtained by prostate biopsy and by biopsies of suspicious imaged lesions. Radiologists onsite read the scans initially; subsequently, three independent radiologists read the same scans in a blinded study.
The second study evaluated the agreement between 96 Axumin and C11 choline (an approved PET scan imaging test) scans in patients with median PSA values of 1.44 ng/mL. Radiologists on-site read the scans, and the same three independent radiologists who read the scans in the first study read the Axumin scans in this second blinded study. The results of the independent scan readings were generally consistent with one another, and confirmed the results of the onsite scan readings. Both studies supported the safety and efficacy of Axumin for imaging prostate cancer in men with elevated PSA levels following prior treatment.
Axumin is a radioactive drug and should be handled with appropriate safety measures to minimize radiation exposure to patients and healthcare providers during administration. Image interpretation errors can occur with Axumin PET imaging. A negative image does not rule out the presence of recurrent prostate cancer and a positive image does not confirm the presence of recurrent prostate cancer. Clinical correlation, which may include histopathological evaluation of the suspected recurrence site, is recommended.
The most commonly reported adverse reactions in patients are injection site pain, redness, and a metallic taste in the mouth.
Axumin is marketed by Blue Earth Diagnostics, Ltd., Oxford, United Kingdom
Patent
http://www.google.com/patents/WO2014023775A1?cl=en
The non-natural amino acid [ F]-l-amino-3-fluorocyclobutane-l-carboxylic acid
([18F]-FACBC, also known as [18F]-Fluciclovine) is taken up specifically by amino acid transporters and has shown promise for tumour imaging with positron emission tomography (PET).
A known synthesis of [18F]-FACBC begins with the provision of the protected precursor compound 1 -(N-(t-butoxycarbonyl)amino)-3 –
[((trifluoromethyl)sulfonyl)oxy]-cyclobutane-l-carboxylic acid ethyl ester. This precursor compound is first labelled with [18F]-fluoride:

II before removal of the two protecting groups:

IT III
EP2017258 (Al) teaches removal of the ethyl protecting group by trapping the [18F]- labelled precursor compound (II) onto a solid phase extraction (SPE) cartridge and incubating with 0.8 mL of a 4 mol/L solution of sodium hydroxide (NaOH). After 3 minutes incubation the NaOH solution was collected in a vial and a further 0.8 mL 4 mol/L NaOH added to the SPE cartridge to repeat the procedure. Thereafter the SPE cartridge was washed with 3 mL water and the wash solution combined with the collected NaOH solution. Then 2.2 mL of 6 mol/L HCl was then added with heating to 60°C for 5 minutes to remove the Boc protecting group. The resulting solution was purified by passing through (i) an ion retardation column to remove Na+ from excess NaOH and Cl~ from extra HCl needed to neutralise excess of NaOH to get a highly acidic solution before the acidic hydrolysis step, (ii) an alumina column, and (iii) a reverse-phase column. There is scope for the deprotection step(s) and/or the
purification step in the production of [18F]-FACBC to be simplified.
Example 1: Synthesis of f FIFACBC
No-carrier- added [18F]fluoride was produced via the 180(p,n)18F nuclear reaction on a GE PETtrace 6 cyclotron (Norwegian Cyclotron Centre, Oslo). Irradiations were performed using a dual-beam, 30μΑ current on two equal Ag targets with HAVAR foils using 16.5 MeV protons. Each target contained 1.6 ml of > 96% [180]water (Marshall Isotopes). Subsequent to irradiation and delivery to a hotcell, each target was washed with 1.6 ml of [160]water (Merck, water for GR analysis), giving approximately 2-5 Gbq in 3.2 ml of [160]water. All radiochemistry was performed on a commercially available GE FASTlab™ with single-use cassettes. Each cassette is built around a one-piece-moulded manifold with 25 three-way stopcocks, all made of polypropylene. Briefly, the cassette includes a 5 ml reactor (cyclic olefin copolymer), one 1 ml syringe and two 5 ml syringes, spikes for connection with five prefilled vials, one water bag (100 ml) as well as various SPE cartridges and filters. Fluid paths are controlled with nitrogen purging, vacuum and the three syringes. The fully automated system is designed for single-step fluorinations with cyclotron-produced [18F]fluoride. The FASTlab was programmed by the software package in a step-by-step time-dependent sequence of events such as moving the syringes, nitrogen purging, vacuum, and temperature regulation. Synthesis of
[18F]FACBC followed the three general steps: (a) [18F]fluorination, (b) hydrolysis of protection groups and (c) SPE purification.
Vial A contained K222 (58.8 mg, 156 μπιοΐ), K2C03 (8.1 mg, 60.8 μπιοΐ) in 79.5% (v/v)
MeCN(aq) (1105 μΐ). Vial B contained 4M HC1 (2.0 ml). Vial C contained MeCN
(4.1ml). Vial D contained the precursor (48.4 mg, 123.5 μιηοΐ) in its dry form (stored at -20 °C until cassette assembly). Vial E contained 2 M NaOH (4.1 ml). The 30 ml product collection glass vial was filled with 200 mM trisodium citrate (10 ml). Aqueous
[18F]fluoride (1-1.5 ml, 100-200 Mbq) was passed through the QMA and into the 180-
H20 recovery vial. The QMA was then flushed with MeCN and sent to waste. The trapped [18F]fluoride was eluted into the reactor using eluent from vial A (730 μΐ) and then concentrated to dryness by azeotropic distillation with acetonitrile (80 μΐ, vial C). Approximately 1.7 ml of MeCN was mixed with precursor in vial D from which 1.0 ml of the dissolved precursor (corresponds to 28.5 mg, 72.7 mmol precursor) was added to the reactor and heated for 3 min at 85°C. The reaction mixture was diluted with water and sent through the tC18 cartridge. Reactor was washed with water and sent through the tC18 cartridge. The labelled intermediate, fixed on the tC18 cartridge was washed with water, and then incubated with 2M NaOH (2.0 ml) for 5 min after which the 2M NaOH was sent to waste. The labelled intermediate (without the ester group) was then eluted off the tC18 cartridge into the reactor using water. The BOC group was hydrolysed by adding 4M HC1 (1.4 ml) and heating the reactor for 5 min at 60 °C. The reactor content with the crude [18F]FACBC was sent through the HLB and Alumina cartridges and into the 30 ml product vial. The HLB and Alumina cartridges were washed with water (9.1 ml total) and collected in the product vial. Finally, 2M NaOH (0.9 ml) and water (2.1 ml) was added to the product vial, giving a purified formulation of [18F]FACBC with a total volume of 26 ml. Radiochemical purity was measured by radio-TLC using a mixture of MeCN:MeOH:H20:CH3COOH (20:5:5: 1) as the mobile phase. The radiochemical yield (RCY) was expressed as the amount of radioactivity in the [18F]FACBC fraction divided by the total used [18F]fluoride activity (decay corrected). Total synthesis time was 43 min.
The RCY of [18F]FACBC was 62.5% ± 1.93 (SD), n=4.
/////FDA, diagnostic imaging agent, recurrent prostate cancer, fda 2016, Axumin, marketed, Blue Earth Diagnostics, Ltd., Oxford, United Kingdom, fluciclovine F 18
C1[C@@](C[C@H]1[18F])(N)C(=O)O
UPDATE
![]()
SEE EMA
| Axumin : EPAR – Summary for the public | EN = English | 06/07/2017 |
The active substance fluciclovine (18F) is prepared from the precursor AH113487 by nucleophilic substitution
of a triflate group by 18F-fluoride, followed by two deprotection steps. Due to the short half-life of the 18Ffluorine
radioisotope, each batch is prepared on the day of clinical use.
The active substance is prepared in a proprietary automated synthesiser unit. The synthesiser module is
computer-controlled. A fluid path for synthesis is provided in the form of a single use cassette (FASTlab). The
cassette contains 3 reagent vials and 3 solid phase cartridges. Two other reagent vials are supplied
separately as they have a recommended storage temperature of 2-8°C. These 2 vials are inserted into the
cassette on the day of production.
Assessment report
EMA/237809/2017 Page 13/90
Fluciclovine (18F) is produced in a continuous operation from the precursor AH113487. Due to the radioactive
nature of the process, and the short half-life of [18F] fluorine, intermediates are not isolated and there is no
opportunity for operator intervention or in-process testing. Control of the synthesis of fluciclovine (18F) from
the precursor is achieved through the automated synthesis platform, which is pre-programmed with
synthesis parameters optimised for the process. On-board detectors record transfers of radioactivity through
the fluid path at critical points and monitor temperature and pressure as appropriate so that the operator
may track the progress of the synthesis.
The active substance fluciclovine (18F) progressses immediately to purification, formulation and dispensing as
the finished product within a single, continuous operation. Validation of the manufacturing process for
fluciclovine (18F) is therefore described as part of finished product validation.
The characterisation of the active substance is in accordance with the EU guideline on chemistry of new
active substances.
As mentioned, the manufacture of the active substance and finished product takes place in a single,
continuous process. The active substance is not isolated at any point. Therefore, relevant information about
impurities is given only for the finished product.
For the same reason, information for the container closure system is provided only for the finished product.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004197/WC500230836.pdf
SETIPIPRANT

Setipiprant, KYTH-105
CAS 866460-33-5
2-(2-(1-naphthoyl)-8-fluoro-1,2,3,4-tetrahydropyrido[4,3-b]indol-5-yl)acetic acid
2-[8-fluoro-2-(naphthalene-1-carbonyl)-3,4-dihydro-1H-pyrido[4,3-b]indol-5-yl]acetic acid
5H-Pyrido(4,3-b)indole-5-acetic acid, 8-fluoro-1,2,3,4-tetrahydro-2-(1-naphthalenylcarbonyl)-
MF C24H19FN2O3
MW 402.4176632
IND FILED BY ALLERGAN FOR Alopecia
ACT-129968, a CRTH2 receptor antagonist, had been in phase II clinical trials at Actelion
Setipiprant; UNII-BHF20LA2GM; ACT-129968; 866460-33-5;
Setipiprant is a prostaglandin D2 (PGD2) antagonist. Essentially, it inhibits PGD2 receptor activity
KYTH-105 had previously been studied as a potential allergic inflammation treatment and had undergone eight clinical trials, resulting in a safety database of more than 1,000 patients. Treatment in all studies was well tolerated across all treatment groups.
Intellectual Property
KYTHERA acquired exclusive worldwide rights to KYTH-105, as well as certain patent rights covering the use of PGD2 receptor antagonists for the treatment of hair loss (often presenting as male pattern baldness, or androgenic alopecia).
Next Steps
KYTHERA plans to file an Investigational New Drug (IND) application and initiate a proof-of-concept study to establish the efficacy of KYTH-105 in male subjects with androgenic alopecia (AGA).
In 2015, Allergan acquired Kythera.

![]()
2-(2-(1-Naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic Acid
mp 224.0 °C.
LC(1)/ESI-MS tR = 0.83 min; m/z [M + H+] = 403.09.
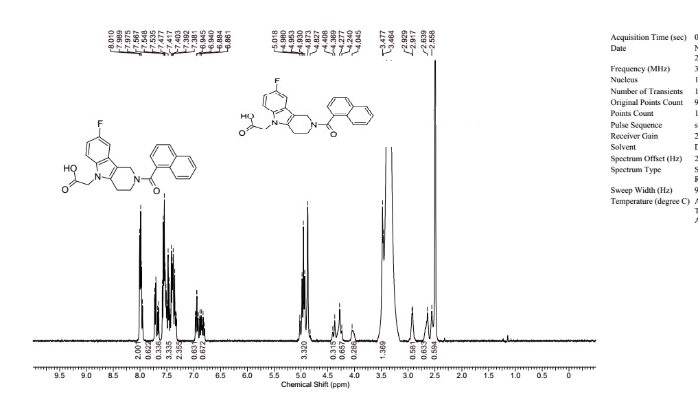
1H NMR (DMSO-d6), 65:35 mixture of two rotamers, δ: 8.02 (m, 2 H), 7.76 (d, J = 7.8 Hz, 0.65 H), 7.72 (m, 0.35 H), 7.49–7.64 (m, 3.35 H), 7.35–7.49 (m, 2.35 H), 6.98 (ddd, JH–F = 9.3 Hz, J1 = 9.3 Hz, J2 = 2.4 Hz, 0.65 H), 6.88 (m, 0.65 H), 4.85–5.14 (m, 3.3 H), 4.42 (m, 0.35 H), 4.32 (m, 0.7 H), 4.06 (m, 0.35 H), 3.50 (t, J = 5.5 Hz, 1.3 H), 2.95 (m, 0.70 H), 2.68 (m, 0.65 H), 2.58 (m, 0.65 H).
13C NMR (DMSO-d6) δ: 170.7, 169.2, 157.7 (d, JC–F = 232 Hz), 157.4 (d, JC–F = 233 Hz), 137.1, 136.2, 135.1, 134.9, 134.0, 133.8, 133.5, 129.6, 129.5, 129.4, 129.3, 128.9, 128.8, 127.5, 127.4, 127.0, 126.9, 126.0, 125.9, 125.7 (d, JC–F = 10 Hz), 125.2, 125.1, 125.0, 124.1, 123.9, 110.9 (d, JC–F = 10 Hz), 110.8 (m), 109.3 (d, JC–F = 26 Hz), 109.1 (d, JC–F = 26 Hz), 106.7 (m), 103.3 (d, JC–F = 23 Hz), 103.0 (d, JC–F = 23 Hz), 44.73, 44.70, 44.5, 44.4, 39.5, 39.3, 23.1, 22.3.
HRMS (ESI): m/zcalcd for C24H20N2O3F [M + H+] 403.1458, found 403.1458.
SYNTHERSIS

Setipiprant (INN) (developmental code names ACT-129,968, KYTH-105) is a drug originally developed by Actelion which acts as a selective, orally available antagonist of the prostaglandin D2 receptor 2 (DP2).[1] It was initially researched as a treatment for allergies and inflammatory disorders, particularly asthma, but despite being well tolerated in clinical trials and showing reasonable efficacy against allergen-induced airway responses in asthmatic patients,[2][3] it failed to show sufficient advantages over existing drugs and was discontinued from further development in this application.[4]
However, following the discovery in 2012 that the prostaglandin D2 receptor (DP/PGD2) is expressed at high levels in the scalp of men affected by male pattern baldness,[5] the rights to setipiprant were acquired by Kythera with a view to potentially developing this drug as a novel treatment for baldness, with a previously unexploited mechanism of action.[6] While it is too early to tell whether setipiprant will be an effective treatment for this condition, the favorable pharmacokinetics and relative lack of side effects seen in earlier clinical trials mean that fresh clinical trials for this new application can be conducted fairly quickly.[7]
Prostaglandin D2 is a known agonist of the thromboxane A2 (TxA2) receptor, the PGD2 (DP) receptor and the recently identified G-protein-coupled “chemoattractant receptor- homologous molecule expressed on Th2 cells” (CRTH2).
The response to allergen exposure in a previously sensitized host results in a cascade effect involving numerous cell types and release of a number of cytokines, chemokines, and multiple mediators. Among these critical initiators are the cytokines interleukin (IL)-4, IL-13, and IL-5, which play critical roles in Th2 cell differentiation, immunoglobulin (Ig)E synthesis, mast cell growth and differentiation, upregulation of CD23 expression, and the differentiation, recruitment, and activation of eosinophils. The stimulated release of the array of mediators, causes end-organ damage, including constriction and hyperresponsi- veness, vascular permeability, edema, mucous secretion, and further inflammation.
Because of the number of responses targeted, corticosteroids have proven to be the most effective therapy. Rather than antagonizing these specific responses in a directed way, another approach is to alter the immune response, that is, to change the nature of the immunological response to allergen. CRTH2 is preferentially expressed on Th2 cells and is a chemoattractant receptor for PGD2 that mediates PGD2-dependent migration of blood Th2 cells. Chemoattractants are responsible for the recruitment of both Th2 cells and other effector cells of allergic inflammation, which can provide the conceptual basis for the development of new therapeutic strategies in allergic conditions.
So far, few compounds having CRTH2 antagonistic activity have been reported in the patent literature. Bayer AG claims the use of Ramatroban ((3R)-3-(4-fluorobenzene- sulfonamido)-l,2,3,4-tetrahydrocarbazole-9-propionic acid) for the prophylaxis and treatment of allergic diseases, such as asthma, allergic rhinitis or allergic conjuvatitis
(GB 2388540). Further, (2-tert.-butoxycarbonyl-l, 2, 3, 4-tetrahydro-pyrido[4,3-b]indol-5- yl)-acetic acid and (2-ethoxycarbonyl-l, 2, 3, 4-tetrahydro-pyrido[4,3-b]indol~5-yl)-acetic acid are disclosed by Kyle F. et al in two patent applications (US 5817756 and WO 9507294, respectively).
Furthermore, oral bioavailability of the Ramatroban and its ability to inhibit prostaglandin D2-induced eosinophil migration in vitro has been reported (Journal of Pharmacology and Experimental Therapeutics, 305(1), p.347-352 (2003)).
Description of the invention:
It has now been found that compounds of the general Formulae (I) and (II) of the present invention are CRTH2 receptor antagonists. These compounds are useful for the treatment of both chronic and acute allergic/immune disorders such as allergic asthma, rhinitis, chronic obstructive pulmonary disease (COPD), dermatitis, inflammatory bowel disease, rheumatoid arthritis, allergic nephritis, conjunctivitis, atopic dermatitis, bronchial asthma, food allergy, systemic mast cell disorders, anaphylactic shock, urticaria, eczema, itching, inflammation, ischemia-reperfusion injury, cerebrovascular disorders, pleuritis, ulcerative colitis, eosinophil-related diseases, such as Churg-Strauss syndrome and sinusitis, basophil- related diseases, such as basophilic leukemia and basophilic leukocytosis.
The compounds of general Formulae (I) and (II), especially those mentioned as being preferred, display high selectivity towards the CRTH2 receptor. No antagonistic effects (IC50 >10 μM) are observed on e.g. prostaglandin D2 receptor DPI; PGI2 receptor (IP), PGE2 receptors (EPl, EP2, EP3, EP4), PGF2 receptor (FP), thromboxane receptor A2 (TxA2), leukotriene receptors (CysLTl, CysLT2, LTB4), complement receptor (C5a), angiotensin receptors (ATI, AT2) or serotonin receptor 5HT2c.
The solubility of compounds of general Formulae (I) and (II) in buffer at pH 7 is generally >800 μg/ml.
In vitro assays with rat and dog liver microsomes, or with rat and human hepatocytes revealed high metabolic stability for compounds of general Foπnulae (I) and (II), especially for those compounds mentioned as being preferred.
The compounds of general Formulae (I) and (II), especially those mentioned as being preferred, do not interfere with cytochrome P-450 enzymes, e.g. they are neither degraded by, nor do they inhibit such enzymes.
Excellent pharmacokinetic profiles have been observed for compounds of general Formulae (I) and (II), especially for those compounds mentioned as being preferred, after oral administration (10 mg/kg) to rats and dogs (bioavailability 20-80%, Tmax 30 min, Cmax 2000- 6000 ng/ml, low clearance, T] 24-8 h). The compounds of general Formulae (I) and (II), especially those mentioned as being preferred, are efficacious in vitro, inhibiting PGD2-induced migration of eosinophils or other CRTH2 expressing cells in a cell migration assay. A number of techniques have been developed to assay such chemotactic migration (see, e.g., Leonard et al., 1995, “Measurement of α- and β-Chemokines”, in Current Protocols in Immunology, 6.12.1- 6.12.28, Ed. Coligan et al, John Wiley & Sons, Inc. 1995). The compounds of the present invention are tested using a protocol according to H. Sugimoto et al. (J Pharmacol Exp Ther. 2003, 305(1), 347-52), or as described hereinafter: Purified eosinophils are labeled with a fluorescent dye, i.e. Calcein-AM and loaded in BD Falcon FluoroBlock upper inserts. Test compounds are diluted and incubated with eosinophils in the BD Falcon
FluoroBlock upper inserts for 30 min at 37 °C in a humidified CO2 incubator. A constant amount of PGD2 is added to BD Falcon FluoroBlock lower chamber, at a concentration known to have a chemotactic effect on CRTH2 cells. As a control, at least one aliquot in the upper well does not contain test compound. The inserts are combined with the chambers and are incubated for 30 min at 37 °C in a humidified CO2 incubator. After an incubation period, the number of migrating cells on the lower chamber is counted using a fluorescent reader, i.e. an Applied Biosystems Cyto Fluor 4000 plate reader. The contribution of a test compound to the chemotactic activity of PGD2 is measured by comparing the chemotactic activity of the aliquots containing only dilution buffer with the activity of aliquots containing a test compound. If addition of the test compound to the solution results in a decrease in the number of cells detected in the lower chamber relative to the number of cells detected using a solution containing only PGD2, then there is identified an antagonist of PGD2 induction of chemotactic activity of eosinophils.
PAPER
Journal of Medicinal Chemistry (2013), 56(12), 4899-4911
http://pubs.acs.org/doi/abs/10.1021/jm400122f
Identification of 2-(2-(1-Naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic Acid (Setipiprant/ACT-129968), a Potent, Selective, and Orally Bioavailable Chemoattractant Receptor-Homologous Molecule Expressed on Th2 Cells (CRTH2) Antagonist

Herein we describe the discovery of the novel CRTh2 antagonist 2-(2-(1-naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid 28 (setipiprant/ACT-129968), a clinical development candidate for the treatment of asthma and seasonal allergic rhinitis. A lead optimization program was started based on the discovery of the recently disclosed CRTh2 antagonist 2-(2-benzoyl-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid 5. An already favorable and druglike profile could be assessed for lead compound 5. Therefore, the lead optimization program mainly focused on the improvement in potency and oral bioavailability. Data of newly synthesized analogs were collected from in vitro pharmacological, physicochemical, in vitro ADME, and in vivo pharmacokinetic studies in the rat and the dog. The data were then analyzed using a traffic light selection tool as a visualization device in order to evaluate and prioritize candidates displaying a balanced overall profile. This data-driven process and the excellent results of the PK study in the rat (F = 44%) and the dog (F = 55%) facilitated the identification of 28 as a potent (IC50 = 6 nM), selective, and orally available CRTh2 antagonist.
PAtent
WO 2005095397
http://www.google.co.in/patents/WO2005095397A1?cl=en
Formula 6.



Scheme 1
Step a)

Step b)

Scheme 2
Formula (I).

References
- Fretz H, Valdenaire A, Pothier J, Hilpert K, Gnerre C, Peter O, Leroy X, Riederer MA. Identification of 2-(2-(1-naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid (setipiprant/ACT-129968), a potent, selective, and orally bioavailable chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) antagonist. J Med Chem. 2013 Jun 27;56(12):4899-911. doi: 10.1021/jm400122f PMID 23721423
- Sidharta PN, Diamant Z, Dingemanse J. Single- and multiple-dose tolerability and pharmacokinetics of the CRTH2 antagonist setipiprant in healthy male subjects. Fundam Clin Pharmacol. 2014 Dec;28(6):690-9. doi: 10.1111/fcp.12079 PMID 24734908
- Diamant Z, Sidharta PN, Singh D, O’Connor BJ, Zuiker R, Leaker BR, Silkey M, Dingemanse J. Setipiprant, a selective CRTH2 antagonist, reduces allergen-induced airway responses in allergic asthmatics. Clin Exp Allergy. 2014 Aug;44(8):1044-52. doi: 10.1111/cea.12357 PMID 24964348
- Norman P. Update on the status of DP2 receptor antagonists; from proof of concept through clinical failures to promising new drugs. Expert Opin Investig Drugs. 2014 Jan;23(1):55-66. doi: 10.1517/13543784.2013.839658 PMID 24073896
- Garza LA, et al. Prostaglandin D2 inhibits hair growth and is elevated in bald scalp of men with androgenetic alopecia. Science Translational Medicine, 21 March 2012; 4(126):126ra34. doi: 10.1126/scitranslmed.3003122
- George Cotsarelis, Garret Fitzgerald, Luis Garza. Compositions and methods for regulating hair growth. US Patent application 2015/0072963
- Pipeline KYTH-105 (setipiprant)
- http://files.shareholder.com/downloads/AMDA-MFNLA/4023632629x0x817836/4E5AC47A-B9EE-4296-9D97-631C0F6B7C97/KYTH-105_setipiprant_.pdf

| Patent ID | Date | Patent Title |
|---|---|---|
| US2015072963 | 2015-03-12 | COMPOSITIONS AND METHODS FOR REGULATING HAIR GROWTH |
| US2014328861 | 2014-11-06 | Combination of CRTH2 Antagonist and a Proton Pump Inhibitor for the Treatment of Eosinophilic Esophagitis |
| US2010234396 | 2010-09-16 | Tetrhydropyridoindole Derivatives |
| US7714132 | 2010-05-11 | Tetrahydropyridoindole derivatives |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-[8-fluoro-2-(naphthalene-1-carbonyl)-3,4-dihydro-1H-pyrido[4,3-b]indol-5-yl]acetic acid
|
|
| Clinical data | |
| Administration | Oral |
| Identifiers | |
| CASRN | 866460-33-5 |
| ATC code | none |
| PubChem | CID 49843471 |
| Chemical data | |
| Formula | C24H19FN2O3 |
| Molar mass | 402.417 g/mol |
///////Setipiprant, KYTH-105, 866460-33-5, ALLERGAN, Alopecia, KYTHERA
c15ccccc5cccc1C(=O)N(CC3)Cc2c3n(CC(O)=O)c(cc4)c2cc4F
Flow synthesis of Fluoxetine
![[1860-5397-11-134-i8]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-134-i8.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Flow synthesis of fluoxetine (46).
PIC CREDIT, The synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry, and , Beilstein J. Org. Chem. 2015, 11, 1194–1219.,doi:10.3762/bjoc.11.134
One of the early published examples of industry-based research on multi-step flow synthesis of a pharmaceutical was reported in 2011 by scientists from Eli Lilly/UK and detailed the synthesis of fluoxetine 46, the API of Prozac[1]. In this account each step was performed and optimised individually in flow, with analysis and purification being accomplished off-line. The synthesis commences with the reduction of the advanced intermediate ketone 47 using a solution of pre-chilled borane–THF complex (48) to yield alcohol 49 (Scheme 1).
Conversion of the pendant chloride into iodide 51 was attempted via Finckelstein conditions, however, even when utilising phase-transfer conditions in order to maintain a homogeneous flow regime the outcome was not satisfactory giving only low conversions. Alternatively direct amination of chloride 49 utilising high temperature flow conditions (140 °C) allowed the direct preparation of amine 50 in excellent yield.
Flow processing using a short residence time (10 min) at the elevated temperature allowed for a good throughput; in addition, the handling of the volatile methylamine within the confines of the flow reactor simplifies the practical aspects of the transformation, however, extra precautions were required in order to address and remove any leftover methylamine that would pose a significant hazard during scaling up.
The final arylation of 50 was intended to be performed as a SNAr reaction, however, insufficient deprotonation of the alcohol 50 under flow conditions (NaHMDS or BEMP instead of using a suspension of NaH as used in batch) required a modification to the planned approach. To this end a Mitsunobu protocol based on the orchestrated mixing of four reagent streams (50, 54 and reagents 52 and 53) was developed and successfully applied to deliver fluoxetine (46) in high yield.
Overall, this study is a good example detailing the intricacies faced when translating an initial batch synthesis into a sequence of flow steps for which several adaptations regarding choice of reagents and reaction conditions are mandatory in order to succeed.

Dr Marcus Baumann
Postdoc
Marcus Baumann studied chemistry at the Philipps-University Marburg/Germany, from where he graduated in 2007. His studies involved a 6 month period as an Erasmus student at the Innovative Technology Centre at the University of Cambridge, UK (with Prof. Steven V. Ley and Dr Ian R. Baxendale), where he developed a new flow-based oxazole synthesis. He soon returned to Cambridge to pursue his doctoral studies with Prof. Steven V. Ley where he developed flow processes for Curtius rearrangements, different fluorination reactions as well as important heterocycle syntheses. Upon completion of his PhD in 2010 Marcus was awarded a Feodor Lynen Postdoctoral Fellowship (Alexander von Humboldt Foundation, Germany) allowing him to join the group of Prof. Larry E. Overman at UC Irvine, USA (2011-2013). During his time in California his research focused on the synthesis of naturally occurring terpenes as well as analogues of ETP-alkaloids. The latter project generated potent and selective histone methyltransferase inhibitors and opened routes towards new probes for epigenetic diseases which are currently under further investigation. In early 2013 Marcus returned to the UK and took up a postdoctoral position with Prof. Ian R. Baxendale at the University of Durham, where his interests concentrate on the development of flow and batch based strategies towards valuable compounds en route for regenerative medicines.

Prof. Ian R. Baxendale
(email at i.r.baxendale@durham.ac.uk)
Research Interests
My general areas of interest are: Organic synthesis (natural products, heterocyclic and medicinal chemistry), Organometallic chemistry, Catalyst design and application, Meso flow chemistry, Microfluidics, Microwave assisted synthesis, Solid supported reagents and scavengers, and facilitated reaction optimisation using Robotics and Automation.
My primary research direction is the synthesis of biologically potent molecules which encompasses the design, development and integration of new processing techniques for their preparation and solving challenges associated with the syntheses of new pharmaceutical and agrochemical compounds. In our work we utilise the latest synthesis tools and enabling technologies such as microwave reactors, solid supported reagents and scavengers, enzymes, membrane reactors and flow chemistry platforms to facilitate the bond making sequence and expedite the purification procedure. A central aspect of our investigations is our pioneering work on flow chemical synthesis and microreactor technology as a means of improving the speed, efficiency, and safety of various chemical transformations. As a part of these studies we are attempting to devise new chemical reactions that are not inherently feasible or would be problematic under standard laboratory conditions. It is our further challenge to enhance the automation associated with these reactor devices to impart a certain level of intelligence to their function so that repetitive wasteful actions currently performed by chemists can be delegated to a machine; for example, reagent screening or reaction optimisation. We use these technologies as tools to enhance our synthetic capabilities but strongly believe in not becoming slaves to any methodology or equipment.
For those interested in our research and wishing to find out more we invite you to visit our website at: http://www.dur.ac.uk/i.r.baxendale/
- Ahmed-Omer, B.; Sanderson, A. J. Org. Biomol. Chem. 2011, 9, 3854–3862. doi:10.1039/C0OB00906G
Paper
Preparation of fluoxetine by multiple flow processing steps
*Corresponding authorsaEli Lilly and Co. Ltd., Lilly Research Centre, Erl Wood Manor, Windlesham, Surrey, UKOrg. Biomol. Chem., 2011,9, 3854-3862DOI: 10.1039/C0OB00906G
http://pubs.rsc.org/en/Content/ArticleLanding/2011/OB/c0ob00906g#!divAbstract
Microflow technology is established as a modern and fashionable tool in synthetic organic chemistry, bringing great improvement and potential, on account of a series of advantages over flask methods. The study presented here focuses on the application of flow chemistry process in performing an efficient multiple step syntheses of (±)-fluoxetine as an alternative to conventional synthetic methods, and one of the few examples of total synthesis accomplished by flow technique.


1 The general method set-up of flow process used for the synthesis of (±)- fluoxetine.

Scheme 1 Synthesis of (±)-fluoxetine in flow: (i) BH3·THF, r.t., 5 min (77%); (ii) NaI, toluene: water, 100 °C, 20 min (43%); (iii); MeNH2 (aq), …
//////////Flow synthesis, fluoxetine
WHO issues revised Guideline on HVAC Systems
DRUG REGULATORY AFFAIRS INTERNATIONAL

The World Health Organization (WHO) recently issued a guideline for commenting which describes the requirements for HVAC systems for the manufacture of non-sterile forms. As most guidelines on this topic address the requirements for sterile dosage forms, the previous version was gladly accepted by industry. Learn more about the revised guideline on HVAC systems.
The World Health Organization (WHO) recently issued a guideline for commenting which describes the requirements for HVAC systems used for the manufacture of non-sterile dosage forms. As most guidelines on this topic address the requirements for sterile forms, the previous version (TRS 961, Annex 1) from 2011 was gladly accepted by industry. Mentioned are non-sterile dosage forms as tablets, capsules, liquids or ointments, but also for the final steps in the manufacture of APIs. The WHO guideline means to provide guidance specifically for the areas design, installation, qualification and maintenance of ventilation systems. For the manufacture of…
View original post 192 more words
EU GMP Annex 1 Revision 2016 – what does the pharmaceutical industry expect?
DRUG REGULATORY AFFAIRS INTERNATIONAL

Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim
Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim talked in his keynote speech at the Pharma Congress 2016 about the revision of Annex 1 of the EU GMP Guide. Read here what the pharmaceutical industry expects form the new Annex 1.
Europe’s biggest Pharma Congress of its kind took place in Düsseldorf on 12 and 13 April. With more than 1000 participants, 90 exhibitors and 10 GMP conferences this Congress 2016 has been the biggest since the first one 18 years ago. 50 lectures, almost exclusively case studies from pharmacuetical companies such as Pfizer, Novartis, Boehringer Ingelheim and many more were discussed. Special attention was paid to the keynotes at the beginning of each congress day.
 Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim talked in his keynote…
Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim talked in his keynote…
View original post 876 more words
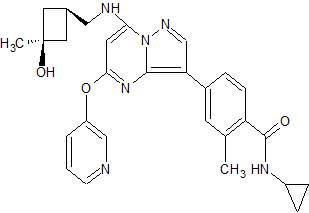
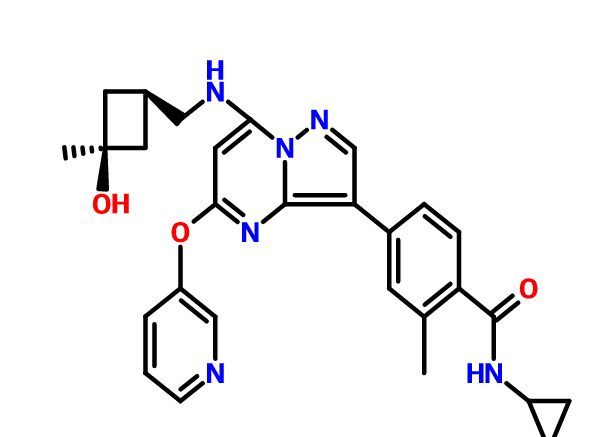
CFI-402257

CFI-402257
N-cyclopropyl-4-(7-((((1s,3s)-3-hydroxy-3-methylcyclobutyl)methyl)amino)-5-(pyridin-2-yloxy)pyrazolo[1,5-a]pyridin-3-yl)-2-methylbenzamide
N-cyclopropyl-4-(7-( (((Is, 3s)-3-hydroxy-3-methylcyclobutyl)methyl)amino)-5- (pyridin-3-yloxy)pyrazolol 1 , 5-a ]pyrimidin-3-yl)-2-methylbenzamide
CAS 1610759-22-2 (free base); 1610677-37-6 (HCl)
MF: C29H31N5O3
MW: 497.2427
CFI-402257 is a highly potent and selective TTK (threonine tyrosine kinase) Inhibitor ((TTK Ki = 0.1 nM) with potential anticancer activity. TTK is an essential chromosomal regulator and is overexpressed in aneuploid tumors. High TTK levels correlate with a high tumor grade11 and poor patient outcomes. TTK inhibition are associated with a disabled mitotic checkpoint, resulting in chromosome segregation errors, aneuploidy, and cell death.
Synthesis
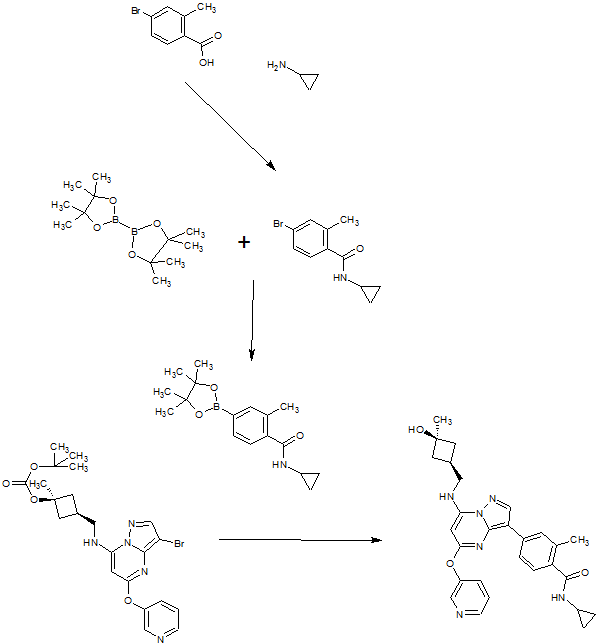
SYN OF INTERMEDIATE
SYNTHESIS COLOUR INDICATED
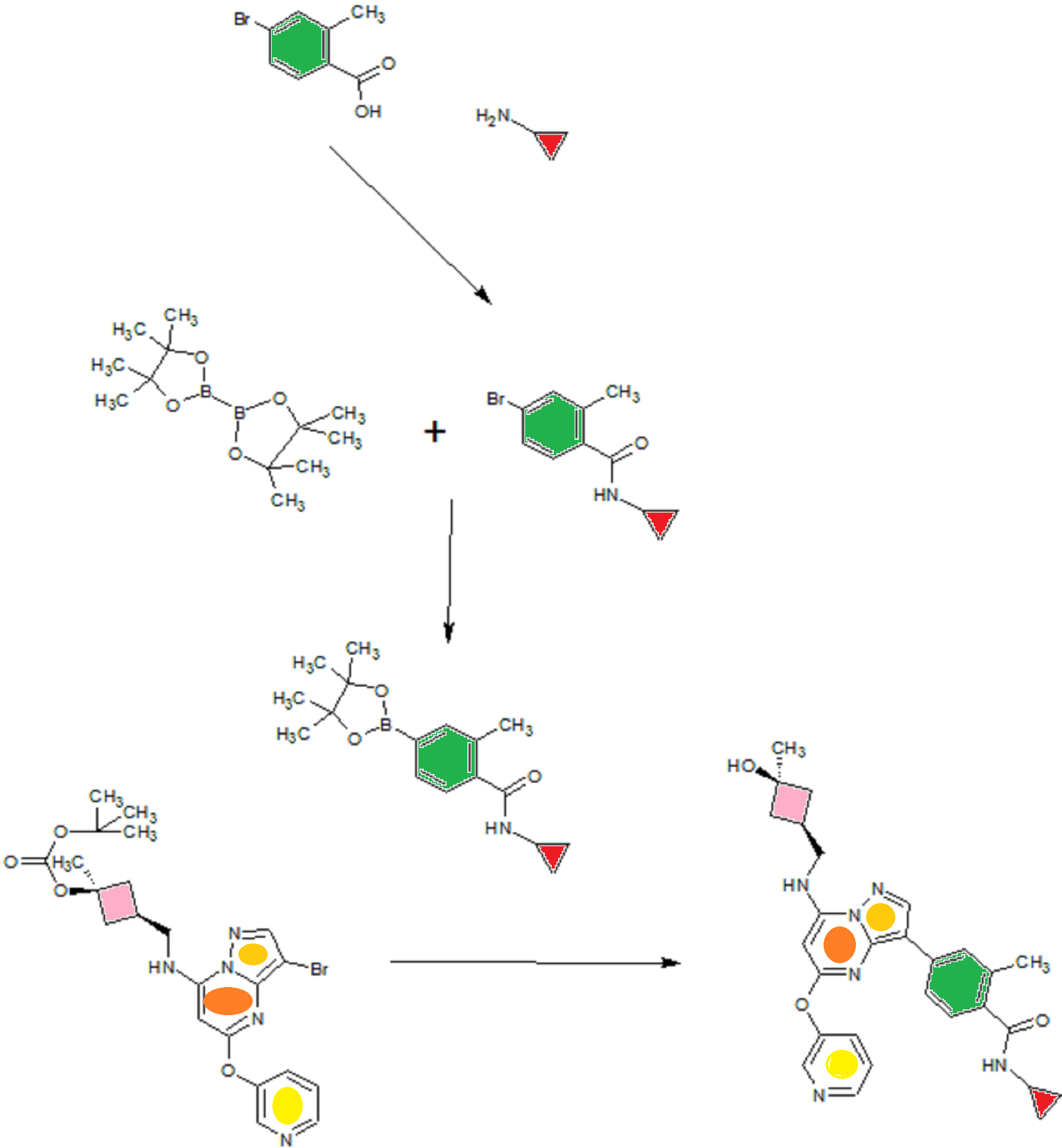
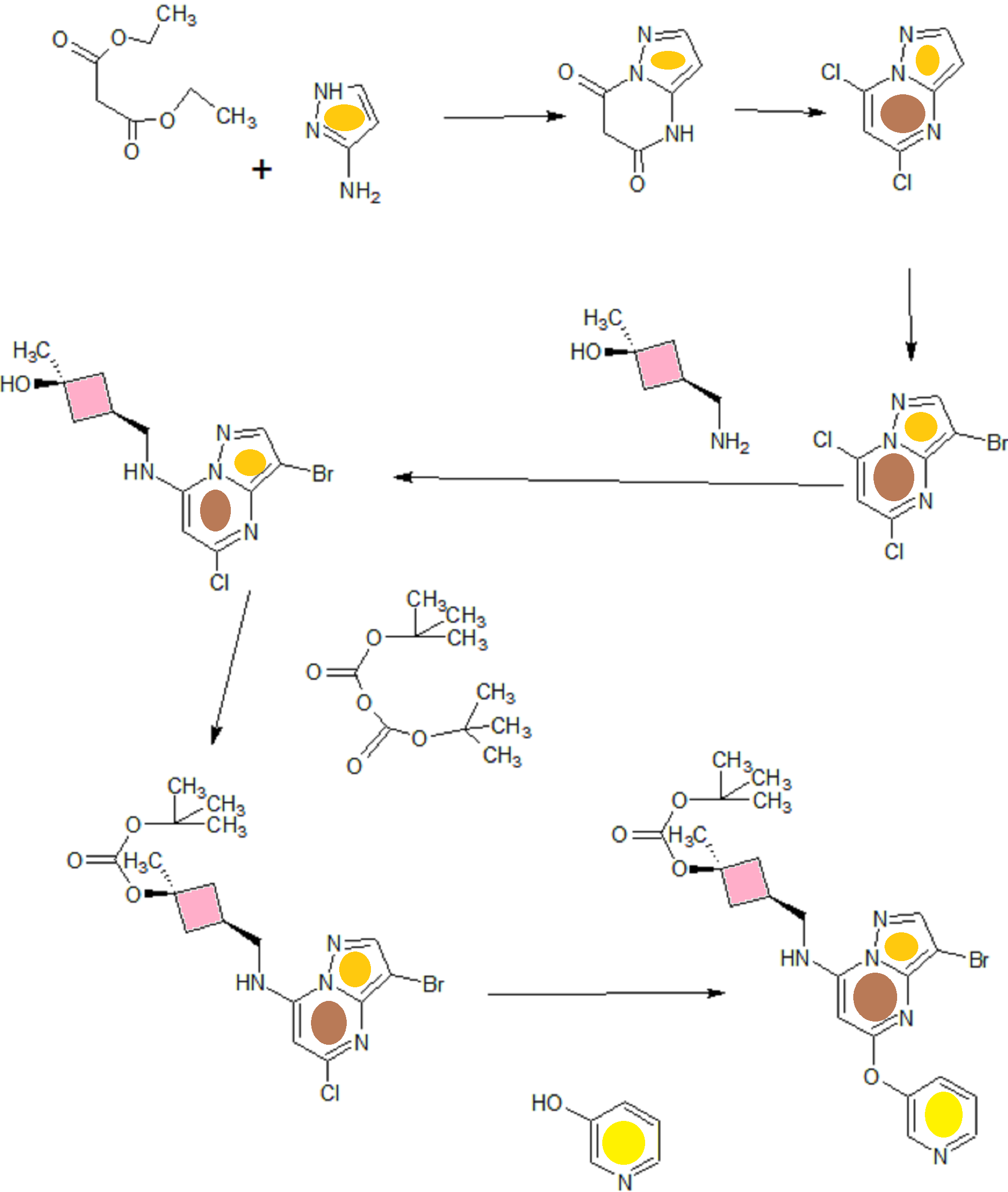
SYN OF INTERMEDIATES
IF YOU HAVE ENJOYED IT ………EMAIL ME amcrasto@gmail.com, +919323115463, India

INDIA FLAG

DR ANTHONY CRASTO , WORLDDRUGTRACKER, HELPING MILLIONS, MAKING INDIA AND INDIANS PROUD
Protein kinases have been the subject of extensive study in the search for new therapeutic agents in various diseases, for example, cancer. Protein kinases are known to mediate intracellular signal transduction by effecting a phosphoryl transfer from a nucleoside triphosphate to a protein acceptor that is involved in a signaling pathway. There are a number of kinases and pathways through which extracellular and other stimuli cause a variety of cellular responses to occur inside the cell.
Human TTK protein kinase (TTK), also known as tyrosine threonine kinase, dual specificity protein kinase TTK, Monopolar Spindle 1 (Mpsl) and Phosphotyrosine -Picked Threonine Kinase (PYT), is a conserved multispecific kinase that is capable of phosphorylating serine, threonine and tyrosine residues when expressed in E. coli (Mills et al., J. Biol. Chem. 22(5): 16000-16006 (1992)). TTK mRNA is not expressed in the majority of physiologically normal tissues in human (Id). TTK mRNA is expressed in some rapidly proliferating tissues, such as testis and thymus, as well as in some tumors (for example, TTK mRNA was not expressed in renal cell carcinoma, was expressed in 50% of breast cancer samples, was expressed in testicular tumors and ovarian cancer samples) (Id). TTK is expressed in some cancer cell lines and tumors relative to normal counterparts (Id.; see also WO 02/068444 Al).
Therefore, agents which inhibit a protein kinase, in particular TTK, have the potential to treat cancer. There is a need for additional agents which can act as protein kinase inhibitors, in particular TTK inhibitors.
In addition, cancer recurrence, drug resistance or metastasis is one of the major challenges in cancer therapies. Cancer patients who responded favorably to the initial anticancer therapy often develop drug resistance and secondary tumors that lead to the relapse of the disease. Recent research evidences suggest that the capability of a tumor to grow and propagate is dependent on a small subset of cells within the tumor. These cells are termed tumor-initiating cells (TICs) or cancer stem cells. It is thought that the TICs are responsible for drug resistance, cancer relapse and metastasis. Compounds that can inhibit the growth and survival of these tumor-initiating cells can be used to treat cancer, metastasis or prevent recurrence of cancer. Therefore, a need exists for new compounds that can inhibit the growth and survival of tumor- imitating cells.
PATENT
WO 2015070349
A4: N-cyclopropyl-4-(7-( (((Is, 3s)-3-hydroxy-3-methylcyclobutyl)methyl)amino)-5- (pyridin-3-yloxy)pyrazolol 1 , 5-a ]pyrimidin-3-yl)-2-methylbenzamide hydrochloride and its free base
A). Through Boc deprotection: A mixture of tert-butyl (3- bromo-5-(pyridin-3-yloxy)pyrazolo[l,5-a]pyrimidin-7- yl)(((ls,3s)-3-((tert-butoxycarbonyl)oxy)-3- methylcyclobutyl)methyl)carbamate (0.23 g, 0.38 mmol), N- cyclopropyl-2-methyl-4-(4,4,5,5-tetramethyl-l,3,2- 
dioxaborolan-2-yl)benzamide (0.15 g, 0.49 mmol), PdC dppfDCM (0.15 g, 0.49 mmol), and 2M K3P04 (0.57 mL, 1.14 mmol) in THF (4 mL) was charged with Ar and heated in the microwave at 130 °C for 3 h. Water and EtOAc were added to separate the phases and the aqueous phase was extracted with EtOAc. The combined organic extracts were dried over NaSC>4, filtered and concentrated. The crude product was purified by flash chromatography (gradient: EtOAc/hex 20-60%) to give a yellow oil.
The above intermediate was dissolved in DCM (10 mL) and treated with TFA (3 mL) at rt for 3 h. After reaction completion, solvent was removed in vacuo and the crude product was dissolved in MeOH (5 mL). The mixture was filtered and purified by prep-HPLC. The compound was passed through a PoraPak cartridge and triturated with Et20 to give the title compound as a free base (white solid). The free base was dissolved in MeOH (5 mL), and HC1 (1 M Et20, 2 equiv) was then added slowly. Solvent was removed in vacuo to give the title compound as a beige solid in HC1 salt (96 mg, 47% over 2 steps). ¾ NMR (400 MHz, CD3OD) δ ppm 9.14 (br. s, 1H), 8.89-8.82 (m, 1H), 8.79-8.71 (m, 1H), 8.40 (s, 1H), 8.31-8.21 (m, 1H), 7.68 (s, 1H), 7.59 (d, J = 9.5 Hz, 1H), 7.23 (d, J= 8.0 Hz, 1H), 6.06 (s, 1H), 3.56 (d, J= 6.5 Hz, 2H), 2.88-2.79 (m, 1H),
2.40-2.31 (m, 1H), 2.29 (s, 3H), 2.26-2.18 (m, 2H), 1.99-1.89 (m, 2H), 1.37 (s, 3H),
0.85-0.76 (m, 2H), 0.63-0.53 (m, 2H); MS ESI [M + H]+ 499.3, calcd for [C^HsoNeOs +
H]+ 499.2. HPLC purity: 99.5% at 254 nm.
B). Through PMB deprotection: A mixture of N- cyclopropyl-4-(7-((((ls,3s)-3-hydroxy-3- methylcyclobutyl)methyl)(4-methoxybenzyl)amino)-5- (pyridin-3-yloxy)pyrazolo[l,5-a]pyrimidin-3-yl)-2- methylbenzamide (9.6 g, 15.5 mmol), TFA (50 mL) in DCE
(70 mL) was heated in an oil bath at 50 °C for 4 h. After reaction completion, solvent was removed in vacuo and the crude product was dissolved in a mixture of MeOH/DCM (100 mL/25 mL). 2M Na2CC (150 mL) was then added and the resulting mixture was stirred at rt for 30 min. The reaction mixture was diluted with DCM and the phases were separated. The aqueous phase was extracted with DCM and the combined organic extracts were washed with water, dried over MgSC , filtered and concentrated. The crude product was triturated and sonicated in a mixture of DCM/Et20 (10 mL/70 mL) to give the title compound as a off white solid in free base (5.9 g, 77%). Ti NMR (400 MHz, CD3OD) δ ppm 8.58-8.53 (m, 1H), 8.50-8.46 (m, 1H), 8.36 (s, 1H), 7.86-7.80 (m, 1H), 7.76-7.72 (m, 1H), 7.61-7.55 (m, 2H), 7.18 (d, J = 8.0 Hz, 1H), 5.92 (s, 1H), 3.52 (d, J = 6.8 Hz, 2H), 2.86-2.77 (m, 1H), 2.38-2.28 (m, 1H), 2.25 (s, 3H), 2.24-2.18 (m, 2H), 1.99-1.88 (m, 2H), 1.37 (s, 3H), 0.84-0.75 (m, 2H), 0.64-0.54 (m, 2H); MS ESI [M + H]+ 499.2, calcd for [CzsHsoNgOs + H]+ 499.2. HPLC purity: 96.1% at 235 nm.
PATENT
WO 2014075168
PAPER
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00485

This work describes a scaffold hopping exercise that begins with known imidazo[1,2-a]pyrazines, briefly explores pyrazolo[1,5-a][1,3,5]triazines, and ultimately yields pyrazolo[1,5-a]pyrimidines as a novel class of potent TTK inhibitors. An X-ray structure of a representative compound is consistent with 11/2 type inhibition and provides structural insight to aid subsequent optimization of in vitro activity and physicochemical and pharmacokinetic properties. Incorporation of polar moieties in the hydrophobic and solvent accessible regions modulates physicochemical properties while maintaining potency. Compounds with enhanced oral exposure were identified for xenograft studies. The work culminates in the identification of a potent (TTK Ki = 0.1 nM), highly selective, orally bioavailable anticancer agent (CFI-402257) for IND enabling studies.
Discovery of Pyrazolo[1,5-a]pyrimidine TTK Inhibitors: CFI-402257 is a Potent, Selective, Bioavailable Anticancer Agent
REFERENCES
Discovery of Pyrazolo[1,5-a]pyrimidine TTK Inhibitors: CFI-402257 is a Potent, Selective, Bioavailable Anticancer Agent
Yong Liu, Radoslaw Laufer, Narendra Kumar Patel, Grace Ng, Peter B. Sampson, Sze-Wan Li, Yunhui Lang, Miklos Feher, Richard Brokx, Irina Beletskaya, Richard Hodgson, Olga Plotnikova, Donald E. Awrey, Wei Qiu, Nickolay Y. Chirgadze, Jacqueline M. Mason, Xin Wei, Dan Chi-Chia Lin, Yi Che, Reza Kiarash, Graham C. Fletcher, Tak W. Mak, Mark R. Bray, and Henry W. Pauls
Publication Date (Web): May 6, 2016 (Letter)
DOI: 10.1021/acsmedchemlett.5b00485
////TTK inhibitors, CFI-402257, pyrazolo[1,5-a]pyrimidines, 11/2 type inhibitors, 1610759-22-2, 1610677-37-6
C[C@]1(O)C[C@H](C1)CNc2cc(nc3c(cnn23)c5ccc(C(=O)NC4CC4)c(C)c5)Oc6cccnc6
Antimycobacterial Agents

Styryl Hydrazine Thiazole Hybrids
Will be updated………kindly email amcrasto@gmail.com
DATA
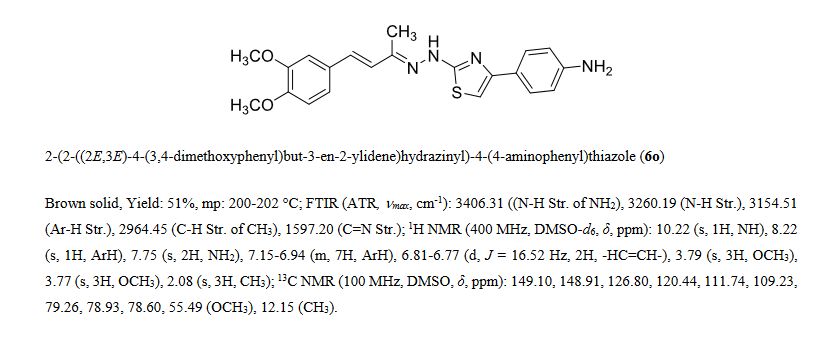
ABOUT Dehydrozingerone

Dehydrozingerone; Feruloylmethane; 1080-12-2; 4-(4-Hydroxy-3-methoxyphenyl)-3-buten-2-one; 4-(4-hydroxy-3-methoxyphenyl)but-3-en-2-one; Vanillalacetone;
http://pubs.acs.org/doi/abs/10.1021/np300465f

Dehydrozingerone (1) is a pungent constituent present in the rhizomes of ginger (Zingiber officinale) and belongs structurally to the vanillyl ketone class. It is a representative of half the chemical structure of curcumin (2), which is an antioxidative yellow pigment obtained from the rhizomes of turmeric (Curcuma longa). Numerous studies have suggested that 2 is a promising phytochemical for the inhibition of malignant tumors, including colon cancer. On the other hand, there have been few studies on the potential antineoplastic properties of 1, and its mode of action based on a molecular mechanism is little known. Therefore, the antiproliferative effects of1 were evaluated against HT-29 human colon cancer cells, and it was found that 1 dose-dependently inhibited growth at the G2/M phase with up-regulation of p21. Dehydrozingerone additionally led to the accumulation of intracellular ROS, although most radical scavengers could not clearly repress the cell-cycle arrest at the G2/M phase. Furthermore, two synthetic isomers of1 (iso-dehydrozingerone, 3, and ortho-dehydrozingerone, 4) were also examined. On comparing of their activities, accumulation of intracellular ROS was found to be interrelated with growth-inhibitory effects. These results suggest that analogues of 1 may be potential chemotherapeutic agents for colon cancer
PAPER

Series of styryl hydrazine thiazole hybrids inspired from dehydrozingerone (DZG) scaffold were designed and synthesized by molecular hybridization approach. In vitro antimycobacterial activity of synthesized compounds was evaluated against Mycobacterium tuberculosis H37Rv strain. Among the series, compound 6o exhibited significant activity (MIC = 1.5 μM; IC50 = 0.48 μM) along with bactericidal (MBC = 12 μM) and intracellular antimycobacterial activities (IC50 = <0.098 μM). Furthermore, 6o displayed prominent antimycobacterial activity under hypoxic (MIC = 46 μM) and normal oxygen (MIC = 0.28 μM) conditions along with antimycobacterial efficiency against isoniazid (MIC = 3.2 μM for INH-R1; 1.5 μM for INH-R2) and rifampicin (MIC = 2.2 μM for RIF-R1; 6.3 μM for RIF-R2) resistant strains of Mtb. Presence of electron donating groups on the phenyl ring of thiazole moiety had positive correlation for biological activity, suggesting the importance of molecular hybridization approach for the development of newer DZG clubbed hydrazine thiazole hybrids as potential antimycobacterial agents.
Dehydrozingerone Inspired Styryl Hydrazine Thiazole Hybrids as Promising Class of Antimycobacterial Agents
IF YOU HAVE ENJOYED IT ………EMAIL ME amcrasto@gmail.com, +919323115463, India

INDIA FLAG

DR ANTHONY CRASTO , WORLDDRUGTRACKER, HELPING MILLIONS, MAKING INDIA AND INDIANS PROUD
///////Antimycobacterial activity, bactericidal, dehydrozingerone, NIAID, thiazole, PRECLINCAL
c1(ccc(c(c1)OC)OC)/C=C/C(C)=N/Nc2nc(cs2)c3ccc(cc3)N
FDA approved a switchover from batch to the new technology for production of HIV drug Prezista, Darunavir on a line at its plant in Gurabo, Puerto Rico

Above is an Illustration example,
FDA urges companies to get on board with continuous manufacturing
The FDA gave Johnson & Johnson’s ($JNJ) Janssen drug unit the thumbs up last week for the continuous manufacturing process that it has been working on for 5 years. The agency approved a switchover from batch to the new technology for production of HIV drug Prezista on a line at its plant in Gurabo, Puerto Rico……http://www.fiercepharma.com/manufacturing/fda-urges-companies-to-get-on-board-continuous-manufacturing
 |
|
 |
SEE……http://www.en-cphi.cn/news/show-29367.html
Just after opening a refurbished manufacturing facility in Cape Town, South Africa earlier this year, pharma giant Johnson & Johnson ($JNJ) recently opened the doors to its Global Public Health Africa Operations office there.
The company has invested $21 million (300 million rand) in the facilities. The global public health facility will focus on HIV, tuberculosis and maternal, newborn and child health, South Africa – The Good News reported.
“This (investment) tells us that South Africa has the capability to provide a facility for world-class manufacturing,” Rob Davies, minister of the Department of Trade and Industry told the publication.
Johnson & Johnson, which has operated in South Africa for more than 86 years, planned to close the Cape Town manufacturing plant by the end of 2008 but was persuaded to keep the facility open for local manufacturing to serve sub-Saharan business. By 2015, the plant was cited by J&J as the most-improved in cost competitiveness from 30 company plants worldwide.
Earlier this month, the FDA gave J&J’s Janssen drug unit the go-ahead to proceed with the continuous manufacturing process it’s been working on for 5 years. The agency approved a switchover from batch to the new technology for production of HIV drug Prezista, Darunavir on a line at its plant in Gurabo, Puerto Rico.

AN EXAMPLE NOT RELATED TO DARUNAVIR
References
International Symposium on Continuous Manufacturing of Pharmaceuticals
Implementation, Technology & Regulatory
![]()
![]()
May 20-21, 2014 (Link to 2016 Meeting Website)
Continuous Bioprocessing
https://iscmp.mit.edu/white-papers/white-paper-4
READ
Achieving Continuous Manufacturing: Technologies and Approaches for Synthesis, Work-Up and Isolation of Drug Substance
https://iscmp.mit.edu/white-papers/white-paper-1
//////
//////FDA, HIV drug, Prezista, Darunavir, Gurabo, Puerto Rico
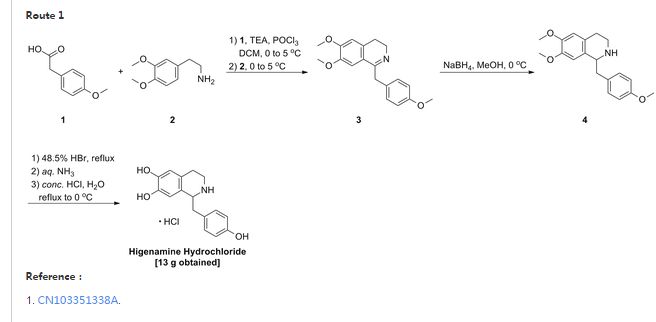
Higenamine Hydrochloride


Higenamine Hydrochloride
- 6,7-Isoquinolinediol, 1,2,3,4-tetrahydro-1-[(4-hydroxyphenyl)methyl]-, hydrochloride (9CI)
- 6,7-Isoquinolinediol, 1,2,3,4-tetrahydro-1-[(4-hydroxyphenyl)methyl]-, hydrochloride, (±)-
- (±)-Demethylcoclaurine hydrochloride
NDA Filed in china
A β-adrenoceptor partial agonist potentially for the treatment of coronary heart disease.
![]()

CAS No.11041-94-4 (Higenamine hydrochloride)

CAS 5843-65-2(free)
Higenamine (norcoclaurine) is a chemical compound found in a variety of plants including Nandina domestica (fruit), Aconitum carmichaelii (root), Asarum heterotropioides, Galium divaricatum (stem and vine), Annona squamosa, and Nelumbo nucifera (lotus seeds).
Legality
Higenamine, also known as norcoclaurine HCl, is legal to use within food supplements in the UK, EU, the USA and Canada. but banned use in The NCAA. Its main is within food supplements developed for weight management, also known as ‘fat burners’. However, products containing (or claiming to contain) pharmacological relevant quantities still require registration as a medicine. The regulatory boundaries for higenamine are unclear as modern formulations have not been clinically evaluated. Traditional formulations with higenamine have been used for thousands of years within Chinese medicine and come from a variety of sources including fruit and orchids. There are no studies comparing the safety of modern formulations (based on synthetic higenamine) with traditional formulations. Nevertheless, it will not be added to the EU ‘novel foods’ catalogue, which details all food supplements that require a safety assessment certificate before use.[1]

Pharmacology
Since higenamine is present in plants which have a history of use in traditional medicine, the pharmacology of this compound has attracted scientific interest. A variety of effects have been observed in in vitro studies and in animal models, but its effects in humans are unknown.
The results of a 2009 study exposed the compound as a β2 adrenergic receptor agonist.[2]
In animal models, higenamine has been demonstrated to be a β2 adrenoreceptor agonist.[2][3][4][5][6] Adrenergic receptors, or adrenoceptors, belong to the class of G protein–coupled receptors, and are the most prominent receptors in the adipose membrane, besides also being expressed in skeletal muscle tissue. These adipose membrane receptors are classified as either α or β adrenoceptors. Although these adrenoceptors share the same messenger, cyclic adenosine monophosphate (cAMP), the specific transduction pathway depends on the receptor type (α or β). Higenamine partly exerts its actions by the activation of an enzyme,adenylate cyclase, responsible for boosting the cellular concentrations of the adrenergic second messenger, cAMP.[7]
In a rodent model, it was found that higenamine produced cardiotonic, vascular relaxation, and bronchodilator effects.[8][9] In particular, higenamine, via a beta-adrenoceptor mechanism, induced relaxation in rat corpus cavernosum, leading to improved vasodilation and erectile function.
Related to improved vasodilatory signals, higenamine has been shown in animal models to possess antiplatelet and antithrombotic activity via a cAMP-dependent pathway, suggesting higenamine may contribute to enhanced vasodilation and arterial integrity.[2][7][9][10]
Toxicity
Regarding toxicity, researchers have suggested that the levels of higenamine reported in food consumption (estimated 47.5 mg in a 9-ounce serving of Lotus) would be comparable to the amount used in food supplements.[citation needed] Higenamine is a beta-adrenergic agonist which has effects on the function of trachea and heart muscles.[11][12]During a study of acute toxicity, mice were orally administered the compound at a dose of 2 g per kg of bodyweight. No mice died during the study.[13] higenamine is one of the main chemicals in a plant called aconite. Aconite has been shown to cause serious heart-related side effects including arrhythmias and even death. in some sources of HIGENAMINE from certain plants that have Aconite
PAPER
Chemical & Pharmaceutical Bulletin (1978), 26(7), 2284-5
https://www.jstage.jst.go.jp/article/cpb1958/26/7/26_7_2284/_pdf
PATENT
CN 103554022
http://google.com/patents/CN103554022B?cl=en
Example 1:
[0024] to the S-necked flask 200mL of anhydrous ammonia clever four furans, lOg instrument crumbs, olive mix was added 0. 5g ship, continue to embrace the mix was added 10 minutes after which 2 drops of 1,2-B burning desert, Continue mixing until the reaction mixture embrace color disappeared, the reaction was cooled to square ° C, and slowly mixed solution thereto 31. 6g4- methoxy Desert Festival and 50mL tetraammine clever furans dropped, about 60min addition was complete, the reaction fluid continues to cool to -65 ° C, to which was slowly dropping 20 percent, 7-dimethoxy-3,4-diamine different wow beep and a mixed solution of ammonia lOOmL four clever furans, the addition was complete continue to maintain – 65 ° C for 2 hours after the embrace slowly warmed 0 ° C, maintaining the internal temperature of 100 ° C 〇 blood slowly added to the reaction mixture, the addition was completed adding 200 blood continues to embrace mixed with ethyl acetate after 0.5 hours, allowed to stand liquid separation, organic phase was separated, dried over anhydrous sulfate steel, concentrated to afford 6, 7-dimethoxy -l- (4- methoxy section yl) -1,2, 3, 4-isopropyl tetraammine wow toot 24. 9g, a yield of 76.1%.
[00 Qiao] to the reaction flask prepared above 6, 7-dimethoxy -l- (4- methoxybenzyl) -1,2, 3, 4 tetraammine different wow beep 24. 9g , 47% aqueous ammonia desert 200 blood acid heated to 130 ° C reflux of cooled to room temperature, precipitation of large amount of solid, filtered higenamine ammonia salt desert, the solid was added 1. of water and continue to add 50 Blood mixed with ammonia football ground, filtered higenamine to higenamine was added lL4mol / L aqueous hydrochloric acid, 80 ° C heat to embrace mixed, cooled to 25 ° C filtration and drying to obtain a final product hydrochloric acid higenamine 11. 7g, a yield of 73.3%.
References
- http://ec.europa.eu/food/food/biotechnology/novelfood/novel_food_catalogue_en.htm
- Tsukiyama, M; Ueki, T; Yasuda, Y; Kikuchi, H; Akaishi, T; Okumura, H; Abe, K (2009). “Beta2-adrenoceptor-mediated tracheal relaxation induced by higenamine from Nandina domestica Thunberg”. Planta Medica 75 (13): 1393–9. doi:10.1055/s-0029-1185743. PMID 19468973.
- Kashiwada, Y; Aoshima, A; Ikeshiro, Y; Chen, YP; Furukawa, H; Itoigawa, M; Fujioka, T; Mihashi, K; et al. (2005). “Anti-HIV benzylisoquinoline alkaloids and flavonoids from the leaves of Nelumbo nucifera, and structure-activity correlations with related alkaloids”.Bioorganic & Medicinal Chemistry 13 (2): 443–8. doi:10.1016/j.bmc.2004.10.020.PMID 15598565.
- Kimura, I; Chui, LH; Fujitani, K; Kikuchi, T; Kimura, M (1989). “Inotropic effects of (+/-)-higenamine and its chemically related components, (+)-R-coclaurine and (+)-S-reticuline, contained in the traditional sino-Japanese medicines “bushi” and “shin-i” in isolated guinea pig papillary muscle”. Japanese journal of pharmacology 50 (1): 75–8.doi:10.1254/jjp.50.75. PMID 2724702.
- Kang, YJ; Lee, YS; Lee, GW; Lee, DH; Ryu, JC; Yun-Choi, HS; Chang, KC (1999). “Inhibition of activation of nuclear factor kappaB is responsible for inhibition of inducible nitric oxide synthase expression by higenamine, an active component of aconite root”. The Journal of Pharmacology and Experimental Therapeutics 291 (1): 314–20.PMID 10490919.
- Yun-Choi, HS; Pyo, MK; Park, KM; Chang, KC; Lee, DH (2001). “Anti-thrombotic effects of higenamine”. Planta Medica 67 (7): 619–22. doi:10.1055/s-2001-17361.PMID 11582538.
- Kam, SC; Do, JM; Choi, JH; Jeon, BT; Roh, GS; Chang, KC; Hyun, JS (2012). “The relaxation effect and mechanism of action of higenamine in the rat corpus cavernosum”.International Journal of Impotence Research 24 (2): 77–83. doi:10.1038/ijir.2011.48.PMID 21956762.
- Bai, G; Yang, Y; Shi, Q; Liu, Z; Zhang, Q; Zhu, YY (2008). “Identification of higenamine in Radix Aconiti Lateralis Preparata as a beta2-adrenergic receptor agonist1”. Acta pharmacologica Sinica 29 (10): 1187–94. doi:10.1111/j.1745-7254.2008.00859.x.PMID 18817623.
- Pyo, MK; Lee, DH; Kim, DH; Lee, JH; Moon, JC; Chang, KC; Yun-Choi, HS (2008). “Enantioselective synthesis of (R)-(+)- and (S)-(-)-higenamine and their analogues with effects on platelet aggregation and experimental animal model of disseminated intravascular coagulation”. Bioorganic & Medicinal Chemistry Letters 18 (14): 4110–4.doi:10.1016/j.bmcl.2008.05.094. PMID 18556200.
- Liu, W; Sato, Y; Hosoda, Y; Hirasawa, K; Hanai, H (2000). “Effects of higenamine on regulation of ion transport in guinea pig distal colon”. Japanese journal of pharmacology 84(3): 244–51. doi:10.1254/jjp.84.244. PMID 11138724.
- Wong, KK; Lo, CF; Chen, CM (1997). “Endothelium-dependent higenamine-induced aortic relaxation in isolated rat aorta”. Planta Medica 63 (2): 130–2. doi:10.1055/s-2006-957628. PMID 9140225.
- Ueki, T; Akaishi, T; Okumura, H; Morioka, T; Abe, K (2011). “Biphasic tracheal relaxation induced by higenamine and nantenine from Nandina domestica Thunberg”. Journal of pharmacological sciences 115 (2): 254–7. doi:10.1254/jphs.10251sc. PMID 21282929.
- Lo, CF; Chen, CM (1997). “Acute toxicity of higenamine in mice”. Planta Medica 63 (1): 95–6. doi:10.1055/s-2006-957619. PMID 9063102.
banned in ncaa https://www.ncaa.org/sites/default/files/2015-16%20NCAA%20Banned%20Drugs.pdf
| CN1539823A * | Oct 27, 2003 | Oct 27, 2004 | 中国医学科学院药物研究所 | Method for preparing new demethyl conclaurine and medinal salt |
| CN1764647A * | Mar 23, 2004 | Apr 26, 2006 | 埃科特莱茵药品有限公司 | Tetrahydroisoquinolyl acetamide derivatives for use as orexin receptor antagonists |
| CN103351338A * | Jun 17, 2013 | Oct 16, 2013 | 张家港威胜生物医药有限公司 | Simple preparation process of higenamine hydrochloride |
| US20060030586 * | Sep 27, 2004 | Feb 9, 2006 | Education Center Of Traditional Chinese Medicine Co. | Method and health food for preventing and/or alleviating psychiatric disorder, and/or for effectuating sedation |
| WO2011038169A2 * | Sep 24, 2010 | Mar 31, 2011 | Mallinckrodt Inc. | One-pot preparation of hexahydroisoquinolines from amides |
 |
|
| Names | |
|---|---|
| IUPAC name
1-[(4-Hydroxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinoline-6,7-diol
|
|
| Other names
norcoclaurine, demethylcoclaurine
|
|
| Identifiers | |
| 5843-65-2 106032-53-5 (R) 22672-77-1 (S) |
|
| ChEBI | CHEBI:18418 |
| ChEMBL | ChEMBL19344 |
| ChemSpider | 102800 |
| Jmol 3D model | Interactive image |
| KEGG | C06346 |
| MeSH | higenamine |
| PubChem | 114840 |
| Properties | |
| C16H17NO3 | |
| Molar mass | 271.32 g·mol−1 |
/////
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

