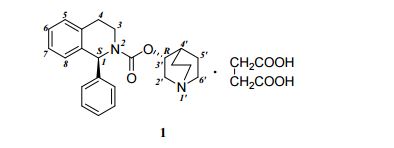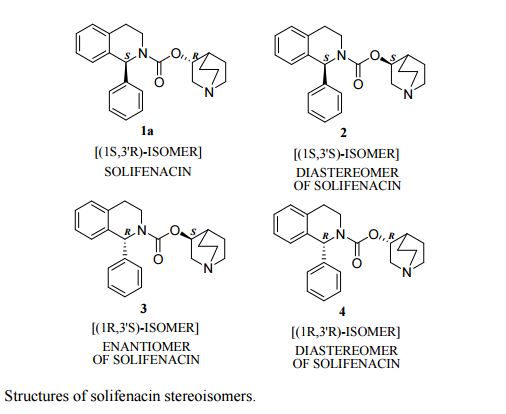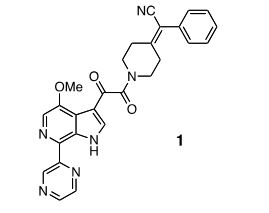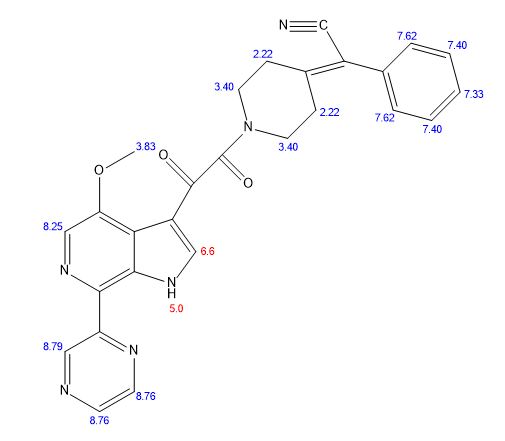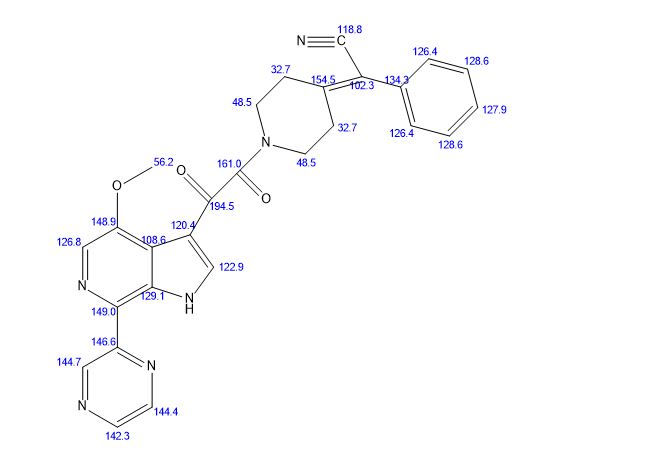
SOLIFENACIN, YM-905
MF C23H26O2, a molecular weigt 362.4647
CAS 242478-37-1
солифенацин [Russian]
سوليفيناسين [Arabic]
索利那新 [Chinese]
242478-37-1 (Solifenacin )
242478-38-2 (Solifenacin Succinate)

Solifenacin succinate, YM-67905
コハク酸ソリフェナシン
| Molecular Formula: |
C11H19NO7S |
| Formula Weight: |
480.56 |
CAS 242478-38-2
Usage Muscarinic M3 receptor antagoinst. Used in treatment of urinary incontinence.
Usage sedative
Usage Solifenacin succinate is a urinary antispasmodic of the antimuscarinic class.
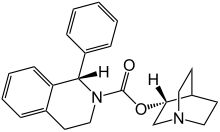
(3R)-l-azabicyclo[2.2.2]oct-3-yl-(lS)-l-phenyl-3,4-dihydroisoquinoline-2-(lH)- carboxylate ((S)-phenyl-l?2,3,4-tetrahydroisoquinoline-2-carboxylic acid 3(R)-quinuclidinyl ester) is known as solifenacin, also known as YM-905 (in its free base form) and YM-67905 (in its succinate form). Solifenacin has the molecular formula C23H26O2, a molecular weight of 362.4647, and the following chemical structure:
C23H26N2O2 Exact Mass: 362.1994
MoI. Wt.: 362.4647 m/e: 362.1994 (100.0%), 363.2028 (25.6%), 364.2061 (3.1%) C, 76.21; H, 7.23; N, 7.73; O, 8.83
Solifenacin succinate is a urinary antispasmodic, acting as a selective antagonist to the M(3)-receptor. It is used as treatment of symptoms of overactive bladder, such as urinary urgency and increased urinary frequency, as may occur in patients with overactive bladder syndrome (OAB), as reviewed in Chilman-Blair, Kim et at., Drugs of Today, 40(4):343 – 353 (2004). Its crystalline powder is white to pale yellowish-white and is freely soluble at room temperature in water, glacial acetic acid, DMSO, and methanol. The commercial tablet is marketed under the trade name VESICAJRE®. As VESICARE®, it was approved by the FDA for once daily treatment of OAB and is prescribed as 5 mg and 10 mg tablets.
The drug was developed by Yamanouchi Pharmaceutical Co. Ltd. and disclosed in US. Patent No. 6,017,927 and its continuation, US. Patent No. 6,174,896.
Solifenacin succinate was first approved by the European Medicines Agency (EMA) on June 8, 2004, then approved by U.S. Food and Drug Administration (FDA) on Nov 19, 2004, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on April 20, 2006. It was developed and marketed as Vesicare® by Astellas.
Solifenacin is a competitive muscarinic receptor antagonist. Muscarinic receptors play an important role in several major cholinergically mediated functions, including contractions of urinary bladder smooth muscle and stimulation of salivary secretion. By preventing the binding of acetylcholine to these receptors, solifenacin reduces smooth muscle tone in the bladder, allowing the bladder to retain larger volumes of urine and reducing the number of micturition, urgency and incontinence episodes. It is indicated for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency, and urinary frequency.
Vesicare® is available as tablet for oral use, containing 5 or 10 mg of Solifenacin succinate. The recommended dose is 5 mg once daily. If the 5 mg dose is well tolerated, the dose may be increased to 10 mg once daily.
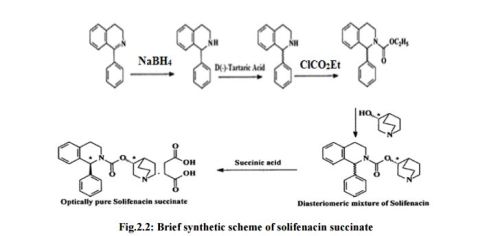
(1S)-3,4-dihydro-1-phenyl-2-(1H)-isoquinolinecarboxylic acid (3R)-1- azabicyclo[2.2.2]oct-3-yl ester. succinate (Solifenacin succinate) (1)
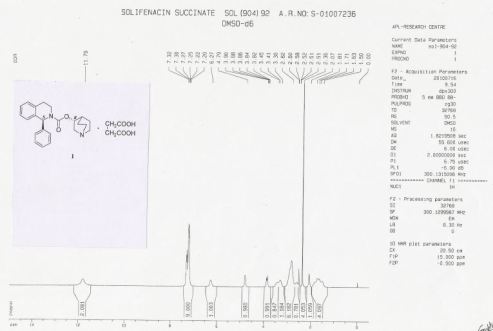
Solifenacin succinate (1) as white crystalline powder. (104.50 g, 87% w/w yield based on in-put).
Chromatographic purity: 99.94 % (by HPLC). Chiral purity: 99.94% (by chiral HPLC). (1S, 3’S)- Diastereomer content: 0.06% (by chiral HPLC).
Mp 145-146 °C.
[α]D 25 (c=1, in Water): + 40.6°.
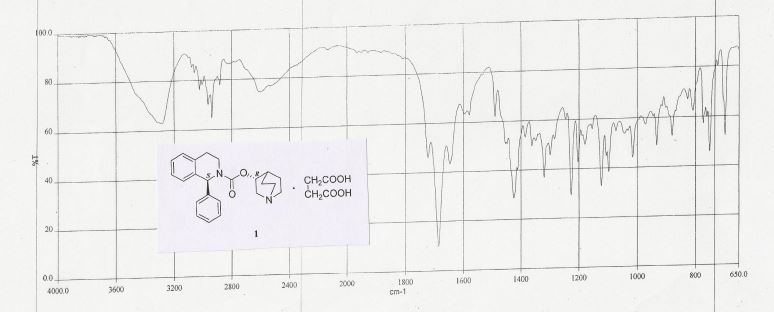
IR (KBr) (cm-1): 3282, 3024, 3007, 2964, 2937, 2881, 2607, 1722, 1685, 1579, 1491, 1227, 761, 751.
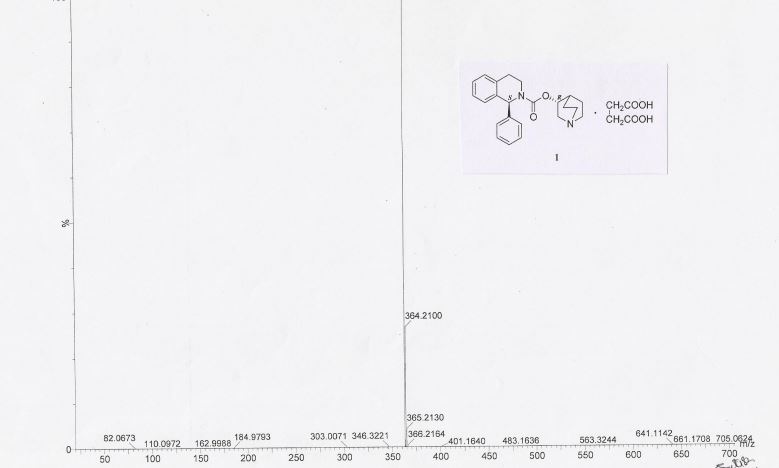
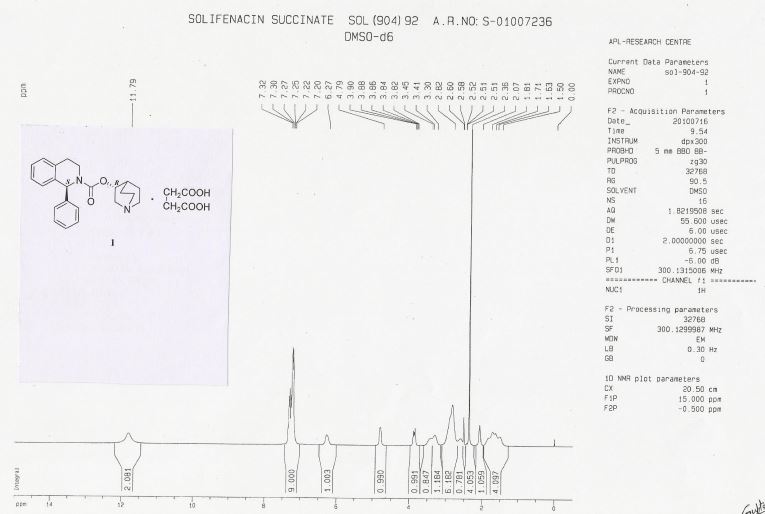
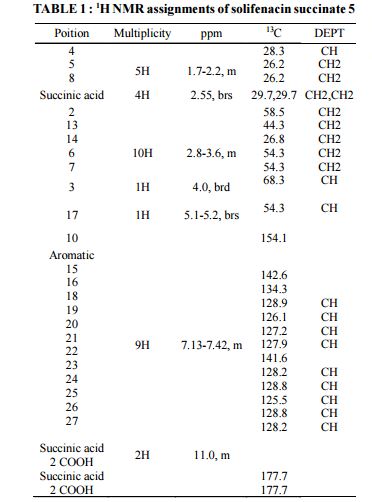
HRMS: m/z = 363.2071 [M + H] + . 1H NMR (DMSO-d6): δ 1.50 – 1.81 (m, 4H), 2.07 (m, 1H), 2.36 (s, 4H), 2.56 – 3.30 (m, 8H), 3.41 & 3.85 (2m, 2H), 4.79 (m, 1H), 6.27 (brs, 1H), 7.20 – 7.32 (m, 9H), 11.79 (brs, 2H).
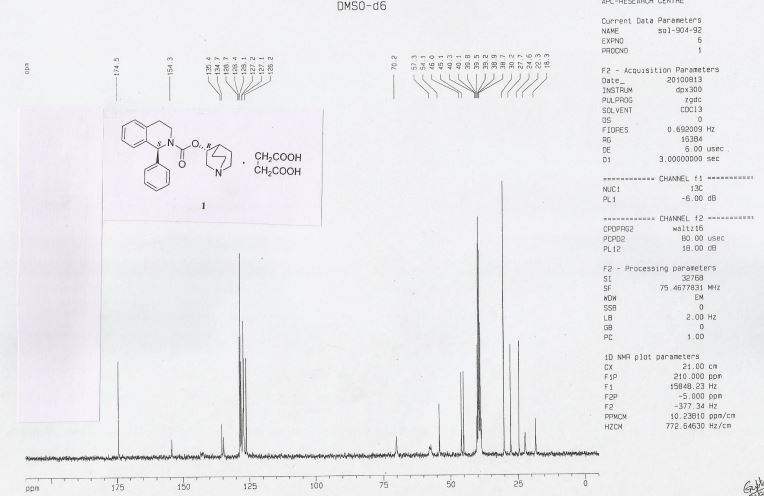
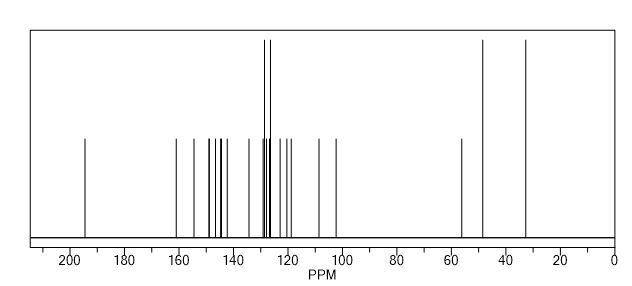
13C NMR (DMSO-d6): δ 18.3 (CH2), 22.3 (CH2), 24.6 (CH), 27.7 (CH2), 30.2 (2xCH2), 38.9 (CH2), 45.1 (CH2), 46.0 (CH2), 54.1 (CH2), 57.3, 70.2, 126.2 (CH), 127.1 (CH), 127.2 (2xCH), 128.1 (CH), 128.4 (2xCH), 128.7 (CH), 134.7, 135.4, 154.3, 174.5.
Solifenacin (INN, trade name Vesicare) is a medicine of the antimuscarinic class and was developed for treating contraction of overactive bladder[1] with associated problems such as increased urination frequency and urge incontinence.[2] It is manufactured and marketed by Astellas, GlaxoSmithKline[3] and Teva Pharmaceutical Industries.
Solifenacin is contraindicated for people with urinary retention, gastric retention, uncontrolled or poorly controlled closed-angle glaucoma, severe liver disease (Child-Pugh class C),[4] and hemodialysis.[2]
Long QT syndrome is not a contraindication although solifenacin, like tolterodine and darifenacin, binds to hERG channels of the heart and may prolong the QT interval. This mechanism appears to be seldom clinically relevant.[5]
Side effects
The most common side effects of solifenacin are dry mouth, blurred vision, and constipation. As all anticholinergics, solifenacin may rarely cause hyperthermia due to decreased perspiration.[4]
Interactions
Solifenacin is metabolized in the liver by the cytochrome P450 enzyme CYP3A4. When administered concomitantly with drugs that inhibit CYP3A4, such as ketoconazole, the metabolism of solifenacin is impaired, leading to an increase in its concentration in the body and a reduction in its excretion.[4]
As stated above, solifenacin may also prolong the QT interval. Therefore, administering it concomitantly with drugs which also have this effect, such as moxifloxacin or pimozide, can theoretically increase the risk of arrhythmia.[3]
Pharmacology
Mechanism of action
Solifenacin is a competitive cholinergic receptor antagonist, selective for the M3 receptor subtype. The binding of acetylcholine to these receptors, particularly M3, plays a critical role in the contraction of smooth muscle. By preventing the binding of acetylcholine to these receptors, solifenacin reduces smooth muscle tone in the bladder, allowing the bladder to retain larger volumes of urine and reducing the number of micturition, urgency and incontinence episodes. Because of a long elimination half life, a once-a-day dose can offer 24-hour control of the urinary bladder smooth muscle tone.[2]
Pharmacokinetics
Peak plasma concentrations are reached 3 to 8 hours after absorption from the gut. In the bloodstream, 98% of the substance are bound to plasma proteins, mainly acidic ones. Metabolism is mediated by the liver enzyme CYP3A4 and possibly others. There is one known active metabolite, 4R-hydroxysolifenacin, and three inactive ones, the N–glucuronide, the N-oxide and the 4R-hydroxy-N-oxide. The elimination half-life is 45 to 68 hours. 69% of the substance, both in its original form and as metabolites, are excreted renally and 23% via the feces.[2]
Chemistry
Like other anticholinergics, solifenacin is an ester of a carboxylic acid containing (at least) an aromatic ring with an alcohol containing a nitrogen atom. While in the prototype anticholinergic atropine the alcohol is tropine, solifenacin has another bicycle, quinuclidinyl alcohol.
The substance is a basic yellow oil, while the form used in tablets, solifenacin succinate, consists of white to slightly yellowish crystals.[6]

Scheme 1 wherein the quinuclidinol reactant is available commercially. The overall synthesis as reported by Mealy, N., et al. in Drugs of the Future, 24 (8): 871-874 (1999) is depicted in Scheme 2:
Scheme 2
U.S. Patent No. 6,017,927 discloses another process for the preparation of solifenacin, wherein 3-quinuclidinyl chloroformate monohydrochloride is admixed with ( IR)-I -phenyl- 1,2,3,4-tetrahydroisoquinoline to obtain solifenacin, as seen below in Scheme 3:
Scheme 3
History
The compound was studied using animal models by the Yamanouchi Pharmaceutical Co., Ltd. of Tokyo, Japan. It was known as YM905 when under study in the early 2000s.[7]
Society and culture
Economics
A 2006 cost-effectiveness study found that 5 mg solifenacin had the lowest cost and highest effectiveness among anticholinergic drugs used to treat overactive bladder in the United States, with an average medical cost per successfully treated patient of $6863 per year.[8]
Chemically, solifenacin succinate is butanedioic acid, compounded with (1S)-(3R)-1-azabicyclo[2.2.2]oct-3-yl 3,4-dihydro-1-phenyl-2(1H)iso-quinolinecarboxylate (1:1) having an empirical formula of C23H26N2O2•C4H6O4, and a molecular weight of 480.55. The structural formula of solifenacin succinate is:
Solifenacin succinate is a white to pale-yellowish-white crystal or crystalline powder. It is freely soluble at room temperature in water, glacial acetic acid, dimethyl sulfoxide, and methanol. Each VESIcare tablet contains 5 or 10 mg of solifenacin succinate and is formulated for oral administration. In addition to the active ingredient solifenacin succinate, each VESIcare tablet also contains the following inert ingredients: lactose monohydrate, corn starch, hypromellose 2910, magnesium stearate, talc, polyethylene glycol 8000 and titanium dioxide with yellow ferric oxide (5 mg VESIcare tablet) or red ferric oxide (10 mg VESIcare tablet).
Paper
http://shodhganga.inflibnet.ac.in/bitstream/10603/71060/10/10_chapter%202.pdf
PAPER
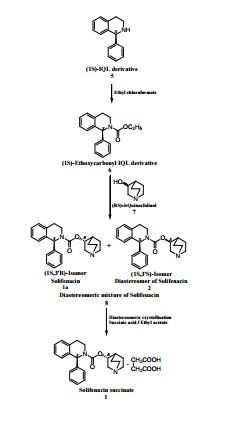
An Improved Process for the Preparation of Highly Pure Solifenacin Succinate via Resolution through Diastereomeric Crystallisation
† Chemical Research and Development, APL Research Center, Aurobindo Pharma Ltd., Survey No. 71 & 72, Indrakaran (V), Sangareddy (M), Medak Dist-502329, Andhra Pradesh, India
‡ Department of Engineering Chemistry, A. U. College of Engineering, Andhra University, Visakhapatnam-530003, Andhra Pradesh, India
Org. Process Res. Dev., 2014, 18 (8), pp 934–940

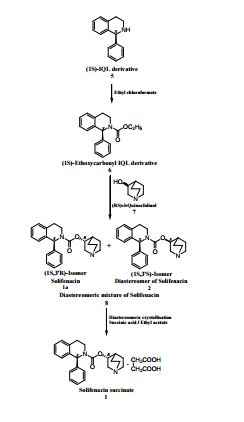
An improved process for the preparation of solifenacin succinate (1) involving resolution through diastereomeric crystallization is described. (1S)-IQL derivative (5) is esterified to form (1S)-ethoxycarbonyl IQL derivative (6) which is condensed with (RS)-3-quinuclidinol (7) to form a solifenacin diastereomeric mixture (8); this is subjected to resolution through diastereomeric crystallization to produce solifenacin succinate (1), which is used for the treatment of an overactive bladder.

CLIP
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
The piperidine scaffold also features in a recently discovered pharmaceutical, namely solifenacin (2.57, Vesicare), a competitive antagonist of the muscarinic acetylcholine receptor used in the treatment of an overactive bladder. This species was co-developed by Astellas and GSK scientists and consists of a chiral hydroisoquinoline linked to a (R)-quinuclidinol unit through a carbamate linkage (Figure 6). Upon protonation the tertiary amine of the quinuclidine is expected to resemble the ammonium substructure of muscarine (2.58) [76].
Figure 6: Structures of solifenacin (2.57) and muscarine (2.58).

This molecule can be prepared by direct coupling of the (R)-quinuclidinol and tetrahydroisoquinoline carbamate partner (Scheme 28). The (R)-quinuclidinol (2.59) itself can be accessed from quinuclidone (2.60), and is most conveniently prepared by alkylation of ethyl isonicotinate (2.61) with ethyl bromoacetate (2.62) followed by full reduction of the pyridine ring therefore yielding the corresponding piperidine 2.63. A base-mediated Dieckmann cyclisation and Krapcho decarboxylation [77] then furnishes 2.60. Traditionally, the reduction of 2.60 to prepare 2.59 can be carried out under fairly mild hydrogenation conditions that ultimately produce racemic quinuclidinol. However, an improved approach makes use of a Noyori-type asymmetric reduction employing a BINAP ligated RuCl2 and a chiral diamine to yield the desired (R)-quinuclidine in high yield and enantioselectivity [78].
The enantioselective synthesis of the tetrahydroisoquinoline fragment is achieved via an asymmetric addition of phenylethylzinc to the imine N-oxide 2.66 yielding the corresponding 3,4-dihydroisoquinoline-N-hydroxide 2.68. Further reductive cleavage of the hydroxylamine moiety followed by activation with 4-nitrophenyl chloroformate [79] yields the intermediate 2.69. In the last step of the sequence the addition of (R)-quinuclidinol generates solifenacin (2.57).
- 76 Broadley, K. J.; Kelly, D. R. Molecules 2001, 6, 142–193.
Return to citation in text:
- 77 Daeniker, H. U.; Grob, C. A. Org. Synth. 1964, 44. doi:10.1002/0471264180.os044.30
Return to citation in text:
- 78 Arai, N.; Akashi, M.; Sugizaki, S.; Ooka, H.; Inoue, T.; Ohkuma, T. Org. Lett. 2010, 12, 3380–3383. doi:10.1021/ol101200z
Return to citation in text:





PATENT
https://www.google.com/patents/EP2489666A2?cl=enhttps://www.google.com/patents/EP2489666A2?cl=en
Solifenacin {1(S)-Phenyl-1,2,3,4-tetrahydroisoquinolin-2-carboxylic acid 3(R)-quinuclidinyl ester or [(3R)-1-azabicyclo[2.2.2]oct-3-yl-(1S)-1-phenyl-3,4-dihydroisoquinoline-2-(1H)-carboxylate]}, also known as YM-905 (in its free base form) has the following structure.

Molecular formula of Solifenacin is C23H26N2O2 and its molecular weight is 362.5. Solifenacin and its salts are used as therapeutic agents for Pollakiuria and incontinence of urine due to hyperactive bladder, not as agents for curing hyperactive bladder itself but as therapeutic agents for suppressing the symptoms thereof.
The drug Solifenacin was first disclosed in US. Patent NoS. 6,017,927and 6,174,896 (CIP of US 6017927 ) (Yamanouchi Pharmaceuticals). Disclosed therein are compounds with the following general formula

A specific method for producing Solifenacin or its HCl salt is also disclosed, as depicted by the following scheme (Scheme-1).

In the reference cited above, the method of preparation of the Solifenacin base using sodium hydride and subsequent conversion of the base to the HCl salt is described, but no data is given for the purity of either the Solifenacin base or the salt. Solifenacin hydrochloride is disclosed particularly in Example-8 of the same patent and crystallization is carried out in a mixture of acetonitrile and diethyl ether. The melting point reported is 212-214 °C. This patent also discloses 3-quinuclidinyl-1-phenyl-1,2,3,4-tetrahydro-2-isoquinoline carboxylate mono oxalate (Example 1) which is the oxalate salt of racemic Solifenacin (M.P. 122-124 °C). The crystallization of the racemate oxalate salt is carried out in a mixture of isopropanol and isopropyl ether.
Polymorphism, the occurrence of different solid state forms, is a property of many molecules and molecular complexes. A single molecular entity may give rise to a variety of solid state forms having distinct crystal structures and physical properties such as melting point, powder X-ray diffraction pattern, infrared (IR) absorption fingerprint and different physicochemical properties. One solid state form may give rise to several polymorphic forms, which are different from one another in all the above properties.
Subsequently, a process for preparation of Solifenacin base and its salts, wherein succinate salt was obtained in high degree of optical purity for medicinal use, was described in EP 1714965 by Astellas Pharma. This document stated that the free base of Solifenacin has the following impurities,


The concentration of the impurities present in the base were as follows:
-
| Compound A |
4.51% |
| Compound B |
2.33% |
| Compound C |
0.14% |
| Compound D |
0.32% |
| Compound E |
1.07% |
This document discloses production of Solifenacin hydrochloride and oxalate-containing composition, respectively in reference examples 2 and 4. It states that the hydrochloride and oxalate salts were also not possible to prepare in pure form and only the succinate salt was obtained in a pure form. It also states that the hydrochloride and oxalate containing composition contains the impurities A and B above (A and B both are chiral impurities), at 0.85% or more and 0.50% or more compared to Solifenacin, respectively, even after salt formation and crystallization steps. Thus, there exists a need to prepare both the HCl and oxalate salts as well as other pharmaceutically acceptable salts of Solifenacin in a form that is chemically and chirally pure, is solid and can be handled on an industrial scale. There also exists a need to prepare chemically pure Solifenacin base.
EP 1726304 more specifically discloses the method of preparation of the Solifenacin using an alkoxide base, which makes the process commercially viable. Compared to the process described in
US 6,017,927 , instead of using sodium hydride having disadvantages like combustion risk and contamination of mineral oil, this method uses an alkoxide base, which overcomes these drawbacks. This document discloses the presence of certain alkylated impurities, which may be present in the Solifenacin base and salts upto 1% concentration.
EP 1757604 discloses four different processes for the preparation of Solifenacin base and the succinate salts.
WO 2008011462 discloses processes for the preparation of Solifenacin base using sodium hydride, and also discloses crystalline form of Solifenacin base and crystalline form of Solifenacin hydrochloride. The salt is prepared as an intermediate step to obtain the succinate salt of Solifenacin in a chemically pure form (purity by HPLC: 99.74%). Nothing is stated about the purity of either the succinate salt or the HCl salt obtained through this process.
WO 2008062282 discloses a process for the preparation of Solifenacin, which is shown below. (Scheme 6).
WO 2008077357 application covers process for preparing Solifenacin using non-nucleophilic base. Reaction of crude Solifenacin base with L-tartaric acid provides crystalline Solifenacin hydrogen tartrate salt, which is then transformed to optically pure base as well as other salts (succinate).
WO2008019055 application discloses process for optical resolution of 1-phenyl-1,2,3,4-tetra hydroisoquinoline, which is one of the key intermediate of Solifenacin.
WO2008019103 discloses amorphous and crystalline forms of Solifenacin base as well as process for preparation of the same. In this document they have disclosed form B
1 of Solifenacin base prepared by slurring amorphous Solifenacin base in DIPE solvent.
WO2008013851 discloses amorphous and crystalline forms-I and II of Solifenacin succinate as well as process for preparation of the same.
WO2008120080 disclosed a process wherein 3(R)-quinuclidinol is activated by reaction with bis [1H-1,2,4-triazol-1-yl]-methanone, and the solution obtained is reacted with 1(S)-phenyl-1,2,3,4-tetrahydroisoquinoline to give Solifenacin.
US 20080114029 discloses new polymorphic form of I(S)-phenyl-1,2,3,4-tetrahydroisoquinoline, a key intermediate for the preparation of Solifenacin base.
Though several processes for preparing both the Solifenacin base as well as hydrochloride and oxalate salts are known, very little is said about the chemical purity of any of them. Most of the processes describe the necessity of formation of the succinate salts for improving the chemical purity. Therefore, there is a need to develop a safe modified process for preparing Solifenacin (free base) which give better yields and improved purity. There also is a requirement to prepare such other new salts of Solifenacin, which not only is chemically and chirally pure but also have superior pharmaceutical properties over one or more of the known salts of Solifenacin. We herein disclose an improved process for preparing Solifenacin base in a pure form (chemically and chirally) and also chemically and chirally pure hydrochloride, oxalate, succinate, gentisate, citrate, hydrobromide, sulphate, nitrate, phosphate, maleate, methane sulphonate, ethane sulphonate, benzene sulphonate, tosylate, α- ketoglutarate, glutarate, nicotinate, malate, 1,5-naphthalene disulfonate and ascorbate salts of Solifenacin. In a preferred embodiment, these salts have atleast 98% purity and may be used to prepare the pure Solifenacin base from the impure base through the intermediate formation of any of these salts. Additionally, several of these salts have superior pharmaceutical properties over one or more known salts of Solifenacin.

Example 1Preparation of (+)-(1S,3’R)-quinuclidin-3′-yl 1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate (Solifenacin)
To the cooled solution of freshly prepared sodium methoxide (1.8 g), (R)-3-quinuclidinol HCl (6.4 g) was added under N2 atmosphere. It was stirred at 5-30 °C for 30 min to 1 hrs. Distilled out the solvent at reduced pressure. To the semi-solid mass dry toluene was added. Reaction mixture was heated to reflux temp. and stirred for 1-3 h. During this process, traces of water and methanol were removed azeotropically by using Dean-Stark apparatus and was cooled to 60-70 °C. (S)- Ethyl 1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-caboxylate (10 g) dissolved in dry toluene and dry DMF were added. It was again heated to reflux temperature and stirred for 5-25 h while distilling off solvent to remove ethanol at intervals with addition of fresh quantity of dry solvent. It was cooled to room temperature.
Workup:
To the reaction mixture water and toluene were added. It was stirred for 10-15 min. and transferred into a separating funnel. Organic layer was collected. The product was extracted with 20 % aqueous HCl solution. It was basified with 40 % aqueous K2CO3 solution at 15-20 °C. The product was extracted with ethyl acetate. Both the extracts were combined and washed with brine solution. Organic layer was collected and dried over anhydrous sodium sulfate and solvent was distilled out at reduced pressure.
(+)-(1S,3’R)-quinuclidin-3′-yl-1-phenyl-1,2,3,4-tetrahydro-isoquinoline-2-carboxylate (Solifenacin) (6.6 g , 51 % yield) was obtained.
% Chemical purity 96.87 %
% Chiral purity by HPLC – 98.83 %.
CLIP

https://www.researchgate.net/figure/235670427_fig2_Figure-6-Scheme5-Synthesis-of-the-chiral-drug-solifenacin
PATENT
WO2014067219A1.

PATENT
https://www.google.com/patents/EP2406257A1?cl=enhttps://www.google.com/patents/EP2406257A1?cl=en

Solifenacin succinate is the international common denomination for butanedioic acid compounded with (l S)-(3R)-l-azabicyclo[2.2.2]oct-3-yl-3,4-dihydro-l-phenyl-2(lH)-isoquinolinecarboxylate (1 : 1), having an empirical formula Of C2SH2ON2O2 .C4H6O4 and the structure is represented in formula VI given below;
Solifeπacin and its pharmaceutically acceptable salts are first reported in US Patent No. 6,017,927 (927′), which disclosed two’ synthetic routes “Route-A and Route-B” for the preparation of (I RS, 3’RS)- Solifenacin and (I S, 3’RS)-Solifenacin as shown in Scheme- 1 :
Scheme 1 : Reported synthetic schemes in US’ 927
Both the routes have several drawbacks such as; a) Use of hazardous and pyrophoric reagent, NaH, in the process which is very difficult to handle and thus makes the process unsafe to handle at industrial level. The use of strong agent NaH also leads to racemization of the products and thus suffers to provide enantiomerically pure Solifencin; b) Use of ethylchloroformate to prepare ethyl carboxylate derivative in route A which is lachrymatory in nature; c) Ethylcarboxylate derivative produces ethanol as a by-product during trans-esterification reaction in the subsequent reaction that interferes in nucleophilic attack against Solifenacin in the presence of a base and hence it is necessary to remove ethanol from the reaction mixture in the form of azeotrope with toluene or the like simultaneously while carrying out the reaction, so as to control the reaction; d) Use of column chromatography for the purification of Solifenacin base, which makes the process industrially not feasible; f) The reaction requires longer time for the completion and hence turn around time of the batch in production makes it less attractive. International Patent Application No WO2005/075474 disclosed another synthetic route for the preparation of Solifenacin and Solifenacin succinate as shown in Scheme-2.

Scheme 2
The above route does not overcome the problems associated with the process disclosed in 927′ as the process described in this scheme also uses ethylchloroformate in the first step and produces ethanol as a by-product in the second step.
Yet another International Patent application no W02005/105795A1 discloses an improved process for preparing Solifenacin as represented in Scheme-3, wherein leaving group (Lv) can be lH-imidazole-1-yl, 2,5-dioxopyrrolidin-l-yloxy, 3-methyl-l H-imidazol-3-ium- l-yl or chloro and further condensation is carried out in the presence of sodium hydride as a base and a mixture of toluene and dimethylformamide or toluene alone as a reaction medium. The process described herein represents few draw backs such as, use of hazardous sodium hydride, use of chromatographic purifications, and use of moisture sensitive leaving groups (Lv) and hence handling of the reaction is difficult. Further the leaving groups used are expensive and thus making the process uneconomic.
Scheme 3 Hence, there is need of efficient process for producing Solifenacin and its succinate salt which is safe to handle, industrially feasible, and economically viable.
Example 1
PREPARATION OF SOLIFENACIN SUCCINATE OF FORMULA (VI);
To a stirred solution of (3R)-quinuclidin-3-ol (25 gm) in dimethylformamide (175 ml) was added bis-(4- dinitrophenyl) carbonate (83.83 gm) with stirring at 25-3O0C under nitrogen atmosphere. The reaction mass was stirred at 25-300C for 2r3. hours. Upon completion of this reaction by HPLC, ( IS)-I -phenyl- 1,2,3,4-tetrahydroisoquinoline (41.0 gm) was added to resultant brown colored reaction solution and further stirred at 25-3O0C for 3-4 hrs. After completion of the reaction (monitored by HPLC), the reaction solution was diluted with water (250 ml) and the pH of the solution was adjusted to 1-2 using concentrated hydrochloric acid. The resulting reaction solution was extracted with diisopropylether (300ml X 2) to separate the nitro-phenol.
The aqueous layer was then extracted with dichloromethane (300 ml) and dichloromethane layer was separated and diluted with 200 ml water. The pH of the biphasic mixture was adjusted to 9-10 with ammonium hydroxide and organic layer was separated, washed with water (200 ml X 2), and concentrated under vacuum to yield 57.0 gm (79%) of compound I as a syrup having HPLC purity of 98.8% and Chiral purity of 99.9%: Compound (I) was further dissolved in acetone (400 ml) and contacted with succinic acid (18.58 gm) at 25-300C. and stirred for 30 min. Precipitated solid was filtered, washed with acetone (57 ml), and dried under vacuum to yield 53.0 gm solifenacin succinate of formula (VI) as a white crystalline solid; HPLC purity 99.93%; Chiral purity : 99.98%;
The ether layer comprising nitro-phenol was subjected to vacuum distillation to recover diisopropylether and nitro-phenol.
Example 2
PREPARATION OF SOLIFENACIN SUCCINATE OF FORMULA (VI);
To a stirred solution of (3R)-quinuclidin-3-ol (5 gm) in dry pyridine (30 ml) was added bis-(4- dinitrophenyl) carbonate (17.5 gm) with stirring at 25-3O0C under nitrogen atmosphere. The reaction mass was stirred at 25-300C for 2-3 hours. Upon completion of the reaction by HPLC, ( IS)-I -phenyl – 1,2,3,4-tetrahydroisoquinoline (7.5 gm) was added to resultant brown colored reaction solution and further stirred at 25-3O0C for 3-4 hrs. After completion of the reaction (monitored by HPLC), the reaction solution was diluted with water (100 ml) and the pH of the solution was adjusted to 1-2 using concentrated hydrochloric acid. The resulting reaction solution was extracted with diisopropylether (60 ml X 2) to separate the nitro-phenol.
The aqueous layer was then extracted with dichloromethane (60 ml), and dichloromethane layer was separated and diluted with 40 ml of water. The pH of the biphasic mixture was adjusted to 9-10 with ammonium hydroxide and organic layer was separated, washed with water (40 ml X 2), and concentrated under vacuum to yield 10.0 gm (70.8%) of solifenacin of formula (I) as a syrup having HPLC purity of 97.9% and Chiral purity of 99.96%: Compound (I) was further dissolved in acetone (70 ml) and contacted with succinic acid (3.25 gm) at 25-300C. and stirred for 30 min. Precipitated solid was filtered, washed with acetone (10 ml), and dried under vacuum to yield 8.5.0 gm solifenacin succinate of formula (VI) as a white crystalline solid; HPLC purity 99.78%; Chiral purity : 99.96%;
Example 3
PREPARATION OF SOLIFENACIN SUCCINATE OF FORMULA (VI):
(3/?)-quinuclidin-3-ol (1.0 gm) of was dissolved in tetrahydrofuran (15 ml) and dry pyridine (1.0 ml) with stirring. Bis-(4-dinitrophenyl) carbonate (3.82 gm) was added to the above solution at 25-300C. After completion of the reaction, (lS)-l-phenyl-l,2,3,4-tetrahydroisoquinoline (1.5 gm) was added to the resulting brown reaction solution and then stirred till completion of the reaction. Upon completion of the reaction, the reaction solution was diluted with water (20 ml) and the pH of the solution was adjusted to 1 -2 using concentrated hydrochloric acid. The resulting solution was extracted with diisopropylether (12.0 ml X 2) to separate the nitro-phenol.
The aqueous layer was separated and further extracted with dichloromethane (12 ml X 2). The dichloromethane layer was diluted with water (8 ml) and pH of the resulting mixture was adjusted to 9-10 using ammonium hydroxide solution. The aqueous layer was separated from organic layer, washed with water (8 ml x 2) and concentrated to yield 1.5 gm (53.5%) solifenacin of Formula (I) having HPLC purity 96.47% ; chiral purity 99.10%; Compound (I) was dissolved in acetone (10.5 ml) and treated with 0.48 gm succinic acid at 25-300C, and stirred for 30 minutes. The precipitated solid was filtered, washed with 1.0 ml acetone, and solid dried under vacuum yield 1.4 gm of compound VI having HPLC purity 99.86%; chiral purity: 99.93%.
Example 4
PREPARATION OF (3^-l-AZABICYCLO[2.2.21OCT-3-YL4-NITROPHENYL CARBONATE
OF FORMULA (TV);
To a stirred solution of (3i?)-quinuclidin-3-ol (1.0 gm) in dichloromethane (10 ml) was added Bis-(4- dinitrophenyl) carbonate (2.87 gm) at 25-300C and the resulting brown solution was stirred at ambient temperature till the completion of reaction by HPLC. Dichloromethane was distilled off to get the residue that was diluted with water (10 ml) and was added concentrated hydrochloric acid till pH of the mixture is I to 2. The acidic solution was extracted with di-isopropylether (10 ml X 2) to separate out the nitro- phenol. The aqueous layer was then extracted with dichloromethane (20 ml) to separate the compound of formula (IV). The dicloromethane layer- comprising the compound of formula (IV) was further mixed with water (10ml) and pH was adjusted to 9-10 with ammonium hydroxide. The organic layer was then separated, washed with water, dried over sodium sulphate, and concentrated under vacuum to yield (3R)-I- azabicyclo[2.2.2]oct-3-yl4-nitrophenyl carbonate of formula (IV) as a syrup with around 46% yield (1.07 gm); HPLC purity: 87.27% by HPLC.
Example 5
PREPARATION OF SOLIFENACIN SUCCINATE OF FORMULA (VI)
To a stirred solution of (3R)-l-azabicyclo[2.2.2]oct-3-yl4-nitrophenyl carbonate (1.0 gm) of formula (IV) obtained as per Example 4 in pyridine (5 ml), (lS)-l-phenyl-l,2,3,4-tetrahydroisoquinoline (0.78 gm) was added and the resulting brown solution was stirred for 6 hrs. After completion of the reaction the solvent was distilled off and the residue obtained was diluted with 10 ml water, the pH of the resulting solution was adjusted to 1-2 using the concentrated hydrochloric acid and extracted with di-isopropylether (10 ml X 2) to separate out the nitro-phenol.
The aqueous layer was separated and further extracted with dichloromethane (20 ml) and obtained dichloromethane layer was mixed with water (10 ml) and pH of the resulting mixture was adjusted to 9- 10 using ammonium hydroxide. Layers were separated, the organic layer was washed with water, dried over sodium sulphate, and concentrated in vacuum to yield the 1.07 gm (89.43%) of compound solifenacin of formula (I) having HPLC purity 97.08% purity
Example 6
PREPARATION OF SOLIFENACIN SUCCINATE OF FORMULA (VD.
To a stirred solution of (3R)-quinuclidin-3-ol (Formula II, 100 gm) in dimethylformamide (400 ml), bis- (4-dinitrophenyl) carbonate (Formula III, 285.04 gm) was added with stirring at 25-30°C under nitrogen atmosphere. The reaction mass was stirred at 25-30°C for 2-3 hours. After completion of the reaction which was monitored by TLC, ( IS)-I -phenyl- 1, 2,3, 4-tetrahydroisoquinoline (Formula V, 171.44 gm) was added to the resultant brown colored reaction solution. The reaction mixture was further stirred at 25- 3O0C for 3-4 hrs. After completion of the reaction (by HPLC), the reaction solution was diluted with water (1000 ml) and the pH of the solution was adjusted to 1-2 using concentrated hydrochloric acid. The resulting reaction solution was extracted with diisopropylether (1000ml X 2) to separate the nitro-phenol. The aqueous layer was then mixed with dichloromethane (1000ml), the content was stirred, and dichloromethane layer was separated. Aqueous layer was re-extracted with dichloromethane (l OOOml).The combined dichloromethane was distilled off completely to obtain the residue. The residue was dissolved in water (1000 ml) and toluene (1000 ml) was added and the pH of the biphasic mixture was adjusted to 9-10 with ammonium hydroxide. The mixture was stirred and toluene layer was separated and aqueous layer was re-extracted with toluene (1000 ml). The combined toluene layer were washed with water (1000 ml) followed by solution of 0.5% sodium hydroxide (1000 ml X 2) and further washed with water (1000 ml). The toluene layer was distilled off completely to obtain the residue which was further dissolved in acetone (800 ml) and toluene 1080 ml). The solution was treated with Succininc acid (88.0 gm) and the mixture obtained was heated at 55-600C for 30 min. The mixture was further cooled to 10-150C, maintained for 60 min and filtered. The product was dried to afford Solifenacin Succinate (Formula VI) as white crystalline solid. Yield of the compound VI270 gm. HPLC purity : 99.85%: Chiral Purity: 99.99%
Example 7
Purification process for Solifenacin Succinate:
The wet material obtained from the example 6 was purified to improve chiral and chemical purity.. The wet material (270 gm) was dissolved in a mixture of water (700 ml) and toluene (700 ml) and stirred for 15 min. The pH of resulting mixture was adjusted to 9-10 using aqueous ammonia, stirred for 15-20 min and separated organic and aqueous layer. Aqueous layer was re-extracted with toluene (700 ml) and combined with the separated organic layer. The combined organic layer was washed with water (700 ml x 2) and distilled off completely to obtain the thick residue. The residue was dissolved in acetone (1600 ml), decolorized with activated charcoal, and treated with succininc acid (75.0 gm). The contents were heated at 55-600C for 30 mih, cooled to 10-15°C, and maintained for 60 min. The crystalline solid obtained was filtered, and dried under vacuum (650-700 mm/Hg to afford Solifenaicn Succinate (Formula VI) as white crystalline solid. Yield: 250 gm (66.6%); HPLC purity: 99.95% and Chiral purity: 100.0%
PAPER
Org. Lett. 2010, 12, 2690-2693.
PATENT
https://www.google.com/patents/WO2007076116A2?cl=en
Scheme 4:
3-Quinuclidinol χ= halogen,
R=alkyl
Scheme 5
EXAMPLES Example 1 : Preparation of solifenacin succinate
A solution of (S)-l-phenyl-l,2,3,4-tetrahydroisoquinoline (C15H15N) (16g), toluene (80ml), and diisopropylethylamine (DIPEA, 13.5g) was cooled to 0°C. Chloroethylchloro formate (C3H4CbO2) (CECF, 13.0gr) was added dropwise, keeping the temperature between 0°-20°C. After stirring at room temperature for 1.5 hours, the mixture was filtered.
The filtrate was added to solution of (R)-quinuclidin-3-ol (C7Hi3NO) (11.6g) in toluene (80ml), DMF (16ml), and NaH (60%, 5.5g) at 80°C during 1 hour, and stirred at 95°-100°C for 17 hours. The mixture was cooled to room temperature, and THF (small amount) was added. A saturated NaCl solution (300ml) was added, and the phases were separated. The organic phase was acidified with 10% HCl solution, and the phases were separated. The aqueous phase was basified with K2CO3solution and extracted with ethyl acetate (EtOAc). The organic phase was filtered and evaporated to obtain solifenacin (SLF) (21.25g). The residue was dissolved in ethanol (EtOH) (100ml) and succinic acid (7.Og) was added. Seeding with SLF-succinate was performed, and the mixture was stirred at RT for 16 hours. The product was isolated by vacuum filtration, washed with EtOH (3x20ml), and dried in vacuum oven at 50° over night to obtain SLF-succinate (10.46g).
Example 2: Preparation of solifenacin succinate
Chloroethylchloroformate (CECF, 13.Og) is added dropwise to solution of (R)- quinuclidin-3-ol (11.6g) and diisopropylethylamine (DIPEA, 13.5g) in THF (150ml), keeping the temperature between 0°-20°C. The mixture is stirred at room temperature for several hours. Then (S)-l-phenyl-l,2,3,4-tetrahydroisoquinoline (16g) is added and the solution is stirred at room temperature for another 16 hours. The solution is diluted with EtOAc (350ml) and washed with a saturated NaCl solution (300ml). The organic phase is acidified with 10% HCl solution, and the phases are separated. The aqueous phase is basified with K.2CO3 solution and extracted with EtOAc. The organic phase is filtered and evaporated to obtain SLF. The residue is dissolved in EtOH (100ml), and succinic acid (7.Og) is added. Seeding with SLF-succinate is performed, and the mixture is stirred at RT for 16 hours. The product is isolated by vacuum filtration, washed with EtOH (3x20ml), and dried in vacuum oven at 50° over night to obtain SLF-succinate. Example 3: Preparation of solifenacin succinate
Chloroethylchloroformate (CECF, 13.Og) is added dropwise to solution of (R)- quinuclidin-3-ol (11.6g) and diisopropylethylamine (DIPEA, 13.5g) in Toluene (150ml), keeping the temperature between 0°-20°C. The mixture is stirred at room temperature for several hours and filtrated. Then (S)-l-phenyl-l,2,3,4-tetrahydroisoquinoline (16g) is added followed by addition of sodium hydride (60%, 5.5g) and the mixture is stirred at reflux for another 16 hours. The solution is diluted with EtOAc (350ml) and washed with a saturated NaCl solution (300ml). The organic phase is acidified with 10% HCl solution, and the phases are separated. The aqueous phase is basified with K2CO3 solution and extracted with EtOAc. The organic phase is filtered and evaporated to obtain SLF. The residue is dissolved in EtOH (100ml), and succinic acid (7.Og) is added. Seeding with SLF-succinate is performed, and the mixture is stirred at RT for 16 hours. The product is isolated by vacuum filtration, washed with EtOH (3x20ml), and dried in vacuum oven at 50° over night to obtain SLF-succinate.
CLIP
PATENT

1. WO9620194A1 / US6017927A.
2. J. Med. Chem. 2005, 48, 6597-6606.
3. WO2007076116A2.
PAPER
High-performance liquidchromatography (HPLC)
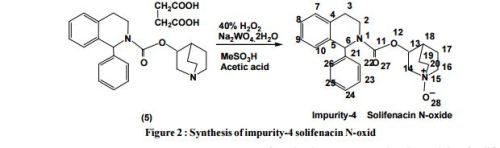
An in house LC Isogradient method was devel- oped for the separation of all possible stereoisomers ofsolifenacinsuccenate. Waters make HPLC system equipped with 515 pump and UV detector was used for betterseparation and quantification of impurities. Used for the preparation of mobile phase wasin the ratio of n-Hexane:Isopropyl alcohol:Ethanol:Diethylamine (85:7.5:7.5:0.02), particle 5 µmsize,Chiraipak AD-H,250X4.6mm column was used with a time 60min isogradient programcolumn overtemperature was 25 º C and column eluent was monitored by UV detector at 215nm. This LC method was able to separate all the process-related chiralsubstanceswith good resolution. An in house LC Isogradient method was developed for the separation of N-oxide impurity and solifenacin succenate. SHIMADZUmake HPLCsystem equipped with 436 pump and UV detector was used for betterseparation and quantification ofimpurities. Used for the preparation of mobile phase wasin the buffer (1.36 gm of potassium dihydrogen ortho- phosphate in 1000ml water containing 1.0 ml of tri- ethylamine), particle5 µm size,kromasil 100- 5C8 ,250X4.6mm columnwas used with a time 30min isogradient program.column overtemperature was 30 ºC and column eluent was monitored byUV detector at 210nm. This LC method was able to separate N- oxide and solifenacinwith good resolution.
REFERENCES FOR ABOVE
[1] R.F.Majewski, K.N.Camphell, S.Dykstra, R.Covington, J.C.Simms; Anticholinergic agents. Esters of 4-alkyl-(or 4- polymethylene)amino-2- butynols, J.Med.Chem., 8, 719-720 (1965).
[2] K.E.Andersson; Current concepts in the treatment
of disorders of micturition, Drugs, 35, 477-494
(1988). [3] I.Masatoshi; Process for producing solifenacin or
its salts, EP 1757604 A1, (2007). [4] P. Jprdo, S. Laura, M. Ester, A. Ignasi, B.Jordi, An
improved process for the synthesis of solifenacin, WO 2008/062282A2, 2008. [5] R.Naito, Y.Yenetoku, Y.Okamoto, A.Toyoshima, K.Ikeda, M.Takeuchi; Synthesis and antimuscarinic
properties of quinuclidin-3-yl 1,2,3,4-
tetrahydroisoquinoline-2-carboxylate derivatives as
novel muscarinic receptor antagonists, J.Med.Chem., 48, 6597-6606 (2005)
PATENT

PATENT
WO2013147458A1.

| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2007076116A2 * |
Dec 21, 2006 |
Jul 5, 2007 |
Teva Pharmaceutical Industries Ltd. |
Intermediates for preparing solifenacin |
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| EP0801067A1 * |
Dec 27, 1995 |
Oct 15, 1997 |
Yamanouchi Pharmaceutical Co. Ltd. |
Novel quinuclidine derivatives and medicinal composition thereof |
| EP1714965A1 * |
Feb 7, 2005 |
Oct 25, 2006 |
Astellas Pharma Inc. |
Composition containing solifenacin succinate |
| EP1726304A1 * |
Mar 11, 2005 |
Nov 29, 2006 |
Astellas Pharma Inc. |
Solifenacin-containing composition |
| EP1757604A1 * |
Apr 25, 2005 |
Feb 28, 2007 |
Astellas Pharma Inc. |
Process for producing solifenacin or its salt |
| Reference |
| 1 |
* |
MEALY N ET AL: “YM-53705 (AS MONOHYDROCHLORIDE) 1(S)-PHENYL-1,2,3,4-TETRAHYDROISOQUIN OLINE-2-CARBOXYLIC AID 3(R)- QUINUCLIDINYL ESTER MONOSUCCINATE” DRUGS OF THE FUTURE, BARCELONA, ES, vol. 24, no. 8, 1999, pages 871-874, XP001061585 ISSN: 0377-8282 cited in the application |
| 2 |
* |
NAITO ET AL: “Synthesis and Antimuscarinic Properties of Quinuclidin-3-yl 1,2,3,4-Tetrahydroisoquinoline-2-carboxyla te Derivatives as Novel Muscarinic Receptor Antagonists” J.MED.CHEM., vol. 48, 20 October 2005 (2005-10-20), pages 6597-6606, XP002435582 |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2008013851A2 * |
Jul 24, 2007 |
Jan 31, 2008 |
Teva Pharmaceutical Industries Ltd. |
Processes for preparing polymorphic forms of solifenacin succinate |
| WO2008013851A3 * |
Jul 24, 2007 |
Dec 24, 2008 |
Mili Abramov |
Processes for preparing polymorphic forms of solifenacin succinate |
| WO2009087664A1 * |
Dec 2, 2008 |
Jul 16, 2009 |
Cadila Healthcare Limited |
Process for preparing chemically and chirally pure solifenacin base and its salts |
| WO2010012459A2 * |
Jul 29, 2009 |
Feb 4, 2010 |
Krka, D.D., Novo Mesto |
A process for the preparation of solifenacin salts and their inclusion into pharmaceutical dosage forms |
| WO2010012459A3 * |
Jul 29, 2009 |
Aug 5, 2010 |
Krka, D.D., Novo Mesto |
A process for the preparation of solifenacin salts and their inclusion into pharmaceutical dosage forms |
| WO2012175119A1 |
Jun 22, 2011 |
Dec 27, 2012 |
Isochem |
Process for the preparation of solifenacin and salts thereof |
| CN102887894A * |
Jul 18, 2011 |
Jan 23, 2013 |
天津市医药集团技术发展有限公司 |
Crystal form of solifenacin succinate and preparation method thereof |
| EP2406257A1 * |
Aug 31, 2009 |
Jan 18, 2012 |
Megafine Pharma (P) Ltd. |
A new method for the preparation of solifenacin and new intermediate thereof |
| EP2406257A4 * |
Aug 31, 2009 |
Nov 14, 2012 |
Megafine Pharma P Ltd |
A new method for the preparation of solifenacin and new intermediate thereof |
| EP2489666A2 * |
Dec 2, 2008 |
Aug 22, 2012 |
Cadila Healthcare Limited |
Chemically and chirally pure solifenacin base and its salts |
| EP3067353A1 |
Jul 29, 2009 |
Sep 14, 2016 |
KRKA, D.D., Novo Mesto |
A process for the preparation of solifenacin salts and their inclusion into pharmaceutical dosage forms |
| US9399624 |
Apr 17, 2015 |
Jul 26, 2016 |
Shanghai Jingxin Biomedical Co., Ltd. |
Process for preparing (1S)-1-phenyl-3,4-dihydro-2(1H)-isoquinoline-carboxylate |
References
////////солифенацин , سوليفيناسين , 索利那新 , Solifenacin, YM 67905, YM 905 , コハク酸ソリフェナシン
コハク酸ソリフェナシン
Solifenacin Succinate

C23H26N2O2▪C4H6O4 : 480.55
[242478-38-2]
Title: Solifenacin
CAS Registry Number: 242478-37-1
CAS Name: (1S)-3,4-Dihydro-1-phenyl-2(1H)-isoquinolinecarboxylic acid (3R)-1-azabicyclo[2.2.2]oct-3-yl ester
Additional Names: (1S,3¢R)-3¢quinuclidinyl-1-phenyl-1,2,3,4-tetrahydro-2-isoquinolinecarboxylate
Molecular Formula: C23H26N2O2
Molecular Weight: 362.46
Percent Composition: C 76.21%, H 7.23%, N 7.73%, O 8.83%
Literature References: Muscarinic M3 receptor antagoinst. Prepn: M. Takeuchi et al., WO 9620194; eidem, US 6017927 (1996, 2000 both to Yamanouchi). Receptor binding studies: K. Ikeda et al., Arch. Pharmacol. 366, 97 (2002); selectivity profile: A. Ohtake et al., Eur. J. Pharmacol. 492, 243 (2004). Clinical study in overactive bladder: C. R. Chapple et al., Br. J. Urol. 93, 303 (2004); F. Habb et al., Eur. Urol. 47, 376 (2005). Clinical comparison with tolterodine: C. R. Chapple et al., ibid. 48, 464 (2005). Review of efficacy and tolerability studies: S. Brunton, L. Kuritzky, Curr. Med. Res. Opin. 21, 71-80 (2005); of clinical development: C. K. Payne, Drugs 66, 175-190 (2006).
Properties: Yellow oil.
Derivative Type: Succinate
CAS Registry Number: 242478-38-2
Manufacturers’ Codes: YM-905
Trademarks: Vesicare (Yamanouchi)
Molecular Formula: C23H26N2O2.C4H6O4
Molecular Weight: 480.55
Percent Composition: C 67.48%, H 6.71%, N 5.83%, O 19.98%
Properties: White to slightly yellowish crystals, mp ~145°. Freely sol in acetic acid, water, methanol, dimethylsulfoxide.
Melting point: mp ~145°
Therap-Cat: In treatment of urinary incontinence.
Keywords: Antimuscarinic.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO







![[1860-5397-9-265-6]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-6.png?scale=2.0&max-width=1024&background=FFFFFF)