
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Direclidine



Direclidine
CAS 1803346-98-6
MF C19H30N4O2 MW346.5 g/mol
ethyl 2-[4-(2-methylpyrazol-3-yl)piperidin-1-yl]-6-azaspiro[3.4]octane-6-carboxylate
ethyl (2r,4s)-2-[4-(1-methyl-1H-pyrazol-5-yl)piperidin-1-yl]-6-azaspiro[3.4]octane-6-carboxylate
muscarinic M4 receptor positive allosteric modulator, TXB4V44U24, NBI-1117568, NBI 1117568
Direclidine (INNTooltip International Nonproprietary Name;[2] developmental code names NBI-1117568, HTL-0016878)[1] is an investigational antipsychotic drug for schizophrenia[3] that was out-licensed from Nxera Pharma to Neurocrine Biosciences, a United States-based pharmaceutical company.[4][1][5] It is an oral small molecule.[6][7]
Direclidine (NBI-1117568) is an investigational, oral drug in Phase 3 trials for schizophrenia, developed by Neurocrine Biosciences, working as a selective M4 muscarinic receptor agonist to treat psychosis by modulating dopamine indirectly. It’s an innovative small molecule with potential for neuropsychiatric disorders, showing promise in early trials and aiming to offer a new treatment approach beyond traditional antipsychotics.
Key aspects of Direclidine:
- Drug Class: Small molecule, muscarinic M4 receptor agonist, antipsychotic.
- Mechanism: Acts as a selective agonist for the M4 receptor, which indirectly helps improve dopamine levels, targeting schizophrenia symptoms.
- Developer: Originally from Nxera Pharma, licensed to Neurocrine Biosciences.
- Status: In Phase 3 clinical trials for schizophrenia.
- Significance: Offers a novel mechanism for treating schizophrenia, potentially with fewer side effects than current treatments.
- Other Uses: Also being explored for bipolar mania and other neuropsychiatric conditions.
Development Timeline & Data:
- Positive Phase 2 data was announced in August 2024, showing promise for its use in schizophrenia.
- Topline data from ongoing Phase 3 studies is expected around 2027.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015118342&_cid=P20-MKJ4D7-04370-1


Typical procedure for the preparation of piperidines via sodium triacetoxyborohydride reductive amination, Boc-deprotection and ethylcarbamate formation as exemplified by the preparation of Example 1 -7, ethyl 2-[4-(1 -methyl-1 H-imidazol-2-yl)piperidin-1 -yl]-6-aza s p i ro [3.4] octa n e -6 -ca rb oxy late

Example 1-7
4-(1 -Methylimidazol-2-yl)piperidine hydrochloride (0.244 g, 1 .21 mmol) and 6-Boc-2-oxo-6-azaspiro[3,4]octane (0.273 g, 1 .21 mmol) were dissolved in DCM (10 mL) at rt and titanium isopropoxide (0.4 mL, 2.42 mmol) was added. The reaction mixture was stirred at rt for 1 h. The reaction mixture was cooled to -5 °C, then STAB (0.513 g, 2.42 mmol) and acetic acid (27 pL, 480 pmol) were added and the reaction mixture was stirred overnight under nitrogen while warming to rt. The reaction mixture was quenched with the addition of NaHC03 (sat aq.) (10 mL) and diluted with DCM then filtered through a pad of celite. The layers were separated and the aqueous layer was extracted with DCM. The combined DCM layers were washed with brine, then dried over MgS04. The solvents were removed in vacuo, and the residue was purified by column chromatography (normal phase, [Biotage SNAP cartridge KP-sil 25g , 40-63 μΠΊ, 60 A, 50 mL per min, gradient 1 % to 10% MeOH in DCM]) to give an inseparable mixture of isomers of terf-butyl 2-[4-(1 -methyl-1 H-imidazol-2-yl)piperidine]-6-azaspiro[3.4]octane-6-carboxylate (0.330 g, 72%) as a yellow gum.
LCMS (Method A): m/z 374 (M+H)+ (ES*), at 1.68 min, UV inactive.
Terf-butyl 2-[4-(1-methyl-1 H-imidazol-2-yl)piperidine]-6-azaspiro[3.4]octane-6-carboxylate (0.326 g, 0.87 mmol) was dissolved in 4 M hydrogen chloride in dioxane (1 .2 mL, 5.2 mmol). The reaction mixture was stirred at rt for 18 h. The volatiles were then removed in vacuo and the residue dissolved DCM (17 mL) and triethylamine (0.49 mL, 3.49 mmol). Ethyl chloroformate (125 μί, 1.31 mmol) was added dropwise and the solution stirred at rt for 18 h. The mixture was then poured into NaHC03 (aq) (75 mL) and DCM (75 mL), extracted (2 x 75 mL) , and the combined DCM extracts washed with brine (20 mL) then dried over MgSO*. After concentration, the residue was purified by column chromatography (normal phase, [Biotage SNAP cartridge KP-sil 25 g, 40-63 μΐη, 60 A, 50 mL per min, gradient 1 % to 10% MeOH in DCM]) to provide ethyl 2-[4-(1 -methyl-1 H-imidazol-2-yl)piperidine]-6-azaspiro[3.4]octane-6-carboxylate as a brown oil as a mixture of diastereomers (0.25 g, 83%). Preparative HPLC was used to separate the diastereomers, using a Phenomenex Gemini-N C18 column, 150 x 21 mm, eluting with 38 to 48% MeCN/H20 at 18 mL/min and collecting fractions by monitoring at 218 nm to give ethyl 2-[4-(1 -methyl-1 H-imidazol-2-yl)piperidine]-6-azaspiro[3.4]octane-6-carboxylate, Example 1-7 Isomer 1 , (0.044 g, 15%) as a colourless oil and ethyl 2-[4-(1 -methyl-1 /-/-imidazol-2-yl)piperidine]-6-azaspiro[3.4]octane-6-carboxylate, Example 1 -7 Isomer 2, (0.031 g, 10%) as a colourless oil. The data for Isomer 2 are in Table 3
PAT
- Pharmaceutical compoundsPublication Number: EP-4413985-A2Priority Date: 2014-02-06
- BICYCLIC AZA COMPOUNDS AS MUSCARINIC RECEPTOR AGONISTSPublication Number: FI-3406609-T3Priority Date: 2014-02-06Grant Date: 2024-09-10
- Bicyclic aza compounds as muscarinic receptor agonistsPublication Number: EP-3406609-B1Priority Date: 2014-02-06Grant Date: 2024-06-26
- Bicyclic aza compounds as muscarinic m1 receptor agonists.Publication Number: WO-2015118342-A1Priority Date: 2014-02-06
- Bicyclic aza compounds as muscarinic m1 receptor and/or m4 receptor agonistsPublication Number: US-2018179184-A1Priority Date: 2014-02-06
- Bicyclic aza compounds as muscarinic m1 receptor and/or m4 receptor agonistsPublication Number: US-2017240530-A1Priority Date: 2014-02-06
- Bicyclic aza compounds as muscarinic m1 receptor and/or m4 receptorPublication Number: US-2020325118-A1Priority Date: 2014-02-06
- Bicyclic aza compounds as muscarinic receptor agonistsPublication Number: ES-2986327-T3Priority Date: 2014-02-06Grant Date: 2024-11-11



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Overview
It is a selective muscarinic acetylcholine M4 receptor agonist that indirectly modulates dopamine as the basis for its putative improvement of schizophrenia.[6] In April 2016, the compound was out-licensed from Nxera Pharma to Allergan, an Irish pharmaceutical company, as part of Nxera’s wider muscarinic agonist portfokio. By September 2017, it had advanced to Phase I clinical trial for the indication of “neuropsychiatric symptoms associated with Alzheimer’s disease and other dementias”[8] Following Allergan’s acquisition by AbbVie, the license was returned to Nxera in January 2021.[9] In November 2021, the compound was newly out-licensed to Neurocrine Biosciences, a U.S. pharmaceutical company.[5] It has been under development as a treatment for schizophrenia, and is currently in Phase III clinical trials.[10][11]
History
- 2016
- April: The rights to develop Nxera Pharma’s muscarinic agonist portfolio, including NBI-1117568 were transferred to Allergan.[9]
- 2017
- September: Allergan initiated Phase I clinical trials for NBI-1117568 to treat “neurobehavioral symptoms related to Alzheimer’s disease and other conditions.”[8]
- 2021
- 2022
- October: Phase II clinical trial of NBI-1117568 for the treatment of adults with schizophrenia was initiated.[12]
- 2024
- 2025
- May: Neurocrine Biosciences initiated a Phase III registrational program for NBI-1117568 for the treatment of adults with schizophrenia.[14]
Clinical trials
Phase II clinical trial
The Phase II clinical trial was conducted in 15 sites across the U.S. with 200 adult patients diagnosed with schizophrenia.[15] The primary endpoint was assessed by the change in the total score of the Positive and Negative Syndrome Scale (PANSS) after six weeks of treatment. The 20 mg once-daily group showed a statistically significant improvement of 7.5 points compared to the placebo group (improvement of 18.2 points from baseline, p = 0.011, effect size = 0.61).[16] However, the 40 mg once-daily group, 60 mg once-daily group, and 30 mg twice-daily group did not show statistically significant differences compared to the placebo group (p-values: 40 mg group: 0.282, 60 mg group: 0.189, 30 mg twice-daily group: 0.090).[16]
Market reaction to phase II clinical trial
With a PANSS improvement of 7.5, NBI-111758 lagged behind xanomeline/trospium (KarXT) (Karuna Therapeutics) with 8.4 and emraclidine (Cerevel Therapeutics) with 12.7, both of which were in clinical trials at the same time. Moreover, the lack of dose-dependency led to disappointment in the stock market.[17] Neurocrine Biosciences’ share price dropped 19% on the day following the announcement of the Phase II clinical trial results.[18]
References
- “Delving into the Latest Updates on Direclidine with Synapse”. Synapse. 30 June 2025. Retrieved 27 July 2025.
- https://cdn.who.int/media/docs/default-source/international-nonproprietary-names-(inn)/pl133.pdf [bare URL PDF]
- Ye N, Wang Q, Li Y, Zhen X (March 2025). “Current emerging therapeutic targets and clinical investigational agents for schizophrenia: Challenges and opportunities”. Medicinal Research Reviews. 45 (2): 755–787. doi:10.1002/med.22086. PMID 39300769.
- “HTL 0016878”. AdisInsight. 2 September 2024. Retrieved 21 October 2024.
- “ニューロクライン社との統合失調症およびその他の精神神経疾患を対象とした新規ムスカリン受容体作動薬に関するライセンス契約締結のお知らせ” [Announcement of License Agreement with Neurocrine for Novel Muscarinic Receptor Agonist for Schizophrenia and Other Neuropsychiatric Disorders] (in Japanese). PR TIMES. 2021-11-22. Retrieved 2024-09-17.
- “ネクセラファーマ株価6.5%高 薬候補で毒性試験成功 – 日本経済新聞” [NexThera Pharma shares rise 6.5% as drug candidate passes toxicity test – Nikkei]. 日本経済新聞 電子版 (Nikkei Electronic Edition) (in Japanese). 日本経済新聞社 (Nikkei Inc.). 2024-04-17. Retrieved 2024-09-17.
- “当社提携先のニューロクライン社が、統合失調症を対象にしたNBI-1117568の第II相臨床試験の開始を発表[そーせいグループ] | NIKKEI COMPASS – 日本経済新聞” [Our partner Neurocrine, Inc. announces initiation of Phase II clinical trial of NBI-1117568 for schizophrenia [Sosei Group] | NIKKEI COMPASS – Nikkei Newspaper]. 日経コンパス (Nikkei Compass) (in Japanese). 日本経済新聞社 (Nikkei Inc.). 2022-10-28. Retrieved 2024-09-17.
- “アルツハイマー病等の主要症状の治療薬として開発中の新薬候補、 選択的ムスカリンM4受容体作動薬の第I相臨床試験で最初の被験者への投与を実施” [First subjects dosed in Phase I clinical trial of selective muscarinic M4 receptor agonist, a potential new drug candidate for treating major symptoms of Alzheimer’s disease] (PDF) (in Japanese). ネクセラファーマ(旧そーせいグループ株式会社). 2017-09-01. Retrieved 2024-09-17.
- “ムスカリン作動薬プログラムのグローバルな研究開発権・販売権が返還” [Global R&D and commercial rights to muscarinic agonist program returned]. プレスリリース・ニュースリリース配信シェアNo.1|PR TIMES (in Japanese). PR Times. 2021-01-05. Retrieved 2024-09-17.
- 日経バイオテクONLINE (2024-09-02). “ネクセラファーマ、統合失調症治療薬候補 NBI-1117568の第II相臨床試験の良好な結果によりニューロクライン社より35百万米ドルのマイルストンを受領” [Nexella Pharma Receives $35 Million Milestone Payment from Neurocrine Following Positive Results of Phase II Clinical Trial of NBI-1117568, a Potential Treatment for Schizophrenia]. 日経バイオテクONLINE (in Japanese). 日本経済新聞社. Retrieved 2024-09-17.
- “ネクセラ—大幅反落、ニューロクラインの株価下落に追随売り | 個別株 – 株探ニュース” [Nexella – Sharp decline, selling follows fall in Neurocrine stock price | Individual stocks – Kabutan News]. kabutan.jp (in Japanese). MINKABU THE INFONOID, Inc. 2024-08-29. Retrieved 2024-09-17.
- “当社提携先のニューロクライン社が、統合失調症を対象にしたNBI-1117568の第Ⅱ相臨床試験の開始を発表” [Our Partner Neurocrine Announces Initiation of Phase 2 Clinical Trial of NBI-1117568 for Schizophrenia]. プレスリリース・ニュースリリース配信シェアNo.1|PR TIMES (in Japanese). PR TIMES. 2022-10-28. Retrieved 2024-09-17.
- 日経バイオテク (Nikkei Biotech) ONLINE (2024-09-02). “ネクセラファーマ、統合失調症治療薬候補 NBI-1117568の第II相臨床試験の良好な結果によりニューロクライン社より35百万米ドルのマイルストンを受領” [Nexella Pharma Receives $35 Million Milestone Payment from Neurocrine Following Positive Results of Phase II Clinical Trial of NBI-1117568, a Potential Treatment for Schizophrenia]. 日経バイオテクONLINE (in Japanese). 日本経済新聞社. Retrieved 2024-09-17.
- “ネクセラファーマの統合失調症薬、最終治験を開始”. 日本経済新聞. 日本経済新聞社. 2025-05-01. Retrieved 2025-05-02.
- “Neurocrine Biosciences Initiates Phase 2 Clinical Study Evaluating NBI-1117568 in Adults with Schizophrenia”. ニューロクライン. 2022-10-27. Retrieved 2024-09-17.
- “ネクセラファーマ[4565]:ニューロクライン社との提携プログラムである統合失調症治療薬候補NBI-1117568の第2相臨床試験で良好な結果 2024年8月28日(適時開示) :日経会社情報DIGITAL:日本経済新聞” [Nexella Pharma [4565]: Positive results in Phase 2 clinical trial of NBI-1117568, a candidate for the treatment of schizophrenia, a collaboration program with Neurocrine, Inc. August 28, 2024 (timely disclosure) : Nikkei Company Information DIGITAL: Nikkei Shimbun]. 日本経済新聞 電子版 (in Japanese). 日本経済新聞社. Retrieved 2024-09-17.
- “Nxera Pharma Official IR Blog 「ニューロクライン社から35百万米ドルのマイルストン受領」” [Received $35 million milestone payment from Neurocrine]. soseiheptares.blogspot.com. ネクセラファーマ. 2024-09-02. Retrieved 2024-09-19.
- “Neurocrine Stock Down 19% on Mixed Schizophrenia Study Results”. Zacks Investment Research. Zacks Investment. 2024-08-29. Retrieved 2024-09-17.
| Clinical data | |
|---|---|
| Other names | HTL-0016878; NBI-1117568; NBI-568[1] |
| Routes of administration | Oral |
| Drug class | Muscarinic acetylcholine M4 receptor agonist |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1803346-98-6 |
| PubChem CID | 118295270 |
| ChemSpider | 133319625 |
| UNII | TXB4V44U24 |
| KEGG | D13221 |
| Chemical and physical data | |
| Formula | C19H30N4O2 |
| Molar mass | 346.475 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
NEXT
ADVERTISEMENT
Advect Process Systems Ltd. https://advectprocess.com/
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
ADVERTISEMENT
BLUE JET HEALTHCARE LTD, https://bluejethealthcare.com
Looking for a Reliable SNAC Manufacturer? Let’s Talk.
At Blue Jet Healthcare Ltd, we specialize in the scalable, high-purity production of SNAC—a critical excipient powering the next generation of oral peptide therapeutics.
With increasing demand for SNAC across global pharma pipelines, choosing the right manufacturing partner is essential. Quality, timelines, and consistency matter.

Phone No. +91 (22) 22075307 / +91 (22) 22071691
Business Development/ Contract Manufacturing: marketing1@bluejethealthcare.com, madhu.gautam71@gmail.com
/////direclidine, ANAX, BLUE JET, ADVECT, muscarinic M4 receptor positive allosteric modulator, TXB4V44U24, NBI-1117568, NBI 1117568
Dezecapavir




Dezecapavir
CAS 2570323-59-8
MF C37H29ClF9N9O5S MW918.19
1H-Cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-1-acetamide, N-[(1S)-1-[(3S)-3-[4-chloro-1-methyl-3-[(methylsulfonyl)amino]-1H-indazol-7-yl]-3,4-dihydro-4-oxo-7-(3,3,3-trifluoropropoxy)pyrido[2,3-d]pyrimidin-2-yl]-2-(3,5-difluorophenyl)ethyl]-3-(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-, (3bS,4aR)-
N-[(1S)-1-[(3P)-3-[4-chloro-3-(methanesulfonamido)-1-methyl-1H-indazol-7-yl]-4-oxo-7-(3,3,3-
trifluoropropoxy)-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl]-2-(3,5-difluorophenyl)ethyl]-2-[(3bS,4aR)-3-
(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-1Hcyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-
yl]acetamide
inhibitor of viral replication, antiviral, SEK9LN2LSM, VH 4011499, VH4011499, VH-4011499, VH 499, GSK2838232, GSK 2838232
Dezecapavir is a potent experimental antiviral compound, specifically a novel HIV-1 capsid inhibitor, developed to block HIV replication by targeting the virus’s capsid protein, showing high effectiveness in lab settings (low nM range EC50) and representing a new class of drugs for HIV treatment, potentially for long-acting injectable therapies. It’s a complex molecule with a unique structure designed to disrupt the HIV capsid assembly, halting the virus’s life cycle early on.
Key Characteristics:
- Mechanism: Inhibits HIV-1 capsid assembly, a crucial step in the viral lifecycle.
- Potency: Very effective in cell cultures, with a low nanomolar EC50 (effective concentration).
- Class: Belongs to a new class of antivirals, distinct from integrase or reverse transcriptase inhibitors, offering a novel approach to HIV treatment.
- Development: Under investigation, often mentioned as a potential candidate for long-acting injectable (LAI) treatments due to its potency.
What it does:
Dezecapavir binds to the HIV capsid, preventing the virus from uncoating and maturing, thereby stopping new infections from forming.
Significance:
It represents a promising new option for HIV treatment, especially in the context of growing resistance to existing drugs, and could be part of future long-acting regimens.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020254985&_cid=P20-MKHOOT-76990-1

Preparation of Example 1: N-((S)-l-((3P)-3-(4-chloro-l-methyl-3-(methylsulfonamido)-lH- indazol-7-yl)-4-oxo-7-(3,3,3-trifluoropropoxy)-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)-2-(3,5- difluorophenyl)ethyl)-2-((3bSr4aR)-3-(difluoromethyl)-5r5-difluoro-3br4r4ar5-tetrahydro-lH- cyciopropa[3, 4]cydopenta[ l,2-c]pyrazoi-l-yi)acetamide

A solution of diisopropyl (E)-diazene-l,2-dicarboxylate (“DIAD”, 0.125 ml, 0.637 mmol) in THF (0.2 mL) was added dropwise to a mixture of N-(l-((3P)-3-(4-chloro-3-(N-(4-methoxybenzyl)methylsulfonamido)-l-methyl-lH-indazol-7-yl)-7-hydroxy-4-oxo-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-3-(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-lH-cyclopropa[3,4]cyclopenta[l,2-c]pyrazol-l-yl)acetamide (0.2 g, 0.212 mmol)), 3,3,3-trifluoropropan-l-ol (0.073 g, 0.637 mmol) and triphenylphosphine (0.178 g, 0.679 mmol) in Tetrahydrofuran (2.1 mL) at rt. The reaction mixture was stirred for 18 h at rt and then was concentrated in vacuo. The residue was purified on silica gel (24 g RediSep Gold column) using a gradient of 0-60 % ethyl acetate in hexanes over 15 CV, and then holding at 60 % ethyl acetate in hexanes for 5 CV. Fractions containing the pure product were pooled and then concentrated to give a yellow solid. This solid was taken up in DCM (1 mL):TFA (0.5 mL); the solution was cooled to 0 °C; and to the solution was added triflic acid (0.057 mL, 0.637 mmol). The mixture was stirred for 1 h and then concentrated in vacuo. The residue was taken up in ethyl acetate; washed with 1 N
NaOH; washed with 0.5M citric acid; dried over Na2SC>4; filtered; and then was concentrated in vacuo. The residue was subjected to silica gel chromatography (24 g RediSep Gold column) using 0-60 % ethyl acetate in hexanes over 20 CV, then at 60 % ethyl acetate for 10 CV. Fractions containing the pure product were pooled and then concentrated in vacuoto give N-(l-((6P)-3-(4-chloro-l-methyl-3-(methylsulfonamido)-lH-indazol-7-yl)-4-oxo-7-(3,3,3-trifluoropropoxy)-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-3-(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-lH-cyclopropa[3,4]cyclopenta[l,2-c]pyrazol-l-yl)acetamide (0.078 g, 0.081 mmol, 38.0 % yield) as a brown solid. *H NMR (500 MHz, METHANOL-d^ d ppm 8.46 – 8.53 (m, 1 H) 7.28 – 7.34 (m, 1 H) 7.19 – 7.24 (m, 1 H) 7.03 – 7.09 (m, 1 H) 6.53 – 6.81 (m, 4 H) 4.80 (dd, J=5.96, 2.98Hz, 3 H) 4.49 – 4.62 (m, 2 H) 3.58 – 3.62 (m, 3 H) 3.40 – 3.49 (m, 1 H) 3.22 – 3.24 (m, 3 H) 3.06 – 3.14 (m, 1 H) 2.80 – 2.89 (m, 2 H) 2.37 – 2.44 (m, 2 H) 1.32 – 1.37 (m, 1 H) 0.96 – 1.01 (m, 1 H). LCMS Analysis Method: Column = Acquity UPLC BEH C18, 2.1 x 100 mm, 1.7 pm particles; Injection Volume = 5.00 pL; Flowrate = 0.80 mL/min; Solvent A = 95:5
WatenMeCN w/ 0.1% v/v formic acid; Solvent B = 5:95 WatenMeCN w/ 0.1% v/v formic acid;
Elution profile = Start %B: 0, End %B: 100, Gradient Time: 3.5 min. then hold at 100% B for 1 min.; Detection wavelength 1 = 220 nm, wavelength 2 = 254 nm. LCMS retention time = 3.097 min; m/z = 918.05 [M+l]+.



PAT
- Pyrido[2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: US-2021323967-A1Priority Date: 2019-06-19
- Pyrido[2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: US-2025019383-A1Priority Date: 2019-06-19
- Pyrido[2,3-D]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: KR-20220024608-APriority Date: 2019-06-19
- Pyrido[2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: EP-3986561-A1Priority Date: 2019-06-19
- Pyrido[2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: EP-3986561-B1Priority Date: 2019-06-19Grant Date: 2024-02-14
- Pyrido [2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: US-12129255-B2Priority Date: 2019-06-19Grant Date: 2024-10-29
- Pyrido[2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: WO-2020254985-A1Priority Date: 2019-06-19
- Inhibitors of human immunodeficiency virus replicationPublication Number: EP-4415685-A1Priority Date: 2021-10-13
- Inhibitors of human immunodeficiency virus replicationPublication Number: WO-2023062559-A1Priority Date: 2021-10-13
- Inhibitors of human immunodeficiency virus replicationPublication Number: US-2024423985-A1Priority Date: 2021-10-13
- Inhibitors of human immunodeficiency virus replicationPublication Number: US-2023149408-A1Priority Date: 2020-04-15
- Pharmaceutical compositions comprising cabotegravirPublication Number: US-2023045509-A1Priority Date: 2019-12-09
ADVERTISEMENT
BLUE JET HEALTHCARE LTD, https://bluejethealthcare.com
Looking for a Reliable SNAC Manufacturer? Let’s Talk.
At Blue Jet Healthcare Ltd, we specialize in the scalable, high-purity production of SNAC—a critical excipient powering the next generation of oral peptide therapeutics.
With increasing demand for SNAC across global pharma pipelines, choosing the right manufacturing partner is essential. Quality, timelines, and consistency matter.

Phone No. +91 (22) 22075307 / +91 (22) 22071691
Business Development/ Contract Manufacturing: marketing1@bluejethealthcare.com, madhu.gautam71@gmail.com
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
NEXT
ADVERTISEMENT
Advect Process Systems Ltd.
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1
Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
ADVERTISEMENT
BLUE JET HEALTHCARE LTD, https://bluejethealthcare.com
Looking for a Reliable SNAC Manufacturer? Let’s Talk.
At Blue Jet Healthcare Ltd, we specialize in the scalable, high-purity production of SNAC—a critical excipient powering the next generation of oral peptide therapeutics.
With increasing demand for SNAC across global pharma pipelines, choosing the right manufacturing partner is essential. Quality, timelines, and consistency matter.
Phone No. +91 (22) 22075307 / +91 (22) 22071691
Business Development/ Contract Manufacturing: marketing1@bluejethealthcare.com, madhu.gautam71@gmail.com
//////////dezecapavir, inhibitor of viral replication, antiviral, SEK9LN2LSM, VH 4011499, VH4011499, VH-4011499, VH 499, GSK2838232, GSK 2838232
Deulorlatinib


Deulorlatinib
CAS 2131126-33-3
MFC21H162H3FN6O2, MW 409.4 g/mol

- (10R)-7-Amino-12-fluoro-10,15,16,17-tetrahydro-10,16-dimethyl-2-(methyl-d3)-15-oxo-2H-4,8-methenopyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile
- 2H-4,8-Methenopyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile, 7-amino-12-fluoro-10,15,16,17-tetrahydro-10,16-dimethyl-2-(methyl-d3)-15-oxo-, (10R)-
(10R)-7-amino-12-fluoro-2-(2H3)methyl-10,16-dimethyl15-oxo-10,15,16,17-tetrahydro-2H-8,4-
(metheno)pyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile
tyrosine kinase inhibitor, antineoplastic, 7PW3UT8C9B, TGRX 326, TGRX-326
Deulorlatinib is an orally bioavailable inhibitor of the receptor tyrosine kinases anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1), with potential antineoplastic activity. Upon oral administration, deulorlatinib targets, binds to and inhibits the activity of ALK and ROS1, which leads to the disruption of ALK- and ROS1-mediated signaling and the inhibition of cell growth in ALK- and ROS1-expressing tumor cells. ALK belongs to the insulin receptor superfamily and plays an important role in nervous system development. ALK is not expressed in healthy adult human tissue but ALK dysregulation and gene rearrangements are associated with a variety of tumor cell types. ROS1, overexpressed in certain cancer cells, plays a key role in cell growth and survival of cancer cells.
- TGRX-326 Chinese Phase III for Advanced Non-small Cell Lung Cancer (NSCLC)CTID: NCT06082635Phase: Phase 3Status: Active, not recruitingDate: 2025-05-18
- TGRX-326 Pharmacokinetic Drug InteractionCTID: NCT06294561Phase: Phase 1Status: CompletedDate: 2024-06-27
- TGRX-326 Chinese Phase I for Advanced Non-small Cell Lung Cancer (NSCLC)CTID: NCT05441956Phase: Phase 1Status: Active, not recruitingDate: 2025-05-18
- TGRX-326 Chinese Phase II for Advanced Non-small Cell Lung Cancer (NSCLC)CTID: NCT05955391Phase: Phase 2Status: Active, not recruitingDate: 2025-05-18
SYN
WO 2017/148325 A1
syn
https://patentscope.wipo.int/search/en/detail.jsf?docId=US348430040&_cid=P11-MKG9AH-82468-1



Example 6: Synthesis of (10R)-7-amino-12-fluoro-2-(methyl-d3)-10,16-dimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile (the Compound of Formula (A))



| To a 250 mL three-necked flask equipped with magnetic stirring were added the compound of formula (J) (7.0 g, 42.2 mmol) and anhydrous dichloromethane (120 mL), and stirred until the solution became clear. The compound of formula (H) (8.77 g, 46.4 mmol) and then triethylamine (4.69 g, 46.4 mmol) were successively added. The mixture was stirred at room temperature under nitrogen atmosphere for 30 minutes to give a pale yellow clear solution for further use. |
| Alkylation of the Compound of Formula (E-a) with the Compound of Formula (F) to Form the Compound of Formula (D-a): |
| To another 250 mL three-necked flask equipped with magnetic stirring were added the compound of formula (E-a) (11.2 g, 59.3 mmol) and acetonitrile (200 mL), and cesium carbonate (25.7 g, 79.0 mmol) was added with stirring. The mixture was heated to 50° C. under nitrogen atmosphere, and stirred at this temperature for 30 min. The above-mentioned solution of the compound of formula (F) in acetonitrile was slowly added dropwise at 50° C. over 10 minutes. After the dropwise addition was completed, the mixture was reacted with stirring at this temperature for 2 hours. By TLC (DCM:MeOH=20:1) and HPLC monitoring, the reaction was completed. After cooling to room temperature, the reaction was quenched by adding water (200 mL). The reaction solution was diluted with ethyl acetate (300 mL), stirred for 5 minutes, and then filtered through Celite to remove insoluble solids. The filter cake was washed with ethyl acetate (50 mL). The organic layer was separated from the filtrate, and the aqueous phase was extracted with ethyl acetate (60 mL×2). The organic phases were combined, washed with a saturated aqueous solution of sodium carbonate (100 mL×3) and then saturated brine (60 mL), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to dryness under reduced pressure to give 17.5 g of a brown solid in a yield of 90.1% and a purity (HPLC) of >85% (ee>95%). LC-MS (APCI): m/z=390.1 (M+1) +. |
| Introduction of Boc Protecting Group into the Compound of Formula (D-a) to Form the Compound of Formula (C): |
| To a 250 mL single-necked flask equipped with magnetic stirring were added the compound of formula (D-a) (17.5 g, 35.8 mmol) and dichloromethane (200 mL), and stirred until the solution became clear. Triethylamine (14.5 g, 143.2 mmol) and then DMAP (850 mg, 7.2 mmol) were successively added. Boc2O (23.4 g, 107.4 mmol) was slowly added dropwise, and the mixture was reacted with stirring at room temperature under nitrogen atmosphere overnight. By TLC (DCM:MeOH=20:1) and HPLC monitoring, the reaction was completed. The reaction solution was evaporated under reduced pressure to remove the solvent, and the residue was purified by silica gel column chromatography (EA/PE=0-35%) to give 15.4 g of a white solid in a yield of 62.4% and a purity (HPLC) of >95% (ee>95%). LC-MS (APCI): m/z=590.1 (M+1−100) +. 1H NMR (300 MHz, CDCl 3) (δ/ppm): 8.06 (d, J=1.8 Hz, 1H), 7.53-7.48 (m, 1H), 7.24-7.20 (m, 2H), 7.04-6.98 (m, 1H), 6.81 (s, 1H), 5.66-5.59 (m, 1H), 4.89-4.69 (m, 2H), 2.97 (s, 3H), 1.58 (d, J=6.0 Hz, 3H), 1.47 (s, 18H). |
| Cyclization of the Compound of Formula (C) Using Palladium Catalyst to Form the Compound of Formula (B): |
| To a 500 mL single-necked flask equipped with magnetic stirring were added the compound of formula (C) (15.4 g, 22.3 mmol) and 2-methyl-2-butanol (300 mL), and stirred until the solution became clear. Potassium acetate (6.56 g, 66.9 mmol) was added. The system was evacuated with suction and purged with nitrogen gas three times. Palladium acetate (0.75 g, 3.35 mmol) and n-butylbis(1-adamantyl)phosphine (1.60 g, 4.46 mmol) were quickly added. The system was evacuated with suction and purged with nitrogen gas three times. The reaction solution was heated to 110° C. under nitrogen atmosphere, and reacted with stirring at this temperature overnight. By TLC (PE:EA=1:1) and HPLC monitoring, the reaction was completed. The reaction solution was cooled to room temperature, diluted with dichloromethane (300 mL), and filtered through Celite to remove insoluble solids. The filter cake was washed with dichloromethane (50 mL). The filtrates were combined, and concentrated to dryness under reduced pressure. To the residue was added acetonitrile (150 mL), and the mixture was heated to reflux for 1 hour. The oil bath was removed, and the mixture was allowed to slowly cool to room temperature. A large amount of a white solid precipitated out, and the precipitated solid was filtered. The filter cake was washed with acetonitrile (10 mL), and dried to give 8.2 g of a white solids in a yield of 60.4% and a purity (HPLC) of >99.5% (ee>99.9%). LC-MS (APCI): m/z=510.1 (M+1−100) +. 1H NMR (300 MHz, CDCl 3) (δ/ppm): 8.22 (d, J=1.8 Hz, 1H), 7.29-7.25 (m, 1H), 7.22-7.16 (m, 2H), 7.03-6.96 (m, 1H), 5.76-5.70 (m, 1H), 4.42 (q, J=14.1 Hz, 2H), 3.15 (s, 3H), 1.76 (d, J=6.0 Hz, 3H), 1.44 (s, 18H). |
| Removal of the Boc from the Compound of Formula (B) Using an Acid to Form the Compound of Formula (A): |

To a 250 mL single-necked flask equipped with magnetic stirring were added the compound of formula (B) (8.2 g, 13.5 mmol) and dichloromethane (100 mL), and stirred until the solution became clear. The mixture was cooled in an ice-water bath, and trifluoroacetic acid (20 mL) was slowly added dropwise. After the addition was completed, the ice bath was removed, and the mixture was reacted with stirring at room temperature for 2 hours. By TLC (DCM:MeOH=20:1) and HPLC monitoring, the reaction was completed. The reaction solution was evaporated under reduced pressure to remove the organic solvent. Dichloromethane (100 mL) and a saturated aqueous solution of sodium bicarbonate (60 mL) were added under cooling, and the mixture was stirred for 10 minutes. The organic phase was separated, and the aqueous layer was extracted with dichloromethane (50 mL×2). The organic phases were combined, washed successively with water (30 mL) and then saturated brine (500 mL), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to give 5.1 g of an amorphous white solid in a yield of 92.6% and a purity (HPLC) of >99.5% (ee>99.9%). LC-MS (APCI): m/z=410.2 (M+1) +. 1H NMR (300 MHz, CDCl 3) (δ) ppm 7.79 (d, J=1.8 Hz, 1H), 7.31-7.27 (m, 1H), 7.23-7.19 (m, 1H), 7.06-6.97 (m, 1H), 6.87 (d, J=1.8 Hz, 1H), 5.75-5.70 (m, 1H), 5.09 (br s, 2H), 4.40 (q, J=14.1 Hz, 2H), 3.12 (s, 3H), 1.78 (d, J=6.6 Hz, 3H).
PAT
Preparation method for deuterated macrocyclic compound
Publication Number: US-2022024908-A1
Priority Date: 2018-11-28
ADVERTISEMENT
Advect Process Systems Ltd.
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////deulorlatinib, tyrosine kinase inhibitor, antineoplastic, 7PW3UT8C9B, TGRX 326, TGRX-326
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
NEXT
ADVERTISEMENT
Advect Process Systems Ltd.
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1
Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
Cenacitinib



Cenacitinib
CAS 2641636-52-2
MF C19H19F2N7O3 MW431.4
Urea, N-[(1R,2S)-2-fluorocyclopropyl]-N′-[5-[(7-fluoro-2,3-dihydro-1,4-benzodioxin-5-yl)amino]-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl]-
N-{5-[(7-fluoro-2,3-dihydro-1,4-benzodioxin-5-yl)amino]-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl}-N′-[(1R,2S)-2-fluorocyclopropyl]urea
Janus kinase inhibitor, anti-inflammatory, VTX958, VTX 958, SB88R8KGL3
VTX958 for the Treatment of Moderately to Severely Active Crohn’s Disease
CTID: NCT05688852
Phase: Phase 2
Status: Terminated
Date: 2025-07-03
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US323750705&_cid=P22-MKEUDK-45432-1
Example 4: Synthesis of 1-(5-((7-fluoro-2,3-dihydrobenzo[b][1,4]dioxin-5-yl)amino)-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl)-3-((1R,2S)-2-fluorocyclopropyl)urea (5)


| Step 1: To a solution of 1E (100 g, 288 mmol) and 2E (57 g, 345 mmol) in dry 1,4-dioxane (3000 mL) under N 2 atmosphere was added Cs 2CO 3 (141 g, 432 mmol), Pd(OAc) 2 (5.2 g, 23.3 mmol) and BINAP (28.6 g, 46.6 mmol). After stirring at 115° C. overnight, the reaction mixture was cooled to rt. and diluted with hexane (3000 mL). The solid was collected by filtration and washed with 2×1500 mL (50% hexane in DCM). The solid was suspended into 5000 mL water and stirred for 1 h. The solid was collected by filtration and dried under vacuum to afford compound 2 (90 g, 65%) as a brown solid. |
PAT
Publication Number: US-2021139486-A1
Priority Date: 2019-11-08
- Tyk2 pseudokinase ligandsPublication Number: EP-4054581-A1Priority Date: 2019-11-08
- Tyk2 pseudokinase ligandsPublication Number: US-2023348478-A1Priority Date: 2019-11-08
- Substituted pyrazolo[1,5-a]pyrimidines as TYK2 pseudokinase ligandsPublication Number: US-11753411-B2Priority Date: 2019-11-08Grant Date: 2023-09-12
- TYK2 pseudokinase ligandsPublication Number: CN-114929226-BPriority Date: 2019-11-08Grant Date: 2024-09-27
- TYK2 pseudokinase ligandPublication Number: CN-114929226-APriority Date: 2019-11-08
- Preparation of a tyk2 inhibitorPublication Number: WO-2024151992-A1Priority Date: 2023-01-13
- Crystalline forms of a tyk2 inhibitorPublication Number: US-2024010654-A1Priority Date: 2022-07-06
- Crystalline forms of TYK2 inhibitorsPublication Number: CN-119816502-APriority Date: 2022-07-06
- Crystalline forms of a tyk2 inhibitorPublication Number: EP-4551576-A1Priority Date: 2022-07-06
- Crystalline forms of a tyk2 inhibitorPublication Number: WO-2024011136-A1Priority Date: 2022-07-06
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions,Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////cenacitinib, cenacitinib, Janus kinase inhibitor, anti-inflammatory, VTX958, VTX 958, SB88R8KGL3
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
Casdatifan


Casdatifan
CAS 2709069-30-5
MF C21H17F4NO3S, 439.4 g/mol
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-(methanesulfonyl)-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methylsulfonyl-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methylsulfonyl-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methanesulfonyl-2, 3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
hypoxia-inducible factor (HIF) inhibitor, antineoplastic, AB 521, DP73UWL6LE
Casdatifan is an orally bioavailable allosteric inhibitor of hypoxia inducible factor (HIF)-2alpha, with potential antineoplastic activity. Upon oral administration, casdatifan targets and allosterically binds to a hydrophobic pocket on HIF-2alpha leading to a confirmational change that prevents HIF-2alpha heterodimerization with HIF-1beta and binding to the hypoxia response element (HRE) binding site on DNA. This results in decreased transcription and expression of HIF-2alpha downstream target genes, many of which regulate tumor cell growth and survival. Blocking HIF-2alpha reduces the proliferation of HIF-2alpha-expressing tumor cells. HIF-2alpha, a heterodimeric transcription factor overexpressed under hypoxic conditions in many cancer cell types, promotes proliferation, progression and metastasis of tumors.
- A Phase 1 Study of AB521 Monotherapy and Combination Therapies in Renal Cell Carcinoma and Other Solid TumorsCTID: NCT05536141Phase: Phase 1Status: RecruitingDate: 2026-01-02
- A Relative Bioavailability Study and Food Effect Study of AB521 in Healthy Adult VolunteersCTID: NCT05999513Phase: Phase 1Status: CompletedDate: 2024-10-17
- A Study to Investigate the Efficacy and Safety of Volrustomig ± Casdatifan vs Nivolumab + Ipilimumab as 1L Treatment for Advanced ccRCCCTID: NCT07000149Phase: Phase 3Status: Active, not recruitingDate: 2025-11-14
- Study of Zanzalintinib (XL092) + AB521 and Zanzalintinib + AB521 + Nivolumab in Participants With Advanced Clear Cell Renal Cell Carcinoma (ccRCC) or Other Advanced Solid Tumors (STELLAR-009)CTID: NCT06191796Phase: Phase 1Status: TerminatedDate: 2025-06-12
- Drug-Drug Interaction Study of Casdatifan in Healthy Adult Participants (ARC-29)CTID: NCT06919991Phase: Phase 1Status: CompletedDate: 2025-11-13
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.4c00497




PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021188769&_cid=P12-MKDEE0-87371-1
Example 215: (5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methanesulfonyl-2, 3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile



ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735
Email : info@anaxlab.com
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US442743749&_cid=P12-MKDEE0-87371-1
Example 2: Synthesis of (5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methanesulfonyl-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile

Step i: Synthesis of Compound 11

Product 10 of step h (37.85 g, 78.28 mmol, 1.0 equiv.) was dissolved in THF (400 mL) at 23° C. A solution of hydrochloric acid (320 mL, 6M) was added dropwise over 20 min, and the mixture was stirred at 30° C. for 4 h. After this time, the reaction reached completion, as shown by LC/MS (MeCN/H 2O—20%→100%, 6 min). The reaction mixture was diluted with water (1 L) and EtOAc (0.6 L), back-extracted twice with EtOAc, and washed with water, sat. sol. NaHCO 3, and brine. The organic layer was dried over Na 2SO 4, filtered, and concentrated. The material (32.25 g, 94%) was triturated with CH 2Cl 2 (45 mL) at 45° C., filtered, and washed with a minimum of cold CH 2Cl 2 and cold hexanes to afford 11 as a white crystalline solid (26.15 g, 76%, 12:1 dr). 1H NMR (400 MHZ, DMSO-d 6) δ 7.96 (ddd, J=8.3, 2.7, 1.3 Hz, 1H), 7.89 (dd, J=8.9, 2.7 Hz, 1H), 7.57 (d, J=8.1 Hz, 1H), 6.66 (d, J=8.1 Hz, 1H), 5.95 (ddd, J=51.2, 13.5, 2.2 Hz, 1H), 5.89 (d, J=5.6 Hz, 1H), 5.47 (ddd, J=10.0, 6.2, 4.9 Hz, 1H), 5.26 (qd, J=52.5, 5.4 Hz, 1H), 5.12 (tddd, J=47.4, 18.7, 10.3, 2.7 Hz, 1H), 4.83 (t, J=5.4 Hz, 1H), 3.30 (s, 3H), 3.28-3.13 (m, 2H), 2.71-2.60 (m, 1H), 2.02-1.85 (m, 1H). 19F NMR (376 MHZ, DMSO-d 6) δ −112.3, −179.6, −196.7, −199.4. ESI MS [M+Na] + for C 21H 17F 4NO 3SNa, calcd 462.0, found 461.9.
PAT
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2023021476-A1Priority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of HIF-2αPublication Number: US-11407712-B2Priority Date: 2020-03-19Grant Date: 2022-08-09
- Tetralin and tetrahydroquinoline compounds as inhibitors of HIF-2alphaPublication Number: US-12103907-B2Priority Date: 2020-03-19Grant Date: 2024-10-01
- Tetralin and tetrahydroquinoline compounds as HIF-2α inhibitorsPublication Number: CN-115298165-APriority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2021317079-A1Priority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: WO-2021188769-A1Priority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2024254079-A1Priority Date: 2020-03-19
- Process for preparing tetralin compoundsPublication Number: US-12145901-B1Priority Date: 2021-09-17Grant Date: 2024-11-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of HIF-2alphaPublication Number: US-11787762-B2Priority Date: 2020-03-19Grant Date: 2023-10-17
- Tetrahydronaphthalene and tetrahydroquinoline compounds as HIF-2 alpha inhibitorsPublication Number: CN-119118872-APriority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as HIF-2α inhibitorsPublication Number: CN-115298165-BPriority Date: 2020-03-19Grant Date: 2024-09-17
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2025214930-A1Priority Date: 2020-03-19
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735
Email : info@anaxlab.com
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////Casdatifan, hypoxia-inducible factor (HIF) inhibitor, antineoplastic, AB 521, DP73UWL6LE
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions,, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
Cambritaxestat


Cambritaxestat
CAS 1979939-16-6
MFC25H22ClF3N4O2 MW502.9 g/mol
N-[(1S)-1-(4-chlorophenyl)ethyl]-3-[3-[[4-(trifluoromethoxy)phenyl]methyl]imidazo[4,5-b]pyridin-2-yl]propanamide
N-[(1S)-1-(4-chlorophenyl)ethyl]-3-(3-{[4-(trifluoromethoxy)phenyl]methyl}-3H-imidazo[4,5-b]pyridin-2-yl)propanamide
autotaxin inhibitor, antineoplastic, Orphan Drug, IOA 289, IOA-289, IOA289, LYY3P2KA27, CRT 0273750
- OriginatorCancer Research Technology; Merck & Co
- DeveloperiOnctura
- ClassAntifibrotics; Antineoplastics; Small molecules
- Mechanism of ActionAngiogenesis inhibitors; Cell proliferation inhibitors; ENPP2 protein inhibitors
- Orphan Drug StatusYes – Pancreatic cancer
- Phase I/IIPancreatic cancer
- Phase ISolid tumours
- PreclinicalNon-alcoholic steatohepatitis
- 14 Oct 2025Efficacy and adverse event data from a phase I/II trial in Pancreatic cancer released by iOnctura
- 04 Oct 2024Cambritaxestat is still in phase-I development in Solid-tumours (In volunteers) in Italy (PO, Capsule) (NCT05027568)
- 31 May 2024Efficacy and adverse event data from a phase I/II trial in Pancreatic cancer presented at the 60th Annual Meeting of the American Society of Clinical Oncology (ASCO-2024)
Cambritaxestat is an autotaxin inhibitor.
Cambritaxestat is an orally bioavailable small molecule inhibitor of autotaxin (ATX; ectonucleotide pyrophosphatase/phosphodiesterase family member 2; ENPP2), with potential antifibrotic and antineoplastic activities. Upon oral administration, cambritaxestat targets and binds to both the substrate pocket and the lysophosphatidic acid (LPA) carrier channel of ATX, thereby inhibiting the activity of ATX. This both directly inhibits the proliferation of tumor cells and reduces fibrosis in the tumor microenvironment (TME), allowing lymphocytes to infiltrate into the tumor and enhancing immune responses against tumor cells. ATX, a secreted glycoprotein with lysophospholipase D activity, hydrolyzes lysophosphatidylcholine (LPC) to LPA. LPA-mediated signaling plays an important role in cellular migration, proliferation and survival in fibrotic response. ATX and LPA are overexpressed in many tumors.
- A Study to Assess an ATX Inhibitor (IOA-289) in Healthy VolunteersCTID: NCT05027568Phase: Phase 1Status: CompletedDate: 2025-03-20
- A Study to Assess an ATX Inhibitor (IOA-289) in Patients with Metastatic Pancreatic CancerCTID: NCT05586516Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-03-20
SYN
WO2016/124939
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016124939&_cid=P22-MKBYYZ-98558-1


SYN

WO2016/124939 describes various ATX inhibitor compounds and their use in the treatment of proliferative disorders in which ATX activity is implicated, including Compound 1.
Compound 1 is example 40 in WO2016/124939, which document is incorporated herein by reference in its entirety. WO2016/124939 describes over 200 examples. Compound 1’s structure is according to Formula I.

PAT
- Autotaxin inhibitory compoundsPublication Number: US-10654846-B2Priority Date: 2015-02-06Grant Date: 2020-05-19
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-B3Priority Date: 2015-02-06Grant Date: 2024-05-29
- Autotaxin inhibitor compoundsPublication Number: ES-2778898-T7Priority Date: 2015-02-06Grant Date: 2024-11-15
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-A1Priority Date: 2015-02-06
- Home chemokine inhibiting compoundsPublication Number: CN-107428752-BPriority Date: 2015-02-06Grant Date: 2021-06-29
- Autotaxin inhibitory compoundsPublication Number: US-11453666-B2Priority Date: 2015-02-06Grant Date: 2022-09-27
- Autotaxin Inhibitory CompundsPublication Number: US-2020283435-A1Priority Date: 2015-02-06
- Autotaxin inhibitory compoundsPublication Number: WO-2016124939-A1Priority Date: 2015-02-06
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: WO-2022207648-A1Priority Date: 2021-03-29
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: EP-4313059-A1Priority Date: 2021-03-29
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: US-2024216385-A1Priority Date: 2021-03-29
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-B1Priority Date: 2015-02-06Grant Date: 2020-01-08
- Autotaxin inhibitory compoundsPublication Number: US-2018016274-A1Priority Date: 2015-02-06
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: WO-2022258693-A1Priority Date: 2021-06-09
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: US-2025057820-A1Priority Date: 2021-06-09
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: EP-4351563-A1Priority Date: 2021-06-09
- Autotaxin (ATX) inhibitors for the treatment of pancreatic cancerPublication Number: CN-117295496-APriority Date: 2021-06-09
- PI3K-δ inhibitors for the treatment of pancreatic cancerPublication Number: CN-116997340-APriority Date: 2021-03-29
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- Characterization and translational development of IOA-289, a novel autotaxin inhibitor for the treatment of solid tumorsPublication Name: Immuno-Oncology and TechnologyPublication Date: 2023-06PMCID: PMC10205783PMID: 37234285DOI: 10.1016/j.iotech.2023.100384
- The IUPHAR Guide to Immunopharmacology: connecting immunology and pharmacologyPublication Name: ImmunologyPublication Date: 2020-03-02PMCID: PMC7160657PMID: 32020584DOI: 10.1111/imm.13175
- Discovery of potent inhibitors of the lysophospholipase autotaxinPublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2016-11-15PMID: 27780639DOI: 10.1016/j.bmcl.2016.10.036
///////Cambritaxestat, autotaxin inhibitor, antineoplastic, Orphan Drug, IOA 289, IOA-289, IOA289, LYY3P2KA27, CRT 0273750
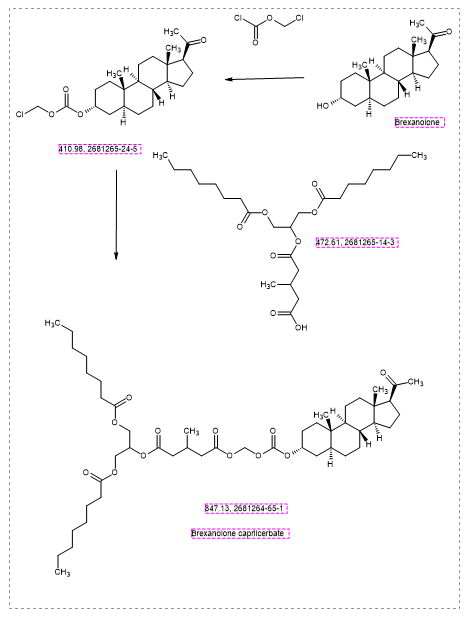
Brexanolone caprilcerbate



Brexanolone caprilcerbate
CAS 2681264-65-1
MFC48H78O12 MW 847.1 g/mol
1-O-[[(3R,5S,8R,9S,10S,13S,14S,17S)-17-acetyl-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl]oxycarbonyloxymethyl] 5-O-[1,3-di(octanoyloxy)propan-2-yl] 3-methylpentanedioate
1-[1,3-bis(octanoyloxy)propan-2-yl] 5-[({[(20-oxo-5α-pregnan3α-yl)oxy]carbonyl}oxy)methyl] 3-methylpentanedioate
GABAA receptor positive allosteric modulator, K3KLQ9T6WM, PHASE 2,
Brexanolone caprilcerbate (INNTooltip International Nonproprietary Name; developmental code names LYT-300, SPT-300) is an orally active prodrug of brexanolone (allopregnanolone) which is under development for the treatment of anxiety disorders.[1][2][3][4] It is a absorbed via the lymphatic system with oral administration.[5] The drug is being developed by Seaport Therapeutics and PureTech Health.[1][2] As of January 2025, it is in phase 2 clinical trials.[1]

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US335021515&_cid=P22-MKAJEO-33027-1
PAT
- Lipid prodrugs of neurosteroidsPublication Number: WO-2021159021-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2023338552-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2022395513-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2021268115-A1Priority Date: 2020-02-05
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Allopregnanolone prodrug”. AdisInsight. 28 January 2025. Retrieved 26 February 2025.
- “Delving into the Latest Updates on Brexanolone caprilcerbate with Synapse”. Synapse. 15 February 2025. Retrieved 26 February 2025.
- “Proposed INN: List 131 International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 38 (2): 270. 2024.
brexanolonum caprilcerbas brexanolone caprilcerbate 1-[1,3-bis(octanoyloxy)propan-2-yl] 5-[({[(20-oxo-5α-pregnan3α-yl)oxy]carbonyl}oxy)methyl] 3-methylpentanedioate GABAA receptor positive allosteric modulator C48H78O12 2681264-65-1
- Carlini SV, Osborne LM, Deligiannidis KM (December 2023). “Current pharmacotherapy approaches and novel GABAergic antidepressant development in postpartum depression”. Dialogues in Clinical Neuroscience. 25 (1): 92–100. doi:10.1080/19585969.2023.2262464. PMC 10557560. PMID 37796239.
- Alashal N, Hussain N (2025). “Approach to the use of rescue medications in children for prolonged epileptic seizures in the community”. Paediatrics and Child Health. 35 (4): 113–117. doi:10.1016/j.paed.2025.01.004.
| Clinical data | |
|---|---|
| Other names | LYT-300; LYT300; SPT-300; SPT300; Allopregnanolone 3-O-caprilcerbate |
| Routes of administration | Oral[1] |
| Drug class | GABAA receptor positive allosteric modulator; Neurosteroid |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2681264-65-1 |
| PubChem CID | 158098654 |
| UNII | K3KLQ9T6WM |
| Chemical and physical data | |
| Formula | C48H76O12 |
| Molar mass | 845.124 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////////Brexanolone caprilcerbate, GABAA receptor positive allosteric modulator, K3KLQ9T6WM, PHASE 2,
Branosotine
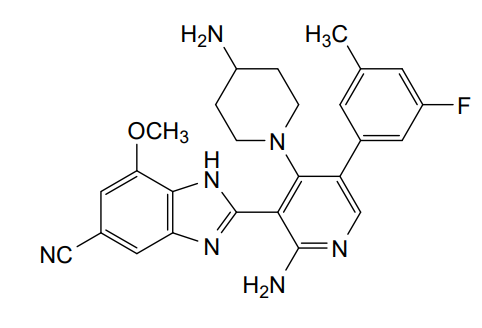
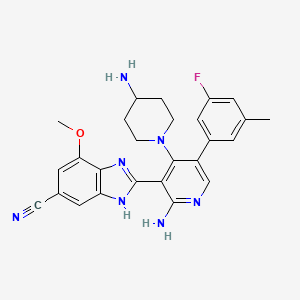
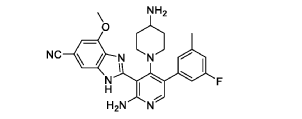
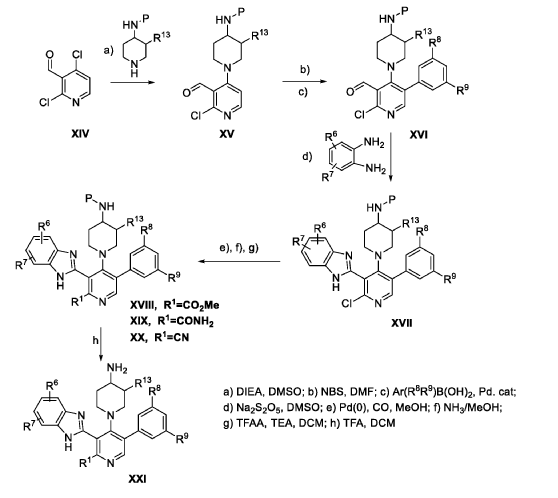
Branosotine
CAS 2412849-26-2
MF C26H26FN7O MW471.5 g/mol
2-[2-amino-4-(4-aminopiperidin-1-yl)-5-(3-fluoro-5-methylphenyl)-3-pyridinyl]-7-methoxy-3H-benzimidazole-5-carbonitrile
2-[2-amino-4-(4-aminopiperidin-1-yl)-5-(3-fluoro-5-
methylphenyl)pyridin-3-yl]-7-methoxy-1H-1,3-benzimidazole-5-
carbonitrile
somatostatin receptor agonist (veterinary use), 4L2VN6D3D8
Branosotine is a small molecule drug. The usage of the INN stem ‘-sotine’ in the name indicates that Branosotine is a non-peptidic somatostatin receptor agonist. Branosotine has a monoisotopic molecular weight of 471.22 Da.
SYN
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020061046&_cid=P21-MK9408-98104-1
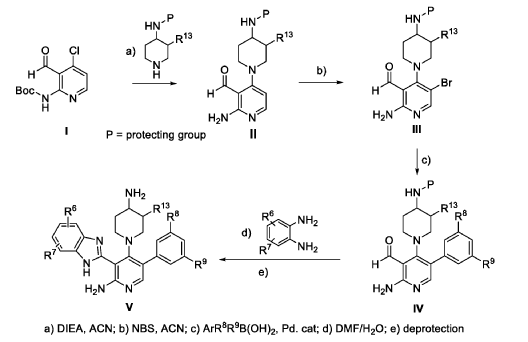
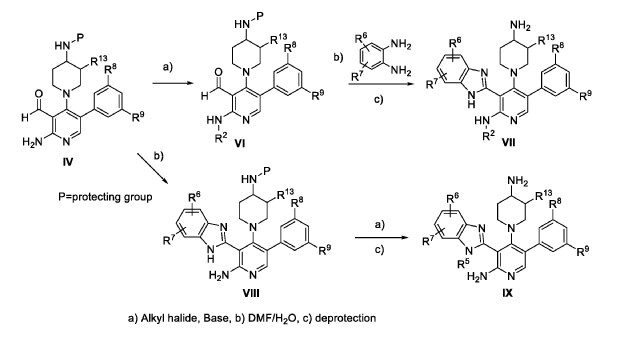
The following examples are provided for illustrative purposes only and not to limit the scope of the claims provided herein.
Example 1 : 2-[2-amino-4-(4-aminopiperidin-1-yl)-5-(3-fluoro-5-methylphenyl)pyridin- 3-yl]-4-methoxy-1H-1,3-benzodiazole-6-carbonitrile (1-1)



PAT
- Somatostatin modulators and uses thereofPublication Number: EP-4548974-A2Priority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: EP-3853218-B1Priority Date: 2018-09-18Grant Date: 2025-02-19
- Somatostatin modulators and uses thereofPublication Number: TW-I852944-BPriority Date: 2018-09-18Grant Date: 2024-08-21
- Somatostatin modulator and its usePublication Number: JP-2022501342-APriority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-11834462-B2Priority Date: 2018-09-18Grant Date: 2023-12-05
- Somatostatin modulators and uses thereofPublication Number: US-2022048924-A1Priority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-2020283453-A1Priority Date: 2018-09-18
- Somatostatin modulators and their usesPublication Number: JP-7431813-B2Priority Date: 2018-09-18Grant Date: 2024-02-15
- Somatostatin modulators and uses thereofPublication Number: US-2023022513-A1Priority Date: 2018-09-18
- Somatostatin modulators for treating pituitary adenomasPublication Number: WO-2021076448-A1Priority Date: 2019-10-14
- Somatostatin modulators and uses thereofPublication Number: US-2020087318-A1Priority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-10696689-B2Priority Date: 2018-09-18Grant Date: 2020-06-30
- Somatostatin modulators and uses thereofPublication Number: TW-202024095-APriority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-11186590-B2Priority Date: 2018-09-18Grant Date: 2021-11-30
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Branosotine, somatostatin receptor agonist (veterinary use), 4L2VN6D3D8
Bosmolisib
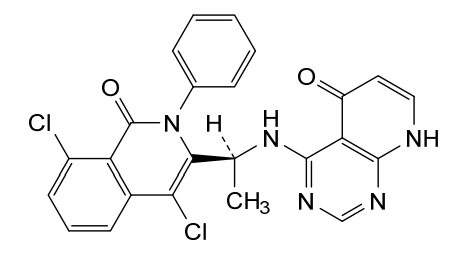
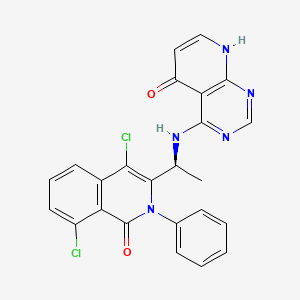
Bosmolisib
CAS 2055765-77-8
MF 2055765-77-8 MW478.3 g/mol
4-{[(1S)-1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl]amino}pyrido[2,3-d]pyrimidin-5(8H)-one
4-[[(1S)-1-(4,8-dichloro-1-oxo-2-phenylisoquinolin-3-yl)ethyl]amino]-8H-pyrido[2,3-d]pyrimidin-5-one
phosphatidylinositol 3-kinase (PI3K) inhibitor, antineoplastic, BR 101801, FJ5CTS1VNJ
- A Study of Bosmolisib (BR101801) in Participants With R/R PTCL.CTID: NCT07180771Phase: Phase 2Status: Not yet recruitingDate: 2025-09-18
- BR101801 in Adult Patients With Advanced Hematologic Malignancies(Phase I)CTID: NCT04018248Phase: Phase 1Status: CompletedDate: 2025-09-10
Bosmolisib is an orally bioavailable inhibitor of phosphoinositide 3-kinase delta (PI3-kinase subunit delta; PI3K-delta; PI3Kdelta) and DNA-dependent protein kinase (DNA-PK), with potential antineoplastic and immunomodulating activities. Upon oral administration, bosmolisib inhibits the activity of both PI3K-delta and DNA-PK. This prevents PI3K-mediated signaling pathways and may lead to the inhibition of cancer cell growth in PI3K-overexpressing tumor cells. Specifically, since PI3K regulates c-myc expression, inhibition of PI3K signaling may lead to a decrease in proliferation of c-myc-expressing tumor cells. Also, by inhibiting the activity of DNA-PK, bosmolisib interferes with the non-homologous end joining (NHEJ) process and prevents the repair of DNA double strand breaks (DSBs) caused by ionizing radiation or chemotherapeutic treatment. This increases chemo- and radiotherapy cytotoxicity by inhibiting the ability of tumor cells to repair damaged DNA. The PI3K pathway is upregulated in a variety of tumors and plays an important role in regulating cancer cell proliferation, growth, and survival. DNA-PK is activated upon DNA damage and plays a key role in repairing double-stranded DNA breaks. The enhanced ability of tumor cells to repair DSBs plays a major role in the resistance of tumor cells to chemo- and radiotherapy. In addition, bosmolisib is able to decrease Tregs and increase CD8 lymphocytes.
- OriginatorBoryung Pharmaceutical
- ClassAntineoplastics; Small molecules
- Mechanism of ActionDNA-activated protein kinase inhibitors; Phosphatidylinositol 3 kinase delta inhibitors; Phosphatidylinositol 3 kinase gamma inhibitors
- Phase IHaematological malignancies
- PreclinicalColorectal cancer
- 18 Sep 2025Boryung Pharmaceutical plans a phase II trial for Peripheral T Cell Lymphoma and Nodal T-follicular helper cell lymphoma (Second-line therapy or greater) in September 2025 (PO, Capsule) (NCT07180771)
- 06 Jan 2025Chemical structure information added.
- 09 Dec 2023Updated efficacy and adverse event data from a phase I trial in Hematological malignancies presented at the 65th American Society of Hematology Annual Meeting and Exposition (ASH-2023
SYN
WO 2016/204429.
SYN
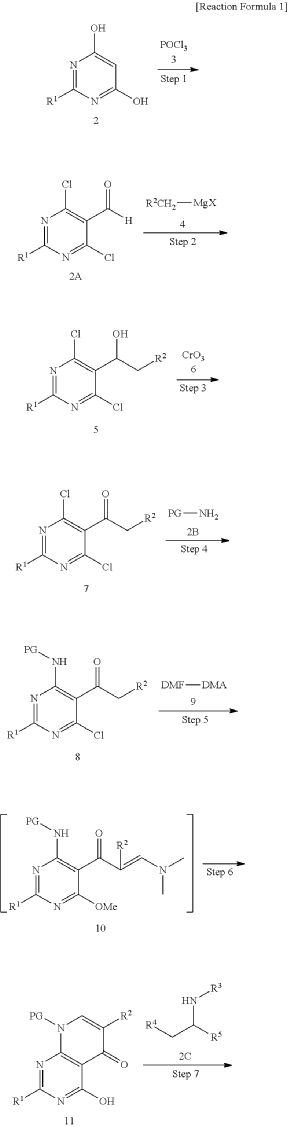
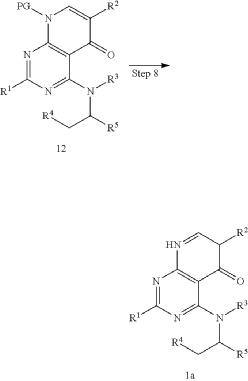
xample 1. Preparation of (S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidin-5(8H)-one
[116](S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidin-5(8H)-one (4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin -3-yl)ethyl)amino)pyrido[2,3-d]pyrimidin-5(8H)-one) represented by the chemical formula 3 above was prepared by the same method as that described in Example 10 of International Patent Publication No.
WO 2016/204429.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016204429&_cid=P22-MK6A2W-95428-1
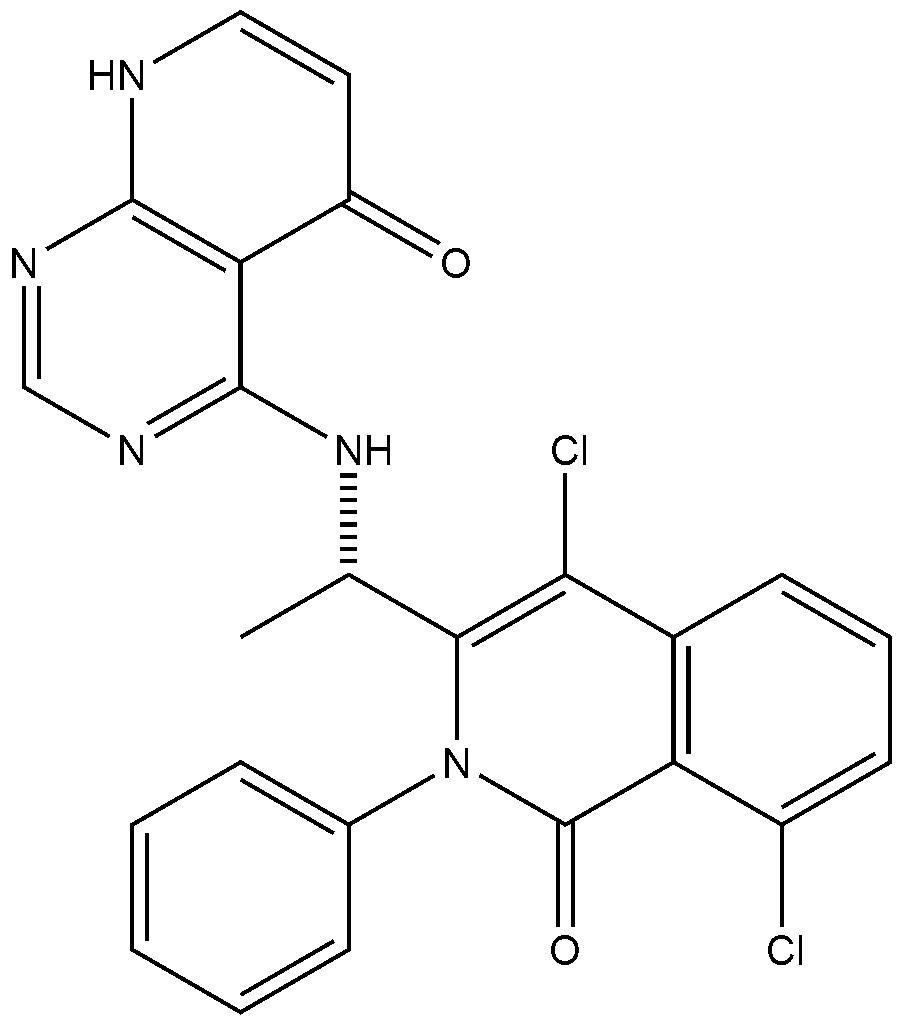
<Example 10> Preparation of (S)-4-((l-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido [2,3-d]pyrimidin-5(8H)-one
In Example 5, 50 mg (0.113 vol) of (S)-4— ((1-(8-chloro-1—oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido [2, 3-d]pyrimidin-5(8H)-one prepared was dissolved in 2 mL of acetic acid, and then 17 mg (0.124 vol) of N—chlorosuccinimide (NCS) was added. The mixture was stirred at 50 ° C for 15 hours, filtered under reduced pressure, neutralized using an aqueous sodium bicarbonate solution, and then the organic layer extracted by adding dichloromethane and water was dried (Na 2 SO 4 ), filtered, concentrated under reduced pressure, and separated by column chromatography (SiO 2 , eluent: dichloromethane/methanol, 30/1 -> dichloromethane/methanol, 10/1) to afford 25 mg (0.052 mmol, 46% yield) of compound (S)— 4-((1— (4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido[2, 3-d]pyramidin-5(8H)-one as a pale yellow solid.
LH NMR (300 MHz, CDC13) δ 10.99 (d, J = 4.8 Hz, 1Ή), 8.25 (s, 1H) , 7.95(dd, JJ = 1.9 Hz, J = 7.5 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H) , 7.46-7.62 (m, 6H), 7.20 (d, J = 6.7 Hz, 1H) , 6.3 (d, J = 7.5 Hz, 1H), 5.04 (t , J = 67.2 Hz, 1H) , 1.67 (d, J = 7.2 Hz, 3H) .
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US214732247&_cid=P22-MK69S5-86256-1
Example 10: Preparation of (S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinoline-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidine-5(8H)-one
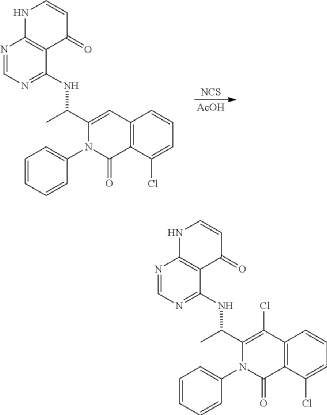
| 50 mg (0.113 mmol) of (S)-4-((1-(8-chloro-1-oxo-2-phenyl-1,2-dihydroisoquinoline-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidine-5(8H)-one prepared in Example 5 was dissolved in 2 mL of acetic acid, to which 17 mg (0.124 mmol) of N-chlorosuccinimide (NCS) was added, followed by stirring at 50° C. for 15 hours. The reaction mixture was filtered under reduced pressure. Saturated sodiumbicarbonate aqueous solution was added thereto, followed by neutralization. Dichloromethane and water were added thereto, followed by extraction. The extracted organic layer was dried (Na 2SO 4), filtered, and concentrated under reduced pressure. The residue was separated by column chromatography (SiO 2, eluent: dichloromethane/methanol, 30/1→dichloromethane/methanol, 10/1) to give 25 mg of the target compound (S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinoline-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidine-5(8H)-one as a pale yellow solid (0.052 mmol, yield: 46%). |
PAT
- A pharmaceutical composition for preventing or treating a heteroaryl derivative or a pharmaceutically acceptable salt thereof, a method for producing the same, and a PI3 kinase-related disease containing the heteroaryl derivative as an active ingredient.Publication Number: JP-6808905-B2Priority Date: 2015-06-18Grant Date: 2021-01-06
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, method of preparation thereof and pharmaceutical composition to prevent or treat diseases associated with PI3 kinases, which contains the same as active principlePublication Number: ES-2816050-T3Priority Date: 2015-06-18Grant Date: 2021-03-31
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method therefor, and pharmaceutical compostion for preventing or treating diseases associated with pi3 kinases, containing same as active ingredientPublication Number: US-2018105527-A1Priority Date: 2015-06-18
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method therefor, and pharmaceutical composition for preventing or treating diseases associated with pi3 kinases, containing same as active ingredientPublication Number: EP-3312175-B1Priority Date: 2015-06-18Grant Date: 2020-07-22
- Heteroaryl derivatives or pharmaceutically acceptable salts thereof, preparation method thereof and pharmaceutical composition for use in preventing or treating pi3 kinase related diseasesPublication Number: TW-I616446-BPriority Date: 2015-06-18Grant Date: 2018-03-01
- HETEROARYL DERIVATIVES OR PHARMACEUTICALLY ACCEPTABLE SALTS THEREOF, PROCESS FOR PRODUCING THE SAME, AND PHARMACEUTICAL COMPOSITIONS FOR PREVENTING OR TREATING PI3-KINASE RELATED DISEASES COMPRISING THE SAME AS THE ACTIVE INGREDIENTPublication Number: JP-2018522852-APriority Date: 2015-06-18
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method therefor, and pharmaceutical composition for preventing or treating diseases associated with PI3 kinases, containing same as active ingredientPublication Number: US-10526337-B2Priority Date: 2015-06-18Grant Date: 2020-01-07
- Heteroaryl derivative or a pharmaceutically acceptable salt thereof, a method for production thereof and a pharmaceutical composition for preventing or treating diseases associated with pi3 kinases, containing said active substancePublication Number: RU-2719367-C2Priority Date: 2015-06-18Grant Date: 2020-04-17
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method thereof, and pharmaceutical composition comprising same as active ingredient for preventing or treating PI3 kinase-associated diseasesPublication Number: CN-107690433-APriority Date: 2015-06-18
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- PI3Kδ/γ inhibitor BR101801 extrinsically potentiates effector CD8+ T cell-dependent antitumor immunity and abscopal effect after local irradiationPublication Name: Journal for ImmunoTherapy of CancerPublication Date: 2022-03PMCID: PMC8921929PMID: 35288465DOI: 10.1136/jitc-2021-003762
- Synergistic radiosensitizing effect of BR101801, a specific DNA-dependent protein kinase inhibitor, in various human solid cancer cells and xenograftsPublication Name: American journal of cancer researchPublication Date: 2021PMCID: PMC8640799PMID: 34873471
/////////bosmolisib, phosphatidylinositol 3-kinase (PI3K) inhibitor, antineoplastic, BR 101801, FJ5CTS1VNJ
Beroterkib


Beroterkib
CAS 2095719-92-7
MF C29H31ClFN5O5 MW584.0 g/mol
(2R)-2-(6-{5-chloro-2-[(oxan-4-yl)amino]pyrimidin-4-yl}-1,3-dihydro-2H-1-oxoisoindol-2-yl) -N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide
(2R)-2-[5-[5-chloro-2-(oxan-4-ylamino)pyrimidin-4-yl]-3-oxo-1H-isoindol-2-yl]-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide
(2R)-2-[5-[5-chloro-2-(oxan-4-ylamino)pyrimidin-4-yl]-3-oxo-1H-isoindol-2-yl]-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide
extracellular signal-regulated kinases (ERK) inhibitor, antineoplastic, ASTX029, ASTX 029, 14FDK6ISC9, Beroterkib anhydrous, AT 35029
Beroterkib Anhydrous is the anhydrous form of beroterkib, an orally bioavailable inhibitor of the extracellular signal-regulated kinases (ERK) 1 and 2, with potential antineoplastic activity. Upon administration, beroterkib specifically binds to and inhibits both ERK 1 and 2, thereby preventing the activation of mitogen-activated protein kinase (MAPK)/ERK-mediated signal transduction pathways. This results in the inhibition of ERK-dependent tumor cell proliferation and survival. The MAPK/ERK pathway is often upregulated in a variety of tumor cell types and plays a key role in the proliferation, differentiation and survival of tumor cells.
- Study of ASTX029 in Subjects With Advanced Solid TumorsCTID: NCT03520075Phase: Phase 1/Phase 2Status: CompletedDate: 2025-07-03
- Phase I/II Study of a Combination of Decitabine and Cedazuridine (ASTX727) and ASTX029, an ERK Inhibitor, for Patients With RAS Pathway Mutant Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative NeoplasmsCTID: NCT06284460Phase: Phase 1/Phase 2Status: WithdrawnDate: 2024-10-24
- A Phase 1 Study to Evaluate the Effect of Food on Pharmacokinetics of ASTX029CTID: NCT04466514Phase: Phase 1Status: CompletedDate: 2024-08-02
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017068412&_cid=P21-MK4TZX-17603-1
Example 685: (2R)-2-(6-{5-chloro-2-[(oxan-4-yl)amino]pyrimidin-4-yl}-1-oxo-2,3-dihydro- 1H-isoindol-2-yl)-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide

A stirred solution of (R)-2-(6-(5-chloro-2-((oxan-4-yl)amino)pyrimidin-4-yl)-1-oxoisoindolin-2- yl)propanoic acid (70 mg, 0.168 mmol), (S)-2-amino-2-(3-fluoro-5-methoxyphenyl)ethanol, HCl (41 mg, 0.185 mmol) and triethylamine (0.094 ml, 0.672 mmol) in DMF (1 ml) was treated with TBTU (65 mg, 0.202 mmol) and stirred at room temperature overnight. The mixture was diluted with ethyl acetate (20 ml), was washed successively with 1M KHSO4 (10 ml), NaHCO3 (10 ml), brine (2x 10 ml) and then water (4x 10 ml), was dried (MgSO4) and evaporated. The residue was purified by chromatography (SiO2, 12 g column, 0- 5% EtOOH in EtOAc) to give a glass, which was triturated with ether (2 ml) to give a solid. The solid was collected by filtration, washed with ether (2x 1 ml) and dried under vacuum at 50°C overnight to give the titlecompound (64.3 mg, 64.3 %) as a cream solid. 1H NMR (DMSO, 400 MHz) δ 8.56 (1H, d), 8.44 (1H, s), 8.07 ‒ 8.00 (1H, m), 7.97 (1H, dd), 7.74 (1H, d), 7.61 (1H, s), 6.76 ‒ 6.64 (3H, m), 4.99 (1H, q), 4.91 (1H, t), 4.86 ‒ 4.70 (2H, m), 4.60 (1H, d), 4.00 ‒ 3.80 (3H, m), 3.76 (3H, s), 3.60 ‒ 3.47 (2H, m), 3.40 ‒ 3.33 (2H, m), 1.84 (2H, d), 1.59 ‒ 1.39 (5H, m). ). LCMS: [M+H]+ = 584.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US237389744&_cid=P21-MK4U5F-21416-1
Example 685: (2R)-2-(6-{5-chloro-2-[(oxan-4-yl)amino]pyrimidin-4-yl}-1-oxo-2,3-dihydro-1H-isoindol-2-yl)-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide

SYN

PAT
- Conjugates comprising hydroxyalkyl starch and a cytotoxic agent and process for their preparationPublication Number: AU-2011276120-B2Priority Date: 2010-07-09Grant Date: 2013-12-19
- Conjugates comprising hydroxyalkyl starch and a cytotoxic agent and process for their preparationPublication Number: AU-2011276120-A1Priority Date: 2010-07-09
- Combustion modified flexible polyurethane foamPublication Number: GB-2124634-APriority Date: 1982-07-26
- Benzolactam compounds as protein kinase inhibitorsPublication Number: ES-2989326-T3Priority Date: 2015-10-21Grant Date: 2024-11-26
- Protein kinase inhibitor benzolactam compoundsPublication Number: CN-114948963-APriority Date: 2015-10-21
- Benzolactam compounds as protein kinase inhibitorsPublication Number: US-2024368136-A1Priority Date: 2015-10-21
- Protein Kinase Inhibitors Benzolactam CompoundsPublication Number: CN-108617166-BPriority Date: 2015-10-21Grant Date: 2022-05-17
- Benzolactam compounds as protein kinase inhibitorsPublication Number: CN-114948963-BPriority Date: 2015-10-21Grant Date: 2025-05-27
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
REF
- Discovery of ASTX029, a clinical candidate which modulates the phosphorylation and catalytic activity of ERK1/2Publication Name: Journal of Medicinal ChemistryPublication Date: 2021-10-06PMID: 34387469DOI: 10.1021/acs.jmedchem.1c00905
- ASTX029, a Novel Dual-mechanism ERK Inhibitor, Modulates Both the Phosphorylation and Catalytic Activity of ERKPublication Name: Molecular Cancer TherapeuticsPublication Date: 2021-07-30PMID: 34330842DOI: 10.1158/1535-7163.mct-20-0909
//////////////Beroterkib, extracellular signal-regulated kinases (ERK) inhibitor, antineoplastic, ASTX029, ASTX 029, 14FDK6ISC9, Beroterkib anhydrous, AT 35029
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}