
Home » Uncategorized (Page 7)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Nedometinib



Nedometinib
CAS 2252314-46-6
NFX-179, K5T4I78IYZ
| Molecular Weight | 470.24 |
|---|---|
| Formula | C17H16FIN4O3 |
2-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1-methylpyrrolo[2,3-b]pyridine-3-carboxamide
- EN300-27122249
- 1H-Pyrrolo[2,3-b]pyridine-3-carboxamide, 2-[(2-fluoro-4-iodophenyl)amino]-N-(2-hydroxyethoxy)-1-methyl-
- 2-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1-methylpyrrolo[2,3-b]pyridine-3-carboxamide
Nedometinib (NFX-179) is a specific MEK1 inhibitor with an IC50 of 135 nM. Nedometinib inhibits p-ERK, MAPK. Nedometinib exerts anticancer activity against squamous cell carcinoma. Nedometinib can be used for research in dermatosis, neurofibromatosis.
Nedometinib is a topical gel formulation composed of an inhibitor of mitogen-activated protein kinase kinase (MAP2K; MAPKK; MEK), with potential antineoplastic activity. Upon topical administration, nedometinib penetrates into the dermis of the skin where it specifically targets, binds to and inhibits the catalytic activity of MEK, thereby inhibiting the activation of MEK-dependent effector proteins including extracellular signal-regulated kinase (ERK) and inhibits the proliferation of tumor cells in which the RAS/RAF/MEK/ERK signaling pathway is overactivated. The threonine/tyrosine protein kinase MEK plays a key role in the RAS/RAF/MEK/ERK signaling pathway, which is frequently upregulated in a variety of tumor cell types and regulates key cellular activities including cell growth, proliferation, survival, differentiation and apoptosis. Rapid degradation of NFX-179 upon reaching the systemic circulation minimizes side effects caused by systemic exposure.
SCHEME

PATENTS
US11161845, https://patentscope.wipo.int/search/en/detail.jsf?docId=US295432044&_cid=P20-MC8HLL-16550-1
Example 2: 2-((2-Fluoro-4-iodophenyl)amino)—N-(2-hydroxyethoxy)-1-methyl-1H-pyrrolo[2,3-b]pyridine-3-carboxamide
2-((2-Fluoro-4-iodophenyl)amino)-1-methyl-1H-pyrrolo[2,3-b]pyridine-3-carbonyl chloride
Alternative 1 for the preparation of 2-((2-Fluoro-4-iodophenyl)amino)—N-(2-hydroxyethoxy)-1-methyl-1H-pyrrolo[2,3-b]pyridine-3-carboxamide
Alternative 2 for the Preparation of 2-((2-Fluoro-4-iodophenyl)amino)—N-(2-hydroxyethoxy)-1-methyl-1H-pyrrolo[2,3-b]pyridine-3-carboxamide
PATENTS
WO2018213810
Science Translational Medicine (2023), 15(717), eade1844
WO2018213810, Nflection Therapeutics, Inc.
WO2023096935
WO2022262797
WO2020106303
WO2020106304
WO2020106303
WO2020106307
WO2020106304
WO2018213810
WO2020106304
WO2020106303
REF
/////////Nedometinib, NFX-179, NFX 179, K5T4I78IYZ, EN300-27122249
Modoflaner



Modoflaner ‘
| Molecular Weight | 715.23 |
|---|---|
| Formula | C23H10F12IN3O2 |
| CAS No. | 1331922-53-2 |
6-fluoro-N-[2-fluoro-3-[[4-(1,1,1,2,3,3,3-heptafluoropropan-2-yl)-2-iodo-6-(trifluoromethyl)phenyl]carbamoyl]phenyl]pyridine-3-carboxamide
- 3-Pyridinecarboxamide, 6-fluoro-N-(2-fluoro-3-(((2-iodo-4-(1,2,2,2-tetrafluoro-1-(trifluoromethyl)ethyl)-6-(trifluoromethyl)phenyl)amino)carbonyl)phenyl)-
- 6-fluoro-N-[2-fluoro-3-[[4-(1,1,1,2,3,3,3-heptafluoropropan-2-yl)-2-iodo-6-(trifluoromethyl)phenyl]carbamoyl]phenyl]pyridine-3-carboxamide
- 6-Fluoro-N-(2-fluoro-3-((2-iodo-4-(perfluoropropan-2-yl)-6-(trifluoromethyl)phenyl)carbamoyl)phenyl)nicotinamide
E583FHZ8C9
Modoflaner is an isophenylamide insecticide. Modoflaner may act through allosteric regulation of gamma-aminobutyric acid-gated chloride channels.
SCHEME

PATENT
WO2019059412
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019059412&_cid=P20-MC71WG-08056-1
SYN

Modoflaner is another isophthalamide insecticide developed by Mitsui Chemicals Agriculture Co., Ltd. in Japan. Its structure is similar to broflanilide and cyproflanilide created in China, except that it introduces iodine and fluoropyridine structures. It is speculated that the mechanism of action of modoflaner is mainly through allosteric regulation of
γ -aminobutyric acid-gated chloride ion channels, which is similar to isoxazoline insecticides and acaricides such as mivolana and eumivolana. Indoor bioassay studies have shown that modoflaner has a killing rate of more than 70% (6 days) against Spodoptera litura, Plutella xylostella and Laodelphax
striatum at a concentration of 100 mg/L. It has a killing rate of 95% (48 hours) against adult
Ctenocephalides felis at a dose of 0.04 μg/
cm2 or 0.0064 mg/ L . It has a killing rate of 90% (48 hours)
against nymphs of American flower ticks, adults of
Ixodes ricinus and adults of
R. sanguineus at a dose of 0.2 μg/cm2. It can prevent female adults of R. sanguineus from laying eggs or hatching eggs after 7 days of in vitro injection at a dose of 0.032 μg/tick. The creation idea and synthetic route of Modoflaner are shown in Figure 2. The synthetic order of iodination and amidation deserves further study.
//////////Modoflaner, E583FHZ8C9
MIVORILANER



MIVORILANER
1414642-93-5
| Molecular Formula | C22H17Cl2F6N3O3S |
| Molecular Weight | 588.35 |
- 3-[(5S)-5-(3,5-Dichloro-4-fluorophenyl)-4,5-dihydro-5-(trifluoromethyl)-3-isoxazolyl]-N-[2-[(2,2-difluoroethyl)amino]-2-oxoethyl]-5,6-dihydro-4H-cyclopenta[c]thiophene-1-carboxamide (ACI)
- 3-[(5S)-5-(3,5-dichloro-4-fluorophenyl)-5-(trifluoromethyl)-4,5-dihydroisoxazol-3-yl]-N-[2-[(2,2-difluoroethyl)amino]-2-oxoethyl]-5,6-dihydro-4H-cyclopenta[c]thiophene-1-carboxamide
- ITABH 19-01
- LY 3116151
- WHO 11674
- XN7QGY28HM
- HI-154
1-[(5S)-5-(3,5-dichloro-4-fluorophenyl)-5-(trifluoromethyl)-4H-1,2-oxazol-3-yl]-N-[2-(2,2-difluoroethylamino)-2-oxoethyl]-5,6-dihydro-4H-cyclopenta[c]thiophene-3-carboxamide
- (S)-3-(5-(3,5-Dichloro-4-fluorophenyl)-5-(trifluoromethyl)-4,5-dihydroisoxazol-3-yl)-N-(2-((2,2-difluoroethyl)amino)-2-oxoethyl)-5,6-dihydro-4H-cyclopenta[c]thiophene-1-carboxamide
- 1-[(5S)-5-(3,5-dichloro-4-fluorophenyl)-5-(trifluoromethyl)-4H-1,2-oxazol-3-yl]-N-[2-(2,2-difluoroethylamino)-2-oxoethyl]-5,6-dihydro-4H-cyclopenta[c]thiophene-3-carboxamide
MIVORILANER is a small molecule drug with a maximum clinical trial phase of I and has 1 investigational indication.
Mivorilaner, an antineoplastic, can be used for the research of veterinary medicine
SCHEME

PATENT
WO2012155676
(S)-3-[5-(3,5-dichloro-4-fluoro-phenyl)-5-trifluoromethyl-4,5-dihydro-isoxazol-3-yl]-5,6-dihydro-4H-cyclopenta[c]thiophene-1-carboxylic acid [(2,2-difluoro-ethylcarbamoyl)-methyl]-amide
3 g of 3-[5-(3,5-dichloro-4-fluoro-phenyl)-5-trifluoromethyl-4,5-dihydro-isoxazol-3-yl]-5,6-dihydro-4H-cyclopenta[c]thiophene-1-carboxylic acid [(2,2-difluoro-ethylcarbamoyl)-methyl]-amide is separated by SFC separation to give desired product (1.4 g, 93%). SFC conditions are as follows: Instrument: Thar 350 Column: AD 250 mm*50 mm, 10 um Mobile phase: A: Supercritical CO2, B: EtOH, A:B=60:40 at 240 ml/min Column Temp: 38° C. Nozzle Pressure: 100 Bar Nozzle Temp: 60° C. Evaporator Temp: 20° C. Trimmer Temp: 25° C. Wavelength: 220 nm. 1H NMR (CDCl3, 400 MHz): δ 7.56 (d, J=6.0, 2H), 6.64 (brs, 1H), 6.40 (brs, 1H), 6.03-5.73 (m, 1H), 4.15 (d, J=5.2, 2H), 4.01 (d, J=17.2, 1H), 3.74-3.65 (m, 1H), 3.62 (d, J=17.2, 1H), 2.97 (t, J=7.6, 2H), 2.89 (t, J=7.6, 2H), 2.56 (m, 2H).
WO2012158396
(WO2012155676, Example 245).
/////////MIVORILANER, ITABH 19-01, LY 3116151, XN7QGY28HM, WHO 11674, HI-154
LEVALBUTEROL TARTRATE


LEVALBUTEROL TARTRATE
Levosalbutamol
cas 661464-94-4
4-[(1R)-2-(tert-butylamino)-1-hydroxyethyl]-2-(hydroxymethyl)phenol;(2R,3R)-2,3-dihydroxybutanedioic acid
MW 628.7, C30H48N2O12
- Xopenex HFA
- Levosalbutamol tartrate
- ADS4I3E22M
- UNII-ADS4I3E22M
- Levosalbutamol tartrate(levalbuterol) is the R-enantiomer of the short-acting β2-adrenergic receptor agonist salbutamol.
Levalbuterol Tartrate is the tartrate salt form of levalbuterol, the R-enantiomer of the short-acting beta-2 adrenergic receptor agonist albuterol, with bronchodilator activity. Levalbuterol selectively binds to beta-2 adrenergic receptors in bronchial smooth muscle, thereby activating intracellular adenyl cyclase, an enzyme that catalyzes the conversion of adenosine triphosphate (ATP) to cyclic-3′,5′-adenosine monophosphate (cAMP). Increased cAMP levels cause relaxation of bronchial smooth muscle, relieve bronchospasms, improve mucociliary clearance and inhibit the release of mediators of immediate hypersensitivity from cells, especially from mast cells.
British patent document GB1298494A firstly discloses synthesis of levosalbutamol, which comprises the steps of carrying out crystallization resolution by using D- (+) -dibenzoyl tartaric acid, carrying out ester reduction reaction, and removing two benzyl protecting groups to obtain levosalbutamol, wherein the process route is as follows:
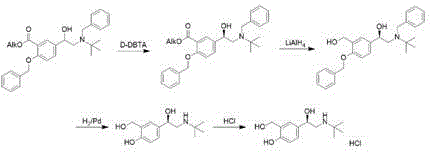
chinese patent CN1705634A, and using rhodium and chiral bidentate phosphine ligand combination, levosalbutamol can be obtained with good yield and good optical purity on a technical scale. The disadvantages are that the toxicity of the reagent is high, the hydrogenation risk is high, and the process route is as follows:
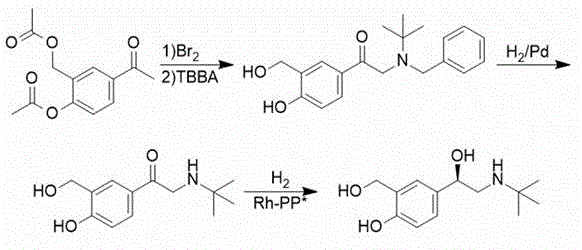
SCHEME

PATENTS
MX2012014342
IN2009MU01097
IN2007CH01847
US20040115136
CN1382685
PATENT
https://patents.google.com/patent/CN114539077A/en
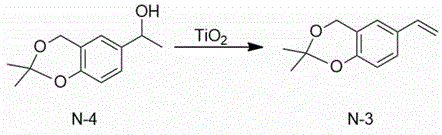
The technical scheme adopted by the invention is as follows: 1) 1- (2, 2-dimethyl-4H-benzo [ d ] [1,3] dioxin-6-yl) ethanol and titanium dioxide are used as initial raw materials, a solvent-free system is adopted, and 2, 2-dimethyl-6-vinyl-4H-benzo [ d ] [1,3] dioxin is synthesized through dehydration.
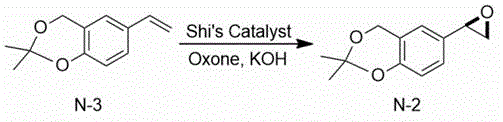
Then, the 2, 2-dimethyl-6-vinyl-4H-benzo [ D ] [1,3] dioxin is subjected to epoxidation under the combined action of 1,2:4, 5-di-O-isopropylidene-BETA-D-erythro-2, 3-dione-2, 6-pyranose (Shi’s Catalyst), Oxone and potassium hydroxide to obtain (R) -2, 2-dimethyl-6- (oxirane-2-yl) -4H-benzo [ D ] [1,3] dioxin.
Reacting and condensing (R) -2, 2-dimethyl-6- (epoxy ethane-2-group) -4H-benzo [ D ] [1,3] dioxin and tert-butylamine in ethanol, and salifying with D- (+) -malic acid to obtain (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-group) ethanol D- (+) -malate.
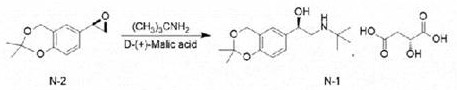
And (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate is subjected to hydroaminolysis and reacts with hydrogen chloride ethanol to prepare the levosalbutamol hydrochloride.
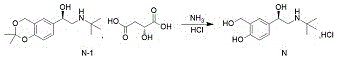
The invention discloses a novel method for synthesizing levalbuterol hydrochloride, wherein the synthesis of a key intermediate is novel without cyclization oxidation, the total yield is 85-90%, and the method is higher than that of the conventional method. The process is convenient to operate, the raw materials are economical, and the method is suitable for large-scale industrial production.
EXAMPLE 12 preparation of 2, 2-dimethyl-6-vinyl-4H-benzo [ d ] [1,3] dioxine
A1000 mL flask was charged with 208g (1.0 mol) of 1- (2, 2-dimethyl-4H-benzo [ d ] [1,3] dioxin-6-yl) ethanol, which was accurately weighed, and stirring was started. Then slowly adding 16g (0.2 mol) of titanium dioxide, installing a water separator and a water flow pipe, starting heating until the internal temperature is kept at 120-130 ℃, and stirring for 12 hours. After the reaction is finished, the temperature is reduced to below 50 ℃, the water separator is removed, the reduced pressure distillation device is changed, and 120 ℃ (less than 100 Pa) fraction is collected to obtain 180.7g of 2, 2-dimethyl-6-vinyl-4H-benzo [ d ] [1,3] dioxin with the yield of 95%.
Mass spectrum: EI (m/z): 190; hydrogen nuclear magnetic resonance spectroscopy:1HNMR(400MHz,CDCl3)δ7.55(d,J=4Hz,1H),7.11(s,1H),6.87(d,J=4Hz,1H),6.65~6.60(m,1H),5.63~5.60(m,1H),5.19~5.5(m,1H),4.59(s,2H),1.49(s,6H)。
EXAMPLE 2 Synthesis of (R) -2, 2-dimethyl-6- (oxiran-2-yl) -4H-benzo [ d ] [1,3] dioxine
A clean 5000mL three-neck flask is taken, 180.5g (0.95 mol) of the compound
2, 2-dimethyl-6-vinyl-4H-benzo [ D ] [1,3] dioxin obtained in the example 1 is added, 2000mL of acetonitrile is added for dissolution, 1,2:4, 5-di-O-isopropylidene-BETA-D-erythro-2, 3-dione-2, 6-pyranose 49.1g (0.19 mol) is added, potassium monopersulfate (Oxone) 876g (1.43 mol) is added under stirring, a proper amount of potassium hydroxide is added after the addition is finished, the pH of the system is kept between 10 and 11, and the stirring reaction is continued at 25 ℃ for 8 to 12 hours. After the reaction, the mixture was slowly poured into 2000ml of purified water prepared in advance, stirred sufficiently for 30min, and then was allowed to stand for layering, and the organic layer was collected. 2000ml of dichloromethane is added for extraction, organic layers are combined and washed by saturated sodium chloride solution, the organic layer is dried by adding anhydrous sodium sulfate and concentrated to dryness to obtain 196g of crude colorless liquid with the yield of 100 percent.
Mass spectrum: EI (m/z): 207; hydrogen nuclear magnetic resonance spectroscopy:1HNMR(400MHz,CDCl3)δ7.25(s,1H),7.18(d,J=4Hz,1H),6.85(d,J=4Hz,1H),4.59(s,2H),3.85~3.81(m,1H),2.96~2.71(m,2H),1.49(s,6H)。
EXAMPLE 3 preparation of (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate salt
A clean 5000mL three-neck flask is taken, the compound (R) -2, 2-dimethyl-6- (oxiranyl-2-yl) -4H-benzo [ d ] [1,3] dioxin obtained in the example 2 is added, 196g (0.95 mol) of the clean 5000mL three-neck flask is taken, 1000mL of ethanol is added for dissolution, 80.4g (1.1 mol) of tert-butylamine is added, stirring is started, heating is carried out till reflux, reaction is carried out for 3H, and the progress of the reaction is detected by TLC. After the reaction is finished, 127g (0.95 mol) of D- (+) -malic acid is added in batches, and stirring and refluxing are continued for 2h after the addition is finished. And then cooling to 5-15 ℃, precipitating a large amount of solid, stirring for 3H, filtering, washing the filter cake with ethanol, collecting the filter cake, and drying to obtain (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate 372g of white solid with the yield of 94.7%.
Mass spectrum: ESI (m/z): 280.1, respectively; hydrogen nuclear magnetic resonance spectroscopy:1HNMR(400MHz,d-DMSO)δ7.25(s,1H),7.18(d,J=4Hz,1H),6.85(d,J=4Hz,1H),4.90~4.76(m,2H),4.59(s,2H),4.44~4.40(m,2H),3.65(br,2H),3.15~2.90(m,2H),2.77~2.52(m,2H),2.03(s,1H),1.50(s,6H),1.27(s,9H)。
EXAMPLE 4 preparation of L-salbutamol hydrochloride
A5000 mL beaker was charged with 372g of the compound (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate salt obtained in example 3, 1500mL of purified water was added, and the mixture was stirred to dissolve it, followed by addition of 1500mL of dichloromethane and cooling in an ice bath. Slowly adding a proper amount of concentrated ammonia water under stirring to adjust the pH value of the water phase to 9-10, continuously stirring for 30min, and standing for layering. Separating and collecting organic layer, adding 1000ml of dichloromethane into water layer, stirring for 10min, standing and demixing. Separating and collecting organic layers, combining the organic layers, adding 2000ml of saturated sodium chloride solution into the organic layers, stirring for 30min, standing for layering, collecting the organic layers, adding a proper amount of anhydrous sodium sulfate, drying, filtering, washing with dichloromethane, and collecting filtrate.
And (3) carrying out rotary evaporation and concentration on the filtrate to about 1500mL, transferring the concentrated filtrate into a 5000mL three-neck bottle, and placing the three-neck bottle in an ice bath to cool the three-neck bottle to 5-15 ℃. About 110g of 30% hydrogen chloride ethanol solution is dropwise added under stirring, and after the dropwise addition is finished, 2000mL of methyl tertiary butyl ether is dropwise added under stirring, so that a large amount of white solid is precipitated. And after the addition is finished, continuously stirring for 3 hours at the temperature of 5-15 ℃, filtering, adding methyl tert-butyl ether into a filter cake for washing, collecting the filter cake, and drying to obtain 241.5g with the yield of 97.3%. Through HPLC analysis, the purity is 99.95%, and the isomer content is not detected, as shown in figures 1-4. The total yield of the four-step reaction is 87.5 percent.
Publication numberPriority datePublication dateAssigneeTitle
CN1413976A *2002-09-132003-04-30苏州君宁新药开发中心有限公司New process for preparing levo-albuterol
US20050261368A1 *2004-05-202005-11-24Valeriano MerliPreparation of levalbuterol hydrochloride
CN103951568A *2014-05-192014-07-30苏州弘森药业有限公司New process for synthesizing salbutamol and sulfate of salbutamol
CN104557572A *2014-12-302015-04-29上海默学医药科技有限公司Levalbuterol intermediate and levalbuterol hydrochloride synthesis method
CN110963929A *2019-11-262020-04-07安徽恒星制药有限公司Preparation method of salbutamol hydrochloride suitable for industrial production
CN113227113A *2018-12-202021-08-06帝斯曼知识产权资产管理有限公司Improved synthesis of epoxidation catalysts
CN113801029A *2020-06-162021-12-17盈科瑞(天津)创新医药研究有限公司Preparation method of levalbuterol hydrochloride
//////////Levosalbutamol, LEVALBUTEROL TARTRATE, Xopenex HFA, Levosalbutamol tartrate, ADS4I3E22M, UNII-ADS4I3E22M
Ibuzatrelvir



Ibuzatrelvir
PF-07817883
CAS 2755812-39-4
| Molecular Weight | 489.49 |
|---|---|
| Formula | C21H30F3N5O5 |
- Ibuzatrelvir
- N-(Methoxycarbonyl)-3-methyl-L-valyl-(4R)-N-[(1S)-1-cyano-2-((3S)-2-oxopyrrolidin-3-yl)ethyl]-4-(trifluoromethyl)-L-prolinamide
- PF 07817883
- methyl N-[(2S)-1-[(2S,4R)-2-[[(1S)-1-cyano-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl]carbamoyl]-4-(trifluoromethyl)pyrrolidin-1-yl]-3,3-dimethyl-1-oxobutan-2-yl]carbamate
- KZ2X7QH2VT
Ibuzatrelvir (development code PF-07817883) is an experimental antiviral drug being developed by Pfizer for the treatment of COVID-19.[1] It is a second-generation improvement over nirmatrelvir which has a similar chemical structure.[2] One of the disadvantages of nirmatrelvir is that it has low metabolic stability and must be given in combination with ritonavir (as Paxlovid) to limit its metabolic degradation in the body.[3] Ibuzatrelvir incorporates modifications to the chemical structure of nirmatrelvir that give it enhanced oral bioavailability, so it does not require coadministration with ritonavir.[3]
Ibuzatrelvir (PF-07817883), a second-generation, orally bioavailable, is SARS-CoV-2 main protease (Mpro and 3CLpro) inhibitor with improved metabolic stability. Ibuzatrelvir has demonstrated pan-human coronavirus antiviral activity and off-target selectivity profile in vitro and in preclinical animal studies. Ibuzatrelvir is well tolerated with a safety profile similar to placebo and prevents viral infection and transmission. Ibuzatrelvir can be used to inhibit COVID-19.
SCHEME
SIDECHAIN

MAIN

PATENT
WO2021250648 PFIZER
WO2023215910
PAPER
The Pfizer scientists described ibuzatrelvir’s medicinal chemistry campaign in a Journal of Medicinal Chemistry paper that was published in April 2024 (DOI: 10.1021/acs .jmedchem.3c02469).
https://pubs.acs.org/doi/10.1021/jacsau.4c00508
Ibuzatrelvir (1) was recently disclosed and patented by Pfizer for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It has received fast-track status from the USA Food and Drug Administration (FDA) and has entered phase III clinical trials as a possible replacement for Paxlovid. Like nirmatrelvir (2) in Paxlovid, this orally active drug candidate is designed to target viral main proteases (Mpro) through reversible covalent interaction of its nitrile warhead with the active site thiol of the chymotrypsin-like cysteine protease (3CL protease). Inhibition of Mpro hinders the processing of the proteins essential for viral replication in vivo. However, ibuzatrelvir apparently does not require ritonavir (3), which is coadministered in Paxlovid to block human oxidative metabolism of nirmatrelvir. Here, we report the crystal structure of the complex of ibuzatrelvir with the active site of SARS-CoV-2 Mpro at 2.0 Å resolution. In addition, we show that ibuzatrelvir also potently inhibits the Mpro of Middle East respiratory syndrome-related coronavirus (MERS-CoV), which is fortunately not widespread but can be dangerously lethal (∼36% mortality). Co-crystal structures show that the binding mode of the drug to both active sites is similar and that the trifluoromethyl group of the inhibitor fits precisely into a critical S2 substrate binding pocket of the main proteases. However, our results also provide a rationale for the differences in potency of ibuzatrelvir for these two proteases due to minor differences in the substrate preferences leading to a weaker H-bond network in MERS-CoV Mpro. In addition, we examined the reversibility of compound binding to both proteases, which is an important parameter in reducing off-target effects as well as the potential immunogenicity. The crystal structures of the ibuzatrelvir complexes with Mpro of SARS-CoV-2 and of MERS-CoV will further assist drug design for coronaviral infections in humans and animals.


General Boc-Deprotection and Coupling Procedure
This procedure was based on a literature procedure.1
The Boc-protected building block (1.0
equiv) was dissolved in 50/50 TFA/DCM and stirred for 1 h at room temperature. The reaction
mixture was then concentrated in vacuo and co-evaporated with DCM (5 × 5 mL). In a separate
RBF the carboxylic acid building block (1.0 equiv) and HATU (1.0 equiv) were dissolved in
DMF. HOAt (0.6 M in DMF) (0.1 equiv) and DIPEA (3.0 equiv) were added and the reaction
mixture was left to incubate at room temperature for 10 mins, as it turned yellow. The previously
concentrated Boc-deprotected building block was dissolved in DMF and added dropwise to the
incubating solution. The reaction mixture was capped under a blanket of argon and stirred at room
temperature for 2–3 h. The reaction mixture was diluted with 5 mL each of water and ethyl acetate
and the layers separated. The aqueous layer was extracted further with ethyl acetate (3 × 5 mL),
and all ethyl acetate layers combined and washed with sat. aq. NaHCO3 (10 mL), 1 M HCl (10
mL), water (2 × 10 mL) and brine (10 mL). It was then dried over Na2SO4, filtered, and
concentrated in vacuo to furnish the product.
Methyl ((S)-1-((2S,4R)-2-(((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)carbamoyl)-4-
(trifluoromethyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (1) Ibuzatrelvir
This known compound was synthesized according to the General Boc-Deprotection and
Coupling Procedure with building blocks 7 and 8. The characterization data matches the literature
report (IPN: WO2021250648A1). The crude material was obtained as a dark yellow sticky residue
that was then purified with flash column chromatography with an eluent of 92:8 EtOAc:MeOH.
The desired compound had an Rf
= 0.40 and was visible with KMnO4 stain. After concentration
of desired fractions, 1 was isolated as a clear, colorless oil that solidified to a white solid (0.051 g,
53%) This compound was isolated and used for all experiments as a mixture of diastereomers in a
ratio of about 2:1 and rotamers present, with only the major set of resonances reported, which are
for the desired isomer. It can be separated using high performance liquid chromatography (HPLC)
methods, as listed in the HPLC Separation of Ibuzatrelvir Diastereomers section.
IR (DCM cast film, vmax / cm–1) 3292, 3053, 2959, 2909, 2875, 1695, 1643, 1550, 1443, 1401,
1370, 1332, 1270, 1236, 1200, 1164, 1130
1H NMR (500 MHz, CDCl3) δH 8.32 (1H, d, J = 7.6 Hz), 6.22 (1H, br), 5.74 (1H, d, J = 9.3 Hz),
4.96 – 4.87 (1H, m), 4.54 (1H, dd, J = 8.6, 3.6 Hz), 4.30 (1H, d J = 9.9 Hz), 3.99 – 3.88 (2H, m),
3.65 (3H, s), 3.42 – 3.26 (2H, m), 2.66 – 2.57 (1H, m), 2.52 – 2.43 (1H, m), 2.40 – 2.28 (3H, m),
1.97 – 1.88 (1H, m), 1.84 – 1.75 (2H, m), 0.99 (9H, s)
13C {1H} NMR (125 MHz, CDCl3) δC 179.1, 171.4, 171.1, 156.9, 126.1 (q, J = 276.3 Hz), 118.3,
59.4, 58.9, 52.4, 47.3, 42.4 (q, J = 29.5 Hz), 40.4, 39.1 37.5, 35.6, 34.2, 28.2, 28.0, 26.3
SR: [α]D
26 = –35.71 (c = 0.21, DCM)
HRMS: (ESI) Calcd for C21H30F3N5NaO5 [M + Na]+
512.2091, found 512.2088



References
- ^ Allerton CM, Arcari JT, Aschenbrenner LM, Avery M, Bechle BM, Behzadi MA, et al. (August 2024). “A Second-Generation Oral SARS-CoV-2 Main Protease Inhibitor Clinical Candidate for the Treatment of COVID-19”. Journal of Medicinal Chemistry. 67 (16): 13550–13571. doi:10.1021/acs.jmedchem.3c02469. PMC 11345836. PMID 38687966.
- ^ Chen P, Van Oers TJ, Arutyunova E, Fischer C, Wang C, Lamer T, et al. (August 2024). “A Structural Comparison of Oral SARS-CoV-2 Drug Candidate Ibuzatrelvir Complexed with the Main Protease (Mpro) of SARS-CoV-2 and MERS-CoV”. JACS Au. 4 (8): 3217–3227. doi:10.1021/jacsau.4c00508. PMC 11350714. PMID 39211604.
- ^ Jump up to:a b Brewitz L, Schofield CJ (July 2024). “Fixing the Achilles Heel of Pfizer’s Paxlovid for COVID-19 Treatment”. Journal of Medicinal Chemistry. 67 (14): 11656–11661. doi:10.1021/acs.jmedchem.4c01342. PMC 11284777. PMID 38967233.
| Clinical data | |
|---|---|
| Other names | PF-07817883 |
| Routes of administration | Oral |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2755812-39-4 |
| PubChem CID | 163362000 |
| DrugBank | 111 |
| ChemSpider | 128942571 |
| UNII | KZ2X7QH2VT |
| Chemical and physical data | |
| Formula | C21H30F3N5O5 |
| Molar mass | 489.496 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- [1]. Owen, et al. Preparation of peptidomimetic nitriles as SARS-CoV-2 3CL protease inhibitors and methods for the treatment of COVID-19. World Intellectual Property Organization, WO2021250648 A1. 2021-12-16.[2]. Mahta Mortezavi, et al. Virologic Response and Safety After Oral Administration of Ibuzatrelvir, a Novel SARS-CoV-2 Mpro Inhibitor, in Non-Hospitalized Adults With Symptomatic COVID-19. European Congress of Clinical Microbiology and Infectious Disease (ECCMID) 2024; 2024 April 27-30.[3]. Westberg M, et al. An orally bioavailable SARS-CoV-2 main protease inhibitor exhibits improved affinity and reduced sensitivity to mutations[J]. Sci Transl Med. 2024 Mar 13;16(738):eadi0979.[4]. Allerton CMN, et al. A Second-Generation Oral SARS-CoV-2 Main Protease Inhibitor Clinical Candidate for the Treatment of COVID-19[J]. J Med Chem. 2024 Apr 30. [Content Brief]
////Ibuzatrelvir, PF 07817883, PF-07817883, PF07817883, KZ2X7QH2VT

{kind=link}