
Home » Phase2 drugs (Page 25)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
New Drug Application for Belinostat in Relapsed or Refractory PTCL Submitted to the FDA

Belinostat (PXD101)
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
| MW 318.07 | |
| Molecular Formula: | C15H14N2O4S |
414864-00-9 cas no
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

- Plumb, Jane A.; Finn, Paul W.; Williams, Robert J.; Bandara, Morwenna J.; Romero, M. Rosario; Watkins, Claire J.; La Thangue, Nicholas B.; Brown, Robert (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101″. Molecular Cancer Therapeutics 2 (8): 721–728. PMID 12939461.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


…………
US20100286279


SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
Traxoprodil mesylate

Traxoprodil mesylate
MF:C23-H35-N-O2.C-H4-O3-S
MW:453.6401
CAS:189894-57-3 mesylate
134234-12-1 (free base)
Traxoprodil mesylate, CP-101606-27,
(S, S) -1 – (4-Hydroxyphenyl) -2 – [4-hydroxy-4-phenylpiperidin-1-yl)-1-propanol methanesulfonate trihydrate

this exhibits activity as NMDA (N-methyl-D-aspartic acid) receptor antagonists and are useful in the treatment of epilepsy, anxiety, cerebral ischemia, muscular spasms, multiinfarct dementia, traumatic brain injury, pain, AIDS related dementia, hypoglycemia, migraine, amyotrophic lateral sclerosis, drug and alcohol addiction, drug and alcohol withdrawal symptoms, psychotic conditions, urinary incontinence and degenerative CNS (central nervous system) disorders such as stroke, Alzheimer’s disease, Parkinson’s disease and Huntington’s disease.
The free base, the anhydrous mesylate and methods of preparing them are referred to, generically, in United States Patent 5,185,343, which issued on February 9, 1993. They and their use in treating certain of the above disorders are referred to, specifically, in United States Patent 5,272,160, which issued on December 21 , 1993. Their use in treating the above disorders is referred to in lntemational Patent Application PCT/IB 95/00380, which designates the United States and was filed on May 18, 1995. Their use in combination with a compound capable of enhancing and thus restoring the balance of excitatory feedback from the ventral lateral nucleus of the thalamus into the cortex to treat Parkinson’s disease is referred to in International Patent Application PCT/IB 95/00398, which designates the United States and was filed on May 26, 1995. The foregoing U.S. patents and patent applications are incoφorated herein by reference in their entireties.
NMDA is an excitatory amino acid. The excitatory amino acids are an important group of neurotransmitters that mediate excitatory neurotransmission in the central nervous system. Glutamic acid and aspartic acid are two endogenous ligands that activate excitatory amino acid (EAA) receptors. There are two types of EAA receptors, ionotropic and metabotropic, which differ in their mode of signal transduction. There are at least three distinct ionotropic EAA receptors characterized by the selective agonist that activates each type: the NMDA, the AMPA (2-amino-3-(5-methyl-3- hdyroxyisoxazol-4-yl)propanoic acid) and the kainic acid receptors. The ionotropic EAA receptors are linked to ion channels that are permeable to sodium and, in the case of NMDA receptors, calcium. Metabotropic receptors, linked to phosphoinositide hydrolysis by a membrane associated G-protein, are activated by quisqualic acid, ibotenic acid, and (1S, 3R)-1-aminocyclopentane 1 ,3-dicarboxyiic acid.
The NMDA receptor is a macromolecular complex consisting of a number of distinct binding sites that gate on ion channels permeable to sodium and calcium ions. Hansen and Krogsgaard-Larson, Med. Res. Rev.. .10, 55-94 (1990). There are binding sites for glutamic acid, glycine, and polyamines, and a site inside the ion channel where compounds such as phencyclidine (PCP) exert their antagonist effects.
Competitive NMDA antagonists are compounds that block the NMDA receptor by interacting with the glutamate binding site. The ability of a particular compound to competitively bind to the NMDA glutamate receptor may be determined using a radioligand binding assay, as described by Murphy e l., British J. Pharmacol.. 95, 932- 938 (1988). The antagonists may be distinguished from the agonists using a rat cortical wedge assay, as described by Harrison and Simmonds, British J. Pharmacol.. 84, 381- 391 (1984). Examples of competitive NMDA antagonists include D-2 amino 5- phosphonopentanoic acid (D-AP5), and D-2-amino-7-phosphonoheptanoic acid, Schoepp et a]. , J. Neur. Transm.. 85, 131-143 (1991).

4-Hydroxypropiophenone (I) was protected as the triisopropylsilyl ether (II) and subsequently brominated with elemental bromine in CCl4. The resultant bromo ketone (III) was subsequently coupled with 4-hydroxy-4-phenylpiperidine (IV) to afford the racemic amino ketone (V). This was stereoselectively reduced with NaBH4 in EtOH yielding the threo-amino alcohol (VI). Then, desilylation of (VI) with tetrabutylammonium fluoride furnished the racemic phenol compound. Resolution into the enantiomers has been reported by formation of the . corresponding D-tartaric acid salts Finally, the title product was obtained by dissolving D-(-)-tartaric salt (VII) in water in the presence of methanesulfonic acid
EXAMPLE 1 Enantiomeric (1S,2S)- and (1R,2R)-1-(4-Hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidin-1-yl)-1-propanols
(+)-Tartaric acid (300 mg, 2 mmol) was dissolved in 30 mL warm methanol. Racemic 1S*,2S*-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidin-1-yl)-1-propanol (655 mg, 2 mmol) was added all at once. With stirring and gentle warming a colorless homogeneous solution was obtained. Upon standing at ambient temperature 24 hours, 319 mg (66%) of a fluffy white precipitate was obtained. This product was recrystallized from methanol to give 263 mg of the (+)-tartrate salt of levorotatory title product as a white solid; mp 206.5-207.5.degree. C.; [alpha].sub.D =-36.2.degree.. This salt (115 mg) was added to 50 mL of saturated NaHCO.sub.3. Ethyl acetate (5 mL) was added and the mixture was vigorously stirred 30 minutes. The aqueous phase was repeatedly extracted with ethyl acetate. The organic layers were combined and washed with brine, dried over calcium sulfate, and concentrated. The tan residue was recrystallized from ethyl acetate-hexane to give 32 mg (39%) of white, levorotatory title product; mp 203-204 C.sub.20 H.sub.25 NO.sub.3 : C, 73.37; H, 7.70; N. 4.28. Found: C, 72.61; H, 7.45; N. 4.21.
The filtrate from the (+)-tartrate salt preparation above was treated with 100 mL saturated aqueous NaHCO.sub.3 and extracted well with ethyl acetate. The combined organic extracts were washed with brine, dried over calcium sulfate and concentrated to give 380 mg of recovered starting material (partially resolved). This material was treated with (-)-tartaric acid (174 mg) in 30 mL of methanol as above. After standing for 24 hours, filtration gave 320 mg (66%) of product which was further recrystallized from methanol to produce 239 mg of the (-)-tartrate salt of dextrorotatory title product; mp 206.5-207.5.degree. C. [alpha].sub.D =+33.9.degree.. The latter was converted to dextrorotatory title product in the manner above in 49% yield; mp 204-205 Found: C, 72.94; H. 7.64; N, 4.24.
EXAMPLE 2 (1S,2S)-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidin-yl)-1-propanol Methanesulfonate Trihydrate
STEP 1

A 50 gallon glass lined reactor was charged with 17.1 gallons of acetone, 8.65 kilograms (kg) (57.7 mol) of 4′-hydroxypropiophenone, 9.95 kg (72.0 mol) of potassium carbonate and 6.8 liters (l) (57.7 mol) of benzylbromide. The mixture was heated to reflux (56 hours. Analysis of thin layer chromatography (TLC) revealed that the reaction was essentially complete. The suspension was atmospherically concentrated to a volume of 10 gallons and 17.1 gallons of water were charged. The suspension was granulated at 25 product was filtered on a 30″ Lapp and washed with 4.6 gallons of water followed by a mixture of 6.9 gallons of hexane and 2.3 gallons of isopropanol. After vacuum drying at 45 (96.4%) of the above-depicted product.
A second run was carried out with 9.8 kg (65.25 mol) of 4′-hydroxypropiophenone using the procedure described above. After drying 15.1 kg (96.3%) of the above-depicted product was obtained.
STEP 2

Under a nitrogen atmosphere, a 100 gallon glass lined reactor was charged with 75 gallons of methylene chloride and 28.2 kg (117.5 mol) of the product from step 1. The solution was stirred five minutes and then 18.8 kg of bromine was charged. The reaction was stirred for 0.5 hours at 22 complete. To the solution was charged 37 gallons of water and the mixture was stirred for 15 minutes. The methylene chloride was separated and washed with 18.5 gallons of saturated aqueous sodium bicarbonate. The methylene chloride was separated, atmospherically concentrated to a volume of 40 gallons and 60 gallons of isopropanol was charged. The concentration was continued until a pot temperature of 80 40 gallons were obtained. The suspension was cooled to 20 granulated for 18 hours. The product was filtered on a 30″ Lapp and washed with 10 gallons of isopropanol. After vacuum drying at 45 yielded 29.1 kg (77.6%) of the above-depicted product.
STEP 3

Under a nitrogen atmosphere, a 20 gallon glass lined reactor was charged with 4.90 kg (15.3 mol) of the product from step 2, 7.0 gallons of ethyl acetate, 2.70 kg (15.3 mol) of 4-hydroxy-4-phenylpiperidine and 1.54 kg of triethylamine (15.3 mol). The solution was heated to reflux (77 C.) for 18 hours. The resulting suspension was cooled to 20 Analysis by TLC revealed that the reaction was essentially complete. The byproduct (triethylamine hydrobromide salt) was filtered on a 30″ Lapp and washed with 4 gallons of ethyl acetate. The filtrate was concentrated under vacuum to a volume of 17 liters. The concentrate was charged to 48 liters of hexane and the resulting suspension granulated for 2 hours at 20 gallons of hexane. After vacuum drying at 50 kg (77%) of the above-depicted product.
A second run was carried out with 3.6 kg (11.3 mol) of the product from step 2 using the procedure described above. After drying 4.1 kg (87%) of the above-depicted product was obtained.
STEP 4

Under a nitrogen atmosphere, a 100 gallon glass lined reactor was charged with 87.0 gallons of 2B ethanol and 1.7 kg (45.2 mol) of sodium borohydride. The resulting solution was stirred at 25 kg (22.6 mol) of the product from step 3 was charged. The suspension was stirred for 18 hours at 25-30 reaction was essentially complete to the desired threo diastereoisomer. To the suspension was charged 7.8 liters of water. The suspension was concentrated under vacuum to a volume of 40 gallons. After granulating for 1 hour, the product was filtered on a 30″ Lapp and washed with 2 gallons of 2B ethanol. The wet product, 9.4 gallons of 2B-ethanol and 8.7 gallons of water were charged to a 100 gallon glass lined reactor. The suspension was stirred at reflux (78 cooled to 25 water followed by 4 gallons of 2B ethanol. After air drying at 50 C., this yielded 8.2 kg (86.5%) of the above-depicted product. This material was recrystallized in the following manner.
A 100 gallon glass lined reactor was charged with 7.9 kg (18.9 mol) of the product from step 3, 20 gallons of 2B ethanol and 4 gallons of acetone. The suspension was heated to 70 solution was concentrated atmospherically to a volume of 15 gallons. The suspension was cooled to 25 product was filtered on a 30″ Lapp. The wet product and 11.7 gallons of 2B ethanol was charged to a 100 gallon glass lined reactor. The suspension was heated to reflux (78 cooled to 25 of 2B ethanol. After air drying at 50 (70.6%) of the above-depicted product.
STEP 5

Under a nitrogen atmosphere, a 50 gallon glass lined reactor was charged with 825 g of 10% palladium on carbon (50% water wet), 5.5 kg (13.2 mol) of the product from step 4 and 15.5 gallons of tetrahydrofuran (THF). The mixture was hydrogenated between 40-50 time, analysis by TLC revealed that the reduction was essentially complete. The reaction was filtered through a 14″ sparkler precoated with Celite and washed with 8 gallons of THF. The filtrate was transferred to a clean 100 gallon glass lined reactor, vacuum concentrated to a volume of 7 gallons and 21 gallons of ethyl acetate were charged. The suspension was atmospherically concentrated to a volume of 10 gallons and a pot temperature of 72 filtered on a 30″ Lapp and washed with 2 gallons of ethyl acetate. After air drying at 55 above-depicted product (i.e., the free base).
STEP 6

A 100 gallon glass lined reactor was charged with 20 gallons of methanol and 3.7 kg (11.4 mol) of the product from step 5 (i.e., the free base). The suspension was heated to 60 D-(-)-tartaric acid were charged. The resulting solution was heated to reflux (65 suspension was cooled to 35 with 1 gallon of methanol. The wet solids were charged to a 100 gallon glass lined reactor with 10 gallons of methanol. The suspension was stirred for 18 hours at 25 Lapp and washed with 2 gallons of methanol. After air drying at 50 C. this yielded 2.7 kg (101%) of the above-depicted product (i.e., the tartaric acid salt of the free base (R-(+)-enantiomer)). This material was purified in the following manner:
A 100 gallon glass lined reactor was charged with 10.6 gallons of methanol and 2.67 kg (5.6 mol) of the above tartaric acid salt. The suspension was heated to reflux (80 to 30 methanol. After air drying at 50 of the above-depicted product (i.e., the tartaric acid salt of the free base).
STEP 7

•Tar tar i c Rc i d
A 55 liter nalgene tub was charged with 30 liters of water and 1056 g (12.6 mol) of sodium bicarbonate at 20 charged 2.0 kg (4.2 mol) of the product from step 6 (i.e., the tartaric acid salt of the free base). The suspension was stirred for 4 hours during which a great deal foaming occurred. After the foaming ceased, the suspension was filtered on a 32 cm funnel and washed with 1 gallon of water. After air drying at 50 the above-depicted product (i.e., the free base).
STEP 8

A 22 liter flask was charged with 1277 g (3.9 mol) of product from step 7 and 14 liters of water. The suspension was warmed to 30 g (3.9 mol) of methane sulfonic acid were charged. The resulting solution was warmed to 60 washed with 2 liters of water. The speck-free filtrate was concentrated under vacuum to a volume of 6 liters. The suspension was cooled to 0-5 18″ filter funnel and washed with 635 ml of speck-free water. After air drying at 25 above-depicted product (i.e., the mesylate salt trihydrate).
Proton and Carbon Nuclear Magnetic Resonance (NMR) Spectra of the Mesylate Salt Trihydrate
The proton and carbon NMR spectra of the mesylate salt trihydrate are described below. Chemical shift assignments in CD3OD (relative to tetramethylsilane (TMS) were made on the basis of ‘HJH Correlated Spectroscopy (COSY), ‘H-‘O Distortionless Enhancement by Polarization Transfer (DEPT), and ‘HJ’C Heteronuclear Chemical Shift Correlation (HETCOR) two-dimensional NMR experiments. The tentative proton and carbon peak assignments are given below and are consistent with the structure of the mesylate salt trihydrate. III

assignment, 13 C (δ, ppm), Protons, 1 H (δ, ppm) 4' 159.2 0 -- 1"' 148.2 0 -- 1' 132.6 0 -- 2' 129.8 2 7.30 (m) 3"' 129.5 m 2 7.38 (t) 4"' 128.4 1 7.30 (m) 2"' 125.6 2 7.56 (d) 3' 116.5 2 6.84 (d) 1 73.5 1 4.66 (d) 4" 69.8 0 -- 2 68.3 1 3.58 (m) 6"(1) 48.8 2 3.32 (d),3.72(t) 2"(1) 43.2 2 3.58 (m) 4 39.5 3 2.70 (s) 5"(2) 36.6 2 2.64 (t),1.98(d) 3"(2) 36.5 2 2.42 (t),1.98(d) 3 9.7 3 1.12(d)
(1) The 6″ and 2″ positions are not chemically equivalent; the assignments may be interchangeable. (2) The 5″ and 3″ positions are not chemically equivalent the assignments may be interchangeable. The proton splitting pattem at 1.96-2.06 ppm appears as two doublets when acquired on a high-field instrument (500 MHz), but only as a triplet when acquired with a lower field (300 MHz) instrument. This is believed to be due to a salt effect arising from the mesylate.
VBL Therapeutics announced FDA has granted Fast Track designation to its lead oncology drug VB-111

VB-111
VBL Therapeutics announced today that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation to its lead oncology drug VB-111, for prolongation of survival in patients with recurrent glioblastoma multiforme (rGBM).
VB-111 – highly targeted anti-angiogenic agent for the specific inhibition of tumor vascular growth
VB-111 is the first highly targeted anti-angiogenic agent for the specific inhibition of tumor vascular growth to use VTS™™, our proprietary platform technology, for cancer therapy. VB-111 is an IV-administered anti angiogenic agent that works in a manner akin to a “biological knife” to destroy tumor vasculature, thus cutting off blood vessels feeding the tumor.
Preclinical Insights
VB-111 has shown significant promise as a targeted cancer treatment with the potential to work synergistically in combination with conventional chemotherapy treatments to provide an effective treatment regimen for cancer patients. Pharmacological and toxicology studies of VB-111 have showed tissue specificity for the tumor tissue, no significant damage to normal non-cancerous tissues or to the normal vasculatures in the body and more than 90 percent tumor burden reduction in a metastatic lung cancer model with only one injection. Similar efficacy was shown in other tumor models.
Completed Clinical Trials
Phase 1 Clinical Trial – in a Phase 1 “all comers” dose escalation study in 33 patients with advanced metastatic cancer, therapeutic doses of VB-111 demonstrated antitumor activity and was found to be safe and well tolerated with no effect on liver function or major changes in complete blood count. Findings have been presented at the American Association of Cancer Research (AACR) and the American Society of Clinical Oncology (ASCO) annual meetings.
Pimavanserin …New Drug Shows Early Promise in Treating Parkinson’s Psychosis

Pimavanserin, ACP 103
N-(4-fluorophenylmethyl)-N-(1-methylpiperidin-4-yl)-N’-(4-(2-methylpropyloxy)phenylmethyl)carbamide, 706779-91-1 cas
 706782-28-7 (tartrate) |
THURSDAY Oct. 31, 2013 — Many people living with Parkinson’s disease suffer from hallucinations and delusions, but an experimental drug might offer some relief without debilitating side effects.
READ ALL AT
http://www.drugs.com/news/new-shows-early-promise-treating-parkinson-s-psychosis-48630.html

The drug — pimavanserin — appears to significantly relieve these troubling symptoms, according to the results of a phase 3 trial to test its effectiveness.
Pimavanserin (ACP-103) is a drug developed by Acadia Pharmaceuticals which acts as an inverse agonist on the serotonin receptor subtype 5-HT2A, with 40x selectivity over 5-HT2C, and no significant affinity or activity at 5-HT2B or dopamine receptors.[1] As of September 3 2009, pimavanserin has not met expectations for Phase III clinical trials for the treatment of Parkinson’s disease psychosis,[2] and is in Phase II trials for adjunctive treatment of schizophrenia alongside an antipsychotic medication.[3] It is expected to improve the effectiveness and side effect profile of antipsychotics.[4][5][6]
3-D MODEL OF DRUG PIMAVANSERIN, THE DEVELOPMENT OF WHICH HAS BEEN EXPEDITED BY THE FDA

- Friedman, JH (October 2013). “Pimavanserin for the treatment of Parkinson’s disease psychosis”. Expert Opinion on Pharmacotherapy 14 (14): 1969–1975.doi:10.1517/14656566.2013.819345. PMID 24016069.
- ACADIA Pharmaceuticals. “Treating Parkinson’s Disease – Clinical Trial Pimavanserin – ACADIA”. Retrieved 2009-04-11.[dead link]
- “ACADIA Announces Positive Results From ACP-103 Phase II Schizophrenia Co-Therapy Trial” (Press release). ACADIA Pharmaceuticals. 2007-03-19. Retrieved 2009-04-11.
- Gardell LR, Vanover KE, Pounds L, Johnson RW, Barido R, Anderson GT, Veinbergs I, Dyssegaard A, Brunmark P, Tabatabaei A, Davis RE, Brann MR, Hacksell U, Bonhaus DW (August 2007). “ACP-103, a 5-hydroxytryptamine 2A receptor inverse agonist, improves the antipsychotic efficacy and side-effect profile of haloperidol and risperidone in experimental models”. J Pharmacol Exp Ther 322 (2): 862–70.doi:10.1124/jpet.107.121715. PMID 17519387.
- Vanover KE, Betz AJ, Weber SM, Bibbiani F, Kielaite A, Weiner DM, Davis RE, Chase TN, Salamone JD (October 2008). “A 5-HT2A receptor inverse agonist, ACP-103, reduces tremor in a rat model and levodopa-induced dyskinesias in a monkey model”. Pharmacol Biochem Behav 90 (4): 540–4. doi:10.1016/j.pbb.2008.04.010. PMC 2806670.PMID 18534670.
- Abbas A, Roth BL (December 2008). “Pimavanserin tartrate: a 5-HT2A inverse agonist with potential for treating various neuropsychiatric disorders”. Expert Opin Pharmacother9 (18): 3251–9. doi:10.1517/14656560802532707. PMID 19040345.
Psychiatrist Herb Meltzer sadly watched the agitated woman accuse her son of trying to poison her. Although not her physician, Dr. Meltzer certainly recognized the devastating effects of his mother-in-law’s Parkinson’s disease psychosis (PDP). Occurring in up to half of all patients with Parkinson’s, symptoms of the psychotic disorder may include hallucinations and delusions. The development of PDP often leads to institutionalization and increased mortality.
“I was on the sidelines,” explains Dr. Meltzer, professor of psychiatry and physiology and director of the Translational Neuropharmacology Program at Northwestern University Feinberg School of Medicine. “I told my brother-in-law it was the disease talking, not his mother.”
Ironically, Dr. Meltzer has been far from the sidelines and right on the PDP playing field for quite a while. In fact, he may soon see a drug he helped develop become the first approved treatment for the disorder. In early April, Dr. Meltzer celebrated, along with colleagues at ACADIA Pharmaceuticals in San Diego for which he has been a clinical advisor, the stunning announcement: the Food and Drug Administration (FDA) had expedited the company’s path to filing a new drug application (NDA) for pimavanserin, a selective serotonin 5-HT2Areceptor blocker. Typically, the FDA requires data from two successful pivotal Phase III clinical studies affirming a drug candidate’s safety and efficacy before the agency will even consider an NDA. Just as ACADIA was planning to launch another Phase III study this spring to fulfill this requirement, the FDA decided the company had amassed enough data to support an NDA filing.

HERBERT MELTZER, MD, DESIGNED ACADIA PHARMACEUTICAL’S INITIAL PROOF OF CONCEPT TRIAL OF THE DRUG PIMAVANSERIN TO TREAT PARKINSON’S DISEASE PSYCHOSIS.
“This action on the part of the FDA is extremely unusual,” says Dr. Meltzer, who designed ACADIA’s initial proof-of-concept trial of pimavanserin, a drug he had initially suggested ACADIA develop to treat schizophrenia, with PDP as a secondary indication. “The FDA staff decided that results from my small clinical study and the first successful Phase III study were sufficient to establish efficacy and safety.”
Bringing a safe and effective drug to market is a monumental achievement. Pimavanserin is not yet there but has significantly moved within striking distance with this recent nod from the regulatory agency.
24 YEARS IN THE MAKING
The neuropharmacologist’s collaboration with ACADIA began in 2000. The company wanted to develop a drug targeting the serotonin 5-HT 2A receptor, a neurotransmitter ACADIA believed played a key role in schizophrenia based upon basic research from Meltzer and their own studies. A distinguished schizophrenia investigator, then at Case Western Reserve University, he welcomed ACADIA’s offer to translate his ideas about developing safer and more effective drug treatments for psychosis. Through his provocative and groundbreaking research, Dr. Meltzer originally championed the idea that blocking the 5-HT2A receptor would lead to better antipsychotic drugs with fewer side effects. Existing drugs often impaired motor function because they targeted the dopamine D2 receptor. Of the 14 different types of serotonin receptors in this complex area of study, Dr. Meltzer zeroed in on the 5-HT2A type—the same receptor that leads to hallucinogenic properties of LSD and mescaline. It was an ideal target to complement weak D2 receptor blockade in schizophrenia and as a standalone treatment for PD psychosis.
………………………………………….
Bristol-Myers Squibb announced promising results from phase 2b study of its rheumatoid arthritis drug clazakizumab
NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL
CLAZAKIZUMAB
PRONUNCIATION klaz” a kiz’ ue mab
THERAPEUTIC CLAIM Autoimmune diseases, rheumatoid arthritis
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human interleukin 6) (human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 heavy chain), disulfide with human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 κ-chain, dimer
2. Immunoglobulin G1, anti-(human interleukin-6 (B-cell stimulatory factor 2, CTL differentiation factor, hybridoma growth factor, interferon beta-2)); humanized rabbit monoclonal BMS-945429/ALD518 [300-alanine(CH2-N67>A67)]1 heavy chain (223-217′)-disulfide with humanized rabbit monoclonal BMS-945429/ALD518 light chain dimer (229-229”:232-232”)-bisdisulfide, O-glycosylated
MOLECULAR FORMULA C6426H9972N1724O2032S42
MOLECULAR WEIGHT 145.2 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION BMS-945429, ALD518
CAS REGISTRY NUMBER 1236278-28-6
Monoclonal antibody
Type Whole antibody
Source Humanized
Target IL6
CAS number 1236278-28-6
Clazakizumab is a humanized monoclonal antibody designed for the treatment of rheumatoid arthritis.[1]
Clazakizumab was developed by Alder Biopharmaceuticals and Bristol-Myers Squibb.
gamma1 heavy chain (1-450) [humanized VH (Homo sapiens IGHV3-66*01 (83.50%) -(IGHD)-IGHJ3*02 M123>L (115)) [8.8.14] (1-120) -Homo sapiens IGHG1*03 CH

Molidustat (BAY 85-3934), Bayer’s drug under initiation in patients with anemia associated with chronic kidney disease and/or end-stage renal disease.
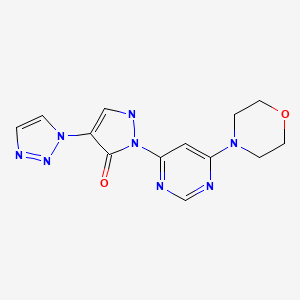
Molidustat
UNII-9JH486CZ13, cas no 1154028-82-6, MW: 314.3076
2-(6-morpholin-4-ylpyrimidin-4-yl)-4-(triazol-1-yl)-1H-pyrazol-3-one
Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors
For the cardio-renal syndrome, a Phase IIb program with the investigational new drug Molidustat (BAY 85-3934) is under initiation in patients with anemia associated with chronic kidney disease and/or end-stage renal disease. Molidustat is a novel inhibitor of hypoxia-inducible factor (HIF) prolyl hydroxylase (PH) which stimulates erythropoietin (EPO) production and the formation of red blood cells. Phase I data have shown that inhibition of HIF-PH by Molidustat results in an increase in endogenous production of EPO.
About Bayer HealthCare
The Bayer Group is a global enterprise with core competencies in the fields of health care, agriculture and high-tech materials. Bayer HealthCare, a subgroup of Bayer AG with annual sales of EUR 18.6 billion (2012), is one of the world’s leading, innovative companies in the healthcare and medical products industry and is based in Leverkusen, Germany. The company combines the global activities of the Animal Health, Consumer Care, Medical Care and Pharmaceuticals divisions. Bayer HealthCare’s aim is to discover, develop, manufacture and market products that will improve human and animal health worldwide. Bayer HealthCare has a global workforce of 54,900 employees (Dec 31, 2012) and is represented in more than 100 countries. More information at www.healthcare.bayer.com.
molidustat
…………………………………………………………..
molidusat sodium
Sodium 1-[6-(morpholin-4-yl)pyrimidin-4-yl]-4-(1H-1,2,3-triazol-1-yl)-1H-pyrazol-5-olate
Molidustat sodium is an orally-available hypoxia-inducible factor prolyl hydroxylase inhibitor in phase I clinical trials at Bayer for the treatment of patients suffering from renal anemia due to chronic kidney disease.
WO 2008067871
WO 2012065967
WO 2013167552
…………………………
2-Heteroaryl-4-aryl-1,2-dihydropyrazolones having a bactericidal and/or fungicidal action are disclosed in EP 165 448 and EP 212 281. The use of 2-heteroaryl-4-aryl-1,2-dihydropyrazolones as lipoxygenase inhibitors for treatment of respiratory tract, cardiovascular and inflammatory diseases is claimed in EP 183 159. 2,4-Diphenyl-1,2-dihydropyrazolones having a herbicidal activity are described in DE 2 651 008.
The preparation and pharmacological properties of certain 2-pyridyl-1,2-dihydropyrazolones are reported in Helv. Chim. Acta 49 (1), 272-280 (1966). WO 96/12706, WO 00/51989 and WO 03/074550 claim compounds having a dihydropyrazolone partial structure for treatment of various diseases, and hydroxy- or alkoxy-substituted bipyrazoles for treatment of neuropsychiatric diseases are disclosed in WO 2006/101903.
Heteroaryl-substituted pyrazole derivatives for treatment of pain and various CNS diseases are furthermore described in WO 03/051833 and WO 2004/089303. WO 2006/114213 has meanwhile disclosed 2,4-dipyridyl-1,2-dihydropyrazolones as inhibitors of HIF prolyl 4-hydroxylases.
The x-ray crystal structure of the compound 3-methyl-1-(pyridin-2-yl)-4-(1-pyridin-2-yl-3-methyl-1H-pyrazol-5-yl)-2H-3-pyrazolin-5 (114)-one (other name: 5,5′-dimethyl-2,2′-di-pyridin-2-yl-1′,2′-dihydro-2H,3′H-3,4′-bipyrazol-3′-one) is reported inActa Crystallogr., Section E: Structure Reports Oμline E57 (11), o1126-o1127 (2001) [Chem. Abstr. 2001:796190].
The synthesis of certain 3′,5-dimethyl-2-phenyl-1′-(1,3-thiazol-2-yl)-1′H,2H-3,4′-bipyrazol-5′-ol derivatives is described inIndian J. Heterocyclic Chem. 3 (1), 5-8 (1993) [Chem. Abstr. 1994:323362].
The preparation and tautomerism of individual 4-(pyrazol-5-yl)-pyrazolin-5-one derivatives is reported in J. Heterocyclic Chem. 27 (4), 865-870 (1990) [Chem. Abstr. 1991:428557]. A therapeutic use has not hitherto been described for the compounds mentioned in these publications. The compound 2-tert-butyl-1′-[4-(4-chlorophenyl)-1,3-thiazol-2-yl]-3′,5-dimethyl-1′H,2H-3,4′-bipyrazol-5′-ol is listed as a test example in WO 2007/008541.
………………………….
https://www.google.co.in/patents/US20100305085
Example 3A 3-(Dimethylamino)-2-(1H-1,2,3-triazol-1-yl)acrylic acid ethyl ester

The preparation of the starting compound is carried out analogously to 2A starting from 1.00 g (6.45 mmol) 2-(1H-1,2,3-triazol-1-yl)acetic acid ethyl ester.
Yield: 1.4 g (100% of th.)
1H-NMR (400 MHz, DMSO-d6): δ=8.10 (d, 1H), 7.78 (d, 1H), 7.65 (s, 1H), 4.03 (q, 2H), 3.06 (br. s, 3H), 2.10 (br. s, 3H), 1.12 (t, 3H).
LC-MS (Method 5): Rt=1.40 min; MS (ESIpos): m/z=211 [M+H]+.
Example 16A 4-(6-Hydrazinopyrimidin-4-yl)morpholine

Stage a): 4-(6-Chloropyrimidin-4-yl)morpholine

45.0 g (302.1 mmol) 4,6-dichloropyrimidine are initially introduced into 450 ml water. 26.3 g (302.1 mmol) morpholine are added and the mixture is stirred at 90° C. for 16 h. Thereafter, it is cooled to 0° C. and the precipitate formed is filtered off. The precipitate is washed once with 50 ml water and dried in air.
Yield: 51.0 g (85% of th.)
LC-MS (Method 4): Rt=1.09 min; MS (ESIpos): m/z=200 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=8.35 (s, 1H), 6.95 (s, 1H), 3.62 (s, 8H).
Stage b) 4-(6-Hydrazinopyrimidin-4-yl)morpholine
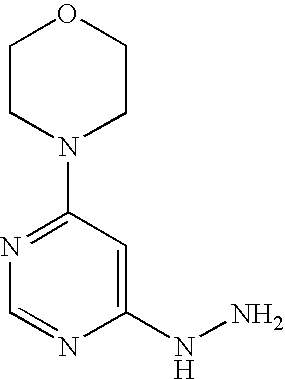
53.0 g (2.7 mmol) 4-(6-chloropyrimidin-4-yl)morpholine are initially introduced into 260 ml ethanol. 132.9 g (2.7 mol) hydrazine hydrate are added and the mixture is stirred under reflux for 16 h. Thereafter, it is cooled to RT and approx. half of the solvent is removed by distillation. The mixture is cooled to 0° C. and the solid formed is filtered off. It is rinsed with cold ethanol and the solid is dried first in air and then in vacuo.
Yield: 35.0 g (68% of th.)
LC-MS (Method 1): Rt=0.17 min; MS (ESIpos): m/z=196 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=7.94 (s, 1H), 7.70 (s, 1H), 5.91 (s, 1H), 4.15 (s, 2H), 3.66-3.60 (m, 4H), 3.45-3.37 (m, 4H).
Example 71 2-(6-Morpholin-4-ylpyrimidin-4-yl)-4-(1H-1,2,3-triazol-1-yl)-1,2-dihydro-3H-pyrazol-3-one

1.9 g (8.8 mmol) of the compound from Example 3A and 1.9 g (9.7 mmol) of the compound from Example 16A are initially introduced into 25 ml ethyl acetate and 504 mg (4.4 mmol) TFA are added at RT. The mixture is stirred under reflux for 16 h, then cooled to 5° C. and subsequently stirred for a further 2 h. The solid formed is filtered off, washed with ethyl acetate and dried first in air and thereafter under a high vacuum. 1.7 g of product are obtained.
The mother liquor is combined with the wash solution and the solvent is removed. According to LC-MS, the residue (2.4 g) still contains the intermediate 3-[2-(6-morpholin-4-ylpyrimidin-4-yl)hydrazino]-2-(1H-1,2,3-triazol-1-yl)prop-2-enoic acid ethyl ester (intermediate stage of the cyclization), which is used directly for the preparation of Example 72 (see there).
Yield: 1.7 g (61% of th.)
LC-MS (Method 9): Rt=0.90 min; MS (ESIpos): m/z=315 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=8.42 (s, 1H), 8.38 (s, 1H), 8.01 (s, 1H), 7.73 (s, 1H), 7.70 (s, 1H), 3.71-3.65 (m, 4H), 3.57-3.51 (m, 4H).
hydrochloride
Example 72 2-(6-Morpholin-4-ylpyrimidin-4-yl)-4-(1H-1,2,3-triazol-1-yl)-1,2-dihydro-3H-pyrazol-3-one hydrochloride

Batch 1: 7.5 ml of a 4 N solution of hydrogen chloride in dioxane are added to 1.7 g (5.4 mmol) of the compound from Example 71. The mixture is stirred at RT, 5 ml dioxane are added and the mixture is stirred at RT for 16 h. The solid is filtered off and washed with 5 ml dioxane. The mixture is dried under a high vacuum for 16 h, 10 ml methanol are then added and the mixture is stirred at RT for 1 h. The solid is filtered off, washed with 4 ml methanol and dried under a high vacuum. 1.6 g of the title compound are obtained.
Batch 2: A further amount of the title compound is obtained as follows: The residue (2.4 g) obtained from the mother liquor during the synthesis of Example Compound 71, which contains the open-ring intermediate state of the cyclization, 3-[2-(6-morpholin-4-ylpyrimidin-4-yl)hydrazino]-2-(1H-1,2,3-triazol-1-yl)prop-2-enoic acid ethyl ester, is dissolved in 12 ml ethanol and 1.5 ml 30% strength sodium methylate solution in methanol are added at RT, while stirring. The mixture is subsequently stirred at RT for 45 min, then adjusted to pH 5 with 2 N hydrochloric acid and subsequently stirred at RT for a further 16 h. The mixture is cooled to 10° C. and the solid is filtered off and washed with 3.5 ml dioxane. The mixture is dried under a high vacuum for 16 h, 5 ml methanol are then added and the mixture is subsequently stirred at RT for 1 h. The solid is filtered off, washed with 2 ml methanol and dried under a high vacuum to give a further 997 mg of the title compound in this way.
Yield: together 2.6 g (83% of th.)
LC-MS (Method 6): Rt=0.89 min; MS (ESIpos): m/z=315 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=8.54 (s, 1H), 8.39 (s, 1H), 8.28 (s, 1H), 7.88 (s, 1H), 7.42 (s, 1H), 3.71 (s, 8H).


amcrasto@gmail.com
email me if u like my posts
Copanlisib (BAY 80-6946), Bayer’s novel, oral phosphatidylinositol-3 kinases (PI3K) inhibitor

Copanlisib (BAY 80-6946)
1032568-63-0, cas no
MW: 480.5262
In oncology, Copanlisib (BAY 80-6946), a novel, oral phosphatidylinositol-3 kinases (PI3K) inhibitor, was selected for accelerated development. Copanlisib demonstrated a broad anti-tumor spectrum in preclinical tumor models and promising early clinical signals in a Phase I study in patients with follicular lymphoma. A Phase II study in patients with Non-Hodgkin’s lymphoma is currently ongoing.
PI3K inhibitor BAY 80-6946
A phosphoinositide 3-kinase (PI3K) inhibitor with potential antineoplastic activity. PI3K inhibitor BAY 80-6946 inhibits the activation of the PI3K signaling pathway, which may result in inhibition of tumor cell growth and survival in susceptible tumor cell populations. Activation of the PI3K signaling pathway is frequently associated with tumorigenesis and dysregulated PI3K signaling may contribute to tumor resistance to a variety of antineoplastic agents.
Finerenone (BAY 94-8862), BAYER’S next generation oral, non-steroidal Mineralocorticoid Receptor antagonist which blocks the deleterious effects of aldosterone
CAS Number: 1050477-31-0, UNII-DE2O63YV8R
MW: 378.4298, C21-H22-N4-O3
Finerenone (BAY 94-8862) is a next generation oral, non-steroidal Mineralocorticoid Receptor antagonist which blocks the deleterious effects of aldosterone.
Currently available steroidal MR antagonists have proven to be effective in reducing cardiovascular mortality in patients with heart failure but have significant side effects that limit their utilization.
Finerenone is currently in clinical Phase IIb development for the treatment of worsening chronic heart failure, as well as diabetic nephropathy.
Aerial Biopharma announce positive Phase II results for narcolepsy drug
ADX-N05, ARL-N05, SKL-N05
Aerial Biopharma announce positive Phase II results for narcolepsy drug
A new drug to treat excessive daytime sleepiness associated with narcolepsy has shown positive results from a phase 2b clinical trial, US-based Aerial Biopharma has announced this week.
read all at
Big boost for Incyte as Jakafi shines in PhII


ruxolitinib
Top-line results from a Phase II trial showed that its JAK inhibitor Jakafi (ruxolitinib), in combination with Roche’s Xeloda (capecitabine), improved survival in some patients with recurrent or treatment refractory advanced pancreatic cancer
http://www.pharmatimes.com/Article/13-08-
22/Big_boost_for_Incyte_as_Jakafi_shines_in_PhII.aspx
Ruxolitinib (trade names Jakafi and Jakavi, by Incyte Pharmaceuticals and Novartis) is a drug for the treatment of intermediate or high-risk myelofibrosis, a type of bone marrow cancer.It is also being investigated for the treatment of other types of cancer (such as lymphomas and pancreatic cancer), for polycythemia vera, and for plaque psoriasis.
The phase III Controlled Myelofibrosis Study with Oral JAK Inhibitor-I (COMFORT-I) and COMFORT-II trials showed significant benefits by reducing spleen size, relieving debilitating symptoms, and improving overall survival.
Mechanism of action
Ruxolitinib is a Janus kinase inhibitor with selectivity for subtypes 1 and 2 of this enzyme.
Side effects
Immunologic side effects have included herpes zoster (1.9%) and case reports of opportunistic infections.[10] Metabolic side effects have included weight gain (7.1%). Laboratory abnormalities have included alanine transaminase (ALT) abnormalities (25.2%), aspartate transaminase (AST) abnormalities (17.4%), and elevated cholesterol levels (16.8%).
Legal status
In November 2011, ruxolitinib was approved by the USFDA for the treatment of intermediate or high-risk myelofibrosis based on results of the COMFORT-I and COMFORT-II Trials.
Some analysts believe this to be a potential blockbuster drug.[3] As of the end of March 2012, and according to an Incyte spokesman, approximately 1000 physicians had prescribed the drug in the United States, out of a total 6500 hematologists and oncologists nationwide.

The US Food and Drug Administration had approved Incyte’s Jakafi (ruxolitinib) to treat patients with the bone marrow disease myelofibrosis (MF). Jakafi is the first and only drug granted license specifically for the treatment of the rare blood cancer.
Jakafi approved by FDA to treat rare bone marrow disease
Posted By Edward Su On November 17th, 2011
MF is a rare, potentially life-threatening blood cancer with limited treatment methods. Patients with the bone marrow disoder, characterized by bone marrow failure, enlarged spleen (splenomegaly), suffer from the symptoms of fatigue, night sweats and pruritus, poor quality of life, weight loss and shortened survival. The US drug firm Incyte estimates the disease affects about 16,000-18,500 people in the USA. Currently, the disease is treated with chemotherapy or bone marrow transplant.
Incyte’s Jakafi, the first drug to reach market from the Wilmington-based drug company, was approved by the FDA as a twice-a-day pill for the treatment of patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF and post-essential thrombocythemia MF. The US regulators reviewed Jakafi under its priority review program for important new therapies.
The approval of Jakafi was based on the results from two clinical studies involved 528 patients with the disease. Patients in the Jakafi treatment arm experienced a significant reduction in the size of their spleen as well as a 50 percent decrease in symptoms, including pain, discomfort and night sweats.
Jakafi, generically known as ruxolitinib, works by blocking JAK1 and JAK2 enzymes associated with the disease. The company has co-developed the drug with Novartis as part of their collaboration signed in 2009. The Swiss drug firm has the rights to market Jakafi in other countries.
“The availability of Jakafi is a significant medical advancement for people living with myelofibrosis, a debilitating disease,” said Paul A. Friedman, M.D., President and Chief Executive Officer of Incyte. “This milestone marks a tremendous achievement for Incyte because a scientific discovery from our research laboratories has become the first JAK inhibitor to reach the market and provide a clinical benefit to patients.”
Richard Pazdur, director of the Office of Hematology and Oncology Drug Products in the FDA’s Center for Drug Evaluation and Research, said that Jakafi “represents another example of an increasing trend in oncology where a detailed scientific understanding of the mechanisms of a disease allows a drug to be directed toward specific molecular pathways”.
Incyte says Jakafi will be available next week, and the drug will cost $7,000 per month, or $84,000 for a year’s supply for insured patients. The company plans to provide Jakafi free to uninsured patients and will offer co-pay assistance to patients with financial need.
(JAK1, JAK2) inhibitor, developed by the Incyte Corporation, trade name Jakafi.
Ruxolitinib synthetic route as shown below. 4 – bromo-pyrazole ( 1 ) with ethyl vinyl ether ( 2 ) to protect, and then with a Grignard reagent to a halogen – exchanged with isopropyl magnesiumpinacol ester ( 3 ) quenching to obtain 4 . Compound 5 is obtained consisting of hydrogen is protected 6 , and then with a boronic acid ester 4 Suzuki coupling occurs under acidic conditions after removal of the protecting group pyrazolyl 7 , 7 and α, β-unsaturated aldehyde 8 chiral catalyst 9 of under the catalysis of asymmetric Michael addition to give ( R ) -10 (90% EE). ( R) -10 , after reaction with ammonia to obtain an imine oxidation with iodine nitrile 11 , respectively, with different conditions for the final removal of the protecting group to afford Ruxolitinib.
