







| Patent ID | Date | Patent Title |
|---|---|---|
| US2015182533 | 2015-07-02 | 5-HT3 RECEPTOR ANTAGONISTS |
| US2014024644 | 2014-01-23 | 5-HT3 RECEPTOR ANTAGONISTS |
Home » PHASE1 (Page 7)
Category Archives: PHASE1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GSK-2879552

GSK-2879552
CAS 1401966-69-5 (ABS), 1401966-63-9(REL)
C23 H28 N2 O2, 364.48
Benzoic acid, 4-[[4-[[[(1R,2S)-2-phenylcyclopropyl]amino]methyl]-1-piperidinyl]methyl]-
4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid
- 4-[[4-[[[(1R,2S)-2-Phenylcyclopropyl]amino]methyl]-1-piperidinyl]methyl]benzoic acid
- 4-[[4-[[((1R,2S)-2-Phenylcyclopropyl)amino]methyl]piperidin-1-yl]methyl]benzoic acid
4-((4-((((1R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-1-yl)methyl)benzoic acid
Phase I
Glaxosmithkline Llc INNOVATOR
Neil W. Johnson, Jiri Kasparec, William Henry Miller, Meagan B. Rouse, Dominic Suarez, Xinrong Tian,
A LSD1 inhibitor potentially for the treatment of small cell lung cancer and acute myeloid leukemia.
![]()
GSK2879552 is an orally available, irreversible, inhibitor of lysine specific demethylase 1 (LSD1), with potential antineoplastic activity. Upon administration, GSK2879552 binds to and inhibits LSD1, a demethylase that suppresses the expression of target genes by converting the dimethylated form of lysine at position 4 of histone H3 (H3K4) to mono- and unmethylated H3K4. LSD1 inhibition enhances H3K4 methylation and increases the expression of tumor-suppressor genes. This may lead to an inhibition of cell growth in LSD1-overexpressing tumor cells. LSD1, overexpressed in certain tumor cells, plays a key role in tumor cell growth and survival. Check for active clinical trials or closed clinical trials using this agent.

Formula: C23H29ClN2O2
M.Wt: 400.94

CAS 1902123-72-1
| Molecular Weight: | 437.41 |
| Formula: | C23H28N2O2.2HCl |
Chromatin modification plays an essential role in transcriptional regulation (T. Kouzarides, 2007, Cell 128: 693-705). These modifications, which include DNA methylation, histone acetylation and hsitone methylation, are disregulated in tumors. This epigenetic disregulation plays an important role in the silencing of tumor suppressors and overexpression of oncogenes in cancer (M. Esteller, 2008, N Engl J Med 358: 1148-59. P. Chi et al, 2010, Nat Rev Cane 10:457-469.). The enzymes that regulate histone methylation are the histone methyl transferases and the histone demethylases.
Lysine-specific demethylase 1 (LSDl; also known as BHC110) is a histone lysine demethylase reported to demethylate H3K4mel/2 (Y. Shi et al, 2004, Cell 119: 941-953) and H3K9mel/2 (R. Schule et al.,2005, Nature 437: 436-439). LSDl is overexpressed in multiple human cancers, including prostate where it is associated with more frequent relapse (P. Kahl et al, 2006, Cane. Res. 66: 11341-11347), breast (J. Kirfel et al, 2010, Carcinogenesis 31: 512-520) neuroblastoma (J. Kirfel et al, 2009, Cane. Res. 69: 2065-2071. G. Sun et al, 2010, Mol. Cell. Biol. 28: 1997-2000). LSDl is essential for transcriptional regulation mediated by a number of nuclear hormone receptors, including androgen receptor in prostate cancer (R. Schuele et al, 2005, Nature 437: 436-439. R. Schuele et al, 2007, Nat. Cell Biol. 9: 347-353. R. Schuele et al, 2010, Nature 464: 792-796), estrogen receptor in breast carcinomas (M.G. Rosenfeld et al, 2007, Cell 128: 505-518), and TLX receptor in neuorblastoma (S. Kato et al, 2008, Mol. Cell. Biol. 28: 3995-4003). These studies have shown that knockdown of LSDl expression results in decreased cancer cell proliferation. Additionally, LSDl is overexpressed in multiple cancer types that are nuclear hormone receptor-independent. Those tumors include ER-negative breast (J. Kirfel et al, 2010, Carcinogenesis 31: 512-520), small-cell lung, bladder, head & neck, colon, serous ovary, and kidney Wilm’s tumor. Therefore, potent selective small molecule inhibitors of LSDl may be useful for treatment of cancers that are nuclear hormone receptor-dependent and/or nuclear hormone receptor-independent.
The compositions and methods provided herein can potentially be useful for the treatment of cancer including tumors such as skin, breast, brain, cervical carcinomas, testicular carcinomas, etc. More particularly, cancers that may be treated by the compositions and methods of the invention include, but are not limited to tumor types such as astrocytic, breast, cervical, colorectal, endometrial, esophageal, gastric, head and neck, hepatocellular, laryngeal, lung, oral, ovarian, prostate and thyroid carcinomas and sarcomas. More specifically, these compounds can potentially be used to treat: Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma, mesothelioma; Gastrointestinal: esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma), stomach (carcinoma, lymphoma, leiomyosarcoma), pancreas (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, vipoma), small bowel (adenocarcinoma, lymphoma, carcinoid tumors, Kaposi’s sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, fibroma), large bowel (adenocarcinoma, tubular adenoma, villous adenoma, hamartoma, leiomyoma); Genitourinary tract: kidney (adenocarcinoma, Wilm’s tumor
(nephroblastoma), lymphoma, leukemia), bladder and urethra (squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma), prostate (adenocarcinoma, sarcoma), testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma); Liver: hepatoma (hepatocellular carcinoma), cholangiocarcinoma, hepatoblastoma,angiosarcoma, hepatocellular adenoma, hemangioma; Bone: osteogenic sarcoma(osteosarcoma), fibrosarcoma, malignant fibrous histiocytoma, chondrosarcoma, Ewing’s sarcoma, malignant lymphoma (reticulum cell sarcoma), multiple myeloma, malignant giant cell tumor chordoma, osteochronfroma (osteocartilaginous exostoses), benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma and giant cell tumors; Nervous system: skull (osteoma, hemangioma, granuloma, xanthoma, osteitis deformans), meninges (meningioma, meningiosarcoma, gliomatosis), brain (astrocytoma, meduUoblastoma, glioma, ependymoma, germinoma (pinealoma), glioblastoma multiform, oligodendroglioma, schwannoma, retinoblastoma, congenital tumors), spinal cord neurofibroma, meningioma, glioma, sarcoma); Gynecological: uterus (endometrial carcinoma), cervix (cervical carcinoma, pre -tumor cervical dysplasia), ovaries (ovarian carcinoma (serous cystadenocarcinoma, mucinous cystadenocarcinoma, unclassified carcinoma), granulosa-thecal cell tumors, Sertoli-Leydig cell tumors, dysgerminoma, malignant teratoma), vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma), vagina (clear cell carcinoma, squamous cell carcinoma, botryoid sarcoma (embryonal rhabdomyosarcoma), fallopian tubes
(carcinoma); Hematologic: blood (myeloid leukemia (acute and chronic), acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplasia syndrome), Hodgkin’s disease, non-Hodgkin’s lymphoma (malignant lymphoma); Skin: malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Kaposi’s sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, keloids, psoriasis; and Adrenal glands: neuroblastoma. Thus, the term “cancerous cell” as provided herein, includes a cell afflicted by any one of or related to the above identified conditions.
SYNTHESIS
GSK-2879552
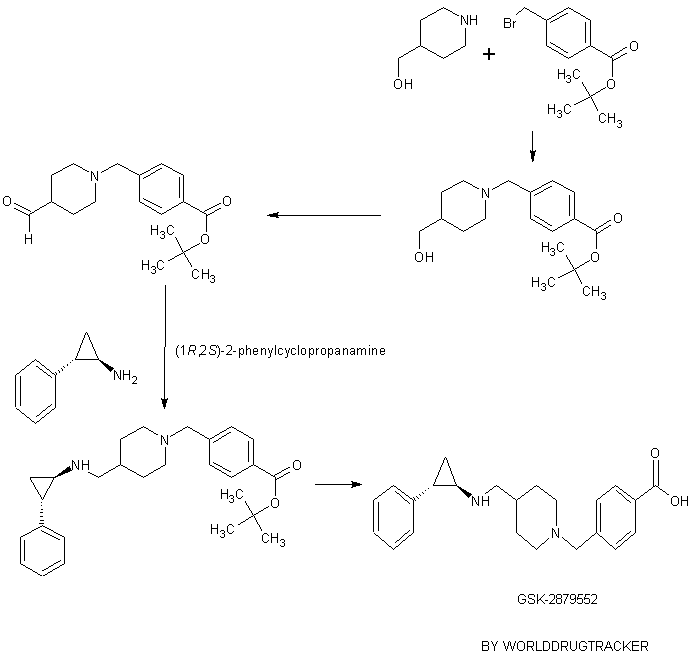
PATENT
WO 2012135113
https://www.google.co.in/patents/WO2012135113A2?cl=en
Example 2
1 , 1 -Dimethylethyl 4-( { \( 1 R,2S)-2-phenylcyclopropyl] amino I methyl)- 1 -piperidinecarboxylate

Following a procedure analogous to the procedure described in Example 1 using [(1R,2S)-2-phenylcyclopropyl]amine ((-) isomer) (94 mg, 0.703 mmol) afforded 1,1 -dimethylethyl 4-({[(lR,2S)-2-phenylcyclopropyl]amino}methyl)-l-piperidinecarboxylate (92 mg, 0.264 mmol, 56.4 % yield) as white solid. 1H NMR (400 MHz, METHANOL-d4) δ 7.29 – 7.37 (m, 2H), 7.23 – 7.28 (m, 1H), 7.17 – 7.22 (m, 2H), 4.14 (d, J= 12.63 Hz, 2H), 3.14 (d, J = 7.07 Hz, 2H), 3.01 (dt, J= 4.14, 7.64 Hz, 1H), 2.81 (br. s., 2H), 2.53 (ddd, J= 3.54, 6.63, 10.29 Hz, 1H), 1.97 (ddd, 1H), 1.80 (d, J= 12.13 Hz, 2H), 1.55 (ddd, J= 4.29, 6.63, 10.55 Hz, 1H), 1.47 (s, 9H), 1.36 – 1.45 (m, 1H), 1.23 (qd, J= 4.29, 12.38 Hz, 2H); LC-MS Rt = 0.78 min; MS (ESI): 331.3 [M+H]+.
Example 6
[(lR,2S)-2-Phenylcyclopropyll(4-piperidinylmethyl)amine

Following a procedure analogous to the procedure described in Example 4 using 1,1-dimethylethyl 4-({[(lR,2S)-2-phenylcyclopropyl]amino}methyl)-l-piperidinecarboxylate (Example 2, 60 mg, 0.182 mmol) afforded [(lR,2S)-2-phenylcyclopropyl](4-piperidinylmethyl)amine (41 mg, 0.146 mmol, 80 % yield)as white solid. 1H NMR (400 MHz, METHANOLS) δ 7.29 – 7.38 (m, 2H), 7.23 – 7.29 (m, 1H), 7.18 – 7.23 (m, 2H), 3.47 (d, J= 13.39 Hz, 2H), 3.21 (d, 2H), 2.89 – 3.13 (m, 3H), 2.60 (ddd, J= 3.79, 6.57, 10.36 Hz, 1H), 2.13 – 2.28 (m, J= 3.85, 3.85, 7.61, 11.21 Hz, 1H), 1.99 – 2.13 (m, 2H), 1.49 – 1.71 (m, 3H), 1.35 – 1.48 (m, 1H); LC-MS Rt = 0.44 min; MS (ESI): 231.2
Example 26
4-((4-(((trans-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoic acid

To the solution of 2,2,2-trifluoro-N-(trans-2-phenylcyclopropyl)-N-(piperidin-4-ylmethyl)acetamide (200 mg, 0.613 mmol, Example l ib) and 4-(bromomethyl)benzoic acid (198 mg, 0.919 mmol) in acetonitrile (6 mL) was added potasium carbonate (254 mg, 1.838 mmol). The reaction mixture was stirred for 3 hours at the 90 °C. The reaction mixture was then filtered and evaporated. The crude oil was mixed with 10 mL of 10 % acetic acid and 10 mL of ethyl acetate. Layers were separated, and the organic layer was discharged. Aqueous layer was neutralized with 1 M Na2C03, and the product was extracted into 10 mL of ethyl acetate. The organic layer was washed with brine, dried over MgS04, filtered and evaporated. The oil was dissolved in 6 ml of EtOH and 3 ml of 1 M NaOH. The reaction mixture was stirred for 20 min, and then it was concentrated. The solution was then partioned between 2 ml of water and 5 mL of ethyl acetate. The organic layer was separated and evaporated. The oil was purified on preparatory HPLC (2 to 10 % AcCN: H20 with 0.1 % formic acid modifier). The fractions were collected. To each
fraction was added 1 ml of 1 M HCl, and the fractions were evaporated to dryness. 4-((4-(((trans-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid (50 mg, 0.118 mmol, 19.33 % yield) was isolated as a white solid. 1H NMR (400 MHz,
METHANOLS) δ 8.16 (d, J= 8.34 Hz, 2H), 7.70 (d, J= 8.34 Hz, 2H), 7.30 – 7.37 (m, 2H), 7.23 – 7.29 (m, 1H), 7.20 (d, J= 7.33 Hz, 2H), 4.44 (br. s., 2H), 3.57 (d, J= 11.62 Hz, 2H), 3.07 – 3.27 (m, 4H), 3.04 (dt, J= 3.95, 7.52 Hz, 1H), 2.59 (ddd, J= 3.54, 6.57, 10.11 Hz, lH), 2.12 (d, J= 13.89 Hz, 3H), 1.54 – 1.81 (m, 3H), 1.42 (q, 1H); LC-MS Rt = 0.47 min; MS (ESI): 365.3 [M+H]+.
[M+H]+.
Example 29
4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid

Step 1.
tert-Butyl 4-((4-(hydroxymethyl)piperidin-l-yl)methyl)benzoate
tert-Butyl 4-(bromomethyl)benzoate (1 g, 3.13 mmol) and piperidin-4-ylmethanol (0.361 g, 3.13 mmol) were dissolved in acetonitrile (25 mL). K2CO3 (1.300 g, 9.40 mmol) was added and the reaction mixture was heated to reflux for 20 min. The reaction mixture was cooled down to room temperature, filtered and evaporated. The resulting solid was partitioned between ethyl acetate (50mL) and 1 M HC1 (50 mL). The layers were separated and the aqueous layer was washed with ethyl acetate and the organic layers were discarded. The aqueous layer was basified with 8 M NaOH to pH -10 and extracted 2 times with 50 mL of ethyl acetate. The organic layers were combined, washed with brine and dried over MgSC^, filtered and evaporated. tert-Butyl 4-((4- (hydroxymethyl)piperidin-l-yl)methyl)benzoate (0.95 g, 2.99 mmol, 95 % yield) was isolated as yellow oil. 1H NMR (400 MHz, CHLOROFORM-d) δ 7.95 (d, J= 8.34 Hz, 2H), 7.39 (d, J = 8.08 Hz, 2H), 3.56 (s, 2H), 3.51 (d, J = 6.57 Hz, 2H), 2.90 (d, J= 11.37 Hz, 2H), 1.94 – 2.04 (m, 2H), 1.73 (d, J= 14.15 Hz, 2H), 1.61 (s, 9H), 1.40 – 1.56 (m, 2H), 1.30 – 1.37 (m, 2H); LC-MS Rt = 0.67 min; MS (ESI): 306.2 [M+H]+.
Step 2.
tert-Butyl 4-((4-formylpiperidin- 1 -yl)methyl)benzoate
To a solution of oxalyl chloride (0.408 mL, 4.67 mmol) in dichloromethane (5 mL) at -60 °C was added a solution of DMSO (0.508 mL, 7.15 mmol) in 15 mL of dichloromethane over 30 minutes. The reaction was stirred for 30 minutes at -60 °C A solution of tert-butyl 4-((4-(hydroxymethyl)piperidin-l-yl)methyl)benzoate (950 mg, 3.11 mmol) in 5 mL of dichloromethane was added over 10 minutes at -60 °C. The reaction mixture was stirred for 3 hours at – 60 °C, then triethylamine (2.168 mL, 15.55 mmol) was added and after 10 minutes 10 mL of water was added. The reaction mixture was allowed to warm up to the room temperature. The layers were separated. The pH of the water layer was adjusted to ~7 with 1 M HC1 and then extracted with 20 mL of dichloromethane. The combined organic layers were washed with water and brine, then dried over MgSO, filtered and evaporated. The resulting oil was purified on a silica column eluting with EtOAc to yield tert-butyl 4-((4-formylpiperidin-l-yl)methyl)benzoate (550 mg, 1.722 mmol, 55.4 % yield) as a yellow oil. 1H NMR (400 MHz, CHLOROFORM-d) δ 9.67 (d, J= 1.26 Hz, 1H), 7.96 (d, J= 8.34 Hz, 2H), 7.38 (d, J= 8.34 Hz, 2H), 3.56 (s, 2H), 2.75 – 2.92 (m, 2H), 2.21 – 2.35 (m, 1H), 2.14 (t, J= 10.48 Hz, 2H), 1.91 (dd, J= 2.78, 13.14 Hz, 2H), 1.65 – 1.81 (m, 2H), 1.58 – 1.64 (m, 9H); LC-MS Rt = 0.69 min; MS (ESI): 304.2
[M+H]+, 322.2 [M+H20]+, 336.6 [M+Na]+
Step 3.
tert-Butyl 4-((4-(((( 1 R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoate
To a solution of tert-butyl 4-((4-formylpiperidin-l-yl)methyl)benzoate (6.7 g, 22.08 mmol) in methanol (50 mL) was added (lR,2S)-2-phenylcyclopropanamine (3.53 g, 26.5 mmol). The reaction mixture was refluxed for 5 minutes then cooled down to the room temperature. Sodium cyanotrihydroborate (2.082 g, 33.1 mmol) was added. The reaction mixture was stirred 1 hour at room temperature. Water (50 mL) was added. The reaction was concentrated and 50 mL of dichloromethane was added. The layers were separated. The organics were washed with 10 % acetic acid (50 mL). The layers were separated and 50 mL of brine was added slowly as a solid crashed out. The solid was filtered and suspended in isopropanol. The suspension was sonicated and filtered. tert-Butyl 4-((4-(((( 1 R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoate (5.8 g, 13.65 mmol, 61.8 % yield) was isolated as a white solid. 1H NMR (400 MHz,
METHANOLS) δ 8.07 (d, J= 8.34 Hz, 2H), 7.70 (d, J= 8.08 Hz, 2H), 7.28 – 7.37 (m, 2H), 7.10 – 7.28 (m, 3H), 4.43 (br. s., 2H), 3.54 (d, J= 10.86 Hz, 2H), 3.08 – 3.26 (m, 4H), 3.03 (dt, J= 3.76, 7.39 Hz, 1H), 2.54 – 2.71 (m, 1H), 2.03 – 2.29 (m, 3H), 1.67 – 1.84 (m, 2H), 1.58 – 1.67 (m, 10H), 1.40 (q, J = 6.82 Hz, lH); LC-MS Rt = 0.76 min; MS (ESI): 421.4 [M+H]+.
Step 4.
4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid
A suspension of tert-butyl 4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoate (5.8 g, 13.79 mmol) in HCL – 1 M (80 ml, 80 mmol) was heated to 89 °C (internal temperature) for 2 hr. The solution was cooled down to the room temperature and held in an ice -bath for 1 hour and then filtered. 4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid (3.8 g, 8.25 mmol, 59.8 % yield) was isolated as white solid. 1H NMR (400 MHz, METHANOL-d4) 5 8.15 (d, J= 8.34 Hz, 2H), 7.72 (d, J= 8.59 Hz, 2H), 7.29 – 7.37 (m, 2H), 7.14 – 7.28 (m, 3H), 4.45 (br. s., 2H), 3.55 (d, J= 10.36 Hz, 2H), 3.07 – 3.29 (m, 4H), 3.04 (dt, J= 3.98, 7.71 Hz, 1H), 2.61 (ddd, J= 3.66, 6.57, 10.23 Hz, 1H), 1.98 – 2.31 (m, 3H), 1.72 (br. s., 2H), 1.62 (ddd, J= 4.42, 6.51, 10.55 Hz, 1H), 1.41 (q, J= 6.82 Hz, lH); LC-MS Rt = 0.49 min; MS (ESI): 365.3 [M+H]+.

Neil Johnson
US Lead of Chemistry Talent Development, External Engagement and Recruitment at GSK
https://www.linkedin.com/in/neil-johnson-6628894
Experience
US Lead of Chemistry Talent Development, External Engagement and Recruitment
GSK
March 2016 – Present (4 months)Greater Philadelphia Area
Investgator
GlaxoSmithKline
1999 – Present (17 years)
Senior Scientist
Cephalon
September 1994 – June 1999 (4 years 10 months)
Education
///////////GSK-2879552, 1401966-63-9, Phase I , A LSD1 inhibitor, small cell lung cancer, acute myeloid leukemia, 1401966-69-5, 1902123-72-1
O=C(O)C1=CC=C(CN2CCC(CN[C@H]3[C@H](C4=CC=CC=C4)C3)CC2)C=C1
O=C(O)c1ccc(cc1)CN2CCC(CC2)CN[C@@H]4C[C@H]4c3ccccc3
GSK-2816126
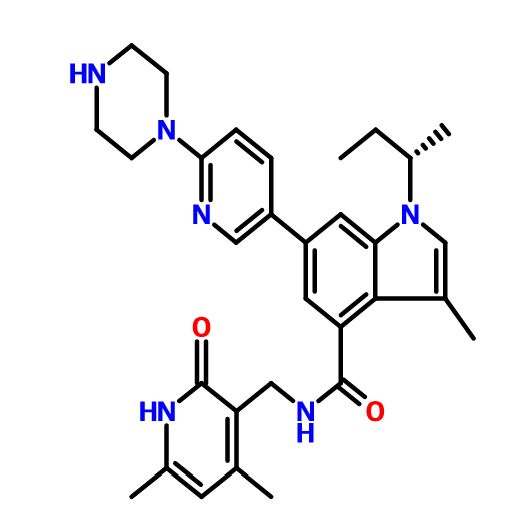

GSK-2816126
N-[(1,2-Dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl]-3-methyl-1-[(1S)-1-methylpropyl]-6-[6-(1-piperazinyl)-3-pyridinyl]-1H-indole-4-carboxamide, GSK 126, GSK 2816126, GSK 2816126A
N-[(4,6-Dimethyl-2-oxo-1,2-dihydro-3-pyridinyl)methyl]-3-methyl-1-((1S)-1-methylpropyl)-6-[6-(1-piperazinyl)-3-pyridinyl]-1H-indole-4-carboxamide
Phase I
| Formula |
C31H38N6O2
|
| Formula Wt. |
526.67
|
An histone-lysine n-methyltransferase EZH2 inhibitor potentially for the treatment of B-cell lymphoma.
![]()
Research Code GSK-2816126; GSK-126; 2816126
CAS No. 1346574-57-9
- Originator GlaxoSmithKline
- Class Antineoplastics
- Mechanism of Action EZH2 enzyme inhibitors; Histone modulators
- Phase I Diffuse large B cell lymphoma; Follicular lymphoma
- Preclinical Acute myeloid leukaemia
Most Recent Events
- 31 Mar 2014 Phase-I clinical trials in Follicular lymphoma (Second-line therapy or greater) in USA and United Kingdom (IV)
- 31 Mar 2014 Phase-I clinical trials in Diffuse large B cell lymphoma (Second-line therapy or greater) in USA and United Kingdom (IV)
- 16 Jan 2014 Preclinical trials in Diffuse large B cell lymphoma & Follicular lymphoma in United Kingdom (IV)
GSK-126 is an inhibitor of mutant EZH2, a histone methyltransferase (HMT) that exhibits point mutations at key residues Tyr641 and Ala677; this compound does not appreciably affect WT EZH2. EZH2 is responsible for modulating expression of a variety of genes. GSK-126 competes with cofactor S-adenylmethionine (SAM) for binding to EZH2. GSK-126 displays anticancer chemotherapeutic activity by inhibiting proliferation in in vitro and in vivo models of diffuse large B-cell lymphoma.
SYNTHESIS
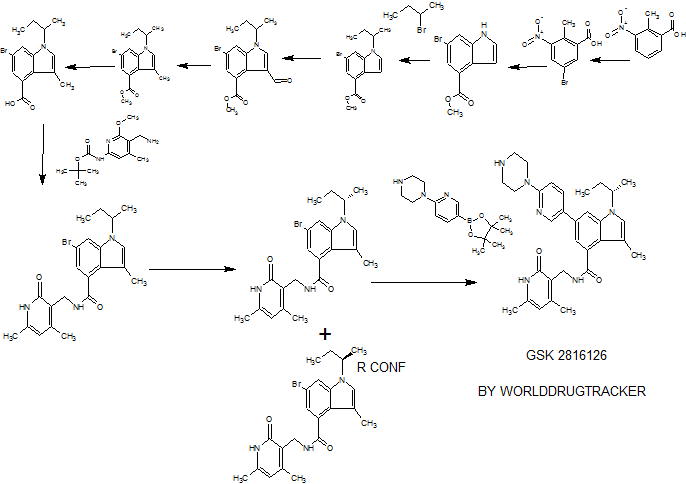
PATENT
CN 105541801
https://www.google.com/patents/CN105541801A?cl=zh
Example 79: Add (S) in a three-necked flask 100 Qiu – bromo – Shu – (isobutyl) – N – ((4,6-dimethyl-2-oxo -l, 2- dihydropyridin-3-yl) methyl) -3-methyl-1 hydrogen – indole carboxamide (365mg, 0.82mmol), 2- (piperazin-1-yl) pyridine-5-boronic acid pinacol ester (309mg, 1.07mmol, 1 · 3eq), potassium phosphate (522mg, 2.46mmol, 3eq), water, and I, 4- diepoxy-hexadecane as the solvent. Then, under nitrogen was added [I, Γ- bis (diphenylphosphino) ferrocene] dichloropalladium (II) dichloromethane complex (53.9mg, 0.066mmo 1), and at 90 ° C reaction, to give the desired product after purification 400mg (92% yield). Goo NMR (500MHz, DMSO- (I6) JO.70-0 · 78 (ιή, 3H), 1.37-1.44 (m, 4H), 1.75-1.87 (m, 2H), 2.11 (s, 3H), 2.16 ( s, 3H), 2.22-2.27 (m, 3H), 2.77-2.85 (m, 4H), 3.41-3.49 (m, 4H), 4.35 (d, J = 5.31Hz, 2H), 4.56-4.68 (m, lH), 5.87 (s, 1H), 6.88 (d, J = 8.84Hz, 1H), 7.17 (d J = 1.52Hz, 1H), 7.26 (s, lH), 7.73 (d J = 1.26Hz, 1H) , 7.91 (dd, J = 8.84Hz, lH), 8.16 (t, J = 5.05Hz, lH), 8.50 (d, J = 2.53Hz, lH); 13C NMR (125MHz, DMSO- (I6) Sll .6 , 12.6,19.1, 19.9,21.7,30.4,35.9,46.3,46.9,52.4,107.6,108.2,108.5,110.6,116.9,122.6,123.8, 130.6,131.5,136.7,138.6,143.5,146.4,150.2,159.2,164.0 , 169.6.
PATENT
Examples 267 and 268
(S)-6-bromo-1 -(sec-butyl)-N-((4,6-dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3- methyl-1 H-indole-4-carboxamide (Ex 267) and (R)-6-Bromo-1 -(sec-butyl)-N-((4,6- dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (Ex 268)

6-Bromo-1-(sec-butyl)-N-((4,6-dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methy methyl-1 H-indole-4-carboxamide (racemic mixture, 1.9 g) was resolved by chiral HPLC (column : Chiralpak AD-H, 5 microns, 50 mm x 250 mm, UV detection :240 nM, flow rate: 100 mL/min, T = 20 deg C, eluent: 60:40:0.1 n-heptane:ethanol:isopropylamine
(isocratic)). For each run, 100 mg of the racemic compound was dissolved in 30 volumes (3.0 ml.) of warm ethanol with a few drops of isopropylamine added. A total of 19 runs were performed. Baseline resolution was observed for each run. The isomer that eluted at 8.3-10.1 min was collected (following concentration) as a white solid, which was dried at 50 °C (< 5 mm Hg) to afford 901 mg, and was determined to be the S isomer* (Ex. 267; chiral HPLC: >99.5% ee (no R isomer detected). The isomer that eluted at 10.8-13.0 min was collected as a white solid, which was dried at 50 °C (< 5 mm Hg) to afford 865 mg, and was determined to be the R isomer* (Ex. 268; chiral HPLC: 99.2% ee; 0.4% S isomer detected). 1H NMR and LCMS were consistent with the parent racemate.
* The absolute configuration was determined by an independent synthesis of each enantiomer from the corresponding commercially available homochiral alcohols via Mitsunobu reaction. The sterochemical assignments were also consistent by vibrational circular dichroism (VCD) analysis.
Example 269
1-(sec-butyl)-N-((4,6-dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-6-(6- (piperazin-1 -yl)pyridin-3-yl)-1 -indole-4-carboxamide

Added sequentially to a reaction vial were 6-bromo-1 -(sec-butyl)-N-((4,6-dimethyl- 2-OXO-1 , 2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (0.15 g, 0.338 mmol), 1-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (0.127 g, 0.439 mmol), and potassium phosphate (tribasic) (0.287 g, 1.350 mmol), followed by 1 ,4- Dioxane (3 mL) and water (0.75 mL). The suspension was stirred under N2 degassing for 10 min., and then added PdCI2(dppf)-CH2CI2adduct (0.028 g, 0.034 mmol). The reaction vial was sealed, placed into a heat block at 95 °C, and stirred for 1.5 h. The contents were removed from heating and allowed to cool to room temperature. The aq layer was removed from bottom of the reaction vial via pipette. The reaction mixture was diluted into EtOAc (20 mL) followed by addition of 0.2 g each of Thiol-3 silicycle resin and silica gel. The volatiles were removed in vacuo and the residue dried on hi-vac for 1 h. The contents were purified by silica gel chromatography (dry loaded, eluent : A:
Dichloromethane, B: 10% (2M Ammonia in Methanol) in Chloroform, Gradient B: 8- 95%). The obtained solid was concentrated from TBME and dried in vacuum oven at 45 °C for 18 h. The product was collected as 129 mg (70%). 1H NMR (400 MHz, DMSO-d6) δ ppm 0.73 (t, J=7.33 Hz, 3H), 1.40 (d, J=6.57 Hz, 3H), 1.80 (dq, J=10.07, 7.08 Hz, 2H), 2.1 1 (s, 3H), 2.14 – 2.19 (m, 3H), 2.24 (s, 3H), 2.76 – 2.85 (m, 4H), 3.41 – 3.49 (m, 4H), 4.35 (d, J=5.05 Hz, 2H), 4.54 – 4.67 (m, 1 H), 5.87 (s, 1 H), 6.88 (d, J=8.84 Hz, 1 H), 7.17 (d, J=1.26 Hz, 1 H), 7.26 (s, 1 H), 7.73 (d, J=1.26 Hz, 1 H), 7.91 (dd, J=8.84, 2.53 Hz, 1 H), 8.16 (t, J=5.05 Hz, 1 H), 8.50 (d, J=2.53 Hz, 1 H), 1 1.48 (br. s.,1 H) ; LCMS MH+ =527.3.
Example 270
A/-[(4,6-dimethyl-2-oxo-1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl-1 -[(1 S)-1 -methylpropyl]-6- [6-(1-piperazinyl)-3-pyridinyl]-1 H-indole-4-carboxamide

To a 30 mL microwave vial were added (S)-6-bromo-1 -(sec-butyl)-N-((4,6- dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (100 mg, 0.225 mmol), 1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (85 mg, 0.293 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1.000 mL) and sodium carbonate (0.338 mL, 0.675 mmol), and the mixture was degassed for 5 min by bubbling nitrogen. PdCI2(dppf)-CH2CI2 adduct (14.70 mg, 0.018 mmol) was added and the tube was sealed. The mixture was irradiated (microwave) at 140 °C for 10 min. The mixture was concentrated and the residue was taken up into MeOH and filtered. The filtrate was purified using reverse-phase HPLC (eluent: 25%ACN/H20, 0.1 % NH4OH to 60%
ACN/H20, 0.1 % NH4OH ) to give 91 mg of product as off-white solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 0.70 – 0.78 (m, 3H), 1.37 – 1.44 (m, 3H), 1 .75 – 1.87 (m, 2H), 2.1 1 (s, 3H), 2.16 (s, 3H), 2.22 – 2.27 (m, 3H), 2.77 – 2.85 (m, 4H), 3.41 – 3.49 (m, 4H), 4.35 (d, J=5.31 Hz, 2H), 4.56 – 4.68 (m, 1 H), 5.87 (s, 1 H), 6.88 (d, J=8.84 Hz, 1 H), 7.17 (d, J=1.52 Hz, 1 H), 7.26 (s, 1 H), 7.73 (d, J=1.26 Hz, 1 H), 7.91 (dd, J=8.84, 2.53 Hz, 1 H), 8.16 (t, J=5.05 Hz, 1 H), 8.50 (d, J=2.53 Hz, 1 H); LCMS: 527.8 (MH+).
Example 271
A/-[(4,6-dimethyl-2-oxo-1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl-1 -[(1 /?)-1-methylpropyl]- 6-[6-(1 -piperazinyl)-3-pyridinyl]-1 -indole-4-carboxamide

To a 30 mL microwave vial were added (R)-6-bromo-1-(sec-butyl)-N-((4,6- dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (100 mg, 0.225 mmol), 1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (85 mg, 0.293 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1.000 mL) and sodium carbonate (0.338 mL, 0.675 mmol), and the mixture was degassed for 5 min by bubbling nitrogen. PdCI2(dppf)-CH2Cl2 adduct (14.70 mg, 0.018 mmol) was added and the tube was sealed. The mixture was irradiated (microwave) at 140 °C for 10 min. The mixture was concentrated and the residue was taken up into MeOH and filtered. The filtrate was purified using reverse-phase HPLC (eluent: 25%ACN/H20, 0.1 % NH4OH to 60%
ACN/H20, 0.1 % NH4OH ) to give 90 mg of product as off-white solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 0.73 (m, 3H), 1.41 (d, J=6.57 Hz, 3H), 1.81 (td, J=7.14, 2.91 Hz, 2H), 2.1 1 (s, 3H), 2.15 – 2.20 (m, 3H), 2.24 (s, 3H), 2.77 – 2.83 (m, 4H), 3.41 – 3.49 (m, 4H), 4.35 (d, J=5.05 Hz, 2H), 4.54 – 4.68 (m, 1 H), 5.87 (s, 1 H), 6.88 (d, J=8.84 Hz, 1 H), 7.17 (d, J=1.52 Hz, 1 H), 7.26 (s, 1 H), 7.73 (d, J=1.26 Hz, 1 H), 7.91 (dd, J=8.84, 2.53 Hz, 1 H), 8.16 (t, J=5.05 Hz, 1 H), 8.50 (d, J=2.27 Hz, 1 H); LCMS: 527.7 (MH+)
PATENT
Example 270
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-[(15)-l-methylpropyl]-6-[6-(l-piperazinyl)-3-pyridinyl]-lH-indole-4-carboxamide

To a 30 niL microwave vial were added (S)-6-bromo-l-(sec-butyl)-N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-3 -methyl- lH-indole-4-carboxamide (100 mg, 0.225 mmol), l-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (85 mg, 0.293 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1.000 mL) and sodium carbonate (0.338 mL, 0.675 mmol), and the mixture was degassed for 5 min by bubbling nitrogen. PdCi2(dppf)-CH2Ci2 adduct (14.70 mg, 0.018 mmol) was added and the tube was sealed. The mixture was irradiated (microwave) at 140 °C for 10 min. The mixture was concentrated and the residue was taken up into MeOH and filtered. The filtrate was purified using reverse-phase HPLC (eluent: 25%ACN/H20, 0.1% NH4OH to 60% ACN/H20, 0.1% NH4OH ) to give 91 mg of product as off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.70 – 0.78 (m, 3H), 1.37 – 1.44 (m, 3H), 1.75 – 1.87 (m, 2H), 2.11 (s, 3H), 2.16 (s, 3H), 2.22 – 2.27 (m, 3H), 2.77 – 2.85 (m, 4H), 3.41 – 3.49 (m, 4H), 4.35 (d, J=5.31 Hz, 2H), 4.56 – 4.68 (m, IH), 5.87 (s, IH), 6.88 (d, J=8.84 Hz, IH), 7.17 (d, J=1.52 Hz, IH), 7.26 (s, IH), 7.73 (d, J=1.26 Hz, IH), 7.91 (dd, J=8.84, 2.53 Hz, IH), 8.16 (t, J=5.05 Hz, IH), 8.50 (d, J=2.53 Hz, IH); LCMS: 527.8 (MH+).
Example 271
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-[(li?)-l-methylpropyl]-6-[6-(l-piperazinyl)-3-pyridinyl]-l -indole-4-carboxamide

To a 30 mL microwave vial were added (R)-6-bromo-l-(sec-butyl)-N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-3 -methyl- lH-indole-4-carboxamide (100 mg, 0.225 mmol), l-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (85 mg, 0.293 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1.000 mL) and sodium carbonate (0.338 mL, 0.675 mmol), and the mixture was degassed for 5 min by bubbling nitrogen. PdCl2(dppf)-CH2Cl2 adduct (14.70 mg, 0.018 mmol) was added and the tube was sealed. The mixture was irradiated (microwave) at 140 °C for 10 min. The mixture was concentrated and the residue was taken up into MeOH and filtered. The filtrate was purified using reverse-phase HPLC (eluent: 25%ACN/H20, 0.1% NH4OH to 60% ACN/H20, 0.1% NH4OH ) to give 90 mg of product as off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.73 (m, 3H), 1.41 (d, J=6.57 Hz, 3H), 1.81 (td, J=7.14, 2.91 Hz, 2H), 2.11 (s, 3H), 2.15 – 2.20 (m, 3H), 2.24 (s, 3H), 2.77 – 2.83 (m, 4H), 3.41 – 3.49 (m, 4H), 4.35 (d, J=5.05 Hz, 2H), 4.54 -4.68 (m, 1H), 5.87 (s, 1H), 6.88 (d, J=8.84 Hz, 1H), 7.17 (d, J=1.52 Hz, 1H), 7.26 (s, 1H), 7.73 (d, J=1.26 Hz, 1H), 7.91 (dd, J=8.84, 2.53 Hz, 1H), 8.16 (t, J=5.05 Hz, 1H), 8.50 (d, J=2.27 Hz, 1H); LCMS: 527.7 (MH+).
REF
| WO2005034845A2 * | Jul 13, 2004 | Apr 21, 2005 | Supergen, Inc. | Compositions and methods for treatment of cancer |
| WO2007053114A1 * | Oct 30, 2006 | May 10, 2007 | S*Bio Pte Ltd | Method of predicting a response to hdac inhibitors |
| WO2010090723A2 * | Feb 2, 2010 | Aug 12, 2010 | University Of Georgia Research Foundation, Inc. | Methods of inhibiting fibrogenesis and treating fibrotic disease |
| US20110035336 | May 1, 2008 | Feb 10, 2011 | Yigang Cai | Rating change for a prepaid session based on movement of a mobile device |
| US20110035340 | Aug 7, 2009 | Feb 10, 2011 | Fibre-Craft Materials Corp. | Decorating system and method of marketing and enhancing a surface area using a decorating system |
| US20110035344 | Aug 6, 2009 | Feb 10, 2011 | International Business Machines Corporation | Computing mixed-integer program solutions using multiple starting vectors |
| US20110064664 * | Oct 8, 2008 | Mar 17, 2011 | The Board Of Regents Of The University Of Texas System | Methods and compositions involving chitosan nanoparticles |
| WO2015077194A1 * | Nov 18, 2014 | May 28, 2015 | Bristol-Myers Squibb Company | Inhibitors of lysine methyl transferase |
| WO2015132765A1 * | Mar 6, 2015 | Sep 11, 2015 | Glaxosmithkline Intellectual Property (No.2) Limited | Enhancer of zeste homolog 2 inhibitors |
| WO2015141616A1 * | Mar 16, 2015 | Sep 24, 2015 | 第一三共株式会社 | 1,3-benzodioxole derivative |
| WO2016066697A1 * | Oct 28, 2015 | May 6, 2016 | Glaxosmithkline Intellectual Property (No.2) Limited | Enhancer of zeste homolog 2 inhibitors |
| US9051269 | Nov 19, 2012 | Jun 9, 2015 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US9085583 | Feb 11, 2013 | Jul 21, 2015 | Constellation—Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US20150344459 * | Dec 20, 2013 | Dec 3, 2015 | Epizyme, Inc. | 1,4-pyridone bicyclic heteroaryl compounds |
/////////GSK-2816126, GSK-126, 2816126, 1346574-57-9, GSK 126, GSK 126, GSK 2816126, GSK 2816126A
CC=5C=C(C)NC(=O)C=5CNC(=O)c1cc(cc2c1c(C)cn2[C@@H](C)CC)c3cnc(cc3)N4CCNCC4
TD 1607
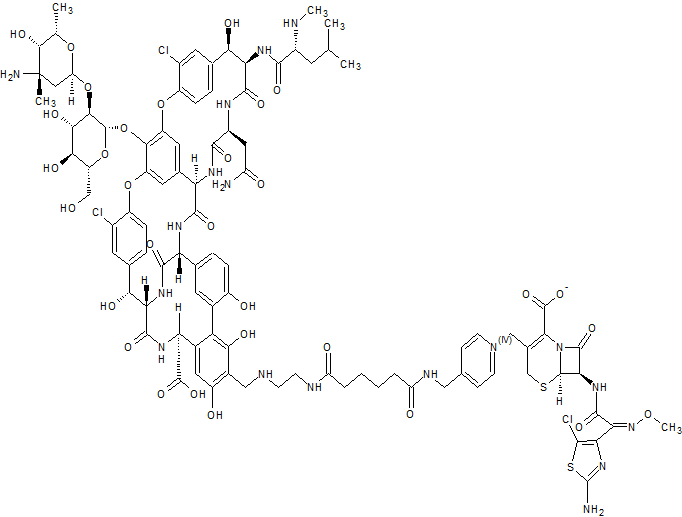
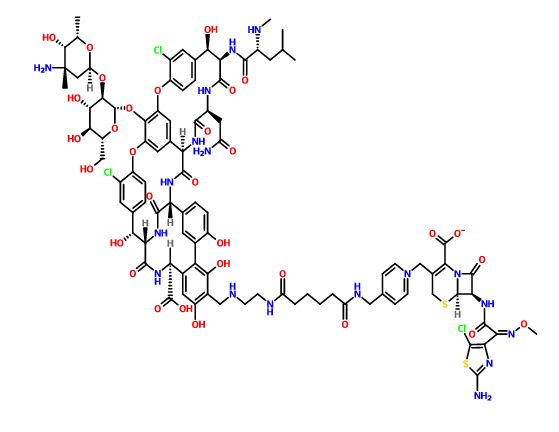
TD-1607
Phase I
A glycopeptide-cephalosporin heterodimer potentially for the treatment of gram-positive bacterial infection.
![]()
CAS No. 827040-07-3
C95 H109 Cl3 N18 O31 S2,
Molecular Weight, 2169.47
Vancomycin, 29-[[[2-[[6-[[[1-[[(6R,7R)-7-[[(2Z)-2-(2-amino-5-chloro-4-thiazolyl)-2-(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]pyridinium-4-yl]methyl]amino]-1,6-dioxohexyl]amino]ethyl]amino]methyl]-, inner salt
Vancomycin, 29-[[[2-[[6-[[[1-[[(6R,7R)-7-[[(2Z)-(2-amino-5-chloro-4-thiazolyl)(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]pyridinium-4-yl]methyl]amino]-1,6-dioxohexyl]amino]ethyl]amino]methyl]-, inner salt
- Originator Theravance
- Developer Theravance Biopharma
- Class Antibacterials; Cephalosporins; Glycopeptides
- Mechanism of Action Cell wall inhibitors
-
- Phase I Gram-positive infections
Most Recent Events
- 21 Apr 2016 Phase I development is ongoing in USA
- 01 Jul 2014 Theravance completes a phase I trial in Healthy volunteers in in USA (NCT01949103)
- 02 Jun 2014 Theravance Biopharma is formed as a spin-off of Theravance
-
Company Theravance Biopharma Inc. Description Glycopeptide cephalosporin heterodimer antibiotic Molecular Target Mechanism of Action Therapeutic Modality Small molecule: Combination Latest Stage of Development Phase I Standard Indication Gram-negative bacterial infection Indication Details Treat Gram-positive bacterial infections



PATENT
WO 2005005436
The present invention provides novel cross-linked glycopeptide – cephalosporin compounds that are useful as antibiotics. The compounds of this invention have a unique chemical structure in which a glycopeptide group is covalently linked to a pyridinium moiety of a cephalosporin group. Among other properties, compounds of this invention have been found to possess surprising and unexpected potency against Gram-positive bacteria including methicillin-resistant Staphylococci aureus (MRSA). Accordingly, in one aspect, the invention provides a compound of formula I:

////////Theravance Biopharma, TD 1607, phase 1, glycopeptide-cephalosporin heterodimer , gram-positive bacterial infection
TAK-058 (ENV-8058)

TAK-058 , ENV-8058
5-HT 3 receptor antagonist
1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide
l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5 .7S)-9-methyl-3-oxa-9-azabicyclo[3.3.11nonan-7-yl)-lH-indole-3-carboxamide
1-(1-methyl-1H- pyrazol-4-yl)-N- ((1R,5S,7S)- 9-methyl-3- oxa-9-azabicyclo [3.3.1]nonan-7- yl)-1H-indole-3- carboxamide, 2,2,2- trifluoroacetic acid salt
N-(9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1-(1-methylpyrazol-4-yl)indole-3-carboxamide
| Molecular Formula: | C21H25N5O2 |
|---|---|
| Molecular Weight: | 379.4555 g/mol |
https://clinicaltrials.gov/ct2/show/NCT02153099
Phase I Schizophrenia
| Latest Stage of Development | Phase I |
| Standard Indication | Schizophrenia |
| Indication Details | Treat schizophrenia |
- 01 Dec 2015 Phase-I clinical trials in Schizophrenia (Combination therapy) in USA (PO)
- 01 Dec 2015 Takeda completes a phase I trial in Healthy volunteers in USA (NCT02389881)
- 28 Nov 2015 Takeda plans a phase I trial in Schizophrenia (Combination therapy) in USA (NCT02614586)

1 -( 1 -methyl- 1 H-pyrazol-4-yl)-N-((lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-lH-indole-3-carboxamide, free base, which is an antagonist of the 5-HT3 receptor. 1 -(1 -Methyl- 1 H-pyrazol-4-yl)-N-((lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-lH-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt, is disclosed in PCT Publication No. WO
2014/014951, published January 23, 2014.
1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide a 5-HT3 receptor antagonist, useful for treating anxiety, depression, eating disorder, schizophrenia, cognitive dysfunction, Parkinson’s disease, Huntington’s Chorea, presenile dementia, Alzheimer’s disease and atherosclerosis.
This compound was originally claimed in WO2014014951, Takeda, following its acquisition of Envoy Therapeutics, is developing TAK-058 (ENV-8058), a 5-HT3 receptor antagonist, as an oral solution for treating schizophrenia, especially cognitive impairment associated with schizophrenia.
In July 2015, the drug was listed as being in phase I development. TAK-058 may have emerged from a schizophrenia therapy program which used Envoy’s bacTRAP translational profiling technology to identify a protein target in the brain.
PATENT
Example 5
Synthesis of l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5 .7S)-9-methyl-3-oxa-9-azabicyclo[3.3.11nonan-7-yl)-lH-indole-3-carboxamide. 2.2.2-trifluoroacetic acid salt
Step 1 : methyl 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylate. TFA
To a sealed tube was added copper(I) iodide (65.2 mg, 0.342 mmol), methyl 1H-indole-3-carboxylate (200 mg, 1.142 mmol) and potassium phosphate (509 mg, 2.397 mmol), then the reaction vessel was evacuated and purged with nitrogen (3x). Next, 4-bromo-l-methyl-lH-pyrazole (184 mg, 1.142 mmol) and (lR,2R)- ,N2-dimethylcyclohexane-l,2-diamine (109 μΐ, 0.685 mmol) were added, followed by toluene (1 142 μΐ). The reaction tube was evacuated and purged with nitrogen, then sealed and heated at 1 10 °C for 24 h. HPLC purification provided the title compound as a colorless oil.
Step 2: 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylic acid hydrochloride
To a solution of methyl 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylate, TFA
(3.5 mg, 9.48 μιηοΐ) in MeOH (95 μΐ) was added a solution of aq. KOH (33.2 μΐ, 0.066 mmol, 2 M). The reaction mixture was stirred at RT overnight, then acidified with IN HC1.
The solvent was evaporated under reduced pressure and the residue was dried under vacuum overnight. The title compound was used without further purification.
Step 3 : l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5 .7S)-9-methyl-3-oxa-9-azabicyclor3.3.11nonan-7-yl)-lH-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt
To a mixture of 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylic acid hydrochloride (2.6 mg, 9.36 μιηοΐ) in DMF (187 μΐ) was added HATU (4.27 mg, 0.01 1 mmol) and DIPEA (8.18 μΐ, 0.047 mmol). After the reaction mixture was stirred at RT for 15 min, (lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-amine, TFA (3.04 mg, 0.01 1 mmol) was added and stirring was continued for 2 h. HPLC purification afforded the title compound as a white solid. MS (ESI, pos. ion) m/z: 380.30 (M+l).
PATENT
EXAMPLE 1 : l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1 ]nonan-7-yl)- lH-indole-3-carboxamide
l-(l-Methyl-lH-pyrazol-4-yl)-lH-indole-3-carboxylic acid (128.7 g, 0.53 mol,) and anhydrous THF (645 mL) was heated to about 43°C. Oxalyl chloride (137.7 g, 92 mL, 1.08 mol) was added dropwise between 40 and 50°C. Gas evolution ceased in approximately 30 minutes. The resulting suspension was stirred for 2 hours at 50°C, allowed to cool to room temperature, and then stirred overnight. The suspension was diluted with heptane (1.5 L), stirred for 10 minutes, and allowed to settle. The supernatant was removed. The addition of heptane (1.5 L), followed by stirring, settling, and decanting was repeated two more times.
The resulting suspension was diluted with anhydrous THF (645 mL) and the ratio between THF and heptane was determined by NMR to be 3:2. The reaction mixture was cooled to 5°C and to the mixture was added DIPEA base (138 g, 1.07 mol) at such a rate that the temperature did not exceed 20°C. Next (li?,55*,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-amine (101.4 g, 0.63 mol) in 500 mL of anhydrous THF was added. The reaction mixture was warmed to ambient temperature and stirred at 20 to 23°C overnight to give a suspension.
The suspension was filtered and the cake was dissolved in IN HC1 (2.6 L). The aqueous layer was washed with EtOAc (3 x 2.6 L). The aqueous layer was cooled to 5°C and was basified to pH 12 with aqueous potassium hydroxide (230 g) solution in water (500 mL). The mixture was stirred at 5 to 10°C overnight to give a solid. The product was filtered, washed with water (2 x 1.2 L), followed by MTBE (2 x 1.2 L), and then dried to give 128 g (64%) of the (crude) title compound.
Patent
https://www.google.co.in/patents/US20140024644
1-(1-methyl-1H- pyrazol-4-yl)-N- ((1R,5S,7S)- 9-methyl-3- oxa-9-azabicyclo [3.3.1]nonan-7- yl)-1H-indole-3- carboxamide, 2,2,2- trifluoroacetic acid salt
Synthetic Procedures Reference 1 Synthesis of (1R,5S,7S)-tert-butyl 7-hydroxy-3-oxa-9-azabicyclo[3.3.1]nonane-9-carboxylate
-

-
Sodium borohydride (259 mg, 6.84 mmol) was added portion-wise to a solution of (1R,5S)-tert-butyl 7-oxo-3-oxa-9-azabicyclo[3.3.1]nonane-9-carboxylate (550 mg, 2.279 mmol) in MeOH (4559 μl) at 0° C. After 5 min, the reaction mixture was allowed to warm to RT then stirred for 30 min. The mixture was concentrated under reduced pressure, dissolved in EtOAc and washed with brine. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford the title compound as a white solid, which was used without further purification.
Example 4 Synthesis of N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1-(1H-pyrazol-4-yl)-1H-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt
-

-
A mixture of 1-((1-benzyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide 2,2,2-trifluoroacetate (85 mg, 0.149 mmol) and 10% Pd—C (120 mg) in MeOH (1.0 ml) was stirred at RT under H2 for 2 days. Filtration and concentration afforded the title compound as a white solid. MS (ESI, pos. ion) m/z: 366.20 (M+1).
Example 5 Synthesis of 1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt
Step 1: methyl 1-(1-methyl-1H-pyrazol-4-yl)-1H-indole-3-carboxylate, TFA
-
To a sealed tube was added copper(I) iodide (65.2 mg, 0.342 mmol), methyl 1H-indole-3-carboxylate (200 mg, 1.142 mmol) and potassium phosphate (509 mg, 2.397 mmol), then the reaction vessel was evacuated and purged with nitrogen (3×). Next, 4-bromo-1-methyl-1H-pyrazole (184 mg, 1.142 mmol) and (1R,2R)—N1,N2-dimethylcyclohexane-1,2-diamine (109 μl, 0.685 mmol) were added, followed by toluene (1142 μl). The reaction tube was evacuated and purged with nitrogen, then sealed and heated at 110° C. for 24 h. HPLC purification provided the title compound as a colorless oil.
Step 2: 1-(1-methyl-1H-pyrazol-4-yl)-1H-indole-3-carboxylic acid hydrochloride
-
To a solution of methyl 1-(1-methyl-1H-pyrazol-4-yl)-1H-indole-3-carboxylate, TFA (3.5 mg, 9.48 μmol) in MeOH (95 μl) was added a solution of aq. KOH (33.2 μl, 0.066 mmol, 2 M). The reaction mixture was stirred at RT overnight, then acidified with 1N HCl. The solvent was evaporated under reduced pressure and the residue was dried under vacuum overnight. The title compound was used without further purification.
Step 3: 1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt
-
To a mixture of 1-(1-methyl-1H-pyrazol-4-yl)-1H-indole-3-carboxylic acid hydrochloride (2.6 mg, 9.36 μmol) in DMF (187 μl) was added HATU (4.27 mg, 0.011 mmol) and DIPEA (8.18 μl, 0.047 mmol). After the reaction mixture was stirred at RT for 15 min, (1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-amine, TFA (3.04 mg, 0.011 mmol) was added and stirring was continued for 2 h. HPLC purification afforded the title compound as a white solid. MS (ESI, pos. ion) m/z: 380.30 (M+1).
| 15 |
 |
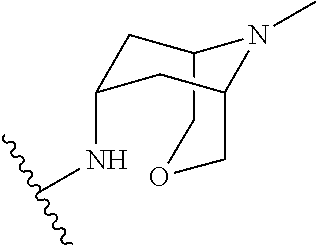 |
TFA |
| 379.456 MW | 380.30 MS +1
|
/////////TAK-058 , ENV-8058, phase I, takeda, 5-HT 3 receptor antagonist, Envoy Therapeutics, Inc., Phase I, Schizophrenia
C12CC(CC(N1C)COC2)NC(c4c3ccccc3n(c4)c5cnn(c5)C)=O
CN1C=C(C=N1)N2C=C(C3=CC=CC=C32)C(=O)NC4CC5COCC(C4)N5C
Zamicastat
CAS 1080028-80-3 BASE
1383828-47-4 OF HCL SALT
C21 H21 F2 N3 O S BASE
2H-Imidazole-2-thione, 1-[(3R)-6,8-difluoro-3,4-dihydro-2H– 1-benzopyran-3-yl]-1,3-dihydro-5-[2-[(phenylmethyl)amino] ethyl] -(R)-5-(2-(Benzylamino)ethyl)-1-(6,8-difluorochroman-3-yl)-1H-imidazole-2(3H)-thione
(R)-5-(2-(Benzylamino)ethyl)-1-(6,8-difluorochroman-3-yl)-1H-imidazole-2(3H)-thione
Molecular Weight, 401.47 BASE

- BIAL – PORTELA & CA., S.A. [PT/PT]; À Avenida da Siderurgia Nacional P-4745-457 S. Mamede do Coronado (PT)

- Zamicastat is a dopamine beta-monooxygenase inhibitor in phase I clinical studies at BIAL for the treatment of hypertension and heart failure.
- Zamicastat is a potent and selective dopamine β-mono-oxygenase inhibitor. Zamicastat Prevents the Deterioration of Cardiometabolic and Inflammatory Biomarkers in a Genetic Model of Salt-sensitive Hypertension. Chronic high salt intake deteriorates several cardiometabolic and inflammatory biomarkers in Dahl/SS rats, which can be prevented by dopamine β-hydroxylase inhibition with zamicastat.
- crystalline forms of l-[(3R)-6,8-difluoro- 3,4-dihydro-2H-l-benzopyran-3-yl]-l,3-dihydro-5-[2-[(phenylmethyl)amino]ethyl]-2H- imidazole-2-thione, i.e. the Renantiomer of

and processes for preparing the same. Background and prior art:Interest in the development of inhibitors of dopamines-hydroxylase (ϋβΗ) has centred on the hypothesis that inhibition of this enzyme may provide significant clinical improvements in patients suffering from cardiovascular disorders such as hypertension or chronic heart failure. The rationale for the use of ϋβΗ inhibitors is based on their capacity to inhibit the biosynthesis of noradrenaline, which is achieved via enzymatic hydroxylation of dopamine. Activation of neurohumoral systems, chiefly the sympathetic nervous system, is the principal clinical manifestation of congestive heart failure (Parmley, W.W., Clinical Cardiology, 18: 440-445, 1995). Congestive heart failure patients have elevated concentrations of plasma noradrenaline (Levine, T.B. et al., Am. J. Cardiol., 49: 1659-1666, 1982), increased central sympathetic outflow (Leimbach, W.N. et al., Circulation, 73: 913- 919, 1986) and augmented cardiorenal noradrenaline spillover (Hasking, G.J. et al., Circulation, 73:615-621, 1966). Prolonged and excessive exposure of the myocardium to noradrenaline may lead to down-regulation of cardiac β] -adrenoceptors, remodelling of the left ventricle, arrhythmias and necrosis, all of which can diminish the functional integrity of the heart. Congestive heart failure patients who have high plasma concentrations of noradrenaline also have the most unfavourable long-term prognosis (Cohn, J.N. et al., N. Engl. J. Med., 311 :819-823, 1984). Of greater significance is the observation that plasma noradrenaline concentrations are already elevated in asymptomatic patients with no overt heart failure and can predict ensuing mortality and morbidity (Benedict, C.R. et al., Circulation, 94:690-697, 1996). An activated sympathetic drive is not therefore merely a clinical marker of congestive heart failure, but may contribute to progressive worsening of the disease.
Potent dopamines-hydroxylase inhibitors having high potency and significantly reduced brain access are disclosed in WO 2008/136695. WO 2008/136695 describes compounds of formula I:

I where Rls R2 and R3 are the same or different and signify hydrogens, halogens, alkyl, nitro, amino, alkylcarbonylamino, alkylamino or dialkylamino group; R4 signifies -alkylaryl or – alkylheteroaryl; X signifies CH2, oxygen atom or sulphur atom; n is 2 or 3; including the individual (R)- and (S)-enantiomers or mixtures of enantiomers thereof; and including pharmaceutically acceptable salts and esters thereof, wherein the term alkyl means hydrocarbon chains, straight or branched, containing from one to six carbon atoms, optionally substituted by aryl, alkoxy, halogen, alkoxycarbonyl or hydroxycarbonyl groups; the term aryl means a phenyl or naphthyl group, optionally substituted by alkyl, alkyloxy, halogen or nitro group; the term halogen means fluorine, chlorine, bromine or iodine; the term heteroaryl means heteroaromatic group. In particular, WO 2008/136695 describes l-[(3R)-6,8-difluoro-3,4-dihydro-2H-l-benzopyran-3-yl]-l,3-dihydro-5-[2- [(phenylmethyl)amino]ethyl]-2H-Imidazole-2-thione.
Processes for the preparation of compounds of formula I, and in particular l-[(3R)-6,8- difluoro-3,4-dihydro-2H-l-benzopyran-3-yl]-l,3-dihydro-5-[2-[(phenylmethyl)amino] ethyl] -2H-Imidazole-2-thione, are described in WO 2008/136695 and are incorporated by reference herein. It is known that polymorphic forms of the same drug may have substantially different pharmaceutically important properties such as dissolution characteristics and bioavailability as well as stability of the drug. Furthermore, different forms may have different particle size, hardness and glass transition temperature. Thus, one form may provide significant advantages over other forms of the same drug in solid dosage form manufacture processes, such as accurate measurement of the active ingredients, easier filtration, or improved stability during granulation or storage. Furthermore, a particular process suitable for one form may also provide drug manufacturers several advantages such as economically or environmentally suitable solvents or processes, or higher purity or yield of the desired product.


PATENT
http://www.google.com/patents/WO2012087174A2?cl=en
Preparation of compound 2
[0090] Six lots of compound 2 (designated as lots 1, 2, 3, 4, 5 and 6) were prepared. The starting materials were prepared according to the following experimental protocols.
Lot 1 (Form A)
To a suspension of (R)-5-(2-aminoethyl)-l-(6,8-difluorochroman-3-yl)-lH- imidazole-2(3H)-thione (6.23 g, 20 mmol) in a mixture of Dichloromethane (DCM – 40 ml) and Methanol (40.0 ml) was added BENZALDEHYDE (2.230 ml, 22.00 mmol). To the resulting clear solution SODIUM CYANOBOROHYDRIDE (1.9 g, 28.7 mmol) was added in portions at 20-25°C to avoid intensive foaming and the solution was stirred at 20- 25°C for 40 h. The solution was quenched at 20-25°C with IN HC1 (35 ml), neutralised with 3N NaOH (35 ml), the mixture was extracted with DCM (200 ml). The organic phase was washed with brine, dried (MgS04), evaporated to dryness. The oily residue crystallised from 2-propanol (40 ml) at 20-25°C over a week-end. The crystals were collected, washed with 2-propanol, dried to give 5.2 g of the crude product. Re- crystallisation from 2-propanol-DCM hasn’t removed all impurities. Everything collected, evaporated with silica, applied on a column, eluted with Ethyl Acetate (EA)->EA-MeOH 9:1->4: 1, fractions 8-25 collected to give 3.8 g. Re-crystallised from 2-propanol (45 ml) and DCM (120 ml, removed on a rotavap) to give 2.77 g => initial lot (a) (HPLC 98.3% area) and 0.3 g of undissolved filtered off, by TLC right product. Initial lot (a) re- crystallised from 2-propanol (35 ml) and DCM (95 ml, removed on a rotavap) to give 2.51 g => initial lot (b) (HPLC 98.3% area). Combined with the above undissolved, re- crystallised from acetonitrile (200 ml, reflux to ice bath) to give 2.57 g => initial lot (c) (HPLC 98.8% area). Re-crystallised from acetonitrile (180 ml, reflux to 15°C) to give 2.25 g => Lot 1 (HPLC 99.2% area), mp 190-92°C. Lot 2 (Form A)
[0092] (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole- 2(3H)-thione (12 g, 29.9 mmol) was dissolved with heating to reflux in Tetrahydrofuran (300 ml), the solution was cooled to 5-10°C, Water (510 ml) was added slowly (approx 10 min) with stirring. The mixture was stirred for 1 h, solid was collected, washed with water, dried to give 11.73 g of product, by HPLC 1% of (R)-5-(2-Aminoethyl)-l-(6,8- difluorochroman-3-yl)-l,3-dihydroimidazole-2-thione hydrochloride and 1% of less polar impurity. The product was dissolved in Tetrahydrofuran (300 ml) with heating to reflux, 2- Propanol (150 ml) was added, the solution was concentrated to approx 100 ml (crystallisation occured), stirred in ice for 1.5 h. Solid was collected, washed with 2- propanol, dried to give 11.2 g of product, by HPLC 0.8% of (R)-5-(2-aminoethyl)-l-(6,8- difluorochroman-3-yl)-lH-imidazole-2(3H)-thione hydrochloride and 0.5% of less polar impurity. The product was dissolved in Tetrahydrofuran (300 ml) with heating to reflux, 2- Propanol (150 ml) was added, the solution was concentrated to approx 100 ml (crystallisation occured), stirred at 20-25°C for 1 h. Solid was collected, washed with 2- propanol, dried to give (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH- imidazole-2(3H)-thione (10.22 g, 25.5 mmol, 85 % yield).,
Lot 3 (form B)
To (R)-5-(2-aminoethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)- thione (2.36 g, 7.58 mmol) in a mixture of Methanol (15.00 ml) and Dichloromethane (15 ml) was added BENZALDEHYDE (0.845 ml, 8.34 mmol). To the resulting clear solution SODIUM CYANOBOROHYDRIDE (0.702 g, 10.61 mmol) was added in portions at 20- 25°C to avoid intensive foaming and the solution was stirred at 20-25°C for 40 h. The solution was quenched at 20-25°C with IN HC1 (12 ml), neutralised with 3N NaOH (12 ml), the mixture was extracted with DCM (100 ml). The organic phase was washed with brine, dried (MgS04), evaporated to dryness. The residue was purified on a column with EA-MeOH 9: 1 as eluent, fractions collected, concentrated to approx 20 ml, cooled in ice. The precipitate collected, washed with Ethyl Acetate-Petroleum Ether 1 : 1, dried on air to give (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)- thione (1.55 g, 3.86 mmol, 50.9 % yield). Lot 4 (Form A)
To a 500 mL flask set up for atmospheric distillation was added (R)-5-(2- (benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)-thione (20 g, 49,8 mmol) and Tetrahydrofuran (400 ml) to afford a suspension. The suspension was heated until full dissolution was achieved (61°C) whereupon it was filtered. The resulting solution was then heated to 66°C in order to commence the distillation. A mixture of Water (125 ml) & 2-Propanol (125 ml) was added at the same rate as the distillate was collected. The distillation was continued until 400 mL of distillate was collected. Crystallisation commenced after ~320 mL of distillate was collected. The suspension was cooled to 20°C and aged for 45 min. before filtering and washing with additional 2- propanol (80 mL) and then dried under vacuum at 50°C overnight to give (R)-5-(2- (benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)-thione (18.79 g, 94%). Lot 5 (Form A)
To a mixture of Methanol (66 L) and Water (10 L) at 20°C was added purified (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)-thione hydrochloride (4.37 kg, 9.98 mol) to afford a suspension. The reaction mixture was then heated to 67°C to affect complete dissolution, whereupon IN Sodium hydroxide (10.48 Ls 10.48 mol, 1.05 eq) was added in a single portion. The reaction mixture was adjusted back to 67°C and held at 67°C for 30 min. The reaction mixture was then cooled to 20°C and aged at 20°C for at least 30 min. The reaction was then filtered and the filter cake washed with aqueous Methanol (1 : 1 v/v, 20 L), sucked down for 15 min. and then dried at 45°C under vacuum, to afford (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH- imidazole-2(3H)-thione (3.855 kg, 96%) as a pale tan crystalline solid.
PATENT
WO 2015038022
http://www.google.com/patents/WO2015038022A1?cl=en
processes .
(J?) -5- (2-Aminoethyl) -1- (6, 8-difluorochroman-3-yl) -1, 3-dihydroimidazole-2 -thione hydrochloride (the compound of formula 1, below) is a potent, non-toxic and peripherally selective inhibitor of ϋβΗ, which can be used for treatment of certain cardiovascular disorders. Compound 1 is disclosed in WO2004/033447 , along with processes for its preparation.

1
The process disclosed in WO2004/033447 involves the reaction of ( R) – 6 , 8 -difluorochroman-3 -ylamine hydrochloride (the structure of ( R) -6, 8-difluorochroman-3 -ylamine is shown below as compound QA) , [4 – ( tert-butyldimethylsilanyloxy) -3 -oxobutyl] carbamic acid tert-butyl ester and potassium thiocyanate .

QA
(R) -6 , 8-difluorochroman- 3 -ylamine (compound QA) is a key intermediate in the synthesis of compound 1. The stereochemistry at the carbon atom to which the amine is attached gives rise to the stereochemistry of compound 1, so it is advantageous that compound QA is present in as pure enantiomeric form as possible. In other words, the (R) -enantiomer of compound QA should be in predominance, with little or no (S) enantiomer present. Thus, the process for preparing compound QA will advantageously produce compound QA with as high enantiomeric excess (ee) as possible.
Advantageous processes for preparing, for example, the compound of formula QA have now been found. In one aspect, the processes involve a biotransformation step. In another aspect, the processes involve chemical transformation. The processes may also be employed in the preparation of similar precursors useful in the production of other peripherally-selective inhibitors of dopamine -β -hydroxylase .
WO2008/136695 discloses a compound of formula YA, its (R) or (S) enantiomer, a mixture of its (R) and (S) enantiomers, or pharmaceutically acceptable salts thereof.

YA
The (R) -enantiomer of the compound of formula YA has been found to be a potent dopamines-hydroxylase inhibitor having high potency and significantly reduced brain access.
As disclosed in WO2008/136695 , the compound of formula YA may be prepared by reacting the compound of formula 1 with benzaldehyde under reductive alkylation conditions. In particular, (R) -5- (2 -aminoethyl ) -1- (6 , 8-difluorochroman-3 -yl) – 1 , 3 -dihydroimidazole-2 -thione and benzaldehyde may be reacted in the presence of a solvent or mixture of solvents, and a reducing agent such as sodium cyanoborohydride or sodium triacetoxyborohydride .
process comprises the following steps:

The route from 2 , 4-difluorophenol may be as described 9/064210.
Preferably, the reagents and conditions are:
(i) H2S04, acetic acid
(ii) NaOCl, MeOH/water
(iii) Ru-based catalyst, H2, 30 bars, MeOH
(iv) aqueous KOH, MeOH, L-tartaric acid
(v) KSCN, AcOH/lPA
(vi) NaBH4, BF3.THF complex, THF then IPA
n one aspect, the process comprises the following steps


i. KOH, Thioglycolic acid or cysteine
ii. MEK
According to an aspect of the present invention, there is provided the following 2 -part synthetic route from the starting material 2 , 4 -difluorophenol to (R) -5- (2 -aminoethyl ) -1- (6 , 8-difluorochroman-3 -yl) -1 , 3 -dihydroimidazole-2 – thione
hydrochloride :
Part (1)


Preferred reagents and conditions:
a) HMTA, CF3COOH, 115°C, 18 hours
b) CH2CHCN, DABCO, DMF, water, 70°C, 16 hours
c) H2S04, AcOH, 100°C, 1 hour
d) NaClO, NaOH, MeOH, 25°C, 24 hours
e) (R) -C3 -TunePhosRu (acac) 2 S/C 3000, 30 bar H2, MeOH, 80°C, 20 hours
f) Water, 2-propanol, reflux to 20°C
g) 40% KOH, MeOH, reflux, 24 hours
h) L-tartaric acid, ethanol, water, RT, 1 hour
Part (2)

![]()

Preferred reagents and conditions
a’) methyl vinyl ketone, t-BuONa, EtOAc, EtOH, 40-50°C, 2-3 hours
Br2, MeOH, 20-25°C, 5 hours
water, reflux, 1 hour
KOH, AcOH, reflux, 1 hour
HCl, water, 2-propanol, 75 °C, 4 hours
KSCN, AcOH, 100°C, 2-4 hours
NaHC03, water, EtOH
NaBH4, 2-propanol, THF, water, 20-25°C, 16 hours
HCl, 2-propanol, water, reflux, 1-2 hours
The ( R ) -5- (2-Aminoethyl) -1- (6, 8-difluorochroman-3 -yl) -1,3-dihydroimidazole-2 – thione hydrochloride may then be used to
prepare (R) -5- (2- (benzylamino) ethyl) -1- (6, 8-difluorochroman-3 -yl) -lH-imidazole-2 (3H) -thione as follows.

Preferred reaction conditions/reagents:
q) NaBH(OAc)3, PhCHO, IPA;
t) NaOH, MeOH , H20
Either r) and s) :
r) HCI aq;
s) MeOH/Toluene;
Or n) , o) and p) :
n) HCI aq;
o) MeOH, toluene;
p) IPA.
EXAMPLES
Example 1
Nitro chromene synthesis

To 3 , 5-difluoro-2-hydroxybenzaldehyde (lOg, 63mmol, leq) , di-n-butylamine (4.1g, 32mmol, 0.5eq) , phtalic anhydride (18.7g, 126mmol, 2eq) in toluene (500mL) was added nitroethanol (5.75g, 63mmol, leq) . The round bottomed flask fitted with a dean stark apparatus was refluxed for 18h. The mixture was cooled and nitroethanol (5.75g, 63mmol, leq) was added. The resulting reaction mixture was then reflux for 12h. After cooling, the solution was evaporated down to approximately 150mL and purified over silica gel (eluent ethyl acetate : hexane 1:1) this gave several fractions that contained only the product by TLC, these was evaporated under reduced pressure to yield 1.8g which was 100% pure by HPLC aera. Several more fractions were collected containing a mixture of product and starting material. These were combined and washed with 2% NaOH solution (2x50mL) to remove starting material. The organic layer was washed with water (50mL) , dried over sodium sulfate and evaporated under reduced pressure to give 2.49g of brown solid ( 100% pure by HPLC aera) . More fractions were collected. These were combined, washed with 2% NaOH solution (3xl00mL) , water (lOOmL) and dried over sodium sulfate. This was then filtered and evaporated down in vacuum to yield 6.14g of a brown solid which was 91.3% pure by HPLC aera. 6 , 8 -difluoro-3 -nitro-2H-chromene (9.90g, 73.4%) was obtained as a brown solid.
Example 2
Nitro chromene synthesis with column purification
To a solution of isobenzofuran-1 , 3 -dione (4,68 g, 31,6 mmol) , 3 , 5-difluoro-2 -hydroxybenzaldehyde (2,5 g, 15,81 mmol) in Toluene (25 ml) was added 2 -nitroethanol (2,88 g, 31,6 mmol). The resulting mixture was heated to reflux overnight (Dean stark) .
The reaction conversion was checked by TLC (eluent PE/EtOAc 9:1) . A yellow spot was observed and corresponds to the expected product .
Reaction was cooled to room temperature and a plug of silica gel was performed. A pale brown solid (3.9g) was obtained. “””H-NMR showed presence of product and starting material. The solid was dissolved in diethylether and the organic layer was washed with aqueous sodium carbonate, dried over Na2S04, filtered and concentrated under reduced pressure. A pale brown solid (1.7g,) was obtained. The 1H-NMR was indicated no starting material but still polymer from nitroethanol and residue of phtalic anhydride. A second silica plug (eluent: PE/EtOAc 95:5) was done. A pale yellow solid (1.5g) was obtained. 1H-NMR of solid showed only product and polymer. The solid was recrystallized from methanol/water . A pale yellow solid (1.05g, 31.2%) was obtained.
Example 3
Nitro chromene synthesis without column purification
To a solution of isobenzofuran- 1 , 3 -dione (18,74 g, 127 mmol) , 3 , 5-difluoro-2 -hydroxybenzaldehyde (10 g, 63,3 mmol) in Toluene (100 ml) was added 2 -nitroethanol (6,86 ml, 95 mmol) . The resulting mixture was heated to reflux for 24h (Dean stark) .
The reaction conversion was checked by HPLC and by 1H-NMR. Only 50% conversion was obtained.
The reaction mixture was cooled to room temperature and diluted with DCM (lOOmL) and 1M NaOH solution (200mL) .
The biphasic system was stirred for 30 minutes and then separated (very difficult to see phase separation) . The aqueous layer was washed with DCM (50mL) and the combined organic layers were washed twice with water (2x50ml) , dried over sodium sulfate. The filtered organic layer was concentrated under reduced pressure. To the residue was added methanol (50mL) . The methanol was then removed by distillation under reduced pressure. A brown solution precipitated when most of the methanol was removed. More methanol was added and more solid crushed out then few drops of water was added to increase the product precipitation. The brown slurry was stirred for 30 minutes and filtered. The brown solid was washed with methanol/water (1:9, 5mL) and dried in a vacuum oven at 40°C for 12h.6, 8-difluoro-3 -nitro-2H-chroraene (4,9 g, 22,99 mmol,) was obtained as brown solid in 36.3% yield.
HPLC showed a purity of 98% and 1H-NMR confirmed the structure and purity around 95%
Example 4
Reduction of nitro chromene to nitro-alkane (racemic mixture)

To a suspension of 6 , 8 -difluoro-3 -nitro-2H-chromene (213mg, 0,999 mmol) and silica (0,8 g, 0,999 mmol) in a mixture of CHC13 (10 ml) and IPA (3,4 ml) at 0°C was added portion wise sodium borohydride (95 mg, 2,498 mmol). The resulting mixture was stirred at 0°C for 45 minutes. Reaction conversion was checked by HPLC. 1 mL of acetic acid was added at 0°C and the resulting mixture was stirred for 30 minutes at room temperature. The slurry was filtered and the silica was washed with DCM. The filtrate was diluted with ethyl acetate and water and the biphasic system was separated. The aqueous layer was back extracted with ethyl acetate. The combined organic layers were washed with brine, dried over MgS04, filtered and concentrated under reduced pressure.
6 , 8-difluoro-3 -nitrochroman (196mg, 0,911 mmol, 91 % yield) was obtained as a pale yellow oil.
Example 5
Preparation of 6 , 8 -difluorochroman-3 -one from nitro chromene

A solution of 6, 8-difluoro-3 -nitro-2H-chromene (lOOmg, 0,469 mmol) in acetic acid (0.5 ml) is added slowly to a stirred slurry of iron (262 mg, 4,69 mmol) in acetic acid (1 ml) at 60.deg. C. The reaction mixture is stirred at 60. °C for 2 hour then allowed to cool to room temperature and stirred overnight. The reaction mixture is poured onto ice-water (30 ml) and filtered through Celite. The solid was wash with dichloromethane (DCM) (50 ml) . The organic portion is separated and washed with water (2 x 30 ml) and brine (30 ml) , dried over MgS04, filtered and concentrated in vacuo to give a brown oil. 6,8-difluorochroman-3 -one (75 mg, 0,407 mmol, 87 % yield) was obtained as a brown oil.
Example 6
Preparation of 6 , 8-difluorochroman-3 -one from methyl 6,8-difluoro-2H-chromen-3 -yl-carbamate

Methanol (1000m ml) was added to a slurry of methyl fluoro-2H-chromen-3 -yl -carbamate (250 g, 1.037 mol) hydrogen chloride 6N (2000 ml, 12 mol) at room temperature. The resulting mixture was reflux and stirred for 2 hours. Reaction monitored by HPLC.
Reaction was not complete but was stopped in order to avoid degradation of the product. The yellow solution was cooled to room temperature. A slurry (two type of solid) was observed and diluted with diethyl ether (300mL) . The resulting slurry was stirred at 5°C for 1 hour then filtered. The yellow solid was washed with water. The resulting wet yellow solid was suspended in diethylether (400mL) and petroleum ether (PE) (400mL) was added. Slight yellow solid was stirred at room temperature overnight, filtered and washed with PE (300mL) , dried in a vacuum oven at 30 °C for 4h. The wet sample was checked by NMR. No starting material was detected. A pale yellow solid (72.5g, solid 1) was obtained. The mother liquors were concentrated to dryness. A yellow solid was obtained, suspended in diethyl ether and PE. The slurry was then stirred for 4 hours, filtered, washed with PE . A dark yellow solid (4.5g, solid 2) was obtained. Solid 1 (2g) was diluted in DCM and washed with water (pH =6). The organic layer was then dried over Na2S04, filtered, concentrated to dryness. A crystalline pale yellow solid (1.9g, solid 3) was obtained. NMR showed the same purity for solid 3 as for solid 1. The remaining part of solid 1 was then diluted in DCM. The resulting organic layer was washed with water, dried over Na2S04, filtered and then concentrated to dryness. Slight yellow crystalline solid (68.5g, solid 4) was obtained. NMR confirmed high quality material.
Loss on Drying (LOD) : 1.03% .
Example 7
Biotransformation: Transaminases

Codexis transaminases ATA-025, ATA-251 and ATA-P2-A07 recognized 6 , 8 -difluorochroman-3 -one as the substrate and produced the corresponding 6 , 8 -difluorochroman-3 -amine .
PATENT
WO 2014077715
WO 2013002660
WO 2008136695
REFERNCES
International Journal of Pharmaceutics (Amsterdam, Netherlands) (2016), 501(1-2), 102-111.
| WO2012087174A2 | Dec 21, 2011 | Jun 28, 2012 | BIAL – PORTELA & Cª., S.A. | Crystalline forms and processes for their preparation |
| WO2012087174A3 * | Dec 21, 2011 | May 10, 2013 | BIAL – PORTELA & Cª., S.A. | Crystalline forms and processes for their preparation |
| WO2013002660A2 | Jun 29, 2012 | Jan 3, 2013 | BIAL – PORTELA & Cª, S.A. | Process |
| WO2014077715A1 * | Nov 14, 2013 | May 22, 2014 | BIAL – PORTELA & Cª, S.A. | 1,3-dihydroimidazole-2-thione derivatives for use in the treatment of pulmonary arterial hypertension and lung injury |
| US8481582 | May 6, 2008 | Jul 9, 2013 | Bial-Portela & Ca, S.A. | 1,3-dihydroimidazole-2-thione derivatives as inhibitors of dopamine-beta-hydroxylase |
| US8865913 | Jun 19, 2013 | Oct 21, 2014 | Bial-Portela & Ca, S.A. | Crystalline forms and processes for their preparation |
| WO1995007284A1 * | Aug 29, 1994 | Mar 16, 1995 | Smithkline Beecham Plc | Phosphinic acid derivatives with anti-hyper glycemic and/or anti-obesity activity |
| WO2006044293A2 * | Oct 11, 2005 | Apr 27, 2006 | Pharmacopeia Drug Discovery, Inc. | Bicyclic compounds as selective melanin concentrating hormone receptor antagonists for the treatment of obesity and related disorders |
| WO2012007548A1 * | Jul 14, 2011 | Jan 19, 2012 | Dsm Ip Assets B.V. | (r)-selective amination |
| WO2013002660A2 * | Jun 29, 2012 | Jan 3, 2013 | BIAL – PORTELA & Cª, S.A. | Process |
| GR1005093B * | Title not available |
///////Zamicastat, BIA-5-1058, dopamine beta-monooxygenase inhibitor, phase I, clinical studies, BIAL, treatment of hypertension , heart failure.
S=C4NC=C(CCNCc1ccccc1)N4[C@@H]2Cc3cc(F)cc(F)c3OC2
DS 2330 by Daiichi Sankyo
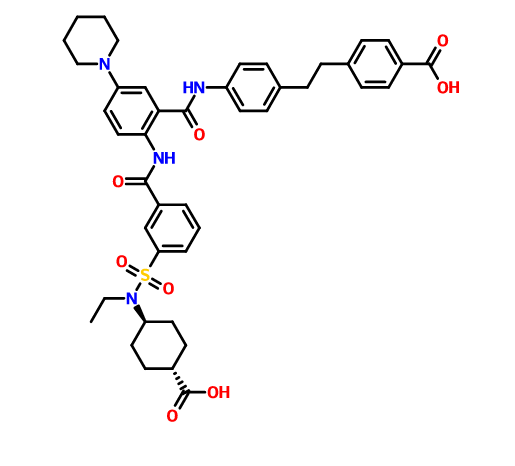
DS 2330
a trans compd
4-[2-(4-{[2-({3-[(trans-4-carboxy-cyclohexyl)(ethyl)sulfocarbamoyl]benzoyl}amino)-5-(piperidin-1-yl)benzoyl]amino}phenyl)ethyl]benzoic acid,
4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] benzoate
CAS 1634680-81-1
C43 H48 N4 O8 S, 780.9
Benzoic acid, 4-[2-[4-[[2-[[3-[[(trans-4-carboxycyclohexyl)ethylamino]sulfonyl]benzoyl]amino]-5-(1-piperidinyl)benzoyl]amino]phenyl]ethyl]-
CIS isomer CAS 1634681-85-8
DISODIUM SALT 1634681-00-7
- Originator Daiichi Sankyo Inc
- Class Hyperphosphataemia therapies
useful for treating hyperphosphatemia, DS-2330, a phosphorous lowering agent, being developed by Daiichi Sankyo, for treating hyperphosphatemia in chronic kidney disease. In April 2016, DS-2330 was reported to be in phase 1 clinical development.
- Phase IHyperphosphataemia
- 31 Oct 2015Phase-I clinical trials in Hyperphosphataemia in USA (unspecified route)
![]()
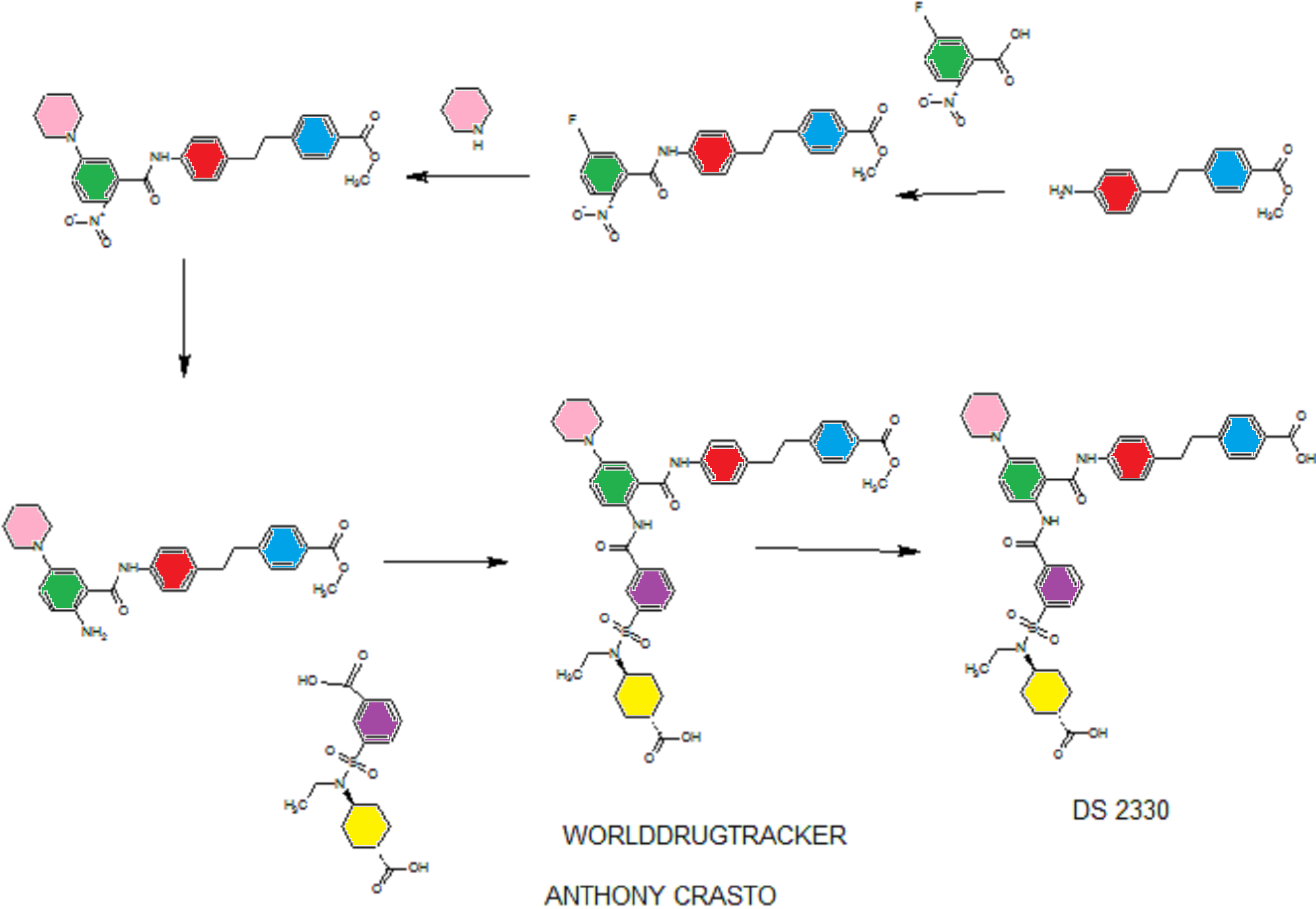
SEE WO2015108038,
PATENT
WO2014175317
http://www.google.com/patents/EP2990400A1?cl=en
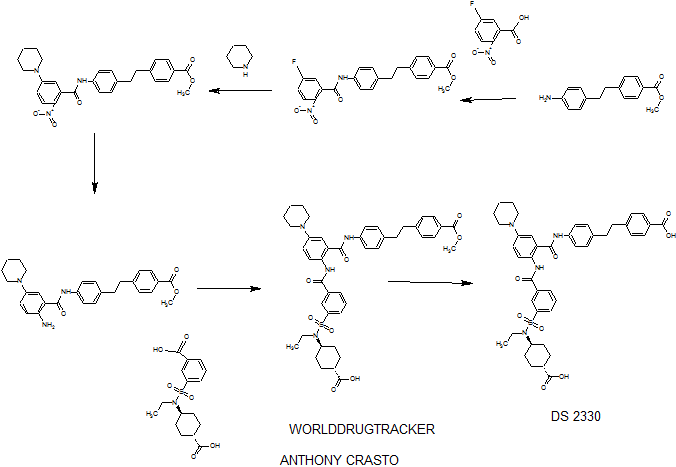
PATENT
he problem is to provide a pharmaceutical for the prevention or treatment of hyperphosphatemia. The solution is a salt of a compound including formula (I), or a crystal of a hydrate thereof.
(Example 1)
disodium 4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl ) benzoyl] amino} phenyl) ethyl] benzoic acid trihydrate
Disodium 4- [2- (4 – { [2 – ({3 – [(trans-4-carboxylatocyclohexyl) (ethyl) sulfamoyl] benzoyl} amino) – 5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] benzoate trihydrate
of α crystal
disodium 4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl ) benzoyl] amino} phenyl) ethyl] benzoic acid trihydrate
Disodium 4- [2- (4 – { [2 – ({3 – [(trans-4-carboxylatocyclohexyl) (ethyl) sulfamoyl] benzoyl} amino) – 5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] benzoate trihydrate
of α crystal
[Formula 7] crystal of disodium salt trihydrate of (α crystal)

(1)
4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] 1 mol / L NaOH aqueous solution to benzoic acid (1.2 g) (3.1 mL) was added and dissolved completely. After stirring at room temperature for 1 day was added acetonitrile (60 mL), at 40 ° C.
and stirred for further 1 day. The precipitated solid was collected by filtration, and 3 hours drying under reduced pressure at room temperature to give the title compound 1.1 g (85%).
4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] 1 mol / L NaOH aqueous solution to benzoic acid (1.2 g) (3.1 mL) was added and dissolved completely. After stirring at room temperature for 1 day was added acetonitrile (60 mL), at 40 ° C.
and stirred for further 1 day. The precipitated solid was collected by filtration, and 3 hours drying under reduced pressure at room temperature to give the title compound 1.1 g (85%).
(2)
4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] benzoate (40.0 g)
in water (46.4 mL), 1-PrOH (72 mL), 4 mol / L NaOH aqueous solution (25.54 mL) was added, then filtered after stirring insolubles at room temperature, water / 1-PrOH: was washed with (3 7, 80 mL). The filtrate was heated up to 40 ℃, 1-PrOH the (160 mL) was added, and further seed crystal (α crystals, 0.2g) was added. Then the temperature was raised to 50 ℃, 1-PrOH (96 ml) was added, and the mixture was stirred overnight.Thereafter, 1-PrOH (480 ml) was added and after overnight stirring, was collected by filtration the precipitated solid was cooled to room temperature.Thereafter, and vacuum dried overnight at 40 ° C., to give the title compound 39.4 g (96%).
in water (46.4 mL), 1-PrOH (72 mL), 4 mol / L NaOH aqueous solution (25.54 mL) was added, then filtered after stirring insolubles at room temperature, water / 1-PrOH: was washed with (3 7, 80 mL). The filtrate was heated up to 40 ℃, 1-PrOH the (160 mL) was added, and further seed crystal (α crystals, 0.2g) was added. Then the temperature was raised to 50 ℃, 1-PrOH (96 ml) was added, and the mixture was stirred overnight.Thereafter, 1-PrOH (480 ml) was added and after overnight stirring, was collected by filtration the precipitated solid was cooled to room temperature.Thereafter, and vacuum dried overnight at 40 ° C., to give the title compound 39.4 g (96%).
REFERENCES
////////////DS 2330, DS-2330, DAIICHI SANKYO, phase 1
O=C(O)[C@@H]1CC[C@H](CC1)N(CC)S(=O)(=O)c2cccc(c2)C(=O)Nc5ccc(cc5C(=O)Nc4ccc(CCc3ccc(cc3)C(=O)O)cc4)N6CCCCC6
OR
O=C(O)[C@@H]1CC[C@H](CC1)N(CC)S(=O)(=O)c2cccc(c2)C(=O)Nc5ccc(cc5C(=O)Nc4ccc(CCc3ccc(cc3)C(=O)O)cc4)N6CCCCC6
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin
Join me on Facebook 
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P7435 from Piramal Enterprises Mumbai, India
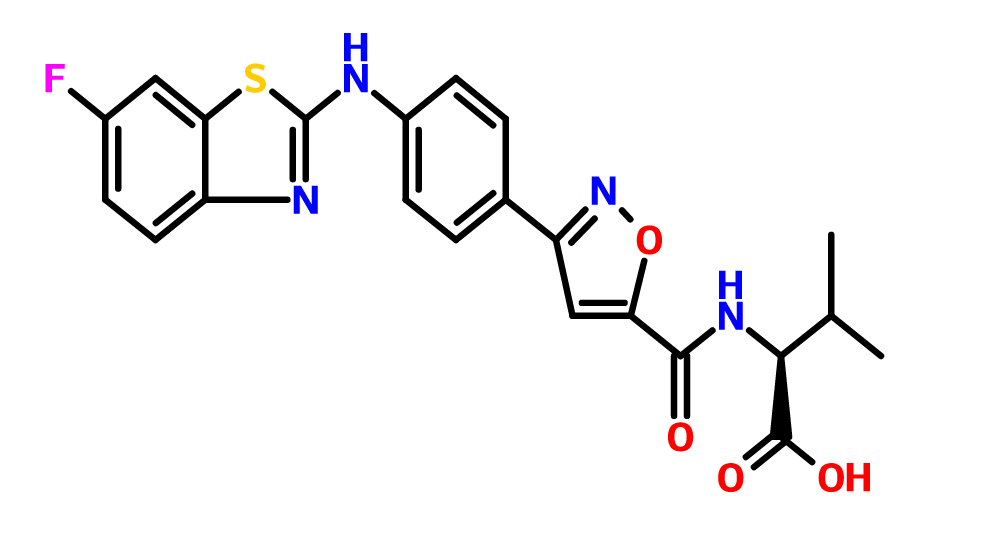
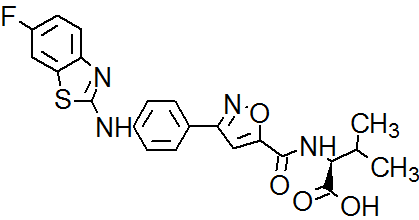
P7435
Piramal Enterprises Mumbai, India
P-7435; P7435-DGAT1, P7435, P 7435
CAS 1210756-48-1,
C22 H19 F N4 O4 S
L-Valine, N-[[3-[4-[(6-fluoro-2-benzothiazolyl)amino]phenyl]-5-isoxazolyl]carbonyl]-
- Molecular Weight, 454.47
GDAT1 inhibitor
- Phase IDiabetes mellitus; Lipid metabolism disorders
- ClassAntihyperglycaemics; Antihyperlipidaemics; Small molecules
- Mechanism of ActionDiacylglycerol O acyltransferase inhibitors

| Latest Stage of Development | Phase I |
| Standard Indication | Metabolic (unspecified) |
| Indication Details | Treat metabolic disorders |
https://clinicaltrials.gov/ct2/show/NCT01910571
https://clinicaltrials.gov/ct2/show/NCT01764425
- 24 Nov 2014Piramal Enterprises completes a phase I trial in healthy, overweight or obese subjects in USA (NCT01910571)
- 17 Jun 2014Adverse events and pharmacokinetics data from a phase I trial in healthy male volunteers presented at the 74th Annual Scientific Sessions of the American Diabetes Association (ADA-2014)
- 17 Jun 2014Pharmacodynamics data from preclinical studies in Dyslipidaemia and obesity presented at the 74th Annual Scientific Sessions of the American Diabetes Association (ADA-2014)

Chairman Ajay Piramal

Swati Piramal-The Vice Chairperson of Piramal Enterprises Ltd

Nandini Piramal, Executive Director, Piramal Enterprises
Piramal Enterprises gets US FDA approval for P7435 IND
http://www.pharmabiz.com/NewsDetails.aspx?aid=76992&sid=2
Our Bureau, Mumbai
Tuesday, August 06, 2013, 12:25 Hrs [IST]
Piramal Enterprises Ltd has received US Food and Drug Administration (FDA) approval for its Investigational New Drug (IND) P7435. This is a novel, potent and highly selective, oral diacylglycerolacyltransferase 1 (DGAT1) inhibitor.
P7435 has been developed by the NCE Research Division of PEL for the management of metabolic disorders such as lipid abnormalities and diabetes. It is well-established that increased lipid levels’ (including triglycerides) is one of the major risk factors for cardiovascular disease (CVD). It has been reported by the World Health Organisation, that CVD, is the number one cause of deaths globally, representing approximately 30 per cent of all deaths. Currently, there is a significant medical need for effective and safe drugs for the management of lipid abnormalities and metabolic disorders.
P7435 has demonstrated its lipid lowering potential in various preclinical studies by showing significant reduction in triglyceride levels, glucose and insulin levels,and decrease in food intake and body weight gain -factors which are associated with lipid abnormalities and metabolic disorders.
PEL has established the safety and tolerability of P7435 in a phase I trial recently completed in India. This extension trial in the US will further evaluate the safety and efficacy of P7435 in a larger population.
Dr Swati Piramal, vice chairperson, Piramal Enterprises, said, “The NCE Research division of PEL continues its ambitious diabetes/metabolic disorders programme to discover and develop NCEs to fight against diseases like diabetes and lipid disorders. With P7435 we are looking at addressing a serious need for effective and well-tolerated drugs that treat lipid disorders, which are commonly associated with diabetes and CVDs. Expansion of this trial will allow testing this NCE in a wider population,which is critical to the development of this drug and will provide therapeutic solutions not just to India but also to the rest of the world.”
The NCE Research division of Piramal Enterprises focuses on the discovery and development of innovative small molecule medicines to improve the lives of patients suffering from cancer, metabolic disorders and inflammatory conditions. The key elements of its strategy include capitalizing on Piramal’s strengths, in particular the India advantage, and leveraging external partnerships to achieve high levels of R&D productivity. Piramal’s state-of-the-art Research Centre in Mumbai has comprehensive capabilities spanning target identification all the way through clinical development. Its robust pipeline, including 8 compounds in clinical development, bears testimony to its innovative and rigorous drug discovery process.
PAPER
European Journal of Medicinal Chemistry (2012), 54, 324-342
http://www.sciencedirect.com/science/article/pii/S0223523412003133
PATENT
WO 2010023609
http://www.google.co.in/patents/WO2010023609A1?cl=en
/////////Piramal Enterprises, Mumbai, India, P-7435, P7435-DGAT1, P7435, P 7435, GDAT1 inhibitor
O=C(O)[C@@H](NC(=O)c1cc(no1)c2ccc(cc2)Nc3nc4ccc(F)cc4s3)C(C)C
RP 6503, Novartis to develop and commercialize Rhizen’s inhaled dual PI3K-delta gamma inhibitor

RP 6503
phase 1

RP 6503
| Molecular Formula: | C30H24F2N6O5S |
|---|---|
| Molecular Weight: | 618.610566 g/mol |
Mass: 619.1 (M++l). MP: 175-178° C Specific optical rotation (C=l in chloroform, at 25°C) : [a]D = + 147.16.

A1
RP 6503
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl) ethyl)-lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
(S)-N-[5-[4-amino-1-[1-[5-fluoro-3-(3-fluorophenyl)-4-oxochromen-2-yl]ethyl]pyrazolo[3,4-d]pyrimidin-3-yl]-2-methoxyphenyl]methanesulfonamide

![]()
Novartis to develop and commercialize Rhizen’s inhaled dual PI3K-delta gamma inhibitor and related compounds worldwide
The immune pipeline includes ‘dual PI3K inhibitors for various indications’ licensed to Novartis
‘inhaled dual inhibitor’,
Phosphoinositide-3 kinase delta inhibitor; Phosphoinositide-3 kinase gamma inhibitor
WO2011055215A2 and WO2012151525A1 and U.S. Publication Nos. US20110118257 and US20120289496
Rhizen Pharmaceuticals Sa INNOVATOR
| Incozen Therapeutics Pvt. Ltd., Rhizen Pharmaceuticals Sa |
PATENT
http://www.google.com/patents/WO2011055215A2?cl=en
PATENT
http://www.google.com/patents/WO2012151525A1?cl=en
scheme 1A:
Ste -1

Step-2

Scheme 2

SCHEME 3

SCHEME4

List of Intermediates





Intermediate 27: 2-( l -(4-amino-3-iodo-lH-pyrazolo[3,4-d]pyrimidin- l – yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: To a solution of 3-iodo- l H- pyrazolo[3,4-d]pyrimidin-4-amine (0.800 g, 2.88 mmol) in DMF (5 ml), potassium carbonate (0.398 g, 2.88 mmol) was added and stirred at RT for 30 min. To this mixture intermediate 22 (0.500 g, 1.44 mmol) was added and stirred for 12h. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a off-white solid (0.300 g, 38%). Ή-NMR (5 ppm, DMSO-d63, 400 MHz): 8.02 (s, 1 H), 7.94 (s, 1 H), 7.84 (dt, J = 8.4,5.7 Hz, 1H), 7.47 (d, 7 = 8.6 Hz, 1H), 7.29 (m, 3H), 7.09 (dt, 7 = 8.8,2.3 Hz, 1 H), 6.87 (s, 2H), 5.88 (q, 7 = 7.0 Hz, 1H), 1.82 (d, 7 = 7.0 Hz, 3H).
SYNTHESIS
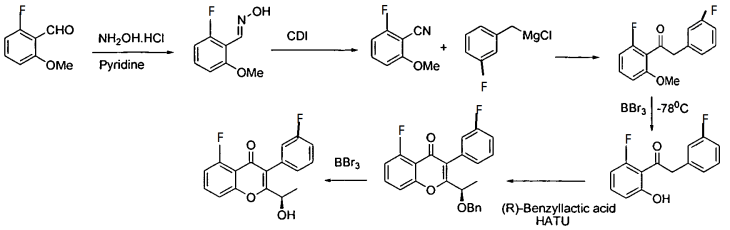
MAIN PART
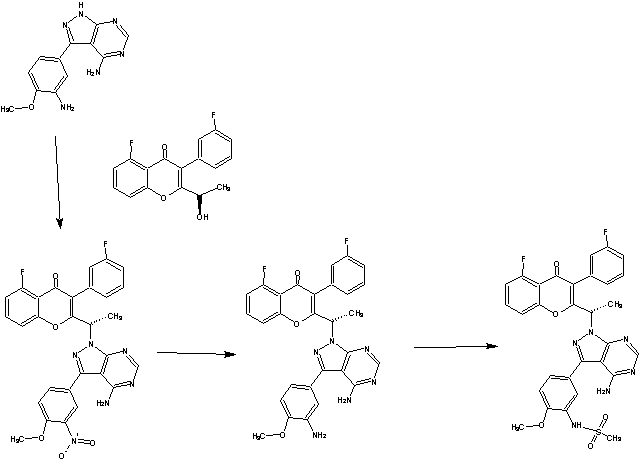
PATENT
http://www.google.com/patents/WO2015198289A1?cl=en
Prashant Kashinath Bhavar, Swaroop Kumar Venkata Satya VAKKALANKA
The present invention relates to a selective dual delta (δ) and gamma (γ) PI3K protein kinase modulator (S)-N-(5-(4-amino-1-(1-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H- chromen-2-yl)ethyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl) methane sulfonamide, methods of preparing them, pharmaceutical compositions containing them and methods of treatment, prevention and/or amelioration of PI3K kinase mediated diseases or disorders with them.
compound of formula (Al):
(Al).
The process comprises the steps of:
(a) subjecting (R)-5-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one:

to a Mitsunobu reaction with 3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-amine:

(for example, in the presence of triphenylphosphine and diisopropylazodicarboxylate) to give (S)-2-(l-(4-amino-3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (Intermediate 3):

Intermediate 3;
(b) reducing Intermediate 3, for example with a reducing agent such as Raney Ni, to give (S)-2-(l-(4-amino-3-(3-amino-4-methoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin- l-yl)ethyl)-5-fluoro-3-( -fluorophenyl)-4H-chromen-4-one (Intermediate 4):

Intermediate 4;
The intermediates described herein may be prepared by the methods described in International Publication Nos. WO 11/055215 and WO 12/151525, both of which are hereby incorporated by reference.
Intermediate 1: N-(5-bromo-2-methoxyphenyl)methanesulfonamide:
To a solution of 5-bromo-2-methoxyaniline(1.00 g, 4.94 mmol) in dichloromethane (10 ml), pyridine (0.800 ml, 9.89 mmol) was added and cooled to 0°C. Methane sulphonyl chloride (0.40 ml, 5.19 mmol) was added and stirred for 30 min. The reaction mixture was quenched with water, extracted with ethyl acetate, dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was chromatographed with ethyl acetate : petroleum ether to afford the title compound as a reddish solid (1.20 g, 87%).
Intermediate 2: N-(2-methoxy-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)methanesulfonamide: Potassium acetate (0.841 g, 8.57 mmol) and bis(pinacolato)diboron (1.190 g, 4.71 mmol) were added to a solution of intermediate 1 (1.20 g, 4.28 mmol) in dioxane (17.5 ml) and the solution was degassed for 30 min.[l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Ci2 (0.104 g, 0.128 mmol) was added under nitrogen atmosphere and heated to 80°C. After 2h the
reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as a yellow solid (1.00 g, 71%).JH-NMR (δ ppm, CDCb, 400 MHz): 7. 91 (d, / = 1.2Hz, 1H), 7. 62 (dd, / = 8.1, 1.2Hz, 1H), 6. 92 (d, / = 8.1Hz, 1H), 6.73 (s, 1H), 3.91 (s, 3H), 2.98 (s, 3H), 1.32 (s, 12H).
Intermediate 3: (S)-2-(l-(4-amino-3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: (S)-2-(l-(4-amino-3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: To a solution of (R)-5-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one (0.500 g, 1.64 mmol) in THF (5 ml), 3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-amine (0.564 g, 1.97 mmol) and triphenylphosphine (0.649 g, 2.47 mmol) were added followed by the addition of diisopropylazodicarboxylate (0.50 ml, 2.47 mmol). ((R)-5-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one can be prepared as described for Intermediates 23, 25, and 26 in International Publication No. WO 2012/0151525.). After 4h at room temperature, the mixture was concentrated and the residue was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as a brown solid (0.270 g, 29%). JH-NMR (δ ppm, DMSO-d6, 400 MHz): 8.04 (s, 1H), 7.83 (m, 1H), 7.63-7.50 (m, 3H), 7.29 (m, 2H), 7.06 (dt, J = 8.7,2.2Hz, 1H), 6.94 (m, 2H), 6.75 (dd, J = 8.1,2.1Hz, 1H), 5.95 (q, J = 7.0Hz, 1H), 4.98 (s, 2H), 3.81 (s, 3H), 1.86 (d, J = 7.0 Hz, 3H).
[109] Intermediate 4: (S)-2-(l-(4-amino-3-(3-amino-4-methoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one:
(S)-2-(l-(4-amino-3-(3-amino-4-methoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one : To a solution of Intermediate 3 (0.260 g, 0.455 mmol) in ethanol (5 ml), Raney Ni (0.130 g) was added and hydrogeneated at 20psi at 50°C for 24h. The reaction mixture was passed through celitepad and concentrated to afford the title compound as a brown solid (0.150 g, 60%). Mass : 540.8 (M+).
Example A
N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)-lH- pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
To a solution of 2-(l-(4-amino-3-iodo-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (0.200 g, 0.366 mmol) in DME (2.1 ml) and water (0.67 ml), intermediate 2 (0.179 g, 0.550 mmol) and sodium carbonate (0.116 g, 1.10 mmol) were added and the system was degassed for 30 min. (2-(l-(4-amino-3-iodo-lH^yrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one can be prepared as described for Intermediates 23, 25, and 26 in International Publication No. WO 2012/0151525). Bis(diphenylphosphino) ferrocene]dichloropalladium(II) (0.059 g, 0.075 mmol) was added and kept under microwave irradiation (microwave power = 100W, temperature = 100 °C) for 45 min. The reaction mixture was Celite filtered, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a brown solid (0.080 g, 35%). MP: 216-218 °C. ¾-NMR (δ ppm, CDCb, 400 MHz): 8.20 (s, 1H), 7.73 (s, 1H), 7.53 (m, 2H), 7.31 (m, 2H), 7.07-6.73 (m, 6H), 6.07 (q, / = 6.2 Hz, 1H), 3.98 (s, 3H), 3.14 (s, 3H), 2.01 (d, / = 6.0Hz, 3H).
Example Al and A2
Method A
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)- lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
and (R)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2- yl)ethyl)-lH-p anesulfonamide

The two enantiomerically pure isomers were separated by preparative SFC (supercritical fluid) conditions from N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)-lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide (0.500 g) on a CHIRALPAK AS-H column (250 x 30 mm; 5μπι) using methanol : CO2 (55:45) as the mobile phase at a flow rate of 80g / min.
Example Al (S-isomer): Brown solid (0.247 g). Enantiomeric excess: 97.4%. Retention time: 2.14 min. Mass: 619.1 (M++l). MP: 156-158° C.
Example A2 (R-isomer): Brown solid (0.182 g). Enantiomeric excess: 99.3%. Retention t: 3.43 min. Mass: 619.1 (M++l). MP: 168-171° C.
Method Al
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)- lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
and (R)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2- yl)ethyl)-lH-p anesulfonamide

The two enantiomerically pure isomers were separated by preparative SFC (supercritical fluid) conditions from N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)-lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl) methanesulfonamide (15.0 g) on a CHIRALPAK AS-H column (250 x 20 mm; 5μπι) using methanol : CO2 (45:55) as the mobile phase at a flow rate of 120g / min.
Example Al (S-isomer): Enantiomeric excess: 100 %. Retention time: 2.21 min. Mass: 619.1 (M++l). MP: 175-178° C Specific optical rotation (C=l in chloroform, at 25°C) : [a]D = + 147.16.
Example A2 (R-isomer): Enantiomeric excess: 99.3%. Retention t: 3.72 min. Mass: 619.1 (M++l). MP: 154-157° C. Specific optical rotation (C=l in chloroform, at 25°C) : [a]D = – 159.54.
Method B
Example Al
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)- lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
To a solution of Intermediate 4 (0.500 g, 0.923 mmol) in dichloromethane (5 ml) cooled to 0°C, pyridine (0.200 ml, 1.84 mmol) was added and stirred for 10 min. Methanesulphonyl chloride (0.100 ml, 0.923 mmol) was added stirred for 30 min. The reaction mixture was quenched with water, extracted with dichloromethane and dried over sodium sulphate. The crude product was column chromatographed with methanol : dichloromethane to afford the title compound as an off-white solid (0.240 g, 42%). MP: 211-213°C. ¾-NMR (δ ppm, DMSO-d6, 400 MHz): 9.15 (s, 1H), 8.06 (s, 1H), 7.83 (m, 1H), 7.49 (m, 4H), 7.28 (m, 4H), 7.08 (dt, / = 8.6, 1.7 Hz, 1H), 6.92 (s, 2H), 5.98 (q, / = 6.9 Hz, 1H), 3.88 (s, 3H), 2.99 (s, 3H), 1.88 (d, / = 7.0 Hz, 3H). Enantiomeric excess: 85.4% as determined by HPLC on a chiralpak AS-3R column, enriched in the fast eluting isomer (retention time = 7.46 min.).

CLIPS
La Chaux-de-Fonds, Switzerland, Sept. 6, 2013 — La Chaux-de-Fonds, Switzerland (6 September 2013): Rhizen Pharmaceuticals S.A. announces a scientific poster presentation on the pre-clinical characterization of its lead calcium release activated channel (CRAC) inhibitor, RP3128, for the treatment of respiratory disorders and an oral presentation on the pharmacological profile of its novel, dual Phosphoinositide-3 kinase (PI3K) delta/gamma inhibitor, RP6503, in the pulmonary disease systems, at the European Respiratory Society Annual Congress (ERS), to be held from 7-11 September 2013, at Barcelona, Spain.
RP6503 is a novel, potent and selective inhibitor of the delta and gamma isoforms of PI3K. It is to be delivered via the inhalation route and has a long duration of action along with excellent PI3K isoform selectivity, which is expected to result in better safety. RP3128 has been optimized with high potency for CRAC channel inhibition, selectivity over the other voltage gated channels and excellent oral bioavailability. Rhizen intends to move both these compounds to the clinic in 2014.
Details of the presentations:
1. Abstract of the Poster Presentation: “Pre-clinical characterization of RP3128, a novel and potent CRAC channel inhibitor for the treatment of respiratory disorders”
Time and Location- 8 September 2013 between 14.45-16.45 in Room 3.6, at Poster Discussion: New drugs in respiratory medicine, at FIRA BARCELONA, Convention Centre de Gran Via, Barcelona, Spain
2. Abstract of Oral Presentation: “In vitro and in vivo pharmacological profile of RP6503, a novel dual PI3K delta/gamma inhibitor, in pulmonary disease systems”
Time and Location- 11 September 2013 at 8.45 in Room 3.9; Session 8.30-10.30, at the Oral Presentation: Emerging new targets for the treatment of respiratory diseases, at FIRA BARCELONA, Convention Centre de Gran Via, Barcelona, Spain
CLIPS
La Chaux-de-Fonds, Switzerland , Dec. 09, 2015 — Rhizen Pharmaceuticals S.A. announced today that they have entered into an exclusive, worldwide license agreement with Novartis for the development and commercialization of Rhizen’s, inhaled dual PI3K-delta gamma inhibitor and its closely related compounds for various indications.
Under the terms of the agreement, Rhizen will receive an upfront payment and is eligible to receive development, regulatory and sales milestones payments. In addition Rhizen is also eligible to receive tiered royalties on annual nets sales.
The lead compound is a novel, potent, and selective dual PI3K-delta gamma inhibitor with demonstrated anti-inflammatory and immuno-modulatory activity in pre-clinical systems and models representative of respiratory diseases. With a favorable ADME and PK profile and high therapeutic index in animals, the inhaled dual PI3K-delta gamma inhibitor holds promise in the treatment of human airway disorders.
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, http://www.rhizen.com.
SEE
http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2013.187.1_MeetingAbstracts.A3880
| WO2011055215A2 | Nov 3, 2010 | May 12, 2011 | Incozen Therapeutics Pvt. Ltd. | Novel kinase modulators |
| WO2012008302A1 | Jun 28, 2011 | Jan 19, 2012 | National University Corporation Tottori University | Method for preparing novel hipsc by means of mirna introduction |
| WO2012121953A1 | Feb 29, 2012 | Sep 13, 2012 | The Trustees Of Columbia University In The City Of New York | Methods and pharmaceutical compositions for treating lymphoid malignancy |
| WO2012151525A1 | May 4, 2012 | Nov 8, 2012 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of protein kinases |
| WO2013164801A1 | May 3, 2013 | Nov 7, 2013 | Rhizen Pharmaceuticals Sa | Process for preparation of optically pure and optionally substituted 2- (1 -hydroxy- alkyl) – chromen – 4 – one derivatives and their use in preparing pharmaceuticals |
| US20110118257 | May 19, 2011 | Rhizen Pharmaceuticals Sa | Novel kinase modulators | |
| US20120289496 | May 4, 2012 | Nov 15, 2012 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of protein kinases |
///////RP 6503, Novartis, develop, commercialize, Rhizen, inhaled dual PI3K-delta gamma inhibitor, PHASE 1, RP-6503
c21c(cccc1O/C(=C(\C2=O)c3cc(ccc3)F)C(C)n4c6ncnc(c6c(n4)c5cc(c(cc5)OC)NS(=O)(=O)C)N)F
CC(C1=C(C(=O)C2=C(O1)C=CC=C2F)C3=CC(=CC=C3)F)N4C5=C(C(=N4)C6=CC(=C(C=C6)OC)NS(=O)(=O)C)C(=NC=N5)N
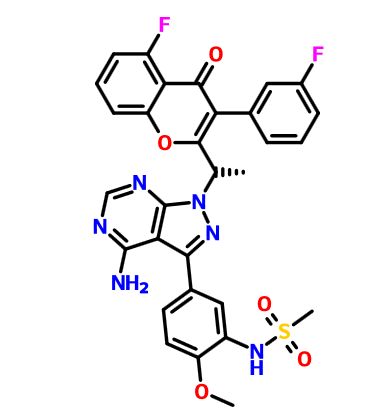
/////
BMS 986120

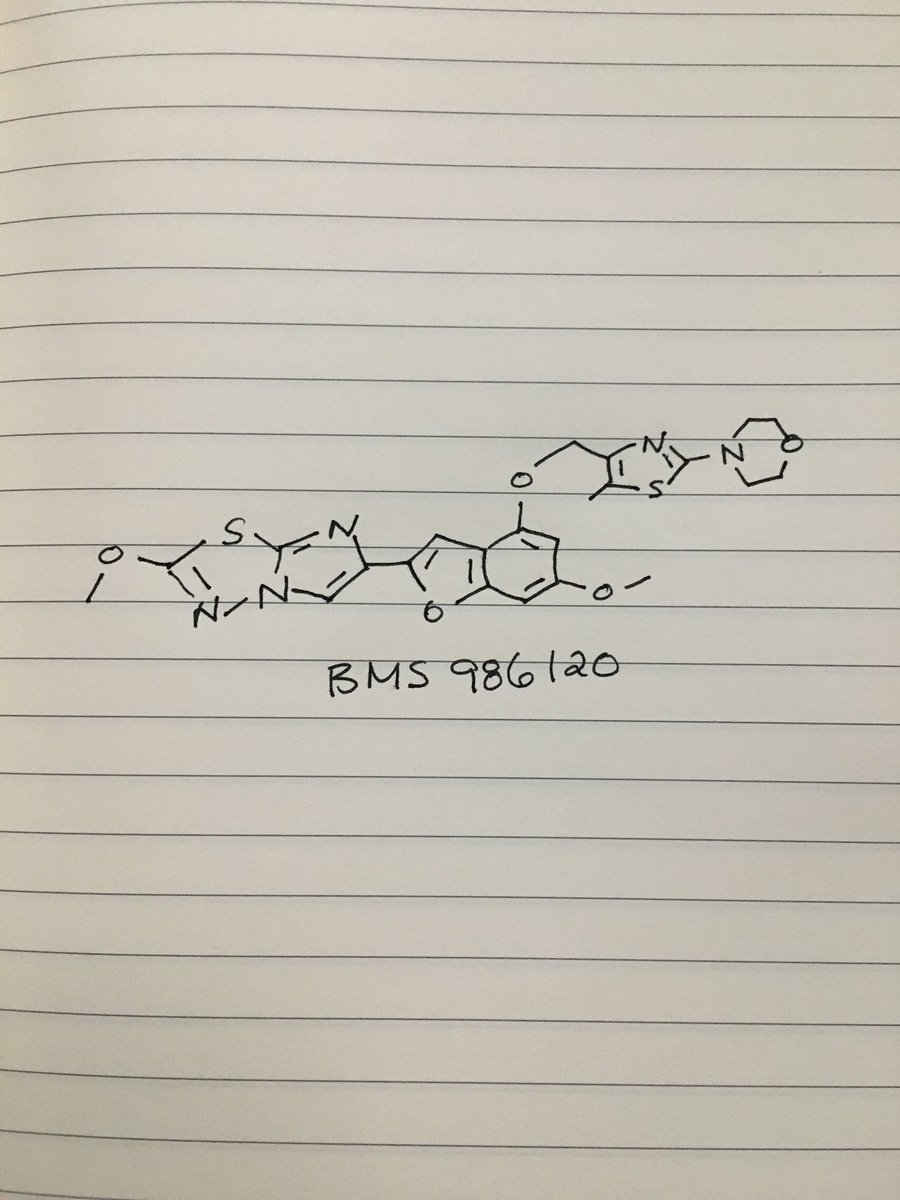 .
.
Picture credit….Bethany Halford
BMS 986120
Originator Bristol-Myers Squibb
Bristol-Myers Squibb Company, Université de Montréal
| Molecular Formula: | C23H23N5O5S2 |
|---|---|
| Molecular Weight: | 513.58922 g/mol |
4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-yl]morpholine
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine
Imidazo[2,1-b] -1,3,4-thiadiazole, 2-methoxy-6-[6-methoxy-4-[[5-methyl-2-(4-morpholinyl)-4- thiazolyl]methoxy]-2-benzofuranyl]-
CAS 1478712-37-6
Phase I Thrombosis
- 02 Apr 2015 Bristol-Myers Squibb plans a phase I trial in Thrombosis (In volunteers) in United Kingdom (NCT02439190)
- 01 Aug 2014 Preclinical trials in Thrombosis in USA (PO)
https://clinicaltrials.gov/ct2/show/NCT02208882
https://clinicaltrials.gov/ct2/show/NCT02439190
Class Imidazoles; Small molecules; Thiadiazoles
$BMY antithrombic compound
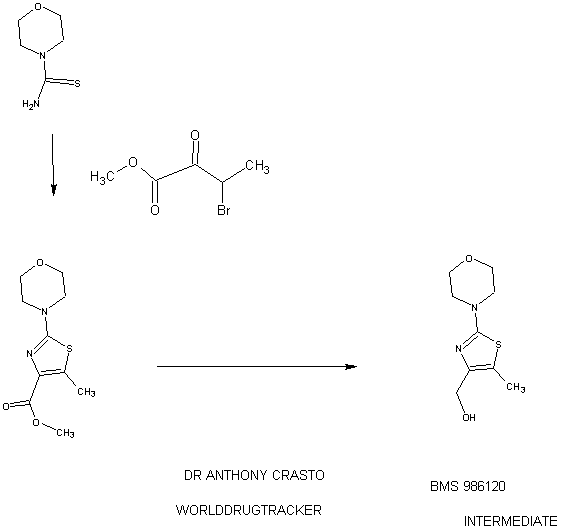
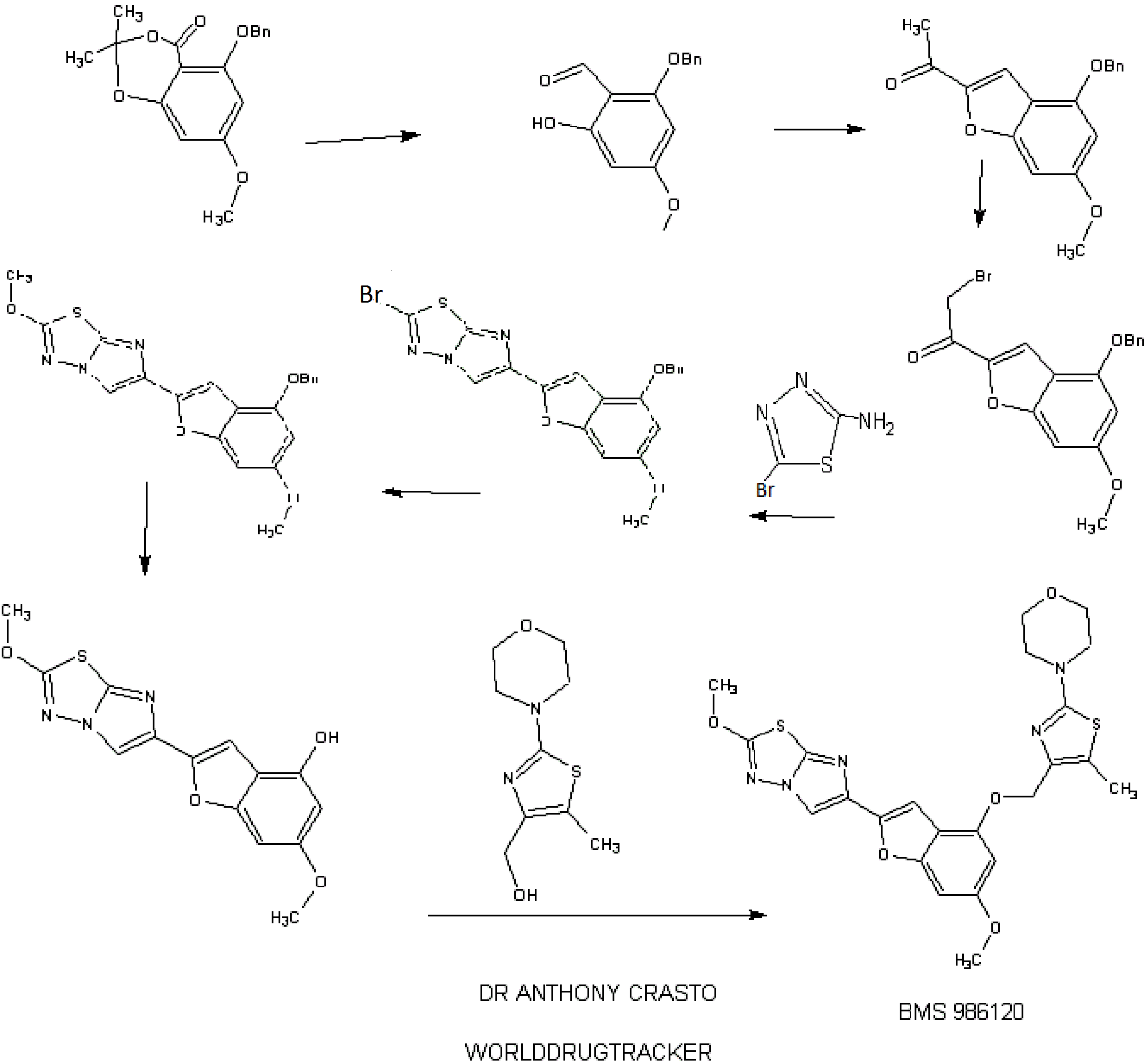
PATENT
http://www.google.com/patents/WO2013163279A1?cl=en
Thromboembolic diseases remain the leading cause of death in developed countries despite the availability of anticoagulants such as warfarin (COUMADIN®), heparin, low molecular weight heparins (LMWH), synthetic pentasaccharides, and antiplatelet agents such as aspirin and clopidogrel (PLAVIX®).
Current anti-platelet therapies have limitations including increased risk of bleeding as well as partial efficacy (relative cardiovascular risk reduction in the 20 to
30% range). Thus, discovering and developing safe and efficacious oral or parenteral antithrombotics for the prevention and treatment of a wide range of thromboembolic disorders remains an important goal.
Alpha-thrombin is the most potent known activator of platelet aggregation and degranulation. Activation of platelets is causally involved in atherothrombotic vascular occlusions. Thrombin activates platelets by cleaving G-protein coupled receptors termed protease activated receptors (PARs). PARs provide their own cryptic ligand present in the N-terminal extracellular domain that is unmasked by proteolytic cleavage, with subsequent intramolecular binding to the receptor to induce signaling (tethered ligand mechanism; Coughlin, S.R., Nature, 407:258-264 (2000)). Synthetic peptides that mimic the sequence of the newly formed N-terminus upon proteolytic activation can induce signaling independent of receptor cleavage. Platelets are a key player in atherothrombotic events. Human platelets express at least two thrombin receptors, commonly referred to as PARI and PAR4. Inhibitors of PARI have been investigated extensively, and several compounds, including vorapaxar and atopaxar have advanced into late stage clinical trials. Recently, in the TRACER phase III trial in ACS patients, vorapaxar did not significantly reduce cardiovascular events, but significantly increased the risk of major bleeding (Tricoci, P. et al, N. Eng. J. Med., 366(l):20-33 (2012). Thus, there remains a need to discover new antiplatelet agents with increased efficacy and reduced bleeding side effects.
There are several early reports of preclinical studies of PAR4 inhibitors. Lee, F-Y. et al., “Synthesis of l-Benzyl-3-(5′-hydroxymethyl-2′-furyl)indazole Analogues as Novel Antiplatelet Agents”, J. Med. Chem., 44(22):3746-3749 (2001) discloses in the abstract that the compound

58
“was found to be a selective and potent inhibitor or protease-activated receptor type 4 (PAR4)-dependent platelet activation. ”
Compound 58 is also referred to as YD-3 in Wu, C-C. et al, “Selective Inhibition of Protease-activated Receptor 4-dependent Platelet Activation by YD-3”, Thromb. Haemost., 87: 1026-1033 (2002). Also, see Chen, H.S. et al, “Synthesis and platelet activity”, J. Bioorg. Med. Chem., 16: 1262-1278 (2008).
EP1166785 Al and EP0667345 disclose various pyrazole derivatives which are useful as inhibitors of platelet aggregation.\
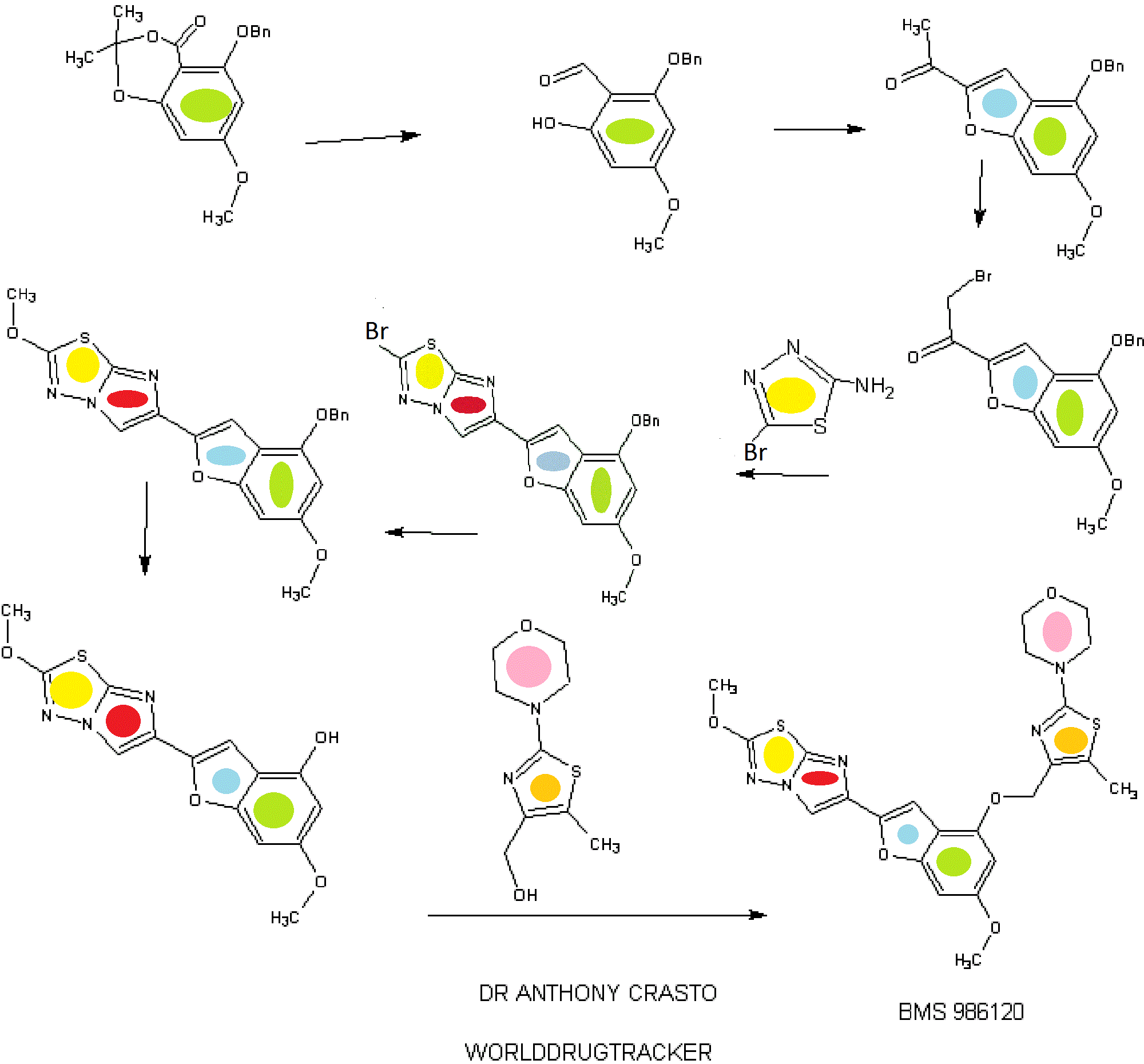
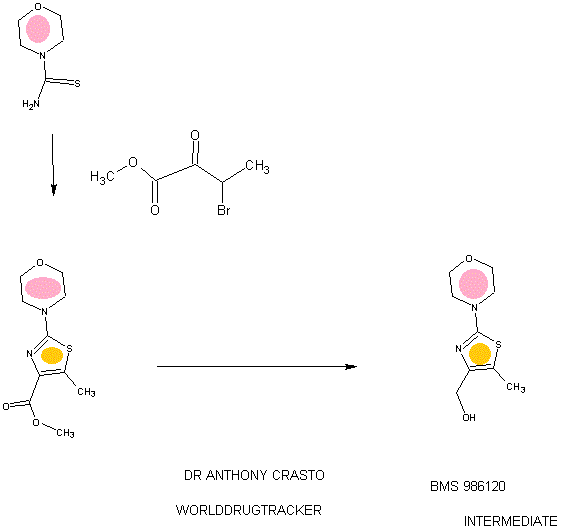
IB. 5-(Benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4-one

A solution of 5-hydroxy-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4- one (30.00 g, 0.134 mol, see Kamisuki, S. et al, Tetrahedron, 60:5695-5700 (2004) for preparation) in N,N-dimethylformamide (400 mL) was treated with powdered anhydrous potassium carbonate (19.41 g, 0.14 mol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with benzyl bromide (24.03 g, 0.14 mol) added dropwise over 15 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting material left by tic). The solid was filtered and washed with N,N- dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (500 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Crystallization form ethyl acetate (50 mL) and hexane (150 mL) gave 35.17 g of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H- benzo[d][l ,3]dioxin-4-one as large colorless prisms. Chromatography of the mother liquors on silica gel (4 x 13 cm, elution toluene – ethyl acetate 0-5%) gave 6.64 g of additional material to afford a total yield of 41.81 g (99%). HRMS(ESI) calcd for
Ci8Hi905 [M+H]+ m/z 315.1227, found 315.1386. 1H NMR (CDC13, 600 MHz) δ 1.68 (s, 6H), 3.77 (s, 3H), 5.19 (s, 2H), 5.19 (s, 2H), 6.04 (d, J = 2.03 Hz, 1H), 6.15 (d, J = 2.03 Hz, 1H), 7.27 (broad t, 1H), 7.36 (broad t, 2H), 7.52 (broad d, 2H).
1 C. 2-(Benzyloxy)-6-hydroxy-4-methoxybenzaldehyde

A solution of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l ,3]dioxin- 4-one (Example IB, 6.76 g, 21.5 mmol) in dichloromethane (120 mL) was cooled to -78 °C and treated with 43 mL (64.5 mmol) of a 1.5 M solution of diisobutylaluminum hydride in toluene added dropwise over 20 min. The resulting mixture was then stirred at -78 °C for 3 h. The reaction mixture was quenched by the careful addition of methanol (5 mL) added dropwise over 15 min, followed by IN hydrochloric acid (50 mL) added dropwise over 15 min. The cooling bath was then removed and an additional 150 mL of IN hydrochloric acid was added over 20 min. The mixture was then stirred at 22 °C for 2 h and diluted with dichloromethane (400 mL). The organic phase was collected and the aqueous phase (pH ~1) was extracted with dichloromethane (3 x 50 mL). The combined organic extracts were washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residual oil was diluted with tetrahydrofuran (70 mL), treated with 10 mL of 0.1N hydrochloric acid and stirred at 20 °C for 2 h. The reaction mixture was diluted with ethyl acetate (300 mL), washed with brine, dried over anhydrous magnesium sulfate, evaporated in vacuo to give a clear oil. Chromatography on silica gel (4 x 13 cm, elution toluene) gave 4.08 g (73% yield) of the title aldehyde as a clear oil which solidified on standing. LC (Method C): 2.237 min. HRMS(ESI) calcd for Ci5Hi504 [M+H]+ m/z 259.0965, found 259.1153. 1H NMR (CDC13, 600 MHz) δ 3.80 (s, 3H), 5.07 (s, 2H), 5.97 (d, J= 2.1 Hz, 1H), 6.01 (d, J= 2.1 Hz, 1H), 7.3 – 7.4 (m, 5 H), 10.15 (s, 1H), 12.49 (s, 1H).
ID. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)ethanone

A solution of 2-(benzyloxy)-6-hydroxy-4-methoxybenzaldehyde (Example 1C, 3.46 g, 13.4 mmol) in N,N-dimethylformamide (50 mL) was treated with powdered anhydrous cesium carbonate (4.58 g, 14.05 mmol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with chloroacetone (1.74 g, 18.7 mmol) added dropwise over 5 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting aldehyde left by tic and formation of the intermediate alkylated aldehyde). The solid was filtered and washed with N,N-dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (300 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. This syrup was diluted with tetrahydrofuran (50 mL) and ethyl acetate (50 mL), treated p- toluenesulfonic acid monohydrate (0.2 g) and stirred at 20 °C for 1 h (tic indicated complete cyclization of the intermediate alkylated aldehyde to the benzofuran). The reaction mixture was diluted with ethyl acetate (300 mL), washed with saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 2-4%) gave 3.51 g (88% yield) of the title benzofuran as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave the title material as large yellow prisms (3.15 g). LC (Method D): 2.148 min. HRMS(ESI) calcd for Ci8Hiv04 [M+H]+ m/z 297.1121, found 297.1092. 1H NMR (CDC13, 600 MHz) δ 2.51 (s, 3H), 3.82 (s, 3H), 5.13 (s, 2H), 6.37 (d, J= 1.77 Hz, 1H), 6.63 (broad s, 1H), 7.34 (broad t, 1H), 7.39 (broad t, 2H), 7.44 (broad d, 2H), 7.55 (d, J = 0.7 Ηζ,ΙΗ). IE. l-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone

A 250-mL, three-necked flask is equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with anhydrous tetrahydrofuran (25 mL) followed by 9.3 mL (9.3 mmol) of a 1M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran. The mixture was cooled to -78 °C and treated with a solution of l-(4- (benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 2.40 g, 8.1 mmole) in tetrahydrofuran (20 mL) added dropwise over 10 min. The resulting mixture was then stirred at -78 °C for 45 min. Then chlorotrimethylsilane (1.18 mL, 9.31 mmol) was added dropwise over 5 min and the resulting solution was stirred at -78 °C for another 20 min. The cooling bath was then removed and the mixture is allowed to warm to room temperature over 30 min. The reaction mixture was then quenched by addition to a cold solution of ethyl acetate (200 mL), saturated sodium bicarbonate (30 mL) and ice. The organic phase was rapidly dried over anhydrous magnesium sulfate (magnetic stirring) and evaporated in vacuo to give the silyl enol ether as an oil which is co-evaporated with toluene (20 mL). The silyl enol ether was then dissolved in dry tetrahydrofuran (40 mL), cooled to -20 °C and treated with solid sodium bicarbonate (0.10 g) followed by N- bromosuccinimide (1.44 g, 8.1 mmol) added in small portions over 15 min. The reaction mixture was allowed to warm to 0 °C over 2h and then quenched by addition of ethyl acetate (300 mL) and saturated sodium bicarbonate. The organic phase was washed with brine, dried over anhydrous magnesium sulfate and evaporated to give an orange oil. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 0-5%) gave 2.62 g (86% yield) of the title bromomethylketone as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave yellow prisms (2.30 g). LC (Method E): 1.977 min. HRMS(ESI) calcd for Ci8Hi6Br04 [M+H]+ m/z 375.0226, found 375.0277. 1H NMR (CDCls, 600 MHz) δ 3.84 (s, 3H), 4.33 (s, 2H), 5.14 (s, 2H), 6.38 (d, J = 1.76 Hz, 1H), 6.64 (broad s, 1H), 7.35 (broad t, 1H), 7.40 (broad t, 2H), 7.44 (broad d, 2H), 7.70 (s, 1H). 1 EE. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone

Benzyltrimethylammonium dichloroiodate (117 g, 169 mmol) was added to a solution of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 50 g, 170 mmol) in THF (500 mL) in a 1 L multineck round bottom flask under nitrogen atmosphere. The reaction mixture was stirred at RT for 6 h, cooled to 0 °C and quenched with 10% NaHCC”3 solution. The organic layer was washed with 1 M sodium thiosulphate solution, water, and brine, dried over Na2S04, and concentrated in vacuo (bath temperature <45 °C). The residue was triturated with 5% EtOAc in pet. ether and dried to obtain the title chloromethylketone as a pale yellow solid (48 g, 130 mmol, 78%). 1H NMR (300 MHz, DMSO-d6) δ 3.84-3.82 (d, J =4.5Hz, 3H) 4.98 (s, 2H), 5.27(s, 2H), 6.62 -6.61 (d, J = 1.8Hz, 1H), 6.92-6.93 (m, 1H), 7.54-7.36 (m, 5H), 8.10-8.09 (d, J = 3Hz, 1H); MS m/z: [M+H]+ 331.0. IF. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoimidazo[2, 1 – b] [ 1 ,3 ,4]thiadiazole

A mixture of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone (Example IE, 3.00 g, 8.0 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (1.65 g, 9.16 mmol) in isopropanol (100 mL) was heated in a pressure flask equipped with a magnetic stirring bar at 78-80 °C for 18 h (homogeneous after 20 min and then formation of a precipitate after 2 h). The cooled mixture is then transferred into five 20 mL microwave vials and then heated in a microwave apparatus to 150 °C for 30 min. Each vial was then diluted with dichloromethane (250 mL) washed with saturated sodium bicarbonate (25 mL) and brine (25 mL), dried over anhydrous magnesium sulfate. The fractions were combined and concentrated in vacuo. Chromatography of the orange-brown residual solid on silica gel (4 x 10 cm, slow elution with dichloromethane due to poor solubility) gave 2.96 g of the title imidazothiadiazole contaminated with some l-(4-(benzyloxy)-6- methoxybenzofuran-2-yl)ethanone. The solid material was triturated with ethyl acetate (20 mL), filtered, washed with ethyl acetate (10 ml) and dried in vacuo to give 2.34 g (64% yield) of pure title imidazothiadiazole as an off white solid which is used as such for the next step. LC (Method E): 2.188 min. HRMS(ESI) calcd for C2oHi5BrN303S [M+H]+ m/z 456.00175, found 456.00397. 1H NMR (CDC13, 600 MHz) δ 3.82 (s, 3H), 5.16 (s, 2H), 6.38 (d, J= 1.67 Hz, 1H), 6.66 (broad s, 1H), 7.15 (s, 1H), 7.31 (broad t, 1H), 7.38 (broad t, 2H), 7.45 (broad d, 2H), 8.02 (s, 1H).
Alternatively, Example IF, 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole, was prepared as follows:
A 1000-mL, three-necked flask equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with dry NMP (200 mL) followed by 1- (4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone (Example 1EE, 50 g, 150 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (27.2 g, 151 mmol). The resulting mixture was stirred at 80 °C for 8h. TLC (8:2 dichloromethane/pet. ether) and LC/MS showed intermediate uncyclized material (m/z 476) and the reaction mixture was stirred at 120 °C for 3h. The reaction mixture was cooled to RT, quenched with water and extracted with EtOAc (3X). The combined organic layers were washed with brine, dried over Na2S04, and concentrated in vacuo. The thick brown residue was purified by silica gel chromatography (0 to 100% dichloromethane in pet. ether) to give a brown solid. This material was triturated with EtOAc and dried to obtain the title imidazothiadiazole (24 g, 50 mmol, 33%>) as a light brown solid. (See the procedure set forth above for analytical data).
1 G. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-methoxyimidazo[2, 1 – b][l,3,4]thiadiazole

A solution of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole (Example IF, 2.30 g, 5.04 mmol) in a mixture of dichloromethane (180 mL) and methanol (45 mL) was treated at 22 °C with 4.2 mL of a 25 wt.% solution of sodium methoxide in methanol (0.2 mmol) added in one portion. More methanol (45 mL) was added and the mixture was stirred for 1 h. The reaction mixture was quenched by the addition of 25 mL of IN hydrochloric acid followed by 20 ml of saturated sodium bicarbonate. The solvent was evaporated under reduced pressure and the residue was diluted with dichloromethane (400 mL), washed with brine, dried over anhydrous magnesium sulfate and evaporated in vacuo. Chromatography of the residue on silica gel (3 x 10 cm, elution with dichloromethane – ethyl acetate 0-4%) gave 1.70 g (83% yield) of the title compound as a white solid. This material was recrystallized from ethyl acetate (30 mL per gram, 80% recovery) to give white needles. LC (Method
D): 2.293 min. HRMS(ESI) calcd for C21H18N3O4S [M+H]+ m/z 408.1013, found 408.1024. 1H NMR (CDC13, 600 MHz) δ 3.81 (s, 3H), 4.18 (s, 3H), 5.16 (s, 2H), 6.37 (d, J = 1.75 Hz, 1H), 6.67 (broad s, 1H), 7.07 (s, 1H), 7.31 (broad t, 1H), 7.37 (broad t, 2H), 7.45 (broad d, 2H), 7.81 (s, 1H).
1H. 6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-ol

A mixture of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- methoxyimidazo[2,l-b][l,3,4]thiadiazole (Example 1G, 1.250 g, 3.06 mmol) and pentamethylbenzene (3.17 g, 21.4 mmol) in dichloromethane (200 mL) was cooled to -78 °C under a nitrogen atmosphere and then treated immediately (to avoid crystallization) with 8 mL (8 mmol) of a 1 M solution of boron trichloride in dichloromethane added dropwise over 3 min. The resulting mixture was stirred at -78 °C for 1 h. The reaction mixture was then quenched by the addition of a solution of sodium bicarbonate (6 g) in water (100 mL) added in one portion. The cooling bath was removed and the resulting mixture was stirred at room temperature for 1 h. The solid formed was filtered, washed successively with water (50 m) and dichloromethane (50 mL). The filter cake was allowed to soak with anhydrous ethanol (15 ml) and then sucked dry. The white solid obtained was then dried under vacuum for 24 h to give 0.788 g (80%> yield) of pure title material (> 95% by hplc). The combined filtrate and washings were diluted with dichloromethane (600 mL) and stirred in a warm water bath till the organic phase was clear with no apparent solid in suspension. The organic phase was collected, dried over anhydrous magnesium sulfate and rapidly filtered while still warm. The filtrate was evaporated and the residue (product and pentamethylbenzene) was triturated with toluene (20 mL), the solid collected and washed with toluene (20 mL) to give 0.186 g (19% yield, 99% combined yield) of title material as a tan solid (> 95% by hplc). LC (Method E): 1.444 min. HRMS(ESI) calcd for C14H12N3O4S [M+H]+ m/z 318.0543, found 318.0578. 1H NMR (DMSO-de, 600 MHz) 5 3.71 (s, 3H), 4.16 (s, 3H), 6.21 (d, J = 1.87 Hz, 1H), 6.61 (broad s, 1H), 6.95 (s, 1H), 8.29 (s, 1H), 9.96 (s, 1H).
Example 94
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine

94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate [00258] A solution of methyl 2-bromo-5-methylthiazole-4-carboxylate (2.80 g, 11.86 mmol) and morpholine (4.5 mL, 51.7 mmol) in THF (10 mL) was heated at reflux under nitrogen for 18 h. The volatiles were then removed under reduced pressure and the crude product was purified on the ISCO using a REDISEP® 40 g column (0 to 40% EtOAc- DCM), to give the title compound (2.20 g, 77%) as a yellow solid. LCMS (APCI): calcd for CioHisNzOsS [M+H]+ m/z 243.07, found 243.1. 1H NMR (CDC13, 400 MHz) δ ppm: 3.89 (s, 3H), 3.77-3.83 (m, 4H), 3.41-3.47 (m, 4H), 2.64 (s, 3H). [00259] Alternatively, Example 94A, methyl 5-methyl-2-morpholinothiazole-4- carboxylate, was prepared as follows:
94AA. Methyl 3-bromo-2-oxobutanoate

A 5L 4-neck round bottom flask equipped with a mechanical stirrer, temperature thermocouple, condenser and a 1L addition funnel, was charged copper(II) bromide (962 g, 4310 mmol) and ethyl acetate (2 L). A solution of methyl 2-ketobutyrate (250 g, 2150 mmol) in CHC13 (828 mL) was added dropwise. A scrubber (400 mL 1 N NaOH) was connected and the reaction mixture was heated to reflux (75 °C). The reaction started as a dark green color and as heating progressed, it became a light green with a white precipitate forming. NMR after one hour at reflux indicated that the reaction was complete. The reaction was cooled to RT and filtered through a pad of CELITE®. The filtrate was concentrated to an oil, dissolved in methylene chloride (500 mL) and filtered again through CELITE®. The filtrate was then passed through a pad of silica gel and eluted with ethyl acetate. Concentration of the filtrate provided the title bromoketoester (399 g, 2040 mmol, 95%) as a yellow oil. 1H NMR (400MHz, CDC13) δ 5.18 (q, J = 6.7 Hz, 1H), 3.94 (s, 3H), 1.83 (d, J = 6.8 Hz, 3H). 94AAA. Morpholine-4-carbothioamide

To a solution of morpholine (199 g, 2280 mmol) in CHC13 (1 L) was added isothiocyanatotrimethylsilane (150 g, 1140 mmol) dropwise. A white precipitate formed almost immediately, and the reaction was stirred for 1 h at RT. The reaction was then filtered and the resulting solid was washed with additional CHC13 and dried in vacuo to give the title thiourea as a white solid. (137 g, 937 mmol, 82%). 1H NMR (400MHz, DMSO-de) δ 3.81 – 3.71 (m, 2H), 3.17 – 3.08 (m, 2H).
94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate

To a solution of morpholine-4-carbothioamide (Example 94 AAA, 175 g, 1200 mmol) in methanol (500 mL) was charged methyl 3-bromo-2-oxobutanoate (Example 94AA, 233 g, 1200 mmol). The reaction was then heated to reflux for 1 hour, cooled to RT, and filtered. The filtrate was concentrated and the crude product was purified on by silica gel chromatography. The title thiazole (206g, 850 mmol, 71%) was isolated as a yellow oil. (See the procedure set forth above for analytical data).
(5-Methyl-2-morpholinothiaz l-4-yl)methanol

The compound was prepared according to the protocol described for Example 92B. The crude product was purified on the ISCO using a REDISEP® Gold 24 g column (0 to 50% EtOAc-DCM) to give the title compound as a white solid (0.086 g, 51%). LCMS (APCI): calcd for C9Hi5N202S [M+H]+ m/z 215.08, found 215.1. 1H NMR (CDCI3, 400 MHz) δ ppm: 4.48 (d, J= 4.7 Hz, 2H), 3.77-3.83 (m, 4H), 3.37-3.43 (m, 4H), 2.30 (t, J= 4.7 Hz, 1H), 2.28 (s, 3H).
Example 94. 4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2, 1 -b] [ 1 ,3,4]thiadiazol-6-yl) benzofuran-4-yl)oxy)methyl)-5 -methylthiazol-2-yl)morpholine

The title compound was prepared according to the protocol described for Example 86. The crude product was purified on the ISCO using a REDISEP® 4 g column (0 to 40% EtOAc-DCM) and the obtained solid was suspended in MeOH, sonicated, filtered and dried to give the title compound as an off-white solid (0.094 g, 53%). LC (Method C): 2.314 min. HRMS(ESI): calcd for C23H24N505S2 [M+H]+ m/z 514.122, found 514.126. 1H NMR (CDC13, 400 MHz) δ ppm: 7.83 (s, 1H), 7.06 (d, J = 0.8 Hz, 1H), 6.69 (d, J= 0.8 Hz, 1H), 6.50 (d, J= 2.0 Hz, 1H), 5.05 (s, 2H), 4.21 (s, 3H), 3.85 (s, 3H), 3.78- 3.84 (m, 4H), 3.39- 3.46 (m, 4H), 2.37 (s, 3H).
ABSTRACT
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 263


| Patent ID | Date | Patent Title |
|---|---|---|
| US2015094297 | 2015-04-02 | IMIDAZOTHIADIAZOLE AND IMIDAZOPYRAZINE DERIVATIVES AS PROTEASE ACTIVATED RECEPTOR 4 (PAR4) INHIBITORS FOR TREATING PLATELET AGGREGATION |
////////BMS 986120, phase 1, Bristol-Myers Squibb , Imidazoles, Small molecules, Thiadiazoles, 1478712-37-6
c1(sc2nc(cn2n1)c3cc4c(cc(cc4o3)OC)OCc5nc(sc5C)N6CCOCC6)OC
CC1=C(N=C(S1)N2CCOCC2)COC3=C4C=C(OC4=CC(=C3)OC)C5=CN6C(=N5)SC(=N6)OC
GDC 0853, Fenebrutinib
 .
.
Picture credit….Bethany Halford
GDC 0853, Fenebrutinib
GDC-0853; RG 7845
| Molecular Formula: | C37H44N8O4 |
|---|---|
| Molecular Weight: | 664.79646 g/mol |
2-[3-(hydroxymethyl)-4-[1-methyl-5-[(7-methyl-6,8-dihydro-5H-[1,2,4]triazolo[1,5-a]pyrazin-2-yl)amino]-6-oxo-3-pyridyl]-2-pyridyl]-3,4,6,7,8,9-hexahydropyrazino[1,2-a]indol-1-one
3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
2H-Cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one, 2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S) -2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino] -6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7- dimethyl-
s ISoMER 1434048-34-6 desired
r iSoMER 1434048-57-3 undesired
Phase 1
Patients with Patients with Resistant B-Cell Lymphoma or Chronic Lymphocytic Leukemia..
@genentech‘s Btk inhibitor
https://clinicaltrials.gov/ct2/show/NCT01991184
Bruton tyrosine kinase inhibitor
- 01 Sep 2015 Phase-I clinical trials in Autoimmune disorders (In volunteers) in USA (PO, Capsule and Tablet) (NCT02699710)
- 16 Oct 2014 Discontinued – Phase-I for Non-Hodgkin’s lymphoma (Second-line therapy or greater) in USA (unspecified route)
- 16 Oct 2014 Discontinued – Phase-I for Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (unspecified route)

GDC-0853; RG 7845; RO 7010939
2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S)-2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino]-6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7-dimethyl-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one
GDC-0853 is an orally bioavailable, selective, and reversible Bruton’s tyrosine kinase (BTK) inhibitor with IC50s ranging from 2-9 nM for basophil activation, B cell receptor activation, and constitutive p-BTK activity in whole blood lysates.1,2 In rats, treatment for longer than 7 days leads to pancreatic toxicity but it does not occur in mice or dogs, even at higher doses.3 Formulations containing GDC-0853 were well-tolerated in Phase I clinical trials and are in additional clinical trials for rheumatoid arthritis, lupus erythematosus, lymphoma, and leukemia.
- Originator Genentech
- Class Antineoplastics; Antirheumatics; Piperazines; Pyrazines; Pyridines
- Mechanism of Action Agammaglobulinaemia tyrosine kinase inhibitors
Highest Development Phases
- Phase II Rheumatoid arthritis; Systemic lupus erythematosus; Urticaria
- Phase I Autoimmune disorders
- Discontinued Chronic lymphocytic leukaemia; Non-Hodgkin’s lymphoma
Most Recent Events
- 01 Jun 2018 Chemical structure information added
- 07 Nov 2017 Genentech initiates enrolment in a phase II extension trial for Systemic Lupus Erythematosus in Spain (EudraCT2017-001764-37)
- 13 Sep 2017 Genentech initiates enrolment in a phase I trial in Healthy volunteers in United Kingdom (NCT03290703)
BTK inhibitor GDC-0853 An orally available inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, GDC-0853 inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
SCHEME

MAIN

Patent
WO 2013067274
https://www.google.co.in/patents/WO2013067274A1?cl=en
part
Example 271a (S)-tert-Butyl 4-(6-(5-Chloro-2-methoxypyridin-3-ylamino)pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271a
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (40 mL), (S)-tert-butyl 4-(6-amino pyridin-3-yl)-3-methylpiperazine-1-carboxylate 101h (2.04 g, 7.0 mmol), 3-bromo-5-chloro-2-methoxypyridine (2.8 g, 12.6 mmol), Pd2(dba)3 (640 mg, 0.70 mmol), XantPhos (404.6 mg, 0.70 mmol), and cesium carbonate (4.56 g, 14.0 mmol). After three cycles of vacuum/argon flush, the mixture was heated at 100 °C for 4 h. After this time the reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 1:3 ethyl acetate/petroleum ether to afford 271a (1.7 g, 57%) as a yellow solid. MS-ESI: [M+H]+ 434.2
Example 271btert-Butyl (3S)-4-(6-{[5-(2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}-3-(hydroxymethyl)pyridin-4-yl)-2-methoxypyridin-3-yl] amino}pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271b
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 271a (650 mg, 1.50 mmol), {3-[(acetyloxy)methyl]-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridin-4-yl}boronic acid 199e (1.79 g, 4.5 mmol), Pd2(dba)3 (137.2 mg, 0.15 mmol), P(cy)3(167.4 mg, 0.60 mmol), Cs2CO3 (978 mg, 3.0 mmol), dioxane (20 mL), and water (0.5 mL). After three cycles of vacuum/argon flush, the mixture was heated at 110°C for 16 h. After this time the reaction was cooled to room temperature. Lithium hydroxide monohydrate (1.89 g, 45 mmol) and water (2.0 mL) were added. The resulting mixture was stirred at 45°C for 4 h. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 3:1 ethyl acetate/petroleum ether to afford 271b (290 mg, 27%) as a yellow solid. MS-ESI: [M+H]+ 709.3
Example 271c 10-[3-(Hydroxymethyl)-4-[5-({5-[(2S)-2-methylpiperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridin-2-yl]-4,4-dimethyl-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-9-one 271c
A solution of 271b (286.6 mg, 0.40 mmol) in dioxane/HCl (30 mL) was stirred at 50 °C for 2 h. It was evaporated under reduced pressure to afford 271c (450 mg, crude) as a black solid. MS-ESI: [M+H]+ 595.3
Example 271 3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one 271
To a solution of 271c (450 mg, 0.75 mmol) in methanol (10 mL) was added oxetan-3-one (162 mg, 2.25 mmol), NaBH3CN (141.8 mg, 2.25 mmol), and ZnCl2 (306 mg, 2.25 mmol). The reaction was stirred at room temperature for 3 h. The mixture was evaporated under reduced pressure and the residue was diluted with water (5 mL). It was then extracted with dichloromethane (3 X 10 mL) and the combined dichloromethane extract was concentrated under reduced pressure. The residue was purified by reverse-phase prep-HPLC to afford 271 (23.0 mg, 8.8%, over two steps) as a yellow solid. MS-ESI: [M+H]+651.3. 1H NMR (500 MHz, CDCl3) δ 9.76 (s, 1H), 8.74 (d, J = 2.0 Hz, 1H), 8.53 (d, J = 5.0 Hz, 1H), 7.99 (d, J = 3.0 Hz, 1H), 7.84 (s, 1H), 7.73 (s, 1H), 7.41 (d, J = 4.5 Hz, 1H), 7.35 (dd, J = 2.5 Hz, 8.5 Hz, 1H), 6.87 (s, 1H), 6.85 (d, J = 9.0 Hz, 1H), 5.16-5.13 (m, 1H), 4.72-4.69 (m, 5H), 4.54-4.53 (m, 1H), 4.36-4.35 (m, 1H), 4.19-4.17 (m, 2H), 3.89-3.87 (m, 1H), 3.56-3.49 (m, 2H), 3.11-3.09 (m, 2H), 2.60-2.48 (m, overlap, 7H), 2.24-2.21 (m, 1H), 1.29 (s, 6H), 1.02 (d, J = 6.0 Hz, 3H)
271
………………………..
syn of 191 j
is intermediatenot product, is acid
To a mixture of 4-chloro-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridine-3-carbaldehyde 108a (500 mg, 1.46 mmol), tert-butyl alcohol (20 mL), and dichloromethane (5 mL) was added 2-methyl-2-butene (3066 mg, 43.8 mmol). An aqueous solution (8 mL) of NaClO2 (263 mg, 2.92 mmol) and NaH2PO4·2water (683 mg, 4.38 mmol) was added dropwise at -10°C and the reaction mixture was stirred at -10 °C for overnight. It was concentrated under reduced pressure and the residue was extracted with ethyl acetate (4 × 20 mL). The combined organic extract was dried over MgSO4 and concentrated. The residue was purified with reverse-phase prep-HPLC to afford 210a (315 mg, 60%) as a pale yellow solid. MS-ESI: [M+H]+ 360.1
Example 210b 2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl} -4-[1-methyl-5-({5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridine-3-carboxylic Acid 210b
A 25-mL round-bottomed flask equipped with a reflux condenser was charged with 210a (400 mg, 1.1 mmol), (S)-1-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-ylamino)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2(1H)-one 191j (536 mg, 1.1 mmol), PdCl2(dppf) (81 mg, 0.11 mmol), K3PO4 (466 mg, 2.2 mmol), sodium acetate (216 mg, 2.2 mmol), acetonitrile (10 mL), and water (0.2 mL). After three cycles of vacuum/argon flush, the mixture was heated at 100°C for 3 h. It was then filtered and the filtrate was evaporated in vacuo. The residue was purified by silica-gel column chromatography eluting with 1:3 petroleum/ethyl acetate to afford 210b as a yellow solid (306 mg, 41%). MS-ESI: [M+H]+ 679.3
construction, use your discretion
Example 130a (3S)-tert- utyl 3-methyl-4-(6-nitropyridin-3-yl)piperazine-l-carboxylate 130a

130a
Following the procedures as described for compound lOlg, reaction of 5-bromo-2-nitropyridine (10.5 g, 50 mmol), and (JS)-tert-butyl-3 -methylpiperazine- 1 -carboxylate (10.0 g, 50 mmol) afforded 130a as a yellow solid (8.05 g, 50%). LCMS: [M+H]+ 323
Example 130b (3 S)-tert-butyl-4-(6-aminopyridin-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130b

130b
Following the procedures as described for compound lOlh, hydrogenation of 130a (5.8 g) afforded 130bas a brown solid (4.9 g, 96%). LCMS: [M+H]+ 293
Example 130c (3 S)-tert-Butyl-4-(6-(5 -bromo- 1 -methyl -2 -oxo- 1,2-dihydropyridin-3 -yl amino) pyridine-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130c
N

Following the procedures as described for compound lOli, reaction of 130b (4.0 g) and 3,5-dibromo-l-methylpyridin-2(lH)-one (5.5 g) afforded 130c as a yellow solid (5.4 g, 83%). LCMS: [M+H]+ 478
Example 130d (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- 2-ylamino)pyridine-2(lH)-one 130d

Following the procedures as described for compound lOlj, acidic hydrolysis of the Boc group of 130c (3.1 g) afforded 130d as a yellow solid (2.3 g, 95%). LCMS: [M+H]+ 380.
Example 130e (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2 -methyl-4-(ox etan-3-yl)piperazin-l-yl) pyridine -2-ylamino)pyridin-2(lH)-one 130e

Following the procedures as described for compound 101k, reductive amination of 130d (2.35 g) with oxetan-3-one (0.4 mL) afforded 130e as a yellow solid (2.6 g, 98%). LCMS: [M+H]+ 434.
Example 13 Of (3S)-l-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)pyridin-2-ylamino) -5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 130f
 check pyridine ring position
check pyridine ring position
A 100 mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 130e (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), AcOK (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90 °C for 4 hrs, then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3: 1 PE/EA (80 mL) to afford 130f as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
check pyridine ring position, use your discretion
Example 191h ( 3S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- -ylamino)pyridine-2(lH)-one 191h

Following the procedure described for compound lOlj and starting with (3S)-tert-butyl 4-(6-(5 -bromo- 1 -methyl-2-oxo- 1 ,2-dihydropyridin-3 -ylamino)pyridine-3 -yl)-3 -methyl-piperazine-l-carboxylate 191g (3.1 g, 6.5 mmol) afforded 191h as a yellow solid (2.3 g, 94%). MS-ESI: [M+H]+ 378.
Example 1 1 i (S)-5 -Bromo- 1 -methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin- 1 -yl)pyridin-2-ylamino)pyridin-2(lH)-one 191i
A mixture of (5)-5-bromo-l-methyl-3-(5-(2-methylpiperazin-l-yl)pyridin-2-ylamino)pyridin-2(lH)-one 191h (40.0 g, 106 mmol), oxetan-3-one (1 1.4 g, 159 mmol), NaBH3CN (10.0 g, 159 mmol), and zinc chloride (21.3 g, 159 mmol) in methanol (700 mL) was stirred at 50°C for 5 hours. The mixture was added to water (100 mL) and concentrated under reduced pressure. The residue was extracted with dichloromethane (200 mL x 3). The combined organic layer was concentrated under reduced pressure and the residue was purified by silica-gel column chromatography eluting with 40: 1 dichloromethane /methanol to afford 191i (35 g, 73%). MS: [M+H]+ 434.
Example 191j (J5)-l-Methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)-pyridin- -ylamino) -5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 191j

191 i 191j
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with (5)-tert-butyl-4-(6-(5-bromo-l-methyl-2-oxo-l ,2-dihydropyridin-3-ylamino)pyridine-3-yl)-3-methylpiperazine-l-carboxylate 191i (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), potassium acetate (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90°C for 4 h. It was then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3 : 1 petroleum ether/ethyl acetate (80 mL) to afford 191j as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
pipeline
http://www.gene.com/medical-professionals/pipeline

Pictrelisib, GDC-0941, RG7321 and GNE0941
| Patent ID | Date | Patent Title |
|---|---|---|
| US8921353 | 2014-12-30 | Heteroaryl pyridone and aza-pyridone compounds |
| US2014378432 | 2014-12-25 | HETEROARYL PYRIDONE AND AZA-PYRIDONE COMPOUNDS |
| US8716274 | 2014-05-06 | Heteroaryl pyridone and aza-pyridone compounds |
Development of an Efficient Manufacturing Process for Reversible Bruton’s Tyrosine Kinase Inhibitor GDC-0853
Haiming Zhang*†  , Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin†
, Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin†
, Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin† †Department of Small Molecule Process Chemistry, Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States
‡Department of Process Chemistry and Catalysis and ¶Department of Drug Substance Scale-up and Supply, F. Hoffmann-La Roche AG, Grenzacherstrasse 124, 4070 Basel, Switzerland
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. https://pubs.acs.org/doi/10.1021/acs.oprd.8b00134

Efforts toward the process development of reversible Bruton’s tyrosine kinase (BTK) inhibitor GDC-0853 (1) are described. A practical synthesis of GDC-0853 was accomplished via a key highly regioselective Pd-catalyzed C–N coupling of tricyclic lactam 5 with 2,4-dichloronicotinaldehyde (6) to afford the C–N coupling product 3, a Suzuki–Miyaura cross-coupling of intermediate 3 with boronic ester 4 derived from a Pd-catalyzed borylation of tetracyclic bromide 7, to generate penultimate aldehyde intermediate 2 and subsequent aldehyde reduction and recrystallization. Process development of starting materials 5, 6, and 7 is also discussed.
(S)-2-(3′-(Hydroxymethyl)-1-methyl-5-((5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-yl)amino)-6-oxo-1,6-dihydro-[3,4′-bipyridin]-2′-yl)-7,7-dimethyl-2,3,4,6,7,8-hexahydro-1H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1-one (crude GDC-0853, 1)
GDC-0853 (1, 196 kg, 81% yield, >99 A%, Pd < 10 ppm): mp 271 °C (DSC);
FTIR (cm–1, neat) 3430, 3313, 2945, 2865, 1606, 1573;
1H NMR (400 MHz, CDCl3) δ 8.65 (d, J = 2.2 Hz, 1H), 8.48 (d, J = 5.1 Hz, 1H), 7.96 (d, J = 2.7 Hz, 1H), 7.83 (d, J = 2.3 Hz, 2H), 7.36 (d, J = 5.1 Hz, 1H), 7.31 (dd, J = 8.9, 2.8 Hz, 1H), 6.87–6.76 (m, 2H), 5.18–4.98 (m, 1H), 4.77–4.58 (m, 5H), 4.50 (m, 1H), 4.33 (m, 1H), 4.16 (m, 2H), 3.86 (m, 1H), 3.71 (s, 3H), 3.61–3.38 (m, 2H), 3.07 (m, 2H), 2.67–2.39 (m, 7H), 2.20 (dd, J = 10.8, 6.3 Hz, 1H), 1.27 (s, 6H), 0.98 (d, J = 6.3 Hz, 3H);
13C NMR (101 MHz, CDCl3) δ 161.7, 157.6, 154.3, 150.3, 148.4, 141.9, 140.0, 131.4, 131.1, 129.7, 128.8, 127.7, 125.8, 123.9, 117.2, 116.3, 112.4, 111.3, 75.5, 75.5, 59.4, 59.1, 56.3, 52.9, 50.0, 49.2, 48.2, 45.9, 42.7, 40.9, 39.6, 38.5, 30.3, 15.3.
HRMS (ESI+) calcd for C37H45N8O4 ([M + H]+), 665.3564; found, 665.3588.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00134/suppl_file/op8b00134_si_001.pdf


/////////////
O=C1N(C)C=C(C2=CC=NC(N3CCN4C(C3=O)=CC5=C4CC(C)(C)C5)=C2CO)C=C1NC(N=C6)=CC=C6N7CCN(C8COC8)C[C@@H]7C
//////GDC 0853, genentech, Btk inhibitor, phase 1, Patients with Resistant B-Cell Lymphoma, Chronic Lymphocytic Leukemia, Bruton tyrosine kinase inhibitor, GDC-0853, RG 7845, 1434048-34-6, Fenebrutinib
N1(CCN(CC1C)C2COC2)c3cnc(cc3)NC=4C(N(\C=C(/C=4)c5c(c(ncc5)N6CCn7c(C6=O)cc8CC(Cc78)(C)C)CO)C)=O
CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus Googleplus
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
SIDE CHAIN
MAIN
