
Home » PATENTS (Page 6)
Category Archives: PATENTS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
WO 2016015596, Omarigliptin, Sunshine Lake Pharma Co Ltd, New patent


(WO2016015596) PROCESS FOR PREPARING 2, 3-DISUBSTITUTED-5-OXOPYRAN COMPOUND
SUNSHINE LAKE PHARMA CO., LTD. [CN/CN]; Northern Industrial Area, Songshan Lake Dongguan, Guangdong 523000 (CN)
SUN, Guodong; (CN).
LIU, Yongjun; (CN).
WEI, Mingjie; (CN).
LAI, Cailang; (CN).
LI, Dasheng; (CN).
ZHANG, Shouhua; (CN).
WANG, Zhongqing; (CN)
To a mixture of methanol (42 mL) and tert-butyl ( (1R, 2S) -1- (2, 5-difluorophenyl) -1-hydroxypent-4-yn -2-yl) carbamate (7.0 g) cooled to-5℃ was added a solution of KOH (3.2 g) in methanol (28 mL) dropwise. After dropwise addition, the resulting mixture was stirred for 30 minutes, then iodine (5.7 g) was added to the mixture. The reaction mixture was stirred at 0 ℃ for 10 minutes, followed by 25 ℃ for 6 hours, and then quenched with water (140 mL) . Then the mixture was stirred at 25 ℃ for 2 hours. The precipitate was collected by filtration and washed sequentially with methanol/water (40 mL, v: v=1: 1) . The resulting solid was dried at 45 ℃ in vacuo to give the title compound as awhite solid (8.8 g, purity: 95.0%)

//////////WO 2016015596, Omarigliptin, Sunshine Lake Pharma Co Ltd, NEW PATENT
WOCKHARDT, WO 2016016766, ISAVUCONAZONIUM SULPHATE, NEW PATENT

![]()
(WO2016016766) A PROCESS FOR THE PREPARATION OF ISAVUCONAZONIUM OR ITS SALT THEREOF
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
KHUNT, Rupesh Chhaganbhai; (IN).
RAFEEQ, Mohammad; (IN).
MERWADE, Arvind Yekanathsa; (IN).
DEO, Keshav; (IN)
The present invention relates to a process for the preparation of stable Isavuconazonium or its salt thereof. In particular of the present invention relates to process for the preparing of isavuconazonium sulfate, Isavuconazonium iodide hydrochloride and Boc-protected isavuconazonium iodide has purity more than 90%. The process is directed to preparation of solid amorphous form of isavuconazonium sulfate, isavuconazonium iodide hydrochloride and Boc-protected isavuconazonium iodide. The present invention process of Isavuconazonium or its salt thereof is industrially feasible, simple and cost effective to manufacture of isavuconazonium sulfate with the higher purity and better yield.
Habil Khorakiwala, chairman of Indian generic drugmaker Wockhardt
Isavuconazonium sulfate is chemically known l-[[N-methyl-N-3-[(methylamino) acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl)thiazol-2-yl]butyl]-lH-[l,2,4]-triazo-4-ium Sulfate and is structurally represented by formula (I):

Formula I
Isavuconazonium sulfate (BAL8557) is indicated for the treatment of antifungal infection. Isavuconazonium sulfate is a prodrug of Isavuconazole (BAL4815), which is chemically known 4-{2-[(lR,2R)-(2,5-Difluorophenyl)-2-hydroxy-l-methyl-3-(lH-l ,2,4-triazol-l-yl)propyl]-l ,3-thiazol-4-yl}benzonitrile compound of Formula II

Formula II
US Ppatent No. 6,812,238 (referred to herein as ‘238); 7,189,858 (referred to herein as ‘858); 7,459,561 (referred to herein as ‘561) describe Isavuconazonium and its process for the preparation thereof.
The US Pat. ‘238 patent describes the process of preparation of Isavuconazonium chloride hydrochloride.
The US Pat. ‘238 described the process for the Isavuconazonium chloride hydrochloride, involves the condensation of Isavuconazole and [N-methyl-N-3((tert-butoxycarbonyl methylamino) acetoxymethyl) pyridine-2-yl]carbamic acid 1 -chloro-ethyl ester. The prior art reported process require almost 15-16 hours, whereas the present invention process requires only 8-10 hours. Inter alia prior art reported process requires too many step to prepare isavuconazonium sulfate, whereas the present invention process requires fewer steps.
Moreover, the US Pat. ‘238 describes the process for the preparation Isavuconazonium hydrochloride, which may be used as the key intermediate for the synthesis of isavuconazonium sulfate, compound of formula I. There are several drawbacks in the said process, which includes the use of anionic resin to prepare Isavuconazonium chloride hydrochloride, consequently it requires multiple time lyophilization, which makes the said prior art process industrially, not feasible.
The inventors of the present invention surprisingly found that Isavuconazonium or a pharmaceutically acceptable salt thereof in yield and purity could be prepared by using substantially pure intermediates in suitable solvent.
Thus, an object of the present invention is to provide simple, cost effective and industrially feasible processes for manufacture of isavuconazonium sulfate. Inventors of the present invention surprisingly found that isavuconazonium sulfate prepared from isavuconazonium iodide hydrochloride, provides enhanced yield as well as purity.
The process of the present invention is depicted in the following scheme:

Formula I
Formula-IA
The present invention is further illustrated by the following example, which does not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present application.
Examples
Example-1: Synthesis of l-[[N-methyl-N-3-[(t-butoxycarbonylmethylamino) acetoxymethyl]pyridin-2-yl]carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3 – [4-(4-cyanophenyl)thiazol-2-yl]butyl] – 1 H-[ 1 ,2,4] -triazo-4-ium iodide
Isavuconazole (20 g) and [N-methyl-N-3((tert-butoxycarbonylmethylamino)acetoxy methyl)pyridine-2-yl]carbamic acid 1 -chloro-ethyl ester (24.7 g) were dissolved in acetonitrile (200ml). The reaction mixture was stirred to add potassium iodide (9.9 g). The reaction mixture was stirred at 47-50°C for 10-13 hour. The reaction mixture was cooled to room temperature. The reaction mass was filtered through celite bed and washed acetonitrile. Residue was concentrated under reduced pressure to give the crude solid product (47.7 g). The crude product was purified by column chromatography to get its pure iodide form (36.5 g).
Yield: 84.5 %
HPLC Purity: 87%
Mass: m/z 817.4 (M- 1)+
Example-2: Synthesis of l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide hydrochloride
l-[[N-methyl-N-3-[(t-butoxycarbonylmethylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide (36.5 g) was dissolved in ethyl acetate (600 ml). The reaction mixture was cooled to -5 to 0 °C. The ethyl acetate hydrochloride (150 ml) solution was added to reaction mixture. The reaction mixture was stirred for 4-5 hours at room temperature. The reaction mixture was filtered and obtained solid residue washed with ethyl acetate. The solid dried under vacuum at room temperature for 20-24 hrs to give 32.0 gm solid.
Yield: 93 %
HPLC Purity: 86%
Mass: m/z 717.3 (M-HC1- 1)
Example-3: Preparation of Strong anion exchange resin (Sulfate).
Indion GS-300 was treated with aqueous sulfate anion solution and then washed with DM water. It is directly used for sulfate salt.
Example-4: Synthesis of l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium Sulfate
Dissolved 10.0 g l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide hydrochloride in 200 ml deminerahzed water and 30 ml methanol. The solution was cooled to about 0 to 5°C. The strong anion exchange resin (sulfate) was added to the cooled solution. The reaction mixture was stirred to about 60-80 minutes. The reaction was filtered and washed with 50ml of demineralized water and methylene chloride. The aqueous layer was lyophilized to obtain
(8.0 g) white solid.
Yield: 93 %
HPLC Purity: > 90%
Mass: m/z 717.4 (M- HS04) +

////////WOCKHARDT, WO 2016016766, ISAVUCONAZONIUM SULPHATE, NEW PATENT
LUPIN, SOFOSBUVIR, NEW PATENT, WO 2016016865
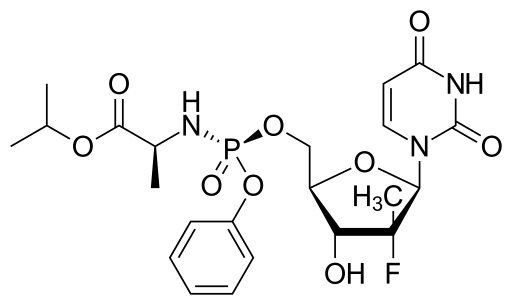
![]()
(WO2016016865) A PROCESS FOR THE PREPARATION OF NUCLEOSIDE PHOSPHORAMIDATE
LUPIN LIMITED [IN/IN]; 159 CST Road, Kalina, Santacruz (East), State of Maharashtra, Mumbai 400 098 (IN)
ROY, Bhairab, Nath; (IN).
SINGH, Girij, Pal; (IN).
SHRIVASTAVA, Dhananjai; (IN).
MEHARE, Kishor, Gulabrao; (IN).
MALIK, Vineet; (IN).
DEOKAR, Sharad, Chandrabhan; (IN).
DANGE, Abhijeet, Avinash; (IN)
The present invention pertains to process for preparing nucleoside phosphoramidates and their intermediates. Phosphoramidates are inhibitors of RNA-dependent RNA viral replication and are useful as inhibitors of HCV NS5B polymerase, as inhibitors of HCV replication and for treatment of hepatitis C infection in mammals. One of the recently approved phosphoramidate by USFDA is Sofosbuvir [1190307-88-0]. Sofosbuvir is a component of the first all-oral, interferon-free regimen approved for treating chronic hepatitis C. The present invention provides novel intermediate, its process for preparation and use for the preparation of Sofosbuvir. The present invention also gives one pot process for preparation of Sofosbuvir.
Hepatitis C virus (HCV) infection is a major health problem that leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals. There are limited treatment options for individuals infected with hepatitis C virus. The current approved therapeutic option is the use of immunotherapy with recombinant interferon- [alpha] alone or in combination with the nucleoside analog ribavirin.
US 7964580 (‘580) is directed towards novel nucleoside phosphoramidate prodrug for the treatment of hepatitis C virus infection.
US’580 patent claims Sofosbuvir and rocess for preparation of Sofosbuvir of Formula 1.

Formula 1
Process for preparation of Sofosbuvir as per US ‘580 patent involve reaction of compound of Formula 4″ with a nucleoside 5’

Compound 4″ nucleoside 5′
Wherein X’ is a leaving group, such as CI, Br, I, tosylate, mesylate, trifluoroacetate, trifluroslfonate, pentafluorophenoxide, p-nitro-phenoxide.
Objects of the invention
The object of the present invention is to provide a novel intermediate of Formula 2

Formula 2
wherein X’ is a leaving group selected from 1-hydroxybenzotriazole, 5-(Difluoromethoxy)-lH-benzimidazole-2-thiol, 2-Mercapto-5-methoxybenzimidazole, cyanuric acid, 2-oxazolidinone, 2-Hydroxy Pyridine. The above leaving group can be optionally substituted with n-alkyl, branched alkyl, substituted alkyl; cycloalkyl; halogen; nitro; or aryl, which includes, but not limited to, phenyl or naphthyl, where phenyl or naphthyl are further optionally substituted with at least one of Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 alkoxy, F, CI, Br, I, nitro, cyano, Ci-C6 haloalkyl, -N(Rr)2, Ci-C6 acylamino, -NHS02Ci-C6 alkyl, -S02N(Rr)2, COR1″, and -S02Ci-C6 alkyl; (Rr is independently hydrogen or alkyl, which includes, but is not limited to, Ci-C2o alkyl, Ci-Cio alkyl, or Ci-C6 alkyl, R1” is -OR1 or -N(Rr)2).
Another object of the present invention is to provide a process to prepare the intermediate of Formula 2.
Another object of the present invention is use of the intermediate of Formula 2 in the preparation of Sofosbuvir of Formula 1.

Formula 1

Example 1:
Process for the preparation of S-oxazolidinone derivative of Formula 2

Step-1 Preparation of phosphorochloridate solution:
Dichloromethane (DCM 400ml) was charged in round bottom flask flushed with nitrogen. Phenyl phosphodichloridate (18.30ml) was added in one portion in the flask. The flask was cooled to -60°-70°C with a dry ice-acetone bath. Solution of L-alanine isopropyl ester hydrochloride (20.6gm)) in DCM (50ml) was added to the reaction flask. To this was added a solution of triethylamine (11.20ml) in MDC (100 ml) was added over a course of 60 minutes, while maintaining internal temperature below -70 °C throughout the addition. After completion of reaction, temperature of reaction mass was raised to room temperature.
100ml THF was charged in another round bottom flask flushed with nitrogen followed by the addition of S-4-phenyloxazolidnone (lOgm). Triethyl-amine (11.2ml) & LiCl (2.85gm) were added to the above flask. The reaction mass was stirred for 15-30 min at room temperature and was cooled to 0-5 °C. Phosphorochloridate solution from step-1 was added drop- wise to the reaction flask in 15-45 min maintaining reaction temperature at 0-5 °C. The reaction mass was stirred for 30-60min at 0°-5°C. The reaction progress was monitored on thin layer chromatography. After completion of the reaction, the reaction temperature was raised to room temperature. Agitation was resumed for an additional 30min. The reaction mass was filtered and concentrated under reduced pressure. To this was added diisopropyl ether (400ml) and aqueous saturated ammonium chloride solution and reaction mass was stirred for 10-15 minutes. Organic layer was separated and was washed with water (100ml) & dried over sodium sulfate and concentrated under vacuum. Cyclohexane (50ml) was charged to the obtained oily mass and reaction mass was stirred till solid precipitated out. Solid was filtered and washed with cyclohexane and dried under vacuum (8.80gm MP 56.5°-56.6°C). The obtained product was characterized by mass, NMR & IR. 1H NMR (DMSO-d6) δ 1.142 -1.18
(m, 9H), 3.85-3.92 (m, 1H), 4.72-4.89(m, 2H), 5.31-5.32(d, 1H), 6.25-6.3 (m, 1H), 6.95-7.31 (m, 10H); MS, m/e 433 (M+l) +
Example 2: Process for the preparation of 2-hydroxy pyridine derivatives of formula 2:

Anhydrous dichloromethane (DCM) 700ml was charged in round bottom flask flushed with nitrogen. The flask was cooled to -60° to -70°C in a dry ice acetone bath. Phenyl phosphodichloridate (76.04 gm) was added in one portion in the flask at -65°C. Solution of L-alanine isopropyl ester hydrochloride (60.56 gm) in DCM (50 ml) was added to the reaction mass. Solution of triethylamine (72.44gm) in DCM (50ml) was added to the reaction mass over a course of 60 minutes, while maintaining internal temperature below -70°C throughout the addition. The resulting white slurry was agitated for additional 60 minutes. Then the temperature of reaction mass was raised to room temperature. Reaction mass was stirred for 60 min & TLC was checked. Reaction mass was filtered and rinsed with anhydrous dichloromethane (2 XI 00 mL). The filtrate was concentrate under vacuum to 20 V and reaction mass was filtered, washed with DCM (15ml). The filtrate was transferred to RBF. The reaction mass was cooled to 0°-10°C. A solution of 2-hydroxy-3-nitro-5- (trifluoromethyl) pyridine (15.gm) in DCM (100ml) & triethyl amine (21.89gm) was added to the reaction mass. Temperature of reaction mass was raised to 20-30°C. Reaction mass was stirred overnight. Reaction was monitored using TLC. After completion, the reaction mass was filtered and washed with DCM (30ml). Filtrate was washed with water (150 ml x 2). Organic layer was concentrated under vacuum and degased. Diisopropyl ether (200ml) was charged to reaction mass and reaction mass was stirred for 15 minutes , filtered and washed with methyl ter-butyl ether (MTBE 30ml). Filtrate was concentrated under vacuum and dried. (8.68gm, MP-125.5°-131.5°C). Obtained compound was characterized by Mass, NMR & IR. 1H NMR (DMSO-d6) δ 1.07 -1.27 (m, 9H), 4.04-4. l l(m, 1H), 4.73-4.79(m, 1H), 6.76-7.43 (m, 5H), 9.00-9.02 (d, 2H); MS, m/e 478 (M+l) +; FTIR, 1203, 1409, 1580, 1732, 3217.
Other 2-hydroxy pyridine derivatives of Formula 2 were prepared by following the process disclosed in example 2-
2-Hydroxy-5-fluoropyridine derivative of Formula 2;-1H NMR (DMSO-d6) δ 1.09 -1.23 (m, 9H), 3.02-3.06 (m, lH), 3.85-4.01 (m,lH), 4.79-4.87(m, 1H), 6.4-6.52 (m,lH), 7.10-7.89 (m,6H); MS, m/e 383 (M+l) +,
2-Hydroxy-5-nitropyridine derivative of Formula 2:- 1H NMR (DMSO-d6) δ 1.06 -1.22 (m, 9H),4.0-4.02 (m,lH), 4.7-4.8(m,lH), 6.5-6.6 (m,lH),7.12-7.42 (m,6H),8.66-8.68 (d, lH),9.07-9.13(d,lH); MS, m/e 410 (M+l) +
2-Hydroxy-3, 5-dinitropyridine derivative of Formula 2:- 1H NMR (DMSO-d6) δ 1.11 -1.24 (m, 9H), 3.04-3.09(m,lH), 4.8-4.86(m,lH), 7.09-7.39 (m,5H),8.97-9.06 (d,2H)
Example 3: Process for the preparation of Sofosbuvir by coupling of isopropyl(((3-nitro-5-(trifluromethyl)pyridin-2-yl)oxy)phenoxy)phosphoryl-L-alaninate with 1-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-3-methyltetrahydrofuran-2-yl)pyrimidine-2,4(lH,3H)-dione :

To a solution of l-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-3-methyltetrahydrofuran-2-yl)pyrimidine-2,4(lH,3H)-dione (0.2gm) in THF (4 ml), tert- butylmagnesium chloride (0.80ml, 1.7 M solution in THF) was added dropwise at room temperature and reaction mass was stirred for 30 minutes. A solution of pyridine derivative from example 2 (0.36gm) in THF (4ml) was added dropwise to the reaction mass at room temperature. Completion of reaction was monitored using TLC. After completion of reaction, reaction mass was quenched by using saturated ammonium chloride solution (10ml). Reaction mass was extracted with ethyl acetate (50ml). Organic layer was separated, dried over magnesium sulfate and concentrated under vacuum. The resulting residue was purified by column chromatography on silica gel & obtained solid product was characterized. MS, m/e 530.2 (M+l) +.
/////////LUPIN, SOFOSBUVIR, NEW PATENT, WO 2016016865
Canagliflozin , New patent, WO 2016016774, SUN PHARMACEUTICAL INDUSTRIES LIMITED


WO2016016774, CRYSTALLINE FORMS OF CANAGLIFLOZIN
SUN PHARMACEUTICAL INDUSTRIES LIMITED [IN/IN]; Sun House, Plot No. 201 B/1 Western Express Highway Goregaon (E) Mumbai, Maharashtra 400 063 (IN)
SANTRA, Ramkinkar; (IN).
NAGDA, Devendra, Prakash; (IN).
THAIMATTAM, Ram; (IN).
ARYAN, Satish, Kumar; (IN).
SINGH, Tarun, Kumar; (IN).
PRASAD, Mohan; (IN).
GANGULY, Somenath; (IN).
WADHWA, Deepika; (IN)
The present invention relates to crystalline forms of canagliflozin, processes for their preparation, and their use for the treatment of type 2 diabetes mellitus. A crystalline Form R1of canagliflozin emihydrate. The crystalline Form R1 of canagliflozin hemihydrate of claim 1, characterized by an X-ray powder diffraction peaks having d-spacing values at about 3.1, 3.7, 4.6, and 8.9 A

The present invention relates to crystalline forms of canagliflozin, processes for their preparation, and their use for the treatment of type 2 diabetes mellitus.
Canagliflozin hemihydrate, chemically designated as (l<S)-l,5-anhydro-l-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-D-glucitol hemihydrate, is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. Its chemical structure is represented by Formula I.

Formula I
U.S. Patent Nos. 7,943,582 and 8,513,202 disclose crystalline forms of canagliflozin hemihydrate.
PCT Publication No. WO 2009/035969 discloses a crystalline form of
canagliflozin, designated as I-S.
PCT Publication No. WO 2013/064909 discloses crystalline complexes of canagliflozin with L-proline, D-proline, and L-phenylalanine, and the processes for their preparation.
PCT Publication No. WO 2014/180872 discloses crystalline non-stoichiometric hydrates of canagliflozin (HxA and HxB), and the process for their preparation.
PCT Publication No. WO 2015/071761 discloses crystalline Forms B, C, and D of canagliflozin.
Chinese Publication Nos. CN 103980262, CN 103936726, CN 103936725, CN 103980261, CN 103641822, CN 104230907, CN 104447722, CN 104447721, and CN 104130246 disclose different crystalline polymorphs of canagliflozin.
In the pharmaceutical industry, there is a constant need to identify critical physicochemical parameters of a drug substance such as novel salts, polymorphic forms, and co-crystals, that affect the drug’s performance, solubility, and stability, and which may play a key role in determining the drug’s market acceptance and success.
The discovery of new forms of a drug substance may improve desirable processing properties of the drug, such as ease of handling, storage stability, and ease of purification. Accordingly, the present invention provides novel crystalline forms of canagliflozin having enhanced stability over known crystalline forms of canagliflozin.

EXAMPLES
Example 1 : Preparation of a crystalline Form Rl of canagliflozin hemihydrate
Amorphous canagliflozin (5 g) was suspended in an aqueous solution of sodium formate (80 mL of a solution prepared by dissolving 137.7 g of sodium formate in 180 mL of de-ionized water). The suspension was stirred at room temperature for 20 hours to obtain a reaction mixture. De-ionized water (100 mL) was added to the reaction mixture, and then the reaction mixture was stirred for 1.5 hours. De-ionized water (50 mL) was added to the reaction mixture, and then the reaction mixture was stirred for 30 minutes. The reaction mixture was filtered, then washed with de-ionized water (300 mL), and then dried under vacuum for 12 hours to obtain a solid. The solid was further dried under vacuum at 60°C for 6 hours.
Yield: 4.71 g
Example 2: Preparation of a crystalline Form R2 of canagliflozin monohydrate
Amorphous canagliflozin (5 g) was suspended in an aqueous solution of sodium formate (80 mL of a solution prepared by dissolving 137.7 g of sodium formate in 180 mL of de-ionized water). The suspension was stirred at room temperature for 20 hours to obtain a reaction mixture. De-ionized water (100 mL) was added to the reaction mixture, and then the reaction mixture was stirred for 1.5 hours. De-ionized water (50 mL) was added to the reaction mixture, and then the reaction mixture was stirred for 30 minutes. The reaction mixture was filtered, then washed with de-ionized water (300 mL), and then dried under vacuum for 12 hours at room temperature.
Yield: 4.71 g
Example 3 : Preparation of a crystalline Form R2 of canagliflozin monohydrate
Canagliflozin hemihydrate (0.15 g; Form Rl obtained as per Example 1) was suspended in de-ionized water (3 mL). The suspension was stirred at room temperature for 24 hours. The reaction mixture was filtered, then dried at room temperature under vacuum for 5 hours.
Yield: 0.143 g
Example 4: Preparation of a crystalline Form R3 of canagliflozin hydrate
Amorphous canagliflozin (100 g) was suspended in an aqueous solution of sodium formate (1224 g of sodium formate in 1600 mL of de-ionized water). The suspension was stirred at room temperature for 20 hours to obtain a reaction mixture. De-ionized water
(2000 mL) was added to the reaction mixture, and then the reaction mixture was stirred for one hour. De-ionized water (1000 mL) was added to the reaction mixture, and then the reaction mixture was stirred for another one hour. The reaction mixture was filtered, then washed with de-ionized water (6000 mL), and then dried under vacuum for 30 minutes to obtain a solid. The solid was then dried under vacuum at 30°C to 35°C until a water content of 8% to 16% was attained.
Yield: 100 g

./////////////Canagliflozin , New patent, WO 2016016774, SUN PHARMACEUTICAL INDUSTRIES LIMITED
WO 2016012938, New patent, LINACLOTIDE, DR. REDDY’S LABORATORIES LIMITED,
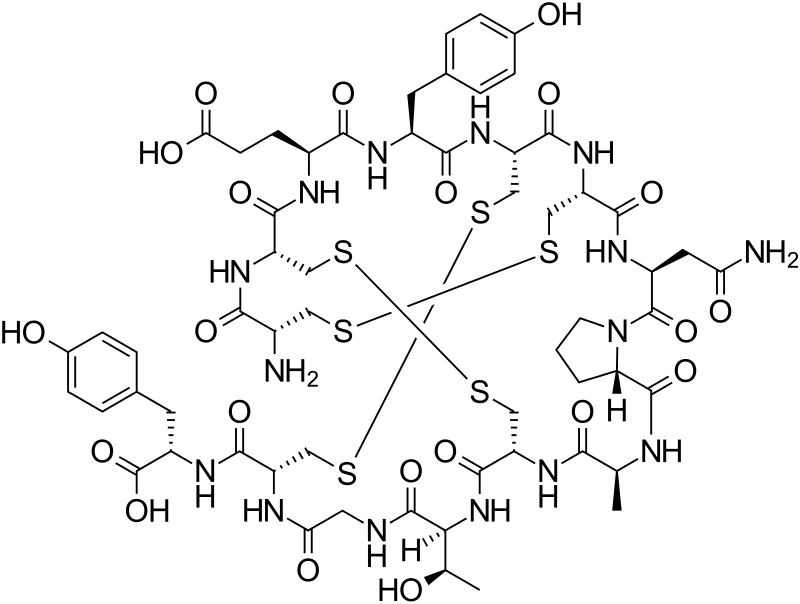
WO2016012938, IMPROVED PROCESS FOR PREPARATION OF AMORPHOUS LINACLOTIDE
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No 3, Banjara Hills, Telangana, INDIA Hyderabad 500034 (IN)
KALITA, Dipak; (IN).
NIVRUTTI, Ramrao Jogdand; (IN).
BALAKUMARAN, Kesavan; (IN).
DESHMUKH, Shivshankar; (IN).
VUTUKURU, Naga Chandra Sekhar; (IN).
KASINA, Vara Prasad; (IN).
NALAMOTHU, Sivannarayana; (IN).
VILVA, Mohan Sundaram; (IN).
KHAN, Rashid Abdul Rehman; (IN).
TIRUMALAREDDY, Ramreddy; (IN).
MUSTOORI, Sairam; (IN)
![]()
The present application relates to an improved process for the formation of disulfide bonds in linaclotide. The present application also relates to an improved process for the purification of linaclotide.
The present application relates to an improved process for the preparation of amorphous linaclotide. Specifically, the present application relates to an improved process for the formation of disulfide bonds in linaclotide. The present application further relates to a purification process for the preparation of amorphous linaclotide.
INTRODUCTION
Linaclotide is a 14-residue peptide which is an agonist of the guanylate cyclase type-C receptor. Linaclotide may be used for the treatment of chronic constipation and irritable bowel syndrome. Structurally, linaclotide has three disulfide bonds and they are present between Cys1-Cys6, Cys2-Cys-10 and Cys5-Cys13. The structure of linaclotide is shown below:
1 2 3 4 5 6 7 8- 9 10 11 12 13 14

Benitez et al. Peptide Science, 2010, Vol. 96, No. 1 , 69-80 discloses a process for the preparation of linaclotide. The process involves the use of 2-chlorotrityl (CTC) resin and 9-fluorenylmethoxycarbonyl (Fmoc) chemistry. The Cys residues are protected by Trt (trityl) group. The amino acids are coupled to one another using 3 equivalents of 1 -[bis(dimethylamino)methylene]-6-chloro-1 H-benzotriazolium hexafluorophosphate 3-oxide (HCTU) as coupling agent and 6 equivalents of diisoprpylethylamine (DIEA) as base in dimethylformamide (DMF). The Fmoc group is removed using piperidine-DMF (1 :4). The Cys residues are incorporated using 3 equivalents of Ν,Ν’-diisopropylcarbodiimide (DIPCDI) as coupling agent and 3 equivalents of 1 -hydroxybenzotriazole (HOBt) as an activating agent. After the elongation of the peptide chain, the peptide was cleaved from the solid support (CTC resin) by first treating with 1 % trifluoroacetic acid (TFA) and then with a mixture of TFA, triisoprpylsilane (TIS) and water in the ratio of 95:2.5:2.5. The disulfide bonds are prepared by subjecting the linear peptide to air oxidation in sodium dihydrogen phosphate (100 mM) and guanidine hydrochloride buffer (2 mM).
US2010/261877A1 discloses a process for purification of linaclotide. The process involves first purification of crude peptide by reverse-phase chromatographic purification followed by concentrating the purified pools and dissolving the purified linaclotide in aqueous-isopropanol or aqueous-ethanol and spray-drying the solution to afford pure Linaclotide.
The synthesis of a peptide containing disulfide bridges is difficult for two main reasons; one is potential risk of racemization during the formation of linear chain and the other is mis-folding of the disulfide bridges. Hence, there is a need in the art to a cost-effective process for the preparation of pure linaclotide.
EXAMPLES
Example 1 : Preparation of Crude Linaclotide using polyvinyl polymer bound complex of sulfur trioxide-pyridine
The linear chain of peptide of formula (I) (0.1 g) and polyvinyl polymer bound complex of sulfur trioxide-pyridine (0.062 g) was charged in water (100 mL). The pH of the reaction mass was adjusted to 8.5 to 9 by addition of ammonium hydroxide. The reaction mass was stirred at 25 °C for 15 hours and trifluoroacetic acid (2 mL) was added to the reaction mass to adjust the pH up to 2-2.5. The reaction mass was stirred for 3 hours at the same temperature to afford crude linaclotide.
HPLC Purity: 59.92%
Example 2: Preparation of Crude Linaclotide using DMSO in water
The pH of water (100 ml_) was adjusted to 9.1 by the addition of aqueous ammonia. DMSO (1 ml_) and linear chain of peptide of formula (I) (100 mg) were charged. The reaction mass was stirred for 17 hours at 25 °C and acidified with trifluoroacetic acid to pH 1 .9 and stirred for 8 hours at the same temperature to afford crude linaclotide.
HPLC Purity: 57%
Example 3: Preparation of Crude Linaclotide using DMSO in water
The pH of water (1500 ml_) was adjusted to 9 by the addition of aqueous ammonia. DMSO (15 ml_) and linear chain of peptide of formula (I) (15 g) were charged. The reaction mass was stirred for 17 hours at 25 °C and acidified with acetic acid to pH 1 .9 and stirred for 8 hours at the same temperature to obtain crude linaclotide.
HPLC Purity: 46.02%
Example 4: Preparation of Crude Linaclotide in water
To a mixture of water (1900 mL) and ammonium sulfate (26.4 g), ammonium hydroxide was added drop wise to adjust the pH up to 8.5. Linear chain of peptide of formula (I) (26.4 g) was added and the reaction mass was stirred for 8 hours at 25 °C. Trifluoroacetic acid (20 mL) was added drop wise and the reaction mixture was stirred for 15 hours at 25 °C to afford crude linaclotide.
HPLC Purity: 63.38%
Example 5: Preparation of Crude Linaclotide using a complex of pyridine-sulfur trioxide
Linear chain of peptide of formula (I) (0.2 g) was added to water (250 mL) and the pH of the reaction mass was adjusted to 8.91 by the drop wise addition of aqueous ammonia. A complex of pyridine-sulfur trioxide (0.124 g) was added to the reaction mass and stirred for 16 hours at 25 °C. Another lot of complex of pyridine-sulfur trioxide (0.124 g) was added to the reaction mass and stirred for 5 hours at 25 °C to afford crude linaclotide.
Example 6: Preparation of Crude Linaclotide using guanidine hydrochloride
To a solution of sodium bicarbonate (0.89 g) in water (100 mL), cysteine (0.363 g), cysteine (0.072 g) and guanidine hydrochloride (9.50 g) were charged. Acetonitrile (15 mL) and linear chain of peptide of formula (I) (0.1 g) was added to the reaction mass.
The reaction mass was stirred for 3 hours at 25 °C and trifluoroacetic acid (2 mL) was added. The reaction mass was stirred for 18 hours at the same temperature. Another lot of trifluoroacetic acid (2 mL) was added to the reaction mass and stirred for 18 hours at the same temperature to afford crude linaclotide.
Example 7: Preparation of Crude Linaclotide using Clear-OX™
Pre-conditioned Clear-Ox™ (0.5 g) was added to a solution of ammonium sulfate (1 .32 g) in water (100 mL) of pH 8.5, adjusted by addition of ammonium hydroxide. The linear chain of peptide of formula (I) (0.1 g) was added to the reaction mass and stirred for 3 hours at 25 °C. Another lot of Pre-conditioned Clear-Ox™ (0.5 g) was added to the reaction mass and stirred for 1 .30 hours. Trifluoroacetic acid (2 mL) was added to the reaction mass and stirred for 16 hours at the same temperature to afford crude linaclotide.
HPLC Purity: 67.5%
Example 8: Preparation of Crude Linaclotide using reduced Glutathione
To a mixture of ammonium sulphate (5.28 g) in water (400 mL) and isopropyl alcohol (400 mL), reduced glutathione (0.248 g) was added and the pH was adjusted to 8.5 by using aqueous ammonia. The linear chain of peptide of formula (I) (0.81 g) was added to the reaction mixture and stirred at ambient temperature for 17 hours. Isopropyl alcohol was evaporated under vacuum to afford crude linaclotide.
HPLC Purity: 69.56%%
Example 9: Preparation of Crude Linaclotide using DMSO and air bubbling
To a mixture of water (95 mL) and ammonium sulfate (1 .32 g), ammonium hydroxide was added drop wise to adjust the pH up to 8.5. Linear chain of peptide of formula (I) (0.1 g) and DMSO (5 mL) was added and the reaction mass was stirred for 20 hours at 25 °C with continuous air bubbling. Trifluoroacetic acid (2 mL) was added to the reaction mass and stirred for 19 hours with continuous air bubbling at the same temperature to afford the title product.
HPLC Purity: 59.1 1 %
Example 10: Preparation of Crude Linaclotide using solid supported TEMPO
To a mixture of water (100 mL) and silica bound TEMPO (0.01 g), linear chain of peptide of formula (I) (0.1 g) and sodium hypochlorite solution (1 mL) were added and the reaction mass was stirred 18 hours at 25 °C. Another lot of sodium hypochlorite solution (0.5 mL) was added to the reaction mass and stirred for further 7 hours at the same temperature to afford title product.
HPLC Purity: 42.70%………………see more in patent
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
L-Cysteinyl-L-cysteinyl-L-glutamyl-L-tyrosyl-L-cysteinyl-L-cysteinyl-L-asparaginyl-L-prolyl-L-alanyl-L-cysteinyl-L-threonylglycyl-L-cysteinyl-L-tyrosine cyclo(1-6),(2-10),(5-13)-tris(disulfide)
|
|
| Clinical data | |
| Trade names | Linzess |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 851199-59-2 |
| ATC code | A06AX04 |
| PubChem | CID 16158208 |
| IUPHAR/BPS | 5017 |
| ChemSpider | 17314504 |
| UNII | N0TXR0XR5X |
| KEGG | D09355 |
| Chemical data | |
| Formula | C59H79N15O21S6 |
| Molar mass | 1526.74 g/mol |
///////WO 2016012938, DR. REDDY’S LABORATORIES LIMITED , Telangana, INDIA , Hyderabad, LINACLOTIDE, new patent
smiles O=C(O)[C@@H](NC(=O)[C@H]4NC(=O)CNC(=O)[C@@H](NC(=O)[C@H]2NC(=O)[C@@H](NC(=O)[C@H]5N(C(=O)[C@@H](NC(=O)[C@H]1NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@@H](N)CSSC1)CSSC2)CCC(=O)O)Cc3ccc(O)cc3)CSSC4)CC(=O)N)CCC5)C)[C@H](O)C)Cc6ccc(O)cc6
WO 2016012539, Tadalafil , New patent, KRKA, D.D., NOVO MESTO

WO 2016012539, A PROCESS FOR THE PREPARATION OF CGMP-PHOSPHODIESTERASE INHIBITOR AND ORAL PHARMACEUTICAL FORMULATION COMPRISING TADALAFIL CO-PRECIPITATES
KRKA, D.D., NOVO MESTO [SI/SI]; Smarjeska cesta 6 8000 Novo mesto (SI)
BARIC, Matej; (SI).
BENKIC, Primoz; (SI).
BOMBEK, Sergeja; (SI).
KRASOVEC, Dusan; (SI).
SKRABANJA, Vida; (SI).
VRECER, Franc; (SI).
BUKOVEC, Polona; (SI).
HUDOVORNIK, Grega; (SI).
KROSELJ, Vesna; (SI)
![]()
The present Invention relates to an improved process for preparation of tadalafil and crystallization and/or purification thereof, wherein the processes are conducted at increased pressure. The invention relates also to a process for preparation of tadalafil co-precipitates and to a solid pharmaceutical composition comprising tadalafil co-precipitates and at least one water soluble diluent and/or water insoluble non-swellable diluent, wherein the composition is substantially free of water insoluble swellable diluents
The present invention relates to a process for the preparation of CGMP-phosphodiesterase inhibitor, particularly tadalafil, a method for production co-precipitate thereof and to solid oral pharmaceutical formulations comprising tadalafil co-precipitate.
Tadalafil, chemically known as (6R-trans)-6-(1,3-benzodioxol-5-il)-2,3,6,7,12,12a-hexahydro-2-methyl-pyrazino.1′, 2′:1,6]pyrido[3,4-b]indole-1,4-dione, is a potent and selective inhibitor of the cyclic guanosine monophosphate (cGMP) – specific phosphodiesterase enzyme PDE5. It is shown below as structural formula I:

Tadalafil is marketed under the tradename CIALIS* and is used for the treatment of erectile dysfunction. The product is available as a film-coated tablet for oral administration containing 2.5, 5, 10 and 20 mg of active ingredient and the following inactive ingredients: lactose monohydrate, hydroxypropylcellulose, sodium lauryl sulfate, croscarmellose sodium, microcrystaliine cellulose, magnesium stearate, hypromellose, triacetin, titanium dioxide (E171), iron oxide (E172) and talc.
Tadalafil is practically insoluble in water and very slightly soluble in organic solvent such as ethanol, methanol and acetone.
Problems associated with low solubility of tadalafil in ethanol and most of other organic solvents resulted in the need of large quantities of solvents required to perform synthesis and crystallization of tadalafil at industrial scale, which have unwanted technological, environmental and economical impact.
US Patent No. 5 859 006 describes the synthesis of the tadalafil and its intermediate (A) which involves reacting D-tryptophan methyl ester with a piperonal in the presence of dichloromethane and trifluoroacetic acid which provides a mixture of desired cis and undesired trans isomer of intermediate A with poor selectivity. The isomers are further separated by column chromatography. The cis isomer is further reacted with chloroacetyl chloride in chloroform, providing another intermediate of tadalafil (B) which reacted with methylamine to give tadalafil of formula (1) in methanol slurry requiring an additional purification step by flash chromatography.
An improved process in the synthesis of tadalafil via modified Pictet-Spengler reaction is described in WO 04/011463 in which D-tryptophan methyl ester hydrochloride and piperonal are condensed in anhydrous isopropyl alcohol to provide hydrochloride of intermediate A. After isolation of desirable cis isomer, the product is further reacted with chloroacetyl chloride and then with methylamine in THF to give tadalafil.
Therefore there still exists a need for an improved process for a synthesis and purification of tadalafil, which would overcome the disadvantages of the prior art processes.
Low solubility of tadalafil in aqueous solutions is further disadvantageous because in vivo absorption is typically dissolution rate-limited which may result in poor bioavailability of the drug. Different approaches in the processes of preparation of pharmaceutical compositions have been applied to overcome the poor solubility.
For example, EP 1 200 092 Bl describes a pharmaceutical composition of free drug particulate form of tadalafil wherein at least 90% of the particles have a particle size of less than about 40 μm as well as composition comprising tadalafil, wherein the compound is present as solid particles not embedded in polymeric co-precipitate. Apparently, preferably at least 90% of the particles have a particle size of less than 10 μm. The technological drawback of such small particles is possible chargeability and secondary agglomeration due to increased surface energy which can cause problems during the micronization and further processing.
WO 2008/134557 describes another approach to overcome the low-solubility problem by pharmaceutical composition comprising starch and tadalafil characterized by particle size having d(90) greater than 40 μm wherein the weight ratio of starch to tadalafil is 4.5 to 1 or greater. Apparently, the preferred ratio is at least 15 to 1.
Yet another approach to overcome the low-solubility problem is to use a “co-precipitate” of tadalafil and a carrier or excipient. For example, EP 828 479 Bl describes a solvent based process wherein tadalafil and a carrier are co-precipitated with a medium in which the tadalafil and carrier are substantially insoluble. EP 828 479 describes a solvent based process wherein tadalafil and hydroxypropyl methylcellulose phthalate are co-precipitated in weakly acidic medium from a combination of non-aqueous water miscible solvent and water. However, pharmaceutical composition prepared according to EP 828479 exhibit deviations in release rate of tadalafil which was due to poor reproducibility of a process for preparation of co-precipitate. It was found that precipitation in acidic media causes unwanted degradation of hydroxypropyl methylcellulose phthalate and that precipitation at higher temperatures does not produce desired product.
WO 2008/005039 also describes a solid composite including tadalafil being in intimate contact with a carrier. The carriers include hydrophilic polymers such as povidone, cellulose derivatives, polyethylene glycol and polymethacrylates. The compositions are prepared by combining tadalafil with hydrophilic polymer and removal of the solvent by evaporation.
WO 2010/115886 describes an adsorbate comprising poorly soluble active ingredient with a particulate and/or porous carrier wherein the adsorbate is prepared by using non-polar solvent. Apparently, the solvents used are selected from the group of chlorinated hydrocarbon (dichloromethane or trichloromethane), diisopropylether and hexane, which is also the main drawback of this solution.
Co-precipitates of phosphodiesterase-5-inhibitor and copolymer of different acrylic acid derivatives are described in WO 2011/012217. The procedures described involve the use of tetrahydrofurane.
Poor solubility can also be solved with co-crystals. WO 2010/099323 discloses crystalline molecular complexes of tadalafil with co-former selected from the group of a short to medium chain organic acids, alcohols and amines.
WO 2012/107541 and WO 2012/107092 disclose co-granulate of tadalafil with cyclodextrines.
WO 2014/003677 discloses a pharmaceutical composition comprising solid dispersion particles containing tadalafil and a dispersing component, which composition further comprises a solubilizer.
Based on the above, there is still a need for an improved dosage form containing tadalafil and improved technological process for the preparation thereof.
The process for preparing tadalafil according to a preferred embodiment of the present invention is disclosed in Scheme 1.
Scheme 1

Example 1: Synthesis of tadalafil intermediate B via intermediate A
D-tryptophan methyl ester hydrochloride (9g) and piperonai (6g) was suspended in acetonitrile (60mL). The reaction mixture was stirred and heated at about 105*C for three to five hours in an autoclave. The reaction suspension was cooled to ambient temperature and aqueous solution (60m L) of sodium carbonate (4.1g) was added. The mixture was then cooled in an ice bath and the solution of chloroacetyl chloride (5.1mL) in acetonitrile was slowly added to the reaction mixture. A solid was obtained, filtered and washed twice with aqueous solution of acetonitrile. The crude product was dried, and intermediate B (13.4g) with a purity of 97% (HPLC area%) was obtained.
Example 1A:
D-tryptophan methyl ester hydrochloride (8.2kg) and piperonai (5.1kg) was suspended in acetonitrile (55L). The reaction mixture was stirred and heated at about to 105″C for three hours in the reactor vessel. The reaction suspension was cooled to ambient temperature and aqueous solution (55L) of sodium carbonate (4.8kg) was added. The mixture was then cooled in an ice bath and the solution of chloroacetyl chloride (5.2L) was slowly added to the reaction mixture at 5-10°C. A solid was obtained, centrifuged and washed twice with aqueous solution of acetonitrile (2x 121). The crude product was dried at temperature up to 50″C, and intermediate B (12.3kg) with a purity of 98% (HPLC area%) was obtained.
Comparative example 1:
D-tryptophan methyl ester hydrochloride (9.0g) and piperonai (5.84g) was suspended in acetonitrile (60mL). The reaction mixture was stirred and heated at about to 80-85’C for 15-20 hours in the reactor vessel. The reaction suspension was cooled to 0-10°C. The Intermediate A was then isolated on centrifuge and was dried at temperature up to 60°C.
The isolated dried Intermediate A (12,8g) was charged into reactor and suspended with ethyl acetate. The aqueous solution (60mL) of sodium carbonate (5.3g) was added to precooied suspension of Intermediate A. The chloroacetyl chloride (3.4mL) was slowly added to the above reaction mixture. The solid was obtained, centrifuge and washed twice with water (2x 10mL). The crude product was dried at temperature up to 70°C, and intermediate B (11.8g) with a purity of 99% (HPLC area%) was obtained.
Example 2: Synthesis oftadalafil
Intermediate B (4g) obtained in Example 1 and 40% aqueous methylamine solution (1.6mL) were dissolved in 70% aqueous solution of 2-propanol (120mL) while heating in a closed reaction vessel above the reflux temperature (110-120°C) for two to five hours. The solution was hot filtered and cooled on an ice bath. The precipitated product was filtered and dried. The purity of the product was 99.9% (HPLC area%) and the particle distribution of the product was D(90) of about 144 microns.
Example 2A: Synthesis of tadalaf il
Intermediate B (12.3kg) obtained in Example 1A and 40% aqueous methylamine solution (4.76L) were dissolved in 70% aqueous solution of 2-propanol (402L) while heating in a closed reaction vessel above the reflux temperature (110-120°C) for three hours. The solution was hot filtered and cooled on an ice bath. The precipitated product was filtered and dried. The final product (9.8kg) with a purity of more than 99.99% (HPLC area%) and the particle distribution of the product was D(90) of about 155 microns was obtained.
Comparative example 2:
Intermediate B (10g) obtained in the above comparative example 1 and 31% ethanolic methylamine solution (12.3mL) were suspended in absolute ethanol (150mL). The suspension
was heated up to 55°C for 3 – 6 hours. The suspension was cooled on an ice bath. The product was filtered and dried. The crude product (8.22g) with a purity of more than 99.9% (HPLC area%) was obtained and crystallized from hot DMSO solution. The product Is crystallized with addition of water.
Example 3: Recrystallization of tadalaf il
Tadalafil (700g) (99% purity) was suspended in 70% aqueous solution of 2-propanol (24.6L) and suspension was heated to about 110°C in an autoclave at pressure of 0.31MPa until the material was dissolved. The obtained solution was then hot filtrated and cooled to about 10°C. The isolated tadalafil (660g) has a purity of 99.95% (HPLC area%) and the particle distribution D(90) of about 144 microns.
Example 3A: Recrystallization of tadalafil
Tadalafil (5g) (99% purity) was suspended in 70% aqueous solution of acetone (lOOmL) and suspension was heated to about 90°C in an autoclave at pressure of 0.28MPa until the material was dissolved. The obtained solution was then hot filtrated and cooled to about 10°C. The isolated tadalafil (4.44g) has a purity of 99.99% (HPLC area%).
Example 3B: Recrystallization of tadalafil
Tadalafil (4g) (99% purity) was suspended in 70% aqueous solution of acetonitrile (lOOmL) and suspension was heated to about 85°C in an autoclave at pressure of 0.2MPa until the material was dissolved. The obtained solution was then hot filtrated and cooled to about 10°C. The isolated tadalafil (3g) has a purity of 99.99% (HPLC area%).
Example 3C: Recrystallization of tadalafil
Tadalafil (5g) (99% purity) was suspended in 70% aqueous solution of tetrahydrofuran (60mL) and suspension was heated to about 120″C in an autoclave at pressure of 0.3MPa until the material was dissolved. The obtained solution was then hot filtrated and cooled to about 10°C. The isolated tadalafil has a purity of 99.99% (HPLC area%).
Comparative example 3:
Tadalafil (lg) (99% purity) was suspended in 2-propanol (200mL) and suspension was heated up to reflux temperature until the material was dissolved. The obtained solution was then hot filtrated and cooled to about lO’C. The crystallized tadalafil was centrifuged and dried in an oven at temperature up to 70°C.
Comparative Example 4: Preparation of tadalafil co-precipitate with HPMCP HP-50, Precipitation at higher temperature
Tadalafil (100 g) and hydroxypropyl methylcellulose phthalate (100 g) were dissolved in a mixture of acetone (2430m L) and water (270mL) at reflux temperature. Solution was hot filtered and added to 0.25 M HCI in water (4150mL) at 65°C. Precipitate was collected by vacuum filtration, washed with water and dried in vacuum tray dryer up to 70°C. Dry material was milled by a pin mill. HPLC assay of tadalafil was 48.5 %; average particle size of co-precipitate was 53 μm, specific surface area 2.5 m2/g-
Example 5: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (1 kg) and hydroxypropyl methylcellulose phthalate (1 kg) were dissolved in mixture of acetone (20L) and water (3 L) at 54°C and under pressure O.lMPa. Solution was hot filtered and added to water (42 L) at 2°C. Suspension was heated up to reflux and acetone was distilled off. Tadalafil co-precipitate was collected by pressure filtration and dried in vacuum dryer. Dry material was milled by a pin mill. HPLC assay of tadalafil was 53.5%.
Example 6: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (1 kg) and hydroxypropyl methylcellulose phthalate (1 kg) were dissolved in mixture of acetone (20 L) and water (3 L) at 54°C and under pressure O.lMPa. Solution was hot filtered and added to water (42 L) at 2°C. Suspension was heated up to reflux and acetone was distilled off. Tadalafil co-precipitate was collected by centrifuge and dried in a fluid bed dryer. Dry material was milled by a pin mill. HPLC assay of tadalafil was 52.5 %.
3
Example 7: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (0.786 kg) and hydroxypropyl methylcellulose phthaiate (1.140 kg) were dissolved in a mixture of acetone (24L) and water (2.3 L) at 54°C and under pressure 0.1MPa. Solution was filtered hot and added to water (42 L) at 2°C. Suspension was collected by centrifuge and dried in a vacuum tray dryer up to 70°C. Dry material was milled by a pin mill. HPLC assay of tadalafil was 43.5 %, average particle size of co-precipitate was 49 μm, specific surface area 31.0 m2/g-
Example 8: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (2 g) and hydroxypropyl methylcellulose phthaiate HP 50 (2 g) were dissolved in a mixture of acetone (48.5mL) and water (5.5mL) at reflux temperature. To obtained solution crospovidone (lg) was added. Obtained suspension was co-precipitated in water (83mL) at 2°C. Obtained material was collected with a vacuum filter and dried in vacuum dryer up to 90°C. HPLC assay of tadalafil 39.9%. Yield was 90%.
Example 9: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (2 g) and hydroxypropyl methylcellulose phthaiate HP 50 (2 g) were dissolved in a mixture of acetone (54mL) and methanol (19mL) at reflux temperature. To obtained solution crospovidone (lg) was added. Obtained suspension was co-precipitated in heptane (83mL) at 0°C. Obtained material was collected with a vacuum filter and dried in vacuum dryer up to 50°C. HPLC assay of tadalafil was 36.1 %. Yield was 90%.
Example 10: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (2 g) and hydroxypropyl methylcellulose phthaiate HP 50 (2 g) were dissolved in a mixture of aceton (54mL) and methanol (19mL) at reflux temperature. Obtained solution was co-precipitated in heptane (83mL) at 0°C. Obtained material was collected with a vacuum filter and dried in vacuum dryer up to 50°C. HPLC assay of tadalafil was 36.1 %. Yield was 90%.
Example 11: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadaiafil (1.3 kg) and hydroxypropyl methylcellulose phthalate {1.53 kg) were dissolved in mixture of acetone (32 L) and water (4 L) at 54°C and 1000 mbar. Solution was hot filtered and added to water (54 L) at 2°C. Tadalafil co-precipitate was collected by decanter centrifuge and dried in a vacuum drier. Dry material (2.4kg) was milled in a pin mill. HPLC assay of tadalafil was 48.8 %; average particle size of co-precipitate was 54 μm and specific surface area 26.1 m2/g<
Example 12: Preparation of tadalafil co-precipitate with hydroxypropyl cellulose
Tadalafil (3g) and Klucel ELF (3g) was dissolved in a mixture of acetone (73mL) and water (8mL) at 50°C. Solution was hot filtered and added to 125mL water at 90°C. After that acetone was distilled off at 65°C and suspension was stirred for additional hour. Precipitated material was filtered using preheated filter funnel and dried at 80°C. Yield 3.8 g, HPLC assay was 50.0%.
Example 13: Preparation of tadalafil co-precipitate with hydroxypropyl cellulose
Tadaiafil (3g) and Klucel ELF (3g) was dissolved in a mixture of acetone (73mL) and water (8m L) at 50°C. Solution was hot filtered and added to 125m L water at 90°C with dissolved lactose (14g) at 90°C. After that acetone was distilled off at 65°C and suspension was stirred for additional hour. Precipitated material was filtered using preheated filter funnel and dried at 80°C. Yield 5 g, HPLC assay was 48.8%.
Examples of tablets prepared according to the present Invention
Example Fl: Tablets containing tadalafil co-precipitate with HPMCP HP-50 prepared in accordance with Example 11 with water soluble mannitol and without swellable water insoluble diluents

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with mannitol, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Example F2: Tablets containing tadalafil co-precipitate with HPC prepared in accordance with Example 13 with water soluble mannitol and without swellable water insoluble diluents

Tadalafil co-precipitate with HPC was homogeneously mixed with mannitol, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Example F3: Tablets containing tadalafil co-precipitate with HPMCP with water soluble spray-dried lactose and without swellable water insoluble diluents

Tadaiafil co-precipitate with HPMCP was homogeneously mixed with spray-dried lactose, starch 1500 and sodium lauryi sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets.
Example F4: Tablets containing tadalafil co-precipitate with HPMCP with water insoluble non-swellable anhydrous dibasic calcium phosphate and without swellable water insoluble diluents

Tadalafil co-precipitate with HPMCP was homogeneously mixed with calcium phosphate, croscarmellose sodium and sodium lauryi sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets.
Comparative examples of tablets containing microcrvstalline cellulose
Comparative example F5: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with water soluble mannitol and water insoluble swellable microcrvstalline cellulose as diluent 
Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with mannitol, microcrystalline cellulose, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Comparative example F6: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with water soluble lactose anhydrous and water insoluble swellable microcrystalline cellulose as diluent

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with lactose anhydrous, microcrystalline cellulose, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Comparative example F7: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with water soluble lactose monohydrate and spray dried lactose and water insoluble swellable microcrystalline cellulose as diluent

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with lactose monohydrate, spray dried lactose, microcrystalline cellulose, croscarmeilose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Comparative example F8: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with water insoluble non-swellable calcium phosphate and water insoluble swellable microcrystalline cellulose as diluent

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with calcium phosphate, microcrystalline cellulose, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Comparative example F9: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with only water insoluble swellable microcrystalline cellulose as diluent

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with microcrystalline cellulose, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Comparative example F10: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with water insoluble swellable microcrystalline cellulose and cellactose as diluents

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with microcrystalline cellulose, cellactose, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example F10 is shown in Figure 2, together with dissolution profiles of the same sample, taken after two months at 22°C and 60% RH.
In comparison, dissolution profile of composition according to invention is unaffected by storage at 40°C/75% for one month (Figure 2).
The aforementioned tablet formulations were film-coated with a film-coating dispersion containing:

Figures 1 and 2 show dissolution profiles of tablet formulations comprising tadalafil co-precipitates prepared according to listed examples. Dissolution conditions comprise: basket apparatus (USP I), 100 RPM, 0.1M HCI + 0.2% SDS, 900 mL
![]()
Krka, tovarna zdravil, d.d., Novo mesto


/////////WO 2016012539, KRKA, D.D., NOVO MESTO, tadalafil, new patent
WO 2016011767, New patent, Clopidogrel, SHENZHEN SALUBRIS/ HUIZHOU SALUBRIS


SHENZHEN SALUBRIS PHARMACEUTICALS CO.,LTD [CN/CN]; 37F Main Tower, Lvjing plaza, Che Gong Miao, No. 6009 Shennan Road, Futian District Shenzhen, Guangdong 518040 (CN).
HUIZHOU SALUBRIS PHARMACEUTICALS CO.,LTD. [CN/CN]; No.42, West petrochemical Avenue, West District,Huizhou DayaBay Huizhou, Guangdong 516083 (CN)
LI, Haidong; (CN).
TAN, Duanming; (CN).
WANG, Hai; (CN)
Provided is a preparation method for high purity clopidogrel and salt thereof. In the present method, inorganic acid solution is used to wash an organic phase containing clopidogrel till a specific pH value range is reached; during the post-processing stage, impurities including TTP can be removed from the clopidogrel product. The ensuing refining step can be avoided, thereby simplifying production techniques and ensuring the quality of the clopidogrel product.


//////WO 2016011767, New patent,Clopidogrel, SHENZHEN SALUBRIS, HUIZHOU SALUBRIS
WO 2016014324, New Patent, Omarigliptin, MERCK SHARP & DOHME CORP
 Omarigliptin , MK-3102
Omarigliptin , MK-3102
WO2016014324, PROCESS FOR PREPARING CHIRAL DIPEPTIDYL PEPTIDASE-IV INHIBITORS
MERCK SHARP & DOHME CORP. [US/US]; 126 East Lincoln Avenue Rahway, New Jersey 07065-0907 (US).
CHUNG, John, Y. L.; (US).
PENG, Feng; (US).
CHEN, Yonggang; (US).
KASSIM, Amude Mahmoud; (US).
CHEN, Cheng-yi; (US).
MAUST, Mathew; (US).
MCLAUGHLIN, Mark; (US).
ZACUTO, Michael, J.; (US).
CHEN, Qinghao; (US).
TAN, Lushi; (US).
SONG, Zhiguo Jake; (US).
CAO, Yang; (US).
XU, Feng; (US)
![]()
A process for preparing a compound of structural Formula Ia: comprising Boc deprotection with TFA of, reductive amination of:.
The present invention is directed to a novel process for the preparation of omarigliptin, (2R,35,,5R)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3 -amine, a dipeptidyl peptidase-IV (DPP-4) inhibitor, for the treatment of Type 2 diabetes, and related intermediates.
BACKGROUND OF THE INVENTION
Syntheses of omarigliptin have previously been described in PCT international patent applications numbers WO 2010/056708 and WO2013/003250. The process described in WO 2010/056708 does not result in a favorable yield of the compound of structural Formula la, as it results in a racemic mixture. WO2013/003250 describes the following scheme to make the compound of structural Formula la, an intermediate for synthesizing omarigliptin:

In WO2013/003250, synthesis of the compound of structural Formula la involves using benzenesulfonic acid (BSA) to remove the Boc protecting group of the compound of structural Formula 1, by first forming a BSA salt of the compound of structural Formula la. The BSA salt is then isolated and undergoes reductive amination with Boc -ketone of the compound of structural Formula 7, to produce the compound of structural Formula la, as a 19: 1 diastereomeric mixture. The BSA mediated Boc deprotection requires up to 72 h to reach full conversion.
An alternative process which eliminates the need to isolate the BSA salt of the compound of Formula la and reduces the overall reaction time of the process is desired. The inventors have now discovered a process for making the compound of structural Formula la which eliminates the step of isolating a salt of the compound of structural Formula la and reduces the overall reaction time. The present process also produces an end-of reaction homogeneous solution via reductive amination, which facilitates crystallization of the compound of structural Formula la. The described process also improves the diastereoselectivity, overall yield, cost and cycle time over the process described in WO2013/003250.
WO2013/003250 also describes the Boc deprotection of the compound of Formula la to produce omarigliptin (Formula I) shown below. As described in WO2013/003250, the Boc deprotection of the compound of Formula la involves aging the substrate in aqueous sulfuric acid in DMAc at 30 °C for 15-20 h, then working up with ammonium hydroxide. This work up produces large amounts of poorly soluble ammonium sulfate which co-crystallizes with the desired product. As a result, isolation of the desired product requires a long cycle time for filtration, washing and drying.

Formula I (omarigliptin)
Because the processes described herein use trifluoroacetic acid with or without a co-solvent for the transformation of the compound of Formula la to omarigliptin, which offers good solubility for the compound of Formula la, omarigliptin is achieved with fast reaction kinetics and good purity profiles.

the compound of structural Formula 1 is prepared by the following processes:

reagents
and,

or alternatively

10 R = Ms
X=OAc
SCHEME 3: Synthesis of the Boc Ketone

16 17 18 19
IPA, H2Q ,
1)956
Step 1 : As

A round bottom flask was charged with ligand L (0.829 g), Cu(II) propionate
monohydrate (0.402 g) (or Cu(II) acetate (0.31 g) or CuCl or CuCl2) and EtOH (350 ml) and agitated at room temperature for lh. 2,4-Difluorobenzaldehyde (100.0 g) was added followed by DABCO (2.368 g) (or 2,4-dimethylpiperizine) and the mixture was cooled to -5 – -15 °C. Cold (0°C) nitromethane (190 ml or 215 g) was added slowly to the cold solution and the solution was aged at -5 to -15 °C for 20-24 h and at 0 °C for 2-4h. 5 wt% EDTA»2Na (500 ml) followed by
water (200 mL) and MTBE (1.0 L) was added to the cold solution, and the temperature was raised to 20°C. The layers were separated and the organic layer was washed with additional 5 wt% EDTA»2Na (500 ml), followed by water (50 mL) and brine (250 mL). The organic layer, containing Compound 17, was concentrated to remove nitromethane, then the solvent was switched to THF.
Step 2: Michael-Lactolization – Nitro lactol

To Compound 17 in 2 volumes of THF (258 mL) from Step 1 under 2 and cooling at 0 °C, 1 equivalent of Hunig’s base was added. 1.15 equivalents of acrolein was added over 1 h via syringe pump at 0-5 °C. The reaction was stirred at -10-0 °C overnight. The resulting mixture was used directly in the next step.
Alternatively, the mixture was concentrated at 0-5 °C to remove excess acrolein, then the residue was flushed with acetonitrile until Hunig’s base and water are mostly removed. The residue was taken up in 8 volumes of acetonitrile and used directly in the next step.
Alternatively, at the end of the reaction the mixture was worked up by diluting with MTBE and washing with aqueous citric acid solution, and aqueous NaHCC solution, and the solvent was switched to acetonitrile. Alternatively, the end reaction mixture was taken forward directly to the next step.
Step 3: Dehydration – Nitro dihydropyrans

1.1 Equivalents of TEA was added to the acetonitrile solution of lactol 18 from Step 2 followed by 1.2 equivalents of mesyl chloride and 1.2 equivalents of S-collidine under < +10 °C . The reaction was aged at 10°C for 0.5-1 h. Alternatively, the end of the reaction mixture from Step 2 was cooled to between -20 °C to 0 °C. Two equivalents of S-collidine and 1.4 equivalents of mesyl chloride were then added. The mixture was heated to 36 °C and aged overnight. The mixture was cooled to room temperature. 15 volumes of MTBE was added and the solution was
washed with 3 volumes 10 wt% citric acid and 6 volumes water, 10 volumes water, then 3 volumes of 5% aHC03 solution and 6 volumes water. The organic was concentrated with 20 volumes of MTBE using 10 volumes MTBE. The organic solution was stirred with 20-30 wt% AQUAGUARD for 2 hours at room temperature. The mixture was filtered and washed with 2 volumes of MTBE.
Step 4: Dynamic Kinetic Resolution (DKR) crystallization – rraws-nitro-dihydropyran (19t)

The organic MTBE solution of Step 3 was solvent switched to 2 volumes of IPA and the final volume was -300 mL. 10 Mol% of TEA (or DAB CO or morpholine or DMAP) was added. Then water (1 15 mL) was slowly added over 3 hours. The slurry was filtered, washed with 80/20 IP A/water (2×100 mL) and vacuum dried under N2.
Step 5: Hydroboration/oxidation – Trans-nitro-pyranol

To a vessel charged with /raws-nitro-dihydropyran (10 g), MTBE (100 mL) was added under nitrogen. The mixture was stirred at room temperature to give a clear orange solution. The solution was cooled to +2 °C and borane dimethyl sulfide complex (9.55 ml) was added. The clear solution was aged for 2-5h until >99% conversion by HPLC analysis. The reaction was slowly quenched with water (7.25 ml) keeping at < +9 °C. After the solution was aged at 5°C for 5 min, water (78 mL) was added at < +13 °C. Solid sodium percarbonate (13.26 g, 84 mmol) was added. The suspension was stirred at 5 °C for 15h. The mixture was transferred to a separatory funnel with the aid of 60 mL MTBE and 20 mL water. The mixture was allowed to warm to room temperature. The aqueous phase was back-extracted with 40 mL MTBE. The combined organic phase was washed once with 30 mL half saturated sodium chloride solution, once with 15 mL brine and 15 mL 0.2N HC1, and once with 30 mL half-saturated sodium chloride solution. The organic layer was dried over a2S04. The organic was filtered, washed with 10 mL MTBE and concentrated to an oil. The oil was diluted to 200 mL for a 0.191M solution.
Step 6: Nitro Reduction/Boc protection – Pyranol

A 3 -neck jacketed round bottom flask equipped with overhead stirrier was charged with 0.191M (5R,6S)-5-nitro-pyran-3-ol (119 ml) (Compound 20) in ethanol and ethanol (32 ml). The solution was cooled to 1 1-12 °C. Cold 6N HC1 (19.55 ml, 1 17 mmol) was added at <
+17°C. Zinc dust (12.93 g) was added in five portions (5×2.59g) at < +26 °C. The mixture was stirred at 12 °C for 22 h. 1M K2C03 (76 mL) was added in one portion. MTBE (59 mL) was added then EDTA 2K 2H20 (22.55 g) was added over 10 min at < +14 °C. To the solution 45 wt% KOH (4.86 mL) solution was added. The solution was cooled to 5 °C, and 1.1 equivalents of B0C2O (5.46 g) was added. The solution was rinsed with MTBE (10 mL) and stirred at 5 °C for 2h, then at 12 °C for 16h, and then at 24 °C for lOh until >99.5% conversion. The solution was transferred to a separatory funnel with the aid of MTBE (30 mL) and water (5 mL). The organic layer was filtered and washed with MTBE (20 mL). The organic filtrate was concentrated. MTBE (60 mL), water (30 mL) and saturated sodium chloride solution (15 mL) were added. The mixture was warmed in a 30 °C bath to dissolve solid, and then concentrated. The concentrate was flushed with toluene in a 60 °C bath, then concentrated. Toluene (8.4 mL) was added and the mixture was heated to 80 °C. Heptane (70.8 mL) was added over lh at 80 °C, then cooled slowly to room temperature. The mixture was filtered and washed with 1 :2 toluene/heptane (23.55 mL), filterated and vacuum dried under nitrogen until a constant weight.
The purity could be further upgraded by the following procedure: a round bottom flask was charged with the product of Step 6 (7.069 g) from above. EtOH (21 mL) was added and the mixture was heated to 45 °C. Water (31.5 mL) was slowly added over 1 h at 45 °C. The mixture was aged for lh. Water (31.5 mL) was added in one portion, then cooled slowly to room temperature and aged overnight. The slurry was filtered and washed with 1 :3.5 EtOH/water (23.56 mL). Crystals were vacuum dried under nitrogen until a constant weight.
Alternatively, Compound 20 was reduced with 100 psi hydrogen in 20 volume wet THF in the presence of 10-30 wt% Raney nickel at 50 °C. Then the reaction mixture was basified with 2 equivalent of K2CO3 and a slight execess B0C2O to afford crude Compound 21 after aqueous work up.
Compound 7 was obtained from 21via oxidation as described in WO2013/003250.
S

Boc-mesyl-pyrazole solid 1 was added to 2.5 volumes of TFA at 0-2 °C, over 2-3 minutes under nitrogen, followed by 0.5 volume of TFA rinse. Conversion to TFA salt was complete within 0.5-lh at 1-2 °C. DMAc (14 vol) followed by triethylamine (5 equivalents or 2.3 volumes) were slowly added to the TFA reaction mixture at 0 °C maintaining < +20 °C. Boc-ketone 7 (0.89 equivalent) was then added at -15 °C followed by solid NaBH(OAc)3 (1.4 equivalents) which was added in three portions over lh. The reaction solution was aged at -15 °C overnight. The solution was then warmed to 22 °C, and after aging for 2-5 h. Diastereomeric ratio was > 96.5:3.5.
The solution was seeded with Boc amine 1 wt% at 22 °C and stirred at 22-40 °C for 2-4 h. 0.36 volume 28% ammonium hydroxide was added over 2-4 h, then, 3.64 volumes 28% ammonium hydroxide was added over 4-10h at 22-60 °C. After cooling to 22 °C, the batch was filtered, washed with 5: 1 DMAc/water, then water. The wet cake was vacuum dried under nitrogen at ambient affording the product. Diastereoselectivity was > 30: 1.
Boc Deprotection of Formula la

A reactor was charged with 2.5 X (by volume) of trifluoroacetic acid. The batch was cooled to 5-10 °C. The reactor was then charged with 0.4 X (by volume) water. The batch was cooled to 0-5 °C. The reactor was then charged with 1 equivalent (1 kg) of the compound of Formula la over 0.5-lh while maintaining the temperature between 0 -5°C. The reactor was then charged with 0.5 X (by volume) trifluoroacetic acid to reactor while maintaining the temperature between 0-5°C. The batch was then heated between 15-20°C and aged for 2-2.5 h. The batch was then cooled to between 5-10°C. A crystallizer was charged with water 5.0 X (by volume) and 0.1 X (by volume) of ammonia water and adjusted to between 3-13°C. To generate a seed bed, Compound I seed (lwt% vs la) was added and the temperature as adjusted to between 3-13°C. A solution of ammonia water 3.8 X (by volume) and of the compound of Formula la was added simultaneously to the seed bed over 2.5 – 3.5 hours while maintaining temperature at 3-13°C and pH -9-10. The batch was aged for at least 30 minutes and then filtered. The resulting crystals were washed with 3. OX (by volume) water at 3 – 13°C twice and vacuum dried at < 50°C to afford the compound of formula I.
//////WO 2016014324, New Patent, Omarigliptin, MERCK SHARP & DOHME CORP, MK-3102
Regorafenib, SHILPA MEDICARE LIMITED, New patent, WO 2016005874
![]()
WO2016005874, PROCESS FOR THE PREPARATION OF REGORAFENIB AND ITS CRYSTALLINE FORMS
SHILPA MEDICARE LIMITED [IN/IN]; 10/80,Second Floor,Rajendra Gunj, Raichur, ರಾಯಚೂರು , karnataka 584102 (IN)
RAMPALLI, Sriram; (IN).
UPALLA, Lav Kumar; (IN).
RAMACHANDRULA, Krishna Kumar; (IN).
PUROHIT, Prashant; (IN).
AKSHAY KANT, Chaturvedi; (IN)
The present invention relates to a process for the preparation of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methylpyridine-2- carboxamide or Regorafenib (I): Formula (I). The present invention further relates to a process for the purification of 4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl} amino)-3-fluorophenoxy]-N-methylpyridine-2- carboxamide or Regorafenib (I) to provide highly pure material. The present invention further relates to a process for the preparation stable crystalline material of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]- N-methyl pyridine-2-carboxamide or Regorafenib (I) useful in the preparation of pharmaceutical compositions for the treatment of cancer.
4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide or Regorafenib is low molecular weight, orally available, inhibitor of multiple protein kinases, including kinases involved in tumour angiogenesis (VEGFR1, -2, -3, TIE2), oncogenesis (KIT, RET, RAF-1, BRAF, BRAFV600E), and the tumour microenvironment (PDGFR, FGFR). In preclinical studies regorafenib has demonstrated antitumour activity in a broad spectrum of tumour models including colorectal tumour models which is mediated both by its antiangiogenic and antiproliferative effects. Major human metabolites (M-2 and M-5) exhibited similar efficacies compared to Regorafenib both in vitro and in vivo models.
Regorafenib was approved by USFDA in 2012 and is marketed under the brand name Stivarga®, is an important chemotherapeutic agent useful for the treatment of adult patients with metastatic colorectal cancer (CRC) who have been previously treated with, or are not considered candidates for, available therapies. These include fluoropyrimidine-based chemotherapy, an anti-VEGF therapy and an anti-EGFR therapy.
Regorafenib is chemically known as 4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl} amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide (I). Regorafenib is a white to slightly pink or slightly brownish solid substance with the empirical formula C2iHi5ClF4N403 and a molecular weight of 482.82. Regorafenib is practically insoluble in water, dilute alkaline solution, dilute acid solution, n-heptane, glycerine and toluene. It is slightly soluble in acetonitrile, dichloromethane, propylene glycol, methanol, 2-propanol, ethanol and ethyl acetate. It is sparingly soluble in acetone and soluble in PEG 400 (macrogol). Regorafenib is not hygroscopic.
Regorafenib is generically disclosed in US 7351834, and specifically disclosed in US 8637553. US ‘553 disclose a process for the preparation of Regorafenib starting from 3-fluoro-4-nitrophenol. The process is as demonstrated below:

The present inventors has repeated the above process and found the following disadvantages:
Unwanted reactions are observed during the formation of Regorafenib, due to the involvement of prolonged time in process.
> Incomplete reactions were observed with excessive impurity formations due to incomplete conversion.
Removal of impurities from final product
US 2010173953 disclose Regorafenib monohydrate and crystalline Form I of Regorafenib. This patent application further discloses that crystalline Form I of Regorafenib stated in this application is obtained as per the process disclosed in WO 2005009961 A2 (Equivalent to US ‘553). The compound obtained was having a melting point of 186-206° C.
This patent publication discloses a process for the preparation of Regorafenib monohydrate comprises dissolving Regorafenib Form I obtained as per WO ‘961 in acetone
and the solution is filtered, followed by addition of water until precipitation, which was filtered and dried at room temperature
US 2010/0113533 discloses crystalline Form II of Regorafenib, comprises dissolving Regorafenib Form I obtained as per WO ‘961 in ethyl acetate, the suspension was heated to 40-45°C, addition of isocyanate solution (isocyanate in ethyl acetate) and is cooled to room temperature to yield the crystals, which was filtered, washed with ethyl acetate and dried at room temperature.
US 2010/0063112 discloses Form III of Regorafenib, process comprises of heating
Regorafenib monohydrate at 100°C or 60 min, and further 15 min at 110°C, followed by cooling to room temperature.
As polymorphism has been given importance in the recent literatures owing to its relevance to the drugs having oral dosage forms due to its apparent relation to dose preparation/suitability in composition steps/ bioavailability and other pharmaceutical profiles, stable polymorphic form of a drug has often remained the clear choice in compositions due to various reasons of handling, mixing and further processing including bioavailability and stability.
Exploring new process for these stable polymorphic forms which are amenable to scale up for pharmaceutically active / useful compounds such as 4-[4-({[4-chloro-3-(trifluoro methyl)phenyl]carbamoyl } amino)-3 -fluorophenoxy] -N-methylpyridine-2 -carboxamide or Regorafenib may thus provide an opportunity to improve the drug performance characteristics of such products.
Hence, inventors of the present application report a process for the preparation of a stable and usable form of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluoi phenoxy]-N-methylpyridine-2-carboxamide or Regorafenib, which may be industrially amenable and usable for preparing the corresponding pharmaceutical compositions. The present invention provides an improved process for the preparation of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fiuorophenoxy]-N-methylpyridine-2-carboxamide or Regorafenib crystalline forms specifically for crystalline polymorphic forms Form I and Form III. Crystalline polymorphic forms of 4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl } amino)-3 -fluorophenoxy] -N-methylpyridine-2 -carboxamide or Regorafenib obtained by the process of the present invention is non-hygroscopic and chemically stable and has good dissolution properties.
The process related impurities that appear in the impurity profile of the Regorafenib may be substantially removed by the process of the present invention resulting in the formation of highly pure material. The process of the present invention is as summarized below:

Example 1
Preparation of 4-(4-amino-3-fluorophenoxy) pyridine-2-carboxylic acid methyl amide
4-Amino-3-fiuorophenol (l lg, 0.08 moles) and of 4-Chloro-N-methyl-2-pyridinecarboxamide (8.85 g, 0.05 moles) was added to a reaction flask containing N, N-dimethylacetamide (55 ml) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 110-115°C and then potassium tert-butoxide in tetrahydrofuran (60 ml, 0.06 moles) was added slowly over a period of 3 to 4hours. Distill off solvent at same temperature, cooled the reaction mass to 25-30°Cand water(110 ml) was added slowly over a period of 15min. and cooled the reaction mass to 0-5°C . Adjust the pH of the reaction mass in between 7 and 7.5 by using 10% aqueous hydrochloric acid (~7 ml). Stir the reaction mass for 30min at the same temperature. Filter the product, washed with water (22 mL) and Dried at 50-55 °C for 12hrs. The obtained crude material was added to the flask containing Ethyl acetate (55 mL).The reaction mass was heated to reflux to get a clear solution and stirred for 15min at reflux. Cooled to 0-5°C, stir for 2hrs at the same temperature. Filter the product, washed with Toluene (9 mL) and dried at 50-55°C for 3-5hrs.
Above recrystallized material was added to the reaction flask containing methylene dichloride (270 mL) at 25-30°C and stirred for 10-15 min. Activated carbon (1 g) and silica gel (4.4 g) was added to the reaction mass and stir for lh at the same temperature. Filter the reaction mass through hyflow bed and wash with methylene dichloride (18 mL).Distill off solvent still~l-2 volumes of methylene dichloride remains in the flask and then cooled to 25-30°C. Toluene (20 mL) was added and stirred for 30min at the same temperature. Filtered the product, washed with Toluene (9 mL) and dried at 50-55°C for 12h.
Yield: 9 gm
Chromatographic Purity (By HPLC): 98%
Example 2
Preparation of Regorafenib
4-(4-amino-3-fluorophenoxy) pyridine-2-carboxylic acid methyl amide (4g, 0.01 moles) was added in to a reaction flask containing acetone (20 ml) at 25-30°C and stirred for 15 minutes. 4-chloro-3-trifluoromethylisocyanate (6.1g, 0.02 moles) was added slowly over a period of 5 to 10 minutes and stirred the reaction mixture 3 to 4 hours. Toluene (20 n L) was added to the reaction mass and stirred for 30 min at 25-30°C.The obtained reaction mass was filtered and washed with toluene (8 mL). Dried the material still constant weight appears to yield title product a crystalline material.
Yield: 5.5 gm
Chromatographic Purity (By HPLC): 97%
Example 3
Purification of Regorafenib using acetone and toluene mixture
4- [4-( { [4-chloro-3 -(trifluoromethyl)phenyl] carbamoyl } amino)-3 -fluorophenoxy] -N-methylpyridine-2-carboxamide (I) or Regorafenib (1 g) was added slowly in to the reaction flask containing acetone (2 mL) and toluene (3 mL) at 25-30°C and stirred for 15 minutes.
The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes.
Cooled the reaction mass to 25-30°C and stirred for 1 hour. Filter the material, washed with toluene (2 mL) and suck dried for 15 min, followed by drying at 50-55°C for 10-12h to yield
Pure 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methyl pyridine-2-carboxamide (I) or Regorafenib.
Yield: 0.88gm
Chromatographic Purity (By HPLC): 99.3 %
Example 4
Purification of Regorafenib using acetone
4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl} amino)-3 -fluorophenoxy] -N-methylpyridine-2-carboxamide (I) or Regorafenib (1 g) was added slowly in to the reaction flask containing acetone (5 mL) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 0-5°C and stirred for 1 hour. Filter the material, washed with acetone (1 mL) and suck dried for 15 min. The obtained wet cake was added in to the reaction flask containing acetone (5 mL) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50- 55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 0-5°C and stirred for 1 hour. Filter the material, washed with acetone (1 mL) and dried at 60-65°C for 12 h to yield Pure 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methyl pyridine -2-carboxamide (I) or Regorafenib.
Yield: 0.7 gm
Chromatographic Purity (By HPLC): 99.77%
Example 5
Double – Purification of Regorafenib using acetone and toluene mixture
4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] Carbamoyl} amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide (I) or Regorafenib (1 g) was added slowly in to the reaction flask containing acetone (2 mL) and toluene (3 mL) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 25-30°C and stirred for 1 hour. Filter the material, washed with toluene (2 mL) and suck dried for 15 min. The obtained wet cake was added in to the reaction flask containing acetone (2 mL) and toluene (3 mL) mixture at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 25-30°C and stirred for 1 hour. Filter the material, washed with toluene (2 mL) and dry at 60-65°C for 12h.
Yield: 0.80gm
Chromatographic Purity (By HPLC): 99.79 %
Moisture content: 0.09%
Impurity-A: 0.03%
Impurity-B: Not detected
Impurity-C: 0.02%
Example 6
Preparation of Regorafenib Form I
4-(4-amino-3-fluorophenoxy) pyridine-2-carboxylic acid methyl amide (1.3 g, 0.004 moles) was added in to a reaction flask containing acetone (13 mL) at 25-30°C and stirred for 15 minutes.4-chloro-3-trifluoromethylisocyanate (6.6 g, 0.006 moles) wasadded slowly over a period of 15 to 20 minutes and stirred the reaction mixture 3 to 4 hours. The obtained reaction mass was filtered and washed with acetone. Dried the material still constant weight appears to yield title product a crystalline material.
Yield: 1.9 g
Chromatographic Purity (By HPLC): 98.4 %
XRPD was found to resemble similar to Fig-1.
![]()
Omprakash Inani – Chairman, Vishnukant C Bhutada – Managing Director, Namrata Bhutada – Non Executive Director, Ajeet Singh Karan – Independent Director, Carlton Felix Pereira – Independent Director, Pramod Kasat – Independent Director, Rajender Sunki Reddy – Independent Director, N P S Shinh – Independent Director,
 Mr. Omprakash Inani |
Mr. Omprakash Inani – CHAIRMAN
Mr. Omprakash Inani has more than 30 years of Business experience. He monitors business and functional aspects of the Company along with the operations of all the plants. Additionally, he is member of Audit and Remuneration committee of Shilpa Medicare Group of Companies. Currently he is also a council Member in “Academy of Medical Education, Dental College & V.L. College of Pharmacy”, “Taranath Shikshana Samsthe, Raichur” and a trustee in “Akhil Bhartiya Maheshwari Education Trust, Pune”. Mr. Omprakash Inani is also Managing Committee Member of “Karnataka State Cotton Assn., Hubli”. |
|
|
|
||
 Mr. Vishnukant C. Bhutada Mr. Vishnukant C. Bhutada |
Mr. Vishnukant C. Bhutada – MANAGING DIRECTOR
Mr. Vishnukant has vast and diverse Business experience of API and Intermediates and presently leads the core Business and functional teams which accelerate growth and performance by Innovating for Affordable solutions at Shilpa Medicare Group of Companies. He is the key decision maker with the teams for Shilpa Group for successful API and Generics formulation strategies. His untiring efforts have led the company to a leadership position in the Indian pharmaceutical domain and helped create a prominent presence for Oncology APIs globally. For his efforts on APIs Business, Mr. Vishnukant was awarded “Best Entrepreneur Award” by Late Dr Shankar Dayal Sharma – President of India in 1995. Subsequently, various state honours were conferred upon him -like -“Best Entrepreneur” from Karnataka State Govt. in 1996; “Excellence in Exports” from Vishweshwarayya Industrial Trade Centre, Bangalore 1996; and Export Excellence Award-2006” by FKCCI, Bangalore. Success has never stopped coming his way- as he was awarded “First runner up” at the Emerging India Awards London 2008 by CNBC TV18. Recently, his efforts in the Shilpa Group for environment sustainability, has led to “Best National Energy Conservation Award in Drugs & Pharmaceutical Sector for the year 2012” by Hon’ble President of India, Dr. Pranab Mukherjee. |
|
|
|
||
 Dr. Vimal Kumar Shrawat Dr. Vimal Kumar Shrawat |
Dr. Vimal Kumar Shrawat – CHIEF OPERATING OFFICER
Dr. Shrawat by qualification holds degrees of M.Sc (Organic Chemistry), Ph.D. (from Delhi University) and joined Shilpa Medicare in 2009. He has vast experience of more than 25 years of working in large pharma industries like Ranbaxy/ Dabur Pharma- presently known as Fresenius Kabi Oncology Ltd., spanning across activities of R&D, Pilot and Plant Productions, QA/QC, Administration, CRAMS, Project management etc. Presently, Dr. Shrawat is spearheading the entire Operations/ Control of Shilpa Medicare. His vision of team work and time bound approach always guides and motivates teams at all operational sites. His keen interest and consistent efforts for R&D has led him to become one of key contributor in large number of Patent/applications of Shilpa Medicare. |
|
|
|
||
 Dr. Pramod Kumar |
Dr. Pramod Kumar – MANAGING DIRECTOR(LOBA FEINCHEMIE GMBH AUSTRIA), SENIOR VICE-PRESIDENT (SHILPA MEDICARE LTD)
Dr. Pramod Kumar, who by qualification holds degrees of M.Pharm, Ph.D (Pharmaceutical chemistry) and a PGDBA, joined Shilpa Medicare in 1989. Since 2009 he is Managing Director of Loba FeinchemieGmBH, Austria and driving all R&D driven commercial processes. Dr. Pramod Kumar has more than 25 years of experience in Pharmaceutical industry, spanning across activities of production, QA/QC, administration, import/export, CRAMS etc. His efforts in CRAMS have led to the formation of Joint venture company RAICHEM MEDICARE Pvt LTD with Italian companies ICE SPA / P.C.A SPA. |
|
|
|
||
 Mr. Prashant Purohit |
Mr. Prashant Purohit – VICE-PRESIDENT-CRD
Mr. Prashant Purohit by qualification holds degrees of, M.Sc.(Organic Chemistry) and Diploma in Business Management and joined Shilpa Medicare in 1996. He is presently heading Chemical R&D wings of Shilpa Medicare Group. He has vast experience of handling CRAMS and Generics APIs R&D. His vast experience of nearly 35 years in R & D and production in Pharmaceutical Industry has consistently enriched the portfolio of Shilpa Medicare Group of Companies. He is one of key contributor in large number of Patent/applications of Shilpa Medicare. |
|
|
|
||
 Dr. Akshay Kant Chaturvedi |
Dr. Akshay Kant Chaturvedi – HEAD- CORPORATE IPM & LEGAL AFFAIRS
Dr. Akshay Kant by qualification holds degrees of M.Sc, Organic Chemistry (Univ. Gold Medalist), Ph.D. (Medicinal Chem), LL.B., M.B.A. and joined Shilpa Medicare in Jun 2012. Presently, Dr. Akshay is spearheading the entire IP portfolio management/ Legal Affairs of Contractual Business of Shilpa Medicare Group. His vision of innovative and creative thinking, team work and time bound approach always guide and motivate teams at all locations.His keen interest and consistent efforts for R&D has led him to become one of key contributor in large number of Patent/applications of Shilpa Medicare. |
|
|
|
||
 Dr. Seshachalam U. |
Dr. Seshachalam U. -ASSOCIATE VICEPRESIDENT- QUALITY AND RA
Dr. Seshachalam by qualification holds M.Sc (Chemistry) and Ph.D. (Chemistry) and joined Shilpa Medicare in 2008. He is presently heading Regulatory Affairs wings of Shilpa Medicare Group of Companies. He has vast experience of handling regulatory affairs related to Generics APIs. Being instrumental in Shilpa Medicare’s efforts to achieve recognition of different authorities, his key contribution in successful inspection and audit by various regulatory authorities is one of the core strength to the organization’s aims and objectives. |
|
|
|
||
 Mr. Sharath Reddy |
Mr. Sharath Reddy – VICE-PRESIDENT PROJECTS & OPERATIONS
Mr. Sharath Reddy by qualification holds M.Pharm from BITS Pilani and has overall experience of about 22 years predominately in the field of pharmaceuticals new projects and operations. His expertise of Oncology specialized equipment and Utilities designing has boosted organizations confidence to takeover new endeavors of upcoming projects with faster pace of time. His efforts have led to successfully executing Energy Saving projects of Shilpa Medicare Group of Companies and registration of the project under Clean Development Mechanism with UNFCC (Under Kyoto Protocol). |
|
|
|
||
 Mr. R K Somani |
Mr. R K Somani – VICE-PRESIDENT FORMULATION -BUSINESS DEVELOPMENT
Mr. R. K. Somani is a professional Chartered Accountant and holds a Diploma in Central Excise.He has overall business experience of more than 21 years predominately in the field of pharmaceuticals. Mr. Somani is one of the key drivers of Formulation business besides handling various key Contract Businesses of advanced oncology/ Non-Oncology APIs. He is known for successfully building formulations portfolio and spearheading the Generic sales operation. |
|
Shilpa Medicare Limited
1st Floor, 10/80,
Rajendra Gunj,
RAICHUR ರಾಯಚೂರು – 584 102.
Karnataka, India.
Telephone: +91-8532-236494
Fax: +91-8532-235876
Email: info@vbshilpa.com
RAICHUR, ರಾಯಚೂರು Karnataka, India








Historical Stone Elephants in Malayabad, Raichur Taluk …

View of Raichur city and lake Aam Talab
///Regorafenib, SHILPA MEDICARE LIMITED, new patent, WO 2016005874, raichur, ರಾಯಚೂರು , karnataka, india
Lupin Ltd, Patent, Pitavastatin, WO2014203045
![]()
Lupin Ltd, Patent, Pitavastatin, WO2014203045
A NOVEL, GREEN AND COST EFFECTIVE PROCESS FOR SYNTHESIS OF TERT-BUTYL (3R,5S)-6-OXO-3,5-DIHYDROXY-3,5-O-ISOPROPYLIDENE-HEXANOATE
ROY, Bhairabnath; (IN).
SINGH, Girij, Pal; (IN).
LATHI, Piyush, Suresh; (IN).
AGRAWAL, Manoj, Kunjabihari; (IN).
MITRA, Rangan; (IN).
TRIVEDI, Anurag; (IN).
PISE, Vijay, Sadashiv; (IN).
RUPANWAR, Manoj; (IN)
The present invention describes an eco-friendly and cost effective process for the synthesis of teri-butyl (3R,5S)-6-oxo-3,5-dihydroxy-3,5-0-isopropylidene-hexanoate [I]
 PITAVASTATIN
PITAVASTATIN
![]()
TEXT
tert-b tyl (3R,5S)-6-oxo-3,5-dihydroxy-3,5-0-isopropylidene-hexanoate [I] [CAS No. 124752-23-4] is key intermediate for the preparation of statins such as Atorvastatin (Tetrahedron 63, 2007, 8124 -8134), Cerivastatin (Journal of Labeled Compounds and Radiopharmaceuticals, 49, 2006 311-319), Fluvastatin [WO2007125547; US 4739073], Pitavastatin [WO2007/132482; US2012/22102 Al, WO2010/77062 A2; WO2012/63254 Al ; EP 304063; Tetrahedron Letters, 1993, 34, 513 – 516; Bulletin of the Chemical Society of Japan, 1995, 68, 364 – 372] and Rosuvastatin [WO2007/125547 A2; WO2011/132172 Al ; EP 521471]. Statins are used for treatment of hypercholesterolemia, which reduces the LDL cholesterol levels by inhibiting activity of HMG-CoA reductase enzyme, which is involved in the synthesis of cholesterol in liver.

[I]
Compound [I] is generally obtained by various methods of oxidation of teri-butyl 2- ((4R,65)-6-(hydroxymethyl)-2,2-dimethyl-l,3-dioxan-4-yl)acetate [compound II] and are discussed in details hereinafter. In addition, various methods for synthesis of compound [II] are also elaborated below.

[II]
[II]
A) tert-butyl2-((4«,6.S)-6-(hydroxymethyl)-2,2-dimethyl-l,3-dioxan-4-yl)acetate
[compound II]
US patent Number 5278313 describes a process for synthesis of compound [II]
(Schemel). In the said process, (5)-methyl 4-chloro-3-hydroxybutanoate has been obtained in only 70% yield through whole cell enzymatic reduction of methyl 4-chloro-3- oxobutanoate, which has a necessity of special equipment such as fermenters as well as other microbial facilities such as sterile area, autoclaves, incubator for growing seed culture, etc.
(S)-mefhyl 4-chloro-3-hydroxybutanoate upon reaction with teri-butyl acetate in presence of LiHMDS or LDA at -78°C, yielded (S)-ieri-butyl 6-chloro-5-hydroxy-3- oxohexanoate, which was further transformed to corresponding diol through syn selective reduction in presence of methoxydiethyl borane/sodium borohydride at -78°C. The diol thus obtained was converted to compound [II] .
The overall yield for this process is low and required special equipment such as fermenters, etc and in addition to that, this process is not cost effective due to use of costly reagent such as methoxydiethyl borane.
Moreover, methoxydiethylborane is highly pyrophoric (Encyclopedia for organic synthesis, editor in chief L. Paquette; 2, 5304; Published by John and Wiley Sons;
Organic Process Research & Development 2006, 10, 1292-1295) and hence safety is a major concern.

Scheme 1
EP 1282719 B l (PCT application WO 01/85975 Al ) discloses a process for synthesis of compound ( R, 5S)-tert-bv y\ 3,5,6-trihydroxyhexanoate from (S)-tert-b tyl-5,6-dihydroxy-3-oxohexanoate through a) asymmetric hydrogenation in presence of a chiral catalyst e.g. di-mu-chlorobis-[(p-cymene)chlororuthenium(II)] along with an auxiliary such as (IS, 2S)-(+)-N- (4-toluenesulfonyl)-l ,2-diphenylethylenediamine as ligand, which gave desired product only in 70% diastereomeric excess (de); b) Whole cell enzymatic reduction of (S)-tert- butyl 5,6-dihydroxy-3-oxohexanoate to obtain compound (3R, 5S)-tert-bv y\ 3,5,6-trihydroxyhexanoate in 99% de (80% yield).
It is needless to mention that it has necessity of fermenter and other microbiological equipment (Scheme 2).
Moreover, conversion of (2>R,5S)-tert-bv y\ 6-acetoxy-3,5-dihydroxyhexanoate to tert-bv yl 2-((4R,65)-6-(acetoxymethyl)-2,2-dimethyl-l ,3-dioxan-4-yl)acetate was accomplished in only 25% yield and also required the flash chromatography for isolation of desired product.
Thus, overall yield for this process is poor and process is not operation friendly especially at large scale hence cannot be considered feasible for commercial manufacturing.

Scheme 2
EP1317440 Bl (PCT Application WO 02/06266 Al) has disclosed the process for synthesis of compound [II] from 6-chloro-2,4,6-trideoxy-D-erythro-hexose (Scheme 3) .
In the said patent application 6-chloro-2,4,6-trideoxy-D-erythro-hexose was converted to (4R, 65)-4-hydroxy-6-chloromethyl-tetrahydropyran-2one with excess of bromine in presence of potassium bicarbonate, which liberates environmentally undesired gas i.e. carbon dioxide.
Moreover, starting material i.e. 6-chloro-2,4,6-trideoxy-D-erythro-hexose is not commercially available and conversion efficiency of starting material at large scale towards (4R, 65)-4-hydroxy-6-chloromethyl-tetrahydropyran-2-one is only 67%.

Scheme 3
US Patent No. 6689591 B2 has demonstrated the whole cell enzymatic reduction of teri-butyl 6-chloro-3,5-dioxohexanoate to compound [II] (Scheme 4).
In the said process, whole cell enzymatic reduction is not specific; yield for desired product is only 34% and other partially reduced products are also obtained.
Hence, further purification is required for obtaining the desired compound. Thus, this process is not suitable for commercial scale.

Scheme 4
Tatsuya et al (Tetrahedron Letters; 34, 1993,513 – 516) has reported synthesis of compound [I] from derivative of L-tartatric acid (Scheme 5).
In the said process, tartaric acid di-i‘sopropyl ester is doubly protected by tert-butyldimethylsilyl group, which was reacted with dianion of teri-butyl acetoacetate to give β, δ-diketo ester compound.
β,δ-diketo ester was reacted with 2 equivalent of diisobutylaluminium hydride (which is a pyrophoric reagent) to afford -hydroxy,8-keto ester in only 60% yield.
This process is not industrially viable as overall yield is very low and also because of use of costly and pyrophoric reagents/chemicals.

Scheme 5
US7205418 (PCT application WO03/053950A1) has described the process for synthesis of compound [II] from (S)-ieri-butyl-3,4-epoxybutanoate (Scheme 6).
The overall yield for this process is very low and moreover, it required the diastereomeric separation of teri-butyl 2-(6-(iodomethyl)-2-oxo-l,3-dioxan-4-yl)acetate by flash chromatography.
Since overall requirement of title compound is very high, any operation involving flash chromatography will tend to render the process commercially unviable.

Scheme 6
Fengali et al (Tetrahedron: Asymmetry 17; 2006; 2907-2913) has reported the process for synthesis of compound [II] from racemic epichlorohydrin (Scheme 7).
In this process, racemic epichlorohydrin was converted to corresponding nitrile intermediate through reaction with sodium cyanide; nitrile intermediate thus obtained was further resolved through lipase catalyzed stereo-selective esterification to obtain (5)-4-(benzyloxy)-3-hydroxybutanenitrile and (R)-l-(benzyloxy)-3-cyanopropan-2-yl acetate;
separation of desired product i.e. (S)-4-(benzyloxy)-3-hydroxybutanenitrile having 98% de (40% yield) was done by column chromatography.
Needless to mention a commodity chemical like compound [I] cannot be manufactured by such a laboratory method, which involved number of steps.

Scheme 7
Bode et al (Organic letters, 2002, 4, 619-621) has reported diastereomer- specific hydrolysis of 1,3-diol-acetonides (Scheme 8).
In this publication, duration of the reaction for diastereomer- specific hydrolysis of 1,3, diol-acetonides is reported to be 4 h, however, in our hand it was observed that hardly any reaction took place in 4 h, which made it non-reproducible.
In addition to that, separation of desired product is achieved by flash chromatography and it is needless to mention that any process which involved flash chromatography would render the process to be commercially unviable.
Hence, additional innovation needs to be put in for making the process industrially viable.

Scheme 8
CN 101613341A has reported the process for synthesis of compound [II] (Scheme
9).
In the same patent application tert-b tyl (S)-6-chloro-5-hydroxy-3-oxohexanoate was synthesized through Blaise condensation of (5)-4-chloro-3-hydorxy-butanenitrile with zinc enolate of tert butyl bromo acetate.
In the literature, synthesis of tert-bv yl (S)-6-chloro-5-hydroxy-3-oxohexanoate was reported through Blaise condensation of silyl protected (5)-4-chloro-3-(trimethylsilyl)oxy-butanenitrile with zinc enolate of tert butyl bromo acetate, in good yield (Synthesis 2004, 16, 2629-2632). Thus, protection of hydroxy group in (5)-4-chloro-3-hydorxy-butanenitrile is imperative.
In the said Chinese patent application, in claim 7, it was mentioned that solvent used for conversion of tert-bv yl (5)-6-chloro-5-hydroxy-3-oxohexanoate to ( R,5S)-tert-butyl 6-chloro-3,5-dihydroxyhexanoate is anyone or mixture of more than one from tetrahydrofuran, ether, methanol, ethanol, n-propanol, /so-propanol and ethylene glycol.
However, in enablement the only example using mixture of solvent was that of THF-methanol (Experimental section, Example 4: The preparation of (R,5)-6-chloro-3,5- dihydroxyhexanoate) and same outcome was expected in other individual or mixture of solvents.
Claim 8 of CN 101613341A mentioned that reduction was carried out by any one or mixture of more than one reducing agents such as sodium borohydride, potassium borohydride, lithium aluminum hydride, diethylmethoxy borane, triethyl borane and tributyl borane.
It implies that either any one of the reducing agents or a mixture of the same can be employed. From reaction mechanism it is very much clear that diethylmethoxy borane, triethyl borane and tributyl borane form the six membered complex between optically active hydroxyl and carbonyl group, which gets reduced by sodium borohydride, signifying that individually diethylmethoxy borane, triethyl borane and tributyl borane are not reducing agents
Moreover, in claims 12 and 13 (Experimental section, Example 4: The preparation of (R,S)-6-chloro-3,5-dihydroxyhexanoate), it is mentioned that reduction should be carried out in temperature range -80 °C to -60 °C, implying that reaction would not work beyond this temperature range i.e. it would work in the temperature window of -80 °C to -60 °C only.
Summarizing, the teachings of the application are not workable.

Scheme 9
Wolberg et al (Angewandte Chemie International Edition, 2000, 4306) has reported that diastereomeric excess for syn selective reduction using mixture of diethyl methoxy borane/sodium borohydride of compound [VI] gave 93% de for compound [VIII], which required further re-crystallization to obtain compound [VIII] in 99% de and 70% yield.
Thus, all the reported methods for stereo-selective hydride reduction of compound [VI] were achieved through mixture of trialkyl borane or diethyl methoxy borane & sodium borohydride in THF, at -78°C. As mentioned earlier, trialkyl borane or diethyl methoxy borane are pyrophoric in nature; in addition to that anhydrous THF is costly and moreover, reaction required large dilution.
Hence, there is need for developing efficient, environment friendly, cost effective and green process for stereo-selective reduction compound [VI].
B) The process of Oxidation of compound [II] to compound [I] has been discussed in following literature processes.
1) Swern oxidation (US4970313; Tetrahedron Letters, 1990, 2545
Synthetic Communications, 2003, 2275 – 2284).
2) Parrkh-Doering oxidation (J. Am. Chem. Soc, 1967, 89, 5505-5507)
3) TEMPO/NaOCl oxidization (EP2351762)
4) Trichloroisocyanuric acid/ TEMPO (CN 101747313A)
5) Oxidation of compound [II] to compound [I] through IBX [CN101475558A].
It would be evident that most of the reported methods are not “green” and
environmentally benign; none of the reported methods use molecular oxygen as oxidizing agent in presence of metal catalyst/co-catalyst.
Example 18: Process for synthesis of tert-butyl 2-((4R,6S)-6-formyl-2,2-dimethyl-l,3-dioxan-4-yl)acetate [I]

A reactor was charged with 1.1 g of copper (I) chloride and 10 mL of acetonitrile. 2-2′ Bipyridyl (156 mg) and TEMPO (156 mg) were added to the reactor under oxygen environment at 25°C. A solution of (6-Hydroxymethyl-2,2-dimethyl-[l,3]dioxan-4-yl)-acetic acid tert-butyl ester 2.6 g in 26 mL DCM was added dropwise over a period of 10 min into it. The reaction mass was stirred at 40°C and progress of reaction was monitored on GLC, which shows that 90% conversion for desired product.
Example 19: Process for synthesis of tert-butyl 2-((4R,6S)-6-formyl-2,2-dimethyl-l,3-dioxan-4-yl)acetate [I]
A reactor was charged with 1.1 g of copper (I) chloride and 10 mL of dichlorome thane. 2-2′ Bipyridyl (156 mg) and TEMPO (156 mg) were added to the reactor under oxygen environment at 25°C. A solution of (6-Hydroxymethyl-2,2-dimethyl-[l,3]dioxan-4-yl)-acetic acid tert-butyl ester 2.6 g in 26 mL DCM was added dropwise over a period of 10 min into it. The reaction mass was stirred at 40°C and progress of reaction was monitored on GLC, which shows that 90% conversion for desired product.
AUTHORS

///////
