
Home » NDA (Page 2)
Category Archives: NDA
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Some thing for your chin………FDA accepts Kythera’s ATX-101 new drug application

FDA accepts Kythera’s ATX-101 new drug application
Kythera Biopharmaceuticals’ new drug application (NDA) for its ATX-101, a submental contouring injectable drug, has been accepted for filing by the US Food and Drug Administration (FDA).
According to Kythera Biopharmaceuticals, the ATX-101 NDA will be subject to a standard review and will have a prescription drug user fee act (PDUFA) action date of 13 May 2015. The company submitted the NDA in May 2014.
cas 83-44-3, C24 H40 O4
cas of Na salt….302-95-4
NSC-681065 , NSC 8797
| NAMES | Cholan-24-oic acid, 3,12-dihydroxy-, (3α,5β,12α)- |
- OTHERS
- 5β-Cholan-24-oic acid, 3α,12α-dihydroxy- (8CI); 17β-[1-Methyl-3-carboxypropyl]-etiocholane-3α,12α-diol;
- 3α,12α-Dihydroxy-5β-cholan-24-oic acid;
- 3α,12α-Dihydroxy-5β-cholanic acid;
- 3α,12α-Dihydroxy-5β-cholanoic acid; 3α,12α-Dihydroxycholanic acid;
- 5β-Cholanic acid-3α,12α-diol;
- 5β-Deoxycholic acid; 7-Deoxycholic acid; ATX 101;
- Cholerebic; Cholic acid, deoxy-; Cholorebic; Degalol; Deoxycholatic acid; Deoxycholic acid; Desoxycholic acid; Droxolan; NSC 8797; Pyrochol; Septochol
- Deleted CAS Registry Numbers: 728917-93-9
- University of California, Oakland (Originator)
LA BioMed (Originator) - LICENSE….
Kythera Biopharmaceuticals, Inc.
Rapid removal of body fat is an age-old ideal, and many substances have been claimed to accomplish such results, although few have shown results. ”Mesotherapy”, or the use of injectables for the removal of fat. is not widely accepted among medical practitioners due to safety and efficacy concerns, although homeopathic and cosmetic claims have been made since the 1950’s. Mesotherapy was originally conceived in Europe as a method of utilizing cutaneous injections containing a mixture of compounds for the treatment of local medical and cosmetic conditions. Although mesotherapy was traditionally employed for pain relief, its cosmetic applications, particularly fat and cellulite removal, have recently received attention in the United States. One such reported treatment for localized fat reduction, which was popularized in Brazil and uses injections of phosphatidylcholine, has been erroneously considered synonymous with mesotherapy. Despite its attraction as a purported “fat-dissolving” injection, there is little safety and efficacy data of these cosmetic treatments. See, Rotunda, A.M. and M.
olodney, Dermatologic Surgery 32:, 465-480 (2006) (“Mesotherapy and
Phosphatidy lcholine Injections: Historical Clarification and Review**).
Recently published literature reports that the bile acid, DCA, and salts thereof, have fat removing properties when injected into fatty deposits in vivo. See, WO
2005/1 17900 and WO 2005/1 12942, as well as US2005/0261258; US2005/0267080; US2006/127468; and US20060154906, each of which is incorporated herein by reference in its entirety). Deoxycholate injected into fat tissue degrades fat cells via a cytolytic mechanism. Because deoxycholate injected into fat is rapidly inactivated by exposure to protein and then rapidly returns to the intestinal contents, its effects are spatially contained. As a result of this attenuation effect that confers clinical safety, fat removal typically require 4 – 6 sessions. This localized fat removal without the need for surgery is beneficial not only for therapeutic treatment relating to pathological localized fat deposits (e.g., dyslipidemias incident to medical intervention in the treatment of HIV), but also for cosmetic fat removal without the attendant risk inherent in surgery (e.g., liposuction). See, Rotunda et ai, Dermatol. Surgery 30: 1001-1008 (2004) (“Detergent effects of sodium deoxycholate are a major feature of an injectable phosphatidylcholine formulation used for localized fat dissolution”) and Rotunda et al, J. Am. Acad. Dermatol. (2005 : 973-978) (“”Lipomas treated with subcutaneous deoxycholate injections”), both incorporated herein by reference in their entirety. US Patent Nos. 7,622,130 and
7,754,230 describe using DCA for fat removal.
In addition, many important steroids have a 12- -hydroxy-substituent on the C- ring of the steroid. Such compounds include, by way of example, bile acids such as DCA, cholic acid, lithocholic acid, and the like. Heretofore, such compounds were typically- recovered from bovine and ovine sources which provided a ready source of bile acids on a cost effective basis. However, with the recent discovery that pathogens such as prions can contaminate such sources, alternative methods for the synthesis of bile acids from plant sources or synthetic starting materials have become increasingly important. For example, DCA from animals in New Zealand are a source of bile acids for human use under US regulatory regimes, as long as the animals continue to remain isolated and otherwise free of observable pathogens. Such stringent conditions impose a limitation on the amount of suitable mammalian sourced bile acids and does not preclude the possibility that the bile acid will be free of such pathogens. US Patent Publication No.
8,242,294 relates to DCA containing less than 1 ppt 14C.
ATX-101, sodium deoxycholate for injection, is awaiting for approval in the U.S. for the reduction of localized submental fat. Phase II trials for the treatment of superficial lipomas have been completed at Kythera Biopharmaceuticals and Intendis. Treatment with ATX-101 is expected to significantly reduce the size of or eliminate lipomas and provide an effective non-surgical, minimally invasive treatment option for patients.
Licensed to Kythera from Los Angeles Biomedical Institute at Harbor-UCLA Medical Center in 2007, ATX-101 is also being evaluated by the company for aesthetic applications. Specifically, phase II trials are under way for the reduction of submental fat. In 2010, ATX-101 was licensed to Intendis by Kythera Biopharmaceuticals outside of the U.S. and Canada for the treatment of dermatological disorders. In 2010, the product was licensed by Kythera Biopharmaceuticals to Bayer outside Canada and the U.S., and in 2014, Kythera acquired those same rights from Bayer.
………………………………..
WO 2011075701
http://www.google.com/patents/WO2011075701A2?cl=en
Scheme 2

Conversion of Compound 24 to Compound 33:
The hydrogenation of compound 24 on 10.0 g scale using dry 10 % Pd/C (15 wt %) in ethyl acetate (20 parts) was added and applied about 50 psi hydrogen pressure and temperature raised to 70 °C. After reaching temperature 70 °C, observed increase of hydrogen pressure to about 60 psi, at these conditions maintained for 60 h. After 60 hours 0.6% of compound 24 and 2.75% of allylic alcohol were still observed, so further stirred for additional 12 h (observed 0.16% of allylic alcohol and 0.05% of compound 24). After work-up, the reaction provided 9.5g of residue.
Anther hydrogenation reaction on 25 g of compound 24 with above conditions for 76 h provided 24.5 g of residue.
Method A
10% Pd/C (900 mg) was added to a solution of compound 24 (2.0 g, 4.5 mmol) in EtOAc (150 mL) and the resulting slurry was hydrogenated in a Parr apparatus (50 psi) at 50 °C for 16 h. At this point the reaction was determined to be complete by TLC. The mixture was filtered through a small plug of Celite® and the solvent was removed under vacuum, providing compound 33 (1.6 g, 80% yield) as a white solid.
TLC: p-anisaldehyde charring, Rf for 33 = 0.36 and Rf for 25 = 0.32.
TLC mobile phase: 20% – EtOAc in hexanes. 1H NMR (500 MHz, CDC13): δ = 4.67-4.71 (m, 1H), 3.66 (s, 3H), 2.45-2.50 (t, J = 15 Hz, 2H), 2.22-2.40 (m, 1H), 2.01 (s, 3H), 1.69-1.96 (m, 9H), 1.55 (s, 4H), 1.25-1.50 (m, 8H), 1.07-1.19 (m, 2H), 1.01 (s, 6H), 0.84-0.85 (d, J= 7.0 Hz, 3H).
13C NMR (125 MHz, CDC13): δ = 214.4, 174.5, 170.4, 73.6, 58.5, 57.4, 51.3, 46.4, 43.9, 41.2, 38.0, 35.6, 35.5, 35.2, 34.8, 32.0, 31.2, 30.4, 27.4, 26.8, 26.2, 25.9, 24.2, 22.6,
21.2, 18.5,1 1.6,
Mass (m/z) = 447.0 [M+ + 1], 464.0 [M+ + 18].
IR (KBr) = 3445, 2953, 2868, 1731, 1698, 1257, 1029 cm-1.
m.p. =142.2-144.4 °C (from EtOAc/hexanes mixture).
[α]D = +92 (c = 1 % in CHCl3).
ELSD Purity: 96.6%: Retention time = 9.93 (Inertsil ODS 3 V, 250 χ 4.6 mm, 5um, ACN:
0.1 % TFA in water (90: 10)
Method B
A slurry of 10% Pd/C (9 g in 180 mL of ethyl acetate) was added to a solution of compound 24 (36 g, 81 mmol) in EtOAc (720 mL) and the resulting slurry was treated with hydrogen gas (50 psi) at 45-50 °C for 16 h. (A total of 1080 mL of solvent may be used). At this point the reaction was determined to be complete by HPLC (NMT 1% of compound 24). The mixture was filtered through Celite® (10 g) and washed with ethyl acetate (900 mL). The filtrate was concentrated to 50% of its volume via vacuum distillation below 50 °C. To the concentrated solution was added pyridinium
chlorochromate (20.8 g) at 25-35 °C and the mixture was stirred for 2 h at 25-35 °C, when the reaction completed by HPLC (allylic alcohol content is NMT 1%).
The following process can be conducted if compound 24 content is more than 5%. Filter the reaction mass through Celite® (10 g) and wash with ethyl acetate (360 mL). Wash the filtrate with water (3 x 460 mL) and then with saturated brine (360 mL). Dry the organic phase over sodium sulphate (180 g), filter and wash with ethyl acetate (180 mL). Concentrate the volume by 50% via vacuum distillation below 50 °C. Transfer the solution to a clean and dry autoclave. Add slurry of 10% palladium on carbon (9 g in 180 mL of ethyl acetate). Pressurize to 50 psi with hydrogen and stir the reaction mixture at 45-50 °C for 16 h. Upon complete consumption of compound 24 by HPLC (the content of compound 24 being NMT 1%), the reaction mixture was filtered through Celite® (10 g) and the cake was washed with ethyl acetate (900 mL). The solvent was concentrated to dryness via vacuum distillation below 50 °C. Methanol (150 mL) was added and concentrated to dryness via vacuum distillation below 50 °C. Methanol (72 mL) was added to the residue and the mixture was stirred for 15-20 min at 10-15 °C, filtered and the cake was washed with methanol (36 mL). The white solid was dried in a hot air drier at 45-50 °C for 8 h to LOD being NMT 1 % to provide compound 33 (30 g, 83.1 % yield).
Conversion of Compound 33 to Compound 34:
Method A
A THF solution of lithium tri-tert-butoxyaluminum hydride (1 M, 22.4 mL, 22.4 mmol) was added drop wise to a solution of compound 33 (2.5 g, 5.6 mmol) in THF (25 mL) at ambient temperature. After stirring for an additional 4-5 h, the reaction was determined to be complete by TLC. The reaction was quenched by adding aqueous HCl (1 M, 10 mL) and the mixture was diluted with EtOAc (30 mL). The phases were separated and the organic phase was washed sequentially with water (15 mL) and saturated brine solution (10 mL). The organic phase was then dried over anhydrous Na2S04 (3 g) and filtered. The filtrate was concentrated under vacuum and the resulting solid was purified by column chromatography [29 mm ![]() x 500 mm (L), 60-120 mesh silica, 50 g], eluting with EtOAc/hexane (2:8) [5 mL fractions, monitored by TLC with p- anisaldehyde charring]. The fractions containing the product were combined and concentrated under vacuum to provide compound 34 (2.3 g, 91%) as a white solid.
x 500 mm (L), 60-120 mesh silica, 50 g], eluting with EtOAc/hexane (2:8) [5 mL fractions, monitored by TLC with p- anisaldehyde charring]. The fractions containing the product were combined and concentrated under vacuum to provide compound 34 (2.3 g, 91%) as a white solid.
TLC: p-anisaldehyde charring, Rf for 34 = 0.45 and Rf for 33 = 0.55.
TLC mobile phase: 30% – EtOAc in hexanes.
1H NMR (500 MHz, CDC13): δ = 4.68-4.73 (m, 1H), 3.98 (s, 1H), 3.66 (s, 3H), 2.34-2.40 (m, 1H), 2.21-2.26 (m, 1H), 2.01 (s, 3H), 1.75-1.89 (m, 6H), 1.39-1.68 (m, 16H), 1.00-1.38 (m, 3H), 0.96-0.97 (d, J= 5.5 Hz, 3H), 0.93 (s, 3H), 0.68 (s, 3H).
13C NMR (125 MHz, CDCI3): δ = 174.5, 170.5, 74.1, 72.9, 51.3, 48.1, 47.2, 46.4, 41.7, 35.8, 34.9, 34.7, 34.0, 33.5, 32.0, 30.9, 30.8, 28.6, 27.3, 26.8, 26.3, 25.9, 23.4, 22.9, 21.3, 17.2, 12.6 Mass (m/z) = 449.0 [M+ + 1], 466.0 [M + 18].
IR ( Br) = 3621, 2938, 2866, 1742, 1730, 1262, 1 162, 1041, cm-1.
m.p = 104.2-107.7 °C (from EtOAc).
[α]D = +56 (c = 1% in CHCl3).
ELSD Purity: 97.0%: Retention time = 12.75 (Inertsil ODS 3V, 250 χ 4.6 mm, 5um, ACN: Water (60:40)
Method B
A THF solution of lithium tri-rert-butoxyaluminum hydride (1 M, 107.6 mL, 107.6 mmol) was added over 1 h to a solution of compound 33 (30.0 g, 67 mmol) in dry THF (300 mL) at 0-5 °C. After stirring for an additional 4 h at 5-10 °C, the reaction was determined to be complete by HPLC (NMT 1% of compound 33). The reaction was cooled to 0-5 °C and quenched by adding 4N HCl (473 mL). The phases were separated. The aqueous layer was extracted with DCM (2 x 225 mL) and the combined organic phase was washed sequentially with water (300 mL) and saturated brine solution (300 mL). The organic phase was then was concentrated to dryness by vacuum distillation below 50 °C. Methanol (150 mL) was added to the residue and concentrated to dryness by vacuum distillation below 50 °C. Water (450 mL) was then added to the residue and the mixture was stirred for 15-20 min., filtered and the cake was washed with water (240 mL). The white solid was dried in a hot air drier at 35-40 °C for 6 h to provide compound 34 (30 g, 99.6%).
Conversion of Compound 34 to crude DCA:
Method A
A solution of LiOH (187 mg, 4.4 mmol) in H20 (2.0 mL) was added to a solution of compound 34 (500 mg, 1.1 1 mmol) in THF (8 mL) and MeOH (8 mL). The resulting mixture was stirred for 3-4 h at 50 °C. Upon complete disappearance of the starting material by TLC, the reaction mixture was concentrated under vacuum. A mixture of water (10 mL) and 3 N HCl (1 mL) were combined and cooled to 0 °C and then added to the crude product. After stirring for 1 h at 0 °C, the precipitated solids were filtered and then washed with water (10 mL) and hexane (20 mL). Drying under vacuum at room temperature provided deoxycholic acid (DCA, 400 mg, 91% yield) as a white solid. TLC: -anisaldehyde charring, Rf for DC A = 0.32 and Rf for 2.1a = 0.82.
TLC mobile phase: 10% – Methanol in DCM.
1H NMR (500 MHz, DMSO): δ = 11.92 (s, 1H), 4.44 (s, 1H), 4.19 (s, 1H), 3.77 (s, 1H), 3.35-3.36 (m, 1H), 2.19-2.21 (m, 1H), 2.08-2.10 (m, 1H), 1.73-1.80 (m, 4H), 1.43- 1.63 (m, 6H), 1.15-1.35 (m, 12H), 0.98-1.05 (m, 2H), 0.89-0.90 (d, J = 6.0 Hz, 3H),
0.83 (s, 3H), 0.58 (s, 3H).
13C NMR (125 MHz, DMSO): δ =174.8, 71.0, 69.9, 47.4, 46.1, 46.0, 41.6, 36.3, 35.6, 35.1, 34.9, 33.8, 32.9, 30.8, 30.7, 30.2, 28.6, 27.1, 27.0, 26.1, 23.5, 23.0, 16.9, 12.4.
Mass (m/z) = 393 [M+, + 1].
IR = 3363, 2933, 2863, 1694, 1453, 1372, 1042, cm-1.
m.p. = 171.4-173.6 °C (from ethanol); 174-176 °C (Alfa Aesar) and 171-174 °C (Aldrich)
[<x]D = +47 (c = 1% in EtOH ), +54° (c = 2% in ethanol) [Alfa Aesar]
ELSD Purity: 99.7%: Retention time = 5.25 (Inertsil ODS 3 V, 250 χ 4.6 mm, 5um, ACN:
0.1% TFA in water (90:10).
Method B
A 20% solution of NaOH (40 g, 270 mmol) in H20 (54 mL) was added to a solution of compound 34 (30 g, 67 mmol) in THF (120 mL) and MeOH (120 mL) at 0-5 °C. The resulting mixture was stirred for 4 h at 25-35 °C. Upon completion of reaction by HPLC (NMT 0.5% of compound 34 and intermediates), the solvent was removed via vacuum distillation below 50 °C. The residue was dissolve in water (300 mL) and washed with DCM (2 x 150 mL). The pH of aqueous layer was adjusted to 1-2 with 2N HCl (~ 173 mL). The solids were filtered, washed thoroughly with water (3 L) and dried by a hot air drier at 70-75 °C until the moisture content is less than 2% to provide deoxycholic acid (DCA, 26 g, 99% yield) as a white solid.
EXAMPLE 9
Deoxycholic acid (DCA) Purification
1. Solvent Selection
Two solvent systems were explored for further purification of DCA: • 10% Hexanes in EtOAc
• DCM
The following experiments have been conducted and the experimental results tabulated below.

* The DCA to be purified was dissolved in a mixture of methanol and DCM and then the methanol was removed by azeotropic distillation. The amount of methanol required to dissolve the crude DCA depends on how pure it is to begin with.
Typical crude material was—75% pure and could be dissolved at reflux using 10% methanol-DCA (by volume) using—20 mL per gram. With purer DCA, the percentage of methanol had to be increased to 15%.
Effective purification was achieved by crystallization of the product from DCM following dissolution in a mixture of methanol and DCM and azeotropic removal of the methanol via atmospheric distillation.
2. Solvent Quantity
Experiments have been conducted using different solvent volumes and the experimental results are tabulated below.

Excellent recoveries and product quality were obtained at all solvent levels.
3. Isolation Temperature
The following experiments have been conducted by varying the isolation temperature and the results are tabulated below:

Higher quality product was obtained when isolation is done at 25-30 °C as compared to 10-15 °C. Purification of DCA in 100 g Scale
The final purification procedure for this step is given below:

Crude DCA (110 g) was dissolved in 10% methanol in DCM (2.5 L) at reflux temperature. To this clear solution 2.5 L of dichloromethane was added at reflux temperature and then about 3.0 L of solvent was distilled at atmospheric pressure (GC analysis of reaction mass supernatant revealed the presence of about 3% of methanol). The reaction slurry was cooled to 20-25 °C and then stirred for 3-4 h. The mixture was filtered and the solids were washed with DCM (300 mL). The product was dried in a hot air oven at 50-55 °C for 6-8 h.
The above dried DCA was added to water (1.0 L) and then 10% sodium hydroxide solution (122 mL) was added resulting in a clear solution. This solution was filtered through 5μ filter paper. The filtrate was diluted with water (2.0 L), and the pH was adjusted to 1— 2 with 2N HCl solution (204 mL). The precipitated solids were stirred for 1 h, filtered and the solids were washed with additional water (7.0 L). After drying in a hot air oven at 70-75 °C for 16-20 h, purified DCA (~ 66 g with more than 99% purity by HPLC RI detection) was obtained as a white solid.
TLC: 7-Anisaldehyde charring, Rf for DCA = 0.32 and Rf for compound 34 = 0.82. Eluent = 10% methanol in DCM. 1H NMR (500 MHz, DMSO): δ = 11.92(s, 1H),4.44(s, 1H), 4.19(s, 1H), 3.77 (s, 1H), 3.36-3.35 (m, 1H), 2.21-2.19 (m, 1H), 2.10-2.08 (m, 1H), 1.80-1.73 (m, 4H), 1.63- 1.43(m, 6H), 1.35-1.15(m, 12H), 1.05-0.98(m, 2H), 0.90-0.89 (d, J = 6.0 Hz, 3H), 0.83 (s, 3H), 0.58 (s, 3H).
1 C NMR (125 MHz, DMSO): δ =174.8, 71.0, 69.9, 47.4, 46.1, 46.0, 41.6, 36.3, 35.6, 35.1, 34.9, 33.8, 32.9, 30.8, 30.7, 30.2, 28.6, 27.1, 27.0, 26.1, 23.5, 23.0, 16.9, 12.4.
Mass (m/z) = 393 [M+, + 1].
IR = 3363, 2933, 2863, 1694, 1453, 1372, 1042, cm-1.
m.p. = 171.4-173.6 °C (from ethanol); 174-176 °C (Alfa Aesar) and 171-174 °C (Aldrich).
Recrystallization of Deoxycholic acid (DC A)
DCA obtained from Method B (26 g) above, was charged into a clean and dry flask. Methanol (65 mL) and DCM (585 mL) were added. The mixture was heated to reflux to obtain a clear solution. DCM (650 mL) was charged to the solution and the solvent was distilled atmospherically until 780 mL of solvent was collected. The mixture was assayed by GC to determine the solvent composition. If the methanol content is more than 2%, add DCM (200 mL) and distill atmospherically until 200 mL of distillate have been collected. (Check for the methanol content by GC). The reaction mixture was cooled over 1-2 h to 20-25 °C and stirred at this temperature for 3-4 h. The product was filtered and washed with DCM (81 mL), dried in a hot air drier at 50-55 °C for 8 h. The purity was determined by HPLC. If single max impurity is more than 0.1%, the above process is repeated.
The dried material from the above was charged in to a clean flask. Water (190 mL) was added and followed by 10% aqueous NaOH (3.18 g in 31.8 mL of water). The solution was filtered through 5μ filter paper and the filtrate was diluted with additional water (380 mL). The pH was adjusted to 1-2 with 2 N HCl (53 mL). The resulting solids was filtered, washed thoroughly with water (1.9 L), and dried in a hot air drier at 70-75 °C until the water content is below 1% to give DCA as a white solid (17 g, % of recovery: 65). EXAMPLE 10
Alternate method of Synthesis and purification of DCA from compound 33
Step la— Hydrogenation of methyl 3a-acetoxy-12-oxo—5fi-chol-9(ll)-en-24-oate (24)

Dry Pd/C (75.0 g, 25 wt %) was added to 24 (300.0 g, 0.7 mol) in EtOAc (7.5 L, 25 vol). The reaction mixture was heated to 45°— 50°C and pressurized to 50 psi of H2. HPLC analysis after 21 hours indicated < 1.0% area under the curve (AUC) of 24 remained; 4.6% AUC of the allylic alcohol impurity 86 and 1 1.1% AUC of the 87 formed. The reaction mixture was cooled to 30° – 35°C, filtered over Hyflo® (300 g) and washed with EtOAc (7.5 L) to remove the catalyst. The resulting filtrate was
concentrated to about 6 L and taken forward without further manipulation (67.8% AUC by HPLC, 5.5% AUC of the allylic alcohol impurity 86 and 13.0% AUC of 87).
Step lb/c – Oxidation of allylic alcohol 86 and 87 and rehydrogenation of 24 to methyl 3a-acetoxy-12-oxo-5fi-cholan-24-oate (33)

Step lb – PCC oxidation of allylic alcohol 86 and 87
A slurry of PCC (149.1 g, 1.03 equiv.) in EtOAc (1.5 L) was added to the 33 solution from above at 20°— 25°C. The reaction was allowed to proceed for 3.5 hours where HPLC analysis showed that < 1% AUC of the allylic alcohol 86 and < 1% AUC of 87 remained. The reaction mixture was filtered over Hyflo® (300 g) and washed with EtOAc (3.0 L). The EtOAc filtrate was washed with deionized (DI) water (2 x 3.6 L) and brine (3.6 L), filtered over Hyflo® (300 g) and washed with EtOAc (3.0 L). The resulting filtrate was concentrated to -7.5 L and taken forward without further manipulation (77.7% AUC by HPLC containing 5.3% AUC of 24).
Step lc— Rehydrogenation of 24 to 33
Powder activated carbon DARCO (60 g, 20 wt %) was added to the crude 33 solution from above containing 24. The resulting slurry was heated to 45°— 50°C for 4 hours, cooled to 30°— 35°C and filtered over Celite®. The filter cake was washed with EtOAc (7.5 L), concentrated to -7.5 L and added to dry Pd/C (60.0 g, 20 wt %). The reaction mixture was heated to 45° – 50°C and pressurized to 50 psi of H2 for 6 hours. HPLC analysis indicated < 1.0% AUC of 24 remained; 1.1% AUC of 86 impurity and < 1.0% AUC of 87 formed. The reaction was deemed complete and cooled to 30° – 35°C, filtered over Celite® and washed with EtOAc (7.5 L). The EtOAc filtrate was concentrated to—5 volumes and azeotroped with MeOH (2 x 4.5 L) back down to—5 volumes. The resulting slurry was diluted with DI water (2.4 L) and maintained at 20-25 °C. The slurry was filtered, washed with DI water (2 x 600 mL) and dried under vacuum at 40° – 50°C to yield 266 g (88%) of 33 (66.2% AUC by HPLC).
Step 2— Synthesis of 34
A solution of 33 (245 g, 0.5 mol) in THF (2.5 L) was cooled to 0° – 5°C and 1 M solution of Li(t-BuO)3A1H (822.9 niL, 1.5 equiv.) was added while maintaining the temperature below 5°C. The reaction mixture was stirred at 5° – 10°C for 22 hours. Reaction may be complete in 2-4 hours. HPLC analysis indicated that the reaction was complete with < 1% of 33 remaining. The reaction was quenched with 4 M HCl (3.7 L) while maintaining the temperature below 20°C. The reaction mixture was extracted with CH2CI2 (2 x 2.5 L) and the combined organic phases were washed with DI water (2 x 2.5 L). The CH2C12 phase was concentrated to afford 300 g (122%) of 34 (73.5% AUC by HPLC). 1H NMPv analysis indicated that 9.7 wt % of THF and 0.8 wt % of CH2C12 remained.
Step 3 – Synthesis of DCA
A NaOH solution (87.6 g, 4 equiv.) in DI water (438.6 mL) was added to a solution of 34 (245 g, 0.5 mol) in MeOH (980 mL) and THF (475 mL) at 0° – 5°C. The reaction mixture was allowed to warm to 20° – 25°C. HPLC analysis showed that the reaction was complete after 1 hour with < 0.5% 34 and < 0.5% of the hydrolysis intermediates remaining. The reaction was diluted with DI water (2.5 L) and
concentrated to—10 volumes. The aqueous solution was washed with CH2C12 (2 x 1.3 L) and adjusted to pH 1.7— 2.0 using 2 M HCl (1.6 L). A white slurry formed and was stirred at 20° – 25 °C for 1 hour. The slurry was filtered, washed with DI water (7 x 1 L) and dried under vacuum to yield 195 g (91%) of DCA (82.2% AUC by HPLC).
Step 4 – Purification of DCA
A solution of DCA obtained above (190 g, 0.48 mol) in MeOH (475 mL) and CH2C12 (4275 mL) was heated to 35° – 40°C. The MeOH/CH2Cl2 was distilled out of the mixture while CH2CI2 (4740 mL) was added matching the rate of distillation. Analysis of the solvent composition by Ή NMR indicated 4.5 mol % of MeOH remained relative to CH2C12. The slurry was allowed to cool to 20°— 25°C and held for 16 hours. The solids were isolated by filtration, washed with CH2Cl2 (600 mL) and dried under vacuum to yield 104 g (55%) of DCA (> 99% AUC by HPLC-RID and 98.7% AUC by HPLC- CAD).
The recrystallization was repeated by heating a mixture of DCA (103 g, 0.3 mol) in MeOH (359 mL) and CH2C12 (1751 mL) to 35° – 40°C. The MeOH/CH2Cl2 was distilled out of the mixture while CH2CI2 (3760 mL) was added matching the rate of distillation. Analysis of the solvent composition by 1H NMR indicated 4.7 mol % of MeOH remained relative to CH2C12. The slurry was allowed to cool to 20°— 25°C. After 1 hour, the solids were isolated by filtration, washed with CH2CI2 (309 mL) and dried under vacuum to afford 82 g (79%) of DCA (> 99% AUC by HPLC-RID and 99.3% AUC by HPLC-C AD).
To assess the effect of additional purification and reprocessing, the product was recrystallized a third time prior to the normal final water isolation step. The above sample of DCA (80 g, 0.2 mol) in MeOH (240 mL) and CH2C12 (1400 mL) was heated to 35° – 40°C. The MeOH/CH2Cl2 was distilled out of the mixture while CH2C12 (2000 mL) was added matching the rate of distillation. Analysis of the solvent composition by !H NMR indicated 6.7 mol % of MeOH remained relative to CH2C12. The slurry was allowed to cool to 20° – 25°C. After 1 hour, the solids were isolated by filtration, washed with CH2CI2 (240 mL) and dried under vacuum to afford 72 g (89%) of DCA (99.7% AUC by HPLC-CAD).
The sample was slurried in DI water (840 mL) and diluted with a solution of
NaOH (14.0 g) in DI water (140 mL). The resulting solution was filtered over Celite® and washed with DI water (1.4 L). The filtrate was adjusted to pH 1.6 with 2 M HCl (—300 mL) resulting in a white precipitate which was held for 1 hour at 20°— 25°C. The product was isolated by filtration, washed with DI water (9.0°L) and dried under vacuum to afford 63 g (87%) of DCA (99.7% AUC by HPLC-CAD).
SEE MORE IN PATENT
………………………………..
WO 2013044119
http://www.google.com/patents/WO2013044119A1?cl=en

Scheme 10

Example 4: Converting Compound 129 To DCA
[0125| In Scheme 1 below, there is provided a scheme for the synthesis and purification of DCA from compound 1.
Scheme 10
A. Conversion of Compound 129 to Compound 130:
Method Al
[0126] 10% Pd/C (900 mg) was added to a solution of compound 129 (2.0 g, 4.5 mmol) in EtOAc (150 mL) and the resulting slurry was hydrogenated in a Parr apparatus (50 psi) at 50 °C for 16 h. At this point the reaction was determined to be complete by TLC. The mixture was filtered through a small plug of Celite® and the solvent was removed under vacuum, providing compound 130 (1.6 g, 80% yield) as a white solid.
TLC: -anisaldehyde charring, Rt for 130 = 0.36. TLC mobile phase: 20% – EtOAc in hexanes.
Ή NMR (500 MHz, CDCL): δ = 4.67-4.71 (m, 1 H), 3.66 (s, 3H), 2.45-2.50 (t, J = 15 Hz, 2H ), 2.22-2,40 (m, 1H), 2.01 (s, 3H). 1 ,69- 1 .96 (m, 9H), 1 ,55 (s, 4H), 1 ,25- 1.50 (m, 8H)5 1.07-1 . 19 (m. 2H), 1 .01 (s, 6H), 0.84-0.85 (d, J = 7.0 Hz, 3H).
13C NMR (125 MHz, CDC13): δ = 214.4, 174.5, 170.4, 73.6, 58,5, 57.4, 51.3, 46,4, 43.9, 41.2, 38.0, 35.6, 35.5, 35.2, 34.8, 32.0, 31 .2, 30.4, 27.4. 26.8, 26.2, 25.9, 24.2, 22.6, 21 .2, 18.5, 1 1.6,.
Mass (m/z) = 447.0 | \! + 1 ], 464.0 [Mf + 18]. IR ( Br) = 3445, 2953, 2868, 1731 , 1698, 1257, 1029 cm“1 , m.p. = 142,2- 144.4 °C (from EtOAc/hexanes mixture). [a]D = +92 (c = l % in CHCl3).
ELSD Purity: 96.6%: Retention time = 9.93 (Inertsil ODS 3V, 250 * 4.6 mm, 5 urn, ACN: 0.1 % TFA in water (90: 10)
Method A2
[0127J A slurry of 10% Pd/C (9 g in 180 mL of ethyl acetate) was added to a solution of compound 129 (36 g, 81 mmol) in EtOAc (720 mL) and the resulting slurry was treated with hydrogen gas (50 psi) at 45-50 °C for 16 h. (A total of 1080 mL of solvent may be used). At this point the reaction was determined to be complete by HPLC (NMT 1 % of compound 129). The mixture was filtered through Cclite® (10 g) and washed with ethyl acetate (900 mL). The filtrate was concentrated to 50% of its volume via vacuum distillation below 50 °C. To the concentrated solution was added pyridinium
chlorochromate (20.8 g) at 25-35 °C and the mixture was stirred for 2 h at 25-35 °C, when the reaction completed by LIPLC (allylic alcohol content is NMT 1 %).
[0128] The following process can be conducted if compound 129 content is more than 5%. Filter the reaction mass through Celite® (10 g) and wash with ethyl acetate (360 mL). Wash the filtrate with water (3 x 460 mL) and then with saturated brine (360 mL). Dry the organic phase over sodium sulphate (180 g), filter and wash with ethyl acetate ( 180 mL). Concentrate the volume by 50% via vacuum distillation below 50 °C. Transfer the solution to a clean and dry autoclave. Add slurry of 10% palladium on carbon (9 g in 1 80 mL of ethyl acetate). Pressurize to 50 psi with hydrogen and stir the reaction mixture at 45-50 °C for 16 h.
[0129] Upon complete consumption of compound 129 by HPLC ( the content of compound 129 being NMT 1 %), the reaction mixture was filtered through Celite® ( 10 g) and the cake was washed with ethyl acetate (900 mL). The solvent was concentrated to dryness via vacuum distillation below 50 °C. Methanol (150 mL) was added and concentrated to dryness via vacuum distillation below 50 °C. Methanol (72 mL) was added to the residue and the mixture was stirred for 15-20 min at 10- 15 °C, filtered and the cake was washed with methanol (36 mL). The white solid was dried in a hot air drier at 45-50 °C for 8 h to LOD being NMT 1% to provide compound 230 (30 g, 83.1 % yield).
B. Conversion of Compound 130 to Compound 1 1.a
Method Bl
[0130J A THF solution of lithium tri-te -butoxyaluminum hydride (1 M. 22.4 mL, 22.4 mmol) was added drop wise to a solution of compound 130 (2.5 g, 5.6 mmol) in THF (25 mL) at ambient temperature. After stirring for an additional 4-5 h, the reaction was determined to be complete by TLC. The reaction was quenched by adding aqueous HQ (1 M, 10 mL) and the mixture was diluted with EtOAc (30 mL). The phases were separated and the organic phase was washed sequentially with water (15 mL) and saturated brine solution (10 mL). The organic phase was then dried over anhydrous Na2SO-i (3 g) and filtered. The filtrate was concentrated under vacuum and the resulting solid was purified by column chromatography [29 mm ![]() x 500 mm (L), 60-120 mesh silica, 50 g], eluting with EtOAc/hexane (2:8) [5 mL fractions, monitored by TLC with p- anisaldehyde charring]. The fractions containing the product were combined and concentrated under vacuum to provide compound 131. a (2.3 g, 91 %) as a white solid.
x 500 mm (L), 60-120 mesh silica, 50 g], eluting with EtOAc/hexane (2:8) [5 mL fractions, monitored by TLC with p- anisaldehyde charring]. The fractions containing the product were combined and concentrated under vacuum to provide compound 131. a (2.3 g, 91 %) as a white solid.
TLC: /7-anisaldehyde charring, Rf for 131. a = 0.45 and Rt for 130 = 0.55. TLC mobile phase: 30% – EtOAc in hexanes.
Ή NMR (500 MHz, CDC13): δ = 4.68-4.73 (m, 1 H), 3.98 (s, 1 H), 3.66 (s, 3H), 2.34-2.40 (m, 1H), 2.21-2.26 (m, 1H), 2.01 (s, 3H), 1.75-1.89 (m, 6H), 1.39-1.68 (m, 16H), 1.00-1.38 (m, 3H), 0.96-0.97 (d, J = 5.5 Hz, 3H), 0.93 (s, 3H), 0.68 (s, 3H).
13C NMR (125 MHz, CDCI3): δ = 174.5, 170.5, 74.1 , 72.9, 51.3, 48.1 , 47.2, 46.4, 41.7, 35.8, 34.9, 34.7, 34.0, 33.5, 32.0, 30.9, 30.8, 28.6, 27.3, 26.8, 26.3, 25.9, 23.4. 22.9, 21.3. 17.2, 12.6
Mass (m/z) = 449.0 [M+ + 1 ], 466.0 [M+ + 18].
IR (KBr) = 3621 , 2938, 2866, 1742, 1730, 1262, 1 162, 1041 , cm4. m.p = 104.2-107.7 °C (from EtOAc).
[<x]D = +56 (c = 1% in CHCI3). ELSD Purity: 97.0%: Retention time = 12.75 (Inertsil ODS 3V, 250 χ 4.6 mm, 5 urn, ACN: Water (60:40)
Method B2
[0131 ] A THF solution of lithium tri-/er?-butoxyaluminum hydride (1 M, 107.6 mL, 107.6 mmol) was added over 1 h to a solution of compound 130 (30.0 g, 67 mmol) in dry THF (300 mL) at 0-5 °C. After stirring for an additional 4 h at 5-10 °C, the reaction was determined to be complete by HPLC (NMT 1% of compound 130). The reaction was cooled to 0-5 °C and quenched by adding 4N HC1 (473 mL). The phases were separated. The aqueous layer was extracted with DCM (2 x 225 mL) and the combined organic phase was washed sequentially with water (300 mL) and saturated brine solution (300 mL). The organic phase was then was concentrated to dryness by vacuum distillation below 50 °C. Methanol (150 mL) was added to the residue and concentrated to dryness by vacuum distillation below 50 °C. Water (450 mL) was then added to the residue and the mixture was stirred for 15-20 min., filtered and the cake was washed with water (240 mL). The white solid was dried in a hot air drier at 35-40 °C for 6 h to provide compound 131.a (30 g, 99.6%).
C. Conversion of Compound 131.a to crude DCA:
[01321 To a solution of 131. a in MeOH (4 vol) and THF (4 vol) was added a solution of NaOH (4.0 equiv) in DI water (5 M) maintaining the temperature below 20 °C. HPLC analysis after 20 hours at 20-25 °C indicated <0.5% AUC of 131.a and the two
intermediates remained. The reaction was deemed complete, diluted with DI water (10 vol) and concentrated to -10 volumes. The sample was azeotroped with 2-MeTHF (2 x 10 vol) and assayed by Ή NMR to indicate MeOH was no longer present. The rich aqueous phase was washed with 2-MeTHF (2 x 10 vol) and assayed by HPLC to indicate 0.3% AUC of the alcohol impurity remained. The aqueous phase was diluted with 2- MeTHF (10 vol ) and adjusted to pH = 1 .7-2.0 using 2 M HC1 (~4 vol ). The phases were separated and the 2-MeTHF phase was washed with DI water (2 x 10 vol). The 2- MeTHF phase was filtered over Celite and the filter cake was washed with 2-MeTHF (2 vol). The 2-MeTHF filtrate was distillated to -10 volumes and azeotroped with ^-heptane containing Statsafe™ 5000 (3 x 10 vol) down to -10 vol. The mixture was assayed by Ή N MR to indicate <5 mol% of 2-MeTHF remained relative to o-heptane. The slurry was held for a minimum of 2 hours at 20-25 °C and filtered. The filter cake was washed with //-heptane (2 x 10 vol) and conditioned under vacuum on the Niitsche filter with N2 for a minimum of 1 hour to afford DCA-crude as white solids. Purity = 94.6% (by HPLC). HPLC analysis for DS-DCA (NMT 5% AUC).
D. Recrystallization of DCA
|0133] DCA-crude was diluted with 2 mol% MeOH in CH2C12 (25 vol) and heated to 35—37 °C for 1 hour. The slurry was allowed to cool to 28-30 °C and filtered. The filter cake was washed with CITC (5 vol) and dried under vacuum at 40 °C to afford DCA. HPLC analysis for DS-DCA (NMT 0.15% AUC).
[0134] DCA was dissolved in 10% DI water/ EtOH (12 vol), polish filtered over Celite and washed with 10% DI water/ EtOH (3 vol). The resulting 15 volume filtrate was added to DI water (30 vol) and a thin white slurry was afforded. The slurry was held for 24 hours, filtered, washed with DI water (20 vol) and dried under vacuum at 40 °C to afford pure DCA. OVI analysis for CH2C12. EtOH. ^-heptane, MeOH and MeTHF was conducted to ensure each solvent was below ICH guideline. KF analysis conducted (NMT 2.0%). Purity = 99.75% (by HPLC). Yield from DCA-crude = 59%.
……………………………
WO 2012174229
http://www.google.com/patents/WO2012174229A2?cl=en
In Scheme 1 below, there is provided a scheme for the synthesis and purification of deoxycholic acid from compound 1.
Scheme 1


Conversion of Compound 1 to Compound 2:
[0043] The hydrogenation of compound 1 on 10.0 g scale using dry 10 % Pd/C (15 wt %) in ethyl acetate (20 parts) was added and applied about 50 psi hydrogen pressure and temperature raised to 70 °C. After reaching temperature 70 °C, observed increase of hydrogen pressure to about 60 psi, at these conditions maintained for 60 h. After 60 hours 0.6% of compound 2 and 2.75%> of allylic alcohol were still observed, so further stirred for additional 12 h (observed 0.16% of allylic alcohol and 0.05% of compound 2). After work-up, the reaction provided 9.5 g of residue.
[0044] Anther hydrogenation reaction on 25 g of compound 1 with above conditions for 76 h provided 24.5 g of residue.
Method A
[0045] 10% Pd/C (900 mg) was added to a solution of compound 1 (2.0 g, 4.5 mmol) in EtOAc (150 mL) and the resulting slurry was hydrogenated in a Parr apparatus (50 psi) at 50 °C for 16 h. At this point the reaction was determined to be complete by TLC. The mixture was filtered through a small plug of Celite® and the solvent was removed under vacuum, providing compound 2 (1.6 g, 80%> yield) as a white solid.
TLC: /?-anisaldehyde charring, Rf for 2 TLC mobile phase: 20% – EtOAc in hexanes.
1H NMR (500 MHz, CDC13): δ = 4.67-4.71 (m, 1H), 3.66 (s, 3H), 2.45-2.50 (t, J = 15 Hz, 2H), 2.22-2.40 (m, 1H), 2.01 (s, 3H), 1.69-1.96 (m, 9H), 1.55 (s, 4H), 1.25-1.50 (m, 8H), 1.07-1.19 (m, 2H), 1.01 (s, 6H), 0.84-0.85 (d, J= 7.0 Hz, 3H).
13C NMR (125 MHz, CDC13): δ = 214.4, 174.5, 170.4, 73.6, 58.5, 57.4, 51.3, 46.4, 43.9, 41.2, 38.0, 35.6, 35.5, 35.2, 34.8, 32.0, 31.2, 30.4, 27.4, 26.8, 26.2, 25.9, 24.2, 22.6, 21.2, 18.5,11.6,.
Mass (m/z) = 447.0 [M+ + 1], 464.0 [M+ + 18].
IR (KBr) = 3445, 2953, 2868, 1731, 1698, 1257, 1029 cm“1.
m.p. =142.2-144.4 °C (from EtO Ac/hex anes mixture).
[a]D = +92 (c = 1% in CHC13).
ELSD Purity: 96.6%: Retention time = 9.93 (Inertsil ODS 3V, 250 4.6 mm, 5 urn, ACN: 0.1% TFA in water (90: 10)
Method B
[0046] A slurry of 10%> Pd/C (9 g in 180 mL of ethyl acetate) was added to a solution of compound 1 (36 g, 81 mmol) in EtO Ac (720 mL) and the resulting slurry was treated with hydrogen gas (50 psi) at 45-50 °C for 16 h. (A total of 1080 mL of solvent may be used). At this point the reaction was determined to be complete by HPLC (NMT 1% of compound 1). The mixture was filtered through C elite® (10 g) and washed with ethyl acetate (900 mL). The filtrate was concentrated to 50% of its volume via vacuum distillation below 50 °C. To the concentrated solution was added pyridinium
chlorochromate (20.8 g) at 25-35 °C and the mixture was stirred for 2 h at 25-35 °C, when the reaction completed by HPLC (allylic alcohol content is NMT 1%).
[0047] The following process can be conducted if compound 1 content is more than 5%>. Filter the reaction mass through Celite® (10 g) and wash with ethyl acetate (360 mL). Wash the filtrate with water (3 x 460 mL) and then with saturated brine (360 mL). Dry the organic phase over sodium sulphate (180 g), filter and wash with ethyl acetate (180 mL). Concentrate the volume by 50% via vacuum distillation below 50 °C. Transfer the solution to a clean and dry autoclave. Add slurry of 10% palladium on carbon (9 g in 180 mL of ethyl acetate). Pressurize to 50 psi with hydrogen and stir the reaction mixture at 45-50 °C for 16 h.
[0048] Upon complete consumption of compound 1 by HPLC (the content of compound 1 being NMT 1%), the reaction mixture was filtered through Celite® (10 g) and the cake was washed with ethyl acetate (900 mL). The solvent was concentrated to dryness via vacuum distillation below 50 °C. Methanol (150 mL) was added and concentrated to dryness via vacuum distillation below 50 °C. Methanol (72 mL) was added to the residue and the mixture was stirred for 15-20 min at 10-15 °C, filtered and the cake was washed with methanol (36 mL). The white solid was dried in a hot air drier at 45-50 °C for 8 h to LOD being NMT 1% to provide compound 2 (30 g, 83.1 % yield).
Conversion of Compound 2 to Compound 3:
Method A
[0049] A THF solution of lithium tri-tert-butoxyaluminum hydride (1 M, 22.4 mL, 22.4 mmol) was added drop wise to a solution of compound 2 (2.5 g, 5.6 mmol) in THF (25 mL) at ambient temperature. After stirring for an additional 4-5 h, the reaction was determined to be complete by TLC. The reaction was quenched by adding aqueous HCl (1 M, 10 mL) and the mixture was diluted with EtO Ac (30 mL). The phases were separated and the organic phase was washed sequentially with water (15 mL) and saturated brine solution (10 mL). The organic phase was then dried over anhydrous Na2S04 (3 g) and filtered. The filtrate was concentrated under vacuum and the resulting solid was purified by column chromatography [29 mm ![]() x 500 mm (L), 60-120 mesh silica, 50 g], eluting with EtO Ac/hex ane (2:8) [5 mL fractions, monitored by TLC with p- anisaldehyde charring]. The fractions containing the product were combined and concentrated under vacuum to provide compound 3 (2.3 g, 91%) as a white solid.
x 500 mm (L), 60-120 mesh silica, 50 g], eluting with EtO Ac/hex ane (2:8) [5 mL fractions, monitored by TLC with p- anisaldehyde charring]. The fractions containing the product were combined and concentrated under vacuum to provide compound 3 (2.3 g, 91%) as a white solid.
TLC: /?-anisaldehyde charring, Rf for 3 = 0.45 and Rf for 2 = 0.55.
TLC mobile phase: 30% – EtO Ac in hexanes.
1H NMR (500 MHz, CDC13): δ = 4.68-4.73 (m, 1H), 3.98 (s, 1H), 3.66 (s, 3H), 2.34-2.40 (m, 1H), 2.21-2.26 (m, 1H), 2.01 (s, 3H), 1.75-1.89 (m, 6H), 1.39-1.68 (m, 16H), 1.00-1.38 (m, 3H), 0.96-0.97 (d, J= 5.5 Hz, 3H), 0.93 (s, 3H), 0.68 (s, 3H). ljC NMR (125 MHz, CDC13): δ = 174.5, 170.5, 74.1, 72.9, 51.3, 48.1, 47.2, 46.4, 41.7, 35.8, 34.9, 34.7, 34.0, 33.5, 32.0, 30.9, 30.8, 28.6, 27.3, 26.8, 26.3, 25.9, 23.4, 22.9, 21.3, 17.2, 12.6
Mass (m/z) = 449.0 [M+ + 1], 466.0 [M+ + 18].
IR (KBr) = 3621, 2938, 2866, 1742, 1730, 1262, 1162, 1041, cm“1.
m.p = 104.2-107.7 °C (from EtOAc).
[a]D = +56 (c = 1% in CHC13).
ELSD Purity: 97.0%: Retention time = 12.75 (Inertsil ODS 3V, 250 4.6 mm, 5 urn, ACN: Water (60:40)
Method B
[0050] A THF solution of lithium tri-tert-butoxyaluminum hydride (1 M, 107.6 mL, 107.6 mmol) was added over 1 h to a solution of compound 2 (30.0 g, 67 mmol) in dry THF (300 mL) at 0-5 °C. After stirring for an additional 4 h at 5-10 °C, the reaction was determined to be complete by HPLC (NMT 1% of compound 2). The reaction was cooled to 0-5 °C and quenched by adding 4N HC1 (473 mL). The phases were separated. The aqueous layer was extracted with DCM (2 x 225 mL) and the combined organic phase was washed sequentially with water (300 mL) and saturated brine solution (300 mL). The organic phase was then was concentrated to dryness by vacuum distillation below 50 °C. Methanol (150 mL) was added to the residue and concentrated to dryness by vacuum distillation below 50 °C. Water (450 mL) was then added to the residue and the mixture was stirred for 15-20 min., filtered and the cake was washed with water (240 mL). The white solid was dried in a hot air drier at 35-40 °C for 6 h to provide compound 3 (30 g, 99.6%).
Conversion of Compound 3 to crude DCA:
[0051] To a solution of 3 in MeOH (4 vol) and THF (4 vol) was added a solution of NaOH (4.0 equiv) in DI water (5 M) maintaining the temperature below 20 °C. HPLC analysis after 20 hours at 20-25 °C indicated <0.5% AUC of 3 and the two intermediates remained. The reaction was deemed complete, diluted with DI water (10 vol) and concentrated to ~10 volumes. The sample was azeotroped with 2-MeTHF (2 x 10 vol) and assayed by 1H NMR to indicate MeOH was no longer present. The rich aqueous phase was washed with 2-MeTHF (2 x 10 vol) and assayed by HPLC to indicate 0.3% AUC of the alcohol impurity remained. The aqueous phase was diluted with 2-MeTHF (10 vol) and adjusted to pH = 1.7-2.0 using 2 M HC1 (~4 vol). The phases were separated and the 2-MeTHF phase was washed with DI water (2 x 10 vol). The 2- MeTHF phase was filtered over Celite and the filter cake was washed with 2-MeTHF (2 vol). The 2-MeTHF filtrate was distillated to ~10 volumes and azeotroped with n-heptane containing Statsafe™ 5000 (3 x 10 vol) down to ~10 vol. The mixture was assayed by 1H NMR to indicate <5 mol% of 2-MeTHF remained relative to n-heptane. The slurry was held for a minimum of 2 hours at 20-25 °C and filtered. The filter cake was washed with n-heptane (2 x 10 vol) and conditioned under vacuum on the Nutsche filter with N2 for a minimum of 1 hour to afford DCA-crude as white solids. Purity = 94.6% (by HPLC). HPLC analysis for DS-DCA (NMT 5% AUC).
Recrystallization of Deoxycholic acid (DCA)
[0052] DCA-crude was diluted with 2 mol% MeOH in CH2C12 (25 vol) and heated to 35-37 °C for 1 hour. The slurry was allowed to cool to 28-30 °C and filtered. The filter cake was washed with CH2C12 (5 vol) and dried under vacuum at 40 °C to afford DCA. HPLC analysis for DS-DCA (NMT 0.15% AUC).
[0053] DCA was dissolved in 10% DI water/ EtOH ( 12 vol), polish filtered over Celite and washed with 10% DI water/ EtOH (3 vol). The resulting 15 volume filtrate was added to DI water (30 vol) and a thin white slurry was afforded. The slurry was held for 24 hours, filtered, washed with DI water (20 vol) and dried under vacuum at 40 °C to afford pure DCA. OVI analysis for CH2C12, EtOH, n-heptane, MeOH and MeTHF was conducted to ensure each solvent was below ICH guideline. KF analysis conducted (NMT 2.0%). Purity = 99.75% (by HPLC). Yield from DCA-crude = 59%.
……………………………..
| WO2011075701A2 * | Dec 17, 2010 | Jun 23, 2011 | Kythera Biopharmaceuticals, Inc. | Methods for the purification of deoxycholic acid |
| EP0336521B1 * | Apr 7, 1989 | Apr 1, 1992 | Roussel-Uclaf | 9-alpha-hydroxy-17-methylene steroids, process for their preparation and their use in the preparation of corticosteroids |
| US20100179337 * | May 16, 2008 | Jul 15, 2010 | Kythera Biopharmaceuticals, Inc. | Preparation of bile acids and intermediates thereof |
old cut paste
http://clinicaltrials.gov/ct2/show/NCT01426373
The drug is sodium deoxycholate for injection, code-named ATX-101 was developed for the treatment of lipomas – benign tumors of subcutaneous adipose tissue, as well as other unwanted fatty growths, such as a double chin. This substance, which is a salt of one of the bile acids, emulsifies fats, destroying their excess deposits


ATX-101 (a first-in-class injectable drug being studied for the reduction of localized fat. ATX-101 is a proprietary formulation of deoxycholate a well-studied endogenous compound that is present in the body), a facial injectable drug for the reduction of unwanted fat under the chin, or submental fat. V. Leroy Young, MD, FACS, presented the initial results at the American Society for Aesthetic Plastic Surgery (ASAPS) 45th Annual Aesthetic Meeting in Vancouver, British Columbia, on May 4, 2012.
In August 2010 Bayer Consumer Care AG signed a licensing and development collaboration agreement with KYTHERA, thereby obtaining commercialization rights to ATX-101 outside the US and Canada. KYTHERA and Bayer are collaborating on the development of ATX-101 in Europe.
KYTHERA Biopharmaceuticals Inc. 02 MAR 3013, announced positive interim results from a Phase IIIb multi-center open-label study (ATX-101-11-26) to evaluate the safety and efficacy of ATX-101 an investigational injectable drug for the reduction of unwanted submental fat (SMF) commonly known as double chin. The results presented at the Late Breaking Research Symposium at the 71st American Academy of Dermatology (AAD) Annual Meeting in Miami Beach Fla. found that ATX-101 is well-tolerated and may be effective in reducing SMF by both clinician and patient reported outcome measures. The ATX-101 global clinical development program has enrolled more than 2500 total patients of which more than 1500 have been treated with ATX-101.
“In my practice patients often request a non-surgical way to treat their submental fat or undesirable double chin” said investigator Susan Weinkle MD FAAD a board certified dermatologist and affiliate clinical professor at the University of South Florida. “For these patients double chin is often resistant to diet and exercise. The results of this study suggest that microinjections of ATX-101 can reduce submental fat without worsening skin laxity.”
ATX-101 is a proprietary synthetically-derived formulation of deoxycholic acid (DCA) a naturally-occurring molecule found in the body that aids in fat metabolism. In this open-label Phase IIIb study interim results three months after the last ATX-101 treatment found:
- Reduction of submental fat
- 87 percent of patients achieved at least a one-grade improvement from baseline on the Clinician-Reported Submental Fat Rating Scale (CR-SMFRS)
- Similarly 83 percent of patients achieved at least a one-grade improvement on the Patient-Reported Submental Fat Rating Scale (PR-SMFRS)
- 96 percent of patients had unchanged or improved skin laxity based on the clinician rated Submental Skin Laxity Grading Scale (SMSLG)
- 95 percent of patients were satisfied with treatment based on the Global Post Treatment Satisfaction Scale
- Adverse events were of mild to moderate intensity transient and primarily associated with the treatment area
Topline results from this study were announced in November 2012. As previously announced 71.3 percent of subjects had at least a one-grade improvement on the CR-SMFRS / PR-SMFRS composite and 14.0 percent had at least a two-grade improvement on the same composite measure.
These results are based on a multicenter 12-month open-label Phase IIIb study conducted at 21 sites across the United States evaluating 165 adults who received injections of ATX-101 for up to six treatments at four-week intervals. Patients received ATX-101 (2 mg/cm2) by subcutaneous microinjections directly into their SMF and were evaluated three months after their last treatment. The study population includes females (77.6 percent) and males (22.4 percent) with a mean age of 47 who report at least moderate SMF and dissatisfaction with the appearance of their chin. All Fitzpatrick Skin Types an industry standard scale to categorize skin tone are represented.
“We are pleased with these ATX-101 study results” said Patricia S. Walker M.D. Ph.D. chief medical officer KYTHERA Biopharmaceuticals Inc. “These results along with efficacy analyses in double-blind placebo-controlled studies support ATX-101 entering the market as potentially the first medical aesthetic drug approved for the reduction of submental fat.”
About ATX-101
ATX-101 is a potential first-in-class injectable drug candidate under clinical investigation for the reduction of unwanted submental fat. ATX-101 is a proprietary formulation of synthetic deoxycholic acid a well-characterized endogenous compound that is present in the body to promote the natural breakdown of dietary fat. ATX-101 is designed to be a locally-injected drug that causes proximal preferential destruction of adipocytes or fat cells with minimal effect on surrounding tissue. Based on clinical trials conducted to date ATX-101 has exhibited significant meaningful and durable results in the reduction of submental fat which commonly presents as an undesirable “double chin.” These results correspond with subject satisfaction measures demonstrating meaningful improvement in perceived chin appearance.
In August 2010 Bayer signed a licensing and collaboration development agreement with KYTHERA thereby obtaining development and commercialization rights to ATX-101 outside of the U.S. and Canada. Bayer recently completed two pivotal Phase III trials of ATX-101 in Europe for the reduction of submental fat. Topline results from these trials were reported in the second quarter of 2012. KYTHERA completed enrollment in its pivotal Phase III clinical program for ATX-101 in more than 1000 subjects randomized to ATX-101 or placebo in 70 centers across the United States and Canada in August 2012. The Company expects to release topline results in mid-2013.
About KYTHERA Biopharmaceuticals Inc.
KYTHERA Biopharmaceuticals Inc. is a clinical-stage biopharmaceutical company focused on the discovery development and commercialization of novel prescription products for the aesthetic medicine market. KYTHERA initiated its pivotal Phase III clinical program for ATX-101 in March 2012 and completed enrollment of more than 1000 patients randomized to ATX-101 or placebo in 70 centers across the U.S. and Canada in August 2012. KYTHERA also maintains an active research interest in hair and fat biology. Find more information at www.kytherabiopharma.com.

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX,


FDA Accepts Filing of NDA for IV Antibiotic Oritavancin with Priority Review

Oritavancin
(4R)-22-O-(3-Amino-2,3,6-trideoxy-3-C-methyl-alpha-L-arabinohexopyranosyl)-N3-(p-(p-chlorophenyl)benzyl)vancomycin
(3S, 6R, 7R, 22R, 23S, 26S, 36R, 38aR) -22 – (3-Amino-2 ,3,6-trideoxy-3-C-methyl-alpha-L-mannopyranosyloxy) -3 – (carbamoylmethyl ) -10,19-dichloro-44-[2-O-[3 – (4′-chlorobiphenyl-4-ylmethylamino) -2,3,6-trideoxy-3-C-methyl-alpha-L-mannopyranosyl] – beta-D-glucopyranosyloxy] –
| CAS No. | 171099-57-3 |
| CBNumber: | CB92451283 |
| Molecular Formula: | C86H97Cl3N10O26 |
| Formula Weight: | 1793.12 |
Also known as NDISACC-(4-(4-chlorophenyl)benzyl)A82846B and LY333328,N-(4-(4-chlorophenyl)benzyl)A82846B
Abbott (Supplier), Lilly (Originator), InterMune (Licensee)
The medicines company—
-
the Oritavancin Program Results.pdf
 phx.corporate-ir.net/External.File?item…t=1
phx.corporate-ir.net/External.File?item…t=1Jul 2, 2013 – Inhibits two key steps of cell wall synthesis: – Transglycosylation. – Transpeptidation. • Disrupts bacterial membrane integrity. Differentiated from …
FDA Accepts Filing of NDA for IV Antibiotic Oritavancin with Priority Review
PARSIPPANY, NJ — (Marketwired) — 02/19/14 — The Medicines Company (NASDAQ: MDCO) today announced that the U.S. Food and Drug Administration (FDA) has accepted the filing of a new drug application (NDA) for oritavancin, an investigational intravenous antibiotic, with priority review. The Medicines Company is seeking approval of oritavancin for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), administered as a single dose.
In December 2013, the FDA designated oritavancin as a Qualified Infectious Disease Product (QIDP). The QIDP designation provides oritavancin priority review, and an additional five years of exclusivity upon approval of the product for the treatment of ABSSSI. Priority review means the FDA’s goal is to take action on the application within six months, compared to 10 months under standard review. The FDA action date (PDUFA date) for oritavancin is August 6, 2014.
Oritavancin (INN, also known as LY333328) is a novel semi-synthetic glycopeptide antibiotic being developed for the treatment of serious Gram-positive infections. Originally discovered and developed by Eli Lilly, oritavancin was acquired by InterMune in 2001 and then by Targanta Therapeuticsin late 2005.[1]
In Dec 2008 the FDA declined to approve it, and an EU application was withdrawn.
In 2009 the development rights were acquired by The Medicine Co. who are running clinical trials for a possible new FDA application in 2013.[2]
Its structure is similar to vancomycin[3] It is a lipoglycopeptide
About Oritavancin
Oritavancin is an investigational intravenous antibiotic for which The Medicines Company is seeking approval in the treatment of ABSSSI caused by susceptible gram-positive bacteria, including MRSA. In clinical trials, the most frequently reported adverse events associated with oritavancin were nausea, headache, vomiting and diarrhea. Hypersensitivity reactions have been reported with the use of antibacterial agents including oritavancin.

Oritavancin shares certain properties with other members of the glycopeptide class of antibiotics, which includes vancomycin, the current standard of care for serious Gram-positive infections in the United States and Europe.[4] Data presented at the 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) in September 2007 demonstrated that oritavancin possesses potent and rapid bactericidal activity in vitro against a broad spectrum of both resistant and susceptible Gram positive bacteria, including Staphylococcus aureus, methicillin-resistant Staphylococcus aureus, Enterococci, and Streptococci.[5] Two posters presented at the meeting also demonstrated that oritavancin was more active than either metronidazole or vancomycin against strains of Clostridium difficile tested.[6]
Anthrax : Research presented at the American Society for Microbiology (ASM) 107th Annual General Meeting in May 2007, suggested oritavancin’s potential utility as a therapy for exposure to Bacillus anthracis, the gram-positive bacterium that causes anthrax, having demonstrated efficacy in a mouse model both pre- and post-exposure to the bacterium[7]
 oritavancin
oritavancin
The 4′-chlorobiphenylmethyl group disrupts the cell membrane of gram positive bacteria.[8] It also acts by inhibition of transglycosylation and inhibition of transpeptidation.[9]
Results have been presented (in 2003) but possibly not yet published from two pivotal Phase 3 clinical trials testing the efficacy of daily intravenous oritavancin for the treatment of complicated skin and skin-structure infections (cSSSI) caused by Gram-positive bacteria. The primary endpoints of both studies were successfully met, with oritavancin achieving efficacy with fewer days of therapy than the comparator agents (vancomycin followed by cephalexin). In addition, oritavancin showed a significantly improved safety profile with a 19.2 percent relative reduction in the overall incidence of adverse events versus vancomycin/cephalexin (p<0.001) in the second and larger pivotal trial.[10]
A Phase 2 clinical study was planned to run until May 2008 entitled “Single or Infrequent Doses for the Treatment of Complicated Skin and Skin Structure Infections (SIMPLIFI),” evaluating the efficacy and safety of either a single dose of oritavancin or an infrequent dose of oritavancin compared to the previously studied dosing regimen of 200 mg oritavancin given once daily for 3 to 7 days.[11] Results published May 2011.[12]
Regulatory submissions
USA
On February 11, 2008, Targanta submitted a New Drug Application (NDA) to the US FDA seeking approval of oritavancin;[13] in April 2008, the FDA accepted the NDA submission for standard review.[14] On 9 Dec 2008 the FDA said insufficient data for approval of oritavancin had been provided and they requested a further phase 3 clinical study to include more patients with MRSA.[15]
Europe
June 2008, Targanta’s Marketing Authorization Application (MAA) for oritavancin was submitted and accepted for review by the European Medicines Agency (EMEA),[16] but the company later withdrew the application in Aug 2009.[17]
About The Medicines Company
The Medicines Company’s purpose is to save lives, alleviate suffering, and contribute to the economics of healthcare by focusing on 3,000 leading acute/intensive care hospitals worldwide. Its vision is to be a leading provider of solutions in three areas: acute cardiovascular care, surgery and perioperative care, and serious infectious disease care. The company operates in the Americas, Europe and the Middle East, and Asia Pacific regions with global centers today in Parsippany, NJ, USA and Zurich, Switzerland.
“We look forward to working with the FDA during the review process, and sharing the knowledge we have gained in our studies of oritavancin,” said Matthew Wikler, MD, Vice President and Medical Director, Infectious Disease Care for The Medicines Company. “We believe that upon approval, oritavancin, administered as a single dose for the treatment of ABSSSI, will offer new options for both physicians and their patients for the treatment of these infections.”
The oritavancin NDA is based on data from two Phase 3 clinical trials, SOLO I and SOLO II, which were conducted under a Special Protocol Assessment (SPA) agreement with the FDA. These Phase 3 trials evaluated the efficacy and safety of a single 1200mg dose of oritavancin compared to 7 to 10 days of twice-daily vancomycin in adults with ABSSSI, including infections caused by MRSA. The combined SOLO studies were conducted in 1,959 patients (modified intent-to -treat population, or mITT), with 405 of the patients suffering from an ABSSSI with a documented MRSA infection.
 oritavancin
oritavancin
Drug substance
Oritavancin diphosphate
CLINICAL TRIALS..http://clinicaltrials.gov/search/intervention=oritavancin
- LY 333328 diphosphate
- LY333328 diphosphate
- Oritavancin diphosphate
- UNII-VL1P93MKZN
- 192564-14-0 CAS NO
INTRODUCTION
Oritavancin
Oritavancin inhibits cell wall synthesis by complexing with the terminal D-Ala-D-Ala of a nascent peptidoglycan chain and also to the pentaglycine bridge, thus inhibiting transglyco- sylation and transpeptidation. Unlike other glycopeptides, oritavancin is able to bind to depsipeptides including D-Ala-D-Lac, which fa- cilitates its inhibition of cell wall synthesis even in organisms exhibiting VanA-type resistance. Oritavancin forms homodimers prior to binding to D-Ala-D-Ala or D-Ala-D-Lac, which increases its binding affinity for the target site.The p-chloro-phenylbenzyl side chain of oritavancin interacts with the cell membrane, exerting two beneficial effects. This binding acts to main- tain the antibacterial in a prime position for peptidoglycan interactions and it also imparts oritavancin with the ability to disrupt the bac- terial membrane potential and thus increase membrane permeability.[22,23] Oritavancin has been shown to dissipate membrane potential in both stationary and exponential phase growing bacteria, which is rare and may carry clinical implications in terms of its activity against slowly growing organisms and biofilms. The dual mechanism of action could also theoretically increase effectiveness and reduce the risk of resist- ance selection. In addition to the aforemen- tioned mechanisms, it has also been hypothesized that oritavancin inhibits RNA synthesis.


vancomycin, desmethylvancomycin, eremomycin, teicoplanin (complex of five compounds), dalbavancin, oritavancin, telavancin, and A82846B (LY264826) having structures A, B, C, D, E, F, G and H:

R = B-2-Acetylamido-glucopyraπosyl- Attorney Docket No 33746-704 602


Dalbavancin, oritavancin and telavancin are semisynthetic lipoglycopeptides that demonstrate promise for the treatment of patients with infections caused by multi-drug-resistant Gram-positive pathogens. Each of these agents contains a heptapeptide core, common to all glycopeptides, which enables them to inhibit transglycosylation and transpeptidation (cell wall synthesis). Modifications to the heptapeptide core result in different in vitro activities for the three semisynthetic lipoglycopeptides. All three lipoglycopeptides contain lipophilic side chains, which prolong their half-life, help to anchor the agents to the cell membrane and increase their activity against Gram-positive cocci. In addition to inhibiting cell wall synthesis, telavancin and oritavancin are also able to disrupt bacterial membrane integrity and increase membrane permeability; oritavancin also inhibits RNA synthesis. Enterococci exhibiting the VanA phenotype (resistance to both vancomycin and teicoplanin) are resistant to both dalbavancin and telavancin, whileoritavancin retains activity. Dalbavancin, oritavancin and telavancin exhibit activity against VanB vancomycin-resistant enterococci.
All three lipoglycopeptides demonstrate potent in vitro activity against Staphylococcus aureus and Staphylococcus epidermidis regardless of their susceptibility to meticillin, as well as Streptococcus spp. Both dalbavancin and telavancin are active against vancomycin-intermediate S. aureus (VISA), but display poor activity versus vancomycin-resistant S. aureus (VRSA). Oritavancin is active against both VISA and VRSA. Telavancin displays greater activity against Clostridium spp. than dalbavancin, oritavancin or vancomycin. The half-life of dalbavancin ranges from 147 to 258 hours, which allows for once-weekly dosing, the half-life of oritavancin of 393 hours may allow for one dose per treatment course, while telavancin requires daily administration. Dalbavancin and telavancin exhibit concentration-dependent activity and AUC/MIC (area under the concentration-time curve to minimum inhibitory concentration ratio) is the pharmacodynamic parameter that best describes their activities.
Oritavancin’s activity is also considered concentration-dependent in vitro, while in vivo its activity has been described by both concentration and time-dependent models; however, AUC/MIC is the pharmacodynamic parameter that best describes its activity. Clinical trials involving patients with complicated skin and skin structure infections (cSSSIs) have demonstrated that all three agents are as efficacious as comparators. The most common adverse effects reported with dalbavancin use included nausea, diarrhoea and constipation, while injection site reactions, fever and diarrhoea were commonly observed withoritavancin therapy. Patients administered telavancin frequently reported nausea, taste disturbance and insomnia. To date, no drug-drug interactions have been identified for dalbavancin, oritavancin or telavancin. All three of these agents are promising alternatives for the treatment of cSSSIs in cases where more economical options such as vancomycin have been ineffective, in cases of reduced vancomycin susceptibility or resistance, or where vancomycin use has been associated with adverse events.
Oritavancin diphosphate (oritavancin) is a semi-synthetic lipoglycopeptide derivative of a naturally occurring glycopeptide. Its structure confers potent antibacterial activity against gram-positive bacteria, including vancomycin-resistant enterococci (VRE), methicillin- and vancomycin-resistant staphylococci, and penicillin-resistant streptococci. The rapidity of its bactericidal activity against exponentially-growing S. aureus (≧3-log reduction within 15 minutes to 2 hours against MSSA, MRSA, and VRSA) is one of the features that distinguishes it from the prototypic glycopeptide vancomycin (McKay et al., J Antimicrob Chemother. 63(6):1191-9 (2009), Epub 2009 Apr. 15).
Oritavancin inhibits the synthesis of peptidoglycan, the major structural component of the bacterial cell wall by a mechanism that is shared with glycopeptides, such as vancomycin (Allen et al., Antimicrob Agents Chemother 41(1):66-71 (1997); Cegelski et al., J Mol Biol 357:1253-1262 (2006); Arhin et al., Poster C1-1471: Mechanisms of action of oritavancin in Staphylococcus aureus [poster]. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007; Chicago, Ill.). Oritavancin, like vancomycin, binds to the Acyl-D-Alanyl-D-Alanine terminus of the peptidoglycan precursor, lipid-bound N-acetyl-glucosamine-N-acetyl-muramic acid-pentapeptide (Reynolds, Eur J Clin Microbiol Infect Dis 8(11):943-950 (1989); Nicas and Allen, Resistance and mechanism of action.
In: Nagarajan R, editor. Glycopeptide antibiotics. New York: Marcel Dekker 195-215 (1994); Allen et al., Antimicrob Agents Chemother 40(10):2356-2362 (1996); Allen and Nicas, FEMS Microbiology Reviews 26:511-532 (2003); Kim et al., Biochemistry 45:5235-5250 (2006)). However, oritavancin inhibits cell wall biosynthesis even when the substrate is the altered peptidoglycan precursor that is present in VRE and vancomycin-resistant S. aureus (VRSA). Thus, the spectrum of oritavancin antibacterial activity extends beyond that of vancomycin to include glycopeptide-resistant enterococci and staphylococci (Ward et al., Expert Opin Investig Drugs 15:417-429 (2006); Scheinfeld, J Drugs Dermatol 6:97-103 (2007)). Oritavancin may inhibit resistant bacteria by interacting directly with bacterial proteins in the transglycosylation step of cell wall biosynthesis (Goldman and Gange, Curr Med Chem 7(8):801-820 (2000); Halliday et al., Biochem Pharmacol 71(7):957-967 (2006); Wang et al., Poster C1-1474: Probing the mechanism of inhibition of bacterial peptidoglycan glycotransferases by glycopeptide analogs. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007). Oritavancin also collapses transmembrane potential in gram positive bacteria, leading to rapid killing (McKay et al., Poster C1-682: Oritavancin disrupts transmembrane potential and membrane integrity concomitantly with cell killing in Staphylococcus aureus and vancomycin-resistant Enterococci. 46th Intersci Conf Antimicro Agents Chemo, San Francisco, Calif., Sep. 27-30, 2006). These multiple effects contribute to the rapid bactericidal activity of oritavancin.
Vancomycin (U.S. Patent 3,067,099); A82846A, A82846B, and A82846C (U.S. Patent 5,312,738, European Patent Publication 256,071 A1); PA-42867 factors A, C, and D (U.S. Patent4,946,941 and European Patent Publication 231,111 A2); A83850 (U.S. Patent No. 5,187,082); avoparcm (U.S. Patent 3,338,786 and U.S. Patent 4,322,343); actmoidin, also known as K288 (J. Antibiotics Series A 14:141 (1961); helevecardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 86/157,397); galacardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 89/221,320); and M47767 (European Patent Publication 339,982).
Oritavancin is in clinical development against serious gram-positive infections, where administration of the drug is via intravenous infusion using several dosages administered over a series of days. The development of alternative dosing regimens for the drug could expand treatment options available to physicians. The present invention is directed to novel dosing regimens.
Means for the preparation of the glycopeptide antibiotics, including oritavancin and analogs thereof, may be found, for example, in U.S. Pat. No. 5,840,684,
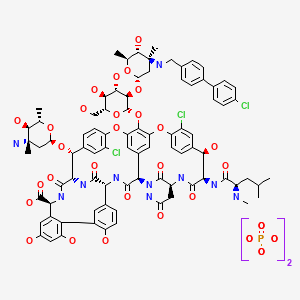
SYNTHESIS

LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
J Antibiot1996, 49, (6) :575-81
(3S,6R,7R,22R,23S,26S,36R,38aR)-3-(Carbamoylmethyl)-10,19-dichloro-7,28,30,32-tetrahydroxy-6-(N-methyl-D-leucylamido)-2,5,24,38,39-pentaoxo-22-(L-vancosaminyloxy)-44-[2-O-(L-vancosaminyl)-beta-D-glucopyranosyloxy]-2,3,4,5,6,7,23,24,25,26,36,37,38,38a-tetradecahydro-1H,22H-8,11:18,21-dietheno-23,36-(iminomethano)-13,16:31,36-dimetheno-[1,6,9]oxadiazacyclohexadecino[4,5-m][10,2,16]benzoxadiazacyclotetracosine-26-carboxylic acid; A82846B (I)
4′-chloro[1,1′-biphenyl]-4-carbaldehyde (II)
LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
…………………..
EXAMPLE 4
Preparation of Compound 229
A three liter 3-necked flask was fitted with a
condenser, nitrogen inlet and overhead mechanical stirring apparatus. The flask was charged with pulverized A82846B acetate salt (20.0 g, 1.21 × 10-3 mol) and methanol (1000 mL) under a nitrogen atmosphere. 4′-chlorobiphenylcarboxaldehyde (2.88 g, 1.33 × 10-2 mol, 1.1 eq.) was added to this stirred mixture, followed by methanol (500 mL). Finally, sodium cyanoborohydride (0.84 g, 1.33 × 10-2 mol, 1.1 eq.) was added followed by methanol (500 mL). The resulting mixture was heated to reflux (about 65°C).
After 1 hour at reflux, the reaction mixture attained homogeneity. After 25 hours ac reflux, the heat source was removed and the clear reaction mixture was measured with a pH meter (6.97 at 58.0°C). 1 N NaOH (22.8 mL) was added
dropwise to adjust the pH to 9.0 (at 54.7°C). The flask was equipped with a distillation head and the mixture was concentrated under partial vacuum to a weight of 322.3 grams while maintaining the pot temperature between 40-45°C.
The distillation head was replaced with an addition funnel containing 500 mL of isopropanol (IPA). The IPA was added dropwise to the room temperature solution over 1 hour. After approximately 1/3 of the IPA was added, a granular precipitate formed. The remaining IPA was added at a faster rate after precipitation had commenced. The flask was weighed and found to hold 714.4 grams of the IPA/methanol slurry.
The flask was re-equipped with a still-head and
distilled under partial vacuum to remove the remaining methanol. The resulting slurry (377.8 g) was allowed to chill in the freezer overnight. The crude product was filtered through a polypropylene pad and rinsed twice with 25 mL of cold IPA. After pulling dry on the funnel for 5 minutes, the material was placed in the vacuum oven to dry at 40°C. A light pink solid (22.87 g (theory = 22.43 g) ) was recovered. HPLC analysis versus a standard indicated 68.0% weight percent of Compound 229 (4- [4-chlorophenyl] benzyl-A82846B] in the crude solid, which translated into a
corrected crude yield of 69.3%.
The products of the reaction were analyzed by reverse-phase HPLC utilizing a Zorbax SB-C18 column with ultraviolet light (UV; 230 nm) detection. A 20 minute gradient solvent system consisting of 95% aqueous buffer/5% CH3CN at time=0 minutes to 40% aqueous buffer/60% CH3CN at time=20 minutes was used, where the aqueous buffer was TEAP (5 ml CH3CN, 3 ml phosphoric acid in 1000 ml water).
………………….
Oritavancin (also termed N-(4-(4-chlorophenyl)benzyl)A82846B and LY333328) has the following Formula III:

References
- Targanta Revives Oritavancin: Next Weapon Against cSSSI? BioWorld Today, November 26, 2007
- “Biotechs pick up slack in antibiotics development”. 17 May 2011.
- http://www.farm.ucl.ac.be/Full-texts-FARM/Domenech-2009-1.pdf “Interactions of oritavancin, a new lipoglycopeptide derived from vancomycin, with phospholipid bilayers: Effect on membrane permeability and nanoscale lipid membrane organization” 2009
- Scheinfeld, N (2007). “A comparison of available and investigational antibiotics for complicated skin infections and treatment-resistant Staphylococcus aureus and enterococcus“.J Drugs Dermatol. 6 (4): 97–103. PMID 17373167.
- 2007 ICAAC Posters: E-1612 “In Vitro Activity Profile of Oritavancin against a Broad Spectrum of Aerobic and Anaerobic Bacterial Pathogens”/E -1613 “In Vitro Activity Profile of Oritavancin (ORI) Against Organisms Demonstrating Key Resistance Profiles to Other Antimicrobial Agents”/E-1614 “In vitro Time Kill Studies of Oritavancin against Drug-resistant Isolates ofStaphylococcus aureus and Enterococci”/E-1615 “Anti-Enterococcal Activity Profile of Oritavancin, a Potent Lipoglycopeptide under Development for Use Against Gram-Positive Infections”/E-1616 “Anti-Streptococcal Activity Profile of Oritavancin, a Potent Lipoglycopeptide under Development for Use Against Gram-Positive Infections”/E-1617 “In Vitro Activity Profile of Oritavancin (ORI) Against Resistant Staphylococcal Populations From a Recent Surveillance Initiative”/E-1620 “Pharmacokinetic Concentrations of Oritavancin Kill Stationary-Phase and Biofilm Staphylococcus aureus In Vitro.” / Targanta Press Release September 19, 2007
- ICAAC 2007 Posters: “In Vitro Susceptibility of Genotypically Distinct Clostridium difficileStrains to Oritavancin” and “Activity of Metronidazole, Vancomycin and Oritavancin Against Epidemic Clostridium difficile Spores” / Targanta Press Release September 19, 2007
- ASM 2007 Poster: “Efficacy of Oritavancin in a Murine Model of Bacillus anthracis Spore Inhalation Anthrax” / Targanta Press Release May 24, 2007
- Belley; McKay, GA; Arhin, FF; Sarmiento, I; Beaulieu, S; Fadhil, I; Parr Jr, TR; Moeck, G (2010).“Oritavancin Disrupts Membrane Integrity of Staphylococcus aureus and Vancomycin-Resistant Enterococci To Effect Rapid Bacterial Killing”. Antimicrobial agents and chemotherapy 54(12): 5369–71. doi:10.1128/AAC.00760-10. PMC 2981232. PMID 20876372.
- Zhanel et al. (2012). “Oritavancin: Mechanism of Action”. Clin Infect Dis.doi:10.1093/cid/cir920.
- ICAAC 2003 Late-breaker poster: “Phase III Trial Comparing 3-7 days of Oritavancin vs. 10-14 days of Vancomycin/Cephalexin in the Treatment of Patients with Complicated Skin and Skin Structure Infections (cSSSI)” / InterMune Press Release September 15, 2003
- ClinicalTrials.gov NCT00514527
- Comparison of the Efficacy and Safety of Oritavancin Front-Loaded Dosing Regimens to Daily Dosing: An Analysis of the SIMPLIFI Trial. May 2011. doi:10.1128/AAC.00029-11.
- “Drugs.com, Targanta Submits Oritavancin New Drug Application”. Retrieved 2008-02-12.
- “FDA News, Targanta to Get FDA Decision by December”. Retrieved 2008-04-10.
- http://www.fiercebiotech.com/press-releases/fda-issues-complete-response-letter-oritavancin Dec 2008.
- “Pharmaceutical Business Review, EMEA accepts Targanta’s oritavancin MAA for review”. Retrieved 2008-06-26.
- http://www.nelm.nhs.uk/en/NeLM-Area/News/2009—August/24/European-application-for-investigational-antibiotic-oritavancin-withdrawn-/
- http://onlinelibrary.wiley.com/doi/10.1111/j.1574-6976.2003.tb00628.x/pdf
- http://www.pjps.pk/wp-content/uploads/pdfs/26/5/Paper-30.pdf
- Antimicrobial Agents and Chemotherapy, 2003 , vol. 47, 5 p. 1700 – 1706
- Antimicrobial Agents and Chemotherapy, 1999 , vol. 43, 1 p. 115 – 120
- Antimicrobial Agents and Chemotherapy, 1997 , vol. 41, 10 p. 2165 – 2172
- Tetrahedron, 2004 , vol. 60, 47 p. 10611 – 10618………… NMRhttp://www.sciencedirect.com/science/article/pii/S0040402004015108
Cooper, R.D.G.; Snyder, N.J.; Zweifel, M.J.; et al.; Reductive alkylation of glycopeptide antibiotics: Synthesis and antibacterial activity. J Antibiot 1996, 49, 6, 575-81.
Cooper, R.D.G.; Huff, B.E.; Nicas, T.I.; Quatroche, J.T.; Rodriguez, M.J.; Snyder, N.J.; Staszak, M.A.; Thompson, R.C.; Wilkie, S.C.; Zweifel, M.J. (Eli Lilly and Company); Glycopeptide antibiotic derivs. EP 0667353; EP 1016670; EP 1031576 .
| EP0435503A1 * | Dec 11, 1990 | Jul 3, 1991 | Eli Lilly And Company | Improvements in or relating to gylcopeptide derivatives |
| US4639433 * | Aug 14, 1985 | Jan 27, 1987 | Eli Lilly And Company | Glycopeptide derivatives |
| US4698327 * | Apr 18, 1986 | Oct 6, 1987 | Eli Lilly And Company | Novel glycopeptide derivatives |
| US20040106590 * | Aug 29, 2003 | Jun 3, 2004 | Barry Eisenstein | Methods and reagents for treating infections of clostridium difficile and diseases associated therewith |
| US20050197333 | Dec 22, 2004 | Sep 8, 2005 | Van Duzer John H. | Rifamycin analogs and uses thereof |
| US20070014849 | Sep 20, 2006 | Jan 18, 2007 | Daniela Jabes | Use of ramoplanin to treat diseases associated with the use of antibiotics |
| US20030176327 * | Oct 18, 2002 | Sep 18, 2003 | Cassell Gail Houston | Antibiotics for treating biohazardous bacterial agents |
| US20040147441 | Aug 25, 2003 | Jul 29, 2004 | Leach Timothy S. | Methods and reagents for preventing bacteremias |
| WO1999010006A1 | Aug 18, 1998 | Mar 4, 1999 | Lilly Co Eli | Therapy for staphylococcus aureus |
| WO2000066144A2 | Apr 19, 2000 | Nov 9, 2000 | Lilly Co Eli | Monthly doses of glycopeptide antibiotics for treatment of streptococcus pneumoniae infections |
| WO2008097364A2 | Sep 24, 2007 | Aug 14, 2008 | Targanta Therapeutics Corp | Use of oritavancin for prevention and treatment of anthrax |
| WO1998052592A1 * | May 5, 1998 | Nov 26, 1998 | Lilly Co Eli | Urea and thiourea derivatives of glycopeptides |
| WO2002036612A1 * | Nov 2, 2001 | May 10, 2002 | Univ Cambridge Tech | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane-associating elements |
| WO2007138999A1 | May 25, 2007 | Dec 6, 2007 | Shionogi & Co | Glycopeptide antibiotic derivative |
| WO2009081958A1 | Dec 25, 2008 | Jul 2, 2009 | Shionogi & Co | Glycosylated glycopeptide antibiotic derivative |
| EP2314599A1 | Nov 24, 2005 | Apr 27, 2011 | National University Corporation Nagoya University | Glycopeptide antibiotic monomer derivatives |
| US5919756 * | May 1, 1997 | Jul 6, 1999 | Eli Lilly And Company | Amides |
| US5919771 * | Apr 30, 1998 | Jul 6, 1999 | Eli Lilly And Company | Urea and thiourea derivatives of glycopeptides |
| US7078380 | Nov 2, 2001 | Jul 18, 2006 | Cambridge University Technical Services Limited | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane associating elements |
| US8481696 | Dec 25, 2008 | Jul 9, 2013 | Shionogi & Co., Ltd. | Glycosylated glycopeptide antibiotic derivatives |


SUMATRIPTAN …Avanir files new drug application for migraine drug

SUMATRIPTAN, GR-43175
1-[3-(2-dimethylaminoethyl)-1H-indol-5-yl]- N-methyl-methanesulfonamide
3-[2-(Dimethylamino)ethyl]-N-methyl-1H-indole-5-methanesulfonamide
| Formula | C14H21N3O2S |
|---|---|
| Mol. mass | 295.402 g/mol |
| CAS number | 103628-46-2 |
|---|
Avanir Pharmaceuticals has filed a new drug application (NDA) with the US Food and Drug Administration (FDA) for approval of its new breath-powered investigational drug-device combination product, ‘AVP-825’, for the acute treatment of migraines. click on title Avanir files new drug application for migraine drug
 SUMATRIPTAN
SUMATRIPTAN
SUMATRIPTAN SUCCINATE

AVP-825 is an investigational drug-device combination product consisting of low-dose sumatriptan powder delivered intranasally utilizing a novel Breath Powered delivery technology. If approved, AVP-825 would be the first and only fast-acting, dry-powder intranasal form of sumatriptan for the treatment of migraine.
The Breath Powered delivery technology is activated by user’s breath to propel medications deep into the nasal cavity where absorption is more efficient and consistent than through most other routes. A user exhales into the device, automatically closing the soft palate and sealing off the nasal cavity completely. Through a sealing nosepiece placed into the nostril, the exhaled breath carries medication from the device directly into one side of the nose. Narrow nasal passages are gently expanded and medication is dispersed deep into the nasal cavity reaching areas where it can be rapidly absorbed. As the medication is delivered, the air flows around to the opposite side of the nasal cavity and exits through the other nostril. Closure of the soft palate helps prevent swallowing or inhalation of sumatriptan powder into the lungs.
| Canada | 2469019 | APPROVED 2005-09-13 | EXP 2022-12-04 |
| United States | 6135979 | 1997-03-21 | 2017-03-21 |
| United States | 5705520 | 1994-12-10 | 2011-12-10 |
| Canada | 2098302 | 2001-10-16 | 2011-12-10 |
| Patent No | PatentExpiry | use code |
|---|---|---|
| 5307953 | Dec 2, 2012 | |
| 5307953*PED | Jun 2, 2013 | |
| 5554639 | Sep 10, 2013 | U-232…METHOD OF TREATING MIGRAINE |
| 5554639*PED | Mar 10, 2014 |
Sumatriptan is a synthetic drug belonging to the triptan class, used for the treatment of migraine headaches. Structurally, it is an analog of the naturally occurring neuro-active alkaloids dimethyltryptamine (DMT), bufotenine, and 5-methoxy-dimethyltryptamine, with an N-methyl sulfonamidomethyl- group at position C-5 on the indole ring.[1]
Sumatriptan is produced and marketed by various drug manufacturers with many different trade names such as Sumatriptan, Imitrex, Treximet, Imigran, Imigran recovery.
Large doses of sumatriptan can cause sulfhemoglobinemia, a rare condition in which the blood changes from red to greenish-black, due to the integration of sulfur into the hemoglobin molecule.[2] If sumatriptan is discontinued, the condition reverses within a few weeks.
Serious cardiac events, including some that have been fatal, have occurred following the use of sumatriptan injection or tablets. Events reported have included coronary artery vasospasm, transient myocardial ischemia, myocardial infarction, ventricular tachycardia, and ventricular fibrillation.
The most common side-effects[3] reported by at least 2% of patients in controlled trials of sumatriptan (25, 50, and 100 mg tablets) for migraine are atypical sensations (paresthesias and warm/cold sensations) reported by 4% in the placebo group and 5–6% in the sumatriptan groups, pain and other pressure sensations (including chest pain) reported by 4% in the placebo group and 6–8% in the sumatriptan groups, neurological events (vertigo) reported by less than 1% in the placebo group and less than 1% to 2% in the sumatriptan groups. Malaise/fatigue occurred in less than 1% of the placebo group and 2–3% of the sumatriptan groups. Sleep disturbance occurred in less than 1% in the placebo group to 2% in the sumatriptan group.
 SUMATRIPTAN
SUMATRIPTAN
Sumatriptan is structurally similar to serotonin (5HT), and is a 5-HT (types 5-HT1D and 5-HT1B[4]) agonist. The specific receptor subtypes it activates are present on the cranial arteries and veins. Acting as an agonist at these receptors, sumatriptan reduces the vascular inflammation associated with migraines.
The specific receptor subtype it activates is present in the cranial and basilar arteries. Activation of these receptors causes vasoconstriction of those dilated arteries. Sumatriptan is also shown to decrease the activity of the trigeminal nerve, which, it is presumed, accounts for sumatriptan’s efficacy in treating cluster headaches. The injectable form of the drug has been shown to abort a cluster headache within fifteen minutes in 96% of cases.[5]
Sumatriptan is administered in several forms; tablets, subcutaneous injection, and nasal spray. Oral administration (as succinate) suffers from poorbioavailability, partly due to presystemic metabolism—some of it gets broken down in the stomach and bloodstream before it reaches the target arteries. A new rapid-release tablet formulation has the same bioavailability, but the maximum concentration is achieved on average 10–15 minutes earlier. When injected, sumatriptan is faster-acting (usually within 10 minutes), but the effect lasts for a shorter time. Sumatriptan is metabolised primarily by monoamine oxidase A into an indole acetic acid analogue, part of which is further conjugated with glucuronic acid. These metabolites are excreted in the urine and bile. Only about 3% of the active drug may be recovered unchanged.
There is no simple, direct relationship between sumatriptan concentration (pharmacokinetics) per se in the blood and its anti-migraine effect (pharmacodynamics). This paradox has, to some extent, been resolved by comparing the rates of absorption of the various sumatriptan formulations, rather than the absolute amounts of drug that they deliver.[6][7]
Sumatriptan was the first clinically available triptan (in 1991). In the United States, it is available only by medical prescription. However, it can be bought over the counter in the UK and Sweden in 50 mg dosage. Several dosage forms for sumatriptan have been approved, including tablets, solution for injection, and nasal inhalers.
On April 15, 2008, the US FDA approved Treximet, a combination of sumatriptan and naproxen, an NSAID.[8] This combination has shown a benefit over either medicine used separately.[9]
In July 2009, the US FDA approved a single-use jet injector formulation of sumatriptan. The device delivers a subcutaneous injection of 6 mg sumatriptan, without the use of a needle.Autoinjectors with needles have been previously available in Europe and North America for several years.[10]
Phase III studies with a iontophoretic transdermal patch (Zelrix/Zecuity) started in July 2008.[11] This patch uses low voltage controlled by a pre-programmed microchip to deliver a single dose of sumatriptan through the skin within 30 minutes.[12][13]Zecuity was approved by the US FDA in January 2013.[14]

Sumatriptan vials 100 5509
On November 6, 2008, Par Pharmaceutical announced that it would begin shipping generic versions of sumatriptan injection (sumatriptan succinate injection) 4 mg and 6 mg starter kits and 4 mg and 6 mg pre-filled syringe cartridges to the trade immediately. In addition, Par anticipates launching the 6 mg vials early in 2009.[15]
Mylan Laboratories Inc., Ranbaxy, Sandoz, Dr. Reddy’s Pharmaceuticals and other companies have received FDA approval for generic versions of Imitrex tablets in 25-, 50-, and 100-milligram doses since 2009. The drug is available in U.S. and European markets, since Glaxo’s patent protections have expired in those jurisdictions. However, sales of a generic delivered via nasal spray are still restricted in the United States.
See also Sumavel DosePro (above).[10]
Chemistry
hydrogenation of nitrile with pd/c in presence of dimethyl amine
…………………


The diazotation of 4-amino-N-methylbenzenemethanesulfonamide (I) with NaNO2-HCl followed by reduction with SnCl2 gives the 4-hydrazino compound (II), which is condensed with (phenylthio)acetaldehyde (III) in ethanol yielding the ethylideneamino compound (IV). The cyclization of (IV) with HCl in ethanol affords N-methyl-3-(phenylthio)-1H-indole-5-methansulfonamide (V), which is desulfurized with RaNi in refluxing ethanol-water to give N-methyl-1H-indole-5-methanesulfonamide (VI). The reaction of (VI) with oxalyl chloride and dimethylamine yields the oxalyl derivative (VII), which is finally reduced with LiAlH4 in refluxing THF.

The condensation of hydrazine (II) with 4,4-dimethoxy-N,N-dimethylbutylamine (VIII) by means of HCl in water gives the butylidenehydrazino compound (IX), which is cyclized with polyphosphate ester (PPE) in CHCl3.

……………………
Beilstein J. Org. Chem. 2011, 7, 442–495.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-7-57#S9
ref are below article
Indoles
The neuroamine transmitter serotonin contains an indole ring, so it is not surprising that indoles are a recurring theme in many drugs affecting central nervous system (CNS) function including antidepressants, antipsychotics, anxiolytics and antimigraine drugs, as well as psychedelic agents. Indole is also one of the best represented heterocyclic motifs present in the top selling pharmaceuticals, being found in eight of the top 200 drugs, with five of these belonging to the triptan family of antimigraine treatments. The classical Fischer indole synthesis is usually reported as one of the first choice routes to prepare these scaffolds. Drugs such as GSK’s serotonin receptor modulators sumatriptan (49, Imitrex) and zolmitriptan (50, Zomig) use the Fischer indole synthesis at a late stage in order to form the desired compound albeit in only low to moderate yields (Scheme 9).
![[1860-5397-7-57-i9]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i9.png)
However, in sumatriptan the indole product resulting from the Fischer synthesis can still react further which leads to the formation of by-products and significantly reduced yields. One way to minimise this was to protect the nitrogen of the sulfonamide group prior to indole formation [11]. This leads not only to an increased yield in the indole forming step (to 50%) but also facilitates chromatographic purification. The dimethylamino group can be present from the beginning of the synthesis or can be introduced via displacement of chloride or reduction of a cyano moiety. Alternatively, the dimethyl ethylene amine side chain can be introduced in position 3 via a Friedel–Crafts-type acylation. The resulting acid chloride is transformed in situ to the corresponding amide which on reduction with lithium aluminium hydride affords sumatriptan (Scheme 10) [12].
![[1860-5397-7-57-i10]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i10.png)
In the standard Fischer indole synthesis a hydrazine, which is most commonly derived from the corresponding diazonium salt, is reacted with a suitable carbonyl compound. Alternatively, the Japp–Klingemann reaction can be used to directly couple the diazonium salt with a β-ketoester to obtain a hydrazone which can then undergo indole ring formation (Scheme 11) [13].
![[1860-5397-7-57-i11]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i11.png)
As can be seen from Scheme 11 the indole 59 prepared via the Japp–Klingemann reaction is substituted at position 2 by an ester group which prevents reaction with electrophiles, thereby reducing the amount of undesired by-products. A simple sequence of hydrolysis and decarboxylation then affords sumatriptan [14].
All the reported methods for the synthesis of sumatriptan begin with the sulfonamide group already present on the aromatic ring and several routes are possible to introduce this functional group. The scalable route to the sulfonamides inevitably involves the preparation of the sulfonyl chloride intermediate which is then trapped with the desired amine. The sulfonyl chloride can also be prepared from the corresponding hemithioacetal 61 by treatment with NCS in wet acetic acid (Scheme 12). This efficient oxidation produces only methanol and formaldehyde as by-products [15].
![[1860-5397-7-57-i12]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i12.png)
- 11. Pete, B.; Bitter, I.; Szántay, C., Jr.; Schön, I.; Töke, L. Heterocycles 1998, 48, 1139–1149. doi:10.3987/COM-97-8087
- 12…Oxford, A. W. Indole Derivative. U.S. Patent 5,037,845, Aug 6, 1991.
- 13…Japp, F. R.; Klingemann, F. Chem. Ber. 1887, 20, 2942–2944. doi:10.1002/cber.188702002165
- Pete, B.; Bitter, I.; Harsányi, K.; Töke, L. Heterocycles 2000, 53, 665–673. doi:10.3987/COM-99-8815
- Kim, D.-W.; Ko, Y. K.; Kim, S. H. Synthesis 1992, 12, 1203–1204. doi:10.1055/s-1992-26333
[15
References for full article
- The presence of the sulfonamide group in the molecule does not make sumatriptan a “sulfa drug”, since it does not have any anti-microbial properties.
- “Patient bleeds dark green blood”. BBC News. 8 June 2007. Retrieved 6 March 2010.
- Tablets
- Razzaque Z, Heald MA, Pickard JD, et al. (1999). “Vasoconstriction in human isolated middle meningeal arteries: determining the contribution of 5-HT1B- and 5-HT1F-receptor activation”.Br J Clin Pharmacol 47 (1): 75–82. doi:10.1046/j.1365-2125.1999.00851.x. PMC 2014192.PMID 10073743.
- Treatment of acute cluster headache with sumatriptan. The Sumatriptan Cluster Headache Study Group. N Engl J Med 1991;325:322-6.
- Fox, A. W. (2004). “Onset of effect of 5-HT1B/1D agonists: a model with pharmacokinetic validation”. Headache 44 (2): 142–147. doi:10.1111/j.1526-4610.2004.04030.x.PMID 14756852. edit
- Freidank-Mueschenborn, E.; Fox, A. (2005). “Resolution of concentration-response differences in onset of effect between subcutaneous and oral sumatriptan”. Headache 45 (6): 632–637. doi:10.1111/j.1526-4610.2005.05129a.x. PMID 15953294. edit
- GSK press release – Treximet (sumatriptan and naproxen sodium) tablets approved by FDA for acute treatment of migraine
- Brandes JL, Kudrow D, Stark SR, et al. (April 2007). “Sumatriptan-naproxen for acute treatment of migraine: a randomized trial”. JAMA 297 (13): 1443–54.doi:10.1001/jama.297.13.1443. PMID 17405970.
- Brandes, J.; Cady, R.; Freitag, F.; Smith, T.; Chandler, P.; Fox, A.; Linn, L.; Farr, S. (2009). “Needle-free subcutaneous sumatriptan (Sumavel DosePro): bioequivalence and ease of use.”. Headache 49 (10): 1435–1444. doi:10.1111/j.1526-4610.2009.01530.x.PMID 19849720. edit
- ClinicalTrials.gov NCT00724815 The Efficacy and Tolerability of NP101 Patch in the Treatment of Acute Migraine (NP101-007)
- SmartRelief -electronically assisted drug delivery (iontophoresis)
- Pierce, M; Marbury, T; O’Neill, C; Siegel, S; Du, W; Sebree, T (2009). “Zelrix: a novel transdermal formulation of sumatriptan”. Headache 49 (6): 817–25. doi:10.1111/j.1526-4610.2009.01437.x. PMID 19438727.
- Zecuity Approved by the FDA for the Acute Treatment of Migraine
- “PAR PHARMACEUTICAL BEGINS SHIPMENT OF SUMATRIPTAN INJECTION”. Par Pharmaceutical. 2008-11-06. Retrieved 2008-11-25.
- Serotonin 5HT1-receptor agonist. Prepn: M. D. Dowle, I. H. Coates, DE 3320521; eidem, US 4816470; A. W. Oxford, GB 2162522 (1983, 1989, 1986 all to Glaxo).
- Receptor binding studies: P. P. A. Humphrey et al., Br. J. Pharmacol.94, 1123 (1988); P. Schoeffter, D. Hoyer, Arch. Pharmacol. 340, 135 (1989).
- LC-MS determn in plasma: J. Oxford, M. S. Lant, J. Chromatogr. 496, 137 (1989).
- Clinical evaluations in migraine: A. Doenicke et al., Lancet 1, 1309 (1988);
- Subcutaneous Sumatriptan International Study Group, N. Engl. J. Med. 325, 316 (1991); in acute cluster headache: Sumatriptan Cluster Headache Study Group, ibid. 322.
- Review of pharmacology and clinical experience: S. J. Peroutka, Headache 30 (Suppl. 2), 554-560 (1990).
- Drugs Fut 1989,14(1),35
|
1-4-2012
|
Noncardiotoxic pharmaceutical compounds
|
|
|
7-9-2010
|
NON-MUCOADHESIVE FILM DOSAGE FORMS
|
|
|
1-22-2010
|
Fixed Combination Dosage Forms for the Treatment of Migraine
|
|
|
12-11-2009
|
ACTIVE AGENT DELIVERY SYSTEMS AND METHODS FOR PROTECTING AND ADMINISTERING ACTIVE AGENTS
|
|
|
10-9-2009
|
PHARMACEUTICAL COMPOSITIONS COMPRISING A TRIPTAN AND A NONSTEROIDAL ANTI-INFLAMMATORY DRUG
|
|
|
10-9-2009
|
ACTIVE AGENT DELIVERY SYSTEMS AND METHODS FOR PROTECTING AND ADMINISTERING ACTIVE AGENTS
|
|
|
5-7-2009
|
Patient controlled drug delivery device
|
|
|
3-20-2009
|
DEUTERIUM-ENRICHED SUMATRIPTAN
|
|
|
3-13-2009
|
Rapid dissolution of combination products
|
|
|
2-19-2009
|
A METHOD OF IDENTIFYING MODULATORS OF CELL SURFACE MEMBRANE RECEPTORS USEFUL IN THE TREATMENT OF DISEASE
|
|
4-8-1992
|
PREPARATION OF INDOLE DERIVATIVES
|
|
|
1-10-1992
|
PHARMACEUTICAL PREPARATIONS
|
|
|
10-32-1991
|
SYSTEM AND METHOD FOR DETERMINING THREE-DIMENSIONAL STRUCTURES OF PROTEINS
|
|
|
8-7-1991
|
Indole derivative
|
|
|
7-4-1990
|
Pharmaceutical formulations
|
|
|
8-8-1984
|
Fuel and water homogenizer
|
Avanir Pharmaceuticals, Inc. is a biopharmaceutical company focused on bringing innovative medicines to patients with central nervous system disorders of high unmet medical need. As part of our commitment, we have extensively invested in our pipeline and are dedicated to advancing medicines that can substantially improve the lives of patients and their loved ones. For more information about Avanir, please visit http://www.avanir.com.
AVANIR® is a trademark or registered trademark of Avanir Pharmaceuticals, Inc. in the United States and other countries. All other trademarks are the property of their respective owners.
Avanir Pharmaceuticals, Inc. licensed exclusive rights for the development and commercialization of AVP-825, a novel Breath Powered intranasal system containing a low-dose sumatriptan powder from OptiNose Inc. of Yardley, PA.
IMITREX Tablets contain sumatriptan succinate, a selective 5-HT1B/1D receptor agonist. Sumatriptan succinate is chemically designated as 3-[2-(dimethylamino)ethyl]-N-methyl-indole- 5-methanesulfonamide succinate (1:1), and it has the following structure:
IMITREX Tablets contain sumatriptan succinate, a selective 5-HT1B/1Dreceptor agonist. Sumatriptan succinate is chemically designated as 3-[2-(dimethylamino)ethyl]-N-methyl-indole- 5-methanesulfonamide succinate (1:1), and it has the following structure:
 |
The empirical formula is C14H21N3O2S•C4H6O4, representing a molecular weight of 413.5. Sumatriptan succinate is a white to off-white powder that is readily soluble in water and in saline.
Each IMITREX Tablet for oral administration contains 35, 70, or 140 mg of sumatriptan succinate equivalent to 25, 50, or 100 mg of sumatriptan, respectively. Each tablet also contains the inactive ingredients croscarmellose sodium, dibasic calcium phosphate, magnesium stearate, microcrystalline cellulose, and sodium bicarbonate. Each 100-mg tablet also contains hypromellose, iron oxide, titanium dioxide, and triacetin.
Idelalisib ….US FDA Accepts NDA for Gilead’s Idelalisib for the Treatment of Refractory Indolent Non-Hodgkin’s Lymphoma

An antineoplastic agent and p110delta inhibitor
(S)-2-(1-(9H-purin-6-ylamino)propyl)-5-fluoro-3-phenylquinazolin-4(3H)-one
Icos (Originator)
- CAL-101
- GS-1101
- Idelalisib
- UNII-YG57I8T5M0
M.Wt: 415.43
Formula: C22H18FN7O
CAS No.: 870281-82-6
CAL-101 Solubility: DMSO ≥80mg/mL Water <1.2mg/mL Ethanol ≥33mg/mL
5-Fluoro-3-phenyl-2-[(1S)-1-(7H-purin-6-ylamino)propyl]-4(3H)-quinazolinone
idelalisib
Idelalisib (codenamed GS-1101 or CAL-101) is a drug under investigation for the treatment of chronic lymphocytic leukaemia. It is in Phase III clinical trials testing drug combinations with rituximab and/or bendamustine as of 2013. The substance acts as aphosphoinositide 3-kinase inhibitor; more specifically, it blocks P110δ, the delta isoform of the enzyme phosphoinositide 3-kinase.[1][2]
GDC-0032 is a potent, next-generation beta isoform-sparing PI3K inhibitor targeting PI3Kα/δ/γ with IC 50 of 0.29 nM/0.12 nM/0.97nM,> 10 fold over Selective PI3K [beta].
GS-1101 is a novel, orally available small molecule inhibitor of phosphatidylinositol 3-kinase delta (PI3Kdelta) develop by Gilead and is waiting for registration in U.S. for the treatment of patients with indolent non-Hodgkin’s lymphoma that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy and for the treatment of chronic lymphocytic leukemia. The compound is also in phase III clinical evaluation for the treatment of elderly patients with previously untreated small lymphocytic lymphoma (SLL) and acute myeloid leukemia. Clinical trials had been under way for the treatment of inflammation and allergic rhinitis; however, no recent development has been reported. Preclinical studies have shown that GS-1101 has desirable pharmaceutical properties. The compound was originally developed by Calistoga Pharmaceuticals, acquired by Gilead on April 1, 2011.
clinical trials, click link
http://clinicaltrials.gov/search/intervention=CAL-101%20OR%20GS-1101%20OR%20Idelalisib
FOSTER CITY, Calif.–(BUSINESS WIRE)–Jan. 13, 2014– Gilead Sciences, Inc. (Nasdaq: GILD) announced today that the U.S. Food and Drug Administration (FDA) has accepted for review the company’s New Drug Application (NDA) for idelalisib, a targeted, oral inhibitor of PI3K delta, for the treatment of refractory indolent non-Hodgkin’s lymphoma (iNHL). FDA has granted a standard review for the iNHL NDA and has set a target review date under the Prescription Drug User Fee Act (PDUFA) of September 11, 2014.
The NDA for iNHL, submitted on September 11, 2013, was supported by a single arm Phase 2 study (Study 101-09) evaluating idelalisib in patients with iNHL that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy. Following Gilead’s NDA submission for iNHL, FDA granted idelalisib a Breakthrough Therapy designation for relapsed chronic lymphocytic leukemia (CLL). The FDA grants Breakthrough Therapy designation to drug candidates that may offer major advances in treatment over existing options. Gilead submitted an NDA for idelalisib for the treatment of CLL on December 6, 2013.
About Idelalisib
Idelalisib is an investigational, highly selective oral inhibitor of phosphoinositide 3-kinase (PI3K) delta. PI3K delta signaling is critical for the activation, proliferation, survival and trafficking of B lymphocytes and is hyperactive in many B-cell malignancies. Idelalisib is being developed both as a single agent and in combination with approved and investigational therapies.
Gilead’s clinical development program for idelalisib in iNHL includes Study 101-09 in highly refractory patients and two Phase 3 studies of idelalisib in previously treated patients. The development program in CLL includes three Phase 3 studies of idelalisib in previously treated patients. Combination therapy with idelalisib and GS-9973, Gilead’s novel spleen tyrosine kinase (Syk) inhibitor, also is being evaluated in a Phase 2 trial of patients with relapsed or refractory CLL, iNHL and other lymphoid malignancies.
Additional information about clinical studies of idelalisib and Gilead’s other investigational cancer agents can be found at http://www.clinicaltrials.gov. Idelalisib and GS-9973 are investigational products and their safety and efficacy have not been established.
About Indolent Non-Hodgkin’s Lymphoma
Indolent non-Hodgkin’s lymphoma refers to a group of largely incurable slow-growing lymphomas that run a relapsing course after therapy and can lead ultimately to life-threatening complications such as serious infections and marrow failure. Most iNHL patients are diagnosed at an advanced stage of disease, and median survival from time of initial diagnosis for patients with the most common form of iNHL, follicular lymphoma, is 8 to 10 years. The outlook for refractory iNHL patients is significantly poorer.
About Gilead Sciences
Gilead Sciences is a biopharmaceutical company that discovers, develops and commercializes innovative therapeutics in areas of unmet medical need. The company’s mission is to advance the care of patients suffering from life-threatening diseases worldwide. Headquartered in Foster City, California, Gilead has operations in North and South America, Europe and Asia Pacific.
The delta form of PI3K is expressed primarily in blood-cell lineages, including cells that cause or mediate hematologic malignancies, inflammation, autoimmune diseases and allergies. By specifically inhibiting only PI3K delta, a therapeutic effect is exerted without inhibiting PI3K signalling that is critical to the normal function of healthy cells. Extensive studies have shown that inhibition of other PI3K forms can cause significant toxicities, particularly with respect to glucose metabolism, which is essential for normal cell activity.
In 2011, orphan drug designation was assigned to GS-1101 in the U.S. for the treatment of CLL. In 2013, several orphan drug designations were assigned to the compound in the E.U. and U.S.: for the treatment of follicular lymphoma, for the treatment of mucosa-associated lymphoid tissue lymphoma (MALT), for the treatment of nodal marginal zone lymphoma, for the treatment of splenic marginal zone lymphoma, and for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma. Orphan drug designation was also assigned in the U.S. for the treatment of lymphoplasmacytic lymphoma with or without Walenstom’s macroglobulinemia and, in the E.U., for the treatment of Waldenstrom’s macroglobulinemia (lymphoplasmacytic lymphoma).
Later in 2013, some of these orphan drug designations were withdrawn in the E.U.; for the treatment of chronic lymphocytic leukemia / small lymphocytic lymphoma, for the treatment of extranodal marginal-zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma), for the treatment of of nodal marginal-zone lymphoma and for the treatment of splenic marginal-zone lymphoma. In 2013, the FDA granted a breakthrough therapy designation for the treatment of chronic lymphocytic leukemia.
- H. Spreitzer (13 May 2013). “Neue Wirkstoffe – Ibrutinib und Idelalisib”. Österreichische Apothekerzeitung (in German) (10/2013): 34.
- Wu, M.; Akinleye, A.; Zhu, X. (2013). “Novel agents for chronic lymphocytic leukemia”.Journal of Hematology & Oncology 6: 36. doi:10.1186/1756-8722-6-36.PMC 3659027. PMID 23680477.
CAL-101 is an Oral Delta Isoform-Selective PI3 Kinase Inhibitor.
| US8207153 | 6-27-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2012015964 | 1-20-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2011306622 | 12-16-2011 | METHODS OF TREATING HEMATOLOGICAL DISORDERS WITH QUINAZOLINONE COMPOUNDS IN SELECTED SUBJECTS |
| US7932260 | 4-27-2011 | Quinazolinones as Inhibitors of Human Phosphatidylinositol 3-Kinase Delta |
| US2011044942 | 2-25-2011 | METHODS OF TREATMENT FOR SOLID TUMORS |
| US2010256167 | 10-8-2010 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2010202963 | 8-13-2010 | THERAPIES FOR HEMATOLOGIC MALIGNANCIES |
| WO2005113556A1 * | 12 May 2005 | 1 Dec 2005 | Icos Corp | Quinazolinones as inhibitors of human phosphatidylinositol 3-kinase delta |
| WO2005117889A1 * | 12 Nov 2004 | 15 Dec 2005 | Didier Bouscary | Methods for treating and/or preventing aberrant proliferation of hematopoietic |
| WO2005120511A1 * | 4 Jun 2005 | 22 Dec 2005 | Joel S Hayflick | Methods for treating mast cell disorders |
| WO2006089106A2 * | 16 Feb 2006 | 24 Aug 2006 | Icos Corp | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| US20060106038 * | 25 May 2005 | 18 May 2006 | Icos Corporation | Methods for treating and/or preventing aberrant proliferation of hematopoietic cells |
The synthesis of a compound in accordance with formula I is first exemplified using steps A-E below, which provide a synthetic procedure for compound 107, the structure of which is shown below.

(107) is idelalisib
……………….
Synthesis of 2-fluoro-6-nitro-N-phenyl-benzamide (108)
Step A: A solution of 2-fluoro-6- nitrobenzoic acid (100 g, 0.54 mol) and dimethylformamide (5 mL) in dichloromethane (600 mL) was treated dropwise with oxalyl chloride (2 M in dichloromethane, 410 mL, 0.8 mol, 1.5 eq) over 30 min. After stirring 2 h at room temperature, the reaction was concentrated to an orange syrup with some solids present. The syrup was dissolved in dry dioxane (80 mL) and slowly added to a suspension of aniline (49 mL, 0.54 mol, 1 eq) and sodium bicarbonate (90 g, 1.08 mol, 2 eq) in a mixture of dioxane (250 mL) and water (250 mL) at 6 0C. The temperature reached 27°C at the end of the addition. After 30 min, the reaction mixture was treated with water (1.2 L). The precipitate was collected by vacuum filtration, washed with water (300 mL) , air dried in the funnel, and dried in vacuo at 50°C for 24 h to afford an off-white solid product (139 g, 99%). 1H NMR (300 MHz, DMSO-d6) δ 10.82 (s, IH), 8.12 (d, J = 7.7 Hz, IH), 7.91-7.77 (m, 2H), 7.64 (d, J = 7.7 Hz, 2H), 7.38 (t, J = 7.9 Hz, 2H), 7.15 > (t, J = 7.4 Hz, IH), ESI-MS m/z 261 (MH+). The reaction described above and compound 108 are shown below.

………………………..
Synthesis of(S) – [1- (2-fluoro-6-nitro-benzoyl) -phenyl-aminocarbonyl] – propyl-carbamic acid tert-butyl ester (109)
Step B: A suspension of compound 108 (0.5 mol) and dimethylformamide (5 mL) in thionyl chloride (256 mL, 2.5 mol, 5 eq) was stirred at 85°C for 5 hours. The reaction mixture was concentrated in vacuo to a brown syrup. The syrup was dissolved in dichloromethane (200 mL) and was slowly added to a solution of N-BOC-L-2-aminobutyric acid (112 g, 0.55 mol, 1.1 eq) and triethylamine (77 mL, 0.55 mol, 1.1 eq) in dichloromethane (600 mL) at 10 0C. After stirring at room temperature for 3 h, salts were removed by filtration, and the solution was washed with 100 mL of water, saturated sodium bicarbonate, water, 5% citric acid, and saturated sodium chloride. The organic phase was dried with magnesium sulfate and concentrated to a red syrup. The syrup was dissolved in dichloromethane (450 mL) and purified by flash chromatography on a silica gel plug (15 x 22 cm, 4 L dry silica) eluted with hexanes/ethyl acetate (10%, 8 L; 15%, 8 L; 20%, 8 L; 25%, 4 L) to yield the compound 109 as an off-white solid (147 g, 66%). 1H NMR (300 MHz, DMSO-d6) δ 8.13 (d, J = 8.0 Hz, IH), 7.84 (t, J = 8.6 Hz, IH), 7.78- 7.67 (m, IH), 7.65-7.49 (m, 3H), 7.40-7.28 ( m, 2H), 7.19 (d, J = 7.5 Hz, IH), 4.05 (broad s, IH), 1.75- 1.30 (m, 2H), 1.34 (s, 9H), 0.93 (broad s, 3H). ESI- MS m/z 446.3 (MH+) . The reaction described above and compound 109 are shown below.

Synthesis of(S) – [1- (5-fluoro-4-oxo-3-phenyl-3 , 4-dihydro-quinazolin-2- yl) -propyl] -carbamic acid tert-butyl ester (110)
Step C: A solution of compound 109 (125 mmol, 1 eq) in acetic acid (500 mL) was treated with zinc dust (48.4 g, 740 mmol, 6 eq) added in 3 portions, and the reaction mixture was allowed to cool to below 35°C between additions. After stirring for 2 h at ambient temperature, solids were filtered off by vacuum filtration and washed with acetic acid (50 mL) . The filtrate was concentrated in vacuo, dissolved in EtOAc (400 mL) , washed with water (300 mL) , and the water layer was extracted with EtOAc (300 mL) . The combined organic layers were washed with water (200 mL) , sat’d sodium bicarbonate (2 x 200 mL) , sat’d NaCl (100 mL) , dried with MgSO4, and concentrated to a syrup. The syrup was dissolved in toluene (200 mL) and purified by flash chromatography on a silica gel plug (13 x 15 cm, 2 L dry silica) eluted with hexanes/ethyl acetate (10%, 4 L; 15%, 4 L; 17.5%, 8 L; 25%, 4 L) to yield compound 110 as an off-white foamy solid (33.6 g, 69%). 1H NMR (300 MHz, DMSO-d6) δ 7.83 (td, J = 8.2, 5.7 Hz, IH), 7.64-7.48 (m, 5H), 7.39 (broad d, J = 7.6 Hz, IH), 7.30 (dd, J = 8.3 Hz, IH), 7.23 (d, J = 7.6 Hz, IH), 4.02-3.90 (m, IH), 1.76-1.66 (m, IH), 1.62-1.46 (m, IH), 1.33 (s, 9H), 0.63 (t, J= 7.3 Hz, 3H). ESI-MS m/z 398.3 (MH+). The reaction described above and compound 110 are shown below.

…………..
Syn of (S) -2- (1-amino-propyl) -5-fluoro-3-phenyl-3H-quinazolin-4- one (111)
Step D: A solution of compound 110 (85 mmol) in dichloromethane (60 mL) was treated with trifluoroacetic acid (60 mL) . The reaction mixture was stirred for 1 h, concentrated in vacuo, and partitioned between dichloromethane (150 mL) and 10% K2CO3 (sufficient amount to keep the pH greated than 10) . The aqueous layer was extracted with additional dichloromethane (100 raL) , and the combined organic layers were washed with water (50 mli) and brine (50 mL) . After drying with Mg SO4, the solution was concentrated to provide compound 111 as an off-white solid (22 g, 88%) . 1H NMR (300 MHz,
CDCl3) δ 7.73-7.65 (m, IH), 7.62-7.49 (m, 4H), 7.32- 7.22 (m, 2H), 7.13-7.06 (m, IH), 3.42 (dd, J= 7.5, 5.2 Hz, IH), 1.87-1.70 (m, IH), 1.58-1.43 (m, IH), 0.80 (t, J = 7.4 Hz, 3H) . ESI-MS m/z 298.2 (MH+) . The reaction described above and compound 111 are shown below.

………………
syn of (S) -5-fluoro-3-phenyl-2- [1- (9H-purin-6-ylamino) -propyl] – 3H-quinazolin-4-one (107)
Step E: A suspension of compound 111(65.6 mmol, 1 eq) , 6-bromopurine (14.6 g, 73.4 mmol, 1.1 eq) , and DIEA (24.3 mL, 140 mmol, 2 eq) in tert- butanol (40 mL) was stirred for 24 h at 800C. The reaction mixture was concentrated in vacuo and treated with water to yield a solid crude product that was collected by vacuum filtration, washed with water, and air dried. Half of the obtained solid crude product was dissolved in MeOH (600 mL) , concentrated onto silica gel (300 mL dry) , and purified by flash chromatography (7.5 x 36 cm, eluted with 10 L of 4% MeOH/CH2Cl2) to yield a solid product. The solid product was then dissolved in EtOH (250 mL) and concentrated in vacuo to compound 107 idelalisib as a light yellow solid (7.2 g, 50%).
1H NMR (300 MHz, 80 0C, DMSO-d5) δ 12.66 (broad s, IH), 8.11 (s, IH), 8.02 (broad s, IH), 7.81-7.73 (m, IH),7.60-7.42 (m, 6H), 7.25-7.15 (m, 2H), 4.97 (broad s, IH), 2.02-1.73 (m, 2H), 0.79 (t, J= 7.3 Hz, 3H).
ESI-MS m/z 416.2 (MH+).
C, H, N elemental analysis (C22Hi8N7OF-EtOH- 0.4 H2O).
Chiral purity 99.8:0.2 (S:R) using chiral HPLC (4.6 x 250 mm Chiralpak ODH column, 20 °C, 85:15 hexanes : EtOH, 1 rnL/min, sample loaded at a concentration of 1 mg/mL in EtOH) . The reaction described above and compound 107 idelalisib are shown below.

| WO2001030768A1 * | 26 Oct 2000 | 3 May 2001 | Gustave Bergnes | Methods and compositions utilizing quinazolinones |
| WO2001081346A2 * | 24 Apr 2001 | 1 Nov 2001 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2003035075A1 * | 27 Aug 2002 | 1 May 2003 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2005016348A1 * | 13 Aug 2004 | 24 Feb 2005 | Jason Douangpanya | Method of inhibiting immune responses stimulated by an endogenous factor |
| WO2005016349A1 * | 13 Aug 2004 | 24 Feb 2005 | Thomas G Diacovo | Methods of inhibiting leukocyte accumulation |
| WO2005067901A2 * | 7 Jan 2005 | 28 Jul 2005 | Carrie A Northcott | Methods for treating and preventing hypertension and hypertension-related disorders |
Aeterna Zentaris Submits New Drug Application to FDA for Macimorelin Acetate (AEZS-130) for Evaluation of AGHD

Macimorelin
CAS 381231-18-1
Chemical Formula: C26H30N6O3
Exact Mass: 474.23794
Molecular Weight: 474.55480
Elemental Analysis: C, 65.80; H, 6.37; N, 17.71; O, 10.11

945212-59-9 (Macimorelin acetate)
AEZS-130
ARD-07
D-87875
EP-01572
EP-1572
JMV-1843
USAN (ab-26)
MACIMORELIN ACETATE
THERAPEUTIC CLAIM
Diagnostic agent for adult growth hormone deficiency (AGHD)
CHEMICAL NAMES
1. D-Tryptophanamide, 2-methylalanyl-N-[(1R)-1-(formylamino)-2-(1H-indol-3-yl)ethyl]-, acetate (1:1)
2. N2-(2-amino-2-methylpropanoyl-N1-[(1R)-1-formamido-2-(1H-indol-3-yl)ethyl]- D-tryptophanamide acetate
MOLECULAR FORMULA
C26H30N6O3.C2H4O2
MOLECULAR WEIGHT
534.6
SPONSOR
Aeterna Zentaris GmbH
CODE DESIGNATIONS
D-87575, EP 1572, ARD 07
CAS REGISTRY NUMBER
945212-59-9
Macimorelin (also known as AEZS-130, EP-1572) is a novel synthetic small molecule, acting as a ghrelin agonist, that is orally active and stimulates the secretion of growth hormone (GH). Based on results of Phase 1 studies, AEZS-130 has potential applications for the treatment of cachexia, a condition frequently associated with severe chronic diseases such as cancer, chronic obstructive pulmonary disease and AIDS. In addition to the therapeutic application, a Phase 3 trial with AEZS-130 as a…
View original post 2,777 more words
New Drug Application for Belinostat in Relapsed or Refractory PTCL Submitted to the FDA

Belinostat (PXD101)
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
| MW 318.07 | |
| Molecular Formula: | C15H14N2O4S |
414864-00-9 cas no
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

- Plumb, Jane A.; Finn, Paul W.; Williams, Robert J.; Bandara, Morwenna J.; Romero, M. Rosario; Watkins, Claire J.; La Thangue, Nicholas B.; Brown, Robert (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101″. Molecular Cancer Therapeutics 2 (8): 721–728. PMID 12939461.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


…………
US20100286279


SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
Durata Therapeutics Announces FDA’s Acceptance for Priority Review of NDA for Dalvance (dalbavancin hydrochloride)

| CAS No. | 171500-79-1 |
| Chemical Name: | Dalbavancin |
| Synonyms: | MDL 63397;Dalbavancin |
| CBNumber: | CB41028737 |
| Molecular Formula: | C88H100Cl2N10O28 |
| Formula Weight: | 1816.71 |
CHICAGO, Nov 26, 2013 (GLOBE NEWSWIRE via COMTEX) — Durata Therapeutics, Inc. DRTX +6.48% today announced that the New Drug Application (NDA) for its investigational drug, Dalvance (dalbavancin hydrochloride) for injection, has been accepted for priority review by the U.S. Food and Drug Administration (FDA) with an action date of May 26, 2014. Durata is seeking FDA approval of Dalvance(TM) for the treatment of patients with acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible Gram-positive microorganisms, including MRSA (methicillin resistant Staphylococcus aureushttp://www.drugs.com/nda/dalbavancin_131126.html

Dalbavancin is prepared by chemical modification of the natural glycopeptide complex A-40,926 as described in Malabarba and Donadio (1999) Drugs of the Future 24(8):839-846. The predominant component of dalbavancin is Factor Bo, which accounts for >75% of the whole complex.
[0042] The amount of each of the components present in a dalbavancin composition is dictated by a variety of factors, including, for example, the fermentation conditions employed in the preparation of the natural glycopeptide complex A-40926, which is the precursor to dalbavancin (see, e.g., U.S. Pat. No.5,843,679), the conditions employed to recover A-40926 from the fermentation broth, the chemical reactions employed to selectively esterify the caxboxyl group of the sugar moiety of A-40926, the conditions employed to amidate the peptidyl carboxyl group, the conditions employed to saponify the ester of the carboxyl group of the N-acylaminoglucuronic acid function, the conditions employed to recover dalbavancin from the synthetic mixture, and the like.
the semisynthetic glycopeptide dalbavancin was synthesized from the natural antibiotic A 40926, originally isolated from an Actinomadura culture (Malabarba et al., 1998, U.S. Pat. No. 5,750,509). Dalbavancin has shown greater efficacy against various bacterial strains than vancomycin or the antibiotic linezolid and represents a promising new treatment for skin and soft tissue infections (see, e.g., Jabés et al., 2004, Antimicrob. Agents Chemother. 48:1118-1123). According to U.S. Pat. No. 5,750,509, dalbavancin is a glycopeptide antibiotic with a monomethyl moiety at its N15 amino (see FIG. 1 for numbering), and this N15-monomethyl amino could be free (i.e. —NHCH3) or protected with an amino protecting group such as t-butoxycarbonyl, carbobenzyloxy, arylalkyl or benzyl. The method for making certain of the dalbavancin components reported in the ‘509 patent also produced N15,N15-dialkyl analogs of dalbavancin in minor-quantities, but these molecules were not characterized.
Dalbavancin (INN, trade name Zeven) is a novel second-generation lipoglycopeptideantibiotic. It belongs to the same class as vancomycin, the most widely used and one of the few treatments available to patients infected with methicillin-resistant Staphylococcus aureus (MRSA).[1]
Dalbavancin (BI397) is a novel semisynthetic lipoglycopeptide that was designed to improve upon the natural glycopeptides currently available, vancomycin and teicoplanin.[2]
It possesses in vitro activity against a variety of Gram-positive pathogens[3][4] includingMRSA and MRSE.[5] It is a once-weekly, two-dose antibiotic that Pfizer acquired when it bought Vicuron Pharmaceuticals in 2005.[6]
Dalbavancin has undergone a phase III clinical trial for adults with complicated skin infections, but in Dec 2007 the FDA said more data was needed before approval.[6] On September 9, 2008, Pfizer announced that it will withdraw all marketing applications in order to conduct another Phase 3 clinical trial.[7] Durata Therapeutics acquired the rights to dalbavancin in December 2009 and has initiated two new Phase III clinical trials for treatment of acute bacterial skin and skin structure infections.[8] Preliminary results in Dec 2012 looked good.[9]
- Vicuron Pharmaceuticals Submits New Drug Application for Dalbavancin to U.S. Food and Drug Administration
- Scheinfeld NS. (May 2006). “Dalbavancin: A review for dermatologists.”. Dermatology Online Journal 12 (6). Unknown parameter
|numero=ignored (help) PMID 17083861 - Chen AY, Zervos MJ, Vazquez JA (2007). “Dalbavancin: a novel antimicrobial”. Int. J. Clin. Pract. 61 (5): 853–63. doi:10.1111/j.1742-1241.2007.01318.x. PMC 1890846.PMID 17362476.
- Das B, Sarkar C, Biswas R, Pandey S (2008). “Review: dalbavancin-a novel lipoglycopeptide antimicrobial for gram positive pathogens”. Pak J Pharm Sci 21 (1): 78–88. PMID 18166524.
- Dalbavancin: A Novel Lipoglycopeptide Antibacterial
- UPDATE 1-Pfizer says US FDA wants more data on antibiotic. Dec 2007
- “Pfizer Will Withdraw Global Marketing Applications for Dalbavancin to Conduct a New Trial” (Press release). Pfizer Inc. 2008-09-09. Retrieved 2008-09-11.
- Durata Begins Dalbavancin Study Enrollment. Drug Discovery & Development – October 05, 2011.
- Durata Therapeutics Announces Phase 3 Clinical Trial Results for Dalbavancin in the Treatment of ABSSSI

DAPTOMYCIN

VANCOMYCIN
Aeterna Zentaris Submits New Drug Application to FDA for Macimorelin Acetate (AEZS-130) for Evaluation of AGHD

Macimorelin
CAS 381231-18-1
Chemical Formula: C26H30N6O3
Exact Mass: 474.23794
Molecular Weight: 474.55480
Elemental Analysis: C, 65.80; H, 6.37; N, 17.71; O, 10.11

945212-59-9 (Macimorelin acetate)
AEZS-130
ARD-07
D-87875
EP-01572
EP-1572
JMV-1843
USAN (ab-26)
MACIMORELIN ACETATE
THERAPEUTIC CLAIM
Diagnostic agent for adult growth hormone deficiency (AGHD)
CHEMICAL NAMES
1. D-Tryptophanamide, 2-methylalanyl-N-[(1R)-1-(formylamino)-2-(1H-indol-3-yl)ethyl]-, acetate (1:1)
2. N2-(2-amino-2-methylpropanoyl-N1-[(1R)-1-formamido-2-(1H-indol-3-yl)ethyl]- D-tryptophanamide acetate
MOLECULAR FORMULA
C26H30N6O3.C2H4O2
MOLECULAR WEIGHT
534.6
SPONSOR
Aeterna Zentaris GmbH
CODE DESIGNATIONS
D-87575, EP 1572, ARD 07
CAS REGISTRY NUMBER
945212-59-9
Macimorelin (also known as AEZS-130, EP-1572) is a novel synthetic small molecule, acting as a ghrelin agonist, that is orally active and stimulates the secretion of growth hormone (GH). Based on results of Phase 1 studies, AEZS-130 has potential applications for the treatment of cachexia, a condition frequently associated with severe chronic diseases such as cancer, chronic obstructive pulmonary disease and AIDS. In addition to the therapeutic application, a Phase 3 trial with AEZS-130 as a diagnostic test for growth hormone deficiencies in adults has been completed.
http://www.ama-assn.org/resources/doc/usan/macimorelin-acetate.pdf
QUEBEC, Nov. 5, 2013 /PRNewswire/ – Aeterna Zentaris Inc. (the “Company”) today announced that it has submitted a New Drug Application (“NDA”) to the U.S. Food and Drug Administration (“FDA”) for its ghrelin agonist, macimorelin acetate (AEZS-130). Phase 3 data have demonstrated that the compound has the potential to become the first orally-approved product that induces growth hormone release to evaluate adult growth hormone deficiency (“AGHD”), with accuracy comparable to available intravenous and intramuscular testing procedures. read at
http://www.drugs.com/nda/macimorelin_acetate_131105.html
http://www.ama-assn.org/resources/doc/usan/macimorelin-acetate.pdf
macimorelin (JMV 1843), a ghrelin-mimetic growth hormone secretagogue in Phase III for adult growth hormone deficiency (AGHD)

Macimorelin, a growth hormone modulator, is currently awaiting registration in the U.S. by AEterna Zentaris as an oral diagnostic test of adult growth hormone deficit disorder. The company is also developing the compound in phase II clinical trials for the treatment of cancer related cachexia. The compound was being codeveloped by AEterna Zentaris and Ardana Bioscience; however, the trials underway at Ardana were suspended in 2008 based on a company strategic decision. AEterna Zentaris owns the worldwide rights of the compound. In 2007, orphan drug designation was assigned by the FDA for the treatment of growth hormone deficit in adults.
New active series of growth hormone secretagogues
J Med Chem 2003, 46(7): 1191
WO 2001096300
WO 2007093820
…………………………
J Med Chem 2003, 46(7): 1191
http://pubs.acs.org/doi/full/10.1021/jm020985q


Synthetic Pathway for JMV 1843 and Analoguesa
a Reagents and conditions: (a) IBCF, NMM, DME, 0 °C; (b) NH4OH; (c) H2, Pd/C, EtOH, HCl; (d) BOP, NMM, DMF, Boc-(d)-Trp-OH; (e) Boc2O, DMAP cat., anhydrous CH3CN; (f) BTIB, pyridine, DMF/H2O; (g) 2,4,5-trichlorophenylformate, DIEA, DMF; (h) TFA/anisole/thioanisole (8:1:1), 0 °C; (i) BOP, NMM, DMF, Boc-Aib-OH; (j) TFA/anisole/thioanisole (8:1:1), 0 °C; (k) RP preparative HPLC.
TFA, H-Aib-(d)-Trp-(d)-gTrp-CHO (7). 6 (1 g, 1.7 mmol) was dissolved in a mixture of trifluoroacetic acid (8 mL), anisole (1 mL), and thioanisole (1 mL) for 30 min at 0 °C. The solvents were removed in vacuo, the residue was stirred in ether, and the precipitated TFA, H-Aib-(d)-Trp-(d)-gTrp-CHO was filtered. 7 was purified by preparative HPLC and obtained in 52% yield. 1H NMR (400 MHz, DMSO-d6) + correlation 1H−1H: δ 1.21 (s, 3H, CH3 (Aib)), 1.43 (s, 3H, CH3 (Aib)), 2.97 (m, 2H, (CH2)β), 3.1 (m, 2H, (CH2)β‘), 4.62 (m, 1H, (CH)αA and (CH)αB), 5.32 (q, 0.4H, (CH)α‘B), 5.71 (q, 0.6H, (CH)α‘A), 7.3 (m, 4H, H5 and H6 (2 indoles)), 7.06−7.2 (4d, 2H, H2A and H2B (2 indoles)), 7.3 (m, 2H, H4 or H7 (2 indoles)), 7.6−7.8 (4d, 2H, H4A and H4B or H7A and H7B), 7.97 (s, 3H, NH2 (Aib) and CHO (formyl)), 8.2 (d, 0.4H, NH1B (diamino)), 8.3 (m,1H, NHA and NHB), 8.5 (d, 0.6H, NH1A (diamino)), 8.69 (d, 0.6H, NH2A (diamino)), 8.96 (d, 0.4H, NH2B (diamino)), 10.8 (s, 0.6H, N1H1A (indole)), 10.82 (s, 0.4H, N1H1B (indole)), 10.86 (s, 0.6H, N1H2A (indole)), 10.91 (s, 0,4H, N1H2B (indole)). MS (ES), m/z: 475 [M + H]+, 949 [2M + H]+. HPLC tR: 16.26 min (conditions A).
…………………………..
http://www.google.com/patents/US8192719
The inventors have now found that the oral administration of growth hormone secretagogues (GHSs) EP 1572 and EP 1573 can be used effectively and reliably to diagnose GHD.
EP 1572 (Formula I) or EP 1573 (Formula II) are GHSs (see WO 01/96300, Example 1 and Example 58 which are EP 1572 and EP 1573, respectively) that may be given orally.

EP 1572 and EP 1573 can also be defined as H-Aib-D-Trp-D-gTrp-CHO and H-Aib-D-Trp-D-gTrp-C(O)NHCH2CH3. Wherein, His hydrogen, Aib is aminoisobutyl, D is the dextro isomer, Trp is tryptophan and gTrp is a group of Formula III:

…………………………….
http://www.google.com/patents/US6861409
H-Aib-D-Trp-D-gTrp-CHO: 
Example 1 H-Aib-D-Trp-D-gTrp-CHO
Total synthesis (percentages represent yields obtained in the synthesis as described below):

Z-D-Tr-NH2
Z-D-Trp-OH (8.9 g; 26 mmol; 1 eq.) was dissolved in DME (25 ml) and placed in an ice water bath to 0° C. NMM (3.5 ml; 1.2 eq.), IBCF (4.1 ml; 1.2 eq.) and ammonia solution 28% (8.9 ml; 5 eq.) were added successively. The mixture was diluted with water (100 ml), and the product Z-D-Trp-NH2 precipitated. It was filtered and dried in vacuo to afford 8.58 g of a white solid.
Yield=98%.
C19H19N3O3, 337 g.mol−1.
Rf=0.46 {Chloroform/Methanol/Acetic Acid (180/10/5)}.
1H NMR (250 MHZ, DMSO-d6): δ 2.9 (dd, 1H, Hβ, Jββ′=14.5 Hz; Jβα=9.8 Hz); 3.1 (dd, 1H, Hβ′, Jβ′β=14.5 Hz; Jβ′α=4.3 Hz); 4.2 (sextuplet, 1H, Hα); 4.95 (s, 2H, CH2 (Z); 6.9-7.4 (m, 11H); 7.5 (s, 1H, H2); 7.65 (d, 1H, J=7.7 Hz); 10.8 (s, 1H, N1H).
Mass Spectrometry (Electrospray), m/z 338 [M+H]+, 360 [M+Na]+, 675 [2M+H]+, 697 [2M+Na]+.
Boc-D-Trp-D-Trp-NH2
Z-D-Trp-NH2 (3 g; 8.9 mmol; 1 eq.) was dissolved in DMF (100 ml). HCl 36% (845 μl; 1.1 eq.), water (2 ml) and palladium on activated charcoal (95 mg, 0.1 eq.) were added to the stirred mixture. The solution was bubbled under hydrogen for 24 hr. When the reaction went to completion, the palladium was filtered on celite. The solvent was removed in vacuo to afford HCl, H-D-Trp-NH2 as a colorless oil.
In 10 ml of DMF, HCl, H-D-Trp-NH2 (8.9 mmol; 1 eq.), Boc-D-Trp-OH (2.98 g; 9.8 mmol; 1.1 eq.), NMM (2.26 ml; 2.1 eq.) and BOP (4.33 g; 1.1 eq.) were added successively. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (100 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo to afford 4.35 g of Boc-D-Trp-D-Trp-NH2 as a white solid.
Yield=85%.
C27H31N5O4, 489 g.mol−1.
Rf=0.48 {Chloroform/Methanol/Acetic Acid (85/10/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.28 (s, 9H, Boc); 2.75-3.36 (m, 4H, 2 (CH2)β; 4.14 (m, 1H, CHα); 4.52 (m, 1H, CHα′); 6.83-7.84 (m, 14H, 2 indoles (10H), NH2, NH (urethane) and NH (amide)); 10.82 (d, 1H, J=2 Hz, N1H); 10.85 (d, 1H, J=2 Hz, N1H).
Mass Spectrometry (Electrospray), m/z 490 [M+H]+, 512 [M+Na]+, 979 [2M+H]+.
Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2
Boc-D-Trp-D-Trp-NH2 (3 g; 6.13 mmol; 1 eq.) was dissolved in acetonitrile (25 ml).
To this solution, di-tert-butyl-dicarbonate (3.4 g; 2.5 eq.) and 4-dimethylaminopyridine (150 mg; 0.2 eq.) were successively added. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate/hexane {5/5} to afford 2.53 g of Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2 as a white solid.
Yield=60%.
C37H47N5O8, 689 g.mol−1.
Rf=0.23 {ethyl acetate/hexane (5/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.25 (s, 9H, Boc); 1.58 (s, 9H, Boc); 1.61 (s, 9H, Boc); 2.75-3.4 (m, 4H, 2 (CH2)β); 4.2 (m, 1H, CHα′); 4.6 (m, 1H, CHα); 7.06-8 (m, 14H, 2 indoles (10H), NH (urethane), NH and NH2 (amides)).
Mass Spectrometry (Electrospray), m/z 690 [M+H]+, 712 [M+Na]+, 1379 [2M+H]+, 1401 [2M+Na]+.
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-H
Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2 (3 g; 4.3 mmol; 1 eq.) was dissolved in the mixture DMF/water (18 ml/7 ml). Then, pyridine (772 μl; 2.2 eq.) and Bis(Trifluoroacetoxy)IodoBenzene (2.1 g; 1.1 eq.) were added. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and aqueous saturated sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. Boc-D-NiBoc)Trp-D-g(NiBoc)Trp-H was used immediately for the next reaction of formylation.
Rf=0.14 {ethyl acetate/hexane (7/3)}.
C36H47N5O7, 661 g.mol−1.
1H NMR (200 MHZ, DMSO-d6): δ 1.29 (s, 9H, Boc); 1.61 (s, 18H, 2 Boc); 2.13 (s, 2H, NH2 (amine)); 3.1-2.8 (m, 4H, 2 (CH2)β); 4.2 (m, 1H, CHα′); 4.85 (m, 1H, CHα); 6.9-8 (m, 12H, 2 indoles (10H), NH (urethane), NH (amide)).
Mass Spectrometry (Electrospray), m/z 662 [M+H]+, 684 [M+Na]+.
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-H (4.3 mmol; 1 eq.) was dissolved in DMF (20 ml). Then, N,N-diisopropylethylamine (815 μl; 1.1 eq.) and 2,4,5-trichlorophenylformate (1.08 g; 1.1 eq.) were added. After 30 minutes, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate/hexane {5/5} to afford 2.07 g of Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO as a white solid.
Yield=70%.
C37H47N5O8, 689 g.mol−1.
Rf=0.27 {ethyl acetate/hexane (5/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.28 (s, 9H, Boc); 1.6 (s, 9H, Boc); 1.61 (s, 9H, Boc); 2.75-3.1 (m, 4H, 2 (CH2)β); 4.25 (m, 1H, (CH)αA&B); 5.39 (m, 0.4H, (CH)α′B); 5.72 (m, 0.6H, (CH)α′A); 6.95-8.55 (m, 14H, 2 indoles (10H), NH (urethane), 2 NH (amides), CHO (formyl)).
Mass Spectrometry (Electrospray), m/z 690 [M+H]+, 712 [M+Na]+, 1379 [2M+H]+.
Boc-Aib-D-Trp-D-gTrp-CHO
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO (1.98 g; 2.9 mmol; 1 eq.) was dissolved in a -mixture of trifluoroacetic acid (16 ml), anisole (2 ml) and thioanisole (2 ml) for 30 minutes at 0° C. The solvents were removed in vacuo, the residue was stirred with ether and the precipitated TFA, H-D-Trp-D-gTrp-CHO was filtered.
TFA, H-D-Trp-D-gTrp-CHO (2.9 mmol; 1 eq.), Boc-Aib-OH (700 mg; 1 eq.), NMM (2.4 ml; 4.2 eq.) and BOP (1.53 g; 1.2 eq.) were successively added in 10 ml of DMF. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate to afford 1.16 g of Boc-Aib-D-Trp-D-gTrp-CHO as a white solid.
Yield=70%.
C31H38N6O5, 574 g.mol−1.
Rf=0.26 {Chloroform/Methanol/Acetic Acid (180/10/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.21 (s, 6H, 2 CH3(Aib)); 1.31 (s, 9H, Boc); 2.98-3.12 (m, 4H, 2 (CH2)β); 4.47 (m, 1H, (CH)αA&B); 5.2 (m, 0.4H, (CH)α′B); 5.7 (m, 0.6H, (CH)α′A); 6.95-8.37 (m, 15H, 2 indoles (10H), 3 NH (amides), 1 NH (urethane) CHO (formyl)); 10.89 (m, 2H, 2 N1H (indoles)).
Mass Spectrometry (Electrospray), ml/z 575 [M+H]+, 597 [M+Na]+, 1149 [2M+H]+, 1171 [2M+Na]+.
H-Aib-D-Trp-D-gTrT-CHO
Boc-Aib-D-Trp-D-gTrp-CHO (1 g; 1.7 nmmol) was dissolved in a mixture of trifluoroacetic acid (8 ml), anisole (1 ml) and thioanisole (1 ml) for 30 minutes at 0° C. The solvents were removed in vacuo, the residue was stirred with ether and the precipitated TFA, H-Aib-D-Trp-D-gTrp-CHO was filtered.
The product TFA, H-Aib-D-Trp-D-gTrp-CHO was purified by preparative HPLC (Waters, delta pak, C18, 40×100 mm, 5 μm, 100 A).
Yield=52%.
C26H30N6O3, 474 g.mol−1.
1H NMR (400 MHZ, DMSO-d6)+1H/1H correlation: δ 1.21 (s, 3H, CH3 (Aib)); 1.43 (s, 3H, CH3 (Aib)); 2.97 (m, 2H, (CH2)β); 3.1 (m, 2H, (CH2)β′); 4.62 (m, 1H, (CH)αA&B); 5.32 (q, 0.4H, (CH)α′B); 5.71 (q, 0.6H, (CH)α′A); 7.3 (m, 4H5 and H6 (2 indoles)); 7.06-7.2 (4d, 2H, H2A et H2B (2 indoles)); 7.3 (m, 2H, H4 or H7 (2 indoles)); 7.6-7.8 (4d, 2H, H4A and H4B or H7A et H7B); 7.97 (s, 3H, NH2 (Aib) and CHO (Formyl));8.2 (d, 0.4H, NH1B (diamino)); 8.3 (m,1H, NHA&B); 8.5 (d, 0.6H, NH1A (diamino)); 8.69 (d, 0.6H, NH2A (diamino)); 8.96 (d, 0.4H, NH2B (diamino)); 10.8 (s, 0.6H, N1H1A (indole)); 10.82 (s, 0.4H, N1H1B (indole)); 10.86 (s, 0.6H, N1H2A (indole)); 10.91 (s, 0.4, N1H2B (indole)).
Mass Spectrometry (Electrospray), m/z 475 [M+H]+, 949 [2M+H]+.
………………………………
UPDATED INFO AS ON JAN 6 2014
Aeterna Zentaris NDA for Macimorelin Acetate in AGHD Accepted for Filing by the FDA
Quebec City, Canada, January 6, 2014 – Aeterna Zentaris Inc. (NASDAQ: AEZS) (TSX: AEZS) (the “Company”) today announced that the U.S. Food and Drug Administration (“FDA”) has accepted for filing the Company’s New Drug Application (“NDA”) for its ghrelin agonist, macimorelin acetate, in Adult Growth Hormone Deficiency (“AGHD”). The acceptance for filing of the NDA indicates the FDA has determined that the application is sufficiently complete to permit a substantive review.
The Company’s NDA, submitted on November 5, 2013, seeks approval for the commercialization of macimorelin acetate as the first orally-administered product that induces growth hormone release to evaluate AGHD. Phase 3 data have demonstrated the compound to be well tolerated, with accuracy comparable to available intravenous and intramuscular testing procedures. The application will be subject to a standard review and will have a Prescription Drug User Fee Act (“PDUFA”) date of November 5, 2014. The PDUFA date is the goal date for the FDA to complete its review of the NDA.
David Dodd, President and CEO of Aeterna Zentaris, commented, “The FDA’s acceptance of this NDA submission is another significant milestone in our strategy to commercialize macimorelin acetate as the first approved oral product for AGHD evaluation. We are finalizing our commercial plan for this exciting new product. We are also looking to broaden the commercial application of macimorelin acetate in AGHD for use related to traumatic brain injury victims and other developmental areas, which would represent significant benefit to the evaluation of growth hormone deficiency, while presenting further potential revenue growth opportunities for the Company.”
About Macimorelin Acetate
Macimorelin acetate, a ghrelin agonist, is a novel orally-active small molecule that stimulates the secretion of growth hormone. The Company has completed a Phase 3 trial for use in evaluating AGHD, and has filed an NDA to the FDA in this indication. Macimorelin acetate has been granted orphan drug designation by the FDA for use in AGHD. Furthermore, macimorelin acetate is in a Phase 2 trial as a treatment for cancer-induced cachexia. Aeterna Zentaris owns the worldwide rights to this novel patented compound.
About AGHD
AGHD affects about 75,000 adults across the U.S., Canada and Europe. Growth hormone not only plays an important role in growth from childhood to adulthood, but also helps promote a hormonally-balanced health status. AGHD mostly results from damage to the pituitary gland. It is usually characterized by a reduction in bone mineral density, lean mass, exercise capacity, and overall quality of life.
About Aeterna Zentaris
Aeterna Zentaris is a specialty biopharmaceutical company engaged in developing novel treatments in oncology and endocrinology. The Company’s pipeline encompasses compounds from drug discovery to regulatory approval.

back to home for more updates

DR ANTHONY MELVIN CRASTO Ph.D
Cubist Pharmaceuticals, Inc. announced that it has submitted a NDA to the U.S. FDA for approval of its investigational antibiotic tedizolid phosphate (TR-701).
TEDIZOLID PHOSPHATE
PRONUNCIATION ted” eye zoe’ lid
THERAPEUTIC CLAIM Treatment of complicated skin and skin structure infections
CHEMICAL NAMES
1. 2-Oxazolidinone, 3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5- [(phosphonooxy)methyl]-, (5R)-
2. [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxooxazolidin-5- yl]methyl hydrogen phosphate
http://www.ama-assn.org/resources/doc/usan/tedizolid-phosphate.pdf
MOLECULAR FORMULA C17H16FN6O6P
MOLECULAR WEIGHT 450.3
TRADEMARK None as yet
SPONSOR Trius Therapeutics
CODE DESIGNATION TR-701 FA
CAS REGISTRY NUMBER 856867-55-5
Note: This adoption statement supersedes the USAN torezolid phosphate (N09/81), which is hereby rescinded and replaced by the USAN tedizolid phosphate (N10/118).
Cubist Announces Submission of New Drug Application for Investigational Antibiotic Tedizolid for Treatment of Serious Skin Infections
LEXINGTON, Mass.–(BUSINESS WIRE)– Cubist Pharmaceuticals, Inc. today announced that it has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of its investigational antibiotic tedizolid phosphate (TR-701). Cubist is seeking approval of tedizolid phosphate for the treatment of acute bacterial skin and skin structure infections (ABSSSI). Tedizolid phosphate is a once daily oxazolidinone being developed for both intravenous (I.V.) and oral administration for the treatment of serious Gram-positive infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
http://www.drugs.com/nda/tedizolid_131023.html
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistance. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in the discovery of antibacterial agents.
Several antibacterial agents have been described in the prior art (for example, see PCT International Application Nos. PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178 and PCT/US2009/041200). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.
my old article cut paste
Tedizolid, 856866-72-3
(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-5-(hydroxymethyl)-1,3-oxazolidin-2-one
- Molecular Formula: C17H15FN6O3
- Average mass: 370.337799
Torezolid (also known as TR-701 and now tedizolid[1]) is an oxazolidinone drug being developed by Trius Therapeutics (originator Dong-A Pharmaceuticals) for complicated skin and skin-structure infections (cSSSI), including those caused by Methicillin-resistantStaphylococcus aureus (MRSA).[2]
As of July 2012, tedizolid had completed one phase III trial, with another one under way. [3]Both trials compare a six-day regimen of tedizolid 200mg once-daily against a ten-day regimen of Zyvox (linezolid) 600mg twice-daily.
The prodrug of tedizolid is called “TR-701″, while the active ingredient is called “TR-700″.[4][5]

March 5 2013
Trius Therapeutics will soon be reporting data from its second phase III trial (ESTABLILSH-2) and the recently announced publication of the data from its first phase III trial (ESTABLISH-1) in the Journal of the American Medical Association (JAMA)
- “Trius grows as lead antibiotic moves forward”. 31 Oct 2011.
- “Trius Completes Enrollment In Phase 2 Clinical Trial Evaluating Torezolid (TR-701) In Patients With Complicated Skin And Skin Structure Infections”. Jan 2009.
- http://clinicaltrials.gov/ct2/results?flds=Xf&flds=a&flds=b&term=tedizolid&phase=2&fund=2&show_flds=Y
- PMID 19528279 In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent.
- PMID 19218276 TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens.
BioDelivery Sciences Announces FDA Acceptance of Bunavail NDA for Filing


buprenorphine

naloxone
RALEIGH, N.C., Oct. 9, 2013 /PRNewswire/ — BioDelivery Sciences International, Inc. announced today that its New Drug Application (NDA) for Bunavail (buprenorphine naloxone buccal film) for the maintenance treatment of opioid dependence has been accepted for filing by the U.S. Food and Drug Administration (FDA), indicating that the application is sufficiently complete to permit a substantive review. Based on timelines established by the Prescription Drug User Fee Act (PDUFA), the review of the Bunavail NDA is expected to be completed by early June 2014.
