WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
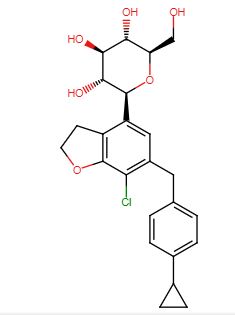
CAS: 1415472-28-4 Chemical Formula: C24H27ClO6 Molecular Weight: 446.92 Elemental Analysis: C, 64.50; H, 6.09; Cl, 7.93; O, 21.48
Green Cross Corp INNOVATOR
Daewoong Pharmaceutical Co Ltd
Enavogliflozin is an antidiabetic (hypoglycemic).
Daewoong is investigating DWJ-304 , a sodium/glucose cotransporter 2 (SGLT-2) inhibitor, for treating type 2 diabetes. By February 2017, preclinical development was underway. Daewoong is developing DWP-16001 , presumed to be enavogliflozin, a SGLT-2 inhibitor, for treating type 2 diabetes. In September 2019, launch was expected in 2023.
The present invention relates to a method for producing an intermediate useful for the synthesis of a diphenylmethane derivative that can be used as a SGLT inhibitor. A method for synthesizing a compound of formula 7 according to the present invention has solved the problem of an existing synthesis process which requires an additional process due to the synthesis of Grignard reagent and the management of a related substance. In addition, the process can be simplified by minimizing the formation of the related substance and eliminating the need for reprocessing of reaction products, thereby becoming capable of maximizing a yield of a diphenylmethane derivative.
Process for preparing intermediates of SGLT inhibitor and their use for the synthesis of diphenyl-methane derivative, which can be used as SGLT inhibitors.
Sodium-dependent glucose cotransporters (SGLT) allow the transport of Na + along the concentration gradient simultaneously with the transport of glucose across the concentration gradient. Currently two important SGLT isoforms have been cloned, known as SGLT1 and SGLT2. SGLT1 is located in the intestine, kidney and heart and regulates cardiac glucose transport. SGLT1 is a high affinity low dose transporter and therefore only accounts for a portion of renal glucose reuptake. In contrast, SGLT2 is a low affinity, high dose transporter located primarily in the apica domain of epithelial cells in the early proximal manure tubules. In healthy individuals, over 99% of the plasma glucose filtered out of the renal glomeruli is reabsorbed and less than 1% of the total filtered glucose is excreted in the urine. It is estimated that 90% of renal glucose reuptake is promoted by SGLT2 and the remaining 10% is mediated by SGLT1 in the late proximal canal. Genetic mutations in SGLT2 do not have a particular adverse effect on carbohydrate metabolism but cause increased kidney glucose secretion of about 140 g / day following mutation. Human mutation studies have been the subject of therapeutic studies because SGLT2 is believed to be responsible for most renal glucose resorption.
[3]
Korean Unexamined Patent Publication No. 2017-0142904 discloses a method for producing a diphenylmethane derivative having inhibitory activity against SGLT2. Since the above document prepares diphenylmethane derivatives by a convergent synthesis method in which each group is individually synthesized and then coupled, the synthesis route is more concise and yield is higher than the linear synthesis method disclosed in the prior art. It is disclosed that it can increase and reduce the risks inherent in sequential synthesis pathways.
[4]
However, the preparation method of the diphenylmethane derivative according to Korean Patent Publication No. 2017-0142904 uses a heavy metal such as pyridinium chlorochromate (PCC) to burden safety management, and the Grignard reagent. In addition to the need for a separate manufacturing process, the cost of the additional process is not only incurred, but also the management of the flexible material is necessary because the flexible material from the Grignard reagent manufacturing process is included in the final product. In addition, since the product generated after the reaction between the intermediate and the Grignard reagent includes additional flexible materials, there is a problem that a reprocessing process of such flexible materials is required.
(4-bromo-7-chloro-2,3-dihydrobenzofuran-6-yl) (4-cyclopropylphenyl) methanone (Compound 5) in a mixture of dichloromethane (9.7 mL) and acetonitrile (9.7 mL) at -15 ° C. g, 2.57 mmol) was added Et 3 SiH (1.2 mL, 7.71 mmol) and BF 3 -Et 2 O (0.79 mL, 6.42 mmol) in this order. The reaction mixture was allowed to warm to room temperature and then stirred for 4 hours. After completion of the reaction by TLC, the reaction solution was added with saturated NaHCO 3aqueous solution (40 mL) to terminate the reaction, and extracted with ethyl acetate. The organic layer obtained by extraction was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. The concentrated residue was purified by silica gel chromatography to give the title compound 6 (0.84 g, 89.9%) as an off-white solid.
Kim, Byungwook; Huh, Ki Young; Hwang, Jun Gi; Nah, JaeJin; Huh, Wan; Cho, Jae Min; Jang, In-Jin; Yu, Kyung-Sang; Kim, Yun; Lee, SeungHwan (April 2023). “Pharmacokinetic and pharmacodynamic interaction between DWP16001, an sodium–glucose cotransporter 2 inhibitor and metformin in healthy subjects”. British Journal of Clinical Pharmacology. 89 (4): 1462–1470. doi:10.1111/bcp.15613. PMID36422809. S2CID253838705.
Yoon, Sukyong; Park, Min Soo; Jin, Byung Hak; Shin, Hyobin; Na, Jaejin; Huh, Wan; Kim, Choon Ok (3 July 2023). “Pharmacokinetic and pharmacodynamic interaction of DWP16001, a sodium-glucose cotransporter-2 inhibitor, with phentermine in healthy subjects”. Expert Opinion on Drug Metabolism & Toxicology. 19 (7): 479–485. doi:10.1080/17425255.2023.2249397. PMID37593838. S2CID265846294.
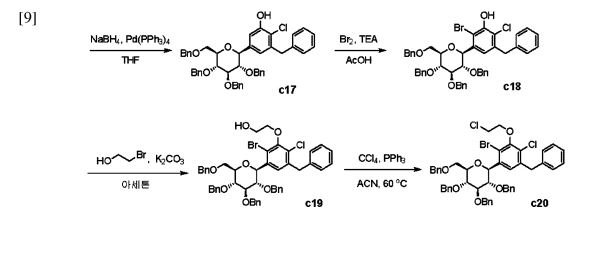
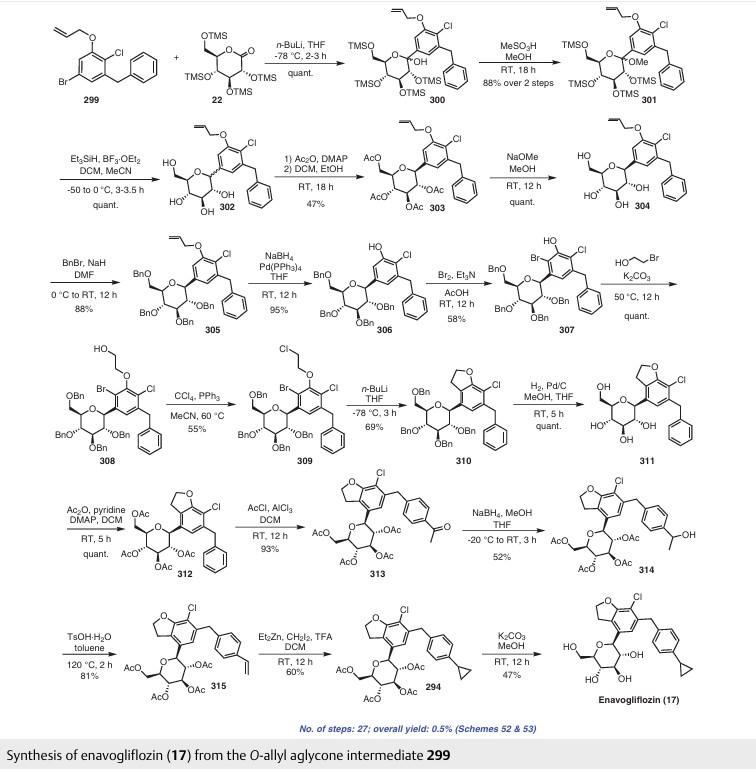
Enavogliflozin (17) (DWP-16001) was developed by Green Cross Corp. and Daewoong Pharmaceuticals Co. Ltd.with the intention of addressing type 2 diabetes. Its chemical name is (2S,3R,4R,5S,6R)-2-(7-chloro-6-(4-cyclopropylbenzyl)-2,3-dihydrobenzofuran-4-yl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol. In clinical trials, this medication exhibited remarkable efficacy as both an antidiabetic agent and an SGLT inhibitor. The initial synthetic pathway for producing envogliflozin (17), in addition to other C-aryl-glycoside-type derivatives, was documented in the United States, specifically through patent application number US9034921B2.75 Enavogliflozin (17) belongs to the category of C-aryl glycoside derivatives and its synthesis encompasses 19 steps, ultimately achieving an overall yield of 12%. The process starts with the preparation of aglycone key intermediate 290 (Scheme 50), and involves a series chemical transformations starting from the commercially available 3-methoxy-2-nitrobenzoic acid (275). Following the successful synthesis of the aglycone intermediate 290, the process was advanced by employing n-BuLi for lithium–halogen exchange on 290 and subsequent addition of lithiated 290 to O-silyl-protected compound 22 at –78 °C (Scheme51). This sequence yielded the TMS-protected lactol inter mediate 291 in quantitative yield. By subjecting this intermediate to treatment with MsOH/MeOH, the desired product 292 was obtained, achieving a 2-step overall yield of 88%. During these reactions, the O-silyl groups of the C-glucoside 292 were cleaved. Furthermore, the reduction of intermediate 292 was executed in the presence of triethylsilane and boron trifluoride–diethyl etherate complex, lead ing to the formation of desmethoxy intermediate 293 in 100% yield. Subsequent acetylation of the four hydroxy groups was performed using acetic anhydride and a catalytic quantity of DMAP, producing the tetra-acetyl intermediate 294 in a yield of 59%. Ultimately, removal of the acetyl groups was achieved in the presence of NaOH, culminating in the generation of the final product, enovagliflozin (17), with a 100% yield (Scheme 51). This synthetic procedure is plagued by significant limitations, including an extended route to obtain the aglycone intermediate 290, the application of protection and deprotection chemistry, and the necessity of cryogenic conditions to obtain the lactol intermediate 291. According to the reaction sequences given in Schemes 50 and 51, the overall yield of the final compound is calculated to be 12% via a total of 19 steps. In another approach, enavogliflozin (17) was synthesized in 27 steps with an overall yield of 0.5% (Schemes 52and 53).76 Initially, the synthesis of O-allyl aglycone intermediate 299 was achieved in nine steps starting from 3 methoxy-2-nitrobenzoic acid (275) (see Scheme 51). Methylation of compound 275 using methyl iodide and potassium carbonate in DMF afforded methyl ester 276 in 98% yield. Reduction of the NO2 group of 276 was carried out using Pd/C to afford aryl amine 277 in excellent yield. Next, bromination of 277 was facilitated by using NBS in DMF and ethyl acetate to afford the brominated compound 278 in 86% yield. Chlorination was then carried out on intermediate 278 under diazotization reaction conditions to afford 279. The ester group of 279 was hydrolyzed under basic conditions to afford the aryl carboxylic acid 295. Subsequently, the preparation of acid chloride 296 from 295 was achieved using oxalyl chloride and a catalytic amount of DMF, which was coupled with benzene under Friedel Crafts acylation conditions to give the aryl benzophenone intermediate 297. Reduction of the keto group of 297 was achieved by using triethylsilane and TFA/TfOH. Finally, the O-allyl aglycone intermediate 299 was obtained, when in termediate 298 was subjected to O-allylation using allylbromide and potassium carbonate in acetone. Next, the O-allyl aglycone intermediate 299 was subjected to a Br/Li exchange reaction using n-BuLi and addition of the obtained lithiated compound was carried out on gluconolactone 22 to afford the lactol intermediate 300 in quantitative yield (Scheme 53). The hydroxy group of 300 was methylated using methanesulfonic acid in methanol to give 301 in 88% yield over two steps from 299. Demethoxylation and TMS cleavage was carried out on 301 using triethylsilane and BF3·Et2O to furnish intermediate 302. This hydroxy intermediate was protected using acetic anhydride and DMAP to afford the acetylated compound 303, deprotection of which with sodium methoxide gave product 304 in 100% yield. Benzylation of 304 was carried out using BnBr and NaH to give tetra-O-benzylated compound 305. Next, the O-allyl group of 305 was reduced to give alcohol 306 in 95% yield. Bromination followed by O-alkylation of the intermediate 306 then furnished compound 308. The hydroxy group of 308 was replaced by a chlorine atom under treatment with CCl4 and PPh3 to afford 309. Using n-BuLi, intramolecular cyclization was carried out to give com pound 310 in 69% yield and subsequent debenzylation by treatment with Pd/C and H2 afforded compound 311. Again, acetylation of the free hydroxy groups of 311 was achieved using acetic anhydride and DMAP to give the O-acetylated intermediate 312. A Friedel–Crafts reaction on the aryl moiety of intermediate 312 gave acylated product 313 in 93% yield. The keto group of 313 was reduced using sodium borohydride in methanol to give the 314, which was further reduced under acidic conditions to give the alkene intermediate 315. The Simmons–Smith cyclopropanation was achieved on the alkene intermediate 315 to give compound 294 in 60% yield. Finally, the acetyl groups were removed from the sugar moiety of 294 to give enavogliflozin (17) in 47% yield. This synthetic route also contains major disadvantages in terms of the use of protection/deprotection strategies, a lengthy linear process and employs several harmful reagents.
(75) Choi, S.; Song, K. S.; Lee, S. H.; Kim, M. J.; Seo H. J.; Park, E.-J.; Kong, Y.; Park, S. O.; Kang, H.; Jung, M. E.; Lee, K.; Kim, H. J.; Lee, J. S.; Lee, M. W.; Kim, M.-S.; Hong, D. H.; Kang, M. US9034921B2, 2015. (76) Yoon, H.-K.; Park, S.-H.; Yoon, J.-S.; Choi, S.; Seo, H. J.; Park, E.-J.; Kong, Y.; Song, K.-S.; Kim, M. J.; Park S. O. WO2017217792A1, 2017.
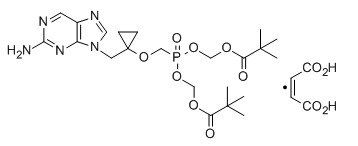
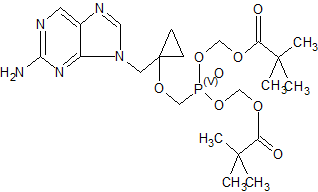
Besifovir, also known as ANA-380; LB-80380; PMCDG dipivoxil, is a reverse transcriptase inhibitor potentially for treatment of hepatitis B infection. LB80380 is a prodrug and an oral nucleotide analogue that inhibits viral replication by incorporation into the viral DNA. Antiviral activity against wild-type virus and virus with drug-resistant mutations was demonstrated in Phase II trials, with significant reduction of viral load in patients treated with LB80380. LB80380 was also shown to be safe and well tolerated.
CAS 441785-26-8
Chemical Formula: C22H34N5O7P
Molecular Weight: 511.5158
Ildong Pharma to release 1st chronic hepatitis B treatment next month
By Constance Williams
Published 2017.10.26 16:51
Updated 2017.10.26 19:12
Ildong Pharmaceutical will release its first chronic hepatitis B therapy of nucleotide series “Besivo” as an insurance benefit drug next month, the company said Thursday.
Besivo is a treatment for chronic hepatitis B based on the nucleotide sequence, which is composed of besifovir.
The price of the insurance is 3,403 won ($3.02) per tablet, which was recently confirmed by the Ministry of Health and Welfare보건복지부. Insurance benefits also cover El-carnitine medications used in combination, and the insurance price for one tablet (330mg) is 111 won.
According to the results of clinical trials, Besivo has demonstrated comparable levels of therapeutic efficacy in a randomized, double-blind trial compared with traditional therapies such as Entecavir (trade name: Baraclude) and Tenofovir (trade name: Viread). Besivo improved the prospects as a valid option for the treatment of chronic hepatitis B by improving the side effects found in the existing medications.
In particular, further analysis of clinical trials has shown that typical side effects such as decreased renal function and decreased bone density, which was a problem in the existing Tenofovir. Knodell necro-inflammatory score, was also superior to the control group regarding the histological improvement of the liver.
For the deterioration of renal function, the rate of increase in serum creatinine — a test that measures kidney function – was significantly lower than that of Tenofovir.
With the case of measuring bone mineral density, the proportion of patients showing bone turnover increased and the percentage of patients showing average bone mineral density decreased in the case of Tenofovir. With Besivo, the rate of patients with bone loss decreased, and the percentage of patients with average bone mineral density increased.
Ildong Pharma 일동제약 (CEO: Yun Woong-sup윤웅섭) plans to go on marketing with the idea that Besivo is a domestic drug that secures safety by improving side effects of existing medicines as well as treatment effects that are comparable to that of foreign pharmaceutical companies. In particular, the company expects the cost of pharmaceuticals to be 25 percent lower than that of Viread, the leading drug in the market.
“Due to the nature of chronic hepatitis B treatment for long-term use, safety is critical, and there are few side effects, so Besivo is highly valuable as a few nucleotide drugs existing in consideration of cross-resistance,” said Professor Ahn Sang-hoon안상훈 of Severance Hospital세브란스병원who participated in the clinical study.
“There is a strong competitive edge regarding the advantages of Besivo in connection with the entry into the Asian market, where the demand for therapeutic drugs is increasing as a major outbreak of hepatitis B,” he added.
Il Dong, under license from LG Life Sciences , has developed and launched Besivo (besifovir dipivoxil maleate), a phosphonate nucleoside inhibitor of HBV polymerase, for treating HBV infection. In October 2012, Il Dong was planning on seeking to outlicense the drug outside of Korea.
Besifovir dipivoxil maleate is a DNA polymerase inhibitor discovered and developed by LG Chem. The product was launched in Korea in 2017 by codeveloper ILDONG for the treatment of hepatitis B.
In April 2004, Anadys (acquired by Roche in 2011) obtained an exclusive license from LG Chem for the commercialization of LB-80380 worldwide excluding China, Korea, India and Southeast Asia. In August 2007, Anadys reported that it had discontinued development of LB-80380 and returned all rights to LG Chem in order to focus on other key compounds.
PAPER
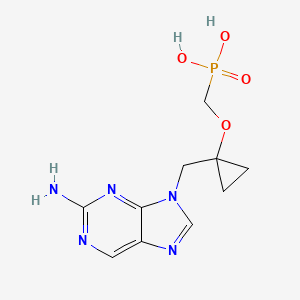
A Novel Class of Phosphonate Nucleosides. 9-[(1-Phosphonomethoxycyclopropyl)methyl]guanine as a Potent and Selective Anti-HBV Agent
9-[1-(Phosphonomethoxycyclopropyl)methyl]guanine (PMCG, 1), representative of a novel class of phosphonate nucleosides, blocks HBV replication with excellent potency (EC50 = 0.5 μM) in a primary culture of HepG2 2.2.15 cells. It exhibits no significant cytotoxicity in several human cell lines up to 1.0 mM. It does not inhibit replication of human immunodeficiency virus (HIV-1) or herpes simplex virus (HSV-1) at 30 μM. Many purine base analogues of 1 also exhibit inhibitory activity against HBV, but at 30 μM, pyrimidine analogues do not. 1 is 4 times more potent than 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA), which was used as a positive control (EC50 = 2.0 μM). The characteristic cyclopropyl moiety at the 2‘-position of 1 was prepared by titanium-mediated Kulinkovich cyclopropanation. 1 was modified to give the orally available drug candidate, PMCDG Dipivoxil (2). Compound 2 exhibited excellent efficacy when administered at 5 mg per kg per day in a study with woodchucks infected with woodchuck hepatitis B virus (WHBV). Drug candidate 2 has successfully completed phase I clinical trials and is currently undergoing phase II clinical studies for evaluation of efficacy.
({1-[(2-Amino-9H-purin-9-yl)methyl]cyclopropyl}oxy)methylphosphonic Acid (PMCDG, 8). 8 (89.5% yield) as yellowish solids. The compound was recrystallized from water for X-ray crystallography. 1H NMR (400 MHz, DMSO-d6): δ 0.92 (br q, 4H), 3.76 (d, J = 12.0 Hz, 2H), 4.33 (s, 2H), 8.0 (br s, 2H), 8.74 (s, 1H), 9.00 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 11.6 (2C), 45.9, 62.9 (d, J = 15.0 Hz), 63.0 (d, J = 161 Hz), 125.6, 139.1, 149.8, 154.2, 157.1. HRMS (MH+): 300.0862 calcd for C10H14N5O4P, found 300.0872. Anal. (C10H14N5O4P·H2O) C, H, N.
Novel crystalline polymorphic forms of 3-[({1-[(2-amino-9H-furyn-9-yl) methyl] cyclopropyl}oxy) methyl]-8,8-dimethyl-3,7-dioxo-2,4,6-trioxa-3λ5-phosphanon-1-yl-pivalate orotate (Besifovir dipivoxil), a process for its preparation, and composition comprising the salt for treating viral infections are claimed.
[({1 – [(2-amino -9 H -purin-9-yl) methyl] cyclopropyl} oxy) methyl] -8,8- dimethyl-3,7-dioxo Orotate of bis-4,6-trioxa-3? 5 -phosphonon-1-yl-pivalate (besifovir dipivoxil), a process for its preparation and a pharmaceutical composition comprising said salt Lt; / RTI & gt;
[Formula 1]
The free compound of the above formula (1) is a novel anti-viral substance disclosed in Korean Patent No. 0441638 and WO 02/057288. However, these compounds are very unstable against heat and moisture, and are difficult to be used as raw materials for pharmaceutical compositions.
[6]
In Korean Patent No. 0935904, various pharmaceutically acceptable salts have been prepared to solve such problems. In this process, some of the salts have proven to be difficult to obtain as crystalline solids and have been successfully obtained as crystalline solids only in the case of maleate, p-toluenesulfonate, methanesulfonate, naphthalenesulfonate and ethanesulfonate, Bessyfovir dipivicum maleic acid mono-salt is remarkably excellent in thermal stability compared to its free compounds and other salts.
[7]
However, bissipovid epipixyl maleate is very unstable at high temperatures of 100 ° C or higher and is mostly decomposed in 6 hours and still insufficient in stability. In case of contact, it may cause severe irritation accompanied by redness, pain, Due to the characteristics of maleic acid, symptoms similar to those of conjunctivitis or acute exposure may occur during manufacture (see the Toxicological Information of the Food and Drug Administration), and caution is also required for safety.
FIG.5 shows the infrared spectroscopy (FT-IR) of the bisphosphonite pyrophosphorate of Example 1. Fig.
6 shows the 1 H nuclear magnetic resonance spectrum (NMR) of the bisporovir diplyl orthoate of Example 1. Fig.
The free compound of the above formula (1) is a novel anti-viral substance disclosed in Korean Patent No. 0441638 and WO 02/057288. However, these compounds are very unstable against heat and moisture, and are difficult to be used as raw materials for pharmaceutical compositions.
[6]
In Korean Patent No. 0935904, various pharmaceutically acceptable salts have been prepared to solve such problems. In this process, some of the salts have proven to be difficult to obtain as crystalline solids and have been successfully obtained as crystalline solids only in the case of maleate, p-toluenesulfonate, methanesulfonate, naphthalenesulfonate and ethanesulfonate, Bessyfovir dipivicum maleic acid mono-salt is remarkably excellent in thermal stability compared to its free compounds and other salts.
[7]
However, bissipovid epipixyl maleate is very unstable at high temperatures of 100 ° C or higher and is mostly decomposed in 6 hours and still insufficient in stability. In case of contact, it may cause severe irritation accompanied by redness, pain, Due to the characteristics of maleic acid, symptoms similar to those of conjunctivitis or acute exposure may occur during manufacture (see the Toxicological Information of the Food and Drug Administration), and caution is also required for safety.
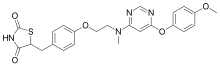
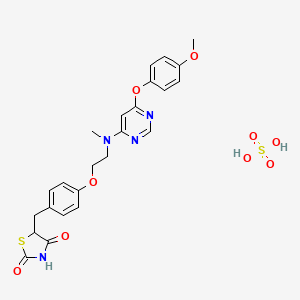
Lobeglitazone sulfate was approved by the Ministry of Food and Drug Safety (Korea) on July 4, 2013. It was developed and marketed as Duvie® by Chong Kun Dang Corporation.
Lobeglitazone is an agonist for both PPARα and PPARγ, and it works as an insulin sensitizer by binding to the PPAR receptors in fat cells and making the cells more responsive to insulin. It is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes.
Duvie® is available as tablet for oral use, containing 0.5 mg of free Lobeglitazone. The recommended dose is 0.5 mg once daily.
Lobeglitazone which was reported in our previous works belongs to the class of potent PPARα/γ dual agonists (PPARα EC50: 0.02 μM, PPARγ EC50: 0.018 μM, rosiglitazone; PPARα EC50: >10 μM, PPARγ EC50: 0.02 μM, pioglitazone PPARα EC50: >10 μM, PPARγ EC50: 0.30 μM). Lobeglitazone has excellent pharmacokinetic properties and was shown to have more efficacious in vivo effects in KKAy mice than rosiglitazone and pioglitazone.17 Due to its outstanding pharmacokinetic profile, lobeglitazone was chosen as a promising antidiabetes drug candidate.
Medical uses
Lobeglitazone is used to assist regulation of blood glucose level of diabetes mellitus type 2 patients. It can be used alone or in combination with metformin.[4]
Lobeglitazone was approved by the Ministry of Food and Drug Safety (Korea) in 2013, and the postmarketing surveillance is on progress until 2019.[4][5]
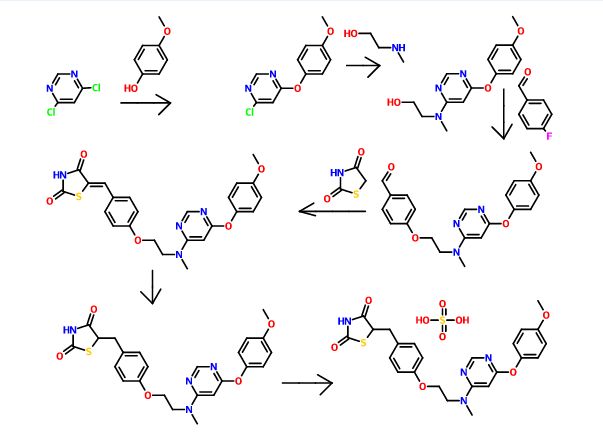
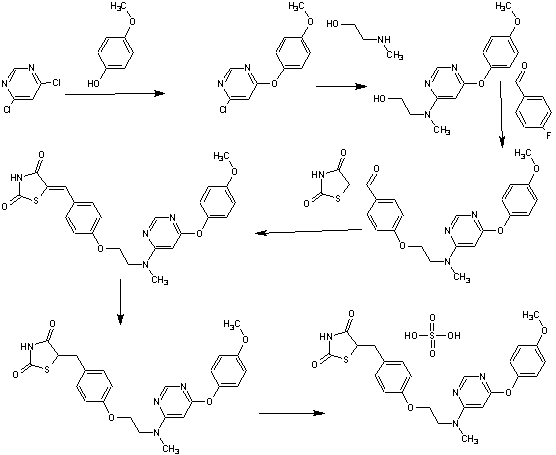
SYNTHESIS
PAPER
Org. Process Res. Dev.2007, 11, 190-199.
Process Development and Scale-Up of PPAR α/γ Dual Agonist Lobeglitazone Sulfate (CKD-501)
Process Research and Development Laboratory, Chemical Research Group, Chong Kun Dang Pharmaceutical Cooperation, Cheonan P. O. Box 74, Cheonan 330-831, South Korea, and Department of Chemistry, Korea University, 5-1-2, Anam-Dong, Seoul 136-701, Korea
A scaleable synthetic route to the potent PPARα/γ dual agonistic agent, lobeglitazone (1), used for the treatment of type-2 diabetes was developed. The synthetic pathway comprises an effective five-step synthesis. This process involves a consecutive synthesis of the intermediate, pyrimidinyl aminoalcohol (6), from the commercially available 4,6-dichloropyrimidine (3) without the isolation of pyrimidinyl phenoxy ether (4). Significant improvements were also made in the regioselective 1,4-reduction of the intermediate, benzylidene-2,4-thiazolidinedione (10), using Hantzsch dihydropyridine ester (HEH) with silica gel as an acid catalyst. The sulfate salt form of lobeglitazone was selected as a candidate compound for further preclinical and clinical study. More than 2 kg of lobeglitazone sulfate (CKD-501, 2) was prepared in 98.5% purity after the GMP batch. Overall yield of 2 was improved to 52% from 17% of the original medicinal chemistry route.
Lee JH, Noh CK, Yim CS, Jeong YS, Ahn SH, Lee W, Kim DD, Chung SJ. (2015). “Kinetics of the Absorption, Distribution, Metabolism, and Excretion of Lobeglitazone, a Novel Activator of Peroxisome Proliferator-Activated Receptor Gamma in Rats.”.Journal of Pharmaceutical sciences104 (9): 3049–3059.doi:10.1002/jps.24378. PMID25648999.
Kim JW, Kim JR, Yi S, Shin KH, Shin HS, Yoon SH, Cho JY, Kim DH, Shin SG, Jang IJ, Yu KS. (2011). “Tolerability and pharmacokinetics of lobeglitazone (CKD-501), a peroxisome proliferator-activated receptor-γ agonist: a single- and multiple-dose, double-blind, randomized control study in healthy male Korean subjects.”. Clinical therapeutics33 (11): 1819–1830.doi:10.1016/j.clinthera.2011.09.023. PMID22047812.
Lee JH, Woo YA, Hwang IC, Kim CY, Kim DD, Shim CK, Chung SJ. (2009). “Quantification of CKD-501, lobeglitazone, in rat plasma using a liquid-chromatography/tandem mass spectrometry method and its applications to pharmacokinetic studies.”. Journal of Pharmaceutical and Biomedical Analysis50 (5): 872–877.doi:10.1016/j.jpba.2009.06.003. PMID19577404.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....