
Home » GENERIC DRUG (Page 4)
Category Archives: GENERIC DRUG
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Glenmark Generics receives final ANDA approval for Telmisartan Tablets

Glenmark Generics receives final ANDA approval for Telmisartan Tablets
Mumbai, India, July 8, 2014
Glenmark Generics Inc., USA a subsidiary of Glenmark Generics Limited has been granted final abbreviated new drug approval (ANDA) from the United States Food and Drug Administration (US FDA) for Telmisartan Tablets. Glenmark will commence distribution of the product immediately.
Telmisartan Tablets are Glenmark’s generic version of Boehringer Ingelheim’s Micardis®. Telmisartan is indicated for the treatment of hypertension.
The approval is for the 20mg, 40mg and 80mg tablets. For the 12 month period ending March 2014, Telmisartan garnered annual sales of USD 250 Million according to IMS Health.
Glenmark receives USFDA approval for telmisartan tablets
Telmisartan, which is the generic version of Boehringer Ingelheim’s Micardis, garnered annual sales of $ 250 million for the 12 month period ending March 2014
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor…….

AMPRENAVIR
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available.

| Systematic (IUPAC) name | |
|---|---|
| (3S)-oxolan-3-yl N-[(2S,3R)-3-hydroxy-4-[N-(2-methylpropyl)(4-aminobenzene)sulfonamido]-1-phenylbutan-2-yl]carbamate | |
| Clinical data | |
| Trade names | Agenerase |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699051 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. | C (US) |
| Routes | oral |
| Pharmacokinetic data | |
| Protein binding | 90% |
| Metabolism | hepatic |
| Half-life | 7.1-10.6 hours |
| Excretion | <3% renal |
| Identifiers | |
| CAS number | 161814-49-9 |
| ATC code | J05AE05 |
| PubChem | CID 65016 |
| DrugBank | DB00701 |
| ChemSpider | 58532 |
| UNII | 5S0W860XNR |
| KEGG | D00894 |
| ChEBI | CHEBI:40050 |
| ChEMBL | CHEMBL116 |
| NIAID ChemDB | 006080 |
| Chemical data | |
| Formula | C25H35N3O6S |
| Mol. mass | 505.628 g/mol |
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available
………………….
New approaches to the industrial synthesis of HIV protease inhibitors
http://pubs.rsc.org/en/content/articlelanding/2004/ob/b404071f/unauth#!divAbstract
Efficient and industrially applicable synthetic processes for precursors of HIV protease inhibitors (Amprenavir, Fosamprenavir) are described. These involve a novel and economical method for the preparation of a key intermediate, (3S)-hydroxytetrahydrofuran, from L-malic acid. Three new approaches to the assembly of Amprenavir are also discussed. Of these, a synthetic route in which an (S)-tetrahydrofuranyloxy carbonyl is attached to L-phenylalanine appears to be the most promising manufacturing process, in that it offers satisfactory stereoselectivity in fewer steps.

AGENERASE (amprenavir) is an inhibitor of the human immunodeficiency virus (HIV) protease. The chemical name of amprenavir is (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulfonamido)-1-benzyl-2-hydroxypropyl]carbamate. Amprenavir is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H35N3O6S and a molecular weight of 505.64. It has the following structural formula:
 |
Amprenavir is a white to cream-colored solid with a solubility of approximately 0.04 mg/mL in water at 25°C.
AGENERASE Capsules (amprenavir capsules) are
available for oral administration. Each 50- mg capsule contains the inactive ingredients d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), polyethylene glycol 400 (PEG 400) 246.7 mg, and propylene glycol 19 mg. The capsule shell contains the inactive ingredients d-sorbitol and sorbitans solution, gelatin, glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink. Each 50- mg AGENERASE Capsule contains 36.3 IU vitamin E in the form of TPGS. The total amount of vitamin E in the recommended daily adult dose of AGENERASE is 1,744 IU.
See also
- Fosamprenavir, a prodrug of amprenavir
External links
- Amprenavir bound to proteins in the PDB

Vinorelbine …For the treatment of non-small-cell lung carcinoma.


4-(acetyloxy)- 6,7-didehydro- 15-((2R,6R,8S)-4-ethyl- 1,3,6,7,8,9-hexahydro- 8-(methoxycarbonyl)- 2,6-methano- 2H-azecino(4,3-b)indol-8-yl)- 3-hydroxy- 16-methoxy- 1-methyl- methyl ester,
3′,4′-Didehydro-4′-deoxy-8′-norvincaleukoblastine
71486-22-1 cas
(2R,3R)-2,3-Dihydroxysuccinic acid – methyl (2ξ,3β,4β,5α,12β,19α)-4-acetoxy-15-[(12S,14R)-16-ethyl-12-(methoxycarbonyl)-1,10-diazatetracyclo[12.3.1.03,11.04,9]octadeca-3(11),4,6, 8,15-pentaen-12-yl]-3-hydroxy-16-methoxy-1-methyl-6,7-didehydroaspidospermidine-3-carboxylate (2:1)
Vinorelbine (trade name Navelbine) is an anti-mitotic chemotherapy drug that is given as a treatment for some types of cancer, including breast cancer and non-small cell lung cancer.
Vinorelbine i.v. is a semi-synthetic derivative of a vinca alkaloid launched in 1989 by Pierre Fabre for the treatment of non-metastatic breast cancer and non-small cell lung cancer (NSCLC). In 2011, a complete response letter was assigned by the FDA for an NDA filed by Adventrx Pharmaceuticals seeking approval for the treatment of non-small cell lung cancer (NSCLC). Pierre Fabre and licensee GlaxoSmithKline had been evaluating the potential of the drug for the treatment of breast cancer, prostate cancer and NSCLC with an oral formulation, but no recent developments have been reported. The evaluation of an injectable emulsion formulation developed by Adventrx Pharmaceuticals is in phase I clinical development for the potential treatment of these indications. Several trials are ongoing to evaluate vinorelbine in combination with other chemotherapy for the treatment of metastatic breast cancer. The University of California, Davis is evaluating vinorelbine in combination with lapatinib for the treatment of solid tumors.
Clinicians sometimes use the abbreviation “NVB” for vinorelbine, although (like many medical abbreviations) it is not a unique identifier.
The antitumor activity is due to inhibition of mitosis through interaction with tubulin.[2] Vinorelbine is the first 5´NOR semi-synthetic vinca alkaloid. It is obtained by semi-synthesis from alkaloids extracted from the rosy periwinkle, Catharanthus roseus. It is marketed in India by Abbott Healthcare under the brand name Navelbine.
History
Vinorelbine was invented by the pharmacist Pierre Potier and his team from the CNRS in France in the 1980s and was licensed to the oncology department of the Pierre Fabre Group. The drug was approved in France in 1989 under the brand name Navelbine for the treatment of non-small celllung cancer. It gained approval to treat metastatic breast cancer in 1991. Vinorelbine received approval by the United States Food and Drug Administration (FDA) in December 1994 sponsored by Burroughs Wellcome Company. Pierre Fabre Group now markets Navelbine in the U.S., where the drug went generic in February 2003.
Vinorelbine interferes with microtubule assembly, particularly that of mitotic microtubules. Like other vinca alkaloids, vinorelbine may also interfere with the metabolism of amino acid, cyclic AMP, and glutathione, the activity of calmodulin-dependent Ca+2 transport ATPase, cellular respiration, and the biosynthesis of lipids and nucleic acid.
Originally developed at Pierre Fabre, vinorelbine i.v. was first licensed to GlaxoSmithKline in the U.S., Canada and Europe and to Kyowa Hakko in Japan. In July 2005, Pierre Fabre licensed the U.S. and Canadian rights to an oral formulation of vinorelbine to Novacea (acquired by Transcept Pharmaceuticals in 2009), while Pierre Fabre will continue to develop and commercialize this formulation in Europe and other countries. In October 2005, SD Pharmaceuticals granted Adventrx an exclusive license to certain rights to the emulsion formulation of the drug. In 2009, the company filed a regulatory application seeking approval for an injectable emulsion for the treatment of patients with stage III or IV NSCLC. Vinorelbine i.v. is currently registered in over 80 countries worldwide. In 2010, this application was withdrawn upon receipt of a refuse to file (FTF) decision from the FDA. The product is available for outlicensing.
In most European countries, vinorelbine is approved to treat non-small cell lung cancer and breast cancer. In the United States it is approved only for non-small cell lung cancer.
NAVELBINE (vinorelbine tartrate) Injection is for intravenous administration. Each vial contains vinorelbine tartrate equivalent to 10 mg (1-mL vial) or 50 mg (5-mL vial) vinorelbine in Water for Injection. No preservatives or other additives are present. The aqueous solution is sterile and nonpyrogenic. Vinorelbine tartrate is a semi-synthetic vinca alkaloid with antitumor activity. The chemical name is 3′,4′-didehydro-4′-deoxy-C’-norvincaleukoblastine [R-(R*,R*)-2, 3-dihydroxybutanedioate (1:2)(salt)]. Vinorelbine tartrate has the following structure:
 |
vinorelbine tartrate is a white to yellow or light brown amorphous powder with the molecular formula C45H54N4O8•2C4H6O6 and molecular weight of 1079.12. The aqueous solubility is > 1,000 mg/mL in distilled water. The pH of NAVELBINE (vinorelbine tartrate) Injection is approximately 3.5.
Uses
As stated above, Vinorelbine is approved for the treatment of non small cell lung cancer and metastatic breast cancer. It is also active inrhabdomyosarcoma.[3]
Oral formulation
An oral formulation has been marketed and registered in most European countries for the same settings. It has similar efficacy as the intravenous formulation, avoids venous toxicities of an infusion and is easier to take.

Side effects
Vinorelbine has a number of side-effects that can limit its use:
Chemotherapy-induced peripheral neuropathy (a progressive, enduring and often irreversible tingling numbness, intense pain, and hypersensitivity to cold, beginning in the hands and feet and sometimes involving the arms and legs[4]), lowered resistance to infection, bruising or bleeding, anaemia,constipation, diarrhea, nausea, tiredness and a general feeling of weakness (asthenia), inflammation of the vein into which it was injected (phlebitis). Seldom severe hyponatremia is seen.
Less common effects are hair loss and allergic reaction.
vinorelbine (trade name: Navelbine (Navelbing)) is a novel semi-synthetic vinca alkaloids anticancer drugs, chemical name 3 ‘, 4’ – didehydro-4 ‘- deoxy _8 ‘- vinorelbine, developed by the French PieerFabre company and was listed first in France in 1989, it is mainly through inhibition of centromere tubulin polymerization, to stop cell division in mitotic metaphase, is a cell cycle-specific antineoplastic agents. A change in the structure, it has a strong and specific anti-mitotic properties, and exhibit antineoplastic vinca alkaloids than other low neurotoxicity, characteristics of strong anti-tumor activity.
vinorelbine complex chemical structure, synthesis is difficult, and the difficulty of separating large, making the synthesis of low yield and high cost.Published patent, if the application Patent CN101037446A, CN101284842A, CN1552715A, which are made of boron tetrafluoride shrink ring silver as reagents, but boron tetrafluoride is the price of silver more expensive chemical reagents, increased production costs.
http://www.google.com/patents/EP2135872A1?cl=en


……………………
(1) or a salt thereof to dehydration vinblastine (I) as starting material, the reaction of bromo-condensed ring integrated crude vinorelbine (III):

Example 1
(a) by the dehydration hydrochloride ⑴ vinblastine vinorelbine crude preparation (III)
In a dry round bottom flask, dehydrated hydrochloric vinblastine 20g (laboratory preparation, HPLC purity 92.5%), in the dark and under nitrogen was added IOOOml dry dichloromethane, stirred and dissolved, add 20ml of pyridine , cooled in a dry ice-acetone bath to _50 ° C below bromosuccinimide was added dropwise 6g, trifluoroacetic acid and 13ml of dry methylene IOOOml mixed solution after the addition was complete, stirring below -50 ° C maintaining the reaction 2 hours.After completion of the reaction, adding silver nitrate 12g, 12g and IOOOml ammonium acetate and 800ml of deionized water mixed solution of tetrahydrofuran, stirred rapidly, and gradually heated to 20 ~ 30 ° C, maintaining this temperature, the reaction was stirred for 16 hours. After completion of the reaction, stirring was added dropwise 10% aqueous sodium carbonate aqueous phase PH8 ~ 9, filtered through celite after phase separation, the aqueous phase discarded, and the organic phase was dried, filtered and concentrated to dryness to give crude 14 vinorelbine. 3g (HPLC purity San 85%), 75% yield.
(2) Purification
The above crude product was 14.3g vinorelbine the column of basic alumina with 300 mesh, and with an eluent of 4% methanol – methylene chloride, collecting rich eluate was concentrated to dryness to give product vinorelbine First pure Bin 10. 7g (HPLC purity ^ 97%); then pure product obtained in the beginning of a C18 reversed phase column packing 50μπι, and with an eluent of 40% water – ethanol solution eluted, collected and washed pure deliquored product was extracted with dichloromethane and concentrated to dryness to give 8. lg, and then recrystallized from methanol to obtain pure vinorelbine 6. lg (HPLC purity San 99.5%). Relative to the total dewatering vinblastine hydrochloride 32% yield.
Example 2
(a) by the dehydration hydrochloride ⑴ vinblastine vinorelbine crude preparation (III)
In a dry round bottom flask, dehydrated hydrochloric vinblastine 20g (laboratory preparation, HPLC purity 92.5%), in the dark and under nitrogen was added IOOOml dry dichloromethane, stirred to dissolve, add 2,6 – lutidine 20ml, cooled in a dry ice-acetone bath to _50 ° C or less, is added dropwise bromosuccinimide and 5. 5g, 15ml of trifluoroacetic acid and a mixed solution of dry methylene IOOOml, dropping After stirring below -50 ° C maintaining the reaction 1.5 hours. After completion of the reaction, adding silver nitrate 12g, 12g and IOOOml ammonium acetate and 800ml of deionized water mixed solution of tetrahydrofuran, stirred rapidly, and gradually heated to 20 ~ 30 ° C, maintaining this temperature, the reaction was stirred M hours. After completion of the reaction, stirring was added dropwise 10% aqueous sodium carbonate aqueous phase PH8 ~ 9, filtered through celite and phase separation, the aqueous phase discarded, and the organic phase was dried, filtered and concentrated to dryness to give crude 14 vinorelbine. 7g (HPLC purity> 85%), yield 77.8%.
(2) Purification
The above crude vinorelbine 14. 7g on a column of basic alumina column with 300 mesh, and with an eluent of 4% methanol – methylene chloride, collecting rich eluate was concentrated to dryness to give product vinorelbine early pure llg (HPLC purity ^ 97%); Then get in early on 50 μ m pure product of C18 reverse phase column packing, and then with an eluent of 40% water – ethanol elution fractions containing pure product eluate product is extracted with dichloromethane and concentrated to dryness to give 8. 4g, and then recrystallized from methanol to obtain pure vinorelbine 6. 3g (HPLC purity> 99.5%) relative to the dewatering 0 Vinblastine Hydrochloride total yield of 33.3%.
…………….
Bioorganic and Medicinal Chemistry, 2008 , vol. 16, 11 p. 6269 – 6285
http://www.sciencedirect.com/science/article/pii/S0968089608003532?via=ihub

……………………
Journal of Heterocyclic Chemistry, 1995 , vol. 32, 4 p. 1255 – 1260
http://onlinelibrary.wiley.com/doi/10.1002/jhet.5570320427/abstract
During the development of the bis-indole alkaloid anticancer drug Navelbine® (vinorelbine), several chemical degradants of the drug were isolated and identified. These included 7′-nor-6′,9′-secovinorelbine (7′,8′-bisnor-6′,9′-secoanhydrovinblastine) and 4-deacetyl-8′-vinorelbine (4-deacetyl-8′-noranhydrovinblastine). The elucidation of the structure of 7′-nor-6′,9′-secovinorelbine is described; the assignment of the proton and carbon spectra of both compounds is contrasted to the shift assignments of Navelbine.
References
- Marty M, Fumoleau P, Adenis A, Rousseau Y, Merrouche Y, Robinet G, Senac I, Puozzo C (2001). “Oral vinorelbine pharmacokinetics and absolute bioavailability study in patients with solid tumors”. Ann Oncol 12 (11): 1643–9. doi:10.1023/A:1013180903805. PMID 11822766.
- Jordan, M.A.; Wilson, L. (2004). “Microtubules as a target for anticancer drugs.”. Nature Reviews. Cancer 4 (4): 253–65. doi:10.1038/nrc1317.PMID 15057285.
- Casanova, M; Ferrari, A; Spreafico, F; Terenziani, M; Massimino, M; Luksch, R; Cefalo, G; Polastri, D et al. (2002). “Vinorelbine in previously treated advanced childhood sarcomas: Evidence of activity in rhabdomyosarcoma”. Cancer 94 (12): 3263–8. doi:10.1002/cncr.10600. PMID 12115359.
- del Pino BM. Chemotherapy-induced Peripheral Neuropathy. NCI Cancer Bulletin. Feb 23, 2010;7(4):6.
NADIFLOXACIN, Jinofloxacin

-
(+-)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidino)-5-methyl-1-oxo-1H,5H-benzo(ij)quinolizine-2-carboxylic acid
- CCRIS 4066
- Jinofloxacin
- Nadifloxacin
- Nadifloxacine
- Nadifloxacine [INN-French]
- Nadifloxacino
- Nadifloxacino [INN-Spanish]
- Nadifloxacinum
- Nadifloxacinum [INN-Latin]
- Nadixa
- OPC-7251
- S-Nadifloxacin
- UNII-6CL9Y5YZEQ
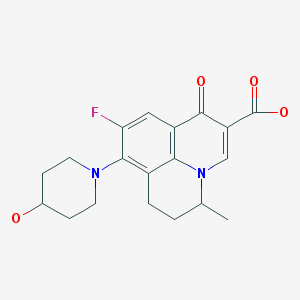
Nadifloxacin is chemically, 9-fluoro-6,7-dihydro-8-(4-hydroxy-l-pyperidinyl)-5-methyl- l-oxo-lH,5H-benzo(I,j)quinolizine-2-carboxylic acid of Formula I provided below.

FORMULA I Nadifloxacin is a synthetic quinolone with potent broad-spectrum anti-bacterial activity. Nadifloxacin inhibits the enzyme DNA gyrase that is involved in bacterial DNA synthesis and replication, thus inhibiting the bacterial multiplication. RS-nadifloxacin and S-nadifloxacin, in particular, exhibit strong antibacterial activity against Gram-positive, Gram-negative and anaerobic bacteria, resistant Gram-positive organisms such as methicillin-resistant Staphylococcus aureus (MRSA), quinolone-resistant Staphylococcus aureus, coagulase negative staphylococci, such as methicillin-resistant Staphylococcus epidermidis (MRSE), enterococci, betahemolytic streptococci and viridans group of streptococci, mycobacteria and newly emerging nosocomial pathogens such as Chryseobacterium meninges epticum, and Gram-negative pathogens such as E.coli, Klebsiella, Proteus, Serratia, Citrobacter and Pseudomonas. Recently, it has also been shown that S-(-)-nadifloxacin, in particular exhibits potent antibacterial activity against glycopeptide intermediate S. aureus (GISA), vancomycin intermediate S. aureus (VISA) and vancomycin-resistant S. aureus (VRSA). Nadifloxacin is also active against quinolone-resistant Staphylococci.
Nadifloxacin is marketed in the form of cream for topical application for the treatment of acne vulgaris, folliculitis and sycosis vulgaris. It is also indicated for the treatment of topical bacterial infections with susceptible bacteria.
The use of quinolone antibiotics to treat infections is known art in the field of ophthalmic pharmaceutical compositions and methods of treatment. Several quinolone antibacterial agents available in the market include gatifloxacin (available as Zymar®), Levofloxacin (available as Quixin® or Iquix®), Ciprofloxacin (available as Ciloxan®), Ofloxacin (available as Ocuflox®), Lomefloxacin (available as Lomeflox®), Moxifloxacin (available as Vigamox®) and Norfloxacin (available as Chibroxin®).
U.S. Patent No. 4,844,902 discloses a topically applicable formulation comprising by weight about 0.01 to 30% of an anti-bacterially active compound, 0.01 to 10% of a corticosteroid and a carrier. U.S. Patent No. 6,333,045 discloses liquid pharmaceutical compositions of gatifloxacin or salt thereof and disodium edetate.
U.S. Patent No. 6,716,830 discloses ophthalmic dosage forms of moxifioxacin or salts thereof in a concentration of 0.1% to 1% (w/w) and pharmaceutically acceptable vehicle.
U.S. Patent No. 6,359,016 relates to topical suspension formulations containing ciprofloxacin and dexamethasone.
U.S. Patent No 4,399,134 discloses processes for the preparation of nadifloxacin or salts thereof and antibacterially effective pharmaceutical compositions of nadifloxacin. Typical dosage forms include tablets, pills, powders, liquid preparations, suspensions, emulsions, granules, capsules, suppositories, and injectable preparations (solutions, suspensions, etc).
U.S. Patent No 6,884,768 discloses solid oral pharmaceutical compositions that includes nadifloxacin, an absorbefacient and taurine compounds.
U.S. Patent Application 20060183698 describes topical ophthalmic formulation that includes serum electrolytes; an antimicrobial compound and an anti-inflammatory or steroidal compound. Several antimicrobial agents have been disclosed including nadifloxacin.
U.S. Patent Application 20040176337 discloses topical . compositions of benzoquinolizine-2-carboxylic acid antimicrobial drug.
U.S. Patent Application 20040176321 discloses injectable pharmaceutical composition for intravenous delivery of an active agent that includes RS-(±)-nadifloxacin; S-(-)- nadifloxacin and hydrates thereof; or S~(-)-nadifloxacin arginine and salts thereof. PCT Publication WO 04/00360 describes pharmaceutical compositions of several active ingredients including nadifloxacin for topical use for treatment of dermatosis.
European Patent EP 275,515 and U.S. Patent No. 4,923,862 disclose aqueous pharmaceutical compositions of levofloxacin and ofloxacin or salts thereof.
PCT application WO 02/39993 discloses a stable pharmaceutical preparation of a combination drug, comprising an anti-infective agent, selected from the group consisting of quinolone derivatives, amino-glycoside derivatives and their pharmaceutically acceptable salts; an ant-inflammatory agent which is a corticosteroid; a complexation enhancing polymer; a solubilizer exhibiting an inclusion phenomena; pharmaceutically acceptable excipients within a suitable carrier system.
Journal of Ocular Pharmacology and Therapeutics, vol 23(3): 243-256, 2007 discloses (7- [(3R)-3 -aminohexahydro- 1 H-azepine- 1 -yl]-8-chloro- 1 -cyclopropyl-6-fluoro- 1 ,4-dihydro- 4-oxo-3-quinolinecarboxylivc acid as the topical agent for the treatment of ophthalmic infections.

5-Bromo-6-fluoro-2-methylquinoline (II)
5-Bromo-6-fluoro-2-methyl-1,2,3,4-tetrahydroquinoline (III)
diethyl 2-[(E)-ethoxymethylidene]succinate (IV)
8-Bromo-9-fluoro-5-methyl-1-oxo-6,7-dihydro-1H,5H-pyrido[3,2,1-ij]quinoline-2-carboxylic acid (V)
4-Piperidinol; 4-Hydroxypiperidine

______________________________________Elemental Analysis for C.sub.19 H.sub.21 N.sub.2 O.sub.4 F C H N______________________________________Calc'd (%): 63.32 5.87 7.78Found (%): 63.28 5.76 7.89______________________________________

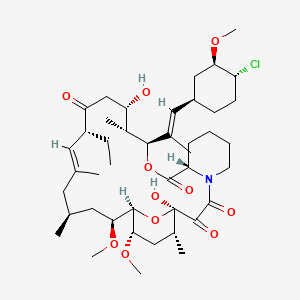
Pimecrolimus Пимекролимус…For treatment of mild to moderate atopic dermatitis.

Pimecrolimus
137071-32-0 cas
(3S,4R,5S,8R,9E,12S,14S,15R,16S,18R,19R,26aS)- 3-{(E)-2-[(1R,3R,4S)-4-Chloro-3-methoxycyclohexyl]- 1-methylvinyl}-8-ethyl-5,6,8,11,12,13,14,15,16,17,
18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy- 14,16-dimethoxy-4,10,12, 18-tetramethyl-15,19-epoxy- 3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1, 7,20,21(4H,23H)-tetrone
The systematic name of pimecrolimus is (lR,9S,12S,13R,14S,17R,18E,21S,23S,24R,25S,27R)-12-[(lE)-2- {(1 R,3R,4S)-4-chloro-3-methoxycyclohexyl} – 1 -methylvinyl] – 17-ethyl- 1,14- dihydroxy-23,25-dimethoxy-13,19,21,27-tetramethyl-ll,28-dioxa-4-aza- tricyclo[22.3.1.04‘9]octacos-18-ene-2,3,10,16-tetraone.
Pimecrolimus is the 32 epichloro derivative of ascomycin.
|
4-11-2008
|
Pharmaceutical Composition
|
| Canada | 2200966 | 2006-12-19 | expiry 2015-10-26 |
| United States | 6423722 | 1998-12-26 | 2018-12-26 |
PATENT AND EXPIRY DATE
| 5912238 | Jun 15, 2016 | |
| 5912238*PED | Dec 15, 2016 | |
| 6352998 | Oct 26, 2015 | |
| 6352998*PED | Apr 26, 2016 | |
| 6423722 | Jun 26, 2018 | |
| 6423722*PED | Dec 26, 2018 |
Viktor Gyollai, Csaba Szabo, “Methods of preparing pimecrolimus.” U.S. Patent US20060142564, issued June 29, 2006.

NDA..021302, 13 DEC 2001… VALEANT BERMUDA..ELIDEL1% TOPICAL CREAM
Pimecrolimus is an immunomodulating agent used in the treatment of atopic dermatitis (eczema). It is currently available as a topical cream, once marketed by Novartis, (however Galderma will be promoting the molecule in Canada in early 2007) under the trade name Elidel.
NMR…http://file.selleckchem.com/downloads/nmr/S500401-Pimecrolimus-NMR-Selleck.pdf
HPLC…….http://file.selleckchem.com/downloads/hplc/S500401-Pimecrolimus-HPLC-Selleck.pdf
http://file.selleckchem.com/downloads/hplc/S500401-Pimecrolimus-HPLC-Selleck.pdf
Pimecrolimus is an immunomodulating agent used in the treatment of atopic dermatitis (eczema). It is available as a topical cream, once marketed by Novartis (however, Galderma has been promoting the compound in Canada since early 2007) under the trade name Elidel.

Pimecrolimus is an ascomycin macrolactam derivative. It has been shown in vitro that pimecrolimus binds to macrophilin-12(also referred to as FKBP-12) and inhibits calcineurin. Thus pimecrolimus inhibits T-cell activation by inhibiting the synthesis and release of cytokines from T-cells. Pimecrolimus also prevents the release of inflammatory cytokines and mediators from mast cells.
Pimecrolimus is a chemical that is used to treat atopic dermatitis (eczema). Atopic dermatitis is a skin condition characterized by redness, itching, scaling and inflammation of the skin. The cause of atopic dermatitis is not known; however, scientists believe that it may be due to activation of the immune system by various environmental or emotional triggers. Scientists do not know exactly how pimecrolimus reduces the manifestations of atopic dermatitis, but pimecrolimus reduces the action of T-cells and mast cells which are part of the immune system and contribute to responses of the immune system. Pimecrolimus prevents the activation of T-cells by blocking the effects of chemicals (cytokines) released by the body that stimulate T-cells. Pimecrolimus also reduces the ability of mast cells to release chemicals that promote inflammation.
Pimecrolimus, like tacrolimus, belongs to the ascomycin class of macrolactam immunosuppressives, acting by the inhibition of T-cell activation by the calcineurin pathway and inhibition of the release of numerous inflammatory cytokines, thereby preventing the cascade of immune and inflammatory signals.[1] Pimecrolimus has a similar mode of action to that of tacrolimus but is more selective, with no effect on dendritic (Langerhans) cells.[2] It has lower permeation through the skin than topical steroids or topical tacrolimus[3] although they have not been compared with each other for their permeation ability through mucosa. In addition, in contrast with topical steroids, pimecrolimus does not produce skin atrophy.[4] It has been proven to be effective in various inflammatory skin diseases, e.g., seborrheic dermatitis,[5] cutaneous lupus erythematosus,[6]oral lichen planus,[7] vitiligo,[8] and psoriasis.[9][10] Tacrolimus and pimecrolimus are both calcineurin inhibitors and function as immunosuppressants.[11]
Ascomycin macrolactams belong to a new group of immunosuppressive, immunomodulatory and anti-inflammatory agents and include, e.g., ascomycin (FK520), tacrolimus (FK506) and pimecrolimus (ASM 981). The main biological effect of ascomycin macrolactams appears to be the inhibition of the synthesis of both Th1 and Th2-type cytokines in target cells.
As used herein, the term “ascomycin macrolactam” means ascomycin, a derivative of ascomycin, such as, e.g., tacrolimus and pimecrolimus, or a prodrug or metabolite of ascomycin or a derivative thereof.
Ascomycin, also called immunomycin, is a structurally complex macrolide produced by Streptomyces hygroscopicus. Ascomycin acts by binding to immunophilins, especially macrophilin-12. It appears that ascomycin inhibits the production of Th1 (interferon- and IL-2) and Th2 (IL-4 and IL-10) cytokines. Additionally, ascomycin preferentially inhibits the activation of mast cells, an important cellular component of the atopic response. Ascomycin produces a more selective immunomodulatory effect in that it inhibits the elicitation phase of allergic contact dermatitis but does not impair the primary immune response when administered systemically. The chemical structure of ascomycin is depicted below.

Tacrolimus (FK506) is a synthetic derivatives of ascomycin. As a calcineurin inhibitor, it works through the FK-binding protein and inhibits the dephosphorylation of nuclear factor of activated T cells (NFAT), thereby preventing the transport of the cytoplasmic component of NFAT to the cell nucleus. This leads to transcriptional inhibition of proinflammatory cytokine genes such as, e.g., interleukin 2, which are dependent on the nuclear factor of activated NFAT. The chemical structure of tacrolimus is depicted below.

Pimecrolimus, an ascomycin derivative, is a calcineurin inhibitor that binds with high affinity to the cytosolic receptor macrophilin-12, inhibiting the calcium-dependent phosphatase calcineurin, an enzyme required for the dephosphorylation of the cytosolic form of the nuclear factor of the activated T cell (NF-AT). It thus targets T cell activation and proliferation by blocking the release of both TH1 and TH2 cytokines such as IF-g, IL-2, -4, -5, and -10.3 It also prevents the production of TNF-a and the release of proinflammatory mediators such as histamine, hexosaminidase, and tryptase from activated mast cells.3 It does not have general antiproliferative activity on keratinocytes, endothelial cells, and fibroblasts, and in contrast to corticosteroids, it does not affect the differentiation, maturation, functions, and viability of human dendritic cells. The chemical structure of pimecrolimus is depicted below.

Pimecrolimus is an anti-inflammatory compound derived from the macrolactam natural product ascomycin, produced by certain strains of Streptomyces.

In January 2006, the United States Food and Drug Administration (FDA) announced that Elidel packaging would be required to carry a black box warning regarding the potential increased risk of lymph node or skin cancer, as for the similar drug tacrolimus. Whereas current practice by UKdermatologists is not to consider this a significant real concern and they are increasingly recommending the use of such new drugs.[12]
Importantly, although the FDA has approved updated black-box warning for tacrolimus and pimecrolimus, the recent report of the American Academy of Dermatology Association Task Force finds that there is no causal proof that topical immunomodulators cause lymphoma or nonmelanoma skin cancer, and systemic immunosuppression after short-term or intermittent long-term topical application seems an unlikely mechanism.[13] Another recent review of evidence concluded that postmarketing surveillance shows no evidence for this systemic immunosuppression or increased risk for any malignancy.[14] However, there are still some strong debates and controversies regarding the exact indications of immunomodulators and their duration of use in the absence of active controlled trials.[15] Dermatologists’ and Allergists’ professional societies, the American Academy of Dermatology[1], and the American Academy of Allergy, Asthma, and Immunology, have protested the inclusion of the black box warning. The AAAAI states “None of the information provided for the cases of lymphoma associated with the use of topical pimecrolimus or tacrolimus in AD indicate or suggest a causal relationship.”[2].

ELIDEL® (pimecrolimus) Cream 1% contains the compound pimecrolimus, the immunosuppressant 33-epi-chloro-derivative of the macrolactam ascomycin.
Chemically, pimecrolimus is (1R,9S,12S,13R,14S,17R,18E,21S,23S,24R,25S,27R)-12-[(1E)-2{(1R,3R,4S)-4-chloro-3-methoxycyclohexyl}-1-methylvinyl]-17-ethyl-1,14-dihydroxy-23,25 dimethoxy-13,19,21,27-tetramethyl-11,28-dioxa-4-aza-tricyclo[22.3.1.0 4,9]octacos-18-ene2,3,10,16-tetraone.
The compound has the empirical formula C43H68CINO11 and the molecular weight of 810.47. The structural formula is
 |
Pimecrolimus is a white to off-white fine crystalline powder. It is soluble in methanol and ethanol and insoluble in water.
Each gram of ELIDEL Cream 1% contains 10 mg of pimecrolimus in a whitish cream base of benzyl alcohol, cetyl alcohol, citric acid, mono- and di-glycerides, oleyl alcohol, propylene glycol, sodium cetostearyl sulphate, sodium hydroxide, stearyl alcohol, triglycerides, and water.
The second representative of the immunosuppressive macrolides for topical application – after tacrolimus (Protopic ®) – has 21 October in the trade. Pimecrolimus is approved for short-term and intermittent long-term treatment for patients aged two years who suffer from mild to moderate atopic dermatitis.
Pimecrolimus is a lipophilic derivative of macrolactam Ascomycin. The macrolides inhibit the production and release of pro-inflammatory cytokines by blocking the phosphatase calcineurin.The anti-inflammatory effect unfolds the drug in the skin. Since he is only minimally absorbed to not measurable, it hardly affects the local or systemic immune response. Therefore, the authorization neither restricts nor a maximum daily dose treatable area or duration of therapy.The cream can also be applied on the face, head and neck, and in skin folds, but not simultaneously with other anti-inflammatory topical agents such as glucocorticoids.
In studies in phases II and III patients aged three months and treated a maximum of one year.In two six-week trials involving 186 infants and young children as well as 403 children and adolescents, the verum symptoms and itching decreased significantly better than the cream base. Already in the first week of itching in 44 percent of children and 70 percent of the infants improved significantly. In adults, pimecrolimus was less effective than 0.1 percent betamethasone 17-valerate.
In the long-term treatment the verum significantly reduced the incidence of flares, revealed two studies with 713 and 251 patients. About a half and one year each about twice as many of the small patients were free of acute disease exacerbations than with the cream base (example: 61 versus 34 per cent of children, 70 versus 33 percent of infants older than six months). Moreover, the use of topical corticosteroids decreased significantly.
In a study of 192 adults with moderate to severe eczema half suffered six months no relapses more (24 percent with placebo). In the long-term therapy pimecrolimus was less effective than 0.1 percent triamcinolone acetonide cream and 1 percent hydrocortisone cream in adults.
The new topicum is-apart from burning and irritation at the application site – relatively well tolerated. It is neither kontaktsensibilisierend still phototoxic or sensitizing and does not cause skin atrophy. As in atopic Ekzen but usually a long-term therapy is necessary studies can reveal long-term adverse effects of the immunosuppressant on the skin only beyond one year.Also available from direct comparative studies between tacrolimus and pimecrolimus. They could help to delineate the importance of the two immunosuppressants.
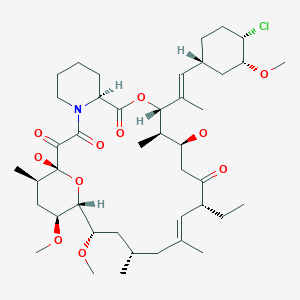

Pimecrolimus (registry number 137071-32-0; Figure 1) is a macro lide having anti-inflammatory, antiproliferative and immunosuppressive properties. This substance is present as an active ingredient in the Elidel ® drug recently approved in Europe and in the USA for topical treatment of inflammatory conditions of the skin such as atopic dermatitis.

Figure 1: structural formula of pimecrolimus
19th Ed., vol. π, pg. 1627, spray-drying consists of bringing together a highly dispersed liquid and a sufficient volume of hot air to produce evaporation and drying of the liquid droplets. Spray-drying however is often limited to aqueous solutions unless special expensive safety measures are taken. Also, in spite of the short contact time, certain undesirable physical and chemical characteristics of the emerging solids are in particular cases unavoidable. The turbulence present in a spray-drier as a result of the moving air may alter the product in an undesirable manner. Modifications to the spray-drying technique are disclosed in WO 03/063821 and WO 03/063822. [00012] European Patent EP 427 680 Bl discloses a method of synthesizing amorphous pimecrolimus (Example 66a). The method yields amorphous pimecrolimus as a colorless foamy resin.
U.S. Patent No. US 6,423,722 discloses crystalline forms of pimecrolimus, such as form A, form B, etc. US 722 also contend that by performing example 66a from the European Patent EP 427 680 Bl, amorphous pimecrolimus is obtained.
The preparation of pimecrolimus was described for the first time in the patent application EP427680 on behalf of Sandoz. Used as raw material in such document is ascomycin (compound identified by registry number 11011-38-4), a natural product obtained through fermentation from Streptomyces strains (such as for example Streptomyces hygroscopicus var ascomyceticus, or Streptomyces hygroscopicus tsukubaensis N°9993). Pimecrolimus is obtained from the ascomycin through a sequence of four steps of synthesis (scheme 1)

Scheme 1 : synthesis process described in EP427680
From a structural point of view, pimecrolimus is the 33-epi-chloro derivative of ascomycin. As described in EP427680, the simultaneous presence – in the structure of ascomycin – of two secondary hydroxyl groups in position 24 and in position 33, requires the protection of the hydroxyl in position 24 before substituting the second hydroxyl in position 33 with an atom of chlorine.
In order to obtain the monoprotection of the hydroxyl in position 24 of ascomycin, such synthesis process provides for the preparation of 24,33-disilyl derivative and the subsequent selective removal of the silyl ester in position 33.
The high ratio between the silylating agent and the substrate and the non-complete selectivity of the subsequent step of deprotection requires carrying out two chromatographic purifications on the column of silica gel (Baumann K., Bacher M., Damont A., Hogenauer K., Steck A. Tetrahedron, (2003), 59, 1075-1087). The general yields of such synthesis process are not indicated in literature; an experiment by the applicant revealed that such yields amount to about 16% molar starting from ascomycin.
Other synthesis processes were recently proposed as alternatives to the synthesis of EP427680.
In particular, the International patent application WO2006040111 on behalf of Novartis provides for the direct substitution of the hydroxyl in position 33 of ascomycin with an atom of chlorine and a second alternative, described in the international patent application WO2006060614 on behalf of Teva, uses – as a synthetic intermediate – a sulfonate derivative in position 33 of ascomycin. Both the proposed synthetic alternatives are not entirely satisfactory in that in WO2006040111 the proposed halogenating agents (chlorophosphorane and N- chlorosuccinimide) are not capable, according to the same authors, of regioselectively substituting the hydroxyl function in position 33, while in WO2006060614 the quality characteristics of the obtained product are, even after chromatographic purification and/or crystallisation, low for a product to be used for pharmaceutical purposes (i.e. purity of 96% as described in the experimental part).
Generally, purified enzymatic systems may be used for the organic synthesis of polyfunctional molecules (Wang Y-F, Wong C-H. J Org Chem (1988) 53, 3127- 3129; Santaniello E., Ferraboschi P., Grisenti P., Manzocchi A. Chem. Rev. (1992), 92(5), 1071-140; Ferraboschi P., Casati S., De Grandi S., Grisenti P., Santaniello E. Biocatalysis (1994), 10(1-4), 279-88); WO2006024582). WO2007103348 and WO2005105811 describe the acylation of rapamycin in position 42 in the presence of lipase from Candida antartica.
…………………….

Scheme 2: synthesis of pimecrolimus for enzymatic transesterification of ascomycin.

Scheme 3. Synthesis of pimecrolimus for enzyme-catalyzed alcoholysis from 33,24- diacetate of ascomycin
Example 1
Preparation of the 33-acetyl derivative of ascomvcin (compound I of scheme II)
Lipase from Candida antarctica (CAL B, Novozym 435) [0.140 g (2 U/mg)
FLUKA] was added to a solution of ascomycin (100 mg; 0.126 mmol) in toluene (8 ml) and vinyl acetate (4.5 eq; 0.473 g). The reaction is kept under stirring at the temperature of 30° C for 80 hrs then the enzyme is taken away for filtration and the filtrate is concentrated at low pressure to obtain 105 mg of 33-acetyl ascomycin.
A sample of such intermediate was purified for analytical purposes by chromatography on silica gel (n-hexane/acetone = 8/2 v/v as eluents) and thus crystallised by acetone/water.
The following analysis were carried out on such sample: 1H-NMR (500MHz) δ:
2.10 (CH3CO), 3.92 and 4.70 (24CH and 33CH); IR (cm-1): 3484.245, 2935.287,
1735.331, 1649.741, 1450.039,
1372.278; DSC: endotherm at 134.25° C; [α]D=-74,0° (c=0.5 CHCl3).
Spectrum of MS (ESI +): m/z: 856.4 (M+23; 100.0%)
Elementary analysis calculated for C45H7iNO13: C 64.80%; H, 8.58%; N, 1.68%;
O, 24.94%
Elementary analysis found: C 64.78%; H, 8.54%; N, 1.59%; O, 24.89%
Preparation of the 24-tgrt-butyldimethylsilylether-33 -acetyl derivative of ascomvcin (intermediate 24-silyl-33-Oac; compound II of scheme 2)
2,6-lutidine (0.29Og; 2.7 mmolels) and tert-butyldimethylsilyl triflate (0.238g; 0.9 mmoles) are added to a solution of 33-acetyl derivative of ascomycin (150 mg;
0.18 mmoles) in dichloromethane (5ml). The reaction is left under stirring at ambient temperature for 30 minutes. After this period the reaction mixture is washed with a solution saturated with sodium bicarbonate (5 ml) and organic phase obtained is washed in sequence with HCl 0.1N (5 ml 3 times) and with a solution at 30% of NaCl (5ml). The organic phase is anhydrified on sodium sulphate, filtered and concentrated to residue under vacuum to obtain 128 mg of product.
Spectrum of MS (ESI +): m/z: 970.5 (M+23; 100.0%)
1H-NMR (500 MHz) δ: 0.05 and 0.06 ((CHs)2Si), 0.90 ((CH3)3C-Si), 2.10
(CH3CO), 4.70 (33CH)
IR (cm-‘): 3462.948, 2934.450, 1739.236, 1649.937
Elementary analysis calculated for C51H85NOi3Si: C 64.59%; H, 9.03%; N, 1.48%; O, 21.93%
Elementary analysis found: C 64.50%; H, 9.05%; N, 1.41%; O, 21.88%
DSC= endoderma a 236,43° C. [α]D=-81,4° (c=0.5 CHCl3).
Preparation of 24-tert-butyldimethylsilylether of ascomycin (intermediate 24- silyl-33-OH; compound III of scheme 2) n-octan-1-ol (0.035g; 0.265 mmoles) and CAL B (Novozym 435) [0.100 g (2
U/mg) FLUKA] are added to a solution of 24-tert-butyldimethylsilylether-33- acetyl derivative of ascomycin (50 mg; 0.053 mmoles) in tert-butylmethylether (4 ml). The reaction is kept under stirring at the temperature of 40° C for 120 hours.
After this period the reaction mixture is filtered and the filtrate is evaporated to residue under vacuum to obtain a reaction raw product which is purified by chromatography on silica gel: 44 mg of product (0.048 mmoles) are recovered through elution with petroleum ether/acetone 7/3.
The chemical/physical properties of the obtained product match those of a reference sample obtained according to patent EP427680.
Preparation of 24-tert-butyldimethylsilylether-33-epi-chloro ascomycin
(intermediate 24-silyl-33-chloro; compound IV of scheme 2)
A solution of 24-silyl FR520, i.e. 24-silyl ascomycin (165 g; 0.18 moles) in anhydrous toluene (1.4 litres) and pyridine (50 ml) is added to a suspension of dichlorotriphenylphosphorane (99.95g) in anhydrous toluene (1.1 litres), under stirring at ambient temperature (20-25 °C) in inert atmosphere.
After adding, the reaction mixture is heated at the temperature of 60° C for 1 hour.
After this period the temperature of the reaction mixture is taken to 25° C and thus the organic phase is washed in sequence with water (1 time with 1 L) and with an aqueous solution of NaCl at 10% (4 times with 1 L each time), then it is anhydrified on sodium sulphate, filtered and concentrated under vacuum to obtain about 250 g of a moist solid of toluene. Such residue product is retaken with n- hexane (500 ml) and then evaporated to dryness (in order to remove the toluene present). The residue product is diluted in n-hexane (500 ml) under stirring at ambient temperature for about 45 minutes and then the undissolved solid taken away for filtration on buckner (it is the sub-product of dichlorophosphorane).
The filtrate is concentrated at low pressure to obtain 148.6 g of a solid which is subsequently purified by chromatography on silica gel (elution with n- heptane/acetone = 9/1) to obtain 123 g (0.13 moles) of product.
The chemical/physical properties of the obtained product match those described in literature (EP427680).
Preparation of the pimecrolimus from 24-fert-butyldimethylsilylether-33-epi- chloro ascomycin
The intermediate 24-silyl-33 chloro (123g; 0.13 Moles; compound IV of scheme
2) is dissolved under stirring at ambient temperature in a dichloromethane/methanol mixture=l/l=v/v (1.1 litres) then p-toluenesulfonic acid monohydrate (10.11 g) is added.
The reaction is kept under stirring at the temperature of 20-25° C for 72 hours, thus a solution of water (600 ml) and sodium bicarbonate (4.46 g) is added to the reaction mixture. The reaction mixture is kept under stirring at ambient temperature for 10 minutes, the organic phase is then prepared and washed with an aqueous solution at 10% of sodium chloride (600 ml).
The organic phase is anhydrified on sodium sulphate, filtered and concentrated under vacuum to obtain 119 g of raw pimecrolimus. Such raw product is purified by chromatography on silica gel (n-hexane/acetone as eluents) and thus crystallised by ethyl acetate, cyclohexane/water to obtain 66 g (81.5 mmoles) of purified pimecrolimus.
The chemical/physical data obtained matches the data indicated in literature.
Example 2
Preparation of ascomvcin 24.33-diacetate (intermediate 24, 33-diacetate; compound V of scheme 3)
DMAP (4.5 eq; 0.136 g) and acetic anhydride (4.5 eq; 0.114 g) are added to a solution of ascomycin (200 mg; 0.25 mmoles) in pyridine (2.5 ml), under stirring at the temperature of 0° C.
The reaction is kept under stirring for 1.5 hours at the temperature of 0° C then it is diluted with water and it is extracted with ethyl acetate (3 times with 5 ml). The organic extracts are washed with HCl 0.5 N (5 times with 10 ml), anhydrified on
Na2SO4 concentrated under vacuum.
The residue product was purified by chromatography on silica gel (n- hexane/acetone 8/2 v/v as eluent) to obtain ascomycin 24,32-diacetate (210 mg;
0.24 mmoles).
We carried out the following analysis on such purified sample:
1H-NMR (500 MHz) δ: 2.02 and 2.06 (2 CH3CO), 5.20 and 4.70 (24CH and
33CH);
IR (Cm-1): 3462.749, 2935.824, 1734.403, 1650.739, 1449.091, 1371.079.
DSC: endothermic peak at 234.10° C ; [α]D=- 100.0° (C=0.5 CHCl3).
Spectrum of MS (ESI+): m/z: 898.4 (100.0%; m+23).
Elementary analysis calculated for C47H73NO14: C 64.44%; H 8.40%; N 1.60%; O
25.57%
Elementary analysis found: C 64.55%; H 8.44%; N 1.61%; O 25.40%
Preparation of the 24-acetyl ascomycin (intermediate 24-acetate-33-OH; compound VI of scheme 3)
Lipase from Candida antartica (CAL B Novozym 435) [1.1 g (2 U/mg) FLUKA] is added to a solution of ascomycin 33,24-diacetate (500 mg; 0.57 mmol) in
TBDME (25 ml) and n-octan-1-ol (4.5 eq; 0.371 g). The reaction is kept under stirring at 30° C for 100 hours, then the enzyme is taken away for filtration and the obtained filtrate is concentrated under low pressure to obtain 425 mg (0.51 mmoles) of product.
A sample was purified for analytical purposes by chromatography on silica gel (n- hexane/acetone = 7:3 v/v as eluents) and thus crystallised by acetone/water.
We carried out the following analysis on such purified sample: 1H-NMR
(500MHz) δ: 2.05 (CH3CO); IR (an 1): 3491.528, 2935.860, 1744.728, 1710.227,
1652.310, 1448.662, 1371.335. DSC: endothermic peak at 134.68° C; [α]D=-
102.7° (c=0.5 CHCl3)
Spectrum of MS (ESI +): m/z: 856.4 (M+23; 100.0%)
Elementary analysis calculated for C45H71NO13: C 64.80%; H, 8.58%; N, 1.68%;
0, 24.94%
Elementary analysis found: C 64.71%; H, 8.49%; N, 1.60%; O, 24.97%
Preparation of the 24-acetyl-33epi-chloro ascomycin (intermediate 24-Acetate-33- chloro; compound VII of scheme 3) Supported triphenylphosphine (0.335 g; 1.1 mmoles) is added to a solution of 24- acetyl ascomycin (400 mg; 0.48 mmoles) in carbon tetrachloride (5 ml). The reaction mixture is kept under reflux for 3 hours then it is cooled at ambient temperature. The obtained suspension is filtered and the filtrate is concentrated to residue under vacuum to obtain 0.45g of reaction raw product which is purified by chromatography on silica gel: 163mg (0.19 mmoles) of product are obtained by elution with petroleum ether/acetone = 90/10.
1H-NMR δ: 2.08 (CH3CO); 4.60 (33CH); IR (Cm“1)= 3464.941, 2934.360,
1738.993, 1650.366, 1450.424, 1371.557; DSC: endothermic peak at 231.67° C
[α]D=-75.2° (c=0.5 CHCl3)
Spectrum of MS (ESI +): m/z: 874.3 (M+23; 100.0%)
Elementary analysis calculated for C45H70ClNO12: C 63.40%; H, 8.28%; Cl,
4.16%; N, 1.64%; O, 22.52%
Elementary analysis found: C 63.31%; H, 8.30%; Cl, 4.05%; N, 1.58%; O,
22.42%.
Preparation of pimecrolimus from 24-acetyl-33-epi-chloro ascomycin
A solution of 24-acetyl-33-epi-chloro ascomycin (200 mg; 0.23 mmoles; compound VII) in methanol (2 ml) and HCl 3N (1 ml) is stirred at ambient temperature for 40 hours. After this period, the reaction is neutralised with an aqueous bicarbonate solution, the methanol evaporated under vacuum. The mixture is extracted with dichloromethane (3 times with 5 ml), anhydrified on sodium sulphate, filtered and concentrated to residue to obtain a residue product which is purified by chromatography on silica gel (n-hexane/acetone as eluents) and thus crystallised by ethyl acetate, cyclohexane/water to obtain 78 mg of purified pimecrolimus (0.096 mmoles).
The chemical/physical characteristics of the obtained product matches the data indicated in literature for pimecrolimus.
Example 4 (comparative*)
Verification of the method of synthesis of pimecrolimus described in EP427680 Imidazole (508 mg) and tert-Butyldimethylsilylchloride (1.125 g) are added in portions to a solution of 2g (2.53 mmoles) of ascomycin in anhydrous N,N- dimethylformamide (40 ml). The reaction mixture is kept under stirring at ambient temperature for 4.5 days. The reaction is thus processed diluting it with ethyl acetate (200 ml) and processing it using water (5 x 100 ml). The organic phase is separated, anhydrified on sodium sulphate, filtered and evaporated to residue under vacuum to obtain a foamy raw product which is subsequently purified by chromatography on silica gel (1:30 p/p): 2.1 g (2.05 mmoles; yields 81% molars) of ascomycin 24,33 disilyl intermediate are obtained by elution with n- hexane/ethyl acetate 3/1. The chemical/physical data of such intermediate matches that indicated in EP427680.
2.1 g (2.05 mmoles) of ascomycin 24,33 disilyl intermediate are dissolved in a solution under stirring at the temperature of 0°C composed of acetonitrile (42 ml) and aqueous HF 40% (23.1 ml). The reaction mixture is kept under stirring at the temperature of 0°C for 2 hours then it is diluted with dichloromethane (30 ml). Then the reaction is washed in sequence with a saturated aqueous solution using sodium bicarbonate (30 ml) and water (30 ml). The separated organic phase is anhydrified on sodium sulphate, filtered and evaporated to residue under vacuum to obtain a foamy residue which is subsequently purified by chromatography on silica gel (1:30 p/p): 839 mg (0.92 mmoles; yields 45% molars) of ascomycin 24 monosilyl intermediate are obtained by elution with dichloromethane/methanol 9/1. The chemical/physical data of such intermediate matches that obtained on the compound III scheme 2 and matches the data of literature indicated in EP427680. A mixture of 839 mg (0.92 mmoles; yields 45% molars) of ascomycin 24 monosilyl intermediate, triphenylphosphine (337 mg) in carbon tetrachloride (36.4 ml) is heated under stirring under reflux for 15 hours. After this period the reaction mixture is evaporated to residue under vacuum to obtain a solid product purified by chromatography on silica gel (1:30 p/p): 535 mg (0.57 mmoles; yields 63% molars) of ascomycin 24 monosilyl intermediate, 33-chloro derivative are obtained by elution with n-hexane/ethyl acetate 2/1. The chemical/physical data of such intermediate matches those we obtained on compound IV scheme 2 and matches the data of literature indicated in EP427680.
535 mg (0.57 mmoles) of ascomycin 24 monosilyl intermediate, 33-chloro derivative are dissolved under stirring at ambient temperature in acetonitrile (16.4 ml) and aqueous HF 40% (0.44 ml). The reaction mixture is kept under stirring at ambient temperature for 45′ and then it is diluted with ethyl acetate (100 ml). The organic phase is thus washed in sequence with an aqueous solution of sodium bicarbonate (70 ml) with water (2 x 70 ml) and thus it is anhydrified on sodium sulphate, filtered and evaporated under vacuum to obtain a solid which is subsequently purified by chromatography on silica gel (1 :30 p/p): 323 mg (0.399 mmoles; yields 70% molars) of pimecrolimus is obtained by elution with n- hexane/ethyl acetate 2/3. The chemical/physical characteristics of the obtained product matches the data indicated in literature regarding pimecrolimus; the overall yield of the process is 16%.
………………………..
POLYMORPHS…….WO2006060615A1
Example 7: Preparation of amorphous pimecrolimus by precipitation [00094] 19,5 g purified pimecrolimus (colorless resin) was dissolved in 217 ml acetone at 4O0C and concentrated. Residue: 38,76 g. The residue was diluted with 6 ml distilled water with stirring. Finally 1 ml acetone was added. This solution was added slowly to 2 L chilled distilled water that was stirred efficiently. After the addition had been completed, the suspension was stirred 20 min at O0C. Then the solid was filtered and dried at 450C in vacuum oven overnight. Product: 15,65 g yellowish solid. Amorphous (XRD, DSC).
Example 8: Preparation of amorphous pimecrolimus by grinding
[00095] Procedure of grinding: 200 mg of Pimecrolimus sample was ground gently in an agate mortar using a pestle for half a minute. ,
References
- Allen BR, Lakhanpaul M, Morris A, Lateo S, Davies T, Scott G, Cardno M, Ebelin ME, Burtin P, Stephenson TJ (2003). “Systemic exposure, tolerability, and efficacy of pimecrolimus cream 1% in atopic dermatitis patients”. Arch Dis Child 88 (11): 969–973. doi:10.1136/adc.88.11.969.PMC 1719352. PMID 14612358.
- Meingassner JG, Kowalsky E, Schwendinger H, Elbe-Bürger A, Stütz A (2003). “Pimecrolimus does not affect Langerhans cells in murine epidermis”. Br J Dermatol 149 (4): 853–857.doi:10.1046/j.1365-2133.2003.05559.x. PMID 14616380.
- Billich A, Aschauer H, Aszódi A, Stuetz A (2004). “Percutaneous absorption of drugs used in atopic eczema: pimecrolimus permeates less through skin than corticosteroids and tacrolimus”. Int J Pharm 269 (1): 29–35. doi:10.1016/j.ijpharm.2003.07.013.PMID 14698574.
- Firooz A, Solhpour A, Gorouhi F, Daneshpazhooh M, Balighi K, Farsinejad K, Rashighi-Firoozabadi M, Dowlati Y (2006). “Pimecrolimus cream, 1%, vs hydrocortisone acetate cream, 1%, in the treatment of facial seborrheic dermatitis: a randomized, investigator-blind, clinical trial”. Archives of Dermatology 142 (8): 1066–1067. doi:10.1001/archderm.142.8.1066.PMID 16924062.
- Firooz A, Solhpour A, Gorouhi F, Daneshpazhooh M, Balighi K, Farsinejad K, Rashighi-Firoozabadi M, Dowlati Y (2006). “Pimecrolimus cream, 1%, vs hydrocortisone acetate cream, 1%, in the treatment of facial seborrheic dermatitis: a randomized, investigator-blind, clinical trial”. Archives of Dermatology 142 (8): 1066–1067. doi:10.1001/archderm.142.8.1066.PMID 16924062.
- Kreuter A, Gambichler T, Breuckmann F, Pawlak FM, Stücker M, Bader A, Altmeyer P, Freitag M (2004). “Pimecrolimus 1% cream for cutaneous lupus erythematosus”. J Am Acad Dermatol 51(3): 407–410. doi:10.1016/j.jaad.2004.01.044. PMID 15337984.
- Gorouhi F, Solhpour A, Beitollahi JM, Afshar S, Davari P, Hashemi P, Nassiri Kashani M, Firooz A (2007). “Randomized trial of pimecrolimus cream versus triamcinolone acetonide paste in the treatment of oral lichen planus”. J Am Acad Dermatol 57 (5): 806–813.doi:10.1016/j.jaad.2007.06.022. PMID 17658663.
- Boone B, Ongenae K, Van Geel N, Vernijns S, De Keyser S, Naeyaert JM (2007). “Topical pimecrolimus in the treatment of vitiligo”. Eur J Dermatol 17 (1): 55–61. doi:10.1111/j.1610-0387.2006.06124.x. PMID 17081269.
- Kreuter A, Sommer A, Hyun J, Bräutigam M, Brockmeyer NH, Altmeyer P, Gambichler T (2006). “1% pimecrolimus, 0.005% calcipotriol, and 0.1% betamethasone in the treatment of intertriginous psoriasis: a double-blind, randomized controlled study”. Arch Dermatol 142 (9): 1138–1143. doi:10.1001/archderm.142.9.1138. PMID 16983001.
- Jacobi A, Braeutigam M, Mahler V, Schultz E, Hertl M (2008). “Pimecrolimus 1% cream in the treatment of facial psoriasis: a 16-week open-label study”. Dermatology 216 (2): 133–136.doi:10.1159/000111510. PMID 18216475.
- Scheinfeld N (2004). “The use of topical tacrolimus and pimecrolimus to treat psoriasis: a review”. Dermatol. Online J. 10 (1): 3. PMID 15347485.
- N H Cox and Catherine H Smith (December 2002). “Advice to dermatologists re topical tacrolimus” (DOC). Therapy Guidelines Committee. British Association of Dermatologists.
- Berger TG, Duvic M, Van Voorhees AS, VanBeek MJ, Frieden IJ; American Academy of Dermatology Association Task Force (2006). “The use of topical calcineurin inhibitors in dermatology: safety concerns Report of the American Academy of Dermatology Association Task Force”. J Am Acad Dermatol 54 (5): 818–823. doi:10.1016/j.jaad.2006.01.054.PMID 16635663.
- Spergel JM, Leung DY (2006). “Safety of topical calcineurin inhibitors in atopic dermatitis: evaluation of the evidence”. Curr Allergy Asthma Rep 6 (4): 270–274. doi:10.1007/s11882-006-0059-7. PMID 16822378.
- Stern RS (2006). “Topical calcineurin inhibitors labeling: putting the “box” in perspective”.Archives of Dermatology 142 (9): 1233–1235. doi:10.1001/archderm.142.9.1233.PMID 16983018.
| WO2005105811A1 | Apr 12, 2005 | Nov 10, 2005 | Ping Cai | Regiospecific synthesis of rapamycin 42-ester derivatives |
| WO2006024582A1 | Jul 26, 2005 | Mar 9, 2006 | Poli Ind Chimica Spa | A method for the preparation of mycophenolate mofetil by enzimatic transesterification |
| WO2006040111A2 | Oct 10, 2005 | Apr 20, 2006 | Novartis Ag | Heteroatoms-containing tricyclic compounds |
| WO2006060614A1 | Dec 1, 2005 | Jun 8, 2006 | Teva Gyogyszergyar Zartkoeruen | Methods for preparing pimecrolimus |
| WO2007103348A2 | Mar 5, 2007 | Sep 13, 2007 | Wyeth Corp | Process for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants |
| EP0427680A1 | Nov 7, 1990 | May 15, 1991 | Sandoz Ltd. | Heteroatoms-containing tricyclic compounds |
- Elidel official homepage
- FDA News
- NPS RADAR
- Article about American Academy of Dermatology speaking out against black box warning
- Report of the Calcineurin Task Force of the ACAAI and AAAAI


| WO2005117837A1 * | Jun 1, 2005 | Dec 15, 2005 | Lorant Gyuricza | Process for preparation of amorphous form of a drug |
| EP0427680A1 * | Nov 7, 1990 | May 15, 1991 | Sandoz Ltd. | Heteroatoms-containing tricyclic compounds |
| EP0480623A1 * | Oct 2, 1991 | Apr 15, 1992 | Merck & Co., Inc. | New halomacrolides and derivatives having immunosuppressive activity |
| US6423722 * | Oct 17, 2000 | Jul 23, 2002 | Novartis Ag | Crystalline macrolides and process for their preparation |
Cidofovirסידופוביר سيدوفوفير


CIDOFOVIR
(S)-1-(3-Hydroxy-2-phosphonylmethoxypropyl)cytosine
[(S)-2-(4-Amino-2-oxo-1,2-dihydropyrimidin-2-yl)-1-(hydroxymethyl)ethoxymethyl]phosphonic acid
113852-37-2 CAS
120362-37-0 (Na salt)
149394-66-1 (dihydrate)
launched 1996 Gilead
SYNTHESIS.. CHEMDRUG
Rega Instituut (Originator)
For the treatment of CMV retinitis in patients with acquired immunodeficiency syndrome (AIDS)
US5142051 PATENT
| Canada | 1340856 | 1999-12-21 | EXPIRY 2016-12-21 |
| United States | 5142051 | 1993-06-26 | 2010-06-26 |
Cidofovir is a DNA polymerase inhibitor that was launched in 1996 by Gilead for the intravenous treatment of cytomegaloviral (CMV) retinitis in AIDS patients. Early clinical trials are underway at the National Institute for Allergy & Infectious Disease (NIAID) for the treatment of BK virus nephropathy (BKVN) in patients who have undergone kidney transplants.
Cidofovir suppresses CMV replication by selective inhibition of viral DNA synthesis. Biochemical data support selective inhibition of CMV DNA polymerase by cidofovir diphosphate, the active intracellular metabolite of cidofovir. Cidofovir diphosphate inhibits herpesvirus polymerases at concentrations that are 8- to 600-fold lower than those needed to inhibit human cellular DNA polymerases alpha, beta, and gamma1, 2, 3. Incorporation of cidofovir into the growing viral DNA chain results in reductions in the rate of viral DNA synthesis.
Cidofovir was originally developed under a collaboration between the Academy of Sciences of the Czech Republic and the Rega Institute for Medical Research. In 1991 and 1992, Gilead entered into license agreements with the Rega Institute that covered a large number of nucleotide analogue compounds and structures, including cidofovir. The drug became the subject of a marketing collaboration between Gilead and Pfizer (formerly Pharmacia & Upjohn) in August 1996 that covers all countries outside the U.S.
Cidofovir (brand name Vistide) is an injectable antiviral medication primarily used as a the treatment for cytomegalovirus (CMV) retinitis (an infection of the retina of the eye) in patients with AIDS.[1][2]
Its only indication that has received regulatory approval worldwide is cytomegalovirus retinitis.[1][2] Cidofovir has also shown efficacy in the treatment ofaciclovir-resistant HSV infections.[3] Cidofovir has also been investigated as a treatment for progressive multifocal leukoencephalopathy with successful case reports of its use.[4] Despite this meta-analyses have failed to demonstrate any efficacy in AIDS patients,[5] and the limited data in non-AIDS patients fail to demonstrate any efficacy either.[6] Cidofovir might have anti-smallpox efficacy and might be used on a limited basis in the event of a bioterror incident involving smallpox cases.[7] A cidofovir derivative with much higher activity against smallpox that can be taken orally has been developed.[8] It has inhibitory effects on varicella-zoster virus replication in vitro although no clinical trials have been done to date, likely due to the abundance of safer alternatives such as aciclovir.[9] Cidofovir shows anti-BK virus activity in a subgroup of transplant patients.[10] Cidofovir is being investigated as a complementary intralesional therapy against papillomatosis caused by HPV.[11][12]
It first received FDA approval on the 26th of June 1996,[13] TGA approval on the 30th of April 1998[2] and EMA approval on the 23rd of April 1997.[14]
Other
It has been suggested as an antitumour agent, due to its suppression of FGF2.[15][16]
Cidofovir was discovered at the Institute of Organic Chemistry and Biochemistry, Prague, by Antonín Holý, and developed by Gilead Sciences[20] and is marketed with the brand name Vistide by Gilead in the USA, and by Pfizerelsewhere.
The chemical name of cidofovir is 1-[(S)-3-hydroxy-2-(phosphonomethoxy)propyl]cytosine dihydrate (HPMPC), with the molecular formula of C8H14N3O6P•2H2O and a molecular weight of 315.22 (279.19 for anhydrous). The chemical structure is:

Cidofovir is a white crystalline powder with an aqueous solubility of ≥ 170 mg/mL at pH 6 to 8 and a log P (octanol/aqueous buffer, pH 7.1) value of -3.3.
Cidofovir Injection is a sterile, hypertonic aqueous solution for intravenous infusion only. The solution is clear and colorless. It is supplied in clear glass vials, each containing 375 mg of anhydrous cidofovir in 5 mL aqueous solution at a concentration of 75 mg/mL.
The formulation is pH-adjusted to 7.4 (range 7.1 to 7.7) with sodium hydroxide and/or hydrochloric acid and contains no preservatives. The appropriate volume of Cidofovir Injection must be removed from the single-use vial and diluted prior to administration
INTRODUCTION
Cidofovir’s chemical formula is C8H14N3O6P and its IUPAC name is ({[(S)-1-(4-amino-2-oxo-1,2-dihydropyrimidin-1-yl)-3-hydroxypropan-2-yl]oxy}methyl)phosphonic acid. Cidofovir has also been described as (S)-(1-(4-amino-2-oxopyrimidin-1(2H)-yl)-3-hydroxypropan-2-yloxy)methylphosphonic acid as well as possibly by other chemical names. Its chemical structure is:

Cidofovir was discovered at the Institute of Organic Chemistry and Biochemistry, Prague, and developed by Gilead Sciences. Today, cidofovir is an injectable antiviral medication for the treatment of cytomegalovirus (CMV) retinitis in patients with AIDS. It suppresses CMV replication by selective inhibition of viral DNA polymerase and therefore prevention of viral replication and transcription. It is an acyclic nucleoside phosphonate, and is therefore independent of phosphorylation by viral enzyme, in contrast to, for instance, acyclovir.
Cidofovir is marketed with the brand name Vistide® by Gilead in the United States and by Pfizer in other parts of the world. Vistide® is a sterile, hypertonic aqueous solution for intravenous infusion only. The solution is clear and colorless. It is supplied in clear glass vials, each containing 375 mg of anhydrous cidofovir in 5 mL aqueous solution at a concentration of 75 mg/mL. The formulation is pH-adjusted to 7.4 with sodium hydroxide and/or hydrochloric acid and contains no preservatives. Renal impairment is the major toxicity of Vistide®.
Presently, there are no Orange Book patents listed as having claims which cover Vistide®, although previously U.S. Pat. No. 5,142,051 was listed in the Orange Book for Vistide®. The ‘051 patent is not directed specifically to cidofovir or its crystalline forms. Instead, it broadly discloses N-phosphonylmethoxyalkyl derivatives of pyrimidine and purine bases.
Cytomegalovirus (Cytomegaoviyns, CMV) is one of the biggest dangers of the herpes virus, the body’s infection rates as high as 50% to 80% of the current adult prevalence rate of more than 95%, generally showed a latent infection, most infections had no clinical symptoms, but under certain conditions, the invasion of organs and systems to produce more severe disease. The virus can invade the lung, liver, kidney, salivary gland, mammary gland and other polymorphonuclear leukocytes and lymphocytes, and, since the long-term or intermittent saliva, milk sweat, blood, urine, semen, exclude uterine secretions of the virus. Spread through a variety of ways in the mouth, genital tract, placenta, blood transfusion or organ transplantation.
When the body’s immune dysfunction, such as infected with HIV, cancer patients undergoing radiotherapy, chemotherapy, organ or bone marrow transplantation immunosuppressive anti-rejection etc will stimulate active infection, can cause acute retinitis, interstitial pneumonia, gastroenteritis and encephalitis, blindness or death without treatment rate of over 70%. With the rise in HIV infection rates and organ transplants extensively for anti-CMV drugs is also increasing demand.
cidofovir (cidofovir, HPMPC) are novel ether derivatives of cytidine phosphono chemical name
[5]-NL [(3 – hydroxy-2 – methoxy-phosphonic acid) glycerol]-N4-cytosine, Molecular structure of the formula (I):

Gilead developed by the United States, in May 1996 the FDA approved injectable celecoxib Duofu Wei listed, France and Canada also continued with the approval of the use of the trade name Vistide. Its CAS number is 113852-37-2, formula C8H14N3O6P, the structure of formula (I). Cidofovir for CMV is highly inhibitory activity of certain ganciclovir or foscarnet resistant strains of the virus are also active. And herpes simplex virus (HSV), herpes zoster virus (VZV), human papillomavirus (HPV), also has a strong activity.
Its mechanism of action: cidofovir having a phosphoric acid group, a ring-opening mechanism of the antiviral nucleoside phosphonate compound (ANP) and the consistent cyclic nucleoside analogues are nucleosides or virus in vivo kinase activation into triphosphate metabolite, thereby inhibiting viral replication by DNA polymerase and reverse transcriptase. Unlike the three-step cyclic nucleoside analogues must phosphorylation reaction, ring opening nucleoside phosphonate group containing phosphorus compound itself, eliminating the first step of the phosphorylation reaction speed, and thus a higher activity. Cidofovir is absorbed when the cells in the cell pyrimidine nucleoside phosphorylase kinase (P bandit kinase and NDP kinase) to effect conversion of the active metabolite monophosphate (HPMPCp), diphosphate (HPMPCpp) and a bile acid base adducts. Cidofovir diphosphate inhibits viral DNA polymerase or reverse transcriptase activity, and its corresponding natural dNTP incorporated into the viral DNA chain competition, since no 3 – hydroxy end, continue to extend the DNA chain termination. Can slow the synthesis of DNA, viral DNA and to the loss of stability, thereby inhibiting viral replication, transcription of the ability to reduce viral DNA to exert antiviral activity. Compared with other anti-CMV drugs, cidofovir characteristics: significant and lasting effect, started the first two weeks administered once a week, then only administered once every two weeks, easy to use, and to reduce its toxicity side effects.
Several major techniques are based on the synthesis of cidofovir cytosine as starting material, mainly carried out to improve the synthesis of the side chain.
(I) J. Med Chem, 1989,32,1457 ~ 1463 discloses a synthetic process:

The route to cytosine as the raw material, with a chiral side chain by condensation, deprotection and reduction can be obtained in three steps cidofovir.However, chiral side chain subject to a six-step reaction system. The total yield is low, adverse side. And using Me3SiBr, so that the costs and the risk of surge, is not conducive to industrial production.
(2) US 5591852,1995-1-7; US 2005/023833 & WO 2006/014429 and US 2009/0270618, Tetrahedron Lett 1994,35,3243-3246 and “Chinese Journal of New Drugs”, 2007,16. , 1272-1274 for the synthesis of a lot of improvements:

Benzoyl cytosine with a chiral starting material and trityloxymethyl ethylene oxide condensation, deprotection and hydrolysis was then prepared by deprotection cidofovir group. The synthetic steps to make some shorter, but still use expensive Me3SiBr, adverse ones, the low yield of the security at the cost of industrial production is still unfavorable. (Several different patent protection only in the order of the amino cytosine different!)
(3) Patent Publication No. CN1690065A, CN1690066A, CN1690067A (2005 年 11 月 2 Publication Date) and the “Chinese Journal of Medicinal Chemistry” 2007,17,41-46, reported a new synthetic route:

The route of process steps is too long, the total yield is low, side effects side. But not conducive to industrial production.
(4) Patent No. CN 101205215A (25 June 2008 publicly) announced a halogen epoxy propane as a starting material for the synthesis route:
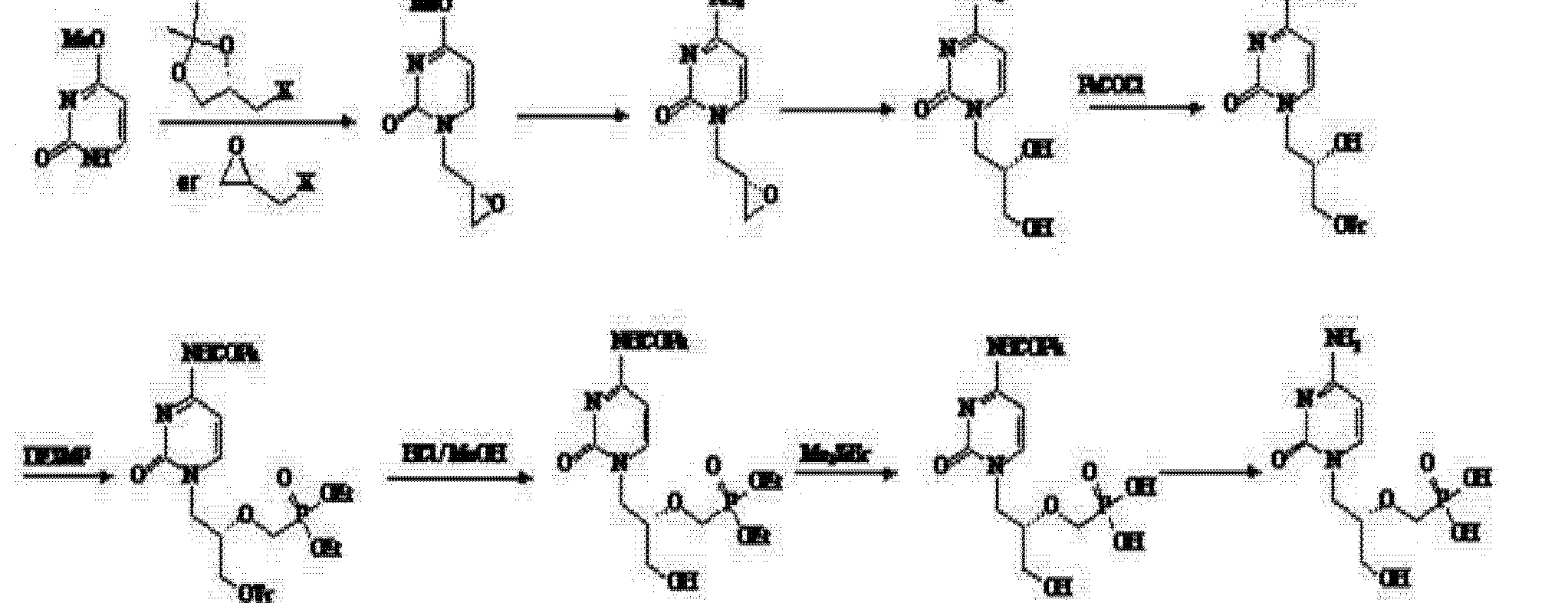
Use of the route (R) – epihalohydrin reaction with cytosine, cytosine ring because alkaline easily cause epoxy ring-opening reaction of the ring, but side reactions, the purified product is not, nor is suitable for industrial production.
Subsequently, the patent number CN 101525352A (2009 年 9 月 9 Publication Date) discloses (4) based on the modified route through epoxypropionate alkane ether in the form of a direct reaction with cytosine, after a series of similar steps obtain the final product cidofovir.
In view of the clinical application of cidofovir more favorable therapeutic effect in, looking for a high yield and because of economic and practical, easy to control, the risk of small synthetic methods and technology is now more urgent needs.
Synthesis

Brodfuehrer, P; Howell, Henry G.; Sapino, Chester; Vemishetti, Purushotham (1994). “A practical synthesis of (S)-HPMPC”. Tetrahedron Letters 35 (20): 3243. doi:10.1016/S0040-4039(00)76875-4.
………………………………………
CN 102268040

, Example 1:
1 Synthesis of 4,4 ‘- dimethoxytrityl methyl – (R) – glycidol (Compound III): The 5 04 g (15 mmoDDMT-Cl grain port 0 20 g (1 52 mmol… ) 4_ dimethylaminopyridine (DMAP) was dissolved in 100 mL CH2C12 cooled to 0 ° C, was added dropwise 10 mL TEA was slowly added 2. 00 g (27mmol) hydroxymethyl chiral oxirane (Compound II ) addition was completed, the reaction warmed to room temperature naturally. fly 4 h, until TLC until the disappearance of the detection DMT-Cl, the reaction was stopped by filtration, the filtrate was washed with saturated NaHC03 solution (50mLX2), saturated NaCl solution (50 mLX2), anhydrous Na2S04 dried, filtered, and concentrated to a viscous colorless directly, i.e., 5 08 g of 4,4 ‘-dimethoxy-triphenylmethyl _ -.. (R) – glycidol (Compound III), yield 90 %, HPLC purity 99%.
2, Synthesis (S)-N1_ [(2 – hydroxy-3 – (dimethoxytrityl) propyl] cytosine (Compound IV):. Under nitrogen to 3 56 g (32 mmol) of cytosine was added 150 mL of anhydrous N, N-dimethylformamide (DMF), and at room temperature, was added portionwise 1. 24 g (31 mmol, molar concentration of 60%) NaH, 0. 5 h after adding 11 92 g (31 mmol) 4,4 ‘-. dimethoxytrityl methyl – (R) – glycidol (Compound III), plus finished warming up to 10 (Tll (TC reaction . 6-8 h and then filtered, and the filtrate evaporated under reduced pressure DMF, the remaining solid phase was added 500 mL of ethyl acetate and 50 mL of water, separated and the organic layer was washed with saturated NaHC03 solution (50 mL X 2), saturated NaCl solution (50 mL X 2), dried over anhydrous Na2S04 filtered and dried, and concentrated to give 13 90 g of a white solid, S Jie (S)-Nl-[(2 -.. hydroxy-3 – (methoxy-dimethoxytrityl ) propyl] cytosine (Compound IV), yield 92%, HPLC purity 98%.
3 Synthesis ⑶-Nl-{[2_ (phosphonic acid methoxy diethoxy) -3 – (methoxy-dimethoxytrityl)] propyl} cytosine (Compound V):
75 ~ 80 ° C under the conditions, 48 76 g (0 100 mol.) (S) _N1_ [(2 – hydroxy-3 – (dimethoxytrityl) propyl]. Cytosine (Compound IV) was added to 150 mL anhydrous DMF, and then inputs 8. 5g (0. 050 mol) tert-butoxide, magnesium reaction 0.5-1 h, tosyloxy added diethyl 32 methylsulfinyl . 2 g (0. 100 mol), the reaction epileptic 8 h, p-toluenesulfonic acid was added to neutralize the excess alkali to neutral distilled DMF, ethyl acetate (300 mLX 3) washing the combined ethyl acetate phase was concentrated to give a solid, i.e., synthetic 58 18 g (S)-Nl-. {[2 – (diethoxy-phosphono-methoxy) -3 – (methoxy-dimethoxytrityl)] propyl} cytosine (Compound V), yield 89%, HPLC purity greater than 95%.
4 Synthesis of (S)-Nl-{[2_ (phosphonic acid methoxy diethoxy) -3 – hydroxy] propyl} cytosine (Compound VI): The 10 g (S)-Nl- {[2 – (phosphono-methoxy ethoxy) -3 – (methoxy-dimethoxytrityl)] propyl}-cell
Pyrimidine (compound V) was dissolved in a concentration of 70 mL of 80% acetic acid solution, 90 ° C reaction. After 5 h, cooled to room temperature, 50 mL of water and 30 mL of dichloromethane, and the organic phase washed with water (30 mL X2) and the combined aqueous phase was concentrated to give crude 9. 5 g, can be performed directly in the next reaction.
can also be separated by flash column chromatography (CH2C12 = MeOH = 10: 1), 4.6 g obtained as a pale yellow oil, i.e. (S)-Nl-{[2 – (methoxy diethoxy phosphono ) -3 – hydroxy] propyl} cytosine (Compound VI), yield 90%.
5 was synthesized ⑶-Nl-{[2_ (diphosphonic acid methoxy) -3 – hydroxy] propyl} cytosine (Compound I):
The 9.5g (S)-Nl-{[2 – (methoxy diethoxy phosphonomethyl) -3 – hydroxy] propyl} cytosine (Compound VI) into a crude product containing 5 76 g (0.. 045 mol) solution of hydrogen iodide, hydroiodic acid, and after reflux for 4-5 h. (50 mLX 2) wash solution was separated with ethyl acetate. The aqueous phase was added sodium hydroxide to adjust pH between 3 Γ3 6, filtered, recrystallized from methanol to give 3.81 g of white crystalline solid, S Jie (S)-Ni-{[2 -.. (Diphosphonic acid methoxy yl) -3 – hydroxy] propyl} cytosine (Compound I), yield 88% (containing two crystal water), HPLC purity greater than 99%.
…………………………………………
POLYMORPHS
Example 7 Amorphous Cidofovir
Intermediate 5 (FIG. 7; 0.5 g, 0.054 mol) was heated with a solution of sodium methoxide in methanol (0.5 M, 15 mL, 7.5 mmol) at 72° C. for 14.5 h then at 90° C. for 5.5 h. The reaction mixture was quenched with water (10 mL) and filtered through a bed of ion exchange resin Dowex® 50WX8 100-200 (H). The filtrate was cycled through the ion exchange bed (2 times) then washed successively with 1:1 methanol:water (40 mL), methanol (40 mL) and 4% triethylamine:methanol (50 mL). This ion-exchange bed was further washed with 48:48:4 methanol:water:triethylamine (100 mL) until no UV absorbance was detected in the filtrate. This reaction produced intermediate 7 (FIG. 7) together with cyclic cidofovir impurity. This mixture was then dissolved in 6 N HCl and heated to 65° C. After cooling the reaction mixture to room temperature, ethyl acetate was charged and stirred and the aqueous layer separated. The aqueous was stirred with ethanol (50 mL). The precipitated material was filtered and the solid was washed with ethanol. The ethanol filtrate was concentrated. The concentrated material was taken up in acetonitrile and stirred with trimethylsilyl bromide (19 mL) at room temperature for 18 h. The reaction mixture was filtered and the filtrate concentrated. The residue was taken up in toluene (30 mL) and ammonium hydroxide (28%, 50 mL) was charged and stirred at room temperature. The organic phase was separated and the aqueous phase was concentrated to dryness. Water (20 mL) and ethanol (15 mL) were added to the residue. The mixture pH was 6 and was adjusted to pH 3 with concentrated HCl (2 mL) then adjusted to pH 4 to 4.5 with 28% NH4OH. After stirring for 0.5 h, the mixture was cooled, filtered and the solids washed with 2:1 EtOH:H2O and dried under vacuum for 18 h. The isolated solid was taken up in water (10 mL) and 28% NH4OH added to give a solution. Concentrated HCl was added to the solution until pH 4 was reached. Ethanol (13 mL) was charged and the mixture stirred at −17° C. for 18 h, filtered and the solids washed with 2:1 EtOH:water (2×8 mL), dried under vacuum at 35° C. The cidofovir isolated in this manner was determined to be in the amorphous form by XRPD.

……………………………..
Journal of the American Chemical Society, 2011 , vol. 133, 7 p. 2264 – 2274
http://pubs.acs.org/doi/abs/10.1021/ja109823e

………………………………………………..
READ ALSO
Synthesis and antiviral activity of the nucleotide analogue (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine
J Med Chem 1989, 32(7): 1457
http://pubs.acs.org/doi/abs/10.1021/jm00127a010
References
- “Vistide (cidofovir) dosing, indications, interactions, adverse effects, and more”.Medscape Reference. WebMD. Retrieved 4 February 2014.
- “Product Information VISTIDE®”. TGA eBusiness Services. Gilead Sciences Pty Ltd. 3 September 2013. Retrieved 5 February 2014.
- Chilukuri, S; Rosen, T (2003 Apr). “Management of acyclovir-resistant herpes simplex virus.”.Dermatologic clinics 21 (2): 311–20. doi:10.1016/S0733-8635(02)00093-1.PMID 12757254.
- Segarra-Newnham M, Vodolo KM (June 2001). “Use of cidofovir in progressive multifocal leukoencephalopathy”. Ann Pharmacother 35 (6): 741–4. doi:10.1345/aph.10338.PMID 11408993.
- De Luca, A; Ammassari, A; Pezzotti, P; Cinque, P; Gasnault, J; Berenguer, J; Di Giambenedetto, S; Cingolani, A; Taoufik, Y; Miralles, P; Marra, CM; Antinori, A; Gesida 9/99, IRINA, ACTG 363 Study, Groups (September 2008). “Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: a multicohort analysis.”. AIDS (London, England) 22 (14): 1759–67.doi:10.1097/QAD.0b013e32830a5043. PMID 18753934.
- Langer-Gould, A; Atlas, SW; Green, AJ; Bollen, AW; Pelletier, D (28 July 2005). “Progressive Multifocal Leukoencephalopathy in a Patient Treated with Natalizumab”. New England Journal of Medicine 353 (4): 375–381. doi:10.1056/NEJMoa051847. PMID 15947078.
- De Clercq E (July 2002). “Cidofovir in the treatment of poxvirus infections”. Antiviral Res. 55(1): 1–13. doi:10.1016/S0166-3542(02)00008-6. PMID 12076747.
- Bradbury, J (March 2002). “Orally available cidofovir derivative active against smallpox.”.Lancet 359 (9311): 1041. doi:10.1016/S0140-6736(02)08115-1. PMID 11937193.
- Magee, WC; Hostetler, KY; Evans, DH (August 2005). “Mechanism of Inhibition of Vaccinia Virus DNA Polymerase by Cidofovir Diphosphate”. Antimicrobial Agents and Chemotherapy49 (8): 3153–3162. doi:10.1128/AAC.49.8.3153-3162.2005. PMC 1196213.PMID 16048917.
- Araya CE, Lew JF, Fennell RS, Neiberger RE, Dharnidharka VR (February 2006).“Intermediate-dose cidofovir without probenecid in the treatment of BK virus allograft nephropathy”. Pediatr Transplant 10 (1): 32–7. doi:10.1111/j.1399-3046.2005.00391.x.PMID 16499584.
- Broekema FI, Dikkers FG (August 2008). “Side-effects of cidofovir in the treatment of recurrent respiratory papillomatosis”. Eur Arch Otorhinolaryngol 265 (8): 871–9. doi:10.1007/s00405-008-0658-0. PMC 2441494. PMID 18458927.
- Soma MA, Albert DM (February 2008). “Cidofovir: to use or not to use?”. Curr Opin Otolaryngol Head Neck Surg 16 (1): 86–90. doi:10.1097/MOO.0b013e3282f43408.PMID 18197029.
- “Cidofovir Monograph for Professionals – Drugs.com”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 5 February 2014.
- “Vistide : EPAR -Product Information” (PDF). European Medicines Agency. Gilead Sciences International Ltd. 7 November 2013. Retrieved 5 February 2014.
- Liekens S, Gijsbers S, Vanstreels E, Daelemans D, De Clercq E, Hatse S (March 2007). “The nucleotide analog cidofovir suppresses basic fibroblast growth factor (FGF2) expression and signaling and induces apoptosis in FGF2-overexpressing endothelial cells”. Mol. Pharmacol.71 (3): 695–703. doi:10.1124/mol.106.026559. PMID 17158200.
- Liekens S (2008). “Regulation of cancer progression by inhibition of angiogenesis and induction of apoptosis”. Verh. K. Acad. Geneeskd. Belg. 70 (3): 175–91. PMID 18669159.
- Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3. edit
- “Vistide (cidofovir)” (package insert). Gilead Sciences. September 2010. DOSAGE AND ADMINISTRATION: Dosage.
- Safrin, S; Cherrington, J; Jaffe, HS (September 1997). “Clinical uses of cidofovir”. Reviews in Medical Virology 7 (3): 145–156. doi:10.1002/(SICI)1099-1654(199709)7:3<145::AID-RMV196>3.0.CO;2-0. PMID 10398479.
- “Press Releases: Gilead”. Retrieved 2007-12-05.
- Synthesis and antiviral activity of the nucleotide analogue (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine
J Med Chem 1989, 32(7): 1457 - Synthesis and antiherpesvirus activity of (S)-1-((3-hydroxy-2-phosphonylmethoxy)propyl)cytosine (HPMPC) and related nucleotide analoguesNucleosides Nucleotides 1989, 8(5-6): 923
- Journal of Pharmaceutical Sciences, 2012 , vol. 101, 9 p. 3249 – 326
- BRODFUEHRER P R ET AL: “A Practical Synthesis of (S)-HPMPC“, 19940101, vol. 35, no. 20, 1 January 1994 (1994-01-01), pages 3243-3246, XP002012084
- http://www.chemdrug.com/databases/8_0_nhnpseknoegbdisx.html CHEMDRUG SYNTHESIS
| 2 | * | BRONSON, JOANNE J. ET AL: “Synthesis and antiviral activity of nucleotide analogs bearing the (S)-(3-hydroxy-2-phosphonylmethoxy)propyl moiety attached to adenine, guanine, and cytosine“, ACS SYMPOSIUM SERIES , 401(NUCLEOTIDE ANALOGUES ANTIVIRAL AGENTS), 88-102 CODEN: ACSMC8; ISSN: 0097-6156, 1989, XP002677024, |
| 3 | * | BRONSON, JOANNE J. ET AL: “Synthesis and antiviral activity of the nucleotide analog (S)-1-[3-hydroxy-2-(phosphonylmethoxy)prop yl]cystosine“, JOURNAL OF MEDICINAL CHEMISTRY , 32(7), 1457-63 CODEN: JMCMAR; ISSN: 0022-2623, 1989, XP002677026, |
| 4 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; JIANG, XING-KAI ET AL: “Practical synthesis of a broad-spectrum antiviral drug cidofovir”, XP002677148, retrieved from STN Database accession no. 2009:1568897 -& JIANG XING-KAI ET AL: “Practical synthesis of a broad-spectrum antiviral drug cidofovir“, JIEFANGJUN YAOXUE XUEBAO – PHARMACEUTICAL JOURNAL OF CHINESE PEOPLE’S LIBERATION ARMY, ZHONGGUO RENMIN JIEFANGJUN, ZONGHOUQINBU, WEISHENGBU, YAOPIN YIQI JIANYANSUO, CN, vol. 25, no. 5, 1 January 2009 (2009-01-01), pages 395-397, XP008152443, ISSN: 1008-9926 |
| 5 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LIU, JIANFENG ET AL: “Improved synthesis of cidofovir”, XP002677149, retrieved from STN Database accession no. 2008:1052727 -& LIU JIANFENG ET AL: “Improved synthesis of cidofovir“, ZHONGGUO YAOWU HUAXUE ZAZHI – CHINESE JOURNAL OF MEDICINAL CHEMISTRY, GAI-KAI BIANJIBU, SHENYANG, CN, vol. 17, no. 1, 1 February 2007 (2007-02-01), pages 41-43, XP008152444, ISSN: 1005-0108 |
| 6 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LU, NING ET AL: “Synthesis of cidofovir”, XP002677151, retrieved from STN Database accession no. 2007:1124844 -& LU NING ET AL: “Synthesis of cidofovir“, ZHONGGUO XIN YAO ZAZHI – CHINESE NEW DRUGS JOURNAL, GAI-KAN BIANJIBU, BEIJING, CN, vol. 16, no. 16, 1 January 2007 (2007-01-01), pages 1272-1274, XP008152436, ISSN: 1003-3734 |
| 7 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; YI, HONG ET AL: “Synthesis of antiviral agent cidofovir”, XP002677150, retrieved from STN Database accession no. 2007:1249882 -& YI HONG ET AL: “Synthesis of antiviral agent cidofovir“, ZHONGGUO KANGSHENGSU ZAZHI/ CHINESE JOURNAL OF ANTIBIOTICS, SICHUAN, CN, vol. 31, no. 7, 1 January 2006 (2006-01-01), pages 412-413, XP008152413, ISSN: 1001-8689 |
| 8 | * | FROMTLING R A ET AL: “Cidofovir. HPMPC. GS-504. GS-0504. Viside“, DRUGS OF THE FUTURE, PROUS SCIENCE, ES, vol. 21, no. 10, 1 January 1996 (1996-01-01), pages 1003-1013, XP008152410, ISSN: 0377-8282 |
| 9 | * | HOLY A: “SYNTHESES OF ENANTIOMERIC N-(3-HYDROXY-2-PHOSPHONOMETHOXYPROPYL) DERIVATIVES OF PURINE AND PYRIMIDINE BASES“, COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, INSTITUTE OF ORGANIC CHEMISTRY & BIOCHEMISTRY, PRAGUE; CZ, vol. 58, no. 3, 1 January 1993 (1993-01-01), pages 649-674, XP009042514, ISSN: 0010-0765, DOI: 10.1135/CCCC19930649 |
| 10 | * | JOANNE J BRONSONA ET AL: “A New Synthesis of the Potent and Selective Anti-Herpesvirus Agent (S)-1-[3-Hydroxy-2-(Phosphonylmethoxy)Prop yl]Cytosine“, NUCLEOSIDES, NUCLEOTIDES AND NUCLEIC ACIDS, TAYLOR & FRANCIS, PHILADELPHIA, PA , vol. 9, no. 6 1 January 1990 (1990-01-01), pages 745-769, XP008152412, ISSN: 1525-7770, DOI: 10.1080/15257779008043142 Retrieved from the Internet: URL:http://www.tandfonline.com/doi/abs/10.1080/15257779008043142 [retrieved on 2006-10-04] |
| 11 | * | PETR ALEXANDER AND ANTONÍN HOL: “General Method of Preparation of N-[(S)-(3-Hydroxy-2-phosphonomethoxypropyl )] Derivatives of Heterocyclic Bases“, COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, INSTITUTE OF ORGANIC CHEMISTRY & BIOCHEMISTRY, PRAGUE; CZ, vol. 58, no. 5, 1 May 1993 (1993-05-01), pages 1151-1163, XP008152404, ISSN: 0010-0765, DOI: 10.1135/CCCC19931151 |
| 12 | * | SNOECK, ROBERT ET AL: “(S)-1-(3-Hydroxy-2-phosphonylmethoxypropy l)cytosine, a potent and selective inhibitor of human cytomegalovirus replication“, ANTIMICROBIAL AGENTS AND CHEMOTHERAPY , 32(12), 1839-44 CODEN: AMACCQ; ISSN: 0066-4804, 1988, XP002677023, |
| 13 | * | SYMERSK AND A HOL J: “Structure of 1-(S)-(3-hydroxy-2-phosphonylmethoxypropyl )cytosine; an antiviral agent“, ACTA CRYSTALLOGRAPHICA SECTION C. CRYSTAL STRUCTURE COMMUNICATIONS, MUNKSGAARD, COPENHAGEN, DK, vol. 47, no. 10, 15 October 1991 (1991-10-15), pages 2104-2107, XP008152403, ISSN: 0108-2701, DOI: 10.1107/S0108270190013257 |
| 14 | * | ULRIKA ERIKSSON: “Synthesis and biological evaluation of novel cidofovir prodrugs targeting HPepT1“, SYNTHESIS AND BIOLOGICAL EVALUATION OF NOVEL CIDOFOVIR PRODRUGS TARGETING HPEPT1- A DISSERTATION PRESENTED TO THE FACULTY OF THE GRADUATE SCHOOL UNIVERSITY OF SOUTHERN CALIFORNIA IN PARTIAL FULFILLMEN , 1 January 2005 (2005-01-01), pages 1-287, XP008152406, Retrieved from the Internet: URL:http://search.proquest.com/pqdtscieng/docview/305421788/fulltextPDF/1371C222DB27F96BE2D/2?accountid=29404 |
| 15 | * | WEBB, ROBERT R., II ET AL: “Synthesis of (S)-N1-(3-hydroxy-2-phosphonylmethoxy)prop ylcytosine, (S)-HPMPC“, TETRAHEDRON LETTERS , 29(43), 5475-8 CODEN: TELEAY; ISSN: 0040-4039, 1988, XP002677025 |
| CN1559429A * | Feb 18, 2004 | Jan 5, 2005 | 肖广常 | 注射用西多福韦冻干粉针剂 |
| CN1690066B * | Apr 19, 2004 | Apr 28, 2010 | 横店集团成都分子实验室有限公 | 抗病毒剂西多福韦新衍生物 |
| CN101525352A * | Feb 16, 2009 | Sep 9, 2009 | 苏州凯达生物医药技术有限公司 | 西多福韦及其中间体的制备方法 |
| CN102268040A * | Sep 5, 2011 | Dec 7, 2011 | 扬州三友合成化工有限公司 | 抗病毒药物西多福韦的一种合成方法 |
| US5142051 | Jul 17, 1987 | Aug 25, 1992 | Ceskoslovenska Akademie Ved | N-phosphonylmethoxyalkyl derivatives of pyrimidine and purine bases and a therapeutical composition therefrom with antiviral activity |
RAMELTEON, TAK 375 ..Melatonin MT1/MT2 receptor agonist

RAMELTEON
- HSDB 7787
- Ramelteon
- Rozerem
- TAK-375
- UNII-901AS54I69
| CAS number | 196597-26-9 |
|---|
| (S)-N-[2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)ethyl]propionamide |
(5)-N-[2-(l,6,7,8-tetrahydro-2H-indeno-[5,4-ό]furan-8- yl)ethyl]propionamide
| United States | US 6034239 | 1999-07-22 | expiry 2019-07-22 |
EP885210B1 , EP1792899A1 and J. Med Chem. 2002, 45, 4222-4239
read all at
http://www.allfordrugs.com/2014/02/23/ramelteon-tak-375-melatonin-mt1mt2-receptor-agonist/
MOXIFLOXACIN, Bay-12-8039
 MOXIFLOXACIN Bay-12-8039 US FDA:link CAS 354812-41-2 186826-86-8 HYDROCHLORIDE
MOXIFLOXACIN Bay-12-8039 US FDA:link CAS 354812-41-2 186826-86-8 HYDROCHLORIDE
| 1-cyclopropyl-7-[(1S,6S)-2,8-diazabicyclo[4.3.0]non-8-yl]-6-fluoro-8-methoxy-4-oxo- quinoline-3-carboxylic acid |

Moxifloxacin is a fourth-generation synthetic fluoroquinolone antibacterial agent developed by Bayer AG (initially called BAY 12-8039). It is marketed worldwide (as the hydrochloride) under the brand names Avelox, Avalox, and Avelon for oral treatment. In most countries, the drug is also available in parenteral form for intravenous infusion. Moxifloxacin is also sold in an ophthalmic solution (eye drops) under the brand names Vigamox, Moxezafor the treatment of conjunctivitis (pink eye). A United States patent application was submitted on 30 June 1989, for Avelox (moxifloxacin hydrochloride).[1] In 1999 Avelox was approved by theU.S. Food and Drug Administration (FDA) for use in the United States.[2] In the United States, moxifloxacin is licensed for the treatment of acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, community acquired pneumonia, complicated and uncomplicated skin and skin structure infections, and complicated intra-abdominal infections.[3] In the European Union, it is licensed for acute bacterial exacerbations of chronic bronchitis, non-severe community-acquired pneumonia, and acute bacterial sinusitis. Based on its investigation into reports of rare but severe cases of liver toxicity and skin reactions, the European Medicines Agency recommended in 2008 that the use of the oral (but not the IV) form of moxifloxacin be restricted to infections in which other antibacterial agents cannot be used or have failed.[4] In the US, the marketing approval does not contain these restrictions, though the label contains prominent warnings against skin reactions. 
- Acute Exacerbations of Chronic Bronchitis (AECB)
- Acute Bacterial Sinusitis (ABS)
- Community Acquired Pneumonia (CAP)
Additional indications were approved by the FDA as follows:
- April 2001: Uncomplicated Skin and Skin Structure Infections (uSSSI)[8]
- May 2004: Community Acquired Pneumonia caused by multi-drug resistant Streptococcus pneumoniae.[9]
- June 2005: Complicated Skin and Skin Structure Infections (cSSSI)[10]
- November 2005: Complicated Intra-Abdominal Infections (cIAI).[11]
The European Union requires that moxifloxacin only be prescribed when other antibiotics that have been initially recommended for treatment cannot be used or have failed.[12][13] At the current time,[when?] there are no approved uses within the pediatric population for Oral and I.V. moxifloxacin. A significant number of drugs found within this class, including moxifloxacin, are not licensed by the FDA for use in children due to the risk of permanent injury to the musculoskeletal system.[14][15][16] In ophthalmology, moxifloxacin is approved for the treatment of conjunctival infections caused by susceptible bacteria.[17] Note: Moxifloxacin may be licensed for other uses, or restricted, by the various regulatory agencies worldwide Marketing authorisations for the tablet and injectable forms of Moxifloxacin are held by Bayer, while Alcon (now a subsidiary of Novartis) produces ophthalmic solutions for treating conjunctivitis under the brand names of Moxeza, Vigamox, and Moxivig. Avelox generated sales of USD320 million in the first 9 months of 2013. Moxifloxacin is available in three distinct administration forms. Formulated as a salt, Moxifloxacin hydrochloride is sold as an oral 400 mg film-coated tablet and as an injectable solution for infusion by Bayer; although in the US it is distributed by Merck Sharp and Dohme under license from Bayer. Alcon has formulated Moxifloxacin hydrochloride as a 0.5% ophthalmic solution under license from Bayer. Moxifloxacin was first discovered in 1988 and received the first market authorisation eleven years later in 1999 in the US. Moxifloxacin hydrochloride is a synthetic broad-spectrum antibacterial agent. The active moiety, moxifloxacin has been shown to be clinically active against most strains of microorganisms such as aerobic gram-positive microorganisms including staphylococcus aureus, streptococcus pneumonia (penicillin-susceptible strains) and streptococcus pyogenes, aerobic gram-negative microorganisms including haemophilus influenza hemophilus parainfluenzae, klebisiella pneumonia. Moxifloxacin is commercially available under the brand name of AVELOX® marketed by Bayer pharms. VIGAMOX® (moxifloxacin hydrochloride ophthalmic solution) 0.5% is a sterile solution for topical ophthalmic use. Moxifloxacin hydrochloride is an 8-methoxy fluoroquinolone anti-infective, with a diazabicyclononyl ring at the C7 position.
 |
C21H24FN304•HC1 Mol Wt 437.9 Chemical Name: l-Cyclopropyl-6-fluoro-l,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolol[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid, monohydrochloride. Moxifloxacin hydrochloride is a slightly yellow to yellow crystalline powder. Each mL of VIGAMOX® solution contains 5.45 mg moxifloxacin hydrochloride, equivalent to 5 mg moxifloxacin base. Contains: Active: Moxifloxacin 0.5% (5 mg/mL); Inactives: Boric acid, sodium chloride, and purified water. May also contain hydrochloric acid/sodium hydroxide to adjust pH to approximately 6.8. VIGAMOX® solution is an isotonic solution with an osmolality of approximately 290 mOsm/kg. syn…………. http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html Market Considerations Amongst the US approvals, Dr. Reddy’s, Teva, Torrent, and Aurobindo have received tentative approvals for the 400 mg oral tablet formulation. Akorn, Teva and Apotex have received tentative approvals for a Moxifloxacin hydrochloride ophthalmic solution. No 180 day period of exclusivity has been awarded since all patents were found to be valid. In the UK, Teva, Rivopharm, and Double-E Pharma have received marketing authorisations for the 400 mg Moxifloxacin tablets, while Noridem has received a market authorisation for the equivalent 400mg/250ml solution for infusion. A similar trend of generic competition, for tablets and infusions, following molecule patent expiry is expected throughout Europe. Currently no generic market authorisations for ophthalmic formulations have been granted in major European countries. However, Sandoz and Hexal have gained market authorisations in some European markets for the ophthalmic dosage form following Novartis’ acquisition of Alcon. In Canada the only generic manufacturer holding a marketing authorisation is Sandoz, however, this was granted as a New Drug Submission rather than as an ANDS. Following patent expiries from mid-2014, the European and North American markets are likely to have significant competition if the numbers of companies filing litigation, ANDS, ANDAs and the like in the northern hemisphere is anything to go by.  MOXIFLOXACIN
MOXIFLOXACIN
History
Moxifloxacin was first patented (United States patent) in 1991 by Bayer A.G., and again in 1997.[47] Avelox was subsequently approved by the U.S. Food and Drug Administration (FDA) for use in the United States in 1999 to treat specific bacterial infections.[2] Ranking 140th within the top 200 prescribed drugs in the United States for 2007[48] moxifloxacin, in the same manner asciprofloxacin, has proven to be a blockbuster drug for Bayer A. G., generating billions of dollars in additional revenue. In 2007 alone, Avelox generated sales of $697.3 million dollars worldwide.[26] Moxifloxacin is also manufactured by Alcon as Vigamox. syn………http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html
Patent
A United States patent application was made on 30 June 1989, for Avelox (moxifloxacin hydrochloride),(Bayer A.G. being the assignee), which was subsequently approved on 5 February 1991. This patent was scheduled to expire on 30 June 2009. However, this patent was extended for an additional two and one half years on 16 September 2004, and as such is not expected to expire until 2012.[49] Moxifloxacin was subsequently (ten years later) approved by the U.S. Food and Drug Administration (FDA) for use in the United States in 1999. There have been at least four additional United States patents filed regarding moxifloxacin hydrochloride since the 1989 United States application,[47][50] as well as patents outside of the USA.
Additional regulatory history
6/12/2002 Changes made to minimize the impact of warnings concerning adverse reactions.[51] 26 June 2003 New Zealand Pharmacovigilance warns of moxifloxacin induced respiratory insufficiency.[52] 10/6/2003 Changes made to minimize the impact of post marketing reports as well as the risk of tendon injuries.[53] 29 December 2008 Addition of numerous adverse reactions associated with the use of moxifloxacin.[54] 27 April 2009 Issuance of a Medication Guide and revisions to include new safety information including the addition of the Black Box Warning to the Medication Guide. The FDA had determined that Moxifloxacin poses a serious and significant public health concern, requiring the distribution of a Medication Guide.[55] 24 June 2009 Updating of the carton and container labels to include a statement to let dispensers know that a Medication Guide must be dispensed with the product.(emphasis added)[56] Patent related
|
As indicated by the Key Patent Indicator (Fig. 2), patents in the families with priority DE3824072A, 15/07/1988 (‘072), and DE4200414A, 10/01/1992, (‘414) provide protection for the Moxifloxacin molecule and are considered to be the main constraint to generic entry. As patents in the ‘414 family have expired or were never granted, the only remaining constraint to generic entry is the ‘072 family. The term of the Australian patent of this family have been extended to 19 June 2014 while the Canadian member will enjoy the longer term of 17 years from grant, expiring in November 2015. Supplementary protection certificates (SPCs) have been granted in France, Germany, Spain and the UK, and will expire in June 2014. Given that there are less than 2 years until these SPCs expire, and that as yet no applications for paediatric extension of the SPCs have been published in Europe, they are unlikely to be extended by 6 months on the basis of the approved Paediatric Investigation Plans. The US member, 4,990,517 (‘517), protecting the general structure of the Moxifloxacin molecule, expired in June 2012, after enjoying 6 month paediatric extension on top of a 901 day s156 extension. However, the absence of generics on the US market is due to Bayer securing a divisional patent, 5,607,942 (‘942). This patent claims the Moxifloxacin molecule specifically and is due to expire in September 2014, after being awarded a 6 month paediatric extension. Members of the ‘072 family from both Canada and the US have been the subject of litigation after generic manufacturers identified these patents in paragraph IV filings, and the equivalent in Canada, as early as 2006. After filing an infringement suit in the US against Teva in relation to US ‘517, US ‘942, Bayer enjoyed a satisfying validification of their patents when Teva agreed that it would be infringing two of the patents, while the third was decided by the court to be equally valid. In Canada, Novopharm, Cobalt, Apotex, Mylan and Apotex have also all tested the litigation waters relating to the equivalent patent with no success noted so far. A third patent family that promises to be a constraint for generic ophthalmic formulations is Alcon’s 1998 patent, US10250498P (Fig. 2), which identifies an ophthalmic formulation of Moxifloxacin and its use in the treatment and prevention of eye infections. Patents in this family are set to expire in August 2019. US members 6,716,830 (‘830) and 7,671,070 (‘070) have been awarded a 6 month paediatric extension, extending their expiration until March 2020. The validity of US ‘830 was upheld following Teva filing paragraph IV certifications to manufacture generic Vigamox. Teva has since appealed this ruling. In addition, Alcon has filed patent infringement suits against Watson, Lupin and Apotex in relation to US ‘070 after they submitted Abbreviated New Drug Applications (ANDAs)with paragraph IV filings in preparation for commercialisation of a Moxeza/Vigamox generic equivalent. There has been no outcome from these suits to date. In addition, applications for Orders of Prohibition against Cobalt, Apotex and Teva have also been noted for the equivalent Canadian patent following the filing of Abbreviated New Drug Submissions (ANDS) by these companies. The equivalent European patent 1,117,401 was revoked following opposition by Teva filed in the European patent office. Its divisional patents, 1,384,478 (granted) and 2,301,541 (accepted) have restricted claims to the use of Moxifloxacin in the topical treatment of ophthalmic infections caused by P. aeruginosa and H. influenza, respectively. This may provide a prepared generic competitor an opportunity to launch their Moxifloxacin ophthalmic equivalent in Europe soon after the expiry of patents protecting the molecule, subject to legal review of the remaining claims of the patent. Alcon have secured additional protection for their Moxeza ophthalmic formulation by way of patents in the family with priority US5987708P (09/06/2008). Patent claims specify ratios of Moxifloxacin to inactive ingredients and additional inactive ingredients and therefore generic competitors are likely to circumvent the patent by reformulation. Lupin has filed paragraph IV certifications to US8450311, which is currently subject of a patent infringement suit. Families with priorities DE19546249A (12/12/1995), DE19751948A (24/11/1997), DE19855758A (10/11/1998) and US36433499A (30/07/1999) are not considered to be a constraint to generic entry because the protected technologies are likely to be circumvented. Generic equivalents In 2007, the U.S. District Court for the District of Delaware held that two Bayer patents on Avelox (moxifloxacin hydrochloride) are valid and enforceable, and infringed by Dr. Reddy’s ANDA for a generic version of Avelox.[70][71] The district court sided with Bayer, citing the Federal Circuit’s prior decision in Takeda v. Alphapharm[72] as “affirming the district court’s finding that defendant failed to prove a prima facie case of obviousness where the prior art disclosed a broad selection of compounds, any one of which could have been selected as a lead compound for further investigation, and defendant did not prove that the prior art would have led to the selection of the particular compound singled out by defendant.” According to Bayer’s press release[70] announcing the court’s decision, it was noted that Teva had also challenged the validity of the same Bayer patents at issue in the Dr. Reddy’s case. Within Bayer’s first quarter 2008 stockholder’s newsletter[73] Bayer stated that they had reached an agreement with Teva Pharmaceuticals USA, Inc., the adverse party, to settle their patent litigation with regard to the two Bayer patents. Under the settlement terms agreed upon, Teva would obtain a license to sell its generic moxifloxacin tablet product in the U.S. shortly before the second of the two Bayer patents expires in March 2014.
- Economic impact: adverse reactions:
The advocacy group Public Citizen has lobbied for increasing safety warnings and for the removal of some fluoroquinolone drugs from clinical practice.[74][75][76][77][78][79][80][81]
|
3-5-1997
|
7-(1-pyrrolidinyl)-3-quinolone- and – naphthyridone-carboxylic acid derivatives as antibacterial agents and feed additives
|
|
6-9-2000
|
INHIBITORS OF MULTIDRUG TRANSPORTERS INHIBITORS OF MULTIDRUG TRANSPORTERS
|
|
|
5-19-2000
|
PHARMACEUTICAL MOXIFLOXACIN PREPARATION
|
|
|
5-12-2000
|
AQUEOUS DRUG FORMULATION FOR ORAL APPLICATION AQUEOUS DRUG FORMULATION FOR ORAL APPLICATION
|
|
|
4-7-2000
|
ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE
|
|
|
4-7-2000
|
ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE
|
|
|
4-7-2000
|
ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE
|
|
|
1-20-2000
|
COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS
|
|
|
12-17-1999
|
NOVEL THERAPEUTIC AGENTS THAT MODULATE ENZYMATIC PROCESSES
|
|
|
4-2-1999
|
MEDICAMENT FORMULATION WITH A CONTROLLED RELEASE OF AN ACTIVE AGENT
|
|
|
8-28-1998
|
COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS
|
|
11-10-2006
|
Amorphous moxifloxacin hydrochloride
|
|
|
10-4-2006
|
Method for producing 8-methoxy-quinolinecarboxylic acids
|
|
|
7-15-2005
|
Pharmaceutical composition
|
|
|
7-13-2005
|
Aqueous pharmaceutical composition containing moxifloxacin or salts thereof
|
|
|
5-25-2005
|
Method for producing 8-methoxy-quinolinecarboxylic acids
|
|
|
2-6-2004
|
Medicinal composition
|
|
|
5-21-2003
|
Method for the enantiomer separation of cis-8-benzyl-7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane
|
|
|
11-31-2000
|
OPTICALLY ACTIVE QUINOLINE CARBOXYLIC ACID DERIVATIVES HAVING 7-PYRROLIDINE SUBSTITUTES CAUSING OPTICAL ACTIVITY AND A PROCESS FOR PREPARING THEREOF
|
|
|
11-17-2000
|
ANTIBACTERIAL OPTICALLY PURE BENZOQUINOLIZINE CARBOXYLIC ACIDS, PROCESSES, COMPOSITIONS AND METHODS OF TREATMENT (S)-BENZOQUINOLIZINE CARBOXYLIC ACIDS AND THEIR USE AS ANTIBACTERIAL AGENTS
|
|
|
8-32-2000
|
COMPOSITIONS AND METHODS FOR IMPROVED DELIVERY OF HYDROPHOBIC THERAPEUTIC AGENTS
|
|
6-18-2010
|
MULTI-ARM POLYMER PRODRUGS
|
|
|
4-9-2010
|
COMPOSITION COMPRISING AN ANTIBIOTIC AND A CORTICOSTEROID
|
|
|
7-22-2009
|
Tri-, tetra-substituted-3-aminopyrrolidine derivative
|
|
|
7-3-2009
|
NOVEL HYDRATE FORM
|
|
|
3-20-2009
|
Multi-Arm Polymer Prodrugs
|
|
|
1-16-2009
|
Sulfonamide Derivatives for the Treatment of Bacterial Infections
|
|
|
11-21-2008
|
Phosphonated Fluoroquinolones, Antibacterial Analogs Thereof, and Methods for the Prevention and Treatment of Bone and Joint Infections
|
|
|
8-15-2008
|
Multi-arm polymer prodrugs
|
|
|
8-24-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-29-2007
|
PHARMACEUTICAL COMPOSITION
|
|
7-4-2012
|
TRI-, TETRA-SUBSTITUTED-3-AMINOPYRROLIDINE DERIVATIVE
|
|
|
6-13-2012
|
Process for the Synthesis of Moxifloxacin Hydrochloride
|
|
|
12-2-2011
|
COMPACTED MOXIFLOXACIN
|
|
|
10-12-2011
|
Treatment of bacterial diseases of the respiratory organs
|
|
|
9-16-2011
|
Novel Hydrate Form
|
|
|
9-2-2011
|
NOVEL POLYMORPH OF MOXIFLOXACIN HYDROCHLORIDE
|
|
|
6-31-2011
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-10-2011
|
SYNTHESIS OF (4aS,7aS)-OCTAHYDRO-1H-PYRROLO[3,4-b]PYRIDINE
|
|
|
7-30-2010
|
MULTI-ARM POLYMER PRODRUGS
|
|
|
6-30-2010
|
Multi-arm polymer prodrugs
|
DESCRIPTION
-
Moxifloxacin is a therapeutic agent that shows a broad spectrum antibacterial action. Moxifloxacin is the international non-proprietary name (INN) for 1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid.
-
The structure of moxifloxacin corresponds to formula (I):

-
Racemic moxifloxacin was firstly described in EP-A-350733 and, particularly, moxifloxacin having a (S,S)-configuration is described inEP-A-550903 .
-
In experimental example 19 of EP-A-550903 and example Z19 of EP-A-591808 , a method for preparing and isolating moxifloxacin base is described. The same method is described in patent document EP-A-592868 ). These published patent applications neither describe nor suggest the possible existence of a crystalline form of moxifloxacin base. In WO-A-2008059521 it is disclosed that by performing the mentioned examples an acetonitrile solvated form of moxifloxacin with low purity is obtained, and so the obtained solvate form can not be used as such in pharmaceutical formulations. In the above mentioned examples, the obtained moxifloxacin base crude is purified and isolated by chromatography using methylene chloride/methanol/17% aqueous ammonia as the solvent system. The purification process disclosed in said documents has been reproduced by the authors of the present invention, but only an amorphous form of moxifloxacin was obtained. This process for the purification and isolation of moxifloxacin base is complex and difficult to perform on industrial scale due to the need for purifying the product by column chromatography.
-
WO-A-9926940 discloses a process for the preparation of moxifloxacin from a difluoro precursor comprising the step of adjusting the pH to 6.8- 7.0. However, the reproduction of this example shows that at this pH moxifloxacin hydrochloride or a mixture of moxifloxacin hydrochloride and moxifloxacin base is obtained, since the X-ray diffractogram of the isolated compound corresponds with the hydrochloride moxifloxacin. This fact has also been confirmed by the reproduction of the subsequent crystallization described in the international patent application of the previous compound in ethanol/water. The behaviour of this solid was very different regarding its solubility in respect what was described in the international patent application.
-
WO-A-2004091619 and WO-A-2007010555 disclose the preparation of moxifloxacin base as a solid. Nevertheless, they do not describe nor suggest the preparation of a crystalline form of moxifloxacin base. The authors of the present invention have proved that the X-ray diffractogram of the product obtained by reproducing the reference example disclosed in WO-A-2004091619 , based on a pH adjustment to 7.0-7.2, corresponds to the X-ray diffractogram of the monohydrate of moxifloxacin hydrochloride disclosed in US 5849752 . Similarly, by reproducing the reference example of WO-A-2007010555 , also for obtaining moxifloxacin base but based on a pH adjustment to 5.0-6.0, it is neither expected to obtain moxifloxacin base due to the adjustment of the solution within a range of acidic pH values.
-
[0008]WO-A-2008059521 discloses a process for the preparation of a crystalline form of moxifloxacin base, designated as Form I, by recrystallization in a ketosolvent, such as acetone. This form is characterized by its X-ray diffraction pattern corresponding to a hemihydrate. The best yield obtained is a 78.2% starting from moxifloxacin hydrochloride in example 18. This crystalline form has a tendency to occlude solvent molecules within the crystalline network in amounts very superiors to the allowed ones by for Guidelines Residual Solvents (CPMP/ICH/283/95) and can be difficult to impossible to remove by drying, what forces to carry out laborious treatments, either physical, or chemical to reach allowed solvent levels. The presence of any non-aqueous solvents in amounts over the allowed ones would not make this crystalline form suitable for the preparation of pharmaceutical formulations.
-
The existence of polymorphs is unpredictable and there is no a priori established procedure to prepare an unknown polymorph. The difference in the physical properties of different morphological forms results from the orientation and intermolecular interactions of adjacent molecules are complexes in the bulk solid. Furthermore, the different solid forms of a pharmaceutically active ingredient can have different characteristics, and offer certain advantages in methods of manufacture and also in pharmacology. Thus, the discovery of new solid forms can contribute to clear improvements in the efficiency of methods of production and/or improvements in the characteristics of the pharmaceutical formulations of the active ingredients, since some forms are more adequate for one type of formulation, and other forms for other different formulations.

Moxifloxacin Hydrochloride namely (4aS-Cis) -l-cyclopropyl-7- (2, 8- diazabicyclo [4.3.0] non-8-yl) -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylic acidhydrochloride has the formula

Moxifloxacin Hydrochloride Moxifloxacin is a fluoroquinolone broad spectrum antibacterial particularly against Gram-positive bacteria significantly better than those of Sparfloxacin and Ciprofloxacin that was disclosed in EP No 350,733 and EP No 550,903. Moxifloxacin has activity against Gram- negative and Gram-positive organisms, including Streptococcus pneumonia, Staphylococcus aureus, Pseudomonas aeruginosa, particularly against the respiratory disease-causing pathogens like Mycoplasma pneumonia, Mycobacterium tuberculosis, Chlamydia pneumoniae and the activity shown to be unaffected by B-lactamases . US Patent No 5,157,117 discloses (l-cyclopropyl-6, 7-difluoro-8-methoxy- 4-oxo-l, 4-dihydro-3-quinoline carboxylic acid-O3, 04)bis (acyloxy-O) borate and process for its preparation by reacting ethyl-1- cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, -dihydro-3-quinoline carboxylate with Boric acid and acetic anhydride in presence of zinc chloride and its conversion to Gatifloxacin hydrochloride. WO 2005/012285 discloses the process for the preparation of moxifloxacin hydrochloride using a novel intermediate namely (4aS-Cis)-(1-cyclopropyl-7-(2,8-diazabicyclo[4,3,0]non-8-yl)-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-3-quinoline carboxylic acid-O3,O4)bis(acycloxy-O)borate. Hydrates of Moxifloxacin hydrochloride known are the anhydrous and monohydrate. US Patent No. 5,849,752 discloses the monohydrate of Moxifloxacin hydrochloride and its preparation by treating the anhydrous crystalline form with ethanol/ water mixtures. The prior art disclosed in European Patent No’s EP 350,733, EP 550,903 and EP 657,448 discloses the preparation of Moxifloxacin hydrochloride involving the condensation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid or its esters with (S,S) 2,8-Diaza bicyclo [4.3.0] nonane in presence of a base and its conversion to hydrochloride at higher temperatures leading to the desired Moxifloxacin along with its positional isomer namely (4aS-Cis)-l- cyclopropyl-6- (2, 8-diazabicyclo [4.3.0] non-8-yl) -7-fluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid as a major impurity. As the impurity and the Moxifloxacin are positional isomers they are difficult to separate. Purification of Moxifloxacin to remove this isomer results in lower yields thereby increasing the product cost. Similarly methods described in the prior art involves the preparation of Moxifloxacin and then its conversion to its hydrochloride thereby incorporating an additional step in the manufacturing process also leading to lowering of yields. Moxifloxacin and its pharmacologically acceptable salts are disclosed in European patents EP 350733, EP 550903 and EP 657,448. The disclosed process for the preparation of moxifloxacin hydrochloride comprises of condensing l-cyclopropyl-6,7- difluoro-8-methoxy-4-oxo-l,4-dihydro-3-quinoline carboxylic acid or its esters with (S,S)2,8-diazobicyclo[4.3.0]nonane, in presence of a base at high temperature followed by conversion into hydrochloride salt . This process not only produces desired moxifloxacin hydrochloride but also its positional isomer namely l-cyclopropyl-7-fluoro- 1,4- dihydro -8- methoxy -6- (4aS,7aS)- octahydro- 6H -pyrrolo [3,4-b] pyridine-6-yl] -4- oxo-quinolinecarboxylic acid as a major impurity which is difficult to separate. The purification of moxifloaxcin to remove this isomer results in lower yields thereby increasing the product cost. The International publication WO 2005/012285 discloses an improved process for the preparation of moxifloxacin hydrochloride incorporated herein by reference. The disclosed process involves the preparation of moxifloxacin hydrochloride from the ethyl 1 -cyclopropyl-6,7-difluoro-8-methoxy-4-oxo- 1 ,4-dihydro-3-quinolme carboxylate through a novel intermediate (4aS-cis)-l-cyclopropyl-7-(2,8-diazabicyclo[4.3.0]non-8- yl)-6-fluoro-8-methoxy-4-oxo- 1 ,4-dihydro-3 -quinolinecarboxylicacid-03,04)bis(acyloxy -0)-borate. US patent application 6897315 discloses a process for the preparation of 8-methoxy-3-quinoline carboxylic acid especially moxifloxacin incorporated herein by reference. The disclosed process involves the preparation of moxifloxacin from 8-halo moxifloxacin derivative using methanol and potassium tertiary butoxide. US patent 5639886 discloses one-pot process for the preparation of 3-quinoline carboxylic acid derivatives including moxifloxacin. WO 2004 091619 claims anhydrous crystalline form-Ill of moxifloxacin hydrochloride and WO 2004/039804 claims amorphous form of moxifloxacin hydrochloride. US Pat.No.5, 849,752 discloses specific crystalline forms of anhydrous moxifloxacin mono hydrochloride and monohydrated moxifloxacin mono hydrochloride. Anhydrous moxifloxacin mono hydrochloride disclosed in US Pat. No.5, 849,752 has been designated as “Form-I” and the hydrated form as “Form-II” in US Pat. No.7,230,006. It also discloses a novel crystalline Form-Ill of anhydrous moxifloxacin mono hydrochloride. US patent US 5,480,879 discloses the melting range of moxifloxacin in example part as 203-208°C and does not speaks about polymorphism of moxifloxacin. Experiment executed as per the procedure given in example Zl 9 of US 5,480,879 and resulted in acetonitrile solvated form of moxifloxacin with low purity and the obtained solvated form can not used for formulations U.S. Pat. No. 5,849, 752 (“the ‘752 patent”), incorporated by reference, described two crystalline forms of moxifloxacin hydrochloride namely, anhydrous moxifloxacin hydrochloride and monohydrated moxifloxacin hydrochloride. For convenience, the anhydrous crystalline form described in the 752 patent is designated as “Form I”, and the hydrated form as “Form II”. According to U.S. Pat. No. ‘752’, moxifloxacin hydrochloride monohydrate Form II was obtained by stirring a suspension of the anhydrous moxifloxacin hydrochloride in aqueous media until hydration. Moxifloxacin hydrochloride monohydrate of ‘752’ was also prepared by crystallizing moxifloxacin hydrochloride from a media having a water content which is stoichiometrically sufficient but limited to 10%. WO patent application publication No. 04/091619 disclosed anhydrous Form III of moxifloxacin hydrochloride. WO patent application publication No. 04/039804 disclosed amorphous form of moxifloxacin hydrochloride. WO 2005/054240 disclosed two novel crystalline forms which were designated as Form A and Form B of moxifloxacin hydrochloride. WO patent application publication No. 07/010555 disclosed two crystalline forms which were Form X and Form Y of moxifloxacin hydrochloride. According to WO Publication No. 2007/010555, Form Y was obtained by crystallization of moxifloxacin hydrochloride from the mixture of methanol and water in the ratio of about 8:1 by volume. WO patent application publication No. 07/148137 disclosed hydrate form of moxifloxacin hydrochloride. According to WO Publication No. 2007/148137, moxifloxacin hydrochloride monohydrate was obtained by crystallization moxifloxacin hydrochloride by humidification of moxifloxacin hydrochloride at 50-90% relative humidity at 25-60° C. for 8 to 24 hours. WO patent application publication No. 08/028959 disclosed crystalline form of moxifloxacin hydrochloride. According to WO Publication No. 2008/028959, moxifloxacin hydrochloride was obtained by dissolving moxifloxacin hydrochloride in a mixture of methanol and water and adding acetone and recovering moxifloxacin hydrochloride crystalline form. WO patent application publication No. 08/059521 disclosed process for the preparation of anhydrous crystalline form I of moxifloxacin hydrochloride. WO patent application publication No. 08/095964 disclosed crystalline form of moxifloxacin base.  …………………… SYNTHESIS Drugs Fut 1997,22(2),109 36th Intersci Conf Antimicrob Agents Chemother (Sept 15-18, New Orleans) 1996,Abst. F1.
…………………… SYNTHESIS Drugs Fut 1997,22(2),109 36th Intersci Conf Antimicrob Agents Chemother (Sept 15-18, New Orleans) 1996,Abst. F1.  The anhydrization of pyridine-2,3-dicarboxylic acid (I) with acetic anhydride gives the corresponding anhydride (II), which by treatment with benzylamine (III) is converted into the benzylimide (IV). The hydrogenation of (IV) with H2 over Pd/C yields 8-benzyl-2,8-diazabicyclo[4.3.0]nonane-7,9-dione (V), which is further hydrogenated with LiAlH4, affording (?-cis-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VI) (1). The optical resolution of (VI) by separation of the cis-(R,R)-isomer as crystalline L-(+)-tartrate and further purification of the cis-(S,S)-isomer (VII) as the D-(-)-tartrate affords enantiomerically pure (S,S)-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VII). The debenzylation of (VII) by hydrogenolysis with H2 over Pd/C gives (S,S)-2,8-diazabicyclo[4.3.0]nonane (VIII), which is condensed with 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) in basic medium and finally salified with HCl. The 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) has been obtained as follows: The reaction of 2,4,5-trifluoro-3-methoxybenzoyl chloride (X) with malonic acid monoethyl ester monopotassium salt (XI) by means of triethylamine gives 2-(2,4,5-trifluoro-3-methoxybenzoyl)acetic acid ethyl ester (XII), which is condensed with triethyl orthoformate yielding the corresponding ethoxymethylene derivative (XIII). The reaction of (XIII) with cyclopropylamine affords the cyclopropylaminomethylene derivative (XIV), which is finally cyclized to (IX) by means of NaF in DMF. ……………………….. J Label Compd Radiopharm 2000,43(8),795
The anhydrization of pyridine-2,3-dicarboxylic acid (I) with acetic anhydride gives the corresponding anhydride (II), which by treatment with benzylamine (III) is converted into the benzylimide (IV). The hydrogenation of (IV) with H2 over Pd/C yields 8-benzyl-2,8-diazabicyclo[4.3.0]nonane-7,9-dione (V), which is further hydrogenated with LiAlH4, affording (?-cis-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VI) (1). The optical resolution of (VI) by separation of the cis-(R,R)-isomer as crystalline L-(+)-tartrate and further purification of the cis-(S,S)-isomer (VII) as the D-(-)-tartrate affords enantiomerically pure (S,S)-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VII). The debenzylation of (VII) by hydrogenolysis with H2 over Pd/C gives (S,S)-2,8-diazabicyclo[4.3.0]nonane (VIII), which is condensed with 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) in basic medium and finally salified with HCl. The 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) has been obtained as follows: The reaction of 2,4,5-trifluoro-3-methoxybenzoyl chloride (X) with malonic acid monoethyl ester monopotassium salt (XI) by means of triethylamine gives 2-(2,4,5-trifluoro-3-methoxybenzoyl)acetic acid ethyl ester (XII), which is condensed with triethyl orthoformate yielding the corresponding ethoxymethylene derivative (XIII). The reaction of (XIII) with cyclopropylamine affords the cyclopropylaminomethylene derivative (XIV), which is finally cyclized to (IX) by means of NaF in DMF. ……………………….. J Label Compd Radiopharm 2000,43(8),795  The condensation of 2,4,5-trifluoro-3-methoxybenzoyl chloride (I) with 14C-labeled diethyl malonate (II) by means of MgCl2 and TEA gives the benzoylmalonate (III), which is monodecarboxylated with TsOH in refluxing water, yielding the benzoylacetate (IV). The reaction of (IV) with triethyl orthoformate and Ac2O at 140 C affords the benzoylacrylate (V), which is treated with cyclopropylamine (VI) in cyclohexane to provide ethyl 3-(cyclopropylamino)-2-(2,4,5-trifluoro-3-methoxybenzoyl)acrylate (VII). The cyclization of (VII) by means of K2CO3 in hot N-methylpyrrolidone gives the quinolone carboxylate (VIII), which is hydrolyzed with NaOH in hot methanol, affording the carboxylic acid (IX). Finally, this compound is condensed with (S,S)-2,8-diazabicyclo[4.3.0]octane (X) by means of 1,4-diazabicyclo[2.2.2]octane (DABCO) in refluxing acetonitrile. …………………….
The condensation of 2,4,5-trifluoro-3-methoxybenzoyl chloride (I) with 14C-labeled diethyl malonate (II) by means of MgCl2 and TEA gives the benzoylmalonate (III), which is monodecarboxylated with TsOH in refluxing water, yielding the benzoylacetate (IV). The reaction of (IV) with triethyl orthoformate and Ac2O at 140 C affords the benzoylacrylate (V), which is treated with cyclopropylamine (VI) in cyclohexane to provide ethyl 3-(cyclopropylamino)-2-(2,4,5-trifluoro-3-methoxybenzoyl)acrylate (VII). The cyclization of (VII) by means of K2CO3 in hot N-methylpyrrolidone gives the quinolone carboxylate (VIII), which is hydrolyzed with NaOH in hot methanol, affording the carboxylic acid (IX). Finally, this compound is condensed with (S,S)-2,8-diazabicyclo[4.3.0]octane (X) by means of 1,4-diazabicyclo[2.2.2]octane (DABCO) in refluxing acetonitrile. ……………………. 
 …………………… EP1651630A1 The reaction scheme is given below: Stage-I Acetic anhydride
…………………… EP1651630A1 The reaction scheme is given below: Stage-I Acetic anhydride


Ethyl-l-cyclopropyl-6, 7- ( l-cyclopropyl-6, 7-difluoro-l, 4- difluoro-1, 4-dihydro-8- dihydro-8- methoxy-4-oxo-3- methoxy-4-oxo-3-quinoline quinoline carboxylic acid-03, 04) carboxylate . Bis ( acetate-O) -borate (Borate complex) Stage-II Triet yl amine Acetonitrile


l-cyclo propyl-6, 7-difluoro- [S, S] -2, 8-diazabicyclo- (1- cyclo propyl-6, fluoro-7 (2, 8- 1, 4-dihydro-8- methoxy-4-oxo [4 ,3.0]nonane Diazabicyclo-nonane) 1,4- -3-quinoline carboxylic acid- dihydro-8-methoxy-4-oxo-3 03,04)Bis ( acetate-O) -borate- quinoline carboxylic acid- (Borate complex) (03,04) bis (acetate-O) -borate itage-III

(1- cyclo propyl-6, fluoro- (2, 8- Moxifloxacin HCI pseudohydrate Diazabicyclo-nonane) 1,4- dihydro-8-methoxy-4-oxo-3 -quinolinecarboxylicacid- (O^O4) bis (acetate-O) -borate Stage-TV

Moxifloxacin HCI pseudohydrate Moxifloxacin HCI monohydrate EXAMPLE – I Stage-1: Preparation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo- l,4-dihydro-3-quinoline carboxylic acid-O3,O*)bis (acyloxy-O)borate Acetic anhydride (175 g) is heated to 70°C and boric acid (30 g) is slowly added lot wise in a temperature range of 70°C to 90°C. The temperature is then raised, maintained under reflux for 1 hr followed by cooling to about 70°C. Ethyl-l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylate (100 g) is added under stirring. The temperature is then raised and maintained for 1 hr in the range of 100°C to 105°C. The reaction mass is cooled to 0°C, chilled water (400 ml) is added slowly followed by cold water (600 ml) at temperature 0°C to 5°C and maintained for 2 hrs at 0°C to 5°C. The product which is a boron acetate complex is filtered, washed with water (500 ml) and dried at 55°C to 60°C under vacuum to constant weight. The dry wt is 130.0 g corresponding to yield of 95.2%. Stage-2: Preparation of (4aS-Cis) -l-Cyclopropyl-7- (2, 8-diazabicyclo [4.3.0]non-8-yl) -6-fluoro-8-methoxy-4-oxo-l , 4-dihydro-3-quinoline carboxylicacid-03,0*)bis (acyloxy-O)borate The boron acetate complex (130 g) prepared in stage 1 is suspended in acetonitrile (650 ml), and [S, S] -2, 8-diazabicyclo [4.3.0] nonane (47 g) and triethyl amine (72.9 g) are added. The temperature is raised to reflux and maintained for 1 hr. at reflux, followed by cooling to about 40°C. The solvent is removed under vacuum at temperature below 40°C, and n-hexane (200 ml) is added. After maintaining the reaction mass for 1 hr at room temperature the product is isolated by filtration followed by washing of the wet cake with n-hexane . The product is dried at about 45°C to about 50°C to constant weight. Dry wt of the novel intermediate is 117.0 g corresponding to yield of 71.5%. Elemental analysis: C: 56.42%, H: 5.62%, N: 7.76% and the calculated values for the intermediate, formula C25H29BFN308C: 56.6%, H: 5.47%, N: 7.92% IR Spectrum (KBr, cm-1) : 3415, 3332, 2936, 1718, 1630, 1573, 1526, 1445, 1273, 1042, 935, 860, 798, 682 ^ NMR (200 MHz, CDC13, ppm) : 9.00 (1H), 7.82 (1H), 4.12 (4H), 3.57 (3H), 3.43 (4H), 3.07 (2H) , 2.75 (2H), 2.4 (1H),’ 2.1 (6H), 1.84 (2H) , 1.6 (1H), 1.31 (2H) Mass Spectrum (MJ : 530.3 [M+H] , 470.2 [M+ – CH3COOH] , 428.2 [M+– (CH3CO)20, 100%], 402.2, 388.2 Stage -3: Preparation of Moxifloxacin Hydrochloride pseudohydrate The intermediate (117 g) prepared stage-2 is dissolved in ethanol (600 ml) by stirring for about 30 min. at room temperature and the insolubles if any are filtered off. pH of the filtrate is adjusted to about 0.5 by addition of hydrochloric acid at room temperature and maintained for 2 hrs. The reaction mass is cooled, and maintained for two hrs, at about 0°C to about 5°C. The product is filtered, washed with chilled ethanol (50 ml) and dried at about 50°C to about 55°C till constant weight. The dry weight of the Moxifloxacin hydrochloride pseudohydrate is 87.5g corresponding to yield of 91.0%. Water content of the product by KF is 0.64% w/w. X-ray diffraction pattern data are given in Table-1 EXAMPLE – II Stage- 2 : Preparation of Moxifloxacin pseudohydrate with out isolating (4aS-Cis) -l-Cyclopropyl-7- (2 , 8-diazabicyclo [4.3.0] on-8-yl) -6-fluoro- 8-methoxy-4-oxo-l,4-dihydro-3-quinolinecarboxylicacid-03,04)bis (acyloxy-O) borate The boron acetate complex (130 g) prepared in stage-1 of Example-1 is suspended in acetonitrile (650ml) and [S, S] -2, 8-Diazabicyclo [4.3.0]nonane (47 g) & triethyl amine (72.9 g) are added. Temperature of the reaction mass is raised to reflux, maintained for 1 hr. at reflux and cooled to room temperature. Methanol (600 ml) is added and maintained for 30 min at room temperature to obtain a clear solution. The solution is filtered to remove insolubles if any and pH of the filtrate is adjusted to about 0.5 with hydrochloric acid (57.5 g) . The reaction mass is maintained for 2 hrs at temperature in the range of about 20°C to about 25°C, cooled to 0°C followed by maintaining the reaction mass at about 0°C to about 5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at about 50°C to 55°C until constant weight. Dry wt of the Moxifloxacin hydrochloride pseudohydrate is 88g corresponding to yield of 68.7%. EXAMPLE – III : Preparation of Moxifloxacin Hydrochloride monohydrate Moxifloxacin hydrochloride (50 g) prepared as above is suspended in a mixture of ethanol (250 ml) and hydrochloric acid (25 ml) . Raised the temperature, maintained for two hrs at 40°C to 45°C followed by cooling to about 25°C. The product is filtered and dried under vacuum at 50-55°C until become constant weight. Dry wt of Moxifloxacin hydrochloride monohydrate is 46 g corresponding to yield of 90.5%. The IR spectral data and XRD pattern are identical with available Moxifloxacin hydrochloride monohydrate.


Formula-2a Formula-2b

Formula-3a Formula-3b Formula-3c Formula-3d

Formula-4b

Formula-4c

SCHEME-5:


![[1860-5397-9-265-2]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Due to the elaborate substitution pattern of the parent quinolone ring systems these compounds are usually prepared via a linear consecutive sequence. In the case of moxifloxacin, an intramolecular base catalysed nucleophilic aromatic substitution is used to prepare the bicyclic ring system of the highly substituted aromatic1.104 (Scheme 19). A SNAr reaction is then used to introduce the saturated piperidinopyrrolidine appendage 1.105to furnish the desired structure [57-60]. In order to obtain a high yield for the substitution reaction a one-pot procedure was developed, initial masking of the acid (1.104) is achieved by silylation with subsequent borane chelate formation. Addition of the amine nucleophile 1.105 under basic conditions then renders the desired product in high yield. The available patent literature however does not comment on regioselectivity issues of the SNAr reaction due to the presence of the second fluoride substituent in the substrate, although not necessarily as electronically favourable for displacement it is certainly more accessible.
![[1860-5397-9-265-i19]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i19.png?scale=2.0&max-width=1024&background=FFFFFF)
The saturated (S,S)-2,8-diazabicyclo[4.3.0]nonane (1.105) used in the final step can be prepared by a double nucleophilic substitution between tosylamine and 2,3-bis-chloromethylpyridine (1.112) followed by catalytic reduction of the resulting bicycle using palladium on carbon in acetic acid (Scheme 20). As the corresponding sulfonamide 1.113 was found to be a crystalline solid a resolution using (D)-(+)-O,O-dibenzoyltartaric acid was reported to separate the enantiomers [Petersen, U.; Schenke, T.; Krebs, A.; Schenke, T.; Philipps, T.; Grohe, K.; Bremm, K.-D.; Endermann, R.; Metzger, K. G. New Quinoline and Naphthyridinonecarboxylic Acid Derivatives. Ger. Patent DE 4 208 792 A1, March 23, 1993.].
![[1860-5397-9-265-i20]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i20.png?scale=2.0&max-width=1024&background=FFFFFF)
| First way of enantioselective synthesis of moxifloxacin intermediate LI GuangXun, WU Lei, FU QingQuan, TANG Zhuo, ZHANG XiaoMei
A new method of enantioselective synthesis of (S,S)-2,8-diazobicyclo [4.3.0] nonane was found by using (R)-2-amino-2-phenyl-ethanol as chiral induction reagent. The entire synthetic process included 8 steps which were easy to operate with high yield. The purification method was only simple recrystallization or even used directly in the next step without further purification. The total yield was 29%.
|
|||
2013 Vol. 56 (3): 307-311 [Abstract] ( 22 ) [ PDF (518 KB) ] ( 92 ) [Supporting Information] DOI: 10.1007/s11426-012-4803-7
|


- Drugwatch. “Avelox Patent Family”. Retrieved 16 September 2012.
- “Details for NDA:021085”. DrugPatentWatch. Retrieved 17 July 2009.
- “www.accessdata.fda.gov”.
- “www.emea.europa.eu”.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021085s047,021277s041lbl.pdf
- “Avelox”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/1999/21085ltr.pdf
- http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21-085S010_Avelox_Approv.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2004/21085se1-022,21277se1-017ltr.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/021085s026,021277s022ltr.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/021085s027,029,021277s024,025ltr.pdf.
- “Moxifloxacin: restricted use : MHRA”.
- European Medicines Agency (24 July 2008). “European Medicines Agency recommends restricting the use of oral moxifloxacin-containing medicines”. Retrieved 20 July 2009.
- “SYNOPSIS”. Retrieved 29 January 2009.
- Karande SC, Kshirsagar NA (February 1992). “Adverse drug reaction monitoring of ciprofloxacin in pediatric practice”. Indian Pediatr 29 (2): 181–8. PMID 1592498.
- Dolui SK, Das M, Hazra A (2007). “Ofloxacin-induced reversible arthropathy in a child”. Journal of Postgraduate Medicine 53 (2): 144–5. doi:10.4103/0022-3859.32220 .PMID 17495385.
- “Center for drug evaluation and research Application number 21-598”. Food and Drug Administration (FDA). 15 April 2005. Retrieved 21 July 2009.
- Jump up^ Babar, S. (October 2013). “SIADH Associated With Ciprofloxacin.”. The Annals of Pharmacotherapy (Sage Publiahing) 47 (10): 1359–1363.doi:10.1177/1060028013502457 . ISSN 1060-0280.PMID 24259701. Retrieved November 139,2013.
- Renata Albrecht (28 July 2004). “NDA 21-085/S-024, NDA 21-277/S-019”. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Renata Albrecht (31 May 2007). “NDA 21-085/S-036, NDA 21-277/S-030”. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Renata Albrecht (15 February 2008). “NDA 21-085/S-038, NDA 21-277/S-031”. Division of Special Pathogen and Transplant Products. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- DEPARTMENT OF HEALTH & HUMAN SERVICES (28 February 2008). “NDA 21-085/S-014, S-015, S-017”. Food and Drug Administration (FDA). Retrieved 17 July 2009.
- Nea Zealand Government (26 June 2003). “Adverse Reaction Reporting and IMMP”. New Zealand: Medsafe. Retrieved 30 January 2009.
- http://www.mhra.gov.uk/Safetyinformation/DrugSafetyUpdate/CON087781, retrieved 2012-11-10 Missing or empty
|title=(help) - http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf, retrieved 2012-11-10 Missing or empty
|title=(help) - “EU agency recommends restricting moxifloxacin use”. Reuters. 24 July 2008. Retrieved 21 July 2009.
- Renata Albrecht (3 October 2008). “NDA 21-085/S-040, NDA 21-277/S-034”. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Updated Labeling for Antibiotic Avelox (Moxifloxacin) Regarding Risk of Severe Liver Injury / Information Update: 2010-42 For immediate release 22 March 2010http://www.hc-sc.gc.ca/ahc-asc/media/advisories-avis/_2010/2010_42-eng.php
- Saint F, Gueguen G, Biserte J, Fontaine C, Mazeman E (September 2000). “[Rupture of the patellar ligament one month after treatment with fluoroquinolone]”. Rev Chir Orthop Reparatrice Appar Mot (in French) 86 (5): 495–7.PMID 10970974.
- http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf, retrieved 2012-11-10 Missing or empty
|title=(help) - Bayer (December 2008). “AVELOX (moxifloxacin hydrochloride) Tablets AVELOX I.V. (moxifloxacin hydrochloride in sodium chloride injection)”. Food and Drug Administration (FDA). p. 19. Retrieved 2 November 2010. Unknown parameter
|line=ignored (help) - “Moxifloxacin”. University of Maryland Medical Center. 2009. Retrieved 22 July 2009.
- http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf
- Shin HC, Kim JC, Chung MK (September 2003). “Fetal and maternal tissue distribution of the new fluoroquinolone DW-116 in pregnant rats”. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 136 (1): 95–102.doi:10.1016/j.cca.2003.08.004 . PMID 14522602.
- Dan M, Weidekamm E, Sagiv R, Portmann R, Zakut H (February 1993). “Penetration of fleroxacin into breast milk and pharmacokinetics in lactating women” (PDF).Antimicrob. Agents Chemother. 37 (2): 293–6.doi:10.1128/AAC.37.2.293 . PMC 187655.PMID 8452360.
- Noel GJ, Bradley JS, Kauffman RE (October 2007). “Comparative safety profile of levofloxacin in 2523 children with a focus on four specific musculoskeletal disorders”.Pediatr. Infect. Dis. J. 26 (10): 879–91.doi:10.1097/INF.0b013e3180cbd382 . PMID 17901792.
- Joette Meyer; Renata Albrecht (16 March 2004).“Division of Special Pathogen and Immunologic Drug Products Summary of Clinical Review of Studies Submitted in Response to a Pediatric Written Request”. Food and Drug Administration (FDA). Retrieved 22 July 2009.
- Elbe DH, Chang SW (February 2005). “Moxifloxacin-warfarin interaction: a series of five case reports”. Ann Pharmacother 39 (2): 361–4. doi:10.1345/aph.1E179 .PMID 15632222.
- Karen E. Brown. “Top Ten Dangerous Drug Interactions in Long-Term Care”. Medication Management Project. Retrieved 1 August 2009.
- Stass HH (29 June – 3 July 1997). “Bay 12-8039 (BA) does not interact with theophylline (TH)”. Presented at the 20th International Congress of Chemotherapy.
- Medscape: Medscape Access
- http://69.20.19.211/cder/warn/dec99/wl121599.pdf
- Renata Albrecht (16 May 2002). “NDA 21-085/S-012”.Food and Drug Administration (FDA). Retrieved 17 July 2009.
- Drlica K, Zhao X (1 September 1997). “DNA gyrase, topoisomerase IV, and the 4-quinolones”. Microbiol Mol Biol Rev. 61 (3): 377–92. PMC 232616. PMID 9293187.
- “Drug card for Moxifloxacin (DB00218)”. Canada: DrugBank. 19 February 2009. Retrieved 3 August 2009.
- Alffenaar J. W. C., van Altena R., Bökkerink H. J (2009). “Pharmacokinetics of moxifloxacin in cerebrospinal fluid and plasma in patients with tuberculous meningitis”. Clinical Infectious Diseases 49 (7): 1080–2. doi:10.1086/605576 .PMID 19712035.
- “Inventors/Applicants”. patentlens.net. 3 October 2006. Retrieved 17 July 2009.
- Ed Lamb (1 May 2008). “Top 200 Prescription Drugs of 2007”. Pharmacy Times. Retrieved 21 July 2009.
- http://www.uspto.gov/web/offices/pac/dapp/opla/term/certs/4990517.pdf
- US 4990517 A (Feb 1991) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+4990517#list US 5051509 A (Sep 1991) Nagano et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5051509#list US 5059597 A (Oct 1991) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5059597#list US 5395944 A (Mar 1995) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5395944#list US 5416096 A (May 1995) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5416096#list
- Renata Albrecht (12 June 2002). “NDA 21-085/S-006, S-007 – NDA 21-277/S-002”. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- The 114Th Medicines Adverse Reactions Committee Meeting; Professor P. Ellis, Professor D.C.G. Skegg, Dr M. Rademaker, Dr F. McClure, Dr N. Rafter, Dr M. Tatley (26 June 2003). “Adverse Reaction Reporting and IMMP”. New Zealand: Medsafe. Retrieved 12 August 2009.
- Renata Albrecht, (6 October 2003). “NDA 21-085/S-019 – NDA 21-277/S-011”. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- Renata Albrecht (6 October 2003). “NDA 21-085/S-039 NDA 21-277/S-033”. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- Bayer HealthCare Pharmaceuticals Inc. (August 2008).“MEDICATION GUIDE AVELOX (AV-eh-locks) (moxifloxacin hydrochloride) Tablets AVELOX I.V. (AV-eh-locks) (moxifloxacin hydrochloride in sodium chloride injection)”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- Ozlem Belen (24 June 2009). “NDA 21-085/S-041 NDA 21-277/S-035”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- Bailey RR, Natale R, Linton AL (October 1972). “Nalidixic acid arthralgia”. Can Med Assoc J 107 (7): 604 passim.PMC 1940945. PMID 4541768.
- Bailey RR, Kirk JA, Peddie BA (July 1983). “Norfloxacin-induced rheumatic disease”. N Z Med J 96 (736): 590.PMID 6223241.
- Szarfman A, Chen M, Blum MD (January 1995). “More on fluoroquinolone antibiotics and tendon rupture” (PDF). N Engl J Med 332 (3): 193.doi:10.1056/NEJM199501193320319 . PMID 7800023.
- “Petition to Require a Warning on All Fluoroquinolone Antibiotics (HRG Publication #1399)”. Public Citizen. 1 August 1996. Retrieved on 27 December 2008.
- “Reports of adverse events with fluoroquinolones”. FDA Medical Bulletin 26 (3). October 1996.[dead link] Retrieved on 27 December 2008.
- “Madigan, Public Citizen, petition FDA for “black box” warning regarding potential adverse effects of certain popular antibiotics” (Press release). Office of the Illinois Attorney General. 29 August 2006. Retrieved 27 December 2008.
- “Public Citizen Petitions the FDA to Include a Black Box Warning on Fluoroquinolone Antibiotics (HRG Publication #1781)”. Public Citizen. 29 August 2006. Retrieved 27 December 2008.
- “Public Citizen v. Food and Drug Administration (FDA) (Fluoroquinolone)”. Public Citizen. 3 January 2008. Retrieved 27 December 2008.
- Ravn, Karen (18 August 2008). “Behind the FDA’s ‘black box’ warnings”. Los Angeles Times. Retrieved 27 December 2008.
- “FDA Requests Boxed Warnings on Fluoroquinolone Antimicrobial Drugs” (Press release). U.S. Food and Drug Administration. 8 July 2008. Retrieved 11 October 2008.
- “Drugs@FDA”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- “MedWatch: The FDA Safety Information and Adverse Event Reporting Program”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- MacCarthy, Paul (22 October 2008). “Important Change in the Avelox (moxifloxacin hydrochloride) and Cipro (ciprofloxacin) Complete Prescribing Information – Addition of Boxed Warning and Medication Guide Regarding Tendinitis and Tendon Rupture”. Bayer HealthCare Pharmaceuticals. Retrieved 27 December 2008.
- Bayer AG (6 November 2007). “Ruling in Bayer’s favor over Avelox patents”. Bayer. Retrieved 29 August 2009.
- http://www.orangebookblog.com/Bayer_20v._20Dr._20Reddy_27s.pdf
- “United States Court of Appeals for the Federal Circuit”. uscourts.gov. 28 June 2007. Retrieved 29 August 2009.
- Bayer AG (24 April 2008). “Risk Report”. Bayer. Retrieved 29 August 2009.
- In The United States District Court For The District Of Columbia Public Citizen, Inc. VS. Food And Drug Administration 3 January 2008
- Office of the Attorney General State of Illinois Lisa Madigan Citizen Petition to Include a Black Box Warning on Fluoroquinolone Antibiotics 18 May 2005
- Public Citizen’s Petition to Include a Black Box Warning on Fluoroquinolone Antibiotics (HRG Publication #1781) 29 August 2006
- Public Citizen’s Petition to Require a Warning on All Fluoroquinolone Antibiotics (HRG Publication #1399) 1 August 1996
- Public Citizen’s Petition to Ban the Antibiotic Gatifloxacin (Tequin) (HRG Publication #1768)
- Public Citizen’s Petition to immediately ban the antibiotic Trovafloxacin (Trovan). (HRG Publication #1485) Date: 3 June 1999
- Public Citizen’s Petition to immediately stop the distribution of dangerous, misleading prescription drug information to the public. HRG Publication #1442 Date: 9 June 1998
- June 2004, A petition To the United States Congress to immediately take action to protect consumers from the reckless and negligent abuses of the FDA and the following Pharmaceutical Companies: Bayer, Ortho-McNeill, Pfizer, Merck, Bristol-Myers Squibb, Sanofi Winthrop, Bertek Pharmaceuticals – Rhone-Poulenc Rorer and Barr. These companies manufacture and distribute fluoroquinolone antibiotics in the United States in a manner that fails to warn of serious adverse event risks, and downplays and fails to warn physicians of the serious risks associated with fluoroquinolone therapy.
| EP0350733A2 | Jun 30, 1989 | Jan 17, 1990 | Bayer Ag | 7-(1-Pyrrolidinyl)-3-quinolone- and -naphthyridone-carboxylic-acid derivatives, method for their preparation and for substituted mono- and bi-cyclic pyrrolidine intermediates, and their antibacterial and feed additive compositions |
| EP0550903A1 | Dec 28, 1992 | Jul 14, 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| EP0591808A1 | Sep 27, 1993 | Apr 13, 1994 | Bayer Ag | Quinolonecarboxylic acids |
| EP0592868A1 | Sep 29, 1993 | Apr 20, 1994 | Bayer Ag | Quinolonecarboxylic acids |
| US5849752 | Dec 5, 1996 | Dec 15, 1998 | Bayer Aktiengesellschaft | Crystal modification of CDCH a process for its preparation and pharmaceutical formulations comprising this modification |
| WO1999026940A2 | Nov 12, 1998 | Jun 3, 1999 | Bayer Ag | Method for producing 8-methoxy-quinoline carboxylic acids |
| WO2004091619A1 | Apr 9, 2004 | Oct 28, 2004 | Reddys Lab Ltd Dr | A crystalline form iii of anhydrous moxifloxacin hydrochloride and a process for preparation thereof |
| WO2007010555A2 | Jul 13, 2006 | Jan 25, 2007 | Ramprasad Achampeta Kodanda | Novel crystalline forms of moxifloxacin hydrochloride and process for preparation thereof |
| WO2008059521A2 | Sep 27, 2007 | May 22, 2008 | Ramprasad Achampeta Kodanda | Novel process for the preparation of moxifloxacin hydrochloride and a novel polymorph of moxifloxacin |
| CN102030751B | Dec 1, 2010 | Nov 21, 2012 | 上虞京新药业有限公司 | Process for crystallizing moxifloxacin hydrochloride |
| EP2154137A1 | Aug 4, 2008 | Feb 17, 2010 | Chemo Ibérica, S.A. | Crystalline form of moxifloxacin base |
| US20110212990 * | Nov 6, 2008 | Sep 1, 2011 | Hetero Research Foundation | Novel polymorph of moxifloxacin hydrochloride |
| WO2005012285A1 * | Aug 5, 2004 | Feb 10, 2005 | Satyanarayana Chava | An improved process for the preparation of moxifloxacin hydrochloride |
| EP0443498A1 * | Feb 18, 1991 | Aug 28, 1991 | Kyorin Pharmaceutical Co., Ltd. | Isoindoline derivatives |
| EP0550903A1 * | Dec 28, 1992 | Jul 14, 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| US6323213 * | Feb 12, 1997 | Nov 27, 2001 | Bayer Aktiengesellschaft | Possibly substituted 8-cyano-1-cyclopropyl-7-(2,8-diazabicyclo-[4.3.0]-nonan-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinolin carboxylic acids and their derivatives |
| US5849752 * | Dec 5, 1996 | Dec 15, 1998 | Bayer Aktiengesellschaft | Crystal modification of CDCH a process for its preparation and pharmaceutical formulations comprising this modification |
| WO2007010555A2 * | Jul 13, 2006 | Jan 25, 2007 | Ramprasad Achampeta Kodanda | Novel crystalline forms of moxifloxacin hydrochloride and process for preparation thereof |
| WO2008059521A2 * | Sep 27, 2007 | May 22, 2008 | Ramprasad Achampeta Kodanda | Novel process for the preparation of moxifloxacin hydrochloride and a novel polymorph of moxifloxacin |
| WO2008138759A1 * | Apr 30, 2008 | Nov 20, 2008 | Sandoz Ag | Process for the preparation of moxifloxacin hydrochloride |
| WO2007148137A1 * | Jun 22, 2007 | Dec 27, 2007 | Generics Uk Ltd | Novel hydrate form of moxifloxacin monohydrochloride |
| WO2008028959A1 * | Sep 7, 2007 | Mar 13, 2008 | Sint Quimica Sa | Crystalline form of moxifloxacin hydrochloride |
| WO2008059223A2 * | Nov 13, 2007 | May 22, 2008 | Cipla Ltd | Process for the synthesis of moxifloxacin hydrochloride |
| WO2008095964A1 * | Feb 6, 2008 | Aug 14, 2008 | Sint Quimica Sa | Crystalline form of moxifloxacin base |
| WO2009087151A1 * | Jan 7, 2009 | Jul 16, 2009 | Chemo Iberica Sa | Polymorphic forms of moxifloxacin hydrochloride and processes for preparation thereof |
| CN102603738B | Feb 24, 2012 | Dec 11, 2013 | 天津市汉康医药生物技术有限公司 | 一种稳定的盐酸莫西沙星化合物 |
| EP2083010A1 | Jan 8, 2008 | Jul 29, 2009 | Chemo Ibérica, S.A. | Polymorphic Forms of Moxifloxacin hydrochloride and processes for preparation thereof |
| EP2154137A1 | Aug 4, 2008 | Feb 17, 2010 | Chemo Ibérica, S.A. | Crystalline form of moxifloxacin base |
| US8198451 | Nov 13, 2007 | Jun 12, 2012 | Cipla Limited | Process for the synthesis of moxifloxacin hydrochloride |
| US20110212990 * | Nov 6, 2008 | Sep 1, 2011 | Hetero Research Foundation | Novel polymorph of moxifloxacin hydrochloride |
 syn…………. http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html spectroscopy…………. http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html
syn…………. http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html spectroscopy…………. http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html
Temozolomide 替莫唑胺

Temozolomide 替莫唑胺
Temozolomide is a DNA damage inducer.
4-methyl-5-oxo-2,3,4,6,8-pentazabicyclo[4.3.0]nona-2,7,9-triene-9-carboxamide
3,4-dihydro-3-methyl-4-oxoimidazo(5,1-d)-1,2,3,5-tetrazine-8-carboxamide
Methazolastone, Temodar, Temodal
CAS NO 85622-93-1
Molecular Weight: 194.15
MF C6H6N6O2
Cancer Research UK (Originator), Schering-Plough (Licensee), National Cancer Institute (Codevelopment)
NMR..http://file.selleckchem.com/downloads/nmr/S123702-Methazolastone-NMR-Selleck.pdf
HPLC.http://file.selleckchem.com/downloads/hplc/S123702-Methazolastone-HPLC-Selleck.pdf
Temozolomide is an antitumor agent indicated for treating patients with malignant glioma such as cancer, breast cancer, refractory anaplastic astrocytoma, i.e., patients at first relapse who have experienced disease progression in malignant glioma, glioblastoma multiform and anaplastic astrocytoma, on a drug regimen containing a nitrosourea and procarbazine.
Temozolomide preparations are sold on the US market as hard capsules containing 5 mg, 20 mg, 100 mg or 250 mg Temozolomide (marketed as Temodar® by Schering Corporation, Kenilworth, N.J., USA). In other markets it is sold as Temodal®.
Temozolomide (brand names Temodar and Temodal and Temcad) is an oral chemotherapy drug. It is an alkylating agent used for the treatment of Grade IV astrocytoma — an aggressive brain tumor, also known as glioblastoma multiforme — as well as for treating melanoma, a form of skin cancer.
Temozolomide is also indicated for relapsed Grade III anaplastic astrocytoma and not indicated for, but as of 2011 used to treatoligodendroglioma brain tumors in some countries, replacing the older (and less well tolerated) PCV (Procarbazine–Lomustine–Vincristine) regimen.
Temozolomide, 3-methyl-8-aminocarbonyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one, is a known antitumor drug; see for example Stevens et al., J. Med. Chem. 1984, 27, 196-201, and Wang et al., J. Chem. Soc., Chem. Commun.,1994,1687-1688. Temozolomide, the compound of formula 1:

is described in U.S. Pat. No. 5,260,291 (Lunt et al.).
The synthesis of 1 by the process described in J. Med. Chem. 1984, 27, 196-201 is depicted in the scheme I below.

In this process, 5-amino-1H-imidazole-4-carboxamide (A) is converted into 5-diazo-1H-imidazole-4-carboxamide (B), which is then cyclized with methylisocyanate in dichloromethane to provide a high yield of temozolomide. However, this process requires isolation of the unstable and potentially dangerous 5-diazo-1H-imidazole-4-carboxamide (B). Moreover, methylisocyanate is a difficult reagent to handle and ship, especially on the industrial scale, and indeed is better avoided in industrial manufacture. Furthermore, the cycloaddition of methylisocyanate requires a very long reaction time: Table I in J. Med Chem.1984, 27,196-201, suggests 20 days. Additionally, Stevens et al mention that the cycloaddition of the methylisocyanate to the compound of the formula (B) can proceed through two different intermediates:
The production of I by the two processes described in J. Chem. Soc., Chem. Commun., 1994, 1687-1688 provides a low overall yield from 5-amino-1H-imidazole-4-carboxamide (A): less than 20% (unoptimized—about 17% through 5-diazo-1H-imidazole-4-carboxamide (B) and about 15% through 5-amino-N1-(ethoxycarbonylmethyl)-1H-imidazole-1,4-dicarboxamide (C)); Scheme II below

The agent was developed by Malcolm Stevens[1] and his team at Aston University in Birmingham,[2][3] Temozolomide is a prodrug and animidazotetrazine derivative of the alkylating agent dacarbazine. It has been available in the US since August 1999, and in other countries since the early 2000s.
The therapeutic benefit of temozolomide depends on its ability to alkylate/methylate DNA, which most often occurs at the N-7 or O-6 positions ofguanine residues. This methylation damages the DNA and triggers the death of tumor cells. However, some tumor cells are able to repair this type of DNA damage, and therefore diminish the therapeutic efficacy of temozolomide, by expressing a protein O6-alkylguanine DNA alkyltransferase (AGT) encoded in humans by the O-6-methylguanine-DNA methyltransferase (MGMT) gene.[4] In some tumors, epigenetic silencing of the MGMT gene prevents the synthesis of this enzyme, and as a consequence such tumors are more sensitive to killing by temozolomide.[5] Conversely, the presence of AGT protein in brain tumors predicts poor response to temozolomide and these patients receive little benefit from chemotherapy with temozolomide.[6]
- Nitrosourea- and procarbazine-refractory anaplastic astrocytoma
- Newly diagnosed glioblastoma multiforme
- Malignant prolactinoma
Temozolomide (sometimes referred to as TMZ) is an imidazotetrazine derivative of the alkylating agent dacarbazine. It undergoes rapid chemical conversion in the systemic circulation at physiological pH to the active compound, 3-methyl-(triazen-1-yl)imidazole-4-carboxamide (MTIC). Temozolomide exhibits schedule-dependent antineoplastic activity by interfering with DNA replication. Temozolomide has demonstrated activity against recurrent glioma. In a recent randomized trial, concomitant and adjuvant temozolomide chemotherapy with radiation significantly improves, from 12.1 months to 14.6 months, progression free survival and overall survival in glioblastoma multiforme patients.
Formulations
Temozolomide is available in the United States in 5 mg, 20 mg, 100 mg, 140 mg, 180 mg & 250 mg capsules. Now also available in an IV form for people who can not swallow capsules or who have insurance that does not cover oral cancer agents.
A generic version is available in the UK.
Further improvement of anticancer potency
Laboratory studies and clinical trials are investigating whether it might be possible to further increase the anticancer potency of temozolomide by combining it with other pharmacologic agents. For example, clinical trials have indicated that the addition of chloroquine might be beneficial for the treatment of glioma patients.[8] In laboratory studies, it was found that temozolomide killed brain tumor cells more efficiently when epigallocatechin gallate (EGCG), a component of green tea, was added; however, the efficacy of this effect has not yet been confirmed in brain tumor patients.[9]More recently, use of the novel oxygen diffusion-enhancing compound trans sodium crocetinate (TSC) when combined with temozolomide and radiation therapy has been investigated in preclinical studies [10] and a clinical trial is currently underway.[11]
Because tumor cells that express the MGMT gene are more resistant to killing by temozolomide, it was investigated[according to whom?] whether the inclusion of [[O6-benzylguanine]] (O6-BG), an AGT inhibitor, would be able to overcome this resistance and improve the drug’s therapeutic effectiveness. In the laboratory, this combination indeed showed increased temozolomide activity in tumor cell culture in vitro and in animal models in vivo.[12] However, a recently completed phase-II clinical trial with brain tumor patients yielded mixed outcomes; while there was some improved therapeutic activity when O6-BG and temozolomide were given to patients with temozolomide-resistant anaplastic glioma, there seemed to be no significant restoration of temozolomide sensitivity in patients with temozolomide-resistant glioblastoma multiforme.[13]
There are also efforts to engineer hematopoietic stem cells expressing the MGMT gene prior to transplanting them into brain tumor patients. This would allow for the patients to receive stronger doses of temozolomide, since the patient’s hematopoietic cells would be resistant to the drug.[14]
High doses of temozolomide in high grade gliomas have low toxicity, but the results are comparable to the standard doses.[15]
A case report suggests that temozolomide may be of use in relapsed primary CNS lymphoma.[16] Confirmation of this possible use seems indicated.
Temozolomide, 3-methyl-8-aminocarbonyl-imidazo[5,1-d]- 1 ,2,3,5-tetrazin- 4(3H)-one, is a known antitumor drug; see for example Stevens et al., J. Med. Chem. 1984, 27, 196-201 , and Wang et al., J. Chem. Soc, Chem. Commυn., 1994, 1687-1688. Temozolomide, the compound of formula 1 :

1 is described in U.S. Patent No. 5,260,291 (Lunt et al.).
The synthesis of 1 by the process described in J. Med. Chem. 1984, 27, 196- 201 is depicted in the scheme I below. Scheme I:

In this process, 5-amino-1 H-imidazole-4-carboxamide (A) is converted into 5- diazo-1 H-imidazole-4-carboxamide (B), which is then cyclized with methylisocyanate in dichloromethane to provide a high yield of temozolomide.
However, this process requires isolation of the unstable and potentially dangerous 5-diazo-1 H-imidazole-4-carboxamide (B). Moreover, methylisocyanate is a difficult reagent to handle and ship, especially on the industrial scale, and indeed is better avoided in industrial manufacture.
Furthermore, the cycloaddition of methylisocyanate requires a very long reaction time: Table I in J. Med Chem. 1984, 27,196-201 , suggests 20 days. Additionally, Stevens et al mention that the cycloaddition of the methylisocyanate to the compound of the formula (B) can proceed through two different intermediates:
The production of I by the two processes described in J. Chem. Soc, Chem.
Commun., 1994, 1687-1688 provides a low overall yield from 5-amino-1 H- imidazole-4-carboxamide (A): less than 20% (unoptimized – about 17% through 5- diazo-1 H-imidazole-4-carboxamide (B) and about 15% through 5-amino-N1– (ethoxycarbonylmethyl)- 1 H-imidazole- 1 ,4-dicarboxamide (C)); Scheme II below
Scheme II:

Moreover, the unstable 5-diazo-1 H-imidazole-4-carboxamide (B) still has to be isolated in the branch of this process that uses it as an intermediate. Clearly, therefore, there is a need for synthetic methods that: a) are more convenient and higher yielding, especially on commercial scale; b) approach the synthesis of the temozolomide nucleus in novel ways; or c) improve the preparation or use of intermediates for the processes.
Temozolomide of formula I, is an antitumor drag and is chemically known as 3-methyl-8- aminocarbonyl-imidazole[5,l-d]-l,2,3,5-tetrazin-4(3H)-one.

Formula I
It is indicated for treating patients with malignant glioma such as cancer, breast cancer, refractory anaplastic, astrocytoma, i.e. patient at first relapse who have experienced disease progression in malignant glioma, glioblastoma multiform and anaplastic astrocytoma, on a drug containing a nitrosourea and procarbazine. It is sold in the US market as hard capsules containing 5 mg, 20 mg, 100 mg or 250 mg as Temodar® by Schering corporation.
Temozolomide and compounds having similar activity (higher alkyl analogues at the 3 -position) were first disclosed in US patent 5,260,291. According to said patent, temozolomide is prepared by the reaction of 5-diazoimidazole-4-carboxamide with methyl isocyanate in the presence of N- methylpyrrolid-2-one in dichloromethane at room temperature for three to four weeks. Melting point of temozolomide reported in above patent is 200 0C (recrystallized from acetonitrile); 21O0C with effervescence (recrystallized from acetone and water), and 2150C with effervescence and darkening (recrystallized from hot water). Major drawback of process is the longer reaction duration of three to four weeks for completion of reaction.
Further, the process described in the patent involves use of low boiling and extremely toxic, methyl isocyanate, which is very difficult to handle, especially on industrial scale, as its use should be avoided in the industrial synthesis. Further, cycloaddition reaction requires a very long period of 21 to 28 days, which makes the process unattractive for industrial scale.
US patent 5,003,099 discloses a process for preparation of aminocyanoacetamide, a key intermediate for the synthesis of temozolomide. According to the patent, aminocyanoacetamide is synthesized in two steps by the reaction of cyanoacetic acid alkyl ester using sodium nitrite in the presence of glacial acetic acid to form a hydroxyimino intermediate, which is then reduced in the presence of platinum on carbon to yield aminocyanoacetic acid alkyl ester, which is unstable.
The alkyl ester intermediate is then in situ reacted with aqueous ammonia to give the desired product. The main drawback of the above mentioned process is the use of aqueous ammonia, since aminocyanoacetamide, generated in reaction, is soluble in aqueous solution and hence difficult to extract from the reaction mass which results in lower yields. The patent is silent about the purity of intermediate and process needs extraction of the above mentioned intermediate from filtrate.
US patent 6,844,434 describes synthesis of temozolomide by cyclization of 5-amino-l-(N-rnethyl- hydrazinocarbonyl)-lH-imidazole-4-carboxylic acid in the presence of tetrabutyl nickel and periodic acid to form a reaction mixture which is concentrated under reduce pressure and resulting residue was treated with acetonitrile and filtered. The filtrate was concentrated and chromatographed on a column of silica gel to give temozolomide.
Use of time consuming and cumbersome technique i.e. column chromatography for isolation of product makes the process not suitable to employ at industrial level. US patent 7,087,751 discloses a process for the preparation of temozolomide from protected imidazole intermediate.
The process involves reaction of l-methyl-3-carbamoyliminomethyl-urea with JV- protected aminocyanoacetamide in the presence of acetic acid in a suitable solvent to form an JV- protected imidazole intermediate which is then cyclized in the presence of lithium chloride to minimize undesired cyclisation product. After cyclisation, the protected group has to be removed which makes the process more laborious with more number of steps.
As exemplified in example 1 of the above patent, yield of the JV-protected imidazole intermediate obtained is very low, almost half of the product goes in the filtrate which further needs extraction from the filtrate. After extraction of inteπnediate from the filtrate, the combined yield is only 67 %. The intermediate obtained is only 93 to 94% pure and requires additional purifications, crystallization using ethyl acetate and slurry wash with mixture of methyl tertiary butyl ether and isopropanol. These additional purification further takes away around 20 % yield of the inteπnediate thus yield of the pure intermediate, which is suitable for the further reaction, remains around 53 % which is very low from commercial point of view.
The patent also describes condensation of l-methyl-3-carbamoyliminomethyl-urea with unprotected aminocyanoacetamide in presence of acetic acid to give an imidazole intermediate. This patent fails to disclose the process of conversion of above imidazole intermediate to temozolomide, but only up to hydrolysis to prepare 5-amino-lH-imidazole-4-carboxamide hydrochloride is reported.
Another US patent no. 6,844,434 of same applicant (Schering) discloses a process for the conversion of 5-amino- lH-imidazole-4-carboxamide hydrochloride, which is prepared by the hydrolysis of above imidazole intermediate, to temozolomide. By combining the above two processes, this adds further four additional steps to the synthesis of temozolomide. The process of preparation of temozolomide is described by the following scheme:

It has been observed that for the preparation of unprotected imidazole intermediate as exemplified in US 7,087,751, use of excess amount of the acetic acid (around 21 times with respect to aminocyanoacetamide) is reported. Thereafter acetic acid is removed by distillation.
The inventors of the present invention have repeated example 2 as described in US 7,087,751 for the preparation of unprotected imidazole intermediate. As per the process, after the completion of the reaction, acetic acid has to be removed from the reaction mixture. It is noticed that removal of acetic acid is a very tedious move so as on commercial scale and leads to decomposition.
In a publication namely, Journal of Organic Chemistry, volume 62, no. 21, 7288-7294, a process is disclosed for the preparation of temozolomide by the hydrolysis of 8-cyano-3-methyl-[3H]-imidazole~ [5,l-d]-tetrazin-4-one in the presence of hydrochloric acid to give hydrochloride salt of temozolomide, which has to be neutralized to obtain temozolomide. In the same Journal, another process for the preparation of temozolomide is also described. Temozolomide is prepared by the nitrosative cyclization of imidazole intermediate using aqueous solution of sodium nitrite and tartaric acid to give temozolomide in 45 % yield in solution.
US patent publication 2007/0225496 exemplified a process for preparation of temozolomide by pyrolising N’-methyl-N,N-diphenyl urea to form vapor of methyl isocyanate which is then reacted with 5-diazo-5H-imidazole-4-carboxylic acid amide to form temozolomide.
The above described process involves use of methyl isocyanate, which is highly flammable and makes the process unsuitable for industrial synthesis, hi addition to this, isolation of temozolomide from the reaction mixture requires addition of large amount of ethyl acetate followed by addition of hexane and again ethyl acetate to isolate compound.
US patent publication 2009/0326028 describes a process for preparation of temozolomide by diazotization of imidazole intermediate in the presence of at least one metal halide, a source of nitrous acid and an acid to form acidic solution of temozolomide, wherein temozolomide forms a salt with acid. The desired product i.e. temozolomide is then isolated from the acidic solution by extraction with a solvent.
The process requires very strict reaction parameters including the addition of metal halide during diazotization as well as addition of pre-cooled reaction mixture to sodium nitrite solution to achieve desired level of selective cyclization. Patent application also describes two methods for the extraction of temozolomide.
US patent publication 2010/0036121 discloses a process for the preparation of temozolomide by reaction of 5-aminoimidazole-4-carboxamide with N-succinimidyl-N’-methylcarbamate to form carbamoyl 5~aminoimidazole-4-carboxamide which is then reacted with alkali or alkaline earth nitrile to give reaction mass containing temozolomide
-
Temozolomide, is a known antitumour drug, and is represented by formula I:

3-methyl-8-aminocarbonyl-imidazo [5,1-d]-1,2,3,5-tetrazin-4(3H)-one
-
It is described in US 5,260,291 together with compounds of broadly similar activity such as higher alkyl analogs at the 3-position.
-
J.Med.Chem. 1984, 27, 196-201 describes a process wherein 5-amino-1H-imidazole-4-carboxamide is converted into 5-diazo-1H-imidazole-4-carboxamide, which is then cyclised with methylisocyanate in dichloromethane to provide a high yield of temozolomide.
-
This process requires isolation of the unstable and potentially dangerous 5-diazo-1H-imidazole-4-carboxamide, methyl isocyanate is a difficult reagent to handle and ship, especially on the industrial scale. Furthermore, the cycloaddition of methylisocyanate requires a long reaction time (Table I in J.Med.Chem. 1984, 27, 196-201, suggests 20 days).
-
The product obtained by this process contains, high residual dichloromethane. It is essential to limit dichloromethane content in the final API below 600 ppm as per ICH guideline. Dichloromethane content can be reduced if one follows technique of US 5,260,291 .
-
US 5,260,291 discloses acetone-water recrystallisation of temozolomide, which results in low yield (60% recovery) due to decomposition of temozolomide to impurities like 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide, compound of formula V

and 5-amino-1H-imidazole-4-carboxamide.
-
The production of compound of formula I by the two processes described in J.Chem.Soc., Chem.Commun., 1994, 1687-1688 provides a low overall yield from 5-amino-1H-imidazole-4-carboxamide: less than 20% (about 17% through 5-diazo-1H-imidazole-4-carboxamide and about 15% through 5-amino-N1-(ethoxy carbonylmethyl)-1H-imidazole-1,4-dicarboxamide).
-
The unstable 5-diazo-1H-imidazole-4-carboxamide has to be isolated in the branch of this process that uses it as an intermediate.
-
US 2002/0133006 discloses a process for the preparation of compound of formula I using methyl hydrazine which is a toxic and flammable liquid, hence not feasible on industrial scale and the final isolation involves tedious workup including column chromatography.
-
J.Org.Chem. 1997, 62, 7288-7294 describes a process wherein the final step of diazotization provides equi-formation of aza-hypoxanthine and temozolomide, resulting in low yield. This literature does not provide the experimental procedure for work up.
-
US 2005/0131227 describes a process involving the use of a bulky protecting group on nitrogen of the primary amide for cyclisation in presence of LiCl to minimize the undesired cyclization product. After cyclization the protecting group has to be removed which makes the process more laborious with more number of steps (Scheme I).

U.S. Pat. No. 6,844,434 describes the preparation of Temozolomide, alkyl analogs and intermediates thereof. The process, which is depicted in Scheme 3 below, comprises reacting 5-amino-1H-imidazole-4-carboxamide hydrochloride (II) with 4-nitrophenyl chloroformate to afford compound (III), which is subsequently reacted with methyl hydrazine to obtain the corresponding compound (IV), which is cyclized to yield Temozolomide.

Another process for preparing Temozolomide is described in U.S. patent application having the Publication No. 2002/0095036 (see Scheme 4 below). In this process, the imine (V) is converted to 2-cyano-N-(1,1-dimethylethyl)-2-[(diphenyl-methylene)amino]-acetamide, which is converted to 2-amino-2-cyano-N-(1,1-dimethyl-ethyl)-acetamide hydrochloride.
The latter is reacted with compound (VI) to obtain 5-amino-N4-(1,1-dimethylethyl)-N1-methyl-1H-imidazole-1,4-dicarboxamide, which is converted to 3,4-dihydro-N-(1,1-dimethylethyl)-3-methyl-imidazo-[5,1-d]-1,2,3,5-tetrazine-8-carboxamide (tert-butyl-Temozolomide), which yields Temozolomide under acidic treatment with concentrated sulfuric acid.

Yet another synthesis of Temozolomide is described by Stevens et al. in J. Org. Chem., Vol. 62, No. 21, 7288-7294, 1997, wherein Temozolomide hydrochloride salt is obtained in 65% yield by the hydrolysis of 8-cyano-3-methyl-[3H]-imidazo-[5,1-d]-tetrazin-4-one with hydrochloric acid, as shown in Scheme 5.

The main disadvantage of this process is the low yield in which Temozolomide hydrochloride is obtained (65%). It is assumed that the relatively elevated temperature of 60° C. used in the process increases the content of decomposition products.






…………………………
Synthesis
US Patent 8,232,392
Temozolomide (1) is a drug that was discovered more than 30 years ago. In the past 10 years, it has been used to treat aggressive brain tumors. S. Turchetta and co-inventors summarize several processes for preparing temozolomide, all of which use toxic reagents such as MeNCO or MeNHNH2or generate large amounts of chemical waste. They describe a safer route to 1.
The inventors’ method starts with the preparation of carbamoyl compound 4 from amide 2 by treating it with succinimidyl reagent 3 in the presence of a base. The product is isolated in 88% yield and 96.9% purity by HPLC. Reagent 3 is a nonexplosive, crystalline solid with comparatively low toxicity and is much safer than MeNCO for this reaction.

In the next stage, the amine group in 4 is converted to diazonium salt 5 via a diazotization reaction. The details of this reaction are not described, but reference is made to a method reported in 1997 (Wang, Y., et al. J. Org. Chem. 1997, 62, 7288–7294). Compound 5 is not isolated; when acid is added, it cyclizes by the reaction of the diazonium group with one of the two amide groups to give products 1 and 6 in approximately equal amounts. The desired product 1 is formed by the reaction of the secondary amide group; when the primary amide reacts, the product is its isomer, 6.
Products 1 and 6 are separated by passing the acidified reaction mixture from the diazotization reaction over a column of a polymeric adsorbent resin. The material used in the example is XAD 1600 from Rohm & Haas; other resins are covered in the claims. Compound 6 elutes from the column first; then 1 is eluted with acidified aq EtOH. After separation, 1 is recrystallized from acidified acetone and isolated in 30% yield with 99.9% purity.
The process provides an alternative, safer route to temozolomide, but half of intermediate 4 is lost as unwanted product 6. [Chemi S.p.A. [Cinisello Balsamo, Italy]. US Patent 8,232,392, July 31, 2012; )
………………..
SYNTHESIS
http://www.google.com/patents/WO2002057268A1?cl=en

EXAMPLE 1
Preparation of Temozolomide (1 ) Step A Preparation compound (3)

5-Amino-1 H-imidazole-4-carboxamide*HCI (4) (25 g, 0.154 mol) (Aldrich 16,496-8), CH2CI2 (0.6 L) and Et3N (45 mL) (Aldrich, 13,206-3) were placed into a dry 2-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen at ambient temperature. The mixture was stirred, and a solution of 400 mL of 4-nitrophenyl chloroformate (34 g, 0.169 mol) (Aldrich, 16,021-0) in CH2CI2was added dropwise.
The reaction mixture was stirred vigorously for 4 hours and then left to stand for 18 hours at room temperature. The precipitate was collected by vacuum filtration and washed with H20 (1.5 L) to afford the product (3) as a pale yellow solid (42 g, 0.144 mol). 1H NMR (400MHz, DMSO-d6, δ): 8.40 (d, 2H), 7.83 (s, 1 H), 7.74 (d, 2H), 7.08 (bs, 1 H), 6.95 (bs, 1 H), 6.52 (s, 2H). Step B Preparation of compound (2)

Compound (3) (42 g, 0.144 mol) and DMF (0.27 L) were placed into a dry
1 -liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was cooled to 0°C, and methylhydrazine (10 mL, 0.188 mol) (Aldrich, M5.000-1 ) was added dropwise.
The reaction mixture was stirred vigorously for 1 hour at 0°C and was then poured into EtOAc (2.1 L). The precipitate was collected by vacuum filtration and was dried under vacuum (20 mm Hg, room temperature, 18 hours) to afford (2) as a tan solid (27.1 g, 0.137 mol). 1H NMR (400MHz, DMSO-d6, δ): 7.62 (s, 1 H), 6.85 (bs, 1 H), 6.75 (bs,1 H), 6.00 (s, 2H), 5.10 (s, 2H), 3.15, s, 3H).mp: 188°C (dec).
Analysis: Calcd for C6H10N6O2: C, 36.36; H, 5.09; N, 42.41.
Found: C, 36.46; H, 4.99; N, 42.12.
Step C Preparation of Temozolomide (1 )

2 1 (Temozolomide)
Compound (2) (500 mg, 2.5 mmol), Bu4NI (95 mg, 0.25 mmol), THF (250 mL) and CH3CN (250 mL) were placed into a dry 1 -liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen.
The reaction mixture was heated at 60°C for 20 mm and then cooled to room temperature. H5lθ6 (1.14 g, 5 mmol) was added and the reaction mixture was stirred vigorously at room temperature for 1 hour. The resulting solution was treated with saturated aqueous Na2S2O3 (5 mL) and was then concentrated under reduced pressure to dryness. The residue was treated with CH3CN (200 mL) and was filtered. The filtrate was concentrated and chromatographed on a column of silica gel (1.5% to 2% AcOH/EtOAc) to afford temozolomide (1 ) (280 mg). 1H NMR (400MHz, DMSO-d6, δ): 8.80 (s, 1 H), 7.80 (bs, 1 H), 7.66 (bs, 1 H), 3.43 (s,3H).
………………
SYNTHESIS

…………………
SYNTHESIS
http://www.google.com/patents/WO2010140168A1?cl=en
Accordingly, the present invention provides an improved process for the preparation of temozolomide of formula I,

Formula I which proves to be efficient and industrially advantageous.
The process comprises the step of: a), condensing compound of formula II,

Formula II with compound of formula III,
CH3 H CH3 Formula III in the presence of an acid in an alcoholic solvent to form a compound of formula IV;

Formula IV b). isolating the compound of formula IV from the reaction mixture by filtration; c). diazotizing and cyclizing the compound of formula IV in the presence of source of nitrous acid and a suitable acid; d). isolating temozolomide therefrom; and e). optionally purifying temozolomide of formula I.
Accordingly, the present invention provides an improved process for the preparation of temozolomide of formula I, process comprises the steps of: a), diazotizing and cyclizing the compound of formula IV in the presence of a source of nitrous acid and a suitable acid; b). optionally, cooling the reaction mixture; c). isolating precipitate of temozolomide from the reaction mixture; and d). purifying temozolomide of formula I with a suitable solvent
REFERENCE EXAMPLE:
Preparation* of S-Aøiino-N’-methyl-lH-imidazole-ljΦdicarboxamide (US 7,087,751) 2-Amino-2-cyanoacetamide (10 g), l-methyl-3-methylcarbamoyliminomethyl urea (19 g) and acetic acid (120 ml) were stirred together at ambient temperature under the positive pressure of nitrogen for 2 hours. Excess acetic acid was removed under reduced pressure and methyl tertiary butyl ether (25 ml) was added to the concentrated reaction mass, cooled to obtained crude solid.
The mixture was stirred for 30 minutes and the precipitate was collected by vacuum filtration. The solid was dried under vacuum at 20-250C for 18 hours to obtain 13 g of title compound as grayish solid. The crude product was stirred with water (66 ml) for 1 hour at 20-250C, filtered, suck dried and dried under vacuum at2O0C for 18 hours to obtain 11.2 g of title compound as greyish solid.
EXAMPLES
Example 1: Preparation of hydroxylirainocyano acetic acid ethyl ester
To a suspension of ethyl cyanoacetate (1.0 Kg, 8.84 mol) and sodium nitrite (0.735 kg, 10.65 mol) in water (0.80 L), acetic acid (0.70 kg, 11.66 mol) was added at 0-50C over a period of one hour.
Temperature was slowly raised to 23-270C and the reaction mixture was stirred for one hour at that temperature. After the complete consumption of ethyl cyanoacetate (monitored by TLC/GC), the reaction mixture was extracted with ethyl acetate (5 x 1.5 L). The combined organic layer was successively washed with 10% sodium bicarbonate (2 x 1.25 L) and brine solution (1.25 L), dried over sodium sulfate and filtered through hyflow bed. Solvent was removed under reduced pressure at 40-
450C. The resulting solid was stirred with cyclohexane (3.0 L) for 30 minutes at 25-300C, filtered and dried at 40-450C under vacuum to afford 1.14 kg (91.2 %) of title compound having purity 99.82% by
HPLC.
Example 2: Preparation of aminocyanoacetic acid ethyl ester
To a solution hydroxyliminocyano acetic acid ethyl ester (1.14 Kg, 8.02 mol) in methanol (11.4 L) was added 5% platinum on carbon (91.2 g, 50 % wet) and the mixture was hydrogenated at hydrogen gas pressure of 6.2-6.4 kg/cm2 over a period of 12 hours and the completion of reaction was checked by
TLC. The reaction mixture was filtered under nitrogen atmosphere to recover the catalyst. The filtrate was used as such for the next stage.
Example 3: Preparation of amimøcyanoacetamide
The solution of aminocyanoacetic acid ethyl ester (as prepared above) in methanol was cooled to 0-5
0C and ammonia gas was purged into it approximately for 1 hour. After the completion of the reaction
(monitored by TLC), the reaction mass was concentrated to 2.5-3.0 L under reduced pressure at 40-
45°C, cooled to 0-50C and stirred for 1 hour. The precipitated solid was filtered, washed with chilled methanol (200 ml) and dried at 35-400C under vacuum for 6 hours to obtain 572 g of title compound.
The resulting product was added to methanol (4.57 L) and heated to reflux till the solution become clear. Activated charcoal (25g) was added to the reaction mixture and refluxed for 15 minutes. The solution was filtered through hyflow bed, the bed was washed with methanol (500 ml) and the filtrate was concentrated to half of its original volume (approx 2.0 L). The mixture was cooled to 0-50C and stirred for 45 minutes. The resulting solid was filtered, washed with chilled methanol (250 ml) and dried at 40-450C under vacuum to obtain 425g (53.6%) of pure title compound having purity 99.46% by HPLC. Example 4: Preparation of l-methyl-3-methylcarbamoyliminomethyl urea
A suspension of monomethyl urea (1.5 kg, 20.27 mol) in triethyl orthoformate (4.5 L, 30.40 mol) was heated to reflux at 150-1600C for 12 hours. The reaction mixture was cooled to 5-100C, and stirred for 1 hour to ensure complete precipitation, of the product. The resulting solid was filtered, washed with ethyl acetate (350ml) and dried under vacuum at 45-5O0C to yield 1.08 kg (67.9%) of title compound having purity 93.82% by HPLC.
Exainple-5: Preparation of S-amino-N^methyl-lH-imidazole-l^-dicarboxamide Acetic acid (200 ml, 3.53 mol) was added to a suspension of aminocyanoacetamide (40Og, 4.04 mol) and l-methyl-3-methylcarbamoyliminomethyl urea (76Og, 4.8 mol) in methanol (2.0 L) at 20-250C and the mixture was stirred at 20-250C for 18 hours till completion of the reaction (monitored by HPLC). The reaction mixture was cooled to 0-50C, stirred for 1 hour and the resulting solid was filtered, washed with chilled methanol (450 ml), suck dried and finally dried under vacuum at 30-350C to afford 648 g (88.04%) of title compound as an off white colored solid having purity 99.21 % by HPLC. Example 6: Preparation of temozolomide
Acetic acid (450 ml, 7.95 mol) was added to a suspension of S-amino-N^methyl-lH-imidazole-l^- dicarboxamide (500g, 2.73mol) and sodium nitrite (25Og, 3.62mol) in water (5.0 L) at -5 to 00C at such a rate so that temperature does not rise above 5°C. The reaction mixture was stirred at 0 to 5°C for one hour and absence of starting material was checked by HPLC analysis. Ice bath was removed and powdered calcium chloride (1.25Kg) was added in small lots to the reaction mass and stirred at 25- 300C for 2 hours. The reaction mass was extracted with a 2.5% solution of dimethylsulfoxide in dichloromethane (5 X 50 L). Combined organic layer was dried over sodium sulfate and filtered through a hyflow bed. Solvent was removed under reduced pressure below 4O0C and residual dimethylsulfoxide layer was degassed completely. The dimethylsulfoxide layer was cooled to 0 to – 100C and stirred for 1 hour. The resulting solid was filtered, washed with ethyl acetate (25OmL), and suck dried for 2 hours to afford 32Og of the title compound having purity 78.5% by HPLC. Example 7: Preparation of temozolomide
Acetic acid (9ml, 0.159mol) was added to a suspension of 5-ammo-N1 -methyl- lH-imidazole- 1,4- dicarboxamide (1Og, 0.054mol) and sodium nitrite (5g, 0.072mol) in water (100ml) at -5 to 00C at a rate so that temperature does not rise above 0-50C. The reaction mixture was stirred at 0-50C for one and half hour. Brine (30g) was added to the reaction mixture and stirred at room temperature for two hours to saturate the reaction mixture. The reaction mass was extracted with a 2.5% solution of dimethylsulfoxide in dichloromethane (5 X 1 L). Combined organic layer was dried over sodium sulfate and filtered through a hyflow bed. Solvent was removed under reduced pressure and residual dimethylsulfoxide layer was degassed completely. The dimethylsulfoxide layer was cooled to 0 to -5°C and stirred for 1 hour. The resulting solid was filtered, washed with ethyl acetate (2x 5 ml), and suck dried for 2 hours to afford 5.0 g of the title compound having purity 81.6% by HPLC. Example 8: Preparation of temozolomide
Acetic acid (450ml) was added to a suspension of 5 -amino-N1 -methyl- lH-imidazole- 1,4- dicarboxamide (500g) and sodium nitrite (25Og) in water (5.0 L) at -5 to O0C at a rate so that temperature does not rise above 0-50C. The reaction mixture was stirred at 0-50C for one and half hour and the absence of starting material was checked by HPLC analysis. Ice bath was removed and powdered calcium chloride (1.25 kg) was added to the reaction mixture and stirred at room temperature for two hours. The reaction mass was extracted with a 2.5% solution of dimethylsulfoxide in dichloromethane (5 X 50 L). Combined organic layer was dried over sodium sulfate and filtered through a hyflo bed. Solvent was removed under reduced pressure at below 400C and residue at 35- 400C was filtered through a candle filter to remove suspended particles and the filtrate was then degassed completely. The residual dimethylsulfoxide layer was cooled to 0±2°C and stirred for one hours. The resulting solid was filtered and sucked dried. The solid was then washed with ethyl acetate (2x 250 ml), and suck dried for 1 hours to afford 240 g of the title compound.
………………………………….
SYNTHESIS
http://www.google.com/patents/US20020133006
Example 1
Preparation of Temozolomide (1)

5-Amino-1H-imidazole-4-carboxamide.HCl (4) (25 g, 0.154 mol) (Aldrich 16,496-8), CH2Cl2(0.6 L) and Et3N (45 mL) (Aldrich, 13,206-3) were placed into a dry 2-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen at ambient temperature. The mixture was stirred, and a solution of 400 mL of 4-nitrophenyl chloroformate (34 g, 0.169 mol) (Aldrich, 16,021-0) in CH2Cl2 was added dropwise. The reaction mixture was stirred vigorously for 4 hours and then left to stand for 18 hours at room temperature. The precipitate was collected by vacuum filtration and washed with H2O (1.5 L) to afford the product (3) as a pale yellow solid (42 g, 0.144 mol).
1H NMR (400 MHz, DMSO-d6, δ): 8.40 (d, 2H), 7.83 (s, 1H), 7.74 (d, 2H), 7.08 (bs, 1H), 6.95 (bs, 1H), 6.52 (s, 2H).

Compound (3) (42 g, 0.144 mol) and DMF (0.27 L) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was cooled to 0° C., and methylhydrazine (10 mL, 0.188 mol) (Aldrich, M5,000-1) was added dropwise. The reaction mixture was stirred vigorously for 1 hour at 0° C. and was then poured into EtOAc (2.1 L). The precipitate was collected by vacuum filtration and was dried under vacuum (20 mm Hg, room temperature, 18 hours) to afford (2) as a tan solid (27.1 g, 0.137 mol).
1H NMR (400 MHz, DMSO-d6, δ): 7.62 (s, 1H), 6.85 (bs, 1H), 6.75 (bs,1H), 6.00 (s, 2H), 5.10 (s, 2H), 3.15, s, 3H).mp: 188° C. (dec.).
Analysis: Calcd for C6H10N6O2: C, 36.36; H, 5.09; N, 42.41.
Found: C, 36.46; H, 4.99; N, 42.12.

Compound (2) (500 mg, 2.5 mmol), Bu4NI (95 mg, 0.25 mmol), THF (250 mL) and CH3CN (250 mL) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was heated at 60° C. for 20 mm and then cooled to room temperature. H5I06 (1.14 g, 5 mmol) was added and the reaction mixture was stirred vigorously at room temperature for 1 hour. The resulting solution was treated with saturated aqueous Na2S2O3 (5 mL) and was then concentrated under reduced pressure to dryness. The residue was treated with CH3CN (200 mL) and was filtered. The filtrate was concentrated and chromatographed on a column of silica gel (1.5% to 2% AcOH/EtOAc) to afford temozolomide (1) (280 mg).
1H NMR (400 MHz, DMSO-d6, δ): 8.80 (s, 1H), 7.80 (bs, 1H), 7.66 (bs, 1H), 3.43 (s, 3H).
…………………….
EXAMPLES
Example 1:
- Preparation of 3-Methyl-8-aminocarbonyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (Temozolomide).
-
Glacial acetic acid (25 ml), water (250 ml) and LiCl (225 g) were charged and the contents were stirred for 30 minutes and cooled to room temperature. 5-Amino-1-(N-methylcarbamoyl) imidazole-4-carboxamide (II) (25 g) was added and stirred the contents for further 30 minutes. The reaction mixture was cooled to 0°C and then added drop wise to NaNO2 solution (12.5 g in 50 ml water) at -10 to 5 °C. The reaction mass was stirred for 1 hr at 0-5 °C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution (25 g in 250 ml of water) was added slowly and stirred for 20 minutes (solution A). This process yielded an acidic solution containing temozolomide.
……………………..
SYNTHESIS
EXAMPLES Example 1
A 250 ml reaction vessel equipped with a magnetic stirrer and a reflux condenser was charged with 8-cyano-3-methyl-[3H]-imidazo-[5,1-d]-tetrazin-4-one (10 grams, 0.0568 mol) and hydrochloric acid (36.5-38%, 50 ml). The reaction mixture was heated to 32-35° C. and stirring was maintained at this temperature for about 3 hours. A sample was withdrawn and analyzed by HPLC to verify that the high conversion was received. (If the content of the starting material 8-cyano-3-methyl-[3H]-imidazo-[5,1-d]-tetrazin-4-one is more than 2.5% by area according to HPLC, the stirring may be continued for additional one hour).
The reaction mixture was then cooled to 20° C. and 50 ml of acetone were added drop-wise while maintaining the temperature at 20° C. Stirring was continued for 15-30 minutes. The precipitated white crystals were washed with cold acetone (20 ml) and dried at 40° C. in vacuum to obtain 11.7 grams (0.0507 mol) of Temozolomide hydrochloride (89.3% yield). Purity (by HPLC): 99.6%.
…………………………
SYNTHESIS
EXAMPLES
The following Examples illustrate but do not in any way limit the present invention. Chemicals obtained from Aldrich Chemical Company (Milwaukee, Wis.) are identified by their catalog number. It should be noted that nomenclature may differ slightly between this specification and the Aldrich catalog.
Example 1 Preparation of Temozolomide (1)
Step A Preparation Compound (3)

5-Amino-1H-imidazole-4-carboxamide.HCl (4) (25 g, 0.154 mol) (Aldrich 16,496-8), CH2Cl2(0.6 L) and Et3N (45 mL) (Aldrich, 13,206-3) were placed into a dry 2-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen at ambient temperature. The mixture was stirred, and a solution of 400 mL of 4-nitrophenyl chloroformate (34 g, 0.169 mol) (Aldrich, 16,021-0) in CH2Cl2was added dropwise. The reaction mixture was stirred vigorously for 4 hours and then left to stand for 18 hours at room temperature. The precipitate was collected by vacuum filtration and washed with H2O (1.5 L) to afford the product (3) as a pale yellow solid (42 g, 0.144 mol).
1H NMR (400 MHz, DMSO-d6, δ): 8.40 (d, 2H), 7.83 (s, 1H), 7.74 (d, 2H), 7.08 (bs, 1H), 6.95 (bs, 1H), 6.52 (s, 2H).
Step B Preparation of Compound (2)

Compound (3) (42 g, 0.144 mol) and DMF (0.27 L) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was cooled to 0° C., and methylhydrazine (10 mL, 0.188 mol) (Aldrich, M5,000-1) was added dropwise. The reaction mixture was stirred vigorously for 1 hour at 0° C. and was then poured into EtOAc (2.1 L). The precipitate was collected by vacuum filtration and was dried under vacuum (20 mm Hg, room temperature, 18 hours) to afford (2) as a tan solid (27.1 g, 0.137 mol).
1H NMR (400 MHz, DMSO-d6, δ): 7.62 (s, 1H), 6.85 (bs, 1H), 6.75 (bs,1H), 6.00 (s, 2H), 5.10 (s, 2H), 3.15, s, 3H).mp: 188° C. (dec.). Analysis: Calcd for C6H10N6O2: C, 36.36; H, 5.09; N, 42.41. Found: C, 36.46; H, 4.99; N, 42.12.
Step C Preparation of Temozolomide (1)

Compound (2) (500 mg, 2.5 mmol), Bu4NI (95 mg, 0.25 mmol), THF (250 mL) and CH3CN (250 mL) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was heated at 60° C. for 20 mm and then cooled to room temperature. H5IO6 (1.14 g, 5 mmol) was added and the reaction mixture was stirred vigorously at room temperature for 1 hour. The resulting solution was treated with saturated aqueous Na2S2O3 (5 mL) and was then concentrated under reduced pressure to dryness. The residue was treated with CH3CN (200 mL) and was filtered. The filtrate was concentrated and chromatographed on a column of silica gel (1.5% to 2% AcOH/EtOAc) to afford temozolomide (1) (280 mg).
1H NMR (400 MHz, DMSO-d6, δ): 8.80 (s, 1H), 7.80 (bs, 1H), 7.66 (bs, 1H), 3.43 (s, 3H).

TEMOZOLOMIDE
References
- Malcolm Stevens – interview, Cancer Research UK impact & achievements page
- Newlands ES, Stevens MF, Wedge SR, Wheelhouse RT, Brock C (January 1997). “Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials”. Cancer Treat. Rev. 23 (1): 35–61. doi:10.1016/S0305-7372(97)90019-0. PMID 9189180.
- Stevens MF, Hickman JA, Langdon SP, Chubb D, Vickers L, Stone R, Baig G, Goddard C, Gibson NW, Slack JA et al. (November 1987). “Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine”. Cancer Res. 47 (22): 5846–52.PMID 3664486.
- Jacinto, FV; Esteller, M (August 2007). “MGMT hypermethylation: a prognostic foe, a predictive friend.”. DNA Repair 6 (8): 1155–60. doi:10.1016/j.dnarep.2007.03.013. PMID 17482895.
- Hegi ME, R, Hau, Mirimanoff et al. (March 2005). “MGMT gene silencing and benefit from temozolomide in glioblastoma”. N. Engl. J. Med. 352 (10): 997–1003. doi:10.1056/NEJMoa043331.PMID 15758010. More than one of
|last1=and|author=specified (help) - National Cancer Institute Of Canada Clinical Trials, Group; Hegi, ME; Mason, WP; Van Den Bent, MJ; Taphoorn, MJ; Janzer, RC; Ludwin, SK; Allgeier, A et al. (May 2009). “Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial”. Lancet Oncology 10 (5): 459–466. doi:10.1016/S1470-2045(09)70025-7. PMID 19269895.
- Sitbon Sitruk, L.; Sanson, M.; Prades, M.; Lefebvre, G.; Schubert, B.; Poirot, C. (2010). “Chimiothérapie à gonadotoxicité inconnue et préservation de la fertilité : Exemple du témozolomide☆”.Gynécologie Obstétrique & Fertilité 38 (11): 660–662. doi:10.1016/j.gyobfe.2010.09.002. PMID 21030284. edit
- Gilbert MR (March 2006). “New treatments for malignant gliomas: careful evaluation and cautious optimism required”. Ann. Intern. Med. 144 (5): 371–3. PMID 16520480.
- Pyrko P, Schönthal AH, Hofman FM, Chen TC, Lee AS (October 2007). “The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas”.Cancer Res. 67 (20): 9809–16. doi:10.1158/0008-5472.CAN-07-0625. PMID 17942911.
- Sheehan J, Cifarelli C, Dassoulas K, Olson C, Rainey J, Han S (2010). “Trans-sodium crocetinate enhancing survival and glioma response on magnetic resonance imaging to radiation and temozolomide”. Journal of Neurosurgery 113 (2): 234–239. doi:10.3171/2009.11.JNS091314. PMID 20001586.
- “Safety and Efficacy Study of Trans Sodium Crocetinate (TSC) With Concomitant Radiation Therapy and Temozolomide in Newly Diagnosed Glioblastoma (GBM)”. ClinicalTrials.gov. November 2011.
- Ueno T, Ko SH, Grubbs E et al. (March 2006). “Modulation of chemotherapy resistance in regional therapy: a novel therapeutic approach to advanced extremity melanoma using intra-arterial temozolomide in combination with systemic O6-benzylguanine”. Mol. Cancer Ther. 5 (3): 732–8. doi:10.1158/1535-7163.MCT-05-0098. PMID 16546988.
- Friedman, HS; Jiang, SX; Reardon, DA; Desjardins, A; Vredenburgh, JJ; Rich, JN; Gururangan, S; Friedman, AH et al. (March 2009). “Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma”. J. Clin. Oncol. 27 (8): 1262–7. doi:10.1200/JCO.2008.18.8417. PMC 2667825. PMID 19204199.
- http://labs.fhcrc.org/kiem/Hans-Peter_Kiem.html
- Dall’oglio S, D’Amico A, Pioli F, Gabbani M, Pasini F, Passarin MG, Talacchi A, Turazzi S, Maluta S (December 2008). “Dose-intensity temozolomide after concurrent chemoradiotherapy in operated high-grade gliomas”. J Neurooncol 90 (3): 315–9. doi:10.1007/s11060-008-9663-9. PMID 18688571.
- Osmani AH, Masood N; Masood (2012). “Temozolomide for relapsed primary CNS lymphoma”. J Coll Physicians Surg Pak 22 (9): 594–595. PMID 22980617.
- Chemotherapy Drug Shrinks Brain Tumors American Academy of Neurology, May 21, 2007
- Information for people undergoing treatment with temozolomide Cancer Research UK (CancerHelp UK)
Wang, et al., “Alternative Syntheses of the antitumor drug temozolomide avoiding the use of methyl isocyanates”, Journal of Chemical Society, Chemical Communication, Chemical Society, Letchworth, GB, p. 1687-1688 (1994).
Wang, et al., “Antitumor imidazotetrazines. Part 33. new syntheses of the antitumor drug temozolomide using ‘masked’ methyl isocyanates”, J. Chem. Soc., Perkin Trans. 1(21):2783-2787 (1995).
Wang, et al., “Synthetic studies of 8-carbamoylimidzo-‘5, 1-D!-1, 2, 3, 5-tetrazi n-4(3H)- one: a key derivative of antitumor drug temozolomide”, Bioorg. Med Chem. Lett., 6(2):185-188 (1996).
Yongfeng Wang, “A new route to the antitumor drug temozolomide, but not thiotemozolomide”, Chem. Commun., 4:363-364 (1997).
Wang, et al., “Antitumor Imidazotetrazines. 35. New Synthetic Routes to the Antitumor Drug Temozolomide”, J. org. Chem. 62(21):7228-7294 (1997).
Newlands, E.S., et al., “Temozolomide: a review of its discovery, chemical properties, pre-clinica development and clinical trials”, Cancer Treat. Rev. , 23(1):35-61 (1997).
Wang, et al., Antitumor Imidazotetrazines. Part 36. Conversion of 5-Amino-Imidazole-4-Carboxamide to . . . Journal of the Chemical Society, Perkin Transactions 1, Chemical Society, Letchworth, GB, 10:1669-1675 (1998).
1 Catapano CV, et al. Cancer Res. 1987, 47(18), 4884-4889.
[2] Sun S, et al. J Neurooncol. 2012.
[3] Bauer M, et al. PLoS One. 2012, 7(6):e39956.
[4] Wong ST, et al. Anticancer Res. 2012, 32(7), 2835-2841.
[5] Lin CJ, et al. PLoS One. 2012, 7(6), e38706.
[6] Gori JL, et al. Cancer Gene Ther. 2012.
| US5260291 | Oct 18, 1991 | Nov 9, 1993 | Cancer Research Campaign Technology Limited | Tetrazine derivatives |
| US20020133006 | Jan 16, 2002 | Sep 19, 2002 | Schering Corporation | Synthesis of temozolomide and analogs |
| US20050131227 | Jan 21, 2005 | Jun 16, 2005 | Schering Corporation | Synthesis of temozolomide and analogs |
| US20060183898 * | Feb 16, 2006 | Aug 17, 2006 | Olga Etlin | Process for preparing temozolomide |
| CN1487941A * | Jan 16, 2002 | Apr 7, 2004 | 先灵公司 | Synthesis of temozolomide and analogs |
| CN1706843A * | Apr 8, 2005 | Dec 14, 2005 | 江苏天士力帝益药业有限公司 | Temozolomide refining process |
| US20060183898 * | Feb 16, 2006 | Aug 17, 2006 | Olga Etlin | Process for preparing temozolomide |
| US20070225496 * | Mar 23, 2007 | Sep 27, 2007 | Palle Raghavendracharyulu Venk | rocess for preparing temozolomide |
| US8258294 * | Sep 28, 2007 | Sep 4, 2012 | Cipla Limited | Process for the preparation of temozolomide and analogs |
| EP2151442A2 | Jul 22, 2009 | Feb 10, 2010 | Chemi SPA | Process for preparing temozolomide |
| EP2374807A2 * | Sep 28, 2007 | Oct 12, 2011 | Cipla Limited | An improved process for the isolation of temozolomide |
| WO2008038031A1 | Sep 28, 2007 | Apr 3, 2008 | Cipla Ltd | An improved process for the preparation of temozolomide and analogs |
| WO2010140168A1 * | Jun 2, 2010 | Dec 9, 2010 | Ind-Swift Laboratories Limited | Improved process for preparing temozolomide |
| WO2011036676A2 | Sep 14, 2010 | Mar 31, 2011 | Ashwini Nangia | Stable cocrystals of temozolomide |
