
Home » FDA 2019 (Page 5)
Category Archives: FDA 2019
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ferric Maltol, マルトール第二鉄
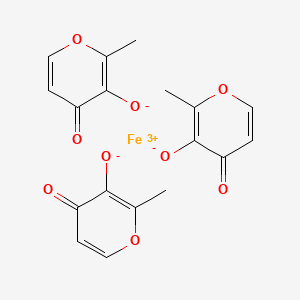
Ferric Maltol
Iron, tris(3-hydroxy-2-methyl-4H-pyran-4-onato-O3,O4)-
| Molecular Formula: | C18H15FeO9 |
|---|---|
| Molecular Weight: | 431.154 g/mol |
iron(3+);2-methyl-4-oxopyran-3-olate
RN: 33725-54-1
UNII: MA10QYF1Z0
Feraccru
Ferric maltol; UNII-MA10QYF1Z0; MA10QYF1Z0; Ferric maltol (INN); Ferric maltol [INN]; 33725-54-1
Shield Therapeutics, under license from Vitra Pharmaceuticals
UPDATE
- Originator University of Cambridge; University of London
- Developer Shield Therapeutics
- Class Antianaemics; Ferric compounds; Pyrones; Small molecules
- Mechanism of Action Iron replacements
- Marketed Iron deficiency anaemia
Most Recent Events
- 26 Jul 2019 Registered for Iron deficiency anaemia (In adults) in USA (PO)
- 24 Apr 2019 Swissmedic approves a extension of the approved indication for ferric maltol to include treatment of all adults with iron deficiency (ID) with or without anaemia
- 14 Mar 2019 The European Patent Office decides in favour of Shield Therapeutics in relation to the group’s patent No. 2 668 175
The US Food and Drug Administration (FDA) has approved oral ferric maltol(Accrufer, Shield Therapeutics) AUG 2019 for the treatment of iron deficiency in adults.
The product is already approved in the European Union and Switzerland for the treatment of iron deficiency in adults, where it is sold as Feraccru.
OLD DATA NDA filing expected in US in 2H 2018, Ph 3 trial is planned in 2018/19 for treatment of iron deficiency anemia (IDA) in children., Expected dose form: Oral Capsule; 30 mg
Treatment of iron deficiency anemia (IDA) associated with inflammatory bowel disease (IBD) and Chronic Kidney disease.
Iron deficiency anaemia (IDA) occurs when iron levels are insufficient to support red blood cell production and is defined – according to the WHO – as haemoglobin levels below 13 g/dL in men over 15 years, below 12 g/dL in non-pregnant women over 15 years, and below 11 g/dL in pregnant women. Iron is absorbed at the apical surface of enterocytes to be transported by ferroportin, the only known iron exporter, across the basolateral surface of the enterocyte into circulation. Inflammation from IBD interferes with iron absorption by causing an increase in hepcidin, a peptide hormone synthesized in the liver that inhibits ferroportin activity. Anaemia is the most common extra-intestinal complication of inflammatory bowel disease (IBD) and although it often involves a combination of IDA and anaemia of chronic disease, IDA remains an important contributor in this condition due to chronic intestinal bleeding and decreased iron intake (from avoidance of foods that may exacerbate symptoms of IBD). In a variety of populations with IBD, the prevalence of iron deficiency anaemia ranges from 36%-76%. The serum markers of iron deficiency are low ferritin, low iron, raised total iron binding capacity, raised red cell protoporhyrin and increased transferrin binding receptor (sTfR). Serum ferritin is the most powerful test for iron deficiency. The cut-off level of ferritin which is diagnostic varies between 12-15 µg/L. Higher levels of serum ferritin do not exclude the possibility of iron deficiency, and a serum ferritin level of <100 μg/L may still be consistent with iron deficiency in patients with IBD. A transferrin saturation of <16% is indicative of iron deficiency, either absolute or functional. Other findings on a complete blood count panel that are suggestive of iron deficiency anaemia, but are not considered diagnostic, include microcytosis, hypochromia, and elevation of red cell distribution width.
A deficiency of iron can have a significant impact on a patient’s quality of life. Appropriate diagnosis and treatment of iron deficiency anaemia are important to improve or maintain the quality of life of patients. The goals of treatment are to treat the underlying cause, limit further blood loss or malabsorption, avoid blood transfusions in haemodynamically stable patients, relieve symptoms, and improve quality of life. More specifically, therapeutic goals of treatment include normalizing haemoglobin levels within 4 weeks (or achieving an increase of >2 g/dL) and replenishing iron stores (transferrin saturation >30%). Oral iron supplementation has been considered standard treatment because of an established safety profile, lower cost, and ease of administration. It has been shown to be effective in correcting anaemia and repleting iron stores. One concern with higher doses of daily oral iron is intolerance due to GI side effects. Symptoms include nausea, vomiting, diarrhea, abdominal pain, constipation, and melena-like stools. Guidelines on the Diagnosis and Management of Iron Deficiency and Anaemia in Inflammatory Bowel Diseases recommend IV iron therapy over oral iron supplementation in the treatment of iron deficiency anaemia in patients with IBD, citing faster and prolonged response to treatment, decreased irritation of existing GI inflammation, improved patient tolerance, and improved quality of life. Patients with severe anaemia (haemoglobin level of <10 g/dL), failure to respond or intolerance to oral iron therapy, severe intestinal disease or patients receiving concomitant erythropoietin are recommended indications for IV iron therapy. Other conditions where patients should be considered for first-line IV therapy over oral therapy include congestive heart failure, upper GI bleeding, and in situations where rapid correction of anaemia may be required.
Across EU there are several iron (Fe+2) oral preparations as ferrous fumarate, ferrous gluconate, ferrous sulphate and ferrous glycine sulfate, formulated as tablet, solution or gastroresistent capsules. All ferrous compounds are oxidised in the lumen of the gut or within the mucosa with release of activated hydroxyl radicals, which may attack the gut wall and can effect a range of gastrointestinal symptoms and discomfort. Ferric preparations also exist but with less bioavailability. Across EU there are also several IV products on the market: iron (III) hydroxide dextran complex, iron sucrose, ferric carboxymaltose, iron isomaltoside. IV iron therapy, however, is inconvenient, invasive and associated with the risk of rare but serious hypersensitivityreactions; it is used in those situations when oral preparations cannot be used or when there is a need to deliver iron rapidly. Feraccru is a trivalent iron, oral iron replacement preparation. The active substance of Feraccru is ferric maltol (also known as 3-hydroxy-2-methyl-4H-pyrane-4-one iron (III) complex, or ST10, or ferric trimaltol or ferric maltol) an oral ferric iron/maltol complex. It is presented as red hard gelatine capsules containing 30 mg iron (ferric iron). Maltol is a sugar derivative that strongly chelates iron in the ferric form (FeIII) rendering the iron stable and available for absorption. Upon dissociation of the ferric maltol complex, the maltol molecules are absorbed and glucuronidated in the intestinal wall, and within the liver during first pass metabolism, and subsequently eliminated from the body in the urine. The iron is absorbed via the endogenous dietary iron uptake system. The indication finally agreed with the CHMP was: Feraccru is indicated in adults for the treatment of iron deficiency anaemia (IDA) in patients with inflammatory bowel disease (IBD) (see section 5.1). The proposed dosage is one 30 mg capsule twice daily on an empty stomach, corresponding to 60 mg ferric iron per day. There was agreement in the paediatric investigation plan to grant a deferral and a waiver for iron as iron (III)-maltol complex (EMEA-001195-PIP01-11).The PDCO granted a waiver in infants under 6 months of age and a referral for the completion of the planned paediatric studies (ST10-021 PK-PED/ST10-01-102, an open label, randomised, multiple-dose, parallel PK study and ST10-01-303, a randomised, open label comparative safety and efficacy study of ST10 and oral ferrous sulphate as comparator) until the adult studies are completed.

SYN
Patent
https://patents.google.com/patent/WO2017167963A1/en
The sugar derivative maltol is a hydroxypyrone (IUPAC name: 3- hydroxy-2-methyl-4£f-pyran-4-one) and it strongly chelates iron and the resulting complex (ferric trimaltol) is well absorbed, unlike many other ferric iron therapies. Ferric trimaltol appears well tolerated even in populations highly susceptible to gastrointestinal side-effects, such as IBD patients (Harvey et al . , 1998), and as such it provides a valuable alternative to patients who are intolerant of oral ferrous iron products, notably in place of intravenous iron. Clinical trials using ferric trimaltol have been carried out, see for example, Gasche et al., 2015.
However, despite the evidence of bioavailability and tolerability for ferric trimaltol, its clinical development has been limited by the absence of adequate synthetic routes. In particular, most manufacturing processes require the use of organic solvents, which increase manufacturing costs, for example to deal with post-synthesis solvent removal, and require additional safety measures, for example to deal with flammability . Critically, solvent-based syntheses are not robust and often generate ferric hydroxide, described in the prior art to be an unwanted impurity of the synthesis.
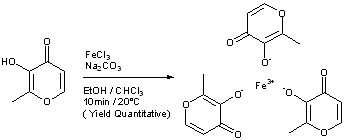
WO 03/097627 (Vitra Pharmaceuticals Limited) describes the synthesis of ferric trimaltol from iron salts of carboxylic acids in aqueous solution at a pH greater than 7. In a first
synthesis, ferric citrate is added to a solution of sodium hydroxide at room temperature and maltol is added to a second solution of sodium hydroxide at pH 11.6. The ferric citrate solution is added to the maltol solution, leading to the
production of a deep red precipitate. This composition is then evaporated until dryness and the material is powdered and dried. Alternative syntheses are described using ferrous fumarate or ferrous gluconate as the iron carboxylate salt starting material, and by dissolving maltol in sodium carbonate solution in place of sodium hydroxide. However, despite the fact that this process is fully aqueous, several of the iron carboxylate salts employed are expensive, especially as they need to be pharmaceutical grade if the ferric trimaltol is to be suitable for human administration. More importantly, this process introduces high levels of
carboxylates (equimolar to iron or greater) to the synthesis that are not easily removed by filtration or centrifugation of the ferric trimaltol cake. Instead these water soluble contaminants must be washed off (e.g. water washed), but this would result in considerable losses of the product due to the amphipathic nature of ferric trimaltol.
WO 2012/101442 (Iron Therapeutic Holdings AG) describes the synthesis of ferric trimaltol by reacting maltol and a non- carboxylate iron salt in an aqueous solution at alkaline pH .
However, despite the lower cost of non-carboxylate iron salts, pharmaceutically appropriate grades are still required if the ferric trimaltol is to be suitable for human administration and hence are comparatively expensive starting materials.
Importantly, the use of non-carboxylate iron salts (e.g. ferric chloride) results in the addition of considerable levels of the respective counter-anion (e.g. three moles of chloride per every mole of iron) of which a significant part is retained in the filtration (or centrifugation) cake and thus must be washed off. As such, WO 2012/101442 does not address the problem of product losses in WO 03/097627. Furthermore, the addition of a non- carboxylate iron salt (e.g. ferric chloride) to a very alkaline solution, as described in WO 2012/101442, promotes the formation of stable iron oxides, which is an unwanted contaminant in ferric trimaltol . As a consequence, further costly and time-consuming processing of the material would be required for manufacturing .
Overall, the cost of the current aqueous syntheses is driven by regulatory demands for low levels of toxic heavy metals and residual reagents in the final pharmaceutical formulation, which force the use of highly purified, and thus expensive, iron salts as well as thorough washing of the final product (resulting in significant losses of product) . This will impact on the final price of ferric trimaltol and potentially limits patient access to this therapy. As such, there is a need for a process that can use lower iron grades and limited wash cycles, whilst producing ferric trimaltol of adequate purity.
Ferric maltols are a class of compounds that include ferric trimaltol, a chemical complex formed between ferric iron (Fe3+) and the hydroxypyrone, maltol (IUPAC name: 3-Hydroxy-2-methyl-4£f- pyran-4-one) , in a molar ratio of ferric iron to maltol of 3:1. Maltol strongly chelates the ferric iron and the resulting complex (ferric trimaltol which may also be written as ferric tri-maltol) is well absorbed, in contrast to some other ferric iron supplements, fortificants and therapies. Maltol binds metal cations mainly in the form of a dioxobidentate ligand in a similar manner proposed for other 4 ( 1H) -pyranones :

Structure of maltol (3-hydroxy-2-methyl-4 (H) -pyran-4-one) and dioxo-chelation to metal cations (M) such as iron. For ferric trimaltol three maltol groups surround one iron.
Examples
Example 1: Ferric trimaltol from L-lyslne coated ferric hydroxide
Synthesis of lysine-coated ferric hydroxide colloid
14.87g FeCl3. 6H20 was added to 25 mL UHP water and stirred until dissolved. 14.9g NaOH 5M was then added drop-wise to this solution with constant stirring, during which a ferric hydroxide colloid was gradually produced. This colloidal suspension was then added to a L-Lysine suspension (5.02g in 25mL ddH2<D) .
Ferric trimaltol synthesis
7 g NaOH pellets was added to 25 mL UHP water and stirred until dissolved. Next, 24.5g maltol was added and stirred until dissolved. Then, the suspension of lysine-coated ferric
hydroxide colloids was gradually added to the maltol with vigorous stirring, producing a dark red precipitate (with a significant brown hue) . This suspension was incubated overnight during which time it became lighter and the brown hue
disappeared. This precipitate was then recovered by
centrifugation (4500 rpm x 5min) and dried overnight (50°C) .
Example 2: Ferric trimaltol from L-lysine modified ferric hydroxide
Synthesis of lysine-modified ferric hydroxide gel
14.87g FeCl3.6H20 and 5.02g L-Lysine were added to 25 mL UHP water and stirred until dissolved. 32 mL NaOH 5M was then gradually added to this solution producing a ferric hydroxide gel .
Ferric trimaltol synthesis
7 g NaOH pellets was added to 25 mL UHP water and stirred until dissolved. Next, 24.5g maltol was added and stirred until dissolved. Next, the lysine-modified ferric hydroxide gel was gradually added to this solution with vigorous stirring. A 1.2 M HC1 solution was then used to drop the pH of the solution to 10, which was then incubated for 70 min. Finally, a dark red precipitate (i.e., ferric trimaltol) was recovered by
centrifugation (4500 rpmx5min) and dried overnight (45°C) .
Example 3: Absence of ferric hydroxide in ferric trimaltol
Ferric trimaltol is soluble in ethanol whereas ferric hydroxide (a potential contaminant) is not. As such ferric trimaltol powders produced as per Examples 1 and 2 were dissolved in ethanol. The material from Example 2 dissolved completely confirming the absence of iron hydroxides whereas the material from Example 1 did not. This supported the preference in the present invention for ligand modification, rather just surface coating, to ensure full conversion to ferric trimaltol .
Example 4: Ferric trimaltol from tartrate-modified ferric hydroxide
Synthesis of tartrate-modified ferric hydroxide gel
14.87g FeCl3.6H20 (0.055 mol) was added to 25 mL UHP water and stirred until dissolved. 4.12 g tartaric acid (0.0275 mol) was added to this solution and stirred until dissolved. 38 mL NaOH 5M was then gradually added to this solution producing a ferric hydroxide gel .
Ferric trimaltol synthesis
2 g NaOH pellets was added to 25 mL UHP water and stirred until dissolved. Next, 24.5g maltol was added and stirred. This produced a slurry in which most of the maltol remained
undissolved. Next, the tartrate-modified ferric hydroxide gel was gradually added to this solution with vigorous stirring during which the remainder of maltol dissolved. After 15 min a dark red precipitate (i.e. ferric trimaltol) had been formed and pH had stabilised at 8.5. The material was then washed by (1)
centrifuging, (2) disposing of the supernatant and (3)
resuspending in water back to its original volume. Finally, the material was recovered by centrifugation (4500 rpm x 5min) and dried overnight (50°C) . Previously disclosed synthetic processes for the production of ferric trimaltol under aqueous conditions require the addition of NaOH (or other suitable bases) for conversion of maltol from its protonated form to its deprotonated form prior to complexation of iron. However this results in the formation of unwanted sodium ions which must be washed off. In contrast, the use of ferric hydroxides according to the methods of the present invention reduces the requirements for base and associated counter cation (e.g. sodium), which is a favourable feature. Note that ferric hydroxides are represented above as Fe (OH) 3 for illustrative purposes only. Different iron hydroxides possess different structures and elemental compositions (see Cornell & Schwertmann, The Iron Oxides Structure, Properties, Reactions, Occurrence and Uses. 2nd edition, 1996, VCH Publishers, New York) . Example 5: Ferric trimaltol from tartrate-modified ferric hydroxide (with removal of contaminants from ferric hydroxide)
Material prepared as in Example 4, except excess reactants and reaction products (e.g. unbound tartaric acid, sodium chloride) were removed from the ferric hydroxide gel. This was achieved by centrifuging the ferric hydroxide gel after its synthesis and discarding the supernatant, which contained unwanted soluble species. Finally, the ferric hydroxide gel was re-suspended in water back to its original volume prior to being added to a maltol slurry.
Example 6: Ethanolic clean up for ferric trimaltol produced from ligand coated ferric hydroxide
Ferric trimaltol precipitate was purified as it contained an unwanted iron oxide fraction. Part of the wet pellet recovered by centrifugation (4.5 g) was dissolved in 1L ethanol. The iron oxide fraction (which remained undissolved) was then removed by filtration, producing a turbidity-free solution. Next, ethanol was evaporated (40°C in a rotavapor under vacuum) producing a concentrated ferric trimaltol slurry. This was then recovered and oven dried overnight at 50°C.
PATENT
https://patents.google.com/patent/WO2012101442A1/en
Comparative Example 1
Preparation of Iron Trimaltol from Pure Maltol Maltol was dissolved in an aqueous solution of ferric chloride and ferric trimaltol was precipitated upon the addition of sodium hydroxide.
An accurate mass of ferric chloride hexahydrate granules (330g) was dissolved in distilled water to yield a pH of 0.6. To this solution, an equimolar amount of maltol was added (490g in total, initially 250g) and allowed to dissolve with continuous stirring. The pH of this solution was found to be zero and the colour of this solution was deep- purple. Spectroscopy showed that the initial solution was mainly a 1 :1 Fe/maltol mixture with some 1 :2 component. The remaining maltol was added. After an hour of stirring, sodium hydroxide (147g NaOH in 750 ml water) was added dropwise to the solution until a pH of 8.3 was achieved. The solution and precipitate were red. The precipitate was collected using a Buchner funnel under vacuum. The precipitate was dried at 40°C under vacuum.
Maltol is only slightly soluble in an aqueous acidic reaction medium. After an hour of stirring, traces of undissolved maltol were visible on the surface of the ferric chloride/maltol solution, on the walls of the reaction vessel and on the stirrer. Upon addition of sodium hydroxide, there appeared to be lumps of a brownish-black substance on the walls of the reaction vessel and on the stirrer which seemed to add to the impurities in the desired product.
An attempt to heat the ferric chloride/maltol solution so as to assist the maltol to dissolve in the ferric chloride solution resulted in a burnt, off spec, colour iron maltol sample. This method also produces two by-products which consume expensive maltol namely Fe(OH)2 (Maltol) and Fe (OH) (Maltol)2.
The sodium hydroxide solution has to be added extremely slowly to prevent “gumming up” and formation of undesirable lumps at the bottom of the reaction vessel. A yield of about 78% ferric trimaltol was obtained using this method of preparation.
When maltol is added to a ferric chloride solution at a low pH, no ferric trimaltol is formed and ferric hydroxide is generated with ferric monomaltol and a small percentage of ferric dimaltol species. The charge neutralisation of these complexes is either the hydroxy! functional group or the chloride anion. This addition also results in the formation of black deposits and gums consisting of ferric chloride/ferric hydroxide polymers. These black deposits are also produced if the solutions are heated. Therefore it is not possible to obtain the correct stoichiometry for the formation of ferric trimaltol and manufacture a pharmaceutically acceptable product using this method.
The addition of maltol to an aqueous solution of ferric or ferrous chloride was deemed impractical for scale up and manufacturing purposes and Examples 2 to 4 investigate the addition of the iron chlorides to maltol in solution.
The problem of working in an aqueous environment
Ferric chloride as a hydrated ion in aqueous solution is a strong Lewis acid with a Ka of 7x 103 and ferrous chloride as a hydrated ion in aqueous solution is also a strong Lewis acid with a Ka of 5 x 10“9. Over the desired range for using iron chlorides as starting materials for the synthesis of ferric trimaltol, ferric chloride in aqueous solution has a pH value in the range of 1-3 and ferrous chloride has a pH in the range of 3-5. Furthermore, commercial solutions of iron chlorides have a pH circa 1 because they are stabilised by the addition of hydrochloric acid to prevent the precipitation of ferric hydroxide species.
The present invention recognises that maltol is virtually insoluble at these low pH values and has limited solubility when dissolved in water in the pH range 6-8. The maximum aqueous solubility is 1g/100m! at 20°C. However, the solubility of maltol can be increased to 10g/100ml by heating to near boiling temperatures. Maltol is stable in aqueous solution at these temperatures and this property has been employed in Example 4 to synthesise ferric trimaltol. At low pH values ferric trimaltol is not the preferred species due to disproportionation. In order to obtain significant amounts of ferric trimaltol using a stoichiometric ratio of iron salt to hydroxypyrone of 1 :3, the eventual pH of the solution must exceed 7 since below that pH ferric dimaltol and monomaltol species will exist. Therefore two methods of increasing the pH were researched 1) using sodium carbonate and 2) using sodium hydroxide. Other alkali hydroxides could be used such as potassium hydroxide. The sodium carbonate neutralisation was found to be less preferable due to C02 generation. This research lead to an improved synthesis of ferric trimaltol.
Example 2
Maltol was dissolved in an aqueous solution of sodium hydroxide and iron maltol was precipitated upon the addition of ferric chloride.
In view of some of the difficulties experienced in Example 1 , and the fact that maltol is very soluble in aqueous alkali hydroxide solutions, it was decided to change the manufacturing procedure.
The initial work using this method of preparation showed that a 90% yield was achieved. Various operating parameters were then optimised and the following procedure outlines the final method chosen. A yield of 95% was then achieved. An accurate mass of sodium hydroxide pellets (20g) was dissolved in distilled water to yield a pH of 13.50. An equimolar amount of maltol (63g) was added to this aqueous solution of NaOH to give a clear yellow coloured solution with a pH of 11.6. Almost immediately a stoichiometric amount of ferric chloride (45g) was added slowly to this solution to give a pH of 7.1 and a red precipitate formed, which was then collected using a Buchner funnel under vacuum. The precipitate was then dried at 40°C under vacuum.
Adding the maltol solution in sodium hydroxide to ferric chloride as in method 1 is not preferred since it gives an off spec product and gums and a black precipitate.
Maltol is very soluble in aqueous alkali hydroxide solutions giving a yellow solution. The concentration of the hydroxide solution preferably does not exceed 20%.
This method is advantageous since it has the potential to produce only one by-product viz, ferric hydroxide Fe(OH)3 which consumes some of the iron intended to complex with the maltol. This is not easily measurable in the presence of iron maltol and so the following method was used to measure the ferric hydroxide. Fe(OH)3 is insoluble in ethanol and so the iron maltol product was dissolved in ethanol. It was found that small amounts of Fe(OH)3 may be present in the batches of iron maltol synthesized according to Example 2.
Taking the extremes of the specification, in one embodiment, the amount of Fe(OH)3 present in the active material may not exceed 2 wt. % Fe(OH)3 based on the total weight of the composition. In view of its well known inert characteristics the level of this compound is adequately controlled and a final specification including controlled ferric hydroxide should be acceptable.
The mass balance for maltol and iron was closed at 99%.
A yield of 95% iron maltol was obtained using this method of preparation.
Example 3
Maltol was dissolved in an aqueous solution of Sodium Carbonate and Iron Maltol was precipitated upon the addition of Ferric Chloride.
An accurate mass of sodium carbonate (Na2C03) (53g) was dissolved in distilled water to give a solution having pH = 11.5. An equimolar amount of maltol (65g) was added to this aqueous alkali solution to give a murky creme coloured solution of pH = 9.9. A stoichiometric amount of a ferric chloride solution was added drop wise to this solution to a pH of 8.00. A further 15 grams of Na2C03 was added to this solution to increase the pH to 9.00. The remainder of the ferric chloride solution was then added to give a solution pH = 8.77 and a red coloured precipitate appeared.
The precipitate was collected using a Buchner funnel under vacuum. The precipitate was then dried at 40°C under vacuum. The release of C02 during the reaction tends to make this process less desirable due to foaming on the surface. The final product is a gellike solid when wet and the removal of moisture during drying can therefore be time consuming. The process may not be preferred but the ferric trimaltol produced could be acceptable.
Example 4 Maltol was dissolved in water and heated to a near boiling temperature and ferric or ferrous chloride was added to form a 1 :1/1:2 mixture of ferric maltol. The solution was allowed to cool and was added to maltol dissolved in sodium hydroxide. Stage 1
Depending on the batch size required, the ferric chloride was added slowly to a maltol solution in water at a pH of 6-7. The solubility of maltol is greatly enhanced up to 10g/100ml by heating to temperatures above 60°C. Addition of ferric chloride or ferrous chloride and monitoring the pH of the solution and maintaining the pH> 3 mainly produces ferric dimaltol species but very little ferric trimaltol. Above pH 3, no ferric hydroxide appeared to be generated. Ferric monomaltol and dimaltol species either with hydroxy or chloride giving the charge neutralisation are very soluble and a concentrated solution in excess of 30g/100ml can be generated. In order to obtain the correct stoichiometry for the formation of ferric trimaltol, further maltol is required and the pH needs to be corrected to values higher than 7.
As anhydrous ferrous or ferric chloride either 126g or 162g in 200ml of water can be added to a litre of water containing 120g of maltol. This ratio of iron to maltol does not provide sufficient maltol to produce any significant amounts of ferric trimaltol which does not precipitate at this stage.
Stage 2 Maltol in alkaline solution has been described as set out above. Conveniently, because maltol solutions up to 20% in sodium hydroxide have a pH circa 11.6, mixing of this solution with the ferric mono/dimaltol solutions from stage 1 yields a precipitate of ferric trimaltol with a deep characteristic burgundy red colour of high purity as determined by UV-vis spectroscopy. The filtrate yields product which is suitable for a GMP (good manufacturing process). The sodium chloride which is generated by this process is found in the supernatant since it has a much higher solubility at 35g/100ml than ferric trimaltol. The small amounts of sodium chloride in the ferric trimaltol can be reduced, if required, by washing in water. A further, surprising feature of the research resulted from work on ferrous chloride. Ferrous chloride may be substituted in stage 1 to form ferric dimaltol since the maltol was found to auto-oxidise the ferrous to ferric during the process of chelation. One aspect of this work which was considered to be potentially very useful if larger batch sizes were required arose from the finding that being a weaker Lewis acid than ferric chloride the pH of the starting solution was in excess of 3. Therefore the risk of generating ferric hydroxide was lower than with the use of ferric chloride at higher concentrations.
Ferrous and ferric chloride in solution or as a solid may be added to an alkaline solution of maltol in sodium hydroxide, combining stages 1 & 2. Providing a small excess of maltol up to about 10% is added then a precipitate of ferric trimaltol with a small amount of maltol is obtained. Such a preparation would be satisfactory as a GMP ferric trimaltol product.
PATENT
https://patents.google.com/patent/WO2017167963A1/en
AMPLE 1
Synthesis of ferric trimaltol using ferric citrate
NaOH (12g, 0.3 moles) is dissolved in water (50 ml) to form a sodium hydroxide solution. 20 ml of the sodium hydroxide solution is placed in a separate vessel.
Ferric citrate (30g, 0.11 moles) is slowly added to the sodium hydroxide solution in the separate vessel at room temperature with gentle stirring. Further portions of the sodium hydroxide solution are added to the solution of ferric citrate, as necessary, in order to ensure that all of the ferric citrate is dissolved.
Maltol (49g, 0.39 moles) is added to the remaining volume of sodium hydroxide solution and dissolved. The pH of the maltol solution is 11.6.
The ferric citrate solution is slowly added to the maltol solution with gentle stirring. A deep red precipitate forms; the supernatant is a deep red colour.
The solution is slowly evaporated to dryness at 60 to 80° C until the material is suitable for powdering. The material is powdered and the powder is then dried to a constant weight.
The yield of the final product is 87g. The final product comprises ferric trimaltol and sodium citrate. The product was assayed, using elemental analysis, for iron and sodium content. The iron content is 7.89% (theoretical 7.8%) and the sodium content is 13.45%.
The pH of a solution of the final product in water was measured. The pH of a 1% solution of the product by total weight of aqueous solution is 9.9 at 20°C.
EXAMPLE 2
Synthesis of ferric trimaltol using ferrous fumarate
NaOH (40g, 1 mole) is dissolved in water (100 ml) to form a sodium hydroxide solution. The pH of the solution is approximately 13.0.
Ferrous fumarate (170g, 1 mole) is slowly added to the sodium hydroxide solution at room temperature with gentle stirring.
Maltol (408g, 3.23 moles) is added to a separate volume of sodium hydroxide (40g, 1 mole) dissolved in water (100 ml) and dissolved. The pH of the solution is approximately 11.
The ferrous fumarate solution is slowly added to the maltol solution with gentle stirring. A deep red precipitate forms; the supernatant is a deep red colour.
The solution is slowly evaporated to dryness at 60 to 80° C until the material is suitable for powdering. The material is powdered and the powder is then dried to a constant weight. The yield of the final product is 615g.
The final product comprises ferric trimaltol and sodium fumarate.
EXAMPLE 3
Synthesis of ferric trimaltol using sodium carbonate to vary pH
Sodium carbonate (2.5g) is dissolved in 10ml of distilled water at room temperature. The pH of the solution is 11.6. Maltol (9.6g – three molar equivalents of sodium carbonate) is added to the sodium carbonate solution to give a cream coloured solution having a pH of 10.0.
A stoichiometric amount of ferric citrate (5g, allowing for a small excess of maltol) in an aqueous solution of sodium hydroxide (lg in 5ml of distilled water) is added slowly to the solution of maltol. The pH of the combined solutions is about 9. A red precipitate appears which is separated by decantation and dried at 80°C in an oven.
The red precipitate is ferric trimaltol, as confirmed by UV-Vis spectrometry.
EXAMPLE 4
Synthesis of ferric trimaltol using ferrous gluconate
Potassium hydroxide (5.5g) is dissolved in 50ml of distilled water at room temperature. To 25ml of this solution, maltol (16.5g, 0.13 moles) is added and gently heated to form a clear solution. To the other 25ml aliquot of the potassium hydroxide solution ferrous gluconate (22.5g) is added. This is gently heated to form a dark green saturated solution. The ferrous gluconate solution is added to the maltol solution and immediately a colour change to dark brown is noted.
On cooling, a deep brown precipitate forms (which is ferric trimaltol). The supernatant is a deep brown solution containing ferric trimaltol and potassium gluconate. The precipitate and the supernatant are dried separately at 80°C in an oven. The ferric trimaltol is a deep red brown powder with a characteristic caramel odour and UV-vis spectrum in aqueous solution.
EXAMPLE 5
Synthesis of ferric trimaltol using solid ferrous gluconate
Example 4 was repeated with the modification that the maltol is added to all of the 50 ml solution of potassium hydroxide and then solid ferrous gluconate is added directly to the maltol solution. This method gives similar end products to Example 4.
EXAMPLE 6
Synthesis of ferric trimaltol using sodium ferrous citrate
A 20% solution w/v of sodium ferrous citrate in distilled water is prepared from 7.5g of sodium ferrous citrate in 37.5ml of water. The solution of sodium ferrous citrate is dark green with an iron content of about 20%. A solution of maltol (containing 10g/50ml) in 20% sodium hydroxide is added to the solution of sodium ferrous citrate. A characteristic deep red/brown iron complex of ferric trimaltol is formed.
EXAMPLE 7
Synthesis of ferric trimaltol using solid sodium ferrous citrate
Example 6 was repeated using the same amounts and concentrations of components but the method is varied in that solid sodium ferrous citrate (7.5g) is added directly to the maltol solution (containing lOg of maltol in 50ml). Ferric trimaltol is formed using this alternative method.
EXAMPLE 8
Synthesis of ferric trimaltol using sodium ferric citrate
A 20% solution w/v of sodium ferric citrate in distilled water is prepared from 7.5g of sodium ferric citrate in 37.5ml of water. The solution of sodium ferric citrate is dark brown with an iron content of about 20%.
A solution of maltol (containing 10g/50ml) in 20% sodium hydroxide is added to the solution of sodium ferric citrate. A characteristic deep red/brown iron complex of ferric trimaltol is formed. EXAMPLE 9
Example 8 was repeated using the same amounts and concentrations of components but the method is varied in that solid sodium ferric citrate (7.5g) is added directly to the maltol solution (containing lOg of maltol in 50ml). Ferric trimaltol is formed using this alternative method.
If any of Examples 3 to 9 are repeated using maltol in a neutral or acidic aqueous medium, such as for example in buffered citric acid, brown/black impurities appear and insoluble fractions are formed (probably of ferric hydroxide) and the UN-vis spectra of the solutions are not correct. In particular, there is a peak shift towards 510nm indicating the formation of mono or dimaltol complexes or compounds.
PATENT
WO 2017167970
POLYMORPH
GB 2531742
PATENT
WO 2016066555
https://patents.google.com/patent/WO2016066555A1/en
An adequate supply of iron to the body is an essential requirement for tissue growth and the maintenance of good health in both man and animals. Moreover, in certain pathological conditions where there is an insidious blood loss, or where there is a mal-distribution of iron in the body, there may be a state of low iron stores in the body leading to an iron deficiency and a concomitant chronic anaemia. This is seen in inflammatory diseases of the gastrointestinal tract, such as gastric and peptic ulcers, reflux oesophagitis, ulcerative colitis and Crohn’s disease.
Anaemia can also follow operations that result in serious blood loss and can be associated with gastrointestinal infections, such as those caused by Helicobacter pylori.
Ferric maltol comprises a complex of one ferric iron and three maltol anions and has the following molecular formula: (C6H503)3Fe. Maltol is also known as 3-hydroxy-2-methyl-4- pyrone.
Polymorphic forms occur where the same composition of matter crystallises in a different lattice arrangement, resulting in different thermodynamic properties and stabilities specific to the particular polymorphic form. WO 03/097627 A1 discloses a method of forming iron hydroxypyrone compounds.
EP 0 159 917 A3 describes a pharmaceutical composition containing a hydroxypyrone-iron complex. WO 2012/101442 A1 discloses a method of forming iron hydroxypyrone compounds.
Schlindwein et al (Dalton Transactions, 2006, Vol. 10, pages 1313-1321) describes lipophilic 3-hydroxy-4-pyridinonate iron(lll) complexes. Ferric maltol has been known for about 100 years but no polymorphs have been identified or studied prior to this invention.
We have now found that it is possible to produce different polymorphs of ferric maltol, which crystalline forms may be referred to herein as the “compounds of the invention”. One polymorph form can be preferable in some circumstances when certain aspects, such as ease of preparation and stability, such as thermodynamic stability are required. In other situations, a different polymorph may be preferred for greater solubility and/or superior pharmacokinetics. The polymorphs of the invention can provide advantages in terms of improved or better bioavailability or improved or better stability or solubility.
The term “ferric maltol” as used herein refers to both ferric trimaltol and the designation INN ferric maltol. In one aspect of the invention there is provided a Form I polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising characteristic crystalline peaks expressed in degrees 2-theta at each of 15.6 and 22.5 ± 0.25 or 0.2 degrees, optionally wherein the Form I polymorph comprises greater than about 92 wt.% ferric maltol based on the weight of the polymorph, such as greater than about 95 wt.%, preferably greater than about 96 wt.%, or about 98 wt.%, or about 99 wt.% such as about 99.8 wt.%.
In a further aspect of the invention there is provided a Form II polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising a peak expressed in degrees 2-theta at 8.3 ± 0.25 degrees.
In a yet further aspect of the invention there is provided a Form III polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising a peak expressed in degrees 2-theta at 7.4 ± 0.25 degrees. In a still further aspect of the invention there is provided a Form IV polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising peaks expressed in degrees 2-theta at 9.5 and 14.5 ± 0.2 degrees.
The measurements of degrees 2-theta generally refer to measurements at ambient temperature, such as from about 5 to about 40°C, preferably about 10 to about 30°C. The relative intensities of the peaks can vary, depending on the sample preparation technique, the sample mounting procedure, the particular instrument employed, and the morphology of the sample. Moreover, instrument variation and other factors can affect the 2-theta values. Therefore, XRPD peak assignments for the polymorphs of the invention, as defined herein in any embodiment, can vary by, for example, ± 0.2, such as ±0.1 or ±0.05. The term “about” in relation to XRPD peak values may include for example, ±0.25 or ± 0.2, such as ±0.1 or ±0.05. These ranges may apply to any of the peak values in degrees referred to herein.
In another embodiment of the invention, there is provided a process for the preparation of a ferric maltol polymorph, such as Form I or Form II polymorph, which comprises combining ferric citrate with maltol anions to form a mixture comprising ferric maltol and wherein the process comprises the use of a ferric maltol seed crystal. The seed crystal may comprise a Form I and/or Form II polymorph as described herein and these polymorphs may be prepared using the methods described herein.
In another aspect of the invention, there is provided a process for the preparation of Form I polymorph, which comprises combining ferric citrate with maltol anions to form a mixture comprising ferric maltol polymorph Form I wherein the process comprises the use of a ferric maltol seed crystal comprising Form I and/or Form II polymorph and preferably wherein the polymorph formed is washed (typically with water) prior to drying.
In a further aspect of the invention, there is provided a process for the preparation of Form II polymorph, which comprises combining ferric citrate with maltol anions in solution to form a mixture comprising ferric maltol polymorph Form II, wherein the process preferably comprises the use of a ferric maltol seed crystal comprising Form I and/or Form II polymorph and preferably wherein the polymorph formed is washed (typically with water) prior to drying.
The invention also provides a pharmaceutical composition comprising a polymorph according to the invention, or mixtures thereof, and a pharmaceutically acceptable adjuvant, diluent or carrier. In addition, the invention provides a composition comprising Form I and Form II polymorphs as defined herein.
In an aspect of the invention, the polymorph of the invention is for use in the prevention or treatment of iron deficiency with or without anaemia in a subject. The anaemia is preferably iron deficiency anaemia.
In a further aspect of the invention there is provided the use of a polymorph of the invention for the manufacture of a medicament for the prevention or treatment of iron deficiency with or without anaemia in a subject. The anaemia is preferably iron deficiency anaemia.
The invention further provides a method for the prevention or treatment of iron deficiency with or without anaemia which method comprises the administration of a polymorph according to the invention to a subject in need of such treatment. The anaemia is preferably iron deficiency anaemia.
Preferably the polymorphs of the invention are obtained in forms that are greater than about 90%, such as greater than about 95%, crystalline (e.g. greater than about 98% crystalline and, particularly, 100%, or nearly 100%, crystalline). By “substantially crystalline” we include greater than about 60%, preferably greater than about 75%, and more preferably greater than about 80% (such as about 90%) crystalline. The degree (%) of crystallinity may be determined by the skilled person using X-ray powder diffraction (XRPD). Other techniques, such as solid state NMR, FT-IR, Raman spectroscopy, differential scanning calorimetry (DSC) microcalorimetry and calculations of the true density may also be used.
The polymorphs of the invention may be characterised by an X-ray powder diffraction pattern comprising the following characteristic crystalline peaks with approximate 2-Theta values (in degrees) as well as an indication of the relative intensity of those peaks in brackets, where a percentage relative intensity of approximately 25- 00% is termed “vs” (very strong), approximately 10-25% is termed “s” (strong), approximately 3-10% is termed “m” (medium) and approximately 1-3% is termed “w” (weak).
Form I: The Form I polymorph preferably comprises characteristic crystalline peaks with 2-Theta values (in degrees) of around (i.e. at about or at approximately) 15.6 and 22.5 ± 0.25, or 0.2 degrees. The diffraction pattern typically does not comprise peaks at one or more, or all, or each of, about 6.9, 7.4, 8.3, 9.3, 10.5, or about 11.8 degrees, such as 8.3 or 11.8 ± 0.25, or ± 0.2, or ±0.1 such as about ±0.05 degrees.
Form II:
The form II polymorph preferably comprises a characteristic crystalline peak with 2-Theta value (in degrees) of around (i.e. at about or at approximately) 8.3 ± 0.25, or ± 0.2, or +0.1 such as about ±0.05 degrees. The diffraction pattern typically does not comprise peaks at one or more, or all, or each of, about 6.9, 7.4, 9.3, 9.5, 10.5, 11.4 or about 13.7 degrees, such as 11.4 or 13.8 ±0.25, or ±0.2, or ±0.1 such as about ±0.05 degrees.
The Form III polymorph preferably comprises a characteristic crystalline peak with 2-Theta value (in degrees) of around (i.e. at about or at approximately) 7.4 ±0.3, ±0.25, or 0.2, or ±0.1 such as about ±0.05 degrees. The diffraction pattern typically does not comprise peaks at one or more, or two or more, or three or more or each of, about 6.9, 8.3, 9.5, 11.3, 12.0, 12.5, 12.9, 14.5, or about 15.8 degrees, such as 6.9, 9.5, 11.3 ±0.25, or ±0.2, or ±0.1 such as about ±0.05 degrees.
The form IV polymorph preferably comprises a characteristic crystalline peaks with 2-Theta values (in degrees) of around (i.e. at about or at approximately) 9.5 and 14.5 +0.2, or ±0.1 such as about ±0.05 degrees. The diffraction pattern typically does not comprise peaks at one or more, or two or more, or three or more or each of, about 6.9, 8.3, 10.5, 11.7, 12.0, 12.2, 12.5, 13.0, 13.4, and about 15.8 degrees, such as 6.9, 8.3, 11.7 ±0.25, or ±0.2, or ±0.1 such as about ±0.05 degrees.
Example 1 : Form I 9.04 kg ferric citrate was combined with 29 litres of purified water. Separately, 12.2 kg of maltol was combined with 15.2 litres of sodium hydroxide solution (20 % w/w). The ferric citrate and sodium hydroxide were charged into a vessel with the addition of 4 litres of water and then stirred at 20 to 25°C. A seed was then added. The seed was 65g of ferric maltol polymorph in 12 litres of water. The seed crystal was prepared by the same process as described in Example 1 but without the use of a seed crystal. The seed was added to the vessel to aid a consistent crystallisation/precipitation. The mixture was held in the vessel, as a suspension, to allow crystal growth and then filtered and washed three times, each time with 13 litres of water. The resulting solid was dried at less than 80°C and produced 13.25 kg of dried ferric maltol.
The ferric maltol in Example 1 was produced on a scale of 12 to 15 kg in different batches. The analysis of the ferric maltol produced showed the % w/w of iron present was about 12.8 to 13.0 and the % w/w of maltol present was about 87.6 to 89.3.
Patent
//////////////Ferric Maltol, マルトール第二鉄 , Feraccru, FDA 2019
CC1=C(C(=O)C=CO1)[O-].CC1=C(C(=O)C=CO1)[O-].CC1=C(C(=O)C=CO1)[O-].[Fe+3]
CC1=C(C(=O)C=CO1)[O-].CC1=C(C(=O)C=CO1)[O-].CC1=C(C(=O)C=CO1)[O-].[Fe+3]
LASMIDITAN

LASMIDITAN, COL-144 , LY-573144
- Molecular FormulaC19H18F3N3O2
- Average mass377.360 Da
-
ласмидитанلاسميديتان
613677-28-4 HYDROCHLORIDE
439239-90-4 (free base)
SUCCINATE 439239-92-6
2,4,6-Trifluoro-N-[6-(1-methylpiperidin-4-ylcarbonyl)pyridin-2-yl]benzamide
2,4,6-trifluoro-N-{6-[(1-methylpiperidin-4-yl)carbonyl]pyridin-2-yl}benzamide
CoLucid Pharmaceuticals, PHASE 3, MIGRAINE
UNII:760I9WM792
Lasmiditan is an oral medication used in the termination of migraine headaches that was first approved for use in the United States in October 2019.
A high-affinity, highly selective serotonin 5-HT(1F) receptor agonist.
Lasmiditan, also known as COL-144 and LY573144, is a novel, centrally acting, highly selective 5-HT(1F) receptor agonist (K1=2.21 μM) without vasoconstrictor activity that seemed effective when given as an intravenous infusion in a proof-of-concept migraine study. Lasmiditan showed efficacy in its primary endpoint, with a 2-hour placebo-subtracted headache response of 28.8%, though with frequent reports of dizziness, paresthesias, and vertigo.
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Lasmiditan succinate | W64YBJ346B | 439239-92-6 | MSOIHUHNGPOCTH-UHFFFAOYSA-N |
| Lasmiditan succinate; UNII-W64YBJ346B; Lasmiditan succinate [USAN]; W64YBJ346B; 439239-92-6; Lasmiditan succinate (USAN)
|
|
| Molecular Formula: | C42H42F6N6O8 |
|---|---|
| Molecular Weight: | 872.822 g/mol |

Patent and Exclusivity for: N211280
Exclusivity Data
Lasmiditan, sold under the brand name Reyvow, is a medication used for the acute (active but short-term) treatment of migraine with or without aura (a sensory phenomenon or visual disturbance) in adults.[2] It is not useful for prevention.[2] It is taken by mouth.[2]
Common side effects include sleepiness, dizziness, tiredness, and numbness.[3][4]
Lasmiditan was approved in the United States in October 2019[3] and became available in February 2020.[5] It was developed by Eli Lilly.[3] The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[6]
WO-2018010345, from Solipharma and the inventor on this API. Eli Lilly , following its acquisition of CoLucid Pharmaceuticals , is developing lasmiditan, a 5-HT 1f agonist, for treating acute migraine.
SYNTHESIS
SYN1
Synthetic Reference
Sheng, Xiaohong; Sheng, Xiaoxia; Jiang, Xiawei. Preparation of crystalline form of lasmiditan and its pharmaceutical composition. Assignee SoliPharma LLC, Peop. Rep. China. WO 2018010345. (2018).

SYN2
Synthetic Description
Reference: Carniaux, Jean-Francois; Cummins, Jonathan. Compositions and methods of synthesis of pyridinoylpiperidine derivatives as 5-HT1F agonists for treating and preventing migraine. Assignee Colucid Pharmaceuticals, Inc., USA. WO 2011123654. (2011).

SYN3
Synthetic Description
Reference: Cohen, Michael Philip; Kohlman, Daniel Timothy; Liang, Sidney Xi; Mancuso, Vincent; Victor, Frantz; Xu, Yao-Chang; Ying, Bai-Ping; Zacherl, Deanna Piatt; Zhang, Deyi. Preparation of pyridinoylpiperidines as 5-HT1F agonists. Assignee Eli Lilly and Company, USA. WO 2003084949. (2003).

SYN
SYN 2
REF
https://www.sciencedirect.com/science/article/abs/pii/S0223523420306395
5 Lasmiditan (Reyvow). Lasmiditan, developed by Eli Lilly, is a highly selective
agonist of 5-HT1F receptors [87]. The FDA approved lasmiditan as the first
neutrally-acting medication to treat migraine headaches [88]. Lasmiditan is a selective
5-HT1F agonist, but inactive against other 5-HT receptors or monoamine receptors
[89,90]. Unlike the triptan class of anti-migraine medications that lead to blood
pressurelability and other cardiovascular side effects, lasmiditan could terminate
migraines but without vasoconstriction [91]. However, lasmiditan may cause
significant driving impairment due to the CNS depression. And it is not a preventive
medication for migraine [92].
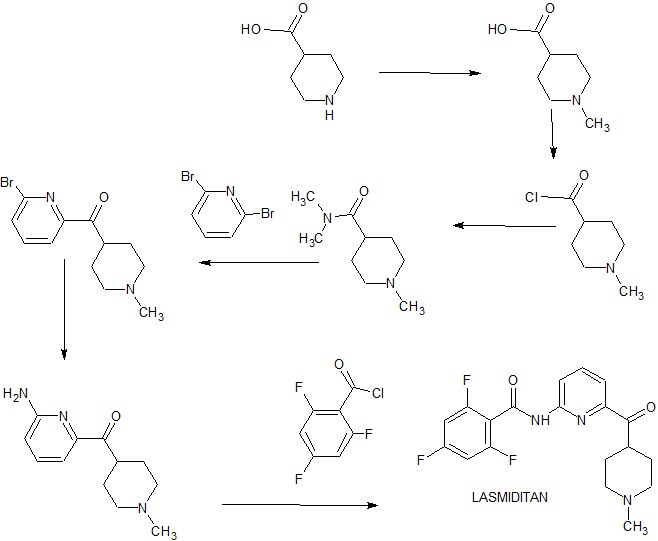
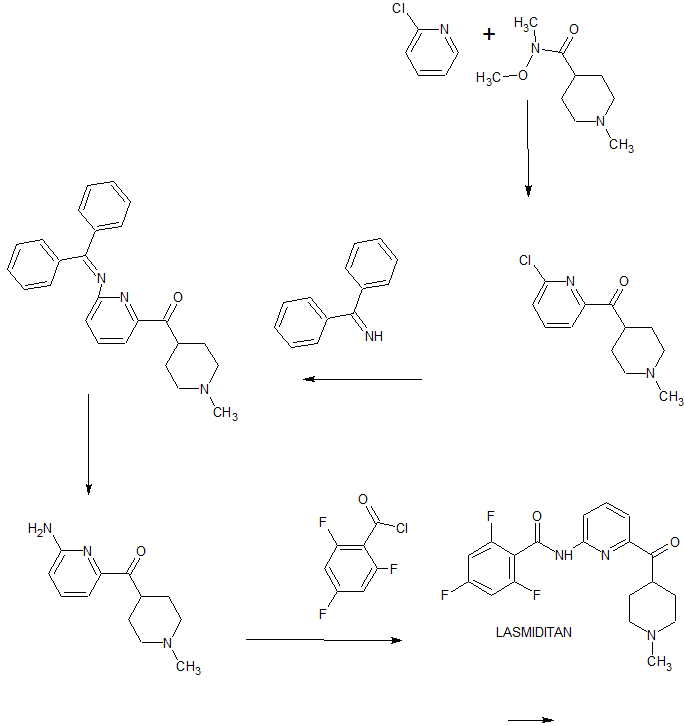
Eli Lilly has disclosed a kilogram-scale procedure to lasmiditan, which is described in
Scheme 14 [93]. Borch reduction of piperidine-4-carboxylic acid 84 gave 85. Further
chlorination with oxalyl chloride formed acyl chloride 86 in good yield. Next,
substitution with commercial dimethylamine, followed by coupling with
2,6-dibromopyridine 88 with assistance of tert-butyllithium gave rise to coupling
product 89 in 84% yield within the two-step sequence. Copper-catalyzed amination
between 89 and NH3 in ethylene glycol gave aminate product 90, which then treated
with acyl chloride 91 to give lasmiditan (XI) in 91% yield
CLICK ON IMAGE TO EXPAND
[87] T.V. Dupre, D.P. Jenkins, R.C. Muise-Helmericks, R.G. Schnellmann, The
5-hydroxytryptamine receptor 1F stimulates mitochondrial biogenesis and
angiogenesis in endothelial cells, Biochem. Pharmacol. 169 (2019) 113644.
[88] Y.N. Lamb, Lasmiditan: first approval, Drugs 79 (2019) 1989-1996.
[89] E. Rubio-Beltran, A. Labastida-Ramirez, K.A. Haanes, A. van den Bogaerdt, A.
Bogers, E. Zanelli, L. Meeus, A.H.J. Danser, M.R. Gralinski, P.B. Senese, K.W.
Johnson, J. Kovalchin, C.M. Villalon, A. MaassenVanDenBrink,
Characterization of binding, functional activity, and contractile responses of the
selective 5-HT1F receptor agonist lasmiditan, Br. J. Pharmacol. 176 (2019)
4681-4695.
[90] G.M. Dubowchik, C.M. Conway, A.W. Xin, Blocking the CGRP pathway for
acute and preventive treatment of migraine: the evolution of success, J. Med.
Chem. 63 (2020) 6600–6623.
[91] B. Kuca, S.D. Silberstein, L. Wietecha, P.H. Berg, G. Dozier, R.B. Lipton, C.M.S.
Group, Lasmiditan is an effective acute treatment for migraine: a phase 3
randomized study, Neurology 91 (2018) 2222-2232.
[92] P.J. Goadsby, L.A. Wietecha, E.B. Dennehy, B. Kuca, M.G. Case, S.K. Aurora, C.
Gaul, Phase 3 randomized, placebo-controlled, double-blind study of lasmiditan
for acute treatment of migraine, Brain 142 (2019) 1894-1904.
[93] M.P. Cohen, D.T. Kohlman, S.X. Liang, V. Mancuso, F. Victor, Y.-C. Xu, B.-P.
Ying, D.P. Zacherl, D. Zhang, Preparation of pyridinoylpiperidines as 5-HT1F
agonists, 2003. WO2003084949.
PATENT
https://patents.google.com/patent/WO2020095171A1/en
In one embodiment, the present invention provides a process as depicted in scheme III for the preparation of lasmiditan, a compound of formula I.







[0192] Example-31:

Mechanism of action
Lasmiditan is a serotonin receptor agonist that, like the unsuccessful LY-334,370, selectively binds to the 5-HT1F receptor subtype. A number of triptans have been shown to act on this subtype as well, but only after their affinity for 5-HT1B and 5-HT1D has been made responsible for their anti-migraine activity.[7] The lack of affinity for these receptors might result in fewer side effects related to vasoconstriction compared to triptans in susceptible people, such as those with ischemic heart disease, Raynaud’s phenomenon or after a myocardial infarction,[8] although a 1998 review has found such side-effects to rarely occur in people taking triptans.[9][10]
Adverse effects
There is a risk of driving impairment while taking lasmiditan. People are advised not to drive or operate machinery for at least eight hours after taking lasmiditan, even if they feel well enough to do so. People who cannot follow this advice are advised not to take lasmiditan. The drug causes central nervous system (CNS) depression, including dizziness and sedation. It should be used with caution if taken in combination with alcohol or other CNS depressants.[2]
History
Lasmiditan was discovered by Eli Lilly and Company and was then relicensed to CoLucid Pharmaceuticals in 2006, until CoLucid was bought by Eli Lilly in 2017, to allow Eli Lilly to reacquire the drug’s intellectual property.[11] The drug is protected by patents until 2031.[12]
Phase II clinical trials for dose finding purposes were completed in 2007, for an intravenous form[13] and in early 2010, for an oral form.[14] Eli Lilly submitted a new drug application to the U.S. Food and Drug Administration (FDA) in November 2018.[15]
Three Phase III clinical trials were completed. The SPARTAN trial compared placebo with 50, 100, and 200 mg of lasmiditan.[16] SAMURAI compared placebo with 100 and 200 mg doses of lasmiditan. GLADIATOR is an open-label study that compared 100 and 200 mg doses of lasmiditan in subjects that received the drug as part of a prior trial.[17]
Topline results from the SPARTAN trial showed that the drug induced met its primary and secondary endpoints in the trial. The primary result showed a statistically significant improvement in pain relief relative to placebo 2 hours after the first dose. The secondary result showed a statistically significantly greater percentage of subjects were free of their most bothersome symptom (MBS) compared with placebo at two hours following the first dose.[18]
The FDA approved lasmiditan primarily based on data from two clinical trials, Trial 1 (# NCT02439320) and Trial 2 (#NCT02605174) of 4439 subjects with migraine headaches with or without aura.[19] Trials were conducted at 224 sites in the United States, the United Kingdom, and Germany.[19]
The FDA approved the drug in October 2019.[19] However, as of October 2019, the drug was awaiting Drug Enforcement Administration (DEA) scheduling before it was made available in the United States.[20] It was placed into Schedule V in January 2020.[21][1]
Dosage
Lasmiditan is delivered in 50 & 100 mg tablet form.[22]
Novel crystalline forms of a 5-HT1F receptor agonist, particularly lasmiditan – designated as Forms 1-3 and A-D – processes for their preparation and compositions comprising them are claimed. Also claim is their use for treating anxiety, fatigue, depression, premenstrual syndrome, trauma syndrome, memory loss, dementia (including Alzheimer’s), autism, schizophrenia, attention deficit hyperactivity disorder, obsessive-compulsive disorder, epilepsy, anorexia nervosa, alcoholism, tobacco abuse, mutism and trichotillomania.
Biological Activity
Lasmiditan (also known as COL-144 and LY573144) is a high-affinity, highly selective serotonin (5-HT) 5-HT(1F) receptor agonist.
In vitro binding studies show a K(i) value of 2.21 nM at the 5-HT(1F) receptor, compared with K(i) values of 1043 nM and 1357 nM at the 5-HT(1B) and 5-HT(1D) receptors, respectively, a selectivity ratio greater than 470-fold. Lasmiditan showed higher selectivity for the 5-HT(1F) receptor relative to other 5-HT(1) receptor subtypes than the first generation 5-HT(1F) receptor agonist LY334370.
In two rodent models of migraine, oral administration of lasmiditan potently inhibited markers associated with electrical stimulation of the trigeminal ganglion (dural plasma protein extravasation, and induction of the immediate early gene c-Fos in the trigeminal nucleus caudalis).
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
PATENT
WO 03084949
https://www.google.co.in/patents/WO2003084949A1?cl=en
8. 2,4,6-Trifluoro-N-[6-(l -methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide mono-hydrochloride salt

Combine 2-amino-6-(l-methylpiperidin-4-ylcarbonyl)pyridine (0.20 g, 0.92 mmol), 2,4,6-Trifluorobenzoyl chloride (0.357 g, 1.84 mmol), and 1 ,4-Dioxane (10 mL), and stir while heating at reflux. After 3 hr., cool the reaction mixture to ambient temperature and concentrate. Load the concentrated mixture onto an SCX column (lOg), wash with methanol, and elute with 2M ammonia in methanol. Concentrate the eluent to obtain the free base of the title compound as an oil (0.365 g (>100%)). Dissolve the oil in methanol (5 mL) and treat with ammonium chloride (0.05 g, 0.92 mmol). Concentrate the mixture and dry under vacuum to obtain the title compound. HRMS Obs. m/z 378.1435, Calc. m/z 378.1429; m.p. 255°C (dec).
Examples
21. 2,4,6-Trifluoro-N-[6-(l-methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide

Add triethylamine (10.67 mL, 76.70 mmol, 2.4 eq) to a solution of 2-amino-(6-(l- methylpiperidin-4-ylcarbonyl)-pyridine (7g, 31.96 mmol, 1 eq) in anhydrous THF (100 mL) under a nitrogen atmosphere. Add 2,4,6-triflubenzoylchloride (7.46g, 5 mL, 38.35 mmol, 1.20 eq) dropwise at room temperature. After 2 hrs., add additional 2,4,6- triflubenzoylchloride (0.75 mL, 0.15 eq) and triethylamine (1.32 mL, 0.3 eq) to the reaction mixture and agitate the mixture for an additional 3 hrs. Quench the reaction with distilled water (10 mL) and 30%o NaOH (15 mL). Stir the resulting biphasic system for 1 hour and then separate the phases. Extract the organic fraction by adding H2O (75 mL) and acetic acid (12 mL), followed by cyclohexane (70 mL). Wash the organic fraction with H2O (50 mL) containing acetic acid (1 mL). Combine all the aqueous fractions and washes and neutralize the mixture with 30% NaOH (15 mL). Extract with methyl-tert- butyl ether (MTBE) (3×50 mL). Combine the organic fractions and dry with MgSO4, filter, concentrate under reduce pressure, and vacuum dry at room temperature, to obtain the title compound as a light-brown solid (11.031 g, 91 % yield).
Mass spectrum, (Electrospray) m/z = 378 (M+l); Η NMR (250 MHz, Chloroform-D) ppm 1.54 (m, 2 H) 2.02 (m, 2 H) 2.13 (t, J=l 1.48 Hz, 2 H) 2.29 (s, 3 H) 2.80 (m, J=l 1.96 Hz, 1 H) 3.56 (m, 1 H) 4.26 (d, J=7.87 Hz, 1 H) 6.17 (d, J=8.50 Hz, 1 H) 6.75 (m, 2 H) 7.45 (t, J=7.87 Hz, 1 H) 7.53 (m, 1 H) 7.95 (s, 1 H); 13C-NMR: (62.90 MHz, Chloroform-D) ppm 202.78; 162.6 (dm C-F-couplings); 162.0 (m C-F-couplings); 160.1 (m C-F-couplings); 158.1 ; 150.0; 139.7; 1 19.3; 1 17.9; 1 10.2 (m C-F-couplings); 100.9 (m C-F-couplings); 55.2; 46.5; 41.9; 28.1
22. 2,4,6-Trifluoro-N-[6-(l-methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide mono-hydrochloride salt

Dissolve 2,4,6-trifluoro-N-[6-(l-methylpiperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide – free base (5g, 23.26mmol) in isopropanol (50 mL) at room temperature and add a solution of 3.3 M diethylether/HCl (8 mL). Heat the reaction mixture under reflux for 30 minutes. Cool the reaction mixture to room temperature and agitate for 2 hrs. Filter the resulting white precipitate and rinse with isopropanol (5 mL). Dry the residual solid under reduce pressure at 40°C overnight to obtain the title compound (5.12 g, 93% yield). M.p. 223-224°C (sublimation); Η NMR (400 MHz, d6-DMSO) d ppm 1.94 (m, 2 H) 2.14 (m, J=11.15 Hz, 2 H) 2.74 (s, 3 H) 2.99 (m, J=9.19 Hz, 2 H) 3.49 (m, J=1 1.15 Hz, 2 H) 3.77 (m, 1 H) 7.41 (t, J=8.71 Hz, 2 H) 7.78 (d, J=7.43 Hz, 1 H) 8.10 (t, J=7.92 Hz, 1 H) 8.37 (d, J=6.85 Hz, 1 H) 10.50 (s, 1 H) 1 1.51 (s, 1 H); 13C-NMR: (100.61 MHz, Chloroform-D) ppm 200.7; 130.6-158.0 (m, C-F-couplings); 150.4; 150.1; 140.2; 118.5; 1 18.2; 11 1.9; 101.3 (t, C-F couplings); 52.8; 42.6; 25.2
23. 2,4,6-Trifluoro-N-[6-(l-methyl-piperidine-4-carbonyl)-pyridin-2-yl]- benzamide hemi-succinate salt

Add succinic acid (0.25g, 2.148 mmol, 0.5eq) to a solution of 2,4,6-trifluoro-N-[6-
(l-methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]-benzamide – free base (1.62g, 4.297 mmol, leq) in acetone (16.2 mL), at room temperature. Warm the solution under reflux for 30 minutes. Cool the solution to room temperature and filter off the resulting white precipitate. Rinse the precipitate with acetone (0.2 mL) and dry under vacuum at 50°C for 16 hours to provide the title compound (1.5g, 80% yield). M.p. 198.5°C; mass spectrum (Electrospray) m/z = 495.45
The following examples are prepared by combinatorial chemistry techniques as follows:
Examples 24-54

Combine R-acid (300 μL of 0.5M solution in dimethylformamide (DMF)), HATU (57 mg, 0.15 mmol), collidine (19 μL, 0.15 mmol), 2-amino-(6-(l-methylpiperidin-4- ylcarbonyl)-pyridine and DMF (1.5 mL), and agitate for 48 hr. Dilute the reaction mixture with 10% acetic acid in methanol (0.5 L). Load the resulting reaction mixture onto a 2 g SCX column. Wash the column thoroughly with methanol and then elute with 1 M ammonia in methanol. Concentrate the eluent and further purify the product by high- throughput mass guided chromatography. This procedure is repeated in parallel for examples 24-54.
Examples 55-58

Heat R-acid chloride (300 μL of 0.5M solution in pyridine) to 55°C, add 2-amino- (6-(l-methylpiperidin-4-ylcarbonyl)-pyridine (200 μL of 0.5M solution in pyridine), and continue heating the reaction mixture for 24 hr. Concentrate the reaction mixture and then dilute with 10% Acetic acid in methanol (0.5 mL) and methanol (0.5 mL). Load the resulting reaction mixture directly onto a 2 g SCX column. Thoroughly wash the column with methanol and then elute the column with 1 M ammonia in methanol. Concentrate the eluent and then further purify the product by high- throughput mass guided chromatography. This procedure is repeated in parallel for examples 55-58.
Examples 59-71

Heat 2-amino-(6-(l-methylpiperidin-4-ylcarbonyl)-pyridine (200 μL of 0.5M solution in pyridine) to 55°C then add R-acid chloride (0.10 mmol), heat for 2 hr. Concentrate the reaction mixture and then dilute with 10% Acetic acid in methanol (0.5 mL) and methanol (0.5 mL). Load the resulting reaction mixture directly onto a 2 g SCX column. Thoroughly wash the column with methanol and then elute the column with 1 M ammonia in methanol. Concentrate the eluent and then further purify the product by high-throughput mass guided chromatography. This procedure is repeated in parallel for examples 59-71.
PATENT
References
- ^ Jump up to:a b “2020 – Placement of Lasmiditan in Schedule V”. DEA Diversion Control Division. 31 January 2020. Retrieved 31 January 2020.
- ^ Jump up to:a b c d “Reyvow- lasmiditan tablet”. DailyMed. 11 October 2019. Retrieved 15 November 2019.
- ^ Jump up to:a b c “FDA approves new treatment for patients with migraine”. U.S. Food and Drug Administration (FDA) (Press release). 11 October 2019. Archived from the original on 16 November 2019. Retrieved 17 October 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Lasmiditan (Professional Patient Advice)”. Drugs.com. 4 June 2019. Retrieved 23 February 2020.
- ^ “Lilly’s Reyvow (lasmiditan) C-V, the First and Only Medicine in a New Class of Acute Treatment for Migraine (ditan), Now Available for Prescription”. Eli Lilly and Company. 31 January 2020. Retrieved 23 February 2020.
- ^ “New Drug Therapy Approvals 2019”. U.S. Food and Drug Administration. 31 December 2019. Retrieved 15 September 2020.
- ^ Rissardo, JamirPitton; Fornari Caprara, AnaLetícia (2020). “The ditans, a new class for acute migraine: Minireview”. Journal of Current Research in Scientific Medicine. 6 (1): 11. doi:10.4103/jcrsm.jcrsm_45_19. ISSN 2455-3069.
- ^ “Molecule of the Month July 2010: Lasmiditan hydrochloride”. Prous Science. Archived from the original on 28 July 2011. Retrieved 3 August 2011.
- ^ Dahlöf CG, Mathew N (October 1998). “Cardiovascular safety of 5HT1B/1D agonists–is there a cause for concern?”. Cephalalgia. 18 (8): 539–45. doi:10.1046/j.1468-2982.1998.1808539.x. PMID 9827245. S2CID 30125923.
- ^ Mutschler E, Geisslinger G, Kroemer HK, Schäfer-Korting M (2001). Arzneimittelwirkungen (in German) (8th ed.). Stuttgart: Wissenschaftliche Verlagsgesellschaft. p. 265. ISBN 978-3-8047-1763-3. OCLC 47700647.
- ^ “Lilly buys migraine biotech CoLucid, and the drug it outlicensed, for $960M”.
- ^ “Lasmiditan – Eli Lilly and Company – AdisInsight”.
- ^ “A Placebo-Controlled Adaptive Treatment Assignment Study of Intravenous COL-144 in the Acute Treatment of Migraine”. ClinicalTrials.gov. 8 November 2019. Retrieved 23 February 2020.
- ^ “Dose-ranging Study of Oral COL-144 in Acute Migraine Treatment”. ClinicalTrials.gov. 20 December 2019. Retrieved 23 February 2020.
- ^ “Lilly Submits New Drug Application to the FDA for Lasmiditan for Acute Treatment of Migraine, Receives Breakthrough Therapy Designation for Emgality (galcanezumab-gnlm) for Prevention of Episodic Cluster Headache”. Eli Lilly and Company. 14 November 2018. Retrieved 12 October 2019 – via PR Newswire.
- ^ Clinical trial number NCT02605174 for “Three Doses of Lasmiditan (50 mg, 100 mg and 200 mg) Compared to Placebo in the Acute Treatment of Migraine (SPARTAN)” at ClinicalTrials.gov
- ^ Clinical trial number NCT02565186 for “An Open-label, Long-term, Safety Study of Lasmiditan for the Acute Treatment of Migraine (GLADIATOR)” at ClinicalTrials.gov
- ^ “Lilly Announces Positive Results for Second Phase 3 Study of Lasmiditan for the Acute Treatment of Migraine”. Archived from the original on 5 August 2017. Retrieved 5 August 2017.
- ^ Jump up to:a b c “Drug Trials Snapshots: Reyvow”. U.S. Food and Drug Administration (FDA). 11 October 2019. Retrieved 26 January 2020.
- ^ Vinluan F (11 October 2019). “FDA OKs Lilly’s Lasmiditan, First New Acute Migraine Drug in Decades”. Xconomy. Retrieved 12 October 2019.
- ^ “Schedules of Controlled Substances: Placement of Lasmiditan in Schedule V”. Federal Register. 31 January 2020.
- ^ “Reyvow (Lasmiditan Tablets): Uses, Dosage, Side Effects, Interactions, Warning”. RxList. Retrieved 20 August 2020.
-
Lasmiditan Clinical data Trade names Reyvow Other names COL-144 AHFS/Drugs.com Monograph MedlinePlus a620015 License data - US DailyMed: Lasmiditan
Routes of
administrationBy mouth, intravenous ATC code Legal status Legal status - US: Schedule V [1]
Identifiers CAS Number PubChem CID IUPHAR/BPS DrugBank ChemSpider UNII KEGG CompTox Dashboard (EPA) Chemical and physical data Formula C19H18F3N3O2 Molar mass 377.367 g·mol−1 3D model (JSmol) 
 (what is this?) (verify)
(what is this?) (verify)

External links
- “Lasmiditan”. Drug Information Portal. U.S. National Library of Medicine.
- /////////////LASMIDITAN, phase III, LILY, COL-144 , LY-573144, CoLucid Pharmaceuticals, PHASE 3, MIGRAINE, ласмидитан, لاسميديتان , FDA 2019
- Capi M, de Andres F, Lionetto L, Gentile G, Cipolla F, Negro A, Borro M, Martelletti P, Curto M: Lasmiditan for the treatment of migraine. Expert Opin Investig Drugs. 2017 Feb;26(2):227-234. doi: 10.1080/13543784.2017.1280457. [Article]
- Nelson DL, Phebus LA, Johnson KW, Wainscott DB, Cohen ML, Calligaro DO, Xu YC: Preclinical pharmacological profile of the selective 5-HT1F receptor agonist lasmiditan. Cephalalgia. 2010 Oct;30(10):1159-69. doi: 10.1177/0333102410370873. Epub 2010 Jun 15. [Article]
- Lupi C, Benemei S, Guerzoni S, Pellesi L, Negro A: Pharmacokinetics and pharmacodynamics of new acute treatments for migraine. Expert Opin Drug Metab Toxicol. 2019 Mar;15(3):189-198. doi: 10.1080/17425255.2019.1578749. Epub 2019 Feb 12. [Article]
- Vila-Pueyo M: Targeted 5-HT1F Therapies for Migraine. Neurotherapeutics. 2018 Apr;15(2):291-303. doi: 10.1007/s13311-018-0615-6. [Article]
- Rubio-Beltran E, Labastida-Ramirez A, Haanes KA, van den Bogaerdt A, Bogers AJJC, Zanelli E, Meeus L, Danser AHJ, Gralinski MR, Senese PB, Johnson KW, Kovalchin J, Villalon CM, MaassenVanDenBrink A: Characterization of binding, functional activity and contractile responses of the selective 5-HT1F receptor agonist lasmiditan. Br J Pharmacol. 2019 Aug 16. doi: 10.1111/bph.14832. [Article]
- Reuter U, Israel H, Neeb L: The pharmacological profile and clinical prospects of the oral 5-HT1F receptor agonist lasmiditan in the acute treatment of migraine. Ther Adv Neurol Disord. 2015 Jan;8(1):46-54. doi: 10.1177/1756285614562419. [Article]
- FDA Approved Drugs: Reyvow [Link]
- AChemBlock: Lasmiditan hemisuccinate MSDS [Link]
- FDA News Release: Lasmiditan Approval [Link]
CN1CCC(CC1)C(=O)C2=NC(=CC=C2)NC(=O)C3=C(C=C(C=C3F)F)F.CN1CCC(CC1)C(=O)C2=NC(=CC=C2)NC(=O)C3=C(C=C(C=C3F)F)F.C(CC(=O)O)C(=O)O
Pitolisant
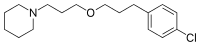
Pitolisant
CAS 362665-56-3
FDA APPROVED 2019 AUG
1-(3-(3-(4-Chlorophenyl)propoxy)propyl)piperidine
MF C17H26ClNO
MW 295.1703
- HBS-101
- Pitolisant
- Tiprolisant
- UNII-4BC83L4PIY
(Wakix®)Approved EU 31/3/2016, Narcolepsy
A histamine H3 receptor antagonist/inverse agonist used to treat narcolepsy.
![]()
BF-2649; BF-2.649; FUB-649, Ciproxidine, Tiprolisant
CAS 362665-56-3, 362665-57-4 (oxalate)
CAS 903576-44-3(Pitolisant Hydrochloride)
APPROVED IN EU, European Medicine Agency (EMA) on Mar 31, 2016.
- BF 2.649
- BF 2649
- BF2.649
- Ciproxidine
- Pitolisant hydrochloride
- Tiprolisant
- UNII-YV33CH63HI
1-{3-[3-(4-Chlorophenyl)propoxy]propyl}piperidine hydrochloride (1:1)
| Molecular Weight | 332.31 |
| Formula | C17H26ClNO ● HCl |
![]()
Bioprojet INNOVATOR
Jean-Charles Schwartz, Jeanne-Marie Lecomte
- OriginatorBioprojet; Ferrer
- DeveloperAlium Medical; AOP Orphan Pharmaceuticals AG; Bioprojet; Ferox Therapeutics; Harmony Biosciences
- ClassNootropics; Piperidines; Sleep disorder therapies
- Mechanism of ActionHistamine H3 receptor antagonists; Histamine H3 receptor inverse agonists
- Orphan Drug StatusYes – Narcolepsy
- New Molecular EntityYes
Highest Development Phases
- MarketedNarcolepsy
- Phase IIIHypersomnia
- Phase IDrug abuse; Type 1 diabetes mellitus
- 15 Aug 2019Registered for Narcolepsy in USA (PO)
- 15 Aug 2019Harmony Biosciences intends to market pitolisant for excessive daytime sleepiness in patients with Narcolepsy in USA, in 4Q of 2019
- 19 Jun 2019Phase-I clinical trials in Type 1 diabetes mellitus in USA (PO) (NCT04026750)
- Pitolisant (INN), also known as tiprolisant (USAN),[1] is a medication in the United States that was approved by the FDA in August 2019. It was granted orphan designation for the treatment of narcolepsy, Fast Track designation for the treatment of excessive daytime sleepiness (EDS) and cataplexy in patients with narcolepsy, and Breakthrough Therapy designation for the treatment of cataplexy in patients with narcolepsy. Pitolisant, a first-in-class medication, is a potent and highly selective Histamine 3 (H₃) receptorantagonist/inverse agonist; it enhances the activity of histaminergic neurons in the brain that function to improve a patient’s wakefulness and inhibit attacks of cataplexy. It was designed and developed by Bioprojet, who has marketed the product in Europe since its approval by the European Medicines Agency in 2016. Pitolisant represents the first new therapy in the U.S. in over 15 years for the treatment of both EDS and cataplexy in adult patients with narcolepsy.The NDA (New Drug Submission) submission is based on results from the clinical development program in narcolepsy, which included over 300 patients, some of whom were treated for up to five years. It also included safety data in over 1500 patients across multiple patient populations. [1]It was developed by Jean-Charles Schwartz, Walter Schunack, and colleagues after the former discovered the H₃ receptor.[2] It was the first H₃ receptor inverse agonist to be tested in humans or introduced for clinical use.[2]
Pitolisant (INN) or tiprolisant (USAN) is a histamine receptor inverse agonist/antagonist selective for the H3 subtype.[1] It hasstimulant and nootropic effects in animal studies,[2] and may have several medical applications, having been researched for the treatment of narcolepsy, for which it has been granted orphan drug status in the EU and US.[3][4] It is currently in clinical trials forschizophrenia and Parkinson’s disease.[4][5][6]
Pitolisant hydrochloride was approved by European Medicine Agency (EMA) on Mar 31, 2016. It was developed and marketed as Wakix® by Bioprojet in EU.
Pitolisant is being developed by Bioprojet for the oral treatment of central nervous system disorders. Pitolisant is a selective histamine H3-receptor antagonist/inverse agonist which enhances the activity of histaminergic neurons. Pitolisant has been launched in several countries for the treatment of narcolepsy, and is approved in the US, EU, Iceland and Liechtenstein. Clinical development is underway for type-1 diabetes, hypersomnia and drug abuse in countries worldwide.
Phase III development was also conducted for the treatment of hypersomnia in Switzerland. Phase II development for attention-deficit hyperactivity disorder was conducted in France. However, there were no recent reports on development identified. Development in epilepsy and obesity has been discontinued.
Ferrer and Bioprojet appeared to have a co-development agreement for pitolisant that allowed the mutual use of both companies’ technical and scientific resources; however, as per Ferrer’s communication dated June 2016, the drug is no longer in its portfolio.

Pitolisant hydrochloride is an antagonist/inverse agonist of the histamine H3 receptor, which is indicated in adults for the treatment of narcolepsy with or without cataplexy.
Wakix® is available as tablet for oral use, containing 4.5 mg and 18 mg of Pitolisant hydrochloride. The initial dose of 9 mg (two 4.5 mg, tablets) per day, and it should be used at the lowest effective dose, depending on individual patient response and tolerance, according to an up-titration scheme, without exceeding the dose of 36 mg/day.
Pitolisant was developed by Jean-Charles Schwartz, Walter Schunack and colleagues after the former discovered H3 receptors.[7]Pitolisant was the first clinically used H3 receptor inverse agonist.
Pitolisant, also known as Tiprolisant, is a histamine receptor inverse agonist/antagonist selective for the H3 subtype. It has stimulant and nootropic effects in animal studies, and may have several medical applications, having been researched for the treatment of narcolepsy, for which it has been granted orphan drug status in the EU and US. It is currently in clinical trials for schizophrenia and Parkinson’s disease. Pitolisant was the first clinically used H3 receptor inverse agonist.

The European Medicines Agency (EMA) has recommended granting marketing authorization for pitolisant (Wakix, Bioprojet Pharma) for narcolepsy with or without cataplexy, the agency announced today.
Narcolepsy is a rare sleep disorder that affects the brain’s ability to regulate the normal sleep-wake cycle, leading to excessive daytime sleepiness, including the sudden urge to sleep, and disturbed night-time sleep. Some patients also experience sudden episodes of cataplexy, potentially causing dangerous falls and increasing the risks for accidents, including car accidents. Symptoms of narcolepsy can be severe and significantly reduce quality of life.
Pitolisant “will add to the available treatment options for narcolepsy. It is a first-in-class medicine that acts on histamine H3 receptors in the brain. This leads to increased histamine release in the brain, thereby enhancing wakefulness and alertness,” the EMA notes in a news release.
The EMA recommendation for approval of pitolisant is based on an evaluation of all available safety and efficacy data conducted by the Committee for Medicinal Products for Human Use (CHMP). The data include two pivotal placebo-controlled trials involving 259 patients, as well as one uncontrolled, open-label study involving 102 patients with narcolepsy and one supportive study in 105 patients.
The studies showed that pitolisant was effective in reducing excessive daytime sleepiness in patients with narcolepsy. The beneficial effect of the drug on cataplexy was demonstrated in one of the pivotal studies as well as in the supportive study.
No major safety concerns with pitolisant emerged in testing. Insomnia, headache, and nausea were among the most common adverse effects observed in the clinical trials, and the CHMP decided on measures to mitigate these risks, the EMA said. The CHMP also requested the company conduct a long-term safety study to further investigate the safety of the drug when used over long periods.
Pitolisant for narcolepsy received orphan designation from the Committee for Orphan Medicinal Products in 2007. Orphan designation provides medicine developers access to incentives, such as fee reductions for scientific advice, with the aim of encouraging the development of treatments for rare disorders.
The CHMP opinion will now be sent to the European Commission for the adoption of a decision on a European Union–wide marketing authorization. Once that has been granted, each member state will decide on price and reimbursement based on the potential role/use of this medicine in the context of its national health system.

Narcolepsy-cataplexy.
Narcolepsy-cataplexy, or Gelineau syndrome, is a rare but serious disorder characterized by excessive daytime sleepiness which can be an extreme hindrance to normal professional and social activities, and which is accompanied by more or less frequent attacks of cataplexy (a sudden loss of muscle tone triggered by emotions as varied as laughter or fear) and erratic episodes of REM sleep (during wakefulness and during sleep), sometimes associated with hypnagogic hallucinations. Moreover, individuals with narcolepsy have various degrees of cognitive impairment and tend to be obese (reviewed by Dauvilliers et al., Clin. Neurophysiol., 2003, 114, 2000; Baumann and Bassetti, Sleep Med. Rev., 2005, 9, 253).
The disorder is caused by the loss of a group of neurons in the brain which produce two peptides, orexins, also known as hypocretins, located in the anterior hypothalamus and projecting to the main groups of aminergic neurons which regulate wakefulness and sleep. Patients with the disorder generally have very low levels of orexins in cerebrospinal fluid. Orexin knock-out mice display many of the symptoms seen in narcoleptic subjects, confirming the role of these peptides and thereby providing an excellent animal model of the disease (Chemelli et al., Cell, 1999, 98, 437).
Several types of treatments which can improve the symptoms of narcolepsy already exist, although they do not completely relieve symptoms and, furthermore, can cause significant side effects limiting their usefulness.
For instance, amphetamines or analogues such as methylphenidate which release catecholamines are used to treated daytime sleepiness, but these agents induce a state of excessive excitation as well as cardiovascular disturbances and also carry a potential for drug addiction.
Modafinil, a drug whose mechanism of action is unclear, also improves daytime sleepiness without causing as many side effects as amphetamines. Nonetheless, its efficacy is limited and it can cause headaches and nausea, particularly at high doses. Moreover amphetamines and/or modafinil do not appear to improve some of the most disabling symptoms of the disease, particularly cataplexy attacks, cognitive deficits and weight gain. With regard to cataplexy, treatments include antidepressants and oxybate. Effectiveness of the former has not been demonstrated (Cochrane Database Syst. Rev., 2005, 20, 3), and the latter is a drug of illegal abuse and its use is restricted.
It has also been shown that histamine H3 receptor antagonists induce the activation of histaminergic neurons in the brain which release histamine, a neurotransmitter with a crucial role in maintaining wakefulness (Schwartz et al., Physiol. Rev. 1991, 71, 1).
PATENT
Pharmaceutical products with histamine H3 receptor ligand properties and 0 subsequent pharmacological activities thereof are described in EP-980300. An especially important product among those disclosed is 1-[3-[3-(4- chlorophenyl)propoxy] propyl]-piperidine. This compound is disclosed as the free base and as the oxalate salt.
5 The use of 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine as the free base is limited because of its oily nature. On the contrary, 1-[3-[3-(4- chlorophenyl)propoxy]propyl]-piperidine oxalate is a crystalline substance but its low aqueous solubility (0.025 g/ml at 230C) also limits its use as a
pharmaceutical ingredient.
0
Subsequent patents EP-1100503 and EP-1428820 mention certain salts of 1- [3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine. However, the only one specifically described is the oxalate salt. The crystalline monohydrochloride salt is not described.
Example 1 : 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine
According to the method disclosed in EP-982300, Example 78, sodium 3-piperidinopropanolate (2.127 kg; 12.88 mol), 3-(4-chlorophenyl)propyl mesylate (1.121 kg; 4.51 mol) and 0.322 mol of 15-crown-5 in 4.5 kg of dry toluene were refluxed for 4 hours. The solvent was evaporated and the residue purified by column chromatography on silica gel (eluent: methylene chloride/methanol (90/10)). The obtained oil was distilled in a fractionating equipment at reduced pressure (0.3-0.7 mmHg) and with a heating jacket at 207-2100C. The head fractions and the distilled fraction at 0.001-0.010 mmHg with a jacket temperature of 180-2000C were collected. The obtained oil (1.0 kg; 3.38 mol) corresponds to 1-[3-[3-(4-chlorophenyl)propoxy] propyl]-piperidine. Yield 75%.
Example 2: 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine
monohydrochloride
Preparation
Distilled 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine (1.0 kg) and anhydrous ethyl acetate (4.5 kg) are transferred to a 10-L glass vessel fitted with a cooling bath and a gas inlet. A stream of gaseous hydrogen chloride is bubbled in the reaction mixture at 20-250C.
The pH of the solution is checked by taking a 0.5 mL sample of the reaction mixture and diluting it with 5 mL of deionized water. The final pH must be about 3-4.
The mixture is cooled to -10°C-(-12°C) and stirred at this temperature for 1 h. The precipitate is filtered by using a sintered glass filter and washed with 0.5 L of anhydrous ethyl acetate previously cooled to 0-50C. The product is dried in a vacuum oven at 5O0C for a minimum period of 12 hours. The resulting crude 1 -[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine monohydrochloride weighs 1.10 kg.
Purification
A mixture of the above-described crude, 3.98 kg of anhydrous ethyl acetate and 0.35 kg of /-propanol is heated slowly at 55-6O0C in a 10-L glass vessel fitted with a heating and cooling system. When the solution has been completed, it is filtered through a heat-isolated sintered glass filter, keeping the temperature at 55-6O0C. The solution is transferred to a 10 L glass vessel and the mass is slowly cooled to 0-50C for about 1 hour. The mixture is stirred at this temperature for 1 hour and the precipitate is filtered through a sintered glass filter. The solid is washed with a mixture of 1.6 kg of anhydrous ethyl acetate and 0.14 kg of /-propanol cooled at 0-50C. The solid is dried in a vacuum oven at 5O0C for a minimum period of 12 hours. M. p. 117-1190C. Yield 80%.
IR spectrum (KBr): bands at 1112 and 1101 (C-O Ether/ St. asym), 2936 and 2868 (Alkane CH(CH2)) / St.), 1455 (Alkane CH(CH2)) / Deform.), 2647 and 2551 (Amine Salt / St.), 1492 (Amine / St.), 802 (Aromatic / Deform.) cm“1.
SEE
Eur. J. Pharm. Sci. 2001, 13, 249–259.
References
- Celanire S, Wijtmans M, Talaga P, Leurs R, de Esch IJ (December 2005). “Keynote review: histamine H3 receptor antagonists reach out for the clinic”. Drug Discov. Today. 10 (23-24): 1613–27. doi:10.1016/S1359-6446(05)03625-1. PMID 16376822.
- Ligneau X, Perrin D, Landais L, Camelin JC, Calmels TP, Berrebi-Bertrand I, Lecomte JM, Parmentier R, Anaclet C, Lin JS, Bertaina-Anglade V, la Rochelle CD, d’Aniello F, Rouleau A, Gbahou F, Arrang JM, Ganellin CR, Stark H, Schunack W, Schwartz JC. BF2.649 [1-{3-[3-(4-Chlorophenyl)propoxy]propyl}piperidine, hydrochloride], a nonimidazole inverse agonist/antagonist at the human histamine H3 receptor: Preclinical pharmacology. Journal of Pharmacology and Experimental Therapeutics. 2007 Jan;320(1):365-75. PMID 17005916
- Lin JS, Dauvilliers Y, Arnulf I, Bastuji H, Anaclet C, Parmentier R, Kocher L, Yanagisawa M, Lehert P, Ligneau X, Perrin D, Robert P, Roux M, Lecomte JM, Schwartz JC. An inverse agonist of the histamine H(3) receptor improves wakefulness in narcolepsy: studies in orexin-/- mice and patients. Neurobiology of Disease. 2008 Apr;30(1):74-83. PMID 18295497
- ^ Jump up to:a b Prous Science: Molecule of the Month September 2011
- Ligneau X, Landais L, Perrin D, Piriou J, Uguen M, Denis E, Robert P, Parmentier R, Anaclet C, Lin JS, Burban A, Arrang JM, Schwartz JC. Brain histamine and schizophrenia: potential therapeutic applications of H3-receptor inverse agonists studied with BF2.649. Biochemical Pharmacology. 2007 Apr 15;73(8):1215-24. PMID 17343831
- Stocking EM, Letavic MA (2008). “Histamine H3 antagonists as wake-promoting and pro-cognitive agents”. Current Topics in Medicinal Chemistry. 8 (11): 988–1002. doi:10.2174/156802608784936728. PMID 18673168.
- Schwartz, Jean-Charles (May 2011). “The histamine H3 receptor: from discovery to clinical trials with pitolisant”. BPJ. doi:10.1111/j.1476-5381.2011.01286.x.
References
- ^ http://adisinsight.springer.com/drugs/800029451
- ^ Jump up to:a b Schwartz JC (2011). “The histamine H3 receptor: from discovery to clinical trials with pitolisant”. Br. J. Pharmacol. 163(4): 713–21. doi:10.1111/j.1476-5381.2011.01286.x. PMC 3111674. PMID 21615387.
External links
REFERENCES
1: Leu-Semenescu S, Nittur N, Golmard JL, Arnulf I. Effects of pitolisant, a histamine H3 inverse agonist, in drug-resistant idiopathic and symptomatic hypersomnia: a chart review. Sleep Med. 2014 Jun;15(6):681-7. doi: 10.1016/j.sleep.2014.01.021. Epub 2014 Mar 18. PubMed PMID: 24854887.
2: Dauvilliers Y, Bassetti C, Lammers GJ, Arnulf I, Mayer G, Rodenbeck A, Lehert P, Ding CL, Lecomte JM, Schwartz JC; HARMONY I study group. Pitolisant versus placebo or modafinil in patients with narcolepsy: a double-blind, randomised trial. Lancet Neurol. 2013 Nov;12(11):1068-75. doi: 10.1016/S1474-4422(13)70225-4. Epub 2013 Oct 7. PubMed PMID: 24107292.
3: Nirogi R, Ajjala DR, Kandikere V, Pantangi HR, Jonnala MR, Bhyrapuneni G, Muddana NR, Vurimindi H. LC-MS/MS method for the determination of pitolisant: application to rat pharmacokinetic and brain penetration studies. Biomed Chromatogr. 2013 Nov;27(11):1431-7. doi: 10.1002/bmc.2939. Epub 2013 Jun 13. PubMed PMID: 23760876.
4: Kasteleijn-Nolst Trenité D, Parain D, Genton P, Masnou P, Schwartz JC, Hirsch E. Efficacy of the histamine 3 receptor (H3R) antagonist pitolisant (formerly known as tiprolisant; BF2.649) in epilepsy: dose-dependent effects in the human photosensitivity model. Epilepsy Behav. 2013 Jul;28(1):66-70. doi: 10.1016/j.yebeh.2013.03.018. Epub 2013 May 8. PubMed PMID: 23665640.
5: Uguen M, Perrin D, Belliard S, Ligneau X, Beardsley PM, Lecomte JM, Schwartz JC. Preclinical evaluation of the abuse potential of Pitolisant, a histamine H₃ receptor inverse agonist/antagonist compared with Modafinil. Br J Pharmacol. 2013 Jun;169(3):632-44. doi: 10.1111/bph.12149. PubMed PMID: 23472741; PubMed Central PMCID: PMC3682710.
6: Brabant C, Charlier Y, Tirelli E. The histamine H₃-receptor inverse agonist pitolisant improves fear memory in mice. Behav Brain Res. 2013 Apr 15;243:199-204. doi: 10.1016/j.bbr.2012.12.063. Epub 2013 Jan 14. PubMed PMID: 23327739.
7: Zhang DD, Sisignano M, Schuh CD, Sander K, Stark H, Scholich K. Overdose of the histamine H₃ inverse agonist pitolisant increases thermal pain thresholds. Inflamm Res. 2012 Nov;61(11):1283-91. doi: 10.1007/s00011-012-0528-5. Epub 2012 Jul 21. PubMed PMID: 22820944.
8: Inocente C, Arnulf I, Bastuji H, Thibault-Stoll A, Raoux A, Reimão R, Lin JS, Franco P. Pitolisant, an inverse agonist of the histamine H3 receptor: an alternative stimulant for narcolepsy-cataplexy in teenagers with refractory sleepiness. Clin Neuropharmacol. 2012 Mar-Apr;35(2):55-60. doi: 10.1097/WNF.0b013e318246879d. PubMed PMID: 22356925.
9: Schwartz JC. The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br J Pharmacol. 2011 Jun;163(4):713-21. doi: 10.1111/j.1476-5381.2011.01286.x. Review. PubMed PMID: 21615387; PubMed Central PMCID: PMC3111674.
 |
|
| Clinical data | |
|---|---|
| Trade names | Wakix |
| Synonyms | Tiprolisant; Ciproxidine; BF2.649 |
| License data | |
| Routes of administration |
Oral |
| Drug class | Histamine H3 receptor inverse agonists |
| ATC code | |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C17H26ClNO |
| Molar mass | 295.851 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////Pitolisant Hydrochloride, Wakix, histamine H3 receptor antagonist/inverse agonist, narcolepsy, orphan drug, tiprolisant, EU 2016, FDA 2019

Trifarotene
Trifarotene
CAS 895542-09-3
3”-Tert-butyl-4′-(2-hydroxyethoxy)-4”-(pyrrolidin-1-yl)(1,1′:3′,1”)terphenyl-4-carboxylic acid
3′-[3-tert-butyl-4-(pyrrolidin-1-yl)phenyl]-4′-(2-hydroxyethoxy)-[1,1′-biphenyl]-4-carboxylic acid
UNII-0J8RN2W0HK,
Galderma Research & Development
459.5766
C29 H33 N O4
- CD-5789
- CD5789
Trifarotene, sold under the brand name Aklief, is a medication for the topical treatment of acne vulgaris in those nine years of age and older.[1] It is a retinoid;[2] more specifically, it is a fourth generation selective retinoic acid receptor (RAR)-γ agonist.[3]
It was approved for medical use in the United States in 2019,[1][4][5] but is not approved in the European Union as of January 2021.[6] Trifarotene was granted orphan drug designation for the treatment of congenital ichthyosis by both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA).[7][8]
State Solid
Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 245C | FDA Label |
| pKa | 5.69 (pKa1) | FDA Label |
USFDA
The drug substance, trifarotene, a terphenyl acid derivative, is a retinoic
acid receptor (RAR) aQonist and is classified as a rotenoid. Trifarotene
intended as a drug for the treatment of acne vulgaris. Since trifarotene
has not been previously approved as an active ingredient in any drug
product in the United States, it is classified as a new molecular entity
(NME).
Trifarotene is produced as a white to off-white to slightly yellow crystalline
powder. It is slightly soluble in acetone, ethanol, and toluene, very slight
soluble in isopropanol, and practically insoluble in water (tiJT4
1
Cb><“JTrifarotene is nonhygroscopic and has pKa1 of 5.69 and pKa2 of 4.55. The chemical name
for trifarotene is 4-{3-[3-tert-butyl-4-(pyrrolidin-1-yl) phenyl]-4-(2-
hydroxyethoxy) phenyl} benzoic acid. It has the chemical formula of
C29H33NQ4, the molecular weiQht of 459.59, …………https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211527Orig1s000ChemR.pdf
Prescription Products
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Aklief | Cream | 50 mcg | Topical | Galderma | 2019-11-28 | Not applicable |  |
|
| Aklief | Cream | 50 ug/1g | Topical | Galderma Laboratories, L.P. | 2019-10-04 | Not applicable |  |
For treatment of congenital ichthyosis, PRECLINICAL, Galderma Res & Dev,
Galderma announced that the U.S. Food and Drug Administration (FDA) granted Orphan Drug Designation status for the company’s trifarotene molecule for the treatment of congenital ichthyosis. Based on this decision, Galderma plans to implement a clinical development plan, reinforcing its commitment to exploring new treatment options for rare diseases, as well as meeting the needs of all patients with skin diseases over the course of their lives.
Galderma治療先天性魚鱗癬的Trifarotene分子取得FDA的孤兒藥資格認定
http://news.msn.com.tw/market3773054.aspx

The company’s molecule trifarotene is a selective agonist of the gamma retinoic acid receptor (RAR-gamma), which is currently in clinical development for use in other more common dermatological conditions. It is the drug’s retinoid functionality and potent keratolytic properties that make it a potentially viable treatment of the lamellar ichthyosis pathology. Galderma has already initiated the program for investigating the treatment of lamellar ichthyosis with trifarotene and is currently working in collaboration with regulatory authorities to implement an innovative and expedient clinical development plan.
Ichthyoses comprise a large group of skin scaling disorders with diverse etiologies. The stereotypic pathophysiology is epidermal hyperplasia and abnormal desquamation, leading to visible accumulation of squames (scales) on the skin’s surface. Congenital ichthyosis is a term used to refer to a specific group of rare inherited forms of ichthyoses that are generally more severe than non-inherited forms of the disease. Lamellar ichthyosis is one such disorder that falls within the congenital ichthyosis category. Lamellar ichthyosis is recognized as a severe disease which persists throughout life. After birth, during the first post-natal weeks, the hyperkeratotic (colloidion) membrane patients are typically born with, is gradually shed and is replaced by scaling and lichenification that involves the entire body, including face, scalp, palms and soles. While usually not life threatening, lamellar ichthyosis can result in disability, partial deafness, poor adaptation to environmental conditions (due to hypohydrosis), severe discomfort (pruritus, fissuring of the skin), and significant psycho-social impact. The estimated prevalence of LI in the US is in the range of 1 per 100,000 to 1 per 200,000 persons.
Synthesis Reference
Thoreau, E. et. al. Structure-based design of Trifarotene (CD5789), a potent and selective RARγ agonist for the treatment of acne. Bioorganic & Medicinal Chemistry Letters, Volume 28, Issue 10. 2018. Pages 1736-1741
https://www.sciencedirect.com/science/article/abs/pii/S0960894X18303482
Trifarotene – Synthetic Route 1

Synthetic Description
Reference: Biadatti, Thibaud; Dumais, Laurence; Soulet, Catherine; Talano, Sandrine; Daver, Sebastien. Preparation of [1,1′:3′,1”]terphenyl-4-carboxylic acid and esters a novel ligands modulating retinoic acid receptors (RAR), and use thereof in human medicine and in cosmetics. Assignee Galderma Research & Development, S.N.C., Fr. WO 2006066978. (2016).
PATENT
WO 2006066978
http://www.google.com/patents/WO2006066978A1?cl=en
Example 25 – 3″-ter.-Butyl-4′-(2-hvdroxyethoxy)-4″-pyrrolidin-1-ylM,1′:3′,1″1- terphenyl-4-carboxylic acid

In a manner similar to that of Example 6b, by reacting 500 mg (0.9 mmol) of ethyl 4′-(2- acetoxyethoxy)-3″-terf-butyl-4″-pyrrolidin-1 -yl[1 , 1 ‘;3’, 1 “]terphenyl-4-carboxylate with
300 mg (8 mmol) of sodium hydroxide, 242 mg of 3″-tert-butyl-4′-(2-hydroxyethoxy)-4″- pyrrolidin-1-yl[1l1′;3′,1″]terphenyl-4-carboxylic acid are obtained (yield = 55 %) in the form of a white solid (m.p. = 2230C).
1H NMR (DMSO. 400 MHz): 1.43 (s, 9H); 1.90 (m, 4H); 3.0 (m, 4H); 3.73 (d, J=4.7Hz, 2H); 4.1 (m, 2H); 4.7 (s, 1H); 7.2 (d, 1H, J=8.6Hz); 7.48 (m, 2H); 7.59 (d, J=1.6Hz, 1H); 7.64 (d, J=UHz, 1H); 7.68 (dd, J=2Hz, 7.8Hz, 1H); 7.82 (d, J=8.3Hz, 2H); 7.99 (d, J=8.4Hz, 2H).
PATENT
WO 2013178759
http://www.google.com/patents/WO2013178759A1?cl=en
PATENT
WO 2013178758
http://www.google.com/patents/WO2013178758A1?cl=en
PATENT
WO 2013178760
http://www.google.com/patents/WO2013178760A1?cl=en
The details of skin application are given in the table below.

SYN
New Drug Approvals for 2019: Synthesis and Clinical Applications
New Drug Approvals for 2019: Synthesis and Clinical Applications
Shuo Yuan, Bin Yu, Hong-Min Liu
PII: S0223-5234(20)30639-5
DOI: https://doi.org/10.1016/j.ejmech.2020.112667
Reference: EJMECH 112667
To appear in: European Journal of Medicinal Chemistry
Trifarotene (Aklief). In October 2019, trifarotene, a topical retinoid that
selectively targets retinoic acid receptor gamma (RAR-γ), was approved by the FDA
for the treatment of acne vulgaris [142]. The drug was developed and marketed by
Galderma Pharmaceutical in Switzerland. Trifarotene is considered as the first of the
‘fourth-generation’ retinoids due to its uniquely selective agonism at RAR-γ. The
selective agonism leads to downstream alterations, confering improved efficacy and
reduced side effects [143]. In two phase 3 clinical trials of 2420 patients with
moderate acne on the face and trunk, trifarotene was well tolerated and significantly
reduced inflammatory lesions as early as two weeks on the face and four weeks on the
back, shoulders and chest compared to vehicle (p<0.05) [144].
The synthetic approach of this drug was disclosed by Galderma Research &
Development (Scheme 25) [145]. Bromination of commercially available
2-(tert-butyl)aniline 171 gave 4-bromo-2-(tert-butyl)aniline 172 in quantitative yield,
which then reacted with 1-dibromobutane 173 to give phenylpyrrolidine 174 in 52%
yield. Miyaura reaction of 174 was realized by employing n-BuLi and triisopropyl
borate (TIPB) followed by washed with aqueous HCl, resulting in arylboronic acid
adduct 175 in 66% yield. Treatment of 175 with aromatic bromide 176 in the presence
of Pd(PPh3)4 gave the coupling product 177 in 47% yield, which then underwent
hydrolysis delivering trifarotene (XIX) in 55% yield.
The preparation of coupling partner 176 is depicted in Scheme 26. Esterification of
4-hydroxy-4-biphenylcarboxylic acid 178 gave ethyl benzoate derivative 179 upon
treatment with catalytic H2SO4 in the refluxing EtOH [145]. The resulting ester was
subjected to treatment with tetrabutylammonium bromide (TBAB) in THF, resulting
in bromide 180 in good yields, further NaH-mediated Williamson ether synthesis with
2-bromoethyl acetate 181 gave 176 in 95% yield.

[142] L.J. Scott, Trifarotene: first approval, Drugs 79 (2019) 1905-1909.
[143] E. Thoreau, J.M. Arlabosse, C. Bouix-Peter, S. Chambon, L. Chantalat, S.
Daver, L. Dumais, G. Duvert, A. Feret, G. Ouvry, J. Pascau, C. Raffin, N.
Rodeville, C. Soulet, S. Tabet, S. Talano, T. Portal, Structure-based design of
trifarotene (CD5789), a potent and selective RARγ agonist for the treatment of
acne, Bioorg. Med. Chem. Lett. 28 (2018) 1736-1741.
[144] J. Tan, D. Thiboutot, G. Popp, M. Gooderham, C. Lynde, J.D. Rosso, J. Weiss,
U. Blume-Peytavi, J. Weglovska, S. Johnson, L. Parish, D. Witkowska, N.S.
Colon, A.A. Saenz, F. Ahmad, M. Graeber, L.S. Gold, Randomized phase 3
evaluation of trifarotene 50 µg/g cream treatment of moderate facial and truncal
acne, J. Am. Acad. Dermatol. 80 (2019) 1691-1699.
[145] T. Biadatti, L. Dumais, C. Soulet, S. Talano, S. Daver, Novel ligands that
modulate rar receptors, and use thereof in human medicine and in cosmetics,
2006. WO2006066978.
| WO2006066978A1 * | Dec 21, 2005 | Jun 29, 2006 | Galderma Res & Dev | Novel ligands that modulate rar receptors, and use thereof in human medicine and in cosmetics |
| EP0826366A2 | Aug 1, 1997 | Mar 4, 1998 | Unilever N.V. | Cosmetic compositions containing hydroxy acid or retinoid |
| EP0989846A2 | Sep 22, 1998 | Apr 5, 2000 | E-L Management Corp. | Non-irritating cosmetic and pharmaceutical compositions |
| EP1831149A1 | Dec 21, 2005 | Sep 12, 2007 | Galderma Research & Development | Novel ligands that modulate rar receptors and use thereof in human medicine and in cosmetics |
| FR2915682A1 * | Title not available | |||
| US5851538 | Dec 29, 1995 | Dec 22, 1998 | Advanced Polymer Systems, Inc. | Retinoid formulations in porous microspheres for reduced irritation and enhanced stability |
| WO1999010308A1 * | Aug 21, 1998 | Mar 4, 1999 | Bernardon Jean Michel | Biphenyl derivatives substituted by an aromatic or heteroaromatic radical and pharmaceutical and cosmetic compositions containing same |
| US6150413 * | May 26, 1998 | Nov 21, 2000 | Centre International De Recherches Dermatologiques | Treatment of dermatological, rheumatic, respiratory, cardiovascular, bone and ophthalmological disorders, as well as mammalian skin and hair conditions; 4-(4-(biphenyl-2-yl)but-3-en-1-ynyl)benzoic acid, for example |
 |
|
| Clinical data | |
|---|---|
| Trade names | Aklief |
| Other names | CD5789 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620004 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Topical |
| Drug class | Skin and mucous membrane agents |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.278.901 |
| Chemical and physical data | |
| Formula | C29H33NO4 |
| Molar mass | 459.586 g·mol−1 |
| 3D model (JSmol) | |
References
- ^ Jump up to:a b “Drug Trials Snapshots: Aklief”. U.S. Food and Drug Administration (FDA). 11 October 2019. Archived from the original on 19 November 2019. Retrieved 18 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Trifarotene Monograph
- ^ Scott LJ (November 2019). “Trifarotene: First Approval”. Drugs. 79 (17): 1905–1909. doi:10.1007/s40265-019-01218-6. PMID 31713811.
- ^ “Aklief (trifarotene) FDA Approval History”. Drugs.com. 7 October 2019. Retrieved 19 November 2019.
- ^ “Drug Approval Package: Aklief”. U.S. Food and Drug Administration (FDA). 21 October 2019. Archived from the original on 19 November 2019. Retrieved 18 November 2019.
- ^ “Trifarotene”. European Medicines Agency. Retrieved 17 June 2020.
- ^ “Trifarotene Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 19 August 2020.
- ^ “EU/3/20/2264”. European Medicines Agency (EMA). 12 August 2020. Retrieved 19 August 2020.
External links
- “Trifarotene”. Drug Information Portal. U.S. National Library of Medicine (NLM).
- Aubert J, Piwnica D, Bertino B, Blanchet-Rethore S, Carlavan I, Deret S, Dreno B, Gamboa B, Jomard A, Luzy AP, Mauvais P, Mounier C, Pascau J, Pelisson I, Portal T, Rivier M, Rossio P, Thoreau E, Vial E, Voegel JJ: Nonclinical and human pharmacology of the potent and selective topical retinoic acid receptor-gamma agonist trifarotene. Br J Dermatol. 2018 Aug;179(2):442-456. doi: 10.1111/bjd.16719. Epub 2018 Jul 4. [PubMed:29974453]
- Balak DMW: Topical trifarotene: a new retinoid. Br J Dermatol. 2018 Aug;179(2):231-232. doi: 10.1111/bjd.16733. [PubMed:30141539]
- Blume-Peytavi U, Fowler J, Kemeny L, Draelos Z, Cook-Bolden F, Dirschka T, Eichenfield L, Graeber M, Ahmad F, Alio Saenz A, Rich P, Tanghetti E: Long-term safety and efficacy of trifarotene 50 mug/g cream, a first-in-class RAR-gamma selective topical retinoid, in patients with moderate facial and truncal acne. J Eur Acad Dermatol Venereol. 2019 Jul 15. doi: 10.1111/jdv.15794. [PubMed:31306527]
- Tan J, Thiboutot D, Popp G, Gooderham M, Lynde C, Del Rosso J, Weiss J, Blume-Peytavi U, Weglovska J, Johnson S, Parish L, Witkowska D, Sanchez Colon N, Alio Saenz A, Ahmad F, Graeber M, Stein Gold L: Randomized phase 3 evaluation of trifarotene 50 mug/g cream treatment of moderate facial and truncal acne. J Am Acad Dermatol. 2019 Jun;80(6):1691-1699. doi: 10.1016/j.jaad.2019.02.044. Epub 2019 Feb 22. [PubMed:30802558]
- Chien A: Retinoids in Acne Management: Review of Current Understanding, Future Considerations, and Focus on Topical Treatments J Drugs Dermatol. 2018 Dec 1;17(12):s51-55. [PubMed:30586483]
- FDA Approved Drugs: Aklief® [Link]
| FORM | ROUTE | STRENGTH |
|---|---|---|
| Cream | Topical | 50 mcg |
| Cream | Topical | 50 ug/1g |
| Cream | Topical | 50 MICROGRAMMI/G |
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 4 | Enrolling by Invitation | Treatment | Acne Vulgaris | 1 |
| 3 | Completed | Treatment | Acne Vulgaris | 4 |
| 2 | Completed | Treatment | Acne Vulgaris | 1 |
| 2 | Recruiting | Treatment | Lamellar Ichthyosis | 1 |
| 1 | Completed | Treatment | Malignant Lymphomas | 1 |
