
Home » FAST TRACK FDA (Page 6)
Category Archives: FAST TRACK FDA
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
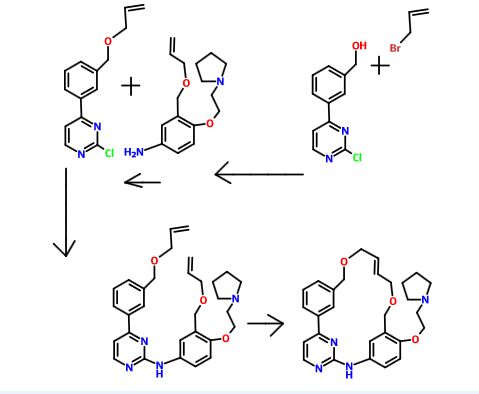
Pacritinib

Pacritinib
パクリチニブ;
| Formula |
C28H32N4O3
|
|---|---|
| CAS |
937272-79-2
|
| Mol weight |
472.5787
|
UPDATE FDA APPROVED 2/28/2022, Vonjo
To treat intermediate or high-risk primary or secondary myelofibrosis in adults with low platelets
A Jak2 inhibitor potentially for the treatment of acute myeloid Leukemia and myelofibrosis.
UNII-G22N65IL3O
пакритиниб
باكريتينيب
帕瑞替尼
ONX-0803; SB-1518
CAS No. 937272-79-2
472.57868 g/mol, C28H32N4O3
S*Bio Pte Ltd. and concert innovator
11-(2-pyrrolidin-1-ylethoxy)-14,19-dioxa-5,7,26-triazatetracyclo(19.3.1.1(2,6).1(8,12))heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
| Pacritinib (SB1518) is a potent and selective inhibitor of Janus Kinase 2 (JAK2) and Fms-Like Tyrosine Kinase-3 (FLT3) with IC50s of 23 and 22 nM, respectively. | ||||||
UPDATED
Pacritinib, sold under the brand name Vonjo, is an anti-cancer medication used to treat myelofibrosis.[1][2] It is a macrocyclic Janus kinase inhibitor. It mainly inhibits Janus kinase 2 (JAK2) and Fms-like tyrosine kinase 3 (FLT3).
Common side effects include diarrhea, low platelet counts, nausea, anemia, and swelling in legs.[2]
Medical uses
Pacritinib in indicated to treat adults who have a rare form of a bone marrow disorder known as intermediate or high-risk primary or secondary myelofibrosis and who have platelet (blood clotting cells) levels below 50,000/µL.[1][2]
History
The effectiveness and safety of pacritinib were demonstrated in a study that included 63 participants with intermediate or high-risk primary or secondary myelofibrosis and low platelets who received pacritinib 200 mg twice daily or standard treatment.[2] Effectiveness was determined based upon the proportion of participants who had a 35% or greater spleen volume reduction from baseline to week 24.[2] Nine participants (29%) in the pacritinib treatment group had a 35% or greater spleen volume reduction, compared to one participant (3%) in the standard treatment group.[2]
The U.S. Food and Drug Administration (FDA) granted the application for pacritinib priority review, fast track, and orphan drug designations.[2]
Society and culture
Names
Pacritinib is the International nonproprietary name (INN).[3][4]
References
- ^ Jump up to:a b c “Enforcement Reports”. Accessdata.fda.gov. Retrieved 5 March 2022.
- ^ Jump up to:a b c d e f g h “FDA approves drug for adults with rare form of bone marrow disorder”. U.S. Food and Drug Administration. 1 March 2022. Retrieved 3 March 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ World Health Organization (2010). “International nonproprietary names for pharmaceutical substances (INN). proposed INN: list 104” (PDF). WHO Drug Information. 24 (4): 386. hdl:10665/74579.
- ^ World Health Organization (2011). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 66”. WHO Drug Information. 25 (3). hdl:10665/74683.
External links
- “Pacritinib”. Drug Information Portal. U.S. National Library of Medicine.
OLD—
Pacritinib (INN[1]) is a macrocyclic Janus kinase inhibitor that is being developed for the treatment of myelofibrosis. It mainly inhibits Janus kinase 2 (JAK2). The drug is in Phase III clinical trials as of 2013.[2] The drug was discovered in Singapore at the labs of S*BIO Pte Ltd. It is a potent JAK2 inhibitor with activity of IC50 = 23 nM for the JAK2WT variant and 19 nM for JAK2V617F with very good selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively).[3][4] The drug is acquired by Cell Therapeutics, Inc. (CTI) and Baxter international and could effectively address an unmet medical need for patients living with myelofibrosis who face treatment-emergent thrombocytopenia on marketed JAK inhibitors.[5]
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.
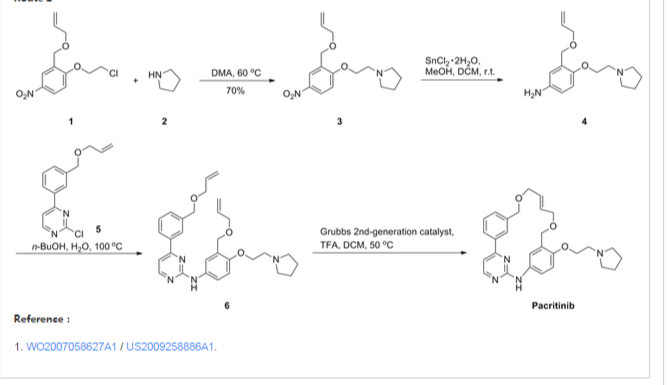
Synthesis Reference
A245943 — William AD, Lee AC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Tan E, Chen D, Williams M, Sun ET, Goh KC, Ong WC, Goh SK, Hart S, Jayaraman R, Pasha MK, Ethirajulu K, Wood JM, Dymock BW: Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem. 2011 Jul 14;54(13):4638-58. doi: 10.1021/jm200326p. Epub 2011 Jun 15.
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.


The compound 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) was first described in PCT/SG2006/000352 and shows significant promise as a pharmaceutically active agent for the treatment of a number of medical conditions and clinical development of this compound is underway based on the activity profiles demonstrated by the compound.

-
In the development of a drug suitable for mass production and ultimately commercial use acceptable levels of drug activity against the target of interest is only one of the important variables that must be considered. For example, in the formulation of pharmaceutical compositions it is imperative that the pharmaceutically active substance be in a form that can be reliably reproduced in a commercial manufacturing process and which is robust enough to withstand the conditions to which the pharmaceutically active substance is exposed.
-
In a manufacturing sense it is important that during commercial manufacture the manufacturing process of the pharmaceutically active substance be such that the same material is reproduced when the same manufacturing conditions are used. In addition it is desirable that the pharmaceutically active substance exists in a solid form where minor changes to the manufacturing conditions do not lead to major changes in the solid form of the pharmaceutically active substance produced. For example it is important that the manufacturing process produce material having the same crystalline properties on a reliable basis and also produce material having the same level of hydration.
-
In addition it is important that the pharmaceutically active substance be stable both to degradation, hygroscopicity and subsequent changes to its solid form. This is important to facilitate the incorporation of the pharmaceutically active substance into pharmaceutical formulations. If the pharmaceutically active substance is hygroscopic (“sticky”) in the sense that it absorbs water (either slowly or over time) it is almost impossible to reliably formulate the pharmaceutically active substance into a drug as the amount of substance to be added to provide the same dosage will vary greatly depending upon the degree of hydration. Furthermore variations in hydration or solid form (“polymorphism”) can lead to changes in physico-chemical properties, such as solubility or dissolution rate, which can in turn lead to inconsistent oral absorption in a patient.
-
Accordingly, chemical stability, solid state stability, and “shelf life” of the pharmaceutically active substance are very important factors. In an ideal situation the pharmaceutically active substance and any compositions containing it, should be capable of being effectively stored over appreciable periods of time, without exhibiting a significant change in the physico-chemical characteristics of the active substance such as its activity, moisture content, solubility characteristics, solid form and the like.
-
In relation to 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene initial studies were carried out on the hydrochloride salt and indicated that polymorphism was prevalent with the compound being found to adopt more than one crystalline form depending upon the manufacturing conditions. In addition it was observed that the moisture content and ratio of the polymorphs varied from batch to batch even when the manufacturing conditions remained constant. These batch-to-batch inconsistencies and the exhibited hygroscopicity made the hydrochloride salt less desirable from a commercial viewpoint.
-
Accordingly it would be desirable to develop one or more salts of 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene which overcome or ameliorate one or more of the above identified problems.
PATENT
US 2011263616
http://www.google.com/patents/US20110263616
11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26triaza-tetra-cyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) which have been found to have improved properties. In particular the present invention relates to the maleate salt of this compound. The invention also relates to pharmaceutical compositions containing this salt and methods of use of the salt in the treatment of certain medical conditions.

PATENT
http://www.google.com/patents/US8415338
Representative Procedure for the Synthesis of Compounds Type (XVIIId) [3-(2-Chloro-pyrimidin-4-yl)-phenyl]-methanol (XIIIa2)

Compound (XIIIa2) was obtained using the same procedure described for compound (XIIIa1); LC-MS (ESI positive mode) m/z 221 ([M+H]+).
4-(3-Allyloxymethyl-phenyl)-2-chloro-pyrimidine (XVa2)

Compound (XVa2) was obtained using the same procedure described for compound (XVa1); LC-MS (ESI positive mode) m/z 271 ([M+H]+).
[4-(3-Allyloxymethyl-phenyl)-pyrimidin-2-yl]-[3-allyloxymethyl-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-amine (XVIId1)

Compound (XVIId1) was obtained using the same procedure described for compound (XVIIb1); LC-MS (ESI positive mode) m/z 501.
Macrocycle Example 3 Compound 13

Compound (13) was obtained using the same procedure described for compound (1) HPLC purity at 254 nm: 99%; LC-MS (ESI positive mode) m/z 473 ([M+H]+); 1H NMR (MeOD-d4) δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34-8.31 (m, 1H), 7.98-7.96 (m, 1H), 7.62-7.49 (m, 2H), 7.35 (d, 1H), 7.15-7.10 (m, 1H), 7.07-7.02 (m, 1H), 5.98-5.75 (m, 2H, 2×=CH), 4.67 (s, 2H), 4.67 (s, 2H), 4.39-4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88-3.82 (m, 2H), 3.70 (t, 2H), 2.23-2.21 (m, 2H), 2.10-2.07 (m, 2H).
PAPER
J MC 2011, 54 4638
http://pubs.acs.org/doi/abs/10.1021/jm200326p

Discovery of the activating mutation V617F in Janus Kinase 2 (JAK2V617F), a tyrosine kinase critically involved in receptor signaling, recently ignited interest in JAK2 inhibitor therapy as a treatment for myelofibrosis (MF). Herein, we describe the design and synthesis of a series of small molecule 4-aryl-2-aminopyrimidine macrocycles and their biological evaluation against the JAK family of kinase enzymes and FLT3. The most promising leads were assessed for their in vitro ADME properties culminating in the discovery of 21c, a potent JAK2 (IC50 = 23 and 19 nM for JAK2WT and JAK2V617F, respectively) and FLT3 (IC50 = 22 nM) inhibitor with selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively). Further profiling of 21c in preclinical species and mouse xenograft and allograft models is described. Compound 21c(SB1518) was selected as a development candidate and progressed into clinical trials where it is currently in phase 2 for MF and lymphoma.
 Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
S*BIO Pte. Ltd., 1 Science Park Road, #05-09, The Capricorn, Singapore Science Park II, Singapore 117528
J. Med. Chem., 2011, 54 (13), pp 4638–4658
DOI: 10.1021/jm200326p
Publication Date (Web): May 23, 2011
Copyright © 2011 American Chemical Society
(21c)
The title compound was synthesized from 21a and pyrrolidine (yield, 83%; mixture of trans/cis85:15 by NMR). LC-MS (ESI positive mode) m/z 473 ([M + H]+). HRMS: theoretical C28H32N4O3MW, 472.2474; found, 473.2547. 1H NMR (MeOD-d4): δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34–8.31 (m, 1H, CH), 7.98–7.96 (m, 1H), 7.62–7.49 (m, 2H), 7.35 (d, 1H), 7.15–7.10 (m, 1H), 7.07–7.02 (m, 1H), 5.98–5.75 (m, 2H), 4.67 (s, 2H), 4.67 (s, 2H), 4.39–4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88–3.82 (m, 2H), 3.70 (t, 2H), 2.23–2.21 (m, 2H), 2.10–2.07 (m, 2H); chloride content (titration) 7.7% (1.18 equivs); water content (Karl Fischer) 6.1% (1.85 equivs); Anal. Calcd. for C28H32N4O3·1.18HCl·1.85H2O: C, 61.46; H, 6.46; N, 10.24; Cl, 7.65. Found: C, 61.99; H, 6.91; N, 10.25; Cl, 7.45.
References
1 “International Nonproprietary Names for Pharmaceutical Substances (INN) List 104” (PDF). WHO Drug Information 24 (4): 386. 2010.
2“JAK-Inhibitoren: Neue Wirkstoffe für viele Indikationen”. Pharmazeutische Zeitung (in German) (21). 2013.
3William, A. D.; Lee, A. C. -H.; Blanchard, S. P.; Poulsen, A.; Teo, E. L.; Nagaraj, H.; Tan, E.; Chen, D.; Williams, M.; Sun, E. T.; Goh, K. C.; Ong, W. C.; Goh, S. K.; Hart, S.; Jayaraman, R.; Pasha, M. K.; Ethirajulu, K.; Wood, J. M.; Dymock, B. W. (2011). “Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma”. Journal of Medicinal Chemistry 54 (13): 4638–58. doi:10.1021/jm200326p. PMID 21604762.
4Poulsen, A.; William, A.; Blanchard, S. P.; Lee, A.; Nagaraj, H.; Wang, H.; Teo, E.; Tan, E.; Goh, K. C.; Dymock, B. (2012). “Structure-based design of oxygen-linked macrocyclic kinase inhibitors: Discovery of SB1518 and SB1578, potent inhibitors of Janus kinase 2 (JAK2) and Fms-like tyrosine kinase-3 (FLT3)”. Journal of Computer-Aided Molecular Design 26 (4): 437–50. doi:10.1007/s10822-012-9572-z. PMID 22527961.
5http://www.pmlive.com/pharma_news/baxter_licenses_cancer_drug_from_cti_in_$172m_deal_519143
| US8153632 * | Nov 15, 2006 | Apr 10, 2012 | S*Bio Pte Ltd. | Oxygen linked pyrimidine derivatives |
| US8415338 * | Apr 4, 2012 | Apr 9, 2013 | Cell Therapeutics, Inc. | Oxygen linked pyrimidine derivatives |
| US20110294831 * | Dec 9, 2009 | Dec 1, 2011 | S*Bio Pte Ltd. | 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene citrate salt |
| Patent | Submitted | Granted |
|---|---|---|
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US8153632] | 2009-03-19 | 2012-04-10 |
| ANTIVIRAL JAK INHIBITORS USEFUL IN TREATING OR PREVENTING RETROVIRAL AND OTHER VIRAL INFECTIONS [US2014328793] | 2012-11-30 | 2014-11-06 |
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US2013172338] | 2013-02-20 | 2013-07-04 |
| METHOD OF SELECTING THERAPEUTIC INDICATIONS [US2014170157] | 2012-06-15 | 2014-06-19 |
| CYCLODEXTRIN-BASED POLYMERS FOR THERAPEUTIC DELIVERY [US2014357557] | 2014-05-30 | 2014-12-04 |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE MALEATE SALT [US2011263616] | 2011-10-27 | |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE CITRATE SALT [US2011294831] | 2011-12-01 | |
| BIOMARKERS AND COMBINATION THERAPIES USING ONCOLYTIC VIRUS AND IMMUNOMODULATION [US2014377221] | 2013-01-25 | 2014-12-25 |
| Oxygen linked pyrimidine derivatives [US8415338] | 2012-04-04 | 2013-04-09 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(16E)-11-[2-(1-Pyrrolidinyl)ethoxy]-14,19-dioxa-5,7,26-triazatetracyclo[19.3.1.12,6.18,12]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 46216796 |
| ChemSpider | 28518965 |
| ChEMBL | CHEMBL2035187 |
| Synonyms | SB1518 |
| Chemical data | |
| Formula | C28H32N4O3 |
| Molecular mass | 472.58 g/mol |


S*Bio Pte Ltd
Address: 1 Science Park Rd, Singapore 117528
Phone:+65 6827 5000

S*BIO Pte Ltd. provides research and clinical development services for small molecule drugs for the treatment of cancer in Singapore. The company’s products include JAK2 inhibitors, such as SB1518 for leukemia/myelofibrosis, lymphoma, and polycythemia; and SB1578 for RA/psoriasis. The company also offers SB939, a histone deacetylases for MDS/AML+combo, prostate cancer, sarcoma, pediatric tumor, and myelofibrosis; SB2602, a mTOR inhibitor; SB2343, a mTOR/PI3K inhibitor; and SB1317, a CDK/Flt3 inhibitor. The company was founded in 2000 and is based in Singapore. S*BIO Pte Ltd. operates as a subsidiary of Chiron Corporation Limited.
SEE……..http://apisynthesisint.blogspot.in/2016/01/pacritinib.html

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
|
|
| Clinical data | |
|---|---|
| Trade names | Vonjo |
| Other names | SB1518 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C28H32N4O3 |
| Molar mass | 472.589 g·mol−1 |
| 3D model (JSmol) | |
///////Vonjo, FDA APPTOVESD 2022, APPROVALS 2022, PACRITINIB, パクリチニブ, priority review, fast track, orphan drug, UNII-G22N65IL3O, пакритиниб , باكريتينيب , 帕瑞替尼 , SB 1518
c1cc2cc(c1)-c3ccnc(n3)Nc4ccc(c(c4)COC/C=C/COC2)OCCN5CCCC5
C1CCN(C1)CCOC2=C3COCC=CCOCC4=CC=CC(=C4)C5=NC(=NC=C5)NC(=C3)C=C2
Uridine triacetate, ウリジントリアセタート FDA approves first emergency treatment for overdose of certain types of chemotherapy

12/11/2015 12:05 PM EST
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
UPDATED IN SEPT 2016…………..
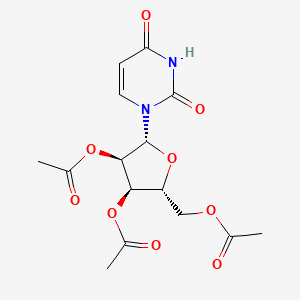
Uridine triacetate
Uridine, 5-hydroxy-, 2′,3′,5′-triacetate
Orphan drug
FAST TRACK
MF C15H18N2O9, MW 370.314
|
[(2R,3R,4R,5R)-3,4-bis(acetyloxy)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)oxolan-2-yl]methyl acetate
|
Vistogard [Trade name]
Xuriden [Trade name]
(2R,3R,4R,5R)-2-(acetoxymethyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3,4-diyl diacetate
Wellstat (Originator)
PN-401; RG-2133; TAU
MOA:Pyrimidine analog
Indication:Hereditary orotic aciduria; Chemotherapy induced poisoning
To treat patients with hereditary orotic aciduria
| Drug Name(s) | XURIDEN |
| FDA Application No. | (NDA) 208169 |
| Active Ingredient(s) | URIDINE TRIACETATE |
| Company | WELLSTAT THERAP |
| Original Approval or Tentative Approval Date | September 4, 2015 |
FDA APPROVAL SUMMARY
Chemotherapy induced poisoning, VISTOGARD, FDA 2015-12-11
Hereditary orotic aciduria, Xuriden, FIRST APPROVAL, 2015-09-04
|
|||||||
| External Identifiers |
|
|---|
Uridine triacetate is a drug used in the treatment of hereditary orotic aciduria[1] and to treat patients following an overdose ofchemotherapy drugs 5-fluorouracil or capecitabine, or in patients exhibiting early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of 5-fluorouracil or capecitabine administration.[2][3]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics and it is marketed in USA by BTG. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine triacetate is a prodrug of uridine.[4]
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTP incorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [3]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidine metabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
Marketed as the product Xuriden (FDA), uridine triacetate is indicated for the treatment of hereditary orotic aciduria. Marketed as the product Vistogard (FDA), uridine triacetate is indicated for the emergency treatment of adult and pediatric patients in the following situations: following a fluorouracil or capecitabine overdose regardless of the presence of symptoms; or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration.

Uridine Triacetate was approved by the U.S. Food and Drug Administration (FDA) on Sep 4, 2015. It was developed by Wellstat Therapeutics, then marketed as Xuriden® by Wellstat Therapeutics in US. Then it was also approved by FDA for overdose of certain types of chemotherapy on Dec 11, 2015 and marketed as Vistogard®.
Uridine Triacetate is a prodrug of the nucleoside uridine used to treat hereditary orotic aciduria. Hereditary orotic aciduria is inherited from a recessive gene. The disease is due to a defective or deficient enzyme, which results in the body being unable to normally synthesize uridine, a necessary component of ribonucleic acid (RNA). Signs and symptoms of the disease include blood abnormalities (anemia, decreased white blood cell count, decreased neutrophil count), urinary tract obstruction due to the formation of orotic acid crystals in the urinary tract, failure to thrive, and developmental delays.
Xuriden® is approved as oral granules that can be mixed with food or in milk or infant formula, and is administered once daily. The starting dosage is 60 mg/kg once daily; the dose may be increased to 120 mg/kg (not to exceed 8 grams) once daily for insufficient efficacy.
Mechanism Of Action
Uridine triacetate is an acetylated form of uridine. Following oral administration, uridine triacetate is deacetylated by nonspecific esterases present throughout the body, yielding uridine in the circulation (Figure 1).
Figure 1: Uridine Triacetate Conversion to Uridine

URIDEN provides uridine in the systemic circulation of patients with hereditary orotic aciduria who cannot synthesize adequate quantities of uridine due to a genetic defect in uridine nucleotide synthesis.
Uridine triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578] such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Uridine triacetate is also used for replacement therapy in the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase (UMPS) deficiency. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria.
Route 1


Reference:1. J. Am. Chem. Soc. 1953, 75, 2017-2019.
2. Angew. Chem. internat. Edit. 1971, 10, 75.
3. US3116282.
PATENT
Production Example 1

5.6 g of uracil and 0.1 g of ammonium sulfate were dissolved in 22.4 ml of 1,1,1,3,3,3-hexamethyldisilazane and reacted at 120° C. for 2.5 hours. After the completion of the reaction, the reaction mixture was distilled to give 11.8 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine. 1H-NMR (400 MHz, in C2D6CO): δ=0.29 (s, 9H), 0.31 (s, 9H), 6.35 (d, J=5.6 Hz, 1H), 8.19 (d, J=5.5Hz, 1H)
Referential Example 11.21 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 1.15 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.8 ml of acetonitrile and cooled to 5° C. Next, 0.94 g of SnCl4 was added dropwise thereinto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted for 3 hours. The reaction mixture was analyzed by HPLC. Thus, β-uridine triacetate was obtained with a reaction yield of 83%.
Example 1

0.93 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 0.92 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.7 ml of acetonitrile and cooled to 4° C. Then 0.49 g of FeCl3 was added thereto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted. The reaction was monitored by HPLC. After the completion of the reaction, the reaction mixture was added dropwise at 4° C. into a cold aqueous solution of sodium hydrogencarbonate which had been preliminarily prepared. After filtering off the catalyst residue, the filtrate was separated and the aqueous layer was extracted with 20 ml portions of ethyl acetate thrice. The organic layers were combined, washed with a saturated aqueous solution of sodium chloride and dried over sodium sulfate. After distilling off the solvent, 1.2 g (purity 80%) of the target compound was obtained as a viscous white solid.
Namely, the target compound could be obtained at a yield comparable to REFERNTIAL EXAMPLE 1 wherein SnCl4 was employed as the catalyst. 1H-NMR (400 MHz, in CDCl3): δ=2.11 (s, 3H), 2.14 (s, 3H), 2.15 (s, 3H), 4.35 (m, 3H), 5.33 (m, 2H), 5.79 (d, J=8.2 Hz, 1H), 6.04 (d, J=4.9 Hz, 1H), 7.39 (d, J=8.2 Hz, 1H)

CLIP
12/11/2015 12:05 PM EST
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
CLIP
With support from Almac, Wellstat delivers for a rare disease.
Proximity of API and finished drug development helps uridine triacetate to market for two indications
By Rick Mullin
“The initial contact was a cold call by Almac in 2010 or 2011,” recalls Mike Bamat, senior vice president of R&D at Wellstat Therapeutics, a small drug company in Gaithersburg, Md. “There were probably a couple of calls. It was one of those things where timing is everything.”
Almac, a Craigavon, Northern Ireland-based pharmaceutical services company, was looking to get in on Wellstat’s development of uridine triacetate, a synthetic pyrimidine analog, as an antidote for fluorouracil and capecitabine toxicity and overdose in cancer patients receiving those chemotherapies. And the calls, which Almac records indicate followed some communication between the companies, happened to come just when Wellstat was looking to change service partners as it moved toward commercial development of the drug.

Uridine triacetate
Discovery: Wellstat Therapeutic’s research on the therapeutic potential of exogenous uridine leads to a determination that uridine triacetate is a safe means of delivering the agent
Applications: Treatment of hereditary orotic aciduria (HOA), an extremely rare disease in which the body does not produce uridine, causing overproduction of orotic acid; emergency treatment of toxic reaction to or overdose of the cancer treatments fluorouracil and capecitabine
Methods of action: Treating HOA, uridine triacetate restores intracellular nucleotide concentrations, normalizing orotic acid production; as a chemotherapy antidote, it increases intracellular levels of uridine to dilute fluorouracil and capecitabine
Years in development: Since 2008 for chemotherapy antidote, and 2013 for HOA
Approved: Xuriden for HOA, Sept. 4, 2015; Vistogard for chemotherapy antidote, Dec. 11, 2015
The job went to Almac, as did work that sprang up as the result of another phone call to Wellstat—this one from the U.S. Food & Drug Administration.
As Bamat explains, uridine triacetate caught FDA’s attention regarding another potential indication—an extremely rare and life-threatening disease called hereditary orotic aciduria, or HOA. A consequence of the body’s inability to produce uridine, a necessary component of ribonucleic acid, HOA can manifest in a range of symptoms including blood abnormalities, developmental delays, and urinary tract obstruction caused by overproduction of orotic acid. There have been 20 reported cases of HOA since the 1950s. Only four cases are currently known in the U.S., Bamat says, and likely fewer than 20 in the world.
Wellstat landed approvals for Xuriden, the HOA treatment, in September of last year and Vistogard, the chemotherapy antidote, in December.
The story of Xuriden centers on a raft of FDA incentives for super-rare diseases that enabled Wellstat to move forward on an expedited application for a drug that will never be made in any great volume. But bringing Xuriden and Vistogard to market may also be viewed as the story of a drug discovery firm becoming a commercial enterprise thanks to its partnership with a service provider.
As Wellstat began late-stage development of the chemotherapy antidote, its research partner at the time, QS Pharma, was acquired by the service firm WIL Research. The look and feel of the partnership changed, according to Bamat.
“We kind of lost the small, easy-to-work-with relationship we had with them,” he says. Wellstat also needed support on development and manufacturing of a finished drug product composed of granules delivered in packets or sachets. The drug is administered orally, usually sprinkled on food such as applesauce or yogurt.
Almac was deemed a good fit because of its experience with developing drugs in granule form for “sachet presentation,” a packaging method more common in Europe than in the U.S. The Northern Ireland firm’s ability to develop and manufacture the active pharmaceutical ingredient (API) and the drug product in one location—at its headquarters—would also prove to be a significant advantage.
The distance between Gaithersburg and Craigavon, however, was a concern, according to Bamat. “We debated it. Especially those of us who knew we would be going there,” he says. “We couldn’t just jump in a car and go. But we looked at a variety of things, including cost and value, and it was all very positive at Almac.”
According to David Downey, vice president of commercial operations at Almac, bringing Wellstat’s work on uridine triacetate to commercial production posed several challenges, the first being to secure supply of uridine starting material, which is extracted from sugar beets by Euticals, an Italian firm. Next was developing a method to control particle size in both the API and the finished product. Almac also had to validate process equipment as it scaled up production.
“Uridine triacetate is Wellstat’s first commercial product,” Downey says. “So we were provided with a process more fit for development than for commercial production.”
The basic formulation of a granule drug product is simple, according to Downey: The API and excipient are mixed in a dry blender. The challenge is developing an analytical regimen to assure the granules are blended uniformly. Meeting the challenge required a high level of coordination between API and drug product process development.
“Wellstat needed a partner that could support them from the API to the drug product,” Downey says. The physical proximity between the Almac facilities in Craigavon conducting API and drug product work was a key advantage, he claims.

Uridine triacetate is formulated into granules presented in packets and sprinkled on food.
Credit: Wellstat Therapeutics
“If you listen to our business development people, you’ll hear them use the term, ‘crossing car parks as opposed to crossing oceans,’ ” Downey says, explaining that many competitors who offer API and finished drug services run these operations thousands of kilometers apart from each other, sometimes on different continents.
Before it signed on with Almac, Wellstat had been working with uridine triacetate for about 10 years. Its focus on developing the antidote drug started in 2008. Branching into the HOA treatment, however, upped the stakes.
Clinical study development for an HOA therapy was expedited via a full house of regulatory incentives from FDA, according to Bamat. “We had orphan drug designation, rare pediatric designation, breakthrough therapy designation, and priority review,” he says. “So they really went all out in helping us develop this.”
Although Wellstat was interested in developing a life saving drug for children, it was concerned about paying for it, given the tiny market. “At that time, the rare pediatric disease priority review voucher program was just on the radar,” Bamat says. “FDA said, ‘Consider this new program. Maybe it’s a way that at some risk you could recoup some of your costs.’ We looked at it and were willing to take the risk.”
It paid off. Wellstat was able to sell its priority review voucher—which entitles a company that brings a rare pediatric drug to market to receive expedited review of a subsequent drug—to AstraZeneca last year for an undisclosed amount. Other vouchers sold in 2015 brought high sums, including $350 million for one that AbbVie bought from United Therapeutics in August.
Bamat says Wellstat is not likely to change focus after its success with uridine triacetate. It continues to investigate new indications for the compound and will likely work with Almac on anything going into commercial development.
He emphasizes the importance of maintaining an effective working relationship with an outsourcing partner. “My main consideration is that these are people we can really work with on a day-to-day, week-to-week basis,” Bamat says. “Will the communication be good? Will they be honest and transparent with us, and will we be the same for them? That was a key factor, and we felt it was a plus with Almac.”
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2-3 hours |
| Biological half-life | 2-2.5 hours |
| Excretion | Renal |
| Identifiers | |
| DrugBank | DB09144 |
| Chemical data | |
| Formula | C15H18Cl0N2O9S0 |
| Molar mass | 370.31 g·mol−1 |
References
- HIGHLIGHTS OF PRESCRIBING INFORMATION OF XURIDEN
- Jump up^ BTG Announces FDA Approval of VISTOGARD® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine
- Jump up^ “FDA Approved Drugs:Uridine Triacetate”. FDA. 2015-12-11. Retrieved 2016-04-29.
- “Uridine triacetate”. DrugBank.
| Patent ID | Date | Patent Title |
|---|---|---|
| US7807654 | 2010-10-05 | Compositions and methods for treatment of mitochondrial diseases |
| US2010222296 | 2010-09-02 | PYRIMIDINES, SUCH AS URIDINE, IN TREATMENTS FOR PATIENTS WITH BIPOLAR DISORDER |
| US7737128 | 2010-06-15 | Pyrimidines, such as uridine, in treatments for patients with bipolar disorder |
| US2010098678 | 2010-04-22 | Methods of Treatment of Mitochondrial Disorders |
| US2010041620 | 2010-02-18 | METHODS FOR IMPROVING FRONTAL BRAIN BIOENERGETIC METABOLISM |
| US2010041621 | 2010-02-18 | METHODS AND COMPOSITIONS FOR IMPROVING COGNITIVE PERFORMANCE |
| US7582619 | 2009-09-01 | Compositions and methods for treatment of mitochondrial diseases |
| US2008226684 | 2008-09-18 | METHOD AND PROCESS FOR THE PRODUCTION OF MULTI-COATED RECOGNITIVE AND RELEASING SYSTEMS |
| US7105498 | 2006-09-12 | Acylated uridine and cytidine and uses thereof |
| US6956028 | 2005-10-18 | Compositions and methods for treatment of mitochondrial diseases |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015307542 | 2015-10-29 | MODIFIED NUCLEIC ACID MOLECULES AND USES THEREOF |
| US2015167017 | 2015-06-18 | ALTERNATIVE NUCLEIC ACID MOLECULES AND USES THEREOF |
| US8821899 | 2014-09-02 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8771713 | 2014-07-08 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8741316 | 2014-06-03 | Highly porous, recognitive polymer systems |
| US2012294869 | 2012-11-22 | Methods for Treating Fatty Liver Disease |
| US2012078529 | 2012-03-29 | DETERMINING THE SEVERITY OF 5-FLUOROURACIL OVERDOSE |
| US8067392 | 2011-11-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7915233 | 2011-03-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7884202 | 2011-02-08 | Nucleobase Having Perfluoroalkyl Group and Process for Producing the Same |
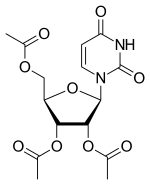
Uridine triacetate
- Molecular FormulaC15H18N2O9
- Average mass370.311 Da
ウリジントリアセタート
[(2R,3R,4R,5R)-3,4-diacetyloxy-5-(2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl acetate
Uridine, 2′,3′,5′-triacetate
uridini triacetas
Vistogard [Trade name]
Xuriden [Trade name]
(2R,3R,4R,5R)-2-(acetoxymethyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3,4-diyl diacetate
[(2R,3R,4R,5R)-3,4-bis(acetyloxy)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)oxolan-2-yl]methyl acetate
FDA APPROVED2015/9/4 . AS Xuriden
Uridine triacetate (INN),[1] formerly known as vistonuridine, is an orally active tri-acetylated prodrug of uridine[2] used:
- in the treatment of hereditary orotic aciduria (brand name Xuriden /ˈzʊərədɛn/ ZOOR-ə-den);[3]
- to treat patients following an overdose of chemotherapy drugs 5-fluorouracil (5-FU) or capecitabine regardless of the presence of symptoms, or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration (brand name Vistogard).[4][5][6]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine Triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. Uridine, a pyrimidine nucleotide, has been used in a variety of diseases including depressive disorders and inherited myopathies. (NCI04)
Pharmacology from NCIt
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTPincorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidinemetabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
from DrugBank
Uridine triacetate is an acetate ester that is uracil in which the three hydroxy hydrogens are replaced by acetate group. A prodrug for uridine, it is used for the treatment of hereditary orotic aciduria and for management of fluorouracil toxicity. It has a role as a prodrug, a neuroprotective agent and an orphan drug. It is a member of uridines and an acetate ester.
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 65” (PDF). World Health Organization. p. 92. Retrieved 12 March 2017.
- ^ “Uridine triacetate — DrugBank Page”. 12 March 2017.
- ^ “Xuriden (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “Vistogard (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “BTG Announces FDA Approval of Vistogard® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine”. BTG International Ltd. 11 December 2015. Retrieved 12 March 2017.
- ^ “Approved Drugs — Uridine Triacetate”. U.S. Food and Drug Administration. Retrieved 12 March 2017.
External links
Patents
- US7776838
- US5968914
- US6258795
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7776838 |
| Expiration | Aug 17, 2027 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
from FDA Orange Book
| FDA Orange Book Patents: 2 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6258795 |
| Expiration | Jul 10, 2019 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
from FDA Orange Book
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2–3 hours |
| Elimination half-life | 2–2.5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.021.710 |
| Chemical and physical data | |
| Formula | C15H18N2O9 |
| Molar mass | 370.31 g·mol−1 |
| 3D model (JSmol) | |
////////////Uridine triacetate, ウリジントリアセタート , FDA 2015, breakthrough therapy designation ,
//////////174105-38-8, Priority review drug , Orphan drug, FDA 2015, Vistogard, uridine triacetate, fast track designations, PN-401, RG-2133, TAU, XURIDEN
|
CC(=O)OC[C@H]1O[C@H]([C@H](OC(C)=O)[C@@H]1OC(C)=O)N1C=CC(=O)NC1=O
|
MK 7655, RELEBACTAM, a β-Lactamase inhibitor
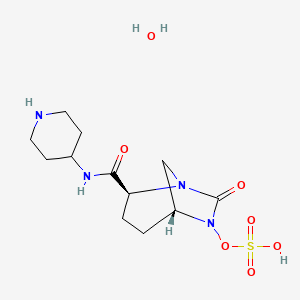
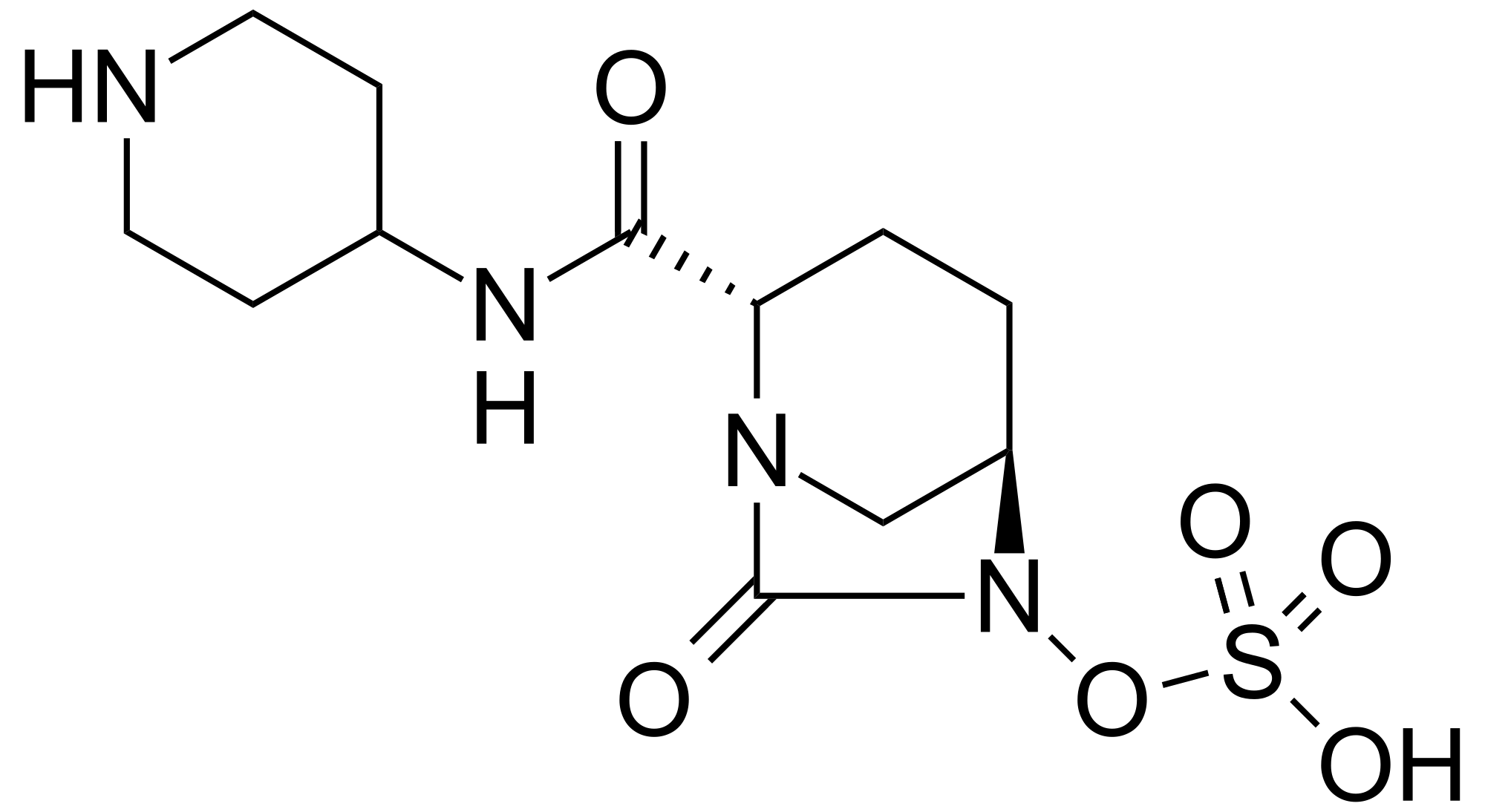
MK 7655, RELEBACTAM
(1R,2S,5R)-7-Oxo-N-(4-piperidinyl)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
| MF C12H22N4O7S | |
| MW | 366.39068 g/mol |
|---|
CAS 1174020-13-3
β-Lactamase inhibitor
MK-7655 is a beta-lactamase inhibitor in phase III clinical studies at Merck & Co for the treatment of serious bacterial infections…….See clinicaltrials.gov, trial identifier numbers NCT01505634 and NCT01506271.
In 2014, Qualified Infectious Disease Product (QIDP) and Fast Track designations were assigned by the FDA for the treatment of complicated urinary tract infections, complicated intra-abdominal infections and hospital-acquired bacterial pneumonia/ventilator-associated bacterial pneumonia.

PAPER
A concise synthesis of a beta-lactamase inhibitor
Org Lett 2011, 13(20): 5480
http://pubs.acs.org/doi/abs/10.1021/ol202195n
http://pubs.acs.org/doi/suppl/10.1021/ol202195n/suppl_file/ol202195n_si_001.pdf

MK-7655 (1) is a β-lactamase inhibitor in clinical trials as a combination therapy for the treatment of bacterial infection resistant to β-lactam antibiotics. Its unusual structural challenges have inspired a rapid synthesis featuring an iridium-catalyzed N–H insertion and a series of late stage transformations designed around the reactivity of the labile bicyclo[3.2.1]urea at the core of the target.
H NMR (400 MHz, DMSO-d6): δ 8.30 (br s, 2H), 8.20 (d, J = 7.8 Hz, 1H), 4.01 (s, 1H), 3.97-3.85 (m, 1H), 3.75 (d, J = 6.5 Hz, 1H), 3.28 (dd, J = 12.9, 2.5 Hz, 2H), 3.05-2.93 (m, 4H), 2.08-1.97 (m, 1H), 1.95-1.79 (m, 3H), 1.73-1.59 (m, 4H);
13C NMR (DMSO-d6, 100 MHz) δ 169.7, 166.9, 59.8, 58.3, 46.9, 44.3, 42.9, 28.5, 28.3, 20.8, 18.9;
HRMS calculated for C12H20N4O6S (M+H): 349.1182, found: 349.1183.
[α]D 25 = -23.3 (c = 1.0, CHCl3)


PATENT
WO 2009091856
http://www.google.com/patents/WO2009091856A2?cl=en
EXAMPLE IA
(2S ,5 R)-7-Oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo [3.2.1 ]octane-2-carboxamide

Step 1 : tert-butyl 4-({[(2S,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]oct-2- yljcarbonyl } amino)piperidine- 1 -carboxylate : To a solution of (2S,5R)-6-(phenylmethoxy)-7-oxo-l,6-diazabicyclot3.2.1]octane-
2-carboxylic acid (1.484 g, 5.37 mmol) in dry dichloromethane (60 ml) was added triethylamine (1.88 ml, 13.49 mmol), 2-chloro-l-methylpyridinium iodide (1.60 g, 6.26 mmol), and 4-amino-l- BOC-piperidine (1.30 g, 6.49 mmol) sequentially at room temperature under nitrogen. The reaction was then heated to 500C for 1 hour. The reaction mixture was concentrated under vacuum and purified by silica gel chromatography on an Isco Combiflash (40 g silica gel, 40 mL/min, 254 nM, 15% to 100% EtOAc/hexane over 14 column volumes then 100% EtOAc for 4 column volumes; title compuond eluted at 65% ethyl acetate/hexane) to afford the title compound as a pale orange solid.
Step 2: tert-butyl 4-({[(2S,5R)-6-hydroxy-7-oxo-l ,6-diazabicyclo[3.2.1]oct-2- yl] carbonyl } amino)piρeridine- 1 -carboxylate:
Palladium on carbon (394 mg; 10% Pd/C) was added to a solution of the product of step 1 (1.81 g, 3.95 mmol) in methanol (50.6 mL) and the resulting mixture was stirred under hydrogen (balloon) overnight. LC/MS analysis indicated the reaction was not complete. Acetic acid (6 drops) and additional catalyst (159 mg of 10% Pd/C) were added to the reaction and the resulting mixture was stirred under hydrogen (balloon) for an additional 90 minutes. Additional catalyst (0.2085 g of 10% Pd/C) was added to the reaction and stirring under hydrogen was continued for an additional 2.5 hours at which time the reaction was judged complete by LC-MS analysis. The reaction was filtered through a celite pad and the collected solid was washed well wtih MeOH. The filtrate was concentrated under vacuum to afford the title compound as a colorless oil which was used without purification in the next step.
Step 3 : tert-butyl-4-({ [(2S,5R)-7-oxo-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]oct-2- yl] carbonyl } amino)ρiperidine- 1 -carboxylate:
To a solution of the product of step 2 (1.455 g, 3.95 mmol; theoretical yield of step 2) in dry pyridine (30 mL) was added sulfur trioxide pyridine complex (3.2 g, 20.11 mmol) at room temperature under nitrogen. The resulting thick mixture was stirred over the weekend.
The reaction was filtered and the white insoluble solids were washed well with dichloromethane. The filtrate was concentrated in vacuo. The residue was further azeotroped with toluene to remove excess pyridine to afford the title compound which was used without purification in the next step.
Step 4: (2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2- carboxamide:
To a mixture of the product of step 3 (1.772 g, 3.95 mmol; theoretical yield of step 3) in dry dichloromethane (30 ml) at 00C under nitrogen was slowly added trifluoroacetic acid (6.1 ml, 79 mmol). Immediately the reaction became a solution. After 1 hour, additional trifluoroacetic acid (8 ml) was added to the reaction. The reaction was stirred at 00C until judged complete by LC-MS analysis then concentrated in vacuo. The residue was triturated with ether (3X) to remove excess TFA and organic impurities. The resulting white insoluble solid was collected via centrifugation, dried in vacuo, then purified by preparative HPLC (250X21.2 mm Phenomenex Synergi Polar-RP 80A column; 10 micron; 35 mL/min.; 210 nM; 0% to 30% methanol/water over 15 minutes; title compound eluted at 10% methanol/water). Fractions containing the title compound were combined and Iyophilized overnight to afford the title compound as a white solid. LC-MS (negative ionization mode) m/e 347 (M-H).
PAPER
Discovery of MK-7655, a beta-lactamase inhibitor for combination with Primaxin
Bioorg Med Chem Lett 2014, 24(3): 780
http://www.sciencedirect.com/science/article/pii/S0960894X13014856

PATENT
WO 2014200786
http://www.google.dj/patents/WO2014200786A1?cl=en


![]()




Exemplary Scheme

– 50% isolated yield overall from 1 to 5

O via crystallization
XAMPLE 1
(2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide 
Preparation of (15′,45)-5-((2-nitrophenyl)sulfonyl)-2-oxa-5-azabicyclo[2.2.2]octan-3 one (2)
To a reactor (R-1) equipped with an additional funnel, nitrogen inlet and agitator was charged (2S,5S)-5-hydroxypiperidine-2-carboxylic acid (77.3 wt%) (50.0 g, 344 mmol), and water (150 mL). Agitation was begun, the pH adjusted to 10-11 by addition of 10 N NaOH (~ 46.5 mL) and the reactor charged with acetone (50.0 mL).
In a separate reactor (R-2) equipped with an agitator and nitrogen inlet was charged 2-nitrobenzene-l-sulfonyl chloride (97%) (106.0 g, 478 mmol) and acetone (80 mL). The contents of R-2 were transferred to R-1 at 23-30 °C while the pH of the solution was maintained at 10-11 by simultaneously addition of 10 N NaOH. After 15 to 30 min, the pH was adjusted to about 6 by addition of 12 N HC1. The solution was charged with EtOAc (500 mL) and the pH adjusted to 3.0 by addition of 12 N HC1. The layers were separated and the aqueous back-extracted with EtOAc (150 mL x 2).
To a separate reactor (R-3) was charged product la in the combined organic layers, 2-nitrobenzene-l-sulfonyl chloride (73.0 g, 329 mmol), and triethylamine (130 mL). The batch in R-3 was agitated at 20-28°C for 30 min. The solution was charged with water (100 mL), the layers separated, and the aqueous back extracted with EtOAc (150 mL x 2). The combined EtOAc layer was washed with 10% NaHC03 (100 mL) and brine (100 mL). The organic phase was concentrated to 150 mL upon which a crystalline slurry was formed. The concentrated solution was agitated at 13-18°C for 2-3 hours followed by filtration of crystalline solids. The resulting wet cake was washed with EtOAc (60 mL) and then dried under vacuum oven at 25-30°C to afford 2 (65.6 g, 79% yield), m.p. 126.0-126.7 °C. 1H NMR (CDC13, 400 MHz) δ: 8.02 (m, 1 H), 7.80-7.71 (m, 2 H), 7.66 (m, 1 H), 4.88 (m, 1 H), 4.55 (dd, J= 3.8, 2.7 Hz, 1 H), 3.78 (dt, J= 11.2, 3.0 Hz, 1 H), 3.66 (dd, J = 11.2, 1.1 Hz, 1 H), 2.44 (m, 1 H), 2.11 (m, 2 H), 1.91 (m, 1 H); 13C NMR (CDC13, 100 MHz) δ: 168.4, 148.3, 134.4, 132.1, 131.0, 130.7, 124.2, 73.5, 51.4, 48.0, 25.1, 23.2
Preparation oftert-butyl 4-((25*,55)-l-((2-nitrophenyl)sulfonyl)-5-(((2- nitrophenyl)sulfony l)oxy)piperidine-2-carboxamido)piperidine- 1 -carboxylate (3)
To a reactor (R-l) was charged lactone 2 (65.5 g, 210 mmol), THF (131 mL) and tert-butyl 4-aminopiperidine-l -carboxylate (44.5 g, 222 mmol). The stirred solution was heated to reflux (typical temperature 72 °C) for ~18 hr. The reaction was cooled to 25-35 °C and then charged with THF (325 mL) and 4-dimethylaminopyridine (40.1 g, 328 mmol) followed by agitation for 30 minutes.
To a separate reactor (R-2) was charged 2-nitrobenzene-l-sulfonyl chloride (60.9 g,
275 mmol) and THF (200 mL). The contents of R-2 were added to R-l over the course of 45 to 75 minutes maintaining batch temperature of 20 to 30°C. The batch in R-l was agitated for 2 to 4 hours at a temperature of 20 to 30°C.
To a separate reactor (R-3) was charged water (600 mL) and methanol (600 mL). The contents of R-3 were charged to the main batch over the course of 45 to 75 minutes with agitation while maintaining temperature of 20 to 30°C. The batch was cooled to 5 to -5°C and then agitated at 5 to -5°C for at least 4 hours. The solids were filtered and then washed twice with methanol (130 mL x 2). The wet cake was dried in a vacuum oven at 40 to 50°C to afford 3 (144.0 g, 98% yield), m.p. 131.8-133.1 °C. 1H NMR (CDC13, 400 MHz) δ: 8.14 (m, 2 H), 7.83-7.74 (m, 6 H), 6.50 (d, J= 7.9 Hz, 1 H), 4.69 (m, 1 H), 4.43 (s, 1H), 4.11 (dd, , J= 13.7, 4.9 Hz, 1H), 3.95 (m, 2H), 3.83 (m, 1H), 3.47 (s, 1H), 3.10 (dd, J= 13.7, 11.0 Hz, 1H), 2.81 (m, 2H), 2.51 (m, 1H), 2.12 (m, 1H), 1.85-1.72 (m, 4H), 1.45 (s, 9H), 1.26 (m, 1H); 13C NMR (CDC13, 100 MHz) δ: 166.9, 154.6, 148.2, 147.6, 135.2, 134.8, 132.6, 132.5, 131.9, 131.6, 131.4, 129.7, 124.9, 124.7, 79.8, 76.5, 55.0, 47.1, 46.0, 31.8, 31.5, 28.4, 27.3, 24.4.
Preparation of N-4-nitrobenzene sulfonyl-O-benzylhydroxylamine
![]()
To a reactor (R-l) was charged O-benzylhydroxylamine hydrochloride (61.0g, 382 mmol) and pyridine (400 mL). The solution cooled to 5 to -5°C.
To a separate reactor (R-2) was charged 4-nitrobenzenesulfonyl chloride (89.0 g, 402 mmol) and pyridine (200 mL). The contents of R-2 were transferred to R-l at a rate to maintain temperature range of -5 to -5°C. The batch in R-l was agitated at 5 to -5 °C for 15 to 45 minutes then warmed to 20 to 30°C for 45 to 75 minutes. Water (250 mL) was then added at a rate to maintain 20 to 30°C and agitated 5 to 15 minutes. The solids were filtered and the wet cake washed with water (100 mL x 3). The wet cake was dried in vacuum oven at 50°C to afford N-4-nitrobenzenesulfonyl-O-benzylhydroxylamine (113.3 g, 96% yield), m.p. 128.4-130.0 °C. 1H NMR (CDCls, 400 MHz) δ: 8.36 (d, J = 8.9 Hz, 2 H), 8.11 (d, J = 8.9 Hz, 2 H), 7.36 (m, 5H), 7.11 (s, 1H), 5.02 (s, 2H); 13C NMR (CDC13, 100 MHz) δ: 151.0, 142.5, 134.9, 130.2, 129.7, 129.3, 128.9, 124.5, 80.2.
Step C. Preparation of tert-butyl 4-((2S,5R)-5-((benzyloxy)amino)piperidine -2-carboxamido)piperidine- 1 -carboxylate (4)
Boc
To a reactor (R-l) was charged tert-butyl 4-((2R,5R)-l-((2-nitrophenyl)sulfonyl)-5-(((2-nitrophenyl)sulfonyl)oxy)piperidine-2-carboxamido)piperidine-l -carboxylate (3) (110 g, 158 mmol), N-4-nitrobenzene sulfonyl-O-benzylhydroxylamine (58 g, 188 mmol), potassium carbonate (25.9 g, 187 mmol) and dimethylacetamide (440 mL). The stirred solution was heated to 60 to 70°C for 24 – 32 hours. The batch was cooled to 20 to 30°C and charged with toluene (660 mL). The batch was extracted with 1 N sodium hydroxide (3×220 mL) then washed with water (220 mL).
The toluene solution was azotropically distilled at ~50°C to about 1/3 volume. The solution was solvent-switched to MeOH at 45-55°C, adjusted to 237 mL.
The batch was cooled to 20-25°C, charged with thioglycolic acid (57.9 g, 629 mmol) at 10 °C, and then charged with K2CO3 anhydrous (172.0 g, 1225 mmol). The batch was agitated at 10-15°C for 0.5 h, warmed to 20-25°C, agitated at 20-25°C for 10-15 h, and heated at 48-53°C for 3-6 h.
The batch was charged with 10 wt% sodium chloride (1.10 L) and toluene (880 mL) at about 40°C. The layers were separated and the aq. layer back-extracted with toluene (3 x440 mL). The combined organic layer was washed with 10% NaHC03 (2 x220 mL). The batch was concentrated at 40-50°C to 165 mL, then cooled to 35-40°C. The batch was charged with seed (50 mg) and agitated for 1 h at 35-40°C. The batch was charged with heptanes (110 mL) at 35-40°C over 1 h, then slowly cooled to 15-20°C over 1 h. The batch was agitated for 3 h and the solids filtered. The wet cake was washed with toluene/heptanes (137.5 mL) then dried in vacuum oven at 30 °C for 3-8 h to affored 4. (47.3 g, 70% overall yield from 3), m.p. 117.5-118.0 °C. 1H NMR (CDC13, 500 MHz) δ: 7.37-7.29 (m, 5 H), 6.64 (d, J= 8.2 Hz, 1 H), 5.36 (brs, 1 H), 4.67 (s, 2 H), 4.00 (m, 2 H), 3.90 (m, 1 H), 3.28 (ddd, J= 11.8, 4.0, 1.7 Hz, 1 H), 3.12 (dd, J= 10.2, 3.2 Hz, 1 H), 2.95 (m, 1 H), 2.86 (m, 2 H), 2.46 (dd, J= 11.8, 9.5 Hz, 1 H), 2.10 (m, 1 H), 1.93-1.83 (m, 3 H), 1.58 (brs, 1 H), 1.45 (s, 9 H), 1.41 (m, 1 H), 1.35-1.23 (m, 3 H); 13C NMR (CDC13, 125 MHz) δ: 172.8, 154.7, 137.7, 128.4 (4 C), 127.9, 79.6, 76.9, 59.8, 57.0, 49.2, 46.1, 42.8 (br, 2 C), 32.0 (2 C), 28.4 (3 C), 28.3, 27.2.
Step D: Preparation of tert-butyl 4-((lR,2S,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1 ]octane-2-carboxamido)piperidine- 1 -carboxylate (5)
To a reactor (R-l) was charged tert-butyl 4-((2S,5R)-5-((benzyloxy)amino)piperidine-2-carboxamido)piperidine-l-carboxylate (4) (46.3 g, 107 mmol), dichloromethane (463 mL), and Hunig’s base (58.0 mL). The batch was cooled to -18°C and then charged with triphosgene in four portions (25.1 g total; 85 mmol) at <-8°C. The batch was agitated at -5 to 0°C for 0.5 h then charged with 11.4 wt% aqueous H3P04 at -5 to 0 °C (347 g, 3541 mmol). The batch was agitated at 20-25°C for 15-20 h then phase cut. The aqueous layer was back-extracted with dichloromethane (138 mL). The combined organic layer was washed with 10% NaHC03 (115 mL), then water (115 mL). The organic solution was concentrated at atmospheric pressure to ~80
mL, then charged with MTBE (347 mL) at 35-45 °C over 0.5 h, then concentrated at 35-45 °C to 231 mL two times to form a slurry.
The slurry was charged with heptanes (139 mL) at 35-45 °C over 2 h, then slowly cooled to 15-20°C over 1 h. The batch was agitated at 15-20°C for 6-8 h. Solids were filtered and the wet cake washed with MTBE/heptanes (1.4 : 1 , 185 mL) then dried under vacuum at 25-30°C for 5-10 hours to afford 5 (43.7 g, 92% yield), m.p. 161.3-161.8 °C. 1H NMR (CDC13, 500 MHz) δ: 7.45-7.32 (m, 5 H), 6.55 (d, J= 8.2 Hz, 1 H), 5.05 (d, J= 11.6 Hz, 1 H), 4.90 (d, J= 11.6 Hz, 1 H), 4.02 (m, 2 H), 3.90 (m, 2 H), 3.30 (m, 1 H), 2.99 (dt, J= 11.7, 1.1 Hz, 1 H), 2.86 (m, 2 H), 2.64 (d, J = 11.7 Hz, 1 H), 2.37 (dd, J= 14.6, 6.9 Hz, 1 H), 2.04-1.82 (m, 4 H), 1.58 (m, 1 H), 1.45 (s, 9 H), 1.30 (m, 2 H); 13C NMR (CDC13, 125 MHz) δ: 168.3, 167.5, 154.7, 135.6, 129.2 (2 C), 128.8, 128.6 (2 C), 79.7, 78.3, 60.4, 57.8, 47.5, 46.8, 42.5 (br, 2 C), 32.0, 31.7, 28.4 (3 C), 20.8, 17.2.
Step E: Preparation of tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1“|octane- 2-carboxamido) iperidine- 1 -carboxylate
tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-l -carboxylate (9.2 g, 20.1 mmol) was charged to a glass bottle, and the solids were dissolved in THF (150 mL). The solution was then charged to a hydrogenation reactor along with Pd/Al203 (10 wt%, 1.5 g). The reaction was purged three times with hydrogen and then set to a hydrogen pressure of 50 psi. The reaction temperature was adjusted to 25°C and the reaction was allowed to agitate for 22 hours. After the reaction was complete as determined by HPLC analysis, the solution was filtered through SOLKA-FLOC® (Interational Fiber Corporation, North Tonawanda, NY) to remove the catalyst and the filter cake was washed with THF. The filtrate and washes were then solvent switched by vacuum distillation to iPrOAc to a final volume of 40 mL. The resulting iPrOAc slurry was aged at room temperature for 1 hour. The solids were then filtered and washed with iPrOAc (20 mL) and dried under vacuum and N2 at 40°C to afford the title product (6.62 g., 17.97 mmol, 90% isolated yield). Spectral data matched the reference compound.
Preparation of (2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide
tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-l-carboxylate (20 g, 54.3 mmol), THF (200 mL), 2-picoline (10.9 mL, 309 mmol) and pyridine-S03 complex (30.2 g, 190 mmol) were charged to a flask under nitrogen. The heterogeneous mixture was allowed to stir overnight (~15 h). The reaction mixture was cooled to -10°C then DCM (200 mL) was added. 0.5 M K2HP04 (168 mL, 84 mmol) was added over 10 minutes. Bu4NHS04 (19.4 g, 57 mmol) was then added over 10 minutes. The biphasic mixture was stirred for 30 minutes, phase cut and the water layer was back extracted with 40 ml of DCM. The combined DCM solution was washed with water (120 ml), phase cut and the organic solution was solvent-switched to MeCN (320 ml) by vacuum distillation with 3 bed volumes of MeCN (total 1.0 L) and used as is in the next step. The solution of Bu4N+ OSO3 salt 7 in MeCN solution was used with an assumed yield of 100% (37.5 g, 54.3 mmol). The reaction mixture was cooled in an ice bath, and TMSI (10.26 ml, 70.7 mmol) was added via addition funnel over 30 minutes between 0°C and 5°C. The resulting mixture was agitated for 1-2 h and then quenched with H20:MeCN (1 :1, 6 ml) to afford a slurry. The slurry was warmed to room temperature and agitated for 12 h and after this time the pH of the supernatant was about 3.0. Tetrabutylammonium acetate (13.6 ml, 13.59 mmol) was slowly added over 30 min. The slurry was agitated for 1 h and pH of the supernatant was about 4.0. Solids were collected by filtration. The solid was washed with 60 mL of aqueous MeCN to afford 19.5 g of the crude product 8 in a 93% isolated yield from compound 6 .
At this stage, all byproducts (including hydro lyzation products of TMS-carbonate) and impurities were soluble in the organic phase.
The product was dissolved back into 140 ml of MeCN:H20 (1 :2) at room temperature. 1-Butanol (390 ml) as antisolvent was slowly added into the solution to afford a slurry. The slurry was agitated overnight. The white crystalline solid was filtered and washed with 3:1 IPA: water (40 ml) and dried under vacuum and nitrogen at room temperature to afford the title product in the form of a crystalline hydrate. (Yield = 16.3 g, 82%). Spectral data matched reference compound.
Preparation of (2S,5R)-7-oxo-2-(piperidin- 1 -ium-4-ylcarbamoyl)- 1 ,6-diazabicyclo[3.2.1 ]octan-6-yl sulfate (1).
tert-Butyl 4-( {[(25*,5i?)-6-hydroxy-7-oxo- 1 ,6-diazabicyclo[3.2.1 ]oct-2-yl]carbonyl}amino)piperidine-l-carboxylate 16 (0.54 g, 1.5 mmol), THF (5.4 mL), 2-picoline (0.29 mL, 2.9 mmol) and pyridine-S03 complex (0.70 g, 4.4 mmol) were charged to a vial under nitrogen. The heterogeneous mixture was allowed to stir overnight (~15 hr). The reaction mixture was cooled to -10°C then dichloromethane (5.4 mL) was added. 0.5 M K2HPO4 (4.5 mL, 2.3 mmol) was added over 10 minutes. BU4NHSO4 (0.53 g, 1.54 mmol) was then added over 10 min. The biphasic mixture was stirred for 30 min, phase cut and the water layer was back extracted with 1 ml of DCM. The combined DCM solution was washed with water (2.0 mL), phase cut and the organic solution was solvent-switched to MeCN (3.2 mL) by vacuum distillation with 3 bed volumes of MeCN. The product was used as is in the next step (water content less than 1000 ppm).
The solution of Bu4N+S04~~ salt 8 in MeCN solution was used with an assumed yield of 100% (1.0 g, 1.47 mmol). The reaction mixture was cooled in an ice bath, and Ν,Ο-bis(trimethylsilyl)trifluoroacetamide (BSTFA) (0.4 lg, 1.59 mmol) was added into the reaction and was allowed to stir for 10 min. TMSI (0.06g, 0.27 mmol) was added between 0°C and 5°C. The resulting mixture was allowed to agitate for 2 hr and then quenched with H2O (0.07g, 4.1 mmol) and acetic acid (0.08g, 1.5 mmol) to afford a slurry. The slurry was warmed to room temperature and agitated for 12 hr. Filter to collect the solid. The solid was washed with MeCN/water (94:6, 1 mL X 4) to afford the crystalline product 1 (0.38 g) in a 75% yield.
If NO-bis(trimethylsilyl)acetamide (BSA) (0.32g, 1.59 mmol) was applied, the reaction needed 24 hr to achieve full conversion.
Patent
WO2015033191
Scheme 1.

Formula (V)
Formula (VI)

Formula (I)
Scheme – 1
Example -1
Preparation of (2S, 5R)-Sulfuric acid mono-{2-[N’-(4-aminopiperidinyl)-carbonyl]-7-oxo- l,6-diaza-bicyclo[3.2.1]oct-6-yl} ester (I).
Step-1: Preparation of (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (IV):
To a 250 ml round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-sodium 6-benzyloxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane-2-carboxylate (11.1 gm, 0.037 mol, prepared using a method disclosed in Indian Patent Application No 699/MUM/2013) in water (180 ml) followed by l-tert-butoxycarbonyl-4-amino-piperidine (7.8 gm, 0.039 mol), EDC hydrochloride (11 gm, 0.055 mol) and 1 -hydro ybenzotriazole (4.8 gm, 0.037 mol) at 30°C successively under stirring. The reaction mixture was stirred for 24 hours at 30°C to provide a suspension. The suspension was filtered under suction and washed with 45°C warm water (40 ml) to provide (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 12.7 gm quantity in 74% yield after drying under vacuum.
Analysis
NMR: (CDC13,) = 7.36-7.44 (m, 5H), 6.56 (d,lH), 5.06 (d,lH), 4.91 (d, 1H), 4.03 (br s, 1H), 3.88-3.97 (m, 2H), 3.29 (s, 1H), 3.00 (d, 1H), 2.86 (t, 2H), 2.64 (d, 1H), 2.37 (dd, 1H), 1.85-2.01 (m, 4H), 1.54-1.62 (m, 2H), 1.45 (s, 9H), 1.25-1.36 (m, 2H).
MS (ES+) C24H34N405 = 459.5 (M+l).
Step-2: Preparation of (2S, 5R)-tert-butyl { (6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (V):
To a 100 ml single neck round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (9 g, 19.5 mmol) in methanol (90 ml) followed by 10% palladium on carbon (2.7 g) at 35°C. The reaction mixture was stirred under 1 atm hydrogen pressure at 35°C for 2 hours. The catalyst was removed by filtering the reaction mixture under suction over a celite bed. The celite bed was washed with dichloromethane (50 ml). The combined filtrate was evaporated under vacuum below 35°C to provide (2S, 5R)-tert-butyl {(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 8.45 g quantity; it was used as such for the next reaction.
Analysis
NMR: (CDC13,) = 6.60 (d, 1H), 3.88-4.10 (m, 4H), 3.78 (s, 1H), 3.20 (d, 1H), 3.90 (t, 2H), 2.80 (d, 1H), 2.46 (dd, 1H), 2.1-2.2 (m, 1H), 2.85-2.20 (m, 4H), 1.70-1.80 (m, 1H), 2.47 (s, 9H), 1.30-1.41 (m, 3H).
MS (ES+) C17H28N405 = 369.4 (M+l).
Step-3: Preparation of Tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (VI):
To a 100 ml single neck round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-tert-butyl {(6-hydroxy-7-oxo-l,6-diaza-bicyclo [3.2.1 ]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (6.40 g, 7.6 mmol) in dichloromethane (90 ml), triethyl amine (9.3 ml), followed by pyridine – sulfur trioxide complex (5.4 g, 34.2 mmol) at 35°C under stirring. The reaction mixture was stirred for additional 4 hours at 35°C. The solvent was evaporated under vacuum below 40°C to provide a residue. The residue was stirred with 0.5N aqueous potassium dihydrogen phosphate solution (90 ml) for 1 hour. The resulting solution was extracted with dichloromethane (2 x 100 ml) to remove impurities. To the aqueous layer was added tetrabutyl ammonium hydrogen sulfate (6.9 g, 20.52 mmol) and the reaction mixture was stirred for 14 hours at 35°C. It was extracted with dichloromethane (3 x 30 ml). Combined organic layer was dried over sodium sulfate and evaporated under vacuum to provide tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 8.0 g quantity in 62% yield.
Analysis
NMR: (CDC13,) – 6.64 (d, 1H), 4.36 (br s, 1H), 4.05(br s, 2H), 3.90-4.00 (m, 1H), 3.87 (d, 1H), 2.28-3.34 (m, 10H), 3.80-3.95 (m, 2H), 3.74 (d, 1H), 2.42 (dd, 1H), 2.15-2.24 (m, 1H), 1.82-1.97 (m, 4H), 1.61-1.74 (m, 14 H), 1.41-1.52 (m, 10 H), 1.02 (t, 12H).
MS (ES-) C17H27N408S. N(C4H9)4 = 447.4 (M-l) as a free sulfonic acid.
Step-4: Synthesis of (2S, 5R)- Sulfuric acid mono-{ [(4-aminopiperidin-4-yl) carbonyl]-7-oxo-l,6-diaza-bicyclo[3.2.1]-oct-6-yl} ester (I):
To a 100 ml round bottom flask equipped with magnetic stirrer was charged a solution of tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (6.0 g) in dichloromethane (15 ml). The solution was cooled to -10°C under stirring and to it was added trifluoro acetic acid (15 ml) drop wise. The reaction mixture was stirred at -10°C for 1 hour. Solvents were evaporated under vacuum below 30°C to its 1/3 volume to provide a thick residue. The thick residue was stirred twice with diethyl ether (60 ml each time) to provide a precipitation. The solid obtained was filtered at suction and suspended in acetone (90 ml). To the suspension was added 10% solution of sodium-2-ethyl-hexanoate in acetone to adjust pH between 4.5 to 5.5. The suspension was stirred for 10 minutes and filtered under suction. The wet cake was washed with acetone and dried under vacuum below 40°C to provide 3 gm crude compound. The crude compound was stirred with aqueous isopropanol (3ml water: 21 ml iospropanol) for overnight to purify further. The resulting suspension was filtered under suction and washed with aqueous isopropanol (1 ml water: 7 ml IPA mixture). Finally the cake was dried under vacuum below 40°C to provide the title compound as a off-white solid in 1.8 g quantity in 65% yield.
Analysis
H1NMR (DMSO-d6, D20 exchange) = 8.19 (d, exchanges with D20), 3.99 (s, 1H), 3.82-3.92 (m, 1H), 3.72 (d, 1H), 2.24 (br d, 3H), 2.90-3.04 (m, 5H), 1.96-2.06 (m, 1H), 1.80-1.94 (m, 3H), 1.58-1.72 (m, 4H).
MS (ES+) C12H20N4O6S = 349.2 (M+l) as a free sulfonic acid;
Purity by HPLC: 99.2%
Specific rotation: [a] D -45.25 °, (c 0.3%, water)
SEE BACTAM SERIES…………..http://apisynthesisint.blogspot.in/p/bactam-series.html
//////
C1CC(N2CC1N(C2=O)OS(=O)(=O)O)C(=O)NC3CCNCC3.O
UPDATE,,,,,,,,,,
Improved Preparation of a Key Hydroxylamine Intermediate for Relebactam: Rate Enhancement of Benzyl Ether Hydrogenolysis with DABCO
Jianguo Yin, Mark Weisel, Yining Ji, Zhijian Liu  , Jinchu Liu, Debra J. Wallace, Feng Xu , Benjamin D. Sherry, and Nobuyoshi Yasuda*
, Jinchu Liu, Debra J. Wallace, Feng Xu , Benjamin D. Sherry, and Nobuyoshi Yasuda*
, Jinchu Liu, Debra J. Wallace, Feng Xu , Benjamin D. Sherry, and Nobuyoshi Yasuda* Process R&D Department, MRL, Merck & Co., Inc., Rahway, New Jersey 07065, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00381
Publication Date (Web): February 1, 2018
Copyright © 2018 American Chemical Society
*E-mail: nobuyoshi_yasuda@merck.com

Previous methods to prepare a bicyclic N-hydroxyl urea intermediate in the synthesis of the potent β-lactamase inhibitor relebactam were effective, but deemed unsuitable for long-term use. Therefore, we developed an in situ protection protocol during hydrogenolysis and a robust deprotection/isolation sequence of this unstable intermediate employing a reactive crystallization. During the hydrogenation studies, we discovered a significant rate enhancement of O-benzyl ether hydrogenolysis in the presence of organic amine bases, especially DABCO. The broader utility of the application of organic bases on the hydrogenolysis of a range of O– and N-benzyl-containing substrates was demonstrated.

5 could be isolated by concentrating the filtrate and storing the solution at 5 °C overnight. 1H NMR (500 MHz, CDCl3): δ 6.58 (d, J = 7.9 Hz, 1H), 4.10–3.86 (m, 4H), 3.55 (bs, 1H), 3.14 (bd, J = 11.5 Hz, 1H), 2.86 (bt, J = 12.0 Hz, 2H), 2.76 (d, J = 11.5 Hz, 1H), 2.36 (dd, J = 15.1, 7.1 Hz, 1H), 2.12 (m, 1H), 2.00–1.82 (m, 3H), 1.66 (m, 1H), 1.44 (s, 9H), 1.31 (m, 2H), 0.25 (S, 9H). 13C NMR (125 MHz, CDCl3): δ 169.2, 168.3, 154.8, 79.8, 60.7, 60.0, 47.3, 46.9, 42.6 (br, 2C), 32.2, 31.9, 28.5 (3C), 20.5, 17.5, −0.75 (3C). (+)-ESI HRMS: calcd for C20H36N4NaO3Si (M + Na)+, 463.2347; found, 463.2348.
Ciraparantag, Aripazine
Ciraparantag
PER977, Aripazine
CAS Number:1438492-26-2
Chemical Name:N1,N1-[piperazine-1,4-diylbis(propane-1,3-diyl)]bis-L-argininamide
(2S,2’S)-N,N’-(Piperazine-1,4-diyldipropane-3,1-diyl)bis(2-amino-5-carbamimidamidopentanamide)
2S,2’S)-N,N’-(piperazine-1,4-diylbis(propane-3,1-diyl))bis(2-amino-5-guanidinopentanamide)
C22H48N12O2
Mw: 512.40232
Mechanism of Action: an intravenously administered anticoagulant Reversal Agent
Blood coagulation factor modulators; Factor Xa inhibitors
Indication: Anticoagulant Reversal
Development Stage: Phase II
Developer:Perosphere, Inc..Perosphere Inc.
Highest Development Phases
- Phase IIHaemorrhage
Most Recent Events
- 02 Apr 2015Ciraparantag receives Fast Track designation for Haemorrhage [IV] (In volunteers) in USA
- 05 Nov 2014Efficacy and adverse events data from a phase I/II trial in Haemorrhage released by Perosphere
- 06 Oct 2014Aripazine is available for licensing as of 06 Oct 2014. http://www.perosphere.com/
Aripazine(PER977, ciraparantag)

Ciraparantag, also known as PER977, is a A Small Molecule Reversal Agent for New Oral Anticoagulants and Heparins. PER977 is water-soluble, cationic molecule that is designed to bind specifically to unfractionated heparin and low-molecular-weight heparin through noncovalent hydrogen bonding and charge–charge interactions.
PER-977 is an intravenous heparin neutralizer in phase II clinical trials at Perosphere to reverse edoxaban’s induced anticoagulation.
In April 2015, fast track designation was assigned in the U.S. as an investigational anticoagulant reversal agent.
WO 2013082210
http://www.google.com/patents/WO2013082210A1?cl=en
In one scheme, the compound of Formula V (DAP)

is synthesized by reacting excess equivalents (e.g., at least about two equivalents) of compound 1

with one equivalent of compound 2

in the presence of a peptide coupling reagent, to obtain a compound 3

wherein PI is a protecting group and P2 is a protecting group or is a hydrogen.
the coupling involved reacting compound 1, wherein PI was Boc and P2 was a hydrogen (depicted as Boc-Arg-OH HCl below), with compound 2 as depicted below:

The resultant crude product was more than 95% pure by thin layer
chromatography (TLC).
Subsequently, the deprotection step was carried out as depicted below:

The deprotected product was purified by preparative HPLC using 1% acetic acid buffer. Product purity of >98% was observed. Residual TFA was removed by low quantity of DOWEX resin. The molecular weight of DAP (the compound of Formula V) is 512.4, and the compound synthesized according to the above scheme exhibited the following primary peak by mass spectroscopy: [M+H]+=513.4.
|
References |
1: Dzik WH. Reversal of oral factor Xa inhibitors by prothrombin complex concentrates: a re-appraisal. J Thromb Haemost. 2015 Jun;13 Suppl 1:S187-94. doi: 10.1111/jth.12949. PubMed PMID: 26149022.
2: Crowther M, Crowther MA. Antidotes for Novel Oral Anticoagulants: Current Status and Future Potential. Arterioscler Thromb Vasc Biol. 2015 Aug;35(8):1736-45. doi: 10.1161/ATVBAHA.114.303402. Epub 2015 Jun 18. PubMed PMID: 26088576.
3: Sullivan DW Jr, Gad SC, Laulicht B, Bakhru S, Steiner S. Nonclinical Safety Assessment of PER977: A Small Molecule Reversal Agent for New Oral Anticoagulants and Heparins. Int J Toxicol. 2015 Jun 15. pii: 1091581815590667. [Epub ahead of print] PubMed PMID: 26079256.
4: Mo Y, Yam FK. Recent advances in the development of specific antidotes for target-specific oral anticoagulants. Pharmacotherapy. 2015 Feb;35(2):198-207. doi: 10.1002/phar.1532. Epub 2015 Feb 3. PubMed PMID: 25644580.
5: Yates SW. Interrupting anticoagulation in patients with nonvalvular atrial fibrillation. P T. 2014 Dec;39(12):858-80. PubMed PMID: 25516695; PubMed Central PMCID: PMC4264672.
6: Vanden Daelen S, Peetermans M, Vanassche T, Verhamme P, Vandermeulen E. Monitoring and reversal strategies for new oral anticoagulants. Expert Rev Cardiovasc Ther. 2015 Jan;13(1):95-103. doi: 10.1586/14779072.2015.987126. Epub 2014 Nov 28. PubMed PMID: 25431993.
7: Costin J, Ansell J, Laulicht B, Bakhru S, Steiner S. Reversal agents in development for the new oral anticoagulants. Postgrad Med. 2014 Nov;126(7):19-24. doi: 10.3810/pgm.2014.11.2829. Review. PubMed PMID: 25387210.
8: Ansell JE, Bakhru SH, Laulicht BE, Steiner SS, Grosso M, Brown K, Dishy V, Noveck RJ, Costin JC. Use of PER977 to reverse the anticoagulant effect of edoxaban. N Engl J Med. 2014 Nov 27;371(22):2141-2. doi: 10.1056/NEJMc1411800. Epub 2014 Nov 5. PubMed PMID: 25371966.
9: Hankey GJ. Intracranial hemorrhage and novel anticoagulants for atrial fibrillation: what have we learned? Curr Cardiol Rep. 2014 May;16(5):480. doi: 10.1007/s11886-014-0480-9. Review. PubMed PMID: 24643903.
///////
Defibrotide


Defibrotide sodium is an oligonucleotide mixture with profibrinolytic properties. The chemical name of defibrotide sodium is polydeoxyribonucleotide, sodium salt. Defibrotide sodium is a polydisperse mixture of predominantly single-stranded (ss) polydeoxyribonucleotide sodium salts derived from porcine intestinal tissue having a mean weighted molecular weight of 13-20 kDa, and a potency of 27-39 and 28-38 biological units per mg as determined by two separate assays measuring the release of a product formed by contact between defibrotide sodium, plasmin and a plasmin substrate. The primary structure of defibrotide sodium is shown below.
DEFITELIO (defibrotide sodium) injection is a clear, light yellow to brown, sterile, preservative-free solution in a single-patient-use vial for intravenous use. Each milliliter of the injection contains 80 mg of defibrotide sodium and 10 mg of Sodium Citrate, USP, in Water for Injection, USP. Hydrochloric Acid, NF, and/or Sodium Hydroxide, NF, may have been used to adjust pH to 6.8-7.8.
Defibrotide is the sodium salt of a mixture of single-stranded oligodeoxyribonucleotides derived from porcine mucosal DNA. It has been shown to have antithrombotic, anti-inflammatory and anti-ischemic properties (but without associated significant systemic anticoagulant effects). It is marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries, but is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide is used to treat or prevent a failure of normal blood flow (occlusive venous disease, OVD) in the liver of patients who have had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others.
In 2012, an IND was filed in Japan seeking approval of the compound for the treatment of veno-occlusive disease.
Approved 3/30/3016 US FDA, defibrotide sodium, (NDA) 208114

To treat adults and children who develop hepatic veno-occlusive disease with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation
Polydeoxyribonucleotides from bovine lung or other mamalian organs with molecular weight between 15,000 and 30,000 Da
CAS 83712-60-1
Defibrotide is a polydisperse mixture of oligonucleotides produced by random, chemical cleavage (depolymerisation) of porcine DNA. It is predominantly single stranded, of varying base sequence, lengths and conformations; unfolded, folded or combined. The mean oligonucleotide length is 50 bases with a mean molecular weight of 17 ± 4 kDa. No individually defined component is at more than femtomolar concentration. The only meaningful scientific information that can be obtained about the biochemical nature of defibrotide (aside from determination of percentage of each nucleobase) is a measurement of its average length and its average percentage double stranded character. Therefore, it can be established that this active substance is of highly heterogenic nature.

Defibrotide (Defitelio, Gentium)[1] is a deoxyribonucleic acid derivative (single-stranded) derived from cow lung or porcine mucosa. It is an anticoagulant with a multiple mode of action (see below).
It has been used with antithrombin III.[2]
Jazz Pharmaceuticals plc announced that the FDA has accepted for filing with Priority Review its recently submitted New Drug Application (NDA) for defibrotide. AS ON OCT 2015
Defibrotide is an investigational agent proposed for the treatment of patients with hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS), with evidence of multi-organ dysfunction (MOD) following hematopoietic stem-cell transplantation (HSCT).
Priority Review status is designated for drugs that may offer major advances in treatment or provide a treatment where no adequate therapy exists. Based on timelines established by the Prescription Drug User Fee Act (PDUFA), FDA review of the NDA is expected to be completed by March 31, 2016.
“The FDA’s acceptance for filing and Priority Review status of the NDA for defibrotide is an important milestone for Jazz and reflects our commitment to bringing meaningful medicines to patients who have significant unmet needs,” said Karen Smith, M.D., Ph.D., Global Head of Research and Development and Chief Medical Officer of Jazz Pharmaceuticals. “We look forward to continuing to work closely with the FDA to obtain approval for defibrotide for patients with hepatic VOD with evidence of MOD in the U.S. as quickly as possible, as there are no other approved therapies for treating this rare, often fatal complication of HSCT.”
The NDA includes safety and efficacy data from three clinical studies of defibrotide for the treatment of hepatic VOD with MOD following HSCT, as well as a retrospective review of registry data from the Center for International Blood and Marrow Transplant Research. The safety database includes over 900 patients exposed to defibrotide in the clinical development program for the treatment of hepatic VOD.
The compound was originally developed under a collaboration between Sanofi and Gentium. In December 2001, Gentium entered into a license and supply agreement with Sigma-Tau Pharmaceuticals, pursuant to which the latter gained exclusive rights to distribute, market and sell the product for the treatment of VOD in the U.S. This agreement was expanded in 2005 to include all of North America, Central America and South America.
Defibrotide was granted orphan drug designations from the FDA in July 1985, May 2003 and January 2007 for the treatment of thrombotic thrombocytopenic purpura (TTP), for the treatment of VOD and for the prevention of VOD, respectively. Orphan drug was also received in the E.U. for the prevention and treatment of hepatic veno-occlusive disease (VOD) in 2004 and for the prevention of graft versus host disease (GvHD) in 2013.
Pharmacokinetics
Defibrotide is available as an oral, intravenous, and intramuscular formulation. Its oral bioavailability is in the range of 58-70% of theparenteral forms. T1/2 alpha is in the range of minutes while T1/2 beta is in the range of hours in studies with oral radiolabelleddefibrotide. These data suggest that defibrotide, in spite of its macromolecular nature, is absorbed well after oral administration. Due to the drug’s short half-life, it is necessary to give the daily dose divided in 2 to 4 doses (see below).
In 2014, Jazz Pharmaceuticals (parent of Gentium) acquired the rights of the product in U.S. and in the Americas
Mode of action
The drug appears to prevent the formation of blood clots and to help dissolve blood clots by increasing levels of prostaglandin I2, E2, and prostacyclin, altering platelet activity, increasing tissue plasminogen activator (tPA-)function, and decreasing activity of tissue plasminogen activator inhibitor. Prostaglandin I2 relaxes the smooth muscle of blood vessels and prevents platelets from adhering to each other. Prostaglandin E2 at certain concentrations also inhibits platelet aggregation. Moreover, the drug provides additional beneficial anti-inflammatory and antiischemic activities as recent studies have shown. It is yet unclear, if the latter effects can be utilized clinically (e.g., treatment of ischemic stroke).
Unlike heparin and warfarin, defibrotide appears to have a relatively mild anticoagulant activity, which may be beneficial in the treatment of patients at high risk of bleeding complications. Nevertheless, patients with known bleeding disorders (e.g., hemophilia A) or recent abnormal bleedings should be treated cautiously and under close medical supervision.
The drug was marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries. It is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide also received fast track designation from the FDA for the treatment of severe VOD in recipients of stem cell transplants. In 2011, the compound was licensed to Medison Pharma by Gentium in Israel and Palestine. The license covers the management of named-patient sales program and local registration, authorization, marketing, reimbursement and medical affairs for the treatment of peripheral vascular disease.
Usual indications
Defibrotide is used to treat or prevent a failure of normal blood flow (Veno-occlusive disease, VOD) in the liver of patients having had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others. Without intensive treatment, VOD is often a fatal condition, leading to multiorgan failure. It has repeatedly been reported that defibrotide was able to resolve the condition completely and was well tolerated.
Other indications are: peripheral obliterative arterial disease, thrombophlebitis, and Raynaud’s phenomenon. In very high doses, defibrotide is useful as treatment of acute myocardial infarction. The drug may also be used for the pre- and postoperative prophylaxis of deep venous thrombosis and can replace the heparin use during hemodialytic treatments.
It has been investigated for use in treatment of chronic venous insufficiency.[3]
Potential indications in the future
Other recent preclinical studies have demonstrated that defibrotide used in conjunction with Granulocyte Colony-Stimulating Factor (rhG-CSF) significantly increases the number of Peripheral Blood Progenitor Cells (Stem cells). The benefit of this increase in stem cells may be crucial for a variety of clinical indications, including graft engineering procedures and gene therapy programs. This would expand the clinical usefulness of defibrotide to a complete distinct area.
Very recently (since early 2006) combination therapy trials (phase I/II) with defibrotide plus melphalan, prednisone, and thalidomide in patients with multiple myeloma have been conducted. The addition of defibrotide is expected to decrease the myelosuppressive toxicity of melphalan. However, is too early for any definitive results at that stage.
Cautions and contraindications
- The efficacy of the drug has been reported to be poorer in patients with diabetes mellitus.
- Pregnancy: The drug should not be used during pregnancy, because adequate and well controlled human studies do not exist.
- Lactation: No human data is available. In order to avoid damage to the newborn, the nursing mother should discontinue either the drug or breastfeeding, taking into account the importance of treatment to the mother.
- Known Bleeding Disorders or Bleeding Tendencies having occurred recently: Defibrotide should be used cautiously. Before initiation of treatment, the usual coagulation values should be obtained as baseline and regularly controlled under treatment. The patient should be observed regularly regarding local or systemic bleeding events.
Side-effects
Increased bleeding and bruising tendency, irritation at the injection site, nausea, vomiting, heartburn, low blood pressure. Serious allergic reactions have not been observed so far.
Drug interactions
Use of heparin with defibrotide may increase the aPTT, reflecting reduced ability of the body to form a clot. Nothing is known about the concomitant application of other anticoagulants than heparin and dextran containing plasma-expanders, but it can be anticipated that the risk of serious bleeding will be increased considerably.
PATENT
WO 2001078761
G-CSF (CAS registry number 143011-2-7/Merck Index, 1996, page 4558) is a haematopoietic growth factor which is indispensable in the proliferation and differentiation of the progenitor cells of granulocytes; it is a 18-22 kDa glycoprotein normally produced in response to specific stimulation by a variety of cells, including monocytes, fibroblasts and endothelial cells. The term defibrotide (CAS registry number 83712-60-1) normally identifies a polydeoxyribonucleotide obtained by extraction (US 3,770,720 and US 3,899,481) from animal and/or vegetable tissue; this polydeoxyribonucleotide is normally used in the form of a salt of an alkali metal, generally sodium. Defibrotide is used principally for its anti- thrombotic activity (US 3,829,567) although it may be used in different applications, such as, for example, the treatment of acute renal insufficiency (US 4,694,134) and the treatment of acute myocardial ischaemia (US 4,693,995). United States patents US 4,985,552 and US 5,223,609, finally, describe a process for the production of defibrotide which enables a product to be obtained which has constant and well defined physico-chemical characteristics and is also free from any undesired side-effects
References
- “Jazz Pharma Acquiring Gentium for $1B”. Gen. Eng. Biotechnol. News (paper) 34 (2). January 15, 2014. p. 10.
- Haussmann U, Fischer J, Eber S, Scherer F, Seger R, Gungor T (June 2006). “Hepatic veno-occlusive disease in pediatric stem cell transplantation: impact of pre-emptive antithrombin III replacement and combined antithrombin III/defibrotide therapy”. Haematologica 91 (6): 795–800. PMID 16769582.
- Coccheri S, Andreozzi GM, D’Addato M, Gensini GF (June 2004). “Effects of defibrotide in patients with chronic deep insufficiency. The PROVEDIS study”. Int Angiol 23 (2): 100–7.PMID 15507885.
External links
- Palmer KJ, Goa KL. Defibrotide: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in vascular disorders. Drugs 1993;45:259-94.
- http://www.globalrx.com/medinfo/Defibrotide.htm
- Fisher J, Holland TK, Pescador R, Porta R, Ferro L (January 1996). “Study on pharmacokinetics of radioactive labelled defibrotide after oral or intravenous administration in rats”. Thromb. Res. 81 (1): 55–63. doi:10.1016/0049-3848(95)00213-8. PMID 8747520.
- http://www.gentium.it/Defibrotide.aspx (information provided by manufacturer)
- “Melphalan: profile and news”. Archived from the original on 2007-09-28. (on cytostatic combination therapy)
- Beşişik SK, Oztürk GB, Calişkan Y, Sargin D (March 2005). “Complete resolution of transplantation-associated thrombotic microangiopathy and hepatic veno-occlusive disease by defibrotide and plasma exchange”. Turk J Gastroenterol 16 (1): 34–7. PMID 16252186.
| WO2003101468A1 * | Jun 2, 2003 | Dec 11, 2003 | Guenther Eissner | Method for the protection of endothelial and epithelial cells during chemotherapy |
| US4985552 | Jul 5, 1989 | Jan 15, 1991 | Crinos Industria Farmacobiologica S.P.A. | Process for obtaining chemically defined and reproducible polydeoxyribonucleotides |
| US5223609 | May 26, 1992 | Jun 29, 1993 | Crinos Industria Farmacobiologica S.P.A. | Process for obtaining chemically defined and reproducible polydeoxyribonucleotides |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1999026639A1 * | 24 Nov 1998 | 3 Jun 1999 | Allegheny University Of The He | Methods for mobilizing hematopoietic facilitating cells and hematopoietic stem cells into the peripheral blood |
| EP0317766A1 * | 20 Oct 1988 | 31 May 1989 | Crinos Industria Farmacobiologica S.p.A. | A method for preventing blood coaguli from being formed in the extra-body circuit of dialysis apparatus and composition useful thereof |
| EP0416678A1 * | 10 Aug 1990 | 13 Mar 1991 | Crinos Industria Farmacobiologica S.p.A. | Topical compositions containing Defibrotide |
| US5199942 * | 26 Sep 1991 | 6 Apr 1993 | Immunex Corporation | Method for improving autologous transplantation |
| US5977083 * | 5 Jun 1995 | 2 Nov 1999 | Burcoglu; Arsinur | Method for using polynucleotides, oligonucleotides and derivatives thereof to treat various disease states |
| Reference | ||
|---|---|---|
| 1 | * | CARLO-STELLA, C. (1) ET AL: “Defibrotide significantly enhances peripheral blood progenitor cell mobilization induced by recombinant human granulocyte colony – stimulating factor ( rhG – CSF.” BLOOD, ( NOVEMBER 16, 2000 ) VOL. 96, NO. 11 PART 1, PP. 553A. PRINT. MEETING INFO.: 42ND ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY SAN FRANCISCO, CALIFORNIA, USA DECEMBER 01-05, 2000 AMERICAN SOCIETY OF HEMATOLOGY. , XP002176349 |
| 2 | * | GURSOY A: “PREPARATION, CHARACTERIZATION AND ANTI-INFLAMMATORY EFFECT OF DEFIBROTIDE LIPOSOMES” PHARMAZIE,DD,VEB VERLAG VOLK UND GESUNDHEIT. BERLIN, vol. 48, no. 7, 1 July 1993 (1993-07-01), pages 549-550, XP000372658 ISSN: 0031-7144 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2005017160A2 * | 12 Aug 2004 | 24 Feb 2005 | Childrens Hosp Medical Center | Mobilization of hematopoietic cells |
| WO2009115465A1 * | 13 Mar 2009 | 24 Sep 2009 | Gentium Spa | Synthetic phosphodiester oligonucleotides and therapeutical uses thereof |
| EP2103689A1 * | 19 Mar 2008 | 23 Sep 2009 | Gentium S.p.A. | Synthetic phosphodiester oligonucleotides and therapeutical uses thereof |
| US7417026 | 12 Aug 2004 | 26 Aug 2008 | Children’s Hospital Medical Center | Mobilization of hematopoietic cells |
| US7915384 | 5 Jan 2009 | 29 Mar 2011 | Children’s Hospital Medical Center | Chimeric peptides for the regulation of GTPases |
| US8242246 | 28 Feb 2011 | 14 Aug 2012 | Children’s Hospital Medical Center | Chimeric peptides for the regulation of GTPases |
| US8674075 | 13 Aug 2012 | 18 Mar 2014 | Children’s Medical Center Corporation | Chimeric peptides for the regulation of GTPases |
| US8980862 | 12 Nov 2010 | 17 Mar 2015 | Gentium S.P.A. | Defibrotide for use in prophylaxis and/or treatment of Graft versus Host Disease (GVHD) |
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
oral, i.m., i.v. |
| Pharmacokinetic data | |
| Bioavailability | 58 – 70% orally (i.v. and i.m. = 100%) |
| Biological half-life | t1/2-alpha = minutes; t1/2-beta = a few hours |
| Identifiers | |
| CAS Registry Number | 83712-60-1 |
| ATC code | B01AX01 |
| DrugBank | DB04932 |
| UNII | 438HCF2X0M |
| KEGG | D07423 |
///////////Approved, 3/30/3016, US FDA, defibrotide sodium, NDA 208114, FDA 2016
Updates……….
FDA approves first treatment for rare disease in patients who receive stem cell transplant from blood or bone marrow
For Immediate Release
March 30, 2016
Release
The U.S. Food and Drug Administration today approved Defitelio (defibrotide sodium) to treat adults and children who develop hepatic veno-occlusive disease (VOD) with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation (HSCT). This is the first FDA-approved therapy for treatment of severe hepatic VOD, a rare and life-threatening liver condition.
HSCT is a procedure performed in some patients to treat certain blood or bone marrow cancers. Immediately before an HSCT procedure, a patient receives chemotherapy. Hepatic VOD can occur in patients who receive chemotherapy and HSCT. Hepatic VOD is a condition in which some of the veins in the liver become blocked, causing swelling and a decrease in blood flow inside the liver, which may lead to liver damage. In the most severe form of hepatic VOD, the patient may also develop failure of the kidneys and lungs. Fewer than 2 percent of patients develop severe hepatic VOD after HSCT, but as many as 80 percent of patients who develop severe hepatic VOD do not survive.
“The approval of Defitelio fills a significant need in the transplantation community to treat this rare but frequently fatal complication in patients who receive chemotherapy and HSCT,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
The efficacy of Defitelio was investigated in 528 patients treated in three studies: two prospective clinical trials and an expanded access study. The patients enrolled in all three studies had a diagnosis of hepatic VOD with liver or kidney abnormalities after HSCT. The studies measured the percentage of patients who were still alive 100 days after HSCT (overall survival). In the three studies, 38 to 45 percent of patients treated with Defitelio were alive 100 days after HSCT. Based on published reports and analyses of patient-level data, the expected survival rates 100 days after HSCT would be 21 to 31 percent for patients with severe hepatic VOD who received only supportive care or interventions other than Defitelio.
The most common side effects of Defitelio include abnormally low blood pressure (hypotension), diarrhea, vomiting, nausea and nosebleeds (epistaxis). Serious potential side effects of Defitelio that were identified include bleeding (hemorrhage) and allergic reactions. Defitelio should not be used in patients who are having bleeding complications or who are taking blood thinners or other medicines that reduce the body’s ability to form clots.
The FDA granted the Defitelio application priority review status, which facilitates and expedites the development and review of certain drugs in light of their potential to benefit patients with serious or life-threatening conditions. Defitelio also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Defitelio is marketed by Jazz Pharmaceuticals based in Palo Alto, California
Beijing Shenogen Granted Fast Track Status for Novel Cancer Drug, Icaritin
Icaritin; 118525-40-9; AC1NSXIV; UNII-UFE666UELY;
3,5,7-trihydroxy-2-(4-methoxyphenyl)-8-(3-methylbut-2-enyl)chromen-4-one
3,5,7-trihydroxy-2-(4-methoxyphenyl)-8-(3-methylbut-2-enyl)chromen-4-one
| C21H20O6 | |
| Molecular Weight: | 368.3799 g/mol |
|---|
The roots of Epimedium brevicornu Maxim

Beijing Shenogen Granted Fast Track Status for Novel Cancer Drug
Written by Richard Daverman, PhD, Executive Editor, Greg B. Scott.
Beijing Shenogen Biomedical announced that Icaritin, a China Class I cancer drug, was granted Fast Track Review status after the company filed its New Drug Approval submission to the Beijing Food & Drug Administration. Icaritin is an oral traditional Chinese medicine, derived from barrenwort, which targets the estrogen receptor α36. Shenogen has conducted clinical trials of Icaritin in patients with liver cancer, though it expects the drug will also prove effective in breast cancer and other estrogen-related cancers as well. More details…. http://www.chinabiotoday.com/articles/20150917
Antiproliferative agent (IC50 values are 8,13 and 18 μM for K562, CML-CP and CML-BC cells respectively). Inhibits H/R-induced PTK activation. Induces G(2)/M cell cycle arrest and mitochondrial transmembrane potential drop. Modulates MAPK/ERK/JNK and JAK2/STAT3 /AKT signaling. Inhibits PPAR-g. Modulates differentiation. Inhibits cytochrome P450 in vivo. Orally active.
Cardiovascular function improvement, hormone regulation and antitumor activity.
2. The anti-MM activity of Icaritin was mainly mediated by inhibiting IL-6/JAK2/STAT3 signaling.
3. The inhibitory activity of Icariside II on pre-osteoclast RAW264.7 growth was synergized by Icaritin, which maybe contribute to the efficiency of Herba Epimedii extract on curing bone-related diseases, such as osteoporosis.
4. The Icaritin at low concentration (4 or 8 μmol/L) can promote rat chondrocyte proliferation and inhibit cell apoptosis, while the effect of Icaritin on rat chondrocyte at high concentration was reversed.
5. Icaritin might be a new potent inhibitor by inducing S phase arrest and apoptosis in human lung carcinoma A549 cells.
6. Icaritin dose-dependently inhibits ENKL cell proliferation and induces apoptosis and cell cycle arrest at G2/M phase. Additionally, Icaritin upregulates Bax, downregulates Bcl-2 and pBad, and activates caspase-3 and caspase-9.
What is Epimedium ?
Herba epimedii (Epimedium, also called bishop’s hat, horny goat weed or yin yang huo), a traditional Chinese medicine, has been widely used as a kidney tonic and antirheumatic medicine for thousands of years. It is a genus of about 60 flowering herbs, cultivated as a ground cover plant and an aphrodisiac. The bioactive components in herba epimedii are mainly prenylated flavonol glycosides, end-products of the flavonoid pathway. Epimedium species are also used as garden plants due to the colorful flowers and leaves. Most of them bloom in the early spring, and the leaves of some species change colors in the fall, while other species retain their leaves year round.

Figure 1 Epimedium
Epimedium Raw Material
The herbs we used to extract icariin is one species of Epimedium, which name is Epimedium brevicornum Maxim. This kind of epimedium only can be abundantly found in Gansu province of China. And because of the growth habit of this kind of herb, which only grows under trees, it can’t to be planted, only can harvest the wild one.
This wild epimedium contains quite a bit of active components, depending on its long growth time and rich nutrient. Usually the content of the icariin is not lower than 1%.
Below photo is the herb specimen which we use. Picking in the epimedium full-bloom stage. And the medicinal value of the herb is the best at this time. The herb we select contains roots, stems, leaves and flowers. And we extract with the whole herb.

Figure 2 Epimedium for extract
Epimedium Extract
Epimedium extract is a herbal supplement claimed to be beneficial for the treatment of sexual problems such as impotence. It is believed to contain a number of active components, including plant compounds that may have antioxidant activity and estrogen-like compounds. The major components of Epimedium brevicornum are icariin, epimedium B and epimedium C. It is reported to have anti-inflammatory, anti-proliferative, and anti-tumor effects. It is also reported to have potential effects on the management of erectile dysfunction.

Figure 3 HPLC spectrum of icariin
Our specification available is Icariin HPLC 50%- 98%. Below please see the the information for reference:

Figure 4 Epimedium Extract(Icariin)
Derivatives
The plant extracts of epimedium traditionally used for male impotence, and the individual compounds is icariin, were screened against phosphodiesterase-5A1 (PDE5A1) activity. Human recombinant PDE5A1 was used as the enzyme source. The E. brevicornum extract and its active principle icariin were active. To improve its inhibitory activity, some derivatives ware subjected to various structural modifications, which include icaritin, icariside II and 3,7-bis(2-hydroxyethyl) icaritin. There have some scientific papers report that the improved pharmacodynamic profile and lack of cytotoxicity on human fibroblasts make such compounds a promising candidate for further development. We hope that our new products can help you to find more commercial opportunity.
In this way, we can introduce those products as below, and we can also provide more details about the products according to your demand. The 1H-NMR of icaritin and 3,7-bis(2-hydroxyethyl) icaritin is as below.
| Product Name | Specification | CAS No. |
| Icariin | HPLC 50%-98% | 489-32-7 |
| icaritin | HPLC 98% | 118525-40-9 |
| icariside II | HPLC 98% | 113558-15-9 |
| 3,7-bis(2-hydroxyethyl) icaritin | HPLC 98% | 1067198-74-6 |

Figure 4 1H-NMR of icaritin and 3,7-bis(2-hydroxyethyl) icaritin
Main Function of Epimedium Extract
horny goat weed; epimedium; Icariin; penis medicine;epimedium p.e;epimedium brevicornum; shorthorned epimedium herb; Icariins; Icaritin; 3,7-Bis(2-Hydroxyethyl)Icaritin; icariin 60%; icariin 98%; epimedium graepimedium; icarisides II;epimedium sagittatum;epimedium leaf; barrenwort.powder extract
Epimedium has been used to treat male erectile dysfunction in Traditional Chinese Medicine for many centuries. The main functions of Epimedium brevicornum in ancient Chinese books focused on the nourishment of kidney viscera and reinforcement of ‘yang’, resulting in the restoration of erectile function in males.
Epimedium contains chemicals which might help increase blood flow and improve sexual function. It also contains phytoestrogens, chemicals that act somewhat like the female hormone estrogen that might reduce bone loss in postmenopausal women.

Figure 5 some products from epimedium extract
………..
PAPER

The novel total synthesis of icaritin (1), naturally occurring with important bioactive 8-prenylflavonoid, was performed via a reaction sequence of 8 steps including Baker-Venkataraman reaction, chemoselective benzyl or methoxymethyl protection, dimethyldioxirane (DMDO) oxidation, O-prenylation, Claisen rearrangement and deprotection, starting from 2,4,6-trihydroxyacetophenone and 4-hydroxybenzoic acid in overall yields of 23%. The key step was Claisen rearrangement under microwave irradiation. MS, 1H and 13C NMR techniques have been used to confirm the structures of all synthetic compounds. – See more at: http://www.eurekaselect.com/124334/article
…….
PAPER
![[1860-5397-11-135-1]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-11-135-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of icariin (1), icariside I (2) and icaritin (3).
Synthesis of icariin from kaempferol through regioselective methylation and para-Claisen–Cope rearrangement
, , , and Guolin Zhang1
1Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, China
2Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu 610041, China
3College of Chemistry and Environmental Protection Engineering, Southwest University for Nationalities, Chengdu 610041, China…http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-11-135
2Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu 610041, China
3College of Chemistry and Environmental Protection Engineering, Southwest University for Nationalities, Chengdu 610041, China…http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-11-135
![[1860-5397-11-135-i1]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: (a) Ac2O, pyridine, 94%; (b) BnBr, KI, K2CO3, acetone, 85%; (c) Me2SO4, K2CO3, acetone, MeOH, 82%; (d) MOMCl, N,N-diisopropylethylamine (DIPEA), CH2Cl2, 93%; (e) 3,3-dimethylallyl bromide, 18-crown-6, K2CO3, acetone, 86%; (f) Eu(fod)3, NaHCO3, PhCl, 85 °C, 61%; (g) MeOH, 3 M HCl (aq), reflux, 95%; (h) Pd/C, 1,4-cyclohexadiene, MeOH, 84%.
![[1860-5397-11-135-i2]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i2.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Decomposition of 8.
![[1860-5397-11-135-i3]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Claisen rearrangement of flavonol 8.
![[1860-5397-11-135-i4]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i4.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reagents and conditions: (a) 15, DMF/CHCl3, Ag2CO3, molecular sieves (4 Å, powder); (b) 16, CH2Cl2, Ag2O, molecular sieves (4 Å powder), 31% for 2 steps; (c) NH3 (g), MeOH, 94%; (d) NH3 (g), MeOH, 63% for 2 steps.
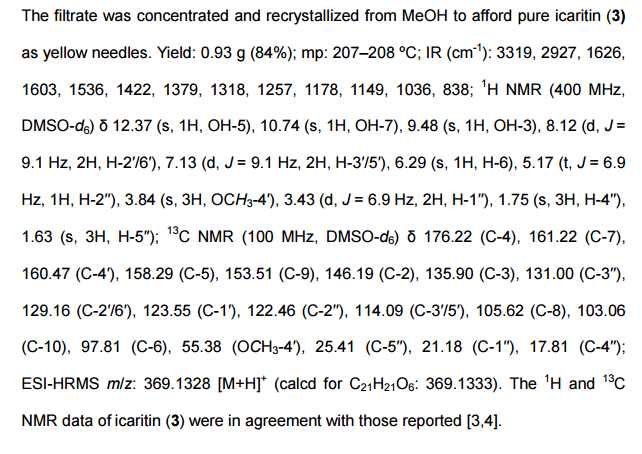
3 Nguyen, V.-S.; Shi, L.; Li, Y.; Wang, Q.-A. Lett. Org. Chem. 2014, 11, 677–681.
4. Dell’Agli, M.; Galli, G. V.; Dal Cero, E.; Belluti, F.; Matera, R.; Zironi, E.; Pagliuca, G.; Bosisio, E. J. Nat. Prod. 2008, 71, 1513–1517.
1H NMR
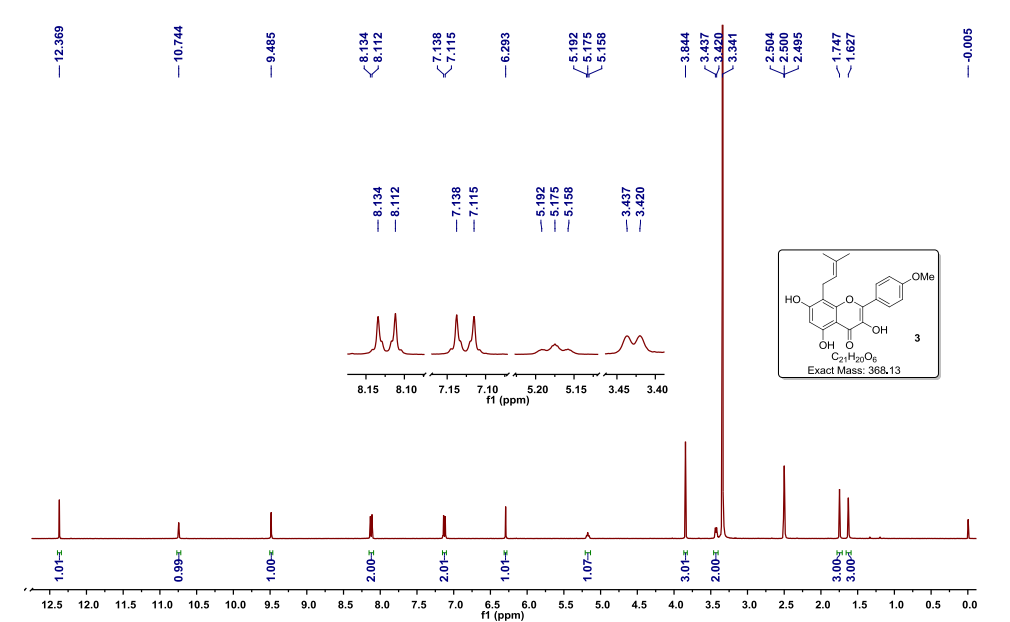
13C NMR
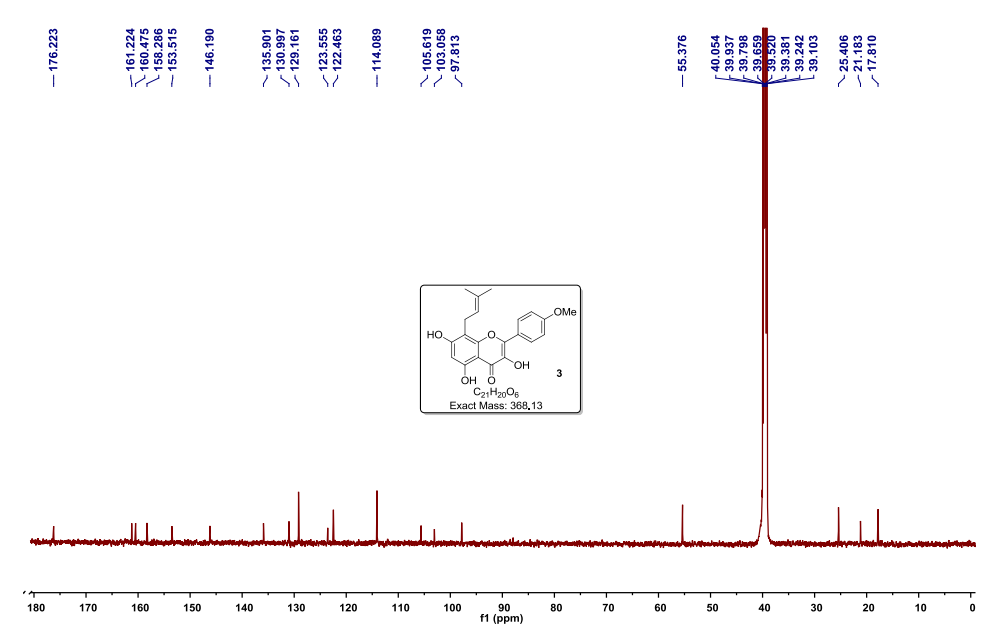
HMBC
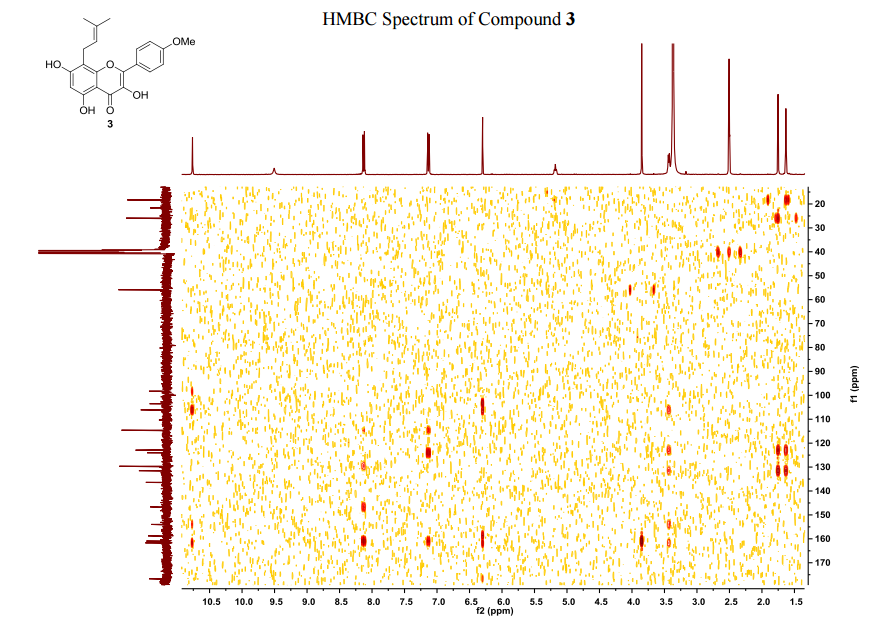
NOESY
………….
The present invention relates to compositions comprising icariside I, and to a novel, one step method of preparing such compositions, comprising converting specific prenylated flavonol glycosides such as epimedium A, epimedium B, epimedium C, icariin, and their corresponding acetate derivatives contained in an Epimedium plant extract to a single compound, namely icariside I shown below as compound I, which was surprisingly discovered to be a strong PDE-5 inhibitor.

This invention further comprises compositions enriched for anhydroicaritin, and to methods of preparing such compositions. One method of this invention for preparing compositions enriched for anhydroicaritin comprises a one-step method of converting prenylated flavonol glycosides, specifically the sagittatoside compounds A, B, and C, and the corresponding acetate derivatives, present in Epimedium plant extracts to a single compound, namely anhydroicaritin shown below as compound II, which was also discovered to be a strong PDE-5 inhibitor.

EXAMPLES Example 1 Acid Hydrolysis of a 50% EtOH Extract and Purification by Reversed Phase ChromatographyWhole Epimedium grandiflorum leaves were extracted with a 1:1 mixture of ethanol and water at 55° C. The resulting extract (referred to as a “50% EtOH extract”) was filtered and the filtrate concentrated at 40-50° C. under vacuum and then dried under vacuum at 60° C. to a dry solid. The dried extract (131 g) containing approximately 5.8 g of total PFG’s was placed in a 2 liter round bottom flask and 1 L of 90% ethanol was added. The mixture was heated to reflux to help dissolve the solids. Concentrated sulfuric acid (28 mL) was added. The mixture refluxed for 2 hr, cooled to room temperature, and 900 mL of water added with stirring. Next the mixture was filtered using vacuum to remove insoluble sulfate salts and other solids and loaded on a 2.5×56 cm (275 mL) column packed with 250-600 micron divinylbenzene cross-linked polystyrene resin (Mitsubishi Chemical). The column was washed with 2 column volumes (CVs) of 60% ethanol and the icariside I was eluted with 2 CVs of 95% ethanol. The product pool was air-dried producing 11.3 g of brown solids. HPLC analysis (FIG. 5) showed that the solids contained 18% icariside I (peak 15.27 min) and 12% anhydroicaritin (peak 25.15 min). The recovery of the icariside I in the product pool was 87% of the amount present in the hydrolyzate.
Example 2 Purification of a Hydrolyzate by Liquid/liquid ExtractionThe ethanolic hydrolyzate (25 mL) prepared in Example 1 was mixed with 62.5 mL of de-ionized water and the pH was adjusted to 7.0 using 50% (w/w) sodium hydroxide solution. The resulting mixture was extracted with three 25 mL portions of ethyl acetate and the combined ethyl acetate extracts were back extracted with 150 mL of water. The ethyl acetate layers were combined, dried, and assayed for icariside I. HPLC analysis (FIG. 6) showed that the dried EtOAc fractions contained 22% icariside I (peak 15.29 min) and 11% anhydroicaritin (peak 25.27 min), and icariside I recovery into the ethyl acetate was 97% of the amount present in the hydrolyzate. The partition coefficient for icariside I between ethyl acetate and water was found to be 16, indicating that the icariside I has a high affinity for ethyl acetate over water.
Example 3 Acid Hydrolysis of a 50% EtOH Extract and Purification by PrecipitationThe dried extract (204 g) described in Example 1 was mixed with 1 L of 90% EtOH and then heated to reflux to help dissolve the solids. Sulfuric acid (25 ML) was added slowly with swirling. The mixture was refluxed 90 minutes and immediately chilled to stop the reaction. After cooling to room temperature, the mixture was filtered under reduced pressure through cellulose paper to remove insoluble sulfates and other materials, and the cake was washed with about 350 mL of 90% ethanol. The resulting ethanolic hydrolyzate (1.34 L) contained 4.1 g of icariside I.
The ethanolic hydrolyzate prepared above (1.32 L) was placed in a 10 L container and 40 g of 50% (w/w) sodium hydroxide solution was added followed by 20 mL of phosphoric acid. Next 3.3 L of deionized water was added with stirring. The pH of this mixture was 2.4. Sodium hydroxide solution (50% w/w ) was added until the pH was 8.25. The mixture was heated to 65° C. to assist with the coagulation of the precipitate. The mixture was cooled to room temperature and stirred for 0.5 hr at room temperature before filtering through a cellulose filter using vacuum. The resulting brown solids were washed with 715 mL of 10% ethanol and dried either under vacuum at room temperature or in air at 55° C. to yield brown solids. HPLC analysis (FIG. 7) showed the solids contained 20% icariside I (peak 15.27 min) and 10% anhydroicaritin. Recovery of icariside I using this precipitation procedure was 94% of the amount present in the hydrolyzate.
Example 4 Acid Hydrolysis of a Water Extract and Purification by PrecipitationGround Epimedium grandiflorum leaves (0.40 kg) were mixed with 5 L water in a 10 L round bottom flask. The flask was placed on a rotary evaporator for two hours at a rotation speed of 120 rpm and a water bath temperature of 90° C. The extract was filtered under reduced pressure through cellulose paper. The resulting filtrate (3.2 L) was evaporated using the rotary evaporator to a volume of 100 mL and dried under vacuum at 50° C.
The dark brown solids prepared above (40.4 g) were mixed with 200 mL of 90% ethanol and 6.0 mL of sulfuric acid in a 500 mL round bottom flask. The mixture was refluxed for 90 minutes and immediately chilled to stop the reaction. This mixture was filtered under reduced pressure through cellulose paper to remove insoluble sulfates and other materials. The cake was washed with 15 mL of 90% ethanol. The resulting ethanolic hydrolyzate (215 mL) contained 0.53 g of icariside I.
The hydrolyzate prepared above (50 mL) was transferred to a 250 mL beaker and 2.5 mL of 50% (w/w) sodium hydroxide solution was added with stirring to adjust the pH of the solution to pH 9, followed by 1.5 mL of concentrated phosphoric acid. Deionized water (125 mL) was added, and the mixture was adjusted to pH 8.2 using 1.5 mL of 50% sodium hydroxide solution. The mixture was heated to 65° C. to assist with coagulation of the precipitate and cooled to room temperature. The mixture was allowed to sit undisturbed at room temperature for 30 minutes prior to filtration under reduced pressure through cellulose paper. The resulting olive-green solids were washed with 25 mL of de-ionized water and dried under vacuum at room temperature or in air at 80° C. to produce olive-green solids. HPLC analysis (FIG. 8) showed the solids contained 60% icariside I (peak 15.33 min) and 2.4% anhydroicaritin (peak 25.40 min). Recovery of icariside I using this precipitation procedure was 92% of the amount present in the hydrolyzate.
Example 5 Enzymatic Hydrolysis of Icariside Ia) The substrate was a partially purified icariside I product with 20% icariside I and 11% anhydroicaritin. About 50 mg was dissolved in 10 mL of ethanol, and water or buffer was added until the mixture became cloudy (about 20% ethanol). The following dry enzymes were added to separate samples: α-amylase, α-glucosidase, β-amylase, β-glucosidase, hesperidinase, lactase, and pectinase. The samples were incubated overnight at 40 ° C. and analyzed by HPLC. The results were only semi-quantitative due to the difficulty in dissolving the anhydroicaritin that precipitated from the samples. However, several of the chromatograms did show a definite reduction in icariside I and increase in the ratio of anhydroicaritin to icariside I. The best results were obtained using hesperidinase, lactase, β-glucosidase and pectinase.
A larger scale experiment was done using hesperidinase in order to isolate pure anhydroicaritin for characterization. Pure icariside I (20 mg )was dissolved in 10 mL of ethanol and 50 mL of water and 200 mg of hesperidinase enzyme was added and the mixture was incubated for 24 hr at 40 ° C. Crude anhydroicaritin was collected via filtration and purified on a 2.5×30 cm semi-prep C-18 HPLC column using a gradient of 50:50 (MeCN/H2O) to 80:20 (MeCN/H2O) in 20 min. The pure anhydroicaritin was analyzed by LC/MS and proton NMR.
b) Enzymatic Hydrolysis of PFG’s: The purified PFG solids (55.3%, purified by reversed-phase chromatography of a 50% EtOH extract) were subjected to enzymatic hydrolysis with the same enzymes and conditions described in part (a). Hesperidinase, lactase, β-glucosidase and pectinase appeared to convert the mixture of PFG’s to a mixture of sagittatosides, but no icariside I or anhydroicaritin were observed. This indicated that these enzymes were specific for the 7-β-glucosyl group and did not hydrolyze the 3-position sugar(s).
Example 6 Preparation of a High Anhydroicaritin-containing ProductA high sagittatosides Epimedium sagittatum extract containing 24.7% total sagittatosides (assayed as icariin) and 8.1% icariin and other expected prenylated flavonol glycosides was obtained from China. A 50 g portion of this extract was mixed with 250 mL of 90% ethanol and 7.5 mL of concentrated sulfuric acid in a 500 mL round bottom flask. The mixture was refluxed for 90 minutes, then allowed to cool to room temperature. The hydrolyzed mixture was filtered under reduced pressure through cellulose paper to remove insoluble sulfates and other materials. The cake was washed with approximately 20 mL of 90% ethanol. The resulting filtered ethanolic hydrolyzate (305 mL) contained 3.75 g of anhydroicaritin and 2.50 g of icariside I.
The filtered hydrolyzate prepared above (200 mL) was transferred to a 1000 mL container and 8.0 mL of 50% (w/w) sodium hydroxide solution was added with stirring, followed by 4.0 mL of phosphoric acid. De-ionized water (500 mL) was then added. This mixture was adjusted to pH 4.9 using 50% sodium hydroxide solution. The mixture was allowed to sit undisturbed at room temperature for 24 hours prior to decanting off the liquid. The resulting solids were macerated using de-ionized water and filtered under reduced pressure through cellulose paper. The resulting dark brown solids (11.9 g) were washed with de-ionized water and dried in air overnight. The dark brown solids contained 20% anhydroicaritin and 12% icariside I and an anhydroicaritin/icariside I ratio of 1.66. The recovery of anhydroicaritin in the precipitation procedure was 94% from the hydrolyzate.
Example 7 Recrystallization of Icariside IIcariside 1 (30 mg) obtained by a method described in Example 1 was dissolved in a minimum of hot tetrahydrofuran (THF). Hot methanol (approximately 10 mL) was then added. The hot THF/MeOH solution was filtered through a PTFE filter into a vial and allowed to evaporate at room temperature to about 5 mL, whereupon crystals began to form, and then placed in a 4° C. refrigerator for 24 hours. The crystals were filtered and washed with cold methanol and dried in a vacuum. Icariside I (21 mg) was isolated as yellow crystals and had a chromatographic purity of 97.4%.
Example 8 Large Scale Acid Hydrolysis of an Epimedium extractAn 800 g portion of an Epimedium sagittatum powder extract obtained from China containing about 13% total prenylflavonol glycosides as icariin was mixed with 4.0 L of 90% ethanol and 120 mL of sulfuric acid in a 10 L round bottom flask. The mixture was refluxed for 90 minutes and immediately chilled to stop the reaction. This mixture was filtered under reduced pressure through cellulose paper to remove insoluble sulfates and other materials. The cake was washed with approximately 200 mL of 90% ethanol. The resulting ethanolic hydrolyzate (4.0 L) contained 33.7 g of icariside I.
The ethanolic hydrolyzate prepared above was transferred to a 34 L container and 200 mL of 50% (w/w) sodium hydroxide solution was added with stirring, followed by 120 mL of phosphoric acid. De-ionized water (10 L) was then added. This mixture was adjusted to pH 8.2 using 120 mL of 50% sodium hydroxide solution. The mixture was stirred for 10 minutes and allowed to sit undisturbed at room temperature for 60 minutes prior to filtration under reduced pressure through cellulose paper. The resulting olive-green solids were washed with 750 mL of de-ionized water and dried under vacuum at 50° C. or in air at 80° C. The olive-green solids contained 44.6% icariside I. Recovery of icariside I in the precipitation procedure was 96% from the hydrolyzate.
Example 9 Large Scale Purification of an Epimedium Extract Containing Prenylflavonoid GlycosidesA 3.7 kg portion of an Epimedium sagittatum powdered extract obtained from China containing approximately 10% total prenylflavonol glycosides (PFG’s) assayed as icariin was stirred with 35 L of 85/15 acetone/water (v/v) in a 50 L mixing tank. The mixture was stirred vigorously for 30 minutes and allowed to sit for 5 minutes. The acetone extract layer (36 L) was decanted from the tank and contained 362 g of PFG’s. Recovery of the PFG’s in this extraction procedure was 96%.
A portion (about 500 mL) of the acetone extract was dried under reduced pressure at 50° C. or less, providing 16.1 g of brown solids which were analyzed to contain 28.6% total PFG’s when assayed as icariin.
| TABLE 1 | |||
| PDE-5 | |||
| IC50 | |||
| Entry | Sample description | % PFG’s | (μg/mL) |
| 1 | Vat extraction of Epimedium leaves, | 8.0 | 5.78 |
| refluxing for 17 hours with methanol | |||
| 2 | Extract prepared by extracting Epimedium | 7.2 | 4.24 |
| leaves with 50% ethanol | |||
| 3 | Extract prepared by extracting Epimedium | 10.2 | 12.50 |
| leaves with 90% ethanol | |||
| 4 | Extract prepared by extracting Epimedium | 16.30 | 5.27 |
| leaves with 50% EtOH and then purifying | |||
| the extract (after removal of EtOH) by | |||
| liq/liq extraction with butanol. Sample | |||
| tested was the butanol fraction. | |||
| 5 | Extract prepared by extracting Epimedium | 19.3 | 3.97 |
| leaves with 50% EtOH and purifying by | |||
| liquid/liquid extraction. Sample tested was | |||
| the aqueous fraction of the liq/liq extraction. | |||
| 6 | Purification of a 90% ethanol extract on | 65.60 | 1.87 |
| a HP-20 reversed phase column | |||
| TABLE 2 | |||
| PDE-5 | |||
| % | IC50 | ||
| Entry | Sample description | icarside I | (μg/mL) |
| 7 | Crude hydrolyzate composition obtained | 2.1 | 24.30 |
| from a 50% EtOH extract of Epimedium | |||
| leaves | |||
| 8 | Crude hydrolyzate composition obtained | 5.3 | 9.39 |
| from a 90% EtOH extract of Epimedium | |||
| leaves | |||
| 9 | Icariside I fraction obtained from | 21.4 | 1.50 |
| purifying hydrolyzate Sample No. 7 on a | |||
| SP-70 reversed-phase column and | |||
| eluting icariside I with alcohol | |||
| 10 | Pure (recrystallized) icariside I | 100 | 0.33 |
| 11 | Pure anhydroicaritin | 0 | 1.50 |
| 12 | icariside I hydrate | 0 | 21.50 |
| 13 | sildenafil | 0 | 0.031 |
- Liang DL & Zheng SL Effects of icaritin on cytochrome P450 enzymes in rats. Pharmazie 69:301-5 (2014).Read more (PubMed: 24791596) »
- Guo Y et al. An anticancer agent icaritin induces sustained activation of the extracellular signal-regulated kinase (ERK) pathway and inhibits growth of breast cancer cells. Eur J Pharmacol 658:114-22 (2011). Read more (PubMed: 21376032) »
- Zhu Jf et al. Icaritin shows potent anti-leukemia activity on chronic myeloid leukemia in vitro and in vivo by regulating MAPK/ERK/JNK and JAK2/STAT3 /AKT signalings. PLoS One 6:e23720 (2011). Read more (PubMed: 21887305) »
- The roots of Epimedium brevicornu Maxim
| Patent | Submitted | Granted |
|---|---|---|
| Compositions comprising icariside I and anhydroicaritin and methods for making the same [US6399579] | 2002-06-04 | |
| COSMETIC COMPOSITION CONTAINING HYDROLYSATES OF ICARIIN [US2009170787] | 2009-07-02 | |
| COMPOUNDS AND METHODS FOR TREATING ESTROGEN RECEPTOR-RELATED DISEASES [US8252835] | 2008-06-19 | 2012-08-28 |



/////////Beijing Shenogen, Granted Fast Track Status, Novel Cancer Drug, Icaritin, New Drug Approval submission, Beijing Food & Drug Administration, oral traditional Chinese medicine, barrenwort
Odalasvir
ACH-3102 , Odalasvir
Odalasvir
ACH-0143102; ACH-3102
CAS : 1415119-52-6
Dimethyl N, N ‘- (tricyclo [8.2.2.24,7] hexadeca-1 (12), 4,6, 10,13,15-hexaene-5,11-diylbis {1H-benzimidazole-5,2-diyl [(2S, 3aS, 7aS) -octahydro-1H-indole-2,1-diyl] [(1S) -1 – (1-methylethyl) -2-oxoethylene]}) biscarbamate
Carbamic acid, N,N’-(tricyclo(8.2.2.24,7)hexadeca-4,6,10,12,13,15-hexaene-5,11-diylbis(1H-benzimidazole-6,2-diyl((2S,3aS,7aS)-octahydro-1H-indole-2,1-diyl)((1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl)))bis-, C,C’-dimethyl ester
Dimethyl N,N’-(1,4(1,4)-dibenzenacyclohexaphane-12,42-diylbis(1hbenzimidazole-5,2-diyl((2S,3aS,7aS)-octahydro-1H-indole-2,1-diyl)((2S)-3-methyl-1-oxobutan-1,2-diyl)))biscarbamate
Mechanism of Action: HCV NS5A Protein inhibitor
Indication: Hepatitis C
Developer: Achillion Pharmaceuticals, Inc.

-
C60-H72-N8-O6
- 1001.2788
Odalasvir[1] is an investigational new drug in development for the treatment hepatitis C.
Achillion Pharmaceuticals Inc’s Odalasvir (ACH-3102) is an investigational new drug in development for the treatment hepatitis C. Achillion’s ongoing study tests its NS5A inhibitor, ACH-3102, with Sovaldi in previously untreated genotype 1 hepatitis C patients over six and eight weeks of therapy. The main goal is to achieve a cure, or sustained virological response, 12 weeks after the completion of therapy.
Odalasvir is a hepatitis C virus (HCV NS5A) inhibitor in phase II clinical studies at Achillion for the treatment of hepatitis C.
In 2012, fast track designation was assigned to the compound in the U.S. for the treatment of chronic hepatitis C.
WILL BE UPDATED………….
WO 2012166716
http://www.google.com/patents/US20120302538

General Considerations
All nonaqueous reactions were performed under an atmosphere of dry argon gas using oven-dried glassware and anhydrous solvents. The progress of reactions and the purity of target compounds were determined using one of the following two HPLC methods: (1) Waters AQUITY HPLC BEH C18 1.7 μm 2.1×50 mm column with an isocratic elution of 0.24 min at 90:10 water:acetonitrile containing 0.05% formic acid followed by a 4.26-min linear gradient elution from 90:10 to 10:90 at a flow rate of 1.0 mL/min with UV (PDA), ELS, and MS (SQ in APCI mode) detection (method 1); and (2) Waters AQUITY HPLC BEH C18 1.7 μm 2.1×50 mm column with an isocratic elution of 0.31 min at 95:5 water:acetonitrile containing 0.05% formic acid followed by a 17.47-min linear gradient elution from 95:5 to 5:95 at a flow rate of 0.4 mL/min with UV (PDA), ELS, and MS (SQ in APCI mode) detection (method 2).
Target compounds were purified via preparative reverse-phase HPLC using a YMC Pack Pro C18 5 μm 150×20 mm column with an isocratic elution of 0.35 min at 95:5 water:acetonitrile containing 0.1% trifluoroacetic acid followed by a 23.3-min linear gradient elution from 95:5 to 5:95 at a flow rate of 18.9 mL/min with UV and mass-based fraction collection.

Example 1
Synthesis of Compound 10
Compound 10 was prepared via bromination of [2.2]paracyclophane as outlined previously (Reich, H. J.; Cram, D. J. J. Am. Chem. Soc. 1969, 91, 3527-3533; Reich, H. J.; Cram, D. J. J. Am. Chem. Soc. 1969, 91, 3534-3543). Compounds 1, 2, 6, 8, and 10 can be obtained from commercial sources. Compounds 3-7 and 9 were prepared using general synthetic methods known in the art.
Example 2Synthesis of Compound 11
A deoxygenated (argon) mixture of 9 (284.2 mg), 10 (52.3 mg), K3PO4 (248.1 mg), and PdCl2dppf.CH2Cl2 (7.4 mg) in dioxane/water (5.5 mL/0.55 mL) was irradiated in a microwave for 2 h at 80° C. The resulting mixture was evaporated under reduced pressure and the remaining solid was extracted with DCM. This crude material was purified by PTLC (20 cm×20 cm×2000 μm glass plates; eluted with 45:50:5 v/v/v DCM:EtOAc:MeOH, Rf 0.28) to give 75.3 mg of 11. The purity of 11 was determined via analytical reverse-phase HPLC using a 3.5-min gradient elution of increasing concentrations of ACN in water (10-90%) containing 0.05% formic acid with a flow rate of 1.0 mL/min on a Waters AQUITY HPLC BEH C18 1.7 μm 2.1×50 mm column with UV (PDA), ELS, and MS (SQ in APCI mode) detection. HPLC: tR 1.57 min (98% purity). MS m/z calculated for C56H64N8O6 ([M]+), 945. found, 946 ([M+1]+).
SEE ALSO
US 2012302538
http://www.google.com/patents/US20120302538
……………
see
US 20150023913
http://www.google.com/patents/US20150023913
…………..
see
WO 2015005901
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Dimethyl N,N′-(1,4(1,4)-Dibenzenacyclohexaphane-12,42-diylbis(1hbenzimidazole-5,2-diyl((2S,3aS,7aS)-octahydro-1H-indole-2,1-diyl)((2S)-3-methyl-1-oxobutan-1,2-diyl)))biscarbamate
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 1415119-52-6 |
| ATC code | None |
| Chemical data | |
| Formula | C60H72N8O6 |
| Molecular mass | 1001.28 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin

Join me on Facebook 
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
//////////
Evofosfamide

Evofosfamide, HAP-302 , TH-302
 |
|
| Names | |
|---|---|
| IUPAC name
(1-Methyl-2-nitro-1H-imidazol-5-yl)methyl N,N’-bis(2-bromoethyl)phosphorodiamidate
|
|
| Other names
TH-302; HAP-302
|
|
| Identifiers | |
| 918633-87-1 |
|
| ChemSpider | 10157061 |
| Jmol-3D images | Image |
| PubChem | 11984561 |
| Properties | |
| C9H16Br2N5O4P | |
| Molar mass | 449.04 g·mol−1 |
| 6 to 7 g/l | |
TH-302 is a nitroimidazole-linked prodrug of a brominated derivative of an isophosphoramide mustard previously used in cancer drugs
evofosfamide (first disclosed in WO2007002931), useful for treating cancer.
Threshold Pharmaceuticals and licensee Merck Serono are codeveloping evofosfamide, the lead in a series of topoisomerase II-inhibiting hypoxia-activated prodrugs and a 2-nitroimidazole-triggered bromo analog of ifosfamide, for treating cancer, primarily soft tissue sarcoma and pancreatic cancer (phase 3 clinical, as of April 2015).
In November 2014, the FDA granted Fast Track designation to the drug for the treatment of previously untreated patients with metastatic or locally advanced unresectable soft tissue sarcoma.
Evofosfamide (INN,[1] USAN;[2] formerly known as TH-302) is an investigational hypoxia-activated prodrug that is in clinical development for cancer treatment. The prodrug is activated only at very low levels of oxygen (hypoxia). Such levels are common in human solid tumors, a phenomenon known as tumor hypoxia.[3]
Evofosfamide is being evaluated in clinical trials for the treatment of multiple tumor types as a monotherapy and in combination with chemotherapeutic agents and other targeted cancer drugs
Discovered at Threshold, TH-302 is a hypoxia-activated prodrug (HAP) designed to exploit low oxygen levels in hypoxic tumor regions. Therapeutics that specifically target resistant hypoxic zones could provide significant additional antitumor activity and clinical benefit over current chemotherapeutic and radiation therapies.
Evofosfamide (TH-302) was developed by Threshold Pharmaceuticals Inc. (Threshold).[4] The company is located in South San Francisco, CA, USA.
In 2012, Threshold signed a global license and co-development agreement for evofosfamide with Merck KGaA, Darmstadt, Germany, which includes an option for Threshold to co-commercialize eofosfamide in the United States. Threshold is responsible for the development of evofosfamide in the soft tissue sarcoma indication in the United States. In all other cancer indications, Threshold and Merck KGaA are developing evofosfamide together.[5] From 2012 to 2013, Merck KGaA paid 110 million US$ for upfront payment and milestone payments to Threshold. Additionally, Merck KGaA covers 70% of all evofosfamide development expenses.[6]
Discovered at Threshold, TH-302 is a hypoxia-activated prodrug (HAP) designed to exploit low oxygen levels in hypoxic tumor regions. Therapeutics that specifically target resistant hypoxic zones could provide significant additional antitumor activity and clinical benefit over current chemotherapeutic and radiation therapies.
History
| Date | Event |
|---|---|
| Jun 2005 | Threshold files evofosfamide (TH-302) patent applications in the U.S.[49] |
| Jun 2006 | Threshold files a evofosfamide (TH-302) patent application in the EU and in Japan[50] |
| Sep 2011 | Threshold starts a Phase 3 trial (TH-CR-406) of evofosfamide in combination with doxorubicin in patients with soft tissue sarcoma |
| Feb 2012 | Threshold signs an agreement with Merck KGaA to co-develop evofosfamide |
| Apr 2012 | A Phase 2b trial (TH-CR-404) of evofosfamide in combination with gemcitabine in patients with pancreatic cancer meets primary endpoint |
SEE
WO2007002931
http://www.google.com/patents/WO2007002931A2?cl=en
Example 8
Synthesis of Compounds 25, 26 [0380] To a solution of 2-bromoethylammmonium bromide (19.4 g) in DCM (90 mL) at – 1O0C was added a solution OfPOCl3 (2.3 mL) in DCM (4 mL) followed by addition of a solution of TEA (14.1 mL) in DCM (25 mL). The reaction mixture was filtered, the filtrate concentrated to ca. 30% of the original volume and filtered. The residue was washed with DCM (3×25 mL) and the combined DCM portions concentrated to yield a solid to which a mixture of THF (6 mL) and water (8 mL) was added. THF was removed in a rotary evaporator, the resulting solution chilled overnight in a fridge. The precipitate obtained was filtered, washed with water (10 mL) and ether (30 mL), and dryed in vacuo to yield 2.1 g of:

Isophosphoramide mustard

can be synthesized employing the method provided in Example 8, substituting 2- bromoethylammmonium bromide with 2-chloroethylammmonium chloride. Synthesis of Isophosphoramide mustard has been described (see for example Wiessler et al., supra).
The phosphoramidate alkylator toxin:

was transformed into compounds 24 and 25, employing the method provided in Example 6 and the appropriate Trigger-OH.
Example 25
Synthesis of l-N-methyl-2-nitroimidazole-5-carboxylis acid

A suspension of the nitro ester (39.2 g, 196.9 rnmol) in IN NaOH (600 mL) and water (200 mL) was stirred at rt for about 20 h to give a clear light brown solution. The pH of the reaction mixture was adjusted to about 1 by addition of cone. HCl and the reaction mixture extracted with EA (5 x 150 mL). The combined ethyl acetate layers were dried over MgS O4 and concentrated to yield l-N-methyl-2-nitroimidazole-5-carboxylis acid (“nitro acid”) as a light brown solid (32.2 g, 95%). Example 26
Synthesis of l-N-methyl-2-nitroimidazole-5-carboxylis acid

A mixture of the nitro acid (30.82 g, 180.23 mmol) and triethylamine (140 niL, 285 mmol) in anhydrous THF (360 mL) was stirred while the reaction mixture was cooled in a dry ice-acetonitrile bath (temperature < -20 0C). Isobutyl chloroformate (37.8 mL, 288 mmol) was added drop wise to this cooled reaction mixture during a period of 10 min and stirred for 1 h followed by the addition of sodium borohydride (36 g, 947 mmol) and dropwise addition of water during a period of 1 h while maintaining a temperature around or less than O0C. The reaction mixture was warmed up to O0C. The solid was filtered off and washed with THF. The combined THF portions were evaporated to yield l-N-methyl-2- nitroimidazole-5-methanol as an orange solid (25 g) which was recrystallized from ethyl acetate.
……………………………………….
WO-2015051921

EXAMPLE 1

1
N-Formylsarcosine ethyl ester 1 (1 ,85 kg) was dissolved in toluene (3,9 kg) and ethyl formate (3,28 kg) and cooled to 10 °C. A 20 wt-% solution of potassium tert-butoxide (1 ,84 kg) in tetrahydrofuran (7,4 kg) was added and stirring was continued for 3h. The reaction mixture was extracted 2x with a solution of sodium chloride in water (10 wt-%) and the combined water extracts were washed lx with toluene.
Aqueous hydrogen chloride (25% wt-%; 5,62 kg) was added to the aqueous solution, followed by ethylene glycol (2,36 kg). The reaction mixture was heated to 55-60 °C for lh before only the organic solvent residues were distilled off under vacuum.
Aqueous Cyanamide (50 wt-%, 2,16 kg) was then added at 20 °C, followed by sodium acetate (3,04 kg). The resulting reaction mixture was heated to 85-90 °C for 2h and cooled to 0-5 °C before a pH of ~ 8-9 was adjusted via addition of aqueous sodium hydroxide (32% wt-%; 4,1 kg). Compound 3 (1,66 kg; 75%) was isolated after filtration and washing with water.
Ή-NMR (400 MHz, d6-DMSO): δ= 1,24 (3H, t, J= 7,1 Hz); 3,53 (3H, s); 4,16 (2H, q, J= 7,0 Hz) ; 6,15 (s, 2 H); 7,28 (s, 1H).
HPLC (Rt = 7,7 min): 97,9% (a/a).
REFERENCES
1
- WHO Drug Information; Recommended INN: List 73
- 2
- Adopted Names of the United States Adopted Names Council
- 3
- Duan J; Jiao, H; Kaizerman, J; Stanton, T; Evans, JW; Lan, L; Lorente, G; Banica, M et al. (2008). “Potent and Highly Selective Hypoxia-Activated Achiral Phosphoramidate Mustards as Anticancer Drugs”. J. Med. Chem. 51 (8): 2412–20. doi:10.1021/jm701028q. PMID 18257544.
- 4
- Website of Threshold Pharmaceuticals Inc.
- 5
- Threshold Pharmaceuticals and Merck KGaA Announce Global Agreement to Co-Develop and Commercialize Phase 3 Hypoxia-Targeted Drug TH-302 – Press release from 3 February 2012
- 6
Threshold Pharmaceuticals Form 8-K from 3 Nov 2014
DHAKA BANGLADESH




 .
.
Steamers and ferries in Sadarghat Port
 Kawran Bazar
Kawran Bazar
 .
.
 Dry fish sellers at the Karwan Dry Fish Market (Bazar), Dhaka, Bangladesh.
Dry fish sellers at the Karwan Dry Fish Market (Bazar), Dhaka, Bangladesh.

BioCryst’s BCX4161 receives FDA fast-track designation to treat HAE
![]()
BioCryst’s BCX4161 receives FDA fast-track designation to treat HAE
BioCryst Pharmaceuticals has received fast-track designation from the US Food and Drug Administration (FDA) for its BCX4161, an orally administered and selective inhibitor of plasma kallikrein in advanced clinical development to treat hereditary angioedema (HAE).
READ HERE……[LINK]
“BCX4161 and our second-generation molecules have the potential to significantly improve HAE patient treatment and their quality of life.”
………………………………………….
PREVIOUS ARTICLE CUT PASTE

(RTTNews.com) – BioCryst Pharmaceuticals Inc. ( BCRX ) will be reporting results from OPuS-1, a phase IIa trial of orally-administered BCX4161 in patients with hereditary angioedema, on Tuesday, May 27, 2014 at 8:30 a.m. Eastern Time.
The OPuS-1 clinical trial is testing 400 mg of BCX4161 administered three times daily for 28 days in up to 25 hereditary angioedema patients who have a high frequency of attacks (≥ 1 per week), in a randomized, placebo-controlled, two-period cross-over design.
Read more: http://www.nasdaq.com/article/bcrx-to-watch-out-for-gtiv-adopts-poison-pill-teva-qgen-drtx-get-fda-nod-20140527-00005#ixzz335Khl0sk
BCX-4161 is a novel, selective inhibitor of plasma kallikrein in development for prevention of attacks in patients with hereditary angioedema (HAE). By inhibiting plasma kallikrein, BCX-4161 suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
……………………………….
old article
BCRX – BioCryst – Entering The HAE Market
BioCryst announced on Monday July 22 the successful completion of a Phase I study on the safety and PK of BCX4161, a candidate for the treatment of Hereditary angioedema (HAE). HAE is a genetic disorder resulting from the loss or dysfunction of complement C1 Inhibitor (C1INH).
Among the functions performed by C1INH is regulation of the hormone bradykinin, which when activated, leads to the dilation of blood vessels. Left unchecked, excess bradykinin can cause painful attacks of swelling, or angioedemas, in any part of the body, including the face, abdomen, hands, and larynx. Death can occur from asphyxiation, particularly in children.
The mechanics involved in HAE are fairly well understood today. There are several approved drugs available today that work at three major points in the pathway. Ultimately, each prevents bradykinin from activating its receptor on endothelial cells.
C1 Inhibitors, of which four have been approved, prevent Factor XIIa activation of Plasma Kallikrein and inhibit Kallikrein itself. The single specific Kallikrein inhibitor is Kalbitor from Dyax. C1INHs and kallikrein inhibitors prevent the formation of bradykinin (labeled “BK” in this diagram). Then there is Firazyr from Shire, a B2 bradykinin receptor antagonist; while not preventing overproduction of the hormone, activation of downstream activity is suppressed.
Interestingly, of all the available therapies, only C1INH Cinryze from Viropharma is approved for prophylactic use- all others are designated strictly for treatment of acute attacks. A key reason for this is Cinryze’s long half-life, allowing sustained activity over longer intervals. As each of these drugs are given by injection, frequent treatment is not practical. Consider, for instance, Kalbitor has a half life of just two hours.
This is where BioCryst comes in. The company is pursuing the less crowded prophylaxis indication. It has the only orally available (although just barely) plasma kallikrein inhibitor. And while PK is not great, requiring three-times daily dosing to ensure adequate drug levels, pills make this a feasible option. As you can see, 800 mg appears optimal, however, 400 mg was selected as the Phase IIa dose due to 3 cases of moderate AEs seen at 800. This study was in healthy volunteers and the drug was otherwise well tolerated [ref].
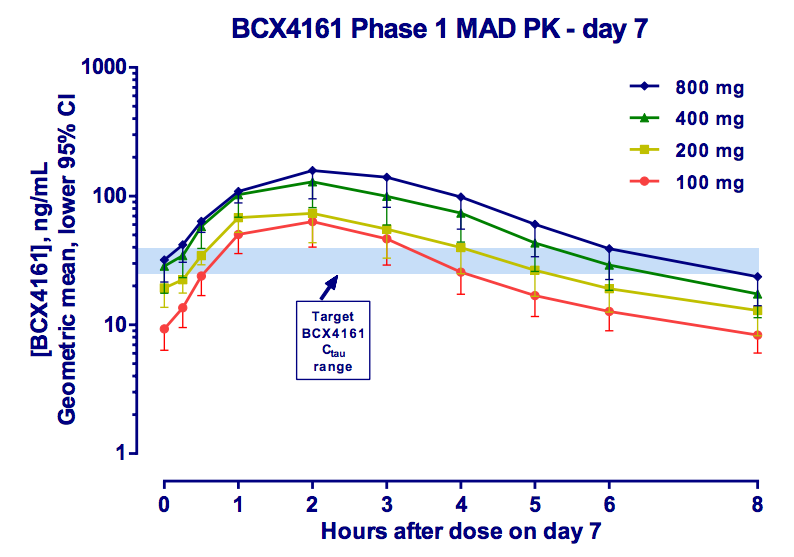
(From Company Presentation)
BCX4161 is an interesting compound. Based on patent literature, we believe the molecule has a similar structure to the one illustrated below:
BCX4161 is not a specific inhibitor of kallikrein, and in fact has near equal potency against Factor XIIa. This dual-activity is also seen with C1INH, setting the compound apart from Kalbitor and Firazyr.
The different profile may improve efficacy, but that is unknown at this point. Along with Factor XIIa, BCX4161 inhibits additional factors involved in coagulation. Bleeding issues has been something the company has been testing and will be certain to monitor. As a drug designed for chronic use, safety will be a major concern.
A 25 patient Phase IIa study set for Q4 will be placebo-controlled double-blind crossover of the following design:
(From Company Presentation)
Individuals with a high frequency of attacks(~1/week) will be enrolled, the primary endpoint is attack frequency. Viropharma conducted a pivotal trial of similar design (but two twelve week dosing periods), reporting ~50% reduction in attacks vs. placebo. We imagine BioCryst would need to achieve results in this range for the drug to be competitive.
A major impedance toward these efficacy goals will likely be individual adherence to dosing every eight hours schedule. Missed doses will mean severe drops in drug levels, potentially putting the patient at risk for an attack. The company noted patients on Cinryze occasionally miss doses with no apparent adverse effect. We will see if this holds true for their own compound.
The Phase IIa is being run in Germany, ostensibly because of the country’s well organized HAE medical treatment system. The study is expected to initiate in 4Q 2013. BioCryst aims to market the drug in the U.S. on their own, likely partnering in the EU.
Handicapping this Phase II is rather difficult with the lack of any prior efficacy results. BioCryst has selected a well-validated target in a fairly well understood disease. The data suggests BCX4161 is an active drug. What we will soon find out is whether the compound is active enough and has a sufficiently clean profile. As attractive as oral dosing is- it has an achilles heel. Regardless of the medication, patients continue to have attacks, only of less frequency and severity. If a patient should suffer major laryngeal swelling, pills may not be an option as a rescue medicine. Cinryze on the hand can serve as both prophylaxis and acute treatment.
Commercially, we believe the compound will have a difficult time competing with Cinryze. True, Cinryze has its own issues, namely a requirement for infusions every 3 to 4 days, but it is difficult to see how a 3-times/day treatment is much of an improvement. In any case, by the time BCX4161 reaches the market, Viropharma should have a much simpler subcutaneous version of its C1INH available, allowing it to maintain a strong monopoly in prophylaxis HAE treatments. Additional competition may come in the form of a follow-up kallikrein inhibitor in development at Dyax; the long acting antibody is designed specifically for the prophylaxis market and is expected to enter the clinic 2H 2013.
Nabriva’s lefamulin, BC 3781 receives FDA fast-track status to treat CABP and ABSSSI
Nabriva’s lefamulin receives FDA fast-track status to treat CABP and ABSSS
Austria-based Nabriva Therapeutics has received qualified infectious disease product (QIDP) and fast-track status designation from the US Food and Drug Administration for its lefamulin (BC 3781).
read

BC-3781
Topical pleuromutilin antibiotic agent
Gram-positive, including MRSA, PHASE 2 COMPLETED,Infection, acute bacterial skin and skin structure (ABSSSI)
Nabriva (Austria), Nabriva Therapeutics AG
BC-3781
cas 1061872-97-6
UNII-61H04Z5F9K
(3aS,4R,5S,6S,8R,9R,9aR,10R)-5-Hydroxy-4,6,9,10-tetramethyl-1-oxo-6-vinyldecahydro-3a,9-propanocyclopenta[8]annulen-8-yl [[(1R,2R,4R)-4-amino-2-hydroxycyclohexyl]sulfanyl]acetate;
14-O-[2-[(1R,2R,4R)-4-Amino-2-hydroxycyclohexylsulfanyl]acetyl]mutilin
BC-3781 is a pleuromutilin antibiotic in early clinical development at Nabriva for the treatment of community acquired pneumonia and for the treatment of patients with acute bacterial skin and skin structure infections (ABSSSI). Pleuromutilin antibiotics interfere with bacterial protein synthesis via a specific interaction with the 23S rRNA of the 50S bacterial ribosome subunit. They have a distinct antibacterial profile and show no cross-resistance with any other class of antibiotics. In 2012, a codevelopment agreement was signed between Forest and Nabriva, but, in 2014, this agreement terminated and Nabriva retained all rights.
The pleuromutilin BC-3781 belongs to the first generation of pleuromutilins to combine excellent oral
bioavailability with substantial activity against Gram-positive pathogens and atypicals as well as some
Gram-negative pathogens. In particular, BC-3781 is highly active against multi-drug resistant (MDR)
pathogens including methicillin resistant Staphylococcus aureus (MRSA), MDR Streptococcus pneumonia
(i.e. macrolide and quinolone resistance), and vancomycin resistant Enterococcus faecium. It is
characterized by excellent in vivo activities (e.g. pneumonia model), outstanding PK/PD parameters,
allowing once a day dosing, and a novel mode of action. BC-3781 is being developed for both oral and IV
administration and is intended for the treatment of serious multi-drug resistant skin & skin structure
infections (CSSI) and moderate to severe pneumonia (CAP, HAP etc).

Pleuromutilins have been known since 1951, but only entered the market![]() in 2007 with the approval of retapamulin for topical use. Until today, there are no pleuromutilins for systemic use approved in human clinical practice.
in 2007 with the approval of retapamulin for topical use. Until today, there are no pleuromutilins for systemic use approved in human clinical practice.
Nabriva is currently working on the development of new compounds is this class. The lead compound, BC-3781, if approved, will be the first pleuromutilin for systemic use in humans.
The compound shows potent in vitro activity against a large collection of staphylococci, streptococci, andE. faecium. When compared to linezolid and vancomycin, the compound shows greater overall potency againstS. aureus [121]. BC-3781 shows improved activity against most bacteria commonly associated with community-acquired respiratory tract infections, the compound is especially potent against S. pneumoniaincluding penicillin resistant strains. It also shows improved activity against H. influenza, M. catarrhalis, M. pneumoniae and C. pneumoniae.
BC-3781 is undergoing Phase I clinical trials for CAP and in March of 2011 has completed a Phase II clinical study comparing it to vancomycin for treatment of aBSSSI [119,120,121,122,123]. Nabriva Therapeutics AG announced that the cooperation with Forest Laboratories to develop the compound had elapsed, and that Nabriva retained all rights in BC-3781. The company informed that the product was Phase III ready and that it was seeking partners to continue further development [203].
Nabriva is also developing BC-7013 for topical use against Gram-positive infections and working on the discovery of new pleuromutilins [119,124].
Dr William Prince, CMO Nabriva Therapeutics commented:
“This is the first patient study with a systemic pleuromutilin. It will be an important proof of concept
for an exciting new class of antibiotics. The phase II study builds on our extensive preclinical and
phase I data which have demonstrated that BC-3781 can achieve therapeutically relevant blood and
tissue levels in man with excellent tolerability when administered by either oral or intravenous
routes.”
Dr. David Chiswell, CEO Nabriva Therapeutics commented:
“With a worldwide problem due to antibiotic resistant bacteria, there is a very significant need for
new classes of antibiotics with unique modes of action such as the pleuromutilins. The commercial
prospects for BC-3781 as the leading compound of an exciting new class are excellent, especially as it
has an ideal anti-bacterial spectrum for both skin and respiratory infections and is being developed
with both oral and intravenous formulations”
BC-3781 is highly active against key pathogens, including MRSA, associated with skin infections and
community and hospital acquired pneumonia and is more potent than Linezolid and vancomycin. The
compound’s novel mode of action ensures that it overcomes resistance mechanisms affecting all
approved classes of antibiotics. BC-378
About Nabriva Therapeutics
Nabriva Therapeutics is a biotechnology company focused on developing a new class of antibiotics for
the treatment of serious infections caused by resistant pathogens. Nabriva’s lead systemic product,
BC-3781, is being developed for the treatment of serious skin infections and bacterial pneumonia
caused by S. aureus, , S. pneumoniae, H. influenza, Mycoplasma, Legionella and other bacteria,
including drug resistant strains such as MRSA and vancomycin resistant E. faecium. In addition,
Nabriva Therapeutics’ topical pleuromutilin product candidate, BC-7013, is in clinical phase I. Nabriva
Therapeutics has a proven track record in world-class medicinal chemistry, clinical expertise, a
seasoned management team and solid IP. Nabriva Therapeutics is located in Vienna, Austria.
For more information on Nabriva please visit http://www.nabriva.com. Nabriva Therapeutics AG
…………………………………………
EP 2390245
http://www.google.com/patents/EP2390245A1?cl=en
……………………………………………..
http://www.google.im/patents/US20090118366?cl=es
The trivial name mutilin refers to the IUPAC systematic name (1S, 2R, 3S, 4S, 6R, 7R, 8R, 14R)-3,6-dihydroxy-2,4,7,14-tetramethyl-4-vinyl-tricyclo[5.4.3.01,8]tetradecan-9-one. In the examples, pleuromutilin derivatives are numbered in analogy to the mutilin numbering system described by H. Berner (Berner, H.; Schulz, G.; Schneider H. Tetrahedron 1980, 36, 1807-1811.):

…………………………………………………….
http://www.google.com/patents/WO2008113089A1?cl=en
Pleuromutilin, a compound of formula A

is a naturally occurring antibiotic, e.g. produced by the basidomycetes Pleurotus mutilus and P. passeckerianus, see e.g. The Merck Index, 13th edition, item 7617. A number of further pleuromutilins having the principle ring structure of pleuromutilin and being substituted at the hydroxy group have been developed, e.g. as antimicrobials.
From WO 02/04414 Al pleuromutilin derivatives, e.g. 14-O-[(Aminocyclohexan-2-yl (and – 3-yl)-sulfanyl)-acetyl]-mutilins; from WO 07/014409 Al e.g. 14-O-[((Mono- or dialkylamino)-cycloalkylsulfanyl)-acetyl]-mutilins and from WO 07/000004 Al e.g. [((Acyl- hydroxy-amino)-cycloalkylsulfanyl)-acetyl]-mutilins, are known.
14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin hydrochloride
Example 1 – 14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin hydrochloride + (IS, 2S, 4S) diastereomer hydrochloride
Step Al. 14-O-{[(1R, 2R, 4R)-4-tert-Butoxycarbonylamino-2-hydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer and 14-O-{[(lR, 2R, 5S)-5-i’eri’-Butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin + (IS, 2S, 5R) diastereomer and
14-0-{[(lR, 2R, 4S)-4-tert-Butoxycarbonylamino-2-hydroxy-cyclohexylsuIfanyl]-acetyl}- mutilin + (IS, 2S, 4R) diastereomer
To a solution of 3,4-epoxycyclohexyl-carbamic acid tert-butyl ester (Gomez-Sanchez, E.; Marco-Contelles J. Tetrahedron 2005, 61, 1207-1219.) (4.27g, 20mmol) and pleuromutilin thiol (Nagarajan, R. Eli Lilly and Company 1978, US4, 130,709) (7.10 g, 18 mmol) in 200 ml of tetrahydrofuran was added aluminum oxide (40 g, Brockmann activity I, neutral) and the resulting mixture was stirred for 40 hours at room temperature. The suspension was filtered and concentrated under reduced pressure. The residue was subjected to chromatography (silica, cyclohcxane / ethyl acetate = 1/1) to yield 14-O-{[(1R, 2R, 4R)-4-ler(- butoxycarbonylamino-2-hydroxy-cyclohcxylsulfanyl]-acctyl}-mutilin + (IS, 2S, 4S) diastereomer (a) (Rf = 0.38, 1.34g, 12%) as well as a mixture of 14-O-{[(1R, 2R, 5S)-5-tert- butoxycarbonylumino-2-hy(lroxy-cyclohcxylsulfnnyl]-ncctyl}-niυtilin + (I S, 2S, 5R) diastereomer and 14-O-{[(1R, 2R, 4S)-4-tert-butoxycarbonylamino-2-hydroxy- cyclυhexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4R) diastereomer (b) (Rf = 0.26, 2.81 g, 25%) as colorless amorphous foams. (a): 1H NMR (400MHz, DMSOd6, δ, ppm, inter alia): 6.74 (d, IH, NH, J = 7Hz), 6.13 (dd, IH, 19-H, J – I lHz and 18Hz), 5.54 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.90 (d, IH, 2′-OH, J = 5Hz), 4.48 (d, IH, 11-OH, J = 6Hz), 3.55 – 3.20 (m, 6H, 1 ‘-H, 2‘-H, 4′-H, 11-H, 22-H), 2.40 (bs, IH, 4-H), 1.36 (s, 3H, 15-CH3), 1.35 (s, 9H, tert-butyl), 1.06 (s, 3H, 18-CH3), 0.81 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 630 (MNa+), 1237 (2MNa+).
(b): 1H NMR (400MHz, DMSO-de, δ, ppm, inter alia): 6.70 (d, IH, NH, J = 7Hz), 6.12 (dd, IH, 19-H, J = HHz and 18Hz), 5.34 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.82, 4.78 (d, IH, 2′-OH, J = 4Hz), 4.48 (d, IH, 11-OH, J = 6Hz), 3.55 – 3.20 (m, 5H, 2′-H, 475′-H, 11- H, 22-H), 2.97 (m, IH, 1 ‘-H), 2.40 (bs, IH, 4-H), 1.35 (s, 12H, 15-CH3, tert-butyl), 1.05 (s, 3H, 18-CH3), 0.82 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 630 (MNa+), 1237 (2MNa+).
or Step A2. 14-O-{[(1R, 2R, 4R)-4-tert-Butoxycarbonylamino-2-liydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer and
14-O-{[(1R, 2R, 5S)-5-tert-Butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin + (IS, 2S, 5R) diastereomer and
14-O-{[(1R, 2R, 4S)-4-rerf-Butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin + (IS, 2S, 4R) diastereomer
To a solution of 3,4-epoxycyclohexyl-carbamic acid tert-butyl ester (10 g, 47 mmol) and pleuromutilin thiol (16.6 g, 42 mmol) in 200 ml of methanol and 20 ml of dioxane was added 2N NaOH (21 ml, 42 mmol) and the resulting mixture was stirred for 16 hours at room temperature. After completion of the reaction the pH was set to 7 with diluted HCl and the reaction mixture was concentrated under reduced pressure. The residue was diluted with water and brine and extracted three times with ethyl acetate. The organic layers were dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure and after chromatography (silica, cyclohexane / ethyl acetate = 1/1) 14-O-{[(1R, 2R, 4R) A-tert- butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl] -acetyl }-mutilin + (IS, 2S, 4S) diastereomer (Rf = 0.40, 3.1g, 12% yield) as well as a mixture of 14-O-{[(1R, 2R, 5S)-5-tert- butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 5R) diastereomer and 14-O-{[(1R, 2R, 4S)-4-tert-butoxycarbonylamino-2-hydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4R) diastereomer (Rf = 0.25, 6.35 g, 25%) were obtained as colorless amorphous foams. or Step A3. 14-O-{[(1R, 2R, 4R)-4-tert-Butoxycarbonylamino-2-hydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer and 14-O-{ [(1R, 2R, 5S)-5-tert-Butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin + (IS, 2S, 5R) diastereomer
To a solution of Pleuromutilin thiol (9.25 g, 23.5 mmol) in 100 ml of acetonitrile (dried over 4A molecular sieve) was added l,5-diazabicyclo[4.3.0]non-5-ene (DBN, 2.9 μl, 23.5 mmol) and after 1 hour of stirring at room temperature under argon atmosphere the mixture was ^ charged with syn-3,4-epoxycyclohexyl-carbamic acid tert-butyl ester (4.17 g, 19.5 mmol) and stirred for further 16 hours at room temperature. The reaction mixture was concentrated under reduced pressure. The residue was charged with water and brine and extracted three times with dichloromethane. The organic layers were dried over sodium sulphate and filtered. The filtrate was concentrated under reduced pressure and subjected to chromatography (silica, cyclohexane / ethyl acetate = 1/1) to yield 14-O-{[(1R, 2R, 4R)-4-teAY-butoxycarbonylamino- 2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer (Rf = 0.38, 5.07g, 43%) as well as 14-O-{[(1R, 2R, 5S)-5-tert-butoxycarbonylamino-2-hydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 5R) diastereomer (Rf = 0.25, 2.95 g, 16.5%) as colorless amorphous foams.
Step B. 14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer
To a solution of 14-O-{[(1R, 2R, 4R)-4-teΛ-t-butoxycarbonylamino-2-hydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer (1.34 g, 2.20 mmol) in 75 ml of dichloromethane was added trifiuoroacetic acid (4 ml) at 4°C and stirred for 5 hours at room temperature. The reaction mixture was diluted with dichloromethane and cautiously poured into a saturated NaHCO3 solution. The phases were separated and the aqueous layer was washed two times with dichloromethane. The combined organic layers are dried over sodium sulfate and filtered. After chromatography (silica, ethyl acetate/methanol/35% ammonia solution = 50/50/1) 14-O-{[(1R, 2R, 4R)-4-amino-2-hydroxy-cyclohexylsulfanyl]- acetyl}-mutilin + (IS, 2S, 4S) diastereomer (745 mg, 67% yield) was obtained as colorless amorphous foam.
1H NMR (400MHz, DMSO-de, δ, ppm, inter alia): 6.14 (dd, IH, 19-H, J = 1 IHz and 18Hz), 5.54 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.50 (d, IH, 11-OH, J = 6Hz), 3.50 – 3.20 (m, 5H, 2′-H, 4′-H, H-H, 22-H), 2.55 (m, IH, l ‘-H), 2.40 (bs, IH, 4-H), 1.35 (s, 3H, 15- CH3), 1.06 (s, 3H, 18-CH3), 0.82 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 508 (MH+), 530 (MNa+), 1015 (2MH+), 1037 (2MNa+).
Step C. 14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsulfanyI]-acetyl}- mutilin hydrochloride + (IS, 2S, 4S) diastereomer hydrochloride
A solution of 14-O-{[(1R, 2R, 4R)-4-amino-2-hydroxy-cyclphexylsulfanyl] -acetyl }-mutilin + (IS, 2S, 4S) diastereomer (325 mg, 0.64 mmol) in 20 ml of dioxane was treated with IN HCl (0.64ml, 0.64 mmol). After stirring at room temperature for 30 minutes the solution was lyophilized to obtain 14-O-{[(1R, 2R, 4R)-4-amino-2-hydroxy-cyclohexylsulfanyl] -acetyl }- mutilin hydrochloride + (IS, 2S, 4S) diastereomer hydrochloride (quantitative yield) as colorless amorphous solid.
1H NMR (500MHz, DMSO-Cl6, δ, ppm, inter alia): 7.6 (bs, 3H, NH3 +), 6.14 (dd, IH, 19-H, J = 1 IHz and 18Hz), 5.55 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.52 (d, IH, H-OH, J = 6Hz), 3.50 – 3.20 (m, 4H, 2′-H, H-H, 22-H), 3.03 (m, IH, 4′-H), 2.53 (m, IH, 1 ‘-H), 2.40 (bs, IH, 4-H), 1.37 (s, 3H, 15-CH3), 1.06 (s, 3H, 18-CH3), 0.82 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 508 (MH+), 530 (MNa+), 1015 (2MH+), 1037 (2MNa+), 542 (MCl“).
Example IA – 14-O-{[(1S, 2S, 4S)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin and 14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsuIfanyl]-acetyl}-mutilin
The mixture of 14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer (12 g, 23.6 mmol) from Example 1 Step B was separated on a cbiral column (250 x 20 mm CHIRALCEL OD-H, n-heptane / ethanol / diethylamine = 80/20/0.1) to yield 14-O-{[(1S*, 2S*, 4S*)-4-amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin (a) (early eluting compound, 4.76 g, 37% yield, uncorrected) and 14-O-{[(1R*, 2R*, 4R*)-4-amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin (b) (late eluting compound, 3.63 g, 30% yield, uncorrected) as colorless amorphous foams.
(a): 1H NMR (400MHz, DMSO-(I6, δ, ppm, inter alia): 6.13 (dd, IH, 19-H, J = 1 IHz and 18Hz), 5.54 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.50 (d, IH, H-OH, J = 6Hz), 3.50 – 3.20 (m, 5H, 2′-H, 4′-H, H-H, 22-H), 2.55 (m, IH, l ‘-H), 2.40 (bs, IH, 4-H), 1.35 (s, 3H, 15- CH3), 1.05 (s, 3H, 18-CH3), 0.82 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 508 (MH+), 530 (MNa+), 1015 (2MH+), 1037 (2MNa+), 506 (M-H) “, 542 (MCl“).
(b): 1H NMR (400MHz, DMSO-d6> δ, ppm, inter alia): 6.13 (dd, IH, 19-H, J = 1 IHz and 18Hz), 5.54 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.50 (d, IH, H-OH, J = 6Hz), 3.50 – 3.20 (m, 5H, 2′-H, 4′-H, 11-H, 22-H), 2.55 (m, IH, 1 ‘-H), 2.40 (bs, IH, 4-H), 1.35 (s, 3H, 15- CH3), 1.05 (s, 3H, 18-CH3), 0.82 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 508 (MH+), 530 (MNa+), 1015 (2MH+), 1037 (2MNa+), 506 (M-H) “, 542 (MCl“).
…………………………………………………….
WO 2011146954
http://www.google.com/patents/WO2011146954A1?cl=en
The present invention relates to crystalline 14-0-{[(4-amino-2-hydroxy-cyclohexyl)- sulfanyl] -acetyl }-mutilin, new processes for its preparation and crystalline salts thereof.
Pleuromutilin, a compound of formula
Pleuromutilin

is a naturally occurring antibiotic, e.g. produced by the basidiomycetes Pleurotus mutilus and P. passeckerianus, see e.g. The Merck Index, 12th edition, item 7694.
A number of further pleuromutilins having the principle ring structure of pleuromutilin and being substituted at the primary hydroxy group have been developed, e.g. as antimicrobials. Due to their pronounced antimicrobial activity, a group of pleuromutilin derivatives, amino- hydroxy-substituted cyclohexylsulfanylacetylmutilins, as disclosed in WO 2008/113089, have been found to be of particular interest. As described in WO2008/11089 14-0-{[(4- Amino-2-hydroxy-cyclohexyl)-sulfanyl] -acetyl }-mutilins are particularly useful compounds because they demonstrate activity against Gram-positive and Gram-negative pathogens e.g. associated with respiratory tract and skin and skin structure infections. For the production of substantially pure isomers/diastereomers of this group of compounds, there is a need for a production process which is convenient for use on an industrial scale and which also avoids the use of costly starting materials, environmentally hazardous reagents and solvents or time consuming and laborious purification steps. The production process described in WO 2008/113089 involves chromatographic purification of the compounds prepared according to individual synthesis steps and the final diastereomers are separated by chiral HPLC chromatography which cannot be used on industrial scale. Surprisingly, crystalline intermediates have been found which on the one hand have unexpected chemical purification potential which is important for the production processes for pure amino-hydroxy-substituted cyclohexylsulfanylacetylmutilins avoiding
chromatographic purification and separation steps.
It has to be pointed out that 14-0-{[(4-amino-2-hydroxy-cyclohexyl)-sulfanyl]-acetyl}- mutilins are potential new drug substances for the human market with regulatory
requirements defined in the corresponding ICH guidelines (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use). The ICH guideline on impurities in new drug substances (Q3 A(R2)) includes the following thresholds:

As can be seen from the ICH thresholds above it is desirable to have all individual unknown impurities below 0.10% area and the structure elucidated impurities below 0.15%, respectively. Processes provided according to the present invention enable to produce APIs (Active Pharmaceutical Ingredients) within the desired specifications and fulfilling ICH requirements.
On the other hand, even more surprisingly, the crystalline intermediates yields to significant chiral enrichment which has a huge benefit in the production of the pure stereoisomers starting from cheaper racemic materials or less chirally pure starting materials. The described processes do not involve any chromatographic purification neither normal nor chiral phase in contrast to the synthetic procedures described in WO2008/113089 wherein is disclosed e.g. in Example 1, Step B that 14-0-{[(4-amino-2-hydroxy-cyclohexyl)-sulfanyl]- acetyl}-mutilins was isolated in the form of diastereomeric mixtures as colorless amorphous foams after normal phase chromatography. The chiral pure diastereomers are described to have been received in WO2008/113089, e.g. in Example 1 A after subjecting the mixture to chiral chromatography whereafter the separated pure diastereomers were isolated in the form of colorless amorphous foams.
Chiral chromatography, however is not a technology which can be applied on industrial large scale, and moreover no crystalline salts of 14-0-{[(4-amino-2-hydroxy-cyclohexyl)- sulfanyl]-acetyl}-mutilins were obtained according to WO2008/113089. In contrast to that, according to the present invention crystalline pharmaceutical acceptable salts of 14-0-{[(4-amino-2-hydroxy-cyclohexyl)-sulfanyl]-acetyl}-mutilins having surprising and superior properties over the amorphic prior art salts disclosed in
WO2008/113089 have been found; e.g. surprisingly the chemical stability of the crystalline salts of the present invention is improved over the amorphic salt forms; and also and in addition the crystalline salts of the present invention show a surprising low hygroscopicity.
Processes for the preparation of such crystalline salts wherein the salts may be obtained in a single stereoisomeric form from 14-0-{[(4-amino-2-hydroxy-cyclohexyl)-sulfanyl]-acetyl}- mutilins and processes for the preparation of stereoisomerically pure 14-0-{[(4-amino-2- hydroxy-cyclohexyl)-sulfanyl]-acetyl}-mutilins in crystalline form as a basis for the crystalline salts have also been found.
In one aspect the present invention provides a process for the preparation of a compound of formula I

in the form of a single stereoisomer in crystalline form, comprising
deprotecting the amine group
either in a compound of formula Ila

in a mixture of a compound of formula Ila with a compound of formula lib

wherein R is an amine protecting group, and isolating a compound of formula I obtained in the form of a single diastereomer in crystalline form either directly from the reaction mixture or via recrystallization in organic solvent.
In another aspect the present invention provides a compound of formula I as defined above in the form of a single stereoisomer in crystalline form.
Compounds of formula Ila are new and also form part of the present invention.
In another aspect the present invention provides a compound of formula Ha.
In a compound of formula I, or Ha, respectively, the carbon atoms of the cyclohexyl ring to which the hydroxy group, the amine group and the sulfanyl-acetyl-mutilin group are attached are all in the R configuration and thus a compound of formula I, or Ila represents an optionally amine protected 14-0-{[(l ?,2i?,4 ?)-4-amino-2-hydroxy-cyclohexylsulfanyl]- acetyl}-mutilin. In contrast to that, in a compound of formula lb

or lib the carbon atoms of the cyclohexyl ring to which the hydroxy group, the amine group and the sulfanyl-acetyl-mutilin group are attached are all in the S configuration and thus a compound of formula lib represents an optionally amino protected 14-0-{[(lS,2S,4S)-4-Amino-2-hydroxy- cyclohexylsulfanyl] -acetyl } -mutilin. An amine protecting group includes protecting groups known to a skilled person and which are removable under acidic, basic, hydrogenating, oxidative or reductive methods, e.g. by hydrogenolysis, treatment with an acid, a base, a hydride, a sulfide. Appropriate amine protecting groups e.g. are described in T. W. Greene, P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley-Interscience, 4th edition, 2007, particularly p. 696-868.
Example 1
tert-Butyl [(lR3Ri4R)-3-hydroxy-4-mercapto-cyclohexyl]-carbamate

3.94 Kg of {(li?,2J?,4i?)-4-[(tert-Butoxycarbonyl)-amino]-2-hydroxy-cyclohexyl}-benzene- carbothioate and 37 L of CH2C12 were charged to a vessel and the mixture obtained was stirred at 15-25°C. 0.39 Kg of 1,4-dithio-DL-threitol (10% wt) was added to the mixture and rinsed through with 2 L of CH2C12. To the mixture obtained 0.84 Kg of hydrazine monohydrate was added. The mixture obtained was stirred at 18 to 22°C for 3 h and the reaction was followed by HPLC. Upon completion of the reaction, 39 L of 1 M phosphoric acid solution was added and the mixture obtained was stirred for a further 15-30 min. Two phases formed were separated and the organic phase obtained was washed with 39 L of of 1 M phosphoric acid solution followed by 39 L 1% aqueous NaCl solution. The organic layer obtained was concentrated in vacuo at <40°C, to the concentration residue 20 L of CH2C12 was added and the mixture obtained again was concentrated. To the concentration residue obtained a further 8 L of CH2C12 was added and the mixture obtained was concentrated to dryness.
2.89 Kg of tert-Butyl [(l ?,3/?,4i?)-3-hydroxy-4-mercapto-cyclohexyl]-carbamate in the form of a white solid was obtained.
1H NMR (200 MHz, DMSO-de, ppm) δ 6.79 (d, J=7.8Hz, 1H), 4.99 (d, J=5.8Hz, 1H), 3.34 – 3.24 (m, 1H), 3.14 – 3.04 (m, 1H), 2.37 (d, J=3.8 Hz, 1H), 2.00 -1.89 (m, 1H), 1.87 – 1.82 (m, 1H), 1.73 – 1.67 (m, 1H), 1.47 – 1.04 (m, 12H)
Example 2
22-0-TosylpIeuromutilin

22-O-Tosylpleuromutilin is a known compound from literature. However a preparation procedure is outlined below.
A solution of 13.0 kg of pleuromutilin and 6.57 kg of 4-toluenesulfonyl chloride in 42.1 L of CH2CI2 at 10 to 15 °C was treated with 9.1 L of 5.7 M aqueous NaOH over 20 min, maintaining a temperature < 25 °C. The resulting off-white suspension was heated to reflux for 20 h and the reaction was followed until completion determined by HPLC. Upon reaction completion the mixture obtained was cooled to 20 to 30 °C, diluted with 52 L of CH2C12, stirred at 15 to 25 °C for 10 min, and the layers obtained were separated. The organic phase obtained was washed several times with 52 L of water until a pH of the aqueous layer was adjusted to < 9. The organic layer obtained was concentrated to 4 volumes and
azeotropically dried twice with 52 L of CH2C12. To the solution obtained 52 L of heptane were added dropwise and the solution obtained was concentrated at < 40 °C to
approximately 4 volumes. To the concentrate obtained 52 L of heptane was added and the resulting suspension was stirred at 20 to 25 °C for 2 to 2.5 h, filtered, the filter cake obtained was washed with 39 L of heptane and pulled dry on the filter.
The solid was dried under vacuum at < 40 °C for at least 12 h.
16.9 kg of 22-O-tosylpleuromutilin in the form of a white solid was obtained.
1H NMR (200 MHz, DMSO-d6, ppm, inter alia) δ 7.81 (d, 2H), 7.47 (d, 2H), 6.14 – 6.0 (m, 1H), 5.54 (d, J=7.8Hz, 1H), 5.08 – 4.99 (m, 2H), 4.70 (AB, J=16.2Hz, 2H), 3.41 (d, J=5.2Hz, 1H), 2.41(s, 4H), 1.04(s, 3H), 0.81 (d, 3H), 0.51 (d, 3H)
Example 3
14-0-{[(l/?,2R,4R)-4-ter/-Butoxycarbonylamino-2-hydroxy-cyclohexyI-sulfanyl]-acetyl}- mutilin

4.75 Kg of Pleuromutilin tosylate (Tos-PLEU) and 44.4 L of MTBE were charged into a vessel and to the mixture obtained 0.31 Kg of benzyl-tri-«-butylammonium chloride was added and rinsed through with 2.4 L of MTBE. To the mixture obtained 20 L of IM aqueous NaOH solution and 2.84 Kg of tert-Butyl [(lif,3i?,4^)-3-hydroxy-4-mercapto-cyclohexyl]- carbamate were added and the mixture obtained was stirred at 17 to 23 °C for 3 h. Upon completion of the reaction (determined by HPLC) two layers formed were separated and the lower aqueous layer was removed. The organic phase obtained was washed with 19 L of IM aqueous NaOH solution, twice with 20 L of 0.1 M phosphoric acid, 20 L of 10% aqueous NaHC03 solution and twice with 20 L of water. The organic liquors obtained were concentrated, the concentrate obtained was taken up in 7.46 Kg of 2-propanol, the mixture obtained was concentrated again and dried in vacuo at <40°C. 6.66 Kg of 14-O- { [( 1 -¾,2i?,4i?)-4-/ert-Butoxycarbonylamino-2-hydroxy-cyclohexyl-sulfanyl]-acetyl } -mutilin in the form of a white foam was obtained.
Ή NMR (200 MHz, DMSO-d6, ppm, inter alia) δ 6.78 (d, J=7.8Hz, 1H), 6.22 – 6.08 (m,lH), 5.55 (d, J=7.8Hz, 1H), 5.13 – 5.02 (m, 2H), 4.95 (d, J=5Hz, 1H), 4.52 (d, J=6Hz, 1H), 3.36 (AB, J=15Hz, 2H), 2.40 (s, broad, 1H), 2.15 – 2.0 (m, 3H), 1.9 – 1.8 (m, 1H), 1.35 (s, 9H), 0.81 (d, J=7Hz, 3H), 0.62 (d, J=6.6Hz, 3H)
MS (ESI, g/mol): m/z 653 [M+2Na] +
Example 4
14-0-{[(lR,2R,4R)-4-Amino-2-hydroxy-cyclohexyIsulfanyI]-acetyl}-mutilin, crystalline Form 2

Step A: 14-O-{[(li?.2i?,4i?)-4-Amino-2Thvdroxy-cyclohexylsulfanvn-acetvU-mutilin in crystalline Form 1
6.6 Kg of 14-O-{[(li?,2if,4/?)-4-tert-Butoxycarbonylamino-2-hydroxy-cyclohexyl-sulfanyl]- acetyl}-mutilin and 13.2 L of isopropanol were charged into a vessel and stirred at 20 to 25°C. 11.20 kg of 85% phosphoric acid was added and the mixture obtained was heated to approximately 50°C for at least 16 h. The mixture obtained was analyzed for reaction completion by HPLC. Upon completion of the reaction the mixture was cooled to 20 to 25°C and 52 L of CH2C12 was added. The mixture obtained was cooled to 0 to 5°C and 51 L of 30% aqueous K2CO3 solution was added over 1 h at <25°C. The mixture obtained was warmed to rt, stirred for 30 min and the pH of the aqueous layer was determined. To the mixture obtained a further 15 L of 30% aqueous K2C03 solution was added at <25°C, the mixture obtained was stirred at 15°C to 25 °C for 30 min and the two phases obtained were separated. The aqueous phase obtained was extracted with 51 L of CH2CI2 and the combined organic phases were washed with 51 L of purified water. The mixture obtained was concentrated to a volume of 25 L, 33.6 Kg of CH2C12 was added and the mixture obtained was concentrated to 25 L. To the concentrate obtained 33.6 Kg of CH2C12 was added and the mixture obtained was concentrated to 10 L. The concentration residue obtained was cooled to 18 to 22°C and 50 L of di-wopropyl ether was added over a period of 1 h. The slurry obtained was stirred at 15 to 25°C for a minimum of 2 h, filtered and the solid obtained was washed with 10 L of di-wopropyl ether and was dried.
3.79 Kg of 14-0-{[(li?,2i?,4if)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin in crystalline Form 1 was obtained.
Step B: 14-O-{r(l-R.2i?,4j?)-4-amino-2-hvdroxy-cvclohexylsulfanyl1-acetv -mutilin. in crystalline Form 2
For further purification 14-O-{[(l ?,2 ?,4i?)-4-Amino-2-hydroxy-cyclohexylsulfanyl]- acetyl}-mutilin from Step A and 18.75 L of n-butanol were heated to 88 to 92°C until complete dissolution and stirred for 30 to 60 min. The mixture obtained was allowed to cool to 40 to 45°C over at least 2 h and further stirred at this temperature for 2 h. The mixture obtained was filtered and the precipitate obtained was washed with 3.75 L of «-butanol followed by 3.75 L of MTBE. That purification procedure was repeated and the resultant product was dried in vacuo at <40°C.
3.27 Kg of crystalline 14-0-{[(li?,2if,4i?)-4-amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin in crystalline Form 2 was obtained in the form of a white solid.
lH NMR (400 MHz, CDC13, ppm, inter alia) δ 6.51 – 6.44 (m, 1H), 5.78 (d, J=8Hz, 1H), 5.38 – 5.20 (m, 2H), 3.48 – 3.40 (m, 1H), 3.36 (d, J=7Hz, 1H), 3.25 (AB, J=15Hz, 2H), 2.92 – 2.82 (m, 1H), 2.6 – 2.5 (m, 1H), 1.45 (s, 3H), 1.20 (s, 3H), 0.88 (d, J=7Hz, 3 H), 0.73 (d, J=8Hz, 3H)
MS (ESI, g/mol): m/z 508 [M+H] +
Example 5
14-0-{[(lR^/f,4R)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin, crystalline

To a solution of 900 g of 14-0-{[(li?,2i?,4i?)-4-tert-butoxycarbonylamino-2-hydroxy- cyclohexyl-sulfanyl]-acetyl}-mutilin in 9 L of CH2C12 at 15 to 25°C was added 1.8 L of TFA at 15 to 25°C and the resulting solution was stirred for 2 h. Following reaction completion the reaction mixture was concentrated under vacuum and the concentration residue obtained was azeo-dried with a total of 9 L of CH2C12. The concentrate obtained was dissolved in 4.5 L of CH2C12, the solution obtained cooled to 0 to 5°C and the pH was adjusted to pH 11 with aqueous 3.6 L 2CO3 (2.5M) solution. The biphasic mixture obtained was warmed to 15 to 20°C and stirred for 5 to 10 minutes. The layers obtained were separated, the aqueous phase obtained was extracted with 1.8 L of CH2C12, the organic phases obtained were combined, washed with 2.3 L of H20, dried over Na2S04 and concentrated to dryness under vacuum at <40°C. Crude 14-0- { [( 1 R,2R,4R)-4- Amino-2-hydroxy-cyclohexyl-sulfanyl]-acetyl } -mutilin was obtained. Yield: 744 g
For further purification the following procedure was applied:
To 744 g of crude 14-O-{[(li?,2i?,4i-)-amino-2-hydroxy-cyclohexyl-sulfanyl]-acetyl}- mutilin was added 2.23 L of THF and the resulting suspension was stirred at 15 to 25°C for 60 min. To the mixture obtained 7.44 L of MTBE was added over 15 to 30 min, the suspension obtained was aged for 60 min and filtered under nitrogen. The collected solids were washed with a total of 3 L of MTBE and pulled dry on the filter under nitrogen for 1.5 h.
626 g of 14-0-{[(li?,2i?,4i?)-4-Amino-2-hydroxy-cyclohexyl-sulfanyl]-acetyl}-mutilin in crystalline Form 1 was obtained.
The Ή NMR pattern confirms the structure of 14-O-{[(li?,2i?,4i?)-4-amino-2-hydroxy- cyclohexylsulfanyl] -acetyl} -mutilin. The NMR pattern for 14-O-{[(l ?,2i?,4/?)-4-amino-2- hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin is described in example 4.
…………………………………………………………
REF
http://www.phase4-partners.com/wp-content/uploads/2013/09/100412.pdf
http://www.glsv-vc.com/downloads/2010-06-02_First%20Patient_PressRelease.pdf
119
Nabriva. Pleuromutilins. Available online: http://www.nabriva.com/programs/pleuromutilins/ (accessed on 7 December 2012).
120
Forest Laboratories. Our pipeline: Solid, and set for further growth. Available online: http://www.frx.com/research/pipeline.aspx (accessed on 13 April 2013).
121
Sader, H.S.; Biedenbach, D.J.; Paukner, S.; Ivezic-Schoenfeld, Z.; Jones, R.N. Antimicrobial activity of the investigational pleuromutilin compound BC-3781 tested against Gram-positive organisms commonly associated with acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 2012,56, 1619–1623, doi:10.1128/AAC.05789-11.
122
Sader, H.S.; Paukner, S.; Ivezic-Schoenfeld, Z.; Biedenbach, D.J.; Schmitz, F.J.; Jones, R.N. Antimicrobial activity of the novel pleuromutilin antibiotic BC-3781 against organisms responsible for community-acquired respiratory tract infections (CARTIs). J. Antimicrob. Chemother. 2012, 67, 1170–1175, doi:10.1093/jac/dks001.
123
Nabriva Therapeutics AG. Study comparing the safety and efficacy of two doses of BC-3781 vs. vancomycin in patients with acute bacterial skin and skin structure infection (ABSSSI). Available online: http://www.clinicaltrials.gov/ct2/show/NCT01119105 (accessed on 13 April 2013).
124
Novak, R. Are pleuromutilin antibiotics finally fit for human use? Ann. NY Acad. Sci. 2011, 1241, 71–81, doi:10.1111/j.1749-6632.2011.06219.x.
 valnemulin
valnemulin
 retapamulin
retapamulin
| WO2002004414A1 * | Jul 9, 2001 | Jan 17, 2002 | Gerd Ascher | Pleuromutilin derivatives having antibacterial activity |
| WO2007000004A1 * | Jun 26, 2006 | Jan 4, 2007 | Nabriva Therapeutics Forschung | Pleuromutilin derivatives containing a hydroxyamino- or acyloxyaminocycloalkyl group |
| WO2007014409A1 * | Jul 26, 2006 | Feb 8, 2007 | Nabriva Therapeutics Forschung | Pleuromutilin derivatives useful as antibacterials |
| WO2008011089A2 | Jul 19, 2007 | Jan 24, 2008 | Mark B Gagner | Wagering game with special-event eligibility feature based on passive game play |
| WO2008113089A1 | Mar 19, 2008 | Sep 25, 2008 | Nabriva Therapeutics Ag | Pleuromutilin derivatives for the treatment of diseases mediated by microbes |
| US20060276503 * | Aug 30, 2004 | Dec 7, 2006 | Glaxo Group Limited | Novel process salts compositions and use |
