
Home » CHINA 2023
Category Archives: CHINA 2023
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Befotertinib
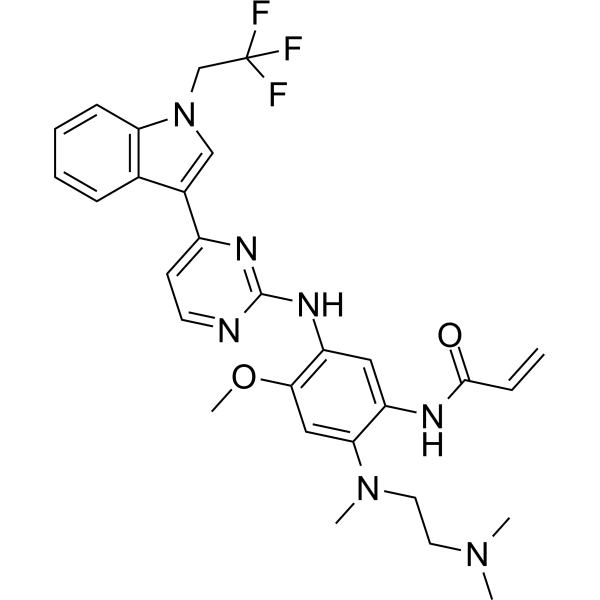
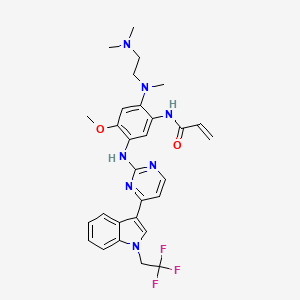
Befotertinib
D-0316, 0XT2CPR891
CAS No. : 1835667-63-4, MESYLATE CAS No. 2226167-02-6
- 2-propenamide, n-(2-((2-(dimethylamino)ethyl)methylamino)-4-methoxy-5-((4-(1-(2,2,2-trifluoroethyl)-1h-indol-3-yl)-2-pyrimidinyl)amino)phenyl)-
- N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1-(2,2,2-trifluoroethyl)-1h-indol-3-yl)pyrimidin-2-yl)amino)phenyl)prop-2-enamide
- N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[1-(2,2,2-trifluoroethyl)indol-3-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide
| Molecular Weight | 567.61 |
|---|---|
| Formula | C29H32F3N7O2 |
Befotertinib (D-0316) is an orally active EGFR tyrosine kinase inhibitor. Befotertinib can inhibit the proliferation of tumor cells. Befotertinib can be used in the research of EGFR T790M-positive non-small cell lung cancer (NSCLC).
Befotertinib is an orally available inhibitor of the epidermal growth factor receptor (EGFR) mutant form T790M, with potential antineoplastic activity. Upon administration, befotertinib specifically binds to and inhibits EGFR T790M, a secondarily acquired resistance mutation, which prevents EGFR-mediated signaling and leads to cell death in EGFR T790M-expressing tumor cells. Compared to some other EGFR inhibitors, befotertinib may have therapeutic benefits in tumors with T790M-mediated drug resistance. EGFR, a receptor tyrosine kinase that is mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization.
PAPER
J. Med. Chem. 2017, 60, 6480−6515.
PATENT
WO 2019218987
https://patentscope.wipo.int/search/en/WO2019218987
[0054]
U.S. Publication No. 2017/0355696 A1 describes a method of preparing Compound 4 and various pharmaceutically acceptable salts thereof. The exemplified synthetic process in U. S. Publication No. 2017/0355696 A1 includes a two-step conversion from the aniline compound, corresponding to Compound 1 of this disclosure, into the bismesylate of Compound 4, which has a low yield.
[0055]
As shown herein, representative methods of preparation of Compound 4, or a pharmaceutically acceptable salt, (or alternatively referred to as synthetic methods) , can provide the desired Compound 4, or a pharmaceutically acceptable salt, in improved yield and high purity and can be adapted for large-scale manufacture.
[0056]
In various embodiments, the present invention provides a novel method of preparing Compound 4, or a pharmaceutically acceptable salt thereof. The method typically includes converting a compound of Formula III, or a salt thereof, into compound 4, typically under an elimination reaction condition:
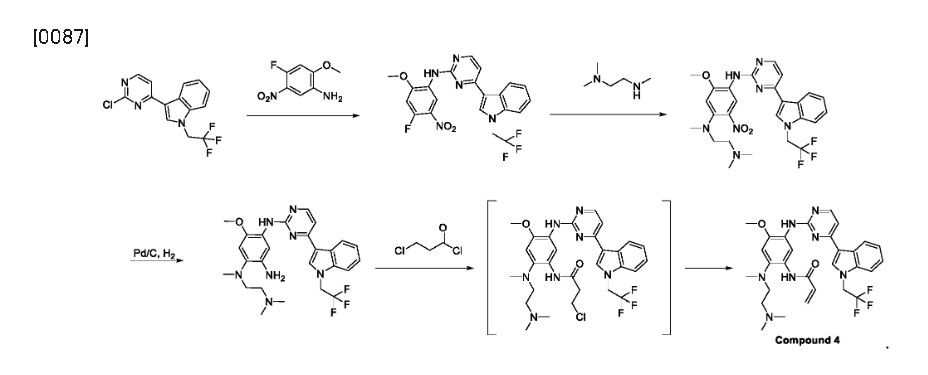
Syn
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Befotertinib (Surmana). Befotertinib (17), an oral, highly selective, third generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) developed by Betta Pharmaceuticals and InventisBio, was approved in China in May 2023 for the second-line treatment of patients
with locally advanced or metastatic nonsmall cell lung cancer (NSCLC) with positive EGFR T790 M mutation who have disease progression on previous EGFR TKI therapy. 140 139 NSCLC
has a high incidence and disease burden in China, which has spurred the development of multiple EGFR TKIs by Chinese companies.
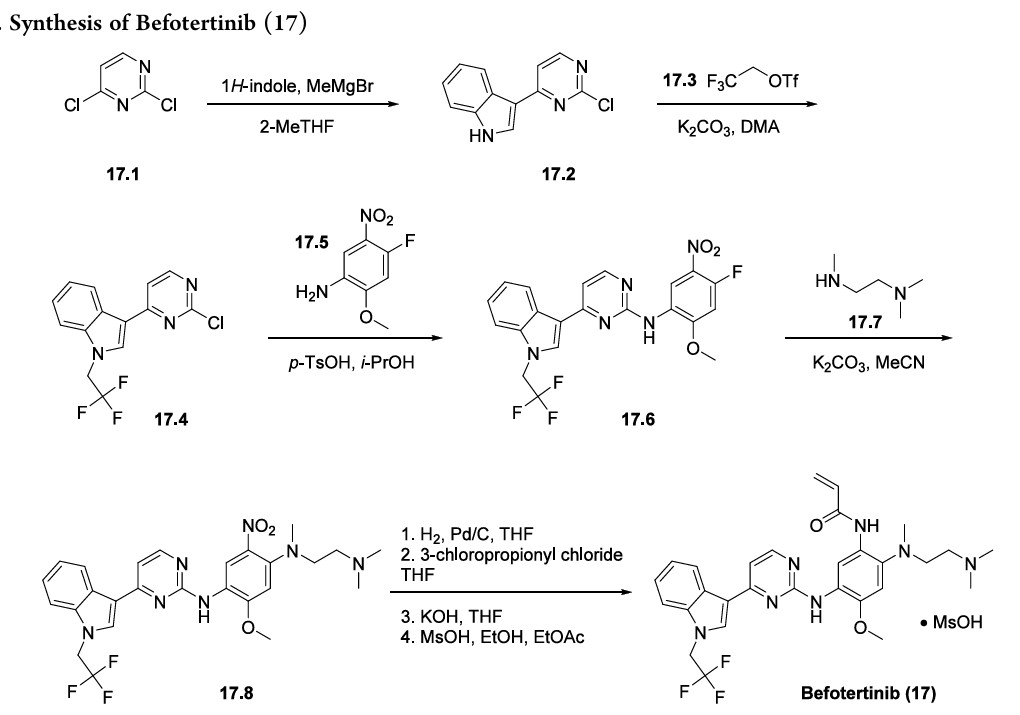
Achromatography-free process route to befotertinib (17) has been reported in the patent literature by researchers at InventisBio (Scheme 29), although details about scale and yields were not provided.
141 142 The reaction sequence closely follows that of osimertinib, a third generation EGFR inhibitor
that was first approved in 2015 and was covered in our previous review.
Osimertinib and befotertinib share a common backbone, differing only in N-substitution on the indole ring.
Friedel−Crafts arylation of 1H-indole with 2,4-dichloropyrimidine (17.1) gave the 3-pyrimidinyl indole 17.2. The trifluoroethyl moiety in indole 17.4 was introduced via Nalkylation of 17.2 with triflate 17.3. This was followed by an SAr reaction with nitroaniline 17.5 to provide amino pyrimidine 17.6. Next, N,N,N′-trimethylethylenediamine (17.7) displaced the electrophilic aryl fluoride in an SNArreaction to generate intermediate 17.8. The acrylamide moiety was installed using a three-step sequence: hydrogenolytic
reduction of the nitro group to the corresponding aniline, acylation with 3-chloropropanoyl chloride, and immediate elimination to the acrylamide. Mesylate salt formation and crystallization furnished befotertinib mesylate (17) in eight steps from 17.1.
(139) Blair, H. A. Befotertinib: first approval. Drugs 2023, 83, 1433−
1437.
(140) Lau, S. C. M.; Ou, S.-H. I. And still they come over troubled
waters: can Asia’s third-generation EGFR tyrosine kinase inhibitors
(Furmonertinib, Aumolertinib, Rezivertinib, Limertinib, Befotertinib,
SH-1028, and Lazertinib) affect global treatment of EGFR+ NSCLC. J.
Thorac. Oncol. 2022, 17, 1144−1154.
(141) Dai, X.; Jiang, Y. Preparation of pyrimidine derivative and its
pharmaceutical salt as EGFR inhibitors for the treatment of cancer and
other diseases. WO 2019218987, 2019.
(142) Flick, A. C.; Ding, H. X.; Leverett, C. A.; Kyne, R. E.; Liu, K. K.
C.; Fink, S. J.; O’Donnell, C. J. Synthetic approaches to the new drugs
approved during 2015. J. Med. Chem. 2017, 60, 6480−6515.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
- [1]. Nagasaka M, et, al. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors For Advanced EGFR+ NSCLC. J Thorac Oncol. 2021 May;16(5):740-763. [Content Brief][2]. Blair HA. Befotertinib: First Approval. Drugs. 2023 Oct;83(15):1433-1437. [Content Brief]
/////////Befotertinib, APPROVALS 2023, CHINA 2023, Betta Pharmaceuticals, InventisBio, CANCER, D-0316, D 0316, 0XT2CPR891
Iruplinalkib
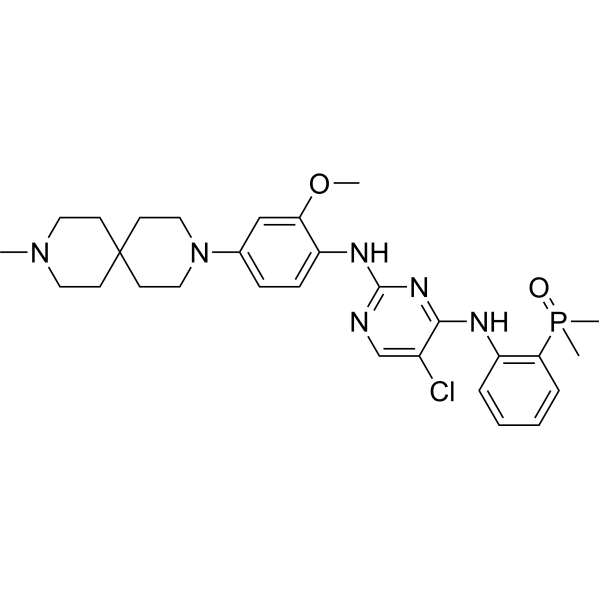
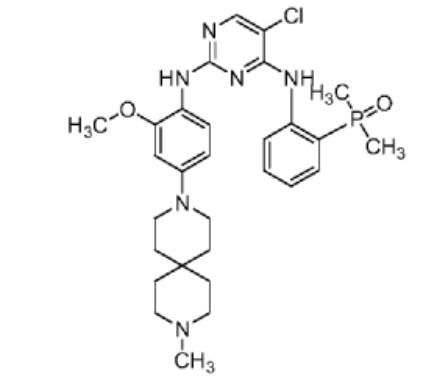
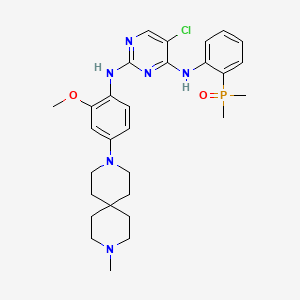
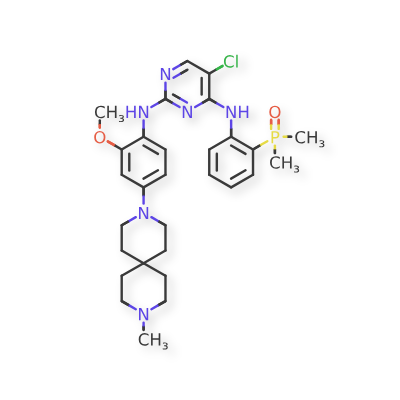
Iruplinalkib
CAS No. : 1854943-32-0
| Molecular Weight | 569.08 |
|---|---|
| Formula | C29H38ClN6O2P |
5-chloro-4-N-(2-dimethylphosphorylphenyl)-2-N-[2-methoxy-4-(9-methyl-3,9-diazaspiro[5.5]undecan-3-yl)phenyl]pyrimidine-2,4-diamine
Iruplinalkib (WX-0593) is an orally active and selective ALK/ROS1 inhibitor. Iruplinalkib can effectively inhibit tyrosine autophosphorylation of ALK and mutant ALK, EGFR, with the IC50 between 5.38 and 16.74 nM. Iruplinalkib is also a suppressive agent of the transporter MATE1, MATE2K, P-gp and BCRP. Iruplinalkib can be used in the study of non-small cell lung cancer.
Iruplinalkib is an orally available, small molecule inhibitor of the receptor tyrosine kinase (RTK) anaplastic lymphoma kinase (ALK), with potential antineoplastic activity. Upon oral administration, iruplinalkib binds to and inhibits ALK tyrosine kinase, ALK fusion proteins, ALK point mutation variants ALK L1196M, ALK C1156Y, and EGFR L858R/T790M. Inhibition of ALK leads to the disruption of ALK-mediated signaling and the inhibition of cell growth in ALK-expressing tumor cells. ALK belongs to the insulin receptor superfamily and plays an important role in nervous system development. ALK is not expressed in healthy adult human tissue but ALK dysregulation and gene rearrangements are associated with a series of tumors. Additionally, ALK mutations are associated with acquired resistance to small molecule tyrosine kinase inhibitors.
SYN
Bioorg. Chem. 2023, 140, No. 106807
Bioorg. Med. Chem. Lett. 2022, 66, No. 128730.
CN106928275, 2017.
EP 3165530 A1, 2017
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US196228122&_cid=P22-ME5G3V-88608-1
Example 9
(2-((5-Chloro-2-((2-methoxy-4-(9-methyl-3,9-diazaspiro[5.5]undecan-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethyl phosphine oxide
This Example was prepared according to the process as described in Example 7, take the place of (2-((5-chloro-2-((2-methoxy-4-(2,6-diazaspiro[3.4]octan-6-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethyl phosphine oxide was replaced with (2-((5-chloro-2-((2-methoxy-4-(3,9-diazaspiro[5.5]undecan-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethyl phosphine oxide to give the title compound as yellow solid, yield 57%. 1H NMR (400 MHz, CD 3OD): δ, 8.28 (s, 1H), 8.12 (br. s., 1H), 7.81-7.68 (m, 3H), 7.65 (d, J=2.0 Hz, 1H), 7.56-7.49 (m, 1H), 7.37 (d, J=8.8 Hz, 1H), 4.02 (s, 3H), 3.74 (br. s., 4H), 3.47 (d, J=12.8 Hz, 2H), 3.24 (t, J=12.8 Hz, 2H), 2.93 (s, 3H), 2.42-1.97 (m, 5H), 1.93-1.75 (m, 9H). LCMS (ESI) (0-60AB): m/z: 569.2 [M+1].
PATENT
CN 110407877, 2019
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN276468167&_cid=P22-ME5FTE-83156-1
| Experimental Example 11 |
| (2-((5-chloro-2-((2-methoxy-4-(9-methyl-3,9-diaza-spiro[5.5]undec-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethylphosphine oxide (Compound I) |
| NMP (N-methylpyrrolidone, 102.6 g) was added to the reaction bottle (room temperature), and 20 g of compound (e) was added to the reaction bottle under stirring. 21.85 g of compound (f) was added to the reaction bottle, and 19.93 g (3 eq) of MeSO 3 H was added dropwise to the reaction flask (temperature was controlled at <40°C) 2 Bubble for 15-20 minutes. Heat the reaction to 85-90°C and react at 85-90°C for 12 hours before sampling and testing (HPLC). Sample testing (test method: dissolve 0.1 ml of the reaction solution in 2 ml of MeOH). Stop the reaction when compound (f) is <2.5%. Add NaOH solution (1M, 287 g) to the reaction solution to adjust the pH to 13. A large amount of solid precipitates and continue stirring for 2 hours. Filter the solid and wash the filter cake with water (40 g) until the filtrate is colorless. Collect the filter cake, filter and dry (55-60°C) to obtain an off-white crude product. Add methanol (268 g) to the reaction flask, then add the obtained solid, heat to 60°C to dissolve, add activated carbon (5.2 g) to the reaction flask, then stir at 60°C for 2.5 hours, cool to 30°C, filter, and concentrate the mother liquor under reduced pressure to obtain a gray-green solid. MeOH (155.5 g) was added to the reaction flask, followed by the solid obtained above. The mixture was heated under reflux until clear. Purified water (388 g) was added to the reaction flask and the temperature was naturally lowered to 15-20°C. A white solid precipitated. The solid was filtered and washed once with purified water (194 g). The filter cake was collected and dried to yield Compound I (32.5 g). 1 H NMR (400MHz, CD3OD): 8.36 (dd, J=8.0, 4.4Hz, 1H), 8.03 (s, 1H), 7.69 (d, J=8.8Hz, 1H), 7.65-7.55 (m, 1H), 7.51 (dd, J=8.0 8.0Hz,1H),7.32-7.20(m,1H),6.66(d,J=2.4Hz,1H),6.45(dd,J=8.8,2.4Hz,1H),3.85(s,3H) ,3.18-3.06(m,4H),2.54-2.38(m,4H),2.30(s,3H),1.85(d,J=13.6Hz,6H),1.74-1.54(m,8H). |
| Example 1: Preparation of Form A of Compound I: |
| 10 g of (2-((5-chloro-2-((2-methoxy-4-(9-methyl-3,9-diaza-spiro[5.5]undec-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethylphosphine oxide (Compound I) was heated to reflux for complete dissolution with 45 ml of ethanol and 20 ml of purified water. The mixture was then cooled to 10-20° C., 50 ml of purified water was added, and the mixture was stirred at this temperature for 2-3 hours. The mixture was filtered and dried in vacuo at 50-60° C. to obtain 9.0 g of an off-white solid with a yield of 90%. X-ray powder diffraction analysis showed an XRPD pattern as shown in FIG1 . DSC-TGA analysis yielded pattern 2. |
Syn
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
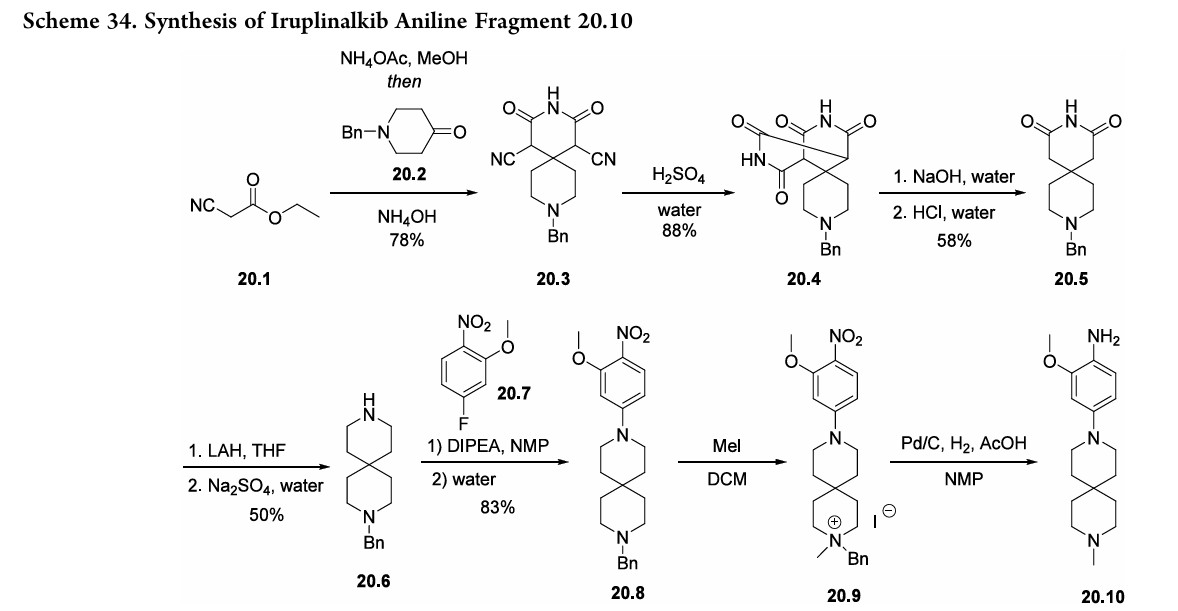
Iruplinalkib. The final NSCLC treatment approved in 2023 was iruplinalkib (20), developed by Qilu Pharmaceutical Co., Ltd. and approved for use in China in June 2023.156,157 Iruplinalkib is a highly selective oral anaplastic kinase (ALK) and ROS1TKI,andis, therefore, an oral treatment for ALK-positive (ALK+) or ROS1-positive (ROS1+) NSCLC.156,157 This treatment is specifically meant for use by patients with locally advanced or metastatic ALK-positive NSCLC whose disease has progressed after crizotinib therapy, often due to the develop ment of crizotinib resistance. Although early syntheses were published 158 further optimized routes were disclosed by Qilu Pharmaceutical Co., Ltd.(Scheme 34 and Scheme 35).First, ethyl 2-cyanoacetate 20.1) and cyclic ketone20.2 were combined to generate spirocyclic imide 20.3 (Scheme 34). The cyano substituents were then removed by treating 20.3 with sulfuric acid followed
by sodiumhydroxide to form 20.5. Subsequent amide reduction via lithium aluminum hydride treatment revealed secondary amine 20.6, which underwent a substitution reaction with fluorophenyl 20.7 to produce intermediate 20.8. Next, addition of methyl iodide generated a quaternary amine (20.9). The
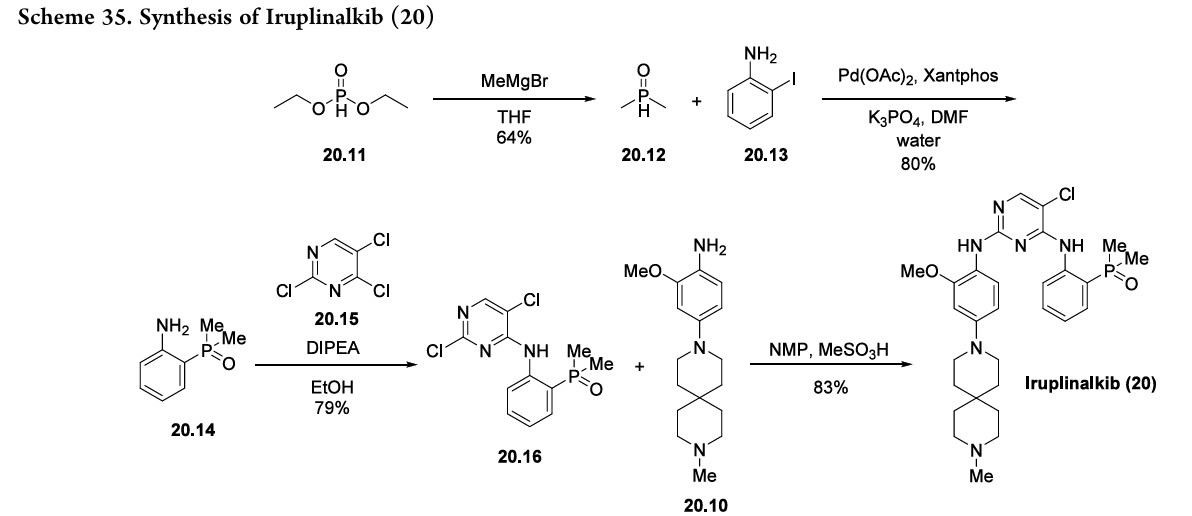
following hydrogenolysis step reduced the nitro group and removed the benzyl substituent using palladium on carbon to produce the final aniline fragment, 20.10. To generate the other fragment (20.16) to construct iruplinalkib, diethylphosphite (20.11) and methyl magnesium bromide were combinedtoproducedimethylphosphineoxide 20.12(Scheme35).Couplingthisproductwith2-iodoaniline
20.13andsubsequent selectiveSNArwithtrichloropyrimidine20.15 generated the resultingdichloropyrimidine20.16.Finally, a selective SNAr reaction yielded the desired product,
iruplinalkib(20).
(156) Yang, Y.; Zheng, Q.; Wang, X.; Zhao, S.; Huang, W.; Jia, L.; Ma,
C.; Liu, S.; Zhang, Y.; Xin, Q.; et al. Iruplinalkib (WX-0593), a novel
ALK/ROS1 inhibitor, overcomes crizotinib resistance in preclinical
modelsfornon-smallcell lung cancer. Invest. New Drugs 2023, 41,254−
266.
(157) Keam, S. J. Iruplinalkib: first approval. Drugs 2023, 83, 1717−
1721.
(158) Liu, X.; Zhang, L.; Wan, H.; Zhu, Z.; Jin, J.; Qin, Y.; Mao, W.;
Yan, K.; Fang, D.; Jiang, W.; et al. Discovery and preclinical evaluations
of WX-0593, a novel ALK inhibitor targeting crizotinib-resistant
mutations. Bioorg. Med. Chem. Lett. 2022, 66, No. 128730.
(159) Ding, Z.; Chen, S.; Liu, X.; Wan, H.; Zhang, L. Preparation,
intermediate and crystal form of spiroamine type arylphosphine oxide.
CN106928275, 2017.
(160) Ding, Z.; Zhang, M.; Chen, S.; Liu, X.; Zhu, Y.; Fan, C.;
Baoping, Z.; Chang, L.; Yang, Y.; Zheng, Q.; et al. Preparation,
intermediate and crystal form of spiroamine type arylphosphine oxide.
EP 3165530 A1, 2017.
(161) Lin, D.; Zhou, G.; Li, S.; Wang, X.; Zhang, Z.; Liu, Z.; Wang, X.
Polymorph of spirocycloaryl phosphorus oxide. CN 110407877, 2019.
(162) Gao, H.; Zhang, J.-Y.; Zhao, L.-J.; Guo, Y.-Y. Synthesis and
clinical application of small-molecule inhibitors and PROTACs of
anaplastic lymphoma kinase. Bioorg. Chem. 2023, 140, No. 106807.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. Yang Y, et al. Iruplinalkib (WX 0593), a novel ALK/ROS1 inhibitor, overcomes crizotinib resistance in preclinical models for non-small cell lung cancer. Invest New Drugs. 2023 Apr;41(2):254-266. [Content Brief][2]. Shi Y, et al. Safety and activity of WX-0593 (Iruplinalkib) in patients with ALK- or ROS1-rearranged advanced non-small cell lung cancer: a phase 1 dose-escalation and dose-expansion trial. Signal Transduct Target Ther. 2022;7(1):25. [Content Brief]
/////////Iruplinalkib, china 2023, approvals 2023, Qilu Pharmaceutical, Z5F65W1YAZ, Qixinke, WX0593, WX 0593, FL006, FL 006, FL-006
Keverprazan
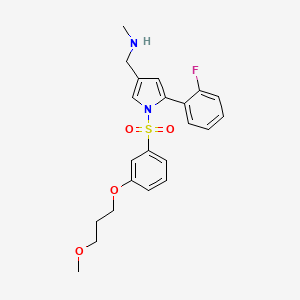
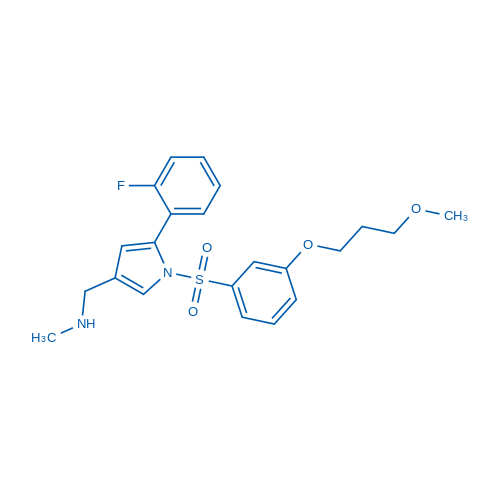
- P-CAB agent 2
- keverprazan
- 1978371-23-1
- Keprason
- SOC12UY3ZP
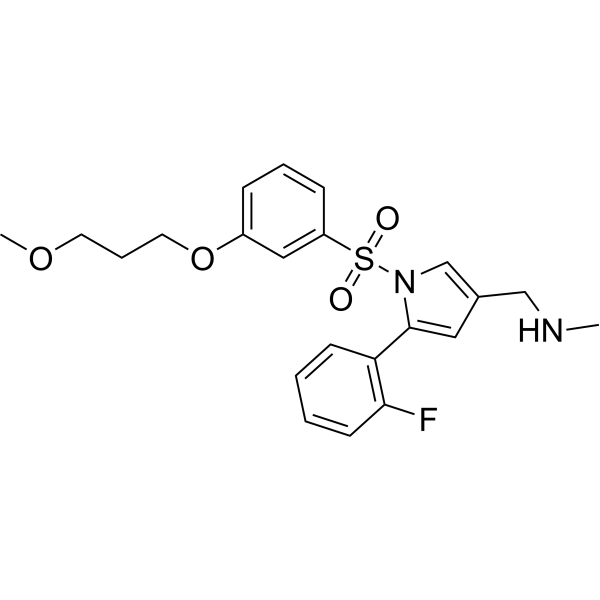
Keverprazan
432.5 g/mol
1-[5-(2-fluorophenyl)-1-[3-(3-methoxypropoxy)phenyl]sulfonylpyrrol-3-yl]-N-methylmethanamine
C22H25FN2O4S
- 1H-Pyrrole-3-methanamine, 5-(2-fluorophenyl)-1-[[3-(3-methoxypropoxy)phenyl]sulfonyl]-N-methyl-
- 5-(2-Fluorophenyl)-1-[[3-(3-methoxypropoxy)phenyl]sulfonyl]-N-methyl-1H-pyrrole-3-methanamine
- Keverprazan Hydrochloride: First ApprovalPublication Name: DrugsPublication Date: 2023-04-19PMID: 37074491DOI: 10.1007/s40265-023-01865-w
- Efficacy of keverprazan for duodenal ulcer: A phase II randomized, double‐blind, parallel‐controlled trialPublication Name: Journal of Gastroenterology and HepatologyPublication Date: 2022-09-16PMID: 36068945DOI: 10.1111/jgh.16000
- The efficacy and safety of keverprazan, a novel potassium‐competitive acid blocker, in treating erosive oesophagitis: a phase <scp>III</scp>, randomised, double‐blind multicentre studyPublication Name: Alimentary Pharmacology & TherapeuticsPublication Date: 2022-05-03PMID: 35505467DOI: 10.1111/apt.16959
- Potassium-competitive acid blockers – are they the next generation of proton pump inhibitors?Publication Name: World Journal of Gastrointestinal Pharmacology and TherapeuticsPublication Date: 2018-12-13PMCID: PMC6305499PMID: 30595950DOI: 10.4292/wjgpt.v9.i7.63
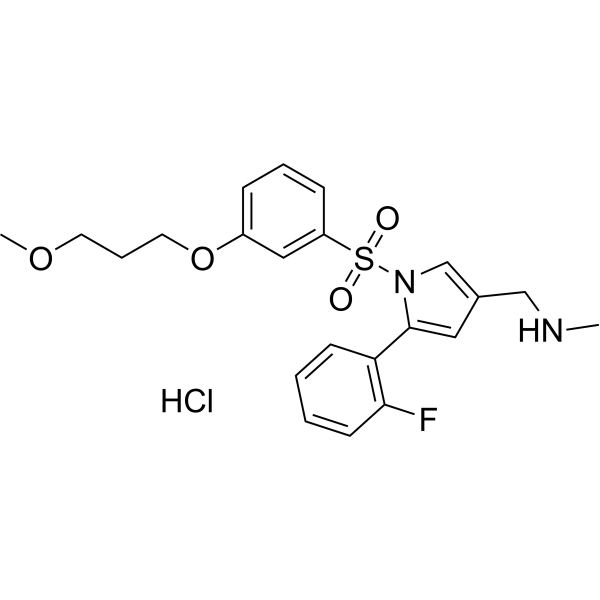
CAS 2209911-80-6
| Molecular Weight | 468.97 |
|---|---|
| Formula | C22H26ClFN2O4S |
Jiangsu Carephar Pharmaceuticals, is
a potassium ion competitive acidblocker (P-CAB) that was approved inFebruary2023inChina for thetreatment of refluxesophagitis or duodenal ulcer inadults
P-CAB agent 2 hydrochloride is a potent and orally active potassium-competitive acid blocker and a gastric acid secretion inhibitor. P-CAB agent 2 hydrochloride inhibits H+/K+-ATPase activity with an IC50 value of <100 nM. P-CAB agent 2 hydrochloride inhibits the hERG potassium channel with an IC50 value of 18.69 M. P-CAB agent 2 hydrochloride shows no acute toxicity and inhibits histamine (HY-B1204)-induced gastric acid secretion.
P-CAB agent 2 is a potent and orally active potassium-competitive acid blocker and a gastric acid secretion inhibitor. P-CAB agent 2 inhibits H+/K+-ATPase activity with an IC50 value of <100 nM. P-CAB agent 2 inhibits the hERG potassium channel with an IC50 value of 18.69 M. P-CAB agent 2 shows no acute toxicity and inhibits histamine (HY-B1204)-induced gastric acid secretion[1].
REF
https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c02079?ref=PDF
KeverprazanHydrochloride. Keverprazan hydrochloride(4),developedby Jiangsu CarepharPharmaceuticals,is
apotassiumioncompetitiveacidblocker (P-CAB) thatwas approvedinFebruary2023inChina forthetreatmentofrefluxesophagitisorduodenal ulcer inadults.36Those illnesses arecaused by gastric acid entering the esophagus, leading toregurgitation and heartburn.37 Current treatments mainly
employ acid-suppressing therapies such as proton pumpinhibitors (PPIs, e.g., lansoprazole); however, PPIs do notacidicenvironments.38Toaddress those issues,P-CABshavebeen developed as a new class of acid suppressants.39
Keverprazan, a novel P-CAB, reversibly binds to gastricH+,K+-ATPase in competition against K+ ions, therebyinhibitingtheenzyme.40Itprovidesstableandsustainedgastricacidinhibitioneffectat20mgdose,whilebeingsafeandwelltoleratedupto60mginasingle-ascendingdosestudy.41Theoriginal synthesisof keverprazanhydrochloridedevelopedbyQinetal. isillustratedinScheme7.42TheroutebeganwithSN2alkylationof3-nitrophenol4.1withalkylbromide4.2
toformphenylether4.3.Thenitrogroupwasreducedtoaniline 4.4underRaney-Ni-mediatedhydrogenation.Aniline4.4wasconverted to sulfonyl chloride 4.5 via copper-mediated
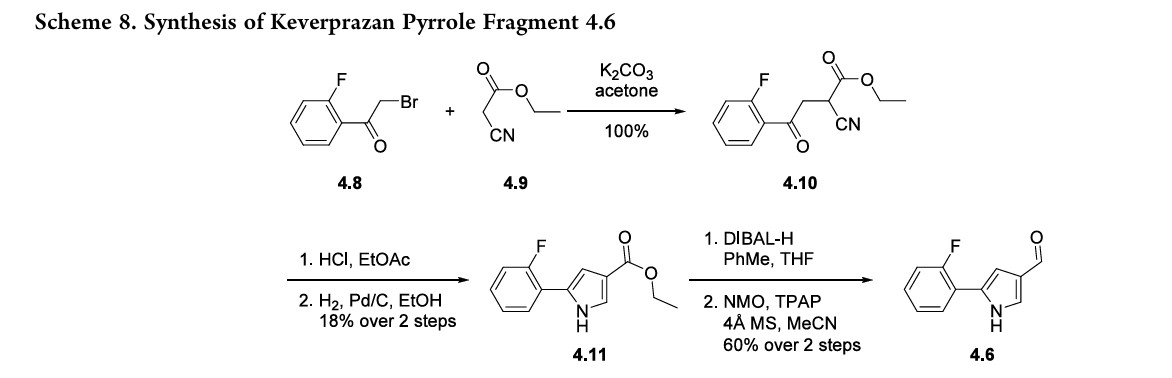
sulfonylationin55%yield.Thesulfonyl chloridewascoupledwithpyrrolederivative4.6toformsulfonamide4.7in60%yield.
Finally, reductiveaminationofthealdehydewithmethylamine and sodium cyanoborohydride, followed by HCl salt formation,43furnishedkeverprazanhydrochloride(4)in18%yield.
The synthesis of pyrrole fragment 4.6was reported byArikawaetal.andillustratedinScheme8.39SN2displacementofα-bromoketone 4.8 with ethyl cyanoacetate 4.9 affordedintermediate 4.10. Treatment of 4.10 with anhydrous HCl in EtOAc followed by hydrogenation effected cyclization to form pyrazole 4.11. The ester was reduced to the corresponding
alcohol with DIBAL-H,andthealcoholoxidized to aldehyde4.6 with NMO and TPAP.
(36) Kang, C.Keverprazan Hydrochloride: first approval. Drugs 2023,
83, 639−643.
(37) Chen, S.; Liu, D.; Chen, H.; Liao, A.; Li, F.; Liu, C.; Li, X.; Li, S.;
Zhang, Y.; Wang, Y.; et al. The efficacy and safety of keverprazan, a
novel potassium-competitive acid blocker, in treating erosive
oesophagitis: a phase III, randomised, double-blind multicentre
study. Aliment. Pharmacol. Ther. 2022, 55, 1524−1533.
(38)Tan,N.-d.; Liu, X.-w.; Liu, C.-x.; Li, S.-b.; Chen, H.-h.; Li, X.; Wu,
H.; Liao, A.-J.; Zhen, Y.-b.; Shen, P.-z.; et al. Efficacy of keverprazan for
duodenal ulcer: a phase II randomized, double-blind, parallel
controlled trial. J. Gastroenterol. Hepatol. 2022, 37, 2060−2066.
(39) Arikawa, Y.; Nishida, H.; Kurasawa, O.; Hasuoka, A.; Hirase, K.;
Inatomi, N.; Hori, Y.; Matsukawa, J.; Imanishi, A.; Kondo, M.; et al.
Discovery of a novel pyrrole derivative 1-[5-(2-fluorophenyl)-1
(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine fuma
rate (TAK-438) as a potassium-competitive acid blocker (P-CAB). J.
Med. Chem. 2012, 55, 4446−4456.
(40) Parsons, M. E.; Keeling, D. J. Novel approaches to the
pharmacological blockade of gastric acid secretion. Expert Opin. Invest.
Drugs 2005, 14, 411−421.
(41) Zhou,S.;Xie, L.; Zhou, C.; Zhao, Y.; Wang,L.;Ding,S.;Chen, J.;
Zhu, B.; Su, M.; Shao, F. Keverprazan, a novel potassium-competitive
acid blocker: single ascending dose safety, tolerability, pharmacoki
netics, pharmacodynamics and food effect in healthy subjects. Eur. J.
Pharm. Sci. 2023, 190, No. 106578.
(42) Qin, Y.; Su, M.; Jin, Q.; Chen, T.; Jiang, J. Pyrrole sulfonyl
derivative, and preparation method and medical use thereof. EP
3248963 B1, 2019.
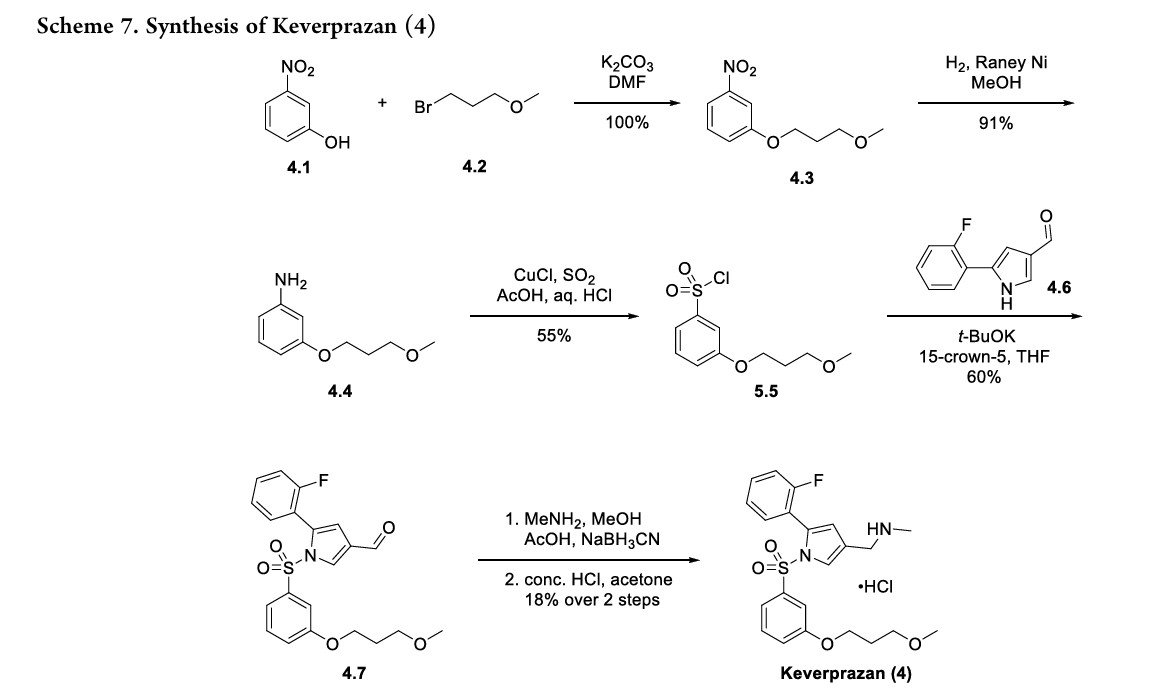
SYN
WO2016119505
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016119505&_cid=P12-MD5D4X-02931-1
[0042]Preparation of 1-(5-(2-fluorophenyl)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrol-3-yl)-N-methylmethanamine (EXP 1)
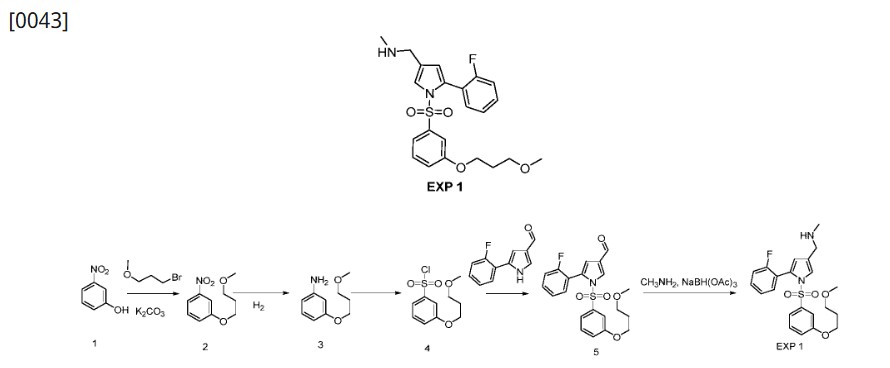
1) Preparation of 1-(3-methoxypropoxy)-nitrobenzene (Compound 2)
[0045]3-Nitrophenol (Compound 1, 1.0 g, 7.19 mmol), potassium carbonate (2.9 g, 21.6 mmol) and 1-bromo-3-methoxypropane (1.65 g, 10.79) were dissolved in anhydrous DMF (20 mL), stirred at 90 °C overnight, added with water (50 mL), and extracted with ethyl acetate (50 mL x 3). The organic phases were combined, dried, and concentrated to give a yellow solid 1-(3-methoxypropoxy)-nitrobenzene (Compound 2, 1.5 g, yield 100%).
[0046]2) Preparation of 1-(3-methoxypropoxy)-aniline (Compound 3)
[0047]1-(3-methoxypropoxy)-nitrobenzene (Compound 2, 1.0 g, 4.74 mmol) and Raney Ni (100 mg) were dissolved in anhydrous methanol (20 mL) and stirred overnight at room temperature under a hydrogen atmosphere. The mixture was filtered and the filtrate was dried to obtain solid 1-(3-methoxypropoxy)-aniline (Compound 3, 0.80 g, 91% yield).
[0048]3) Preparation of 1-(3-methoxypropoxy)-benzenesulfonyl chloride (Compound 4)
[0049]At 0°C, sodium nitrite (571 mg, 8.29 mmol) was added to acetic acid (10 mL) and aqueous hydrochloric acid solution (2N, 10 mL) of 1-(3-methoxypropoxy)-aniline (compound 3, 1.0 g, 5.52 mmol) in batches. After the addition was completed, the mixture was stirred at 0°C for 25 min to obtain solution I. At 0°C, cuprous chloride (190 mg, 1.1 mmol) was added to an acetic acid solution of sulfur dioxide (10 mL, 2N) to obtain solution II. At 0°C, solution I was added dropwise to solution II, and after the addition was completed, the mixture was naturally warmed to room temperature and stirred for 3 hours. The mixture was extracted with ethyl acetate (150 mL x 3), the organic phases were combined, dried, concentrated, and column chromatography (petroleum ether: ethyl acetate = 20:1) was performed to obtain 1-(3-methoxypropoxy)-benzenesulfonyl chloride (compound 4, 800 mg, yield 55%) as a yellow oil.
[0050]4) Preparation of 5-(2-fluorophenyl)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrole-3-carbaldehyde (Compound 5)
[0051]At 0°C, t-BuOK (233 mg, 2.08 mmol) was added to a solution of 5-(2-fluorophenyl)-1H-pyrrole-3-carboxaldehyde (200 mg, 1.04 mmol) in anhydrous THF (5 mL), and the mixture was stirred at 0°C for 30 min. After the reaction was completed, 15-crown-5 (542 mg, 2.08 mmol) and 1-(3-methoxypropoxy)-benzenesulfonyl chloride (compound 4, 412 mg, 2.08 mmol) were added respectively. After the addition was completed, the mixture was naturally heated to room temperature and stirred for 90 min. After the reaction was completed, the mixture was quenched with ice water (50 g) and extracted with ethyl acetate (50 mL x 3). The organic phases were combined, dried, concentrated, and purified by column chromatography (petroleum ether:ethyl acetate=8:1) to give a yellow solid 5-(2-fluorobenzene)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrole-3-carbaldehyde (260 mg, yield 60%).
[0052]5) Preparation of 1-(5-(2-fluorophenyl)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrol-3-yl)-N-methylmethanamine (EXP 1)
[0053]5-(2-Fluorobenzene)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrole-3-carbaldehyde (Compound 5, 500 mg, 1.19 mmol), acetic acid (144 mg, 2.39 mmol), and methylamine alcohol solution (1 mL) were dissolved in 3 mL of anhydrous methanol and stirred at room temperature for 4 h. NaBH
3 CN (212 mg, 3.59 mmol) was added. The mixture was stirred for 60 min. Ice water (30 g) was added to quench the reaction and the mixture was extracted with ethyl acetate (50 mL x 3). The organic phases were combined, dried, concentrated, and subjected to column chromatography to obtain 1-(5-(2-fluorobenzene)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrole-3-yl)-N-methylmethanamine (EXP 1, 100 mg, 20%) as a white solid.
[0054]
HPLC:99.4%;MS(ESI)m/z:[M+H] +=433.0; 1H-NMR(400MHz,DMSO-d6)δ:8.72(s,1H),7.78(d,1H),7.46-7.55(m,2H),7.21-7.32(m,3H),6.85-7.11(m,2H),6.83-6.85(m,1H),6.44(d,1H),3.95-4.02(m,4H),3.47(t,2H),3.32(s,3H),2.52(m,3H),1.94(t,2H)ppm。
[References]
[1] YinLin Qin, et al. Pyrrole sulfonyl derivative, and preparation method and medical use thereof. WO2016119505A1.
///////////KEVERPRAZAN, Keverprazan, CHINA 2023, APPROVAL 2023, Jiangsu Carephar Pharmaceuticals, ULCER
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
DIMDAZENIL


DIMDAZENIL
CAS 308239-86-3
WeightAverage: 372.81
Monoisotopic: 372.1101515
Chemical FormulaC17H17ClN6O2
EVT-201, 308239-86-3, EVT201, 6J8AF7CLE4, EVT 201
7-Chloro-3-[5-[(dimethylamino)methyl]-1,2,4-oxadiazol-3-yl]-4,5-dihydro-5-methyl-6H-imidazo[1,5-a]
[1,4]benzodiazepin-6-one
7-chloro-3-[5-[(dimethylamino)methyl]-1,2,4-oxadiazol-3-yl]-5-methyl-4H-imidazo[1,5-a][1,4]benzodiazepin-6-one
EVT 201 is a novel partial positive allosteric modulator of the GABAA receptor complex which is being developed as a treatment for insomnia. It is being developed by Evotec Inc.
- OriginatorRoche
- DeveloperEvotec SE; Zhejiang Jingxin Pharmaceutical
- ClassBenzodiazepines; Chlorobenzenes; Dimethylamines; Imidazoles; Ketones; Oxadiazoles; Sleep disorder therapies; Small molecules
- Mechanism of ActionGABA A receptor modulators
- RegisteredInsomnia
- 29 Nov 2023Registered for Insomnia in China (PO) – First global approval
- 24 Oct 2023Efficacy and adverse events data from a phase III trial in Insomnia released by Zhejiang Jingxin Pharmaceutical
- 21 Oct 2023Efficacy and adverse events data from a phase II trial in Insomnia released by Zhejiang Jingxin Pharmaceutical
Dimdazenil, sold under the brand name Junoenil, is a medication used in the treatment of insomnia in China.[1] It is a benzodiazepine derivative and a partial positive allosteric modulator of the GABAA receptor[2] with two- to four-fold higher functional affinity for the α1 subunit relative to the α2, α3, and α5 subunits.
Medical use
Dimdazenil shows effectiveness in the treatment of insomnia, but has less intrinsic activity in comparison to currently-marketed benzodiazepines and the Z-drugs;[3] however, it is thought that the lower efficacy may result in fewer side effects, such as motor incoordination.[3] In China, dimdazenil is approved for short-term treatment of insomnia.[4]
History
Dimdazenil was originally developed by Roche, based on preclinical data, as a non-sedating anxiolytic, but was found to produce sedation in humans in phase I clinical trials. For this reason, it was subsequently licensed to Evotec, which is now developing it for the treatment of insomnia.[3] By 2007, dimdazenil completed phase II clinical trials for this indication, with positive findings reported.[5] In China, the drug was developed by Zhejiang Jingxin Pharmaceutical.
SCHEME

PATENT
CN111620834
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN306317338&_cid=P10-MAWAJX-84923-1
| Example 16 |
| |
| 1M lithium bis(trimethylsilyl)amide (320 mL, 0.32 mol, 3 eq, 1 Mol/liter) was added to the flask, nitrogen was passed through, the temperature was lowered to -15°C, and the compound K1 (22.6 g, 0.11 mol, 1 eq) obtained in Example 11 was added dropwise. After the addition, the mixture was kept warm at -15°C to -5°C for 2 hours. After the addition, the compound b (26 g, 0.11 mol, 1 eq) obtained by the method of Example 15 was added dropwise. The mixture was kept warm at -15°C to -5°C for 2 hours. After the addition, the mixture was naturally heated to room temperature, and glacial acetic acid was slowly added dropwise. The temperature was controlled to be below 35°C. After completion, the temperature was raised to 55-60°C, and the reaction was kept warm for 2 hours. Then, the mixture was transferred to a rotary evaporator, and the mixture was concentrated under reduced pressure at 45-50°C in batches. The temperature was lowered to 25-30°C, and water and dichloromethane were added in batches. The layers were stirred and separated, and the organic layer was collected. The aqueous layer was extracted once more with dichloromethane, and the organic layers were combined. The layers were washed with a saturated aqueous solution of sodium bicarbonate and water. After washing, the organic layers were collected and transferred to a rotary evaporator for concentration to obtain a solid. The solid was slurried with ethanol at -15°C to -5°C for 15 minutes, filtered, rinsed with cold ethanol, and dried under reduced pressure at 55-60°C to obtain a compound of formula I (36 g, 96.6%), MS: M ++ 1=373.1, HPLC purity 99.85%. |
| 1 H-NMR data: 1 H NMR (400 MHz, DMSO-d 6 δ8.57(s,1H),7.69(d,J=1.9Hz,3H),4.60(d,J=3.7Hz,2H),3.61(s,2H),3.05(s,3H),2.16(s,6H). |
| 13 C-NMR data: 13 C NMR (101 MHz, DMSO) δ 163.35, 163.25, 161.50, 138.88, 134.17, 133.15, 132.81, 130.95, 128.29, 122.67, 114.56, 110.52, 61.10, 46.6 (2), 41.77, 34.48. |
PATENT
WO2000069858
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2000069858&_cid=P10-MAWAOS-90001-1


EXAMPLE
a) 6-Chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5(lH)-dione (III).
25.0 g 6-chloro-isatoic anhydride (II) and 12.4 g sarcosine were suspended under stirring and argon atmosphere in 100 ml p-xylene and heated at reflux for two hours. The suspension was cooled to room temperature and further stirred 1 hour, then filtered off. The precipitate was washed with 25 ml p-xylene twice and dried at 50°C under vacuum. The solid so obtained (6-chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5( lH)-dione (II)) was digested in 75 ml deionized water at 0°C for 1 hour, filtered off, washed with 25 ml deionized water and dried under vacuum 18 hours at 80°C. Crude product: 25.2 g as a beige powder, m.p. 230-232°C
b) Ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[ l,5-a] [ 1,4] benzodiazepine- 3-carboxylate (V).
25.0 g 6-Chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5( lH)-dione (III) were suspended under stirring and argon atmosphere in 200 ml toluene and 32.1 ml N,N-dimethyl-p-toluidine. The suspension was heated to 100°C and 11.2 ml phosphorus oxychloride were added over 30 minutes and stirring was pursued two and an half hours at 100°C. The dark-orange solution was cooled to 40°C and toluene was removed under reduced pressure to give 82 g of a dark-orange oil.
Meanwhile, 81.2 ml hexamethyldisilazane and 265 ml tetrahydrofuran were mixed and cooled to -35°C. 229.5 ml Butyllithium were added over 45 minutes and, after stirring 30 minutes at -35°C, a solution of 35.2 g ethyl(dimethylamino-methylenamino)acetate in 70.4 ml tetrahydrofuran was added over 30 minutes. The orange solution obtained was stirred one more hour at -35°C and a solution of the crude iminochloride in 100 ml
tetrahydrofuran was added over 1 hour at -15°C. The dark red solution was stirred one hour at -15°C, then 18 hours at room temperature (r.t.). 75 ml Acetic acid were added in 10 minutes, then 75 ml deionized water were added in one portion and the orange suspension was heated at reflux for two hours. Tetrahydrofuran was removed under reduced pressure and the residue was partitioned between 200 ml dichloromethane and 100 ml deionized water. The phases were separated and the organic phase was washed with 100 ml aqueous HC1 IN twice and with 100 ml deionized water. The aqueous phases were extracted twice with 100 ml dichloromethane. The combined organic extracts were dried (Na2S04) and evaporated. The residue was digested in 200 ml n-heptane 30 minutes at r.t. and filtered off. The sticky crystals obtained were digested at reflux for 30 minutes in 213.5 ml ethanol, then stirred 3 hours to r.t. and 2 hours at -20°C. The precipitate (ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3-carboxylate (V)) was filtered off, washed three times with 20 ml ethanol and dried under reduced pressure 16 hours at 60°C. Crude product: 23.4 g as a beige powder, m.p. 225.5-226.5 °C c) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo [ 1 ,5-a] [ 1 ,4]benzodiazepine-3- carboxamide (VI).
22.8 g Ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [l,4]- benzodiazepine-3-carboxylate (V)were suspended under stirring and argon atmosphere in 91.2 ml 1 ,4-dioxane. 14.1 ml Formamide and 13.9 ml sodium methanolate were successively added to yield a clear light-orange solution, which turned to a white suspension after 10 minutes. This suspension was stirred two hours at 30°C. 200 ml deionized water were added in one portion and 1,4-dioxane was distilled off at 40°C under reduced pressure. The remaining white suspension was stirred two hours at 0°C and filtered. The precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[ l,5-a] [ l,4]benzodiazepine-3-carboxamide (VI)) was washed with 50 ml deionized water three times and dried under reduced pressure for 18 hours at 80°C. Crude product: 19.43 g as a white powder. m.p.>250°C
d) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5-a] [l,4]benzodiazepine-3- carbonitrile (VII).
19.0 g 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3- carboxamide (VI) were suspended under stirring and argon atmosphere in 95 ml 1,4- dioxane and 6.58 phosphorous oxychloride were added in one portion. The reaction mixture was heated to reflux for one hour giving a yellow solution, which was concentrated at 50°C under reduced pressure. The residue was digested in 100 ml deionized water for two hours at r.t.. The precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5- a] [ l ,4]benzodiazepine-3-carbonitrile (VII)) was filtered off, washed three times with 30 ml deionized water and dried under vacuum at 80°C for 18 hours. Crude product: 17.3 g as a light yellow powder, m.p. 238.5-239.5°C
_ e) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5-a] [l,4]benzodiazepine-3- carboxamidoxime (VIII).
16.8 g 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3- carbonitrile (VIII) were suspended under stirring and argon atmosphere in 101 ml N,N- dimethylformamide and 13.48 g hydroxylamine hydrochloride was added in one portion. 34.2 ml Sodium methanolate were then added over 60 minutes to the yellow suspension, which turned to a colorless suspension. It was stirred one more hour at r.t., then cooled to 0-2°C and 202 ml deionized water were added over 30 minutes. After stirring one more hour at 0°C, the precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[l,5-a] [l,4]benzodiazepine-3-carboxamidoxime (VIII) was filtered off, washed twice with 40 ml deionized water and dried under vacuum at 70°C for 18 hours Crude product 17.84 g as a white powder m.p.>250°C
f) 7-Chloro-3- (5-chloromethyl- [ 1 ,2,4] oxadiazol-3-yl)-5-methyl-4,5-dihydro- imidazo [ 1 ,5-a] [ 1 ,4] benzodiazepin-6-one (IX).
8.0 g 7-chloro-5,6-dιhydro-5-methyl-6-oxo-4H-ιmιdazo[ 1,5-a] [ l,4]benzodιazepιne-3-carboxamidoxime (VIII) and 1.0 g magnesium oxide were suspended under stirring and argon atmosphere in 160 ml 1,4-dioxane. 2 7 ml Chloracetyl chloride were added in one portion and the white thick gel obtained was stirred 4 hours at r.t. and then 17 hours at reflux to give a lightly orange fluid suspension 100 ml Dioxane were distilled off and the reaction mixture was cooled to room temperature. 180 ml Deionized water were added within 15 minutes and the suspension was stirred 1 hour at r.t . The precipitate was filtered off, washed with 50 ml deionized water twice and dried under vacuum at 80°C for 18 hours Crude product: 8.3 g as a light pink powder. This crude product was dissolved in 120 ml tetrahydrofuran at reflux and 0.83 g active charcoal Darco G 60 were added. The system was refluxed 1 hour, then filtered on 25 g Dicaht-Speedex and the filter cake was washed with three portions of 50 ml warm tetrahydrofuran. The filtrate was concentrated at 40°C under reduced pressure The residue was digested in 80 ml ethanol 1 hour at reflux, then stirred 16 hours at r.t. and finally 2 hours at 2°C. The precipitate (7-chloro-3-(5-chloromethyl- [ l,2,4]oxadιazol-3-yl)-5-methyl-4,5-dιhydro-ιmιdazo [ 1,5-a] [ l,4]benzo-dιazepιn-6-one (IX)) was filtered off, washed with 2 portions of 25 ml cold tert-butyl ethvl- ether and dried under vacuum 5 hours at 80°C Crude product: 7.6 g as a light beige powder, m p. 234-238°C
g) 7-Chloro-3-(5-dimethylaminomethyl-[l,2,4]oxadiazol-3-yl)-5-methyl-4,5- dιhydro-imidazo[l,5-a] [l,4]benzodιazepin-6-one (I).
7.0 g 7-Chloro-3-(5-chloromethyl- [ l,2,4]oxadιazol-3-yl)-5-methyl-4,5-dιhydro-ιmιdazo-[ 1,5-a] [ l,4]benzodιazepιn-6-one (IX) were suspended under stirring and argon
atmosphere in 70 ml 1,4-dioxane and 25.7 ml dimethylamine (33% in ethanol) were added over 60 minutes The reaction mixture was stirred one more hour at r.t. and then the solvents were removed under reduced pressure at 35°C. The residue was partitioned between 50 ml dichloromethane and 20 ml deionized water. The phases were separated and the organic phase was washed twice with 20 ml deionized water. The aqueous phases were extracted separately with the same portion of 25 ml dichloromethane, twice. The combined organic extracts were dried (Na2SO4) and the solvent was removed under reduced pressure Crude product: 8.0 g as a light yellow foam Purification
The crude product was dissolved in 40 ml ethanol at reflux and 400 mg active charcoal Darco G 60 were added. The system was stirred 1 hour at reflux, then filtered on a hot pad of Dicalit Speedex, which was washed with two portions of 40 ml hot ethanol. The filtrate was concentrated to 14 g under reduced pressure, heated to reflux and at this temperature and 40 ml terf-butyl-methylether were added over 5 minutes. The suspension was cooled slowly to r.t., stirred 16 hours, further cooled to 2°C. After stirring 1 hour at 2°C, the precipitate was filtered off, washed with 20 ml tert-butyl-methylether and dried 1 hour at 60°C under vacuum. The so obtained powder was dissolved at reflux in 26 ml ethyl acetate. 6.5 ml Ethyl acetate were then distilled off and the turbid solution obtained was slowly cooled to r.t., then to 0°C. After 1 hour stirring at 0°C, the precipitate was filtered off, washed with 10 ml cold tert-butyl-methylether and dried under vacuum at 60°C for 16 hours. The so obtained powder (7-chloro-3-(5-dimethylaminomethyl-[ 1,2,4] oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[ 1,5-a] [l,4]benzodiazepin-6-one (I)) was crystallized a second time in 24.3 ml ethyl acetate according to the procedure described above. Product: 5.5 g as a white powder, m.p. 151.5-153°C
7-Chloro-3-(5-dimethylaminomethyl-[l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo [ 1 ,5-a] [ 1 ,4] benzodiazepin-6-one maleate (1:1)
373 mg 7-Chloro-3-(5-dimethylaminomethyl-[ l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[ 1,5-a] [ l,4]benzodiazepin-6-one (I) and 116 mg maleic acid were dissloved in 3 ml hot ethanol. The salt crystalized on cooling. The suspension was stirred for 10 min at 0°C. Filtration and drying afforded 460 mg 7-Chloro-3-(5-dimethylaminomethyl-[ l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[l,5-a] [ l,4]benzodiazepin-6-one maleate (1:1) as a white solid, m.p. 182-184°C
References
- ^ Huang Z, Zhan S, Chen C, Zhang R, Zhou Y, He J, et al. (February 2024). “Efficacy and safety of Dimdazenil in adults with insomnia disorder: results from a multicenter, randomized, double-blind, placebo-controlled phase III trials”. Sleep. 47 (2). doi:10.1093/sleep/zsad272. PMC 10851846. PMID 37875349.
- ^ Guilleminault C (2010). Sleep Medicine. Elsevier Health Sciences. pp. 574–. ISBN 978-1-4377-1836-2.
- ^ Jump up to:a b c Monti JM, Pandi-Perumal SR, Möhler H (28 September 2010). GABA and Sleep: Molecular, Functional and Clinical Aspects. Springer Science & Business Media. pp. 50–51. ISBN 978-3-0346-0226-6.
- ^ Syed YY (March 2024). “Dimdazenil: First Approval”. Drugs. doi:10.1007/s40265-024-02020-9. PMID 38546956.
- ^ Plunkett JW (September 2007). Plunkett’s Biotech & Genetics Industry Almanac 2008: Biotech & Genetics Industry Market Research, Statistics, Trends & Leading Companies. Plunkett Research, Ltd. pp. 311–. ISBN 978-1-59392-087-6.
External links
| Clinical data | |
|---|---|
| Trade names | Junoenil |
| Other names | EVT-201; EVT201 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 308239-86-3 |
| PubChem CID | 9885841 |
| DrugBank | DB05721 |
| ChemSpider | 8061514 |
| UNII | 6J8AF7CLE4 |
| ChEMBL | ChEMBL5095096 |
| CompTox Dashboard (EPA) | DTXSID301032055 |
| Chemical and physical data | |
| Formula | C17H17ClN6O2 |
| Molar mass | 372.81 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////DIMDAZENIL, EVT-201, 308239-86-3, EVT201, 6J8AF7CLE4, EVT 201, CHINA 2023, INSOMNIA

{kind=link}