
Home » Anthony crasto (Page 3)
Category Archives: Anthony crasto
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
HELP, Need one time help to pay 10 year concessional subscription to this, your favorite blog to WordPress

Just One viewer please come forward
Dear Kind Viewer’s
WordPress is kind to me and negotiated a one time 10 year concessional subscription of 260 US dollars…….https://newdrugapprovals.org/
I need one time help to pay this one time 10 year concessional subscription to our favorite blog.
This is done to keep this blog running even after my death.
Currently I am paying 99 US Dollars per annum
email me
amcrasto@gmail.com
call +919323115463
Paypal will work for me via email request to you by me, Indian govt does not allow automatic transfer via paypal buttons on the blog
email me at amcrasto@gmail.com and tell me amount, i will request you via paypal

DR ANTHONY CRASTO
LIONEL MY SON, MY MOTIVATION
.
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, He cried bitterly and we had never seen him so depressed
Now I keep Lionel as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son and family happy.

ps
The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent,
///////////
New Drug Approvals Blog has 2 lakh plus viewers in USA alone

New Drug Approvals Blog has 2 lakh plus viewers in USA alone
that is 200 thousand viewers
A record 1170135 views (11 lakh plus)all over the world in 211 countries
that is 1100 thousand plus views on this blog
I suffered a paralytic stroke in dec 2007 and bound to a wheelchair, this seems to have injected feul in me to help chemists around the world, I am more active than before and pushing boundaries, I have 2,5 lakh connections on all networking sites, I am available to all, contact me on +91 9323115463, amcrasto@gmail.com, Twitter @amcrasto

My son lionel was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, He cried bitterly and we had never seen him so depressed
Now I keep Lionel as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son and family happy.


////////////
New Drug Approvals blog by Dr Anthony Crasto hits ten lakh views in 211 countries
New Drug Approvals hits ten lakh views in 211 countries
|
 THANKS AND REGARD’S MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link 
|


//////////
SILICO LINEZOLID, SILINEZOLID, NDS 10024

Therapeutic options for brain infections caused by pathogens with a reduced sensitivity to drugs are limited. Recent reports on the potential use of linezolid in treating brain infections prompted us to design novel compounds around this scaffold. Herein, we describe the design and synthesis of various oxazolidinone antibiotics with the incorporation of silicon.
Our findings in preclinical species suggest that silicon incorporation is highly useful in improving brain exposures. Interestingly, three compounds from this series demonstrated up to a 30-fold higher brain/plasma ratio when compared to linezolid thereby indicating their therapeutic potential in brain associated disorders
Design, Synthesis, and Identification of Silicon Incorporated Oxazolidinone Antibiotics with Improved Brain Exposure


Examples from patent

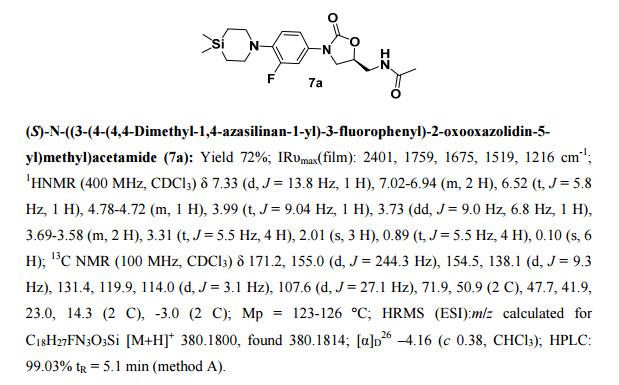
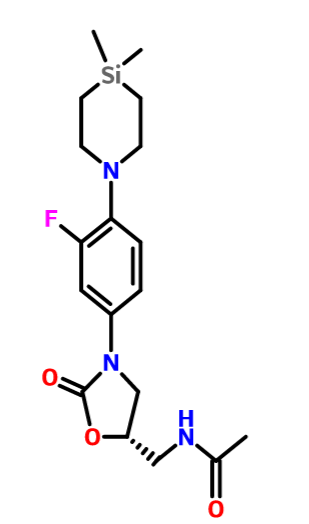
- (S)—N((3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)-2 oxooxazolidin-5-yl)methyl)acetamide
- NDS 10024
- Preparation of (S)—N((3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)-2 oxooxazolidin-5-yl)methyl)acetamide (12)
-
-
To a solution of 8 (50 mg, 0.135 mmol) in dimethylformamide (DMF), lithium-t-butoxide (LiOtBu) (32.3 mg, 0.4 mmol) is added. The mixture is stirred at 25° C. for 15 min, followed by the addition of MeOH (0.01 mL, 0.27 mmol). 6 (52 mg, 0.27 mmol) is then added and the reaction mixture is allowed to stir at 25° C. for 24 h. Glacial acetic acid is then added and the organic phase is extracted with EtOAc and washed with brine solution. The crude material is purified by column chromatography on silica gel using hexane-EtOAC mixtures to furnish the pure product 12. The analogous procedure for the corresponding morpholine analogue was adapted from Lu, C. V.; Chen, J. J.; Perrault, W. R.; Conway, B. G.; Maloney, M. T.; Wang, Y. Org. Pro. Res. and Development. 2006, 10, 272-277.
-
1H NMR (200 MHz, CDCl3): δ 7.33 (d, J=13.8 Hz, 1H), 7.02-6.94 (m, 2H), 6.52 (t, J=5.8 Hz, 1H), 4.77-4.73 (m, 1H), 3.99 (t, J=9.04 Hz, 1H), 3.72 (dd, J=9.0 Hz, 6.8 Hz, 1H), 3.69-3.58 (m, 2H), 3.31 (t, J=5.5 Hz, 4H), 2.01 (s, 3H), 0.89 (t, J=5.5 Hz, 4H), 0.10 (s, 6H). 13C NMR (100 MHz, CDCl3): δ171.2, 155.0 (d, J=244.3 Hz), 154.5, 138.2 (d, J=9.3 Hz), 131.5, 119.9, 114.0 (d, J=3.4 Hz), 107.6 (d, J=27.1 Hz), 71.9, 50.9, 47.7, 41.9, 23.0, 14.3, −2.9.
- Preparation of Bis(bromomethyl)dimethylsilane (2) (as per scheme 2)
-

-
HBr gas is bubbled to a solution of dimethyl divinylsilane 1 (10.0 g, 89.28 mmols), and dibenzoylperoxide (DBP, 100 mg), in heptane (100 mL) at 0° C. for 30 min. The Reaction mixture (RM) is allowed to stir at room temperature (25° C.) for 18 h, water (200 mL) is added to the reaction mixture and the organic layer is separated. The heptane layer is washed with 2N NaOH (2 100 mL), dried and concentrated to obtain the product 2 as a colourless liquid (24.5 g) in 100% yield.
-
1H NMR (200 MHz, CDCl3): δ 3.58-3.49 (m, 4H), 1.45-1.40 (m, 4H), 0.09 (s, 6H).
-

-
Benzylamine (20 mL, 182 mmol) and Et3N (15.2 mL, 109 mmol) are added to a solution of bis-(bromomethyl) dimethylsilane 2 (10 g, 36.5 mmol) in chloroform (100 mL). The mixture is then refluxed for 16 h. 5% sodiumhydroxide solution (150 mL) is then added and the aqueous layer is extracted with dichloromethane (DCM, 2×100 mL). It is then washed with brine (200 mL), dried and concentrated. The product is purified by column chromatography on silica gel using hexane-EtOAc mixtures to obtain the product 3 as a light yellow liquid (4.3 g) in 54% yield.
-
1H NMR (200 MHz, CDCl3): δ 7.23-7.35 (m, 5H), 3.66 (s, 2H), 2.68 (t, J=6.3 Hz, 4H), 0.75 (t, J=6.3 Hz, 4H), 0.04 (s, 6H).
- Preparation of 1-benzyl-4,4-dimethyl-1,4-azasilinane (3)
Preparation of 4,4-dimethyl-1,4-azasilinane hydrochloride (4)
-

-
To a solution of 4,4-dimethyl-1,4-azasilinane 3 (2.3 g, 10.5 mmol) in EtOH (20 mL), 6N hydrochloricacid (1.75 mL, 10.5 mmol) is added and the solvent is removed under reduced pressure. The reaction mixture is co-evaporated with EtOH (2×10 mL) and recrystallized from EtOH-diethyl ether. To a slurry of Pd/C (50 mg) in EtOH (15 mL) an ethanolic solution of above prepared HCl salt is added drop wise and stirred at 25° C. under hydrogen atmosphere for 20 h. The reaction mixture is filtered through celite and washed with 2×20 mL of MeOH. The filtrate is then concentrated under reduced pressure to give viscous oil which was triturated with diethyl ether to obtain the product 4 as a white solid (950 mg) in 70% yield.
Preparation of 1-(2-fluoro-4-nitrophenyl)-4,4-dimethyl-1,4-azasilinane (9)
-

-
To a solution of 4,4-dimethyl-1,4-azasilinane hydrochloride 4 (500 mg, 3.85 mmol) in EtOAc (15 mL), triethylamine (1.3 mL, 9.63 mmol) is added and stirred at 25° C. for 10 min. The reaction mixture is cooled to 0° C. and 3,4-difluoronitrobenzene (612 mg, 3.85 mmol) is added drop wise and allowed to stir at 25° C. for 6 h. Water is then added and the organic layer is separated. The aqueous layer is extracted with EtOAc (2×10 mL) and the solvent is removed under reduced pressure. The product is purified by column chromatography using hexane-EtOAc mixtures and a crystalline yellow solid 9 (721 mg) is obtained in 70% yield.
-
1H NMR (200 MHz, CDCl3): δ 7.93-7.84 (m, 2H), 6.86 (t, J=4 Hz, 1H), 3.70-3.67 (m, 4H), 0.91-0.85 (m, 4H), 0.12 (s, 6H). 13C NMR (50 MHz, CDCl3): δ 151.1 (d, J=246.71 Hz), 144.4 (d, J=7.13 Hz), 137.8 (d, J=8.59 Hz), 121.4, 115.9 (d, J=4.61 Hz), 113.2 (J=27.78 Hz), 49.4, 13.8, −2.8. IR (CHCl3): ν 2948, 2894, 1603, 1523, 1492, 1400, 1342, 1223, 983, 832, 742 cm−1′. M.P: 70-72° C.
Preparation of benzyl 4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenylcarbamate (10)
-

-
To a solution of compound 9 (610 mg, 2.28 mmol) in THF (25 mL), Pd/C (30 mg) is added and hydrogenated under a pressure of 35 psi in a par hydrogenator for 8 h. The reaction mixture is filtered through celite. Celite pad is washed with THF (2×20 mL). To the filtrate, saturated NaHCO3 (420 mg, 5.01 mmol) and CBzCl (427 mg, 2.5 mmol) are added at 0° C. and stirred at 25° C. for 5 h. 10 mL water is added to reaction mixture and the aqueous layer is extracted with EtOAc (2×20 mL). The crude mixture is then subjected to column chromatography on silica gel using hexane-EtOAc mixtures to afford the product as a viscous liquid 10 (690 mg) in 82% yield.
-
1H NMR (200 MHz, CDCl3): δ 7.41-7.37 (m, 5H), 6.94-6.93 (m, 2H), 6.68 (s, 1H), 5.21 (s, 1H), 3.3 (t, J=6.38 Hz, 4H), 0.93 (t, J=6.08 Hz, 4H), −0.13 (s, 6H). 13C NMR (50 MHz, CDCl3): 155.4 (d, 244.4 Hz), 153.6, 136.1, 135.9, 128.6, 128.5, 128.3, 120.4, 117.2 (d, 18.7 Hz), 114.7, 108.3 (20.5 Hz), 67.1, 51.4, 14.4, −3.0. IR (CHCl3): ν 3317, 2953, 2803, 1706, 1594, 1521, 1271, 1221, 1058, 869, 759 cm−1. M.P: 80-82° C.
Preparation of (S)-5-(aminomethyl)-3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)oxazolidin-2-one (11) (NDS-10057)
-

-
To a solution of 10 (1.20 g, 3.23 mmol) and (S)-tert-butyl 3-chloro-2-hydroxypropylcarbamate (1.35 g, 6.47 mmol) in DMF (10 mL), LiOtBu (1.03 g, 12.94 mmol) is added at 0° C. The mixture is stirred at 25° C. for 45 h. The starting material 10 is not consumed completely. Saturated NH4Cl is then added; the organic phase is extracted with EtOAc (2×20 mL), washed with brine solution, dried and concentrated. The crude residue is dissolved in 20 mL of DCM-TFA mixture (8:2) and stirred at 25° C. for 3 h. RM is concentrated and dissolved in water (10 mL), the aqueous layer is washed with diethyl ether (2×50 mL), basified with saturated NaHCO3 and extracted with DCM (2×50 mL). The DCM layer is dried and concentrated. The crude is purified by column chromatography on silica gel using hexane-EtOAc mixtures to obtain the product as an off-white solid (500 mg) in 45% (based on recovery of starting material) over 2 steps.
-
1H NMR (400 MHz, CDCl3): δ 7.36 (dd, J=14.2 Hz, 2.3 Hz, 1H), 7.09 (dd, J=8.8 Hz, 1.7 Hz, 1H), 6.96 (t, J=9.5 Hz, 1H), 4.72-4.59 (m, 1H), 4.00 (t, J=8.3 Hz, 1H), 3.79 (dd, J=8.7 Hz, 6.8 Hz, 1H), 3.30 (t, J=6.2 Hz, 4H), 3.03 (dq, J=13.6 Hz, 4.2 Hz, 2H), 0.90 (t, J=6.2 Hz, 4H), 0.10 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 155.1 (d, J=244.3 Hz), 154.7, 137.9 (d, J=9.0 Hz), 132.1 (d, J=10.3 Hz), 112.0 (d, J=4.3 Hz), 113.8 (d, J=3.2 Hz), 107.4 (d, J=26.9 Hz), 73.8, 51.0, 47.8, 45.01, 14.4, −2.9. IR (CHCl3): ν 3685, 3021, 2955, 2809, 2401, 1747, 1515, 1416, 1219, 1029, 991, 870, 771, 667 cm−1. M.P: 94-96° C. ESI-MS: 360.11 (M+Na).
Preparation of (S)—N-((3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)-2-oxooxazolidin-5-yl)methy)acetamide (12) (NDS 10024)
-

-
To solution of amine 11 (300 mg, 0.9 mmol) and DIPEA (0.3 mL, 1.78 mmol) in dry THF (4.0 mL), acetylchloride (0.08 mL, 1.07 mmol) is added at 0° C., and stirred at 25° C. for 3 h. Further, saturated NaHCO3 (5.0 mL) is added to the reaction mixture and extracted with EtOAc (2×5 mL). The organic layer is washed with brine, dried and concentrated. The product is purified by column chromatography on silica gel using hexane-EtOAc mixtures to obtain the product as an off-white solid (170 mg) in 50% yield.
-
1HNMR (400 MHz, CDCl3): δ 7.33 (d, J=13.8 Hz, 1H), 7.02-6.94 (m, 2H), 6.52 (t, J=5.8 Hz, 1H), 4.77-4.73 (m, 1H), 3.99 (t, J=9.04 Hz, 1H), 3.72 (dd, J=9.0 Hz, 6.8 Hz, 1H), 3.69-3.58 (m, 2H), 3.31 (t, J=5.5 Hz, 4H), 2.01 (s, 3H), 0.89 (t, J=5.5 Hz, 4H), 0.10 (s, 6H). 13C NMR (100 MHz, CDCl3): δ171.2, 155.0 (d, J=244.3 Hz), 154.5, 138.2 (d, J=9.3 Hz), 131.5, 119.9, 114.0 (d, J=3.4 Hz), 107.6 (d, J=27.1 Hz), 71.9, 50.9, 47.7, 41.9, 23.0, 14.3, −2.9. IR (CHCl3): ν 2401, 1759, 1675, 1519, 1216, 759, 669 cm−1 M.P: 123-126° C. ESI-MS: 380.10 (M+H).


SCHEME2


SCHEME 3

SCHEME 4



Dr. D. Srinivasa Reddy of NCL winner Shanti Swarup Bhatnagar Award 2015
see
http://oneorganichemistoneday.blogspot.in/2015/02/dr-d-srinivasa-reddy.html
Dr. Srinivasa Reddy of CSIR-NCL bags the
prestigious Shanti Swarup Bhatnagar Prize

AN INTRODUCTION
Ph.D., University of Hyderabad, 2000 (Advisor: Professor Goverdhan Mehta).
Post-doctoral with Profs. Sergey A. Kozmin(University of Chicago, USA) and Prof.
Jeffrey Aubé (University of Kansas, USA)
Experienced in leading drug discovery programs (Dr. Reddy’s & TATA Advinus – 7
years of pharma experience)
Acquired skills in designing novel small molecules and lead optimization
Experienced in planning and execution of total synthesis of biologically active
molecules with moderate complexity
One of the molecules is currently in human clinical trials.
MYSELF WITH HIM

OTHER AUTHORS




////////
C[Si]1(C)CCN(CC1)c2ccc(cc2F)N3C[C@H](CNC(C)=O)OC3=O
This blog New Drug Approvals will touch 10 lakh views soon……..as on 7 NOV 2015

This blog New Drug Approvals will touch 10 lakh views soon……..as on 7 NOV 2015
////////
7 LAKH VIEWS ON THIS BLOG……NEW DRUG APPROVALS
FLAGS AND HITS
DR ANTHONY MELVIN CRASTO Ph.D
web link
blogs are


 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 
 @amcrasto
@amcrasto Googleplus
GoogleplusMyself recovering from leg swelling

DR ANTHONY CRASTO at Metro hospital Manpada Thane, India
13-16 Apr, 2015
I am back
vladivostok








 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
amcrasto@gmail.com
CDK Inhibitor, MK 7965, DINACICLIB, SCH 727965

CDK Inhibitor, MK 7965, DINACICLIB, SCH 727965
REVIEW…….http://www.mdpi.com/2072-6694/6/4/2224/htm
One of the most popular CDK inhibitor in clinical trials in the recent years was dinaciclib (MK-7965, SCH 727965) (Figure 3), the inhibitor of CDK1, CDK2, CDK5, and CDK9. A Phase I trial on the effect of dinaciclib in combination with aprepitant was performed in patients with advanced malignancies [44]. Aprepitant is used for the prevention of chemotherapy-induced nausea and vomiting, is known as an inhibitor and inducer of CYP3A4, which metabolizes dinaciclib.
Coadministration of dinaciclib with aprepitant resulted in no clinically significant effect on the pharmacokinetics and did not alter the safety profile of dinaciclib. The first Phase I clinical trial on dinaciclib as a single agent was performed on patients with advanced malignancies [68]. Forty-eight patients with various solid tumors were treated and 10 of them achieved prolonged stable disease for at least four treatment cycles. Adverse effects were mild, the most common being nausea, anemia, decreased appetite and fatigue.
A phase II multi-center study of dinaciclib for relapsed and/or refractory AML was performed on 20 patients [69]. Temporary decrease in peripheral blood and/or bone marrow blasts was observed in 60% of patients. Four of 13 (31%) patients with circulating blasts had >50% decrease and 6 (46%) >80% decrease in the absolute blast count within 1–8 days of the first dinaciclib dose. Toxicities included diarrhea, fatigue, transaminitis, and manifestations of tumor lysis syndrome, with one patient who deceased of acute renal failure. Another Phase II study was performed of dinaciclib versus erlotinib in patients with non-small cell lung cancer [70].
Unfortunately, it was found that dinaciclib was not successful as monotherapy in non-small cell lung cancer. Most common toxicities included neutropenia, leukopenia, vomiting, and diarrhea. Yet another Phase II study was performed on dinaciclib versus capecitabine in patients with advanced breast cancer [71]. Dinaciclib treatment demonstrated antitumor activity in two of seven patients with ER-positive and ERBB 2-negative metastatic breast cancer, however efficacy was not superior to capecitabine (p = 0.991).
Toxicities included neutropenia, leukopenia, increase in aspartate aminotransferase, and febrile neutropenia. Phase I nonrandomized dose-escalation trial was performed, where patients with relapsed or refractory chronic lymphocytic leukemia were treated with dinaciclib and rituximab [72]. Four out of six patients achieved stable disease, and one patient achieved complete response. Drug-related adverse events were mostly hematological, digestive and metabolic and no dose-limiting toxicities were observed. Dinaciclib was also moved into Phase III development for refractory chronic lymphocytic leukemia [73]. Phase I/II clinical trial Dinaciclib in patients with relapsed multiple myeloma showed promise as single agent [74]. The overall confirmed response rate was 3 of 27 (11%). Adverse effects included leukopenia, thrombocytopenia, gastrointestinal symptoms, alopecia, and fatigue. –
FOR REF See more at: http://www.mdpi.com/2072-6694/6/4/2224/htm#sthash.amBuLwq1.dpuf

Dinaciclib (SCH-727965) is an experimental drug that inhibits cyclin-dependent kinases (CDKs.[1] It is being evaluated in clinical trials for various cancer indications.[2]
Mechanisms of action
- Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains.[3]
- Dinaciclib (SCH727665) inhibits the unfolded protein response (UPR) through a CDK1 and CDK5-dependent mechanism.[4]
Anti-tumoral action
- In chronic lymphocytic leukemia (CLL)
- In pancreatic cancer
- Dinaciclib inhibits pancreatic cancer growth and progression in murine xenograft models.[7]
- In osteosarcoma
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (S)-3-(((3-Ethyl-5-(2-(2-hydroxyethyl)piperidin-1-yl)pyrazolo[1,5-a]pyrimidin-7-yl)amino)methyl)pyridine 1-oxide | |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS number | 779353-01-4 |
| ATC code | ? |
| PubChem | CID 46926350 |
| ChemSpider | 25027387 |
| ChEMBL | CHEMBL2103840 |
| Synonyms | SCH-727965 |
| Chemical data | |
| Formula | C21H28N6O2 |
Clinical trials

http://www.google.com.tr/patents/US8076479
One example of these inhibitors is the compound of Formula II.

The synthesis of the compound of Formula II is described in the ‘878 publication according to Scheme II:
Scheme II:
Step 1—Amidization to Form Substituted Pyrazole

http://www.google.com.tr/patents/US8076479
Step 2—Formation and Dehalogenation of pyrazolo[1,5a]pyrimidine

Step 3—Amination (Two Separate, Sequential Reactions)

As described in the ‘878 publication, Synthetic Scheme II leading to the compound of Formula II has several disadvantages from the standpoint of commercial scale synthesis. In step 1, the starting material (compound “C”) used in the formation of compound “D” is a sticky, viscous oil which is difficult to process (weigh, transfer, and blend). Moreover, step 1, as described in the ‘878 publication, requires isolation and chromatographic purification of compounds C and D prior to carrying out each subsequent derivatization reaction. In addition, as described in the ‘878 publication, the reaction of compound C with malonate diester is carried out using the diester as a solvent. After isolation and purification of the resultant malonate adduct, compound D, ring closure to form diketone compound E is carried out in methanol. In accordance with the procedure described in the ‘878 publication, compound E is isolated and dried, then converted to the corresponding dichloride in N,N-dimethyl aniline by treatment with phosphorous oxychloride (POCl3). The dichloride thus formed was isolated and purified by chromatography prior to the sequential amination reactions. Additionally, the compounds of Formula G and of Formula II require chromatography purification and isolations, as described in the ‘878 publication.
As further described in the ‘878 publication, each of the amination reactions were run separately with isolation and chromatographic purification between amination reactions. Accordingly, the ‘878 publication describes the preparation of the compound of Formula II utilizing a scheme consisting of five separate reaction steps with intervening isolation and purification of the products, each sequential step being carried out in a different solvent system. The overall yield of the compound of Formula II reported for this synthesis, based on starting compound C (Scheme II) is about 20%.
Example 1Preparation of Diketone Compound E (Scheme VI) 3-Ethylpyrazolo[1,5-a]pyrimidine-5,7(4H,6H)-dione

To a 250 ml, three-necked flask equipped with a thermometer, a reflux condenser and mechanical stirrer was charged 3-amino-4-ethylpyrazole oxalate (10 g, 50 mmole), dimethylmalonate (10 ml, 88 mmole), methyl alcohol (80 ml) and sodium methoxide (50 ml, 245 mmole, 25% in methyl alcohol). The batch was heated at reflux for 16 hours then cooled to room temperature. Celite (5 g) and water (60 ml) were added to the batch and agitated for 10 minutes. The batch was filtered to remove the solid residue. The filtrate was pH adjusted to pH˜3 with aqueous HCl (10 ml) to effect precipitation. The precipitate (compound “E”) was filtered and washed with water (40 ml). The wet cake was dried for 18 hours in vacuum oven maintained in the range of oven at 45° C. to 55° C., to give a solid product (84.3%, 7.5 g). C8H9N3O3, Mp: 200-205° C.; NMR in DMSO-d6: 1.05 (t, 3H), 2.23 (q, 2H), 3.26 (bs, 1H), 3.89 (bs, 1H), 7.61 (s, 1H), 11.50(bs, 1H).
Example 2Preparation of Dichloride Compound F (Scheme VI) 5,7-Dichloro-3-Ethylpyrazolo[1,5-a]pyrimidine

Into a 3-neck flask fitted with an inert gas inlet, a reflux condenser and a mechanical stirring apparatus and containing 83 liters of acetonitrile was placed 3-Ethylpyrazolo[1,5-a]pyrimidine-5,7(4H,6H)-dione (E) prepared as described in Step 1 (11.0 kg, 61.5 mole), N,N-dimethylaniline (8.0 L, 63 mole) and POCl3 (7 kg, 430 mole). With stirring the mixture was brought to reflux and maintained under refluxing conditions for 15 hours. The reaction mixture was sampled periodically to monitor the amount of compound “E” present. After the conversion was complete, the solution was cooled to 15° C. Into the cooled reaction mixture was added water which had been cooled to a temperature of less than 20° C. The product is filtered and washed with 4 aliquots of acetonitrile-water (1:3) which had been cooled to a temperature of 20° C. followed by a wash with 10× water. The wet cake is dried in a vacuum oven maintained at 40° C. for at least 15 hours to yield the compound “F” (86.7%); 1H NMR (CDCl3): 1.32(t, 3H), 2.81 (q, 2H), 6.92 (s, 1H), 8.10 (s, 1H)
mp: 90-95° C.
Example 3Preparation of Compound G (Scheme VI) 5-Chloro-3-Ethyl-N-[(1-oxido-pyridinyl)methyl]pyrazolo-[1,5-a]pyrimidine-5.7(4H,6H)-dion-7-amine

Into a 3-liter, three-necked flask equipped with a thermometer, a reflux condenser and mechanical stirrer was charged an aliquot of the dichloride compound “F” prepared in Step 2 (150 g, 0.69 mole), potassium phosphate tribasic monohydrate (338.0 g, 1.47 mole), the dihydrochloride salt of N-oxide-pyridin-3-yl-methylamine, compound F1a (142.5 g, 0.72 mole), water (1500 ml) and acetonitrile (300 ml). The batch was heated at reflux for 6 hours. At the end of the refluxing period the batch was cooled to room temperature over 2 hours and then held at room temperature for 4 hours. The resulting precipitate was filtered and washed with water (600 ml). The wet cake was returned to the flask with water (1500 ml) and acetonitrile (300 ml), and heated to reflux. Reflux was maintained for 6 hours additional. At the end of the second reflux period the reaction mixture was cooled to room temperature over a 2 hour period and left to stand at room temperature for 4 hours. The resulting precipitate was filtered and washed with water (600 ml). The wet cake was dried in an air draft oven at 50° C. for 18 hours to give the first amine adduct “G” material (179 g, 84.9%). mp: 187-189C; NMR in CDCl3, 1.26(t, 3H), 2.73(q, 2H), 4.60(d, 2H), 5.87(s, 1H), 6.83(bs, 1H), 7.33(t, 1H), 7.70(d, 1H), 7.84(s, 1H), 8.58(d, 1H), 8.64(d, 1H).
Example 4
Preparation of the Compound of Formula II (Scheme VI) 1-[3-Ethyl-7-[(1-oxido-3-pyridinyl)methyl]amino]pyrazolo[1,5-a]pyrimidin-5-yl]-2(s)-piperidinemethanol

Into a three-neck flask fitted with a mechanical stirrer and a reflux condenser were placed the first amine adduct prepared in Step 3, compound “G”, (7 kg, 23 mole), amino-alcohol compound G1a (5.6 kg, 43.3 mole), sodium carbonate (3.5 kg, 33.0 mole), 110 ml of water and 1-methyl-2-pyrrolidinone (NMP) (11 L). The reaction mixture was heated to 150° C. for 4 days. After chromatography indicated that the reaction was complete (90-95% substrate consumed), the reaction mixture was cooled to room temperature and quenched by adding water. The mixture was then extracted with ethyl acetate. The batch was dried by distillation of the water azeotrope under atmospheric pressure and concentrated to about 28 L volume. THF was added and the solution was heated to reflux until all the solids dissolve. Ethyl acetate and trietylamine are added to the hot solution. The batch was cooled to ambient and then agitated with the temperature maintained in the range of from 20° C. to 25° C. for 12 hours. The solids were collected by filtration, washed first with ethyl acetate then water, and dried in the filter under vacuum for 24 hours with the temperature maintained at from 40° C. to 50° C., yielding 4.9 kg, 51.3% of the compound of Formula II.
DSC, 168.6° C.; Specific Rotation (10 mg/ml in MeOH, 20° C.), −117.8 °;
1HNMR (400 MHz, DMSO): 8.31 ppm (1H, s), 8.11-8.13 ppm (1H, td, J=5.7 Hz, J=1.4 Hz), 7.97 ppm (1H, t, J=6.7 Hz), 7.68 ppm (1H, s), 7.41 ppm (1H, s), 7.37-7.43 ppm (1H, dd), 5.55 ppm (1H, s), 4.85 ppm (1H, t, J=5.4 Hz), 4.49-4.59 ppm (3H, m), 4.24-4.28 ppm (1H, broad), 3.27-3.46 ppm (2H, m), 2.76-2.83 ppm (1H, t, J=13.0 Hz), 2.45-2.50 ppm (2H, q, J=7.5 Hz), 1.72-1.79 (1H, m), 1.54-1.68 ppm (6H, m), 1.30-1.34 ppm (1H, m), 1.16 ppm (3H, t, J=7.5 Hz)
References
- Parry, D; Guzi, T; Shanahan, F; Davis, N; Prabhavalkar, D; Wiswell, D; Seghezzi, W; Paruch, K; Dwyer, M. P.; Doll, R; Nomeir, A; Windsor, W; Fischmann, T; Wang, Y; Oft, M; Chen, T; Kirschmeier, P; Lees, E. M. (2010). “Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor”. Molecular Cancer Therapeutics 9 (8): 2344–53. doi:10.1158/1535-7163.MCT-10-0324. PMID 20663931.
- Jump up^ Bose P, Simmons GL, Grant S (2013). “Cyclin-dependent kinase inhibitor therapy for hematologic malignancies”. Expert Opin Investig Drugs 22 (6): 723–38.doi:10.1517/13543784.2013.789859. PMC 4039040. PMID 23647051.
- Martin, M. P.; Olesen, S. H.; Georg, G. I.; Schönbrunn, E (2013). “Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains”. ACS Chemical Biology 8 (11): 2360–5. doi:10.1021/cb4003283. PMC 3846258. PMID 24007471.
- Nguyen, T. K.; Grant, S (2013). “Dinaciclib (SCH727665) inhibits the unfolded protein response (UPR) through a CDK1 and CDK5-dependent mechanism”. Molecular Cancer Therapeutics 13(3): 662–74. doi:10.1158/1535-7163.MCT-13-0714. PMID 24362465.
- Jump up^ Desai, B. M.; Villanueva, J; Nguyen, T. T.; Lioni, M; Xiao, M; Kong, J; Krepler, C; Vultur, A; Flaherty, K. T.; Nathanson, K. L.; Smalley, K. S.; Herlyn, M (2013). “The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling”. PLoS ONE 8 (3): e59588. doi:10.1371/journal.pone.0059588. PMC 3601112. PMID 23527225.
- Jump up^ Johnson, A. J.; Yeh, Y. Y.; Smith, L. L.; Wagner, A. J.; Hessler, J; Gupta, S; Flynn, J; Jones, J; Zhang, X; Bannerji, R; Grever, M. R.; Byrd, J. C. (2012). “The novel cyclin-dependent kinase inhibitor dinaciclib (SCH727965) promotes apoptosis and abrogates microenvironmental cytokine protection in chronic lymphocytic leukemia cells”. Leukemia 26 (12): 2554–7.doi:10.1038/leu.2012.144. PMC 3645353. PMID 22791353.
- Jump up^ Feldmann, G; Mishra, A; Bisht, S; Karikari, C; Garrido-Laguna, I; Rasheed, Z; Ottenhof, N. A.; Dadon, T; Alvarez, H; Fendrich, V; Rajeshkumar, N. V.; Matsui, W; Brossart, P; Hidalgo, M; Bannerji, R; Maitra, A; Nelkin, B. D. (2011). “Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models”.Cancer biology & therapy 12 (7): 598–609. PMC 3218385. PMID 21768779.
- Jump up^ Fu, W; Ma, L; Chu, B; Wang, X; Bui, M. M.; Gemmer, J; Altiok, S; Pledger, W. J. (2011). “The cyclin-dependent kinase inhibitor SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma cells”. Molecular Cancer Therapeutics 10 (6): 1018–27. doi:10.1158/1535-7163.MCT-11-0167. PMID 21490307.
- Jump up^ Fu, W; Sharma, S. S.; Ma, L; Chu, B; Bui, M. M.; Reed, D; Pledger, W. J. (2013). “Apoptosis of osteosarcoma cultures by the combination of the cyclin-dependent kinase inhibitor SCH727965 and a heat shock protein 90 inhibitor”. Cell Death and Disease 4 (3): e566. doi:10.1038/cddis.2013.101. PMC 3613821. PMID 23538447.
- Jump up^ Nemunaitis, J. J.; Small, K. A.; Kirschmeier, P; Zhang, D; Zhu, Y; Jou, Y. M.; Statkevich, P; Yao, S. L.; Bannerji, R (2013). “A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies”. Journal of Translational Medicine 11 (1): 259. doi:10.1186/1479-5876-11-259.PMC 3853718. PMID 24131779.
- Jump up^ Mita, M; Joy, A. A.; Mita, A; Sankhala, K; Jou, Y. M.; Zhang, D; Statkevich, P; Zhu, Y; Yao, S. L.; Small, K; Bannerji, R; Shapiro, C. L. (2013). “Randomized Phase II Trial of the Cyclin-Dependent Kinase Inhibitor Dinaciclib (MK-7965) Versus Capecitabine in Patients with Advanced Breast Cancer”. Clinical Breast Cancer 14 (3): 169–76. doi:10.1016/j.clbc.2013.10.016.PMID 24393852.
- Jump up^ Stephenson, J. J.; Nemunaitis, J; Joy, A. A.; Martin, J. C.; Jou, Y. M.; Zhang, D; Statkevich, P; Yao, S. L.; Zhu, Y; Zhou, H; Small, K; Bannerji, R; Edelman, M. J. (2014). “Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer”. Lung Cancer 83 (2): 219–23.doi:10.1016/j.lungcan.2013.11.020. PMID 24388167.
External links
- dinaciclib at the US National Library of Medicine Medical Subject Headings (MeSH)

Patent Submitted Granted
Process and intermediates for the synthesis of (3-alkyl-5-piperidin-1-yl-3,3a-dihydro-pyrazolo[1,5-a]pyrimidin-7-yl)-amino derivatives and intermediates [US8076479]2008-03-06 GRANT2011-12-13
Process for resolving chiral piperidine alcohol and process for synthesis of pyrazolo[1,5-a] pyrimidine derivatives using same [US7786306]2008-02-28 GRANT2010-08-31
Sequential Administration of Chemotherapeutic Agents for Treatment of Cancer [US2011129456]2011-06-02
TARGETING CDK4 AND CDK6 IN CANCER THERAPY [US2011009353]2011-01-13
Pyrazolopyrimidines as cyclin dependent kinase inhibitors [US2007225270]2007-09-27
PYRAZOLO[1,5-a]PYRIMIDINES [US2007275963]2007-11-29
Novel pyrazolopyrimidines as cyclin dependent kinase inhibitors [US2007281951]2007-12-06
Novel pyrazolopyrimidines as cyclin dependent kinase inhibitors [US2008050384]2008-02-28
Novel pyrazolopyrimidines as cyclin dependent kinase inhibitors [US2007054925]2007-03-08

A Novel and Practical Synthesis of Ramelteon
 RAMELTEON
RAMELTEON
An efficient and practical process for the synthesis of ramelteon 1, a sedative-hypnotic, is described. Highlights in this synthesis are the usage of acetonitrile as nucleophilic reagent to add to 4,5-dibromo-1,2,6,7-tetrahydro-8H-indeno[5,4-b]furan-8-one 2 and the subsequent hydrogenation which successfully implement four processes (debromination, dehydration, olefin reduction, and cyano reduction) into one step to produce the ethylamine compound 13where dibenzoyl-l-tartaric acid is selected both as an acid to form the salt in the end of hydrogenation and as the resolution agent. Then, target compound 1 is easily obtained from13 via propionylation. The overall yield in this novel and concise process is almost twice as much as those in the known routes, calculated on compound 2.
A Novel and Practical Synthesis of Ramelteon
http://pubs.acs.org/doi/abs/10.1021/op500386g





