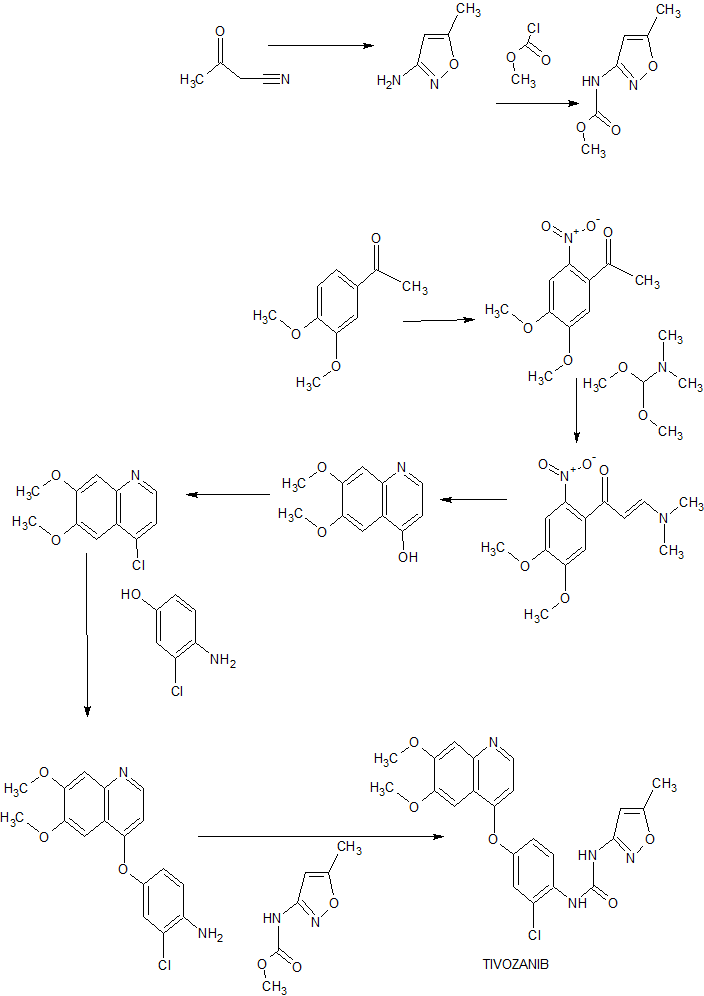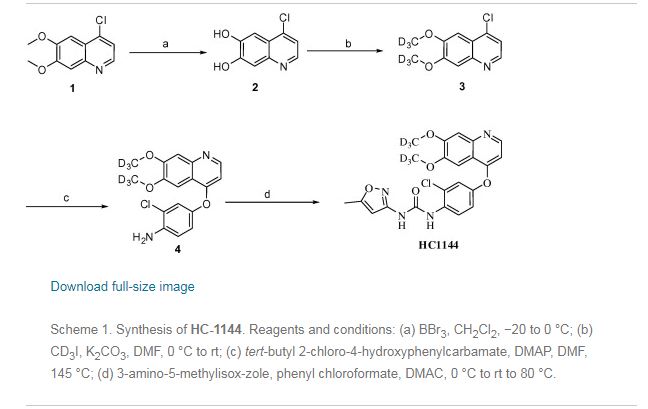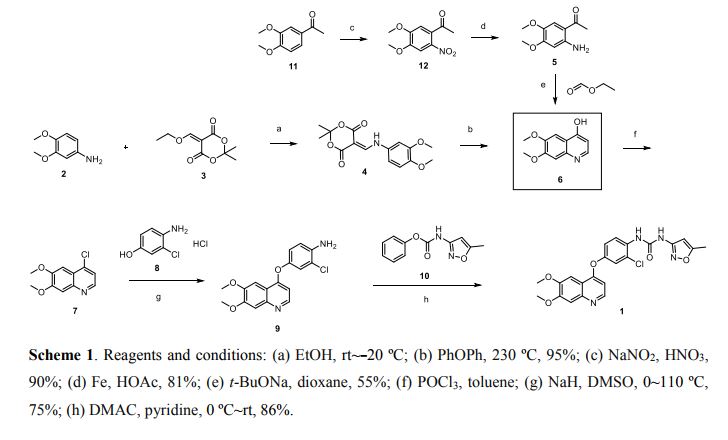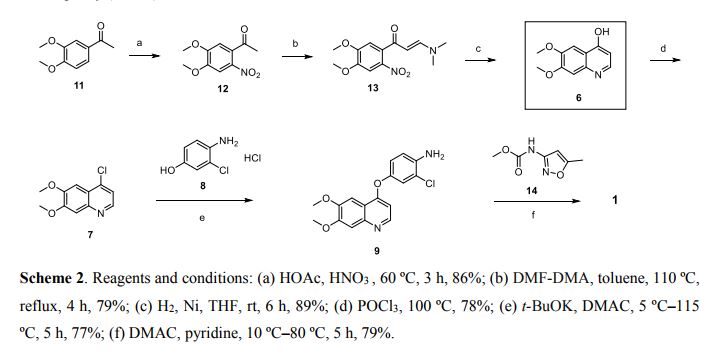
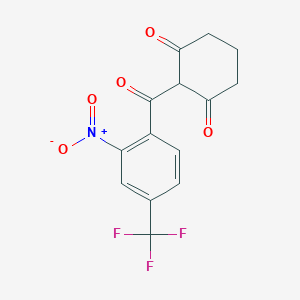
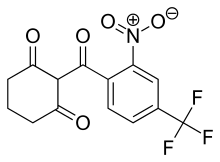
Nitisinone
ニチシノン
Orfadin
Launched – 2002, NTBC
SC-0735
SYN-118
2-(alpha,alpha,alpha-Trifluoro-2-nitro-p-tuluoyl)-1,3-cyclohexanedione
2-(2-Nitro-4-trifluoromethylbenzoyl)cyclohexane-1,3-dione
Priority, Orphan
| Formula |
C14H10F3NO5
|
| CAS |
104206-65-7
|
| Mol weight |
329.2281
|
1,3-Cyclohexanedione, 2-[2-nitro-4-(trifluoromethyl)benzoyl]-
2-(2-Nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione
Orfadin®|SC-0735
QB-0882
SC0735
UNII:K5BN214699
UNII-K5BN214699
Research Code:SC-0735
Trade Name:Orfadin®
MOA:4-hydroxyphenylpyruvate dioxygenase inhibitor
Indication:Hereditary tyrosinemia
Company:Swedish Orphan Biovitrum AB (SOBI) (Originator)
Nitisinone is a synthetic reversible inhibitor of 4-hydroxyphenylpyruvate dioxygenase. It is used in the treatment of hereditary tyrosinemia type 1. It is sold under the brand name Orfadin.
Nitisinone was first approved by the U.S. Food and Drug Administration (FDA) on January 18, 2002, then approved by the European Medicines Agency (EMA) on February 21, 2005. It was developed and marketed as Orfadin® by Swedish Orphan Biovitrum AB (SOBI) in the US .
The mechanism of action of nitisinone involves reversibile inhibition of 4-Hydroxyphenylpyruvate dioxygenase(HPPD). It is indicated for use as an adjunct to dietary restriction of tyrosine and phenylalanine in the treatment of hereditary tyrosinemia type 1 (HT-1).
Orfadin® is available as capsule for oral use, containing 2, 5 or 10 mg of free Nitisinone. The recommended initial dose is 1 mg/kg/day divided into two daily doses. Maximum dose is 2 mg/kg/day.
Nitisinone was launched in 2002 by Swedish Orphan (now Swedish Orphan Biovitrum) in a capsule formulation as an adjunct to dietary restriction of tyrosine and phenylalanine in the treatment of hereditary tyrosinemia type I. In 2015, this product was launched in Japan for the same indication. The same year, an oral suspension formulation for pediatric patients was registered in the E.U., and launch took place in the United Kingdom shortly after. This formulation was approved in 2016 in the U.S. for the same indication. In 2016, nitisinone tablet formulation developed by Cycle Pharmaceuticals was approved in Canada (this formulation is also available also in the U.S.).
Indication
Used as an adjunct to dietary restriction of tyrosine and phenylalanine in the treatment of hereditary tyrosinemia type 1.
Associated Conditions
EU

http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004281/WC500236080.pdf
Nitisinone MendeliKABS
22 June 2017 EMA/CHMP/502860/2017
Product name, strength, pharmaceutical form: Orfadin • Marketing authorisation holder: Swedish Orphan Biovitrum International AB • Date of authorisation: 21/02/2005
Procedure No. EMEA/H/C/004281/0000
During the meeting on 22 June 2017, the CHMP, in the light of the overall data submitted and the scientific discussion within the Committee, issued a positive opinion for granting a Marketing authorisation to Nitisinone MendeliKABS.
The chemical name of nitisinone is 2-[2-Nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione corresponding to the molecular formula C14H10F3NO5. It has a relative molecular mass of 329.23 g/mol and the following structure: Figure 1. Structure of nitisinone.
Nitisinone appears as off-white to yellowish non-hygroscopic fine crystalline powder. It is practically insoluble in unbuffered water. It is freely soluble in dichloromethane, sparingly soluble in ethyl alcohol, slightly soluble in isopropyl alcohol and 70% aqueous isopropyl alcohol and in pH 6.8 phosphate buffer, very slightly soluble in pH 4.5 acetate buffer and practically insoluble at pH 1.1. Solubility in acidified aqueous media depends on the acid counter ion. Solubility increases with increasing pH. Its pKa was found to be around 10. Nitisinone is achiral and does not show polymorphism.
ALSO
2005
http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000555/WC500049192.pdf
Nitisinone is a white to yellowish-white crystalline powder poorly soluble in water. The active substance is a weak acid and it is highly soluble in the pH range 4.5-7.2 in phosphate buffer solutions. Nitisinone has the chemical name 2-(2-nitro-4-trifluoromethylbenzoyl)-cyclohexane-1,3-dione. It does not show polymorphism.
US FDA
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/206356Orig1s000ChemR.pdf
Company: Swedish Orphan Biovitrum AB
Application No.: 206356Orig1
Approval Date: April 22, 2016
Nitisinone (INN), also known as NTBC (an abbreviation of its full chemical name) is a medication used to slow the effects of hereditary tyrosinemia type 1. Since its first use for this indication in 1991, it has replaced liver transplantation as the first-line treatment for this rare condition. It is also being studied in the related condition alkaptonuria. It is marketed under the brand name Orfadin by the company Swedish Orphan Biovitrum (Sobi); it was first brought to market by Swedish Orphan International. It was originally developed as a candidate herbicide.
Uses
Nitisinone is used to treat hereditary tyrosinemia type 1, in combination with restriction of tyrosine in the diet.[1][2][3]
Since its first use for this indication in 1991, it has replaced liver transplantation as the first-line treatment for this rare condition.[4] I It is marketed under the brand name Orfadin.
It has been demonstrated that treatment with nitisinone can reduce urinary levels of homogentisic acid in alkaptonuria patients by 95%.[5] A series of clinical trials run by DevelopAKUre to determine whether nitisinone is effective at treating the ochronosis suffered by patients with alkaptonuria are ongoing.[6] If the trials are successful, DevelopAKUre will try to get nitisinone licensed for use by alkaptonuria patients.[7]
Mechanism of action
The mechanism of action of nitisinone involves reversibile inhibition of 4-Hydroxyphenylpyruvate dioxygenase (HPPD).[8][9] This is a treatment for patients with Tyrosinemia type 1 as it prevents the formation of maleylacetoacetic acid and fumarylacetoacetic acid, which have the potential to be converted to succinyl acetone, a toxin that damages the liver and kidneys.[4] This causes the symptoms of Tyrosinemia type 1 experienced by untreated patients.[10]
Alkaptonuria is caused when an enzyme called homogentisic dioxygenase (HGD) is faulty, leading to a buildup of homogenisate.[11]Alkaptonuria patients treated with nitisinone produce far less HGA than those not treated (95% less in the urine),[5] because nitisinone inhibits HPPD, resulting in less homogenisate accumulation. Clinical trials are ongoing to test whether nitisinone can prevent ochronosisexperienced by older alkaptonuria patients.[6]
Adverse effects
Nitisinone has several negative side effects; these include but are not limited to: bloated abdomen, dark urine, abdominal pain, feeling of tiredness or weakness, headache, light-colored stools, loss of appetite, weight loss, vomiting, and yellow-colored eyes or skin.[12]
Research
Nitisinone is being studied as a treatment for alkaptonuria.[13]
Research at the National Institutes of Health (NIH) has demonstrated that nitisinone can reduce urinary levels of HGA by up to 95% in patients with alkaptonuria. The primary parameter of the NIH trial was range of hip motion, for which the results were inconclusive.[citation needed]
Research done using alkaptonuric mice has shown that mice treated with nitisinone experience no ochronosis in knee joint cartilage. In contrast, all of the mice in the untreated control group developed ochronotic knee joints.[14]
The efficacy of Nitisinone is now being studied in a series international clinical trials called DevelopAKUre.[15] The studies will recruit alkaptonuria patients in Europe.[16] A larger number of patients will be recruited in these trials than in the previous NIH trial.[17] The trials are funded by the European Commission.[18]
Nitisinone has been shown to increase skin and eye pigmentation in mice, and has been suggested as a possible treatment for oculocutaneous albinism.[19][20]
History
Nitisinone was discovered as part of a program to develop a class of herbicides called HPPD inhibitors. It is a member of the benzoylcyclohexane-1,3-dione family of herbicides, which are chemically derived from a natural phytotoxin, leptospermone, obtained from the Australian bottlebrush plant (Callistemon citrinus).[21] HPPD is essential in plants and animals for catabolism, or breaking apart, of tyrosine.[22] In plants, preventing this process leads to destruction of chlorophyll and the death of the plant.[22] In toxicology studies of the herbicide, it was discovered that it had activity against HPPD in rats[23] and humans.[24]
In Type I tyrosinemia, a different enzyme involved in the breakdown of tyrosine, fumarylacetoacetate hydrolase is mutated and doesn’t work, leading to very harmful products building up in the body.[1] Fumarylacetoacetate hydrolase acts on tyrosine after HPPD does, so scientists working on making herbicides in the class of HPPD inhibitors hypothesized that inhibiting HPPD and controlling tyrosine in the diet could treat this disease. A series of small clinical trials attempted with one of their compounds, nitisinone, were conducted and were successful, leading to nitisinone being brought to market as an orphan drug Swedish Orphan International,[8] which was later acquired by Swedish Orphan Biovitrum (Sobi).
Sobi is now a part of the DevelopAKUre consortium. They are responsible for drug supply and regulatory support in the ongoing clinical trials that will test the efficiacy of nitisinone as a treatment for alkaptonuria.[25] It is hoped that if the trials are successful, nitisinone could also be licensed for treatment of alkaptonuria.[7]
Generic versions
There is no generic version of Orfadin in G7 countries. Prior to the market authorization of MDK-Nitisinone in Canada, the only Nitisinone product available globally was Orfadin.[26]Until recently, Nitisinone was not approved in Canada where it was distributed for over 20 years via a Health Canada Special Access Program. In September 2016, MendeliKABS was granted approval of a Priority New Drug Submission (PNDS) by Health Canada for a bioequivalent generic version of Orfadin capsules (MDK-Nitisinone). In November 2016 Cycle Pharma was also granted approval of a PNDS by Health Canada for Nitisinone tablets that are bioequivalent to Orfadin capsules.[27] SOBI was granted approval of a PNDS in December 2016.[28]
PAPER
1H NMR, 13C NMR, and Computational DFT Studies of the Structure of 2-Acylcyclohexane-1,3-diones and Their Alkali Metal Salts in Solution
Faculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warszawa, Poland
J. Org. Chem., 2006, 71 (12), pp 4636–4641
DOI: 10.1021/jo060583g
1H and 13C NMR spectra of 2-acyl-substituted cyclohexane-1,3-diones (acyl = formyl, 1; 2-nitrobenzoyl, 2; 2-nitro-4-trifluoromethylbenzoyl, 3) and lithium sodium and potassium salts of 1have been measured. The compound 3, known as NTBC, is a life-saving medicine applied in tyrosinemia type I. The optimum molecular structures of the investigated objects in solutions have been found using the DFT method with B3LYP functional and 6-31G** and/or 6-311G(2d,p) basis set. The theoretical values of the NMR parameters of the investigated compounds have been calculated using GIAO DFT B3LYP/6-311G(2d,p) method. The theoretical data obtained for compounds 1−3 have been exploited to interpret their experimental NMR spectra in terms of the equilibrium between different tautomers. It has been found that for these triketones an endo-tautomer prevails. The differences in NMR spectra of the salts of 1 can be rationalized taking into account the size of the cation and the degree of salt dissociation. It seems that in DMSO solution the lithium salt exists mainly as an ion pair stabilized by the chelation of a lithium cation with two oxygen atoms. The activation free energy the of formyl group rotation for this salt has been estimated to be 51.5 kJ/mol. The obtained results suggest that in all the investigated objects, including the free enolate ions, all atoms directly bonded to the carbonyl carbons lie near the same plane. Some observations concerning the chemical shift changes could indicate strong solvation of the anion of 1 by water molecules. Implications of the results obtained in this work for the inhibition mechanism of (4-hydroxyphenyl) pyruvate dioxygenase by NTBC are commented upon.
2-(2-Nitro-4-trifluoromethylbenzoyl)cyclohexane-1,3-dione (NTBC; 3). The compound was prepared in the same manner as 2. The synthesis of an appropriate benzoic acid derivative was started from the transformation of commercially available 2-nitro-4-trifluoromethylaniline into benzonitrile by the classical Sandmeyer method. Then the nitrile was hydrolyzed in 65% sulfuric acid to give 2-nitro-4-trifluoromethylbenzoic acid.13 The obtained triketone 3 had a mp of 140−142 °C (lit.14 141−143 °C). For NMR data, see Supporting Information….. https://pubs.acs.org/doi/suppl/10.1021/jo060583g/suppl_file/jo060583gsi20060420_080852.pdf
NMR data for 2-(2-nitro-4-trifluoromethylbenzoyl)cyclohexane-1,3-dione, 3, in CDCl3

1 H NMR: 16.25 (s, 1H, OH), 8.47 (ddq, 1H, H10, J10,12=1.7 Hz, J10,13=0.4 Hz, J10,F=0.7 Hz), 7.94 (ddq, 1H, H12, J12,13=8.0 Hz, J12,F=0.7 Hz), 7.39 (ddq, 1H, H13, J13,F=0.8 Hz), 2.81 (t-like m, 2H, H4, H4’, JH4,H4’= -18.8 Hz, JH4,H5=5.4 Hz, JH4,H5’=7.3 Hz, JH4,H6=0.7 Hz, JH4,H6’= -0.8 Hz), 2.37 (tlike m, 2H, H6, H6’, JH6,H6’= -16.5 Hz, JH6,H5=4.6 Hz, JH6,H5’=8. 5 Hz), 2.04 (pentet-like m, 2H, H5, H5’, JH5,H5’= -13.6 Hz.

13C NMR: 196.3 (s, C(O)Ph), 195.8 (s, C3), 194.1 (s, C1), 145.5 (s, C9), 139.7 (s, C8), 132.0 (q, C11, J11,F=34.3 Hz), 130.8 (q, C12 J12,F=3.5 Hz), 127.7 (s, C13), 122.6 (q, CF3, JC,F=272.9 Hz), 121.1 (q, C10, J10,F=3.9 Hz), 112.7 (s, C2), 37.3 (s, C6) 31.6 (s, C4), 19.1 (s, C5).


PATENT
EP 186118
US 4780127



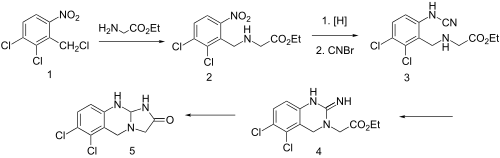
The condensation of cyclohexane-1,3-dione (I) with 2-nitro-4-(trifluoromethyl)benzoyl chloride by means of TEA in dichloromethane gives the target Nitisinone.EP 0186118
JP 1986152642, US 4774360, US 4780127

Nitisinone
-
- Synonyms:NTBC, SC 0735
- ATC:A16AX04
- Use:treatment of inherited tyrosinemia type I
- Chemical name:2-[2-nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione
- Formula:C14H10F3NO5
- MW:329.23 g/mol
- CAS-RN:104206-65-7
Synthesis Path

Substances Referenced in Synthesis Path
| CAS-RN |
Formula |
Chemical Name |
CAS Index Name |
| 504-02-9 |
C6H8O2 |
cyclohexane-1,3-dione |
1,3-Cyclohexanedione |
| 81108-81-8 |
C8H3ClF3NO3 |
2-nitro-4-trifluoromethylbenzoyl chloride |
|
Trade Names
| Country |
Trade Name |
Vendor |
Annotation |
| D |
Orfadin |
Orphan Europe |
|
| USA |
Orfadin |
Swedish Orphan ,2002 |
|
References
-
- WO 9 300 080 (ICI; 7.1.1993; appl. 18.6.1992; GB-prior. 24.6.1991).
- US 4 774 360 (Stauffer Chemical; 27.9.1988; appl. 29.6.1987).
-
synergistic herbicidal combination:
- WO 9 105 469 (Hoechst AG; 2.5.1991; appl. 12.10.1990; D-prior. 18.10.1989).
-
preparation of benzoylcyclohexanedione herbicides:
- US 4 780 127 (Stauffer Chemical; 25.10.1988; appl. 30.6.1986; USA-prior. 25.3.1982).
-
certain 2-(2-nitrobenzoyl)-1,3-cyclohexanediones:
- EP 186 118 (Stauffer Chemical; 2.7.1986; appl. 18.12.1985; USA-prior. 20.12.1984).
-
stable herbicidal compositions:
- WO 9 727 748 (Zeneca; 7.8.1997; appl. 3.2.1997; USA-prior. 2.2.1996).
PATENT
US9783485B1
https://patents.google.com/patent/US9783485B1/en
NTBC is a drug marketed by Swedish Orphan Biovitrum International AB under the brand name Orfadin® and it is used to slow the effects of hereditary tyrosinemia type 1 (HT-1) in adult and pediatric patients. It has been approved by FDA and EMA in January 2002 and February 2005 respectively.
HT-1 disease is due to a deficiency of the final enzyme of the tyrosine catabolic pathway fumarylacetoacetate hydrolase. NTBC is a competitive inhibitor of 4-hydroxyphenylpyruvate dioxygenase (HPPD), an enzyme which precedes fumarylacetoacetate hydrolase. By inhibiting the normal catabolism of tyrosine in patients with HT-1, NTBC prevents the accumulation of the toxic intermediates maleylacetoacetate and fumarylacetoacetate, that in patients with HT-1 are converted to the toxic metabolites succinylacetone and succinylacetoacetate, the former inhibiting the porphyrin synthesis pathway leading to the accumulation of 5-aminolevulinate.
Usefulness of NTBC in the treatment of further diseases has also been documented. A non-comprehensive list is reported hereinafter.
Effectiveness of Orfadin® in the treatment of diseases where the products of the action of HPPD are involved (e.g., HT-1) has been described notably in EP0591275B1 corresponding to U.S. Pat. No. 5,550,165B1. Synthesis of NTBC is also described in this patent.
WO2011106655 reports a method for increasing tyrosine plasma concentrations in a subject suffering from oculocutaneous/ocular albinism, the method comprising administering to the subject a pharmaceutically acceptable composition comprising NTBC in the range of between about 0.1 mg/kg/day to about 10 mg/kg/day.
U.S. Pat. No. 8,354,451B2 reports new methods of combating microbial infections due to fungi or bacteria by means of administration to a subject of a therapeutically active amount of NTBC.
WO2010054273 discloses NTBC-containing compositions and methods for the treatment and/or prevention of restless leg syndrome (RLS).
EP1853241B1 claims the use of NTBC in the treatment of a neurodegenerative disease, notably Parkinson disease.
Introne W. J., et al., disclosed usefulness of nitisinone in the treatment of alkaptonuria (Introne W. J., et al., Molec. Genet. Metab., 2011, 103, 4, 307). The key step of the synthesis reported in EP0591275B1 (now propriety of Swedish Orphan Biovitrum International AB, SE), involves the reaction of 2-nitro-4-trifluromethylbenzoyl chloride and cyclohexane-1,3-dione in the presence of triethylamine and then use of acetone cyanohydrin in order to promote the rearrangement of the key intermediate enol ester. After washing and extraction from CH2Cl2, the crude product is recrystallized from ethyl acetate to get the desired 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione as a solid having a melting point of 88-94° C.
Another patent (U.S. Pat. No. 4,695,673) filed in name of Stauffer Chemical Company disclosed a process of synthesis of acylated 1,3-dicarbonyl compounds in which the intermediate enol ester is isolated prior to its rearrangement into the final product, said rearrangement making use of a cyanohydrin compound derived from alkali metal, methyl alkyl ketone, benzaldehyde, cyclohexanone, C2-C5aliphatic aldehyde, lower alkyl silyl or directly by using hydrogen cyanide.
Yet another patent (U.S. Pat. No. 5,006,158) filed in name of ICI Americas Inc. disclosed a process similar to the one disclosed in U.S. Pat. No. 4,695,673 wherein the intermediate enol ester was isolated prior to its rearrangement into the final product by use of potassium cyanide. Said reaction can optionally be done by concomitant use of a phase transfer catalyst such as Crown ethers. The preferred solvent for conducting such a reaction is 1,2-dichloroethane.
Still a further patent (EP0805791) filed in name of Zeneca Ltd disclosed an alternative synthesis of nitisinone involving the reaction of 1,3-cyclohexanedione and variously substituted benzoyl chloride in the presence of sodium or potassium carbonate in CH3CN or DMF. Best yields were obtained using CH3CN as solvent and sodium carbonate as the base. Reaction was performed at 55-57° C. in 17 hours.
It is well known that one of the problems of the actual drug formulation (i.e., Orfadin® capsules) is its chemical instability. Indeed, even if Orfadin® has to be stored in a refrigerator at a temperature ranging from 2° C. to 8° C., its shelf life is of only 18 months. After first opening, the in-use stability is a single period of 2 months at a temperature not above 25° C., after which it must be discarded. It will be evident that such storage conditions have an impact in the distribution chain of the medicine, in terms of costs and also in terms of logistics for the patient. Therefore, there is an urgent need of more stable formulations, both from a logistic supply chain point of view, and from the patient compliance point of view. Since the formulation of Orfadin® contains only the active ingredient and starch as excipient, relative instability may be attributed to the active pharmaceutical ingredient itself; in other words it can derive from the way it is synthesized and/or the way it is extracted from the reaction mixture, and/or the way it is finally crystallized. Furthermore, some impurities may contribute to render the final product less stable overtime. Consequently, it is of major importance to identify a process of synthesis and/or a crystallization method that enable the reliable production of a highly pure and stable product.
Impurities as herein-above mentioned can derive either from the final product itself (through chemical degradation) or directly from the starting materials/solvents used in the process of synthesis. Regarding the latter option, it is therefore primordial to ascertain that at each step, impurities are completely removed in order not to get them at the final stage, also considering that some of them could potentially be cyto/genotoxic.
The impurities correlated to nitisinone can be either derived from the starting materials themselves (i.e., impurities 1 and 2) or obtained as side products during the process of synthesis and/or under storage conditions (i.e., impurities 3 to 5) and are the following:
-
- 2-nitro-4-(trifluoromethyl) benzoic acid (Impurity no 1),
- 1,3-cyclohexanedione (CHD) (Impurity no 2),
- 4-(trifluoromethyl)salicylic acid (Impurity no 3),
- 2-[3-nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione (Impurity no 4), and
- 6-trifluoromethyl-3,4-dihydro-2H-xanthene-1,9-dione (Impurity no 5).
Impurity-2, impurity-3, and impurity-5 have been previously reported in WO2015101794. Strangely, impurity-4 has never been reported, even if it is an obvious side-product which can easily be formed during the coupling reaction between 1,3-cyclohexanedione and 2-nitro-4-(trifluoromethyl) benzoic acid, the latter being not 100% pure but containing some amount of regioisomer 3-nitro-4-(trifluoromethyl) benzoic acid.
Potential genotoxicity of impurity no 4 which possesses an aromatic nitro moiety was assessed using in-silico techniques and resulted to be a potential genotoxic impurity. According to the FDA ICH M7 guidelines, daily intake of a mutagenic impurity (Threshold of Toxicological Concern, TTC) in an amount not greater than 1.5 μg per person is considered to be associated with a negligible risk to develop cancer over a lifetime of exposure. Consequently, assuming a daily dose of 2 mg/kg, for a person weighing 70 kg, the maximum tolerated impurity content of such a compound would be of about 11 ppm, as calculated according to the equation underneath.
concentration limit ( ppm ) = T T C ( µg / day ) Dose ( g / day )
It is therefore of paramount importance to ensure that the process of synthesis of nitisinone and the purification steps of the same give rise to an API devoid of such impurity no 4, or at least far below the threshold of 11 ppm as indicated above. The skilled person will understand that total absence of said impurity is highly desirable.
It is well known in the pharmaceutical field that investigation of potential polymorphism of a solid API is of crucial importance and is also recommended by major regulatory authorities such as FDA.
Notwithstanding the fact that nitisinone has been used for years to treat HT-1 patients, it appears that no NTBC formulation fully satisfies the requisites of stability and/or compliance standard for the patients. Therefore, there is an unmet medical need of long-term pure and stable formulations.
Example 1
Thionyl chloride (162 g, 1.36 mol) was added dropwise into a suspension of 2-nitro-4-trifluoromethylbenzoic acid (228 g, 0.97 mol) in toluene (630 g) at 80° C. The thus obtained solution was kept under stirring at 80° C. for 20 hours, and then cooled to 50° C. The volatiles were removed under reduced pressure in order to get the expected 2-nitro-4-trifluoromethylbenzoyl chloride as an oil. The latter, cooled to 25° C. was added dropwise to a suspension of 1,3-cyclohexanedione (109 g, 0.97 mol) and potassium carbonate (323 g, 2.33 mol) in CH3CN (607 g). After 18 h the mixture was diluted with water (500 ml) and slowly acidified to about pH=1 with HCl 37%. The mixture was then warmed to about 55° C. and the phases were separated. The organic layer was washed with a 10% aqueous solution of sodium chloride and then, concentrated under reduced pressure at a temperature below 55° C. to reach a volume of 380 ml. The thus obtained mixture was stirred at 55° C. for 1 h and then cooled to 0° C. in 16 to 18 h. The resulting solid was filtered and rinsed several times with pre-cooled (0° C.) toluene. The wet solid was dried at 60° C. under vacuum for 6 h to provide nitisinone (164 g) as a white to yellowish solid with a purity of 98.4% as measured by HPLC and a content of potentially genotoxic impurity no 4 of 6.1 ppm measured by HPLC/MS.
Example 2
Nitisinone as obtained from example 1 (164 g) was added to a 3/1 (w/w) mixture of CH3CN/toluene (volume of solvent: 638 ml). The mixture was warmed gently to about 55° C. under stirring until solids were completely dissolved. The solution was then concentrated under reduced pressure maintaining the internal temperature below 50° C. to reach a volume of 290 ml. Then, more toluene (255 g) was added and the solution was concentrated again under reduced pressure until the residual volume reached 290 ml. The solution was heated to about 55° C. for 1 h and successively cooled slowly in 10 to 12 h to 10° C. The resulting solid was filtered and rinsed several times with pre-cooled (0° C.) toluene. The wet solid was dried at about 60° C. under vacuum for 4 h to provide nitisinone (136 g) as a white to yellowish solid, with a purity of 99.94% and a 99.8% assay measured by HPLC and a d(90) particle size between 310 and 350 μm. The content of potential genotoxic impurity no 4 resulted below 1 ppm.
CLIP
Nitisinone – WikiVisually
WikiVisually
4-Hydroxyphenylpyruvate dioxygenase – Proposed Reaction Mechanism of HPPD

References
- ^ Jump up to:a b National Organization for Rare Disorders. Physician’s Guide to Tyrosinemia Type 1Archived 2014-02-11 at the Wayback Machine.
- Jump up^ “Nitisinone (Oral Route) Description and Brand Names”. Mayoclinic.com. 2015-04-01. Retrieved 2015-06-04.
- Jump up^ Sobi Orfadin® (nitisinone)
- ^ Jump up to:a b McKiernan, Patrick J (2006). “Nitisinone in the Treatment of Hereditary Tyrosinaemia Type 1”. Drugs. 66 (6): 743–50. doi:10.2165/00003495-200666060-00002. PMID 16706549.
- ^ Jump up to:a b Introne, Wendy J.; Perry, Monique B.; Troendle, James; Tsilou, Ekaterini; Kayser, Michael A.; Suwannarat, Pim; O’Brien, Kevin E.; Bryant, Joy; Sachdev, Vandana; Reynolds, James C.; Moylan, Elizabeth; Bernardini, Isa; Gahl, William A. (2011). “A 3-year randomized therapeutic trial of nitisinone in alkaptonuria”. Molecular Genetics and Metabolism. 103(4): 307–14. doi:10.1016/j.ymgme.2011.04.016. PMC 3148330
 . PMID 21620748.
. PMID 21620748.
- ^ Jump up to:a b “About DevelopAKUre | DevelopAKUre”. Developakure.eu. 2014-06-20. Archived from the original on 2015-05-12. Retrieved 2015-06-04.
- ^ Jump up to:a b “A Potential Drug – Nitisinone”. Akusociety.org. Archived from the original on 2015-05-05. Retrieved 2015-06-04.
- ^ Jump up to:a b Lock, E. A.; Ellis, M. K.; Gaskin, P.; Robinson, M.; Auton, T. R.; Provan, W. M.; Smith, L. L.; Prisbylla, M. P.; Mutter, L. C.; Lee, D. L. (1998). “From toxicological problem to therapeutic use: The discovery of the mode of action of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), its toxicology and development as a drug”. Journal of Inherited Metabolic Disease. 21 (5): 498–506. doi:10.1023/A:1005458703363. PMID 9728330.
- Jump up^ Kavana, Michael; Moran, Graham R. (2003). “Interaction of (4-Hydroxyphenyl)pyruvate Dioxygenase with the Specific Inhibitor 2-[2-Nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione†”. Biochemistry. 42 (34): 10238–45. doi:10.1021/bi034658b. PMID 12939152.
- Jump up^ “Newborn Screening”. Newbornscreening.info. 2013-05-14. Retrieved 2015-06-04.
- Jump up^ “What is Alkaptonuria?”. Akusociety.org. Archived from the original on 2015-04-05. Retrieved 2015-06-04.
- Jump up^ “Nitisinone (Oral Route) Side Effects”. Mayoclinic.com. 2015-04-01. Retrieved 2015-06-04.
- Jump up^ Phornphutkul, Chanika; Introne, Wendy J.; Perry, Monique B.; Bernardini, Isa; Murphey, Mark D.; Fitzpatrick, Diana L.; Anderson, Paul D.; Huizing, Marjan; Anikster, Yair; Gerber, Lynn H.; Gahl, William A. (2002). “Natural History of Alkaptonuria”. New England Journal of Medicine. 347 (26): 2111–21. doi:10.1056/NEJMoa021736. PMID 12501223.
- Jump up^ Preston, A. J.; Keenan, C. M.; Sutherland, H.; Wilson, P. J.; Wlodarski, B.; Taylor, A. M.; Williams, D. P.; Ranganath, L. R.; Gallagher, J. A.; Jarvis, J. C. (2013). “Ochronotic osteoarthropathy in a mouse model of alkaptonuria, and its inhibition by nitisinone”. Annals of the Rheumatic Diseases. 73 (1): 284–9. doi:10.1136/annrheumdis-2012-202878. PMID 23511227.
- Jump up^ “DevelopAKUre”. Developakure.eu. 2014-06-20. Retrieved 2015-06-04.
- Jump up^ “2012-005340-24”. Clinicaltrialsregister.eu. Retrieved 2015-06-04.
- Jump up^ “The Programme | DevelopAKUre”. Developakure.eu. 2014-06-20. Archived from the original on 2015-05-12. Retrieved 2015-06-04.
- Jump up^ “European Commission : CORDIS : Search : Simple”. Cordis.europa.eu. 2012-05-30. Retrieved 2015-06-04.
- Jump up^ Onojafe, Ighovie F.; Adams, David R.; Simeonov, Dimitre R.; Zhang, Jun; Chan, Chi-Chao; Bernardini, Isa M.; Sergeev, Yuri V.; Dolinska, Monika B.; Alur, Ramakrishna P.; Brilliant, Murray H.; Gahl, William A.; Brooks, Brian P. (2011). “Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinism”. Journal of Clinical Investigation. 121 (10): 3914–23. doi:10.1172/JCI59372. PMC 3223618 . PMID 21968110. Lay summary – ScienceDaily (September 26, 2011).
- Jump up^ “Nitisinone for Type 1B Oculocutaneous Albinism – Full Text View”. ClinicalTrials.gov. Retrieved 2015-06-04.
- Jump up^ G. Mitchell, D.W. Bartlett, T.E. Fraser, T.R. Hawkes, D.C. Holt, J.K. Townson, R.A. Wichert Mesotrione: a new selective herbicide for use in maize Pest Management Science, 57 (2) (2001), pp. 120–128
- ^ Jump up to:a b Moran, Graham R. (2005). “4-Hydroxyphenylpyruvate dioxygenase”. Archives of Biochemistry and Biophysics. 433 (1): 117–28. doi:10.1016/j.abb.2004.08.015. PMID 15581571.
- Jump up^ Ellis, M.K.; Whitfield, A.C.; Gowans, L.A.; Auton, T.R.; Provan, W.M.; Lock, E.A.; Smith, L.L. (1995). “Inhibition of 4-Hydroxyphenylpyruvate Dioxygenase by 2-(2-Nitro-4-trifluoromethylbenzoyl)-cyclohexane-1,3-dione and 2-(2-Chloro-4-methanesulfonylbenzoyl)-cyclohexane-1,3-dione”. Toxicology and Applied Pharmacology. 133 (1): 12–9. doi:10.1006/taap.1995.1121. PMID 7597701.
- Jump up^ Lindstedt, Sven; Odelhög, Birgit (1987). “4-Hydroxyphenylpyruvate dioxygenase from human liver”. In Kaufman, Seymour. Metabolism of Aromatic Amino Acids and Amines. Methods in Enzymology. 142. pp. 139–42. doi:10.1016/S0076-6879(87)42021-1. ISBN 978-0-12-182042-8. PMID 3037254.
- Jump up^ “Others | DevelopAKUre”. Developakure.eu. 2014-06-20. Retrieved 2015-06-04.
- Jump up^ Pr MDK-Nitisinone Summary Basis of Decisions, Health Canada 2016. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd-smd-2016-mdk-nitisinone-190564-eng.php
- Jump up^ Pr Nitisinone Tablets Regulatory Decision Summary Health Canada, 2016. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/rds-sdr/drug-med/rds-sdr-nitisinone-tab-193770-eng.php
- Jump up^ PrOrfadin Regulatory Decision Summary Health Canada, 2016. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/rds-sdr/drug-med/rds-sdr-orfadin-193226-eng.php
External links
Title: Nitisinone
CAS Registry Number: 104206-65-7
CAS Name: 2-[2-Nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione
Additional Names: NTBC
Trademarks: Orfadin (Swedish Orphan )
Molecular Formula: C14H10F3NO5
Molecular Weight: 329.23
Percent Composition: C 51.07%, H 3.06%, F 17.31%, N 4.25%, O 24.30%
Literature References: Herbicidal triketone that inhibits 4-hydroxyphenylpyruvate dioxygenase (HPPD), an enzyme involved in plastoquinone biosynthesis in plants and in tyrosine catabolism in mammals. Prepn: C. G. Carter, EP 186118 (1986 to Stauffer); idem, US 5006158 (1991 to ICI). Inhibition of HPPD in plants: M. P. Prisbylla et al., Brighton Crop Prot. Conf. – Weeds 1993, 731; in rats: M. K. Ellis et al., Toxicol. Appl. Pharmacol. 133, 12 (1995). LC determn in plasma: M. Bielenstein et al., J. Chromatogr. B 730,177 (1999). Clinical evaluation in hereditary tyrosinemia type I: S. Lindstedt et al., Lancet 340, 813 (1992). Review of toxicology and therapeutic development: E. A. Lock et al, J. Inherited Metab. Dis. 21, 498-506 (1998); of clinical experience: E. Holme, S. Lindstedt, ibid. 507-517.
Properties: Solid, mp 88-94°.
Melting point: mp 88-94°
Therap-Cat: In treatment of inherited tyrosinemia type I.
////////////////Nitisinone, ニチシノン , Orfadin, FDA 2002, NTBC , SC-0735 , SYN-118 , JAPAN 2015, JAP 2015, EU 2005, Priority, Orphan
[O-][N+](=O)C1=C(C=CC(=C1)C(F)(F)F)C(=O)C1C(=O)CCCC1=O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



















 The enantiocontrolled addition of phthalimide (I) to 1,3-butadiene monoepoxide (II) with a chiral palladium catalyst and Na2CO3 in dichloromethane gives N-(2-hydroxy-1(S)-vinylethyl)phthalimide (III), which is treated with triflic anhydride and TEA in dichloromethane to yield the triflate (IV). The condensation of (IV) with dimethyl malonate (V) by means of NaH in THF affords the alkylated malonate (VI), which is finally decarboxylated and deprotected by a treatment with aqueous refluxing HCl. Note that the synthesis of the biologically active (S)-enantiomer simply requires a change in the chirality of the Pd catalyst used in the first step of the synthesis.
The enantiocontrolled addition of phthalimide (I) to 1,3-butadiene monoepoxide (II) with a chiral palladium catalyst and Na2CO3 in dichloromethane gives N-(2-hydroxy-1(S)-vinylethyl)phthalimide (III), which is treated with triflic anhydride and TEA in dichloromethane to yield the triflate (IV). The condensation of (IV) with dimethyl malonate (V) by means of NaH in THF affords the alkylated malonate (VI), which is finally decarboxylated and deprotected by a treatment with aqueous refluxing HCl. Note that the synthesis of the biologically active (S)-enantiomer simply requires a change in the chirality of the Pd catalyst used in the first step of the synthesis.












































")