
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DAZDOTUFTIDE


DAZDOTUFTIDE
- TRS-01
- CAS 2522933-44-2
- 4-((E)-(5-(2-(2-((S)-2-((S)-1-(L-Threonyl-L-lysyl)pyrrolidine-2-carboxamido)-5-guanidinopentanamido)acetamido)-2-carboxyethyl)-2-hydroxyphenyl)diazenyl)phenyl (2-(trimethylammonio)ethyl) phosphate
- L-Tyrosine, L-threonyl-L-lysyl-L-prolyl-L-arginylglycyl-3-((1E)-2-(4-((hydroxy(2-(trimethylammonio)ethoxy)phosphinyl)oxy)phenyl)diazenyl)-, inner salt
- [4-[[5-[(2S)-2-[[2-[[(2S)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S,3R)-2-amino-3-hydroxybutanoyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]acetyl]amino]-2-carboxyethyl]-2-hydroxyphenyl]diazenyl]phenyl] 2-(trimethylazaniumyl)ethyl phosphate
C43H68N13O13P
1006.1 g/mol
L-Tyrosine, L-threonyl-L-lysyl-L-prolyl-L-arginylglycyl-3-[(1E)-2-[4-[[hydroxy[2-(trimethylammonio)ethoxy]phosphinyl]oxy]phenyl]diazenyl]-, inner salt
SQ
| 1 | TKPRGY |
Protein/Peptide Sequence, Sequence Length: 6
modified (modifications unspecified)
- OriginatorTarsius Pharma
- DeveloperTarsier Pharma
- ClassAnti-inflammatories; Eye disorder therapies; Small molecules
- Mechanism of ActionImmunomodulators
- Orphan Drug StatusYes – Uveitis
- Phase IIIUveitis
- Phase I/IIOcular inflammation
- PreclinicalDiabetic macular oedema; Diabetic retinopathy; Dry age-related macular degeneration
- 16 Jan 2024Tarsier Pharma receives an agreement from the US FDA under Special Protocol Assessment for Tarsier-04 phase III trial for TR S01 eye drops for Uveitis
- 13 Nov 2023Tarsier Pharma announces successful outcome of a Type C meeting with the US FDA supporting the advancement of TRS 01 eye drop for Uveitis
- 13 Nov 2023Tarsier Pharma plans a Tarsier-04 phase III registrational trial of TR S01 for Uveitis in USA

| Molecular Formula | C43H68N13O13P.C2HF3O2 |
| Molecular Weight | 1120.0764 |
TRS-01 trifluoroacetate
I35XEI0JIK
CAS 2522933-45-3
4-((E)-(5-(2-(2-((S)-2-((S)-1-(L-Threonyl-L-lysyl)pyrrolidine-2-carboxamido)-5-guanidinopentanamido)acetamido)-2-carboxyethyl)-2-hydroxyphenyl)diazenyl)phenyl (2-(trimethylammonio)ethyl) phosphate, trifluoroacetate salt
Ocular inflammation, an inflammation of any part of the eye, is one of the most common ocular diseases. Ocular inflammation refers to a wide range of inflammatory disease of the eye, one of them is uveitis. These diseases are prevalent in all age groups and may be associated with systemic diseases such as Crohn’s disease, Behcet disease, Juvenile idiopathic arthritis and others. The inflammation can also be associated with other common eye symptoms such as dry eye and dry macular degeneration. Several drugs have the known side effect of causing uveitis and/or dry eye. The most common treatment for ocular inflammation, is steroids and specifically corticosteroids. However, these treatments have several known and sometimes severe side effects.
Phosphorylcholine (PC) is a small zwitterionic molecule secreted by helminths which permits helminths to survive in the host inducing a situation of immune tolerance as well as on the surface of some bacteria and apoptotic cells. Tuftsin-PhosphorylCholine (TRS) is bi-specific small molecule with immunomodulatory activities. TRS (Thr-Lys-Pro-Arg-Gly-Tyr-PC) is an immunomodulating peptide derivative.
Currently, TRS has been synthesized by post-synthesis modification of Thr-Lys-Pro-Arg-Gly-Tyr, so as to couple the PC moiety to the phenol ring of tyrosine. However, this synthetic approach results in very low yield, thus making the synthesis of TRS ineffective and costly. New simple and efficient methods of synthesizing TRS are highly required.
SCHEME

PATENT
WO2022224259
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022224259&_cid=P11-MAOYY3-78105-1
EXAMPLES
EXAMPLE 1
CONJUGATION OF PHOSPHORYLCHOLINE TO BOC-TYR
[0151] 1) Preparation of diazonium salt
[0152] 4-Aminophenyl (2-(trimethylammonio)ethyl) phosphate (50 mg, 0.18 mmol)) was dissolved in 1M aqueous HC1 (1 mL), cooled in an ice-water bath and sodium nitrite (12.6 mg, 0.18 mmol) was added in a single batch. The resulting solution was stirred at 0°C for 30 min.
[0153] 2) Azo coupling
[0154] A new mixture was prepared with BOC-L-tyrosine (107 mg, 0.38 mmol) in NaHC03(lM)+NaOH buffer (pH 10) (3.3 mL) + acetonitrile (1.2 mL). The mixture was cooled in an ice-water bath. The diazonium salt mixture was added drop-wise. A red solution was formed. Stirring of this was continued at 0 °C for 6 minutes. The reaction mixture was acidified with IN aqueous HC1 to pH=~3.
[0155] The obtained solution was lyophilized overnight, and subsequently purified (e.g. by preparative MPLC), to obtain the compound:
, wherein R is Boc.
EXAMPLE 2
PREPARATION OF AN EXEMPLARY COMPOUND OF THE INVENITON
Preparation of diazonium salt:
Fmoc-Tyr-PPC
(compound 10)
[0156] 4-Aminophenyl (2-(trimethylammonio)ethyl) phosphate (250 mg, 0.912 mmol)) was dissolved in 1M aqueous HC1 (5 mL), cooled in an ice-water bath and sodium nitrite (62.9 mg, 0.912 mmol) was added in a single batch. The resulting solution was stirred at 0°C for 30 min. Azo coupling, a new mixture was prepared with Fmoc-Tyr-OH (739 mg, 1.832 mmol) in saturated NaHC03 (17 mL) + acetonitrile (12.5 mL). The resulting suspension/solution was cooled in an ice-water bath. The diazonium salt mixture was added drop-wise. Stirred at 0°C. The reaction mixture slowly turned yellow. After 5.5 h LCMS showed complete conversion. The reaction mixture was acidified with IN HC1 to pH~6, the yellowish suspension turned into a clear orange solution, which was lyophilized. This afforded 2.10 g. Dissolved in a mixture of DMSO/H20/MeCN (-1:1:1) and purified in 5 runs by acidic preparative MPLC. The fractions were combined and lyophilized overnight, to obtain the desired product (compound 10).
EXAMPLE 3
SPPS SYNTHESIS OF TRS
[0157] While facing difficulties with protection of the hydroxy group of compound 10, the inventors explored a novel strategy for SPPS synthesis of TRS :
[0158] The inventors initiated the SPPS synthesis by implementing the N-protected (Fmoc) phosphorylcholine modified tyrosine (e.g. compound 10) 200 mg of compound 10 were loaded onto the CTC resin. In brief, 2-Chlorotrityl chloride resin (1.0 – 1.2 mmol/g, 200 – 400 mesh) (450 mg, 1.441 mmol) was allowed to swell in dichloromethane (12 mL) by rocking for 30 min. The solvent was removed and a solution of (S,E)-4-((5-(2-((((9//-f1uoren-9-yl)methoxy)carbonyl)amino)-2-carboxyethyl)-2-hydroxyphenyl)diazenyl)phenyl(2-(trimethylammonio)-ethyl) phosphate (200 mg, 0.290 mmol) in dichloromethane (12 mL) containing DIPEA (0.177 mL, 1.016 mmol) (substrate did not dissolve in DCM, after addition of DIPEA a solution was obtained) was added.
[0159] After 17 h the solvent was removed and the resin was washed with dichloromethane (3×10 mL, each washing step > 2 minutes). The capping solution (CH2C12:MeOH: DIPEA 9: 1:0.5) was added (10.5 mL) and the resin was rocked for 1 hour. Then the resin was washed with dichloromethane (3×10 mL) and dried in vacuo.
[0160] This resin was then split into equal portions in order to investigate a number of conditions for the subsequent chemistry in parallel, aimed at preventing the formation of the previously found tyrosine O-acylation, as witnessed by the isolation of compound 13 (see Scheme 2). The different reaction conditions were outlined in Table 1 (see below).
Scheme 2: Solid phase peptide synthesis
Table 1: exemplary coupling conditions tested
[0161] As shown in Table 1, various coupling conditions have been tested. Entries a-c resulted in the formation of a substantial amount of the byproduct (13). An improvement was obtained by using Fmoc-Gly-OSu in DMF (entry d). In this case the formation of byproduct (13) was reduced to only 3% relative to the desired compound 12. Nonetheless, neither of these methods was capable of suppressing the formation of 13 completely, therewith still posing a risk for further peptide synthesis, as this may lead to the accumulation of byproducts (compound 13).
[0162] Surprisingly, the inventors found that the byproduct (or phenolic ester byproduct, represented by compound 13 in Scheme 3) can be cleaved under standard Fmoc deprotection conditions with piperidine or with DBU in DMF, affording compound 15 cleanly, as illustrated below:
/////////DAZDOTUFTIDE, PHASE 3, TRS-01, TRS 01
Rezafungin



Rezafungin
CAS 1396640-59-7
WeightAverage: 1226.411
Monoisotopic: 1225.602719729
Chemical FormulaC63H85N8O17
FDA APPROVED 3/22/2023, Rezzayo, To treat candidemia and invasive candidiasis
Drug Trials Snapshot
2-[[(3S,6S,9S,11R,15S,18S,20R,21R,24S,25S,26S)-6-[(1S,2S)-1,2-dihydroxy-2-(4-hydroxyphenyl)ethyl]-11,20,25-trihydroxy-3,15-bis[(1R)-1-hydroxyethyl]-26-methyl-2,5,8,14,17,23-hexaoxo-18-[[4-[4-(4-pentoxyphenyl)phenyl]benzoyl]amino]-1,4,7,13,16,22-hexazatricyclo[22.3.0.09,13]heptacosan-21-yl]oxy]ethyl-trimethylazanium
- Rezafungin ion
- Rezafungin cation
- CD-101
- SP-3025
- G013B5478J
Rezafungin, sold under the brand name Rezzayo (by Melinta Therapeutics), is a medication used for the treatment of invasive candidiasis.[2] It is an echinocandin antifungal[1][4] that acts as a fungal β-glucan synthase inhibitor.[5]
Rezafungin was approved for medical use in the United States in March 2023,[1][6][5] and in the European Union in December 2023.[2][3]

CAS No. : 1631754-41-0
Rezafungin acetate (Synonyms: Biafungin acetate; CD101 acetate; SP-3025 acetate)
Rezafungin acetate (Biafungin acetate) is a next-generation, broad-spectrum, and long-lasting echinocandin. Rezafungin acetate shows potent antifungal activity against Candida spp., Aspergillus spp., and Pneumocystis spp..
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Rezafungin (Rezzayo). Rezafungin (2) is a secondgeneration echinocandin that was discovered by Seachaid
Pharmaceuticals and developed by Cidera Therapeutics. The once weekly intravenously administered drug is used to treat candidemia and invasive candidiasis and to prevent invasive fungal diseases in blood and bone marrow transplant patients.23
Rezafungin was designed to improve the pharmacokinetic properties of the USFDA-approved first-generation echinocandins anidulafungin, caspofungin, and micafungin, enabling less frequent dosing. Mechanistically, echinocandins exert their antifungal activity by inhibiting β-(1→3)-glucan synthase, a
transmembrane protein complex essential for the synthesis of an important polysaccharide component of the fungal cell wall.
This noncompetitive inhibition destabilizes the cell wall, leading to osmotic imbalance and fungal cell death.24 Rezafungin was approved by the USFDA in March 2023 for use in patients 18 years and older.25
An elegant semisynthesis of rezafungin from anidulafungin (2.1) was reported by Cidera Therapeutics that circumvented chemical instability including potential racemization of the
parent compound (Scheme 3).26,27 The semisynthetic sequence26 begins with boronate formation between the 1,2-diol of 2.1 and 3,4-dimethoxyphenylborane (2.2) utilizing azeotropic distillation, maintaining a constant volume of THF. Addition of a solution of choline chloride, TFA, and TFAA in
MeCN to the slurry of boronate ester 2.3 gave the choline ether. Selective ether formation at the hemiaminal hydroxyl group occurred due to its increased reactivity compared to the other free hydroxyls in the compound.27 The specific boronate ester used in this sequence was found to be beneficial at minimizing the amount of a diastereomer impurity (at the hemiaminal) formed in the choline conjugation, though the authors of the patent shared that this was unexpected given the remote boronic
acid from the hemiaminal that participated in conjugation. A 95:5 α:β selectivity of the conjugation was achieved under acidic conditions, and preferential crystallization of the α-isomer while
maintaining solution equilibrium enabled control of the βisomer to less than 2.0%. Work up of the reaction using ammonium acetate and ammonium hydroxide provided crude
rezafungin. Ion exchange chromatography was used to remove3,4-dimethyoxyphenyl boronic acid, eluting with ammonium acetate to afford rezafungin (2). Using this synthetic sequence, a purity of 98.49% was reported with only minor amounts of racemization observed (0.77% undesired diastereomer and
0.51% unwanted epimer at the benzylic center).
(23) Syed, Y. Y. Rezafungin: first approval. Drugs 2023, 83, 833−840.
(24) Denning, D. W. Echinocandins: a new class of antifungal. J.
Antimicrob. Chemother. 2002, 49, 889−891.
(25) Cidara Therapeutics and Melinta Therapeutics announce FDA
approval of RezzayoTM (Rezafungin for injection) for the treatment of
candidemia and invasive candidiasis. Cidera Therapeutics, March 22,
- https://www.cidara.com/news/cidara-therapeutics-andmelinta-therapeutics-announce-fda-approval-of-rezzayo-rezafunginfor-injection-for-the-treatment-of-candidemia-and-invasivecandidiasis/ (accessed February 2024).
(26) Cidara Therapeutics. Synthesis of echinocandin antifungal agent.
WO 2019241626 A1, 2019.
(27) Jamison, J. A.; LaGrandeur, L. M.; Rodriguez, M. J.; Turner, W.
W.; Zeckner, D. J. The synthesis and antifungal activity of nitrogen
containing hemiaminal ethers of LY303366. J. Antibiot. (Tokyo) 1998,
51, 239−42

.
SYN
Hughes, D., et al. (2022). Synthesis of echinocandin antifungal agent. (U.S. Patent No. 11,524,980 B2). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/34/d5/c2/1a8cdcfb3fe3db/US11524980.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=US327113930&_cid=P11-MAORN7-73998-1
Example 9. Synthesis of Compound 1 from the 3,4-dimethoxyphenylboronate Ester of Anidulafungin—Coupling in the Presence of TFAA
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US10702573 | No | 2020-07-07 | 2033-03-14 |  |
| US9526835 | No | 2016-12-27 | 2033-03-14 | |
| US8722619 | No | 2014-05-13 | 2032-03-02 | |
| US11197909 | No | 2021-12-14 | 2038-07-14 | |
| US11654196 | No | 2023-05-23 | 2032-03-02 | |
| US11712459 | No | 2023-08-01 | 2037-03-15 | |
| US11819533 | No | 2023-11-21 | 2038-07-11 | |



Medical uses
In the United States, rezafungin is indicated in adults who have limited or no alternative options for the treatment of candidemia and invasive candidiasis.[1]
In the European Union, rezafungin is indicated for the treatment of invasive candidiasis in adults.[2]
Rezafungin, while remaining a hydrophilic compound, exhibits a volume of distribution more than twice that of caspofungin.[7] This pharmacokinetic property has supported its investigation for the treatment of deep-seated Candida infections, including osteomyelitis.[8][9]
Legal status
Rezafungin was approved for medical use in the United States in March 2023,[1][10][11] The FDA granted the application for rezafungin orphan drug, fast track, and priority review designations.[12]
In October 2023, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Rezzayo, intended for the treatment of invasive candidiasis in adults.[2] The applicant for this medicinal product is Mundipharma GmbH.[2] Rezafungin was approved for medical use in the European Union in December 2023.[3]
Rezafungin is a member of the family of echinocandins that inhibits 1,3-beta-D-glucan synthase. It is developed by Cidara Therapeutics and approved for the treatment of candidaemia and invasive candidiasis in patients aged >= 18 years who have limited or no alternative treatment options. It is an echinocandin, a quaternary ammonium ion, an antibiotic antifungal drug, an azamacrocycle, a homodetic cyclic peptide and an aromatic ether.
Brand names
Rezafungin is the international nonproprietary name.[13]
Rezafungin is sold under the brand name Rezzayo.[2]
References
- ^ Jump up to:a b c d e “Rezzayo- rezafungin injection, powder, lyophilized, for solution”. DailyMed. 8 June 2023. Retrieved 26 December 2023.
- ^ Jump up to:a b c d e f g “Rezzayo EPAR”. European Medicines Agency (EMA). 12 October 2023. Retrieved 27 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c “Rezzayo Product information”. Union Register of medicinal products. 22 December 2023. Retrieved 26 December 2023.
- ^ Zhao Y, Perlin DS (September 2020). “Review of the Novel Echinocandin Antifungal Rezafungin: Animal Studies and Clinical Data”. Journal of Fungi. 6 (4): 192. doi:10.3390/jof6040192. PMC 7712954. PMID 32998224.
- ^ Jump up to:a b Syed YY (June 2023). “Rezafungin: First Approval”. Drugs. 83 (9): 833–840. doi:10.1007/s40265-023-01891-8. PMID 37212966. S2CID 258831091.
- ^ “Rezzayo approved by FDA amid rapid Candida auris spread”. thepharmaletter.com. 23 March 2023.
- ^ Albanell-Fernández M (January 2025). “Echinocandins Pharmacokinetics: A Comprehensive Review of Micafungin, Caspofungin, Anidulafungin, and Rezafungin Population Pharmacokinetic Models and Dose Optimization in Special Populations”. Clinical Pharmacokinetics. 64 (1): 27–52. doi:10.1007/s40262-024-01461-5. PMC 11762474. PMID 39707078.
- ^ Grasselli Kmet N, Luzzati R, Monticelli J, Babich S, Conti J, Bella SD (March 2025). “Salvage therapy of complicated Candida albicans spondylodiscitis with Rezafungin”. European Journal of Clinical Microbiology & Infectious Diseases. doi:10.1007/s10096-025-05117-5. PMID 40163284.
- ^ Viceconte G, Buonomo AR, Esposito N, Cattaneo L, Somma T, Scirocco MM, et al. (April 2024). “Salvage Therapy with Rezafungin for Candida parapsilosis Spondylodiscitis: A Case Report from Expanded Access Program”. Microorganisms. 12 (5): 903. doi:10.3390/microorganisms12050903. PMC 11123963. PMID 38792732.
- ^ “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 22 December 2023. Retrieved 27 December 2023.
- ^ “Drug Approval Package: Rezzayo”. U.S. Food and Drug Administration (FDA). 18 April 2023. Retrieved 27 December 2023.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 79”. WHO Drug Information. 32 (1). hdl:10665/330941.
External links
- “Rezafungin Injection”. U.S. Food and Drug Administration. 18 April 2023.
| Clinical data | |
|---|---|
| Trade names | Rezzayo |
| Other names | Biafungin; CD101 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623021 |
| License data | US DailyMed: Rezafungin |
| Routes of administration | Intravenous |
| Drug class | Antifungal |
| ATC code | J02AX08 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Pharmacokinetic data | |
| Excretion | Feces |
| Identifiers | |
| CAS Number | 1396640-59-7 |
| PubChem CID | 78318119 |
| DrugBank | DB16310 |
| UNII | G013B5478J |
| KEGG | D11197 |
| ChEBI | CHEBI:229680 |
| Chemical and physical data | |
| Formula | C63H85N8O17+ |
| Molar mass | 1226.412 g·mol−1 |
- Lamoth F: Novel Therapeutic Approaches to Invasive Candidiasis: Considerations for the Clinician. Infect Drug Resist. 2023 Feb 22;16:1087-1097. doi: 10.2147/IDR.S375625. eCollection 2023. [Article]
- Miesel L, Lin KY, Ong V: Rezafungin treatment in mouse models of invasive candidiasis and aspergillosis: Insights on the PK/PD pharmacometrics of rezafungin efficacy. Pharmacol Res Perspect. 2019 Nov 20;7(6):e00546. doi: 10.1002/prp2.546. eCollection 2019 Dec. [Article]
- Thompson GR 3rd, Soriano A, Cornely OA, Kullberg BJ, Kollef M, Vazquez J, Honore PM, Bassetti M, Pullman J, Chayakulkeeree M, Poromanski I, Dignani C, Das AF, Sandison T, Pappas PG: Rezafungin versus caspofungin for treatment of candidaemia and invasive candidiasis (ReSTORE): a multicentre, double-blind, double-dummy, randomised phase 3 trial. Lancet. 2023 Jan 7;401(10370):49-59. doi: 10.1016/S0140-6736(22)02324-8. Epub 2022 Nov 25. [Article]
- Ong V, Wills S, Watson D, Sandison T, Flanagan S: Metabolism, Excretion, and Mass Balance of [(14)C]-Rezafungin in Animals and Humans. Antimicrob Agents Chemother. 2022 Jan 18;66(1):e0139021. doi: 10.1128/AAC.01390-21. Epub 2021 Oct 18. [Article]
- FDA Approved Drug Products: REZZAYO (rezafungin) injection for intravenous use (March 2023) [Link]
- Globe News Wire: Cidara Therapeutics and Melinta Therapeutics Announce FDA Approval of REZZAYO (rezafungin for injection) for the Treatment of Candidemia and Invasive Candidiasis [Link]
- EMA Summary of Product Characteristics: REZZAYO (rezafungin) solution for infusion [Link]
//////////Rezafungin, Rezzayo, APROVALS 2023, FDA 2023, Rezafungin ion, Rezafungin cation, CD 101, SP 3025, G013B5478J
Davelizomib



Davelizomib
| Molecular Weight | 481.25 |
|---|---|
| Formula | C21H26BF2N3O7 |
| CAS No. | 2409841-51-4 |
{(4S)-2-[(1R)-1-{2-[(2S)-1-(2,4-difluorophenyl)azetidine-2- carboxamido]acetamido}-3-methylbutyl]-5-oxo-1,3,2- dioxaborolan-4-yl}acetic acid proteasome inhibitor, antineoplastic
2-[(4S)-2-[(1R)-1-[[2-[[(2S)-1-(2,4-difluorophenyl)azetidine-2-carbonyl]amino]acetyl]amino]-3-methylbutyl]-5-oxo-1,3,2-dioxaborolan-4-yl]acetic acid
- 1,3,2-Dioxaborolane-4-acetic acid, 2-[(1R)-1-[[2-[[[(2S)-1-(2,4-difluorophenyl)-2-azetidinyl]carbonyl]amino]acetyl]amino]-3-methylbutyl]-5-oxo-, (4S)-
- 2-[(4S)-2-[(1R)-1-[[2-[[(2S)-1-(2,4-difluorophenyl)azetidine-2-carbonyl]amino]acetyl]amino]-3-methylbutyl]-5-oxo-1,3,2-dioxaborolan-4-yl]acetic acid
T3LN9U6BRF
Davelizomib is proteasome inhibitor with antineoplastic effect.
DAVELIZOMIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
Multiple myeloma (MM) is a malignant proliferative disease of plasma cells, characterized by abnormal proliferation of clonal plasma cells in the bone marrow, destruction of hematopoietic function, stimulation of osteolytic lesions in the bones, detection of monoclonal immunoglobulins or their fragments (M protein) in serum and/or urine, and clinical manifestations of bone pain, anemia, hypercalcemia, renal impairment, infection, and bleeding. Bortezomib is a reversible proteasome inhibitor that achieves the purpose of treating multiple myeloma by promoting apoptosis of myeloma cells. However, in the long-term treatment process, some multiple myeloma patients have developed resistance to bortezomib. Therefore, there is still a need for new, safe, and highly stable drugs for the treatment of multiple myeloma.
SCHEME

PATENT
Borate of azetidine derivative
Publication Number: JP-2021531302-A
Priority Date: 2018-08-02
WO2020025037

Step 1: Synthesis of compound 4-3
[0252]N, N-diisopropylethylamine (22.02 g) was added to a solution of acetonitrile (200 mL) containing compound 4-1 (10 g) and compound 4-2 (20.13 g) at room temperature. The reaction mixture was stirred at 100 ° C for 16 hours, then cooled to room temperature and then added to ethyl acetate. The organic layer was washed with water and saturated brine, respectively, and then the organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated to remove the solvent, and the residue was purified by silica gel column chromatography (mobile phase: petroleum ether: ethyl acetate = 10: 1) to obtain compound 4-3. Compound 4-3: MS (ESI) m/z: 227.9 [M+1].
[0253]Step 2: Synthesis of compound 4-4
[0254]
LiOH·H 2 O (6.65 g) was added to a mixed solution of compound 4-3 (7.2 g) in methanol (20 mL), tetrahydrofuran (20 mL) and water (10 mL) at 0°C. The reaction mixture was stirred at room temperature for 1 hour, then concentrated under reduced pressure, diluted with water and ethyl acetate, and separated. The aqueous layer was adjusted to pH=6 with 1 mol/L hydrochloric acid, and then extracted with ethyl acetate. The organic phases were combined and washed with saturated brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to remove the solvent to obtain compound 4-4, which was directly used in the next step. Compound 4-4: MS (ESI) m/z: 213.9 [M+1].
[0255]Step 3: Synthesis of compound 4-5
[0256]Glycine methyl ester hydrochloride (1.06 g), TBTU (2.71 g) and N,N-diisopropylethylamine (3.64 g) were added to a solution of compound 4-4 (1.5 g) in dichloromethane (50 mL) at -10°C. The reaction mixture was stirred at -10°C to 0°C for 3 hours, then diluted with water (40 mL) and extracted with dichloromethane. The organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to remove the solvent, and the residue was purified by silica gel column chromatography (mobile phase: petroleum ether: ethyl acetate = 5:1) to obtain compound 4-5. Compound 4-5: MS (ESI) m/z: 284.9 [M+1].
[0257]Step 4: Synthesis of Compound 4-6
[0258]To a mixed solution of compound 4-5 (0.5 g) in tetrahydrofuran (2 mL), methanol (2 mL) and water (1 mL) was added LiOH·H
2 O (369.03 mg) at 0°C. The reaction mixture was stirred at 0°C to 20°C for 2 hours, then concentrated, diluted with water (3 mL), and separated. The aqueous layer was adjusted to pH=6 with 1 mol/L hydrochloric acid and extracted with ethyl acetate. The organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to remove the solvent to obtain compound 4-6, which was directly used in the next step. Compound 4-6: MS (ESI) m/z: 270.9 [M+1].
[0259]Step 5: Synthesis of Compound 4-8
[0260]N,N-diisopropylethylamine (273.56 mg) was added to a solution of compound 4-6 (0.26 g), compound 2-6 (437.84 mg) and TBTU (370.71 mg) in dichloromethane (10 mL) at -10 ° C. The reaction mixture was slowly warmed to room temperature and continued to stir for 2 hours, then the reaction mixture was added to water (10 mL) for dilution and extracted with dichloromethane. The organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to remove the solvent, and the residue was purified by silica gel column chromatography (mobile phase: petroleum ether: ethyl acetate = 1: 1) to obtain compound 4-8. Compound 4-8: MS (ESI) m/z: 518.2 [M+1].
[0261]Step 6: Synthesis of Compound 4-9
[0262]Isobutylboric acid (234.45 mg) and 1 mol/L HCl (1.31 mL) were added to a mixed solution of methanol (4 mL) and n-hexane (6 mL) of compound 4-8 (0.17 g) at 0°C. The reaction mixture was slowly warmed to room temperature and stirred for 12 hours, then concentrated under reduced pressure to remove the solvent to obtain a residue. The residue was purified by preparative HPLC and separated by SFC to obtain compound 4-9. Compound 4-9:
1 H NMR (400MHz, METHANOL-d4) δ6.83(br s,2H),6.61(br s,1H),4.49(br s,1H),4.10(br s,3H),3.84(br s,1H),2.75(br s,1H),2.59(br s,1H),2.48(br s,1H),1.62(br s,1H),1.30(br s,2H),0.92(br s,6H). MS(ESI)m/z:366.1[M-17].
[0263]Preparative HPLC separation method of compound 4-9:
[0264]Column: Xtimate C18 150×25mm, 5μm;
[0265]Mobile phase: water (0.225% FA)-MeOH;
[0266]Elution gradient: 61%-85%;
[0268]Preparation of compound 4-9 SFC separation method:
[0269]Chromatographic column: C2 250mm×30mm, 10μm;
[0270]Mobile phase: A: carbon dioxide, B: methanol;
[0271]Elution gradient B%: 30%-30%;
[0273]The elution order of compound 4-9 is the second peak appearing in high performance chiral liquid column chromatography.
[0274]Step 7: Synthesis of Compound I-1
[0275]Method 1: Add L-malic acid (332 mg) to isopropyl acetate (2.5 mL), heat to 70°C and stir, and after 10 minutes, add compound 4-9 (1.0 g) dissolved in 2.5 mL isopropyl acetate solution. Then stop heating, cool to 25°C and continue stirring at this temperature for 5 days. Filter, collect the filter cake, and vacuum dry to obtain compound I-1, which is Form I crystal of compound I-1.
[0276]Method 2: Add compound I-1 (68.9 g) to a reaction flask, then add 440 mL of isopropyl acetate, and stir the mixture at room temperature for 24 h under nitrogen protection. Filter and dry to obtain Form I crystals of compound I-1 (64.4 g). The X-ray powder diffraction pattern of the obtained crystals using Cu Kα rays is shown in Figure 1.
[0277]
化合物I-1: 1H NMR(400MHz,DMSO-d 6)δ12.30(br s,1H),10.65(br s,1H),8.57(br t,J=5.77Hz,1H),7.11(ddd,J=2.64,9.16,12.30Hz,1H),6.91(br t,J=8.16Hz,1H),6.53(dt,J=5.65,9.60Hz,1H),4.44(br t,J=7.91Hz,1H),4.37(dd,J=3.89,7.65Hz,1H),4.10(br s,2H),3.91-4.01(m,1H),3.76(q,J=7.36Hz,1H),2.61(br d,J=10.79Hz,2H),2.19-2.44(m,3H),1.61(td,J=6.71,13.68Hz,1H),1.20-1.36(m,2H),0.86(t,J=6.02Hz,6H)。
///////Davelizomib, T3LN9U6BRF, PHASE 2
Omaveloxolone



Omaveloxolone
CAS
1474034-05-3
N-[(4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-octadecahydropicen-4a-yl]-2,2-difluoropropanamide
N-[(4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,3,4,5,6,7,8,8a,14a,14b-decahydropicen-4a-yl]-2,2-difluoropropanamide
FDA 2023, 2/28/2023, To treat Friedrich’s ataxia
Drug Trials Snapshot
WeightAverage: 554.723
Monoisotopic: 554.331999611
Chemical FormulaC33H44F2N2O3
- RTA 408
- RTA-408
- OriginatorDartmouth College; University of Texas M. D. Anderson Cancer Center
- DeveloperBiogen
- ClassAnalgesics; Anti-inflammatories; Antineoplastics; Eye disorder therapies; Neuroprotectants; Small molecules; Triterpenes
- Mechanism of ActionNF-E2-related factor 2 stimulants
- Orphan Drug StatusYes – Friedreich’s ataxia; Malignant melanoma
- MarketedFriedreich’s ataxia
- Phase IIMitochondrial disorders; Ocular inflammation; Ocular pain
- Phase I/IIMalignant melanoma
- PreclinicalBrain disorders
- DiscontinuedDuchenne muscular dystrophy; Non-small cell lung cancer; Radiation-induced skin damage
- 08 Apr 2025Biogen completes a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT06612879)
- 17 Mar 2025Registered for Friedreich’s ataxia (In adolescents, In adults) in Canada (PO)
- 18 Oct 2024Biogen initiates enrolment in a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT06612879)
Omaveloxolone, sold under the brand name Skyclarys, is a medication used for the treatment of Friedreich’s ataxia.[2][5] It is taken by mouth.[2]
The most common side effects include an increase in alanine transaminase and an increase of aspartate aminotransferase, which can be signs of liver damage, headache, nausea, abdominal pain, fatigue, diarrhea and musculoskeletal pain.[5]
Omaveloxolone was approved for medical use in the United States in February 2023,[2][5][6][7][8] and in the European Union in February 2024.[3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[9]
SYNTHESIS

PATENT
Sheikh, AY et al. (2018). Bardoxolonmethyl-2,2-difluoropropionamide derivatives, polymorphe forms and procedures for use thereof. DK/EP 2989114 T3. Danish Patent and Trademark Office. Available at https://patentimages.storage.googleapis.com/51/87/43/97d0fb3e69ee73/DK2989114T3.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP159939262&_cid=P21-MAKI10-93498-1

[0164] Reagents and conditions: (a) (PhO) 2PON 3 (DPPA), triethylamine, toluene, 0 °C for 5 minutes, then ambient temperature overnight, ∼94%; (b) benzene, 80 °C for 2 hours; (c) HCl, CH 3CN, ambient temperature for 1 hour; (d) CH 3CF 2CO 2H, dicyclohexylcarbodiimide, 4-(dimethylamino)pyridine, CH 2Cl 2, ambient temperature overnight, 73% from RTA 401 (4 steps).
[0165]Compound 1: RTA 401 (20.0 g, 40.6 mmol), triethylamine (17.0 mL, 122.0 mmol), and toluene (400 mL) were added into a reactor and cooled to 0 °C with stirring. Diphenyl phosphoryl azide (DPPA) (13.2 mL, 61.0 mmol) was added with stirring at 0 °C over 5 minutes, and the mixture was continually stirred at room temperature overnight (HPLC-MS check shows no RTA 401 left). The reaction mixture was directly loaded on a silica gel column and purified by column chromatography (silica gel, 0% to 5% ethyl acetate in CH 2Cl 2) to give compound 1 (19.7 g, ∼94%, partially converted into compound 2) as a white foam.
[0166]Compound 2: Compound 1 (19.7 g, ∼38.1 mmol) and benzene (250 mL) were added into a reactor and heated to 80 °C with stirring for 2 hours (HPLC-MS check shows no compound 1 left). The reaction mixture was concentrated at reduced pressure to afford crude compound 2 as a solid residue, which was used for the next step without purification.
[0167]Compound 3: Crude compound 2 (≤38.1 mmol) and CH 3CN (200 mL) were added into a reactor and cooled to 0 °C with stirring. HCl (12 N, 90 mL) was added at 0 °C over 1 minute, and the mixture was continually stirred at room temperature for 1 hour (HPLC-MS check shows no compound 2 left). The reaction mixture was cooled to 0 °C and 10% NaOH (∼500 mL) was added with stirring. Then, saturated NaHCO 3 (1 L) was added with stirring. The aqueous phase was extracted by ethyl acetate (2×500 mL). The combined organic phase was washed by H 2O (200 mL), saturated NaCl (200 mL), dried over Na 2SO 4, and concentrated to afford crude compound 3 (16.62 g) as a light yellow foam, which was used for the next step without purification.
[0168]RTA 408: Crude amine 3 (16.62 g, 35.9 mmol), CH 3CF 2CO 2H (4.7388 g, 43.1 mmol), and CH 2Cl 2 (360 mL) were added into a reactor with stirring at room temperature. Then, dicyclohexylcarbodiimide (DCC) (11.129 g, 53.9 mmol) and 4-(dimethylamino)pyridine (DMAP) (1.65 g, 13.64 mmol) were added and the mixture was continually stirred at room temperature overnight (HPLC-MS check shows no compound 3 left). The reaction mixture was filtered to remove solid by-products, and the filtrate was directly loaded on a silica gel column and purified by column chromatography (silica gel, 0% to 20% ethyl acetate in hexanes) twice to give compound RTA 408 (16.347 g, 73% from RTA 401 over 4 steps) as a white foam: 1H NMR (400 MHz, CD 3Cl) δ ppm 8.04 (s, 1H), 6.00 (s, 1H), 5.94 (s, br, 1H), 3.01 (d, 1H, J = 4.8 Hz), 2.75-2.82 (m, 1H), 1.92-2.18 (m, 4H), 1.69-1.85 (m, 7H), 1.53-1.64 (m, 1H), 1.60 (s, 3H), 1.50 (s, 3H), 1.42 (s, 3H), 1.11-1.38 (m, 3H), 1.27 (s, 3H), 1.18 (s, 3H), 1.06 (s, 3H), 1.04 (s, 3H), 0.92 (s, 3H); m/z 555 (M+1).
SYNTHESIS
J. Med. Chem. 2025, 68, 2147−2182
Omaveloxolone (Skyclarys). Omaveloxolone (6) was approved in February 2023 for the treatment of Friedreich’s Ataxia (FRDA), a genetic, neurodegenerative disease. Patients with FRDA have lowered activity of the frataxin gene (FXN), attributed to an expansion of a guanine-adenine-adenine (GAA)
triplet. The resulting decrease in frataxin limits the production of iron−sulfur clusters, leading to accumulation of iron in the mitochondria and oxidative stress which in turn leads to cell damageanddeath.49
Omaveloxoloneactivates the nuclear factor erythroid 2-related factor 2 (Nrf2), an important pathway in
oxidative stress. It acts by preventing ubiquitination and subsequent degradation of Nrf2, keeping levels high enough to counteract the oxidative stress associated with FRDA. 50
Omaveloxolone was developed by Reata Pharmaceuticals (which was acquired by Biogen in September 2023) and was granted orphan drug, fast track, priority review, and rare pediatric disease designations. 51Omaveloxolone (6) is a semisynthetic triterpenoid based on the oleanolic acid scaffold.52
advanced intermediate 6.1,The synthesis started from the53also known as CDDO orbardoxolone, which has individually been investigated fortherapeutic benefits from Nrf2 activation (Scheme 10).
Treatment of acid 6.1 with DPPA produced the azide, and subsequent heating in benzene generated isocyanate 6.2 via aCurtius rearrangement. Hydrolysis with aqueous acid generated amine 6.3, and an amidation with 2,2-difluoropropanoic acid produced omaveloxolone (6). A yield of 73% over the sequence was reported, and intermediates were used crude with no purification between steps.
(49) Ghanekar, S. D.; Miller, W. W.; Meyer, C. J.; Fenelon, K. J.;
Lacdao, A.; Zesiewicz, T. A. Orphan drugs in development for the
treatment of Friedreich’s ataxia: focus on omaveloxolone. Degener.
Neurol. Neuromuscular Dis. 2019, 9, 103−107.
(50) Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-inducer
prevents mitochondrial defects and oxidative stress in Friedreich’s
ataxia models. Front. Cell. Neurosci. 2018, 12, 188.
(51) Lee,A.Omaveloxolone:first approval. Drugs 2023, 83, 725−729.
(52) Anderson, E.; Decker, A.; Liu, X. Synthesis, pharmaceutical use,
and characterization of crystalline forms of 2,2-difluoropropionamide
derivatives of bardoxolone methyl. WO 2013163344, 2013.
(53) Honda, T.; Rounds, B. V.; Gribble, G. W.; Suh, N.; Wang, Y.;
Sporn, M. B. Design and synthesis of 2-cyano-3,12-dioxoolean-1,9
dien-28-oic acid, a novel and highly active inhibitor of nitric oxide
production in mouse macrophages. Bioorg. Med. Chem. Lett. 1998, 8,
2711−2714.

SYN
European Journal of Medicinal Chemistry 265 (2024) 116124
Omaveloxolone (Skyclarys)
Omaveloxolone was granted FDA approval on February 28, 2023, to treat Friedrich’s ataxia in individuals aged 16 and older [2]. Omaveloxolone possesses antioxidant and anti-inflammatory properties, making it a semi-synthetic triterpenoid compound. It has the ability to function as a stimulator of nuclear factor-erythroid 2 related factor 2(Nrf2), a transcription factor that reduces oxidative stress. In individuals
suffering from FA, a genetic disorder characterized by mitochondrial dysfunction, the Nrf2 pathway is compromised, leading to a decrease in Nrf2 activity. Hence, Omaveloxolone, an Nrf2 activator, can be
employed as a therapeutic option for the management of these in dividuals [23].The process route of Omaveloxolone is described below in Scheme 724]. The substitution reaction of carboxylic acid OMAV-001 with diphenylphosphoryl azide (DPPA) gave the acyl azide OMAV-002,which underwent Curtius-rearrangement under heating conditions to produce isocyanate OMAV-003. The amine OMAV-004 was obtained under acidic conditions. OMAV-004 was condensed with 2,2-difluoro propionic acid to obtain the final product Omaveloxolone.
[23] B.L. Probst, I. Trevino, L. McCauley, R. Bumeister, I. Dulubova, W.C. Wigley, D.
A. Ferguson, RTA 408, A novel synthetic triterpenoid with broad anticancer and
anti-inflammatory activity, PLoS One 10 (2015) e0122942.
[24] E. Anderson, A. Decker, X. Liu Synthesis, Pharmaceutical Use, and
Characterization of Crystalline Forms of 2,2-difluoropropionamide Derivatives of
Bardoxolone Methyl, 2013. WO2013163344.

.
Medical uses
Omaveloxolone is indicated for the treatment of Friedreich’s ataxia.[2][5]
Friedreich’s ataxia causes progressive damage to the spinal cord, peripheral nerves, and the brain, resulting in uncoordinated muscle movement, poor balance, difficulty walking, changes in speech and swallowing, and a shortened lifespan.[5] The condition can also cause heart disease.[5] This disease tends to develop in children and teenagers and gradually worsens over time.[5]
Although rare, Friedreich’s ataxia is the most common form of hereditary ataxia in the United States, affecting about one in every 50,000 people.[5]
Mechanism of action
The mechanism of action of omaveloxolone and its related compounds has been demonstrated to be through a combination of activation of the antioxidative transcription factor Nrf2 and inhibition of the pro-inflammatory transcription factor NF-κB.[10]
Nrf2 transcriptionally regulates multiple genes that play both direct and indirect roles in producing antioxidative potential and the production of cellular energy (i.e., adenosine triphosphate or ATP) within the mitochondria. Consequently, unlike exogenously administered antioxidants (e.g., vitamin E or Coenzyme Q10), which provide a specific and finite antioxidative potential, omaveloxolone, through Nrf2, broadly activates intracellular and mitochondrial antioxidative pathways, in addition to pathways that may directly increase mitochondrial biogenesis (such as PGC1α) and bioenergetics.[11]
History
Omaveloxolone is a second generation member of the synthetic oleanane triterpenoid compounds and in clinical development by Reata Pharmaceuticals. Preclinical studies have demonstrated that omaveloxolone possesses antioxidative and anti-inflammatory activities[10][12] and the ability to improve mitochondrial bioenergetics.[11] Omaveloxolone is under clinical investigation for a variety of indications, including Friedreich’s ataxia, mitochondrial myopathies, immunooncology, and prevention of corneal endothelial cell loss following cataract surgery.
The efficacy and safety of omaveloxolone was evaluated in a 48-week randomized, placebo-controlled, and double-blind study [Study 1 (NCT02255435)] and an open-label extension.[5] Study 1 enrolled 103 individuals with Friedreich’s ataxia who received placebo (52 individuals) or omaveloxolone 150 mg (51 individuals) for 48 weeks.[5] Of the research participants, 53% were male, 97% were white, and the mean age was 24 years at study entry.[5] Nine (18%) patients were younger than age 18.[5] The primary objective was to evaluate the change in the modified Friedreich’s Ataxia Rating Scale (mFARS) score compared to placebo at week 48.[5] The mFARS is a clinical assessment that measures disease progression, namely swallowing and speech (bulbar), upper limb coordination, lower limb coordination, and upright stability.[5] Individuals receiving omaveloxolone performed better on the mFARS than people receiving placebo.[5]
The US Food and Drug Administration (FDA) granted the application for omaveloxolone orphan drug, fast track, priority review, and rare pediatric disease designations.[5][9]
Society and culture
Legal status
Omaveloxolone was approved for medical use in the United States in February 2023.[2][5]
In December 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Skyclarys, intended for the treatment of Friedreich’s ataxia.[3] The applicant for this medicinal product is Reata Ireland Limited.[3] Omaveloxolone was approved for medical use in the European Union in February 2024.[3][4]
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ Jump up to:a b c d e f “Skyclarys- omaveloxolone capsule”. DailyMed. 12 May 2023. Archived from the original on 1 July 2023. Retrieved 16 December 2023.
- ^ Jump up to:a b c d e “Skyclarys EPAR”. European Medicines Agency (EMA). 14 December 2023. Archived from the original on 15 December 2023. Retrieved 16 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Skyclarys product information”. Union Register of medicinal products. 12 February 2024. Retrieved 19 February 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA approves first treatment for Friedreich’s ataxia”. U.S. Food and Drug Administration (FDA). 28 February 2023. Archived from the original on 1 March 2023. Retrieved 28 February 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Reata Pharmaceuticals Announces FDA Approval of Skyclarys (Omavaloxolone), the First and Only Drug Indicated for Patients with Friedreich’s Ataxia”. Reata Pharmaceuticals Inc. (Press release). 28 February 2023. Archived from the original on 1 March 2023. Retrieved 28 February 2023.
- ^ Lee A (June 2023). “Omaveloxolone: First Approval”. Drugs. 83 (8): 725–729. doi:10.1007/s40265-023-01874-9. PMID 37155124. S2CID 258567442. Archived from the original on 9 December 2023. Retrieved 16 December 2023.
- ^ Subramony SH, Lynch DL (May 2023). “A Milestone in the Treatment of Ataxias: Approval of Omaveloxolone for Friedreich Ataxia”. Cerebellum. 23 (2): 775–777. doi:10.1007/s12311-023-01568-8. PMID 37219716. S2CID 258843532.
- ^ Jump up to:a b New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Jump up to:a b Reisman SA, Lee CY, Meyer CJ, Proksch JW, Ward KW (July 2014). “Topical application of the synthetic triterpenoid RTA 408 activates Nrf2 and induces cytoprotective genes in rat skin”. Archives of Dermatological Research. 306 (5): 447–454. doi:10.1007/s00403-013-1433-7. PMID 24362512. S2CID 25733020.
- ^ Jump up to:a b Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, et al. (July 2011). “Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis”. Free Radical Biology & Medicine. 51 (1): 88–96. doi:10.1016/j.freeradbiomed.2011.03.027. PMC 3109235. PMID 21457778.
- ^ Reisman SA, Lee CY, Meyer CJ, Proksch JW, Sonis ST, Ward KW (May 2014). “Topical application of the synthetic triterpenoid RTA 408 protects mice from radiation-induced dermatitis”. Radiation Research. 181 (5): 512–520. Bibcode:2014RadR..181..512R. doi:10.1667/RR13578.1. PMID 24720753. S2CID 23906747.
External links
Clinical trial number NCT02255435 for “RTA 408 Capsules in Patients With Friedreich’s Ataxia – MOXIe” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Skyclarys |
| Other names | RTA 408 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Omaveloxolone |
| Routes of administration | By mouth |
| ATC code | N07XX25 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3][4] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1474034-05-3 |
| PubChem CID | 71811910 |
| IUPHAR/BPS | 7573 |
| DrugBank | DB12513 |
| ChemSpider | 34980948 |
| UNII | G69Z98951Q |
| KEGG | D10964 |
| ChEBI | CHEBI:229661 |
| CompTox Dashboard (EPA) | DTXSID101138251 |
| Chemical and physical data | |
| Formula | C33H44F2N2O3 |
| Molar mass | 554.723 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Zesiewicz TA, Hancock J, Ghanekar SD, Kuo SH, Dohse CA, Vega J: Emerging therapies in Friedreich’s Ataxia. Expert Rev Neurother. 2020 Dec;20(12):1215-1228. doi: 10.1080/14737175.2020.1821654. Epub 2020 Sep 21. [Article]
- Jiang Z, Qi G, Lu W, Wang H, Li D, Chen W, Ding L, Yang X, Yuan H, Zeng Q: Omaveloxolone inhibits IL-1beta-induced chondrocyte apoptosis through the Nrf2/ARE and NF-kappaB signalling pathways in vitro and attenuates osteoarthritis in vivo. Front Pharmacol. 2022 Sep 27;13:952950. doi: 10.3389/fphar.2022.952950. eCollection 2022. [Article]
- Shekh-Ahmad T, Eckel R, Dayalan Naidu S, Higgins M, Yamamoto M, Dinkova-Kostova AT, Kovac S, Abramov AY, Walker MC: KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain. 2018 May 1;141(5):1390-1403. doi: 10.1093/brain/awy071. [Article]
- Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC, Ferguson DA: RTA 408, A Novel Synthetic Triterpenoid with Broad Anticancer and Anti-Inflammatory Activity. PLoS One. 2015 Apr 21;10(4):e0122942. doi: 10.1371/journal.pone.0122942. eCollection 2015. [Article]
- Lynch DR, Farmer J, Hauser L, Blair IA, Wang QQ, Mesaros C, Snyder N, Boesch S, Chin M, Delatycki MB, Giunti P, Goldsberry A, Hoyle C, McBride MG, Nachbauer W, O’Grady M, Perlman S, Subramony SH, Wilmot GR, Zesiewicz T, Meyer C: Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann Clin Transl Neurol. 2018 Nov 10;6(1):15-26. doi: 10.1002/acn3.660. eCollection 2019 Jan. [Article]
- Zighan M, Arkadir D, Douiev L, Keller G, Miller C, Saada A: Variable effects of omaveloxolone (RTA408) on primary fibroblasts with mitochondrial defects. Front Mol Biosci. 2022 Aug 12;9:890653. doi: 10.3389/fmolb.2022.890653. eCollection 2022. [Article]
- FDA Approved Drug Products: SKYCLARYS (omaveloxolone) capsules for oral use (February 2023) [Link]
- EMA Approved Drug Products: Skyclarys (omaveloxolone) Oral Capsules [Link]
- Health Canada Approved Drug Products: SKYCLARYS (Omaveloxolone) Capsules For Oral Use [Link]
///////////Omaveloxolone, Skyclarys, Friedrich’s ataxia, FDA 2023, APPROVALS 2023, RTA 408, RTA-408, omaveloxolona, RTA 408, 63415, PP415, orphan drug, fast track, priority review, rare pediatric disease



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Dasminapant




Dasminapant
CAS 1570231-89-8
| Molecular Weight | 1157.40 |
|---|---|
| Formula | C60H72N10O10S2 |
| APG-1387, SM-1387, E53VN70K2X, INN 12430, APG-1387 UNII-E53VN70K2X APG-1387 (SMAC MIMETIC) SMAC-mimetic APG-1387 IAP Inhibitor APG-1387 |
| (5S,5’S,8S,8’S,10aR,10’aR)-3,3′-[1,3-phenylenebis(sulfonyl)]bis{N-(diphenylmethyl)-5-[(2S)-2-(methylamino)propanamido]-6-oxodecahydropyrrolo[1,2-a][1,5]diazocine-8-carboxamide} |
(5S,8S,10aR)-3-[3-[[(5S,8S,10aR)-8-(benzhydrylcarbamoyl)-5-[[(2S)-2-(methylamino)propanoyl]amino]-6-oxo-1,2,4,5,8,9,10,10a-octahydropyrrolo[1,2-a][1,5]diazocin-3-yl]sulfonyl]phenyl]sulfonyl-N-benzhydryl-5-[[(2S)-2-(methylamino)propanoyl]amino]-6-oxo-1,2,4,5,8,9,10,10a-octahydropyrrolo[1,2-a][1,5]diazocine-8-carboxamide
Dasminapant (APG-1387), a bivalent SMAC mimetic and an IAP antagonist, blocks the activity of IAPs family proteins (XIAP, cIAP-1, cIAP-2, and ML-IAP). Dasminapant induces degradation of cIAP-1 and XIAP proteins, as well as caspase-3 activation and PARP cleavage, which leads to apoptosis. Dasminapant can be used for the research of hepatocellular carcinoma, ovarian cancer, and nasopharyngeal carcinoma.
Dasminapant, also known as APG-1387 and SM-1387, is a IAP inhibitor. APG-1387 promotes the rapid degradation of cIAP1/2 and XIAP, and it exerts an antitumor effect on nasopharyngeal carcinoma cancer stem cells. Further studies show that APG-1387 enhances the chemosensitivity and promotes apoptosis in combination with CDDP and 5-FU of NPC in vitro and vivo.
PATENTS
WO2022012671
PATENT
WO2014031487 …
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014031487&_cid=P11-MAJOJ5-33000-1
PATENT
US20140057924


SCHEME

///////////////Dasminapant, APG-1387, SM-1387, E53VN70K2X, INN 12430, APG 1387, UNII-E53VN70K2X, APG-1387 (SMAC MIMETIC), SMAC-mimetic APG-1387, IAP Inhibitor APG-1387, SM 1387
Crisugabalin



Crisugabalin
Cas 2209104-84-5
2-[(1S,2S,3R,6S,8S)-2-(aminomethyl)-2-tricyclo[4.2.1.03,8]nonanyl]acetic acid
WeightAverage: 209.289
Monoisotopic: 209.141578856
Chemical FormulaC12H19NO2
- HSK 16149
- HSK-16149
- HSK16149
- Q3MK7E8686
- PHASE 2
Tricyclo[4.2.1.03,8]nonane-2-acetic acid, 2-(aminomethyl)-, (1S,2S,3R,6S,8S)-
(1S,2S,3R,6S,8S)-2-(Aminomethyl)tricyclo[4.2.1.03,8]nonane-2-acetic acid
- (1S,2S,3R,6S,8S)-2-(Aminomethyl)tricyclo[4.2.1.03,8]nonane-2-acetic acid
- 2-[(1S,2S,3R,6S,8S)-2-(aminomethyl)-2-tricyclo[4.2.1.03,8]nonanyl]acetic acid
- Tricyclo[4.2.1.03,8]nonane-2-acetic acid, 2-(aminomethyl)-, (1S,2S,3R,6S,8S)-
Crisugabalin (HSK16149) is a selective GABA analog in development for the treatment of chronic pain. It has a wider therapeutic index than pregabalin, which has a similar mechanism of action. In China, it was approved in May 2024 for the treatment of diabetic peripheral neuropathic pain[1] and approved in July 2024 for the treatment of postherpetic neuralgia.[2] In the United States, it is in Phase III trials as of 2023.[3][4] The drug can be administered with or without food.[5]
Crisugabalin is under investigation in clinical trial NCT06490484 (Efficacy and Safety of HSK16149 Capsule in Chinese Patients With Diabetic Peripheral Neuropathic Pain Who Had an Inadequate Response to Pregabalin).
SCHEME

PATENTS
WO2020029762
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020029762&_cid=P10-MAI1TM-34428-1


[0414]2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.0
3,8 ]nonanyl-2-yl)acetate benzenesulfonate (1:1) (Compound 1)
[0415]
2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.0 3,8]nonan-2-yl)acetic acid compound with benzenesulfonic acid(1:1)
[0416]
[0417]
[0418]Step 1: 3-(Cyclohexyl-3-en-1-yl)propanoic acid (1B)
[0419]
3-(cyclohex-3-en-1-yl)propanoic acid
[0420]
[0421]Anhydrous formic acid (18.82kg, 409.09mol) was pumped into a 100-liter reactor. The temperature was lowered to 10°C. Triethylamine (16.53kg, 163.64mol) was added dropwise to the reaction solution. After addition, it was stirred for 20 minutes. When the internal temperature was 10°C, cycloisopropyl malonate (7.86kg, 54.55mol) was added to the reactor. Then 3-cyclohexene-1-carboxaldehyde (6.00kg, 54.55mol) was added dropwise to the reaction solution at an internal temperature of 40°C. After addition, the temperature was raised to 140-150°C and the reaction was continued until no gas was released. The pH of the reaction solution was adjusted to 1-2 with 6N hydrochloric acid (24.0L). The aqueous phase was extracted with dichloromethane (12L×2), and the organic phases were combined and washed with saturated brine (10L×2). The organic phase was dried over anhydrous sodium sulfate (2.0 kg) for 1 hour, filtered, and the filtrate was concentrated and evaporated to dryness to obtain a yellow oil 1B (8.80 kg).
[0422]
1H NMR(400MHz,CDCl 3)δ10.23(s,1H),5.73–5.55(m,2H),2.46–2.30(m,2H),2.09–1.96(m,2H),1.81–1.53(m,6H),1.35–1.17(m,1H)。
[0423]
[0424]Step 2: 3-(Cyclohexyl-3-en-1-yl)-1-(pyrrolidin-1-yl)propyl-1-one (1C)
[0425]
3-(cyclohex-3-en-1-yl)-1-(pyrrolidin-1-yl)propan-1-one
[0426]
[0427]Dissolve 1B (11.20kg, 72.727mol) in dichloromethane (60.0L) and pump into a 100L reactor. Add DMF (3.0mL) and drop oxalyl chloride (9.046kg, 71.272mol) into the reaction solution. After addition, stir at room temperature for 2.0 hours. Add tetrahydropyrrole (5.689kg, 79.999mol) and triethylamine (8.814kg, 87.272mol) dropwise into the reactor. Control the internal temperature below 10℃, after addition, stir at room temperature overnight. Cool the reaction solution to 10℃. Add 3N hydrochloric acid (20.0L) dropwise to adjust the pH of the reaction solution to between 1-2. Let stand, separate the liquids, and extract the aqueous phase with dichloromethane (10.0L×1). The organic phases were combined and washed with 5% sodium hydroxide solution (10.0 L x 1) and saturated ammonium chloride solution (20.0 L x 1) in sequence. The organic phase was dried over anhydrous sodium sulfate (2.0 kg) for 30 minutes, filtered, and the filtrate was concentrated to obtain brown liquid 1C (15.00 kg, yield 99.6%).
[0428]
1H NMR(400MHz,CDCl 3)δ5.73–5.56(m,2H),3.43(dd,4H),2.37–2.22(m,2H),2.16–2.01(m,4H),1.90(dt,4H),1.81–1.51(m,6H),1.30–1.15(m,2H)。
[0429]
[0430]Step 3: Tricyclo[4.2.1.0
3,8 ]nonanyl-2-one (1R,3S,6R,8R and 1S,3R,6S,8S racemate) (1D)
[0431]
tricyclo[4.2.1.0 3,8]nonan-2-one(1R,3S,6R,8R and 1S,3R,6S,8S racemate)
[0432]
[0433]Dissolve 1C (5.64kg, 27.22mol) in dichloromethane (40.0L) and pump it into a 100L reactor. Cool to -10°C and add 2,4,6-trimethylpyridine (4.94kg, 40.83mol). Add a dichloromethane solution (16.0L) of trifluoromethanesulfonic anhydride (11.50kg, 40.83mol) dropwise to the reaction solution until complete. Heat and reflux for 12 hours. After the reaction is complete as detected by the central control, add an aqueous solution (23.0L) of sodium hydroxide (3.10kg, 77.5mol) dropwise to the reaction solution and adjust the pH of the reaction solution to between 10-11. Continue to reflux for 5-6 hours. Stand and separate the liquids, extract the aqueous phase with dichloromethane (5.0L×1), and combine the organic phases. Pump the organic phase into the reactor and cool to 10°C. 2.0N hydrochloric acid solution (20.0L) was added dropwise to adjust the pH of the reaction solution to between 1 and 2. The solution was separated by standing, and the organic phase was washed with saturated brine (20L×1), concentrated, and the residue was dissolved with acetone (20.0L), then pumped into a 100L reactor and stirred, and a solution of concentrated sulfuric acid (4.0L) and water (20.0L) was added dropwise, and refluxed for 2 hours after addition. The temperature was lowered to 15°C, saturated brine (20.0L) was added to the reaction solution, and extracted with n-hexane (15.0L×2). The organic phases were combined, washed with saturated brine (20.0L×1), and the organic phase was dried over anhydrous sodium sulfate overnight. After filtration, the filtrate was concentrated under reduced pressure to obtain a yellow solid crude product 1D (3.00kg, yield 81%) with a purity of 50%.
[0434]1D Further purification steps:
[0435]Method 1: Anhydrous sodium bisulfite (5.735 kg, 55.147 mol) was dissolved in 66 L of purified water and added to a 100 L reactor. A solution of crude 1D (3.00 kg, 22.059 mol) in ethanol (3.0 L) was added under stirring at room temperature. The mixture was stirred overnight at room temperature and extracted with ethyl acetate (20 L × 2). The aqueous phase was added to the reactor, stirred and cooled to 10°C. A solution of sodium hydroxide (2.250 kg, 56.250 mol) in water (10 L) was added dropwise. The pH was adjusted to 10-12. The mixture was stirred at room temperature for 2 hours. The mixture was extracted with n-hexane (20 L × 2). The organic phases were combined and washed with purified water (20 L × 1). The organic phases were dried with anhydrous sodium sulfate (2 kg) for 1 hour, filtered, and the filtrate was evaporated to dryness to obtain 1D as a colorless crystalline solid (2.7 kg, yield 90%) with a purity of 98.3%.
[0436]Method 2: Sodium bisulfite (1529g, 14.706mol) was dissolved in 22L water, and a solution of 1D crude product (1000g, 7.353mol) in anhydrous ethanol (1000mL) was added dropwise under stirring, and stirred overnight at room temperature (24 hours). The reaction solution was extracted with dichloromethane (5L×2) to remove impurities, and sulfuric acid solution (prepared with 6.4L concentrated sulfuric acid and 6kg crushed ice) was added dropwise to the aqueous phase, and stirred at room temperature for 5 hours. The reaction solution was extracted with n-hexane (extracted 3-4 times, 4L each time), the organic phases were combined and washed with saturated sodium chloride aqueous solution (5L×2), the organic phases were dried with 1kg anhydrous sodium sulfate for 2 hours, filtered, and the filtrate was evaporated to dryness to obtain 1D, a white solid (900g, yield: 90%), and the purity was determined to be 98.1%.
[0437]
1H NMR(400MHz,CDCl 3)δ3.39(m,1H),3.19(m,1H),2.77(m,1H),2.38(m,1H),2.05(m,1H),1.93(d,1H),1.77(m,1H),1.45(m,4H),1.20(m,1H)。
[0438]
[0439]Step 4: tert-Butyl 2-(tricyclo[4.2.1.0
3,8 ]nonanyl-2-ylidene) acetate (1R,3S,6R,8R and 1S,3R,6S,8S racemate) (1E)
[0440]
tert-butyl 2-tricyclo[4.2.1.0 3,8]nonan-2-ylidene)acetate(1R,3S,6R,8R and 1S,3R,6S,8S racemate)
[0441]
[0442]Potassium tert-butoxide (742.0g, 6.62mol) and tetrahydrofuran (6.20L) were added to a 20L reactor. The temperature was lowered to 5°C, and tert-butyl dimethoxyphosphonoacetate (1480g, 6.62mol, 1.1eq) was added dropwise to the reaction solution. The reaction temperature was controlled at 10°C-15°C, and stirring was continued for 1 hour. Then, a solution of 1D (820.0g, 6.02mol, 1.0eq) in tetrahydrofuran (2.0L) was added dropwise to the reaction solution. The addition was completed within 1 hour, and the temperature was naturally raised to room temperature for reaction for 2 hours. Saturated ammonium chloride solution (2.0L) and purified water (2.0L) were added to the reactor in sequence. After stirring for 20 minutes, the mixture was allowed to stand for stratification, and the aqueous phase was extracted with methyl tert-butyl ether (1.5L×2). The organic phases were combined, washed with saturated brine (2L×2), and dried over anhydrous sodium sulfate. Filtration and concentration afforded 1E as a yellow liquid (1.50 kg).
[0443]
[0444]Step 5: tert-Butyl 2-(2-(nitromethyl)tricyclo[4.2.1.0
3,8 ]nonanyl-2-yl)acetate (1R,2R,3S,6R,8R and 1S,2S,3R,6S,8S racemate) (1F)
[0445]
tert-butyl 2-(2-(nitromethyl)tricyclo[4.2.1.03,8]nonan-2-yl)acetate(1R,2R,3S,6R,8R and 1S,2S,3R,6S,8S racemate)
[0446]
[0447]1E (1.40 kg, 5.97 mol, 1.0 eq), nitromethane (1.82 kg, 29.85 mol, 5.0 eq) and dimethyl sulfoxide (9.8 L) were added to a 20 L reactor in sequence. Stir and add cesium carbonate (2.34 kg, 7.16 mol, 1.2 eq) to the reaction solution. After the addition, heat to 80°C-85°C, continue to keep the reaction for 5 hours, then cool to room temperature, add purified water (20.0 L) to the reactor, and extract the aqueous phase with methyl tert-butyl ether (8.0 L × 3). Combine the organic phases, wash with saturated brine (8.0 L × 2), and dry over anhydrous sodium sulfate. Filter and concentrate to obtain a brown liquid 1F (1.62 kg, yield: 92%).
[0448]
[0449]Step 6: tert-Butyl 2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.0
3,8 ]nonanyl-2-yl)acetate (S)-2-acetoxy-2-phenylacetic acid (1H)
[0450]
tert-butyl2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.03,8]nonan-2-yl)acetate(S)-2-acetoxy-2-phenylacetate
[0451]
[0452]Add 1F (730.0 g, 2.47 mol) and methanol (7.3 L) to a 50 L reactor. Stir, add nickel chloride hexahydrate (118 g, 0.49 mol, 0.2 eq) to the reaction, cool the reaction solution to 5 ° C, add sodium borohydride (374 g, 9.88 mol, 4.0 eq) in batches, keep the reaction system temperature at 20 ° C-30 ° C, and add it in about 3 hours. After the addition, continue to stir and react for 2 hours. Add ice water (16.4 L) to the reactor, and filter the aqueous phase with diatomaceous earth. Extract the filtrate with dichloromethane (3.0 L × 2) and combine the organic phases, wash with saturated brine (4 L × 1), and dry over anhydrous sodium sulfate. Filter, add (S)-(+)-O-acetyl-L-mandelic acid (384 g, 1.97 mol, 0.8 eq) to the filtrate, and stir for 20 minutes after the addition. The organic phase was concentrated by distillation until no solvent was evaporated, and then stirred with isopropanol (5.9 L) for 2 hours, cooled to 5°C and stirred for 1 hour. Filtered, the filter cake was washed with isopropanol (0.4 L × 1), and dried to obtain a white solid product 1H crude product (422 g, yield: 34.96%). The solid was taken and the ee value was determined to be 48.35% after derivatization.
[0453]First crystallization: Add crude product 1H (420.0 g, 0.92 mol), isopropanol (4.20 L) and water (0.21 L) to the reactor in sequence. Raise the temperature to 82 °C to completely dissolve the solid and keep warm for 0.5 hours. Cool down to 20 °C for crystallization for about 6 hours. When the internal temperature reaches 20 °C, filter and wash the filter cake with isopropanol (0.40 L × 1). Combine the solids and dry them at 60-65 °C for 4 hours to constant weight. Obtain the first crystal of 1H (260 g, yield: 62%). After taking the solid for derivatization, the ee value is 81.25%.
[0454]Second crystallization: Add the first crystal of 1H (177g, 0.39mol), isopropanol (2.53L) and water (0.126L) to the reactor in sequence. Raise the temperature to 82℃ to completely dissolve the solid and keep warm for 0.5 hours. Cool down to 20℃ for crystallization for about 4.5 hours. When the internal temperature reaches 30℃, filter and wash the filter cake with isopropanol (0.10L×1). Combine the solids and dry them at 60-65℃ for 4 hours to constant weight. Obtain pure white solid 1H (128g, yield: 72%). After taking the solid derivative, the ee value is determined to be 99.73%.
[0455]
[0456]Step 7: 2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.0
3,8 ]nonanyl-2-yl)acetic acid benzenesulfonic acid compound (1:1) (Compound 1)
[0457]
2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.0 3,8]nonan-2-yl)acetic acid compound with benzenesulfonic acid(1:1)
[0458]
[0459]Add 1H pure product (100.0g, 0.218mol) and purified water (0.8L) to the reactor in sequence and cool to 0-10℃. When the internal temperature reaches 0-10℃, add 1mol/L NaOH (218mL) aqueous solution to the reactor and adjust the pH of the reaction solution to 9-10. Let stand for stratification and extract the aqueous phase with dichloromethane (0.30L×2). Combine the organic phases and wash with 1mol/L NaOH (0.10L×1) solution and saturated brine (0.15L×1) in sequence. Add activated carbon (5.0g) to the organic phase for decolorization and dry with anhydrous sodium sulfate. Filter, concentrate the filtrate, and dissolve the residue in the concentration kettle with acetonitrile (280mL). Prepare a solution of benzenesulfonic acid monohydrate (77.0g, 0.437mol) with purified water (280mL) and add it dropwise to the above acetonitrile solution until complete. The temperature was raised to 80-85°C and kept for 4-6 hours. The reaction solution was cooled to 10-20°C for crystallization for about 4-6 hours. When the internal temperature reached 10-20°C, the solution was filtered and the filter cake was washed with water (30 mL × 1) and acetonitrile (50 mL × 1) in turn. After drying, compound 1 was obtained as a white solid (72 g, yield: 90%).
[0460]
1H NMR(400MHz,MeOD)δ7.83(m,2H),7.42(m,3H),3.31(dt,4H),2.86(m,1H),2.55(d,2H),2.48(ddd,1H),2.32(dd,1H),2.15(m,1H),2.04(m,1H),1.77(m,1H),1.62(m,4H),1.45(m,1H),1.28(dt,1H)。
[0461]
LCMS m/z=210.1[M+1]。
References
^ “Monthly Report: New Drug Approval in China, May 2024”.
- ^ “海思科苯磺酸克利加巴林胶囊获批新适应症”. PhIRDA. 19 July 2024. Retrieved 26 April 2025.
- ^ Gou, Xiaoli; Yu, Xiaojuan; Bai, Dongdong; Tan, Bowei; Cao, Pingfeng; Qian, Meilin; Zheng, Xiaoxiao; Chen, Lei; Shi, Zongjun; Li, Yao; Ye, Fei; Liang, Yong; Ni, Jia (March 2021). “Pharmacology and Mechanism of Action of HSK16149, a Selective Ligand of α2δ Subunit of Voltage-Gated Calcium Channel with Analgesic Activity in Animal Models of Chronic Pain”. The Journal of Pharmacology and Experimental Therapeutics. 376 (3): 330–337. doi:10.1124/jpet.120.000315. ISSN 1521-0103. PMID 33293377.
- ^ Guo, Xiaohui; Zhang, Tingting; Yuan, Geheng; Yukun, LI; Hua Ma, Jian; Hong-Mei, LI (2023). “224-OR: The Efficacy and Safety of HSK 16149 in Chinese with Diabetic Peripheral Neuropathic Pain—A Randomized, Double-Blinded, Placebo and Pregabalin-Controlled Phase II/III Study”. Diabetes. 72. doi:10.2337/db23-224-OR.
- ^ Wu, Qingqing; Zhu, Huijuan; Song, Rong; Zhang, Mengqi; Li, Fangqiong; Zeng, Weifang; Wang, Wei; Jia, Jingying; Yu, Chen; Liu, Yanmei (June 2023). “Effect of a high-fat and high-calorie food on the pharmacokinetics of a novel, potent GABA analog HSK16149 in healthy subjects”. Pharmacology Research & Perspectives. 11 (3): e01102. doi:10.1002/prp2.1102. PMC 10199234. PMID 37208866.
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| CAS Number | 2209104-84-5 |
| UNII | Q3MK7E8686 |
| Chemical and physical data | |
| Formula | C12H19NO2 |
| Molar mass | 209.289 g·mol−1 |
//////////Crisugabalin, HSK 16149, HSK-16149, HSK16149, Q3MK7E8686, PHASE 2
Elacestrant


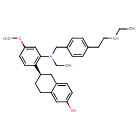
Elacestrant
(6R)-6-[2-[ethyl-[[4-[2-(ethylamino)ethyl]phenyl]methyl]amino]-4-methoxyphenyl]-5,6,7,8-tetrahydronaphthalen-2-ol
(6R)-6-{2-[ethyl({4-[2-(ethylamino)ethyl]phenyl}methyl)amino]-4-methoxyphenyl}-5,6,7,8-tetrahydronaphthalen-2-ol
FDA 1/27/2023, Orserdu
WeightAverage: 458.646
Monoisotopic: 458.293328472
Chemical FormulaC30H38N2O2
To treat estrogen receptor-positive, human epidermal growth factor receptor 2-negative, ESR1-mutated, advanced or metastatic breast cancer with disease progression following at least one line of endocrine therapy
Drug Trials Snapshot
- CAS 722533-56-4
- RAD-1901
- ER-306323
- FM6A2627A8
- WHO 10247
Elacestrant, sold under the brand name Orserdu, is a selective estrogen receptor degrader (SERD) used in the treatment of breast cancer.[1][4] It is taken by mouth.[1][4]
Elacestrant is an antiestrogen that acts as an antagonist of estrogen receptors, which are the biological targets of endogenous estrogens like estradiol.[1] The most common side effects of elacestrant include body pain, nausea and vomiting, increased serum lipids, elevated liver enzymes, fatigue, decreased hemoglobin, raised creatinine, decreased appetite, diarrhea, headache, constipation, abdominal pain, and hot flashes.[2]
Elacestrant was approved for medical use in the United States in January 2023,[1][2][5][6] and in the European Union in September 2023.[3][7]
PATENTS
Cruskie MP, et al. (2019). Polymorphic forms of RAD1901-2HCl (U.S. Patent No. 10,385,008 B2). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/42/82/b6/e9fcbbbd08054e/US10385008.pdf
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US10071066 | No | 2018-09-11 | 2034-10-10 | |
| US10385008 | No | 2019-08-20 | 2038-01-05 | |
| US10420734 | No | 2019-09-24 | 2034-10-10 | |
| US10745343 | No | 2020-08-18 | 2038-01-05 | |
| US11779552 | No | 2023-10-10 | 2034-10-10 | |
| US11819480 | No | 2023-11-21 | 2036-11-29 | |
| US7612114 | No | 2009-11-03 | 2026-08-18 | |
| US8399520 | No | 2013-03-19 | 2023-12-25 | |

PATENT
https://patents.google.com/patent/US10385008B2/en
Medical uses
Elacestrant is indicated for the treatment of postmenopausal women or adult men with estrogen receptor (ER)-positive, human epidermal growth factor receptor 2 (HER2)-negative, ESR1–mutated, advanced or metastatic breast cancer with disease progression following at least one other line of endocrine therapy.[2][4]
Pharmacology
Pharmacodynamics
Elacestrant is an antiestrogen that acts as an antagonist of estrogen receptors, specifically targeting the estrogen receptor alpha (ERα), which is the biological target of endogenous estrogens like estradiol.[1] Additionally, elacestrant is a selective estrogen receptor degrader (SERD), meaning it induces the degradation of ERα.[1][8]
Pharmacokinetics
Elacestrant has an oral bioavailability of approximately 10%.[1] Its plasma protein binding exceeds 99% and remains independent of concentration.[1] Elacestrant is metabolized in the liver, primarily by the cytochrome P450 enzyme CYP3A4 and to a lesser extent by CYP2A6 and CYP2C9.[1] The elimination half-life of elacestrant is 30 to 50 hours.[1] It is excreted primarily in feces (82%) and to a lesser extent in urine (7.5%).[1]
History
The efficacy of elacestrant was evaluated in the EMERALD trial, which was a randomized, open-label, active-controlled, multicenter study involving 478 postmenopausal women and men with ER-positive, HER2-negative advanced or metastatic breast cancer. Among them, 228 participants had ESR1 mutations. Eligible participants had experienced disease progression on one or two prior lines of endocrine therapy, including one line with a CDK4/6 inhibitor, and could have received up to one prior line of chemotherapy in the advanced or metastatic setting.[2]
Participants were randomly assigned in a 1:1 ratio to receive either elacestrant 345 mg orally once daily or investigator’s choice of endocrine therapy. The choices for the control arm included fulvestrant, or an aromatase inhibitor. Randomization was stratified based on whether the ESR1 mutation was detected or not, prior treatment with fulvestrant, and presence of visceral metastasis.[2]
The FDA granted the application for elacestrant priority review and fast track designations.[2]
Research
It is a nonsteroidal combined selective estrogen receptor modulator (SERM) and selective estrogen receptor degrader (SERD) (described as a “SERM/SERD hybrid (SSH)”) that was discovered by Eisai and is under development by Radius Health and Takeda for the treatment estrogen receptor (ER)-positive advanced breast cancer.[9] Elacestrant has dose-dependent, tissue-selective estrogenic and antiestrogenic activities, with biphasic weak partial agonist activity at the ER at low doses and antagonist activity at higher doses.[10] It shows agonistic activity on bone and antagonistic activity on breast and uterine tissues.[11] Unlike the SERD fulvestrant, elacestrant is able to readily cross the blood-brain-barrier into the central nervous system, where it can target breast cancer metastases in the brain,[10][11] and is orally bioavailable and does not require intramuscular injection.[10][11]
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Orserdu- elacestrant tablet, film coated”. DailyMed. 8 February 2023. Archived from the original on 11 February 2023. Retrieved 11 February 2023.
- ^ Jump up to:a b c d e f g “FDA approves elacestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer”. U.S. Food and Drug Administration (FDA). 27 January 2023. Archived from the original on 2 February 2023. Retrieved 1 February 2023. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Orserdu Product information”. Union Register of medicinal products. 18 September 2023. Retrieved 1 October 2023.
- ^ Jump up to:a b c d “Orserdu EPAR”. European Medicines Agency (EMA). 9 October 2023. Retrieved 9 October 2023.
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/217639Orig1s000ltr.pdf Archived 2023-02-02 at the Wayback Machine This article incorporates text from this source, which is in the public domain.
- ^ “Stemline Therapeutics Inc., a wholly owned subsidiary of Menarini Group, Receives Approval from U.S. FDA for Orserdu (elacestrant) as the First and Only Treatment Specifically Indicated for Patients with ESR1 Mutations in ER+, HER2- Advanced or Metastatic Breast Cancer”. Radius (Press release). 31 January 2023. Archived from the original on 2 February 2023. Retrieved 1 February 2023.
- ^ “EC approves Menarini Group’s Orserdu for advanced or metastatic breast cancer”. PMLive. 21 September 2023. Retrieved 22 September 2023.
- ^ Lloyd MR, Wander SA, Hamilton E, Razavi P, Bardia A (2022). “Next-generation selective estrogen receptor degraders and other novel endocrine therapies for management of metastatic hormone receptor-positive breast cancer: current and emerging role”. Therapeutic Advances in Medical Oncology. 14: 17588359221113694. doi:10.1177/17588359221113694. PMC 9340905. PMID 35923930.
- ^ Clinical trial number NCT03778931 for “Phase 3 Trial of Elacestrant vs. Standard of Care for the Treatment of Patients With ER+/HER2- Advanced Breast Cancer” at ClinicalTrials.gov
- ^ Jump up to:a b c Wardell SE, Nelson ER, Chao CA, Alley HM, McDonnell DP (October 2015). “Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader”. Endocrine-Related Cancer. 22 (5): 713–724. doi:10.1530/ERC-15-0287. PMC 4545300. PMID 26162914.
- ^ Jump up to:a b c Garner F, Shomali M, Paquin D, Lyttle CR, Hattersley G (October 2015). “RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models”. Anti-Cancer Drugs. 26 (9): 948–956. doi:10.1097/CAD.0000000000000271. PMC 4560273. PMID 26164151.
| Clinical data | |
|---|---|
| Pronunciation | /ˌɛləˈsɛstrənt/ EL-ə-SES-trənt |
| Trade names | Orserdu |
| Other names | RAD-1901; ER-306323 |
| License data | US DailyMed: Elacestrant |
| Routes of administration | By mouth |
| ATC code | L02BA04 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1][2]EU: Rx-only[3][4] |
| Pharmacokinetic data | |
| Bioavailability | ~10%[1] |
| Protein binding | >99%[1] |
| Metabolism | Liver (major: CYP3A4, minor: CYP2A6, CYP2C9)[1] |
| Elimination half-life | 30–50 hours[1] |
| Excretion | Feces (82%), urine (7.5%)[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 722533-56-4 |
| PubChem CID | 23642301 |
| DrugBank | DB06374 |
| ChemSpider | 57583807 |
| UNII | FM6A2627A8 |
| KEGG | D11671 |
| ChEMBL | ChEMBL4297509 |
| PDB ligand | I0V (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID901045846 |
| ECHA InfoCard | 100.312.890 |
| Chemical and physical data | |
| Formula | C30H38N2O2 |
| Molar mass | 458.646 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Elacestrant, Orserdu, FDA 2023, APPROVALS 2023, FM6A2627A8, WHO 10247, ER 306323, RAD 1901, RAD1901
Clofutriben



Clofutriben
Cas 1204178-50-6
HCL 1203941-88-1
- ASP 3662
- 4-(5-(2-(4-Chloro-2,6-difluorophenoxy)propan-2-yl)-4-methyl-4H-1,2,4-triazol-3-yl)-3-fluorobenzamide
- 4-{5-[2-(4-Chloro-2,6-difluorophenoxy)propan-2-yl]-4-methyl-4H-1,2,4-triazol-3-yl}-3-fluorobenzamide
- 4-[5-[2-(4-chloro-2,6-difluorophenoxy)propan-2-yl]-4-methyl-1,2,4-triazol-3-yl]-3-fluorobenzamide
- 4L1TY1U5VC
| Molecular Weight | 424.80 |
|---|---|
| Formula | C19H16ClF3N4O2 |
Clofutriben (ASP3662) is a 11β-hydroxysteroid dehydrogenase type 1 inhibitor.
Clofutriben is an orally bioavailable selective inhibitor of the enzyme 11beta-hydroxysteroid dehydrogenase type 1 (11b-HSD1; 11bHSD1; HSD11B1; HSD1; HSD-1), with potential protective activity for disorders of corticosteroid excess. Upon oral administration, clofutriben selectively binds to and inhibits the activity of HSD-1. This prevents the conversion of cortisone to the active hormone cortisol and thereby preventing the activation of the glucocorticoid receptors (GRs). By blocking cortisol production in metabolic tissues, clofutriben may inhibit the adverse metabolic effects that are caused by exogenous administration of glucocorticoids or in disorders in which cortisol is secreted in excess. HSD-1 is highly expressed in metabolic tissues, such as liver, skeletal muscle, and adipose tissue. It plays a crucial role in regulating the production of cortisol to activate the GRs.
SCHEME

PATENTS
Clinical and Translational Science (2019), 12(3), 291-301
British Journal of Pharmacology (2018), 175(19), 3784-3796
Sparrow Pharmaceuticals, Inc. WO2020106337
WO2019075394
WO2018117063
WO2010001946
PATENT
PDT PAT FOR HCL SALT, WO2012033070
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012033070
PATENT
PDT PAT FOR BASE, WO2018117063
PATENT
WO2010001946
[1]. Kiso T, et al. Analgesic effects of ASP3662, a novel 11尾-hydroxysteroid dehydrogenase 1 inhibitor, in rat models of neuropathic and dysfunctional pain. Br J Pharmacol. 2018 Oct;175(19):3784-3796. [Content Brief]
////////////Clofutriben, ASP 3662, orphan drug, 4L1TY1U5VC, Sparrow Pharmaceuticals,
Pirtobrutinib



Pirtobrutinib
- CAS 2101700-15-4
- JAYPIRCA
- RXC-005
- LY3527727
- LOXO-305
- WHO 11681
- WeightAverage: 479.436
- Monoisotopic: 479.158052208
- Chemical FormulaC22H21F4N5O3
5-amino-3-[4-[[(5-fluoro-2-methoxybenzoyl)amino]methyl]phenyl]-1-[(2S)-1,1,1-trifluoropropan-2-yl]pyrazole-4-carboxamide
FDA 2023, 1/27/2023, Jaypirca
To treat relapsed or refractory mantle cell lymphoma in adults who have had at least two lines of systemic therapy, including a BTK inhibitor
Drug Trials Snapshot
Pirtobrutinib, sold under the brand name Jaypirca, is an anticancer medication that is used to treat mantle cell lymphoma.[1][2][4] It inhibits B cell lymphocyte proliferation and survival by binding and inhibiting Bruton’s tyrosine kinase (BTK).[5] It is taken by mouth.[1]
The most common adverse reactions include fatigue, musculoskeletal pain, diarrhea, edema, dyspnea, pneumonia, and bruising.[4][6] The most common adverse reactions when used to treat chronic lymphocytic leukemia or small lymphocytic leukemia include fatigue, bruising, cough, musculoskeletal pain, COVID-19, diarrhea, pneumonia, abdominal pain, dyspnea, hemorrhage, edema, nausea, pyrexia, and headache.[7]
Pirtobrutinib was approved for medical use in the United States in January 2023,[4][8][9][10] and in the European Union in November 2023.[2]
PATENTS
Guisot, N. (2017). Compounds useful as kinase inhibitors (WO 2017/103611 A1). World Intellectual Property Organization. https://patentimages.storage.googleapis.com/d7/16/21/9300e49071a21a/WO2017103611A1.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017103611&_cid=P10-MAG7OA-80884-1


[00381] Example 120: 5-amino-3-[4-[[(5-fluoro-2-methoxy-benzoyl)amino]methyl]phenyl]-1- (2,2,2-trifluoro-1 -methyl-ethyl)pyrazole-4-carboxamide
N-[(2,2,2-Trifluoro-1-methyl-ethylidene)aminolbenzamide
General procedure S, benzhydrazide (49.9 mmol) and 1,1,1- trifluoroacetone (74.9 mmol) gave, after washing, the titled compound as a white solid. UPLC-MS (ES + , Short acidic): 1.45 min, m/z 230.9 [M+H] +
-Amino-3-[4-[[(5-fluoro-2-methoxy-benzoyl)amino]methyl]phenyl]-1-(2,2,2-trifluoro-1-methyl-ethyl)pyrazole-4-carboxamide
General procedure M, N-[[4-[5-amino-4-cyano-1-(2,2,2-trifluoro-1-methyl-ethyl)pyrazol-3-yl]phenyl]methyl]-5-fluoro-2-methoxy-benzamide (0.83 mmol) gave, after purification, the titled compound (0.42 mmol) as a white solid. UPLC-MS (ES + , Short acidic): 1.55 min, m/z 480.1 [M+H] + . UPLC-MS (ES + , Long acidic): 3.57 min, m/z 480.1 [M+H] + . 1 H NMR (400 MHz, DMSO-d 6 , δ): 8.84 (t, J = 6.1 Hz, 1H), 7.52 (dd, J = 9.2, 3.3 Hz, 1H), 7.48-7.41 (m, 4H), 7.37-7.32 (m, 1H), 7.19 (dd, J = 9.1, 4.3 Hz, 1H), 6.67 (s, 2H), 5.35-5.24 (m, 1H), 4.56 (d, J = 6.0 Hz, 2H), 3.90 (s, 3H), 1.62 (d, J = 6.9 Hz, 3H).
MORE



Medical uses
In the United States, pirtobrutinib is indicated to treat relapsed or refractory mantle cell lymphoma after at least two lines of systemic therapy, including a Bruton’s tyrosine kinase (BTK) inhibitor.[1][11] In December 2023, the US Food and Drug Administration (FDA) expanded the indication for pirtobrutinib to include the treatment of adults with chronic lymphocytic leukemia or small lymphocytic leukemia.[7][12]
In the European Union, pirtobrutinib is indicated for the treatment of mantle cell lymphoma.[2]
Mechanism of action
B cells are white cells of the lymphocyte subtype that produce antibodies, but when some of them grow uncontrollably they can be a cause of cancer. A key enzyme in B cell stimulation and survival is BTK, and pirtobrutinib inhibits BTK in a way that is different from the prototypical BTK inhibitor ibrutinib by binding in a different way that avoids a genetic change (mutation at active site cysteine residue C481 in BTK) that can make some tumors less responsive to ibrutinib.[5]
History
Pirtobrutinib is manufactured by Eli Lilly and Company and was approved by the US Food and Drug Administration in January 2023, for the treatment of mantle cell lymphoma that has become refractory to other BTK inhibitors.[13]
Efficacy was evaluated in BRUIN (NCT03740529), an open-label, multicenter, single-arm trial of pirtobrutinib monotherapy that included 120 participants with mantle cell lymphoma previously treated with a Bruton’s tyrosine kinase (BTK) inhibitor.[4] Participants had a median of three prior lines of therapy, with 93% having two or more prior lines.[4] The most common prior Bruton’s tyrosine kinase inhibitors received were ibrutinib (67%), acalabrutinib (30%), and zanubrutinib (8%); 83% had discontinued their last Bruton’s tyrosine kinase inhibitor due to refractory or progressive disease.[4] The trial was conducted at 49 sites in 10 countries in the United States, Europe, Australia, and Asia.[6] The same trial was used to assess safety and efficacy.[6]
Efficacy was evaluated in BRUIN (NCT03740529], an open-label, international, single-arm, multicohort trial that included 108 participants with chronic lymphocytic leukemia or small lymphocytic lymphoma previously treated with at least two prior lines of therapy, including a Bruton’s tyrosine kinase (BTK) inhibitor and a B-cell lymphoma-2 (BCL-2) inhibitor.[7] Participants received a median of five prior lines of therapy (range: 2 to 11).[7] Seventy-seven percent of participants discontinued the last BTK inhibitor for refractory or progressive disease.[7] Pirtobrutinib was administered orally at 200 mg once daily and was continued until disease progression or unacceptable toxicity.[7]
Society and culture
Legal status
In April 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Jaypirca, intended for the treatment of relapsed or refractory mantle cell lymphoma (MCL).[14] The applicant for this medicinal product is Eli Lilly Nederland B.V.[14] Pirtobrutinib was approved for medical use in the European Union in November 2023.[2]
References
- ^ Jump up to:a b c d “Jaypirca- pirtobrutinib tablet, coated”. DailyMed. 27 January 2023. Archived from the original on 11 February 2023. Retrieved 11 February 2023.
- ^ Jump up to:a b c d e “Jaypirca EPAR”. European Medicines Agency (EMA). 20 November 2023. Archived from the original on 22 November 2023. Retrieved 22 November 2023.
- ^ “Jaypirca Product information”. Union Register of medicinal products. 31 October 2023. Archived from the original on 22 November 2023. Retrieved 22 November 2023.
- ^ Jump up to:a b c d e f “FDA grants accelerated approval to pirtobrutinib for relapsed or refractory mantle cell lymphoma”. FDA. 27 January 2023. Archived from the original on 28 January 2023. Retrieved 28 January 2023. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b Aslan B, Kismali G, Iles LR, Manyam GC, Ayres ML, Chen LS, et al. (May 2022). “Pirtobrutinib inhibits wild-type and mutant Bruton’s tyrosine kinase-mediated signaling in chronic lymphocytic leukemia”. Blood Cancer Journal. 12 (5): 80. doi:10.1038/s41408-022-00675-9. PMC 9123190. PMID 35595730.
- ^ Jump up to:a b c “Drug Trials Snapshots: Jaypirca”. U.S. Food and Drug Administration (FDA). 27 January 2023. Retrieved 13 May 2024.
- ^ Jump up to:a b c d e f “FDA grants accelerated approval to pirtobrutinib for chronic lymphocytic leukemia and small lymphocytic lymphoma”. U.S. Food and Drug Administration (FDA). 1 December 2023. Archived from the original on 3 December 2023. Retrieved 3 December 2023. This article incorporates text from this source, which is in the public domain.
- ^ “U.S. FDA Approves Jaypirca (pirtobrutinib), the First and Only Non-Covalent (Reversible) BTK Inhibitor, for Adult Patients with Relapsed or Refractory Mantle Cell Lymphoma After at Least Two Lines of Systemic Therapy, Including a BTK Inhibitor” (Press release). Eli Lilly. 27 January 2023. Archived from the original on 30 January 2023. Retrieved 31 January 2023 – via PR Newswire.
- ^ Keam SJ (April 2023). “Pirtobrutinib: First Approval”. Drugs. 83 (6): 547–553. doi:10.1007/s40265-023-01860-1. PMID 37004673. S2CID 257912433. Archived from the original on 19 November 2023. Retrieved 19 November 2023.
- ^ Telaraja D, Kasamon YL, Collazo JS, Leong R, Wang K, Li P, et al. (August 2023). “FDA Approval Summary: Pirtobrutinib for Relapsed or Refractory Mantle Cell Lymphoma”. Clinical Cancer Research. 30 (1): OF1 – OF6. doi:10.1158/1078-0432.CCR-23-1272. PMC 10841293. PMID 37624619. S2CID 265965744.
- ^ De SK (October 2023). “Pirtobrutinib: First Non-covalent Tyrosine Kinase Inhibitor for Treating Relapsed or Refractory Mantle Cell Lymphoma in Adults”. Current Medicinal Chemistry. 31. doi:10.2174/0109298673251030231004052822. PMID 37818564. S2CID 263828536.
- ^ “Jaypirca (pirtobrutinib) Now Approved by U.S. FDA for the Treatment of Adult Patients with Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma Who Have Received at Least Two Lines of Therapy, Including a BTK Inhibitor and a BCL-2 Inhibitor” (Press release). Eli Lilly. 1 December 2023. Archived from the original on 3 December 2023. Retrieved 3 December 2023 – via PR Newswire.
- ^ “FDA approves Eli Lilly’s drug for rare blood cancer”. Reuters. 27 January 2023. Archived from the original on 28 January 2023.
- ^ Jump up to:a b “Jaypirca: Pending EC decision”. European Medicines Agency. 26 April 2023. Archived from the original on 26 April 2023. Retrieved 27 April 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Further reading
- Mato AR, Woyach JA, Brown JR, Ghia P, Patel K, Eyre TA, et al. (July 2023). “Pirtobrutinib after a Covalent BTK Inhibitor in Chronic Lymphocytic Leukemia”. The New England Journal of Medicine. 389 (1): 33–44. doi:10.1056/NEJMoa2300696. hdl:11585/960014. PMID 37407001. S2CID 259358462.
External links
- “Pirtobrutinib”. National Cancer Institute. 16 February 2023.
- “Pirtobrutinib”. NCI Drug Dictionary.
| Clinical data | |
|---|---|
| Trade names | Jaypirca |
| Other names | LOXO-305 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623012 |
| License data | US DailyMed: Pirtobrutinib |
| Routes of administration | By mouth |
| Drug class | Protein kinase inhibitor |
| ATC code | L01EL05 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2101700-15-4 |
| PubChem CID | 129269915 |
| DrugBank | DB17472 |
| ChemSpider | 114875989 |
| UNII | JNA39I7ZVB |
| KEGG | D12050 |
| ChEBI | CHEBI:229212 |
| ChEMBL | ChEMBL4650485 |
| PDB ligand | Y7W (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C22H21F4N5O3 |
| Molar mass | 479.436 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Jensen JL, Mato AR, Pena C, Roeker LE, Coombs CC: The potential of pirtobrutinib in multiple B-cell malignancies. Ther Adv Hematol. 2022 Jun 16;13:20406207221101697. doi: 10.1177/20406207221101697. eCollection 2022. [Article]
- Aslan B, Kismali G, Iles LR, Manyam GC, Ayres ML, Chen LS, Gagea M, Bertilaccio MTS, Wierda WG, Gandhi V: Pirtobrutinib inhibits wild-type and mutant Bruton’s tyrosine kinase-mediated signaling in chronic lymphocytic leukemia. Blood Cancer J. 2022 May 20;12(5):80. doi: 10.1038/s41408-022-00675-9. [Article]
- Alu A, Lei H, Han X, Wei Y, Wei X: BTK inhibitors in the treatment of hematological malignancies and inflammatory diseases: mechanisms and clinical studies. J Hematol Oncol. 2022 Oct 1;15(1):138. doi: 10.1186/s13045-022-01353-w. [Article]
- Mato AR, Shah NN, Jurczak W, Cheah CY, Pagel JM, Woyach JA, Fakhri B, Eyre TA, Lamanna N, Patel MR, Alencar A, Lech-Maranda E, Wierda WG, Coombs CC, Gerson JN, Ghia P, Le Gouill S, Lewis DJ, Sundaram S, Cohen JB, Flinn IW, Tam CS, Barve MA, Kuss B, Taylor J, Abdel-Wahab O, Schuster SJ, Palomba ML, Lewis KL, Roeker LE, Davids MS, Tan XN, Fenske TS, Wallin J, Tsai DE, Ku NC, Zhu E, Chen J, Yin M, Nair B, Ebata K, Marella N, Brown JR, Wang M: Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021 Mar 6;397(10277):892-901. doi: 10.1016/S0140-6736(21)00224-5. [Article]
- Wang E, Mi X, Thompson MC, Montoya S, Notti RQ, Afaghani J, Durham BH, Penson A, Witkowski MT, Lu SX, Bourcier J, Hogg SJ, Erickson C, Cui D, Cho H, Singer M, Totiger TM, Chaudhry S, Geyer M, Alencar A, Linley AJ, Palomba ML, Coombs CC, Park JH, Zelenetz A, Roeker L, Rosendahl M, Tsai DE, Ebata K, Brandhuber B, Hyman DM, Aifantis I, Mato A, Taylor J, Abdel-Wahab O: Mechanisms of Resistance to Noncovalent Bruton’s Tyrosine Kinase Inhibitors. N Engl J Med. 2022 Feb 24;386(8):735-743. doi: 10.1056/NEJMoa2114110. [Article]
- FDA Approved Drug Products: JAYPIRCA (pirtobrutinib) tablets for oral use [Link]
- BioSpace: U.S. FDA Approves Jaypirca (pirtobrutinib), the First and Only Non-Covalent (Reversible) BTK Inhibitor, for Adult Patients with Relapsed or Refractory Mantle Cell Lymphoma After at Least Two Lines of Systemic Therapy, Including a BTK Inhibitor [Link]
//////////////Jaypirca, FDA 2023, APPROVALS 2023, Pirtobrutinib, RXC-005, LY3527727, LOXO-305, LOXO 305, WHO 11681
Clesacostat



Clesacostat
PF 05221304, 752DF9PPPI
CAS 1370448-25-1
WeightAverage: 502.571
Monoisotopic: 502.221620082
Chemical FormulaC28H30N4O5
4-[6-methoxy-4-(7-oxo-1-propan-2-ylspiro[4,6-dihydroindazole-5,4′-piperidine]-1′-carbonyl)pyridin-2-yl]benzoic acid
- Originator Pfizer
- ClassBenzoic acids; Carboxylic acids; Ethers; Hepatoprotectants; Indazoles; Piperidines; Pyridines; Small molecules; Spiro compounds
- Mechanism of ActionAcetyl-CoA carboxylase inhibitors
- Phase IINon-alcoholic fatty liver disease; Non-alcoholic steatohepatitis
- 21 Feb 2024Pfizer completes a phase II trial in Non-alcoholic steatohepatitis (Combination therapy) in Slovakia, Japan, Bulgaria, Canada, China, Hong Kong, India, Poland, Puerto Rico, South Korea, Taiwan (PO) (NCT04321031) (EudraCT2019-004775-39)
- 26 May 2022Clesacostat – Pfizer receives Fast Track designation for Non-alcoholic steatohepatitis [PO] (Combination therapy) in USA
- 28 Apr 2022Pfizer completes a phase II trial for Non-alcoholic fatty liver disease (Combination therapy) in USA and Canada (PO) (NCT04399538)
Clesacostat is under investigation in clinical trial NCT04321031 (Metabolic Interventions to Resolve Non-alcoholic Steatohepatitis (NASH) With Fibrosis (MIRNA)).
CLESACOSTAT is a small molecule drug with a maximum clinical trial phase of II (across all indications) and has 4 investigational indications.
SCHEME
SIDECHAIN

MAIN

PATENT
WO2021171164 89%
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021171164&_cid=P20-MAF4R3-69728-1
4-(4-(1-lsopropyl-7-oxo-1 ,4,6,7-tetrahydrospiro[indazole-5,4′-piperidine]-1′-carbonyl)-6-methoxypyridin-2-yl)benzoic acid,
A preparation of (S)- 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-N-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide is presented in Example 1 of US 2018-0051012A1 , hereby incorporated herein by reference in its entireties for all purposes. A preparation of 4-(4-(1-lsopropyl-7-oxo-1 ,4,6,7-tetrahydrospiro[indazole-5,4′-piperidine]-1 ‘-carbonyl)-6-methoxypyridin-2-yl)benzoic acid is in Example 9 of US 8,859,577, hereby incorporated herein by reference in its entireties for all purposes. Preparation of [(1 R,5S,6R)-3-{2-[(2S)-2-methylazetidin-1-yl]-6-(trifluoromethyl)pyrimidin-4-yl}-3-azabicyclo[3.1 0]hex-6-yl]acetic acid (including a crystalline free acid form thereof) is described in Example 4 of U.S. Patent No. 9,809,579. Preparation of GLP-1 R agonists are described in U.S. Patent No.10,208,019.
Step 6: (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-N-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (Example 1 (DGAT2i Compound))
Oxalyl chloride (13.8 ml_, 160 mmol, 1.2 equiv) and dimethylformamide (0.510 ml_, 6.65 mmol, 0.05 equiv) were added to a suspension of 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid (45.0 g, 133 mmol, 1.0 equiv) in dichloromethane (500 ml_). The suspension was stirred for 2 hours when a solution was achieved. The reaction mixture was concentrated to yield crude acid chloride as a red solid. A solution of (S)-tetrahydrofuran-3-amine (12.2 g, 140 mmol, 1.05 equiv) and diisopropylethylamine (51.0 ml_, 293 mmol, 2.2 equiv) in tetrahydrofuran (100 ml_) was added dropwise to a solution of the crude acid chloride in dichloromethane (200 ml_) at 0 °C. The reaction was allowed to warm to room temperature and stirred for 16 hours. Water (1 .0 L) and ethyl acetate (600 ml_) were added and the organic layer was separated, washed with saturated sodium bicarbonate, dried over magnesium sulfate, and filtered. The filtrate was treated with activated charcoal (20 g) was stirred at 65 °C for 20 minutes. The suspension was filtered warm and filtrate was concentrated to a pale, yellow solid which was recrystallized from methanol in ethyl acetate (1 :4, 1 L) to yield (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-A/-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (43.5 g, 81%) as a colorless solid. The title compound was combined with previous batches (108.7 g, 266.8 mmol) prepared in the same manner and slurried with ethyl acetate (1.0 L) at 80 °C for 4 hours. The suspension was allowed to cool to room temperature and stirred for 4 days. The solid was filtered, washed with ethyl acetate (3×200 ml_) and dried under high vacuum at 50 °C for 24 hours to yield (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-A/-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (100.5 g, 92%) as a colorless solid. 1H NMR (300 MHz, DMSO-d6) 6 1.38 (t, 3H), 1.89-1.98 (m, 1H), 2.15-2.26 (m, 1H), 3.65 (dd, 1H), 3.70-3.78 (m, 1H), 3.85-3.92 (m, 2H), 4.18 (q, 2H), 4.46-4.55 (m, 1H), 7.18 (dd, 1H), 7.58 (dd, 1H), 7.69 (dd, 1H), 8.37 (dd,
1 H), 8.64 (d, 1 H), 8.95 (d, 1 H), 9.28 (s, 2H), 9.39 (d, 1 H). MS (ES+) 408.4 (M+H). Melting point 177.5 °C. Elemental analysis for C21H21N5O4: calculated C, 61.91 ; H, 5.20; N, 17.19; found C, 61.86; H, 5.18; N, 17.30.
PATENT
WO2021171163 65%
WO2020234726 65%
Journal of Medicinal Chemistry (2020), 63(19), 10879-10896
WO2020044266 89%
WO2019102311 89%
//////////Clesacostat, PF 05221304, PHASE 2, 752DF9PPPI

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}