
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Zelicapavir


Zelicapavir, Enanta Pharmaceuticals
Alternative Names: EDP-938; EP 023938
cas 2070852-76-3
RSV-IN-7
549.5 g/mol, C27H22F3N7O3
UNII U4OI721DMD
(3S)-3-[[5-[3-morpholin-4-yl-5-(trifluoromethyl)pyridin-2-yl]-1,3,4-oxadiazol-2-yl]amino]-5-phenyl-1,3-dihydro-1,4-benzodiazepin-2-one
SYN
New England Journal of Medicine (2022), 386(7), 655-666
WO2022157327
WO2018152413
WO2019067864
WO2017015449
PATENT
WO2018152413
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018152413&_cid=P20-M0V26A-30323-1
Step 5 : (<Sf)-3-((5-(3-morpholino-5-(trifluoromethyl)pyridin-2-yl)-1.3.4-oxadiazol-2-

To a mixture of (5)-2-(3-morpholino-5-(trifluoromethyl)picolinoyl)-N-2-oxo-5-phenyl-2,3-dihydro-lH-benzo[e] [l,4]diazepin-3-yl)hydrazine-l-carboxamide (1.4 kg, 1 eq.) in DCM (11.2 L) in a flask was charged with 4A-MS (1.4 kg) and stirred at 20±5 °C for 2hrs. Then, it was cooled to 0°C, charged with triethylamine (0.62 Kg, 2.5 eq.) and stirred for 10 min. /^-Toluenesulfonyl chloride (0.7 kg, 1.5 eq.) in DCM (1.4 L) solution was dropwise added to the reaction mixture with maintaining below 5°C and stirred at at 0±5 °C for 5 hrs. The reaction mixture was filtered and washed with DCM (2 X 4.2 L). The filtrate was treated with water (4.2 L) at 0°C and stirred between 0 and 10°C for 5 min. After separation, the organic phase was washed with 5% aqueous NaHCC solution (7 L), water (7 L) and brine (7 L) successively and separated. The DCM layer was concentrated in vacuo at below 30°C to leave ~7L of organic layer. MTBE (7 L) was added to organic layer and concentrated in vacuo to leave ~ 7 L of organic layer (This step was repeated once). The organic layer was charged with water (7 L) and stirred at 20±5 °C for 4 hrs. The solid was filtered and washed with MTBE (3 X 2.1 L) and purified water (2.8 L). The wet cake was stirred with ethyl acetate (7 L) for 12 hrs, charged with n-heptane (14 L) and stirred at 20±5 °C for 5 hrs. The solid was filtered, washed with n-heptane (2 X 2.8 L) and dried under vacuum at ambient temperature to provide the title compound (0.776 kg, 99.6% purity by HPLC, 97.8%
chiral purity by chiral HPLC) as a pale yellowish solid. LC-MS(ESI, m/z): 550.17 [M+H]+;
¾ NMR: ( DMSO-c 6400 MHz): δ 10.98 (br-s, 1H), 9.40 (d, J=8.0 Hz, 1H), 8.69 (br-d, J=4.0 Hz, 1H), 7.89 (d, J=4.0 Hz, 1H), 7.68 (dt, J=8.0 and 4.0 Hz, 1H), 7.56-7.51 (m, 3H), 7.49-7.45 (m, 2H), 7.38-7.35 (m, 2H), 7.29 (br-t, J=8.0 Hz, 1H)
5.22 (d, J=8.0 Hz, 1H), 3.75-3.72 (m, 4H), 3.09-3.07 (m, 4H); 13C (DMSO-c¾, 100 MHz): δ 167.3, 167.0, 162.8, 156.4, 147.2, 139.2, 138.7, 138.4, 138.3, 138.0, 132.30, 130.7, 130.5, 129.5, 128.4, 126.2, 124.5, 123.4, 121.5, 71.8, 65.9, 51.0.
SCHEME

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US368999603&_cid=P20-M0V2BF-36596-1
Example 253
Example 160 was prepared using a procedure similar to that used to prepare Example 152 where methyl 5-cyano-3-morpholinopicolinate was used in place of ethyl 2-morpholino-4-(trifluoromethyl)benzoate. ESI-MS m/z: 507.2 [M+H] +. 1H NMR (400 MHz, DMSO-d 6) δ 3.02-3.04 (m, 4H), 3.71-3.73 (m, 4H), 5.19-5.21 (d, J=8.0 Hz, 1H), 7.26-7.30 (m, 1H), 7.34-7.36 (m, 2H), 7.44-7.55 (m, 5H), 7.65-7.70 (m, 1H), 8.13 (s, 1H), 8.72 (s, 1H), 9.42-9.45 (m, 1H), 10.98 (s, 1H).
//////////////Zelicapavir, EDP-938, EP 023938, EDP 938, RSV-IN-7, ENANTA
Palopegteriparatide

Palopegteriparatide
Yorvipath , FDA 2024, 8/9/2024, To treat hypoparathyroidism
- G2N64C3385
- 2222514-07-8
- Palopegteriparatide
- UNII-G2N64C3385
- ACP-014
- Mpeg 40000-teriparatide
- Palopegteriparatide [INN]
- Transcon parathyroid hormone (1-34)
- Transcon pth (1-34)
- Palopegteriparatide [USAN]
- TransCon PTH
- WHO 11060


Palopegteriparatide, sold under the brand name Yorvipath, is a hormone replacement therapy used for the treatment of hypoparathyroidism.[1][2] It is a transiently pegylated parathyroid hormone.[4] It is a parathyroid hormone analog.[1]
Palopegteriparatide was approved for medical use in the European Union in November 2023,[2] and in the United States in August 2024.[1][5]
Medical uses
Palopegteriparatide is indicated for the treatment of adults with hypoparathyroidism.[1][2]
Adverse effects
The US Food and Drug Administration (FDA) prescription label for palopegteriparatide includes warnings for a potential risk of risk of unintended changes in serum calcium levels related to number of daily injections and total delivered dose, serious hypocalcemia and hypercalcemia (blood calcium levels that are too high), osteosarcoma (a rare bone cancer) based on findings in rats, orthostatic hypotension (dizziness when standing), and a risk of a drug interaction with digoxin (a medicine for certain heart conditions).[5]
History
The effectiveness of palopegteriparatide was evaluated in a 26-week, randomized, double-blind, placebo-controlled trial that enrolled 82 adults with hypoparathyroidism.[5] Prior to randomization, all participants underwent an approximate four-week screening period in which calcium and active vitamin D supplements were adjusted to achieve an albumin-corrected serum calcium concentration between 7.8 and 10.6 mg/dL, a magnesium concentration ≥1.3 mg/dL and below the upper limit of the reference range, and a 25(OH) vitamin D concentration between 20 to 80 ng/mL.[5] During the double-blind period, participants were randomized to either palopegteriparatide (N = 61) or placebo (N= 21), at a starting dose of 18 mcg/day, co-administered with conventional therapy (calcium and active vitamin D).[5] Study drug and conventional therapy were subsequently adjusted according to the albumin-corrected serum calcium levels.[5] At the end of the trial, 69% of the participants in the palopegteriparatide group compared to 5% of the participants in the placebo group were able to maintain their calcium level in the normal range, without needing active vitamin D and high doses of calcium (calcium dose ≤ 600 mg/day).[5]
The FDA granted the application for palopegteriparatide orphan drug and priority review designations.[5]
Society and culture
Legal status
In September 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Yorvipath, intended for the treatment of chronic hypoparathyroidism in adults.[4][6] The applicant for this medicinal product is Ascendis Pharma Bone Diseases A/S.[4] Palopegteriparatide was approved for medical use in the European Union in November 2023.[2]
Palopegteriparatide was granted an orphan drug designation by the US Food and Drug Administration (FDA) in 2018,[7] and by the EMA in 2020.[8]
Brand names
Palopegteriparatide is the international nonproprietary name.[9][10]
Palopegteriparatide is sold under the brand name Yorvipath.[2]
References
- ^ Jump up to:a b c d e “Yorvipath injection, solution”. DailyMed. 14 August 2024. Retrieved 5 September 2024.
- ^ Jump up to:a b c d e f “Yorvipath EPAR”. European Medicines Agency. 19 October 2020. Archived from the original on 10 December 2023. Retrieved 11 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Yorvipath Product information”. Union Register of medicinal products. 20 November 2023. Archived from the original on 26 November 2023. Retrieved 11 December 2023.
- ^ Jump up to:a b c “Yorvipath: Pending EC decision”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d e f g h “FDA approves new drug for hypoparathyroidism, a rare disorder”. U.S. Food and Drug Administration (FDA) (Press release). 9 August 2024. Archived from the original on 13 August 2024. Retrieved 13 August 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Ascendis Pharma Receives Positive CHMP Opinion for TransCon PTH (palopegteriparatide) for Adults with Chronic Hypoparathyroidism”. Ascendis Pharma (Press release). 14 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023.
- ^ “TransCon Parathyroid Hormone (mPEG conjugated parathyroid hormone 1-34) Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Archived from the original on 24 September 2023. Retrieved 24 September 2023.
- ^ “EU/3/20/2350”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
- ^ World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 89”. WHO Drug Information. 37 (1). hdl:10665/366661.
External links
- Palopegteriparatide Global Substance Registration System
- Palopegteriparatide NCI Thesaurus
- Clinical trial number NCT04701203 for “A Trial Investigating the Safety, Tolerability and Efficacy of TransCon PTH Administered Daily in Adults With Hypoparathyroidism (PaTHway)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Yorvipath |
| Other names | ACP-014, TransCon PTH |
| License data | US DailyMed: Palopegteriparatide |
| Routes of administration | Subcutaneous |
| Drug class | Hormonal agent |
| ATC code | H05AA05 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Identifiers | |
| CAS Number | 2222514-07-8 |
| UNII | G2N64C3385 |
| KEGG | D12395 |
//////Palopegteriparatide, APPRoVALS 2024, FDA 2024, Yorvipath, hypoparathyroidism, UNII-G2N64C3385, ACP-014, TransCon PTH, WHO 11060
Aneratrigine


Aneratrigine
2097163-74-9
5-chloro-2-fluoro-4-[4-fluoro-2-[methyl-[2-(methylamino)ethyl]amino]anilino]-N-(1,3-thiazol-4-yl)benzenesulfonamide
5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
| Benzenesulfonamide, 5-chloro-2-fluoro-4-[[4-fluoro-2-[methyl[2-(methylamino)ethyl]amino]phenyl]amino]-N-4-thiazolyl- |
C19H20ClF2N5O2S2 488.0 g/mol
UNII 6A5ZY5LT78
WHO
SYN

Assignee: Daewoong Pharmaceutical Co., Ltd.
World Intellectual Property Organization, WO2017082688
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017082688&_cid=P11-M0UEPF-95506-1
Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 1) Preparation of tert-butyl (1-(2-amino-5-fluorophenyl)pyridin-3-yl)(methyl)carbamate
2,4-Difluoro-1-nitrobenzene (2.0 g, 12.6 ng/mol) and tert-butyl methyl (pyridin-3-yl)carbamate (2.5 g, 1.0 eq.) were dissolved in DMF (20 mL), and K2C03 ( 2.6 g , 1.5 eq .) was added. The internal temperature was maintained at 60–70 ° C and the mixture was stirred for 2 hours. The completion of the reaction was confirmed by TLC when the reaction solution turned deep yellow. After cooling to room temperature, ethyl acetate (EA)/H20 was added, stirred, and the layers were separated. MgS04 was added to the separated organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was dissolved in EtOH (10 mL) and distilled water (10 mL), and then Na 2 S 2 0 4 (13.0 g, 6 eq.) was added. After stirring for 2 hours while maintaining the internal temperature at 60 to 70 ° C, the completion of the reaction was confirmed by TLC when the yellow color of the reaction solution lightened and became almost colorless. After cooling to room temperature, distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. The filtrate was concentrated under reduced pressure, and the obtained residue was separated by column chromatography (n-Hexane/EA = 3/1) to obtain the title compound (2.0 g, 51. ).
1H NMR (MeOD): 6.73(m, 1H), 6.57(t, 1H), 3.23(m, 1H), 3.10(m, 2H), 2.94(m, 1H), 2.91(s, 3H), 2.25( m, 1H), 1.99(m, 1H)
Step 2) Preparation of tert-butyl thiazol-4-ylcarbamate
Thiazole-4-carboxylic acid (5.0 g, 38.8 vol) was dissolved in t-Bu0H (100 mL), and then TEA (8.1 mL, 1.5 eq.) and DPPA (7.1 mL, 1.5 eq.) were added. The internal temperature was maintained at 90–100 ° C, and the mixture was stirred for 3 days. The completion of the reaction was confirmed by TLC. The product was concentrated under reduced pressure, distilled water (50 mL) was added, and the solution was washed with EA (100 mL).
It was extracted twice. MgSQ 4 was added to the organic layer, stirred, dried, and filtered.
After concentrating the filtrate under reduced pressure, the residue was added to a small amount of EA, slurried, and the resulting solid was filtered to obtain the white title compound (4.0 g, 51.5%).
1H NMR (MeOD): 8.73(s, 1H), 7.24(s, 1H), 1.52(s, 9H)
Step 3) Preparation of tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate
Step 2) The tert-Butyl thiazol-4-ylcarbamate (4.0 g, 20.0 ng ol) prepared in the reaction vessel was placed in a reaction vessel and the interior was replaced with nitrogen gas. After dissolving in THF (32 mL), it was cooled to _78 ° C using dry ice— acetone. After cooling, LiHMDS (22.4 mL, 1.5 eq.) was slowly added and the reaction mass was stirred for 30 minutes. 4-Bromo-5-chloro-2-fluorobenzenesulfonyl chloride (6.0 g, 1.0 eq.) was dissolved in THF (10 mL) and slowly added to the reaction mixture. The reaction mass was stirred overnight and the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was crystallized from THF/n-hexane to obtain the title compound (4.4 g, 59.0%).
1H NMR (MeOD): 9.00(s, 1H), 8.22(d, 1H), 7.90(d, 1H), 7.78(s, 1H), 1.35(s, 9H)
Step 4) Preparation of tert-butyl (l-(2-((4-(N-(tert-butyloxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)-2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate
Tert-butyl (1-(2-amino-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate (0.5 g, 1.1 ng ol) prepared in Step 1) and tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate (0.9 g, 1.2 eq.) prepared in Step 3) were dissolved in 1,4-dioxane (10 mL). Pd(OAc) 2 (0.03 g, 0.1 eq), rac-BINAP (0.19 g, 0.2 eq.), Cs 2 C0 3 (1.5 g, 3.0 eq.) were added to the reaction solution. After reacting at 120 ° C for 30 minutes using a microwave initiator, the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL).
MgS0 4 was added to the organic layer, stirred, filtered and dried. The filtrate was concentrated under reduced pressure, and the residue was separated by column chromatography (EA/n-Hexane = 1/1). This was repeated twice to obtain the title compound (2.0 g, 88.2%).
1H NMR (MeOD): 8.95(s, 1H), 7.94(d, 1H), 7.65(s, 1H), 7.14(t, 1H), 6.70(d, 1H), 6.64(t, 1H), 6.07( d, 1H)ᅳ 3.40(m, 1H), 3.28(m, 2H), 3.16(m, 1H), 2.64(s, 3H), 2.06(m, 1H), 1.89(m, 1H), 1.41(s , 9H), 1.36(s, 9H)
Step 5) Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 4) was prepared by adding 1.25 M HCl in MeOH (15 mL) to tert-butyl (1-(2-((4-(Ν-(tert-butoxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)—2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl) (methyl)carbamate (2.0 g, 2.9 µl). The mixture was heated to 40–50 ° C and stirred overnight, and the completion of the reaction was confirmed by TLC. The product was concentrated, and methylene chloride (15 mL) was added to the residue, which was stirred for 1 hour, and the resulting solid was filtered to obtain the title compound (0.9 g, 58.8%).
1H 證 (MeOD): 8.73(s, 1H), 7.75(d, 1H), 7.12(t, 1H), 7.00(s, 1H), 6.69(d, 1H), 6.67(t, 1H), 6.05( d, 1H), 3.73(m, 1H) , 3.54(m, 1H), 3.45(m, 1H), 3.38(m, 1H), 3.26(m, 1H), 2.63(s, 3H) , 2.31(m , 1H), 1.96(m, 1H)
PATENTS
0002705578SODIUM CHANNEL BLOCKER
20180346459Substituted benzenesulfonamides as sodium channel blockers
2018533606ナトリウムチャネル遮断剤
3375782SODIUM CHANNEL BLOCKER
108349963SODIUM CHANNEL BLOCKER
1020170056461SODIUM CHANNEL BLOCKER

////////////Aneratrigine, DAEWOONG
Seladelpar


Seladelpar
cas 851528-79-5
C21H23F3O5S, 444.47
fda approved 8/14/2024, To treat primary biliary cholangitis (PBC), Livdelzi
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Seladelpar lysine | N1429130KR | 928821-40-3 | WTKSWPYGZDCUNQ-JZXFCXSPSA-N |
- (+)-MBX-8025
- MBX 8025
- MBX-8025
- MBX8025
- RWJ-800025
- ((4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENYL)OXY)ACETIC ACID
- (4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)SULFANYL)-2-METHYLPHENOXY)ACETIC ACID PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR (PPAR) AGONIST,ANTIHYPERLIPIDAEMIC
- (R)-2-(4-((2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)-THIO)-2-METHYLPHENOXY)ACETIC ACID
- ACETIC ACID, (4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENOXY)-
- ACETIC ACID, (4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENOXY)- ((4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENYL)OXY)ACETIC ACID
- ACETIC ACID, 2-(4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENOXY)-
- Seladelpar
Seladelpar, sold under the brand name Livdelzi, is a medication used for the treatment of primary biliary cholangitis.[1] It is used as the lysine dihydrate salt.[1] It is a PPARδ receptor agonist.[1][2][3] The compound was licensed from Janssen Pharmaceutica NV.[4]
Seladelpar was approved for medical use in the United States in August 2024.[1][5]
Seladelpar is a peroxisome proliferator-activated receptor (PPAR)-delta (δ) agonist. Seladelpar is a single enantiomer of the R-configuration.5 On August 14, 2024, seladelpar was granted accelerated approval by the FDA for the treatment of primary biliary cholangitis,6 which is a condition associated with aberrant bile acid metabolism. Seladelpar works to block bile acid synthesis.1
Medical uses
Seladelpar is indicated for the treatment of primary biliary cholangitis in combination with ursodeoxycholic acid in adults who have an inadequate response to ursodeoxycholic acid, or as monotherapy in people unable to tolerate ursodeoxycholic acid.[1]
Clinically, Seladelpar reduces pruritus and IL-31 in patients with primary biliary cholangitis.[6]
- compound 3r [PMID: 17524639]
- Bioorg Med Chem Lett. 2007 Jul 15;17(14):3855-9. doi: 10.1016/j.bmcl.2007.05.007. Epub 2007 May 10.
- 10.1016/j.bmcl.2007.05.007
Drug Discovery, Johnson and Johnson Pharmaceutical Research and Development, LLC, 8 Clarke Drive, Cranbury, NJ 08512, USA
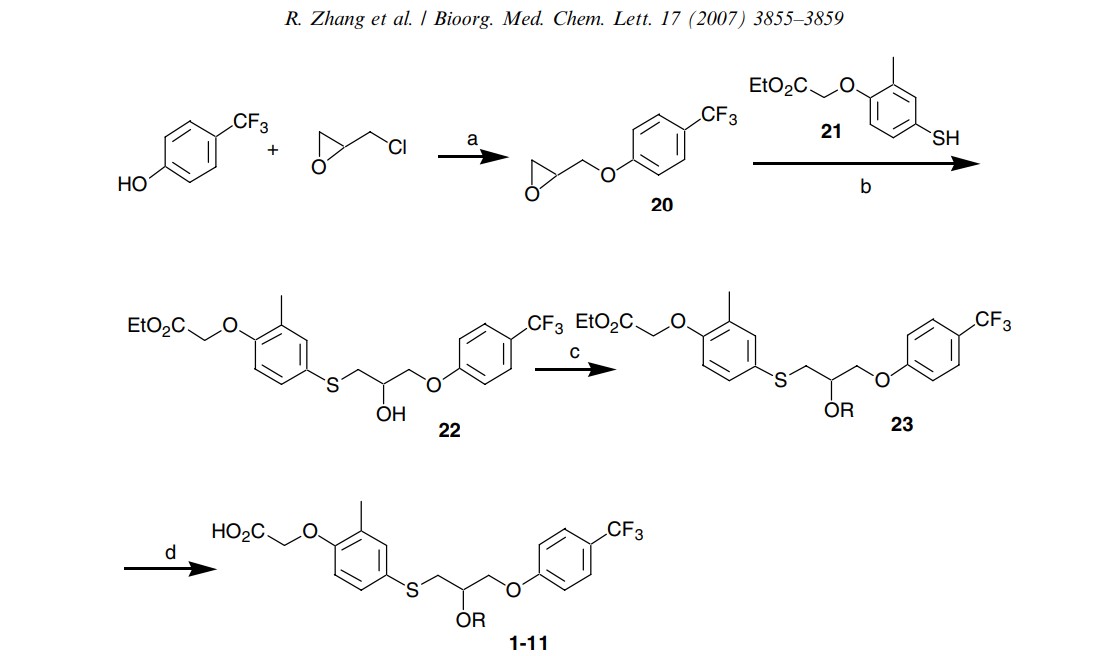
Scheme 1. Reagents and condition: (a) Cs2CO3, dioxane, 100 C 80%; (b) TBAF (cat), THF, 85%; (c) NaH, RI, THF or DMF for esters of 2–5, 8–9, 10–80%; iPr2NEt, RBr or MOMCl, THF for esters of 6–7, 58–79%; ADDP, Ph3P, phenol, CH2Cl2 for esters of 10–11, 68–73%; (d) LiOH, H2O, THF, 90–95%.
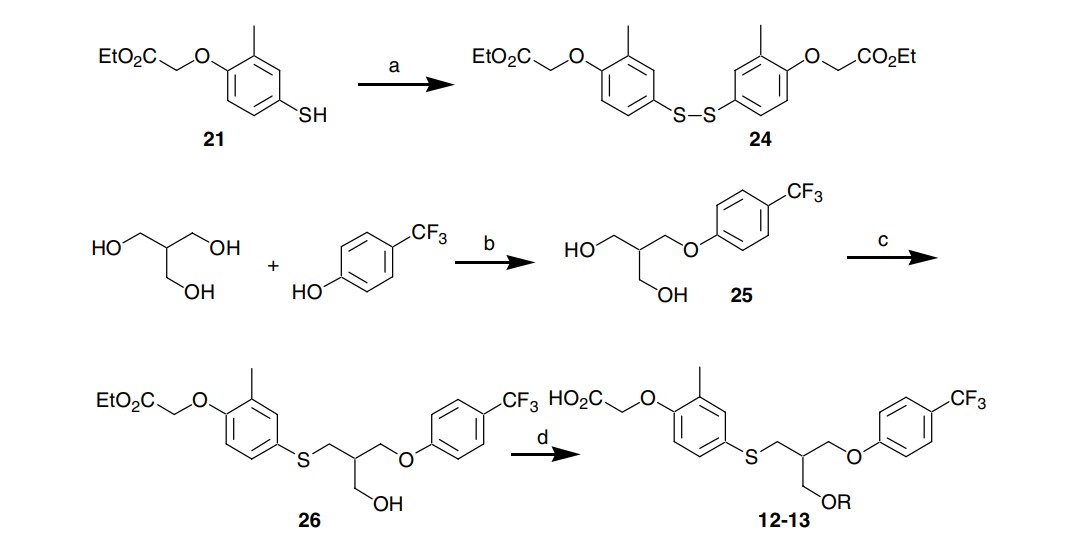
Scheme 2. Reagents: (a) Ba(MnO4)2, CH2Cl2, 89%; (b) DIAD, Ph3P, DMF, THF, 17%; (c) n-Bu3P, 24, Py, 55%; (d) i—NaHMDS, EtOTf, THF for the ethyl ester of 12, 47%; DIAD, Ph3P, para-trifluoromethylphenol for the ethyl ester of 13, 79%; ii—LiOH, H2O, THF, 84–88%.
References
- ^ Jump up to:a b c d e f “Livdelzi- seladelpar lysine capsule”. DailyMed. 14 August 2024. Retrieved 5 September 2024.
- ^ Billin AN (October 2008). “PPAR-beta/delta agonists for Type 2 diabetes and dyslipidemia: an adopted orphan still looking for a home”. Expert Opinion on Investigational Drugs. 17 (10): 1465–1471. doi:10.1517/13543784.17.10.1465. PMID 18808307. S2CID 86564263.
- ^ Bays HE, Schwartz S, Littlejohn T, Kerzner B, Krauss RM, Karpf DB, et al. (September 2011). “MBX-8025, a novel peroxisome proliferator receptor-delta agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin”. The Journal of Clinical Endocrinology and Metabolism. 96 (9): 2889–2897. doi:10.1210/jc.2011-1061. PMID 21752880.
- ^ “Targeting Mixed Dyslipidemia and Metabolic Syndrome”. Metabolex, Inc. 2005. Archived from the original on 17 October 2006.
- ^ “Gilead’s Livdelzi (Seladelpar) Granted Accelerated Approval for Primary Biliary Cholangitis by U.S. FDA” (Press release). Gilead. 14 August 2024. Retrieved 15 August 2024 – via Business Wire.
- ^ Kremer AE, Mayo MJ, Hirschfield GM, Levy C, Bowlus CL, Jones DE, et al. (July 2024). “Seladelpar treatment reduces IL-31 and pruritus in patients with primary biliary cholangitis”. Hepatology. 80 (1): 27–37. doi:10.1097/HEP.0000000000000728. PMC 11191048.
| Clinical data | |
|---|---|
| Trade names | Livdelzi |
| Other names | MBX-8025; RWJ-800025 |
| License data | US DailyMed: Seladelpar |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 851528-79-5 |
| PubChem CID | 11236126 |
| DrugBank | DB12390 |
| ChemSpider | 9411171 |
| UNII | 7C00L34NB9 |
| KEGG | D11256 |
| ChEMBL | ChEMBL230158 |
| CompTox Dashboard (EPA) | DTXSID001045332 |
| Chemical and physical data | |
| Formula | C21H23F3O5S |
| Molar mass | 444.47 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
///////////////Livdelzi, Seladelpar, (+)-MBX-8025, MBX 8025, MBX-8025, MBX8025, RWJ-800025, FDA 2024, APPROVALS 2024
Zevotrelvir, EDP 235
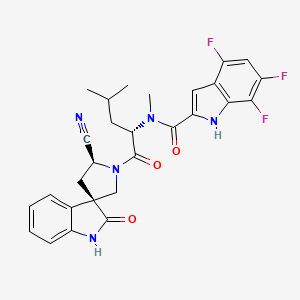
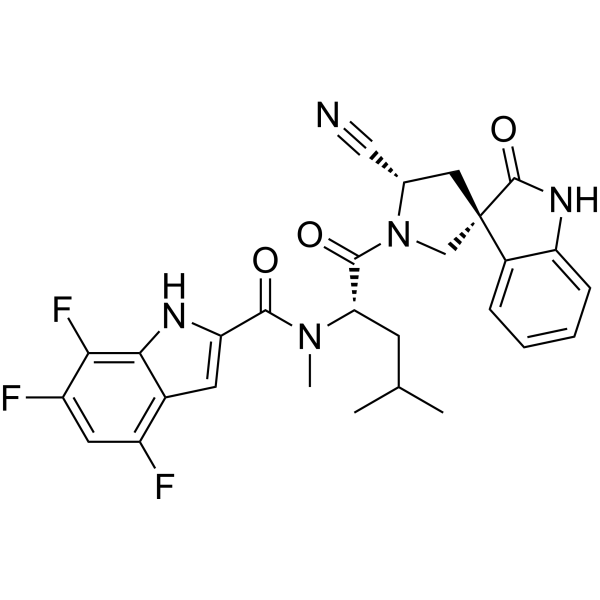
Zevotrelvir, EDP 235
cas 2773516-53-1
N-[(2S)-1-[(2′S,3R)-2′-cyano-2-oxospiro[1H-indole-3,4′-pyrrolidine]-1′-yl]-4-methyl-1-oxopentan-2-yl]-4,6,7-trifluoro-N-methyl-1H-indole-2-carboxamide
C28H26F3N5O3, 537.5
Zevotrelvir (Compound 52) is a coronavirus inhibitor with IC50 ranges of <0.1 μM and <0.1mM for 229E hCoV and SARS-CoV-23C-like (3CL) proteases, respectively. Zevotrelvir has the potential to study viral infections.
Coronaviruses are enveloped, positive-sense, single-stranded RNA viruses. The genomic RNA of CoVs has a 5′-cap structure and 3′-poly-A tail and contains at least 6 open reading frames (ORFs). The first ORF (ORF 1a/b) directly translates two polyproteins: pp1a and pp1ab. These polyproteins are processed by a 3C-Like protease (3CLpro), also known as the main protease (Mpro), into 16 non-structural proteins. These non-structural proteins engage in the production of subgenomic RNAs that encode four structural proteins, namely envelope, membrane, spike, and nucleocapsid proteins, among other accessory proteins. As a result, it is understood that 3C-Like protease has a critical role in the coronavirus life cycle.
3CLpro is a cysteine protease involved in most cleavage events within the precursor polyprotein. Active 3CLpro is a homodimer containing two protomers and features a Cys-His dyad located in between domains I and II.3CLpro is conserved among coronaviruses and several common features are shared among the substrates of 3CLpro in different coronaviruses. As there is no human homolog of 3CLpro, it is an ideal antiviral target. Although compounds have been reported to inhibit 3CLpro activity, they have not been approved as coronavirus therapies. (Refer to
WO 2004101742 A2, US 2005/0143320 Al, US 2006/0014821 Al, US 2009/0137818
Al, WO 2013049382 A2, WO 2013166319 A1, WO2018042343, WO2018023054, WO 2022013684, WO 2021252644, WO2022020711, WO 2022020242, US 11,174,231 B1, US 11,124,497 B1, WO 2005113580, and WO2006061714).
There is a need in the art for novel therapeutic agents that treat, ameliorate or prevent SARS-CoV-2 infection. The present invention provides the process of novel compounds which act in inhibiting or preventing SARS-CoV-2 viral replication and thus are used in the treatment of COVID-19 (see PCT/US21/60247).
Synthesis of substituted spirooxindole and its intermediate has been previously published (Refer to PCT/US21/60247, WO2019086142, WO 2020221811, WO2020221826, J. Med. Chem.2012, 55, 9069). However, the scale-up using previous process is very challenging due to the safety concern associated with certain intermediates, instability of certain intermediates as well as lack of purification process other than column chromatograph. Thus, there is a strong need for developing a safe and efficient processes for the large-scale preparation of these novel substituted spirooxindole derivatives.
SYNTHESIS

https://patents.google.com/patent/US11352363B1/en

PATENT
SYN
[1]. Guoqiang Wang, et al. Novel spiropyrrolidine derived antiviral drugs. Patent CN114524821A.
1.20230295175PROCESSES FOR THE PREPARATION OF SUBSTITUTED SPIROOXINDOLE DERIVATIVES
2.WO/2023/177854PROCESSES FOR THE PREPARATION OF SUBSTITUTED SPIROOXINDOLE DERIVATIVES
3.WO/2022/109363NOVEL SPIROPYRROLIDINE DERIVED ANTIVIRAL AGENTS
Enanta Pharmaceuticals, Inc.
WO2023177854




Example 15. Preparation of Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))

DMF (760 kg, 8V) was added into the reaction at 0 °C (-5~5 °C) followed by compound (j) (63 kg, 1.05 eq) and N-Methylmorpholine (56 kg, 2 eq), HATU
(106 kg, 1.0 eq) and Compound (m-1) (100 kg, 1.0 eq). The reactor was rinsed with DMF (190 kg, 2V) under and warmed up to 25 °C (20~30 °C) and stirred for 5 h (3~6 h) at 25 °C (20~30 °C). After that, additional HATU (0.1 eq) was added and the reaction mixture was stirred for 16-24 h.25% Ammonium hydroxide (38 kg) was added to the reaction mixture at 25 °C (20~30 °C) and stirred for 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was then added to water (5000 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (500 kg, 5 V). The cake was dissolved in ethyl acetate (1350 kg, 15 V) and washed with 10% sodium chloride solution (500 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (660 kg, 5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (137 kg, 2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and the wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H).
Example 16. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF solution of Compound (m-2) (1 kg, 1.0 eq.) was added to a reactor at around 0-10oC. Compound (l) (600 g, 1.0 eq.), NMM (3.00 eq., 850 g) and HATU (1.00 eq., 1.06 kg) was added to the reactor while maintaining the temperature at 0-10oC; The reaction was warmed to 20±5oC, and stirred for at least 6 hours at 20±5oC. HATU (0.20 eq., 210 g) was added to the reactor at 20±5oC and stirred for at least 6 hours at 20±5oC.25% Ammonium hydroxide (390 g, 1.0 eq) was added to the reaction mixture at 20 °C and stirred for 2 h (1~3 h) at 20 °C. EtOAc (14.0 V) and water (14 V) was added at around 25oC over 20 minutes, and the
solution was stirred for at least 30 min. Aqueous phase was extracted with EtOAc for three times and the organic phase was combined, and washed with 10% aq. NaCl for three times at 20±5oC. The organic phase was concentrated to 6 V then EtOH (7.0 V) was charged. The EtOAc-EtOH solvent swap was repeated for three times and concentrated to 5 V before water (7.0 v) was added at 20±5oC. The mixture was cooled to 0-10oC and stirred for 1 h before being filtered. The filter cake was dissolved in ethyl acetate (15 V) and washed with 10% sodium chloride solution for three times. The organic layer was concentrated to 2-3V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in about 70-75% yield over two steps.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H).
Example 17. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF (10.0 v) was added to a reactor at 25 °C followed by Compound (l) (4.4 kg, 1.0 eq.), NMM (3.0 eq.) Compound (m-3) (1.0 eq.) and HATU (1.0 eq) at 20-25oC. The reaction mixture was stirred for at least 12 hours at 20-25 °C. Once reaction was complete, aqueous ammonium hydroxide (1.0 eq.) was to the reaction system at 20-25 °C, then stirred for at least 2 hours at 20-25oC. The reaction mixture was then added to water (220 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (22 kg, 5 V). The cake was dissolved in ethyl acetate (135 g, 15 V) and washed with 10% sodium chloride solution (22 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45 ℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H). Example 18. Preparation of N-((S)-1-((3R,5’S)-5′-cyano-2-oxospiro[indoline-3,3′-pyrrolidin]-1′-yl)-4-methyl-1-oxopentan-2-yl)-4,6,7-trifluoro-N-methyl-1H-indole-2-carboxamide toluene solvate (Compound (I))
(I))

Ethyl acetate (630 kg, 10 V) was added into reactor (R1) followed by Compound (n) (70 kg). Make sure the water content was less than 0.20% (w/w). The reaction was cooled to 0 °C (-5 – 5°C) and then triethylamine (89.6 kg) was added followed by trifluoroacetic anhydride (92.4 kg) at 0 °C (-5 – 5°C). The reaction was stirred for 1 h (0.5~2 h) at 0 °C (-5 – 5°C). Once the reaction was complete, the reaction mixture was added slowly to 0.2 N aqueous HCl solution (700 kg) over 1 h at 0 °C (-5~5 °C). The resulting solution was stirred for 30 min at 0 °C (-5~5 °C) and the organic layer was separated.1% aqueous ammonium hydroxide (700 kg) was added to the organic layer and stirred at 20 °C for 30 min (15~25 °C). The organic layer was separated and washed with 10% brine for four times. Then the organic layer was separated and distilled to 2-3 V. Toluene-EtOAc swap was performed until precipitate was observed at 3-4 V. Then Toluene (5-6 V) was added and the slurry was stirred at 50 oC for 2 h. Then the solution was cooled down to 20 oC over 1-2 h and stirred for 10 hr (6~14 hr) at 20 °C (15~25 °C). The reaction mixture was filtered and the wet cake was rinsed with toluene (120 kg, 2V). The wet cake was then dried at 50˚C (45~55 °C) for 48 hr to provide desired compound (o) as a white solid in 80-85% yield.
1H NMR (400 MHz, Acetone-d6) δ 11.17 (s, 1H), 9.65 (s, 1H), 7.02 (dd, J = 13.7, 7.3 Hz, 2H), 6.94 (dd, J = 6.0, 3.5 Hz, 1H), 6.92 – 6.85 (m, 2H), 6.81 (t, J = 7.5 Hz, 1H), 5.56 (dd, J = 9.4, 5.6 Hz, 1H), 5.21 (t, J = 8.3 Hz, 1H), 4.25 (d, J = 10.7 Hz, 1H), 3.99 (d, J = 10.6 Hz, 1H), 3.43 (s, 3H), 2.79 – 2.61 (m, 2H), 1.93 (ddd, J = 14.4, 9.5, 5.1 Hz, 1H), 1.79 (ddd, J = 14.2, 8.7, 5.6 Hz, 1H), 1.64 (dpd, J = 8.7, 6.6, 5.1 Hz, 1H), 0.98 (dd, J = 18.5, 6.6 Hz, 6H).
US20230103494
CN114524821
SCHEME

MAIN

////////Zevotrelvir, EDP 235
O=C1[C@@]2(CN([C@@H](C2)C#N)C([C@H](CC(C)C)N(C)C(C3=CC4=C(F)C=C(F)C(F)=C4N3)=O)=O)C5=CC=CC=C5N1
Afimetoran
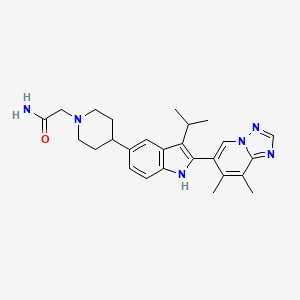

Afimetoran BMS-986256, WHO 11516
cas 2171019-55-7
2-[4-[2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyridin-6-yl)-3-propan-2-yl-1H-indol-5-yl]piperidin-1-yl]acetamide
C26H32N6O,444.583, phase 1
Afimetoran is an immunomodulator and an antagonist of toll-like receptors 7 and 8.1,2 It is also is under investigation in clinical trial NCT04269356 (Study to Assess the Way the Body Absorbs, Distributes, Breaks Down and Eliminates Radioactive BMS-986256 in Healthy Male Participants).

Ref
WO2018005586
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018005586&_cid=P20-M0RQ0D-09010-1
The invention further pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to TLR modulation, such as inflammatory and autoimmune diseases, and methods of inhibiting the activity of TLRs in a mammal.
Toll/IL-1 receptor family members are important regulators of inflammation and host resistance. The Toll-like receptor family recognizes molecular patterns derived from infectious organisms including bacteria, fungi, parasites, and viruses (reviewed in Kawai, T. et al., Nature Immunol., 11:373-384 (2010)). Ligand binding to the receptor induces dimerization and recruitment of adaptor molecules to a conserved cytoplasmic motif in the receptor termed the Toll/IL-1 receptor (TIR) domain. With the exception of TLR3, all TLRs recruit the adaptor molecule MyD88. The IL-1 receptor family also contains a cytoplasmic TIR motif and recruits MyD88 upon ligand binding (reviewed in Sims, J.E. et al., Nature Rev. Immunol., 10:89-102 (2010)).
Toll-like receptors (TLRs) are a family of evolutionarily conserved, transmembrane innate immune receptors that participate in the first-line defense. As pattern recognition receptors, the TLRs protect against foreign molecules, activated by pathogen associated molecular patterns (PAMPs), or from damaged tissue, activated by danger associated molecular patterns (DAMPs). A total of 13 TLR family members have been identified, 10 in human, that span either the cell surface or the endosomal compartment. TLR7-9 are among the set that are endosomally located and respond to single-stranded RNA (TLR7and TLR8) or unmethylated single-stranded DNA containing cytosine-phosphate-guanine (CpG) motifs (TLR9).
Activation of TLR7/8/9 can initiate a variety of inflammatory responses (cytokine production, B cell activation and IgG production, Type I interferon response). In the case of autoimmune disorders, the aberrant sustained activation of TLR7/8/9 leads to worsening of disease states. Whereas overexpression of TLR7 in mice has been shown to exacerbate autoimmune disease, knockout of TLR7 in mice was found to be protective against disease in lupus-prone MRL/lpr mice. Dual knockout of TLR7 and 9 showed further enhanced protection.
As numerous conditions may benefit by treatment involving modulation of cytokines, IFN production and B cell activity, it is immediately apparent that new compounds capable of modulating TLR7 and/or TLR8 and/or TLR9 and methods of using these compounds could provide substantial therapeutic benefits to a wide variety of patients.
The present invention relates to a new class of [1,2,4]triazolo[1,5-a]pyridinyl substituted indole compounds found to be effective inhibitors of signaling through TLR7/8/9. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
EXAMPLE 15
2-(4-(2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyridin-6-yl)-3-isopropyl-1H-indol-5-yl) piperidin-1-yl)acetamide
To a reaction flask were added
6-(3-isopropyl-5-(piperidin-4-yl)-1H-indol-2-yl)-7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyrid ine, 2 HCl (47.66 g, 104 mmol), DCE (220 mL), DBU (62.4 mL, 414 mmol), and 2-bromoacetamide (17.14 g, 124 mmol). The reaction flask was capped. The reaction mixture was stirred overnight at room temperature. The reaction mixture was concentrated, diluted with water, and stirred for 30 minutes then filtered. The solid was recrystallized using ethanol to afford 2-(4-(2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a] pyridin-6-yl)-3-isopropyl-1H-indol-5-yl)piperidin-1-yl)acetamide (42.3 g, 93 mmol,
90% yield) as a white solid. LCMS MH+: 445. HPLC Ret. Time 1.20 min. Method QC-ACN-TFA-XB. 1HNMR (400 MHz, DMSO-d6) δ 10.97-10.86 (m, 1H), 8.78-8.69 (m, 1H), 8.54-8.40 (m, 1H), 7.64-7.49 (m, 1H), 7.30-7.21 (m, 2H), 7.17-7.09 (m, 1H), 7.06-6.93 (m, 1H), 2.99-2.82 (m, 5H), 2.62-2.54 (m, 4H), 2.24-2.12 (m, 5H), 1.92-1.72 (m, 4H), 1.37-1.29 (m, 6H).
ACS Medicinal Chemistry Letters (2022), 13(5), 812-818 83%
References
- Bristol-Myers Squibb: Investor Series [Link]
- Bristol-Myers Squibb: Investor Series [Link]
- MedKoo Biosciences: Afimetoran [Link]
//////////////Afimetoran, BMS-986256, BMS 986256, WHO 11516, phase 1
lazertinib

lazertinib
CAS 1903008-80-9
554.655, C30H34N8O3
FDA APPROVED, 8/19/2024, Lazcluze, To treat non-small cell lung cancer
Drug Trials Snapshot
2-PROPENAMIDE, N-(5-((4-(4-((DIMETHYLAMINO)METHYL)-3-PHENYL-1H-PYRAZOL-1-YL)-2-PYRIMIDINYL)AMINO)-4-METHOXY-2-(4-MORPHOLINYL)PHENYL)-
- N-(5-((4-(4-((DIMETHYLAMINO)METHYL)-3-PHENYL-1H-PYRAZOL-1-YL)PYRIMIDIN-2-YL)AMINO)-4-METHOXY-2-MORPHOLINOPHENYL)ACRYLAMIDE
- C-18112003-G
- GNS 1480
- GNS-1480
- GNS1480
- JNJ-73841937-AAA
- YH 25448
- YH-25448
- YH25448
FDA APPROVED
| 8/19/2024 |
To treat non-small cell lung cancer, Lazcluze
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Lazertinib mesylate monohydrate | WUT449BEG5 | 2411549-88-5 | ZJPNGZUERUYZEG-UHFFFAOYSA-N |
Lazertinib is an oral, third-generation, epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI).2,3 Lazertinib was first approved in South Korea on January 18, 2021, for the treatment of EGFR T790M mutation-positive non-small cell lung cancer (NSCLC) with EGFR mutations.1 It was approved by the FDA on August 19, 2024.5 Lazertinib is used alone or in combination with other chemotherapeutic agents.4
Lazertinib, sold under the brand name Lazcluze and Leclaza, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1][2][3] It is a kinase inhibitor of epidermal growth factor receptor.[1]
The most common adverse reactions include rash, nail toxicity, infusion-related reactions (amivantamab), musculoskeletal pain, edema, stomatitis, venous thromboembolism, paresthesia, fatigue, diarrhea, constipation, COVID-19 infection, hemorrhage, dry skin, decreased appetite, pruritus, nausea, and ocular toxicity.[2]
Lazertinib was approved for medical use in South Korea in January 2021,[4][5] and in the United States in August 2024.[2][6]
Medical uses
Lazertinib is indicated in combination with amivantamab for the first-line treatment of adults with locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 L858R substitution mutations.[2
History
Efficacy was evaluated in MARIPOSA (NCT04487080), a randomized, active-controlled, multicenter trial of 1074 participants with exon 19 deletion or exon 21 L858R substitution mutation-positive locally advanced or metastatic non-small cell lung cancer and no prior systemic therapy for advanced disease.[2] Participants were randomized (2:2:1) to receive lazertinib in combination with amivantamab, osimertinib monotherapy, or lazertinib monotherapy (an unapproved regimen for non-small cell lung cancer) until disease progression or unacceptable toxicity.[2]
Society and culture
Legal status
Lazertinib was approved for medical use in the United States in August 2024.[2]Names
Lazertinib is the international nonproprietary name.[7]
/////////////////////
References
- ^ Jump up to:a b c “Lazcluze- lazertinib tablet, film coated”. DailyMed. 20 August 2024. Retrieved 5 September 2024.
- ^ Jump up to:a b c d e f g “FDA approves lazertinib with amivantamab-vmjw for non-small lung cancer”. U.S. Food and Drug Administration (FDA). 19 August 2024. Retrieved 21 August 2024. This article incorporates text from this source, which is in the public domain.
- ^ Dhillon S (June 2021). “Lazertinib: First Approval”. Drugs. 81 (9): 1107–1113. doi:10.1007/s40265-021-01533-x. PMC 8217052. PMID 34028784.
- ^ “Yuhan wins approval as MFDS clear T790M EGFR TKI drug ‘Lazertinib'”. 바이오스펙테이터. Retrieved 23 August 2024.
- ^ Dhillon S (2021). “Lazertinib: First Approval”. Drugs. 81 (9): 1107–1113. doi:10.1007/s40265-021-01533-x. ISSN 0012-6667. PMC 8217052. PMID 34028784.
- ^ “Rybrevant (amivantamab-vmjw) plus Lazcluze (lazertinib) approved in the U.S. as a first-line chemotherapy-free treatment for patients with EGFR-mutated advanced lung cancer”. Johnson & Johnson (Press release). 20 August 2024. Retrieved 21 August 2024.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 79”. WHO Drug Information. 32 (1). hdl:10665/330941.
External links
- Clinical trial number NCT04487080 for “A Study of Amivantamab and Lazertinib Combination Therapy Versus Osimertinib in Locally Advanced or Metastatic Non-Small Cell Lung Cancer (MARIPOSA)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lazcluze, Leclaza |
| License data | US DailyMed: Lazertinib |
| Routes of administration | By mouth |
| Drug class | EGFR inhibitor |
| ATC code | L01EB09 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1903008-80-9 |
| PubChem CID | 121269225 |
| IUPHAR/BPS | 10136 |
| DrugBank | DB16216 |
| ChemSpider | 64835231 |
| UNII | 4A2Y23XK11 |
| KEGG | D11980D12245 |
| ChEMBL | ChEMBL4558324 |
| Chemical and physical data | |
| Formula | C30H34N8O3 |
| Molar mass | 554.655 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////lazertinib, C-18112003-G, GNS 1480, GNS-1480, GNS1480, JNJ-73841937-AAA, YH 25448, YH-25448, YH25448, Lazcluze, FDA 2024, APPROVALS 2024
COC1=C(NC2=NC=CC(=N2)N2C=C(CN(C)C)C(=N2)C2=CC=CC=C2)C=C(NC(=O)C=C)C(=C1)N1CCOCC1

{kind=link}
{kind=link}
{kind=link}