
Home » 2020 (Page 10)
Yearly Archives: 2020
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
RESMETIROM
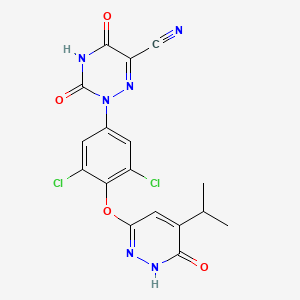
RESMETIROM
| C17H12Cl2N6O4 |
435.2 g/mol
MGL-3196
CAS 920509-32-6, Resmetirom, VIA-3196, UNII-RE0V0T1ES0
FDA APPROVED 3/14/2024, To treat noncirrhotic non-alcoholic steatohepatitis with moderate to advanced liver scarring
Press Release
Phase III, Non-alcoholic fatty liver disease (NAFLD)
2-[3,5-dichloro-4-[(6-oxo-5-propan-2-yl-1H-pyridazin-3-yl)oxy]phenyl]-3,5-dioxo-1,2,4-triazine-6-carbonitrile
2-(3,5-DICHLORO-4-((5-ISOPROPYL-6-OXO-1,6-DIHYDROPYRIDAZIN-3-YL)OXY)PHENYL)-3,5-DIOXO-2,3,4,5-TETRAHYDRO-(1,2,4)TRIAZINE-6-CARBONITRILE
1,2,4-TRIAZINE-6-CARBONITRILE, 2-(3,5-DICHLORO-4-((1,6-DIHYDRO-5-(1-METHYLETHYL)-6-OXO-3-PYRIDAZINYL)OXY)PHENYL)-2,3,4,5-TETRAHYDRO-3,5-DIOXO-
Madrigal Pharmaceuticals , following the merger between Synta and Madrigal Pharmaceuticals (pre-merger) (following the acquisition of VIA Pharmaceuticals ‘ assets (originally under license from Roche )), is developing resmetirom (MGL-3196, VIA-3196), the lead from oral capsule formulation thyroid hormone receptor (THR) beta agonists, cholesterol and triglyceride modulators, for the use in the treatment of metabolic disorders including hypercholesterolemia and other dyslipidemias, and non-alcoholic steatohepatitis.
MGL-3196 is a first-in-class, orally administered, small-molecule, liver-directed, THR β-selective agonist. Preclinical, toxicology and Phase 1 clinical data suggest MGL-3196 has an attractive, differentiated profile as a potential treatment for non-alcoholic steatohepatitis (NASH) and dyslipidemias. THR-β selectivity also enhances the safety profile of MGL-3196, compared to non-selective agents. MGL-3196 has shown no suppression of the central thyroid axis, no THR-α effects on heart rate or bone, and no elevation of liver enzymes. These characteristics make MGL-3196 among the most promising molecules in development in this therapeutic area. MGL-3196 is in a Phase 2 clinical trial for the treatment of non-alcoholic steatohepatitis (NASH).
PATENT
WO-2020010068
Novel crystalline salt of resmetirom as thyroid hormone receptor agonists useful for treating obesity, hyperlipidemia, hypercholesterolemia and diabetes. Appears to be the first filing from the assignee and the inventors on this compound,
Thyroid hormones are critical for normal growth and development and for maintaining metabolic homeostasis (Paul M. Yen, Physiological reviews, Vol. 81(3): pp. 1097-1126 (2001)). Circulating levels of thyroid hormones are tightly regulated by feedback mechanisms in the hypothalamus/pituitary/thyroid (HPT) axis. Thyroid dysfunction leading to hypothyroidism or hyperthyroidism clearly demonstrates that thyroid hormones exert profound effects on cardiac function, body weight, metabolism, metabolic rate, body temperature, cholesterol, bone, muscle and behavior.
[0005] The biological activity of thyroid hormones is mediated by thyroid hormone receptors (TRs or THRs) (M. A. Lazar, Endocrine Reviews, Vol. 14: pp. 348-399 (1993)). TRs belong to the superfamily known as nuclear receptors. TRs form heterodimers with the retinoid receptor that act as ligand-inducible transcription factors. TRs have a ligand binding domain, a DNA binding domain, and an amino terminal domain, and regulate gene expression through interactions with DNA response elements and with various nuclear co-activators and co repressors. The thyroid hormone receptors are derived from two separate genes, a and b. These distinct gene products produce multiple forms of their respective receptors through differential RNA processing. The major thyroid receptor isoforms are aΐ, a2, bΐ, and b2. Thyroid hormone receptors aΐ, bΐ, and b2 bind thyroid hormone. It has been shown that the thyroid hormone receptor subtypes can differ in their contribution to particular biological responses. Recent studies suggest that TIIb 1 plays an important role in regulating TRH (thyrotropin releasing hormone) and on regulating thyroid hormone actions in the liver. T11b2 plays an important role in the regulation of TSH (thyroid stimulating hormone) (Abel et. al, J. Clin. Invest., Vol 104: pp. 291-300 (1999)). TIIb 1 plays an important role in regulating heart rate (B. Gloss et. al. Endocrinology, Vol. 142: pp. 544-550 (2001); C. Johansson et. al, Am. J. Physiol., Vol. 275: pp. R640-R646 (1998)).
[0006] Efforts have been made to synthesize thyroid hormone analogs which exhibit increased thyroid hormone receptor beta selectivity and/or tissue selective action. Such thyroid hormone mimetics may yield desirable reductions in body weight, lipids, cholesterol, and lipoproteins, with reduced impact on cardiovascular function or normal function of the hypothalamus/pituitary/thyroid axis (see, e.g., Joharapurkar et al, J. Med. Chem, 2012, 55 (12), pp 5649-5675). The development of thyroid hormone analogs which avoid the undesirable effects of hyperthyroidism and hypothyroidism while maintaining the beneficial effects of thyroid hormones would open new avenues of treatment for patients with metabolic disease such as obesity, hyperlipidemia, hypercholesterolemia, diabetes and other disorders and diseases such as liver steatosis and NASH, atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer, thyroid diseases, a resistance to thyroid hormone (RTH) syndrome, and related disorders and diseases.
PATENT
WO2018075650
In one embodiment, the metabolite of Compound A comprises a compound
having the following structure:
(“Ml”).
PATENT
WO 2007009913
PATENT
WO 2014043706
https://patents.google.com/patent/WO2014043706A1/en
Example 3: Preparation of (Z)-ethyl (2-cyano-2-(2-(3,5-dichloro-4-((5-isopropyl-6- oxo- l,6-dihydropyridazin-3-yl)oxy)phenyl)hydrazono)acetyl)carbamate (Int. 8)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet was charged with Int. 7 (75.0 g, 0.239 mol, 1 wt), acetic acid (600 mL, 8 vol), water (150 mL, 2 vol), and concentrated HC1 (71.3 mL, 0.95 vol). The resulting thin slurry was cooled to 6 °C and a solution of NaN02 (16.8 g, 0.243 mol, 1.02 equiv) in water (37.5 mL, 0.5 vol) was added over a period of 10 min while maintaining a batch temperature below 10 °C. After an additional 10 min of agitation between 5-10 °C, HPLC analysis showed complete conversion of Int. 7 to the diazonium intermediate. A solution of NaOAc (54.5 g, 0.664 mol, 2.78 equiv) in water (225 mL, 3 vol) was added over a period of 6 min while maintaining a batch temperature below 10 °C. N-cyanoacetylurethane (37.9 g, 0.243 mol, 1.02 equiv) was immediately added, the cooling was removed, and the batch naturally warmed to 8 °C over 35 min. HPLC analysis showed complete consumption of the diazonium intermediate and the reaction was deemed complete. The batch warmed naturally to 21 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (375 mL, 5 vol) twice. The collected orange solid was dried in a 35 °C vacuum oven for 64 h to provide crude Int. 8 (104.8 g, 91%).
A I L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, and N2 inlet/outlet was charged with crude Int. 8 (104.4 g, 1 wt) and acetic acid (522 mL, 5 vol). The resulting slurry was heated to 50 °C and held at that temperature for 1.5 h. The batch cooled naturally to 25 °C over 2 h and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (522 mL, 5 vol) and the cake conditioned under vacuum for 1.75 h. The light orange solid was dried to constant weight in a 40 °C vacuum oven to provide 89.9 g (78% from Int. 7) of the desired product. 1H NMR (DMSO) was consistent with the assigned structure.
Example 4: Preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, N2 inlet/outlet, and reflux condenser was charged with Int. 8 (89.3 g, 0.185 mol, 1 wt), DMAC (446 mL, 5 vol), and KOAc (20.0 g, 0.204 mol, 1.1 equiv). The mixture was heated to 120 °C and held at that temperature for 2 h. HPLC analysis showed complete conversion to Compound A. The batch temperature was adjusted to 18 °C over 1 h, and acetic acid (22.3 mL, 0.25 vol) was added. The batch temperature was adjusted to 8 °C, and water (714 mL, 8 vol) was added over 1 h; an orange slurry formed. The batch was filtered through Sharkskin filter paper and the cake was allowed to condition overnight under N2 without vacuum for convenience. A premixed solution of 1 : 1 acetone/water (445 mL, 5 vol) was charged to the flask and added to the cake as a rinse with vacuum applied. After 2 h of conditioning the cake under vacuum, it was transferred to a clean 1 L, three-neck, round- bottom flask equipped with overhead stirring, a thermocouple, and N2inlet/outlet. Ethanol (357 mL, 4 vol) and acetone (357 mL, 4 vol) were charged and the resulting slurry was heated to 60 °C; dissolution occurred. Water (890 mL, 10 vol) was added over a period of 90 min while maintaining a batch temperature between 55-60 °C. The resulting slurry was allowed to cool to 25 °C and filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with a solution of 1:1 EtOH/water (446 mL, 5 vol). The cake was conditioned overnight under N2 without vacuum for convenience. The cracks in the cake were smoothed and vacuum applied. The cake was washed with water (179 mL, 2 vol) and dried in a 45 °C vacuum oven to a constant weight of 70.5 g (87%, crude Compound A). HPLC analysis showed a purity of 94.8%.
A 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser was charged with crude Compound A (70.0 g) and MIBK (350 mL, 5 vol). The orange slurry was heated to 50 °C and held at that temperature for 2 h. The batch cooled naturally to 23 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with MIBK (35 mL, 0.5 vol) twice. The collected solids were dried in a 45 °C vacuum oven to a constant weight of 58.5 g (84%). This solid was charged to a 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser. Ethanol (290 mL, 5 vol) was added and the slurry was heated to reflux. After 3.5 h at reflux, XRPD showed the solid was consistent with Form I, and heating was removed. Upon reaching 25 °C, the batch was filtered through filter paper, and the reactor and cake were washed sequentially with EtOH (174 mL, 3 vol). The tan solid Compound A was dried in a 40 °C vacuum oven to a constant weight of 50.4 g (87%, 64% from Int. 8). HPLC analysis showed a purity of 99.1%. 1H NMR (DMSO) was consistent with the assigned structure.
Example 5: Scaled up preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A larger scale batch of Compound A was synthesized according to the scheme below. The conditions in the scheme below are similar to those described in Examples 1-4 above.

6A

Compound A
Synthesis of 4: A 50 L jacketed glass vessel (purged with N2) was charged with 3,6- dichloropyridazine (2.00 kg), 4-amino-2,6-dichlorophenol (2.44 kg) and N,N- dimethylacetamide (10.0 L). The batch was vacuum (26 inHg) / nitrogen (1 PSIG) purged 3 times. Cesium carbonate (5.03 kg) was added and the batch temperature was adjusted from 22.3 °C to 65.0 °C over 3.5 hours. The batch was held at 65.0 °C for 20 hours. At this point,
NMR analysis indicated 3.34% 3.6-dichloropyridazine relative to 2. The batch temperature was adjusted to 21.5 °C and ethyl acetate (4.00 L) was added to the batch. The batch was agitated for 10 minutes and then filtered through a 18″ Nutsche filter equipped with polypropylene filter cloth. The filtration took 15 minutes. Ethyl acetate (5.34 L) was charged to the vessel and transferred to the filter as a rinse. The batch was then manually re- suspended in the filter before re-applying vacuum. This process was repeated 2 more times and the filter cake was conditioned for 10 minutes. The filtrate was charged to a 100-L vessel that contained (16.0 L) of a previously prepared 15% sodium chloride in H20. The batch was agitated for 5 minutes and then allowed to separate for 35 minutes. The interface was not visible, so the calculated 23 L of the lower aqueous phase was removed. 16.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 6 minutes and then allowed to separate for 7 minutes. The interface was visible at -19 L and the lower aqueous phase was removed. 17.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 7 minutes and then allowed to separate for 11 minutes. The lower aqueous phase was removed. The vessel was set up for vacuum distillation and the batch was concentrated from 17.0 L to 8.0 L over 2 hours 20 minutes with the batch temperature kept around 21 °C. Benzoic anhydride (3.19 kg) and acetic acid (18.0 L) were charged to the vessel. The vessel was set up for vacuum distillation and the batch was concentrated from 28.0 L to 12.0 L over 2 days (overnight hold at 20 °C) with the batch temperature kept between 20 and 55 °C. At this point, JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.015. Acetic acid (4.0 L) was charged to the batch and the batch was distilled to 12 L. JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.0036. Acetic acid (20.0 L) was charged to the batch and the batch temperature was adjusted to 70.0 °C. The batch was sampled for HPLC analysis and 2 was 0.16%. Sodium acetate (2,20 kg) was added to the batch and the batch temperature was adjusted from 72.4 °C to 110.0 °C. After 18.5 hours, HPLC analysis indicated no Int. B detected. The batch temperature was adjusted from 111.3 to 74.7 °C and DI water (30.0 L) was added to the batch over 2 hours. The batch temperature was adjusted to 20 .5 °C and then filtered using a 24″ Haselloy Nutsche filter equipped with polypropylene filter cloth. A previously prepared solution of 1:1 acetic acid in DI H20 (10.0 L) was charged to the vessel and agitated for 5 minutes. The wash was transferred to the filter and the batch was then manually re- suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged to the vessel and then transferred to the filter. The batch was manually re-suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged directly to the filter and the batch was then manually re-suspended in the filter before re-applying vacuum. The filter cake was allowed to condition for 18 hours to give 14.4 kg of 4. HPLC analysis indicated a purity of 93.7%. This wet cake was carried forward into the purification. A 100 L jacketed glass vessel (purged with N2) was charged with crude 4 (wet cake 14.42 kg), acetic acid (48.8 L) and the agitator was started. DI H20 (1.74 L) was charged. The batch (a slurry) temperature was adjusted from 18.1 to 100.1 °C over 4.25 hours. The batch was held at 100.1 to 106.1 °C for 1 hour and then adjusted to 73.1 °C. DI H20 (28.0 L) was added to the batch over 1 hour keeping the batch temperature between 73.1 and 70.3 °C. The batch temperature was adjusted further from 70.3 °C to 25.0 °C overnight. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with polypropylene filter cloth. The filtration took 13 minutes. A solution of DI H20 (9.00 L) and acetic acid (11.0 L) was prepared and added to the 100 L vessel. The mixture was agitated for 5 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 6 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 9 minutes and then transferred to the filter cake. The batch was allowed to condition for 3 days and then transferred to drying trays for vacuum oven drying. After 3 days at 50 °C and 28’7Hg, the batch gave a 74% yield (3.7 kg) of4 as an off-white solid. The JH NMR spectrum was consistent with the assigned structure, HPLC analysis indicated a purity of 98.87% and KF analysis indicated 0.14% H20. Synthesis of Int. 7: A 100-L jacketed glass vessel (purged with N2) was charged with tetrahydrofuran (44.4 L). The agitator was started (125 RPM) and 4 (3.67 kg) was charged followed by lithium chloride (1.26 kg). The batch temperature was observed to be 26.7 ° C and was an amber solution. Isopropenylmagnesium bromide 1.64 molar solution in 2-methyl THF (21.29 kg) was added over 2 ½ hours keeping the batch between 24.3 and 33.6 °C. The batch was agitated at 24.5 °C for 17 hours at which point HPLC analysis indicated 9% 4. A 2nd 100-L jacketed glass vessel (purged with N2) was charged with 3N hydrogen chloride (18.3 L). The batch was transferred to the vessel containing the 3N HC1 over 25 minutes keeping the batch temperature between 20 and 46 °C. A bi-phasic solution was observed. The quenched batch was transferred back to the 1st 100-L vessel to quench the small amount of residue left behind. THF (2.00 L) was used as a rinse. The batch temperature was observed to be 40.9 ° C and was agitated at 318 RPM for 45 minutes. The batch temperature was adjusted to 21.8 ° C and the layers were allowed to separate. The separation took 10 minutes. The lower aqueous phase was removed (-26.0 L). A solution of sodium chloride (1.56 kg) in DI water (14.0 L) was prepared and added to the batch. This was agitated at 318 RPM for 10 minutes and agitator was stopped. The separation took 3 minutes. The lower aqueous phase was removed (-16.0 L). The batch was vacuum distilled from 58.0 L to 18.4 L using ~24’7Hg and a jacket temperature of 50 to 55 °C. A solution of potassium hydroxide (2.30 kg) in DI water (20.7 L) was prepared in a 72-L round bottom flask. The vessel was set up for atmospheric distillation using 2 distillation heads and the batch was transferred to the 72-L vessel. THF (0.75 L) was used as a rinse. The batch volume was -41.0 L, the temperature was adjusted to 64.1 °C and distillation started with the aid of a N2 sweep. Heating was continued to drive the batch temperature to 85.4 °C while distilling at which point the 72-L vessel was set up for reflux (batch volume was about 28.0 L at the end of the distillation). The batch was held at 85 °C for 13 hours at which point HPLC analysis indicated 0.3% compound 6A. Heating was stopped and the batch was transferred to a 100-L jacketed glass vessel. Solids were observed. The batch temperature was adjusted from 70.6 °C to 56.7 °C. A previously prepared solution of sodium hydrogen carbonate (2.82 kg) in DI water (35.0 L) was added over 80 minutes keeping the batch temperature between 56.7 and 46.7 °C. The batch pH at the end of the addition was 9.8. The batch was held at
46.7 to 49.0 °C for 40 minutes and then cooled to 25.0 °C. The batch was filtered using a 18″ stainless steel Nutsche filter. DI water (18.4 L) was charged to the vessel and transferred to the filter. The filter cake was manually re-suspended in the filter and then the liquors were removed. This process was repeated once more and the filter cake was 3″ thick. The filter cake was conditioned on the filter for 3 days, was transferred to drying trays and dried in a vacuum oven at 45 °C to provide 2.93 kg Int. 7 (95% yield) with an HPLC purity of 87.6%.
Synthesis of Int. 8: A 100 L jacketed glass vessel (purged with N2 and plumbed to a caustic scrubber) was charged with acidic acid (13.0 L). Int. 7 (2.85 kg) was charged to the vessel and the agitator was started. N-Cyanoacetylurethane (1.56 kg) and DI water (5.70 L) were charged to the vessel. The batch temperature was adjusted from 17.0 °C to 5.5 °C and a thin slurry was observed. At this point 37% hydrogen chloride (2.70 L) was added over 10 minutes keeping the batch temperature between 4.8 °C and 8.8 °C. A previously prepared solution of sodium nitrite (638 g) in DI water (1.42 L) was added over 26 minutes keeping the batch temperature between 5.8 °C and 8.7 °C. A brown gas was observed in the vessel head space during the addition. HPLC analysis indicated no Int. 7 detected. At this point a previously prepared solution of sodium acetate (2.07 kg) in DI water (8.50 L) was added over 47 minutes keeping the batch temperature between 5.5 °C and 9.5 °C. After the addition, a thin layer of orange residue was observed on the vessel wall just above the level of the batch. The batch temperature was adjusted from 9.4 °C to 24.5 °C and held at 25 °C (+ 5 °C) for 12 hours. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with tight-weave polypropylene filter cloth. The filtration took 30 minutes. The vessel was rinsed with 14.3 L of a 1 : 1 acidic acid / DI water. The orange residue on the reactor washed away with the rinse. The rinse was transferred to the filter where the batch was manually re-suspended. Vacuum was re-applied to remove the wash. A 2nd 1 : 1 acidic acid / DI water wash was performed as above and the batch was conditioned on the filter for 26 hours. HPLC analysis of the wet filter cake indicated purity was 90.4%. The batch was dried to a constant weight of 3.97 kg (91% yield) in a vacuum oven at 45 °C and 287Hg. Preparation of Compound A DMAC Solvate
A 100 L, jacketed, glass vessel purged with N2 was charged with Int. 8 (3.90 kg) and potassium acetate (875 g). N,N-dimethylacetamide (DMAC, 18.3 L) was charged to the vessel and the agitator was started. The batch temperature was adjusted to 115 °C over 2 h. After 2 h at 115 °C, the batch was sampled and HPLC analysis indicated 0.27% Int. 8 remained. The batch temperature was adjusted to 25.0 °C overnight. Acetic acid (975 mL) was added to the batch and the batch was agitated further for 3 h. The batch was transferred to a carboy and the vessel was rinsed clean with 800 mL of DMAC. The batch was transferred back to the 100 L vessel using vacuum through a 10 μιη in-line filter and a DMAC rinse (1.15 L) was used. The filtration was fast at the beginning but slow at the end, plugging up the filter. The batch temperature was adjusted to 11.1 °C and DI water (35.1 L) was added over 2 h 20 min, keeping the batch temperature between 5-15 °C. The batch was held for 1 h and filtered, using an 18″ Nutsche filter equipped with tight-weave
polypropylene cloth. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel, cooled to 10 °C, and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg to give 89% yield (3.77 kg) of Compound A DMAC solvate as an orange/tan solid. The 1H NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated 0.49% H20. XRPD indicated the expected form, i.e., Compound A DMAC solvate. Thermogravimetric analysis (TGA) indicated 16% weight loss. HPLC analysis indicated a purity of 93.67%.
Preparation of Crude Compound A
A 100 L, jacketed, glass vessel purged with N2 was charged with Compound A
DMAC solvate (3.75 kg) and ethanol (15.0 L). The agitator was started and acetone (15.0 L) was added. The batch temperature was adjusted from 10.6 °C to 60.0 °C over 1 h. At this point, the batch was in solution. DI water was added to the batch over 1.5 h, keeping the batch temperature at 60 + 5 °C. The batch was held at 60 + 5 °C for 1 h and cooled to 23.5 °C. An 18″ Nutsche filter equipped with tight-weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg for five days to give a 94% yield (2.90 kg) of Compound A as a powdery tan solid. The NMR spectrum is consistent with the assigned structure and Karl Fischer analysis indicated 6.6% H20. XRPD indicated the expected form of dihydrate. TGA indicated 6.7% weight loss. HPLC analysis indicated a purity of 96.4% (AUC).
Purification of Crude Compound A
A 50 L, jacketed, glass vessel purged with N2 was charged with Compound A crude
(2.90 kg) and methyl isobutyl ketone (14.5 L). The agitator was started and the batch temperature was adjusted from 20.2 °C to 50.4 °C over 1.5 h. The batch was held at 50 °C (+ 5 °C) for 1 h and cooled to 20-25 °C. The batch was held at 20-25 °C for 2.5 h. An 18″ Nutsche filter equipped with tight- weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 20 min. Methyl isobutyl ketone (MIBK, 1.45 L) was charged to the vessel and transferred to the filter cake. The cake was manually resuspended and the liquors were pulled through with vacuum. Methyl isobutyl ketone (2.90 L) was charged to the filter cake and the cake was manually resuspended. The liquors were pulled through with vacuum and the cake was conditioned with vacuum and nitrogen for 15 h. The filter cake dried into a tan, hard 18″ x 1 ½” disc. This was manually broken up and run through coffee grinders to give a 76% yield (2.72 kg) of MGL-3196 MIBK solvate as a tan, powdery solid. No oven drying was necessary. The NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated <0.1 % H20. XRPD indicated the expected form MIBK solvate. TGA indicated 17.3% weight loss. HPLC analysis indicated a purity of 98.5%.
Example 6: Conversion of Compound A to Form I
Purified Compound A (4802 g) as a 1:1 MIBK solvate which was obtained from Int. 8 as described in Example 5 above was added into a jacketed, 100 L reactor along with 24 liters of ethanol. The resulting slurry was heated to 80 + 5 °C (reflux) over 1 h 25 min; the mixture was stirred at that temperature for 4 h 25 min. Analysis of the filtered solids at 2 h 55 min indicated that the form conversion was complete, with the XRPD spectra conforming to Form I. The mixture was cooled to 20 + 5 °C over 45 min and stirred at that temperature for 15 min. The slurry was filtered and the filter cake was washed twice with prefiltered ethanol (2 x 4.8 L). The wet cake (4.28 kg) was dried under vacuum at 40 + 5 °C for 118 h to afford 3390 g of Compound A form I.
PAPER
Journal of Medicinal Chemistry (2014), 57(10), 3912-3923
https://pubs.acs.org/doi/abs/10.1021/jm4019299
The beneficial effects of thyroid hormone (TH) on lipid levels are primarily due to its action at the thyroid hormone receptor β (THR-β) in the liver, while adverse effects, including cardiac effects, are mediated by thyroid hormone receptor α (THR-α). A pyridazinone series has been identified that is significantly more THR-β selective than earlier analogues. Optimization of this series by the addition of a cyanoazauracil substituent improved both the potency and selectivity and led to MGL-3196 (53), which is 28-fold selective for THR-β over THR-α in a functional assay. Compound 53 showed outstanding safety in a rat heart model and was efficacious in a preclinical model at doses that showed no impact on the central thyroid axis. In reported studies in healthy volunteers, 53 exhibited an excellent safety profile and decreased LDL cholesterol (LDL-C) and triglycerides (TG) at once daily oral doses of 50 mg or higher given for 2 weeks.

//////////////RESMETIROM , MGL-3196, VIA-3196, UNII-RE0V0T1ES0, Phase III
CC(C)C1=CC(=NNC1=O)OC2=C(C=C(C=C2Cl)N3C(=O)NC(=O)C(=N3)C#N)Cl
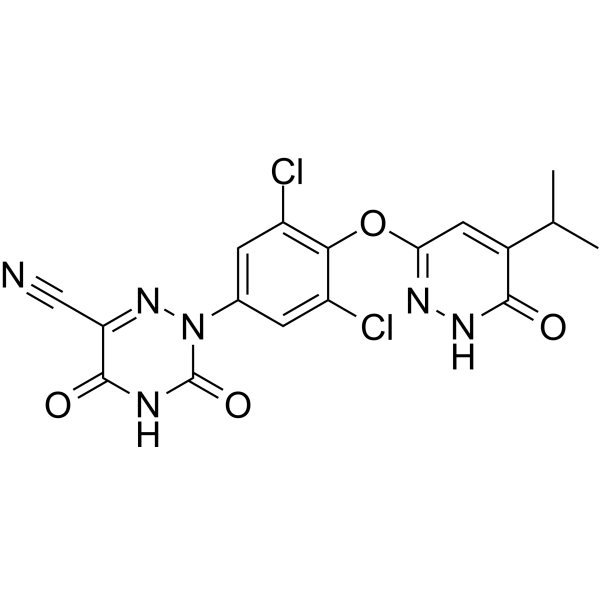
Avapritinib, アバプリチニブ , авапритиниб , أفابريتينيب ,

Avapritinib
BLU-285, BLU285
Antineoplastic, Tyrosine kinase inhibitor
アバプリチニブ
(1S)-1-(4-fluorophenyl)-1-[2-[4-[6-(1-methylpyrazol-4-yl)pyrrolo[2,1-f][1,2,4]triazin-4-yl]piperazin-1-yl]pyrimidin-5-yl]ethanamine
| Formula |
C26H27FN10
|
|---|---|
| CAS |
1703793-34-3
|
| Mol weight |
498.558
|
| No. | Drug Name | Active Ingredient | Approval Date | FDA-approved use on approval date* |
|---|---|---|---|---|
| 1. | Ayvakit | avapritinib | 1/9/2020 | To treat adults with unresectable or metastatic gastrointestinal stromal tumor (GIST) |
PRIORITY; Orphan, NDA 212608
Avapritinib, sold under the brand name Ayvakit, is a medication used for the treatment of tumors due to one specific rare mutation: It is specifically intended for adults with unresectable or metastatic ( y) gastrointestinal stromal tumor (GIST) that harbor a platelet-derived growth factor receptor alpha (PDGFRA) exon 18 mutation.[1]
Common side effects are edema (swelling), nausea, fatigue/asthenia (abnormal physical weakness or lack of energy), cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation (secretion of tears), abdominal pain, constipation, rash. and dizziness.[1]
Ayvakit is a kinase inhibitor.[1]
History
The U.S. Food and Drug Administration (FDA) approved avapritinib in January 2020.[1] The application for avapritinib was granted fast track designation, breakthrough therapy designation, and orphan drug designation.[1] The FDA granted approval of Ayvakit to Blueprint Medicines Corporation.[1]
Avapritinib was approved based on the results from the Phase I NAVIGATOR[2][3] clinical trial involving 43 patients with GIST harboring a PDGFRA exon 18 mutation, including 38 subjects with PDGFRA D842V mutation.[1] Subjects received avapritinib 300 mg or 400 mg orally once daily until disease progression or they experienced unacceptable toxicity.[1] The recommended dose was determined to be 300 mg once daily.[1] The trial measured how many subjects experienced complete or partial shrinkage (by a certain amount) of their tumors during treatment (overall response rate).[1] For subjects harboring a PDGFRA exon 18 mutation, the overall response rate was 84%, with 7% having a complete response and 77% having a partial response.[1] For the subgroup of subjects with PDGFRA D842V mutations, the overall response rate was 89%, with 8% having a complete response and 82% having a partial response.[1] While the median duration of response was not reached, 61% of the responding subjects with exon 18 mutations had a response lasting six months or longer (31% of subjects with an ongoing response were followed for less than six months).[1]
PATENT
WO 2015057873
https://patents.google.com/patent/WO2015057873A1/en
Example 7: Synthesis of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4- yl)pyrrolo[2, 1 -f\ [ 1 ,2,4] triazin-4-yl)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine and (S)- 1 – (4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine (Compounds 43 and 44)


Step 1 : Synthesis of (4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l- f] [ 1 ,2,4] triazin-4-yl)piperazin- 1 -yl)pyrimidin-5-yl)methanone:

4-Chloro-6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/] [l,2,4]triazine (180 mg, 0.770 mmol), (4-fluorophenyl)(2-(piperazin-l-yl)pyrimidin-5-yl)methanone, HC1 (265 mg, 0.821 mmol) and DIPEA (0.40 mL, 2.290 mmol) were stirred in 1,4-dioxane (4 mL) at room temperature for 18 hours. Saturated ammonium chloride was added and the products extracted into DCM (x2). The combined organic extracts were dried over Na2S04, filtered through Celite eluting with DCM, and the filtrate concentrated in vacuo. Purification of the residue by MPLC (25- 100% EtOAc-DCM) gave (4-fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methanone (160 mg, 0.331 mmol, 43 % yield) as an off-white solid. MS (ES+) C25H22FN90 requires: 483, found: 484 [M + H]+.
Step 2: Synthesis of (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-p razol-4-yl)p rrolo[2, l- ] [l,2,4]triazin-4- l)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide:

(S)-2-Methylpropane-2-sulfinamide (110 mg, 0.908 mmol), (4-fluorophenyl)(2-(4-(6-(l- methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5- yl)methanone (158 mg, 0.327 mmol) and ethyl orthotitanate (0.15 mL, 0.715 mmol) were stirred in THF (3.2 mL) at 70 °C for 18 hours. Room temperature was attained, water was added, and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0- 10% MeOH-EtOAc) gave (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)methylene)-2- methylpropane-2-sulfinamide (192 mg, 0.327 mmol, 100 % yield) as an orange solid. MS (ES+) C29H3iFN10OS requires: 586, found: 587 [M + H]+.
Step 3: Synthesis of (lS’)-N-(l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- l)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-

(lS’,Z)-N-((4-Fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide (190 mg, 0.324 mmol) was taken up in THF (3 mL) and cooled to 0 °C. Methylmagnesium bromide (3 M solution in diethyl ether, 0.50 mL, 1.500 mmol) was added and the resulting mixture stirred at 0 °C for 45 minutes. Additional methylmagnesium bromide (3 M solution in diethyl ether, 0.10 mL, 0.300 mmol) was added and stirring at 0 °C continued for 20 minutes. Saturated ammonium chloride was added and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0-10% MeOH-EtOAc) gave (lS’)-N-(l-(4-fluorophenyl)-l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol, 61.5 % yield) as a yellow solid (mixture of diastereoisomers). MS (ES+) C3oH35FN10OS requires: 602, found: 603 [M + H]+. Step 4: Synthesis of l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l- f\ [ 1 ,2,4] triazin-4- l)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine:

(S)-N- ( 1 – (4-Fluorophenyl)- 1 -(2- (4- (6-( 1 -methyl- 1 H-pyrazol-4-yl)pyrrolo [2,1- /] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol) was stirred in 4 M HCl in 1,4-dioxane (1.5 mL)/MeOH (1.5 mL) at room temperature for 1 hour. The solvent was removed in vacuo and the residue triturated in EtOAc to give l-(4-fluorophenyl)- l-(2-(4-(6-(l -methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4- yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine, HCl (110 mg, 0.206 mmol, 103 % yield) as a pale yellow solid. MS (ES+) C26H27FN10requires: 498, found: 482 [M- 17 + H]+, 499 [M + H]+.
Step 5: Chiral separation of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine and (5)-1-(4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin- 1 -yl)pyrimidin- -yl)ethanamine:

The enantiomers of racemic l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (94 mg, 0.189 mmol) were separated by chiral SFC to give (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH- pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine (34.4 mg, 0.069 mmol, 73.2 % yield) and (lS,)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (32.1 mg, 0.064 mmol, 68.3 % yield). The absolute stereochemistry was assigned randomly. MS (ES+)
C26H27FN10 requires: 498, found: 499 [M + H]+.
References
- ^ Jump up to:a b c d e f g h i j k l m “FDA approves the first targeted therapy to treat a rare mutation in patients with gastrointestinal stromal tumors”. U.S. Food and Drug Administration (FDA) (Press release). 9 January 2020. Archived from the original on 11 January 2020. Retrieved 9 January 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Blueprint Medicines Announces FDA Approval of AYVAKIT (avapritinib) for the Treatment of Adults with Unresectable or Metastatic PDGFRA Exon 18 Mutant Gastrointestinal Stromal Tumor”. Blueprint Medicines Corporation (Press release). 9 January 2020. Archived from the original on 11 January 2020. Retrieved 9 January 2020.
- ^ “Blueprint Medicines Announces Updated NAVIGATOR Trial Results in Patients with Advanced Gastrointestinal Stromal Tumors Supporting Development of Avapritinib Across All Lines of Therapy”. Blueprint Medicines Corporation (Press release). 15 November 2018. Archived from the original on 10 January 2020. Retrieved 9 January 2020.
Further reading
- Wu CP, Lusvarghi S, Wang JC, et al. (July 2019). “Avapritinib: A Selective Inhibitor of KIT and PDGFRα that Reverses ABCB1 and ABCG2-Mediated Multidrug Resistance in Cancer Cell Lines”. Mol. Pharm. 16 (7): 3040–3052. doi:10.1021/acs.molpharmaceut.9b00274. PMID 31117741.
- Gebreyohannes YK, Wozniak A, Zhai ME, et al. (January 2019). “Robust Activity of Avapritinib, Potent and Highly Selective Inhibitor of Mutated KIT, in Patient-derived Xenograft Models of Gastrointestinal Stromal Tumors”. Clin. Cancer Res. 25 (2): 609–618. doi:10.1158/1078-0432.CCR-18-1858. PMID 30274985.
External links
- “Avapritinib”. Drug Information Portal. U.S. National Library of Medicine (NLM).
| Clinical data | |
|---|---|
| Trade names | Ayvakit |
| Other names | BLU-285, BLU285 |
| License data | |
| Routes of administration |
By mouth |
| Drug class | Antineoplastic agents |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C26H27FN10 |
| Molar mass | 498.570 g·mol−1 |
| 3D model (JSmol) | |
///////Avapritinib, 2020 APPROVALS, PRIORITY, Orphan, BLU-285, BLU285, FDA 2020, Ayvakit, アバプリチニブ , авапритиниб , أفابريتينيب ,
TERIPARATIDE, テリパラチド , терипаратид , تيريباراتيد , 特立帕肽 ,
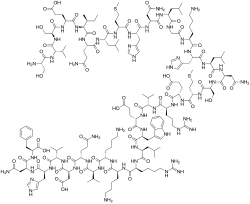
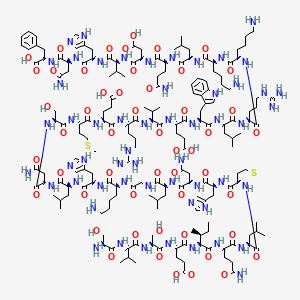


TERIPARATIDE
テリパラチド;
- PTH 1-34
- LY 333334 / LY-333334 / LY333334 / ZT-034
|
Ser Val Ser Glu Ile Gln Leu Met His Asn Leu Gly Lys His Leu Asn
Ser Met Glu Arg Val Glu Trp Leu Arg Lys Lys Leu Gln Asp Val His Asn Phe-OH |
|
| Type |
Peptide
|
|---|
| Formula |
C181H291N55O51S2
|
|---|---|
| CAS |
52232-67-4
99294-94-7 (acetate)
|
| Mol weight |
4117.7151
|
(4S)-4-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S,3S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-3-hydroxypropanoyl]amino]-3-methylbutanoyl]amino]-3-hydroxypropanoyl]amino]-4-carboxybutanoyl]amino]-3-methylpentanoyl]amino]-5-oxopentanoyl]amino]-4-methylpentanoyl]amino]-4-methylsulfanylbutanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-oxobutanoyl]amino]-4-methylpentanoyl]amino]acetyl]amino]hexanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-3-hydroxypropanoyl]amino]-4-methylsulfanylbutanoyl]amino]-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-4-amino-1-[[(1S)-1-carboxy-2-phenylethyl]amino]-1,4-dioxobutan-2-yl]amino]-3-(1H-imidazol-5-yl)-1-oxopropan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-1-oxohexan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-5-oxopentanoic acid
| SVG Image |
 |
|---|---|
| IUPAC Condensed | H-Ser-Val-Ser-Glu-Ile-Gln-Leu-Met-His-Asn-Leu-Gly-Lys-His-Leu-Asn-Ser-Met-Glu-Arg-Val-Glu-Trp-Leu-Arg-Lys-Lys-Leu-Gln-Asp-Val-His-Asn-Phe-OH |
| Sequence | SVSEIQLMHNLGKHLNSMERVEWLRKKLQDVHNF |
| PLN | H-SVSEIQLMHNLGKHLNSMERVEWLRKKLQDVHNF-OH |
| HELM | PEPTIDE1{S.V.S.E.I.Q.L.M.H.N.L.G.K.H.L.N.S.M.E.R.V.E.W.L.R.K.K.L.Q.D.V.H.N.F}$$$$ |
| IUPAC | L-seryl-L-valyl-L-seryl-L-alpha-glutamyl-L-isoleucyl-L-glutaminyl-L-leucyl-L-methionyl-L-histidyl-L-asparagyl-L-leucyl-glycyl-L-lysyl-L-histidyl-L-leucyl-L-asparagyl-L-seryl-L-methionyl-L-alpha-glutamyl-L-arginyl-L-valyl-L-alpha-glutamyl-L-tryptophyl-L-leucyl-L-arginyl-L-lysyl-L-lysyl-L-leucyl-L-glutaminyl-L-alpha-aspartyl-L-valyl-L-histidyl-L-asparagyl-L-phenylalanine |
Other Names
- L-Seryl-L-valyl-L-seryl-L-α-glutamyl-L-isoleucyl-L-glutaminyl-L-leucyl-L-methionyl-L-histidyl-L-asparaginyl-L-leucylglycyl-L-lysyl-L-histidyl-L-leucyl-L-asparaginyl-L-seryl-L-methionyl-L-α-glutamyl-L-arginyl-L-valyl-L-α-glutamyl-L-tryptophyl-L-leucyl-L-arginyl-L-lysyl-L-lysyl-L-leucyl-L-glutaminyl-L-α-aspartyl-L-valyl-L-histidyl-L-asparaginyl-L-phenylalanine
- (1-34)-Human parathormone
- (1-34)-Human parathyroid hormone
- 1-34-Human PTH
- 1-34-Parathormone (human)
- 11: PN: WO0039278 SEQID: 17 unclaimed protein
- 14: PN: WO0181415 SEQID: 16 claimed protein
- 15: PN: WO0123521 SEQID: 19 claimed protein
- 1: PN: EP2905289 SEQID: 1 claimed protein
- 1: PN: WO0198348 SEQID: 13 claimed protein
- 1: PN: WO2011071480 SEQID: 14 claimed protein
- 225: PN: US20090175821 SEQID: 272 claimed protein
- 22: PN: US6110892 SEQID: 22 unclaimed protein
- 2: PN: US20100261199 SEQID: 4 claimed protein
- 31: PN: US20070099831 PAGE: 7 claimed protein
- 32: PN: WO2008068487 SEQID: 32 claimed protein
- 5: PN: WO2008033473 SEQID: 4 claimed protein
- 692: PN: WO2004005342 PAGE: 46 claimed protein
- 69: PN: US20050009742 PAGE: 20 claimed sequence
- 7: PN: WO0031137 SEQID: 8 unclaimed protein
- 7: PN: WO0040611 PAGE: 1 claimed protein
- 93: PN: WO0069900 SEQID: 272 unclaimed protein
- Forsteo
- Forteo
- HPTH-(1-34)
- Human PTH(1-34)
- Human parathormone(1-34)
- Human parathyroid hormone-(1-34)
- LY 333334
- Osteotide
- Parathar
- Parathormone (human)
- Teriparatide
- ZT 034
Product Ingredients
| INGREDIENT | UNII | CAS | |
|---|---|---|---|
| Teriparatide acetate | 9959P4V12N | 99294-94-7 |
Teriparatide is a form of parathyroid hormone consisting of the first (N-terminus) 34 amino acids, which is the bioactive portion of the hormone. It is an effective anabolic (promoting bone formation) agent[2] used in the treatment of some forms of osteoporosis.[3] It is also occasionally used off-label to speed fracture healing. Teriparatide is identical to a portion of human parathyroid hormone (PTH) and intermittent use activates osteoblasts more than osteoclasts, which leads to an overall increase in bone.
Recombinant teriparatide is sold by Eli Lilly and Company under the brand name Forteo/Forsteo. A synthetic teriparatide from Teva Generics has been authorised for marketing in European territories[4]. Biosimilar product from Gedeon Richter plc has been authorised in Europe[5]. On October 4, 2019 the US FDA approved a recombinant teriparatide product, PF708, from Pfenex Inc. PF708 is the first FDA approved proposed therapeutic equivalent candidate to Forteo.
Teriparatide (recombinant human parathyroid hormone) is a potent anabolic agent used in the treatment of osteoporosis. It is manufactured and marketed by Eli Lilly and Company.
Teriparatide is a recombinant form of parathyroid hormone. It is an effective anabolic (i.e., bone growing) agent used in the treatment of some forms of osteoporosis. It is also occasionally used off-label to speed fracture healing. Teriparatide is identical to a portion of human parathyroid hormone (PTH) and intermittent use activates osteoblasts more than osteoclasts, which leads to an overall increase in bone. Teriparatide is sold by Eli Lilly and Company under the brand name Forteo.
Indication
For the treatment of osteoporosis in men and postmenopausal women who are at high risk for having a fracture. Also used to increase bone mass in men with primary or hypogonadal osteoporosis who are at high risk for fracture.
Associated Conditions
Pharmacodynamics
Clinical trials indicate that teriparatide increases predominantly trabecular bone in the lumbar spine and femoral neck; it has less significant effects at cortical sites. The combination of teriparatide with antiresorptive agents is not more effective than teriparatide monotherapy. The most common adverse effects associated with teriparatide include injection-site pain, nausea, headaches, leg cramps, and dizziness. After a maximum of two years of teriparatide therapy, the drug should be discontinued and antiresorptive therapy begun to maintain bone mineral density.
Mechanism of action
Teriparatide is the portion of human parathyroid hormone (PTH), amino acid sequence 1 through 34 of the complete molecule which contains amino acid sequence 1 to 84. Endogenous PTH is the primary regulator of calcium and phosphate metabolism in bone and kidney. Daily injections of teriparatide stimulates new bone formation leading to increased bone mineral density.
Medical uses
Teriparatide has been FDA-approved since 2002.[6] It is effective in growing bone (e.g., 8% increase in bone density in the spine after one year)[7] and reducing the risk of fragility fractures.[6][8] When studied, teriparatide only showed bone mineral density (BMD) improvement during the first 18 months of use. Teriparatide should only be used for a period of 2 years maximum. After 2 years, another agent such a bisphosphonate or denosumab should be used in cases of osteoporosis. [9]
Teriparatide cuts the risk of hip fracture by more than half but does not reduce the risk of arm or wrist fracture.[10]
Other
Teriparatide can be used off-label to speed fracture repair and treat fracture nonunions.[11] It has been reported to have been successfully used to heal fracture nonunions.[12] Generally, due to HIPAA regulations, it is not publicized when American athletes receive this treatment to improve fracture recovery.[11] But an Italian football player, Francesco Totti, was given teriparatide after a tibia/fibula fracture, and he unexpectedly recovered in time for the 2006 World Cup.[11] It has been reported that Mark Mulder used it to recover from a hip fracture Oakland A’s for the 2003 MLB playoffs[13] and Terrell Owens to recover from an ankle fracture before the 2005 Super Bowl.[13]
Administration
Teriparatide is administered by injection once a day in the thigh or abdomen.
Contraindications
Teriparatide should not be prescribed for people who are at increased risks for osteosarcoma. This includes those with Paget’s Diseaseof bone or unexplained elevations of serum alkaline phosphate, open epiphysis, or prior radiation therapy involving the skeleton. In the animal studies and in one human case report, it was found to potentially be associated with developing osteosarcoma in test subjects after over 2 years of use. [14]
Patients should not start teriparatide until any vitamin D deficiency is corrected. [15]
Adverse effects
Adverse effects of teriparatide include headache, nausea, dizziness, and limb pain.[6] Teriparatide has a theoretical risk of osteosarcoma, which was found in rat studies but not confirmed in humans.[2] This may be because unlike humans, rat bones grow for their entire life.[2] The tumors found in the rat studies were located on the end of the bones which grew after the injections began.[15]After nine years on the market, there were only two cases of osteosarcoma reported.[7] This risk was considered by the FDA as “extremely rare” (1 in 100,000 people)[6] and is only slightly more than the incidence in the population over 60 years old (0.4 in 100,000).[6]
Mechanism of action
Teriparatide is a portion of human parathyroid hormone (PTH), amino acid sequence 1 through 34, of the complete molecule (containing 84 amino acids). Endogenous PTH is the primary regulator of calcium and phosphate metabolism in bone and kidney. PTH increases serum calcium, partially accomplishing this by increasing bone resorption. Thus, chronically elevated PTH will deplete bone stores. However, intermittent exposure to PTH will activate osteoblasts more than osteoclasts. Thus, once-daily injections of teriparatide have a net effect of stimulating new bone formation leading to increased bone mineral density.[16][17][18]
Teriparatide is the first FDA approved agent for the treatment of osteoporosis that stimulates new bone formation.[19]
FDA approval
Teriparatide was approved by the Food and Drug Administration (FDA) on 26 November 2002, for the treatment of osteoporosis in men and postmenopausal women who are at high risk for having a fracture. The drug is also approved to increase bone mass in men with primary or hypogonadal osteoporosis who are at high risk for fracture.
Combined teriparatide and denosumab
Combined teriparatide and denosumab increased BMD more than either agent alone and more than has been reported with approved therapies. Combination treatment might, therefore, be useful to treat patients at high risk of fracture by increasing BMD. However, there is no evidence of fracture rate reduction in patients taking a teriparatide and denosumab combination. Moreover, the combination therapy group showed a significant decrease in their bone formation marker, indicating that denosumab, an antiresorptive agent, might actually counteract the effect of teriparatide, a bone formation anabolic agent, in bone formation. [20]
PATENT
KR 2011291
WO 2019077432
CN 109897099
CN 109879955
CN 109879954
CN 108373499
PATENT
WO-2020000555
Process for preparing teriparatide as parathyroid hormone receptor agonist, useful for treating osteoporosis in menopausal women. Appears to be the first filing from the assignee and the inventors on this compound, however, this invention was previously seen as a Chinese national filing published in 12/2013. Daiichi Sankyo , through its subsidiary Asubio Pharma , was developing SUN-E-3001 , a nasally administered recombinant human parathyroid hormone, for the treatment of osteoporosis.
Teriparatide is a 1-34 fragment of human parathyroid hormone, which has the same biological activity as human parathyroid hormone. Hypogonadous osteoporosis and osteoporosis in menopausal women have great market prospects.
In patent CN201410262511, a pseudoproline dipeptide Fmoc-Asn (Trt) -Ser (ψ Me, Me Pro) -OH is used instead of the two amino acids at the original 16-17 positions for coupling one by one, and the final cleavage yields teriparatide. This method adopts the method of feeding pseudoproline dipeptide to avoid the generation of oxidative impurities, but it cannot avoid a variety of missing peptides due to the excessively long peptide chain. At the same time, the pseudoproline dipeptide is expensive and difficult to obtain.
References
- ^ http://www.minsa.gob.pa/sites/default/files/alertas/nota_seguridad_teriparatida.pdf
- ^ Jump up to:a b c Riek AE and Towler DA (2011). “The pharmacological management of osteoporosis”. Missouri Medicine. 108 (2): 118–23. PMC 3597219. PMID 21568234.
- ^ Saag KG, Shane E, Boonen S, et al. (November 2007). “Teriparatide or alendronate in glucocorticoid-induced osteoporosis”. The New England Journal of Medicine. 357 (20): 2028–39. doi:10.1056/NEJMoa071408. PMID 18003959.
- ^ BfArM (2017-05-08). “PUBLIC ASSESSMENT REPORT – Decentralised Procedure – Teriparatid-ratiopharm 20 µg / 80ml, Solution for injection” (PDF).
- ^ “Summary of the European public assessment report (EPAR) for Terrosa”. Retrieved 2019-08-14.
- ^ Jump up to:a b c d e Rizzoli, R.; Reginster, J. Y.; Boonen, S.; Bréart, G. R.; Diez-Perez, A.; Felsenberg, D.; Kaufman, J. M.; Kanis, J. A.; Cooper, C. (2011). “Adverse Reactions and Drug–Drug Interactions in the Management of Women with Postmenopausal Osteoporosis”. Calcified Tissue International. 89 (2): 91–104. doi:10.1007/s00223-011-9499-8. PMC 3135835. PMID 21637997.
- ^ Jump up to:a b Kawai, M.; Mödder, U. I.; Khosla, S.; Rosen, C. J. (2011). “Emerging therapeutic opportunities for skeletal restoration”. Nature Reviews Drug Discovery. 10 (2): 141–156. doi:10.1038/nrd3299. PMC 3135105. PMID 21283108.
- ^ Murad, M. H.; Drake, M. T.; Mullan, R. J.; Mauck, K. F.; Stuart, L. M.; Lane, M. A.; Abu Elnour, N. O.; Erwin, P. J.; Hazem, A.; Puhan, M. A.; Li, T.; Montori, V. M. (2012). “Comparative Effectiveness of Drug Treatments to Prevent Fragility Fractures: A Systematic Review and Network Meta-Analysis”. Journal of Clinical Endocrinology & Metabolism. 97(6): 1871–1880. doi:10.1210/jc.2011-3060. PMID 22466336.
- ^ O’Connor KM. Evaluation and Treatment of Osteoporosis. Med Clin N Am. 2016; 100:807-26
- ^ Díez-Pérez A, Marin F, Eriksen EF, Kendler DL, Krege JH, Delgado-Rodríguez M (September 2018). “Effects of teriparatide on hip and upper limb fractures in patients with osteoporosis: A systematic review and meta-analysis”. Bone. 120: 1–8. doi:10.1016/j.bone.2018.09.020. PMID 30268814.
- ^ Jump up to:a b c Bruce Jancin (2011-12-12). “Accelerating Fracture Healing With Teriparatide”. Internal Medicine News Digital Network. Retrieved 2013-09-20.
- ^ Giannotti, S.; Bottai, V.; Dell’Osso, G.; Pini, E.; De Paola, G.; Bugelli, G.; Guido, G. (2013). “Current medical treatment strategies concerning fracture healing”. Clinical Cases in Mineral and Bone Metabolism. 10 (2): 116–120. PMC 3796998. PMID 24133528.
- ^ Jump up to:a b William L. Carroll (2005). “Chapter 1: Defining the Issue”. The Juice: The Real Story of Baseball’s Drug Problems. ISBN 1-56663-668-X. Retrieved 2013-09-23.
- ^ Harper KD, Krege JH, Marcus R, et al. Osteosarcoma and teriparatide? J Bone Miner Res 2007;22(2):334
- ^ Jump up to:a b https://www.drugs.com/pro/forteo.html
- ^ Bauer, E; Aub, JC; Albright, F (1929). “Studies of calcium and phosphorus metabolism: V. Study of the bone trabeculae as a readily available reserve supply of calcium”. J Exp Med. 49 (1): 145–162. doi:10.1084/jem.49.1.145. PMC 2131520. PMID 19869533.
- ^ Selye, H (1932). “On the stimulation of new bone formation with parathyroid extract and irradiated ergosterol”. Endocrinology. 16 (5): 547–558. doi:10.1210/endo-16-5-547.
- ^ Dempster, D. W.; Cosman, F.; Parisien, M.; Shen, V.; Lindsay, R. (1993). “Anabolic actions of parathyroid hormone on bone”. Endocrine Reviews. 14 (6): 690–709. doi:10.1210/edrv-14-6-690. PMID 8119233.
- ^ Fortéo: teriparatide (rDNA origin) injection Archived 2009-12-27 at the Wayback Machine
- ^ Tsai, Joy N; Uihlein, Alexander V; Lee, Hang; Kumbhani, Ruchit; Siwila-Sackman, Erica; McKay, Elizabeth A; Burnett-Bowie, Sherri-Ann M; Neer, Robert M; Leder, Benjamin Z (2013). “Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: The DATA study randomised trial”. The Lancet. 382 (9886): 1694–1700. doi:10.1016/S0140-6736(13)60856-9. PMC 4010689. PMID 24517156.
External links
 |
|
| Clinical data | |
|---|---|
| Trade names | Forteo/Forsteo, Teribone[1] |
| AHFS/Drugs.com | Monograph |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Subcutaneous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 95% |
| Metabolism | Hepatic (nonspecific proteolysis) |
| Elimination half-life | Subcutaneous: 1 hour |
| Excretion | Renal (metabolites) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.168.733 |
| Chemical and physical data | |
| Formula | C181H291N55O51S2 |
| Molar mass | 4117.72 g/mol g·mol−1 |
| 3D model (JSmol) | |
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| CA2314313 | No | 2005-02-08 | 2018-12-08 |  |
| CA2325371 | No | 2004-08-17 | 2019-08-19 | |
| US6770623 | No | 2004-08-03 | 2018-12-08 |  |
| US6977077 | No | 2005-12-20 | 2019-08-19 | |
| US7163684 | No | 2007-01-16 | 2019-08-19 | |
| US7351414 | No | 2008-04-01 | 2019-08-19 | |
| US7550434 | No | 2009-06-23 | 2018-12-08 | |
| US7144861 | No | 2006-12-05 | 2018-12-08 | |
| US7517334 | No | 2009-04-14 | 2025-03-25 | |
FORTEO (teriparatide [rDNA origin] injection) contains recombinant human parathyroid hormone (1- 34), and is also called rhPTH (1-34). It has an identical sequence to the 34 N-terminal amino acids(the biologically active region) of the 84-amino acid human parathyroid hormone.
Teriparatide has a molecular weight of 4117.8 daltons and its amino acid sequence is shown below:
 |
Teriparatide (rDNA origin) is manufactured using a strain of Escherichia coli modified by recombinant DNA technology. FORTEO is supplied as a sterile, colorless, clear, isotonic solution in a glass cartridge which is pre-assembled into a disposable delivery device (pen) for subcutaneous injection. Each prefilled delivery device is filled with 2.7 mL to deliver 2.4 mL. Each mL contains 250 mcg teriparatide (corrected for acetate, chloride, and water content), 0.41 mg glacial acetic acid, 0.1 mg sodium acetate (anhydrous), 45.4 mg mannitol, 3 mg Metacresol, and Water for Injection. In addition, hydrochloric acid solution 10% and/or sodium hydroxide solution 10% may have been added to adjust the product to pH 4.
Each cartridge, pre-assembled into a delivery device, delivers 20 mcg of teriparatide per dose each day for up to 28 days.
REFERENCES
1: Lindsay R, Krege JH, Marin F, Jin L, Stepan JJ. Teriparatide for osteoporosis: importance of the full course. Osteoporos Int. 2016 Feb 22. [Epub ahead of print] Review. PubMed PMID: 26902094.
2: Im GI, Lee SH. Effect of Teriparatide on Healing of Atypical Femoral Fractures: A Systemic Review. J Bone Metab. 2015 Nov;22(4):183-9. doi: 10.11005/jbm.2015.22.4.183. Epub 2015 Nov 30. Review. PubMed PMID: 26713309; PubMed Central PMCID: PMC4691592.
3: Babu S, Sandiford NA, Vrahas M. Use of Teriparatide to improve fracture healing: What is the evidence? World J Orthop. 2015 Jul 18;6(6):457-61. doi: 10.5312/wjo.v6.i6.457. eCollection 2015 Jul 18. Review. PubMed PMID: 26191492; PubMed Central PMCID: PMC4501931.
4: Lecoultre J, Stoll D, Chevalley F, Lamy O. [Improvement of fracture healing with teriparatide: series of 22 cases and review of the literature]. Rev Med Suisse. 2015 Mar 18;11(466):663-7. Review. French. PubMed PMID: 25962228.
5: Sugiyama T, Torio T, Sato T, Matsumoto M, Kim YT, Oda H. Improvement of skeletal fragility by teriparatide in adult osteoporosis patients: a novel mechanostat-based hypothesis for bone quality. Front Endocrinol (Lausanne). 2015 Jan 30;6:6. doi: 10.3389/fendo.2015.00006. eCollection 2015. Review. PubMed PMID: 25688232; PubMed Central PMCID: PMC4311704.
6: Wheeler AL, Tien PC, Grunfeld C, Schafer AL. Teriparatide treatment of osteoporosis in an HIV-infected man: a case report and literature review. AIDS. 2015 Jan 14;29(2):245-6. doi: 10.1097/QAD.0000000000000529. Review. PubMed PMID: 25532609; PubMed Central PMCID: PMC4438749.
7: Campbell EJ, Campbell GM, Hanley DA. The effect of parathyroid hormone and teriparatide on fracture healing. Expert Opin Biol Ther. 2015 Jan;15(1):119-29. doi: 10.1517/14712598.2015.977249. Epub 2014 Nov 3. Review. PubMed PMID: 25363308.
8: Yamamoto M, Sugimoto T. [Glucocorticoid and Bone. Beneficial effect of teriparatide on fracture risk as well as bone mineral density in patients with glucocorticoid-induced osteoporosis]. Clin Calcium. 2014 Sep;24(9):1379-85. doi: CliCa140913791385. Review. Japanese. PubMed PMID: 25177011.
9: Chen JF, Yang KH, Zhang ZL, Chang HC, Chen Y, Sowa H, Gürbüz S. A systematic review on the use of daily subcutaneous administration of teriparatide for treatment of patients with osteoporosis at high risk for fracture in Asia. Osteoporos Int. 2015 Jan;26(1):11-28. doi: 10.1007/s00198-014-2838-7. Epub 2014 Aug 20. Review. PubMed PMID: 25138261.
10: Eriksen EF, Keaveny TM, Gallagher ER, Krege JH. Literature review: The effects of teriparatide therapy at the hip in patients with osteoporosis. Bone. 2014 Oct;67:246-56. doi: 10.1016/j.bone.2014.07.014. Epub 2014 Jul 15. Review. PubMed PMID: 25053463.
11: Meier C, Lamy O, Krieg MA, Mellinghoff HU, Felder M, Ferrari S, Rizzoli R. The role of teriparatide in sequential and combination therapy of osteoporosis. Swiss Med Wkly. 2014 Jun 4;144:w13952. doi: 10.4414/smw.2014.13952. eCollection 2014. Review. PubMed PMID: 24896070.
12: Krege JH, Lane NE, Harris JM, Miller PD. PINP as a biological response marker during teriparatide treatment for osteoporosis. Osteoporos Int. 2014 Sep;25(9):2159-71. doi: 10.1007/s00198-014-2646-0. Epub 2014 Mar 6. Review. PubMed PMID: 24599274; PubMed Central PMCID: PMC4134485.
13: Nakano T. [Once-weekly teriparatide treatment on osteoporosis]. Clin Calcium. 2014 Jan;24(1):100-5. doi: CliCa1401100105. Review. Japanese. PubMed PMID: 24369286.
14: Yano S, Sugimoto T. [Daily subcutaneous injection of teriparatide : the progress and current issues]. Clin Calcium. 2014 Jan;24(1):35-43. doi: CliCa14013543. Review. Japanese. PubMed PMID: 24369278.
15: Lewiecki EM, Miller PD, Harris ST, Bauer DC, Davison KS, Dian L, Hanley DA, McClung MR, Yuen CK, Kendler DL. Understanding and communicating the benefits and risks of denosumab, raloxifene, and teriparatide for the treatment of osteoporosis. J Clin Densitom. 2014 Oct-Dec;17(4):490-5. doi: 10.1016/j.jocd.2013.09.018. Epub 2013 Oct 25. Review. PubMed PMID: 24206867.
16: Delivanis DA, Bhargava A, Luthra P. Subungual exostosis in an osteoporotic patient treated with teriparatide. Endocr Pract. 2013 Sep-Oct;19(5):e115-7. doi: 10.4158/EP13040.CR. Review. PubMed PMID: 23757619.
17: Borges JL, Freitas A, Bilezikian JP. Accelerated fracture healing with teriparatide. Arq Bras Endocrinol Metabol. 2013 Mar;57(2):153-6. Review. PubMed PMID: 23525295.
18: Thumbigere-Math V, Gopalakrishnan R, Michalowicz BS. Teriparatide therapy for bisphosphonate-related osteonecrosis of the jaw: a case report and narrative review. Northwest Dent. 2013 Jan-Feb;92(1):12-8. Review. PubMed PMID: 23516715.
19: Lamy O. [Bone anabolic treatment with Teriparatide]. Ther Umsch. 2012 Mar;69(3):187-91. doi: 10.1024/0040-5930/a000272. Review. German. PubMed PMID: 22403112.
20: Narváez J, Narváez JA, Gómez-Vaquero C, Nolla JM. Lack of response to teriparatide therapy for bisphosphonate-associated osteonecrosis of the jaw. Osteoporos Int. 2013 Feb;24(2):731-3. doi: 10.1007/s00198-012-1918-9. Epub 2012 Mar 8. Review. PubMed PMID: 22398853.
/////TERIPARATIDE, テリパラチド , терипаратид , تيريباراتيد , 特立帕肽 , PTH 1-34, LY 333334, LY-333334, LY333334, ZT-034, 52232-67-4, PEPTIDES
Coblopasvir

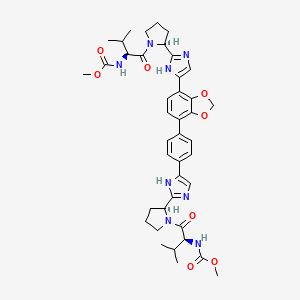
Coblopasvir
CAS: 1312608-46-0
Chemical Formula: C41H50N8O8
Molecular Weight: 782.89
methyl {(2S)-1-[(2S)-2-(4-{4-[7-(2-[(2S)-1-{(2S)-2- [(methoxycarbonyl)amino]-3-methylbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl)-2H-1,3-benzodioxol-4-yl]phenyl}-1Himidazol-2-yl)pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl}carbamate
hepatitis C virus infection
KW-136
Coblopasvir is an antiviral drug candidate.
Coblopasvir dihydrochloride

CAS 1966138-53-3
| C41 H50 N8 O8 . 2 Cl H | |
| Molecular Weight | 855.806 |
| PHASE 3 | Beijing Kawin Technology Share-Holding |
Beijing Kawin Technology Share-Holding, in collaboration with Beijing Fu Rui Tiancheng Biotechnology and Ginkgo Pharma , is developing coblopasvir as an oral capsule formulation of dihydrochloride salt (KW-136), for treating hepatitis C virus infection. In June 2018, an NDA was filed in China by Beijing Kawin Technology and Sichuan Qingmu Pharmaceutical . In August 2018, the application was granted Priority Review in China . Also, Beijing Kawin is investigating a tablet formulation of coblopasvir dihydrochloride.
PATENT
WO2011075607 , claiming substituted heterocyclic derivatives as HCV replication inhibitors useful for treating HCV infection and liver fibrosis, assigned to Beijing Kawin Technology Share-Holding Co Ltd and InterMune Inc ,
PATENT
CN 108675998
PATENT
WO-2020001089
Novel crystalline and amorphous forms of methyl carbamate compound, particularly coblopasvir dihydrochloride , (designated as Forms H) processes for their preparation, compositions and combinations comprising them are claimed. Also claim is an article or kit comprising a container and a package insert, wherein the container contains coblopasvir dihydrochloride.

///////////////Coblopasvir , KW-136, hepatitis C virus infection, CHINA, Beijing Kawin Technology, NDA, Phase III
O=C(OC)N[C@@H](C(C)C)C(N1[C@H](C2=NC(C3=CC=C(C4=C5OCOC5=C(C6=CNC([C@H]7N(C([C@@H](NC(OC)=O)C(C)C)=O)CCC7)=N6)C=C4)C=C3)=CN2)CCC1)=O
