

| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8648077 | SUBSTITUTED HETEROCYCLE FUSED GAMMA-CARBOLINES SOLID |
2011-05-12
|
|
| US9371324 | ORGANIC COMPOUNDS |
2015-02-20
|
2015-06-18
|
| US8993572 | ORGANIC COMPOUNDS |
2011-04-22
|
2013-08-08
|
| US9586960 | SUBSTITUTED HETEROCYCLE FUSED GAMMA-CARBOLINES SOLID |
2015-11-30
|
2016-07-07
|
| US9199995 | SUBSTITUTED HETEROCYCLE FUSED GAMMA-CARBOLINES SOLID |
2014-02-11
|
2014-10-30
|
Home » 2018 (Page 2)
Yearly Archives: 2018
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Anthony Crasto conferred ABPnews award for “Outstanding contribution to Education Sector”
 Conferred prestigious award at event ABP News Presents Healthcare Leadership Awards 26th November, 2018 at Taj Lands End, Mumbai India
Conferred prestigious award at event ABP News Presents Healthcare Leadership Awards 26th November, 2018 at Taj Lands End, Mumbai India
Dedicated to Shobha Crasto Aishal crasto Lionel crasto
Service to education is service to humanity
Society recognises efforts done towards it



/////////////award, event ABP News, Healthcare, Leadership, 26th, November, 2018, Taj Lands End, Mumbai, India, education, anthony crasto
Omidenepag isopropyl, オミデネパグイソプロピル
Omidenepag isopropyl
DE-117
Glycine, N-(6-((((4-(1H-pyrazol-1-yl)phenyl)methyl)(3-pyridinylsulfonyl)amino)methyl)-2-pyridinyl)-, 1-methylethyl ester
[[6-[[[4-(Pyrazol-1-yl)benzyl](pyridin-3-ylsulfonyl)amino]methyl]pyridin-2-yl]amino]acetic acid isopropyl ester
C26H28N6O4S, 520.6033, CAS: 1187451-19-9
APPROVED 2018/9/21 PMDA, JAPAN 2018, Eybelis
Antiglaucoma, Prostaglandin E2 receptor agonist, Treatment of Open-Angle Glaucoma and Ocular Hypertension
- Originator Ube Industries
- Developer Santen Pharmaceutical
- Class Eye disorder therapies; Pyrazoles; Pyridines; Small molecules; Sulfonamides
- Mechanism of Action Prostaglandin E EP2 receptor agonists
- Registered Glaucoma; Ocular hypertension
- 27 Sep 2018 Santen initiates enrolment in the phase III Spectrum 5 trial for Glaucoma and Ocular hypertension in USA (Ophthalmic) (NCT03697811)
- 21 Sep 2018 Santen Pharmaceutical and Ube Industries plan phase III trials for omidenepag isopropyl in USA in the second half of 2018
- 21 Sep 2018 Registered for Ocular hypertension and Glaucoma in Japan (Ophthalmic) – First global approval
SYNTHESIS

PATENT
WO 2009113600
WO 2010113957
JP 2011057633
PATENT
WO 2015190507
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015190507&tab=FULLTEXT&maxRec=1000
[Example 1]
[Formula
 10] 2 – {[6 – ({N-[4-(1H-pyrazol-1-yl) benzyl] pyridin-3-sulfonamido} methyl) pyridin-2-yl] amino} Synthesis of isopropyl acetate
10] 2 – {[6 – ({N-[4-(1H-pyrazol-1-yl) benzyl] pyridin-3-sulfonamido} methyl) pyridin-2-yl] amino} Synthesis of isopropyl acetate
[Formula
To a glass vessel having an inner volume of about 50 ml equipped with a stirring device, a thermometer and an upper cooling device, 3.21 g (10.00 mmol) of N- [4- (1H-pyrazol-1-yl) benzyl] , 2.43 g (10.0 mmol) of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate obtained in Example 6, 6.65 g (20.4 mmol) of cesium carbonate and 17.6 g of acetonitrile was added, and the mixture was heated and stirred at 80 ° C. In the high performance liquid chromatography analysis, the reaction was carried out for 2 hours until the area percentage of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate became 0.03% or less, I went for hours. The reaction conversion ratios of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate after heating and stirring 1 hour and 2 hours were 99.88% and 99.97% . After completion of the reaction, the reaction solution was cooled to room temperature, filtered using Celite (trade name), and the filtrate was washed with acetonitrile. Quantitative analysis of the obtained filtrate by high performance liquid chromatography revealed that 5.08 g of the objective substance was contained (reaction yield: 97.5%). Next, the reaction solution was concentrated under reduced pressure until the weight of the liquid reached 7.85 g, 42.8 g of toluene was added, and the mixture was washed three times with water. 31.5 ml (31.5 mmol) of 1 mol / L hydrochloric acid was added to the obtained organic layer, and the mixture was stirred at room temperature for 20 minutes and then separated. Note that 0.17 g (corresponding to 3.2% yield) of target product was contained in the organic layer after liquid separation. 42.8 g of toluene and 34.6 ml (34.6 mmol) of 1 mol / L sodium hydroxide aqueous solution were added to the obtained aqueous layer, and the mixture was heated to 40 ° C. and stirred for 20 minutes. After filtration at 40 ° C. in the hot state, liquid separation was carried out. The obtained organic layer was washed twice with water. The organic layer was concentrated under reduced pressure until the weight of the liquid reached 8.97 g, and 7.40 g of 2-propanol was added. After warming to 60 ° C., it was slowly cooled and stirred at 33 ° C. for 30 minutes, then slowly cooled to 5 ° C. or less, and further stirred at the same temperature for 1 hour. Precipitated The solid was filtered, washed with chilled 2-propanol and then vacuum dried at 50 ° C. to give 2 – {[6 – ({N- [4- (1 H-pyrazol- 1 – yl) benzyl] pyridine- 3 – sulfonamido} methyl) pyridin-2-yl] amino} acetic acid was obtained as a slightly brown solid (2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate Isolated yield of standard 75.1%). The quantitative purity by HPLC high performance chromatography method was 99.5%, and 0.04% of N- [4- (1H-pyrazol-1-yl) benzyl] pyridine-3-sulfonamide as a raw material was contained It was. Also, in the measurement (wavelength 260 nm) by the HPLC high performance liquid chromatography method, there was no impurity showing an area% of 0.1% or more.
Physical property values of the obtained 2 – {[6 – ({N- [4- (1 H-pyrazol-1 -yl) benzyl] pyridine-3-sulfonamido} methyl) pyridin-2- yl] amino} , It was as follows.
EI-MS (m /
z):. 520 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3, [delta] (ppm)): 1.24 (6H, d, J = 6.3 Hz), 5.07 (1 H, se, J = 5.5 Hz), 3.82 (2 H, d, J = 5.5 Hz), 4.31 (2 H, s), 4.64 (2 H, s), 4.94 J = 6.3 Hz), 6.26 (1 H, d, J = 8.3 Hz), 6.41 (1 H, dd, J = 7.2, 0.5 Hz), 6.46 (1 H, dd, J = 2.5, 1.8 Hz), 7.25 (2H, m), 7.71 (1H, dd, J = 8.3, 7.2 Hz), 7.32 (1H, ddd, J = 8.0, 4.9, 0.8 Hz), 7.37-7.42 J = 1.8, 0.6 Hz), 7.93 (1 H, dd, J = 2.6, 0.6 Hz), 7.94 (1 H, ddd, J = 8.0, 2.4, 1.7 Hz), 8.69 (1 H, dd, J = 4.8, 1.6 Hz ), 8.98 (IH, dd, J = 2.4, 0.8
Hz). 13 C-NMR (CDCl 3, δ (ppm)): 21.8, 43.7, 51.0, 51.1, 68.9, 107.4, 107.7, 112.6, 119.2, 123.3, 126.7, 129.9, 133.8, 134.6, 137.3, 137.6, 139.8, 141.1, 148.0, 152.6, 153.2, 157.3 , 1737 (C = O), (2981, 2933) (CH), 3437 (NH) , 170.5.
IR (KBr cm -1 ): 764 (CH), 1161 (S = O), 1525 .
elemental analysis; Calcd: C, 59.80%; H, 5.31%; N, 16.07%
Found: C, 59.98%; H, 5.42%; N, 16.14%.
EI-MS (m /
z):. 520 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3, [delta] (ppm)): 1.24 (6H, d, J = 6.3 Hz), 5.07 (1 H, se, J = 5.5 Hz), 3.82 (2 H, d, J = 5.5 Hz), 4.31 (2 H, s), 4.64 (2 H, s), 4.94 J = 6.3 Hz), 6.26 (1 H, d, J = 8.3 Hz), 6.41 (1 H, dd, J = 7.2, 0.5 Hz), 6.46 (1 H, dd, J = 2.5, 1.8 Hz), 7.25 (2H, m), 7.71 (1H, dd, J = 8.3, 7.2 Hz), 7.32 (1H, ddd, J = 8.0, 4.9, 0.8 Hz), 7.37-7.42 J = 1.8, 0.6 Hz), 7.93 (1 H, dd, J = 2.6, 0.6 Hz), 7.94 (1 H, ddd, J = 8.0, 2.4, 1.7 Hz), 8.69 (1 H, dd, J = 4.8, 1.6 Hz ), 8.98 (IH, dd, J = 2.4, 0.8
Hz). 13 C-NMR (CDCl 3, δ (ppm)): 21.8, 43.7, 51.0, 51.1, 68.9, 107.4, 107.7, 112.6, 119.2, 123.3, 126.7, 129.9, 133.8, 134.6, 137.3, 137.6, 139.8, 141.1, 148.0, 152.6, 153.2, 157.3 , 1737 (C = O), (2981, 2933) (CH), 3437 (NH) , 170.5.
IR (KBr cm -1 ): 764 (CH), 1161 (S = O), 1525 .
elemental analysis; Calcd: C, 59.80%; H, 5.31%; N, 16.07%
Found: C, 59.98%; H, 5.42%; N, 16.14%.
[Example 2]
[Formula
 11] 2 – ({6 – [(N-benzyl-3-sulfonamido) methyl] pyridin-2-yl} amino) -acetic acid isopropyl
11] 2 – ({6 – [(N-benzyl-3-sulfonamido) methyl] pyridin-2-yl} amino) -acetic acid isopropyl
[Formula
0.253 g (1.02 mmol) of N-benzylpyridine-3-sulfonamide, 0.253 g (1.02 mmol) of 2- { 0.243 g (1.00 mmol) of isopropyl acetate, 0.665 g (2.04 mmol) of cesium carbonate and 1.76 g of acetonitrile were added, and the mixture was heated and stirred at 80 ° C. did. In the high performance liquid chromatography analysis, the reaction was carried out for 2 hours until the area percentage of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate became 0.03% or less, I went for hours. The reaction conversion rates of 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetic acid isopropyl acetate after 1 hour and 2 hours from the start of heating and stirring were 99.81% and 99.99% It was. After completion of the reaction, the reaction solution was cooled to room temperature, filtered using Celite (trade name), and the filtrate was washed with acetonitrile. Quantitative analysis of the obtained filtrate by high-performance liquid chromatography revealed that 0.430 g of the target product was contained (reaction yield: 94.5%). Next, the reaction solution was concentrated under reduced pressure until the weight of the liquid reached 0.785 g, 4.3 g of toluene was added, and the mixture was washed three times with water. At this time, an emulsion containing the desired product was produced, but it was discarded together with the aqueous layer. 3.15 ml (3.15 mmol) of 1 mol / L hydrochloric acid was added to the obtained organic layer, and the mixture was stirred at room temperature for 20 minutes and then separated. To the obtained aqueous layer, 4.27 g of toluene and 3.46 ml (3.46 mmol) of 1 mol / L sodium hydroxide aqueous solution were added, the mixture was heated to 40 ° C. and stirred for 20 minutes. After separation, the obtained organic layer was washed twice with water. The organic layer was concentrated under reduced pressure to a liquid weight of 0.239 g to obtain isopropyl 2 – ({6 – [(N-benzylpyridine-3-sulfonamido) methyl] pyridin-2-yl} amino) acetate as a light brown solid (Obtained as a raw material based on isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate Rate 53.8%). The quantitative purity by HPLC high performance liquid chromatography method was 98.0%. Also, in the measurement (wavelength 260 nm) by the HPLC high performance liquid chromatography method, there was no impurity showing an area% of 0.1% or more.
Physical property values of the obtained 2 – ({6 – [(N-benzylpyridine-3-sulfonamido) methyl] pyridin-2-yl} amino) acetate isopropylate were as follows.
EI-MS (m /
z):. 454 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3 , [delta] (ppm)): 1.27 (6H, d, J = 5.3 Hz), 5.09 (1 H, sep, J = 6.3 Hz), 3.82 (2H, d, J = 5.4 Hz), 4.31 (2H, s), 4.62 7.26 – 7.33 (7 H, m), 7.90 – 7.93 (1 H, m), 8.69 (1 H, m), 6.26 (1 H, d, J = 8.3 Hz)
13 C-NMR (CDCl 3 , δ (ppm)): 21.8, 43.8, 51.1, 51.6, 69.0, 1 H, dd, J = 4.8, 1.6 Hz), 8.95 (1 H, dd, J = 107.2, 112.6, 123.2, 127.9, 128.6, 128.8, 134.7, 135.6, 137.6, 137.7, 148.2, 152.5, 153.6, 157.3, 170.5
IR (KBr cm -1
Calcd: C, 60.77%; H, 5.77%; N, 12.33%
Found (C = : C, 61.03%; H, 5.85%; N, 12.15%.
EI-MS (m /
z):. 454 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3 , [delta] (ppm)): 1.27 (6H, d, J = 5.3 Hz), 5.09 (1 H, sep, J = 6.3 Hz), 3.82 (2H, d, J = 5.4 Hz), 4.31 (2H, s), 4.62 7.26 – 7.33 (7 H, m), 7.90 – 7.93 (1 H, m), 8.69 (1 H, m), 6.26 (1 H, d, J = 8.3 Hz)
13 C-NMR (CDCl 3 , δ (ppm)): 21.8, 43.8, 51.1, 51.6, 69.0, 1 H, dd, J = 4.8, 1.6 Hz), 8.95 (1 H, dd, J = 107.2, 112.6, 123.2, 127.9, 128.6, 128.8, 134.7, 135.6, 137.6, 137.7, 148.2, 152.5, 153.6, 157.3, 170.5
IR (KBr cm -1
Calcd: C, 60.77%; H, 5.77%; N, 12.33%
Found (C = : C, 61.03%; H, 5.85%; N, 12.15%.
Example 3 Synthesis
of
 2 – {[6 – ({N- [4- (1 H-pyrazol-1 -yl) benzyl] pyridine-3-sulfonamido} methyl) pyridin- Synthesis of isopropyl acetate
2 – {[6 – ({N- [4- (1 H-pyrazol-1 -yl) benzyl] pyridine-3-sulfonamido} methyl) pyridin- Synthesis of isopropyl acetate
of
641 mg (2.04 mmol) of N- [4- (1H-pyrazol-1-yl) benzyl] pyridine-3-sulfonamide was added to a glass container having an inner volume of about 30 ml equipped with a stirrer, a thermometer and an upper cooling device, , 485 mg (2.00 mmol) of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate obtained in Example 6, 1.33 g (4.08 mmol) of cesium carbonate and 3.53 g And the mixture was stirred at 30 ° C. The reaction was carried out for 26 hours until the area percentage of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate was 0.3% or less in the high performance liquid chromatography analysis, I went for hours. After completion of the reaction, the reaction solution was filtered, and the filtrate was washed with acetonitrile. Quantitative analysis of the obtained filtrate by high performance liquid chromatography showed that 991 mg of the desired product was contained (reaction yield 95.2%).
Example 4
Synthesis of Isopropyl Acetate of 2 – {[6 – ({N- [4- (1 H-pyrazol-1 -yl) benzyl] pyridine-3-sulfonamido) methyl)
Synthesis of Isopropyl Acetate of 2 – {[6 – ({N- [4- (1 H-pyrazol-1 -yl) benzyl] pyridine-3-sulfonamido) methyl)
To a glass container having an inner volume of about 50 ml equipped with a stirring device, a thermometer and an upper cooling device, 3.21 g (10.00 mmol) of N- [4- (1H-pyrazol-1-yl) benzyl] 2.43 g (10.0 mmol) of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate obtained in Example 6, 2.82 g (20.4 mmol) potassium carbonate obtained in Example 6 and 17.6 g of acetonitrile was added, and the mixture was heated and stirred at 80 ° C. The reaction was carried out for 10 hours in the high performance liquid chromatography analysis until the area percentage of isopropyl 2 – {[6- (chloromethyl) pyridin-2-ylamino] acetate as raw material was 0.03% or less. The reaction conversion rate of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate was 43.9% after 1 hour from the start of heating and stirring. After completion of the reaction, the reaction solution was cooled to room temperature, filtered using Celite (trade name), and the filtrate was washed with acetonitrile. Quantitative analysis of the obtained filtrate by high performance liquid chromatography revealed that 5.00 g of the target product was contained (reaction yield 96.0%). Next, the reaction solution was concentrated under reduced pressure until the weight of the liquid reached 7.85 g, 42.77 g of toluene was added, and then washed three times with water. 31.5 ml (31.5 mmol) of 1 mol / L hydrochloric acid was added to the obtained organic layer, and the mixture was stirred at room temperature for 20 minutes and then separated. Incidentally, 0.62 g (corresponding to a yield of 11.8%) of the target product was contained in the organic layer after liquid separation. 42.77 g of toluene and 34.6 ml (34.6 mmol) of a 1 mol / L sodium hydroxide aqueous solution were added to the obtained aqueous layer, and the mixture was heated to 40 ° C. and stirred for 20 minutes. After filtration at 40 ° C. in the hot state, liquid separation was carried out. The obtained organic layer was washed twice with water. The organic layer was concentrated under reduced pressure until the weight of the liquid reached 8.97 g, and 7.40 g of 2-propanol was added. After heating to 60 ° C., it was slowly cooled and stirred at a temperature at which crystal began to precipitate for 30 minutes, then slowly cooled to 5 ° C. or less, and stirred at the same temperature for 1 hour. The obtained slurry was filtered, and the obtained filtrate was washed with water After washing with cooled 2-propanol and vacuum drying at 50 ° C., 2 – {[6 – ({N- [4- (1 H-pyrazol-1 -yl) benzyl] pyridine- Methyl) pyridin-2-yl] amino} acetic acid 3.90 g as a slightly brown solid (isolation based on isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate Rate 74.9%). The quantitative purity by HPLC high performance chromatography method was 99.0%, and 0.11% of N- [4- (1H-pyrazol-1-yl) benzyl] pyridine-3-sulfonamide as a raw material was contained It was.
Physical property values of the obtained 2 – {[6 – ({N- [4- (1 H-pyrazol-1 -yl) benzyl] pyridine-3-sulfonamido} methyl) pyridin-2- yl] amino} , It was as follows.
EI-MS (m /
z):. 520 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3, [delta] (ppm)): 1.24 (6H, d, J = 6.3 Hz), 5.07 (1 H, se, J = 5.5 Hz), 3.82 (2 H, d, J = 5.5 Hz), 4.31 (2 H, s), 4.64 (2 H, s), 4.94 J = 6.3 Hz), 6.26 (1 H, d, J = 8.3 Hz), 6.41 (1 H, dd, J = 7.2, 0.5 Hz), 6.46 (1 H, dd, J = 2.5, 1.8 Hz), 7.25 (2H, m), 7.71 (1H, dd, J = 8.3, 7.2 Hz), 7.32 (1H, ddd, J = 8.0, 4.9, 0.8 Hz), 7.37-7.42 J = 1.8, 0.6 Hz), 7.93 (1 H, dd, J = 2.6, 0.6 Hz), 7.94 (1 H, ddd, J = 8.0, 2.4, 1.7 Hz), 8.69 (1 H, dd, J = 4.8, 1.6 Hz ), 8.98 (IH, dd, J = 2.4, 0.8
Hz). 13 C-NMR (CDCl 3, δ (ppm)): 21.8, 43.7, 51.0, 51.1, 68.9, 107.4, 107.7, 112.6, 119.2, 123.3, 126.7, 129.9, 133.8, 134.6, 137.3, 137.6, 139.8, 141.1, 148.0, 152.6, 153.2, 157.3 , 1737 (C = O), (2981, 2933) (CH), 3437 (NH) , 170.5.
IR (KBr cm -1 ): 764 (CH), 1161 (S = O), 1525 .
elemental analysis; Calcd: C, 59.80%; H, 5.31%; N, 16.07%
Found: C, 59.98%; H, 5.42%; N, 16.14%.
EI-MS (m /
z):. 520 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3, [delta] (ppm)): 1.24 (6H, d, J = 6.3 Hz), 5.07 (1 H, se, J = 5.5 Hz), 3.82 (2 H, d, J = 5.5 Hz), 4.31 (2 H, s), 4.64 (2 H, s), 4.94 J = 6.3 Hz), 6.26 (1 H, d, J = 8.3 Hz), 6.41 (1 H, dd, J = 7.2, 0.5 Hz), 6.46 (1 H, dd, J = 2.5, 1.8 Hz), 7.25 (2H, m), 7.71 (1H, dd, J = 8.3, 7.2 Hz), 7.32 (1H, ddd, J = 8.0, 4.9, 0.8 Hz), 7.37-7.42 J = 1.8, 0.6 Hz), 7.93 (1 H, dd, J = 2.6, 0.6 Hz), 7.94 (1 H, ddd, J = 8.0, 2.4, 1.7 Hz), 8.69 (1 H, dd, J = 4.8, 1.6 Hz ), 8.98 (IH, dd, J = 2.4, 0.8
Hz). 13 C-NMR (CDCl 3, δ (ppm)): 21.8, 43.7, 51.0, 51.1, 68.9, 107.4, 107.7, 112.6, 119.2, 123.3, 126.7, 129.9, 133.8, 134.6, 137.3, 137.6, 139.8, 141.1, 148.0, 152.6, 153.2, 157.3 , 1737 (C = O), (2981, 2933) (CH), 3437 (NH) , 170.5.
IR (KBr cm -1 ): 764 (CH), 1161 (S = O), 1525 .
elemental analysis; Calcd: C, 59.80%; H, 5.31%; N, 16.07%
Found: C, 59.98%; H, 5.42%; N, 16.14%.
Comparative Example 1
Synthesis of Isopropyl Acetate of 2 – {[6 – ({N- [4- (1 H-pyrazol-1 -yl) benzyl] pyridine-3-sulfonamido} methyl)
Synthesis of Isopropyl Acetate of 2 – {[6 – ({N- [4- (1 H-pyrazol-1 -yl) benzyl] pyridine-3-sulfonamido} methyl)
To a glass vessel having an inner volume of about 50 ml equipped with a stirring device, a thermometer and an upper cooling device, 3.21 g (10.00 mmol) of N- [4- (1H-pyrazol-1-yl) benzyl] , 2.43 g (10.0 mmol) of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate obtained in Example 6, 2.16 g (20.4 mmol) of sodium carbonate and 17.6 g of acetonitrile was added, and the mixture was heated and stirred at 80 ° C. In the high performance liquid chromatography analysis, the reaction was carried out for 110 hours until the area percentage of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate became 0.05% or less. The reaction conversion rate of isopropyl 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate 1 hour after the start of heating and stirring was 0.92%. After completion of the reaction, the reaction solution was cooled to room temperature, filtered using Celite (trade name), and the filtrate was washed with acetonitrile. Quantitative analysis of the obtained filtrate by high performance liquid chromatography revealed that 0.72 g of the target product was contained (reaction yield: 13.8%). Next, the solution was concentrated under reduced pressure until the weight of the solution reached 7.85 g, 42.6 g of toluene was added, and the mixture was washed three times with water. Since the tar component was separated at the time of washing with water, it was discarded together with the aqueous layer. 31.5 ml (31.5 mmol) of 1 mol / L hydrochloric acid was added to the obtained organic layer, and the mixture was stirred at room temperature for 20 minutes and then separated. 42.6 g of toluene and 34.6 ml (34.6 mmol) of 1 mol / L sodium hydroxide aqueous solution were added to the obtained aqueous layer, and the mixture was heated to 40 ° C. and stirred for 20 minutes. After filtration at 40 ° C. in the hot state, liquid separation was carried out, and the obtained organic layer was washed twice with water. The organic layer was concentrated under reduced pressure to give isopropyl acetate (2 – {[6 – ({N- [4- (1 H-pyrazol- 1 – yl) benzyl] pyridine- To obtain a dark brown viscous liquid containing 0.764. The quantitative purity by HPLC high performance chromatography method was 60.2%, the pure content was 0.
Physical property values of the obtained 2 – {[6 – ({N- [4- (1 H-pyrazol-1 -yl) benzyl] pyridine-3-sulfonamido} methyl) pyridin-2- yl] amino} , It was as follows.
EI-MS (m /
z):. 520 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3, [delta] (ppm)): 1.24 (6H, d, J = 6.3 Hz), 5.07 (1 H, se, J = 5.5 Hz), 3.82 (2 H, d, J = 5.5 Hz), 4.31 (2 H, s), 4.64 (2 H, s), 4.94 J = 6.3 Hz), 6.26 (1 H, d, J = 8.3 Hz), 6.41 (1 H, dd, J = 7.2, 0.5 Hz), 6.46 (1 H, dd, J = 2.5, 1.8 Hz), 7.25 (2H, m), 7.71 (1H, dd, J = 8.3, 7.2 Hz), 7.32 (1H, ddd, J = 8.0, 4.9, 0.8 Hz), 7.37-7.42 J = 1.8, 0.6 Hz), 7.93 (1 H, dd, J = 2.6, 0.6 Hz), 7.94 (1 H, ddd, J = 8.0, 2.4, 1.7 Hz), 8.69 (1 H, dd, J = 4.8, 1.6 Hz ), 8.98 (IH, dd, J = 2.4, 0.8
Hz). 13 C-NMR (CDCl 3, δ (ppm)): 21.8, 43.7, 51.0, 51.1, 68.9, 107.4, 107.7, 112.6, 119.2, 123.3, 126.7, 129.9, 133.8, 134.6, 137.3, 137.6, 139.8, 141.1, 148.0, 152.6, 153.2, 157.3 , 1737 (C = O), (2981, 2933) (CH), 3437 (NH) , 170.5.
IR (KBr cm -1 ): 764 (CH), 1161 (S = O), 1525 .
EI-MS (m /
z):. 520 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3, [delta] (ppm)): 1.24 (6H, d, J = 6.3 Hz), 5.07 (1 H, se, J = 5.5 Hz), 3.82 (2 H, d, J = 5.5 Hz), 4.31 (2 H, s), 4.64 (2 H, s), 4.94 J = 6.3 Hz), 6.26 (1 H, d, J = 8.3 Hz), 6.41 (1 H, dd, J = 7.2, 0.5 Hz), 6.46 (1 H, dd, J = 2.5, 1.8 Hz), 7.25 (2H, m), 7.71 (1H, dd, J = 8.3, 7.2 Hz), 7.32 (1H, ddd, J = 8.0, 4.9, 0.8 Hz), 7.37-7.42 J = 1.8, 0.6 Hz), 7.93 (1 H, dd, J = 2.6, 0.6 Hz), 7.94 (1 H, ddd, J = 8.0, 2.4, 1.7 Hz), 8.69 (1 H, dd, J = 4.8, 1.6 Hz ), 8.98 (IH, dd, J = 2.4, 0.8
Hz). 13 C-NMR (CDCl 3, δ (ppm)): 21.8, 43.7, 51.0, 51.1, 68.9, 107.4, 107.7, 112.6, 119.2, 123.3, 126.7, 129.9, 133.8, 134.6, 137.3, 137.6, 139.8, 141.1, 148.0, 152.6, 153.2, 157.3 , 1737 (C = O), (2981, 2933) (CH), 3437 (NH) , 170.5.
IR (KBr cm -1 ): 764 (CH), 1161 (S = O), 1525 .
Example 5
Synthesis of 2 – {[6- (hydroxymethyl) pyridin-2-yl] amino} acetate isopropylate
Synthesis of 2 – {[6- (hydroxymethyl) pyridin-2-yl] amino} acetate isopropylate
948 g of 2-propanol and 76.7 g of concentrated sulfuric acid were added to a glass container having an inner volume of about 2 L and equipped with a stirring device, a thermometer and an upper cooling device, and the mixture was heated to 75 ° C. To this was added 2 – {[(t-butoxycarbonyl) (6-hydroxymethylpyridin-2-yl)] amino} acetic acid tert- butyl ester synthesized by the method described in Reference Example 3- (b) A mixed solution of 135 g of butyl, 45 g of toluene and 311 g of 2-propanol was added dropwise over 40 minutes, followed by heating and stirring at 78 ° C. for 6 hours. After cooling, 677 g of toluene and 406 g of water were added under an internal pressure of 20 hPa and an external temperature of 40 ° C. until the amount of liquid reached 309 g, and the mixture was stirred at room temperature and then separated. The obtained aqueous layer was added dropwise to a mixed solution of 129 g of separately prepared sodium hydrogencarbonate, 812 g of water, and 677 g of toluene over 20 minutes, stirred at room temperature for 1 hour, separated, and the aqueous layer was washed with 338 g . The obtained organic layer was mixed and washed with 426 g of a 5 wt% sodium chloride aqueous solution to obtain 1370 g of an organic layer. Approximately 1356 g of this was taken out, concentrated to a liquid volume of 113 g, and then toluene was added until the liquid amount reached 300 g. 190 g of n-heptane was added to the solution, and the solution was warmed to 45 ° C. to dissolve the crystals, followed by cooling to 35 ° C. A small amount of separately synthesized seed crystals was added in the same way and stirred at 35 ° C. for 1 hour, the crystals gradually increased. 365 g of n-heptane was added dropwise over 30 minutes, cooled for 40 minutes until the internal temperature reached 5 ° C., and stirred at the same temperature for 30 minutes. The precipitated crystals were separated by filtration, washed with n-heptane and then dried under reduced pressure at 50 ° C. to obtain 70.4 g of isopropyl 2 – {[6- (hydroxymethyl) pyridin-2-yl] amino} . The quantitative purity by HPLC high performance chromatography was 94.3%, and the pure content was 66.4 g (raw material 2 – {[(t-butoxycarbonyl) (6-hydroxymethylpyridin-2-yl )] Amino} acetate as t-butyl acetate in an isolated yield of 74.7%).
Physical properties of the obtained 2 – {[6- (hydroxymethyl) pyridin-2-yl] amino} acetate isopropyl were as follows.
EI-MS (m /
z):. 224 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3, [delta] (ppm)): 1.27 (6H, d, J = 6.3 Hz), 3.76 (IH, s), 4.10 (2H, d, J = 5.5 Hz), 4.59 (2H, s), 5.00 (IH, s), 5.10 (IH, m), 6.36
13 C-NMR (CDCl 3, δ (ppm) ), 6.51 (1 H, dd, J = 7.3, 0.7 Hz), 7.41 (1 H, ddd, J = 5.74, 3.88 Hz ) ): 21.8, 44.1, 63.5, 69.0, 106.6, 109.5, 138.0, 156.8, 156.9, 170.7
IR (KBr cm -1): 416, 469, 531, 559, 731, 785, 826, 862, 903, 916, 941, 980, 1014, 1052, 1082, 1106, 1131, 1147, 1182, 1217, 1256, 1276, 1347, 1378,
Calcd: C, 58.91% Calcd: C, 58.91% (C = O) ; H, 7.19%; N, 12.49%
Found: C, 58.99%; H, 7.17%; N, 12.48%.
EI-MS (m /
z):. 224 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3, [delta] (ppm)): 1.27 (6H, d, J = 6.3 Hz), 3.76 (IH, s), 4.10 (2H, d, J = 5.5 Hz), 4.59 (2H, s), 5.00 (IH, s), 5.10 (IH, m), 6.36
13 C-NMR (CDCl 3, δ (ppm) ), 6.51 (1 H, dd, J = 7.3, 0.7 Hz), 7.41 (1 H, ddd, J = 5.74, 3.88 Hz ) ): 21.8, 44.1, 63.5, 69.0, 106.6, 109.5, 138.0, 156.8, 156.9, 170.7
IR (KBr cm -1): 416, 469, 531, 559, 731, 785, 826, 862, 903, 916, 941, 980, 1014, 1052, 1082, 1106, 1131, 1147, 1182, 1217, 1256, 1276, 1347, 1378,
Calcd: C, 58.91% Calcd: C, 58.91% (C = O) ; H, 7.19%; N, 12.49%
Found: C, 58.99%; H, 7.17%; N, 12.48%.
Example 6
Synthesis of 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate isopropylate
Synthesis of 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate isopropylate
To a solution of 35.7 g of isopropyl 2 – {[6- (hydroxymethyl) pyridin-2-yl] amino} acetate obtained in Example 5 in 396 g of methylene chloride was added 19.6 g of thionyl chloride at room temperature Was added dropwise over 20 minutes, and the mixture was stirred at room temperature for 1 hour. The obtained reaction solution was added dropwise to a mixed liquid slurry of 37.8 g of sodium hydrogencarbonate and 149 g of water, and the mixture was stirred at room temperature for 20 minutes. After liquid separation, 6.73 g of magnesium sulfate was added to the organic layer, dehydrated and the filtrate was concentrated to dryness at 50 ° C. to obtain 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate 37 .8 g as a light brown solid.
Physical properties of the obtained 2 – {[6- (chloromethyl) pyridin-2-yl] amino} acetate isopropyl were as follows.
EI-MS (m /
z):. 242 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3, [delta] (ppm)): 1.24 (6H, m) J = 8.3 Hz), 4.7 (2H, d, J = 5.4 Hz), 4.48 (2H, s), 5.03 (IH, s), 5.10 (IH, m), 6.39
13 C-NMR (CDCl 3, δ (ppm)): 21.8, 44.0, 44.7, 68.9, 107.7, 112.2, 138.1, 1 H NMR (CDCl 3, δ (ppm)): 7.43 (1H, dd, J = 7.8, 7.8 Hz)154.6, 157.3, 170.7
IR (KBr cm -1): 415, 446, 530, 560, 627, 735, 804, 827, 874, 903, 939, 952, 982, 1042, 1088, 1108, 1128, 1144, 1167, 1180, 1219, 1269, 1281, 1350,
Elemental analysis: 1378, 1400, 1420, 1434, 1470, 1525 (C = N), 1580, 1613, 1690, 1728 (C = O), 2878, 2934 (CH), 2981 (CH), 3379Calcd: C, 54.44%; H, 6.23%; N, 11.54%
Found: C, 54.46%; H, 6.23%; N, 11.56%.
EI-MS (m /
z):. 242 [M] CI-MS (m /
[Mz):. + 1] 1 H-NMR (CDCl 3, [delta] (ppm)): 1.24 (6H, m) J = 8.3 Hz), 4.7 (2H, d, J = 5.4 Hz), 4.48 (2H, s), 5.03 (IH, s), 5.10 (IH, m), 6.39
13 C-NMR (CDCl 3, δ (ppm)): 21.8, 44.0, 44.7, 68.9, 107.7, 112.2, 138.1, 1 H NMR (CDCl 3, δ (ppm)): 7.43 (1H, dd, J = 7.8, 7.8 Hz)154.6, 157.3, 170.7
IR (KBr cm -1): 415, 446, 530, 560, 627, 735, 804, 827, 874, 903, 939, 952, 982, 1042, 1088, 1108, 1128, 1144, 1167, 1180, 1219, 1269, 1281, 1350,
Elemental analysis: 1378, 1400, 1420, 1434, 1470, 1525 (C = N), 1580, 1613, 1690, 1728 (C = O), 2878, 2934 (CH), 2981 (CH), 3379Calcd: C, 54.44%; H, 6.23%; N, 11.54%
Found: C, 54.46%; H, 6.23%; N, 11.56%.
PAPER
Journal of Medicinal Chemistry (2018), 61(15), 6869-6891.
Identification of a Selective, Non-Prostanoid EP2 Receptor Agonist for the Treatment of Glaucoma: Omidenepag and its Prodrug Omidenepag Isopropyl
Ryo Iwamura*†  , Masayuki Tanaka†, Eiji Okanari†, Tomoko Kirihara‡, Noriko Odani-Kawabata‡, Naveed Shams‡§, and Kenji Yoneda†
, Masayuki Tanaka†, Eiji Okanari†, Tomoko Kirihara‡, Noriko Odani-Kawabata‡, Naveed Shams‡§, and Kenji Yoneda†
, Masayuki Tanaka†, Eiji Okanari†, Tomoko Kirihara‡, Noriko Odani-Kawabata‡, Naveed Shams‡§, and Kenji Yoneda†† Pharmaceuticals Research Laboratory, UBE Industries, Ltd., 1978-5 Kogushi, Ube, Yamaguchi 755-8633, Japan
‡ R&D Division, Santen Pharmaceutical Co., Ltd., Grand Front Osaka Tower A 4-20, Ofukacho, Kita-ku, Osaka 530-8552, Japan
§ R&D Division, Santen Inc., 6401 Hollis Street, Suite 125, Emeryville, California 94608, United States
J. Med. Chem., 2018, 61 (15), pp 6869–6891
DOI: 10.1021/acs.jmedchem.8b00808

EP2 receptor agonists are expected to be effective ocular hypotensive agents; however, it has been suggested that agonism to other EP receptor subtypes may lead to undesirable effects. Through medicinal chemistry efforts, we identified a scaffold bearing a (pyridin-2-ylamino)acetic acid moiety as a promising EP2-selective receptor agonist. (6-((4-(Pyrazol-1-yl)benzyl)(pyridin-3-ylsulfonyl)aminomethyl)pyridin-2-ylamino)acetic acid 13ax (omidenepag, OMD) exerted potent and selective activity toward the human EP2 receptor (h-EP2). Low doses of omidenepag isopropyl (OMDI), a prodrug of 13ax, lowered intraocular pressure (IOP) in ocular normotensive monkeys. OMDI was selected as a clinical candidate for the treatment of glaucoma.
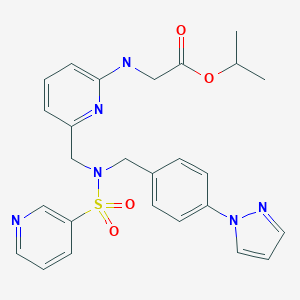
Isopropyl (6-((4-(Pyrazol-1-yl)benzyl)(pyridin-3-ylsulfonyl)aminomethyl)pyridin-2- ylamino)acetate (OMDI)
white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.87 (dd, J = 2.4, 0.7 Hz, 1H), 8.75 (dd, J = 4.8, 1.6 Hz, 1H), 8.48 (dd, J = 2.4, 0.5 Hz, 1H), 8.08 (ddd, J = 8.1, 2.4, 1.6 Hz, 1H), 7.80–7.77 (m, 2H), 7.74 (dd, J = 1.8, 0.5 Hz, 1H), 7.51 (ddd, J = 8.1, 4.8, 0.7 Hz, 1H), 7.36–7.33 (m, 2H), 7.26 (dd, J = 8.3, 7.1 Hz, 1H), 6.89 (t, J = 6.1, 1H), 6.54 (dd, J = 2.4, 1.8 Hz, 1H), 6.38 (d, J = 8.3 Hz, 1H), 6.34 (d, J = 7.1 Hz, 1H), 4.87 (sept, J = 6.3 Hz, 1H), 4.62 (s, 2H), 4.21(s, 2H), 3.76 (d, J = 6.1 Hz, 2H), 1.10 (d, J = 6.3 Hz, 6H). 13C NMR (proton-decoupled spectrum, 500 MHz, DMSO-d6) δ 171.2 (s), 158.1 (s), 153.4 (s), 153.2 (s), 147.6 (s), 141.4 (s), 139.6 (s), 137.5 (s), 137.0 (s), 135.1 (s), 134.4 (s), 129.9 (s), 128.2 (s), 124.4 (s), 118.8 (s), 111.4 (s), 108.3 (assigned for two nonequivalent carbons with identical chemical shift), 68.0 (s), 51.9 (s), 51.2 (s), 43.1 (s), 22.0 (s). MS (CI+) m/z521 (M + H)+. IR wavelength [cm–1] 3437 (N–H), 1736 (C═O), 1608, 1525, and 1511 (C═C and C═N), 1321 (SO2), 1161 (SO2). Elemental analysis [%] (average of three experiments) calculated for C26H28N6O4S: C 59.98, H 5.42, N 16.14. Found: C 59.76, H 5.28, N 16.01. TLC Rf value 0.39 (ethyl acetate).
//////////////Omidenepag isopropyl, JAPAN 2018, オミデネパグイソプロピル , DE-117, UBE, SANTEN
CC(C)OC(=O)CNc1cccc(CN(Cc2ccc(cc2)n3cccn3)S(=O)(=O)c4cccnc4)n1
FDA approves new treatment for patients with acute myeloid leukemia
|
||
The U.S. Food and Drug Administration today approved Daurismo (glasdegib) tablets to be used in combination with low-dose cytarabine (LDAC), a type of chemotherapy, for the treatment of newly-diagnosed acute myeloid leukemia (AML) in adults who are 75 years of age or older or who have other chronic health conditions or diseases (comorbidities) that may preclude the use of intensive chemotherapy.
“Intensive chemotherapy is usually used to control AML, but many adults with AML are unable to have intensive chemotherapy because of its toxicities. Today’s approval gives health care providers another tool to use in the treatment of AML patients with various, unique needs. Clinical trials showed that ..
November 21, 2018
Release
The U.S. Food and Drug Administration today approved Daurismo (glasdegib) tablets to be used in combination with low-dose cytarabine (LDAC), a type of chemotherapy, for the treatment of newly-diagnosed acute myeloid leukemia (AML) in adults who are 75 years of age or older or who have other chronic health conditions or diseases (comorbidities) that may preclude the use of intensive chemotherapy.
“Intensive chemotherapy is usually used to control AML, but many adults with AML are unable to have intensive chemotherapy because of its toxicities. Today’s approval gives health care providers another tool to use in the treatment of AML patients with various, unique needs. Clinical trials showed that overall survival was improved using Daurismo in combination with LDAC compared to LDAC alone for patients who would not tolerate intensive chemotherapy,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of abnormal white blood cells in the bloodstream and bone marrow. The National Cancer Institute at the National Institutes of Health estimates that in 2018, approximately 19,520 people will be diagnosed with AML and approximately 10,670 patients with AML will die of the disease. Almost half of the adults diagnosed with AML are not treated with intensive chemotherapy because of comorbidities and chemotherapy related toxicities.
The efficacy of Daurismo was studied in a randomized clinical trial in which 111 adult patients with newly diagnosed AML were treated with either Daurismo in combination with LDAC or LDAC alone. The trial measured overall survival (OS) from the date of randomization to death from any cause. Results demonstrated a significant improvement in OS in patients treated with Daurismo. The median OS was 8.3 months for patients treated with Daurismo plus LDAC compared with 4.3 months for patients treated with LDAC only.
Common side effects reported by patients receiving Daurismo in clinical trials include low red blood cell count (anemia), tiredness (fatigue), bleeding (hemorrhage), fever with low white blood cell count (febrile neutropenia), muscle pain, nausea, swelling of the arms or legs (edema), low platelet counts (thrombocytopenia), shortness of breath (dyspnea), decreased appetite, distorted taste (dysgeusia), pain or sores in the mouth or throat (mucositis), constipation and rash.
The prescribing information for Daurismo includes a Boxed Warning to advise health care professionals and patients about the risk of embryo-fetal death or severe birth defects. Daurismo should not be used during pregnancy or while breastfeeding. Pregnancy testing should be conducted in females of reproductive age prior to initiation of Daurismo treatment and effective contraception should be used during treatment and for at least 30 days after the last dose. The Boxed Warning also advises male patients of the potential risk of drug exposure through semen and to use condoms with a pregnant partner or a female partner that could become pregnant both during treatment and for at least 30 days after the last dose. Daurismo must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. Patients should also be advised not to donate blood or blood products during treatment. Health care providers should also monitor patients for changes in the electrical activity of the heart, called QT prolongation.
The FDA granted this application Priority Review designation. Daurismo also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Daurismo to Pfizer.
//////////////Daurismo, glasdegib, fda 2018, Priority Review, Orphan Drug
FDA approves first treatment Gamifant (emapalumab) specifically for patients with rare and life-threatening type of immune disease
FDA approves first treatment Gamifant (emapalumab) specifically for patients with rare and life-threatening type of immune disease
The U.S. Food and Drug Administration today approved Gamifant (emapalumab) for the treatment of pediatric (newborn and above) and adult patients with primary hemophagocytic lymphohistiocytosis (HLH) who have refractory, recurrent or progressive disease or intolerance with conventional HLH therapy. This FDA approval is the first for a drug specifically for HLH.
“Primary HLH is a rare and life-threatening condition typically affecting children and this approval fills an unmet medical need for these patients,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to continuing to expedite the development and review of therapies that offer meaningful treatment options for …
November 20, 2018
Release
The U.S. Food and Drug Administration today approved Gamifant (emapalumab-lzsg) for the treatment of pediatric (newborn and above) and adult patients with primary hemophagocytic lymphohistiocytosis (HLH) who have refractory, recurrent or progressive disease or intolerance with conventional HLH therapy. This FDA approval is the first for a drug specifically for HLH.
“Primary HLH is a rare and life-threatening condition typically affecting children and this approval fills an unmet medical need for these patients,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to continuing to expedite the development and review of therapies that offer meaningful treatment options for patients with rare conditions.”
HLH is a condition in which the body’s immune cells do not work properly. The cells become overactive releasing molecules, which leads to inflammation. The immune cells start to damage the body’s own organs, including the liver, brain and bone marrow. It can be inherited, which is known as primary or “familial” HLH. It can also have non-inherited causes. People with primary HLH usually develop symptoms within the first months or years of life. Symptoms may include fever, enlarged liver or spleen and decreased number of blood cells.
The efficacy of Gamifant was studied in a clinical trial of 27 pediatric patients with suspected or confirmed primary HLH with either refractory, recurrent or progressive disease during conventional HLH therapy or who were intolerant of conventional HLH therapy. The median age of the patients in the trial was 1 year old. The study showed that 63 percent of patients experienced a response and 70 percent were able to proceed to stem cell transplant.
Common side effects reported by patients receiving Gamifant in clinical trials included infections, hypertension, infusion-related reactions, low potassium and fever. Patients receiving Gamifant should not receive any live vaccines and should be tested for latent tuberculosis. Patients should be closely monitored and treated promptly for infections while receiving Gamifant.
The FDA granted this application Priority Review and Breakthrough Therapydesignation. Gamifant also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Gamifant to Novimmune SA.
////////////Gamifant, emapalumab, FDA 2018
Lumateperone
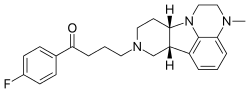
Lumateperone
- Molecular FormulaC24H28FN3O
- Average mass393.497 Da
4-((6bR,10aS)-3-Methyl-2,3,6b,9,10,10a-hexahydro-1H,7H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalin-8-yl)-1-(4-fluorophenyl)-butan-1-one
1-Butanone, 1-(4-fluorophenyl)-4-(2,3,6b,9,10,10a-hexahydro-3-methyl-1H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)-
1-(4-fluorophenyl)-4-{4-methyl-1,4,12-triazatetracyclo[7.6.1.0⁵,¹⁶.0¹⁰,¹⁵]hexadeca-5,7,9(16)-trien-12-yl}butan-1-one
313368-91-1 [RN]
70BSQ12069, Lumateperone, PHASE 3, ITI-007

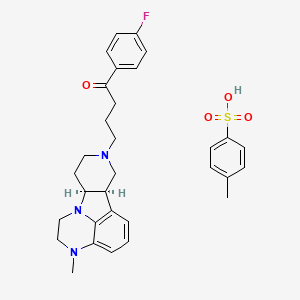
4- methylbenzenesulfonate. SALT
| Molecular Formula: | C31H36FN3O4S |
|---|---|
| Molecular Weight: | 565.704 g/mol |
(6bR,10aS)-8-[4-(4-Fluorophenyl)-4-oxobutyl]-3-methyl-2,3,6b,7,8,9,10,10a-octahydro-1H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalin-8-ium 4-methylbenzenesulfonate
1187020-80-9 [RN]
1-Butanone, 1-(4-fluorophenyl)-4-[(6bR,10aS)-2,3,6b,9,10,10a-hexahydro-3-methyl-1H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl]-, 4-methylbenzenesulfonate (1:1)
ITI-007 tosylate
JIE88N006O
Lumateperone tosylate [USAN]
UNII:JIE88N006O
ITI 007
- Originator Bristol-Myers Squibb
- Develope rIntra-Cellular Therapies
- Class Antidepressants; Antipsychotics; Pyrroles; Quinoxalines; Sleep disorder therapies
- Mechanism of Action Dopamine receptor modulators; NR2B N-Methyl D-Aspartate receptor modulators; Serotonin 2A receptor antagonists; Serotonin plasma membrane transport protein inhibitors; Serotonin uptake inhibitors
- 07 Nov 2018 Intra-Cellular Therapeutics completes enrolment in the phase III Study 401 trial for Bipolar depression (Monotherapy) in USA
- 16 Oct 2018 Intra-Cellular Therapies plans to launch lumateperone for Schizophrenia in USA
- 02 Aug 2018 Intra-Cellular plans a clinical trial for Depressive disorders in 2H of 2018
Highest Development Phases
- Preregistration Schizophrenia
- Phase III Behavioural disorders; Bipolar depression
- Phase II Sleep maintenance insomnia
- Preclinical Mental disorders
- No development reported Mood disorders
Lumateperone (INN; developmental code names ITI-007, ITI-722) is an investigational atypical antipsychotic which is currently under development by Intra-Cellular Therapies, licensed from Bristol-Myers Squibb, for the treatment of schizophrenia.[1][2] It is also being developed by Intra-Cellular Therapies for the treatment of bipolar disorder, depression, and sleep and behavioral disturbance in dementia, autism, and other neuropsychiatric disorders.[3] As of September 2015, lumateperone has passed the first of two phase IIIclinical trials for schizophrenia.[4] In November 2017 the US FDA awarded Intra-Cellular Therapies Fast Track designation for lumateperone.[5]
Pharmacology
Pharmacodynamics
Relative to presently-available antipsychotics, lumateperone possesses a unique and novel mechanism of action.[6][7] It acts as a 5-HT2A receptor antagonist (Ki = 0.54 nM), a partial agonist of presynaptic D2 receptors and an antagonist of postsynaptic D2 receptors (Ki = 32 nM), and a serotonin transporter blocker (Ki = 61 nM).[6][8] It also possesses affinity for the D1 receptor (Ki = 52 nM) and lower affinity for the α1A– and α1B-adrenergic receptors (Ki = 73 nM at α1), 5-HT2C receptor (Ki = 173 nM), and D4 receptor.[6] Lumateperone does not significantly bind to the 5-HT2B, H1 (Ki > 1,000 nM), muscarinic acetylcholine receptors, or many other sites (Ki > 100 nM).[6]
Lumateperone shows a 60-fold difference in its affinities for the 5-HT2A and D2 receptors, which is far greater than that of most or all existing atypical antipsychotics, such as risperidone (12-fold), olanzapine (12.4-fold), and aripiprazole (0.18-fold).[6][9] It is thought that this property may improve the effectiveness and reduce the side effect profile of lumateperone relative to currently-available antipsychotics, a hypothesis which is supported by the observation of minimal catalepsy in mice treated with the drug.[6][9] Moreover, it has been expressed that this property could result in full occupancy and blockade of the 5-HT2A at low doses, with dose-dependent adjustable modulation of the D2 receptor, as well as the SERT, possible with increasing doses, which would uniquely allow for clinical optimization of efficacy and side effect incidence.[6][9]
Unlike most current antipsychotics, such as haloperidol, risperidone, and olanzapine, lumateperone does not disrupt striatal dopamine signaling, a property which is likely due to its partial agonism of presynaptic D2 receptors.[6] In accordance, similarly to aripiprazole, which is also a partial agonist of presynaptic D2 receptors, lumateperone showed no striatum-based motor side effects (i.e., catalepsy) in animals.[6]
Clinical studies
In phase II clinical trials, lumateperone showed statistically-significant efficacy in improvement of psychosis at a dose of 60 mg daily.[2] In addition, it distinguished itself from its comparator risperidone in reducing negative symptoms, including improvement in social function, as well as in alleviating depressive symptoms in schizophrenia patients with comorbid depression, whereas risperidone had no effect.[2][10] Lumateperone also distinguished itself from risperidone in that it produced little or no weight gain, did not negatively affect metabolic parameters (i.e., insulin, glucose, triglyceride, and cholesterol levels), did not increase prolactin levels, and did not show a rate of the side effect of akathisia that differed from placebo.[2][10] In addition, lumateperone did not produce any changes in cardiovascular function, such as QTc prolongation, and unlike risperidone, it did not produce a measurable increase heart rate.[7] Due to its favorable influence on metabolic parameters, it was concluded that lumateperone, unlike many other available antipsychotics such as risperidone, may not cause an increase in the risk of diabetes or cardiovascular disease, and hence may prove to be a significant improvement relative to many existing antipsychotic drugs in terms of long-term safety and tolerability.[2]
Lumateperone, at a dose of 60 mg per day, was not found to be associated with any statistically significant treatment-emergent side effects relative to placebo.[10] At a dose of 120 mg daily, the most frequent adverse effect observed was sedation/somnolence, reported by 32.5% of patients.[10] There was no evidence of extrapyramidal symptoms or increase in suicidal ideation or behavior.[10]
SYNTHESIS
MEDCHEM

PAPER
https://pubs.acs.org/doi/abs/10.1021/jm401958n
dx.doi.org/10.1021/jm401958n | J. Med. Chem. 2014, 57, 2670−2682
5 (367 mg, 53%yield) as a gray solid.
1H NMR (DMSO-d6, 500 MHz) δ 9.10 (br, 1H),8.10−8.01 (m, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.42−7.33 (m, 2H), 7.11 (d, J = 7.8 Hz, 2H), 6.65−6.57 (m, 1H), 6.51 (d, J = 7.3 Hz, 1H), 6.42 (d, J = 7.9 Hz, 1H), 3.59 (dd, J = 12.2, 6.5 Hz, 1H), 3.52−3.37 (m, 3H), 3.37−3.28 (m, 2H), 3.25−3.20 (m, 1H), 3.18−2.99 (m, 5H), 2.81 (s, 3H), 2.71 (td, J = 10.2, 3.0 Hz, 1H), 2.63−2.52 (m, 1H), 2.28 (s, 3H), 2.27−2.22 (m, 1H), 2.15−1.93 (m, 3H).
13C NMR (DMSOd6, 126 MHz) δ 197.2, 165.1 (d, JCF = 252 Hz), 145.6, 137.6, 137.3, 135.2, 133.1, 130.9 (d, JCF = 10 Hz), 128.1, 126.7, 125.5, 120.6, 115.7 (d, JCF = 22 Hz), 112.5, 109.3, 62.2, 55.5, 52.5, 49.8, 47.8, 43.7, 38.6, 37.0, 34.9, 21.7, 20.8, 18.0.
MS (ESI) m/z 394.2 [M + H]+.
HRMS (ESI) m/z calcd for C24H29FN3O [M + H]+, 394.2295; found, 394.2292. UPLC purity, 97.7%; retention time, 2.06 min (method A).

PATENT
WO 2000077002
WO 2000077010
US 20040220178
WO 2008112280
WO 2009114181
WO 2011133224
PATENT
WO 2017172811
0003] l-(4-fluoro-phenyl)-4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-lH,7H- pyrido[3′,4′:4,5]pyrrolo[l,2,3-de]quinoxalin-8-yl)-butan-l-one (sometimes referred to as 4- ((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-lH-pyrido[3′,4′:4,5]pyrrolo[l,2,3- de]quinoxalin-8(7H)-yl)-l-(4-fluorophenyl)-l-butanone, or as ITI-007), has the following structure:

[0004] ITI-007 is a potent 5-HT2A receptor ligand (Ki=0.5 nM) with strong affinity for dopamine (DA) D2 receptors (Ki=32 nM) and the serotonin transporter (SERT) (Ki=62 nM) but negligible binding to receptors (e.g., HI histaminergic, 5-HT2C, and muscarinic) associated with cognitive and metabolic side effects of antipsychotic drugs. ΠΊ-007 is currently in clinical trials, i.a., for treatment of schizophrenia. While ITI-007 is a promising drug, its production and formulation present challenges. In free base form, ITI-007 is an oily, sticky solid, with poor solubility, not only in water but also in many organic solvents. Making salts of the compound has proven to be unusually difficult. A hydrochloride salt form of ITI-007 was disclosed in US 7183282, but this salt is hygroscopic and shows poor stability. A toluenesulfonic acid addition salt (tosylate) of ITI- 007 was finally identified and described in WO 2009/114181.
[0005] There is a need for alternative stable, pharmaceutically acceptable solid forms of ITI-007, which can be readily incorporated into galenic formulations.
XAMPLES
[0027] The following equipment and methods are used to isolate and characterize the exemplified co-crystal forms:
[0028] X-ray powder diffraction (XRPD): The X-ray powder diffraction studies are performed using a Bruker AXS D2 PHASER in Bragg-Brentano configuration, equipment #1549 / #2353. The equipment uses a Cu anode at 30kV, 10 mA; sample stage standard rotating; monochromatization by a Κβ-filter (0.5% Ni). Slits: fixed divergence slits 1.0mm (=0.61°), primary axial Soller slit 2.5°, secondary axial Soller slit 2.5°. Detector: Linear detector LYNXEYE with receiving slit 5° detector opening. The standard sample holder (0.1 mm cavity in (510) silicon wafer) has a minimal contribution to the background signal. Measurement conditions: scan range 5 – 45° 2Θ, sample rotation 5 rpm, 0.5s/step, 0.010°/step, 3.0mm detector slit; and all measuring conditions are logged in the instrument control file. As system suitability, corundum sample A26- B26-S (NIST standard) is measured daily. The software used for data collection is Diffrac. Commander v2.0.26. Data analysis is done using Diffrac.Eva vl.4. No background correction or smoothing is applied to the patterns.
[0029] Simultaneous thermogravimetry (TGA) and differential scanning calorimetry (DSC) or TGA/DSC analysis: The TGA/DSC studies are performed using a Mettler Toledo TGA/DSC 1 Stare System, equipment #1547, auto-sampler equipped, using pin-holed Al- crucibles of 40 μΐ. Measurement conditions: 5 min 30.0 °C, 30.0 – 350.0 °C with 10 °C/min., N2 flow of 40 ml/min. The software used for instrument control and data analysis is STARe vl2.10.
[0030] Differential scanning calorimetry (DSC): The DSC studies are performed using a Mettler Toledo DSC1 STARe System, equipment #1564. The samples are made using Al crucibles (40 μΐ; pierced). Typically 1 – 8 mg of sample is loaded onto a pre- weighed Al crucible and is kept at 30°C for 5 minutes, after which it is heated at 10°C/min from 30°C to 350 °C and kept at 350°C for 1 minute. A nitrogen purge of 40 ml/min is maintained over the sample. As system suitability check Indium and Zinc are used as references. The software used for data collection and evaluation is STARe Software vl2.10 build 5937. No corrections are applied to the thermogram.
[0031] Polarized light microscopy (PLM): The microscopy studies are performed using an Axio Vert 35M, equipped with an AxioCamERc 5s, equipment #1612. The microscope is equipped with four lenses: Zeiss A-Plan 5x/0.12, Zeiss A-Plan lOx/0.25, LD A-Plan 20x/0.30 and Achros TIGMAT 32x/0.40. Data collection and evaluation is performed using Carl Zeiss Zen Axio Vision Blue Edition Lite 2011 vl.0.0.0 software. A small amount of sample is loaded on an object glass and carefully spread until a thin layer is obtained.
[0032] Dynamic Vapour Sorption (DVS): The Dynamic Vapour Sorption studies are performed using a Surface Measurement Systems Ltd. DVS-1 No Video, equipment #2126. The sample is loaded into a balance pan, typically 20-30 mg, and equilibrated at 0% RH. After the material was dried, the RH is increased with 10% per step for 1 hour per increment, ending at 95% RH. After completion of the sorption cycle, the sample was dried using the same method. The software used for data collection is DVSWin v3.01 No Video. Data analysis is performed using DVS Standard Analysis Suite v6.3.0 (Standard).
[0033] Particle size distribution (PSD): The particle size distribution studies are performed using a Malvern Instruments Mastersizer, equipment #1712. The Mastersizer uses a 300RF lens range of 0.05 μηι – 900 mm. Polydisperse is used as analysis model. Measurement conditions: before each sample measurement a background measurement is performed, the background scan time is 12 seconds (12000 snaps). Each sample is dispersed in Multipar G, refractive index of 1.42. The obscuration range on sample dispersion is between 10%-30%. Each sample is measured 6 times at t=0 and t=30 minutes and the measurement scan time is 10 seconds (10000 snaps). The targeted stirring speed of the sample dispersion unit is 2000+10 rpm. Data collection and evaluation is performed using Mastersizer S Version 2.19 software. [0034] Capillary Melting Point: The capillary melting point is determined on a Biichi Melting Point B-545, equipment #000011, conform USP guidelines.
[0035] X-ray fluorescence (XRF): The X-ray fluorescence studies are performed using a Bruker AXS S2 RANGER, equipment #2006. Using an end-window X-ray tube with Palladium anode and an ultra-thin Beryllium window (75 μιη) for superior light element analysis. As detector the Xflash V5 detector with Cr, Ti, Al, Ta collimator (energy resolution < 129 eV FWHM at 100 000 cps Mnka) is used. The S2 Ranger is equipped with an autosampler with integrated 28 position X- Y automatic sample changer with exchangeable tray, which allows maximum sample diameter of 40 mm. Samples are mounted in steel rings of 51.5 mm diameter for automatic operation. Measurement conditions: disposable liquid cups (35 mm inner diameter, 40 mm outer diameter) with polypropylene foil 5 μιη. As system suitability check a copper disk is measured daily and a glass disk, containing several elements, is measured weekly. The software used for data collection is S2 Ranger Control Software V4.1.0. Data analysis is performed using SPECTRA EDX V2.4.3 evaluation software. No background correction or smoothing is applied to the patterns.
[0036] Fourier transform infrared spectroscopy (FT-IR): The FT-IR studies are performed using a Thermo Scientific Nicolet iS50, equipment # 2357. An attenuated total reflectance (ATR) technique was used with a beam splitter of KBr. Experiment setup of the collected sample is used number of scans 16 with a resolution of 4from 400 cm“1 to 4000 cm“1. The software OMNIC version 9.2 is used for data collection and evaluation.
[0037] Thermogravimetric analysis (TGA) with infrared spectroscopy (TGA-IR):
In TGA-IR, the off-gassing materials are directed through a transfer line to a gas cell, where the infrared light interacts with the gases. The temperature ramp and first derivative weight loss information from the TGA is shown as a Gram-Schmidt (GS) profile; the GS profile essentially shows the total change in the IR signal relative to the initial state. In most cases, the GS and the derivative weight loss will be similar in shape, although the intensity of the two can differ. For this experiment are two devices coupled to each other. The TGA studies are performed using a Mettler Toledo TGA/DSCl STARe System with a 34-position auto sampler, equipment #1547. The samples are made using Al crucibles (100 μΐ; pierced). Typically 20-50 mg of sample is loaded into a pre- weighed Al crucible and is kept at 30°C for 5 minutes after which it is heated at 10°C/min from 30°C to 350°C. A nitrogen purge of 40 ml/min is maintained over the sample. The TGA-IR module of the Nicolet iS50 is coupled to the TGA/DSCl. The IR studies were performed using a Thermo Scientific Nicolet iS50, equipment # 2357. Experiment setup of the collected series, the profile Gram-Schmidt is used number of scans 10 with a resolution of 4. The software OMNIC version 9.2 is used for data collection and evaluation.
[0038] High performance liquid chromatography (HPLC): The high performance liquid chromatography analyses are performed on LC-31, equipped with an Agilent 1100 series G1322A degasser equipment #1894, an Agilent 1100 series G1311A quaternary pump equipment #1895, an Agilent 1100 series G1313A ALS equipment #1896, an Agilent 1100 series G1318A column equipment #1897 and an Agilent 1100 series G1314A VWD equipment #1898 / LC-34, equipped with an Agilent 1200 series G1379B degasser equipment #2254, an Agilent 1100 series G1311A quaternary pump equipment #2255, Agilent 1100 series G1367A WPALS equipment #1656, an Agilent 1100 series G1316A column equipment #2257 and an Agilent 1100 series G1315B DAD equipment #2258. Data is collected and evaluated using Agilent ChemStation for LC systems Rev. B.04.02[96]. Solutions are prepared as follows: Mobile phase A: Add 800 ml of MilliQ water to a 1L volumetric flask. Add 1 ml of TFA and homogenize. Fill up to the mark with MilliQ; Mobile phase B: Add 800 ml of Acetonitrile to a 1L volumetric flask. Add 1 ml of TFA and homogenize. Fill up to the mark with Acetonitrile; Diluent: 50/50 MeOH/ACN.
Example 1: Co-crystal screen
[0039] Solubility of free base in various solvents is evaluated, and based on the results of the solubility range, suitable solvents are selected for the co-crystal screen. Co-crystal formation is based on hydrogen bonding and stacking of the molecules, meaning the co-former selection is based on active groups. Grinding is a method to form co-crystals, however the free base itself is an oil/ sticky solid and therefore not suitable for this method. The free base and counter ion are added to a solution in a certain ratio to give the chance to form a co-crystal, similar to salt formation. We found the best method is to add a saturated solution of the co-former to that of the free base to find an optimal ratio for co-crystal formation.
[0040] Three different experiments are performed with each of 26 candidate co-formers, which include sugar alcohols, amino acids, and other compounds identified as having potential to for co- crystals; adding solutions stepwise, slurry experiments and cooling crystallization experiments. The free base and co-former are dissolved prior to adding to each other. Co-formers are added in a 1 : 1 , 2: 1 and 1 :2 ratio to the free base. All experiments are performed using four different solvents, methanol, acetonitrile, ethyl acetate and toluene. All solids are characterized by XRPD. Two different ITI-007 free base co-crystals formed, with nicotinamide and with isonicotinamide. Both co-crystals were obtained by slurry experiments in methanol.
Example 2: Isonicotinamide co-crystal
[0041] Isonicotinamide forms a possible co-crystal with ITI-007 free base by slurrying the mixture in methanol and ethyl acetate, appearing as a red/brown and yellow solid respectively. TGA-DSC analysis of the experiment using isonicotinamide in methanol results in two endothermic events,
Both endothermic events do not correspond to the free base or the co-former, which means ITI-007 free base-isonicotinamide co-crystal is formed. HPLC and Ή-ΝΜΡ analyses confirm both of the free base and the co-former to be present. Using isonicotinamide in ethyl acetate, however, does not result in a co-crystal and, no endothermic event is present in the TGA/DSC analysis.
[0042] The slurry experiment in methanol is repeated at a gram scale. First, ITI-007 free base and isonicotinamide are each dissolved in methanol. Subsequently, the obtained solutions are mixed in a 1: 1 ratio and the resulting mixture is stirred at room temperature for 2 hours. The mixture remains a clear solution, which is evaporated under vacuum to give a brown sticky solid. XRPD analysis shows the brown sticky solid to be crystalline, as shown in Figure 1, ITI-007 free base-isonicotinamide co-crystal has formed. The corresponding peak list is showing in Table 1. The XRPD shows clustered peaks which is likely due to preferred orientation.
PATENT
WO 2018189646
The present application relates to solid state forms of Lumateperone p-Tosylate and processes for preparation thereof.
The drug compound is having the adopted name “Lumateperone” and it has chemical name: l-(4-fluorophenyl)-4-[(6bR,10aS)-2,3,6b,9,10,10a-hexahydro-3-methyl-lH-pyrido[3′,4′:4,5]pyrrolo[l,2,3-de]quinoxalin-8(7H)-yl] 1-Butanone; and a structure depicted by Formula I.
Formula I
International Patent Application Publication Nos. WO2000077002A1, WO2009145900 A 1 and WO2013155504A1 which are incorporated herein in their entirety reported Lumateperone and its related compounds. These compounds have been found to be useful as 5-HT2 receptor agonists and antagonists used in treating disorders of the central nervous system including a disorder associated with 5HT2C or 5HT2A receptor modulation selected from obesity, anorexia, bulemia, depression, a anxiety, psychosis, schizophrenia, migraine, obsessive -compulsive disorder, sexual disorders, depression, schizophrenia, migraine, attention deficit disorder, attention deficit hyperactivity disorder, obsessive-compulsive disorder, sleep disorders, conditions associated with cephalic pain, social phobias, gastrointestinal disorders such as dysfunction of the gastrointestinal tract motility. International Patent Application Publication No. WO2008112280A1 disclose process(es) for preparing Lumateperone and its salts.
International Patent Application Publication No. WO2009114181A2 disclose crystalline forms of the p-Tosylate salt of compound of Formula (I), WO 2017172784 Al disclose oxalate, aminosalicylate, cyclamate salts of Lumateperone, WO 2017172811 Al
disclose co-crystal of Lumateperone with iso-nicotinamide, nicotinatinamide, WO 2018031535 Al disclose crystalline Form Fl of Lumateperone ditosylate.
Crystalline solids normally require a significant amount of energy for dissolution due to their highly organized, lattice like structures. For example, the energy required for a drug molecule to escape from a crystal is more than from an amorphous or a non-crystalline form. It is known that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailability patterns compared to the crystalline form. For some therapeutic indications, one bioavailability pattern may be favored over another. Therefore, it is desirable to have amorphous forms of drugs with high purity to meet the needs of regulatory agencies and also highly reproducible processes for their preparation.
In view of the above, it is therefore, desirable to stable amorphous form of Lumateperone j?-tosylate. The amorphous form provided herein is at least stable under ordinary stability conditions with respect to purity, storage and is free flowing powder.
Amorphous solid dispersions of drugs are generally known to improve the stability and solubility of drug products. However, some of such amorphous solid dispersions are found to be unstable over time. Amorphous solid dispersions of drugs tend to convert to crystalline forms over time, which can lead to improper dosing due to differences of the solubility of crystalline drug material compared to amorphous drug material. The present invention, however provides stable amorphous solid dispersions of Lumateperone j?-tosylate with improved solubility. Moreover, the present invention provides solid dispersions of Lumateperone j?-tosylate which may be reproduced easily and is amenable for processing into a dosage form
EXAMPLE 1 : PREPARATION OF AMORPHOUS LUMATEPERONE p-TOSYLATE
Lumateperone j?-tosylate (500 mg) was dissolved in methanol (25 mL) at room temperature for clear solution and filtered to remove undissolved particles. The resultant filtrate was subjected to fast solvent evaporation using rotavapor at about 55°C to afford the solid compound. The said solid was dried under vacuum at about 45°C to afford the amorphous Lumateperone p-tosylate according to Figure 1.
References
- Jump up^ Sylvain Celanire; Sonia Poli (13 October 2014). Small Molecule Therapeutics for Schizophrenia. Springer. pp. 31–. ISBN 978-3-319-11502-3.
- ^ Jump up to:a b c d e Intra-Cellular Therapies, Inc. (2015). “Intra-Cellular Therapies Announces Further Analyses of the Phase 2 Clinical Trial of ITI-007 in Schizophrenia at the 168th Annual Meeting of the American Psychiatric Association”. GlobeNewswire, Inc.
- Jump up^ Intra-Cellular Therapies. “Product Pipeline – Intra-Cellular Therapies”. Archived from the original on 2015-05-11. Retrieved 2015-05-19.
- Jump up^ Intra-Cellular Therapies. “Intra-Cellular Therapies Announces Positive Top-Line Results From the First Phase 3 Trial of ITI-007 in Patients With Schizophrenia and Confirms the Unique Pharmacology of ITI-007 in a Separate Positron Emission Tomography Study”. intracellulartherapies. Archived from the original on 2016-03-21.
- Jump up^ “Intra-Cellular Therapies Receives FDA Fast Track Designation for Lumateperone for the Treatment of Schizophrenia | Intra-Cellular Therapies Inc”. Intra-Cellular Therapies Inc. Retrieved 2017-11-25.
- ^ Jump up to:a b c d e f g h i Snyder GL, Vanover KE, Zhu H, Miller DB, O’Callaghan JP, Tomesch J, Li P, Zhang Q, Krishnan V, Hendrick JP, Nestler EJ, Davis RE, Wennogle LP, Mates S (2015). “Functional profile of a novel modulator of serotonin, dopamine, and glutamate neurotransmission”. Psychopharmacology. 232 (3): 605–21. doi:10.1007/s00213-014-3704-1. PMC 4302236. PMID 25120104.
- ^ Jump up to:a b Nancy A. Melville (2015). “Novel Drug Promising for Schizophrenia”. Medscape Medical News.
- Jump up^ Li P, Zhang Q, Robichaud AJ, Lee T, Tomesch J, Yao W, Beard JD, Snyder GL, Zhu H, Peng Y, Hendrick JP, Vanover KE, Davis RE, Mates S, Wennogle LP (2014). “Discovery of a tetracyclic quinoxaline derivative as a potent and orally active multifunctional drug candidate for the treatment of neuropsychiatric and neurological disorders”. J. Med. Chem. 57 (6): 2670–82. doi:10.1021/jm401958n. PMID 24559051.
- ^ Jump up to:a b c Davis RE, Vanover KE, Zhou Y, Brašić JR, Guevara M, Bisuna B, Ye W, Raymont V, Willis W, Kumar A, Gapasin L, Goldwater DR, Mates S, Wong DF (2015). “ITI-007 demonstrates brain occupancy at serotonin 5-HT2A and dopamine D 2 receptors and serotonin transporters using positron emission tomography in healthy volunteers”. Psychopharmacology. 232 (15): 2863–72. doi:10.1007/s00213-015-3922-1. hdl:10044/1/24121. PMID 25843749.
- ^ Jump up to:a b c d e Intra-Cellular Therapies, Inc. (2013). “Intra-Cellular Therapies Announces Positive Topline Phase II Clinical Results of ITI-007 for the Treatment of Schizophrenia”. PRNewswire.
External links
- ITI-007 – Intra-Cellular Therapies
- Product Pipeline – Intra-Cellular Therapies
- Lumateperone – AdisInsight
 |
|
| Clinical data | |
|---|---|
| Synonyms | ITI-007; ITI-722 |
| Routes of administration |
By mouth |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C24H28FN3O |
| Molar mass | 393.496 |
| 3D model (JSmol) | |
////// Lumateperone, PHASE 3, ITI-007, ITI-722
FDA approves new drug Aemcolo (rifamycin), to treat travelers’ diarrhea
FDA approves new drug to treat travelers’ diarrhea
The U.S. Food and Drug Administration today approved Aemcolo (rifamycin), an antibacterial drug indicated for the treatment of adult patients with travelers’ diarrhea caused by noninvasive strains of Escherichia coli (E. coli), not complicated by fever or blood in the stool.
“Travelers’ diarrhea affects millions of people each year and having treatment options for this condition can help reduce symptoms of the condition,” said Edward Cox, M.D., M.P.H., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Travelers’ diarrhea is the most common travel-related illness, affecting an estimated 10 to 40 percent of travelers worldwide each year. Travelers’ diarrhea is defined by …
November 16, 2018
Release
The U.S. Food and Drug Administration today approved Aemcolo (rifamycin), an antibacterial drug indicated for the treatment of adult patients with travelers’ diarrhea caused by noninvasive strains of Escherichia coli (E. coli), not complicated by fever or blood in the stool.
“Travelers’ diarrhea affects millions of people each year and having treatment options for this condition can help reduce symptoms of the condition,” said Edward Cox, M.D., M.P.H., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Travelers’ diarrhea is the most common travel-related illness, affecting an estimated 10 to 40 percent of travelers worldwide each year. Travelers’ diarrhea is defined by having three or more unformed stools in 24 hours, in a person who is traveling. It is caused by a variety of pathogens, but most commonly bacteria found in food and water. The highest-risk destinations are in most of Asia as well as the Middle East, Africa, Mexico, and Central and South America.
The efficacy of Aemcolo was demonstrated in a randomized, placebo-controlled clinical trial in 264 adults with travelers’ diarrhea in Guatemala and Mexico. It showed that Aemcolo significantly reduced symptoms of travelers’ diarrhea compared to the placebo.
The safety of Aemcolo, taken orally over three or four days, was evaluated in 619 adults with travelers’ diarrhea in two controlled clinical trials. The most common adverse reactions with Aemcolo were headache and constipation.
Aemcolo was not shown to be effective in patients with diarrhea complicated by fever and/or bloody stool or diarrhea due to pathogens other than noninvasive strains of E. coli and is not recommended for use in such patients. Aemcolo should not be used in patients with a known hypersensitivity to rifamycin, any of the other rifamycin class antimicrobial agents (e.g. rifaximin), or any of the components in Aemcolo.
The FDA granted Aemcolo a Qualified Infectious Disease Product (QIDP)designation. QIDP designation is given to antibacterial and antifungal drug products that treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act. As part of QIDP designation, the Aemcolo marketing application was granted Priority Review under which the FDA’s goal is to take action on an application within an expedited time frame.
The FDA granted approval of Aemcolo to Cosmo Technologies, Ltd.
///////////////// Aemcolo, rifamycin, fda 2018, qidp, priority review
Nemorexant
Nemorexant
ACT-541468, UNII LMQ24G57E9
[(2S)-2-(5-Chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone
1505484-82-1 [RN]
LMQ24G57E9
Methanone, [(2S)-2-(5-chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]-
- Originator Actelion Pharmaceuticals
- Developer Idorsia Pharmaceuticals
- Class Sleep disorder therapies
- Mechanism of Action Orexin receptor type 1 antagonists; Orexin receptor type 2 antagonists
- Phase III Insomnia
- 19 Oct 2018 Idorsia Pharmaceuticals plans a phase I trial for Liver disorders (Hepatic impairment) in November 2018 (PO) (NCT03713242)
- 09 Oct 2018 Idorsia Pharmaceuticals completes a phase I trial in Insomnia (In volunteers) in Netherlands (PO) (NCT03609775)
- 27 Sep 2018 Idorsia Pharmaceuticals plans a phase I trial for Hepatic impairment in November 2018 , (NCT03686995)
Nemorexant (developmental code name ACT-541468) is a dual orexin receptor antagonist (DORA) which was originated by Actelion Pharmaceuticals and is under development by Idorsia Pharmaceuticals for the treatment of insomnia.[1][2] It acts as a selective dual antagonist of the orexin receptors OX1 and OX2.[1][2] As of June 2018, nemorexant is in phase III clinical trials for the treatment of insomnia.[1]
Idorsia is developing nemorexant, a dual orexin receptor antagonist (DORA), for the oral treatment of insomnia and investigating the program for the treatment of COPD. In May 2018, a phase III study was initiated in subjects with insomnia disorder and in September 2018, a phase I trial was initiated in COPD.
SCHEME
SEE AT END OF PAGE
PATENT
WO2013182972 ,
PATENT
WO2015083094 ,
Patent
WO 2015083070

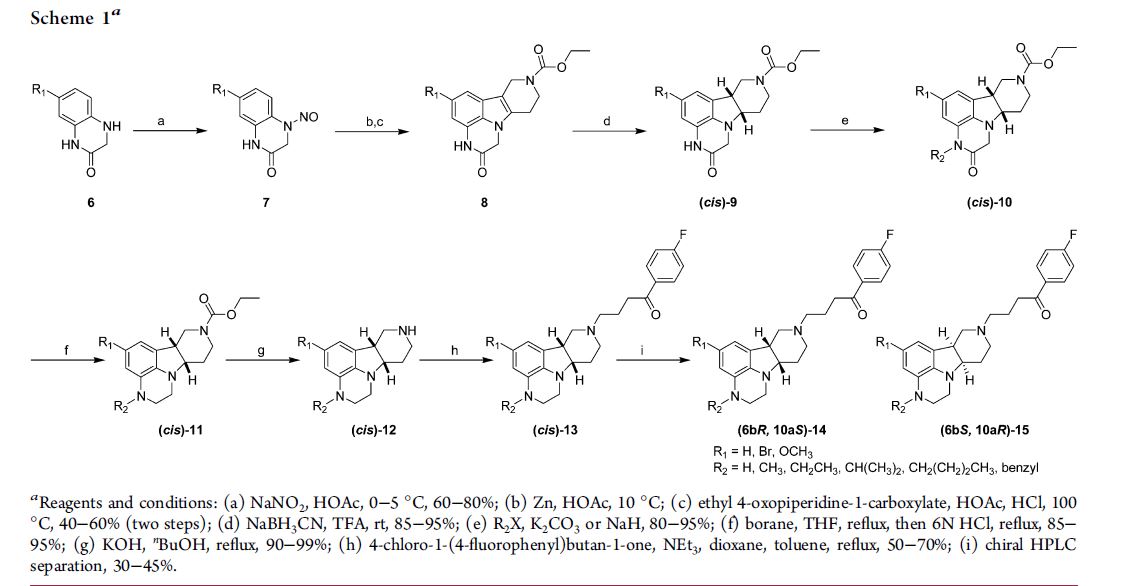
Synthesis of nemorexant, using 2-methyl-L-proline hydrochloride as the starting material
N-Protection of 2-methyl-L-proline hydrochloride with Boc2O gives N-Boc-2-methyl-L-proline,
Which upon condensation with 4-chloro-3-methylbenzene-1,2-diamine using HATU and DIEA in CH2Cl2 affords the corresponding amide.
Cyclization of diamine in the presence of AcOH at 100 °C provides imidazole derivative,
Whose Boc moiety is removed by means of HCl in dioxane to yield 5-chloro-4-methyl-2-[2(S)-methylpyrrolidin-2-yl]benzimidazole hydrochloride.
N-Acylation of pyrrolidine derivative with 5-methoxy-2-(1,2,3-triazol-2-yl)benzoic acid using HATU and DIEA in CH2Cl2 produces Nemorexant
5-methoxy-2-(1,2,3-triazol-2-yl)benzoic acid (prepared by the coupling of 2-iodo-5-methoxybenzoic acid with 1,2,3-triazole using CuI and Cs2CO3 in DMF)
PATENT
WO 2016020403
PATENT
WO 2015083071
Reference Example 1
1) Synthesis of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-lodo-5-methoxy benzoic acid (15.0 g; 53.9 mmol) is dissolved in anhydrous DMF (45 ml) followed by the addition of 1 H-1 ,2,3-triazole (7.452 g; 108 mmol) and cesium carbonate (35.155 g; 108 mmol). By the addition of cesium carbonate the temperature of the reaction mixture increases to 40°C and gas evolved from the reaction mixture. Copper(l)iodide (514 mg; 2.7 mmol) is added. This triggers a strongly exothermic reaction and the temperature of the reaction mixture reaches 70°C within a few seconds. Stirring is continued for 30 minutes. Then the DMF is evaporated under reduced pressure followed by the addition of water (170 ml) and EtOAc (90 ml). The mixture is vigorously stirred and by the addition of citric acid monohydrate the pH is adjusted to 3-4. The precipitate is filtered off and washed with water and EtOAc and discarded. The filtrate is poured into a separation funnel and the phases are separated. The water phase is extracted again with EtOAc. The combined organic layers are dried over MgS04, filtered and the solvent is evaporated to give 7.1 g of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid as a white powder of 94% purity (6 % impurity is the regioisomerically N1-linked triazolo-derivative); tR [min] = 0.60; [M+H]+ = 220.21
2) Synthesis of (S)-1 -(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid
2-Methyl-L-proline hydrochloride (99.7 g; 602 mmol) is dissolved in a 1/1-mixture of MeCN and water (800 ml) and triethylamine (254 ml; 1810 mmol) is added. The temperature of the reaction mixture slightly rises. The reaction mixture is cooled to 10°C to 15°C followed by careful addition of a solution of Boc20 (145 g; 662 mmol) in MeCN (200 ml) over 10 minutes.
Stirring at RT is continued for 2 hours. The MeCN is evaporated under reduced pressure and aq. NaOH solution (2M; 250 ml) is added to the residual aq. part of the reaction mixture. The water layer is washed with Et20 (2x 300 ml) then cooled to 0°C followed by slow and careful addition of aq. HCI (25%) to adjust the pH to 2. During this procedure a suspension forms.
The precipitate is filtered off and dried at HV to give 1 10.9 g of the title compound as a beige powder; tR [min] = 0.68; [M+H]+ = 230.14
3) Synthesis of (S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-
(S)-1-(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid (60 g; 262 mmol) and HATU (100 g; 264 mmol) is suspended in DCM (600 ml) followed by the addition of DIPEA (84.6 g; 654 mmol) and 6-chloro-2,3-diaminotoluene (41 g; 262 mmol). The reaction mixture is stirred at rt for 14 hours then concentrated under reduced pressure and to the residue is added water followed by the extraction of the product with EtOAc (3x). The combined organic layers are washed with brine, dried over MgS04, filtered and the solvent is evaporated under
reduced pressure to give 185 g of the title compound as a dark brownish oil, which is used in the next step without further purification; tR [min] = 0.89; [M+H]+ = 368.01
4) Synthesis of (S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1 -carboxylate
(S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-methylpyrrolidine-1-carboxylate (185 g; 427 mmol) are dissolved in AcOH (100%; 611 ml), heated to 100°C and stirring continued for 90 minutes. The AcOH is evaporated under reduced pressure and the residue is dissolved in DCM followed by careful addition of saturated sodium bicarbonate solution. The phases are separated, the aq. phase is extracted once more with DCM, the combined aq. phases are dried over MgS04, filtered and the solvent is evaporated under reduced pressure to give 142.92 g of the title compound as a dark brown oil which is used in the next step without further purification; tR [min] = 0.69; [M+H]+ = 350.04
5) Synthesis of (S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride
(S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1-carboxylate (355.53 g; 1.02 mol) are dissolved in dioxane (750 ml) followed by careful addition of HCI solution in dioxane (4M; 750 ml; 3.05 mol). The reaction mixture is stirred for 3 hours followed by the addition of Et20 (800 ml) which triggered precipitation of the product. The solid is filtered off and dried at high vacuum to give 298.84 g of the title compound as a redish powder; tR [min] = 0.59; [M+H]+ = 250.23
6) Synthesis of [(S)-2-(5-chloro-4-methyl-1 H-benzoimidazol-2-yl)-2-methyl-pyrrolidin-1- -(5-methoxy-2-[1,2,3]triazol-2-yl-phenyl)-methanone
(S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride (62.8 g; 121 mmol) is dissolved in DCM (750 ml) followed by the addition of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (62.8 g; 121 mmol) and DIPEA (103 ml; 603 mmol). Stirring is continued for 10 minutes followed by the addition of HATU (47 g; 124 mmol). The reaction mixture is stirred for 16 hours at RT. The solvents are evaporated under reduced pressure and the residue is dissolved in EtOAc (1000 ml) and washed with water (3x 750 ml). The organic phase is dried over MgS04, filtered and the solvent is evaporated under reduced pressure. The residue is purified by CC with EtOAc / hexane = 2 / 1to give 36.68 g of the title compound as an amorphous white powder. tR [min] = 0.73; [M+H]+ = 450.96
Table 1 : Characterisation data for COMPOUND as free base in amorphous form
II. Preparation of crystalline forms of COMPOUND
Example 1 :
Preparation of seeding material of COMPOUND hydrochloride in crystalline Form 1
10 mg COMPOUND is mixed with 0.2 mL 0.1 M aq. HCI and 0.8 mL EtOH. The solvent is fully evaporated and 0.05 mL isopropanol is added. Alternatively 0.05 mL methyl-isobutylketone can be added. The sample is stored closed at room temperature for 4 days and crystalline material of COMPOUND hydrochloride in crystalline Form 1 is obtained. This material can be used as seeding material for further crystallization of COMPOUND hydrochloride in crystalline Form 1.
Example 2: Preparation and characterization of COMPOUND hydrochloride in crystalline form 1
5g COMPOUND is mixed with 0.9 mL 1 M aq. HCI and 20 mL EtOH. The solvent is evaporated and 25 mL isopropanol is added. Seeds of COMPOUND hydrochloride are added and the sample is allowed to stand at room temperature. After about 2 days the suspension is filtered and the solid residue is dried at reduced pressure (2 mbar for 1 hour) and allowed to equilibrate open for 2 hours at 24°C/46% relative humidity. The obtained solid is COMPOUND hydrochloride in crystalline Form 1
Table 2: Characterisation data for COMPOUND hydrochloride in crystalline form 1
PATENT
WO-2018202689
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018202689&tab=PCTDESCRIPTION&maxRec=1000
Process for the preparation of a crystalline potassium salt of a 2-(2H-[1,2,3]triazol-2-yl)-benzoic acid derivatives is claimed. Compound is disclosed to be useful for the preparation of pharmaceuticals, especially certain orexin receptor antagonists such as nemorexant .
References
- ^ Jump up to:a b c https://adisinsight.springer.com/drugs/800044843
- ^ Jump up to:a b Equihua-Benítez AC, Guzmán-Vásquez K, Drucker-Colín R (July 2017). “Understanding sleep-wake mechanisms and drug discovery”. Expert Opin Drug Discov. 12 (7): 643–657. doi:10.1080/17460441.2017.1329818. PMID 28511597.
- Muehlan, C.; Heuberger, J.; Juif, P.E.; Croft, M.; van Gerven, J.; Dingemanse, J.
Accelerated development of the dual orexin receptor antagonist ACT-541468: Integration of a microtracer in a first-in-human study
Clin Pharmacol Ther 2018, 104(5): 1022 - A Study to Evaluate the Pharmacokinetics of ACT-541468 in Subjects With Mild, Moderate and Severe Hepatic Impairment (NCT03713242)
ClinicalTrials.gov Web Site 2018, October 24 - Boof, M.-.L.; Ufer, M.; Halabi, A.; Dingemanse, J.
Impact of the dual orexin receptor antagonist ACT-541468 on the pharmacokinetics of the CYP3A4 probe drug midazolam and assessment of the effect of food on ACT-541468
119th Annu Meet Am Soc Clin Pharmacol Ther (ASCPT) (March 21-24, Orlando) 2018, Abst PI-043 - Muehlan, C.; Brooks, S.; Zuiker, R.; van Gerven, J.; Dingemanse, J.
Night-time administration of ACT-541468, a novel dual orexin receptor antagonist: Characterization of its pharmacokinetics, next-day residual effects, safety, and tolerability
32nd Annu Meet Assoc Sleep Soc (SLEEP) (June 2-6, Baltimore) 2018, Abst 0008 - Proposed international nonproprietary names (Prop. INN): List 118
WHO Drug Inf 2017, 31(4): 635
External links
from PubChem
 |
|
| Clinical data | |
|---|---|
| Synonyms | ACT-541468 |
| Routes of administration |
By mouth |
| Drug class | Orexin antagonist |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C23H23ClN6O2 |
| Molar mass | 450.927 g/mol |
| 3D model (JSmol) | |
///////////////Nemorexant, ACT-541468, Phase III, Insomnia

Eflornithine, эфлорнитин , إيفلورنيثين , 依氟鸟氨酸 , エフロルニチン

Eflornithine
Ornithine, 2-(difluoromethyl)-
UNII:ZQN1G5V6SR
ZQN1G5V6SR
эфлорнитин [Russian] [INN]
إيفلورنيثين [Arabic] [INN]
依氟鸟氨酸 [Chinese] [INN]
エフロルニチン
(RS)-2,5-diamino-2-(difluoromethyl)pentanoic acid
- Use:hirsutism treatment inhibitor of ornithine decarboxylase
- Chemical name:2-(difluoromethyl)-dl-ornithine
- Formula:C6H12F2N2O2, MW:182.17 g/mol
- CAS-RN:67037-37-0
- LD50:>3000 mg/kg (M, i.p.); >5000 mg/kg (M, p.o.);
1364 μg/kg (R, intracerebral)
Eflornithine, also known as α-difluoromethylornithine (DFMO), is an Active Pharmaceutical Ingredient (API) on the World Health Organization’s list of essential medicines. DFMO is used to treat the second stage of African trypanosomiasis (sleeping sickness). In addition, DFMO is also used to treat opportunistic infections with Pneumocystis carinii pneumonia, a form of pneumonia found in people with a weak immune system suffering from conditions such as acquired immunodeficiency syndrome (AIDS) It has also been explored as chemopreventive agent in cancer therapy with minor success. Today, its main use is to treat excessive facial hair growth on women (hirsutism). The topical cream (Vaniqa) significantly reduces the psychological burden of those affected.\
Eflornithine is a prescription drug indicated in the treatment of facial hirsutism (excessive hair growth). Eflornithine hydrochloride cream for topical application is intended for use in women suffering from facial hirsutism and is sold by Allergan, Inc. under the brand name Vaniqa. Besides being a non-mechanical and non-cosmetic treatment, eflornithine is the only non-hormonal and non-systemic prescription option available for women who suffer from facial hirsutism. Eflornithine for injection against sleeping sickness was manufactured by Sanofi Aventis and sold under the brand name Ornidyl in the USA. It is now discontinued. Eflornithine is on the World Health Organization’s List of Essential Medicines.
Derivatives
Monohydrochloride
- Formula:C6H12F2N2O2 • HCl
- MW:218.63 g/mol
- CAS-RN:68278-23-9
- EINECS:269-532-0
Monohydrochloride monohydrate
- Formula:C6H12F2N2O2 • HCl • H2O
- MW:236.65 g/mol
- CAS-RN:96020-91-6
Eflornithine, sold under the brand name Vaniqa among others, is a medication used to treat African trypanosomiasis (sleeping sickness) and excessive hair growth on the face in women.[1][2] Specifically it is used for the 2nd stage of sleeping sickness caused by T. b. gambiense and may be used with nifurtimox.[1][3] It is used by injection or applied to the skin.[1][2]
Common side effects when applied as a cream include rash, redness, and burning.[2] Side effects of the injectable form include bone marrow suppression, vomiting, and seizures.[3] It is unclear if it is safe to use during pregnancy or breastfeeding.[3] It is recommended typically for children over the age of 12.[3]
Eflornithine was developed in the 1970s and came into medical use in 1990.[4] It is on the World Health Organization’s List of Essential Medicines, the most effective and safe medicines needed in a health system.[5] There is no generic version as of 2015 in the United States.[6] In the United States the injectable form can be obtained from the Centers for Disease Control and Prevention.[3] In the 1990s the cost of a course of treatment in Africa was 210 USD.[7] In regions of the world where the disease is common eflornithine is provided for free by the World Health Organization.[8]
https://www.google.com/patents/US4330559
Medical uses
Sleeping sickness
Sleeping sickness, or trypanosomiasis, is treated with pentamidine or suramin (depending on subspecies of parasite) delivered by intramuscular injection in the first phase of the disease, and with melarsoprol and eflornithine intravenous injection in the second phase of the disease. Efornithine is commonly given in combination with nifurtimox, which reduces the treatment time to 7 days of eflornithine infusions plus 10 days of oral nifurtimox tablets.[9]
Eflornithine is also effective in combination with other drugs, such as melarsoprol and nifurtimox. A study in 2005 compared the safety of eflornithine alone to melarsoprol and found eflornithine to be more effective and safe in treating second-stage sleeping sickness Trypanosoma brucei gambiense.[10] Eflornithine is not effective in the treatment of Trypanosoma brucei rhodesiense due to the parasite’s low sensitivity to the drug. Instead, melarsoprol is used to treat Trypanosoma brucei rhodesiense.[11] Another randomized control trial in Uganda compared the efficacy of various combinations of these drugs and found that the nifurtimox-eflornithine combination was the most promising first-line theory regimen.[12]
A randomized control trial was conducted in Congo, Côte d’Ivoire, the Democratic Republic of the Congo, and Uganda to determine if a 7-day intravenous regimen was as efficient as the standard 14-day regimen for new and relapsing cases. The results showed that the shortened regimen was efficacious in relapse cases, but was inferior to the standard regimen for new cases of the disease.[13]
Nifurtimox-eflornithine combination treatment (NECT) is an effective regimen for the treatment of second stage gambiense African trypanosomiasis.[14][15]
Trypanosome resistance
After its introduction to the market in the 1980s, eflornithine has replaced melarsoprol as the first line medication against Human African trypanosomiasis (HAT) due to its reduced toxicity to the host.[13] Trypanosoma brucei resistant to eflornithine has been reported as early as the mid-1980s.[13]
The gene TbAAT6, conserved in the genome of Trypanosomes, is believed to be responsible for the transmembrane transporter that brings eflornithine into the cell.[16] The loss of this gene due to specific mutations causes resistance to eflornithine in several trypanosomes.[17] If eflornithine is prescribed to a patient with Human African trypanosomiasis caused by a trypanosome that contains a mutated or ineffective TbAAT6 gene, then the medication will be ineffective against the disease. Resistance to eflornithine has increased the use of melarsoprol despite its toxicity, which has been linked to the deaths of 5% of recipient HAT patients.[13]
Excess facial hair in women
The topical cream is indicated for treatment of facial hirsutism in women.[18] It is the only topical prescription treatment that slows the growth of facial hair.[19] It is applied in a thin layer twice daily, a minimum of eight hours between applications. In clinical studies with Vaniqa, 81% percent of women showed clinical improvement after twelve months of treatment.[20] Positive results were seen after eight weeks.[21] However, discontinuation of the cream caused regrowth of hair back to baseline levels within 8 weeks.[22]
Vaniqa treatment significantly reduces the psychological burden of facial hirsutism.[23]
Chemo preventative therapy
It has been noted that ornithine decarboxylase (ODC) exhibits high activity in tumor cells, promoting cell growth and division, while absence of ODC activity leads to depletion of putrescine, causing impairment of RNA and DNA synthesis. Typically, drugs that inhibit cell growth are considered candidates for cancer therapy, so eflornithine was naturally believed to have potential utility as an anti-cancer agent. By inhibiting ODC, eflornithine inhibits cell growth and division of both cancerous and noncancerous cells.
However, several clinical trials demonstrated minor results.[24] It was found that inhibition of ODC by eflornithine does not kill proliferating cells, making eflornithine ineffective as a chemotherapeutic agent. The inhibition of the formation of polyamines by ODC activity can be ameliorated by dietary and bacterial means because high concentrations are found in cheese, red meat, and some intestinal bacteria, providing reserves if ODC is inhibited.[25] Although the role of polyamines in carcinogenesis is still unclear, polyamine synthesis has been supported to be more of a causative agent rather than an associative effect in cancer.[24]
Other studies have suggested that eflornithine can still aid in some chemoprevention by lowering polyamine levels in colorectal mucosa, with additional strong preclinical evidence available for application of eflornithine in colorectal and skin carcinogenesis.[24][25] This has made eflornithine a supported chemopreventive therapy specifically for colon cancer in combination with other medications. Several additional studies have found that eflornithine in combination with other compounds decreases the carcinogen concentrations of ethylnitrosourea, dimethylhydrazine, azoxymethane, methylnitrosourea, and hydroxybutylnitrosamine in the brain, spinal cord, intestine, mammary gland, and urinary bladder.[25]
Contraindications
Topical
Topical use is contraindicated in people hypersensitive to eflornithine or to any of the excipients.[26]
Throughout clinical trials, data from a limited number of exposed pregnancies indicate that there is no clinical evidence that treatment with Vaniqa adversely affects pregnant women or fetuses.[26]
By mouth
When taken by mouth the risk-benefit should be assessed in people with impaired renal function or pre-existing hematologic abnormalities, as well as those with eighth-cranial-nerve impairment.[27] Adequate and well-controlled studies with eflornithine have not been performed regarding pregnancy in humans. Eflornithine should only be used during pregnancy if the potential benefit outweighs the potential risk to the fetus. However, since African trypanosomiasis has a high mortality rate if left untreated, treatment with eflornithine may justify any potential risk to the fetus.[27]
Side effects
Eflornithine is not genotoxic; no tumour-inducing effects have been observed in carcinogenicity studies, including one photocarcinogenicity study.[28] No teratogenic effects have been detected.[29]
Topical
The topical form of elflornithine is sold under the brand name Vaniqa . The most frequently reported side effect is acne (7–14%). Other side effects commonly (> 1%) reported are skin problems, such as skin reactions from in-growing hair, hair loss, burning, stinging or tingling sensations, dry skin, itching, redness or rash.[30]
Intravenous
The intravenous dosage form of eflornithine is sold under the brand name Ornidyl. Most side effects related to systemic use through injection are transient and reversible by discontinuing the drug or decreasing the dose. Hematologic abnormalities occur frequently, ranging from 10–55%. These abnormalities are dose-related and are usually reversible. Thrombocytopenia is thought to be due to a production defect rather than to peripheral destruction. Seizures were seen in approximately 8% of patients, but may be related to the disease state rather than the drug. Reversible hearing loss has occurred in 30–70% of patients receiving long-term therapy (more than 4–8 weeks of therapy or a total dose of >300 grams); high-frequency hearing is lost first, followed by middle- and low-frequency hearing. Because treatment for African trypanosomiasis is short-term, patients are unlikely to experience hearing loss.[30]
Interactions
Topical
No interaction studies with the topical form have been performed.[26]
Mechanism of action

Figure 1
(A) 3D structure of L-Ornithine
(B) 3D structure of Eflornithine. This molecule is similar to the structure of L-Ornithine, but its alpha-difluoromethyl group allows interaction with Cys-360 in the active site

Eflornithine ODC Reaction Mechanism
Description
Eflornithine is a “suicide inhibitor,” irreversibly binding to ornithine decarboxylase (ODC) and preventing the natural substrate ornithine from accessing the active site (Figure 1). Within the active site of ODC, eflornithine undergoes decarboxylation with the aid of cofactor pyridoxal 5’-phosphate (PLP). Because of its additional difluoromethyl group in comparison to ornithine, eflornithine is able to bind to a neighboring Cys-360 residue, permanently remaining fixated within the active site.[29]
During the reaction, eflornithine’s decarboxylation mechanism is analogous to that of ornithine in the active site, where transamination occurs with PLP followed by decarboxylation. During the event of decarboxylation, the fluoride atoms attached to the additional methyl group pull the resulting negative charge from the release of carbon dioxide, causing a fluoride ion to be released. In the natural substrate of ODC, the ring of PLP accepts the electrons that result from the release of CO2.
The remaining fluoride atom that resides attached to the additional methyl group creates an electrophilic carbon that is attacked by the nearby thiol group of Cys-360, allowing eflornithine to remain permanently attached to the enzyme following the release of the second fluoride atom and transimination.
Evidence

Figure 2
Experimental Evidence for Eflornithine End Product[31]
The reaction mechanism of Trypanosoma brucei‘s ODC with ornithine was characterized by UV-VIS spectroscopy in order to identify unique intermediates that occurred during the reaction. The specific method of multiwavelength stopped-flow spectroscopy utilized monochromatic light and fluorescence to identify five specific intermediates due to changes in absorbance measurements.[32] The steady-state turnover number, kcat, of ODC was calculated to be 0.5 s-1 at 4 °C.[32] From this characterization, the rate-limiting step was determined to be the release of the product putrescine from ODC’s reaction with ornithine. In studying the hypothetical reaction mechanism for eflornithine, information collected from radioactive peptide and eflornithine mapping, high pressure liquid chromatography, and gas phase peptide sequencing suggested that Lys-69 and Cys-360 are covalently bound to eflornithine in T. brucei ODC’s active site.[31] Utilizing fast-atom bombardment mass spectrometry (FAB-MS), the structural conformation of eflornithine following its interaction with ODC was determined to be S-((2-(1-pyrroline-methyl) cysteine, a cyclic imine adduct. Presence of this particular product was supported by the possibility to further reduce the end product to S-((2-pyrrole) methyl) cysteine in the presence of NaBH4 and oxidize the end product to S-((2-pyrrolidine) methyl) cysteine (Figure 2).[31]
Active site

Figure 3
Active Site of ODC Formed by Homodimerization (Green and White Surface Structures)
(A) Ornithine in the Active Site of ODC, Cys-360 highlighted in yellow
(B) Product of Eflornithine Decarboxylation bound to Cys 360 (highlighted in yellow). The pyrroline ring blocks ornithine from entering the active site
Derived from Grishin, Nick V., et al. “X-ray structure of ornithine decarboxylase from Trypanosoma brucei: the native structure and the structure in complex with α-difluoromethylornithine.” Biochemistry 38.46 (1999): 15174-15184. PDB ID: 2TOD
Eflornithine’s suicide inhibition of ODC physically blocks the natural substrate ornithine from accessing the active site of the enzyme (Figure 3).[29] There are two distinct active sites formed by the homodimerization of ornithine decarboxylase. The size of the opening to the active site is approximately 13.6 Å. When these openings to the active site are blocked, there are no other ways through which ornithine can enter the active site. During the intermediate stage of eflornithine with PLP, its position near Cys-360 allows an interaction to occur. As the phosphate of PLP is stabilized by Arg 277 and a Gly-rich loop (235-237), the difluoromethyl group of eflornithine is able to interact and remain fixated to both Cys-360 and PLP prior to transimination. As shown in the figure, the pyrroline ring interferes with ornithine’s entry (Figure 4). Eflornithine will remain permanently bound in this position to Cys-360. As ODC has two active sites, two eflornithine molecules are required to completely inhibit ODC from ornithine decarboxylation.
History
Eflornithine was initially developed for cancer treatment at Merrell Dow Research Institute in the late 1970s, but was found to be ineffective in treating malignancies. However, it was discovered to be highly effective in reducing hair growth,[33] as well as in the treatment of African trypanosomiasis (sleeping sickness),[34] especially the West African form (Trypanosoma brucei gambiense).
Hirsutism[]
In the 1980s, Gillette was awarded a patent for the discovery that topical application of eflornithine HCl cream inhibits hair growth. In the 1990s, Gillette conducted dose-ranging studies with eflornithine in hirsute women that demonstrated that the drug slows the rate of facial hair growth. Gillette then filed a patent for the formulation of eflornithine cream. In July 2000, the U.S. Food and Drug Administration (FDA) granted a New Drug Application for Vaniqa. The following year, the European Commission issued its Marketing Authorisation.
Sleeping sickness treatment
The drug was registered for the treatment of gambiense sleeping sickness on November 28, 1990.[35] However, in 1995 Aventis (now Sanofi-Aventis) stopped producing the drug, whose main market was African countries, because it did not make a profit.[36]
In 2001, Aventis and the WHO formed a five-year partnership, during which more than 320,000 vials of pentamidine, over 420,000 vials of melarsoprol, and over 200,000 bottles of eflornithine were produced by Aventis, to be given to the WHO and distributed by the association Médecins sans Frontières (also known as Doctors Without Borders)[37][38] in countries where sleeping sickness is endemic.
According to Médecins sans Frontières, this only happened after “years of international pressure,” and coinciding with the period when media attention was generated because of the launch of another eflornithine-based product (Vaniqa, for the prevention of facial-hair in women),[36]while its life-saving formulation (for sleeping sickness) was not being produced.
From 2001 (when production was restarted) through 2006, 14 million diagnoses were made. This greatly contributed to stemming the spread of sleeping sickness, and to saving nearly 110,000 lives.
Society and culture
Available forms
Vaniqa is a cream, which is white to off-white in colour. It is supplied in tubes of 30 g and 60 g in Europe.[30] Vaniqa contains 15% w/w eflornithine hydrochloride monohydrate, corresponding to 11.5% w/w anhydrous eflornithine (EU), respectively 13.9% w/w anhydrous eflornithine hydrochloride (U.S.), in a cream for topical administration.
Ornidyl, intended for injection, was supplied in the strength of 200 mg eflornithine hydrochloride per ml.[39]
Cost
In 2000, the cost for the 14-day regimen was US $500; a price that many in countries where the disease is common cannot afford.[13]
Market
Vaniqa, granted marketing approval by the US FDA, as well as by the European Commission[40] among others, is currently the only topical prescription treatment that slows the growth of facial hair.[19] Besides being a non-mechanical and non-cosmetic treatment, it is the only non-hormonal and non-systemic prescription option available for women who suffer from facial hirsutism.[18] Vaniqa is marketed by Almirall in Europe, SkinMedica in the USA, Triton in Canada, Medison in Israel, and Menarini in Australia.[40]
Ornidyl, the injectable form of eflornithine hydrochloride, is licensed by Sanofi-Aventis, but is currently discontinued in the US.[41]
Clip
Scalable Continuous Flow Process for the Synthesis of Eflornithine Using Fluoroform as Difluoromethyl Source
Manuel Köckinger†‡, Christopher A. Hone†‡, Bernhard Gutmann§, Paul Hanselmann§, Michael Bersier§, Ana Torvisco∥, and C. Oliver Kappe*†‡
† Center for Continuous Flow Synthesis and Processing (CC FLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria
‡ Institute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, A-8010 Graz, Austria
§ Microreactor Technology, Lonza AG, CH-3930 Visp, Switzerland
∥ Institute of Inorganic Chemistry, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.8b00318

The development of a scalable telescoped continuous flow procedure for difluoromethylation of a protected amino acid with fluoroform (CHF3, R-23) gas and subsequent high temperature deprotection to provide eflornithine, an important Active Pharmaceutical Ingredient (API), is described. Eflornithine is used for the treatment of sleeping sickness and hirsutism, and it is on the World Health Organization’s list of essential medicines. Fluoroform is produced in large quantities as a side product in the manufacture of polytetrafluoroethylene (PTFE, Teflon). Fluoroform is an ozone-benign and nontoxic gas, but its release into the environment is forbidden under the Kyoto protocol owing to its high global warming potential. The existing manufacturing route to eflornithine uses chlorodifluoromethane (CHClF2, R-22) which will be phased out under the Montreal protocol; therefore, the use of the fluoroform presents a viable cost-effective and more sustainable alternative. The process parameters and equipment setup were optimized on laboratory scale for the two reaction steps to improve product yield and scalability. The telescoped flow process utilizing fluoroform gas was operated for 4 h to afford the target molecule in 86% isolated yield over two steps with a throughput of 24 mmol/h.
1hydrochloride monohydrate as colorless powder. (17.05 g, 72.3 mmol, 86% yield). Mp. 228 °C;
1H NMR (300.36 MHz, D2O): δ = 6.46 (t, 2JHF = 52.8 Hz, 1H), 3.05 (t,3JHH = 7.6 Hz, 2H), 2.25–1.97 (m, 2H), 1.96–1.79 (m, 1H), 1.76–1.59 (m, 1H) ppm.
13C NMR (75 MHz, D2O): δ = 167.8 (d, 3JCF = 6.4 Hz), 114.0 (dd, 1JCF = 249.7 Hz, 1JCF = 247.0 Hz), 64.5 (dd, 2JCF = 20.4 Hz, 2JCF = 18.7 Hz), 38.8 (d, 3JCF = 7.3 Hz), 31.6 (d, 4JCF = 3.2 Hz), 20.8 ppm.
19F NMR (282 MHz, D2O): δ = −126.28 (dd, 2JFF = 283.5 Hz, 2JHF = 52.4 Hz), – 131.76 (dd, 2JFF = 283.5 Hz, 2JHF = 52.4 Hz) ppm.
References
- ^ Jump up to:a b c “19th WHO Model List of Essential Medicines (April 2015)” (PDF). WHO. April 2015. Archived (PDF) from the original on May 13, 2015. Retrieved May 10, 2015.
- ^ Jump up to:a b c “Eflornithine”. The American Society of Health-System Pharmacists. Archivedfrom the original on 20 December 2016. Retrieved 28 November 2016.
- ^ Jump up to:a b c d e “CDC – African Trypanosomiasis – Resources for Health Professionals”. http://www.cdc.gov. 10 August 2016. Archived from the original on 28 November 2016. Retrieved 6 December 2016.
- Jump up^ Marcondes, Carlos Brisola (2016). Arthropod Borne Diseases. Springer. p. 292. ISBN 9783319138848. Archived from the original on 2017-09-10.
- Jump up^ “WHO Model List of Essential Medicines (19th List)” (PDF). World Health Organization. April 2015. Archived (PDF) from the original on 13 December 2016. Retrieved 8 December 2016.
- Jump up^ Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 192. ISBN 9781284057560.
- Jump up^ Grayson, M. Lindsay; Crowe, Suzanne M.; McCarthy, James S.; Mills, John; Mouton, Johan W.; Norrby, S. Ragnar; Paterson, David L.; Pfaller, Michael A. (2010). Kucers’ The Use of Antibiotics Sixth Edition: A Clinical Review of Antibacterial, Antifungal and Antiviral Drugs. CRC Press. p. 2194. ISBN 9781444147520. Archived from the original on 2017-09-10.
- Jump up^ “Trypanosomiasis, human African (sleeping sickness)”. World Health Organization. February 2016. Archived from the original on 4 December 2016. Retrieved 7 December2016.
- Jump up^ Babokhov P; et al. (2013). “A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis”. Pathog Glob Health. 107 (5): 242–52. doi:10.1179/2047773213Y.0000000105. PMC 4001453. PMID 23916333.
- Jump up^ Priotto, Gerardo; et al. (December 2006). “Three drug combinations for late-stageTrypanosoma brucei gambiense sleeping sickness: a randomized clinical trial in Uganda”. PLoS Clinical Trials. 1 (8): e39. doi:10.1371/journal.pctr.0010039. PMC 1687208. PMID 17160135.
- Jump up^ Lutje, Vittoria; Seixas, Jorge; Kennedy, Adrian (2013-06-28). “Cochrane Database of Systematic Reviews”. Cochrane Database of Systematic Reviews (6): CD006201. doi:10.1002/14651858.cd006201.pub3. PMID 23807762.
- Jump up^ Chappuis F, et al. (2005). “Eflornithine is safer than melarsoprol for the treatment of second-stage Trypanosoma brucei gambiense human African trypanosomiasis”. Clinical Infectious Diseases. 41 (5): 748–751. doi:10.1086/432576. PMID 16080099.
- ^ Jump up to:a b c d e Vincent, Isabel M.; et al. (November 2010). “A molecular mechanism for eflornithine resistance in African trypanosomes”. PLoS Pathogens. 6 (11): e1001204. doi:10.1371/journal.ppat.1001204. PMC 2991269. PMID 21124824.
- Jump up^ “Nifurtimox-eflornithine combination treatment for sleeping sickness (human African trypanosomiasis): WHO wraps up training of key health care personnel”. World Health Organization. March 23, 2010.
- Jump up^ Franco, Jose; Pere, Simarro; Diarra; Ruiz-Postigo; Samo; Jannin (2012). “Monitoring the use of nifurtimox-eflornithine combination therapy (NECT) in the treatment of second stage gambiense human African trypanosomiasis” (PDF). Research and Reports in Tropical Medicine. 3: 93–101. doi:10.2147/RRTM.S34399. PMC 6067772. PMID 30100776.
- Jump up^ Sayé M, et al. (2014). “Proline Modulates the Trypanosoma cruzi Resistance to Reactive Oxygen Species and Drugs through a Novel D, L-Proline Transporter”. PLoS ONE. 9 (3): e92028. Bibcode:2014PLoSO…992028S. doi:10.1371/journal.pone.0092028. PMC 3956872. PMID 24637744.
- Jump up^ Barrett, M. P., et al. “Human African trypanosomiasis: pharmacological re‐engagement with a neglected disease.” British Journal of Pharmacology 152.8 (2007): 1155-1171.
- ^ Jump up to:a b “NHS and UKMi New Medicines Profile” (PDF). Archived from the original (PDF)on 2010-02-15.
- ^ Jump up to:a b Balfour JA, McClellan K (June 2001). “Topical Eflornithine”. Am J Clin Dermatol. 2 (3): 197–201. doi:10.2165/00128071-200102030-00009. PMID 11705097.
- Jump up^ Schrode K; Huber F; Staszak J; Altman DJ. “Evaluation of the long-term safety of eflornithine 15% cream in the treatment of women with excessive facial hair. Presented at 58th Annual Meeting of the Academy of Dermatology 2000, 10–15 March, San Francisco; USA, Poster 294”. the Eflornithine Study Group.
- Jump up^ Schrode K, Huber F; Staszak, J; Altman DJ, Shander D & Morton J, the Eflornithine Study Group. “Randomized, double-blind, vehicle-controlled safety and efficacy evaluation of eflornithine 15% cream in the treatment of women with excessive facial hair. Presented at 58th Annual Meeting of the Academy of Dermatology 2000, 10–15 March, San Francisco; USA, Poster 291”.
- Jump up^ Wolf, John E.; Shander, Douglas; Huber, Ferdinand; Jackson, Joseph; Lin, Chen-Sheng; Mathes, Barbara M.; Schrode, Kathy; the Eflornithine HCl Study Group (2007-01-01). “Randomized, double-blind clinical evaluation of the efficacy and safety of topical eflornithine HCl 13.9% cream in the treatment of women with facial hair”. International Journal of Dermatology. 46 (1): 94–98. doi:10.1111/j.1365-4632.2006.03079.x. ISSN 1365-4632. PMID 17214730.
- Jump up^ Jackson J, Caro JJ; Caro G, Garfield F; Huber F, Zhou W; Lin CS, Shander D & Schrode K. “The effect of eflornithine 13.9% cream on the bother and discomfort due to hirsutism. Int J Derm 2007; 46: 976-981”. the Eflornithine HCl Study Group.
- ^ Jump up to:a b c Paul, F. “Revival of 2-(difluoromethyl) ornithine (DFMO), an inhibitor of polyamine biosynthesis, as a cancer chemopreventive agent.” Biochemical Society Transactions 35.Pt 2 (2007): 353-355.
- ^ Jump up to:a b c Gerner EW, Meyskens FL (2004). “Polyamines and cancer: old molecules, new understanding” (Submitted manuscript). Nature Reviews Cancer. 4 (10): 781–792. doi:10.1038/nrc1454. PMID 15510159.
- ^ Jump up to:a b c “Vaniqa Summary of Product Characteristics 2008”. Archived from the original on 2009-12-05.
- ^ Jump up to:a b “Ornidyl Drug Information”. Archived from the original on 2011-06-07.
- Jump up^ Malhotra B, Noveck R, Behr D, Palmisano M (September 2001). “Percutaneous absorption and pharmacokinetics of Eflornithine HCI 13.9% cream in women with unwanted facial hair”. J Clin Pharmacol. 41 (9): 972–978. doi:10.1177/009127000104100907(inactive 2018-09-12). PMID 11549102. Archived from the original on 2016-11-12.
- ^ Jump up to:a b c “Vaniqa Product Monograph”.
- ^ Jump up to:a b c “Vaniqa US Patient Information Leaflet” (PDF). Archived (PDF) from the original on 2010-02-15.
- ^ Jump up to:a b c Poulin, R; Lu, L; Ackermann, B; Bey, P; Pegg, AE (Jan 5, 1992). “Mechanism of the irreversible inactivation of mouse ornithine decarboxylase by alpha-difluoromethylornithine. Characterization of sequences at the inhibitor and coenzyme binding sites”. The Journal of Biological Chemistry. 267 (1): 150–8. PMID 1730582.
- ^ Jump up to:a b Brooks, HB; Phillips, MA (Dec 9, 1997). “Characterization of the reaction mechanism for Trypanosoma brucei ornithine decarboxylase by multiwavelength stopped-flow spectroscopy”. Biochemistry. 36 (49): 15147–55. doi:10.1021/bi971652b. PMID 9398243.
- Jump up^ Wolf JE; Shander D; Huber F; Jackson J; Lin CS; Mathes BM; Schrode K; the Eflornithine Study Group. (January 2007). “Randomized, double-blind clinical evaluation of the efficacy and safety of topical eflornithine HCI 13.9% cream in the treatment of women with facial hair”. Int J Dermatol. 46 (1): 94–8. doi:10.1111/j.1365-4632.2006.03079.x. PMID 17214730.
- Jump up^ Pepin J, Milord F, Guern C, Schechter PJ (1987). “Difluoromethylornithine for arseno-resistant Trypanosoma brucei gambiense sleeping sickness”. Lancet. 2 (8573): 1431–3. doi:10.1016/S0140-6736(87)91131-7. PMID 2891995.
- Jump up^ “New lease of life for resurrection drug”.[permanent dead link]
- ^ Jump up to:a b “Supply of sleeping sickness drugs confirmed”. Archived from the original on 2015-09-21.
- Jump up^ “Sanofi-Aventis Access to Medicines Brochure” (PDF). Archived (PDF) from the original on 2008-11-14.
- Jump up^ “IFPMA Health Initiatives: Sleeping Sickness”. Archived from the original on 2006-08-29.
- Jump up^ “Ornidyl facts”. Archived from the original on 2011-07-20.
- ^ Jump up to:a b “Vaniqa Training Programme Module 5”.
- Jump up^ “Drugs@FDA: FDA Approved Drug Products”. http://www.accessdata.fda.gov. Archivedfrom the original on 2014-09-05. Retrieved 2016-11-17.
External links
References
-
- Bey, P. et al.: J. Org. Chem. (JOCEAH) 44, 2732 (1979).
- Metcalf, B.W. et al.: J. Am. Chem. Soc. (JACSAT) 100, 2551 (1978).
- US 4 413 141 (Merrell-Toraude; 1.11.1983; appl. 17.9.1982; prior. 11.7.1977, 2.7.1979).
- US 4 330 559 (Merrell-Toraude; 18.5.1982; appl. 3.2.1981; prior. 11.7.1977, 10.4.1979).
-
synthesis of (–)-isomer:
- EP 357 029 (Merrell Dow; appl. 30.8.1989; USA-prior. 31.8.1988).
-
pharmaceutical composition:
- BE 881 209 (Merrell-Toraude; appl. 16.5.1980; USA-prior. 10.4.1979).
-
combination with interferon:
- US 4 499 072 (Merrell Dow; 12.2.1985; appl. 24.1.1983; prior. 29.11.1982).
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Vaniqa, others |
| Synonyms | α-difluoromethylornithine or DFMO |
| AHFS/Drugs.com | Monograph |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
intravenous, topical |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 100% (Intravenous) Negligible (Dermal) |
| Metabolism | Not metabolised |
| Elimination half-life | 8 hours |
| Excretion | Kidneys |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C6H12F2N2O2 |
| Molar mass | 182.17 g·mol−1 |
| 3D model (JSmol) | |
/////////////ZQN1G5V6SR, эфлорнитин , إيفلورنيثين , 依氟鸟氨酸 , Eflornithine, エフロルニチン


FDA permits marketing of two devices that detect parathyroid tissue in real-time during surgery
DRUG REGULATORY AFFAIRS INTERNATIONAL
|
|
November 2, 2018
Release
Today, the U.S. Food and Drug Administration permitted marketing of two devices that provide real-time location of parathyroid tissue during surgical procedures such as thyroidectomy (surgery to remove all or part of the thyroid) and parathyroidectomy (surgery to remove one or…
View original post 556 more words
Statement from FDA Commissioner Scott Gottlieb, M.D., on findings from the romaine lettuce E. coli O157:H7 outbreak investigation and FDA’s efforts to prevent future outbreaks
DRUG REGULATORY AFFAIRS INTERNATIONAL
tatement from FDA Commissioner Scott Gottlieb, M.D., on findings from the romaine lettuce E. coli O157:H7 outbreak investigation and FDA’s efforts to prevent future outbreaks
|
November 1, 2018
Statement
Earlier this year, we experienced the largest E. coli
View original post 1,168 more words
