WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
The U.S. Food and Drug Administration today approved Mavyret (glecaprevir and pibrentasvir) to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis, including patients with moderate to severe kidney disease and those who are on dialysis. Mavyret is also approved for adult patients with HCV genotype 1 infection who have been previously treated with a regimen either containing an NS5A inhibitor or an NS3/4A protease inhibitor but not both.
The U.S. Food and Drug Administration today approved Mavyret (glecaprevir and pibrentasvir) to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis, including patients with moderate to severe kidney disease and those who are on dialysis. Mavyret is also approved for adult patients with HCV genotype 1 infection who have been previously treated with a regimen either containing an NS5A inhibitor or an NS3/4A protease inhibitor but not both.
Mavyret is the first treatment of eight weeks duration approved for all HCV genotypes 1-6 in adult patients without cirrhosis who have not been previously treated. Standard treatment length was previously 12 weeks or more.
“This approval provides a shorter treatment duration for many patients, and also a treatment option for certain patients with genotype 1 infection, the most common HCV genotype in the United States, who were not successfully treated with other direct-acting antiviral treatments in the past,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Hepatitis C is a viral disease that causes inflammation of the liver that can lead to diminished liver function or liver failure. According to the Centers for Disease Control and Prevention, an estimated 2.7 to 3.9 million people in the United States have chronic HCV. Some patients who suffer from chronic HCV infection over many years may have jaundice (yellowish eyes or skin) and complications, such as bleeding, fluid accumulation in the abdomen, infections, liver cancer and death.
There are at least six distinct HCV genotypes, or strains, which are genetically distinct groups of the virus. Knowing the strain of the virus can help inform treatment recommendations. Approximately 75 percent of Americans with HCV have genotype 1; 20-25 percent have genotypes 2 or 3; and a small number of patients are infected with genotypes 4, 5 or 6.
The safety and efficacy of Mavyret were evaluated during clinical trials enrolling approximately 2,300 adults with genotype 1, 2, 3, 4, 5 or 6 HCV infection without cirrhosis or with mild cirrhosis. Results of the trials demonstrated that 92-100 percent of patients who received Mavyret for eight, 12 or 16 weeks duration had no virus detected in the blood 12 weeks after finishing treatment, suggesting that patients’ infection had been cured.
Treatment duration with Mavyret differs depending on treatment history, viral genotype, and cirrhosis status.
The most common adverse reactions in patients taking Mavyret were headache, fatigue and nausea.
Mavyret is not recommended in patients with moderate cirrhosis and contraindicated in patients with severe cirrhosis. It is also contraindicated in patients taking the drugs atazanavir and rifampin.
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected adult patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. HBV reactivation in patients treated with direct-acting antiviral medicines can result in serious liver problems or death in some patients. Health care professionals should screen all patients for evidence of current or prior HBV infection before starting treatment with Mavyret.
Glecaprevir (INN,[1] codenamed ABT-493) is a hepatitis C virus (HCV) nonstructural (NS) protein 3/4A protease inhibitor that was identified jointly by AbbVie and Enanta Pharmaceuticals. It is being developed as a treatment of chronic hepatitis C infection in co-formulation with an HCV NS5A inhibitor pibrentasvir. Together they demonstrated potent antiviral activity against major HCV genotypes and high barriers to resistance in vitro.[2]
On December 19, 2016, AbbVie submitted New Drug Application to U.S. Food and Drug Administration for glecaprevir/pibrentasvir (trade name Maviret) regimen for the treatment of all major genotypes (1–6) of chronic hepatitis C.[3]
3,4,6-Tridésoxy-3-(diméthylamino)-β-D-xylo-hexopyranoside de (3aS,4R,7S,9R,10R,11R,13R,15R,15aR)-1-{4-[4-(3-aminophényl)-1H-1,2,3-triazol-1-yl]butyl}-4-éthyl-7-fluoro-11-méthoxy-3a,7,9,11,13,15-hex améthyl-2,6,8,14-tétraoxotétradécahydro-2H-oxacyclotétradécino[4,3-d][1,3]oxazol-10-yle
Solithromycin exhibits excellent in vitro activity against a broad spectrum of Gram-positiverespiratory tractpathogens,[3][4] including macrolide-resistant strains.[5] Solithromycin has activity against most common respiratory Gram-(+) and fastidious Gram-(-) pathogens,[6][7] and is being evaluated for its utility in treating gonorrhea.
May 2011: Solithromycin is in a Phase 2 clinical trial for serious community-acquired bacterial pneumonia (CABP) and in a Phase 1 clinical trial with an intravenous formulation.[8]
September 2011 : Solithromycin demonstrated comparable efficacy to levofloxacin with reduced adverse events in Phase 2 trial in people with community-acquired pneumonia[9]
January 2015: In a Phase 3 clinical trial for community-acquired bacterial pneumonia (CABP), Solithromycin administered orally demonstrated statistical non-inferiority to the fluoroquinolone, Moxifloxacin.[10]
July 2015: Patient enrollment for the second Phase 3 clinical trial (Solitaire IV) for community-acquired bacterial pneumonia (CABP) was completed with results expected in Q4 2015.[11]
Oct 2015: IV to oral solithromycin demonstrated statistical non-inferiority to IV to oral moxifloxacin in adults with CABP.[12]
July 2016: Cempra Announces FDA Acceptance of IV and oral formulations of Solithera (solithromycin) New Drug Applications for in the Treatment of Community-Acquired Bacterial Pneumonia.[13]
Structure
X-ray crystallography studies have shown solithromycin, the first fluoroketolide in clinical development, has a third region of interactions with the bacterial ribosome,[14] as compared with two binding sites for other ketolides.
Macrolide antibiotics, such like erythromycin, azithromycin, and clarithromycin, have proven to be safe and effective for use in treating human infectious diseases such as community-acquired bacterial pneumonia (CABP), urethritis, and other infections.
Because of the importance of macrolide antibiotics, there has been growing recent interest in this area as exemplified by the new fourth-generation macrolide solithromycin , which is developed by Cempra Pharmaceuticals as the first fluoroketolide antibiotic that has recently completed phase III clinical trials and demonstrates potent activity against the pathogens associated with CABP, including macrolide- and penicillin-resistant isolates of S. pneumoniaeis
To date, all macrolide antibiotics are produced by chemical modification (semisynthesis) of erythromycin, a natural product produced on the ton scale by fermentation. Depicted are erythromycin and the approved semisynthetic macrolide antibiotics clarithromycin, azithromycin and telithromycin along with the dates of their FDA approval and the number of steps for their synthesis from erythromycin. The previous ketolide clinical candidate cethromycin and the current clinical candidate solithromycin are also depicted. It is evident that increasingly lengthy sequences are being employed in macrolide discovery efforts.
Nature, 533, 338–345 (19 May 2016), doi:10.1038/nature17967
a, Graphical representation of the convergent synthesis of solithromycin, which was previously only accessible by semisynthesis. This route has been adapted for the synthesis of >30 novel ketolide antibiotic candidates, as well as the FDA-approved ketolide telithromycin. Downward, ‘Y’-shaped arrows signify convergent coupling reactions. b, Synthesis of solithromycin, reagents and conditions (subscripts L and R indicate left and right portions, respectively): (aL) lithium (1S,2R)-1-phenyl-2-(pyrrolidin-1-yl)-1-propanolate, 85%; (bL) CuSO4, sodium L-ascorbate, 95%; (aR) tBuLi, MgBr2, 81%; (bR) KH, MeI, 99%; (cR) H5IO6, 99%; (dR) ZnCl2, 91%; (eR) AgOTf, 70%; (fR) HF (aq.), 95%; (gR) Dess–Martin periodinane, 92%; (h) Cp2TiCl2, cyclopentylmagnesium bromide, 80%; (i) Dess–Martin periodinane, 97%; (j) Bu4NF, 95%; (k) 132 °C, 0.5 mM, PhCl, 66%; (l) KOtBu, FN(SO2Ph)2, 85%; (m) Im2CO, DBU; (n) imidazole hydrochloride, 60 °C, 87% over 2 steps.
SPECTRAL DATA OF AMINE
residue was purified by column chromatography over silica gel (10% methanol–dichloromethane + 1% 30% aqueous ammonium hydroxide) to afford amine 49 as a pale yellow oil (1.11 g, 96%). TLC (10% methanol–dichloromethane + 1% 30% aqueous ammonium hydroxide): Rƒ = 0.12 (UV, anisaldehyde).
The residue was purified by column chromatography (3% methanol–dichloromethane + 0.3% 30% aqueous ammonium hydroxide) to provide solithromycin (170 mg, 87%) as a white powder.
Potential causes for the formation of synthetic impurities that are present in solithromycin (1) during laboratory development are studied in the article. These impurities were monitored by HPLC, and their structures are identified on the basis of MS and NMR spectroscopy. In addition to the synthesis and characterization of these seven impurities, strategies for minimizing them to the level accepted by the International Conference on Harmonization (ICH) are also described.
PAPER
Identification, Characterization, Synthesis, and Strategy for Minimization of Potential Impurities Observed in the Synthesis of Solithromycin
Macrolide antibacterial agents are characterized by a large lactone ring to which one or more deoxy sugars, usually cladinose and desosamine, are attached. The first generation macrolide, erythromycin, was soon followed by second generation macrolides clarithromycin and azithromycin. Due to widespread of bacterial resistance semi-synthetic derivatives, ketolides, were developed. These, third generation macrolides, to which, for example, belongs telithromycin, are used to treat respiratory tract infections. Currently, a fourth generation macrolide, solithromycin (also known as CEM-101 ) belonging to the fluoroketolide class is in the pre-registration stage. Solithromycin is more potent than third generation macrolides, is active against macrolide-resistant strains, is well-tolerated and exerts good PK and tissue distribution.
One route of synthesis of solithromycin is disclosed in WO2004/080391 A2. Said route is based on the synthetic strategy disclosed in Tetrahedron Letters, 2005, 46, 1483-1487. It is formally a 10 step linear synthesis starting from clarithromycin. Its characteristics are a late cleavage of cladinose, a hexose deoxy sugar which is in ketolides replaced with a keto group, and a 3-step building up of the side chain. Most intermediates are amorphous or cannot be purified by crystallization, hence chromatographic separations are required.
WO 2009/055557 A1 describes a process, in which the linker part of the side chain (azidobutyl) is synthesized separately thus making the synthesis more convergent. In addition, the benzoyl protection group instead of the acetyl is used to protect the 3-hydroxy group of the tetrahydropyran moiety. The linker part of the side chain is introduced using 4-azidobutanamine which is prepared through a selective Staudinger monoreduction of 1 ,4-diazidobutane.
WO 2014/145210 A1 discloses several routes of synthesis all based on the use of already fully constructed side chain building blocks, or its protected forms, which are reacted with an imidazoyl carbamate still containing the protected cladinose moiety. After the introduction of the side chain, the cladinose is cleaved and the aniline group protected for the oxidation of the hydroxy group and the fluorination. After fluorination and deprotection or unmasking of the aniline group, solithromycin is obtained.
Synthesis of crude solithromycin
A third aspect of the invention is a process for providing crude solithromycin (the compound of formula 5) through a convergent synthesis that combines both aforementioned building blocks, the macrolide building block (compound of formula 3) and the side chain building block 3-(1 -(4-aminobutyl)-1 H-1 ,2,3-triazol-4-yl)aniline prepared as discussed above.
As shown in Scheme 4, first, the compound of formula 3 and 3-(1-(4-aminobutyl)-1 H-1 ,2,3-triazol-4-yl)aniline are reacted in the presence of a strong base, for example, DBU, in a suitable solvent, for example, MeCN, to give the compound of formula 4. In the last step, the acetyl protecting group of the hydroxy group located on position β to the dimethylamino substituent is cleaved in methanol.
As discussed above, the fluorination is performed in the presence of acetyl group in the pyran part of the molecule and prior to the incorporation of the side chain, which allows that both exchanging the protection group in the pyran part of the molecule and masking or protecting the aniline moiety from oxidation caused under fluorinating conditions can be avoided.
Scheme 4: Representation of the specific embodiment of the present invention.
In another aspect of the present invention a process is provided for purification of crude solithromycin.
Purification of crude solithromycin
Due to its properties, solithromycin is very difficult to purify. The amorphous material obtained after various syntheses is truly a challenge for further processing.
Chromatographic separation is very difficult and of poor resolution. Due to the basic polar functional groups, the compound and its related impurities all tend to “trail” on typical normal stationary phases that are considered suitable for industrial use, such as silica and alumina. Purification by crystallization is just as difficult unless the material is already sufficiently pure. Impurities inhibit its crystallization to such an extent that solutions in alcohols can be stable even in concentrations of several-fold above the saturation levels of the pure material. Such solutions refuse to crystallize even after weeks of stirring or cooling. Seeding with crystalline solithromycin has no effect and the added seeds simply dissolve. In addition, only limited purification is achieved in most solvents. Lower primary alcohols, particularly ethanol, are most efficient for purification by crystallization when this is possible, but give low recovery and the crystallization is most sensitive toward impurities, thus still demanding prior chromatographic separation.
Clearly an alternative method of purification would be advantageous. For this reason we developed a process for purification employing an acidic salt formation, for example solithromycin oxalate salt, freebasing back to solithromycin, and crystallization from ethanol (Scheme 5).
Scheme 5: Representation of a particular embodiment of the present invention.
The formation of crystalline salts from crude solithromycin may be inhibited by impurities and is dependent on the solvent used. Only a limited number of acids gave useful precipitates from solutions of crude solithromycin. Of these, the precipitation of the oxalate salt from isopropyl acetate (‘PrOAc) or 2-methyltetrahydrofuran (MeTHF) was found to be most efficient in regard to yields, reaction times and purification ability. Precipitation of the citrate salt from MeTHF also significantly increased purity. However, impurities strongly inhibited crystal growth rates, the reaction thus required longer times compared to the oxalate salt formation. Crystallization of salts from solutions of impure solithromycin was also found possible with D-(-)-tartaric, dibenzoyl-d-tartaric, 2,4-dihydroxybenzoic, 3,5-dihydroxybenzoic, and (R)-(+)-2-pyrrolidinone-5-carboxylic acids using ethyl acetate, isopropyl acetate or MeTHF as solvents, or their mixtures with methyl f-butyl ether, but the efficiency and purification abilities were inferior to both the oxalate and the citrate salt.
Some acids, such as (R)-(-)-mandelic, L-(+)-tartaric, p-toluenesulfonic, benzoic, malonic, 4-hydroxybenzoic, (-)-malic, and (+)-camphor-10-sulfonic acid formed insoluble amorphous precipitates without improving purity. Many other acids were found unable of forming any precipitate from impure solithromycin under the conditions tested.
A solution of (2S,3R,4S,6R)-4-(dimethylamino)-2-(((3R,5R,6R,7R,9R,13S,14R,Z)-14-ethyl-13-hydroxy-7-methoxy-3,5,7,9,11,13-hexamethyl-2,4,10-trioxooxacyclotetradec-11-en-6-yl)oxy)-6-methyltetrahydro-2H-pyran-3-yl acetate, 1 (313 g) in dichloromethane (2.22 L) was cooled to -25 °C. DBU (115 mL) followed by CDI (125 g) were added and temperature of the reaction was raised to 0°C. The completion of the reaction was followed by HPLC. Upon the completion, the pH of the reaction mixture was adjusted to 6 using 10% aqueous acetic acid. Layers were separated and organic layer was washed twice with water, dried over sodium sulphate and concentrated to afford compound 2 as white foam (HPLC purity: 90 area%).
Example 2: Synthesis of compound 3: (2R,3S,7R,9R, 10R, 11 R, 13S,Z)-10-(((2S,3R,4S,6R)-3- acetoxy-4-(dimethylamino)-6-methyltetrahydro-2H-pyran-2-yl)oxy)-2-ethyl-13- fluoro-9-methoxy-3,5,7,9, 11,13-hexamethyl-6, 12, 14-trioxooxacyclotetradec-4-en- 3-yl 1H-imidazole-1-carboxylate
A solution of (2R,3S,7R,9R,10R,11 R,13R,Z)-10-(((2S,3R,4S,6R)-3-acetoxy-4-(dimethylamino)-6-methyltetrahydro-2H-pyran-2-yl)oxy)-2-ethyl-9-methoxy-3,5,7,9,11,13-hexamethyl-6,12,14-trioxooxacyclotetradec-4-en-3-yl 1H-imidazole-1-carboxylate, 2 in THF (9 L) was cooled to 0 °C. DBU (115 mL) followed by NFSI (211 g) were added and reaction mixture was stirred at 0°C
until completion (followed by HPLC). The reaction mixture was quenched with cold, diluted NaHC03 (3 L). DCM (2.5 L) was added and layers were separated. Aqueous layer was washed with additional DCM (1.5 L). Combined organic layers were washed with brine (2 L), dried over sodium sulphate, filtrated and concentrated. Crude material was suspended in /‘PrOAc (2.5 L). Undissolved material was filtered-off and filtrate was concentrated in vacuo to afford compound 3 as pale yellow foam (475 g, HPLC purity: 85 area%).
A solution of (2S,3R,4S,6R)-4-(dimethylamino)-2-(((3R,5R,6R,7R,9R,13S,14R,Z)-14-ethyl-13-hydroxy-7-methoxy-3,5,7,9,1 1 , 13-hexamethyl-2,4, 10-trioxooxacyclotetradec-1 1-en-6-yl)oxy)-6-methyltetrahydro-2H-pyran-3-yl acetate, 1 (2.45 g) in THF (17 mL) was cooled to 0 °C. DBU (0.9 mL) followed by CDI (0.97 g) were added. The completion of the reaction was followed by HPLC. Upon the completion, reaction was diluted by addition of THF (34 mL). Temperature of the reaction was lowered to -10 °C. DBU (0.72 mL) was added followed by solution of NFSI (1.51 g) in THF (14 mL). Upon completion of the reaction, mixture was diluted with the addition of water/ZPrOAc (1 :4) mixture and layers were separated. Organic phase was washed with water (3 x 25 mL), dried over Na2S04, filtered and concentrated to afford compound 3 as a white foam (3.1 g, HPLC purity: 70 area%).
Example 4: Synthesis of 3-(1 -(4-aminobutyl)-1 H-1 ,2,3-triazol-4-yl)aniline
K2
1 moi% CuS04(aq)
2-(4-Chlorobutyl)isoindoline-1 ,3-dione. A mixture of phthalimide potassium salt (1 134 g, 6.00 mol), potassium carbonate (209 g, 1.50 mol), 1 ,4-dichlorobutane (1555 g, 12.00 mol), and potassium iodide (51 g, 0.30 mol, 5 mol%) in 2-butanone (4.80 L) was stirred 3 days at reflux conditions. The reaction mixture cooled to 40 °C was filtered and the insoluble materials washed with 2-butanone (1.00 L). The filtrate was evaporated at 80 °C under reduced pressure. 2-Propanol (1.00 L) was added to the residue and the solvent removed under reduced pressure. The residue was then crystallized from 2-propanol (4.30 L) at 25 °C. The product was isolated by filtration and washed with 2-propanol (1.00 L). After drying at 40 °C and approximately 50 mbar, there was obtained a white powder (1 1 1 1 g): 95% assay by quantitative 1H NMR; MS (ESI) m/z = 238 [MH]+.
2-(4-(4-(3-Aminophenyl)-1 H-1 ,2,3-triazol-l -yl)butyl)isoindoline-1 ,3-dione. To a solution of 2-(4-chlorobutyl)isoindoline-1 ,3-dione (950 g, 4.00 mol) in DMSO (2.80 L) was added sodium azide (305 g) and the mixture stirred 4 h at 70 °C. The reaction temperature was reduced to 25 °C and there was added in this order water (0.80 L), ascorbic acid (43 g, 0.24 mol, 6 mol%), 0.5M CuS04(aq) (160 ml_, 2 mol%) and m-aminophenylacetylene (493 g, 4.00 mol). The resulting mixture was stirred 18 h at 40 °C, forming a thick yellow slurry, which was then cooled to 0 °C and slowly diluted with water (2.40 L). The product was isolated by filtration, washing the filter cake with water (3 χ 2.00 L) and a 1 : 1 (vol.) mixture of methanol and water (2.00 L). After drying at 50 °C and approximately 50 mbar the product was obtained as a yellow powder (1405 g): 85% assay by quantitative 1H NMR; MS (ESI) m/z = 362 [MH]+.
3-(1-(4-Aminobutyl)-1H-1,2,3-triazol-4-yl)aniline. To a stirred suspension of 2-(4-(4-(3-Aminophenyl)-1 H-1 ,2,3-triazol-1 -yl)butyl)isoindoline-1 ,3-dione (659 g, 1 .55 mol) in 1 -butanol (3.26 L) was added hydrazine hydrate (50-60%, 174 ml_). After stirring for 18 h at 60 °C there was added toluene (0.72 L) and 1 M NaOH(aq) (5.00 L). After stirring for 20 min at 60 °C, the aqueous phase was removed and the organic phase washed at this same temperature with 1 M NaOH(aq) (1 .00 L), saturated NaCI(aq) (2 χ 2.00 L), and concentrated under reduced pressure to 2/3 of the initial quantity, dried over anhydrous sodium sulfate (300 g) in the presence of Fluorisil (30 g), filtered and evaporated under reduced pressure at 70 °C. The residual 1 -butanol is removed azotropically by adding toluene and evaporation under reduced pressure (2 χ 0.50 L). The residue was dissolved in tetrahydrofuran (0.50 L). To this solution kept stirring at 25 °C was slowly added methyl t-butyl ether (0.50 L) at which point the mixture was seeded. Additional methyl t-butyl ether (0.50 L) was slowly added and the product isolated by filtration, washed with methyl t-butyl ether (0.50 L), and dried at 35 °C and approximately 50 mbar to give an amber colored powder (321 g): mp = 70-73 °C (DSC);
A solution of (2R,3S,7R,9R,10R, 1 1 R, 13S,Z)-10-(((2S,3R,4S,6R)-3-acetoxy-4-(dimethylamino)-6-methyltetrahydro-2H-pyran-2-yl)oxy)-2-ethyl-13-fluoro-9-methoxy-3,5,7,9, 1 1 , 13-hexamethyl-6, 12,14-trioxooxacyclotetradec-4-en-3-yl 1 H-imidazole-1-carboxylate (425 g) in acetonitrile (3.1 L) was cooled to 0 °C. 4-(1-(4-aminobutyl)-1 H-1 ,2,3-triazol-4-yl)aniline, 3 (213 g) followed by DBU (83 ml.) were added. The mixture was stirred at 0° C until completion of the reaction (followed by HPLC). Mixture of DCM and water was added (4 L, 1 :1 ) and the pH was adjusted to 6 with 10% aqueous acetic acid. Layers were separated and organic layer was washed with water (2 x 2 L), dried over sodium sulphate and concentrated. The crude material was suspended in EtOAc (2.5 L). Undissolved material was filtered off and filtrate was concentrated in vacuo to afford compound 4 as yellow foam (470 g, HPLC purity: 75 area%).
Example 11 : Purification of crude solithromycin (compound 5) via oxalate (2)
To isopropyl acetate (5.77 L) crude solithromycin (192 g, 72 area%) was added, afterwards the mixture was stirred at reflux and filtered to remove any insoluble material. The filtrate was then stirred at 55 °C and oxalic acid (14.91 g, 164 mmol) was added in one batch. The suspension was cooled to 20 °C in the course of 1 h, stirred for additional 1 h and the product was isolated by filtration, washed with isopropyl acetate (0.5 L), and dried at 40 °C under reduced pressure to give the oxalate salt (106 g): 87.81 area% by HPLC (UV at 228 nm). The evaporation of the filtrate gave a resinous material containing solithromycin that can be recovered by reprocessing (88 g, 61.13 area%).
The above oxalate salt (106 g) was dissolved in water (2.40 L) and washed with MeTHF (2 χ 1.00 L) and ethyl acetate (0.50 L). Aqueous ammonia (25%; 37 mL) was then added to the filtrate while stirring at 25 °C. The precipitated product was extracted with ethyl acetate two times (3.00 L and 0.50 L). The combined extracts were washed with water (0.50 L), dried over Na2S04 and evaporated under reduced pressure. To the residue ethanol (2 χ 0.4 L) was added and again evaporated to affect the solvent exchange. The residue was then dissolved in ethanol (300 mL). After stirring for 24 h at 25 °C, the crystallized product was isolated by filtration and drying at 40 °C under reduced pressure to give solithromycin as an off-white crystalline solid (42 g): 98.61 area% by HPLC (UV at 228 nm).
The filtrate was evaporated under reduced pressure to give a yellowish foam (29 g: 68.65 area% by HPLC (UV at 228 nm) that was used for reprocessing.
Crude solithromycin 5 (106 g, HPLC purity: 71 %) was suspended in /‘PrOAc (3 L) and heated to reflux, then filtered to remove undissolved material. Oxalic acid (8 g) was added to a filtrate at 60°C. Mixture was slowly cooled to RT and left stirring for additional 1 h. Precipitate was filtered off and dried in vacuo. Oxalate salt was suspended in water (1 .3 L) (if needed mixture was first filtered through Celite) then 25% aqueous ammonia (21 mL) was added and mixture was stirred additional 15 minutes. Precipitate was filtered off, rinsing with water. Wet solithromycin was dissolved it EtOAc (1 .8 L)/water (800 mL) solution. Layers were separated and organic phase was washed with water (2 x 250 mL), dried over Na2S04 and filtered. EtOAc was removed in vacuo to afford yellow foam.
Yellow foam was dissolved in EtOH (230 mL) and left stirring at RT until white precipitate fell out. White precipitate was dried in vacuo at room temperature to afford clean material (HPLC purity 97 area%).
A solution of (2S,3R,4S,6R)-2-(((3aS,4R,7S,9R, 10R, 1 1 R, 13R, 15R)-1 -(4-(4-(3-aminophenyl)-1 H-1 ,2,3-triazol-1 -yl)butyl)-4-ethyl-7-fluoro-1 1 -methoxy-3a,7,9, 1 1 , 13,15-hexamethyl-2,6,8, 14-tetraoxotetradecahydro-1 H-[1 ]oxacyclotetradecino[4,3-d]oxazol-10-yl)oxy)-4-(dimethylamino)-6- methyltetrahydro-2H-pyran-3-yl acetate, 4 (470 g) in methanol (4.7 L) was stirred at room temperature and completion of the reaction was followed by HPLC. Upon completion, the reaction mixture was concentrated to afford crude solithromycin 5 as orange foam (402 g, HPLC purity: 73 area%).
Jump up^Putnam, Shannon D.; Castanheira, Mariana; Moet, Gary J.; Farrell, David J.; Jones, Ronald N. (2010). “CEM-101, a novel fluoroketolide: antimicrobial activity against a diverse collection of Gram-positive and Gram-negative bacteria”. Diagnostic Microbiology and Infectious Disease. 66 (4): 393–401. PMID20022192. doi:10.1016/j.diagmicrobio.2009.10.013.
Jump up^Putnam, Shannon D.; Sader, Helio S.; Farrell, David J.; Biedenbach, Douglas J.; Castanheira, Mariana (2011). “Antimicrobial characterisation of solithromycin (CEM-101), a novel fluoroketolide: activity against staphylococci and enterococci”. International Journal of Antimicrobial Agents. 37 (1): 39–45. PMID21075602. doi:10.1016/j.ijantimicag.2010.08.021.
Enasidenib (AG-221) is an experimental drug in development for treatment of cancer. It is a small molecule inhibitor of IDH2 (isocitrate dehydrogenase 2). It was developed by Agios Pharmaceuticals and is licensed to Celgene for further development.
An orally available inhibitor of isocitrate dehydrogenase type 2 (IDH2), with potential antineoplastic activity. Upon administration, AG-221 specifically inhibits IDH2 in the mitochondria, which inhibits the formation of 2-hydroxyglutarate (2HG). This may lead to both an induction of cellular differentiation and an inhibition of cellular proliferation in IDH2-expressing tumor cells. IDH2, an enzyme in the citric acid cycle, is mutated in a variety of cancers; It initiates and drives cancer growth by blocking differentiation and the production of the oncometabolite 2HG.
Isocitrate dehydrogenases (IDHs) catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate (i.e., a-ketoglutarate). These enzymes belong to two distinct subclasses, one of which utilizes NAD(+) as the electron acceptor and the other NADP(+). Five isocitrate dehydrogenases have been reported: three NAD(+)-dependent isocitrate dehydrogenases, which localize to the mitochondrial matrix, and two NADP(+)-dependent isocitrate dehydrogenases, one of which is mitochondrial and the other predominantly cytosolic. Each NADP(+)-dependent isozyme is a homodimer.
IDH2 (isocitrate dehydrogenase 2 (NADP+), mitochondrial) is also known as IDH; IDP; IDHM; IDPM; ICD-M; or mNADP-IDH. The protein encoded by this gene is the
NADP(+)-dependent isocitrate dehydrogenase found in the mitochondria. It plays a role in intermediary metabolism and energy production. This protein may tightly associate or interact with the pyruvate dehydrogenase complex. Human IDH2 gene encodes a protein of 452 amino acids. The nucleotide and amino acid sequences for IDH2 can be found as GenBank entries NM_002168.2 and NP_002159.2 respectively. The nucleotide and amino acid sequence for human IDH2 are also described in, e.g., Huh et al., Submitted (NOV-1992) to the
EMBL/GenBank/DDBJ databases; and The MGC Project Team, Genome Res.
14:2121-2127(2004).
Non-mutant, e.g., wild type, IDH2 catalyzes the oxidative decarboxylation of isocitrate to a-ketoglutarate (a- KG) thereby reducing NAD+ (NADP+) to NADH (NADPH), e.g., in the forward reaction:
It has been discovered that mutations of IDH2 present in certain cancer cells result in a new ability of the enzyme to catalyze the NAPH-dependent reduction of α-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). 2HG is not formed by wild- type IDH2. The production of 2HG is believed to contribute to the formation and progression of cancer (Dang, L et al, Nature 2009, 462:739-44).
The inhibition of mutant IDH2 and its neoactivity is therefore a potential therapeutic treatment for cancer. Accordingly, there is an ongoing need for inhibitors of IDH2 mutants having alpha hydroxyl neoactivity.
Mechanism of action
Isocitrate dehydrogenase is a critical enzyme in the citric acid cycle. Mutated forms of IDH produce high levels of 2-hydroxyglutarate and can contribute to the growth of tumors. IDH1 catalyzes this reaction in the cytoplasm, while IDH2 catalyzes this reaction in mitochondria. Enasidenib disrupts this cycle.[1][2]
Development
The drug was discovered in 2009, and an investigational new drug application was filed in 2013. In an SEC filing, Agios announced that they and Celgene were in the process of filing a new drug application with the FDA.[3] The fast track designation allows this drug to be developed in what in markedly less than the average 14 years it takes for a drug to be developed and approved.[4]
Example 1, Step 1: preparation of 6-trifluoromethyl-pyridine-2-carboxylic acid
Diethyl ether (4.32 L) and hexanes (5.40 L) are added to the reaction vessel under N2 atmosphere, and cooled to -75 °C to -65 °C. Dropwise addition of n-Butyl lithium (3.78 L in 1.6 M hexane) under N2 atmosphere at below -65 °C is followed by dropwise addition of dimethyl amino ethanol (327.45 g, 3.67 mol) and after 10 min. dropwise addition of 2-trifluoromethyl pyridine (360 g, 2.45 mol). The reaction is stirred under N2 while maintaining the temperature below -65 °C for about 2.0-2.5 hrs. The reaction mixture is poured over crushed dry ice under N2, then brought to a temperature of 0 to 5 °C while stirring (approx. 1.0 to 1.5 h) followed by the addition of water (1.8 L). The reaction mixture is stirred for 5-10 mins and allowed to warm to 5-10 °C. 6N HC1 (900 mL) is added dropwise until the mixture reached pH 1.0 to 2.0, then the mixture is stirred for 10-20 min. at 5-10 °C. The reaction mixture is diluted with ethyl acetate at 25-35 °C, then washed with brine solution. The reaction is concentrated and rinsed with n-heptane and then dried to yield 6-trifluoromethyl-pyridine-2-carboxylic acid.
Example 1, Step 2: preparation of 6-trifluoromethyl-pyridine-2-carboxylic acid methyl ester Methanol is added to the reaction vessel under nitrogen atmosphere. 6-trifluoromethyl- pyridine-2-carboxylic acid (150 g, 0.785 mol) is added and dissolved at ambient temperature. Acetyl chloride (67.78 g, 0.863 mol) is added dropwise at a temperature below 45 °C. The reaction mixture is maintained at 65-70 °C for about 2-2.5 h, and then concentrated at 35-45 °C under vacuum and cooled to 25-35 °C. The mixture is diluted with ethyl acetate and rinsed with saturated NaHC03 solution then rinsed with brine solution. The mixture is concentrated at temp 35-45 °C under vacuum and cooled to 25-35 °C, then rinsed with n-heptane and concentrated at temp 35-45 °C under vacuum, then degassed to obtain brown solid, which is rinsed with n-heptane and stirred for 10-15 minute at 25-35 °C. The suspension is cooled to -40 to -30 °C while stirring, and filtered and dried to provide 6-trifluoromethyl-pyridine-2-carboxylic acid methyl ester.
Example 1, Step 3: preparation of 6-(6-Trifluoromethyl-pyridin-2-yl)-lH-l,3,5-triazine-2,4-dione
1 L absolute ethanol is charged to the reaction vessel under N2 atmosphere and Sodium Metal (11.2 g, 0.488 mol) is added in portions under N2 atmosphere at below 50 °C. The reaction is stirred for 5-10 minutes, then heated to 50-55 °C. Dried Biuret (12.5 g, 0.122 mol) is added to the reaction vessel under N2 atmosphere at 50-55 °C temperature, and stirred 10-15 minutes. While maintaining 50-55 °C 6-trifluoromethyl-pyridine-2-carboxylic acid methyl ester (50.0 g, 0.244 mol) is added. The reaction mixture is heated to reflux (75-80 °C) and maintained for 1.5-2 hours. Then cooled to 35-40 °C, and concentrated at 45-50 °C under vacuum. Water is added and the mixture is concentrated under vacuum then cooled to 35-40 °C more water is added and the mixture cooled to 0 -5 °C. pH is adjusted to 7-8 by slow addition of 6N HC1, and solid precipitated out and is centrifuged and rinsed with water and centrifuged again. The off white to light brown solid of 6-(6-Trifluoromethyl-pyridin-2-yl)-lH-l,3,5-triazine-2,4-dione is dried under vacuum for 8 to 10 hrs at 50 °C to 60 °C under 600mm/Hg pressure to provide 6-(6-Trifluoromethyl-pyridin-2-yl)-lH-l,3,5-triazine-2,4-dione.
Example 1, Step 4: preparation of 2, 4-Dichloro-6-(6-trifluoromethyl-pyridin-2-yl)-l, 3, 5-triazine
POCI3 (175.0 mL) is charged into the reaction vessel at 20- 35 °C, and 6-(6-Trifluoromethyl-pyridin-2-yl)-lH-l,3,5-triazine-2,4-dione (35.0 g, 0.1355 mol) is added in portions at below 50 °C. The reaction mixture is de-gassed 5-20 minutes by purging with N2 gas. Phosphorous pentachloride (112.86 g, 0.542 mol) is added while stirring at below 50 °C and the resulting slurry is heated to reflux (105-110 °C) and maintained for 3-4 h. The reaction mixture is cooled to 50-55 °C, and concentrated at below 55 °C then cooled to 20-30 °C. The reaction mixture is rinsed with ethyl acetate and the ethyl acetate layer is slowly added to cold water (temperature ~5 °C) while stirring and maintaining the temperature below 10 °C. The mixture is stirred 3-5 minutes at a temperature of between 10 to 20 °C and the ethyl acetate layer is collected. The reaction mixture is rinsed with sodium bicarbonate solution and dried over anhydrous sodium sulphate. The material is dried 2-3 h under vacuum at below 45 °C to provide 2, 4-Dichloro-6-(6-trifluoromethyl-pyridin-2-yl)-l, 3, 5-triazine. Example 1, Step 5: preparation of 4-chloro-6-(6-(trifluoromethyl)pyridin-2-yl)-N-(2-(trifluoro-methyl)- pyridin-4-yl)-l,3,5-triazin-2-amine
A mixture of THF (135 mL) and 2, 4-Dichloro-6-(6-trifluoromethyl-pyridin-2-yl)-l, 3, 5-triazine (27.0 g, 0.0915 mol) are added to the reaction vessel at 20 – 35 °C, then 4-amino-2-(trifluoromethyl)pyridine (16.31 g, 0.1006 mol) and sodium bicarbonate (11.52 g, 0.1372 mol) are added. The resulting slurry is heated to reflux (75-80 °C) for 20-24 h. The reaction is cooled to 30-40 °C and THF evaporated at below 45 °C under reduced pressure. The reaction mixture is cooled to 20-35 °C and rinsed with ethyl acetate and water, and the ethyl acetate layer collected and rinsed with 0.5 N HC1 and brine solution. The organic layer is concentrated under vacuum at below 45 °C then rinsed with dichloromethane and hexanes, filtered and washed with hexanes and dried for 5-6h at 45-50 °C under vacuum to provide 4-chloro-6-(6-(trifluoromethyl)pyridin-2-yl)-N-(2-(trifluoro-methyl)- pyridin-4-yl)-l,3,5-triazin-2-amine.
Example 1, Step 6: preparation of 2-methyl-l-(4-(6-(trifluoromethyl)pyridin-2-yl)-6-(2-(trifluoromethyl)- pyridin-4-ylamino)-l,3,5-triazin-2-ylamino)propan-2-ol
THF (290 mL), 4-chloro-6-(6-(trifluoromethyl)pyridin-2-yl)-N-(2-(trifluoro-methyl)-pyridin-4-yl)-l,3,5-triazin-2-amine (29.0 g, 0.06893 mol), sodium bicarbonate (8.68 g, 0.1033 mol), and 1, 1-dimethylaminoethanol (7.37 g, 0.08271 mol) are added to the reaction vessel at 20-35 °C. The resulting slurry is heated to reflux (75-80 °C) for 16-20 h. The reaction is cooled to 30-40 °C and THF evaporated at below 45 °C under reduced pressure. The reaction mixture is cooled to 20-35 °C and rinsed with ethyl acetate and water, and the ethyl acetate layer collected. The organic layer is concentrated under vacuum at below 45 °C then rinsed with dichlorom ethane and hexanes, filtered and washed with hexanes and dried for 8-1 Oh at 45-50 °C under vacuum to provide 2-methyl-l-(4-(6-(trifluoromethyl)pyridin-2-yl)-6-(2-(trifluoromethyl)- pyridin-4-ylamino)-l,3,5-triazin-2-ylamino)propan-2-ol.
The U.S. Food and Drug Administration today approved Idhifa (enasidenib) for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) who have a specific genetic mutation. The drug is approved for use with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect specific mutations in the IDH2 gene in patients with AML.
The U.S. Food and Drug Administration today approved Idhifa (enasidenib) for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) who have a specific genetic mutation. The drug is approved for use with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect specific mutations in the IDH2 gene in patients with AML.
“Idhifa is a targeted therapy that fills an unmet need for patients with relapsed or refractory AML who have an IDH2 mutation,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The use of Idhifa was associated with a complete remission in some patients and a reduction in the need for both red cell and platelet transfusions.”
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of abnormal white blood cells in the bloodstream and bone marrow. The National Cancer Institute at the National Institutes of Health estimates that approximately 21,380 people will be diagnosed with AML this year; approximately 10,590 patients with AML will die of the disease in 2017.
Idhifa is an isocitrate dehydrogenase-2 inhibitor that works by blocking several enzymes that promote cell growth. If the IDH2 mutation is detected in blood or bone marrow samples using the RealTime IDH2 Assay, the patient may be eligible for treatment with Idhifa.
The efficacy of Idhifa was studied in a single-arm trial of 199 patients with relapsed or refractory AML who had IDH2 mutations as detected by the RealTime IDH2 Assay. The trial measured the percentage of patients with no evidence of disease and full recovery of blood counts after treatment (complete remission or CR), as well as patients with no evidence of disease and partial recovery of blood counts after treatment (complete remission with partial hematologic recovery or CRh). With a minimum of six months of treatment, 19 percent of patients experienced CR for a median 8.2 months, and 4 percent of patients experienced CRh for a median 9.6 months. Of the 157 patients who required transfusions of blood or platelets due to AML at the start of the study, 34 percent no longer required transfusions after treatment with Idhifa.
Common side effects of Idhifa include nausea, vomiting, diarrhea, increased levels of bilirubin (substance found in bile) and decreased appetite. Women who are pregnant or breastfeeding should not take Idhifa because it may cause harm to a developing fetus or a newborn baby.
The prescribing information for Idhifa includes a boxed warning that an adverse reaction known as differentiation syndrome can occur and can be fatal if not treated. Sign and symptoms of differentiation syndrome may include fever, difficulty breathing (dyspnea), acute respiratory distress, inflammation in the lungs (radiographic pulmonary infiltrates), fluid around the lungs or heart (pleural or pericardial effusions), rapid weight gain, swelling (peripheral edema) or liver (hepatic), kidney (renal) or multi-organ dysfunction. At first suspicion of symptoms, doctors should treat patients with corticosteroids and monitor patients closely until symptoms go away.
Idhifa was granted Priority Review designation, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition. Idhifa also received Orphan Drugdesignation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Idhifa to Celgene Corporation. The FDA granted the approval of the RealTime IDH2 Assay to Abbott Laboratories
The U.S. Food and Drug Administration today approved Idhifa (enasidenib) for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) who have a specific genetic mutation. The drug is approved for use with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect specific mutations in the IDH2 gene in patients with AML.
The U.S. Food and Drug Administration today approved Idhifa (enasidenib) for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) who have a specific genetic mutation. The drug is approved for use with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect specific mutations in the IDH2 gene in patients with AML.
“Idhifa is a targeted therapy that fills an unmet need for patients with relapsed or refractory AML who have an IDH2 mutation,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The use of Idhifa was associated with a complete remission in some patients and a reduction in the need for both red cell and platelet transfusions.”
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of abnormal white blood cells in the bloodstream and bone marrow. The National Cancer Institute at the National Institutes of Health estimates that approximately 21,380 people will be diagnosed with AML this year; approximately 10,590 patients with AML will die of the disease in 2017.
Idhifa is an isocitrate dehydrogenase-2 inhibitor that works by blocking several enzymes that promote cell growth. If the IDH2 mutation is detected in blood or bone marrow samples using the RealTime IDH2 Assay, the patient may be eligible for treatment with Idhifa.
The efficacy of Idhifa was studied in a single-arm trial of 199 patients with relapsed or refractory AML who had IDH2 mutations as detected by the RealTime IDH2 Assay. The trial measured the percentage of patients with no evidence of disease and full recovery of blood counts after treatment (complete remission or CR), as well as patients with no evidence of disease and partial recovery of blood counts after treatment (complete remission with partial hematologic recovery or CRh). With a minimum of six months of treatment, 19 percent of patients experienced CR for a median 8.2 months, and 4 percent of patients experienced CRh for a median 9.6 months. Of the 157 patients who required transfusions of blood or platelets due to AML at the start of the study, 34 percent no longer required transfusions after treatment with Idhifa.
Common side effects of Idhifa include nausea, vomiting, diarrhea, increased levels of bilirubin (substance found in bile) and decreased appetite. Women who are pregnant or breastfeeding should not take Idhifa because it may cause harm to a developing fetus or a newborn baby.
The prescribing information for Idhifa includes a boxed warning that an adverse reaction known as differentiation syndrome can occur and can be fatal if not treated. Sign and symptoms of differentiation syndrome may include fever, difficulty breathing (dyspnea), acute respiratory distress, inflammation in the lungs (radiographic pulmonary infiltrates), fluid around the lungs or heart (pleural or pericardial effusions), rapid weight gain, swelling (peripheral edema) or liver (hepatic), kidney (renal) or multi-organ dysfunction. At first suspicion of symptoms, doctors should treat patients with corticosteroids and monitor patients closely until symptoms go away.
Idhifa was granted Priority Review designation, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition. Idhifa also received Orphan Drugdesignation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Idhifa to Celgene Corporation. The FDA granted the approval of the RealTime IDH2 Assay to Abbott Laboratories
Enasidenib (AG-221) is an experimental drug in development for treatment of cancer. It is a small molecule inhibitor of IDH2 (isocitrate dehydrogenase 2). It was developed by Agios Pharmaceuticals and is licensed to Celgene for further development.
Isocitrate dehydrogenase is a critical enzyme in the citric acid cycle. Mutated forms of IDH produce high levels of 2-hydroxyglutarate and can contribute to the growth of tumors. IDH1 catalyzes this reaction in the cytoplasm, while IDH2 catalyzes this reaction in mitochondria. Enasidenib disrupts this cycle.[1][2]
Development
The drug was discovered in 2009, and an investigational new drug application was filed in 2013. In an SEC filing, Agios announced that they and Celgene were in the process of filing a new drug application with the FDA.[3] The fast track designation allows this drug to be developed in what in markedly less than the average 14 years it takes for a drug to be developed and approved.[4]
MOLECULAR FORMULA C27H31FN4O8
MOLECULAR WEIGHT 558.6
SPONSOR Tetraphase Pharmaceuticals, Inc.
CODE DESIGNATION TP-434
CAS REGISTRY NUMBER 1207283-85-9
WHO NUMBER 9702
Eravacycline (TP-434) is a synthetic fluorocyclineantibiotic in development by Tetraphase Pharmaceuticals. It is closely related to the glycylglycine antibiotic tigecycline and the tetracycline class of antibiotics. It has a broad spectrum of activity including many multi-drug resistant strains of bacteria. Phase III studies in complicated intra-abdominal infections (cIAI) [1] and complicated urinary tract infections (cUTI)[2] were recently completed with mixed results. Eravacylcine has been designated as a Qualified Infectious Disease Product (QIDP), as well as for fast track approval by the FDA.[3]
Eravacylme is a tetracycline antibiotic that has demonstrated broad spectrum activity against a wide variety of multi-drug resistant Gram-negative, Gram-positive and anaerobic bacteria in humans. In Phase I and Phase II clinical trials, eravacycline also demonstrated a favorable safety and tolerability profile. In view of its attractive
pharmacological profile, synthetic routes to eravacycline and, in particular, synthetic routes that result in suitable quantities of eravacycline for drag development and manufacturing, are becoming increasingly important.
As described in International Publication No . WO 2010/017470, eravacycline is conveniently synthesized from 7-fluorosancycline, another tetracycline. 7-Fluorosancycline can be synthesized, in turn, from commercially available 7-ammosancycline or a protected derivative thereof. However, very few procedures for the conversion of (^-ammo-substituted tetracyclines, such as 7-aminosancycline, to C7-fiuoro-substituted tetracyclines, such as 7-fluorosancycline, have been reported, and those that have are not suitable to be deployed at production-scale.
Therefore, there is a need for improved processes, particularly improved production -scale processes, for converting C7-amino-substituted tetracyclines to C7-fluoro-substituted tetracyclines.
Example 3. Preparation of Eravacycline From 9-Aminosancycline Using a Photolytic Fluorination
[00158] Sancycline (0.414 g, 1.0 mmol) was dissolved in trifiuoroacetic acid (TFA). The solution was cooled to 0 °C. To the solution was added N-bromosuccinimide (NBS, 0.356 g, 2.1 mmol). The reaction was complete after stirring at 0 °C for 1 h. The reaction mixture was allowed to warm to rt. Solid NO3 (0.1 Ig, 0.11 mrnoi) was added and the reaction mixture was stirred at rt for 1 h. The reaction solution was added to 75 mL cold diethyl ether. The precipitate was collected by filtration and dried to give 0.46 g of compound 6. Compound 6 can then be reduced to compounds 7, 8, or 9 using standard procedures.
13
[00159] 9-Aminosancycline (7, 1 g, 0233 mmol) was dissolved in 20 mL sulfuric acid and the reaction was cooled using an ice bath. Potassium nitrate (235 mg, 0.233 mmol) was added in several portions. After stirring for 15 min, the reaction mixture was added to 400 mL MTBE followed by cooling using an ice bath. The solid was collected by filtration. The filter cake was dissolved in 10 mL water and the pH of the aqueous solution was adjusted to 5.3 using 25% aqueous NaOH. The resulting suspension was filtered, and the filter cake was dried to give 1 g compound 10: MS (ESI) m/z 475.1 (M+l).
[00160] Compound 10 (1.1 g) was dissolved in 20 mL of water and 10 mL of acetonitrile. To the solution was added acyl chloride 3 (in two portions: 600 mg and 650 mg). The pH of the reaction mixture was adjusted to 3.5 using 25% aqueous NaOH. Another portion of acyl chloride (800 mg) was added. The reaction was monitored by HPLC analysis. Product 11 was isolated from the reaction mixture by preparative HPLC. Lyophilization gave 1.1 g of compound 11: MS (ESI) m/z 586.3 (M+l).
[00161] Compound 11 (1.1 g) was dissolved in methanol. To the solution was added concentrated HC1 (0.5 mL) and 10% Pd-C (600 mg). The reaction mixture was stirred under a hydrogen atmosphere (balloon). After the reaction was completed, the catalyst was removed by filtration. The filtrate was concentrated to give 1 g of compound 12: ‘H NMR (400 MHz, DMSO), 8.37 (s, 1H), 4.38-4.33 (m, 3H), 3.70 (br s, 2H), 3.30-2.60 (m, 1211), 2.36-2.12 (m, 2H), 2.05-1.80 (m, 4H), 1.50-1.35 (m, 1H); MS (ESI) m/z 556.3 (M+l).
[00162] Compound 12 (150 mg) was dissolved in 1 mL of 48% HBF4. To the solution was added 21 mg of NaN02. After compound 12 was completely converted to compound 13 (LC/MS m/z 539.2), the reaction mixture was irradiated with 254 nm light for 6 h while being cooled with running water. The reaction mixture was purified by preparative HPLC using acetonitrile and 0.05 N aqueous HCl as mobile phases to yield the compound 4 (eravacyclme, 33 mg) as a bis-HCl salt (containing 78% of 4 and 10% of the 7-H byproduct, by HPLC): MS (ESI) m/z 559.3 (M+l).
PAPER
Exploring the Boundaries of “Practical”: De Novo Syntheses of Complex Natural Product-Based Drug Candidates
Department of Chemistry and Biochemistry, University of California−Los Angeles, 607 Charles E. Young Drive East, Los Angeles, California 90095-1569, United States
This review examines the state of the art in synthesis as it relates to the building of complex architectures on scales sufficient to drive human drug trials. We focus on the relatively few instances in which a natural-product-based development candidate has been manufactured de novo, rather than semisynthetically. This summary provides a view of the strengths and weaknesses of current technologies, provides perspective on what one might consider a practical contribution, and hints at directions the field might take in the future.
PAPER
Journal of Medicinal Chemistry (2012), 55(2), 597-605
Fluorocyclines. 1. 7-Fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline: A Potent, Broad Spectrum Antibacterial Agent
†Discovery Chemistry, ‡Microbiology, and §Process Chemistry R&D, Tetraphase Pharmaceuticals, 480 Arsenal Street, Watertown, Massachusetts 02472, United States
This and the accompanying report (DOI: 10.1021/jm201467r) describe the design, synthesis, and evaluation of a new generation of tetracycline antibacterial agents, 7-fluoro-9-substituted-6-demethyl-6-deoxytetracyclines (“fluorocyclines”), accessible through a recently developed total synthesis approach. These fluorocyclines possess potent antibacterial activities against multidrug resistant (MDR) Gram-positive and Gram-negative pathogens. One of the fluorocyclines, 7-fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline (17j, also known as TP–434, 50th Interscience Conference on Antimicrobial Agents and Chemotherapy Conference, Boston, MA, September 12–15, 2010, poster F1–2157), is currently undergoing phase 2 clinical trials in patients with complicated intra-abdominal infections (cIAI).
A convergent route to eravacycline (1) has been developed by employing Michael–Dieckmann cyclization between enone 3 and a fully built and protected left-hand piece (LHP, 2). After construction of the core eravacycline structure, a deprotection reaction was developed, allowing for the isoxazole ring opening and global deprotection to be achieved in one pot. The LHP is synthesized from readily available 4-fluoro-3-methylphenol in six steps featuring a palladium-catalyzed phenyl carboxylation in the last step.
Eravacycline di-HCl salt (1) as a yellow solid: purity 97.9%; mp 197–199 °C dec. The spectral data matched those from original sample as reported in our previous publication.(3)
3 Ronn, M.; Zhu, Z.; Hogan, P. C.; Zhang, W.-Y.; Niu, J.; Katz, C. E.; Dunwoody, N.; Gilicky, O.; Deng, Y.;Hunt, D. K.; He, M.; Chen, C.-L.; Sun, C.; Clark, R. B.; Xiao, X.-Y.Org. Process Res. Dev.2013, 17, 838–845, DOI: 10.1021/op4000219
Process research and development of the first fully synthetic broad spectrum 7-fluorotetracycline in clinical development is described. The process utilizes two key intermediates in a convergent approach. The key transformation is a Michael–Dieckmann reaction between a suitable substituted aromatic moiety and a key cyclohexenone derivative. Subsequent deprotection and acylation provide the desired active pharmaceutical ingredient in good overall yield.
Free base of 7. HPLC (248 nm) showed 97.1% purity with 0.80% of the corresponding impurity from 19, 1.2% of epimer of 7, and 0.80% the corresponding impurity from 20 (102g, 88.7% yield) product as a yellow solid.
MS (ES) m/z calcd for +H: 559.2 (100.0), 560.2 (29.9), 561.2 (5.9); found: 559.3, 560.2, 561.3.
Give 7·2HCl (531 g containing 6.9% by weight water, ∼ 784 mmol). HPLC (248 nm) indicated a 96.6% purity with 0.64% of the corresponding impurity from 19, 1.3% of epimer of 7, and 1.3% the corresponding impurity from 20.
aDepartment of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, United States E-mail:pbaran@scripps.edu
bChemical Development, Bristol-Myers Squibb, One Squibb Drive, New Brunswick, United States E-mail:martin.eastgate@bms.com
Tetracycline (Myers/Tetraphase, 2005–2013). Myers’ convergent approach to the tetracyclines is a great example of how a scalable synthesis of a key intermediate en route to a natural product can fuel the discovery of entirely new drug candidates. These broad-spectrum polyketide antibiotics have been widely used in human and veterinary medicine, but, due to the development of tetracycline-resistant strains, there is an unmet need for novel tetracycline drugs. Pioneering work in this eld has been achieved by the Myers’ group, who published a landmark synthetic approach to the tetracycline class of antibiotics in 2005.16 Using this route, over 3,000 fully synthetic tetracyclines have been prepared to date. Central to their strategy was the synthesis of a highly versatile intermediate, AB enone 7, 17 which enabled the convergent construction of novel tetracycline antibiotics (Scheme 3, A).18 Naturally, the route to 7 had to be practical and amenable to large-scale synthesis and consequently, the synthetic approaches to this building block have become more and more practical and efficient with every new generation. In 2007, Myers published their rst practical and enantioselective approach to 7 (Scheme 3, B).17 The route started from 8, which can be accessed in multi-hundred gram amounts from commercially available 3-hydroxy-5-isoxazolecarboxylate (not shown) by O-benzylation followed by DIBAL reduction. In a three-step sequence, 8 was transformed into carbinol 9. In the key step of the sequence, 9 underwent an intramolecular Diels–Alder reaction to give a mixture of 4 diastereomeric cycloadducts, which, aer Swern oxidation, could be readily separated by ash column chromatography to afford 10. Finally, boron trichloride mediated opening of the oxabicyclic ring system and demethylation, followed by TBS protection of the tertiary hydroxyl-group, afforded 40 g of the AB enone 7 in 21% overall yield, over nine steps from commercial material. Slight modications of this route have allowed for the preparation of >20 kg batches of the AB enone. The availability of large-scale batches of 7 has both enabled the discovery and the development of eravacycline (11), the rst fully synthetic tetracycline analog in clinical development, from Tetraphase Pharmaceuticals. In their process route,19 the key Michael– Dieckmann cyclization between 7 and 12 was carried out on kg-scale and afforded 13 in 93.5% yield. This compound was transformed into eravacycline (11) in 3 more steps, including TBS-cleavage, hydrogenolysis and amide bond formation. Using this process, several kg of 11 have been prepared to date to support clinical studies. Finally, a third- and fourth-generation route to 7 has recently been published by Myers that is not only shorter than previous routes, but also amenable of structural modications of the AB-ring enone.20
16 M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395–398. 17 J. D. Brubaker and A. G. Myers, Org. Lett., 2007, 9, 3523–3525. 18 (a) C. Sun, Q. Wang, J. D. Brubaker, P. M. Wright, C. D. Lerner, K. Noson, M. Charest, D. R. Siegel, Y.-M. Wang and A. G. Myers, J. Am. Chem. Soc., 2008, 130, 17913–17927; (b) X.-Y. Xiao, D. K. Hunt, J. Zhou, R. B. Clark, N. Dunwoody, C. Fyfe, T. H. Grossman, W. J. O’Brien, L. Plamondon, M. R¨onn, C. Sun, W.-Y. Zhang and J. A. Sutcliffe, J. Med. Chem., 2012, 55, 597–605; (c) R. B. Clark, M. He, C. Fyfe, D. Loand, W. J. O’Brien, L. Plamondon, J. A. Sutcliffe and X.-Y. Xiao, J. Med. Chem., 2011, 54, 1511–1528; (d) R. B. Clark, D. K. Hunt, M. He, C. Achorn, C.-L. Chen, Y. Deng, C. Fyfe, T. H. Grossman, P. C. Hogan, W. J. O’Brien, L. Plamondon, M. R¨onn, J. A. Sutcliffe, Z. Zhu and X.-Y. Xiao, J. Med. Chem., 2012, 55, 606–622; (e) C. Sun, D. K. Hunt, R. B. Clark, D. Loand, W. J. O’Brien, L. Plamondon and X.-Y. Xiao, J. Med. Chem., 2011, 54, 3704–3731. 19 M. Ronn, Z. Zhu, P. C. Hogan, W.-Y. Zhang, J. Niu, C. E. Katz, N. Dunwoody, O. Gilicky, Y. Deng, D. K. Hunt, M. He, C.-L. Chen, C. Sun, R. B. Clark and X.-Y. Xiao, Org. Process Res. Dev., 2013, 17, 838–845. 20 D. A. Kummer, D. Li, A. Dion and A. G. Myers, Chem. Sci., 2011, 2, 1710–1718. 21 (a) G. R. Pettit, Z. A. Chicacz, F. Gao, C. L. Herald, M. R. Boyd, J. M. Schmidt and J. N. A. Hooper, J. Org. Chem., 1993, 58, 1302–1304; (b) M. Kobayashi, S. Aoki, H. Sakai, K. Kawazoe, N. Kihara, T. Sasaki and I. Kitagawa, Tetrahedron Lett., 1993, 34, 2795–2798. 22 (a) J. Guo, K. J. Duffy, K. L. Stevens, P. I. Dalko, R. M. Roth, M. M. Hayward and Y. Kishi, Angew. Chem., Int. Ed., 1998, 37, 187–190; (b) M. M. Hayward, R. M. Roth, K. J. Duffy, P. I. Dalko, K. L. Stevens, J. Guo and Y. Kishi, Angew. Chem., Int. Ed., 1998, 37, 190–196; (c) I. Paterson, D. Y. K. Chen, M. J. Coster, J. L. Acena, J. Bach, ˜ K. R. Gibson, L. E. Keown, R. M. Oballa, T. Trieselmann, D. J. Wallace, A. P. Hodgson and R. D. Norcross, Angew. Chem., Int. Ed., 2001, 40, 4055–4060; (d) M. T. Crimmins, J. D. Katz, D. G. Washburn, S. P. Allwein and L. F. McAtee, J. Am. Chem. Soc., 2002, 124, 5661–5663; (e) M. Ball, M. J. Gaunt, D. F. Hook, A. S. Jessiman, S. Kawahara, P. Orsini, A. Scolaro, A. C. Talbot, H. R. Tanner, S. Yamanoi and S. V. Ley, Angew. Chem., Int. Ed., 2005, 44, 5433–5438. 23 A. B. Smith, T. Tomioka, C. A. Risatti, J. B. Sperry and C. Sfouggatakis, Org. Lett., 2008, 10, 4359–4362. 24 (a) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496–497; (b) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621; (c) B. List, Tetrahedron, 2002, 58, 5573–5590; (d) A. B. Northrup and D. W. C. MacMillan, Science, 2004, 305, 1752–1755; (e) A. B. Northrup, I. K. Mangion, F. Hettche and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2004, 43, 2152– 2154. 25 A. B. Smith, C. Sfouggatakis, D. B. Gotchev, S. Shirakami, D. Bauer, W. Zhu and V. A. Doughty, Org. Lett., 2004, 6, 3637–3640. 26 A. B. Smith, C. A. Risatti, O. Atasoylu, C. S. Bennett, J. Liu, H. Cheng, K. TenDyke and Q. Xu, J. Am. Chem. Soc., 2011, 133, 14042–14053. 27 A. R. Carroll, E. Hyde, J. Smith, R. J. Quinn, G. Guymer and P. I. Forster, J. Org. Chem., 2005, 70, 1096–1099. 2W. J. O’Brien, L. Plamondon, M. R¨onn, C. Sun, W.-Y. Zhang and J. A. Sutcliffe, J. Med. Chem., 2012, 55, 597–605; (c) R. B. Clark, M. He, C. Fyfe, D. Loand, W. J. O’Brien, L. Plamondon, J. A. Sutcliffe and X.-Y. Xiao, J. Med. Chem., 2011, 54, 1511–1528; (d) R. B. Clark, D. K. Hunt, M. He, C. Achorn, C.-L. Chen, Y. Deng, C. Fyfe, T. H. Grossman, P. C. Hogan, W. J. O’Brien, L. Plamondon, M. R¨onn, J. A. Sutcliffe, Z. Zhu and X.-Y. Xiao, J. Med. Chem., 2012, 55, 606–622; (e) C. Sun, D. K. Hunt, R. B. Clark, D. Loand, W. J. O’Brien, L. Plamondon and X.-Y. Xiao, J. Med. Chem., 2011, 54, 3704–3731. 19 M. Ronn, Z. Zhu, P. C. Hogan, W.-Y. Zhang, J. Niu, C. E. Katz, N. Dunwoody, O. Gilicky, Y. Deng, D. K. Hunt, M. He, C.-L. Chen, C. Sun, R. B. Clark and X.-Y. Xiao, Org. Process Res. Dev., 2013, 17, 838–845. 20 D. A. Kummer, D. Li,
PAPER
Applications of biocatalytic arene ipso,ortho cis-dihydroxylation in synthesis
aDepartment of Chemistry, University of Bath, Bath, UK E-mail:S.E.Lewis@bath.ac.uk
10.1039/C3CC49694E
In 2005, Myers and co-workers reported the first use of 4 in complex natural product total synthesis.13 From their previously reported building block 43, tricyclic diketone 59 was accessible in a further 7 steps (10% overall yield from benzoate, Scheme 7). Diketone 59 serves as a common precursor to the tetracycline AB-ring system and may be coupled with D-ring precursors such as 60 by a Michael–Dieckmann cascade cyclisation that forms the C-ring. Thus, after deprotection, the natural product ()-6-deoxytetracycline 61 is accessible in 14 steps and 7.0% overall yield from benzoate. Several points about the synthesis are noteworthy. The yield represents an improvement of orders of magnitude over the yields for all previously reported total syntheses of tetracyclines. Thus, for the first time, novel tetracycline analogues became accessible in useful quantities; union of 62 with 59 to access 63 is a representative example. Secondly, previous total syntheses of tetracyclines had been bedevilled by the difficulty of installing the C12a tertiary alcohol at a late stage.14c The Myers approach is conceptually distinct in that the C12a hydroxyl group is installed in the very first step, i.e. it is the hydroxyl group deriving from the microbial ipso hydroxylation. Finally, apart from the C12a stereocentre, all other stereocentres in the final tetracyclines are set under substrate control. Thus, all the stereochemical information in the final products may be considered ultimately to be of enzymatic origin. In the years following the Myers group’s initial disclosure, the methodology has been extended and improved to allow for the preparation of a greater diversity of novel tetracycline analogues.14 This has culminated in the development of eravacycline 65 (accessed from 59 and 64) by Tetraphase Pharmaceuticals.15 Eravacycline is indicated for treatment of multidrug-resistant infections and is currently in phase III trials.
13 (a) M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395; (b) M. G. Charest, D. R. Siegel and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 8292.
14 (a) J. D. Brubaker and A. G. Myers, Org. Lett., 2007, 9, 3523; (b) C. Sun, Q. Wang, J. D. Brubaker, P. M. Wright, C. D. Lerner, K. Noson, M. Charest, D. R. Siegel, Y.-M. Wang and A. G. Myers, J. Am. Chem. Soc., 2008, 130, 17913; (c) D. A. Kummer, D. Li, A. Dion and A. G. Myers, Chem. Sci., 2011, 2, 1710; (d) P. M. Wright and A. G. Myers, Tetrahedron, 2011, 67, 9853.
15 (a) R. B. Clark, M. He, C. Fyfe, D. Lofland, W. J. O’Brien, L. Plamondon, J. A. Sutcliffe and X.-Y. Xiao, J. Med. Chem., 2011, 54, 1511; (b) C. Sun, D. K. Hunt, R. B. Clark, D. Lofland, W. J. O’Brien, L. Plamondon and X.-Y. Xiao, J. Med. Chem., 2011, 54, 3704; (c) X.-Y. Xiao, D. K. Hunt, J. Zhou, R. B. Clark, N. Dunwoody, C. Fyfe, T. H. Grossman, W. J. O’Brien, L. Plamondon, M. Ro¨nn, C. Sun, W.-Y. Zhang and J. A. Sutcliffe, J. Med. Chem., 2012, 55, 597; (d) R. B. Clark, D. K. Hunt, M. He, C. Achorn, C.-L. Chen, Y. Deng, C. Fyfe, T. H. Grossman, P. C. Hogan, W. J. O’Brien, L. Plamondon, M. Ro¨nn, J. A. Sutcliffe, Z. Zhu and X.-Y. Xiao, J. Med. Chem., 2012, 55, 606; (e) M. Ronn, Z. Zhu, P. C. Hogan, W.-Y. Zhang, J. Niu, C. E. Katz, N. Dunwoody, O. Gilicky, Y. Deng, D. K. Hunt, M. He, C.-L. Chen, C. Sun, R. B. Clark and X.-Y. Xiao, Org. Process Res. Dev., 2013, 17, 838; ( f ) R. B. Clark, M. He, Y. Deng, C. Sun, C.-L. Chen, D. K. Hunt, W. J. O’Brien, C. Fyfe, T. H. Grossman, J. A. Sutcliffe, C. Achorn, P. C. Hogan, C. E. Katz, J. Niu, W.-Y. Zhang, Z. Zhu, M. Ro¨nn and X.-Y. Xiao, J. Med. Chem., 2013, 56, 8112.
WO 2017125557, Crystalline forms of eravacycline dihydrochloride or its solvates or hydrates. Also claims a process for the preparation of an eravacycline intended for oral or parenteral use, for the treatment of bacterial infections, preferably intra-abdominal and urinary tract infections caused by multidrug resistant gram negative pathogens. Follows on from WO2017097891 .
Tetraphase Pharmaceuticals is developing eravacycline, a fully synthetic fluorocycline antibiotic and the lead from a series of tetracycline analogs which includes TP-221 and TP-170, for treating bacterial infections.
Eravacycline is a tetracycline antibiotic chemically designated (4S,4aS,5aR,l2aS)-4-(Dimethylamino)-7-fluoro-3 ,10,12,12a-tetrahydroxy- 1,11 -dioxo-9-[2-(-pyrrolidin- 1 -yl)acetamido]-l,4,4a,5,5a,6,l l ,12a-octahydrotetracene-2-carboxamide and can be represented by the following chemical structure according to formula (I).
formula (I)
Eravacycline possesses antibacterial activity against Gram negative pathogens and Gram positive pathogens, in particular against multidrug resistant (MDR) Gram negative pathogens and is currently undergoing phase III clinical trials in patients suffering from complicated intraabdominal infections (cIAI) and urinary tract infections (cUTI).
WO 2010/017470 Al discloses eravacycline as compound 34. Eravacycline is described to be prepared according to a process, which is described in more detail only for related compounds.
The last step of this process involves column chromatography with diluted hydrochloric acid/ acetonitrile, followed by freeze drying.
WO 2012/021829 Al discloses pharmaceutically acceptable acid and base addition salts of eravacycline in general and a general process for preparing the same involving reacting eravacycline free base with the corresponding acids and bases, respectively. On page 15, lines 3 to 6, a lyophilized powder containing an eravacycline salt and mannitol is disclosed.
Xiao et. al. “Fluorocyclines. 1. 7-Fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline: A Potent, Broad Spectrum Antibacterial Agent” J. Med. Chem. 2012, 55, 597-605 synthesized eravacycline following the procedure for compounds 17e and 17i on page 603. After preparative reverse phase HPLC, compounds 17e and 17i were both obtained as bis-hydrochloride salts in form of yellow solids.
Ronn et al. “Process R&D of Eravacycline: The First Fully Synthetic Fluorocycline in Clinical Development” Org. Process Res. Dev. 2013, 17, 838-845 describe a process yielding eravacycline bis-hydrochloride as the final product. The last step involves precipitation of eravacycline bis-hydrochloride salt by adding ethyl acetate as an antisolvent to a solution of eravacycline bis-hydrochloride in an ethanol/ methanol mixture. The authors describe in some detail the difficulties during preparation of the bis-hydrochloride salt of eravacycline. According to Ronn et al. “partial addition of ethyl acetate led to a mixture containing suspended salt and a gummy form of the salt at the bottom of the reactor. At this stage, additional ethanol was added, and the mixture was aged with vigorous stirring until the gummy material also became a suspended solid.” In addition, after drying under vacuum the solid contained “higher than acceptable levels of ethanol”. “The ethanol was then displaced by water by placing a tray containing the solids obtained in a vacuum oven at reduced pressure (…) in the presence of an open vessel of water.” At the end eravacycline bis-hydrochloride salt containing about 4 to 6% residual moisture was obtained. The authors conclude that there is a need for additional improvements to the procedure along with an isolation step suitable for large scale manufacturing.
It is noteworthy that eravacycline or its salts are nowhere described as being a crystalline solid and that the preparation methods used for the preparation of eravacycline are processes like lyophilization, preparative column chromatography and precipitation, which typically yield amorphous material.
The cumbersome process of Ronn et al. points towards problems in obtaining eravacycline bis-hydrochloride in a suitable solid state, problems with scaleability of the available production process as well as problems with the isolation and drying steps of eravacycline.
In addition, amorphous solids can show low chemical stability, low physical stability, hygroscopicity, poor isolation and powder properties, etc.. Such properties are drawbacks for the use as an active pharmaceutical ingredients.
Thus, there is a need in pharmaceutical development for solid forms of an active pharmaceutical ingredient which demonstrate a favorable profile of relevant properties for formulation as a pharmaceutical composition, such as high chemical and physical stability, improved properties upon moisture contact, low(er) hygroscopicity and improved powder properties.
The invention relates to crystalline eravacycline bis-hydrochloride and to a process for its preparation. Furthermore, the invention relates to the use of crystalline eravacycline bis-hydrochloride for the preparation of pharmaceutical compositions. The invention further relates to pharmaceutical compositions comprising an effective amount of crystalline eravacycline bis-hydrochloride. The pharmaceutical compositions of the present invention can be used as medicaments, in particular for treatment and/ or prevention of bacterial infections e.g. caused by Gram negative pathogens or Gram positive pathogens, in particular caused by multidrug resistant Gram negative pathogens. The pharmaceutical compositions of the present invention can thus be used as medicaments for e.g. the treatment of complicated intra-abdominal and urinary tract infection
^ Jump up to:abSolomkin, Joseph; Evans, David; Slepavicius, Algirdas; Lee, Patrick; Marsh, Andrew; Tsai, Larry; Sutcliffe, Joyce A.; Horn, Patrick (2016-11-16). “Assessing the Efficacy and Safety of Eravacycline vs Ertapenem in Complicated Intra-abdominal Infections in the Investigating Gram-Negative Infections Treated With Eravacycline (IGNITE 1) Trial: A Randomized Clinical Trial”. JAMA surgery. ISSN2168-6262. PMID27851857. doi:10.1001/jamasurg.2016.4237.
Tetraphase Pharmaceuticals Inc. (NASDAQ:TTPH) today announced that it will present two posters at IDWeek 2013 that examine the potential of its lead antibiotic candidate eravacycline to treat serious multi-drug resistant (MDR) infections. The first will highlight positive results of a Phase 1 study assessing the bronchopulmonary disposition safety and tolerability of eravacycline in healthy men and women; this study represents the first clinical assessment of eravacycline for potential use in treating pneumonia. The second poster will provide the results of a study that examined the activity of eravacycline in vitro against multiple Gram-negative and Gram-positive pathogens to set quality-control limits for monitoring eravacycline activity in future testing programs. http://www.pharmiweb.com/pressreleases/pressrel.asp?ROW_ID=79335#.Uk6MOoanonU
More: http://www.pharmiweb.com/pressreleases/pressrel.asp?ROW_ID=79335#.Uk6MOoanonU#ixzz2gkEMTtQm
Eravacycline (TP-434 or 7-fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline) is a novel fluorocycline that was evaluated for antimicrobial activity against panels of recent aerobic and anaerobic Gram-negative and Gram-positive bacteria. Eravacycline showed potent broad spectrum activity against 90% of the isolates (MIC90) in each panel at concentrations ranging from ≤0.008 to 2 μg/mL for all species panels except Pseudomonas aeruginosa and Burkholderia cenocepacia (MIC90 values of 32 μg/mL for both organisms). The antibacterial activity of eravacycline was minimally affected by expression of tetracycline-specific efflux and ribosomal protection mechanisms in clinical isolates. Further, eravacycline was active against multidrug-resistant bacteria, including those expressing extended spectrum β-lactamases and mechanisms conferring resistance to other classes of antibiotics, including carbapenem resistance. Eravacycline has the potential to be a promising new IV/oral antibiotic for the empiric treatment of complicated hospital/healthcare infections and moderate-to-severe community-acquired infections.
Tetraphase’s lead product candidate, eravacycline, has also received an award from Biomedical Advanced Research and Development Authority (BARDA) that provides for funding to develop eravacycline as a potential counter-measure to certain biothreat pathogens. It is worth up to USD 67 million.
We are developing our lead product candidate, eravacycline, as a broad-spectrum intravenous and oral antibiotic for use as a first-line empiric monotherapy for the treatment of multi-drug resistant (MDR) infections, including MDR Gram-negative bacteria. We developed eravacycline using our proprietary chemistry technology. We completed a successful Phase 2 clinical trial of eravacycline with intravenous administration for the treatment of patients with complicated intra-abdominal infections (cIAI) and have initiated the Phase 3 clinical program.
Eravacycline is a novel, fully synthetic tetracycline antibiotic. We selected eravacycline for development from tetracycline derivatives that we generated using our proprietary chemistry technology on the basis of the following characteristics of the compound that we observed in in vitro studies of the compound:
potent antibacterial activity against a broad spectrum of susceptible and multi-drug resistant bacteria, including Gram-negative, Gram-positive, atypical and anaerobic bacteria;
potential to treat the majority of patients as a first-line empiric monotherapy with convenient dosing; and
potential for intravenous-to-oral step-down therapy.
In in vitro studies, eravacycline has been highly active against emerging multi-drug resistant pathogens like Acinetobacter baumannii as well as clinically important species ofEnterobacteriaceae, including those isolates that produce ESBLs or are resistant to the carbapenem class of antibiotics, and anaerobes.
Based on in vitro studies we have completed, eravacycline shares a similar potency profile with carbapenems except that it more broadly covers Gram-positive pathogens like MRSA and enterococci, is active against carbapenem-resistant Gram-negative bacteria and unlike carbapenems like Primaxin and Merrem is not active against Pseudomanas aeruginosa. Eravacycline has demonstrated strong activity in vitro against Gram-positive pathogens, including both nosocomial and community-acquired methicillin susceptible or resistantStaphylococcus aureus strains, vancomycin susceptible or resistant Enterococcus faecium andEnterococcus faecalis, and penicillin susceptible or resistant strains of Streptococcus pneumoniae. In in vitro studies for cIAI, eravacycline consistently exhibited strong activity against enterococci and streptococci. One of the most frequently isolated anaerobic pathogens in cIAI, either as the sole pathogen or often in conjunction with another Gram-negative bacterium, is Bacteroides fragilis. In these studies eravacycline demonstrated activity against Bacteroides fragilis and a wide range of Gram-positive and Gram-negative anaerobes.
Key Differentiating Attributes of Eravacycline
The following key attributes of eravacycline, observed in clinical trials and preclinical studies of eravacycline, differentiate eravacycline from other antibiotics targeting multi-drug resistant infections, including multi-drug resistant Gram-negative infections. These attributes will make eravacycline a safe and effective treatment for cIAI, cUTI and other serious and life-threatening infections for which we may develop eravacycline, such as ABSSSI and acute bacterial pneumonias.
Broad-spectrum activity against a wide variety of multi-drug resistant Gram-negative, Gram-positive and anaerobic bacteria. In our recently completed Phase 2 clinical trial of the intravenous formulation of eravacycline, eravacycline demonstrated a high cure rate against a wide variety of multi-drug resistant Gram-negative, Gram-positive and anaerobic bacteria. In addition, in in vitro studies eravacycline demonstrated potent antibacterial activity against Gram-negative bacteria, including E. coli; ESBL-producing Klebsiella pneumoniae; Acinetobacter baumannii; Gram-positive bacteria, including MSRA and vancomycin-resistant enterococcus, or VRE; and anaerobic pathogens. As a result of this broad-spectrum coverage, eravacycline has the potential to be used as a first-line empiric monotherapy for the treatment of cIAI, cUTI, ABSSSI, acute bacterial pneumonias and other serious and life-threatening infections.
Favorable safety and tolerability profile. Eravacycline has been evaluated in more than 250 subjects in the Phase 1 and Phase 2 clinical trials that we have conducted. In these trials, eravacycline demonstrated a favorable safety and tolerability profile. In our recent Phase 2 clinical trial of eravacycline, no patients suffered any serious adverse events, and safety and tolerability were comparable to ertapenem, the control therapy in the trial. In addition, in the Phase 2 clinical trial, the rate at which gastrointestinal adverse events such as nausea and vomiting that occurred in the eravacycline arms was comparable to the rate of such events in the ertapenem arm of the trial.
Convenient dosing regimen. In our recently completed Phase 2 clinical trial we dosed eravacycline once or twice a day as a monotherapy. Eravacycline will be able to be administered as a first-line empiric monotherapy with once- or twice-daily dosing, avoiding the need for complicated dosing regimens typical of multi-drug cocktails and the increased risk of negative drug-drug interactions inherent to multi-drug cocktails.
Potential for convenient intravenous-to-oral step-down. In addition to the intravenous formulation of eravacycline, we are also developing an oral formulation of eravacycline. If successful, this oral formulation would enable patients who begin intravenous treatment with eravacycline in the hospital setting to transition to oral dosing of eravacycline either in hospital or upon patient discharge for convenient home-based care. The availability of both intravenous and oral administration and the oral step-down will reduce the length of a patient’s hospital stay and the overall cost of care.
Additionally, in February 2012, Tetraphase announced a contract award from the Biomedical Advanced Research and Development Authority (BARDA) worth up to $67 million for the development of eravacycline, from which Tetraphase may receive up to approximately $40 million in funding. The contract includes pre-clinical efficacy and toxicology studies; clinical studies; manufacturing activities; and associated regulatory activities to position the broad-spectrum antibiotic eravacycline as a potential empiric countermeasure for the treatment of inhalational disease caused by Bacillus anthracis, Francisella tularensis and Yersinia pestis.\
PAPER
Process research and development of the first fully synthetic broad spectrum 7-fluorotetracycline in clinical development is described. The process utilizes two key intermediates in a convergent approach. The key transformation is a Michael–Dieckmann reaction between a suitable substituted aromatic moiety and a key cyclohexenone derivative. Subsequent deprotection and acylation provide the desired active pharmaceutical ingredient in good overall yield.
Process R&D of Eravacycline: The First Fully Synthetic Fluorocycline in Clinical Development
FDA Grants QIDP Designation to Eravacycline, Tetraphase’s Lead Antibiotic Product Candidate
– Eravacycline designated as a QIDP for complicated intra-abdominal infection (cIAI) and complicated urinary tract infection (cUTI) indications –
July 15, 2013 08:30 AM Eastern Daylight Time
WATERTOWN, Mass.–(BUSINESS WIRE)–Tetraphase Pharmaceuticals, Inc. (NASDAQ: TTPH) today announced that the U.S. Food and Drug Administration (FDA) has designated the company’s lead antibiotic product candidate, eravacycline, as a Qualified Infectious Disease Product (QIDP). The QIDP designation, granted for complicated intra-abdominal infection (cIAI) and complicated urinary tract infection (cUTI) indications, will make eravacycline eligible to benefit from certain incentives for the development of new antibiotics provided under the Generating Antibiotic Incentives Now Act (GAIN Act). These incentives include priority review and eligibility for fast-track status. Further, if ultimately approved by the FDA, eravacycline is eligible for an additional five-year extension of Hatch-Waxman exclusivity.
09 Mar 2017 Phase-III clinical trials in Interstitial cystitis in United Kingdom, Poland, Latvia and Canada before March 2017 (PO) (EudraCT2016-000906-12) (NCT02858453)
04 Jan 2017 Aquinox Pharmaceuticals completes a phase I trial in Healthy volunteers in United Kingdom (NCT03185195)
07 Sep 2016 Phase-III clinical trials in Interstitial cystitis in Czech Republic, Hungary, Denmark (PO) (EudraCT2016-000906-12)
Rosiptor, also known as AQX-1125 is a potent and selective SHIP1 activator currently in clinical development.
AQX-1125 inhibited Akt phosphorylation in SHIP1-proficient but not in SHIP1-deficient cells, reduced cytokine production in splenocytes, inhibited the activation of mast cells and inhibited human leukocyte chemotaxis.
AQX-1125 suppresses leukocyte accumulation and inflammatory mediator release in rodent models of pulmonary inflammation and allergy. As shown in the mouse model of LPS-induced lung inflammation, the efficacy of the compound is dependent on the presence of SHIP1. Pharmacological SHIP1 activation may have clinical potential for the treatment of pulmonary inflammatory diseases.
Dysregulated activation of the PI3K pathway contributes to inflammatory/immune disorders and cancer. Efforts have been made to develop modulators of PI3K as well as downstream kinases (Workman et al., Nat. Biotechnol. 24, 794-796, 2006; Simon, Cell 125, 647-649, 2006; Hennessy et al., Nat. Rev. Drug. Discov. 4, 988-1004, 2005; Knight et al., Cell 125, 733-747, 2006; Ong et al., Blood (2007), Vol. 110, No. 6, pp 1942-1949). A number of promising new PI3K isoform specific inhibitors with minimal toxicities have recently been developed and used mouse models of inflammatory disease (Camps et al., Nat. Med. 11, 936-943, 2005; Barber et al., Nat. Med. 11, 933-935, 2005) and glioma (Fan et al., Cancer Cell 9, 341-349, 2006). However, because of the dynamic interplay between phosphatases and kinases in regulating biological processes, inositol phosphatase activators represent a complementary, alternative approach to reduce PIP3 levels. Of the phosphoinositol phosphatases that degrade PIP3, SHIP1 is a particularly ideal target for development of therapeutics for treating immune and hemopoietic disorders because of its hematopietic-restricted expression (Hazen et al., Blood 113, 2924-2933, 2009; Rohrschneider et al., Genes Dev. 14, 505-520, 2000).
Small molecule SHIP1 modulators have been disclosed, including sesquiterpene compounds such as pelorol. Pelorol is a natural product isolated from the tropical marine sponge Dactylospongia elegans (Kwak et al., J. Nat. Prod. 63, 1153-1156, 2000; Goclik et al., J. Nat. Prod. 63, 1150-1152, 2000). Other reported SHIP1 modulators include the compounds set forth in PCT Published Patent Applications Nos. WO 2003/033517, WO 2004/035601, WO 2004/092100, WO 2007/147251, WO 2007/147252, WO 2011/069118, WO 2014/143561 and WO 2014/158654 and in U.S. Pat. Nos. 7,601,874 and 7,999,010.
One such molecule is AQX-1125, which is the acetate salt of (1S,3S,4R)-4-((3aS,4R,5S,7aS)-4-(aminomethyl)-7a-methyl-1-methyleneoctahydro-1H-inden-5-yl)-3-(hydroxymethyl)-4-methylcyclohexanol (AQX-1125). AQX-1125 is a compound with anti-inflammatory activity and is described in U.S. Pat. Nos. 7,601,874 and 7,999,010, the relevant disclosures of which are incorporated in full by reference in their entirety, particularly with respect to the preparation of AQX-1125, pharmaceutical compositions comprising AQX-1125 and methods of using AQX-1125.
AQX-1125 has the molecular formula, C20H36NO2+.C2H3O2−, a molecular weight of 381.5 g/mole and has the following structural formula:
AQX-1125 is useful in treating disorders and conditions that benefit from SHIP1 modulation, such as cancers, inflammatory disorders and conditions and immune disorders and conditions. AQX-1125 is also useful in the preparation of a medicament for the treatment of such disorders and conditions.
Synthetic methods for preparing AQX-1125 are disclosed in U.S. Pat. Nos. 7,601,874 and 7,999,010. There exists, therefore, a need for improved methods of preparing AQX-1125.
Dysregulated activation of the PI3K pathway contributes to
inflammatory/immune disorders and cancer. Efforts have been made to develop modulators of PI3K as well as downstream kinases (Workman et al., Nat. Biotechnol 24, 794-796, 2006; Simon, Cell 125, 647-649, 2006; Hennessy et al., Nat Rev Drug Discov 4, 988-1004, 2005; Knight et al., Cell 125, 733-747, 2006; Ong et al., Blood (2007), Vol. 110, No. 6, pp 1942-1949). A number of promising new PI3K isoform specific inhibitors with minimal toxicities have recently been developed and used in mouse models of inflammatory disease (Camps et al., Nat Med 1 1 , 936-943, 2005; Barber et ai, Nat Med 1 1 , 933-935, 2005) and glioma (Fan et al., Cancer Cell 9, 341-349, 2006). However, because of the dynamic interplay between phosphatases and kinases in regulating biological processes, inositol phosphatase activators represent a complementary, alternative approach to reduce PIP3 levels. Of the phosphoinositol phosphatases that degrade PIP3i SHIP1 is a particularly ideal target for development of therapeutics for treating immune and hemopoietic disorders because of its
hematopietic-restricted expression (Hazen et al., Blood 1 13, 2924-2933, 2009;
Rohrschneider et ai, Genes Dev. 14, 505-520, 2000).
Small molecule SHIP1 modulators have been disclosed, including
sesquiterpene compounds such as pelorol. Pelorol is a natural product isolated from the tropical marine sponge Dactylospongia elegans (Kwak et al., J Nat Prod 63, 1 153-1 156, 2000; Goclik et al., J Nat Prod 63, 1150-1152, 2000). Other reported SHIP1 modulators include the compounds set forth in PCT Published Patent Applications Nos. WO 2003/033517, WO 2004/035601 , WO 2004/092100, WO 2007/147251 , WO 2007/147252, WO 2011/069118, WO 2014/143561 and WO 2014/158654 and in U.S. Patent Nos. 7,601 ,874 and 7,999,010.
While significant strides have been made in this field, there remains a need for effective small molecule SHIP1 modulators.
One such molecule is the acetate salt of (1 S,3S,4 )-4-((3aS,4 ,5S,7aS)-4-(aminomethyl)-7a-methyl-1-methyleneoctahydro-1 /-/-inden-5-yl)-3-(hydroxymethyl)-4-methylcyclohexanol (referred to herein as Compound 1). Compound 1 is a compound with anti-inflammatory activity and is described in U.S. Patent Nos. 7,601 ,874 and 7,999,010, the relevant disclosures of which are incorporated in full by reference in their entirety, particularly with respect to the preparation of Compound 1 ,
pharmaceutical compositions comprising Compound 1 and methods of using
Compound 1.
Compound 1 has the molecular formula, C2oH36N02+ · C2H302“, a molecular weight of 381.5 g/mole
The application is directed to crystalline forms of the acetate salt of (1S,3S,4R)-4-(3aS,4R,5S,7aS)-4-(aminomethyl)-7a- methyl-1-methyleneoctahydro-1H-inden-5-yl)-3-(hydroxymethyl) -4-methylcyclohexanol and processes for their preparation. The compound acts as a SHIP1 modulator and is thus useful in the treatment of cancer or inflammatory and immune disorders and conditions.
Process for the synthesis of substituted indene derivative (particularly AQX-1125 ) as a SH2-containing inositol 5-phosphatase 1 (SHIP1) modulator for treating cancer, inflammatory disorders and immune disorders. Aquinox Pharmaceuticals is developing AQX-1125 (phase III clinical trial in July 2017), a SHIP1 agonist, for the treatment of inflammatory diseases. For a prior filing see WO2016210146 , claiming novel crystalline forms of rosiptor acetate. In July 2017, Seenisamy and Chetia were associated with Syngene
Synthetic Method 1
In one aspect of the invention, AQX-1125 was prepared by the method described below in Reaction Scheme 1 where Pg1 is an oxygen-protecting group, Pg2 is a carbonyl protecting group, Lg1 is a leaving group and X is bromo or chloro:
Reaction Scheme 1A:
Synthetic Example 77
Step 11: Preparation of AQX-1125 from Compound 16
A. To a stirred solution of (1S,3S,4R)-4-((3aS,4R,5S,7aS)-4-(aminomethyl)-7a-methyl-1-methyleneoctahydro-1H-inden-5-yl)-3-(hydroxymethyl)-4-methylcyclohexan-1-ol (Compound 16, 58.0 g, 0.180 mol, 1.0 eq, from Synthetic Example 76) in methanol (174 mL, 3 V) was added acetic acid (23.5 mL, 0.4 V) dropwise at 10° C. under a nitrogen atmosphere over 20 min. The reaction mixture was stirred at room temperature for 1 h. The solution was filtered to remove undissolved particles and washed with methanol (58 mL, 1 V). The filtrate was collected and evaporated at 35° C. to half the volume (˜125 mL). MTBE (348 mL, 6 V) was slowly added to the above concentrated mixture and the reaction stirred at 10° C. for 2 h. During the MTBE addition, slow precipitation of the product was observed. The solids were filtered and washed with MTBE (116 mL, 2V) to afford (1S,3S,4R)-4-((3aS,4R,5S,7aS)-4-(aminomethyl)-7a-methyl-1-methyleneoctahydro-1H-inden-5-yl)-3-(hydroxymethyl)-4-methylcyclohexan-1-ol, acetic acid salt, (AQX-1125) as a white solid (50 g, yield 72.6%). 1H NMR (400 MHz, pyridine-d5): δ 5.85 (br s, 5H), 4.70 (s, 2H), 4.08 (dd, J=10.4, 2 Hz, 1H), 3.95-3.85 (m, 1H), 3.60-3.50 (m, 1H), 3.18 (d, J=14 Hz, 1H), 2.92-2.86 (m, 1H), 2.80 (d, J=13.6 Hz, 1H), 2.50-2.40 (m, 1H), 2.25-1.97 (m, 3H), 2.15 (s, 3H), 1.90-1.65 (m, 4H), 1.56-1.40 (m, 4H), 1.39-1.20 (m, 2H), 1.25 (s, 3H), 0.78 (s, 3H). LCMS: (Method A) 322.4 (M+1), Retention time: 1.95 min, HPLC (Method H): 95.5 area %, Retention time: 16.66 min.
Synthetic Example 66
Preparation of Compound 16 and AQX-1125
A. To a solution of 7a-methyl-5-((1S,2R,5S)-2-methyl-7-oxo-6-oxabicyclo[3.2.1]octan-2-yl)-1-methyleneoctahydro-1H-indene-4-carbaldehyde oxime (Compound 68, 100 mg, 0.30 mmol, from Synthetic Example 65) in 1,4-dioxane (5 mL) in a 25 mL RB flask fitted with reflux condenser was added a solution of lithium aluminum hydride (1 M in THF, 1.51 ml, 1.50 mmol) at RT under nitrogen and the reaction mass was stirred using a magnetic stirrer at 100° C. for 24 hours. Another lot of a solution of lithium aluminum hydride (1 M in THF, 1.51 ml, 1.50 mmol) was added and the reaction was further refluxed for 24 hours. Completion of the reaction was monitored by LCMS analysis.
B. The reaction mass was quenched by the drop-wise addition of saturated aq. sodium sulfate solution, filtered through a CELITE™ bed on glass frit funnel and concentrated by rotary evaporation to get a crude mass which was further purified by preparative HPLC to afford (1S,3S,4R)-4-((4R,5S,7aS)-4-(aminomethyl)-7a-methyl-1-methyleneoctahydro-1H-inden-5-yl)-3-(hydroxymethyl)-4-methylcyclohexan-1-ol (Compound 16, 35 mg, 36% yield) as an off-white solid. 1H-NMR (400 MHz, CD3OD): δ 4.69 (s, 2H), 3.73 (br d, J=10.0 Hz, 1H), 3.52-3.45 (m, 1H), 3.22-3.15 (m, 1H), 3.05-2.98 (m, 1H), 2.62-2.55 (m, 1H), 2.38-2.25 (m, 1H), 2.20-2.15 (m, 1H), 1.95-1.81 (m, 6H), 1.62-1.25 (m, 10H), 1.10 (s, 3H), 0.86 (s, 3H). LCMS (Method A) m/z: 322.5 (M+1), Retention time: 2.06 min, Purity: 98.9 area % (ELSD). HPLC (Method A): Retention time: 2.70 min, Purity: 99.3 area %.
C. AQX-1125 was prepared from Compound 16 in the same manner as described above in Synthetic Example 16.
REFERENCES
1: Nickel JC, Egerdie B, Davis E, Evans R, Mackenzie L, Shrewsbury SB. A Phase II Study of the Efficacy and Safety of the Novel Oral SHIP1 Activator AQX-1125 in Subjects with Moderate to Severe Interstitial Cystitis/Bladder Pain Syndrome. J Urol. 2016 Sep;196(3):747-54. doi: 10.1016/j.juro.2016.03.003. PubMed PMID: 26968644.
2: Chuang YC, Chermansky C, Kashyap M, Tyagi P. Investigational drugs for bladder pain syndrome (BPS) / interstitial cystitis (IC). Expert Opin Investig Drugs. 2016;25(5):521-9. doi: 10.1517/13543784.2016.1162290. PubMed PMID: 26940379.
3: Leaker BR, Barnes PJ, O’Connor BJ, Ali FY, Tam P, Neville J, Mackenzie LF, MacRury T. The effects of the novel SHIP1 activator AQX-1125 on allergen-induced responses in mild-to-moderate asthma. Clin Exp Allergy. 2014 Sep;44(9):1146-53. doi: 10.1111/cea.12370. PubMed PMID: 25040039.
4: Stenton GR, Mackenzie LF, Tam P, Cross JL, Harwig C, Raymond J, Toews J, Wu J, Ogden N, MacRury T, Szabo C. Characterization of AQX-1125, a small-molecule SHIP1 activator: Part 1. Effects on inflammatory cell activation and chemotaxis in vitro and pharmacokinetic characterization in vivo. Br J Pharmacol. 2013 Mar;168(6):1506-18. doi: 10.1111/bph.12039. PubMed PMID: 23121445; PubMed Central PMCID: PMC3596654.
5: Stenton GR, Mackenzie LF, Tam P, Cross JL, Harwig C, Raymond J, Toews J, Chernoff D, MacRury T, Szabo C. Characterization of AQX-1125, a small-molecule SHIP1 activator: Part 2. Efficacy studies in allergic and pulmonary inflammation models in vivo. Br J Pharmacol. 2013 Mar;168(6):1519-29. doi: 10.1111/bph.12038. PubMed PMID: 23121409; PubMed Central PMCID: PMC3596655.
6: Croydon L. BioPartnering North America–Spotlight on Canada. IDrugs. 2010 Mar;13(3):159-61. PubMed PMID: 20191430.
The SHIP1 Pathway – Highlighting the Role of AQX-1125
AQX-1125 is our lead product candidate and has generated positive clinical data from three completed clinical trials, including two proof-of-concept trials, one in COPD and one in allergic asthma, demonstrating a favorable safety profile and anti-inflammatory activity. Overall, more than 100 subjects have received AQX-1125. Importantly, our clinical trial results were consistent with the drug-like properties and anti-inflammatory activities demonstrated in our preclinical studies. AQX-1125 is a once daily oral capsule with many desirable drug-like properties. We are currently investigating AQX-1125 in two Phase 2 clinical trials, one in COPD and one in BPS/IC.
Based on our three completed clinical trials, we have demonstrated that AQX-1125:
has desirable pharmacokinetic, absorption and excretion properties that make it suitable for once daily oral administration;
is generally well tolerated, exhibiting mild to moderate adverse events primarily related to gastrointestinal upset that resolve without treatment or long-term effects and are reduced by taking the drug candidate with food; and
has anti-inflammatory properties consistent with those exhibited in preclinical studies and exhibited activity in two trials using two distinct inflammatory challenges.
AQX-1125 is an activator of SHIP1, which controls the PI3K cellular signaling pathway. If the PI3K pathway is overactive, immune cells can produce an abundance of pro-inflammatory signaling molecules and migrate to and concentrate in tissues, resulting in excessive or chronic inflammation. SHIP1 is predominantly expressed in cells derived from bone marrow tissues, which are mainly immune cells. Therefore drugs that activate SHIP1 can reduce the function and migration of immune cells and have an anti-inflammatory effect. By controlling the PI3K pathway, AQX-1125 reduces immune cell function and migration by targeting a mechanism that has evolved in nature to maintain homeostasis of the immune system.
AQX-1125 has demonstrated compelling preclinical activity in a broad range of relevant inflammatory studies including preclinical models of COPD, asthma, pulmonary fibrosis, BPS/IC and inflammatory bowel disease (IBD). In these studies we have seen a meaningful reduction in the relevant immune cells that are the cells that cause inflammation, such as neutrophils, eosinophils and macrophages, and a reduction in the symptoms of inflammation, such as pain and swelling. The activity, efficacy and potency seen with AQX-1125 in most preclinical studies compare favorably to published results with corticosteroids. In addition, AQX-1125 demonstrated compelling activity in the smoke airway inflammation and Bleomycin Fibrosis models, which are known to be steroid refractory, or in other words, do not respond to corticosteroids. We believe this broad anti-inflammatory profile is not typical amongst drugs in development and supports the therapeutic potential for AQX-1125.
In addition to demonstrating strong in vitro and in vivo activity, AQX-1125 was also selected as a lead candidate based on its many desirable drug-like properties. The drug candidate is highly water soluble and does not require complex formulation for oral administration. AQX-1125 has low plasma protein binding, is not metabolized and is excreted unmetabolized in both urine and feces. After oral or intravenous dosing, AQX-1125 reaches high concentrations in respiratory, urinary and gastrointestinal tracts, all of which have mucosal surfaces of therapeutic interest. In humans, AQX-1125 has shown pharmacokinetic properties suitable for once-a-day dosing. In addition, the absorption of the drug candidate is equivalent whether taken with or without food.
Mechanism of Action Sodium-bile acid cotransporter-inhibitors
Highest Development Phases
Phase II Primary biliary cirrhosis; Pruritus; Type 2 diabetes mellitus
Phase I Cholestasis
Most Recent Events
01 Jan 2017 Phase-II clinical trials in Pruritus in USA (PO) (NCT02966834)
14 Nov 2016 GlaxoSmithKline completes a phase I trial for Cholestasis in Healthy volunteers in Japan (PO, Tablet) (NCT02801981)
11 Nov 2016 Efficacy, safety and pharmacodynamic data from a phase II trial in Primary biliary cirrhosis and Pruritus presented at The Liver Meeting® 2016: 67th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD-2016)
GSK2330672 , an ileal bile acid transport (iBAT) inhibitor indicated for diabetes type II and cholestatic pruritus, is currently under Phase IIb evaluation in the clinic. The API is a highly complex molecule containing two stereogenic centers, one of which is quaternary
GSK-2330672 is highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor for treatment of type 2 diabetes.
More than 200 million people worldwide have diabetes. The World Health Organization estimates that 1 .1 million people died from diabetes in 2005 and projects that worldwide deaths from diabetes will double between 2005 and 2030. New chemical compounds that effectively treat diabetes could save millions of human lives.
Diabetes refers to metabolic disorders resulting in the body’s inability to effectively regulate glucose levels. Approximately 90% of all diabetes cases are a result of type 2 diabetes whereas the remaining 10% are a result of type 1 diabetes, gestational diabetes, and latent autoimmune diabetes of adulthood (LADA). All forms of diabetes result in elevated blood glucose levels and, if left untreated chronically, can increase the risk of macrovascular (heart disease, stroke, other forms of cardiovascular disease) and microvascular [kidney failure (nephropathy), blindness from diabetic retinopathy, nerve damage (diabetic neuropathy)] complications.
Type 1 diabetes, also known as juvenile or insulin-dependent diabetes mellitus (IDDM), can occur at any age, but it is most often diagnosed in children, adolescents, or young adults. Type 1 diabetes is caused by the autoimmune destruction of insulin-producing beta cells, resulting in an inability to produce sufficient insulin. Insulin controls blood glucose levels by promoting transport of blood glucose into cells for energy use. Insufficient insulin production will lead to decreased glucose uptake into cells and result in accumulation of glucose in the bloodstream. The lack of available glucose in cells will eventually lead to the onset of symptoms of type 1 diabetes: polyuria (frequent urination), polydipsia (thirst), constant hunger, weight loss, vision changes, and fatigue. Within 5-10 years of being diagnosed with type 1 diabetes, patient’s insulin-producing beta cells of the pancreas are completely destroyed, and the body can no longer produce insulin. As a result, patients with type 1 diabetes will require daily administration of insulin for the remainder of their lives.
Type 2 diabetes, also known as non-insulin-dependent diabetes mellitus (NIDDM) or adult-onset diabetes, occurs when the pancreas produces insufficient insulin and/or tissues become resistant to normal or high levels of insulin (insulin resistance), resulting in excessively high blood glucose levels. Multiple factors can lead to insulin resistance including chronically elevated blood glucose levels, genetics, obesity, lack of physical activity, and increasing age. Unlike type 1 diabetes, symptoms of type 2 diabetes are more salient, and as a result, the disease may not be diagnosed until several years after onset with a peak prevalence in adults near an age of 45 years. Unfortunately, the incidence of type 2 diabetes in children is increasing.
The primary goal of treatment of type 2 diabetes is to achieve and maintain glycemic control to reduce the risk of microvascular (diabetic neuropathy, retinopathy, or nephropathy) and macrovascular (heart disease, stroke, other forms of cardiovascular disease) complications. Current guidelines for the treatment of type 2 diabetes from the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) [Diabetes Care, 2008, 31 (12), 1 ] outline lifestyle modification including weight loss and increased physical activity as a primary therapeutic approach for management of type 2 diabetes. However, this approach alone fails in the majority of patients within the first year, leading physicians to prescribe medications over time. The ADA and EASD recommend metformin, an agent that reduces hepatic glucose production, as a Tier 1 a medication; however, a significant number of patients taking metformin can experience gastrointestinal side effects and, in rare cases, potentially fatal lactic acidosis. Recommendations for Tier 1 b class of medications include sulfonylureas, which stimulate pancreatic insulin secretion via modulation of potassium channel activity, and exogenous insulin. While both medications rapidly and effectively reduce blood glucose levels, insulin requires 1 -4 injections per day and both agents can cause undesired weight gain and potentially fatal hypoglycemia. Tier 2a recommendations include newer agents such as thiazolidinediones (TZDs pioglitazone and rosiglitazone), which enhance insulin sensitivity of muscle, liver and fat, as well as GLP-1 analogs, which enhance postprandial glucose-mediated insulin secretion from pancreatic beta cells. While TZDs show robust, durable control of blood glucose levels, adverse effects include weight gain, edema, bone fractures in women, exacerbation of congestive heart failure, and potential increased risk of ischemic cardiovascular events. GLP-1 analogs also effectively control blood glucose levels, however, this class of medications requires injection and many patients complain of nausea. The most recent addition to the Tier 2 medication list is DPP-4 inhibitors, which, like GLP-1 analogs, enhance glucose- medicated insulin secretion from beta cells. Unfortunately, DPP-4 inhibitors only modestly control blood glucose levels, and the long-term safety of DPP-4 inhibitors remains to be firmly established. Other less prescribed medications for type 2 diabetes include a-glucosidase inhibitors, glinides, and amylin analogs. Clearly, new medications with improved efficacy, durability, and side effect profiles are needed for patients with type 2 diabetes.
GLP-1 and GIP are peptides, known as incretins, that are secreted by L and K cells, respectively, from the gastrointestinal tract into the blood stream following ingestion of nutrients. This important physiological response serves as the primary signaling mechanism between nutrient (glucose/fat) concentration in the
gastrointestinal tract and other peripheral organs. Upon secretion, both circulating peptides initiate signals in beta cells of the pancreas to enhance glucose-stimulated insulin secretion, which, in turn, controls glucose concentrations in the blood stream (For reviews see: Diabetic Medicine 2007, 24(3), 223; Molecular and Cellular Endocrinology 2009, 297(1-2), 127; Experimental and Clinical Endocrinology & Diabetes 2001 , 109(Suppl. 2), S288).
The association between the incretin hormones GLP-1 and GIP and type 2 diabetes has been extensively explored. The majority of studies indicate that type 2 diabetes is associated with an acquired defect in GLP-1 secretion as well as GIP action (see Diabetes 2007, 56(8), 1951 and Current Diabetes Reports 2006, 6(3), 194). The use of exogenous GLP-1 for treatment of patients with type 2 diabetes is severely limited due to its rapid degradation by the protease DPP-4. Multiple modified peptides have been designed as GLP-1 mimetics that are DPP-4 resistant and show longer half-lives than endogenous GLP-1 . Agents with this profile that have been shown to be highly effective for treatment of type 2 diabetes include exenatide and liraglutide, however, these agents require injection. Oral agents that inhibit DPP-4, such as sitagliptin vildagliptin, and saxagliptin, elevate intact GLP-1 and modestly control circulating glucose levels (see Pharmacology & Therapeutics 2010, 125(2), 328; Diabetes Care 2007, 30(6), 1335; Expert Opinion on Emerging Drugs 2008, 13(4), 593). New oral medications that increase GLP-1 secretion would be desirable for treatment of type 2 diabetes.
Bile acids have been shown to enhance peptide secretion from the
gastrointestinal tract. Bile acids are released from the gallbladder into the small intestine after each meal to facilitate digestion of nutrients, in particular fat, lipids, and lipid-soluble vitamins. Bile acids also function as hormones that regulate cholesterol homeostasis, energy, and glucose homeostasis via nuclear receptors (FXR, PXR, CAR, VDR) and the G-protein coupled receptor TGR5 (for reviews see: Nature Drug Discovery 2008, 7, 672; Diabetes, Obesity and Metabolism 2008, 10, 1004). TGR5 is a member of the Rhodopsin-like subfamily of GPCRs (Class A) that is expressed in intestine, gall bladder, adipose tissue, liver, and select regions of the central nervous system. TGR5 is activated by multiple bile acids with lithocholic and deoxycholic acids as the most potent activators {Journal of Medicinal Chemistry 2008, 51(6), 1831 ). Both deoxycholic and lithocholic acids increase GLP-1 secretion from an enteroendocrine STC-1 cell line, in part through TGR5
{Biochemical and Biophysical Research Communications 2005, 329, 386). A synthetic TGR5 agonist INT-777 has been shown to increase intestinal GLP-1 secretion in vivo in mice {Cell Metabolism 2009, 10, 167). Bile salts have been shown to promote secretion of GLP-1 from colonic L cells in a vascularly perfused rat colon model {Journal of Endocrinology 1995, 145(3), 521 ) as well as GLP-1 , peptide YY (PYY), and neurotensin in a vascularly perfused rat ileum model {Endocrinology 1998, 139(9), 3780). In humans, infusion of deoxycholate into the sigmoid colon produces a rapid and marked dose responsive increase in plasma PYY and enteroglucagon concentrations (Gi/M993, 34(9), 1219). Agents that increase ileal and colonic bile acid or bile salt concentrations will increase gut peptide secretion including, but not limited to, GLP-1 and PYY.
Bile acids are synthesized from cholesterol in the liver then undergo conjugation of the carboxylic acid with the amine functionality of taurine and glycine. Conjugated bile acids are secreted into the gall bladder where accumulation occurs until a meal is consumed. Upon eating, the gall bladder contracts and empties its contents into the duodenum, where the conjugated bile acids facilitate absorption of cholesterol, fat, and fat-soluble vitamins in the proximal small intestine (For reviews see: Frontiers in Bioscience 2009, 74, 2584; Clinical Pharmacokinetics 2002,
41(10), 751 ; Journal of Pediatric Gastroenterology and Nutrition 2001 , 32, 407). Conjugated bile acids continue to flow through the small intestine until the distal ileum where 90% are reabsorbed into enterocytes via the apical sodium-dependent bile acid transporter (ASBT, also known as iBAT). The remaining 10% are deconjugated to bile acids by intestinal bacteria in the terminal ileum and colon of which 5% are then passively reabsorbed in the colon and the remaining 5% being excreted in feces. Bile acids that are reabsorbed by ASBT in the ileum are then transported into the portal vein for recirculation to the liver. This highly regulated process, called enterohepatic recirculation, is important for the body’s overall maintenance of the total bile acid pool as the amount of bile acid that is synthesized in the liver is equivalent to the amount of bile acids that are excreted in feces.
Pharmacological disruption of bile acid reabsorption with an inhibitor of ASBT leads to increased concentrations of bile acids in the colon and feces, a physiological consequence being increased conversion of hepatic cholesterol to bile acids to compensate for fecal loss of bile acids. Many pharmaceutical companies have pursued this mechanism as a strategy for lowering serum cholesterol in patients with dyslipidemia/hypercholesterolemia (For a review see: Current Medicinal Chemistry 2006, 73, 997). Importantly, ASBT-inhibitor mediated increase in colonic bile acid/salt concentration also will increase intestinal GLP-1 , PYY, GLP-2, and other gut peptide hormone secretion. Thus, inhibitors of ASBT could be useful for treatment of type 2 diabetes, type 1 diabetes, dyslipidemia, obesity, short bowel syndrome, Chronic Idiopathic Constipation, Irritable bowel syndrome (IBS), Crohn’s disease, and arthritis.
Certain 1 ,4-thiazepines are disclosed, for example in WO 94/18183 and WO 96/05188. These compounds are said to be useful as ileal bile acid reuptake inhibitors (ASBT).
Patent publication WO 201 1/137,135 dislcoses, among other compounds, the following compound. This patent publication also discloses methods of synthesis of the compound.
The preparation of the above compound is also disclosed in J. Med. Chem, Vol 56, pp5094-51 14 (2013).
Patent publication WO 201 1/137,135 dislcoses general methods for preparing the compound. In addition, a detailed synthesis of the compound is disclosed in Example 26. J. Med. Chem, Vol 56, pp5094-51 14 (2013) also discloses a method for synthesising the compound.
The present invention discloses an improved synthesis of the compound.
The synthetic scheme of the present invention is depicted in Scheme 1 .
Treatment of 2-methoxyphenyl acetate with sulfur monochloride followed by ester hydrolysis and reduction with zinc gave rise to thiophenol (A). Epoxide ring opening of (+)-2-butyl-ethyloxirane with thiophenol (A) and subsequent treatment of tertiary alcohol (B) with chloroacetonitrile under acidic conditions gave chloroacetamide (C), which was then converted to intermediate (E) by cleavage of the chloroacetamide with thiourea followed by classical resolution with dibenzoyl-L-tartaric acid.
Benzoylation of intermediate (E) with triflic acid and benzoyl chloride afforded intermediate (H). Cyclization of intermediate (H) followed by oxidation of the sulfide to a sulphone, subseguent imine reduction and classical resolution with (+)-camphorsulfonic acid provided intermediate (G), which was then converted to intermediate (H). Intermediate (H) was converted to the target compound using the methods disclosed in Patent publication WO 201 1/137,135.
Scheme 1
Dibenzoyl-L-tataric acid
The present invention also discloses an alternative method for construction of the quaternary chiral center as depicted in Scheme 2. Reaction of intermediate (A) with (R)-2-ammonio-2-ethylhexyl sulfate (K) followed by formation of di-p-toluoyl-L-tartrate salt furnished intermediate (L).
The present invention also discloses an alternative synthesis of intermediate (H) as illustrated in Scheme 3. Acid catalyzed cyclization of intermediate (F) followed by triflation gave imine (M), which underwent asymmetric reduction with catalyst lr(COD)2BArF and ligand (N) to give intermediate (O). Oxidation of the sulfide in intermediate (O) gave sulphone intermediate (H).
The present invention differs from the synthesis disclosed in WO 201 1/137,135 and J. Med. Chem, Vol56, pp5094-51 14 (2013) in that intermediate (H) in the present invention is prepared via a new, shorter and more cost-efficient synthesis while the synthesis of the target compound from intermediate (H) remains unchanged.
Intermediate A: 3-Hydroxy-4-methoxythiophenol
A reaction vessel was charged with 2-methoxyphenyl acetate (60 g, 0.36 mol), zinc chloride (49.2 g, 0.36 mol) and DME (600 mL). The mixture was stirred and S2CI2 (53.6 g, 0.40 mol) was added. The mixture was stirred at ambient temperature for 2 h. Concentrated HCI (135.4 mL, 1 .63 mol) was diluted with water (60 mL) and added slowly to the rxn mixture, maintaining the temperature below 60 °C. The mixture was stirred at 60 °C for 2 h and then cooled to ambient
temperature. Zinc dust (56.7 g, 0.87 mol) was added in portions, maintaining the temperature below 60 °C. The mixture was stirred at 20-60 °C for 1 h and then concentrated under vacuum to -300 mL. MTBE (1 .2 L) and water (180 mL) were added and the mixture was stirred for 10 min. The layers were separated and the organic layer was washed twice with water (2x 240 mL). The layers were separated and the organic layer was concentrated under vacuum to give an oil. The oil was distilled at 1 10-1 15 °C/2 mbar to give the title compound (42 g, 75%) as colorless oil, which solidified on standing to afford the title compound as a white solid. M.P. 41 -42 °C. 1 H NMR (500 MHz, CDCI3)$ ppm 3.39 (s, 1 H), 3.88 (s, 3H), 5.65 (br. S, 1 H), 6.75 (d, J – 8.3 Hz, 1 H), 6.84 (ddd, J – 8.3, 2.2, 0.6 Hz, 1 H), 6.94 (d, J – 2.2 Hz).
Intermediate E: (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol, dibenzoyl-L-tartrate salt
A reaction vessel was charged with 3-hydroxy-4-methoxythiophenol (5.0 g, 25.2 mmol), (+)-2-butyl-2-ethyloxirane (3.56 g, 27.7 mmol) and EtOH (30 mL). The mixture was treated with a solution of NaOH (2.22 g, 55.5 mmol) in water (20 mL), heated to 40 °C and stirred at 40 °C for 5 h. The mixture was cooled to ambient temperature, treated with toluene (25 mL) and stirred for 10 min. The layers were separated and the organic layer was discarded. The aqueous layer was neutralized with 2N HCI (27.8 mL, 55.6 mmol) and extracted with toluene (100 mL). The organic layer was washed with water (25 mL), concentrated in vacuo to give an oil. The oil was treated with chloroacetonitrile (35.9 mL) and HOAc (4.3 mL) and cooled to 0 °C. H2SO4 (6.7 mL, 126 mmol, pre-diluted with 2.3 mL of water) was added at a rate maintaining the temperature below 10 °C. After stirred at 0 °C for 0.5 h, the reaction mixture was treated with 20% aqueous Na2CO3 solution to adjust the pH to
7-8 and then extracted with MTBE (70 ml_). The extract was washed with water (35 ml_) and concentrated in vacuo to give an oil. The oil was then dissolved in EOH (50 ml_) and treated with HOAc (10 ml_) and thiourea (2.30 g, 30.2 mmol). The mixture was heated at reflux overnight and then cooled to ambient temperature. The solids were filtered and washed with EtOH (10 ml_). The filtrate and the wash were combined and concentrated in vacuo, treated with MTBE (140 ml_) and washed successively with 10% aqueous Na2C03 and water. The mixture was concentrated in vacuo to give an oil. The oil was dissolved in MeCN (72 ml_), heated to -50 °C and then dibenzoyl-L-tartaric acid (9.0 g, 25.2 mmol) in acetonitrile (22 ml_) was added slowly. Seed crystals were added at -50 °C. The resultant slurry was stirred at 45-50 °C for 5 h, then cooled down to ambient temperature and stirred at ambient temperature overnight. The solids were filtered and washed with MeCN (2x 22 ml_). The wet cake was treated with MeCN (150 ml_) and heated to 50 °C. The slurry was stirred at 50 °C for 5 h, cooled over 1 h to ambient temperature and stirred at ambient temperature overnight. The solids were collected by filtration, washed with MeCN (2 x 20 ml_), dried under vacuum to give the title compound (5.5 g, 34% overall yield, 99.5% purity, 93.9% ee) as a white solid. 1 H NMR (500 MHz, DMSO-d6) δ ppm 0.78 (m, 6H), 1 .13 (m, 4H), 1 .51 (m, 2H), 1 .58 (q, J – 7.7 Hz, 2H), 3.08 (s, 2H), 3.75 (s, 3H), 5.66 (s, 2H), 6.88 (m, 2H), 6.93 (m, 1 H), 7.49 (m, 4H), 7.63 (m, 2H), 7.94 (m, 4H). EI-LCMS m/z 284 (M++1 of free base).
A suspension of (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol, dibenzoyl-L-tartrate salt (29 g, 45.2 mmol) in DCM (435 mL) was treated with water (1 16 mL) and 10% aqueous Na2C03 solution (1 16 mL). The mixture was stirred at ambient temperature until all solids were dissolved (30 min). The layers were separated. The organic layer was washed with water (2 x 60 mL), concentrated under vacuum to give (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol (free base) as an off-white solid (13.0 g, quantitative). A vessel was charged with TfOH (4.68 ml, 52.9 mmol) and DCM (30 mL) and the mixture was cooled to 0 °C. 5 g (17.6 mmol) of (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol (free base) was dissolved in DCM (10 mL) and added at a rate maintaining the temperature below 10 °C. Benzoyl chloride (4.5 mL, 38.8 mmol) was added at a rate maintaining the temperature below 10 °C. The mixture was then heated to reflux and stirred at reflux for 48 h. The mixture was cooled to 30 °C. Water (20 mL) was added and the mixture was concentrated to remove DCM. EtOH (10 mL) was added. The mixture was heated to 40 ° C, treated with 50% aqueous NaOH solution (10 mL) and stirred at 55 °C. After 1 h, the mixture was cooled to ambient temperature and the pH was adjusted to 6-7 with cone. HCI. The mixture was concentrated in vacuo to remove EtOH. EtOAc (100 mL) was added. The mixture was stirred for 5 min and the layers were separated. The organic layer was washed successively with 10% aqueous Na2CO3 (25 mL) and water (25 mL) and then concentrated in vacuo. The resultant oil was treated with DCM (15 mL). The resultant thick slurry was further diluted with DCM (15 mL) followed by addition of Hexanes (60 mL). The slurry was stirred for 5 min, filtered, washed with DCM/hexanes (1 :2, 2 x 10 mL) and dried under vacuum to give the title compound (7.67 g, 80%) as a yellow solid. 1 NMR (500 MHz, DMSO-d6) δ ppm 0.70 (t, 7.1 Hz, 3 H), 0.81 (t, 7.1 Hz, 3H), 1 .04-1 .27 (m, 8H), 2.74 (s, 2H), 3.73 (s, 3H), 6.91 (s, 1 H), 7.01 (s, 1 H), 7.52 (dd, J – 7.8, 7.2 Hz, 2H), 7.63 (t, J = 7.2 Hz, 1 H), 7.67 (d, J = 7.8 Hz, 2H). EI-LCMS m/z 388 (M++1 ).
Intermediate G: (3R,5R)-3-butyl-3-ethyl-8-hydroxy-7-methoxy-5-phenyl-2,3,4,5-tetrahydrobenzo[f][1 ,4]thiazepine 1 ,1 -dioxide, (+)-camphorsulfonate salt
A vessel was charged with (R)-(2-((2-amino-2-ethylhexyl)thio)-4-hydroxy-5-methoxyphenyl)(phenyl)methanone (1 .4 g, 3.61 mmol), toluene (8.4 ml_) and citric acid (0.035 g, 0.181 mmol, 5 mol%). The mixture was heated to reflux overnight with a Dean-Stark trap to remove water. The mixture was concentrated under reduced pressure to remove solvents. Methanol (14.0 ml_) and oxone (2.22 g, 3.61 mmol, 1 .0 equiv) were added. The mixture was stirred at gentle reflux for 2 h. The mixture was cooled to ambient temperature, and filtered to remove solids. The filter cake was washed with small amount of Methanol. The filtrate was cooled to 5 °C, and treated with sodium borohydride (0.410 g, 10.84 mmol, 3.0 equiv.) in small portions. The mixture was stirred at 5 °C for 2 h and then concentrated to remove the majority of solvents. The mixture was quenched with Water (28.0 ml_) and extracted with EtOAc (28.0 ml_). The organic layer was washed with brine, and then concentrated to remove solvents. The residue was dissolved in MeCN (14.0 ml_) and concentrated again to remove solvents. The residue was dissolved in MeCN (7.00 ml_) and MTBE (7.00 ml_) at 40 °C, and treated with (+)-camphorsulfonic acid (0.839 g, 3.61 mmol, 1 .0 equiv.) at 40 °C for 30 min. The mixture was cooled to ambient temperature, stirred for 2 h, and filtered to collect solids. The filter cake was washed with MTBE/MeCN (2:1 , 3 ml_), and dried at 50 °C to give the title compound (0.75 g, 32% overall yield, 98.6 purity, 97% de, 99.7% ee) as white solids. 1 NMR (400 MHz, CDCI3) δ ppm 0.63 (s, 3H), 0.88 (t, J – 6.9 Hz, 3H), 0.97 (m, 6H), 1 .29-1 .39 (m, 5H), 1 .80-1 .97 (m, 6H), 2.08-2.10 (m, 1 H), 2.27 (d, J – 17.3 Hz, 1 H), 2.38-2.44 (m, 3H), 2.54 (b, 1 H), 2.91 (b, 1 H), 3.48 (d, J – 15.4 Hz, 1 H), 3.79 (s, 3H), 4.05 (d, J – 17.2 Hz, 1 H), 6.45 (s, 1 H), 6.56 (s, 1 H), 7.51 -7.56 (m, 4H), 7.68 (s, 1 H), 7.79 (b, 2H), 1 1 .46 (b, 1 H). EI-LCMS m/z 404 (M++1 of free base).
Method 1 : A mixture of (3R,5R)-3-butyl-3-ethyl-8-hydroxy-7-methoxy-5-phenyl-2,3,4,5-tetrahydrobenzo[f][1 ,4]thiazepine 1 ,1 -dioxide, (+)-camphorsulfonate salt (0.5 g, 0.786 mmol), EtOAc (5.0 mL), and 10% of Na2C03 aqueuous solution (5 mL) was stirred for 15 min. The layers were separated and the aqueous layer was discarded. The organic layer was washed with dilute brine twice, concentrated to remove solvents. EtOAc (5.0 mL) was added and the mixture was concentrated to give a pale yellow solid free base. 1 ,4-Dioxane (5.0 mL) and pyridine (0.13 mL, 1 .57 mmol) were added. The mixture was cooled to 5-10 °C and triflic anhydride (0.199 mL, 1 .180 mmol) was added while maintaining the temperature below 15 °C. The mixture was stirred at ambient temperature until completion deemed by HPLC (1 h). Toluene (5 mL) and water (5 mL) were added. Layers were separated. The organic layer was washed with water, concentrated to remove solvents. Toluene (1 .0 mL) was added to dissolve the residue followed by Isooctane (4.0 mL). The mixture was stirred at rt overnight. The solids was filtered, washed with Isooctane (4.0 mL) to give the title compound (0.34 g, 81 %) as slightly yellow solids. 1 NMR (400 MHz, CDCI3) δ ppm 0.86 (t, J – 7.2 Hz, 3H), 0.94 (t, J – 7.6 Hz, 3H), 1 .12-1 .15 (m, 1 H), 1 .22-1 .36 (m, 3H), 1 .48-1 .60 (m, 2H), 1 .86-1 .93 (m, 2H), 2.22 (dt, J = 4.1 Hz, 12 Hz, 1 H), 3.10 (d, J – 14.8 Hz, 1 H), 3.49 (d, J – 14.8 Hz, 1 H), 3.64 (s, 3H), 6.1 1 (s, 1 H), 6.36 (s, 1 H), 7.38-7.48 (m, 5), 7.98 (s, 1 H).
Method 2: A mixture of (R)-3-butyl-3-ethyl-7-methoxy-5-phenyl-2,3-dihydrobenzo[f][1 ,4]thiazepin-8-yl trifluoromethanesulfonate (0.5 g, 0.997 mmol), ligand (N) (0.078 g, 0.1 10 mmol) and lr(COD)2BArF (0.127 g, 0.100 mmol) in DCM (10.0 mL) was purged with nitrogen three times, then hydrogen three times. The mixture was shaken in Parr shaker under 10 Bar of H2 for 24 h. The experiment described above was repeated with 1 .0 g (1 .994 mmol) input of (R)-3-butyl-3-ethyl-7-methoxy-5-phenyl-2,3-dihydrobenzo[f][1 ,4]thiazepin-8-yl
trifluoromethanesulfonate. The two batches of the reaction mixture were combined,
concentrated to remove solvents, and purified by silica gel chromatography
(hexanes:EtOAc =9:1 ) to give the sulfide (O) as slightly yellow oil (0.6 g, 40% yield, 99.7% purity). The oil (0.6 g, 1 .191 mmol) was dissolved in TFA (1 .836 mL, 23.83 mmol) and stirred at 40 °C. H202 (0.268 mL, 2.62 mmol) was added slowly over 30 min. The mixture was stirred at 40 °C for 2 h and then cooled to room temperature. Water (10 mL) and toluene (6.0 mL) were added. Layers were separated and the organic layer was washed successively with aqueous sodium carbonate solution and wate, and concentrated to dryness. Toluene (6.0 mL) was added and the mixture was concentrated to dryness. The residue was dissolved in toluene (2.4 mL) and isooctane (7.20 mL) was added. The mixture was heated to reflux and then cooled to room temperature. The mixture was stirred at room temperature for 30 min. The solid was filtered and washed with isooctane to give the title compound (0.48 g, 75%).
Intermediate L: (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol, di-p-toluoyl-L-tartrate salt
To a mixture of (R)-2-amino-2-ethylhexyl hydrogen sulfate (1 1 .1 g, 49.3 mmol) in water (23.1 mL) was added NaOH (5.91 g, 148 mmol). The mixture was stirred at reflux for 2 h. The mixture was cooled to room temperature and extracted with MTBE (30.8 mL). The extract was washed with brine (22 mL), concentrated under vacuum and treated with methanol (30.8 mL). The mixture was stirred under nitrogen and treated with 3-hydroxy-4-methoxythiophenol (7.70 g, 49.3 mmol). The mixture was stirred under nitrogen at room temperature for 1 h. The mixture was concentrated under vacuum, treated with acetonitrile (154 mL) and then heated to 45 °C. To the stirred mixture was added (2R,3R)-2,3-bis((4-methylbenzoyl)oxy)succinic acid (19.03 g, 49.3 mmol). The resultant slurry was
stirred at 45 °C. After 2 h, the slurry was cooled to room temperature and stirred for 5 h. The solids were filtered, washed twice with acetonitrile (30 mL) and dried to give the title compound (28.0 g, 85%) as white solids. 1 NMR (400 MHz, DMSO-d6) δ (ppm): 0.70-0.75 (m, 6H), 1 .17 (b, 4H), 1 .46-1 .55 (m, 4H), 2.30 (s, 6H), 3.71 (s, 3H), 5.58 (s, 2H), 6.84 (s, 2H), 6.89 (s, 1 H), 7.24 (d, J – 1 1 .6 Hz, 4H), 7.76 (d, J – 1 1 .6 Hz, 4H).
A flask was charged with (R)-(2-((2-amino-2-ethylhexyl)thio)-4-hydroxy-5-methoxyphenyl)(phenyl)methanone (3.5 g, 9.03 mmol), citric acid (0.434 g, 2.258 mmol), 1 ,4-Dioxane (17.50 mL) and Toluene (17.50 mL). The mixture was heated to reflux with a Dean-Stark trap to distill water azetropically. The mixture was refluxed for 20 h and then cooled to room temperature. EtOAc (35.0 mL) and water (35.0 mL) were added and layers were separated. The organic layer was washed with aqueous sodium carbonate solution and concentrated to remove solvents to give crude imine as brown oil. The oil was dissolved in EtOAc (35.0 mL) and cooled to 0-5 °C. To the mixture was added triethylamine (1 .888 mL, 13.55 mmol) followed by slow addition of Tf2O (1 .831 mL, 10.84 mmol) at 0-5 °C. The mixture was stirred at room temperature for 1 h. Water was added and layers were separated. The organic layer was washed with brine, dried over Na2SO4 and concentrated under vacuum. The crude triflate was purified by silica gel chromatography
Journal of Medicinal Chemistry (2013), 56(12), 5094-5114.
The apical sodium-dependent bile acid transporter (ASBT) transports bile salts from the lumen of the gastrointestinal (GI) tract to the liver via the portal vein. Multiple pharmaceutical companies have exploited the physiological link between ASBT and hepatic cholesterol metabolism, which led to the clinical investigation of ASBT inhibitors as lipid-lowering agents. While modest lipid effects were demonstrated, the potential utility of ASBT inhibitors for treatment of type 2 diabetes has been relatively unexplored. We initiated a lead optimization effort that focused on the identification of a potent, nonabsorbable ASBT inhibitor starting from the first-generation inhibitor 264W94 (1). Extensive SAR studies culminated in the discovery of GSK2330672 (56) as a highly potent, nonabsorbable ASBT inhibitor which lowers glucose in an animal model of type 2 diabetes and shows excellent developability properties for evaluating the potential therapeutic utility of a nonabsorbable ASBT inhibitor for treatment of patients with type 2 diabetes.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Example 26: 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl- 2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid
Method 1 , Step 1 : To a solution of (3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-5- phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepine-8-carbaldehyde 1 ,1 -dioxide (683 mg, 1 .644 mmol) in 1 ,2-dichloroethane (20 mL) was added diethyl 3- aminopentanedioate (501 mg, 2.465 mmol) and acetic acid (0.188 mL, 3.29 mmol). The reaction mixture was stirred at room temperature for 1 hr then treated with NaHB(OAc)3 (697 mg, 3.29 mmol). The reaction mixture was then stirred at room temperature overnight and quenched with aqueous potassium carbonate solution. The mixture was extracted with DCM. The combined organic layers were washed with H2O, saturated brine, dried (Na2SO4), filtered, and concentrated under reduced pressure to give diethyl 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5- phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioate (880 mg, 88%) as a light yellow oil: MS-LCMS m/z 603 (M+H)+.
Method 1 , Step 2: To a solution of diethyl 3-({[(3R,5R)-3-butyl-3-ethyl-7- (methyloxy)-l ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioate (880 mg, 1 .460 mmol) in a 1 :1 :1 mixture of
THF/MeOH/H2O (30 mL) was added lithium hydroxide (175 mg, 7.30 mmol). The reaction mixture was stirred at room temperature overnight then concentrated under reduced pressure. H2O and MeCN was added to dissolve the residue. The solution was acidified with acetic acid to pH 4-5, partially concentrated to remove MeCN under reduced pressure, and left to stand for 30 min. The white precipitate was collected by filtration and dried under reduced pressure at 50°C overnight to give the title compound (803 mg, 100%) as a white solid: 1 H NMR (MeOH-d4) δ ppm 8.05 (s, 1 H), 7.27 – 7.49 (m, 5H), 6.29 (s, 1 H), 6.06 (s, 1 H), 4.25 (s, 2H), 3.60 – 3.68 (m, 1 H), 3.58 (s, 3H), 3.47 (d, J = 14.8 Hz, 1 H), 3.09 (d, J = 14.8 Hz, 1 H), 2.52 – 2.73 (m, 4H), 2.12 – 2.27 (m, 1 H), 1 .69 – 1 .84 (m, 1 H), 1 .48 – 1 .63 (m, 1 H), 1 .05 – 1 .48 (m, 5H), 0.87 (t, J = 7.4 Hz, 3H), 0.78 (t, J = 7.0 Hz, 3H); ES-LCMS m/z 547 (M+H) Method 2: A solution of dimethyl 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-
1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioate (~ 600 g) in THF (2.5 L) and MeOH (1 .25 L) was cooled in an ice-bath and a solution of NaOH (206 g, 5.15 mol) in water (2.5 L) was added dropwise over 20 min (10-22°C reaction temperature). After stirring 20 min, the solution was concentrated (to remove THF/MeOH) and acidified to pH~4 with concentrated HCI. The precipitated product was aged with stirring, collected by filtration and air dried overnight. A second 600g batch of dimethyl 3-({[(3R,5R)-3- butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4- benzothiazepin-8-yl]methyl}amino)pentanedioate was saponified in a similar fashion. The combined crude products (~2 mol theoretical) were suspended in CH3CN (8 L) and water (4 L) and the stirred mixture was heated to 65°C. A solution formed which was cooled to 10°C over 2 h while seeding a few times with an authentic sample of the desired crystalline product. The resulting slurry was stirred at 10°C for 2 h, and the solid was collected by filtration. The filter cake was washed with water and air-dried overnight. Further drying to constant weight in a vacuum oven at 55°C afforded crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 – dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioic acid as a white solid (790 g).
Method 3: (3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-5-phenyl-2,3,4,5-tetrahydro- 1 ,4-benzothiazepine-8-carbaldehyde 1 ,1 -dioxide (1802 grams, 4.336 moles) and dimethyl 3-aminopentanedioate (1334 grams, 5.671 moles) were slurried in iPrOAc (13.83 kgs). A nitrogen atmosphere was applied to the reactor. To the slurry at 20°C was added glacial acetic acid (847 ml_, 14.810 moles), and the mixture was stirred until complete dissolution was observed. Solid sodium triacetoxyborohydride (1424 grams, 6.719 moles) was next added to the reaction over a period of 7 minutes. The reaction was held at 20°C for a total of 3 hours at which time LC analysis of a sample indicated complete consumption of the (3R,5R)-3-butyl-3-ethyl- 7-(methyloxy)-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepine-8-carbaldehyde 1 ,1 – dioxide. Next, water (20.36 kgs) and brine (4.8 kgs) were added to the reactor. The contents of the reactor were stirred for 10 minutes and then settled for 10 minutes. The bottom, aqueous layer was then removed and sent to waste. A previously prepared, 10% (wt/wt) aqueous solution of sodium bicarbonate (22.5 L) was added to the reactor. The contents were stirred for 10 minutes and then settled for 10 minutes. The bottom, aqueous layer was then removed and sent to waste. To the reactor was added a second wash of 10% (wt/wt) aqueous, sodium bicarbonate
(22.5 L). The contents of the reactor were stirred for 10 minutes and settled for 10 minutes. The bottom, aqueous layer was then removed and sent to waste. The contents of the reactor were then reduced to an oil under vacuum distillation. To the oil was added THF (7.15 kgs) and MeOH (3.68 kgs). The contents of the reactor were heated to 55°C and agitated vigorously until complete dissolution was observed. The contents of the reactor were then cooled to 25°C whereupon a previously prepared aqueous solution of NaOH [6.75 kgs of water and 2.09 kgs of NaOH (50% wt wt solution)] was added with cooling being applied to the jacket. The contents of the reactor were kept below 42°C during the addition of the NaOH solution. The temperature was readjusted to 25°C after the NaOH addition, and the reaction was stirred for 75 minutes before HPLC analysis indicated the reaction was complete. Heptane (7.66 kgs) was added to the reactor, and the contents were stirred for 10 minutes and then allowed to settle for 10 minutes. The aqueous layer was collected in a clean nalgene carboy. The heptane layer was removed from the reactor and sent to waste. The aqueous solution was then returned to the reactor, and the reactor was prepared for vacuum distillation. Approximately 8.5 liters of distillate was collected during the vacuum distillation. The vacuum was released from the reactor, and the temperature of the contents was readjusted to 25°C. A 1 N HCI solution (30.76 kgs) was added to the reactor over a period of 40 minutes. The resulting slurry was stirred at 25°C for 10 hours then cooled to 5°C over a period of 2 hours. The slurry was held at 5°C for 4 hours before the product was collected in a filter crock by vacuum filtration. The filter cake was then washed with cold (5°C) water (6 kgs). The product cake was air dried in the filter crock under vacuum for approximately 72 hours. The product was then transferred to three drying trays and dried in a vacuum oven at 50°C for 79 hours. The temperature of the vacuum oven was then raised to 65°C for 85 additional hours. The product was off-loaded as a single batch to give 2568 grams (93.4% yield) of intermediate grade 3-({[(3R,5R)-3- butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4- benzothiazepin-8-yl]methyl}amino)pentanedioic acid as an off-white solid.
Intermediate grade 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5- phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid was dissolved (4690 g) in a mixture of glacial acetic acid (8850 g) and purified water (4200 g) at 70°C. The resulting solution was transferred through a 5 micron polishing filter while maintaining the temperature above 30°C. The reactor and filter were rinsed through with a mixture of glacial acetic acid (980 g) and purified water (470 g). The solution temperature was adjusted to 50°C. Filtered purified water (4230 g) was added to the solution. The cloudy solution was then seeded with crystalline 3-({[(3 5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3 ,4,5- tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid (10 g). While maintaining the temperature at 50°C, filtered purified water was charged to the slurry at a controlled rate (1 1030 g over 130 minutes). Additional filtered purified water was then added to the slurry at a faster controlled rate (20740 g over 100 minutes). A final charge of filtered purified water (3780 g) was made to the slurry. The slurry was then cooled to 10°C at a linear rate over 135 minutes. The solids were filtered over sharkskin filter paper to remove the mother liquor. The cake was then rinsed with filtered ethyl acetate (17280 g) then the wash liquors were removed by filtration. The resulting wetcake was isolated into trays and dried under vacuum at 50°C for 23 hours. The temperature was then increased to 60°C and drying was continued for an additional 24 hours to afford crystalline 3-({[(3R,5R)-3-butyl-3-ethyl- 7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioic acid (3740 g, 79.7% yield) as a white solid.
To a slurry of this crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 – dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioic acid (3660 g) and filtered purified water (3.6 L) was added filtered glacial acetic acid (7530 g). The temperature was increased to 60°C and full dissolution was observed. The temperature was reduced to 55°C, filtered, and treated with purified water (3.2 L). The solution was then seeded with crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5- tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid (18 g) to afford a slurry. Filtered purified water was charged to the slurry at a controlled rate (9 L over 140 minutes). Additional filtered purified water was then added to the slurry at a faster controlled rate (18 L over 190 minutes). The slurry was then cooled to
10°C at a linear rate over 225 minutes. The solids were filtered over sharkskin filter paper to remove the mother liquor. The cake was then rinsed with filtered purified water (18 L), and the wash liquors were removed by filtration. The resulting wetcake was isolated into trays and dried under vacuum at 60°C for 18.5 hours to afford a crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl- 2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid (3330 g, 90.8% yield) as a white solid which was analyzed for crystallinity as summarized below.
Paper
Cowan, D. J.; Collins, J. L.; Mitchell, M. B.; Ray, J. A.; Sutton, P. W.; Sarjeant, A. A.; Boros, E. E.Enzymatic- and Iridium-Catalyzed Asymmetric Synthesis of a Benzothiazepinylphosphonate Bile Acid Transporter InhibitorJ. Org. Chem.2013, 78 ( 24) 12726– 12734, DOI: 10.1021/jo402311e
A synthesis of the benzothiazepine phosphonic acid 3, employing both enzymatic and transition metal catalysis, is described. The quaternary chiral center of 3 was obtained by resolution of ethyl (2-ethyl)norleucinate (4) with porcine liver esterase (PLE) immobilized on Sepabeads. The resulting (R)-amino acid (5) was converted in two steps to aminosulfate 7, which was used for construction of the benzothiazepine ring. Benzophenone 15, prepared in four steps from trimethylhydroquinone 11, enabled sequential incorporation of phosphorus (Arbuzov chemistry) and sulfur (Pd(0)-catalyzed thiol coupling) leading to mercaptan intermediate 18. S-Alkylation of 18 with aminosulfate 7 followed by cyclodehydration afforded dihydrobenzothiazepine 20. Iridium-catalyzed asymmetric hydrogenation of 20 with the complex of [Ir(COD)2BArF] (26) and Taniaphos ligand P afforded the (3R,5R)-tetrahydrobenzothiazepine 30 following flash chromatography. Oxidation of 30 to sulfone 31 and phosphonate hydrolysis completed the synthesis of 3 in 12 steps and 13% overall yield.
Paper
Scheme 1. Current Route to Chiral Intermediate 4 in the Synthesis of GSK2330672
Development of an Enzymatic Process for the Production of (R)-2-Butyl-2-ethyloxirane
†Synthetic Biochemistry, Advanced Manufacturing Technologies, ‡API Chemistry, ∥Protein and Cellular Sciences, GlaxoSmithKline, Medicines Research Centre, Gunnels Wood Road, Stevenage SG1 2NY, United Kingdom
§API Chemistry, ⊥Synthetic Biochemistry, Advanced Manufacturing Technologies, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
# Biotechnology and Environmental Shared Service, Global Manufacturing and Supply, GlaxoSmithKline, Dominion Way, Worthing BN14 8PB, United Kingdom
∇ Molecular Design, Computational and Modeling Sciences, GlaxoSmithKline, 1250 S. Collegeville Road, Collegeville, Pennsylvania 19426, United States
An epoxide resolution process was rapidly developed that allowed access to multigram scale quantities of (R)-2-butyl-2-ethyloxirane 2 at greater than 300 g/L reaction concentration using an easy-to-handle and store lyophilized powder of epoxide hydrolase from Agromyces mediolanus. The enzyme was successfully fermented on a 35 L scale and stability increased by downstream processing. Halohydrin dehalogenases also gave highly enantioselective resolution but were shown to favor hydrolysis of the (R)-2 epoxide, whereas epoxide hydrolase from Aspergillus nigerinstead provided (R)-7 via an unoptimized, enantioconvergent process.
REFERENCES
1: Nunez DJ, Yao X, Lin J, Walker A, Zuo P, Webster L, Krug-Gourley S, Zamek-Gliszczynski MJ, Gillmor DS, Johnson SL. Glucose and lipid effects of the ileal apical sodium-dependent bile acid transporter inhibitor GSK2330672: double-blind randomized trials with type 2 diabetes subjects taking metformin. Diabetes Obes Metab. 2016 Jul;18(7):654-62. doi: 10.1111/dom.12656. Epub 2016 Apr 21. PubMed PMID: 26939572.
2: Wu Y, Aquino CJ, Cowan DJ, Anderson DL, Ambroso JL, Bishop MJ, Boros EE, Chen L, Cunningham A, Dobbins RL, Feldman PL, Harston LT, Kaldor IW, Klein R, Liang X, McIntyre MS, Merrill CL, Patterson KM, Prescott JS, Ray JS, Roller SG, Yao X, Young A, Yuen J, Collins JL. Discovery of a highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor (GSK2330672) for treatment of type 2 diabetes. J Med Chem. 2013 Jun 27;56(12):5094-114. doi: 10.1021/jm400459m. Epub 2013 Jun 6. PubMed PMID: 23678871.
Form I may be prepared according to the procedures in WO2011/137135, Example 26.
Method 2
Form I of linerixibat was prepared according to the following procedure at a large scale (> 500 g). All charges were based on input linerixibat.
An intermediate grade linerixibat was dissolved in acetonitrile / Water (12 vols / 8 vols) at reflux (~76 °C). The solution was seeded at 70°C with Form I (2% w/w), cooled to 60 °C over 15 mins and aged at 60 °C for 2 hrs. Water (14 vols) was added over 8 hrs and aged for 1 hr. The suspension was cooled to 20 °C over 1 hr and aged for >30 mins. The slurry was filtered, washed with acetonitrile: water (6:11 v/v) (3.5 vols), then twice with water (2 vols) and blown down with nitrogen 8-18 hrs. Drying was carried out under vacuum at 40-50 °C without agitation until the Karl Fisher measurement (KF) was < 10% w/w. The batch was agitated at 4 rpm for 2 mins every 3 hrs until the KF <1 % w/w to provide Form I.
Form I can also be prepared by the above procedure without the step of seeding. Method 3
Form I of linerixibat was prepared according to the following procedure at a large scale (> 50 kg). A reactor (Reactor 1) was charged with 55.84 kg GSK2330672B (1.0 wt) intermediate grade (IG) followed by acetonitrile (12 vol) and purified water (8 vol). The mixture was heated to reflux (74-79°C), and held until complete dissolution is observed. The solution was then transferred to a reactor (Reactor 2) that had been pre-heated to 74-79°C via filter (0.22pm pipe-line filter). Reactor 1 was rinsed with acetonitrile (MeCN) (0.3 vol) and purified water (0.2 vol), and the solution in Reactor 1 was transferred to Reactor 2 via filter (0.22pm pipe-line filter). The contents of Reactor 2 were held until complete dissolution is observed. The solution in Reactor 2 was cooled to 69-72°C, then seeded with 2 w/w% (based on pure GSK2330672B input). The suspension was cooled to 58-62°C within 10-20 min. The suspension was held at 58-62°C for 2h. Purified water (14 vol) was added over 8 hrs. After the addition was complete, the slurry was held at 58-62°C for 60 min, then cooled to 18-25°C over 50-70 min. The slurry was stirred at 18-25°C for not less than 30 min, then the suspension filtered under vacuum. The reactor was rinsed with MeCN/water (6/11 v:v, 3.5 vol) and the rinse used to wash the cake. The cake was washed twice with water (2 vol). The cake was blown down with nitrogen and dried at 60°C under vacuum to give 46.35 kg GSK2330672B Form 1 solid.
Method 4
10.94g of GSK2330672B was charged and washed into a vessel using 131mL of acetonitrile (MeCN) and 88mL of water. The slurry was then heated to reflux. 4mL of 3:2v/v MeCN/water was charged followed by 5.5mL of 3:2v/v MeCN/water. The contents were cooled to 70 °C, then cooled to 60°C over 15 minutes and held stirring for 2 hours. 153 mL water was added over 8 hours, the contents were cooled to 20°C over 1 hour, and held stirring for 1 hour. The product was isolated, washed with 38mL of 6:11 MeCN/water, then washed twice with 22mL of water. After deliquoring, the product was dried at 45°C under vacuum, to give 9.35g (85.5%w/w) Form I GSK2330672B.
GSK2982772 is a potent and selective receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate for the Treatment of Inflammatory Diseases. GSK2982772 is, currently in phase 2a clinical studies for psoriasis, rheumatoid arthritis, and ulcerative colitis. GSK2982772 potently binds to RIP1 with exquisite kinase specificity and has excellent activity in blocking many TNF-dependent cellular responses. RIP1 has emerged as an important upstream kinase that has been shown to regulate inflammation through both scaffolding and kinase specific functions.
GSK-2982772, an oral receptor-interacting protein-1 (RIP1) kinase inhibitor, is in phase II clinical development at GlaxoSmithKline for the treatment of active plaque-type psoriasis, moderate to severe rheumatoid arthritis, and active ulcerative colitis. A phase I trial was also completed for the treatment of inflammatory bowel disease using capsule and solution formulations.
Originator GlaxoSmithKline
Class Antipsoriatics
Mechanism of Action Receptor-interacting protein serine-threonine kinase inhibitors
Highest Development Phases
Phase II Plaque psoriasis; Rheumatoid arthritis; Ulcerative colitis
Phase I Inflammatory bowel diseases
Most Recent Events
15 Dec 2016 Biomarkers information updated
01 Nov 2016 Phase-II clinical trials in Ulcerative colitis (Adjunctive treatment) in USA (PO) (NCT02903966)
01 Oct 2016 Phase-II clinical trials in Rheumatoid arthritis in Poland (PO) (NCT02858492)
PHASE 2 Psoriasis, plaque GSK
Inflammatory Bowel Disease, Agents for
Rheumatoid Arthritis, Treatment of
Antipsoriatics
Data Science and Informatics Leader | Innovation Advocate
GSK
University of North Carolina at Chapel Hill
He is a data scientist and innovator with experience in both early and late stages of drug development. his current role involves the late stage of drug product development. I’m leading a project to bring GSK’s large molecule process and analytical data onto our big data platform and develop new data analysis and modeling capabilities. Also, working within GSK’s Advanced Manufacturing Technology (AMT) initiative provides plenty of other opportunities to impact how we make medicines.
Previously as a computational chemist (i.e. a data scientist in drug discovery), he worked with scientists from many domains, including chemists, biologists, and other informaticians. he enjoys digging into all the computational aspects of life science research, and solving data challenges by exploiting adjacencies and connections – between diverse fields of knowledge, and the equally diverse scientists trained in them.
He has supported multiple drug discovery projects at GSK starting from target identification (“how should we modulate disease X?”) through to candidate selection and early clinical development (“let’s see if what we discovered can become a medicine”). Deriving insight by custom data integration is one of my specialties; recently he designed and implemented a platform for integrating data sets from multiple experiments that will be used by GSK screening scientists to find and combine hits.
A trained computer scientist and cheminformatician, he is an active member of the algorithms, data science and internal innovation communities at GSK, leading many of these efforts.
His Ph.D. work introduced new computational geometry techniques for structural bioinformatics and protein function prediction. I have touched on several other subject areas:
* data mining/machine learning (predictive modeling and graph mining), * computer graphics and augmented reality (one of the pioneers of projection mapping) * robotics (keen current interest and future aspiration)
Receptor-interacting protein- 1 (RIP1) kinase, originally referred to as RIP, is a TKL family serine/threonine protein kinase involved in innate immune signaling. RIPl kinase is a RHIM domain containing protein, with an N-terminal kinase domain and a C-terminal death domain ((2005) Trends Biochem. Sci. 30, 151-159). The death domain of RIPl mediates interaction with other death domain containing proteins including Fas and TNFR-1 ((1995) Cell 81 513-523), TRAIL-Rl and TRAIL-R2 ((1997) Immunity 7, 821-830) and TRADD ((1996) Immunity 4, 387-396), while the RHIM domain is crucial for binding other RHFM domain containing proteins such as TRIF ((2004) Nat Immunol. 5, 503-507), DAI ((2009) EMBO Rep. 10, 916-922) and RIP3 ((1999) J. Biol. Chem. 274, 16871-16875); (1999) Curr. Biol. 9, 539-542) and exerts many of its effects through these interactions. RIPl is a central regulator of cell signaling, and is involved in mediating both pro-survival and programmed cell death pathways which will be discussed below.
The role for RIPl in cell signaling has been assessed under various conditions
36560-36566), TRAIL ((2012) J .Virol. Epub, ahead of print), FAS ((2004) J. Biol. Chem. 279, 7925-7933)], but is best understood in the context of mediating signals downstream of the death receptor TNFRl ((2003) Cell 114, 181-190). Engagement of the TNFR by TNF leads to its oligomerization, and the recruitment of multiple proteins, including linear K63-linked polyubiquitinated RIPl ((2006) Mol. Cell 22, 245-257), TRAF2/5 ((2010) J. Mol. Biol. 396, 528-539), TRADD ((2008) Nat. Immunol. 9, 1037-1046) and cIAPs ((2008) Proc. Natl. Acad. Sci. USA. 105, 1 1778-11783), to the cytoplasmic tail of the receptor. This complex which is dependent on RIPl as a scaffolding protein (i.e. kinase
independent), termed complex I, provides a platform for pro-survival signaling through the activation of the NFKB and MAP kinases pathways ((2010) Sci. Signal. 115, re4).
Alternatively, binding of TNF to its receptor under conditions promoting the
deubiquitination of RIPl (by proteins such as A20 and CYLD or inhibition of the cIAPs) results in receptor internalization and the formation of complex II or DISC (death-inducing signaling complex) ((2011) Cell Death Dis. 2, e230). Formation of the DISC, which contains RIPl, TRADD, FADD and caspase 8, results in the activation of caspase 8 and the onset of programmed apoptotic cell death also in a RIPl kinase independent fashion ((2012) FEBS J 278, 877-887). Apoptosis is largely a quiescent form of cell death, and is involved in routine processes such as development and cellular homeostasis.
Under conditions where the DISC forms and RJP3 is expressed, but apoptosis is inhibited (such as FADD/caspase 8 deletion, caspase inhibition or viral infection), a third RIPl kinase-dependent possibility exists. RIP3 can now enter this complex, become phosphorylated by RIPl and initiate a caspase-independent programmed necrotic cell death through the activation of MLKL and PGAM5 ((2012) Cell 148, 213-227); ((2012) Cell 148, 228-243); ((2012) Proc. Natl. Acad. Sci. USA. 109, 5322-5327). As opposed to apoptosis, programmed necrosis (not to be confused with passive necrosis which is not programmed) results in the release of danger associated molecular patterns (DAMPs) from the cell.
These DAMPs are capable of providing a “danger signal” to surrounding cells and tissues, eliciting proinflammatory responses including inflammasome activation, cytokine production and cellular recruitment ((2008 Nat. Rev. Immunol 8, 279-289).
Dysregulation of RIPl kinase-mediated programmed cell death has been linked to various inflammatory diseases, as demonstrated by use of the RIP3 knockout mouse (where RIPl -mediated programmed necrosis is completely blocked) and by Necrostatin-1 (a tool inhibitor of RIPl kinase activity with poor oral bioavailability). The RIP3 knockout mouse has been shown to be protective in inflammatory bowel disease (including Ulcerative colitis and Crohn’s disease) ((2011) Nature 477, 330-334), Psoriasis ((2011) Immunity 35, 572-582), retinal-detachment-induced photoreceptor necrosis ((2010) PNAS 107, 21695-21700), retinitis pigmentosa ((2012) Proc. Natl. Acad. Sci., 109:36, 14598-14603), cerulein-induced acute pancreatits ((2009) Cell 137, 1100-1111) and Sepsis/systemic inflammatory response syndrome (SIRS) ((2011) Immunity 35, 908-918). Necrostatin-1 has been shown to be effective in alleviating ischemic brain injury ((2005) Nat. Chem. Biol. 1, 112-119), retinal ischemia/reperfusion injury ((2010) J. Neurosci. Res. 88, 1569-1576), Huntington’s disease ((2011) Cell Death Dis. 2 el 15), renal ischemia reperfusion injury ((2012) Kidney Int. 81, 751-761), cisplatin induced kidney injury ((2012) Ren. Fail. 34, 373-377) and traumatic brain injury ((2012) Neurochem. Res. 37, 1849-1858). Other diseases or disorders regulated at least in part by RIPl -dependent apoptosis, necrosis or cytokine production include hematological and solid organ malignancies ((2013) Genes
Dev. 27: 1640-1649), bacterial infections and viral infections ((2014) Cell Host & Microbe 15, 23-35) (including, but not limited to, tuberculosis and influenza ((2013) Cell 153, 1-14)) and Lysosomal storage diseases (particularly, Gaucher Disease, Nature Medicine Advance Online Publication, 19 January 2014, doi: 10.1038/nm.3449).
A potent, selective, small molecule inhibitor of RIP1 kinase activity would block RIP 1 -dependent cellular necrosis and thereby provide a therapeutic benefit in diseases or events associated with DAMPs, cell death, and/or inflammation.
A mixture of (S)-3-amino-5-methyl-2,3-dihydrobenzo[b][l,4]oxazepin-4(5H)-one, hydrochloride (4.00 g, 16.97 mmol), 5-benzyl-4H-l,2,4-triazole-3-carboxylic acid, hydrochloride (4.97 g, 18.66 mmol) and DIEA (10.37 mL, 59.4 mmol) in isopropanol (150 mL) was stirred vigorously for 10 minutes and then 2,4,6-tripropyl-l,3,5,2,4,6-trioxatriphosphinane 2,4,6-trioxide (T3P) (50% by wt. in EtOAc) (15.15 mL, 25.5 mmol) was added. The mixture was stirred at rt for 10 minutes and then quenched with water and concentrated to remove isopropanol. The resulting crude material is dissolved in EtOAc and washed with 1M HC1, satd. NaHC03 and brine. Organics were concentrated and purified by column chromatography (220 g silica column; 20-90% EtOAc/hexanes, 15 min.; 90%, 15 min.) to give the title compound as a light orange foam (5.37 g, 83%). 1H NMR (MeOH-d4) δ: 7.40 – 7.45 (m, 1H), 7.21 – 7.35 (m, 8H), 5.01 (dd, J = 11.6, 7.6 Hz, 1H), 4.60 (dd, J = 9.9, 7.6 Hz, 1H), 4.41 (dd, J = 11.4, 9.9 Hz, 1H), 4.17 (s, 2H), 3.41 (s, 3H); MS (m/z) 378.3 (M+H+).
Alternative Preparation:
To a solution of (S)-3-amino-5-methyl-2,3-dihydrobenzo[b][l,4]oxazepin-4(5H)-one hydrochloride (100 g, 437 mmol), 5-benzyl-4H-l,2,4-triazole-3-carboxylic acid hydrochloride (110 g, 459 mmol) in DCM (2.5 L) was added DIPEA (0.267 L, 1531 mmol) at 15 °C. The reaction mixture was stirred for 10 min. and 2,4,6-tripropyl-l, 3, 5,2,4,6-trioxatriphosphinane 2,4,6-trioxide >50 wt. % in ethyl acetate (0.390 L, 656 mmol) was slowly added at 15 °C. After stirring for 60 mins at RT the LCMS showed the reaction was complete, upon which time it was quenched with water, partitioned between DCM and washed with 0.5N HCl aq (2 L), saturated aqueous NaHC03 (2 L), brine (2 L) and water (2 L). The organic phase was separated and activated charcoal (100 g) and sodium sulfate
(200 g) were added. The dark solution was shaken for 1 h before filtering. The filtrate was then concentrated under reduced pressure to afford the product as a tan foam (120 g). The product was dried under a high vacuum at 50 °C for 16 h. 1H MR showed 4-5% wt of ethyl acetate present. The sample was dissolved in EtOH (650 ml) and stirred for 30 mins, after which the solvent was removed using a rotavapor (water-bath T=45 °C). The product was dried under high vacuum for 16 h at RT (118 g, 72% yield). The product was further dried under high vacuum at 50 °C for 5 h. 1H NMR showed <1% of EtOH and no ethyl acetate. 1H NMR (400 MHz, DMSO-i¾) δ ppm 4.12 (s, 2 H), 4.31 – 4.51 (m, 1 H), 4.60 (t, J=10.36 Hz, 1 H), 4.83 (dt, 7=11.31, 7.86 Hz, 1 H), 7.12 – 7.42 (m, 8 H), 7.42 – 7.65 (m, 1 H), 8.45 (br. s., 1 H), 14.41 (br. s., 1 H). MS (m/z) 378 (M + H+).
Crystallization:
(S)-5-Benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydrobenzo[b][l,4]oxazepin-3-yl)-4H-l,2,4-triazole-3-carboxamide (100 mg) was dissolved in 0.9 mL of toluene and 0.1 mL of methylcyclohexane at 60 °C, then stirred briskly at room temperature (20 °C) for 4 days. After 4 days, an off-white solid was recovered (76 mg, 76% recovery). The powder X-ray diffraction (PXRD) pattern of this material is shown in Figure 7 and the corresponding diffraction data is provided in Table 1.
The PXRD analysis was conducted using a PANanalytical X’Pert Pro
diffractometer equipped with a copper anode X-ray tube, programmable slits, and
X’Celerator detector fitted with a nickel filter. Generator tension and current were set to 45kV and 40mA respectively to generate the copper Ka radiation powder diffraction pattern over the range of 2 – 40°2Θ. The test specimen was lightly triturated using an agate mortar and pestle and the resulting fine powder was mounted onto a silicon background plate.
Table 1.
Paper
Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases
J Med Chem 2017, 60(4): 1247
RIP1 regulates necroptosis and inflammation and may play an important role in contributing to a variety of human pathologies, including immune-mediated inflammatory diseases. Small-molecule inhibitors of RIP1 kinase that are suitable for advancement into the clinic have yet to be described. Herein, we report our lead optimization of a benzoxazepinone hit from a DNA-encoded library and the discovery and profile of clinical candidate GSK2982772 (compound 5), currently in phase 2a clinical studies for psoriasis, rheumatoid arthritis, and ulcerative colitis. Compound 5 potently binds to RIP1 with exquisite kinase specificity and has excellent activity in blocking many TNF-dependent cellular responses. Highlighting its potential as a novel anti-inflammatory agent, the inhibitor was also able to reduce spontaneous production of cytokines from human ulcerative colitis explants. The highly favorable physicochemical and ADMET properties of 5, combined with high potency, led to a predicted low oral dose in humans.
Synthesis of (<it>S</it>)-3-amino-benzo[<it>b</it>][1,4]oxazepin-4-one via Mitsunobu and S<INF>N</INF>Ar reaction for a first-in-class RIP1 kinase inhibitor GSK2982772 in clinical trials
Tetrahedron Lett 2017, 58(23): 2306 Harris, P.A. Identification of a first-in-class RIP1 kinase inhibitor in phase 2a clinical trials for immunoinflammatory diseases
ACS MEDI-EFMC Med Chem Front (June 25-28, Philadelphia) 2017, Abst
Harris, P. Identification of a first-in-class RIP1 kinase inhibitor in phase 2a clinical trials for immuno-inflammatory diseases
253rd Am Chem Soc (ACS) Natl Meet (April 2-6, San Francisco) 2017, Abst MEDI 313
The hepatitis C virus (HCV), a member of the hepacivirus genera within the Flaviviridae family, is the leading cause of chronic liver disease worldwide (Boyer, N. et al. J Hepatol. 2000, 32, 98-1 12). Consequently, a significant focus of current antiviral research is directed toward the development of improved methods for the treatment of chronic HCV infections in humans (Ciesek, S., von Hahn T., and Manns, MP., Clin. Liver Dis., 201 1 , 15, 597-609; Soriano, V. et al, J. Antimicrob. Chemother., 201 1 , 66, 1573-1686; Brody, H., Nature Outlook, 201 1 , 474, S1 -S7; Gordon, C. P., et al., J. Med. Chem. 2005, 48, 1 -20;
Maradpour, D., et al., Nat. Rev. Micro. 2007, 5, 453-463).
Virologic cures of patients with chronic HCV infection are difficult to achieve because of the prodigious amount of daily virus production in chronically infected patients and the high spontaneous mutability of HCV (Neumann, et al., Science 1998, 282, 103-7; Fukimoto, et al., Hepatology, 1996, 24, 1351 -4;
Domingo, et al., Gene 1985, 40, 1 -8; Martell, et al., J. Virol. 1992, 66, 3225-9). HCV treatment is further complicated by the fact that HCV is genetically diverse and expressed as several different genotypes and numerous subtypes. For example, HCV is currently classified into six major genotypes (designated 1 -6), many subtypes (designated a, b, c, and so on), and about 100 different strains (numbered 1 , 2, 3, and so on).
HCV is distributed worldwide with genotypes 1 , 2, and 3 predominate within the United States, Europe, Australia, and East Asia (Japan, Taiwan, Thailand, and China). Genotype 4 is largely found in the Middle East, Egypt and central Africa while genotype 5 and 6 are found predominantly in South Africa and South East Asia respectively (Simmonds, P. et al. J Virol. 84: 4597-4610, 2010).
The combination of ribavirin, a nucleoside analog, and interferon-alpha (a) (IFN), is utilized for the treatment of multiple genotypes of chronic HCV infections in humans. However, the variable clinical response observed within patients and the toxicity of this regimen have limited its usefulness. Addition of a HCV protease inhibitor (telaprevir or boceprevir) to the ribavirin and IFN regimen improves 12-week post-treatment virological response (SVR12) rates
substantially. However, the regimen is currently only approved for genotype 1 patients and toxicity and other side effects remain.
The use of directing acting antivirals to treat multiple genotypes of HCV infection has proven challenging due to the variable activity of antivirals against the different genotypes. HCV protease inhibitors frequently have compromised in vitro activity against HCV genotypes 2 and 3 compared to genotype 1 (See, e.g., Table 1 of Summa, V. et al., Antimicrobial Agents and Chemotherapy, 2012, 56, 4161 -4167; Gottwein, J. et al, Gastroenterology, 201 1 , 141 , 1067-1079).
Correspondingly, clinical efficacy has also proven highly variable across HCV genotypes. For example, therapies that are highly effective against HCV genotype 1 and 2 may have limited or no clinical efficacy against genotype 3.
(Moreno, C. et al., Poster 895, 61 st AASLD Meeting, Boston, MA, USA, Oct. 29 – Nov. 2, 2010; Graham, F., et al, Gastroenterology, 201 1 , 141 , 881 -889; Foster, G.R. et al., EASL 45th Annual Meeting, April 14-18, 2010, Vienna, Austria.) In some cases, antiviral agents have good clinical efficacy against genotype 1 , but lower and more variable against genotypes 2 and 3. (Reiser, M. et al.,
Hepatology, 2005, 41 ,832-835.) To overcome the reduced efficacy in genotype 3 patients, substantially higher doses of antiviral agents may be required to achieve substantial viral load reductions (Fraser, IP et al., Abstract #48, HEP DART 201 1 , Koloa, HI, December 201 1 .)
Antiviral agents that are less susceptible to viral resistance are also needed. For example, resistance mutations at positions 155 and 168 in the HCV protease frequently cause a substantial decrease in antiviral efficacy of HCV protease inhibitors (Mani, N. Ann Forum Collab HIV Res., 2012, 14, 1 -8;
Romano, KP et al, PNAS, 2010, 107, 20986-20991 ; Lenz O, Antimicrobial agents and chemotherapy, 2010, 54,1878-1887.)
In view of the limitations of current HCV therapy, there is a need to develop more effective anti-HCV therapies. It would also be useful to provide therapies that are effective against multiple HCV genotypes and subtypes.
RELATIVE SIMILAR EXAMPLE WITHOUT DIFLUORO GROUPS, BUT NOT SAME COMPD
Example 1. Preparation of (1 aR,5S,8S,9S,10R,22aR)-5-tert-butyl-N- [(1 R,2R)-2-(difluoromethyl)-1 -{[(1 – methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-9-ethyl-14-methoxy-3,6-dioxo- 1 ,1 a,3,4,5,6,9,10,18,19,20,21 ,22,22a-tetradecahydro-8H-7,10- methanocyclopropa[18,19][1 ,10,3,6]dioxadiazacyclononadecino[1 1 ,12- b]quinoxaline-8-carboxamide.
Step 1 . Preparation of 1-1 : A mixture containing Intermediate B4 (2.03 g, 6.44 mmol), Intermediate E1 (1 .6 g, 5.85 mmol), and cesium carbonate (3.15 g, 9.66 mmol) in MeCN (40 mL) was stirred vigorously at rt under an atmosphere of Ar for 16 h. The reaction was then filtered through a pad of Celite and the filtrate concentrated in vacuo. The crude material was purified by silica gel
chromatography to provide 1-1 as a white solid (2.5 g). LCMS-ESI+ (m/z): [M- Boc+2H]+ calcd for C2oH27CIN3O4: 408.9; found: 408.6.
Step 2. Preparation of 1-2: To a solution 1 -1 (2.5 g, 4.92 mmol) in dioxane
(10 mL) was added hydrochloric acid in dioxane (4 M, 25 mL, 98.4 mmol) and the reaction stirred at rt for 5 h. The crude reaction was concentrated in vacuo to give 1-2 as a white solid (2.49 g) that was used in subsequently without further purification. LCMS-ESI+ (m/z): [M]+ calcd for C2oH26CIN3O4: 407.9; found: 407.9.
Step 3. Preparation of 1-3: To a DMF (35 mL) solution of 1-2 (2.49 g, 5.61 mmol), Intermediate D1 (1 .75 mg, 6.17 mmol) and DIPEA (3.9 mL, 22.44 mmol) was added COMU (3.12 g, 7.29 mmol) and the reaction was stirred at rt for 3 h. The reaction was quenched with 5% aqueous citric acid solution and extracted with EtOAc, washed subsequently with brine, dried over anhydrous MgSO , filtered and concentrated to produce 1 -3 as an orange foam (2.31 g) that was used without further purification. LCMS-ESI+ (m/z): [M]+ calcd for C35H49CIN4O7: 673.3; found: 673.7.
Step 4. Preparation of 1-4: To a solution of 1-3 (2.31 g, 3.43 mmol), TEA (0.72 mL, 5.15 mmol) and potassium vinyltrifluoroborate (0.69 mg, 5.15 mmol) in EtOH (35 mL) was added PdCI2(dppf) (0.25 g, 0.34 mmol, Frontier Scientific). The reaction was sparged with Argon for 15 min and heated to 80 °C for 2 h. The reaction was adsorbed directly onto silica gel and purified using silica gel chromatography to give 1 -4 as a yellow oil (1 .95 g). LCMS-ESI+ (m/z): [M+H]+ calcd for C37H53N4O7: 665.4; found: 665.3.
Step 5. Preparation of 1 -5: To a solution of 1 -4 (1 .95 g, 2.93 mmol) in
DCE (585 ml_) was added Zhan 1 B catalyst (0.215 g, 0.29 mmol, Strem) and the reaction was sparged with Ar for 15 min. The reaction was heated to 80 °C for 1 .5 h, allowed to cool to rt and concentrated. The crude product was purified by silica gel chromatography to produce 1 -5 as a yellow oil (1 .47 g; LCMS-ESI+ (m/z): [M+H]+ calcd for C35H49N4O7: 637.4; found: 637.3).
Step 6. Preparation of 1 -6: A solution of 1 -5 (0.97 g, 1 .52 mmol) in EtOH (15 ml_) was treated with Pd/C (10 wt % Pd, 0.162 g). The atmosphere was replaced with hydrogen and stirred at rt for 2 h. The reaction was filtered through Celite, the pad washed with EtOAc and concentrated to give 1 -6 as a brown foamy solid (0.803 g) that was used subsequently without further purification. LCMS-ESr (m/z): [M+H]+ calcd for C35H5i N4O7: 639.4; found: 639.3.
Step 7. Preparation of 1 -7: To a solution of 1 -6 (0.803 g, 1 .26 mmol) in DCM (10 ml_) was added TFA (5 ml_) and stirred at rt for 3 h. An additional 2 ml_ TFA was added and the reaction stirred for another 1 .5 h. The reaction was concentrated to a brown oil that was taken up in EtOAc (35 ml_). The organic solution was washed with water. After separation of the layers, sat. aqueous NaHCO3 was added with stirring until the aqueous layer reached a pH ~ 7-8. The layers were separated again and the aqueous extracted with EtOAc twice. The combined organics were washed with 1 M aqueous citric acid, brine, dried over anhydrous MgSO4, filtered and concentrated to produce 1 -6 as a brown foamy solid (0.719 g) that was used subsequently without further purification. LCMS-ESr (m/z): [M+H]+ calcd for C3i H43N4O7: 583.3; found: 583.4 .
Step 8. Preparation of Example 1 : To a solution of 1 -7 (0.200 g, 0.343 mmol), Intermediate A10 (0.157 g, 0.515 mmol), DMAP (0.063 g, 0.51 mmol) and DIPEA (0.3 ml_, 1 .72 mmol) in DMF (3 ml_) was added HATU (0.235 g, 0.617 mmol) and the reaction was stirred at rt o/n. The reaction was diluted with MeCN and purified directly by reverse phase HPLC (Gemini, 30-100% MeCN/H2O + 0.1 % TFA) and lyophilized to give Example 1 (1 18.6 mg) as a solid TFA salt. Analytic HPLC RetTime: 8.63 min. LCMS-ESI+ (m/z): [M+H]+ calcd for
The hepatitis C virus (HCV), a member of the hepacivirus genera within the Flaviviridae family, is the leading cause of chronic liver disease worldwide (Boyer, N. et al. J Hepatol. 2000, 32, 98-112). Consequently, a significant focus of current antiviral research is directed toward the development of improved methods for the treatment of chronic HCV infections in humans (Ciesek, S., von Hahn T., and Manns, MP., Clin. Liver Dis., 2011, 15, 597-609; Soriano, V. et al, J. Antimicrob. Chemother., 2011, 66, 1573-1686; Brody, H., Nature Outlook, 2011, 474, S1-S7; Gordon, C. P., et al, J. Med. Chem. 2005, 48, 1-20; Maradpour, D., et al, Nat. Rev. Micro. 2007, 5, 453-463).
Virologic cures of patients with chronic HCV infection are difficult to achieve because of the prodigious amount of daily virus production in chronically infected patients and the high spontaneous mutability of HCV (Neumann, et al, Science 1998, 282, 103-7; Fukimoto, et al, Hepatology, 1996, 24, 1351-4; Domingo, et al, Gene 1985, 40, 1-8; Martell, et al, J. Virol. 1992, 66, 3225-9). HCV treatment is further complicated by the fact that HCV is genetically diverse and expressed as several different genotypes and numerous subtypes. For example, HCV is currently classified into six major genotypes (designated 1-6), many subtypes (designated a, b, c, and so on), and about 100 different strains (numbered 1, 2, 3, and so on).
HCV is distributed worldwide with genotypes 1, 2, and 3 predominate within the United States, Europe, Australia, and East Asia (Japan, Taiwan, Thailand, and China). Genotype 4 is largely found in the Middle East, Egypt and central Africa while genotype 5 and 6 are found predominantly in South Africa and South East Asia respectively (Simmonds, P. et al. J Virol. 84: [0006] There remains a need to develop effective treatments for HCV infections. Suitable compounds for the treatment of HCV infections are disclosed in U.S. Publication No. 2014-0017198, titled “Inhibitors of Hepatitis C Virus” filed on July 2, 2013 including the compound of formula I:
Example 1. Synthesis of (laR,5S,8S,9S,10R,22aR)-5-teri-butyl- V-[(lR,2R)-2-(difluoromethyl)- 1-{ [(1-methylcyclopr opyl)sulfonyl] carbamoyl} cyclopropyl] -9-ethyl- 18,18- difluoro-14-methoxy-3,6-dioxo-l,la,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19] [1,10,3,6] dioxadiazacyclononadecino[ll,12-6]quinoxaline-8- carboxamide (I) by route I
[0195] Compound of formula I was synthesized via route I as shown below:
Synthesis of intermediates for compound of formula I SEE PATENT
The U.S. Food and Drug Administration today approved Vosevi to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis.
The U.S. Food and Drug Administration today approved Vosevi to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis. Vosevi is a fixed-dose, combination tablet containing two previously approved drugs – sofosbuvir and velpatasvir – and a new drug, voxilaprevir. Vosevi is the first treatment approved for patients who have been previously treated with the direct-acting antiviral drug sofosbuvir or other drugs for HCV that inhibit a protein called NS5A.
“Direct-acting antiviral drugs prevent the virus from multiplying and often cure HCV. Vosevi provides a treatment option for some patients who were not successfully treated with other HCV drugs in the past,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Hepatitis C is a viral disease that causes inflammation of the liver that can lead to diminished liver function or liver failure. According to the Centers for Disease Control and Prevention, an estimated 2.7 to 3.9 million people in the United States have chronic HCV. Some patients who suffer from chronic HCV infection over many years may have jaundice (yellowish eyes or skin) and develop complications, such as bleeding, fluid accumulation in the abdomen, infections, liver cancer and death.
There are at least six distinct HCV genotypes, or strains, which are genetically distinct groups of the virus. Knowing the strain of the virus can help inform treatment recommendations. Approximately 75 percent of Americans with HCV have genotype 1; 20-25 percent have genotypes 2 or 3; and a small number of patients are infected with genotypes 4, 5 or 6.
The safety and efficacy of Vosevi was evaluated in two Phase 3 clinical trials that enrolled approximately 750 adults without cirrhosis or with mild cirrhosis.
The first trial compared 12 weeks of Vosevi treatment with placebo in adults with genotype 1 who had previously failed treatment with an NS5A inhibitor drug. Patients with genotypes 2, 3, 4, 5 or 6 all received Vosevi.
The second trial compared 12 weeks of Vosevi with the previously approved drugs sofosbuvir and velpatasvir in adults with genotypes 1, 2 or 3 who had previously failed treatment with sofosbuvir but not an NS5A inhibitor drug.
Results of both trials demonstrated that 96-97 percent of patients who received Vosevi had no virus detected in the blood 12 weeks after finishing treatment, suggesting that patients’ infection had been cured.
Treatment recommendations for Vosevi are different depending on viral genotype and prior treatment history.
The most common adverse reactions in patients taking Vosevi were headache, fatigue, diarrhea and nausea.
Vosevi is contraindicated in patients taking the drug rifampin.
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected adult patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. HBV reactivation in patients treated with direct-acting antiviral medicines can result in serious liver problems or death in some patients. Health care professionals should screen all patients for evidence of current or prior HBV infection before starting treatment with Vosevi.
The U.S. Food and Drug Administration today approved Vosevi to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis.
The U.S. Food and Drug Administration today approved Vosevi to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis. Vosevi is a fixed-dose, combination tablet containing two previously approved drugs – sofosbuvir and velpatasvir – and a new drug, voxilaprevir. Vosevi is the first treatment approved for patients who have been previously treated with the direct-acting antiviral drug sofosbuvir or other drugs for HCV that inhibit a protein called NS5A.
“Direct-acting antiviral drugs prevent the virus from multiplying and often cure HCV. Vosevi provides a treatment option for some patients who were not successfully treated with other HCV drugs in the past,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Hepatitis C is a viral disease that causes inflammation of the liver that can lead to diminished liver function or liver failure. According to the Centers for Disease Control and Prevention, an estimated 2.7 to 3.9 million people in the United States have chronic HCV. Some patients who suffer from chronic HCV infection over many years may have jaundice (yellowish eyes or skin) and develop complications, such as bleeding, fluid accumulation in the abdomen, infections, liver cancer and death.
There are at least six distinct HCV genotypes, or strains, which are genetically distinct groups of the virus. Knowing the strain of the virus can help inform treatment recommendations. Approximately 75 percent of Americans with HCV have genotype 1; 20-25 percent have genotypes 2 or 3; and a small number of patients are infected with genotypes 4, 5 or 6.
The safety and efficacy of Vosevi was evaluated in two Phase 3 clinical trials that enrolled approximately 750 adults without cirrhosis or with mild cirrhosis.
The first trial compared 12 weeks of Vosevi treatment with placebo in adults with genotype 1 who had previously failed treatment with an NS5A inhibitor drug. Patients with genotypes 2, 3, 4, 5 or 6 all received Vosevi.
The second trial compared 12 weeks of Vosevi with the previously approved drugs sofosbuvir and velpatasvir in adults with genotypes 1, 2 or 3 who had previously failed treatment with sofosbuvir but not an NS5A inhibitor drug.
Results of both trials demonstrated that 96-97 percent of patients who received Vosevi had no virus detected in the blood 12 weeks after finishing treatment, suggesting that patients’ infection had been cured.
Treatment recommendations for Vosevi are different depending on viral genotype and prior treatment history.
The most common adverse reactions in patients taking Vosevi were headache, fatigue, diarrhea and nausea.
Vosevi is contraindicated in patients taking the drug rifampin.
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected adult patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. HBV reactivation in patients treated with direct-acting antiviral medicines can result in serious liver problems or death in some patients. Health care professionals should screen all patients for evidence of current or prior HBV infection before starting treatment with Vosevi.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
















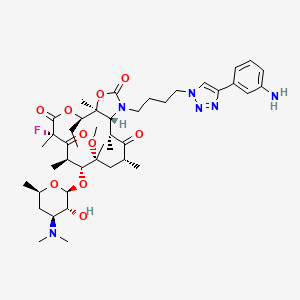








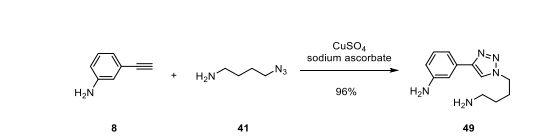























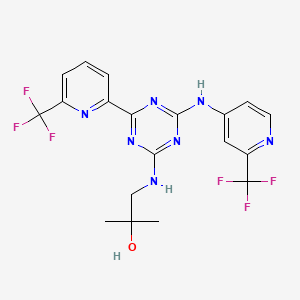


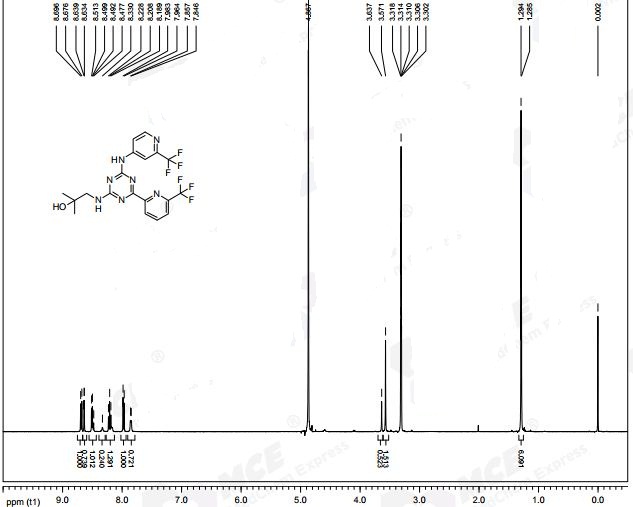









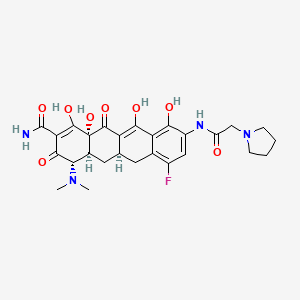
















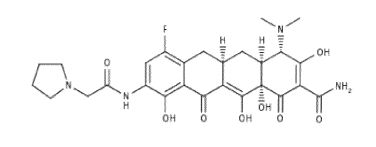






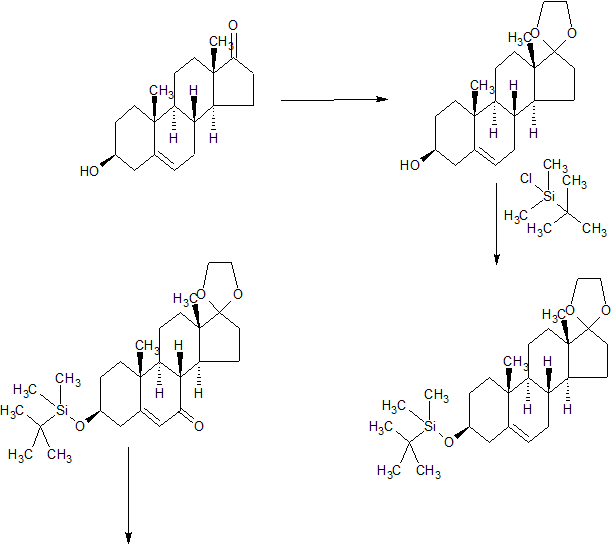







, the key enzymes that control levels of PIP3 are the PI3 kinase (PI3K), and the phosphatases, PTEN and SHIP1 (SH2-containing inositol-5’-phosphatase 1). PI3K generates PIP3, thus initiating the signaling pathway. This signaling is reduced by degradation of PIP3 by PTEN and SHIP1. PTEN is generally considered to be constantly working in the pathway, whereas SHIP1 is dormant until the cell is stimulated. In preclinical models, PTEN has been shown to suppress cancer by controlling cell proliferation, whereas SHIP1, when functioning, has been demonstrated to control inflammation by reducing cell migration and activation.")