
Home » 2017 (Page 10)
Yearly Archives: 2017
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TILOGLIPTIN
PRESENTING 2 MOLECULES………..I AM NOT SURE WHICH IS TITLE MOLECULE
EMAIL ME amcrasto@gmail.com
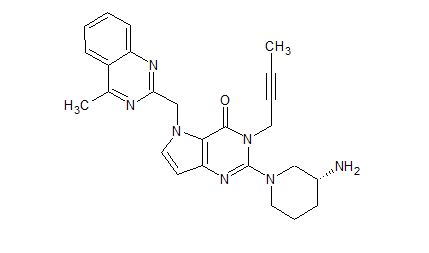
| Molecular Formula: | C25H27N7O |
|---|---|
| Molecular Weight: | 441.539 g/mol |
CAS 1428445-40-2
REF Bioorganic & Medicinal Chemistry (2013), 21(7), 1749-1755.
NEXT ONE………………

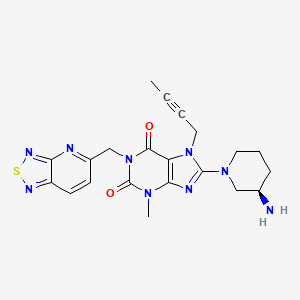
CAS 1415912-31-0
| Molecular Formula: | C21H23N9O2S |
|---|---|
| Molecular Weight: | 465.536 g/mol |
REF CN 102807568, CN 105315301, WO 2016019868
Salt………..
CAS 1874255-95-4
TILOGLIPTIN
HWH-ZGC-2-143
Guangzhou Institutes of Biomedicine and Health
![]()
Chia Tai Tianqing Pharmaceutical Group Co Ltd;

Non-insulin dependent diabetes
Dipeptidyl peptidase IV inhibitor (oral, type 2 diabetes),
DPP-IV inhibitors (oral, type 2 diabetes), Guangzhou Institutes of Biomedicine and Health/Jiangsu Chia Tai Tianqing Pharmaceutical ; HWH-ZGC-2-143 ;
Novel polymorphic forms of thiadiazole derivatives, preferably aglucin, sitagliptin, saxagliptin, vildagliptin, levaratine, useful for treating type II diabetes. Guangzhou Institutes of Biomedicine and Health , in collaboration with Jiangsu Chia Tai Tianqing Pharmaceutical , is investigating tilogliptin , an oral dipeptidyl peptidase IV inhibitor and a pyrrolopyrimidine analog, for treating type 2 diabetes.
As of June 2017, Centaurus BioPharma is developing diabetes therapy, CT-1006 and CT-1005 (in preclinical development) for treating diabetes mellitus.
See WO2016019868, claiming novel citric acid salt of 8-((R)-3-amino-piperidin-1-yl)-1-([1,2,5]-thiadiazolo [3,4-b] pyridine-5methyl)-7-(2-butyn-1-yl)-3-methyl-xanthine, coassigned to Lianyungang Runzhong Pharmaceutical .



PATENT
WO2016019868
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016019868
PATENT
Centaurus BioPharma Co Ltd; Chia Tai Tianqing Pharmaceutical Group Co Ltd
Discovery of potent dipeptidyl peptidase IV inhibitors through pharmacophore hybridization and hit-to-lead optimization
- a Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Science, 190 Kaiyuan Avenue, Guangzhou Science Park, Guangzhou 510530, China
- b Jiangsu Chia-Tai Tianqing Pharmaceutical Co. Ltd, No. 8 Julong North Rd., Xinpu Lianyungang, Jiangsu 222006, China
- c State Key Laboratory of Respiratory Disease, Guangzhou 510120, China
- https://doi.org/10.1016/j.bmc.2013.01.062

- A novel dipeptidyl peptidase IV inhibitor hit (5, IC50 = 0.86 μM) was structurally derived from our recently disclosed preclinical candidate 4 by replacing the cyanobenzyl with a butynyl based on pharmacophore hybridization. A hit-to-lead optimization effort was then initiated to improve its potency. Most N-substituted analogs exhibited good in vitro activity, and compound 18o (IC50 = 1.55 nM) was identified to be a potent dipeptidyl peptidase IV inhibitor with a significantly improved pharmacokinetic properties (bioavailablity: 41% vs 82.9%; T1/2: 2 h vs 4.9 h).
N[C@@H]1CCCN(C1)C5=Nc4ccn(Cc3nc2ccccc2c(C)n3)c4C(=O)N5CC#CC
N[C@@H]1CCCN(C1)c5nc4c(C(=O)N(Cc2ccc3nsnc3n2)C(=O)N4C)n5CC#CC
TEGAFUR
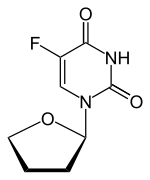
Tegafur
CAS 17902-23-7
- Molecular Weight,200.17, MF C8 H9 F N2 O3
-
172-173 °C
Miyashita, Osamu; Chemical & Pharmaceutical Bulletin 1981, 29(11), PG 3181-90
- Synonyms:Ftorafur
- ATC:L01BC03
- EINECS:241-846-2
- LD50:800 mg/kg (M, i.v.); 775 mg/kg (M, p.o.);
685 mg/kg (R, i.v.); 930 mg/kg (R, p.o.);
34 mg/kg (dog, p.o.)
Derivatives, monosodium salt
- Formula:C8H8FN2NaO3
- MW:222.15 g/mol
- CAS-RN:28721-46-2
Tegafur (INN, BAN, USAN) is a chemotherapeutic prodrug of 5-flourouracil (5-FU) used in the treatment of cancers. It is a component of the combination drug tegafur/uracil. When metabolised, it becomes 5-FU.[1]
Medical uses
As a prodrug to 5-FU it is used in the treatment of the following cancers:[2]
- Stomach (when combined with gimeracil and oteracil)
- Breast (with uracil)
- Gallbladder
- Lung (specifically adenocarcinoma, typically with uracil)
- Colorectal (usually when combined with gimeracil and oteracil)
- Head and neck
- Liver (with uracil)[3]
- Pancreatic
It is often given in combination with drugs that alter its bioavailability and toxicity such as gimeracil, oteracil or uracil.[2] These agents achieve this by inhibiting the enzyme dihydropyrimidine dehydrogenase (uracil/gimeracil) or orotate phosphoribosyltransferase (oteracil).[2]

Adverse effects
The major side effects of tegafur are similar to fluorouracil and include myelosuppression, central neurotoxicity and gastrointestinal toxicity (especially diarrhoea).[2] Gastrointestinal toxicity is the dose-limiting side effect of tegafur.[2] Central neurotoxicity is more common with tegafur than with fluorouracil.[2]

Pharmacogenetics
The dihydropyrimidine dehydrogenase (DPD) enzyme is responsible for the detoxifying metabolism of fluoropyrimidines, a class of drugs that includes 5-fluorouracil, capecitabine, and tegafur.[4] Genetic variations within the DPD gene (DPYD) can lead to reduced or absent DPD activity, and individuals who are heterozygous or homozygous for these variations may have partial or complete DPD deficiency; an estimated 0.2% of individuals have complete DPD deficiency.[4][5] Those with partial or complete DPD deficiency have a significantly increased risk of severe or even fatal drug toxicities when treated with fluoropyrimidines; examples of toxicities include myelosuppression, neurotoxicity and hand-foot syndrome.[4][5]
Mechanism of action
It is a prodrug to 5-FU, which is a thymidylate synthase inhibitor.[2]
Pharmacokinetics
It is metabolised to 5-FU by CYP2A6.[6][7]
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles.[§ 1]

The interactive pathway map can be edited at WikiPathways: “FluoropyrimidineActivity_WP1601”.



MASS SPECTRUM
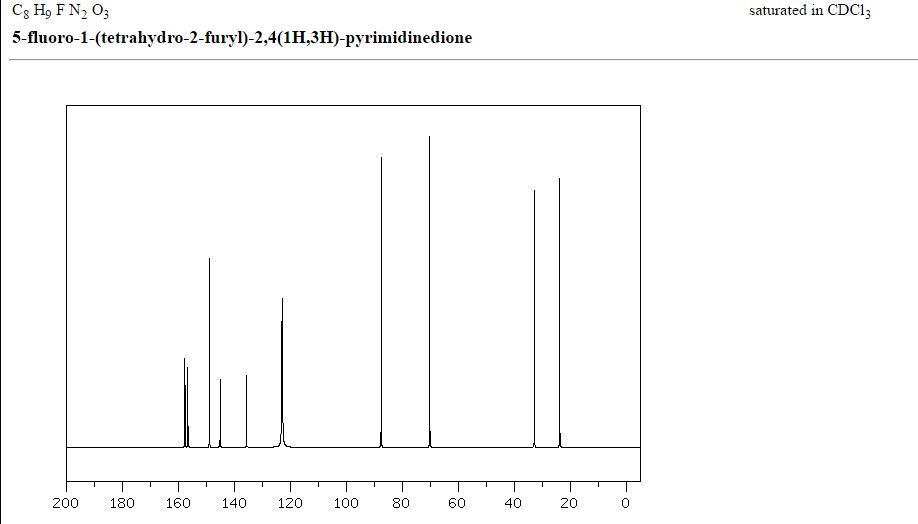
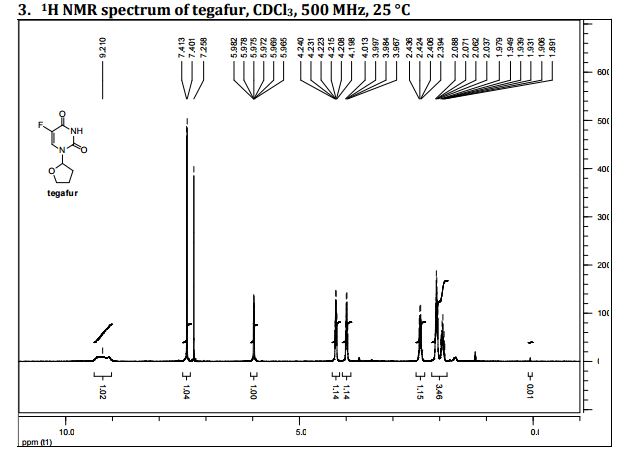
1H NMR
IR
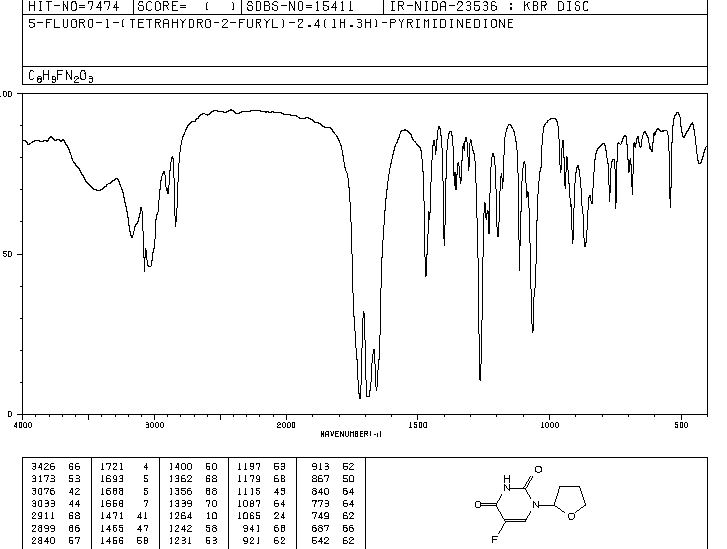
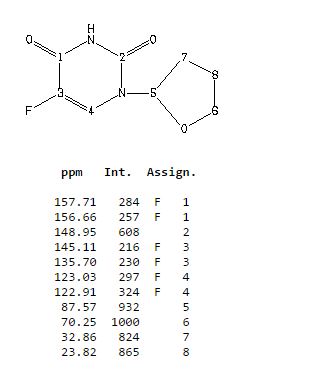
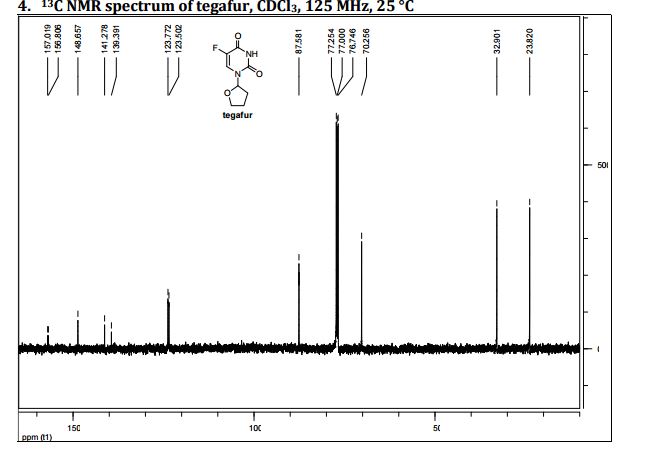
13C NMR
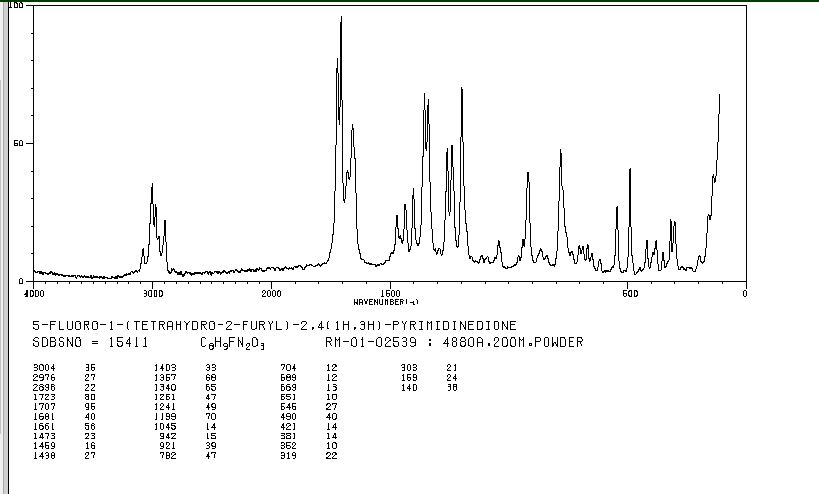
RAMAN
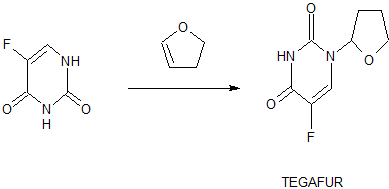
Synthesis
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 58138-78-6 | C10H19FN2O2Si2 | 1,3-bis(trimethylsilyl)fluorouracil | 2,4(1H,3H)-Pyrimidinedione, 5-fluoro-1,3-bis(trimethylsilyl)- |
| 13369-70-5 | C4H7ClO | 2-chlorotetrahydrofuran | Furan, 2-chlorotetrahydro- |
| 1191-99-7 | C4H6O | 2,3-dihydrofuran | Furan, 2,3-dihydro- |
| 51-21-8 | C4H3FN2O2 | 5-fluorouracil | 2,4(1H,3H)-Pyrimidinedione, 5-fluoro- |


SYN1
CN 106397416
SYN 2

Advanced Synthesis & Catalysis, 356(16), 3325-3330; 2014
PATENTS
CN 106397416
CN 104513230
CN 103159746
PATENT
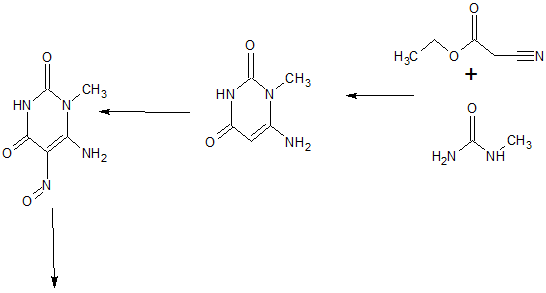
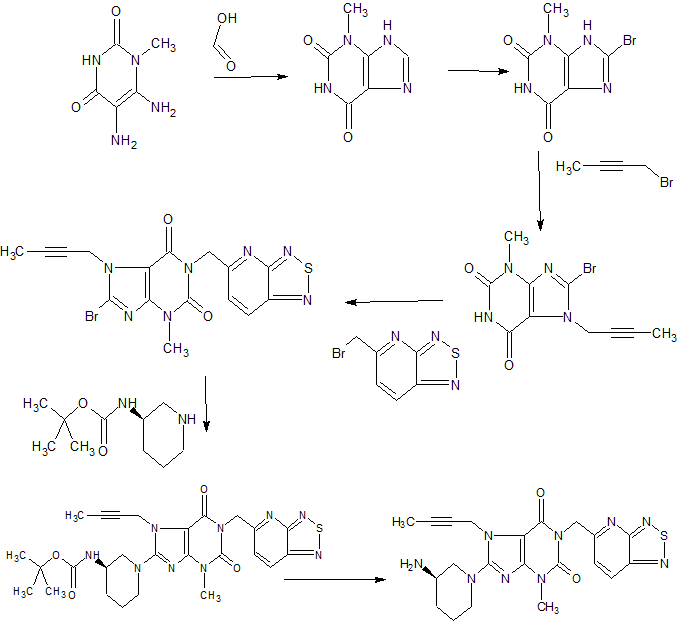
tegafur is a derivative of 5-fluorouracil, and in 1967, Hiller of the former Soviet Union synthesized tegafur (SA Hiller, RA Zhuk, M. Yu. Lidak, et al. Substituted Uracil [ P, British Patent, 1168391 (1969)). In 1974, it was listed in Japan. China was successfully developed by Shandong Jinan Pharmaceutical Factory in 1979. Its present origin is Shanghai and Shandong provinces and cities. The anti-cancer effect of tegafur is similar to that of 5-fluorouracil and is activated in vivo by 5-fluorouracil through liver activation. Unlike 5-fluorouracil, tegafur is fat-soluble, has good oral absorption, maintains high concentrations in the blood for a long time and easily passes through the blood-brain barrier. Clinical and animal experiments show that tegafur on gastrointestinal cancer, breast cancer is better, the role of rectal cancer than 5-fluorouracil good, less toxic than 5-fluorouracil. Teflon has a chemotherapy index of 2-fold for 5-fluorouracil and only 1 / 4-1 / 7 of toxicity. So the addition of fluoride is widely used in cancer patients with chemotherapy.
[0003] The first synthesis of tegafur is Hiller ([SA Hiller, RA Zhuk, Μ. Yu. Lidak, et al. Substituted Uracil [P], British Patent, 1168391 (1969)]. 5-fluorouracil or 2,4-bis (trimethylsilyl) -5-fluorouracil (Me3Si-Fu, 1) and 2-chlorotetrahydrofuran (Thf-Cl), and it is reported that this synthesis must be carried out at low temperature (- 20 to -40 ° C), because Thf-Cl is unstable, and excess Thf-Cl results in a decomposition reaction, thereby reducing the yield of Thf-Fu.
[0004] Earl and Townsend also prepared 1_ (tetrahydro-2-furyl) uracil using Thf-Cl and 2,4-bis (trimethylsilyl) uracil, and then using trifluoromethyl fluorite to product Fluorination. Mitsugi Yasurnoto reacts with the Friedel-Crafts catalyst in the presence of 2,4-bis (trimethylsilyl) -5-fluorouracil (Me3Si-U, 1) 2-acetoxytetrahydrofuran (Thf-OAc, 2) (Kazu Kigasawa et al., 2-tert-Butoxy), & lt; RTI ID = 0.0 & gt;, & lt; / RTI & gt; (K. Kigasawa, M. Hiiragi, K. ffakisaka, et al. J. Heterocyclic Chem. 1977, 14: 473-475) was reacted with 5-Fu at 155-160 ° C. Reported in the literature for the fluoride production route there are the following questions: 1, high energy consumption. In the traditional synthesis method, in order to obtain the product, the second step of the reaction needs to continue heating at 160 ° C for 5-6 hours, high energy consumption; 2, difficult to produce, low yield: 5-fluorouracil as a solid powder The reaction needs to be carried out at a high temperature (160 ° C), which requires the use of a high boiling solvent N, N-dimethylformamide (DMF). But it is difficult to completely remove the fluoride from the addition of fluoride, because DMF can form hydrogen bonds with the fluoride molecules, difficult to separate from each other; 3, in order to unreacted 5-fluorouracil and tegafur separation and recycling , The use of carcinogenic solvent chloroform as a extractant in the conventional method to separate 5-fluorouracil and tegafur. However, the main role of chloroform on the central nervous system, with anesthesia, the heart, liver, kidney damage; the environment is also harmful to the water can cause pollution. Therefore, the use of volatile solvent chloroform, even if the necessary measures to reduce its volatilization, will still cause harm to human health and the environment; 4, low yield. Since both NI and N-3 in the 5-fluorouracil molecule react with 2-tert-butoxytetrahydrofuran, the addition of tegafur is also the addition of 1,3-bis (tetrahydro-2-furyl) -5 – Fluorouracil. Therefore, the improvement of the traditional production process of tegafur is a significant and imminent task.
Example 1 (for example, the best reaction conditions):
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran, 1.9 g (50 mmol) of ethanol was added to a one-necked flask. To this was added 15 ml of tetrahydrofuran (THF). And then weighed 10. 0 mg CuCl2, microwave irradiation 250W at 25 ° C reaction 0. 6h. Cool to room temperature, add 1.95 g (15 mmol) of 5-fluorouracil (5-Fu), and microwave irradiation at 400 ° C for 100 ° C. After distilling off the low boiling solvent, the oil was obtained. Rinsed with ether to give a white solid which was recrystallized from anhydrous ethanol to give 1.34349 g of product. Melting point: 160-165 ° C. The yield was 75%.
[0011] Example 2
Weigh 3,5 g (50 mmol) of 2,3-dihydrofuran and 3.8 g (100 mmol) of ethanol were added to a single-necked flask. To this was added 15 ml of tetrahydrofuran (THF). And then weighed 5mg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cool to room temperature, add 1.95 g (15 mmol) of 5-fluorouracil (5-Fu), microwave irradiation 400W, reaction temperature 60 ° C under the reaction pool. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from absolute ethanol to give the product 0. 46 g. Melting point: 160-165 ° C. The yield was 15%.
[0012] Example 3
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran, 1.9 g (50 mmol) of ethanol was added to a one-necked flask. To this was added 15 ml of tetrahydrofuran (THF). And then weighed 20mg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 2001, reaction temperature 1301: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from anhydrous ethanol to give the product 1.81 g. Melting point: 160-165 ° C. The yield was 61%.
[0013] Example 4
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran and 19 g (500 mmol) of ethanol were added to a single-necked flask. To this was added 20 ml of tetrahydrofuran (THF). And then weighed IOmg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 2001, reaction temperature 1101: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from absolute ethanol to give product U6g. Melting point: 160-165 ° C. The yield was 43%.
[0014] Example 5
Weigh 3,5 g (50 mmol) of 2,3-dihydrofuran and 9.5 g (250 mmol) of ethanol were added to a single-necked flask. To this was added 30 ml of tetrahydrofuran (THF). And then weighed IOmg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 6001, reaction temperature 1001: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from absolute ethanol to give 1.15 g of product. Melting point: 160-165 ° C. The yield was 38%.
[0015] Example 6
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran, 1.9 g (50 mmol) of ethanol was added to a one-necked flask. To this was added 25 ml of tetrahydrofuran (THF). And then weighed 15mg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 5001, reaction temperature 1101: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from anhydrous ethanol to give product 2.10 g. Melting point: 160-165 ° C. The yield was 70%.
Paper
A novel protocol for preparation of tegafur (a prodrug of 5-fluorouracil) is reported. The process involves the 1,8-diazabicycloundec-7-ene-mediated alkylation of 5-fluorouracil with 2-acetoxytetrahydrofuran at 90 °C, followed by treatment of the prepurified mixture of the alkylation products with aqueous ethanol at 70 °C. The yield of the two-step process is 72%.
Synthesis of Tegafur by the Alkylation of 5-Fluorouracil under the Lewis Acid and Metal Salt-Free Conditions
Aleksandra Zasada, Ewa Mironiuk-Puchalska, and Mariola Koszytkowska-Stawińska*
Faculty of Chemistry, Warsaw University of Technology, ul. Noakowskiego 3, 00-664 Warszawa, Poland
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00103
*E-mail: mkoszyt@ch.pw.edu.pl.
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.7b00103
Click to access op7b00103_si_001.pdf
Tegafur, a prodrug of 5-fluorouracil (5-FUra), was discovered in 1967. The compound features high lipophilicity and water solubility compared to 5-FUra. Tegafur is used as a racemate since no significant difference in antitumor activity of enantiomers was observed.
The prodrug is gradually converted to 5-FUra by metabolism in the liver. Hence, a rapid breakdown of the released 5-FUra in the gastrointestinal tract is avoided.(6) In injectable form, tegafur provoked serious side effects, such as nausea, vomiting, or central nervous system disturbances.
The first generation of oral formulation of tegafur , UFT) is a combination of tegafur and uracil in a fixed molar ratio of 1:4, respectively. The uracil slows the metabolism of 5-FUra and reduces production of 2-fluoro-α-alanine as the toxic metabolite. UFT was approved in 50 countries worldwide excluding the USA.
S-1 is the next generation of oral formulation of tegafur.(7) It is a combination of tegafur, gimeracil, and oteracil in a fixed molar ratio of 1:0.4:1, respectively.
Gimeracil inhibits the enzyme responsible for the degradation of 5-FUra. Oteracil prevents the activation of 5-FUra in the gastrointestinal tract, thus minimizing the gastrointestinal toxicity of 5-FUra. S-1 is well-tolerated, but its safety can be influenced by schedule and dose, similar to any other cytotoxic agent. Since common side effects of S-1 can be managed with antidiarrheal and antiemetic medications, the drug can be administered in outpatient settings. S-1 was approved in Japan, China, Taiwan, Korea, and Singapore for the treatment of patients with gastric cancer.
In 2010, the Committee for Medicinal Products for Human Use (CHMP), a division of the European Medicines Agency (EMA), recommended the use of S-1 for the treatment of adults with advanced gastric cancer when given in a combination with cisplatin. Currently, S-1 has not been approved by the FDA in the United States.
There is a great interest in further examination of S-1 as an anticancer chemotherapeutic. Currently, 23 clinical trials with S-1 has been registered in National Institutes of Health (NIH). Combinations of S-1 and other anticancer agents have been employed in a majority of these trials.
5-Fluoro-1-(tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (Tegafur)
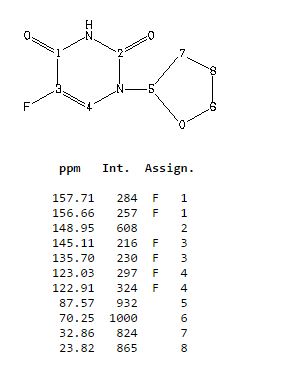
δH 1.89–2.10 (m, 3H), 2.38–2.45 (m, 1H), 3.97–4.01 (q-like m, 1H), 4.20–4.24 (dq-like m), 5.97–5.98 (m, 1H), 7.41 (d, 3JHF 6.1), 9.21 (bs, 1H, NH).
δC 23.82, 32.90, 70.26, 87.58, 123.63 (d, 2JCF 33.89), 140.33 (d, 1JCF 237.20) 148.66, 156.9 (d, 2JCF 26.81).
HRMS m/z calcd for C8H10N2O3F [M – H]+ 201.0670, found 201.0669.
Elemental analysis. Found C%, 46.42; H%, 4.45; N%, 13.35. Calcd for 3(C8H9N2O3F)·H2O: C%, 46.61; H%, 4.73; N%, 13.59.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN85108855A * | Nov 6, 1985 | Sep 24, 1986 | Central Chemical Research Institute | Preparation of 1- (2-tetrahydrofuryl) -5-fluorouracil |
| GB1168391A * | Title not available | |||
| JPS5452085A * | Title not available | |||
| JPS5455581A * | Title not available | |||
| JPS5459288A * | Title not available | |||
| JPS52118479A * | Title not available | |||
| JPS54103880A * | Title not available | |||
| US4256885 * | Dec 10, 1976 | Mar 17, 1981 | Mitsui Toatsu Kagaku Kabushiki Kaisha | Process for the preparation of 1- (2-tetrahydrofuryl) -5-fluorouracil |
| US5075446 * | Oct 12, 1990 | Dec 24, 1991 | Korea Advanced Institute Of Science & Technology | Synthesis of tetrahydro-2-furylated pyrimidine derivatives |
| Reference | ||
|---|---|---|
| 1 | * | KAZUO KIGASAWA, et al .: ” Studies on the Synthesis of Chemotherapeutics. Synthetic of 1- (2-Tetrahydrofuryl) -5-fluorouracil [Ftorafur] (Studies on the Syntheses of Heterocyclic Compound. Part 703) “, “J. HETEROCCLIC CHEM ., Vol. 14, 31 May 1977 (1977-05-31), pages 473 – 475 |
References
1
- El Sayed, YM; Sadée, W (1983). “Metabolic activation of R,S-1-(tetrahydro-2-furanyl)-5-fluorouracil (ftorafur) to 5-fluorouracil by soluble enzymes”. Cancer Research. 43 (9): 4039–44. PMID 6409396.
- 2
- Sweetman, S, ed. (14 November 2011). “Martindale: The Complete Drug Reference”. Pharmaceutical Press. Retrieved 12 February 2014.
- 3
- Ishikawa, T (14 May 2008). “Chemotherapy with enteric-coated tegafur/uracil for advanced hepatocellular carcinoma.” (PDF). World Journal of Gastroenterology : WJG. 14 (18): 2797–2801. doi:10.3748/wjg.14.2797. PMC 2710718
 . PMID 18473401.
. PMID 18473401. - 4
- Caudle, KE; Thorn, CF; Klein, TE; Swen, JJ; McLeod, HL; Diasio, RB; Schwab, M (December 2013). “Clinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing.”. Clinical pharmacology and therapeutics. 94 (6): 640–5. doi:10.1038/clpt.2013.172. PMC 3831181 . PMID 23988873.
- 5
- Amstutz, U; Froehlich, TK; Largiadèr, CR (September 2011). “Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity.”. Pharmacogenomics. 12 (9): 1321–36. doi:10.2217/pgs.11.72. PMID 21919607.
- 6
- Nakayama, T; Noguchi, S (January 2010). “Therapeutic usefulness of postoperative adjuvant chemotherapy with Tegafur-Uracil (UFT) in patients with breast cancer: focus on the results of clinical studies in Japan.” (PDF). The Oncologist. 15 (1): 26–36. doi:10.1634/theoncologist.2009-0255. PMC 3227888 . PMID 20080863.
- 7
Matt P, van Zwieten-Boot B, Calvo Rojas G, Ter Hofstede H, Garcia-Carbonero R, Camarero J, Abadie E, Pignatti F (October 2011). “The European Medicines Agency review of Tegafur/Gimeracil/Oteracil (Teysuno™) for the treatment of advanced gastric cancer when given in combination with cisplatin: summary of the Scientific Assessment of the Committee for medicinal products for human use (CHMP).” (PDF). The Oncologist. 16 (10): 1451–1457. doi:10.1634/theoncologist.2011-0224. PMC 3228070 ![]() . PMID 21963999.
. PMID 21963999.
- (1) Hirose, Takashi; Oncology Reports 2010, V24(2), P529-536
- (2) Fujita, Ken-ichi; Cancer Science 2008, V99(5), P1049-1054
- (3) Tahara, Makoto; Cancer Science 2011, V102(2), P419-424
- (4) Chu, Quincy Siu-Chung; Clinical Cancer Research 2004, V10(15), P4913-4921
- (5) Tominaga, Kazunari; Oncology 2004, V66(5), P358-364
- (6) Peters, Godefridus J.; Clinical Cancer Research 2004, V10(12, Pt. 1), P4072-4076
- (7) Kim, Woo Young; Cancer Science 2007, V98(10), P1604-1608
- Hillers, Solomon; Puti Sinteza i Izyskaniya Protivoopukholevykh Preparatov 1970, VNo. 3, P109-12
- Grishko, V. A.; Trudy Kazakhskogo Nauchno-Issledovatel’skogo Instituta Onkologii i Radiologii 1977, V12, P110-14
- Ootsu, Koichiro; Takeda Kenkyushoho 1978, V37(3-4), P267-77
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
- Yabuuchi, Youichi; Oyo Yakuri 1971, V5(4), P569-84
- Germane, S.; Eksperimental’naya i Klinicheskaya Farmakoterapiya 1970, (1), P85-92
- JP 56046814 A 1981
MORE
- AIST: Integrated Spectral Database System of Organic Compounds. (Data were obtained from the National Institute of Advanced Industrial Science and Technology (Japan))
- ACD-A: Sigma-Aldrich (Spectral data were obtained from Advanced Chemistry Development, Inc.)
- Nomura, Hiroaki; Chemical & Pharmaceutical Bulletin 1979, V27(4), P899-906
- Sakurai, Kuniyoshi; Chemical & Pharmaceutical Bulletin 1978, V26(11), P3565-6
- Miyashita, Osamu; Chemical & Pharmaceutical Bulletin 1981, V29(11), P3181-90
- Lukevics, E.; Zhurnal Obshchei Khimii 1981, V51(4), P827-34
- Needham, F.; Powder Diffraction 2006, V21(3), P245-247
-
- Nomura, Hiroaki; Chemical & Pharmaceutical Bulletin 1979, V27(4), P899-906
- Sakurai, Kuniyoshi; Chemical & Pharmaceutical Bulletin 1978, V26(11), P3565-6
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
- Miyashita, Osamu; Chemical & Pharmaceutical Bulletin 1981, V29(11), P3181-90
- “PhysProp” data were obtained from Syracuse Research Corporation of Syracuse, New York (US)
- Lukevics, E.; Zhurnal Obshchei Khimii 1981, V51(4), P827-34
- Lukevics, E.; Latvijas PSR Zinatnu Akademijas Vestis, Kimijas Serija 1982, (3), P317-20
- Kruse, C. G.; Recueil des Travaux Chimiques des Pays-Bas 1979, V98(6), P371-80
- Lukevics, E.; Latvijas PSR Zinatnu Akademijas Vestis, Kimijas Serija 1981, (4), P492-3
- Kametani, Tetsuji; Heterocycles 1977, V6(5), P529-33
- Kametani, Tetsuji; Journal of Heterocyclic Chemistry 1977, V14(3), P473-5
- Hillers, S.; GB 1168391 1969
 |
|
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Biological half-life | 3.9-11 hours |
| Identifiers | |
| Synonyms | 5-fluoro-1-(oxolan-2-yl)pyrimidine-2,4-dione |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.038.027 |
| Chemical and physical data | |
| Formula | C8H9FN2O3 |
| Molar mass | 200.16 g/mol |
| 3D model (Jmol) | |
///////////TEGAFUR
FC1=CN(C2CCCO2)C(=O)NC1=O
FDA approves first generic Strattera (atomoxetine) for the treatment of ADHD
May 30, 2017
Release
The U.S. Food and Drug Administration today approved the first generic versions of Strattera (atomoxetine) to treat attention-deficit/hyperactivity disorder (ADHD) in pediatric and adult patients.
Apotex Inc., Teva Pharmaceuticals USA Inc., Aurobindo Pharma Limited and Glenmark Pharmaceuticals Limited today gained approval to market atomoxetine in multiple strengths.
“Today’s approvals mark an important step forward in bringing consumers additional treatments that have met the FDA’s rigorous standards,” said Kathleen Uhl, M.D., director of the Office of Generic Drugs in the FDA’s Center for Drug Evaluation and Research. “Quickly bringing generics to market so patients have more options to treat their conditions is a top priority for the FDA.”
Generic prescription drugs approved by the FDA have the same high quality and strength as brand-name drugs. Generic prescription drug manufacturing and packaging sites must pass the same quality standards as those of brand-name drugs.
ADHD is marked by an ongoing pattern of inattention and/or hyperactivity-impulsivity that interferes with functioning or development.
In the clinical trials for atomoxetine in children and adolescents, the most common side effects reported were upset stomach, decreased appetite, nausea or vomiting, dizziness, tiredness, and mood swings. In the clinical trials in adults, the most common side effects reported were constipation, dry mouth, nausea, decreased appetite, dizziness, sexual side effects, and problems passing urine.
Atomoxetine must be dispensed with a patient Medication Guide that describes the drug’s uses and warnings. This medication has a boxed warning for the increased risk of suicidal ideation in children and adolescents. Patients taking this medication should be monitored appropriately and observed closely for clinical worsening, suicidality, and unusual changes in behavior, especially during the initial few months of a course of drug therapy, or at times of dose changes. Other important warnings include the risk of severe liver damage and potential for serious cardiovascular events.
FDA approves first cancer treatment for any solid tumor with a specific genetic feature
May 23, 2017
Release
The U.S. Food and Drug Administration today granted accelerated approval to a treatment for patients whose cancers have a specific genetic feature (biomarker). This is the first time the agency has approved a cancer treatment based on a common biomarker rather than the location in the body where the tumor originated.
Keytruda (pembrolizumab) is indicated for the treatment of adult and pediatric patients with unresectable or metastatic solid tumors that have been identified as having a biomarker referred to as microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR). This indication covers patients with solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options and patients with colorectal cancer that has progressed following treatment with certain chemotherapy drugs.
“This is an important first for the cancer community,” said Richard Pazdur, M.D., acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research and director of the FDA’s Oncology Center of Excellence. “Until now, the FDA has approved cancer treatments based on where in the body the cancer started—for example, lung or breast cancers. We have now approved a drug based on a tumor’s biomarker without regard to the tumor’s original location.”
MSI-H and dMMR tumors contain abnormalities that affect the proper repair of DNA inside the cell. Tumors with these biomarkers are most commonly found in colorectal, endometrial and gastrointestinal cancers, but also less commonly appear in cancers arising in the breast, prostate, bladder, thyroid gland and other places. Approximately 5 percent of patients with metastatic colorectal cancer have MSI-H or dMMR tumors.
Keytruda works by targeting the cellular pathway known as PD-1/PD-L1 (proteins found on the body’s immune cells and some cancer cells). By blocking this pathway, Keytruda may help the body’s immune system fight the cancer cells. The FDA previously approved Keytruda for the treatment of certain patients with metastatic melanoma, metastatic non-small cell lung cancer, recurrent or metastatic head and neck cancer, refractory classical Hodgkin lymphoma, and urothelial carcinoma.
Keytruda was approved for this new indication using the Accelerated Approvalpathway, under which the FDA may approve drugs for serious conditions where there is unmet medical need and a drug is shown to have certain effects that are reasonably likely to predict a clinical benefit to patients. Further study is required to verify and describe anticipated clinical benefits of Keytruda, and the sponsor is currently conducting these studies in additional patients with MSI-H or dMMR tumors.
The safety and efficacy of Keytruda for this indication were studied in patients with MSI-H or dMMR solid tumors enrolled in one of five uncontrolled, single-arm clinical trials. In some trials, patients were required to have MSI-H or dMMR cancers, while in other trials, a subgroup of patients were identified as having MSI-H or dMMR cancers by testing tumor samples after treatment began. A total of 15 cancer types were identified among 149 patients enrolled across these five clinical trials. The most common cancers were colorectal, endometrial and other gastrointestinal cancers. The review of Keytruda for this indication was based on the percentage of patients who experienced complete or partial shrinkage of their tumors (overall response rate) and for how long (durability of response). Of the 149 patients who received Keytruda in the trials, 39.6 percent had a complete or partial response. For 78 percent of those patients, the response lasted for six months or more.
Common side effects of Keytruda include fatigue, itchy skin (pruritus), diarrhea, decreased appetite, rash, fever (pyrexia), cough, difficulty breathing (dyspnea), musculoskeletal pain, constipation and nausea. Keytruda can cause serious conditions known as immune-mediated side effects, including inflammation of healthy organs such as the lungs (pneumonitis), colon (colitis), liver (hepatitis), endocrine glands (endocrinopathies) and kidneys (nephritis). Complications or death related to allogeneic hematopoietic stem cell transplantation after using Keytruda has occurred.
Patients who experience severe or life-threatening infusion-related reactions should stop taking Keytruda. Women who are pregnant or breastfeeding should not take Keytruda because it may cause harm to a developing fetus or newborn baby. The safety and effectiveness of Keytruda in pediatric patients with MSI-H central nervous system cancers have not been established.
The FDA granted this application Priority Review designation, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted accelerated approval of Keytruda to Merck & Co.
///////////Keytruda, pembrolizumab, BIO MARKER, MERCK, FDA 2017
FDA approves first drug Actemra (tocilizumab) to specifically treat giant cell arteritis
May 22, 2017
Release
The U.S. Food and Drug Administration today expanded the approved use of subcutaneous Actemra (tocilizumab) to treat adults with giant cell arteritis. This new indication provides the first FDA-approved therapy, specific to this type of vasculitis.
“We expedited the development and review of this application because this drug fulfills a critical need for patients with this serious disease who had limited treatment options,” said Badrul Chowdhury, M.D., Ph.D., director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Giant cell arteritis is a form of vasculitis, a group of disorders that results in inflammation of blood vessels. This inflammation causes the arteries to narrow or become irregular, impeding adequate blood flow. In giant cell arteritis, the vessels most involved are those of the head, especially the temporal arteries (located on each side of the head). For this reason, the disorder is sometimes called temporal arteritis. However, other blood vessels, including large ones like the aorta, can become inflamed in giant cell arteritis. Standard treatment involves high doses of corticosteroids that are tapered over time.
The efficacy and safety of subcutaneous (injected under the skin) Actemra for giant cell arteritis were established in a double-blind, placebo-controlled study with 251 patients with giant cell arteritis. The primary efficacy endpoint was the proportion of patients achieving sustained remission from Week 12 through Week 52. Sustained remission was defined as the absence of symptoms of giant cell arteritis, normalization of inflammatory laboratory tests, and tapering the use of prednisone (a steroid drug). A greater proportion of patients receiving subcutaneous Actemra with standardized prednisone regimens achieved sustained remission from Week 12 through Week 52 as compared to patients receiving placebo with standardized prednisone regimens. The cumulative prednisone dose was lower in treated patients with Actemra relative to placebo.
The overall safety profile observed in the Actemra treatment groups was generally consistent with the known safety profile of Actemra. Actemra carries a Boxed Warning for serious infections. Patients treated with Actemra who develop a serious infection should stop that treatment until the infection is controlled. Live vaccines should be avoided during treatment with Actemra. Actemra should be used with caution in patients at increased risk of gastrointestinal perforation. Hypersensitivity reactions, including anaphylaxis and death, have occurred. Laboratory monitoring is recommended due to potential consequences of treatment-related changes in neutrophils (type of white blood cell), platelets, lipids and liver function tests.
Subcutaneous Actemra was previously approved for the treatment of moderate to severely active rheumatoid arthritis. Intravenous Actemra was also previously approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis and polyarticular juvenile idiopathic arthritis. Intravenous administration is not approved for giant cell arteritis.
The FDA granted this application a Breakthrough Therapy designation and a Priority Review.
The FDA granted the supplemental approval of Actemra to Hoffman La Roche, Inc.
FDA expands approved use of Kalydeco IVACAFTOR to treat additional mutations of cystic fibrosis
For Immediate Release
May 17, 2017
Release
The U.S. Food and Drug Administration today expanded the approved use of Kalydeco (ivacaftor) for treating cystic fibrosis. The approval triples the number of rare gene mutations that the drug can now treat, expanding the indication from the treatment of 10 mutations, to 33. The agency based its decision, in part, on the results of laboratory testing, which it used in conjunction with evidence from earlier human clinical trials. The approach provides a pathway for adding additional, rare mutations of the disease, based on laboratory data.
“Many rare cystic fibrosis mutations have such small patient populations that clinical trial studies are not feasible,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “This challenge led us to using an alternative approach based on precision medicine, which made it possible to identify certain gene mutations that are likely to respond to Kalydeco.
Cystic fibrosis affects the cells that produce mucus, sweat and digestive juices. These secreted fluids are normally thin and slippery due to the movement of sufficient ions (chloride) and water in and out of the cells. People with the progressive disease have a defective cystic fibrosis transmembrane conductance regulator (CFTR) gene that can’t regulate the movement of ions and water, causing the secretions to become sticky and thick. The secretions build up in the lungs, digestive tract and other parts of the body leading to severe respiratory and digestive problems, as well as other complications such as infections and diabetes.
Results from an in vitro cell-based model system have been shown to reasonably predict clinical response to Kalydeco. When additional mutations responded to Kalydeco in the laboratory test, researchers were thus able to extrapolate clinical benefit demonstrated in earlier clinical trials of other mutations. This resulted in the addition of gene mutations for which the drug is now indicated.
Kalydeco, available as tablets or oral granules taken two times a day with fat-containing food, helps the protein made by the CFTR gene function better and as a result, improves lung function and other aspects of cystic fibrosis, including weight gain. If the patient’s genotype is unknown, an FDA-cleared cystic fibrosis mutation test should be used to detect the presence of a CFTR mutation followed by verification with bi-directional sequencing when recommended by the mutation test instructions for use.
Cystic fibrosis is a rare disease that affects about 30,000 people in the United States.Kalydeco is indicated for patients aged 2 and older who have one mutation in the CFTR gene that is responsive to drug treatment based on clinical and/or in vitro (laboratory) data. The expanded indication will affect another 3 percent of the cystic fibrosis population, impacting approximately 900 patients. Kalydeco serves as an example of how successful patient-focused drug development can provide greater understanding about a disease. For example, the Cystic Fibrosis Foundation maintains a 28,000-patient registry, including genetic data, which it makes available for research.
Common side effects of Kalydeco include headache; upper respiratory tract infection (common cold) including sore throat, nasal or sinus congestion, or runny nose; stomach (abdominal) pain; diarrhea; rash; nausea; and dizziness. Kalydeco is associated with risks including elevated transaminases (various enzymes produced by the liver) and pediatric cataracts. Co-administration with strong CYP3A inducers (e.g., rifampin, St. John’s wort) substantially decreases exposure of Kalydeco, which may diminish effectiveness, and is therefore not recommended.
Kalydeco is manufactured for Boston-based Vertex Pharmaceuticals Inc.
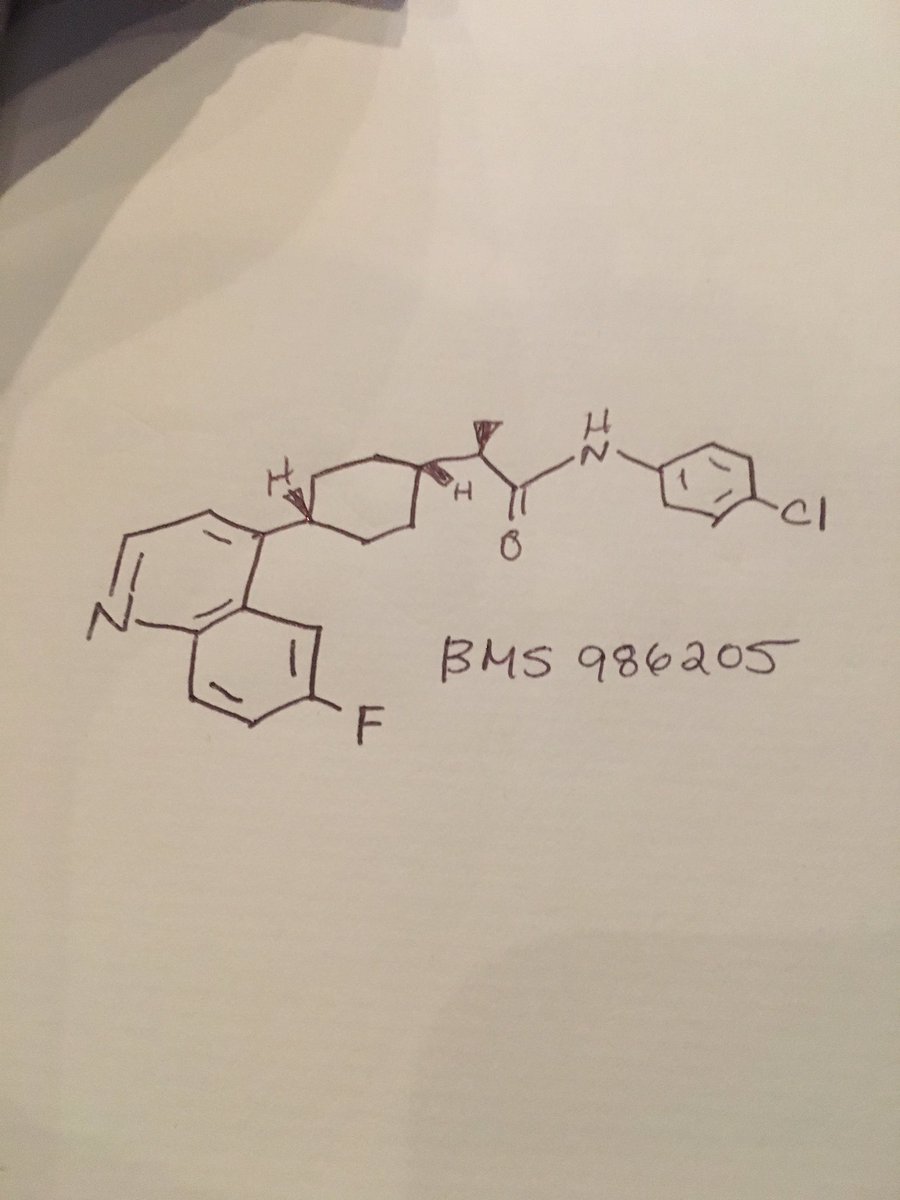
BMS 986205


BMS 986205
CAS: 1923833-60-6
Phase 1 cancer
BMS-986205, ONO-7701, F- 001287
- Molecular Formula C24H24ClFN2O
- Average mass 410.912 Da
- Originator Bristol-Myers Squibb
- Class Antineoplastics
- 01 Feb 2016 Phase-I/II clinical trials in Cancer (Combination therapy, Late-stage disease, Second-line therapy or greater) in Canada (PO) (NCT02658890)
- 31 Jan 2016 Preclinical trials in Cancer in USA (PO) before January 2016
- 01 Jan 2016 Bristol-Myers Squibb plans a phase I/IIa trial for Cancer (Late-stage disease, Combination therapy, Second-line therapy or greater) in USA, Australia and Canada (PO) (NCT02658890)

Hilary Beck
 EX Principal Investigator, Company NameFLX Bio, Inc.,
EX Principal Investigator, Company NameFLX Bio, Inc.,
CURRENTLY Director, Medicinal Chemistry at IDEAYA Biosciences, IDEAYA Biosciences, The University of Texas at Austin
![]()

Brian Wong
Chief Executive Officer at FLX Bio, Inc.
Bristol-Myers Squibb, following its acquisition of Flexus Biosciences, is developing BMS-986205 (previously F- 001287), the lead from an immunotherapy program of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors for the potential treatment of cancer. In February 2016, a phase I/IIa trial was initiated .
BMS-986205 (ONO-7701) is being evaluated at Bristol-Myers Squibb in phase I/II clinical trials for the oral treatment of adult patients with advanced cancers in combination with nivolumab. Early clinical development is also ongoing at Ono in Japan for the treatment of hematologic cancer and for the treatment of solid tumors.
In April 2017, data from the trial were presented at the 108th AACR Annual Meeting in Washington DC. As of February 2017, the MTD had not been reached, but BMS-986205 plus nivolumab treatment was well tolerated, with only two patients discontinuing treatment due to DLTs. The most commonly reported treatment-related adverse events (TRAEs) were decreased appetite, fatigue, nausea, diarrhea, and vomiting. Grade 3 TRAEs were reported in three patients during the combination therapy; however, no grade 3 events were reported during BMS-986205 monotherapy lead-in. No grade 4 or 5 TRAEs were reported with BMS-986205 alone or in combination with nivolumab
Indoleamine 2,3-dioxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO is an oxidoreductase and one of two enzymes that catalyze the first and rate-limiting step in the conversion of tryptophan to N-formyl-kynurenine. It exists as a 41kD monomer that is found in several cell populations, including immune cells, endothelial cells, and fibroblasts. IDO is relatively well-conserved between species, with mouse and human sharing 63% sequence identity at the amino acid level. Data derived from its crystal structure and site-directed mutagenesis show that both substrate binding and the relationship between the substrate and iron-bound dioxygenase are necessary for activity. A homolog to IDO (ID02) has been identified that shares 44% amino acid sequence homology with IDO, but its function is largely distinct from that of IDO. (See, e.g., Serafini P, et al, Semin. Cancer Biol, 16(l):53-65 (Feb. 2006) and Ball, H.J. et al, Gene, 396(1):203-213 (Jul. 2007)).
IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. Importantly, IDO regulates immunity at the T cell level, and a nexus exists between IDO and cytokine production. In addition, tumors frequently manipulate immune function by upregulation of IDO. Thus, modulation of IDO can have a therapeutic impact on a number of diseases, disorders and conditions.
A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res., 12(4): 1144-1151 (Feb. 15, 2006)).
Treatment of cancer commonly entails surgical resection followed by chemotherapy and radiotherapy. The standard treatment regimens show highly variable degrees of long-term success because of the ability of tumor cells to essentially escape by regenerating primary tumor growth and, often more importantly, seeding distant metastasis. Recent advances in the treatment of cancer and cancer-related diseases, disorders and conditions comprise the use of combination therapy incorporating immunotherapy with more traditional chemotherapy and radiotherapy. Under most scenarios, immunotherapy is associated with less toxicity than traditional chemotherapy because it utilizes the patient’s own immune system to identify and eliminate tumor cells.
In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g. , rheumatoid arthritis). Thus, suppression of tryptophan degradation by inhibition of IDO activity has tremendous therapeutic value. Moreover, inhibitors of IDO can be used to enhance T cell activation when the T cells are suppressed by pregnancy, malignancy, or a virus (e.g., HIV). Although their roles are not as well defined, IDO inhibitors may also find use in the treatment of patients with neurological or neuropsychiatric diseases or disorders (e.g., depression).
Small molecule inhibitors of IDO have been developed to treat or prevent IDO-related diseases. For example, the IDO inhibitors 1-methyl-DL-tryptophan; p-(3-benzofuranyl)-DL-alanine; p-[3-benzo(b)thienyl]-DL-alanine; and 6-nitro-L-tryptophan have been used to modulate T cell-mediated immunity by altering local extracellular concentrations of tryptophan and tryptophan metabolites (WO 99/29310). Compounds having IDO inhibitory activity are further reported in WO 2004/094409.
In view of the role played by indoleamine 2,3-dioxygenase in a diverse array of diseases, disorders and conditions, and the limitations (e.g., efficacy) of current IDO inhibitors, new IDO modulators, and compositions and methods associated therewith, are needed.
In April 2017, preclinical data were presented at the 108th AACR Annual Meeting in Washington DC. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively
In April 2017, preclinical data were presented at the 253rd ACS National Meeting and Exhibition in San Francisco, CA. BMS-986205 showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. A good pharmacokinetic profile was seen at oral and iv doses in rats, dogs and monkeys. The compound showed good oral exposure and efficacy in in vivo assays
Preclinical studies were performed to evaluate the activity of BMS-986205, a potent and selective optimized indoleamine 2, 3-dioxygenase (IDO)- 1inhibitor, for the treatment of cancer. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. BMS-986205 was also found to be potent when compared with IDO-1from other species (human < dog equivalent monkey equivalent mouse > rat). In cell-free systems, incubation of inhibitor lead to loss of heme absorbance of IDO-1 which was observed in the presence of BMS-986205 (10 microM), while did not observed with epacadostat (10 microM). The check inhibitory activity and check reversibility (24 h after compound removal) of BMS-986205 was found to be < 1 and 18% in M109 (mouse) and < 1 and 12% SKOV3 (human) cells, respectively. In human whole blood IDO-1, human DC mixed lymphocyte reaction and human T cells cocultured with SKOV3 cells- cell based assays, BMS-986205 showed potent cellular effects (inhibition of kynurenine and T-cell proliferation 3H-thymidine) with IC50 values of 2 to 42 (median 9.4 months), 1 to 7 and 15 nM, respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively. In vivo human-SKOV3 and hWB-xenografts, IC50 values of BMS-986205 were 3.4 and 9.4 NM, respectively. The ADME of BMS-986205 at parameters iv/po dose was 0.5/2, 0.5/1.5 and 0.5/1.2 mg/kg, respectively; iv/clearance was 27, 25 and 19 ml, min/kg, respectively; iv Vss was 3.8, 5.7 and 4.1 l/kg, respectively; t1/2 (iv) was 3.9, 4.7 and 6.6 h, respectively; fraction (po) was 64, 39 and 10%, respectively. At the time of presentation, BMS-986205 was being evaluated in combination with nivolumab.
The chemical structure and preclinical profile was presented for BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.
BMS-986158: a BET inhibitor for cancerAshvinikumar Gavai of Bristol Myers Squibb (BMS) gave an overview of his company’s research into Bromodomian and extra-terminal domain (BET) as oncology target for transcriptional suppression of key oncogenes, such as MYC and BCL2. BET inhibition has been defined as strong rational strategy for the treatment of hematologic malignancies and solid tumors. From crystal-structure guided SAR studies, BMS-986158, 2-{3-(1,4-Dimethyl-1H-1,2,3-triazol-5-yl)-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol, was chosen as a potent BET inhibitor, showing IC50 values for BRD2, BRD3 and BRD4 activity of 1 nM; it also inhibited Myc oncogene (IC50 = 0.5 nM) and induced chlorogenic cancer cell death. In vitro the compound also displayed significant cytotoxicity against cancer cells. When administered at 0.25, 0.5 and 1 mg/kg po, qd to mice bearing human lung H187 SCLC cancer xenograft, BMS-986158 was robust and showed efficacy as a anticancer agent at low doses. In metabolic studies, it showed t1/2 of 36, 40 and 24 min in human, rat and mice, respectively, and it gave an efflux ratio of 3 in Caco-2 permeability assay. In phase 1/II studies, BMS-986158 was well tolerated at efficacious doses and regimens, and drug tolerable toxicity at efficacy doses and regimens. Selective Itk inhibitors for inflammatory disordersThe development of highly selective Itk inhibitors for the treatment of diseases related to T-cell function, such as inflammatory disorders, was described by Shigeyuki Takai (Ono Pharmaceutical). Inhibitory properties of a hit compound, ONO-8810443, were modified via X-ray structure and Molecular Dynamics stimulation to get ONO-212049 with significant kinase selectivity (140-fold) against Lck, a tyrosine kinase operating upstream of Itk in the TCR cascade. Further modifications identified final lead compound ONO-7790500 (N-[6-[3-amino-6-[2-(3-methoxyazetidin-1-yl)pyridin-4-yl]pyrazin-2-yl]pyridin-3-yl]-1-(3-methoxyphenyl)-2,3-dimethyl-5-oxopyrazole-4-carboxamide), which selectively inhibited Itk (IC50 = < 0.004 microM) over Lck (IC50 = 9.1 microM; SI 2000-fold) and suppressed Jurkat T-cell proliferation (IC50 = 0.014 microM). This compound suppressed alphaCD3/CDP28 CD4+T-cell stimulation (IC50 = 0.074 microM) with selectivity over PMA/Ionomycin (IC50 = > 10 microM). ONO-7790500 also exhibited in vivo IL-2 inhibitory properties (62% inhibition at 30 mg/kg po) in mice. In pharmacokinetic studies in balb/c mice, the compound administered orally (10 mg/kg) showed a Cmax of 1420 ng/ml, AUClast of 11,700 ng*h/ml, t1/2 of 5.3 h and oral bioavailability of 68%. Administration iv at 0.3 mg/kg gave an AUC last of 610 ng*h/ml, t1/2 of 3.8 h, Vss of 1260 ml/kg and Cl of 5.1 ml/min/kg. ADMET data showed ONO-7790500 did not have relevant activity in cytochromes and hERG channels (IC50 > 10 microM) in toxicological studies, and gave a PAMPA value of 5.0 x 10(-6) cm/s. Fused imidazole and pyrazole derivatives as TGF-beta inhibitorsDual growth and differentiation factor-8 (GDF-8; also known as myostatin) and TGF-beta inhibitors were described. Both targets belong to TGF-beta superfamily consisting of a large group of structurally related cell regulatory proteins involved in fundamental biological and pathological processes, such as cell proliferation or immunomodulation. Myostatin (GDF8) is a negative regulator negative regulator of skeletal muscle growth and has also been related to bone metabolism. Investigators at Rigel Pharmaceuticals found that compounds designed to be GDF-8 inhibitors were able to inhibit TGF-beta as well, this could be an advantage for the treatment of diseases associated with muscle and adipose tissue disorders, as well as potentially immunosuppressive disorders. Jiaxin Yu from the company described new fused imidazole derivatives, of which the best compound was 6-[2-(2,4,5-Trifluorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl]quinoxaline. This compound was very potent at TGF-beta Receptor Type-1 (ALK5) inhibition with an IC50 value of 1nM. In an in vivo mouse assay this compound showed good activity at 59.7 mg/kg, po, and good plasma exposure; inhibition of GDF-8 and TGFbeta growth factors was 90 and 81.6 %, respectively.Rigel’s Ihab Darwish described a series of fused pyrazole derivatives, with the best compound being 6-[2-(2,4-Difluorophenyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl][1,2,4]triazolo[1,5-a]pyridine. This compound showed an IC50 of 0.06 and 0.23 microM for GDF-8 and TGFbeta, respectively, in the pSMAD (MPC-11) signaling inhibition test. The compound had a good pharmacokinetic profile, with 40% of bioavailability in mice after a 5-mg/kg po dose. An iv dose of 1 mg/kg showed t1/2 of 0.7 h and Vss of 1.0 l/h/kgDiscovery of selective inhibitor of IDO BMS-986205 for cancerIndoleamine-2,3-dioxygenase (IDO)-1 enzyme initiates and regulates the first step of the kynurenine pathway (KP) of tryptophan metabolism, and evidence has shown that overexpression of IDO-1 in cancer tumors is a crucial mechanism facilitating tumor immune evasion and persistence. The chemical structure and preclinical profile of BMS-986205 was presented by Aaron Balog from BMS. BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), is a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.Three further reports have been published from this meeting .The website for this meeting can be found at https://www.acs.org/content/acs/en/meetings/spring-2017.html.
SYNTHESIS
1 Wittig NaH
2 REDUCTION H2, Pd, AcOEt, 4 h, rt, 50 psi
3 Hydrolysis HCl, H2O, Me2CO, 2 h, reflux
4 4-Me-2,6-(t-Bu)2-Py, CH2Cl2, overnight, rt
5 SUZUKI AcOK, 72287-26-4, Dioxane, 16 h, 80°C
6 Heck Reaction, Suzuki Coupling, Hydrogenolysis of Carboxylic Esters, Reduction of Bonds, HYDROGEN
7 Et3N, THF, rt – -78°C , Pivaloyl chloride, 15 min, -78°C; 1 h, 0°C ,THF, 0°C – -78°C, BuLi, Me(CH2)4Me, 15 min, -78°C, R:(Me3Si)2NH •Na, THF, 10 min, -50°C , HYDROLYSIS, (PrP(=O)O)3, C5H5N, AcOEt, 5 min, rt

Patent
WO2016073770
Example 19
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
Example 19 : (i?)-N-(4-chlorophenyl)-2-(cz5-4-(6-fluoroquinolin-4- yl)cyclohexyl)propanamide
[0277] Prepared using General Procedures K, B, E, L, M, N, and O. General Procedure L employed 2-(4-(6-fluoroquinolin-4-yl)-cyclohexyl)acetic acid (mixture of
diastereomers), and ( ?)-2-phenyl-oxazolidinone. General Procedure M employed the cis product and iodomethane. The auxiliary was removed following General Procedure N and the desired product formed employing General Procedure O with 4-chloroaniline.
Purified using silica gel chromatography (0% to 100% ethyl acetate in hexanes) to afford Example 19. 1H NMR of czs-isomer (400 MHz; CDC13): δ 9.14 (s, 1H), 8.70 (d, J= 4.6 Hz, 1H), 8.06 (dd, J= 9.2 Hz, J= 5.6 Hz, 1H), 7.58-7.64 (m, 3H), 7.45 (ddd, J= 9.3 Hz, J= 7.8 Hz, J= 2.7 Hz, 1H), 7.19-7.24 (m, 2H), 7.15 (d, J= 4.6Hz, 1H), 3.16-3.26 (m, 1H), 2.59-2.69 (m, 1H), 2.08-2.16 (m, 1H), 1.66-1.86 (m, 7H), 1.31-1.42 (m, 1H), 1.21 (d, J= 6.8Hz, 3H) ppm. m/z 411.2 (M+H)+.
REFERENCES
23-Feb-2015
Bristol-Myers Squibb To Expand Its Immuno-Oncology Pipeline with Agreement to Acquire Flexus Biosciences, Inc
Bristol-Myers Squibb Co; Flexus Biosciences Inc
17-Dec-2014
Flexus Biosciences, a Cancer Immunotherapy Company Focused on Agents for the Reversal of Tumor Immunosuppression (ARTIS), Announces $38M Financing
Flexus Biosciences Inc
2015106thApril 21Abs 4290
Potent and selective next generation inhibitors of indoleamine-2,3-dioxygenase (IDO1) for the treatment of cancer
American Association for Cancer Research Annual Meeting
Jay P. Powers, Matthew J. Walters, Rajkumar Noubade, Stephen W. Young, Lisa Marshall, Jan Melom, Adam Park, Nick Shah, Pia Bjork, Jordan S. Fridman, Hilary P. Beck, David Chian, Jenny V. McKinnell, Maksim Osipov, Maureen K. Reilly, Hunter P. Shunatona, James R. Walker, Mikhail Zibinsky, Juan C. Jaen
2017108thApril 04Abs 4964
Structure, in vitro biology and in vivo pharmacodynamic characterization of a novel clinical IDO1 inhibitor
American Association for Cancer Research Annual Meeting
John T Hunt, Aaron Balog, Christine Huang, Tai-An Lin, Tai-An Lin, Derrick Maley, Johnni Gullo-Brown, Jesse Swanson, Jennifer Brown
2017253rdApril 05Abs MEDI 368
Discovery of a selective inhibitor of indoleamine-2,3-dioxygenase for use in the therapy of cancer
American Chemical Society National Meeting and Exposition
Aaron Balog
April 2-62017
American Chemical Society – 253rd National Meeting and Exhibition (Part IV) – OVERNIGHT REPORT, San Francisco, CA, USA
Casellas J, Carceller V

Juan Jaen

Jordan Fridman
Chief Scientific Officer at FLX Bio, Inc.

Rekha Hemrajani
Chief Operating Officer at FLX Bio, Inc

Max Osipov
////////////////PHASE 1, BMS 986205, 1923833-60-6, BMS-986205, ONO-7701,Bristol-Myers Squibb, Antineoplastics, F- 001287
C[C@H]([C@H]1CC[C@@H](C2=CC=NC3=CC=C(F)C=C23)CC1)C(NC4=CC=C(Cl)C=C4)=O
Wrapping up #MEDI‘s 1st time disclosures is Aaron Balog of @bmsnews talking about an IOD-1 inhibitor to treat cancer #ACSSanFran
USA Viewership touched 3 lakhs on New Drug Approvals
USA Viewership touched 3 lakhs on New Drug Approvals
Total 16.9 lakhs in 213 countries
Metopimazine

Metopimazine
RP-9965, EXP-999, NG-101
l-(3-[2-(methylsulfonyl)-10H-phenothiazin-10-yl]propyl)-4-piperidinecarboxamide
- Isonipecotamide, 1-[3-[2-(methylsulfonyl)phenothiazin-10-yl]propyl]- (7CI,8CI)
- 1-[3-[2-(Methylsulfonyl)-10H-phenothiazin-10-yl]propyl]-4-piperidinecarboxamide
- 1-[3-[2-(Methylsulfonyl)phenothiazin-10-yl]propyl]-4-piperidinecarboxamide
- 1-[3-[2-(Methylsulfonyl)phenothiazin-10-yl]propyl]isonipecotamide
- 2-Methylsulfonyl-10-[3-(4-carbamoylpiperidino)propyl]phenothiazine
- EXP 999
- Metopimazine
- RP 9965
- Vogalene
- metopimazine (gastroparesis), Neurogastrx
Sanofi (Originator)
Teva
Treatment of Nausea and Vomiting, APPROVED
Dopamine D3 receptor antagonist; Dopamine D2 receptor antagonist
Gastroprokinetic
Metopimazine (INN) is a phenothiazine antiemetic.
Metopimazine is an established antiemetic that has been approved and marketed for many years in Europe for the treatment of acute conditions. The compound does not cross the blood-brain-barrier, and is therefore free from central side effects, and is not associated with cardiovascular side effects
In May 2016, preclinical data were presented at the 2016 DDW in San Diego, CA. In rats, po NG-101 and domperidone did not penetrate the brain at therapeutically relevant concentrations, unlike metoclopramide. In dogs, the amplitude and frequency of antral contractions were increased by NG-101, whereas in rats, po metopimazine resulted in an increase in gastric emptying of solid foods. The blood-brain barrier was not readily crossed and there was no interaction with 5-HT3 or 5-HT4 receptors by NG-101 unlike metoclopramide and domperidone, respectively
Neurogastrx is investigating repurposed metopimazine (NG-101), a selective and peripherally restricted dopamine D2/D3 receptor antagonist, for the potential oral treatment of gastroparesis. By July 2014, preclinical studies were underway . SE BELOW REF
WO-2014105655: Methods for treating GI tract disorders
In May 2016, preclinical data were presented [SEE BELOW].
2016 May 24Abs 1079
NG101: A Potent and Selective Dopamine D2 Receptor Antagonist as a Potential Alternative to Metoclopramide and Domperidone for the Treatment of Gastroparesis
Digestive Disease Week
Cyril De Colle, Marieke van der Hart, Jiande Chen, Arash Rassoulpour, Pankaj J Pasricha
In July 2014, preclinical data were published. Metopimazine at 1mg/kg increased gastric motility in hound dogs. In studies in rodents, metopimazine at 3 and 10 mg/kg increased gastric emptying by 18 and 40%, respectively, compared with vehicle control
There is an increasing demand for antiemetic agents because of the most troublesome adverse effects of chemotherapy-induced nausea and emesis during cancer treatment.
However, the objective of complete prevention of emesis in all patients remains elusive. Therefore, there is a great demand for both development of (i) new antiemetic agents and (ii) new manufacturing processes for existing antiemetic agents. Metopimazine is an existing dopamine D2-receptor antagonist with potent antiemetic properties. It is chemically known as l-(3-[2-(methylsulfonyl)-10H-phenothiazin-10-yl]propyl)-4-piperidinecarboxamide , which belongs to nitrogen- and sulfur-containing tricyclic compounds (phenothiazine class of drugs) with interesting biological and pharmacological activities.
Recently, it has been found that Metopimazine plays a key role as an alternative to Ondansetron in the prevention of delayed chemotherapy-induced nausea and vomiting (CINV) in patients receiving moderate to high emetogenic noncisplatin-based chemotherapy.It has been used in France for many years for the prevention and treatment of nausea and vomiting under the brand name of Vogalene
In 1959, the first synthesis and manufacture process of Metopimazine was reported by Jacob et al.The synthesis starts from the protection of 2-(Methylsulfanyl)-10H-phenothiazine..Jacob, R. M.; Robert, J. G. German Patent No. DE1092476, 1959.
Later, in 1990, Sindelar et al. reported a modified process , which starts from synthesis of 4-(2-fluorophenylthio)-3-nitrophenylmethylsulfone..Sindelar, K.; Holubek, J.; Koruna, I.; Hrubantova, M.; Protiva, M. Collect. Czech. Chem. Commun. 1990, 55, 1586– 1601, DOI: 10.1135/cccc19901586
In 2010, Satyanarayana Reddy et al. reported a modified synthetic route which starts from either N-protection using acetyl chloride or N-alkylation using dihalopropane of 2-(methylsulfanyl)-10H-phenothiazine ..Satyanarayana Reddy, M.; Eswaraiah, S.; Satyanarayana, K. Indian Patent No. 360/CHE/2010 A, Aug 19, 2011.
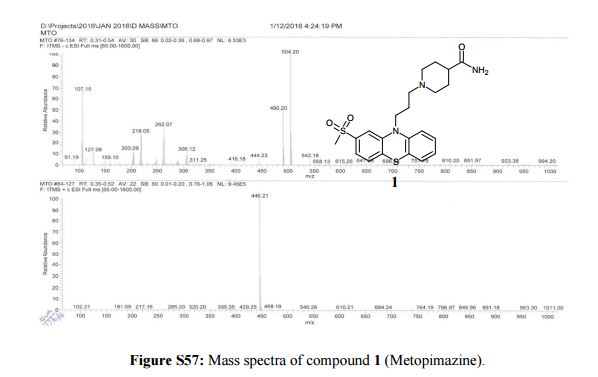
Synthesis of 1-(3-[2-(methylsulfonyl)-10H-phenothiazin-10-yl]propyl)piperidine-4- carboxamide (1)-Metopimazine: Pale yellow color solid, yield. 65% (82 g), DSC 189 °C.
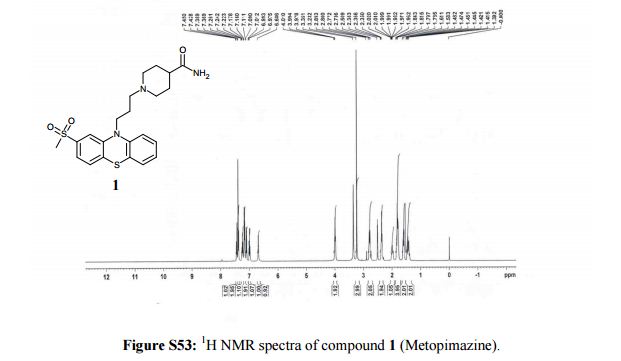
1H NMR (400 MHz, DMSO-d6, δ/ppm): 7.44 (d, 1H, arom H, J = 8.8 Hz), 7.37 (d, 2H, arom H, J = 8.0 Hz), 7.24 (t, 1H, arom H, J = 7.6 Hz), 7.16 (m, 2H, -NH2), 7.1 (d, 1H, arom H, J = 8.4 Hz), 6.99 (t, 1H, arom H, J = 7.6 Hz), 6.68 (s, 1H, arom H), 3.99 (t, 2H, -NCH2, J = 6.4 Hz), 3.23 (s, 3H, -S-CH3), 2.8-2.73 (m, 2H, -CH2-), 2.36 (t, 2H, -CH2-, J = 6.8 Hz), 2.02-1.96 (m, 1H, -CH-), 1.84-1.78 (m, 4H, 2-CH2-), 1.61-1.58 (m, 2H, -CH2-), 1.48-1.44 (m, 2H, – CH2-).
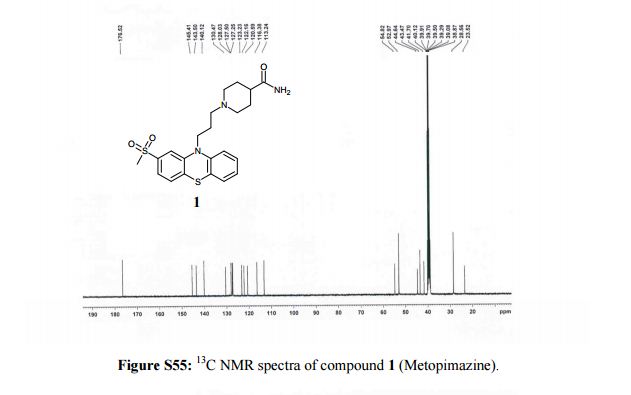
13C NMR (100 MHz, DMSO-d6, δ/ppm): 176.52, 145.41, 143.5, 140.12, 130.47, 128.03, 127.50, 127.25, 123.23, 122.16, 120.59, 116.38, 113.24, 54.82, 52.97, 44.64, 43.47, 41.7, 28.59, 23.52.
MS m/z (ESI): 446.21 (M+H)+.
SYNTHESIS


PATENT
IN 201641043070
IN 2013CH05689
IN 2013CH00361
IN 2010CH00360
DE 1092476/US 3130194
PAPER
A Simple and Commercially Viable Process for Improved Yields of Metopimazine, a Dopamine D2-Receptor Antagonist

http://pubs.acs.org/doi/abs/10.1021/acs.oprd.7b00052

An efficient, practical, and commercially viable manufacturing process was developed with ≥99.7% purity and 31% overall yield (including four chemical reactions and one recrystallization) for an active pharmaceutical ingredient, called Metopimazine (1), an antiemetic drug used to prevent emesis during chemotherapy. The development of two in situ, one-pot methods in the present synthetic route helped to improve the overall yield of 1 (31%) compared with earlier reports (<15%). For the first time, characterization data of API (1), intermediates, and also possible impurities are presented. The key process issues and challenges were addressed effectively and achieved successfully.
Synthesis of 1-(3-[2-(Methylsulfonyl)-10H-phenothiazin-10-yl]propyl)-4-piperidinecarboxamide (1), Metopimazine



- Vogalene
- metopimazina (Italian, Portuguese)
- metopimazin (Danish, Swedish)
- metopimazine (Dutch)
- metopimatsiini (Finnish)
Regulatory List Number
- EC No.: 237-818-4
- EINECS No.: 237-818-4
-
Harmonized Tariff Code
293430
- Moda, Tiago L.; Bioorganic & Medicinal Chemistry 2007, 15(24), PG7738-7745
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
- Julou, Louis; Comptes Rendus des Seances de l’Academie des Sciences, Serie D: Sciences Naturelles 1968, 266(25), PG 2365-8
- Bounoure, Frederic; Journal of Inclusion Phenomena and Macrocyclic Chemistry 2007, 57(1-4), PG191-195
- Sindelar, Karel; Collection of Czechoslovak Chemical Communications 1990, 55(6), PG1586-601
- “PhysProp” data were obtained from Syracuse Research Corporation of Syracuse, New York (US)
- Habibi-Yangjeh, Aziz; Bulletin of the Korean Chemical Society 2008, 29(4), PG833-841
- Metwally, Fadia H.; Bulletin of the Faculty of Pharmacy (Cairo University) 2006, 44(3), PG1-15
- WO-2014105655: Methods for treating GI tract disorders
- 2016 May 24Abs 1079
NG101: A Potent and Selective Dopamine D2 Receptor Antagonist as a Potential Alternative to Metoclopramide and Domperidone for the Treatment of Gastroparesis
Digestive Disease Week
Cyril De Colle, Marieke van der Hart, Jiande Chen, Arash Rassoulpour, Pankaj J Pasricha
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| ECHA InfoCard | 100.034.367 |
| Chemical and physical data | |
| Formula | C22H27N3O3S2 |
| Molar mass | 445.6 g/mol |
| 3D model (Jmol) | |
//////////////Metopimazine, Dopamine D2-Receptor Antagonist, 14008-44-7, sanofi, teva, RP-9965, Nausea and Vomiting, EXP-999, NG-101, metopimazine, gastroparesis, Neurogastrx
NC(=O)C1CCN(CC1)CCCN2c4ccccc4Sc3ccc(cc23)S(C)(=O)=O
FDA approves drug to treat ALS, Radicava (Edaravone) , эдаравон, إيدارافون , 依达拉奉 ,ラジカット,

May 5, 2017
Release
The U.S. Food and Drug Administration today approved Radicava (edaravone) to treat patients with amyotrophic lateral sclerosis (ALS), commonly referred to as Lou Gehrig’s disease.
“After learning about the use of edaravone to treat ALS in Japan, we rapidly engaged with the drug developer about filing a marketing application in the United States,” said Eric Bastings, M.D., deputy director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This is the first new treatment approved by the FDA for ALS in many years, and we are pleased that people with ALS will now have an additional option.”
ALS is a rare disease that attacks and kills the nerve cells that control voluntary muscles. Voluntary muscles produce movements such as chewing, walking, breathing and talking. The nerves lose the ability to activate specific muscles, which causes the muscles to become weak and leads to paralysis. ALS is progressive, meaning it gets worse over time. The Centers for Disease Control and Prevention estimates that approximately 12,000-15,000 Americans have ALS. Most people with ALS die from respiratory failure, usually within three to five years from when the symptoms first appear.
Radicava is an intravenous infusion given by a health care professional. It is administered with an initial treatment cycle of daily dosing for 14 days, followed by a 14-day drug-free period. Subsequent treatment cycles consist of dosing on 10 of 14 days, followed by 14 days drug-free.
The efficacy of edaravone for the treatment of ALS was demonstrated in a six-month clinical trial conducted in Japan. In the trial, 137 participants were randomized to receive edaravone or placebo. At Week 24, individuals receiving edaravone declined less on a clinical assessment of daily functioning compared to those receiving a placebo.
The most common adverse reactions reported by clinical trial participants receiving edaravone were bruising (contusion) and gait disturbance.
Radicava is also associated with serious risks that require immediate medical care, such as hives, swelling, or shortness of breath, and allergic reactions to sodium bisulfite, an ingredient in the drug. Sodium bisulfite may cause anaphylactic symptoms that can be life-threatening in people with sulfite sensitivity.
The FDA granted this drug orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted approval of Radicava to Mitsubishi Tanabe Pharma America, Inc,
Edaravone (brand name ラジカット, Radicut) is a nootropic and neuroprotective agent used for the purpose of aiding neurological recovery following acute brain ischemia and subsequent cerebral infarction.[1] It acts as a potent antioxidant and strongly scavenges free radicals, protecting against oxidative stress and neuronal apoptosis.[2][3][4] It has been marketed solely in Japan by Mitsubishi Pharma since 2001.[1] It is also marketed in India by Edinburgh Pharmaceuticals by the brand name Arone.
On June 26, 2015, Mitsubishi Tanabe Pharma Corporation announced it has received approval to market Radicut for treatment of ALS in Japan. The phase III clinical trial began in 2011 in Japan. The company was awarded Orphan Drug Designation for Radicut by the FDA and EU in 2015. Radicut is an intravenous drug and administrated 14 days followed by 14 days drug holiday.
The biotech company Treeway is developing an oral formulation of edaravone (TW001) and is currently in clinical development. Treeway was awarded orphan drug designation for edaravone by the EMA in November 2014 and FDA in January 2015.
Edaravone has been shown to attenuate methamphetamine– and 6-OHDA-induced dopaminergic neurotoxicity in the striatum and substantia nigra, and does not affect methamphetamine-induced dopamine release or hyperthermia.[5][6] It has also been demonstrated to protect against MPTP-mediated dopaminergic neurotoxicity to the substantia nigra, though notably not to the striatum.[7][8][9]

Edaravone (CAS NO.: 89-25-8), with other name of 3-Methyl-1-phenyl-2-pyrazolin-5-one, could be produced through many synthetic methods.
Following is one of the synthesis routes: By direct cyclization of phenylhydrazine (I) with ethyl acetoacetate (II) in refluxing ethanol.

SYNTHESIS
Edaravone, chemical name: 3-methyl-1-phenyl-2-pyrazoline-5-one, of the formula: Formula: CiciHltlN2O, molecular weight: 174.20, the formula:

[0004] Edaravone is a brain-protecting agent (free radical scavenger). Clinical studies suggest that N- acetyl aspartate (NAA) is a specific sign of the survival of nerve cells, dramatically reducing the initial content of cerebral infarction. In patients with acute cerebral infarction Edaravone suppressed reduce peri-infarct regional cerebral blood flow, so that the first concept of days after the onset of brain NAA glycerol content than the control group significantly increased. Preclinical studies suggest that rats after ischemia / reperfusion of ischemic intravenous edaravone, can prevent the progress of cerebral edema and cerebral infarction, and relieve the accompanying neurological symptoms, suppress delayed neuronal death. Mechanism studies suggest that edaravone can scavenge free radicals, inhibiting lipid peroxidation, thereby inhibiting brain cells, endothelial cells, oxidative damage nerve cells.
For the synthesis of edaravone reported some use of benzene and methyl ethyl ketone amide corpus obtained, but methyl ethyl ketone amide difficult to obtain and slow reaction, which now has basically been abandoned; some use benzene corpus and ethyl acetoacetate in ethanol (see US4857542A, Synthesis Example 1) or water (Dykhanov NN Ethyl and butyl acetoacetates, Med Prom SSSR, 1961,15 (1):. 42-45) refluxing the reaction of the reaction The resulting purity edaravone poor, and the yield is not high, only about 70%.
Edaravone, chemical name: 2,4_-dihydro-5-methyl-2-phenyl pyrazole -3H- – one, of the formula: CiciHltlN2O, molecular weight: 174.20, the formula:

edaravone is a clear cerebral infarction harmful factors (free radicals), protection of new therapeutic agents for cerebral infarction nerve cells. Clinical studies have shown that N- acetyl aspartate (NAA) is a specific sign of the survival of nerve cells, dramatically reducing the initial content of cerebral infarction. When patients with acute cerebral infarction Edaravone, peri-infarct rCBF decrease has improved, and the first 28 days after the onset of brain NAA content was significantly higher than that in the control group glycerol. Mechanism studies suggest that edaravone can clear the brain is highly cytotoxic hydroxyl radicals, inhibiting the synthesis of lipids free radicals, which can suppress brain infarction after reperfusion edema, protecting brain from damage and improve nerve impairment symptoms, and the delayed neuronal death inhibition, to protect the brain.
The first is by phenylhydrazine and methyl ethyl ketone amide (National API process compilation, 1980.737-739) condensation reaction in water at 50 ° C, a yield of up to 97%, but the raw material ketone amide ( CH3C0CH2C0NH2) are not readily available. Formula I
Edaravone synthetic route for the reaction:

[0008] The second is to phenylhydrazine and ethyl acetoacetate in ethanol or water at reflux the reaction, sodium bisulfite as the preparation of the catalyst. From the perspective of the chemical reaction, acetyl ethyl ketone amide more than hydrazine reacted with benzene and ethyl acetoacetate more readily available, the price is cheaper, but lower reaction yield of about 70%. Formula 2 for the synthesis route Edaravone reaction formula:

PATENT
https://www.google.com/patents/CN101830852B?cl=en

1 Edaravone Synthesis Example [0023] Example
[0024] (1) Weigh benzene hydrochloride corpus 13. 5g (94mmol), was added to IOOml water, stirred for 0.5 hours, sodium hydroxide was added an equimolar 3. 76g, stirred for 0.5 hours; [0025] ( 2) To the reaction solution was added dropwise ethyl acetoacetate 11. 7g (90mmol), the reaction exotherm, the reaction was heated to reflux for 2.5 hours, heating was stopped, cooled to room temperature with stirring, filtered and dried to give a pale yellow granular crude 15. 5g;
[0026] (3) The crude product was added 30ml volume ratio of 2: 1 isopropanol – water, 2g of activated carbon was added and refluxed for 1 hour, filtered hot, cooled to room temperature a white solid was precipitated to give 14 a white crystalline powder. 8g, yield 90%, mpU9 ° C, with a purity of 99.9% 0
2 Edaravone Synthesis Example [0027] Example
[0028] (1) Weigh 15g of benzene hydrochloride corpus (I (Mmmol), was added to 120ml of water and stirred for 0.5 hours, sodium hydroxide was added an equimolar 4. 16g, stirred for 0.5 hours;
[0029] (2) To the reaction solution was added dropwise 13g of ethyl acetoacetate (lOOmmol), the reaction exotherm, the reaction was heated to reflux for 2.5 hours, heating was stopped, cooled to room temperature with stirring, filtered and dried to give a pale yellow granular crude 16. 7g;
(3) The crude product was added 40ml volume ratio of 2: 1 isopropanol – water, 2. 5g of activated carbon was added and refluxed for 1 hour, filtered hot, cooled to room temperature to precipitate a white solid, as a white crystalline powder 16. lg, a yield of 88.9%, mpU8 ° C, with a purity of 99.9% 0
3 Edaravone Synthesis Example [0031] Example
[0032] (1) Weigh 22g of benzene hydrochloride corpus (152mm0l), was added to 200ml of water and stirred for 0.5 hours, sodium hydroxide was added an equimolar 6. 08g, stirred for 0.5 hours;
[0033] (2) To the reaction solution was added dropwise 19g of ethyl acetoacetate (146mm0l), the reaction exotherm, the reaction was heated to reflux for 3 hours, heating was stopped, cooled to room temperature with stirring, filtered and dried to give a pale yellow granular crude 24. Sg;
[0034] (3) The crude product was added 50ml volume ratio of 2: 1 isopropanol – water, 3g of activated carbon was added and refluxed for 1 hour, filtered hot, cooled to room temperature a white solid was precipitated to give 23 a white crystalline powder. 2g, a yield of 87. 8%, mpU8 ° C, with a purity of 99.9% 0
[0035] Comparative Example
[0036] The ethyl acetoacetate 65g (0. 5mol) and 180ml of anhydrous ethanol mixed, with stirring at 50 ° C was added dropwise benzyl corpus 54g (0. 5mol) and a solution consisting of 30ml absolute ethanol, dropwise at reflux for 2 Bi hours, ethanol was distilled off 60ml, cooled, suction filtered, washed crystals with cold absolute ethanol twice, and dried in vacuo to give pale yellow crystals 70g. Recrystallized twice from absolute ethanol to give pale yellowish white crystals 56g (yield 65%).
PATENT
https://www.google.com/patents/CN102285920B?cl=en
Example 1: Preparation of phenylhydrazine edaravone.
[0024] a. Weigh 5.1g phenylhydrazine (47mmol), was added under stirring to water containing 45mL round-bottom flask, take appropriate concentrated hydrochloric acid solution was adjusted to pH 6.0 with PH meter.
[0025] b. To the above solution was slowly added dropwise ethyl acetoacetate 5.85g (45mmol), the reaction exotherm, was added 1.5g sodium dithionite (Na2S2O6), heated to 105 ° C to room temperature until reflux After 3h, heating was stopped, and then stirred, cooling, filtration, and dried to give a pale yellow granular edaravone crude.
[0026] c. With anhydrous ethanol recrystallization, filtration, and dried to obtain a white crystalline powder that is refined edaravone, 85% yield, 99.2% purity 0
[0027] Example 2: Preparation of phenylhydrazine hydrochloride edaravone.
[0028] a. Weigh 6.8g phenylhydrazine hydrochloride (47mmol), was added under stirring to water containing 45mL round-bottomed flask, the pH of the solution adjusted to 6.0 with aqueous ammonia.
[0029] b. To the above solution was slowly added dropwise ethyl acetoacetate 5.85g (45mmol), the reaction exotherm, 1.25g was added sodium dithionite (Na2S2O6), heated to 105 ° C to room temperature until reflux After 3h, heating was stopped, and then stirred, cooling, filtration, and dried to give a pale yellow granular edaravone crude.
[0030] c. With anhydrous ethanol recrystallization, filtration, and dried to obtain a white crystalline powder that is refined edaravone, 84% yield, with a purity of 99.2%. [0031] Comparative Example:
Under the [0032] state of agitation will phenylhydrazine 10.2g (94mmol) added to a round bottom flask equipped with IOOmL water in an appropriate amount of concentrated hydrochloric acid was dubbed the volume ratio of 1: 1 aqueous hydrochloric acid, with a PH adjusting pH of the solution was measured 6.0. After weighing Ethylacetoacetate 11.7g (90mmol) added to the reaction mixture, the reaction was exothermic and cooling to room temperature, sodium bisulfite (NaHSO3), heated to 105 ° C under reflux for 3h, the hot solution Water was added into the beaker and mechanical stirring, cooling, filtration, and dried to give the yellow edaravone crude, 73% yield, with a purity of 99.1%.

CLIP
http://www.rsc.org/suppdata/books/184973/9781849739634/bk9781849739634-chapter%204.2.3.pdf



Edaravone:
IR (KBr) max/cm-1 : 3431, 3129, 1602, 1599, 1580;
1 H NMR (300 MHz, CDCl3): δ 7.86 (d, J = 7.5 Hz, 2H, ArH), 7.40 (m, 2H, ArH), 7.18 (m, 1H, ArH), 3.41 (d, J =0.6 Hz, 2H, CH2), 2.19 (s, 3H, CH3);
13C NMR (75 MHz, CDCl3): 170.6, 156.4, 130.1, 128.8, 125.0, 118.9, 43.1, 17.0;
1 H NMR (300 MHz, DMSO-d6): δ 11.5 (bs, 1H, NH), 7.71 (m, 2H, ArH), 7.40 (m, 2H, ArH), 7.22 (m, 1H, ArH), 5.36 (s, 1H, CH), 2.12 (s, 3H, CH3);
13C NMR (75 MHz, DMSO-d6):171.7, 158.9, 148.7, 139.2, 138.6, 129.3,125.4, 124.8, 118.4, 43.5, 17.1, 14.2.
These values are in accordance with the previous published in literature1 .
In the carbon spectrum in DMSO presented in Figure SM 4.2.3.1.8 is evident the presence of the two major tautomeric structures of edaravone, signals are identified by different colours in both structures in the figure. Also in the IR analysis of the solid material (Figure SM 4.2.3.1.9) is possible to see either the NH form (max/cm-1, 3129), the OH form (max/cm- 1 , 3431) and the C=O (max/cm-1, 1599) of the enol and keto tautomeric forms of edaravone.





1. S. Pal, J. Mareddy and N. S. Devi, J. Braz. Chem. Soc., 2008, 19, 1207.
CLIP
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-50532008000600023
We have shown that the short reaction time, in combination with good yields can make microwave assisted reaction of hydrazines with β-ketoesters ideal for a rapid entry to pyrazolones. All the compounds synthesized are characterized by spectroscopic (1H NMR, IR and MS) data. While determination of tautomeric composition of compound 3 is quite challenging as eight possible tautomeric forms need to be considered, interestingly, two major tautomeric forms of compound 3a was observed in two different solvents. For example, it exists as 1,2-dihydro pyrazolone (T-1, Figure 2) in DMSO and 2,4-dihydro form (T-2, Figure 2) in chloroform as indicated by 1H NMR spectra (Figure 3). The olefinic proton of T-1 appeared at 5.36 δ whereas the methylene hydrogens appeared at 3.43 δ in case of T-2. Additionally, the NH proton of T-1 at 11.40 δ was not observed incase of T-2 confirmed the absence of NH in the 2,4-dihydro form. Existence of two major tautomeric forms was also observed in case compound 3b (see 1H NMR data in the experimental section). However, X-ray study on single crystal of 2-(4-chlorophenyl)-5-methyl-1,2-dihydro pyrazol-3-one (3i) indicates that 2-aryl pyrazol-3-ones e.g. 3a-b, 3e-f and 3i exist as 1,2-dihydro form in crystal state. 27 It is mention worthy that the aryl ring of all these 2-aryl pyrazol-3-ones remain twisted with respect to the pyrazole plane as indicated by the crystallographic data of 3i [the dihedral angle between the pyrazole and benzene ring planes was found to be 15.81 (11)º].27


5-Methyl-2-phenyl-1,2-dihydro pyrazol-3-one (3a)
mp 125-127 ºC (lit21 126-130 ºC);
IR (KBr) νmax/cm-1: 3127, 1597, 1525, 1498, 1454;
1H NMR (400 MHz, DMSO-d6) δ 11.40 (bs, 1H), 7.71-7.69 (m, 2H), 7.42-7.38 (m, 2H), 7.21-7.18 (m, 1H), 5.36 (s, 1H), 2.10 (s, 3H);
13C NMR (50 MHz, DMSO-d6) δ 170.6, 156.2, 138.1, 128.8 (2C), 124.9, 118.9 (2C), 43.1, 16.9;
Mass (CI, m/z) 175 (M+1, 100).
1H NMR (400 MHz, CDCl3)δ 7.85 (d, J 8.3 Hz, 2H), 7.40-7.37 (m, 2H), 7.24-7.18 (m, 1H), 3.43 (s, 2H), 2.20 (s, 3H).
21. Makhija, M. T.; Kasliwal, R. T.; Kulkarni, V. M.; Neamati, N.; Bioorg. Med. Chem. 2004, 12, 2317. [ Links ]
| CN101830852A | Mar 22, 2010 | Sep 15, 2010 | 海南美兰史克制药有限公司 | Edaravone compound synthesized by new method |
| CN102060771A | Nov 18, 2009 | May 18, 2011 | 南京长澳制药有限公司 | Edaravone crystal form and preparation method thereof |
| CN102180834A | Mar 24, 2011 | Sep 14, 2011 | 江苏正大丰海制药有限公司 | Preparation method for edaravone |
References
- ^ Jump up to:a b Doherty, Annette M. (2002). Annual Reports in Medicinal Chemistry, Volume 37 (Annual Reports in Medicinal Chemistry). Boston: Academic Press. ISBN 0-12-040537-7.
- Jump up^ Watanabe T, Tanaka M, Watanabe K, Takamatsu Y, Tobe A (March 2004). “[Research and development of the free radical scavenger edaravone as a neuroprotectant]”. Yakugaku Zasshi (in Japanese). 124 (3): 99–111. doi:10.1248/yakushi.124.99. PMID 15049127.
- Jump up^ Higashi Y, Jitsuiki D, Chayama K, Yoshizumi M (January 2006). “Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a novel free radical scavenger, for treatment of cardiovascular diseases”. Recent Patents on Cardiovascular Drug Discovery. 1 (1): 85–93. doi:10.2174/157489006775244191. PMID 18221078.
- Jump up^ Yoshida H, Yanai H, Namiki Y, Fukatsu-Sasaki K, Furutani N, Tada N (2006). “Neuroprotective effects of edaravone: a novel free radical scavenger in cerebrovascular injury”. CNS Drug Reviews. 12 (1): 9–20. doi:10.1111/j.1527-3458.2006.00009.x. PMID 16834755.
- Jump up^ Yuan WJ, Yasuhara T, Shingo T, et al. (2008). “Neuroprotective effects of edaravone-administration on 6-OHDA-treated dopaminergic neurons”. BMC Neuroscience. 9: 75. doi:10.1186/1471-2202-9-75. PMC 2533664 . PMID 18671880.
- Jump up^ Kawasaki T, Ishihara K, Ago Y, et al. (August 2006). “Protective effect of the radical scavenger edaravone against methamphetamine-induced dopaminergic neurotoxicity in mouse striatum”. European Journal of Pharmacology. 542 (1-3): 92–9. doi:10.1016/j.ejphar.2006.05.012. PMID 16784740.
- Jump up^ Kawasaki T, Ishihara K, Ago Y, Baba A, Matsuda T (July 2007). “Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a radical scavenger, prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in the substantia nigra but not the striatum”. The Journal of Pharmacology and Experimental Therapeutics. 322 (1): 274–81. doi:10.1124/jpet.106.119206. PMID 17429058.
- Jump up^ Yokoyama H, Takagi S, Watanabe Y, Kato H, Araki T (June 2008). “Role of reactive nitrogen and reactive oxygen species against MPTP neurotoxicity in mice”. Journal of Neural Transmission (Vienna, Austria : 1996). 115 (6): 831–42. doi:10.1007/s00702-008-0019-6. PMID 18235988.
- Jump up^ Yokoyama H, Yano R, Aoki E, Kato H, Araki T (September 2008). “Comparative pharmacological study of free radical scavenger, nitric oxide synthase inhibitor, nitric oxide synthase activator and cyclooxygenase inhibitor against MPTP neurotoxicity in mice”. Metabolic Brain Disease. 23 (3): 335–49. doi:10.1007/s11011-008-9096-3. PMID 18648914.
External links
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Radicut |
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| Synonyms | MCI-186 |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.001.719 |
| Chemical and physical data | |
| Formula | C10H10N2O |
| Molar mass | 174.20 g/mol |
| 3D model (Jmol) | |
