
Home » 2015 (Page 8)
Yearly Archives: 2015
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
HCV NS5A Inhibitor from Theravance, Inc. to treat hepatitis C virus infection

((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
N-[(1S)-1-[[(2S)-2-[5-[4′-[[[6-[(2R,5S)-2,5-dimethyl-4-[(methylamino)carbonyl]-1-piperazinyl]-3-pyridinyl]carbonyl]amino]-2′-(trifluoromethoxy)[1,1′-biphenyl]-4-yl]-1H-imidazol-2-yl]-1-pyrrolidinyl]carbonyl]-2-methylpropyl]-, Carbamic acid, methyl ester
Carbamic acid, N-[(1S)-1-[[(2S)-2-[5-[4′-[[[6-[(2R,5S)-2,5-dimethyl-4-[(methylamino)carbonyl]-1-piperazinyl]-3-pyridinyl]carbonyl]amino]-2′-(trifluoromethoxy)[1,1′-biphenyl]-4-yl]-1H-imidazol-2-yl]-1-pyrrolidinyl]carbonyl]-2-methylpropyl]-, methyl ester
CAS 1374883-22-3, 819.87, C41 H48 F3 N9 O6
CAS of DIHCl 1480448-59-6
CAS of DIHCl, H2O 1480448-63-2
Theravance, Inc. INNOVATOR
To treat hepatitis C virus infection

-
((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (compound 1):
-
 Recent estimates place the number of people infected with the hepatitis C virus (HCV) worldwide at more than 170 million, including 3 million people in the United States. The infection rate is thought to be roughly 4 to 5 times that of the human immunodeficiency virus (HIV). While in some individuals, the natural immune response is able to overcome the virus, in the majority of cases, a chronic infection is established, leading to increased risk of developing cirrhosis of the liver and hepatocellular carcinomas. Infection with hepatitis C, therefore, presents a serious public health problem.The virus responsible for HCV infection has been identified as a positive-strand RNA virus belonging to the family Flaviviridae. The HCV genome encodes a polyprotein that during the viral lifecycle is cleaved into ten individual proteins, including both structural and non-structural proteins. The six non-structural proteins, denoted as NS2, NS3, NS4A, NS4B, NS5A, and NS5B have been shown to be required for RNA replication. In particular, the NS5A protein appears to play a significant role in viral replication, as well as in modulation of the physiology of the host cell. Effects of NS5A on interferon signaling, regulation of cell growth and apoptosis have also been identified. (Macdonald et al., Journal of General Virology (2004), 85, 2485-2502.) Compounds which inhibit the function of the NS5A protein are expected to provide a useful approach to HCV therapy.Commonly-assigned U.S. Provisional Application Nos. 61/410,267, filed on Nov. 4, 2010, 61/444,046, filed on Feb. 17, 2011, and 61/492,267, filed on Jun. 1, 2011, and U.S. application Ser. No. 13/288,216, filed on Nov. 3, 2012 disclose pyridyl-piperazinyl compounds
Recent estimates place the number of people infected with the hepatitis C virus (HCV) worldwide at more than 170 million, including 3 million people in the United States. The infection rate is thought to be roughly 4 to 5 times that of the human immunodeficiency virus (HIV). While in some individuals, the natural immune response is able to overcome the virus, in the majority of cases, a chronic infection is established, leading to increased risk of developing cirrhosis of the liver and hepatocellular carcinomas. Infection with hepatitis C, therefore, presents a serious public health problem.The virus responsible for HCV infection has been identified as a positive-strand RNA virus belonging to the family Flaviviridae. The HCV genome encodes a polyprotein that during the viral lifecycle is cleaved into ten individual proteins, including both structural and non-structural proteins. The six non-structural proteins, denoted as NS2, NS3, NS4A, NS4B, NS5A, and NS5B have been shown to be required for RNA replication. In particular, the NS5A protein appears to play a significant role in viral replication, as well as in modulation of the physiology of the host cell. Effects of NS5A on interferon signaling, regulation of cell growth and apoptosis have also been identified. (Macdonald et al., Journal of General Virology (2004), 85, 2485-2502.) Compounds which inhibit the function of the NS5A protein are expected to provide a useful approach to HCV therapy.Commonly-assigned U.S. Provisional Application Nos. 61/410,267, filed on Nov. 4, 2010, 61/444,046, filed on Feb. 17, 2011, and 61/492,267, filed on Jun. 1, 2011, and U.S. application Ser. No. 13/288,216, filed on Nov. 3, 2012 disclose pyridyl-piperazinyl compounds

SYNTHESIS
CLICK ON IMAGES FOR CLEAR VIEW
………………………..
……………………………….
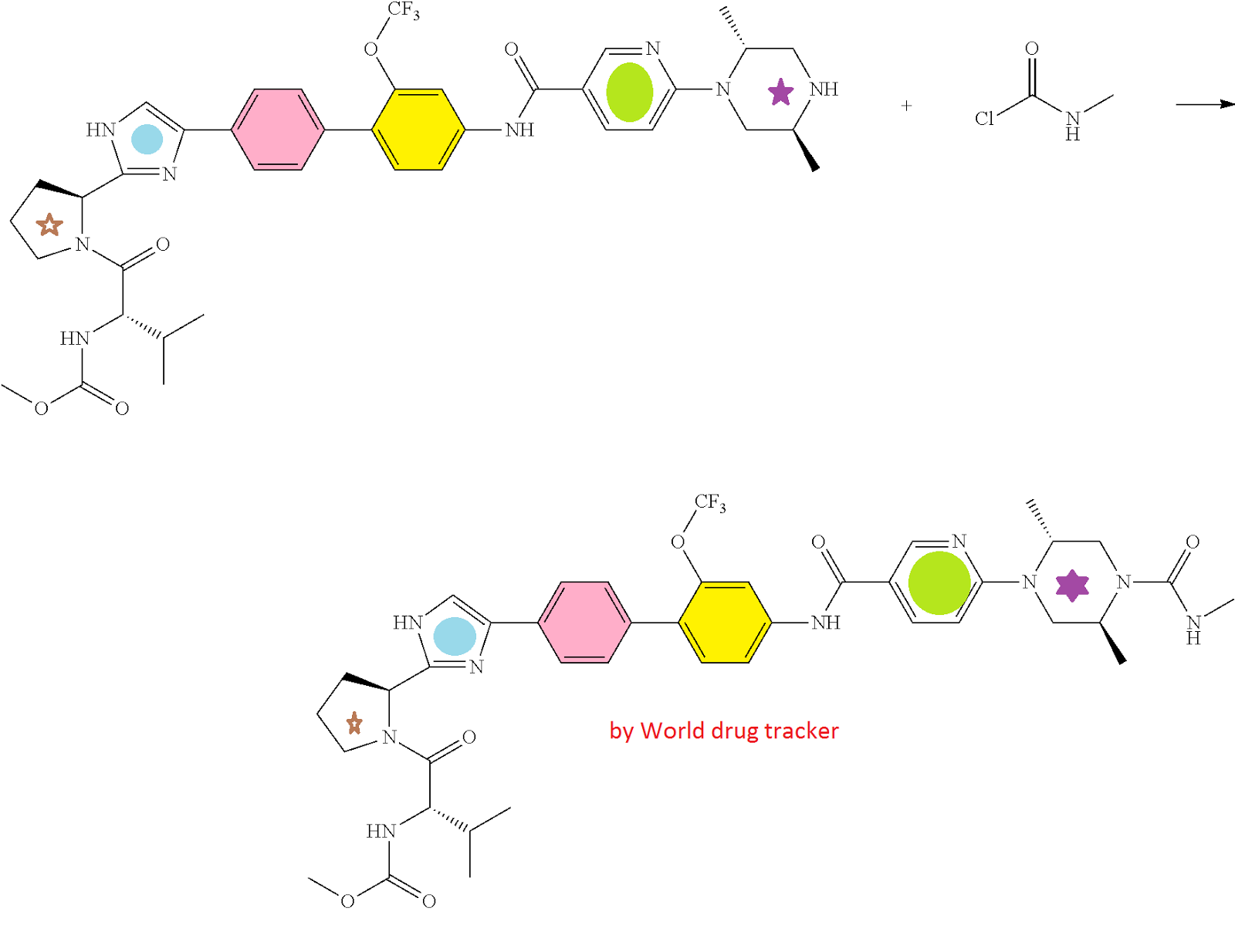
PATENT
WO-2013/165796
https://www.google.co.in/patents/WO2013165796A1?cl=en
PATENT
http://www.google.com/patents/US20130295048
crystalline compound 1 is advantageously prepared directly from the crude product of the final step of the synthesis of compound 1, illustrated in the following scheme, without purification of the amorphous form.

As described in Example 3 below, ((S)-1-{(S)-2-[4-(4′-{[6-(2R,5S)-2,5-dimethyl-piperazin-1-yl)-pyridine-3-carbonyl}-amino]-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (2) is reacted with methylaminoformyl chloride to provide a crude product, which is recovered by conventional extraction and drying. The reaction is typically performed in the presence of an excess of base, in an inert diluent such as dichloromethane. Next, methanol is added to the crude product followed by the slow addition of water in a ratio of methanol:water of about 2.5:1 to about 2.7:1 to form a crystallization mixture. Seeds of crystalline compound 1 are added about halfway through the water addition. The crystallization mixture is stirred for a period of several days to form crystalline compound 1. To increase purity, the product can be recrystallized by a similar process: the crystalline compound is dissolved in methanol, water and seeds are added, such that the ratio of methanol to water in the mixture is about 2.5:1, and the mixture is stirred for a period of at least 12 hours to provide crystalline compound 1, which is recovered conventionally
-

-
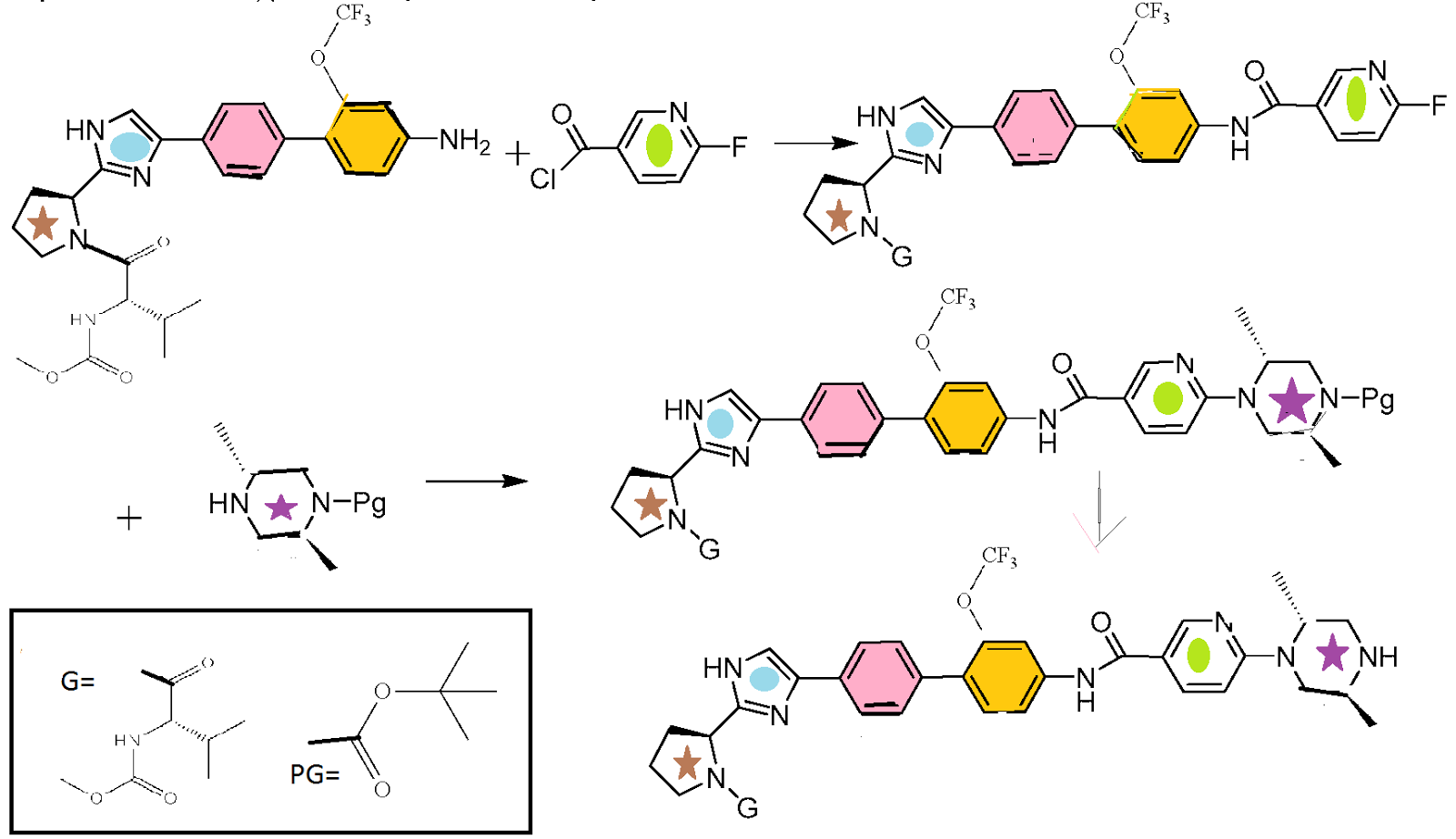
To a solution of 4-bromo-3-trifluoromethoxy-phenylamine (3.15 g, 12.3 mmol) and triethylamine (3.43 mL, 24.6 mmol) in DCM (25 mL) was slowly added a solution of 2-fluoropyridine-5-carbonyl chloride (2.36 g, 14.8 mmol) in DCM (10 mL). After 2 h at RT, MTBE (90 mL) was added and the reaction mixture was washed with water, brine, and saturated sodium carbonate, dried, and evaporated to give a solid (5.4 g). Ethanol (43 mL) was added to the solid and then water (43 mL) was slowly added. The reaction mixture was stirred for 1.5 h, filtered, and washed with 1:4 ethanol:water (2×25 mL) to give the title intermediate as a white solid (3.87 g). Analytical HPLC: Retention time=21.3 min.
- Preparation 1: (2S,5R)-4-[5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester(a) N-(4-Bromo-3-trifluoromethoxy-phenyl)-6-fluoro-nicotinamide

(b) (2S,5R)-4-[5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester
-

-
The product of the previous step (3.86 g, 10.2 mmol) (2S,5R)-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (2.62 g, 12.2 mmol) and N,N-diisopropylethylamine (5.32 mL, 30.5) was dissolved in DMSO (12 mL). The reaction mixture heated at 120° C. for 3 h, diluted with EtOAc (100 mL), washed with water, and saturated NH4Cl, water, and brine. The reaction mixture was evaporated to about 40% volume and 3 M HCl in cyclopentyl methyl ether (4.24 mL, 12.7 mmol) was added slowly. Seeds from a previous run at smaller scale were added and the reaction mixture was stirred for 2 days and filtered to provide the HCl salt of the title intermediate (5.15 g, 83% yield). Analytical HPLC: Retention time=21.1 min.

Preparation 2: (2S,5R)-4-[5-(4′-{2-[(S)-1-((S)-2-Methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester
-

-
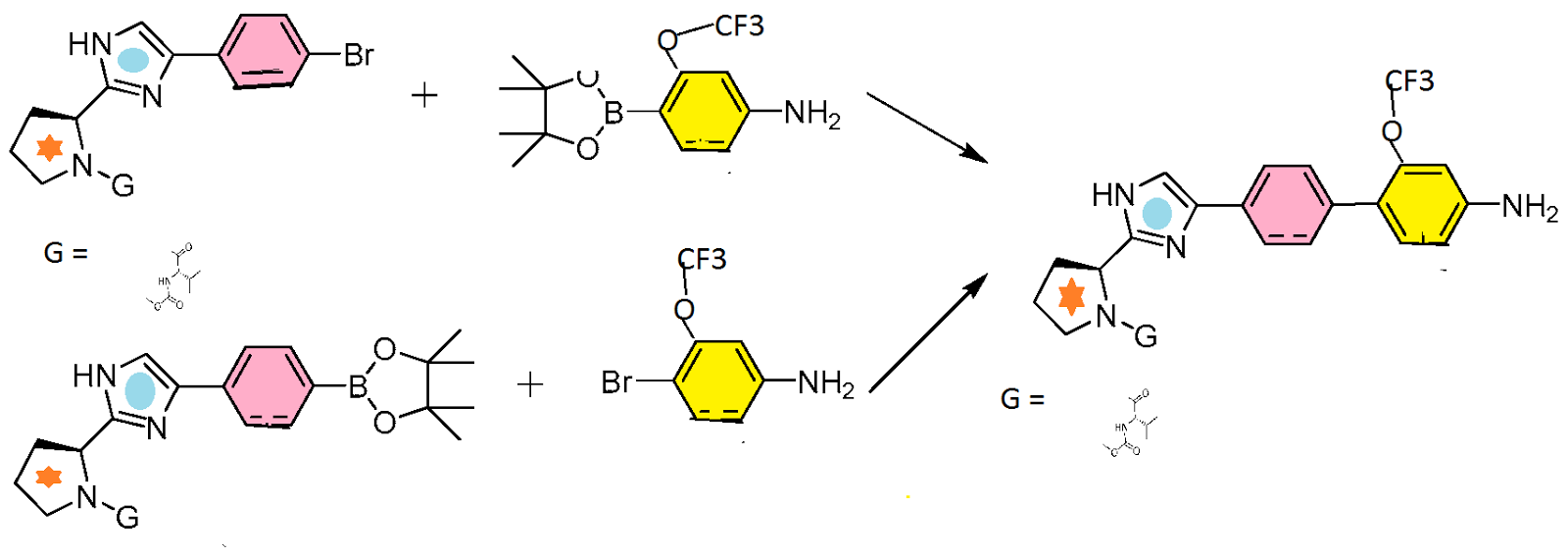
To a solution of ((S)-1-{(S)-2-[4-(4-bromo-phenyl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (3.05 g, 6.8 mmol), bis(pinacolato)diboron (1.81 g, 7.1 mmol) and potassium acetate (1.00 g, 10.2 mmol) was added nitrogen sparged toluene (15 mL). The resulting mixture was sparged with nitrogen and 1,1′-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane (Pd catalyst) (0.17 g, 0.204 mmol) was added. The reaction mixture was stirred at 90° C. overnight.
-
The reaction mixture was cooled to RT and to this mixture was added nitrogen sparged water (7.6 mL), potassium carbonate (5.16 g, 37.3 mmol), and (2S,5R)-4-[5-(4-bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (4.35 g, 7.13 mmol). The reaction mixture was stirred at 95° C. overnight.
-
Another portion of the Pd catalyst used above (0.08 g, 0.10 mmol) was added to the reaction mixture. After 5 h, the reaction mixture was cooled to RT, diluted with EtOAc (150 mL), washed with water (150 mL) and brine (100 mL), dried over sodium sulfate, and evaporated to give a black residue (6.7 g), which was purified by silica gel chromatography (eluted with 50-100% EtOAc/hexane) to provide the title intermediate (5.3 g, 90% yield). Analytical HPLC: Retention time=14.7 min.

Preparation 3: ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
-

-
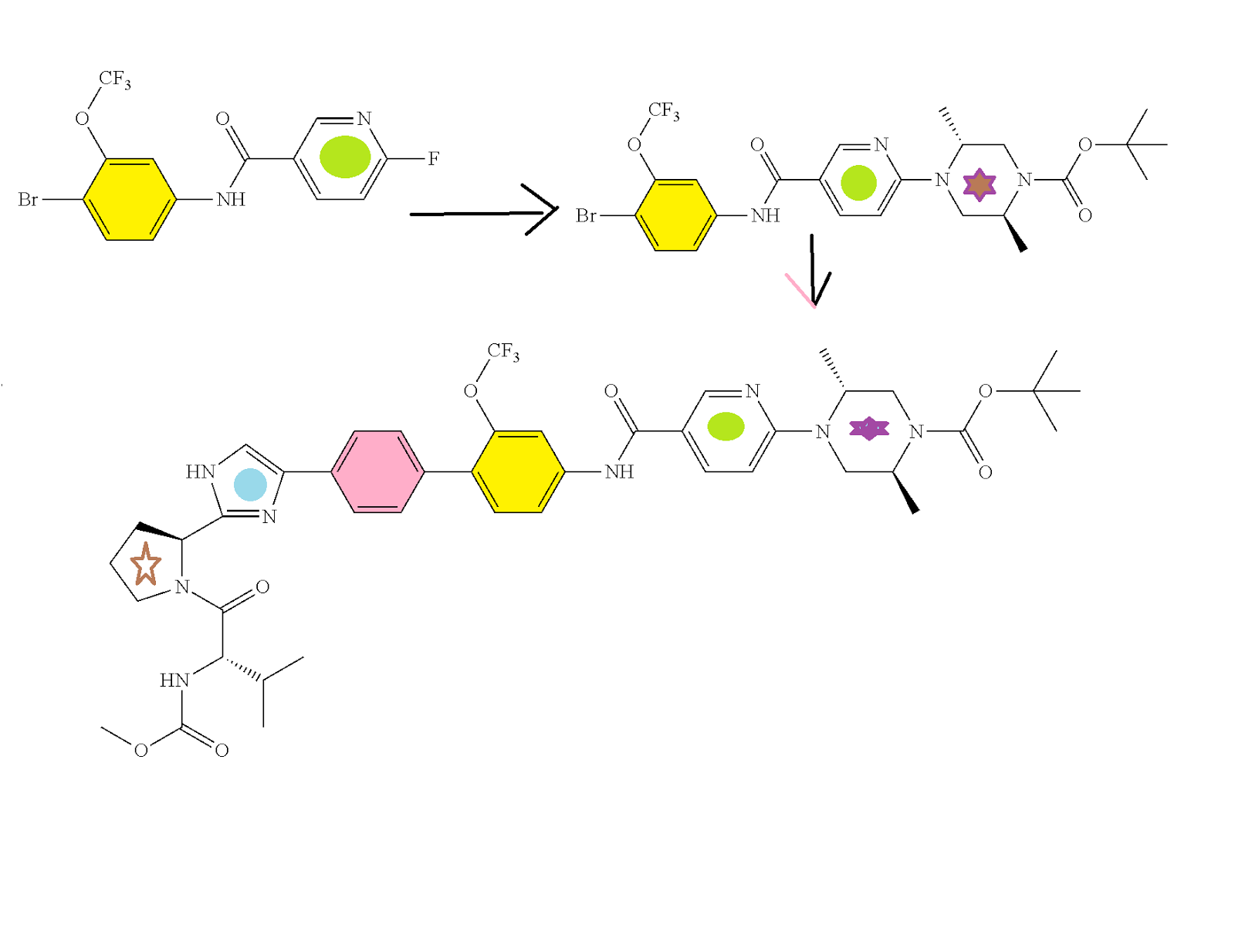
Acetyl chloride (63.2 mL, 888 mmol) was added to ethanol (360 mL) and stirred at RT for 1 h. To the resulting HCl solution was added a solution of (2S,5R)-4-[5-(4′-{2-[(S)-1-((S)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (73 g, 84 mmol) in ethanol (360 mL). The reaction mixture was stirred at RT overnight.
-
The reaction mixture was concentrated to dryness (124 g crude). Water 500 mL) was added and the mixture was extracted with EtOAc (2×500 mL). The aqueous layer was adjusted to pH 4 with 1:1 NaOH:water. Ethyl acetate (400 mL) and sat. aq. Na2CO3 (100 mL) were added and the layers were separated. The organic layer was dried over Na2SO4 and evaporated to give the title intermediate (62.8 g; 88% yield). Analytical HPLC: Retention time=10.0 min.

Example 1Amorphous ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

(a) ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester tri-HCl
-
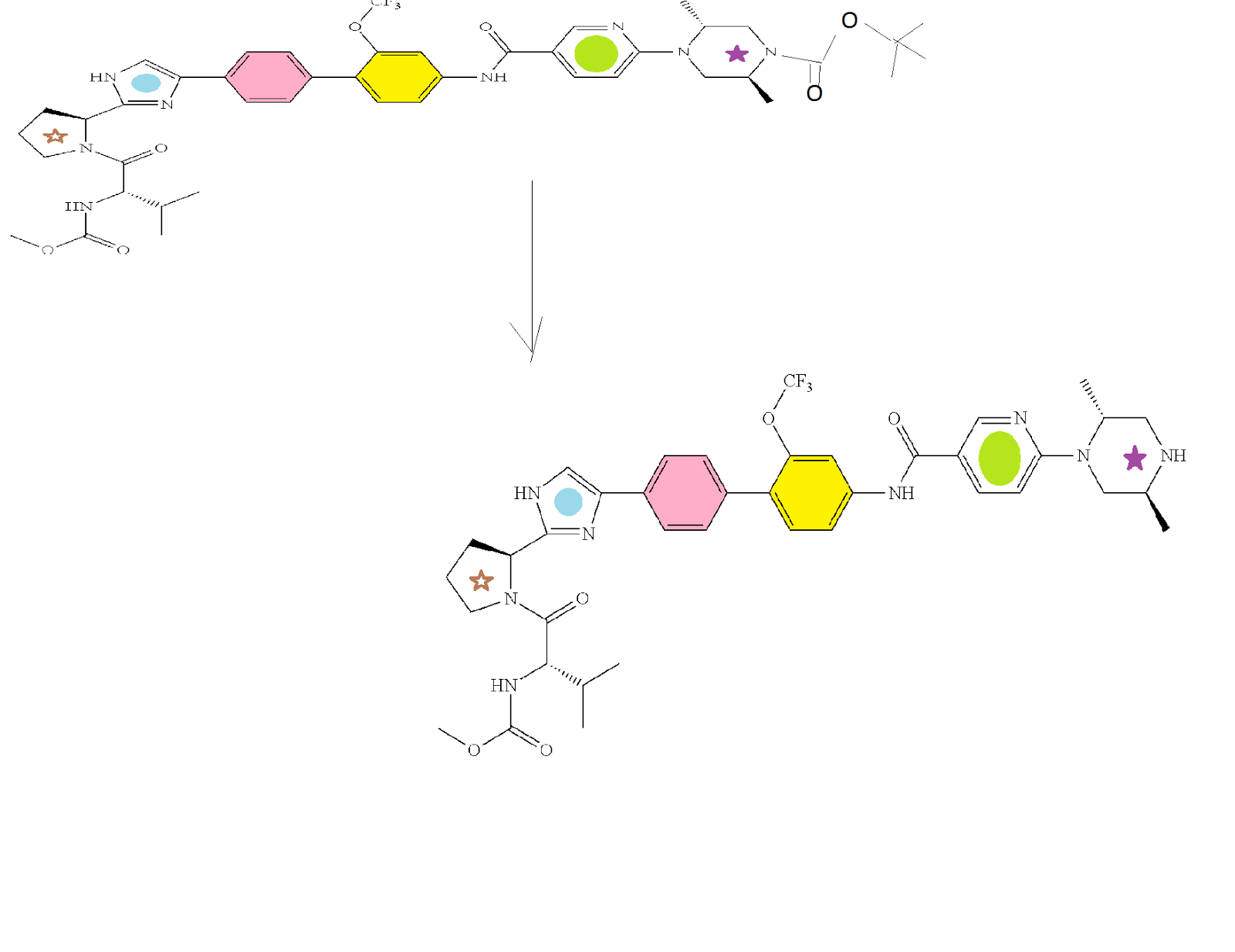
Acetyl chloride (0.71 mL, 10.0 mmol) was added to ethanol (7 mL) and stirred at RT for 1 h. The resulting HCl solution was added to a solution of (2S,5R)-4-[5-(4′-{2-[(5)-1-((S)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (1.55 g, 1.8 mmol) in ethanol (7 mL). The reaction mixture was warmed to 35° C. and stirred overnight. The mixture was concentrated to dryness, and chased with DCM to provide the crude tri-HCl salt of the title intermediate (1.57 g) which was used directly in the next step. HPLC method C: Retention time=10.0 min.
(b) Amorphous ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
-
To a solution of the product of the previous step (1.57 g crude, ca. 1.80 mmol) and N,N-diisopropylethylamine (3.14 mL, 18.0 mmol) in DCM (24 mL) was slowly added 1 M methylaminoformyl chloride in DMA (1.8 mL). The reaction mixture stirred at RT for 1 h, and then 1 M methylaminoformyl chloride in DMA (1.8 mL) was added. The reaction was quenched with sat. aq. NaHCO3 and the reaction mixture was stirred for 20 min. The layers were separated and the organic layer was dried and evaporated to give a residue. To the residue was added methanol (15 mL) followed by 2 N LiOH/water (3 mL). The reaction mixture was stirred at RT for 1 h, diluted with water, extracted with DCM (80 mL), dried, and evaporated to give a crude product which was purified by silica gel chromatography (40 g silica, 2-8% MeOH/DCM) to provide the title compound (0.93 g, 63% yield). Analytical HPLC: Retention time=11.0 min.
HPLC
- Analytical HPLC Method
-
-
- Column: Zorbax Bonus-RP 3.5 μm. 4.6×150 mm
- Column temperature: 35° C.
- Flow rate: 1.0 mL/min
- Mobile Phases: A=Water/ACN (98:2)+0.1% TFA
- B=Water/ACN (10:90)+0.1% TFA,
- Injection volume: 100-1500 μL
- Detector wavelength: 214 nm
- Sample preparation: Dissolve in 1:1 ACN:water
- Gradient: 29 min total (time (min)/% B): 0.5/10, 24/90, 25/90, 26/10, 29/10
-
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010094977A1 * | 22 Feb 2010 | 26 Aug 2010 | Arrow Therapeutics Limited | Novel biphenyl compounds useful for the treatment of hepatitis c |
| WO2012061552A1 * | 3 Nov 2011 | 10 May 2012 | Theravance, Inc. | Novel inhibitors of hepatitis c virus |
| US201113288216 |
About Theravance Biopharma
The mission of Theravance Biopharma (NASDAQ: TBPH) is to create value from a unique and diverse set of assets: an approved product; a development pipeline of late-stage assets; and a productive research platform designed for long-term growth.
Our pipeline of internally discovered product candidates includes potential best-in-class opportunities in underserved markets in the acute care setting, representing multiple opportunities for value creation. VIBATIV® (telavancin), our first commercial product, is a once-daily dual-mechanism antibiotic approved in the U.S., Europe and certain other countries for certain difficult-to-treat infections. Revefenacin (TD-4208) is an investigational long-acting muscarinic antagonist (LAMA) being developed as a potential once-daily, nebulized treatment for COPD. Axelopran (TD-1211) is an investigational potential once-daily, oral treatment for opioid-induced constipation (OIC). Our earlier-stage clinical assets represent novel approaches for potentially treating diseases of the lung and gastrointestinal tract and infectious disease. In addition, we have an economic interest in future payments that may be made by GlaxoSmithKline plc pursuant to its agreements with Theravance, Inc. relating to certain drug development programs, including the combination of fluticasone furoate, umeclidinium and vilanterol (the “Closed Triple”).
With our successful drug discovery and development track record, commercial infrastructure, experienced management team and efficient corporate structure, we believe that we are well positioned to create value for our shareholders and make a difference in the lives of patients.
For more information, please visit www.theravance.com.
THERAVANCE®, the Cross/Star logo, MEDICINES THAT MAKE A DIFFERENCE® and VIBATIV® are registered trademarks of the Theravance Biopharma group of companies.
Journal of Medicinal Chemistry (2014), 57(5), 1643-1672……….

(e)Thalladi, V. R.; Nzerem, J.; Huang, X.; Zhang, W. Crystalline form of a pyridyl-piperazinyl hepatitis C virus inhibitor. World Patent Application WO-2013/165796, November 7, 2013.
PATENT
WO 2012061552
http://www.google.com.ar/patents/WO2012061552A1?cl=en
Preparation 28: ((S)-l-{(S)-2-[4-(4′-{[6-((2JR,5S)-2,5-Dimethyl-piperazin-l- yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2- yl]-pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

A mixture of [(5)-2-methyl-l-((5)-2- {4-[4-(4,4,5,5-tetramethyl- [l,3,2]dioxaborolan-2-yl)-phenyl]-lH-imidazol-2-yl}-pyrrolidine-l-carbonyl)-propyl]- carbamic acid methyl ester (86 mg, 0.17 mmol) and (25′,5R)-4-[5-(4-bromo-3- trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-l-carboxylic acid tert-butyl ester (100 mg, 0.2 mmol, Preparation 27) was dissolved in 1,4-dioxane (1.8 mL, 23 mmol) and water (0.25 mL, 14 mmol). Cesium carbonate (170 mg, 0.52 mmol) was added. The reaction mixture was sparged with nitrogen and then
tetrakis(triphenylphosphine)palladium(0) (12.1 mg, 0.011 mmol) was added. The reaction mixture was sealed under nitrogen and heated at 95 °C overnight. The reaction mixture was extracted with ethyl acetate/water, the organic layer was dried over sodium sulfate and concentrated to produce an orange oil.
The oil from the previous step was treated with 4 M HCl in 1,4-dioxane (2 mL, 7 mmol) and stirred at room temperature for 1 h. The reaction mixture was concentrated and evaporated with ethyl acetate (2 x) to produce the HCl salt of the title compound as a yellow solid which was purified by preparative HPLC to provide the tri-TFA salt of the title compound (150 mg, 30 % overall yield), (m/z): [M+H] calcd for
763.35 found 763.7.
Example 29 ((S)-l-{(S)-2-[4-(4′-{[6-((2JR,5S)-2,5-Dimethyl-4- methylcarbamoyl-piperazin-l-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy- biphenyl-4-yl)-lH-imidazol-2-yl]-pyrrolidine-l-carbonyl}-2-methyl-propyl)- carbamic a

To a solution of ((5)- l – {(5)-2-[4-(4′- { [6-((2R,55)-2,5-dimethyl-piperazin- l-yl)- pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl]- pyrrolidine- l-carbonyl} -2-methyl-propyl)-carbamic acid methyl ester tri-TFA (1 1.4 mg, 0.01 1 mmol; Preparation 28) and N,N-diisopropylethylamine (18 uL, 0.1 1 mmol) dissolved in DMA (0.4 mL, 4 mmol) was added 1.0 M methyl isocyanate in toluene (10 uL, 0.01 mmol). The reaction mixture was stirred at RT overnight, concentrated, dissolved in 1 : 1 acetic acid:water (1.5 mL) and purified by preparative HPLC to provide the di-TFA salt of the title compound (7.1 mg). (m/z): [M+H]+ calcd for C41H48F3N906 820.37 found 820.5.
Alternative synthesis of ((5)-1-{(5)-2-[ -(4′-{[6-((2Λ,ί» 2,5- Dimethyl-4-methylcarbamoyl-piperazin-l-yl)-pyridine-3-carbonyl]-amino}-2′- trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl]-pyrrolidine-l-carbonyl}-2- methyl-propyl)-carbamic acid methyl ester
(a) N-(‘4-Bromo-3-trifluoromethoxy-phenyl -6-fluoro-nicotinamide

To a solution of 4-bromo-3-trifluoromethoxy-phenylamine (3.15 g, 12.3 mmol) and triethylamine (3.43 mL, 24.6 mmol) in DCM (25 mL) was slowly added a solution of 2-fluoropyridine-5-carbonyl chloride (2.36 g, 14.8 mmol) in DCM (10 mL). After 2 h at RT, MTBE (90 mL) was added and the reaction mixture was washed with water, brine, and saturated sodium carbonate, dried, and evaporated to give a solid (5.4 g). Ethanol (43 mL) was added to the solid and then water (43 mL) was slowly added. The reaction mixture was stirred for 1.5 h, filtered, and washed with 1 :4 ethanohwater (2 x 25 mL) to give the title intermediate as a white solid (3.87 g). HPLC method C: Retention time = 21.3 min.
(b) (25,,5R)-4-r5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl) -pyridin-2-yl1-2,5- dimethyl-piperazin – 1 -carboxylic acid fe/t-butyl ester

The product of the previous step (3.86 g, 10.2 mmol) (2S,5R)-2,5-dimethyl- piperazine-1 -carboxylic acid tert-butyl ester (2.62 g, 12.2 mmol) and N,N- diisopropylethylamine (5.32 mL, 30.5) was dissolved in DMSO (12 mL). The reaction mixture heated at 120 °C for 3 h, diluted with EtOAc (100 mL), washed with water, and saturated NH4C1, water, and brine. The reaction mixture was evaporated to about 40% volume and 3 M HCl in cyclopentyl methyl ether (4.24 mL, 12.7 mmol) was added slowly. Seeds from a previous run at smaller scale were added and the reaction mixture was stirred for 2 days and filtered to provide the HCl salt of the title intermediate (5.15 g, 83 % yield). HPLC method C: Retention time = 21.1 min (c) (2 .5R)-4 5-(4′-{2 ffl -((5f)-2-Methoxycarbonylamino-3-methyl-butyrvn- pyiTolidin-2-yl1-lH-imidazol-4-yl}-2-tri
pyridin-2- -2,5-dimethyl-piperazine-l-carboxylic acid fert-butyl ester

To a solution of ((5′)-l-{(5,)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]- pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (3.05 g, 6.8 mmol;), bis(pinacolato)diboron (1.81 g, 7.1 mmol) and potassium acetate (1,00 g, 10.2 mmol) was added nitrogen sparged toluene (15 mL). The resulting mixture was sparged with nitrogen and l,l’-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane (Pd catalyst) (0.17 g, 0.204 mmol) was added. The reaction mixture was stirred at 90 °C overnight.
The reaction mixture was cooled to RT and to this mixture was added nitrogen sparged water (7.6 mL), potassium carbonate (5.16 g, 37.3 mmol). The reaction mixture was stirred at 95°C overnight.
Another portion of the Pd catalyst used above (0.08 g, 0.10 mmol) was added to the reaction mixture. After 5 h, the reaction mixture was cooled to RT, diluted with EtOAc (150 mL), washed with water (150 mL) and brine (100 mL), dried over sodium sulfate, and evaporated to give a black residue (6.7 g), which was purified by silica gel chromatography (eluted with 50-100 % EtOAc/hexane) to provide the title intermediate (5.3 g, 90 % yield). HPLC method C: Retention time = 14.7 min.
(d) (ffl ffl-2 4-(4′ r6-((2R,5^-2,5-Dimethyl-piperazin-l-vn-pyridine-3-carbonyl1- amino}-2′ rifluoromethoxy-biphenyl-4-yl -lH-imidazol-2-yl1-pyrrolidine-l- carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

Acetyl chloride (2.57 mL, 36.2 mmol) was added to ethanol (18 mL) and stirred at RT for 1 h. To the resulting HQ solution was added a solution of the product of the previous step (3.90 g, 4.5 mmol) in ethanol (18 mL). The reaction mixture was warmed to 35 °C and stirred overnight. Acetyl chloride (1.28 mL, 18.1 mmol) was added to ethanol (7.8 mL) and stirred for 30 min. The resulting HC1 solution was added to the reaction mixture at 35 °C. The temperature was raised to 40 °C. The mixture was concentrated to dryness chased by dichloromethane to provide the crude tri-HCl salt of the title intermediate (5.4 g) which was used directly in the next step. HPLC method C: Retention time = 10.1 min.
(e) ((^-l- {(^-2-r4-(4′- {r6-((2R.5^-2.5-Dimethyl-4-methylcarbamoyl-piperazin-l-yl)- pyridine-3-carbonyl1-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl1- pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

To a solution of the product of the previous step (5.4 g crude, ca. 3.96 mmol) and N,N-diisopropylethylamine (6.89 mL, 39.6 mmol) in DCM (52 mL) was slowly added 1 M methylaminoformyl chloride in DMA (4.3 mL). The reaction mixture stirred at room temperature for 1 h, and then water (50 mL) was added. The organic layer was washed with saturated NH4C1 and then brine, dried over Na2S04 and evaporated to give 5.2 g crude product, which was purified by silica gel chromatography (133 g silica, 2 to 8 % methanol/DCM for 15 min then 8 % methanol/DCM for 40 min) to provide the title compound (2.4 g, 74 % yield). HPLC method C: Retention time 1 1.2 min
Synthesis of intermediates
http://www.google.com.ar/patents/WO2012061552A1?cl=en
Preparation 1: 4-(4-bro -phenyl)-2-(S)-pyrrolidin-2-yl-lH-imidazole

(a) 2-Bromo-l-(4-bromo-phenyl)-ethanone
Bromine (80 g, 500 mmol) was added dropwise to a solution of l-(4-bromo- phenyl)-ethanone (100 g, 500 mmol) in dichloromethane (1500 mL) at ambient temperature. The reaction mixture was stirred for 3 h and then concentrated. The residue was washed with dichloromethane (100 mL) to give the crude title compound (120 g, 86
% yield) as a white solid. XH NMR (CDC13, 400 MHz) δ (ppm): 7.78 (d, J=8.4 Hz, 2H), 7.57 (d, J=8.4 Hz, 2H), 4.32 (s, 2H).
(b) (^-pyrrolidine- 1 ,2-dicarboxylic acid 2-r2-(4-bromo-phenyl)-2-oxo-ethyl1 ester \-tert- butyl ester
Diisopropylethylamine (67 g, 518 mmol) was added dropwise to a solution of the product of the previous step (120 g, 432 mmol) and (5)-pyrrolidine-l,2-dicarboxylic acid 1-tert-butyl ester ( -Boc proline) (102 g, 475 mmol) in acetonitrile (2 L) at room temperature. The reaction mixture was stirred overnight and concentrated to dryness. The residue was dissolved in ethyl acetate (2 L) and washed with water (2 L). The organic layer was dried over sodium sulfate and concentrated to give crude title compound (178 g, 100 % yield).
(c) (5f)-2-r4-(4-bromo-phenyl -lH-imidazol-2-yl1-pyrrolidine-l-carboxylic acid fe/t-butyl ester
A solution of the product of the previous step (178 g, 432 mmol) and ammonium acetate (500 g, 6.5 mol) in toluene (2 L) was heated at reflux overnight. The solvent was removed and the residue was dissolved in ethyl acetate (2 L) and washed with water (2 L). The organic layer was dried over sodium sulfate and concentrated. The residue was purified by silica gel column chromatography in 1:3 petroleum ether: ethyl acetate to give the title compound (120 g, 71 % yield) as a yellow solid. ¾ NMR (CDC13, 400 MHz) δ (ppm): 7.56 (s, 1H), 7.39 (d, J=8.0 Hz, 2H), 7.24 (m, 1H), 7.14 (s, 1H), 4.88 (m, 1H), 3.33 (m, 2H), 2.94 (s, 1H), 2.07 (m, 2H), 1.88 (m, 1H), 1.42 (s, 9H).
(d) 4-(4-bromo-phenyl)-2-(5f)-pyrrolidin-2-yl-lH-imidazole
To a solution of (5)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]-pyrrolidine-l- carboxylic acid tert-butyl ester (3 g, 7.6 mmol) in methanol (3 mL) was added 4N HQ in methanol (60 mL) at 0 °C. The reaction mixture was stirred for 2 h and then concentrated to give crude hydrochloride salt of the title compound (2.51 g 100 % yield) as a yellow solid.
Preparation 2: (5)-2-Methoxycarbonylamino-3-methyl-butyric acid

A mixture of (5)-2-amino-3 -methyl-butyric acid (10 g, 85 mmol), NaOH (10.3 g, 255 mmol) in water (100 mL) was treated with methylchloridocarbonate (8 g, 85 mmol) at 0 0 C. The reaction mixture was stirred for 24 h at room temperature and then 5 N aqueous HC1 was added to the reaction mixture to adjust pH to 4. The mixture was filtered through a pad of Celite to give the product (10 g, 67% yield) as a white solid. ¾ NMR (CH3OD, 400 MHz) δ (ppm) 4.05(d, 1H), 3.65(s, 3H), 2.14(m, 1H), 0.95(m, 6H). Preparation 3: ((S)-l-{(S)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]- pyrrolidine-l-carbonyl}-2-met methyl ester

Triethylamine (2.3 g, 11.4 mmol) was added to a solution of 4-(4-bromo-phenyl)- 2-(5)-pyrrolidin-2-yl-lH- imidazole hydrochloride (2 g, 11.4 mol), (5)-2- methoxycarbonylamino-3-methyl-butyric acid (2.5 g, 7.6 mmol), and HATU (4.3 g, 11.4 mmol) in dimethylformamide (50 mL) at 0 °C under nitrogen. The reaction mixture was stirred at room temperature overnight and treated with ethyl acetate (100 mL) and water (1000 mL). The organic layer was washed with water (2 x 100 mL) and brine (100 mL), dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel column chromatography in 1 : 1 petroleum ether: ethyl acetate to give the title compound (2.5 g 74 % yield) as a yellow solid. XH NMR (i¾-DMSO, 400 MHz) δ (ppm) 7.63 (d, J=8.8 Hz, 2H), 7.54 (m, IH), 7.47 (m, 2H), 7.26 (d, J=8.4 Hz, IH), 5.03 (m, IH), 4.02 (t, J =8.4 Hz, IH), 3.76 (m, 2H), 3.51 (s, 3H), 2.10 (m, 2H), 1.93 (m, 3H), 0.85 (d, J=6.8 Hz, 3H), 0.81 (d, J=6.8 Hz, 3H).
I AM NOT SURE OF BELOW DATA, It is a cut paste for TD 6450 , NOT ABLE TO CONNECT CAS 1374883-22-3 WITH TD 6450
IF YOU HAVE A STRUCTURE PIC FOR THE SAME MAIL ME amcrasto@gmail.com, call +919323115463
POSTER
50th Annu Meet Eur Assoc Study Liver (EASL) (April 22-26, Vienna) 2015, Abst P0898
http://ilc-congress.eu/abstract_25_04/ILC2015-abstract-book-25-04-Saturday.pdf
P0898
TD-6450,
A NEXT GENERATION ONCE-DAILY NS5A INHIBITOR, HAS POTENT ANTIVIRAL ACTIVITY FOLLOWING A 3-DAY MONOTHERAPY STUDY IN GENOTYPE 1 HCV INFECTION
E. Lawitz1, M. Rodriguez-Torres2, R. Kohler3, A. Amrite3, C. Barnes3, M.L.C. Pecoraro3, J. Budman3, M. McKinnell3, C.B. Washington3. 1Texas Liver Institute, University of Texas Health Science Center, San Antonio, TX, United States; 2Fundacion de Investigacion, San Juan, Puerto Rico; 3Theravance Biopharma, South San Francisco, CA, United States
E-mail: cwashington@theravance.com
Background and Aims: TD-6450 is a next generation HCV NS5A inhibitor with superior in vitro potency against resistanceassociated variants (RAVs) encountered with first-generation NS5A inhibitors. This study evaluated the safety, pharmacokinetics (PK) and antiviral activity of TD-6450 following multiple oral doses in HCV patients
Theravance Biopharma Announces Positive Results From Phase 1 Proof-of-Concept Study of TD-6450, an NS5A Inhibitor to Treat Hepatitis C
240 mg Achieved a Median Maximal Viral Load Decline of 4.9 Log10 IU/mL Following Three Daily Doses in Genotype 1a Patients
SOUTH SAN FRANCISCO, CA — (Marketwired) — 11/03/14 — Theravance Biopharma (NASDAQ: TBPH), through its U.S. operating subsidiary, Theravance Biopharma US, Inc., today announced positive results from the first three cohorts of Study 0110, a Phase 1 proof-of-concept study of TD-6450, a next-generation investigational NS5A inhibitor in development to treat patients with hepatitis C virus infection (HCV).
TD-6450 was evaluated in three cohorts of eight genotype 1a (GT-1a) patients each at doses of 60, 120 and 240 mg, administered once-daily for three days. TD-6450 demonstrated dose-dependent antiviral activity with median maximal declines of HCV RNA of 3.87, 4.63 and 4.89 log10 IU/mL for doses of 60, 120 and 240 mg, respectively.
In the 120 and 240 mg dose groups, three days of once-daily oral treatment resulted in levels of serum HCV RNA below the limit of detection (LOD) in 43% (3/7) and 57% (4/7) of patients treated with TD-6450, respectively. Three of the seven LOD patients went on to show no measurable virus at Day 14, and two of these patients still had no measurable virus at Day 28. At a two-month time point in a long-term follow-up study, the viral load in these two patients was measurable, but both remained more than three logs below their baseline.
None of the patients in the three dose groups had virologic breakthrough during their three-day treatment course, and 100% of the treated GT-1a patients in the study achieved at least a three log10 IU/mL reduction of HCV RNA. At the 120 and 240 mg doses, 71% (5/7) and 86% (6/7) of treated patients achieved at least a four log10 IU/mL reduction in HCV RNA, respectively.
All doses of TD-6450 were generally well tolerated after three doses and for the 28-day observation period. There were no serious adverse events and no patient discontinuations. There was no pattern of clinical adverse events or laboratory abnormalities related to treatment.
“We see diverse responses to direct antivirals in genotype 1 populations. Despite recent advances in HCV therapy, significant treatment challenges remain, including the required length of drug therapy. The robust activity of TD-6450 in genotype 1a patients suggests that this potentially best-in-class NS5A inhibitor could be a component of short and highly active combination therapy regimens,” said Eric Lawitz, MD, Vice President of Scientific and Research Development at the Texas Liver Institute and Clinical Professor of Medicine, The University of Texas Health Science Center San Antonio, and one of the principal investigators on the Phase 1 study.
“TD-6450, created using the principles of multivalent design, has a heterodimeric structure distinct from other NS5A inhibitors. We believe this unique structure allows it to bind asymmetrically across the NS5A protein interface, providing high in vitro potency against clinically encountered resistance-associated variants. We believe the potency of TD-6450 against both wild type virus and these resistance-associated variants enables the robust antiviral activity that we reported today,” said Mathai Mammen, MD, Senior Vice President, Research and Development, Theravance Biopharma. “We look forward to analyzing the full set of results from this Phase 1 study and evaluating the next steps in the development strategy for TD-6450.”
About the Phase 1 Proof-of-Concept Study (Study 0110)
This Phase 1 study is a double-blind, randomized, placebo-controlled, multiple-dose study to evaluate the safety, tolerability, pharmacokinetics and antiviral activity of orally administered TD-6450 in non-cirrhotic, treatment-naive patients with GT-1, 2, or 3 chronic HCV infection. The study includes seven cohorts. The first three cohorts enrolled eight GT-1a patients each (7 active; 1 placebo) and tested once-daily oral doses of 60, 120 or 240 mg, respectively. Patients were dosed for three days and followed for up to 28 days for viral load quantification. The limit of detection for the viral load quantification assay is 15 IU/mL.
Safety evaluations include monitoring for adverse events, routine laboratory assessments, vital signs and 12-lead ECG tracings.
In cohorts 4 through 6, patients with GT-1b, GT-2 and GT-3 are dosed once-daily at 240 mg. An additional cohort (cohort 7) of GT-1a patients is dosed twice daily with 240 mg. Data generation and analysis of results for cohorts 4 through 7 is ongoing. An interim analysis of those cohorts showed antiviral activity for GT-1b similar to that for GT-1a, but minimal antiviral activity for GT-2 and GT-3.
The Company anticipates presenting further data on all cohorts at a future scientific conference.
About TD-6450
TD-6450 is an internally discovered multivalent NS5A inhibitor designed to have improved antiviral activity against GT-1 resistance-associated variants (RAV) resistant to first generation NS5A inhibitors. TD-6450’s heterodimeric structure permits an asymmetric binding mode to NS5A relative to structurally symmetric inhibitors. TD-6450 has demonstrated additive activity with other classes of anti-HCV agents in replicon assays, and no cross-resistance with RAVs that confer resistance to other anti-HCV agents. The Company believes that the antiviral activity of TD-6450, in combination with other antivirals, may help improve cure rates and/or reduce treatment times for appropriate patients.
TD-6450 was previously evaluated in a single-ascending dose and a 14-day multiple-ascending dose study in healthy subjects (study 0094). This randomized, double-blind, placebo-controlled study evaluated the safety, tolerability and pharmacokinetics of TD-6450. Single doses (up to 500 mg) and multiple doses of TD-6450 (up to 240 mg daily for 14 days) were evaluated in healthy subjects. Following single and multiple doses, TD-6450 was generally well-tolerated and no subjects discontinued due to adverse events. Headache was the most commonly reported adverse event following multiple doses (n=4). TD-6450 pharmacokinetics were linear up to 240 mg following single and multiple doses and its long half-life supports once-daily dosing.
About Hepatitis C and the NS5A Inhibitor Class
Hepatitis C is an infectious disease of the liver. Worldwide, health experts estimate that 130 – 150 million people have chronic hepatitis C, with as many as four million of those cases in the United States. Hepatitis C, like all forms of hepatitis, can damage the liver. Of people infected, 55 to 85 percent will develop chronic infection, and 75 percent of those with chronic infection will develop chronic liver disease.
The hepatitis C non-structural 5A (NS5A) protein of HCV has emerged as an attractive drug target and inhibitors of NS5A have a central role in all-oral HCV therapy. The multi-functional NS5A protein is required for ribonucleic acid (RNA) replication and virion assembly, and a number of investigational and approved NS5A inhibitors have shown antiviral efficacy in HCV-infected patients.
Theravance Biopharma and Trek Therapeutics Announce Initiation of Phase 2a Trial of TD-6450, an NS5A Inhibitor to Treat Hepatitis C
Study Being Conducted by Trek Therapeutics Following Licensing of Worldwide Rights to Drug Candidate From Theravance Biopharma
DUBLIN, IRELAND and CAMBRIDGE, MA — (Marketwired) — 10/27/15 — Theravance Biopharma, Inc. (NASDAQ: TBPH) (“Theravance Biopharma”) and Trek Therapeutics (“TREKtx”) today announced that TREKtx has initiated a Phase 2a clinical trial of TD-6450, a next-generation investigational NS5A inhibitor in development to treat patients with hepatitis C virus (HCV). Theravance Biopharma recently granted TREKtx an exclusive worldwide license for the development, manufacturing, use, marketing and sale of TD-6450 as a component in combination HCV products. Other terms of the transaction have not been disclosed.
The Phase 2a clinical trial will evaluate faldaprevir (FDV), an HCV protease inhibitor, combined with TD-6450 and ribavirin (RBV) in patients infected with HCV genotype 4. The trial is being conducted in the United States.
Mathai Mammen, M.D., Ph.D., Senior Vice President of Research and Development at Theravance Biopharma commented, “We are pleased to see the initiation of this Phase 2a clinical trial with TD-6450. This NS5A inhibitor has shown robust antiviral activity in a Phase 1 trial in patients with HCV genotype 1, as well as preclinical potency against both wild type HCV and resistance-associated variants. We believe that its antiviral activity, in combination with other antivirals, may help improve cure rates and/or reduce treatment times for appropriate patients. We are especially pleased to collaborate with TREKtx and support their commitment to delivering novel and accessible combination HCV treatments to patients worldwide.”
“We are very excited about dosing our first genotype 4 patients in this combination study. If safety and efficacy are demonstrated, the goal is to initiate clinical trials in Egyptnext year, where the need is enormous,” said Dr. Robert Hindes, Chief Medical Officer of Trek Therapeutics.
About TD-6450
Theravance Biopharma discovered TD-6450, a multivalent NS5A inhibitor designed to have improved antiviral activity against genotype 1 resistance-associated variants (RAV) resistant to first generation NS5A inhibitors. TD-6450 has successfully completed Phase 1 studies in both healthy volunteers and HCV patients.
About Faldaprevir
Faldaprevir is a protease inhibitor that TREKtx acquired from Boehringer Ingelheim. FDV has completed Phase 3 studies in combination with pegylated interferon and RBV.
About HCV
Hepatitis C is an infectious disease of the liver. Of people infected, 55 to 85 percent will develop chronic infection, and 75 percent of those with chronic infection will develop chronic liver disease.
The U.S. Centers for Disease Control and Prevention estimates 2.7 million individuals in the United States have active hepatitis C virus (HCV) infection, most of whom are “baby boomers.” In the United States, chronic HCV infection is the leading cause of cirrhosis and liver cancer and the most common reason for liver transplantation. Worldwide, more than 135 million people have chronic HCV infection and most are undiagnosed.
About Trek Therapeutics
TREKtx is a private, clinical stage public benefit corporation developing treatments for serious infections. Its mission is to profitably develop affordable and accessible medicines to treat infectious diseases and to commercialize them for global populations. The company’s founders collectively participated in the development of seven approved antiviral drugs.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008021927A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2010094977A1 * | 22 Feb 2010 | 26 Aug 2010 | Arrow Therapeutics Limited | Novel biphenyl compounds useful for the treatment of hepatitis c |
| US201161492267 |
| WO2013067267A1 * | 2 Nov 2012 | 10 May 2013 | Theravance, Inc. | Rod -like hepatitis c virus inhibitors containing the fragement {2- [4- (bi phenyl – 4 – yl) – 1h – imidazo – 2 – yl] pyrrolidine – 1 – carbonlymethyl} amine |
| WO2013163270A1 * | 24 Apr 2013 | 31 Oct 2013 | Theravance, Inc. | Hepatitis c virus inhibitors |
| WO2013165796A1 * | 25 Apr 2013 | 7 Nov 2013 | Theravance, Inc. | Crystalline form of a pyridyl-piperazinyl hepatitis c virus inhibitor |
//////////////
SOFOSBUVIR HPLC


SEE http://www.google.co.in/patents/US20100298257
Example 28
-
Chemical Purity Determination by HPLC
-
Various HPLC conditions can be used to determine the chemical purity of the compounds disclosed herein. One such example is disclosed above in relation to the thermodynamic aqueous solubility studies. Another example is disclosed below.
-
HPLC Conditions:
-
- LC: Waters Alliance 2695 Separations Module, Waters 2996 PDA detector and Waters Empower 2 Software (Version 6.00)
- Column: Phenomenex Luna C18(2); 4.6×50 mm; 3 μm
- Flow rate: 1.2 mL/min
- Injection Volume: 10 μL
- Mobile phase: Solvent A: 95% Water with 5% Methanol and 10 mM Ammonium Acetate; pH˜5.3
- Gradient Solvent B: MeOH with 10 mM Ammonium Acetate hold at 0% B 3 min
- 0-47% B 3-4 min
- hold at 47% B 4-10 min
- 47%-74% B 10-11 min
- hold at 74% B 11-13.5 min
- return to 0% B 13.5-13.6 min
- hold at 0% B 13.6-15.5 min
-
Under these conditions, the purity of 4, RP-4, and SP-4 was determined to be ˜99.6, ˜99%, and ˜99.5%, respectively. It is noted that higher purities can be realized by optimizing the methods disclosed above.
-
Inspection of the XRPD diffractograms shows that the two crystalline single diastereoisomers gave clearly different XRPD patterns. Additionally, there was a clear difference in the melting point of the two crystalline diastereoisomers, with RP-4 having a considerably higher onset than SP-4 (136° C. vs. 94° C.).
-
Example 29Additional Separation Methods
-
The following SFC separation (conditions listed below) yielded adequate separation of a mixture of the diastereomers, RP-4 and SP-4.
-
Preparative Method: Analytical Method: Chiralpak AS-H (2 × 25 cm) SN# 07-8656 Chiralpak AS-H (25 × 0.46 cm) 20% methanol/CO2 (100 bar) 20% methanol/CO2 (100 bar) 50 ml/min, 220 nm. 3 ml/min, 220 nm. Conc.: 260 mg/30 ml methanol, inj vol.: 1.5 ml -
The following SFC separation (conditions listed below) yielded adequate separation of a mixture of the diastereomers, RP-4 and SP-4.
-
Preparative Method: Analytical Method: Chiralpak IA(2 × 15 cm) 802091 Chiralpak IA(15 × 0.46 cm) 30% isopropanol(0.1% DEA)/CO2, 40% methanol(DEA)/CO2, 100 bar 100 bar 60 mL/min, 220 nm. 3 mL/min, 220 nm. inj vol.: 2 mL, 20 mg/mL methanol -
TABLE 16 Summary of results from the batch characterization of RP-4, 4, and SP-4. Analysis RP-4 4 SP-4 Proton NMR Single diastereoisomer 1:1 Mixture of Single diastereoisomer diastereoisomers XRPD Crystalline – different Amorphous Crystalline – different DSC from SP-4 Endotherm; 59° C. from RP-4 Endotherm; melt – 136° C. Endotherm; melt – 94° C. TGA No wt loss, No wt loss, decomposition No wt loss, decomposition >240° C. >240° C. decomposition >240° C. IR See above See above See above Aq Solubility 1.58 6.11 5.65 (mg · ml−1) HPLC Purity 96.9% 99.6% 99.5% 40° C./75% RH No form change Deliquescence inside 1.5 h Deliquescence inside 4.5 h 25° C./53% RH — Deliquescence No form change GVS Non-hygroscopic up to 90% — Non-hygroscopic up to 60% RH RH
- Example 27Thermodynamic Aqueous Solubility
-
Aqueous solubility was determined by suspending a sufficient amount of compound in water to give a maximum final concentration of ≧10 mg.ml−1 of the parent free-form of the compound. The suspension was equilibrated at 25° C. for 24 hours then the pH was measured. The suspension was then filtered through a glass fiber C filter into a 96 well plate. The filtrate was then diluted by a factor of 101. Quantitation was by HPLC with reference to a standard solution of approximately 0.1 mg.ml−1 in DMSO. Different volumes of the standard, diluted and undiluted sample solutions were injected. The solubility was calculated using the peak areas determined by integration of the peak found at the same retention time as the principal peak in the standard injection.
-
TABLE 14 HPLC Method Parameters for Solubility Measurements Type of method: Reverse phase with gradient elution Column: Phenomenex Luna, C18 (2) 5 μm 50 × 4.6 mm Column Temperature 25 (° C.): Standard Injections (μl): 1, 2, 3, 5, 7, 10 Test Injections (μl): 1, 2, 3, 10, 20, 50 Detection: 260, 80 Wavelength, Bandwidth (nm): Flow Rate (ml · min−1): 2 Phase A: 0.1% TFA in water Phase B: 0.085% TFA in acetonitrile Time (min) % Phase A % Phase B Timetable: 0.0 95 5 1.0 80 20 2.3 5 95 3.3 5 95 3.5 95 5 4.4 95 5 -
[0306]Analysis was performed under the above-noted conditions on an Agilent HP1100 series system equipped with a diode array detector and using ChemStation software vB.02.01-SR1.
-
TABLE 15 Aqueous solubility result for RP-4, 4, and SP-4. pH of Unfiltered Sample ID mixture Solubility/mg · ml−1 Comments RP-4 7.12 1.58 Suspension 4 7.03 6.11 Residual solid SP-4 6.88 5.65 Residual solid

FIG 1
Chemical structures of RBV, BOC, TVR, and VRT-127394. Shown are the chemical structures of the anti-HCV drugs RBV {1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2,4-triazole-3-carboxamide)} (A), BOC {(1R,2S,5S)-N-(4-amino-1-cyclobutyl-3,4-dioxobutan-2-yl)-3-[(2S)-2(tertbutylcarbamoylamino)-3,3-dimethylbutanoyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide} (B), TVR {(3S,3aS,6aR)-2-[(2S)-2-[[(2S)-2-cyclohexyl-2-(pyrazine-2-carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1, 2-dioxohexan-3-yl]-3,3a,4,5,6,6a-hexahydro-1H-cyclopenta[c]pyrrole-3-carboxamide} (C), and VRT-127394 (R diastereoisomer of TVR) (D).
Blank plasma samples used for matrix effect (ME) assessment and for the preparation of calibration and control samples were obtained from citrated blood (1,850 × g, 10 min, +4°C, Beckman J6B centrifuge) collected from Vaquez disease patients on the occasion of their regular phlebotomy.
The blank plasma used for the preparation of the calibration and quality control (QC) samples was acidified with 10% FA (50 μl of 10% FA added to 950 μl of plasma). The acidification of plasma aims at preventing the conversion of TVR to its epimer VRT-127394 that occurs in vivo and in vitro. (Tibotec-Janssen, personal communication).
Equipment.The LC system used consisted of Rheos Allegro quaternary pumps equipped with an online degasser and an HTS PAL autosampler (CTC Analytics AG, Zwingen, Switzerland) controlled by Janeiro-CNS 1.1 software (Flux Instruments AG, Thermo Fischer Scientific Inc., Waltham, MA). Separations were done on a Hypercarb 3-μm column (2.1 mm ID by 100 mm; Thermo Fischer Scientific) placed in a column oven thermostat regulated at +80°C (HotDog 5090; ProLab GmbH, Reinach, Switzerland). The chromatographic system was coupled to a triple-stage quadrupole quantum mass spectrometer (Thermo Fischer Scientific) equipped with an electrospray ionization (ESI) Ion Max interface and operated with the Xcalibur software package (version 2.0; Thermo Fischer Scientific).
READ
http://www.us.edu.pl/uniwersytet/jednostki/wydzialy/chemia/acta/ac14/zrodla/14_AC14.pdf
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2291777/
UPDATE………….DEC2015
SOFOSBUVIR
NEW PATENT WO2015188782,
(WO2015188782) METHOD FOR PREPARING SOFOSBUVIR
CHIA TAI TIANQING PHARMACEUTICAL GROUP CO., LTD [CN/CN]; No. 8 Julong North Rd., Xinpu District Lianyungang, Jiangsu 222006 (CN)
Sofosbuvir synthesis routes currently used include the following two methods:
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015188782&redirectedID=true

![]()
//////
////
AN-2718, a New Borole in the pipeline
AN-2718,
2,1-Benzoxaborole, 5-chloro-1,3-dihydro-1-hydroxy-
5-chloro-1,3-dihydro-l-hydroxy-2,1- benzoxaborole
5-Chloro-2,1-benzoxaborol-1(3H)-ol
CAS 174672-06-1
UNII: 810U6C2DGG
MW 168.3864, MF C7 H6 B Cl O2
MP…150-154 °C [WO 9533754]
MP 142-144 °C [ jmc 2006, 49(15) 447]
M.p. 147- 149 °C. WO2013050591
Anacor Pharmaceuticals Inc, INNOVATOR
Onychomycosis is a disease of the nail caused by yeast, dermatophytes, or other molds, and represents approximately 50% of all nail disorders. Toenail infection accounts for approximately 80% of onychomycosis incidence, while fingernails are affected in about 20% of the cases. Dermatophytes are the most frequent cause of nail plate invasion, particularly in toenail onychomycosis. Onychomycosis caused by a dermatophyte is termed Tinea unguium. Trichophyton rubrum is by far the most frequently isolated dermatophyte, followed by T. mentagrophytes. Distal subungual onychomycosis is the most common presentation of tinea unguium, with the main site of entry through the hyponychium (the thickened epidermis underneath the free distal end of a nail) progressing in time to involve the nail bed and the nail plate. Discoloration, onycholysis, and accumulation of subungual debris and nail plate dystrophy characterize the disease. The disease adversely affects the quality of life of its victims, with subject complaints ranging from unsightly nails and discomfort with footwear, to more serious complications including secondary bacterial infections.
Many methods are known for the treatment of fungal infections, including the oral and topical use of antibiotics (e.g., nystatin and amphotericin B), imidazole anti-fungal agents such as miconazole, clotrimazole, fluconazole, econazole and sulconazole, and non-imidazole fungal agents such as the allylamine derivatives terbinafme and naftifϊne, and the benzylamine butenafine. However, onychomycosis has proven to be resistant to most treatments. Nail fungal infections reside in an area difficult to access by conventional topical treatment and anti-fungal drugs cannot readily penetrate the nail plate to reach the infection sites under the nail. Therefore, onychomycosis has traditionally been treated by oral administration of anti-fungal drugs; however, clearly this is undesirable due to the potential for side effects of such drugs, in particular those caused by the more potent anti-fungal drugs such as itraconazole and ketoconazole. An alternative method of treatment of onychomycosis is by removal of the nail before treating with a topically active anti-fungal agent; such a method of treatment is equally undesirable. Systemic antimycotic agents require prolonged use and have the potential for significant side effects. Topical agents have usually been of little benefit, primarily because of poor penetration of the anti-fungal agents into and through the nail mass.
- 51. Hui X, Baker SJ, Wester RC, In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci 2007;96(10):2622-31 [CrossRef], [PubMed], [Web of Science ®]
- 55. Mao W, Seiradake E, Crepin T, AN2718 has Broad Spectrum Antifungal Activity Necessary for the Topical Treatment of Skin and Nail Fungal Infections. J Am Acad Dermatol 2007:56(2 Suppl):AB124
- 56. ClinicalTrials.gov. Cumulative Irritation Test. Available from: http://clinicaltrials.gov/ct2/show/NCT00781664 [Cited 11 December 2011]
SYNTHESIS

Reduction of 2-bromo-5-chlorobenzoic acid with BH3/THF in THF gives 2-bromo-5-chlorobenzyl alcohol , which is protected as the methoxymethyl derivative by treatment with MOM-Cl in the presence of DIEA in CH2Cl2. Metalation and boronylation of aryl bromide either with t-BuLi or BuLi (1,2,3,4) and (i-PrO)3B (4) or B(OMe)3 in THF affords the title compound .
Alternatively, reaction of 3-chlorobenzaldehyde with p-toluenesulfonylhydrazide in MeOH provides tosyl hydrazone which undergoes thermal decomposition in the presence of BBr3 and catalytic FeCl3 in refluxing CH2Cl2, followed by heating with aqueous NaOH to produce the title oxaborole compound .
You can construct from this…………….
 OH A
OH A
(2-bromo-5-chloro-phenyl)methanol (A)
 B
B
l-bromo-4-chloro-2-(methoxymethoxymethyl)benzene (B)
 1
1
5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
PATENT
WO 9533754
https://www.google.com/patents/WO1995033754A1?cl=en
Example 1 Preparation of 5-chloro-1,3-dihydro-l-hydroxy-2,1- benzoxaborole (Method B).
a)
Preparation of 3-chlorobenzaldehyde tosyl hydrazide
A solution of 3-chlorobenzaldehyde (15.56 parts; 0.109M;
Aldrich) in methylated spirits (40 ml) was added slowly at below 10°C to a stirred suspension of p-toluene-sulphonylhydrazide (20.7 parts;
0.108M) in methylated spirits (150 ml). The reaction mass was then stirred at 20 to 25°C for 1 hour and then heated at 60-70°C for 1M hours when the reactants and products dissolved. The solvent was then removed by rotary evaporation and the product was obtained as a solid which was slurried with ether and washed with n-hexane. Yield = 27.2 parts (81.5% theory) mpt 122-3°C. Elemental analysis
Theory 54.5%C; 4.2%H; 9.1%N
Found 54.5%C; 4.3%H; 9.1%N
Proton NMR (CDCl3:ppm)
8.5, s, 1H(-NH-); 7.9, d, 2H(Tosyl aromatic); 7.7,s, 1H(CH=N); 7.5, s, 1H (aromatic); 7.2-7.4, m, 5H(Tosyl aromatic); 2.3, s, 3H(-CH3) b) Preparation of title compound
A suspension of anhydrous ferric chloride catalyst (0.75 parts, Fisons) in dry dichloromethane (20 ml) was added at 20 to 25°C simultaneously with boron tribromide (25 parts, 0.1M, Aldrich) in dry dichloromethane (100 mis) to a stirred suspension of the hydrazide from a) above (10.18 part, 0.033M) in dry dichloromethane (160 mis) under a nitrogen blanket. The reactants were then stirred under reflux and the evolved hydrogen bromide trapped under aqueous sodium hydroxide. After 3 hours stirring at reflux, the reactants were allowed to stand at 20- 25°C for 48 hours and then stirred under reflux for a further 4 hours. The reaction mass was then cooled and the solvent removed by rotary evaporation. The solid obtained was then stirred under reflux with 2N sodium hydroxide solution (160 ml) for 3 hours. The brown aqueous suspension was extracted with dichloromethane (50 ml), screened and then acidified to about pH 2 by addition of 2N hydrochloric acid. The solid was filtered, slurried with dichloromethane (400 ml) and then washed with a saturated solution of sodium bicarbonate followed by water.
Yield = 24 parts (43% theory). The solid was slurried in hot
dichloromethane and filtered to give 0.36 parts oxaborole mp 140-45°C. The dichloromethane solution was cooled and the solid filtered giving a further 0.35 parts oxaborole mp 146-8°C. The solids were combined and recrystallised from methylated spirits.
Yield = 0.51 parts (9.2% theory) mp 150-4°C.
Elemental Analysis
Theory 49.8%C, 3.5%H, 21.06%C1
Found 49.5%C, 3.5%H, 21.0%C1
Proton NMR (CDCl3) ppm
9.3, s, 1H(-OH); 7.5, d, s, d, 3H(aromatic);
5.0, s, 2H(-CH2-O).
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en

Analytical data for exemplary compounds of structure I are provided below.
4.2. a 5-Chloro-1.3-dihydro-l -hvdroxy-2J-benzoxaborole (Cl) M.ρ. 142-15O0C. MS (ESI): m/z = 169 (M+l, positive) and 167 (M-I, negative). HPLC (220 nm): 99% purity. 1H NMR (300 MHz, DMSO-d6): δ 9.30 (s, IH), 7.71 (d, J = 7.8 Hz, IH), 7.49 (s, IH), 7.38 (d, J = 7.8 Hz, IH) and 4.96 (s, 2H) ppm.
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm0603724?source=chemport
COMPD IS 19d
5-Chloro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (19d) This compound was made from 18d in the same manner as compound 19b (triturated with hexane, 75% yield):: mp 142-144°C. 1 H NMR (300MHz, DMSO-d6) δ (ppm) 4.96 (s, 2H), 7.38 (d, J = 7.8 Hz, 1H), 7.49 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 9.30 (s, 1H); ESI-MS m/z 167 (M-H)- ; HPLC purity 99.0%; Anal (C7H6BClO2 ⋅ 0.1H2O) C, H
precursor 18d
2-Bromo-5-chloro-1-(methoxymethoxymethyl)benzene (18d) To a solution of 2-bromo-5-chlorobenzoic acid (5.49 g, 23.3 mmol) in anhydrous THF (70 mL) under nitrogen was added dropwise a BH3 THF solution (1.0 M, 55 mL) at 0o C and the reaction mixture was stirred overnight at room temperature. Then the mixture was cooled on an ice bath and MeOH (20 mL) was added dropwise to decompose excess BH3. The resulting mixture was stirred until no bubble was released and then 10% NaOH (10 mL) was added. The mixture was concentrated and the residue was S6 mixed with water (200 mL) and extracted with EtOAc. The residue from rotary evaporation was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give 2-bromo-5-chlorobenzyl alcohol as a white solid (4.58 g, 88%): 1 H NMR (300 MHz, DMSO-d6): δ (ppm) 7.57 (d, J = 8.7 Hz, 1H), 7.50-7.49 (m, 1H), 7.28-7.24 (m, 1H), 5.59 (t, J = 6.0 Hz, 1H), 4.46 (d, J = 6.0 Hz, 2H). 2-Bromo-5-chlorobenzyl alcohol obtained above was dissolved in CH2Cl2 (150 mL) and cooled to 0o C on an ice bath. To this solution under nitrogen were added in sequence i-Pr2NEt (5.4 mL, 31 mmol) and chloromethyl methyl ether (2.0 mL, 26 mmol). The reaction mixture was stirred overnight at room temperature and washed with NaHCO3-saturated water and then brine. The residue after rotary evaporation was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give 18d (4.67 g, 85%) as a colorless oil: 1 H NMR (300 MHz, DMSO-d6): δ (ppm) 3.30 (s, 3H), 4.53 (s, 2H), 4.71 (s, 2H), 7.32 (dd, J = 8.4, 2.4 Hz, 1H), 7.50 (dd, J = 2.4, 0.6 Hz, 1H), 7.63 (d, J = 8.7 Hz, 1H).
PATENT
http://www.google.com/patents/WO2013050591A2?cl=en
EXAMPLES Example 1 : Preparation of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
Step 1 : Preparation of (2-bromo-5-chloro-phenyl)methanol (A)
A solution of borane-tetrahydrofuran complex in THF (0.15 L, 1.5 eq) was added dropwise to a solution of 2-bromo-5-chlorobenzoic acid (24 g) in anhydrous tetrahydrofuran (0.24 L) at 0°C and under argon atmosphere. The reaction mixture was stirred at room temperature for 16 h, before being slowly poured onto 0.10 L of a 2N aqueous solution of hydrogen chloride at 0°C. The mixture was stirred for 15 minutes and the volatiles were removed under reduced pressure. The aqueous layer was extracted with ethyl acetate and the combined organic layers were washed with a IN aqueous solution of sodium hydroxide and then water. After drying over sodium sulfate, filtration and concentration under reduced pressure, the crude product was purified by column chromatography; 23.2 g; M.p. 79-80 °C.
Step 2: Preparation of l-bromo-4-chloro-2-(methoxymethoxymethyl)benzene (B)
(2-bromo-5-chloro-phenyl)methanol (A, 12 g) was dissolved in dichloromethane (0.35 mL) and cooled to 0 °C. Under argon atmosphere, diisopropylethylamine (14 mL, 1.5 eq) and chloromethyl methyl ether (5.0 mL, 1.2 eq) were added. After 1 night of stirring at room temperature, the crude reaction mixture was washed with a saturated solution of sodium hydrogen carbonate, dried over sodium sulfate and evaporated under reduced pressure. Purification by column chromatography afforded 10.5 g of l-bromo-4-chloro-2- (methoxymethoxymethyl)benzene (B) as an oil.
Step 3: Preparation of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
To a solution of (B) (6.0 g) in anhydrous tetrahydrofuran (120 mL) at -78°C was added dropwise a solution of butyllithium in hexane (15.6 mL, 1.1 eq). To the resulting yellow- brown solution trimethyl borate (2.5 mL, 1.0 eq) was injected in one portion and the cooling bath was removed. The mixture was warmed gradually for 30 minutes. After stirring at room temperature for 2 hours, 8.0 ml of a 6N aqueous solution of hydrogen chloride were added and the reaction mixture was left stirring overnight at room temperature. Evaporation of the volatiles gave a residue which was taken up in ethyl acetate, washed with water, brine, dried over sodium sulfate and then evaporated. The crude product was crystallized from ethyl acetate to give 1.4 g of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1) as a white powder. Purification of the filtrate by column chromatography afforded 1.2 g more of 1. M.p. 147- 149 °C.
PATENT
http://www.google.fr/patents/WO2010110400A1?cl=en&hl=fr
Reference Example 187
5-chloro-2,1-benzoxazine ball roll -1 (3H) – All

5-chloro-2- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) benzaldehyde 8.50g synthesized in Reference Example 185 was dissolved in methanol 100ml, borohydride Sodium 2.40g was added, and after stirring for 30 minutes at room temperature and stirred for 2 hours at 60 ℃. The reaction solution was concentrated, and the organic layer after the layers were separated with ethyl acetate and saturated aqueous ammonium chloride and then concentrated under reduced pressure. The residue was added 100ml of tetrahydrofuran, and 6N hydrochloric acid 60ml, and stirred for 8 hours at room temperature.After the reaction mixture was dried and the organic layer was extracted with ethyl acetate, and concentrated. The residue was purified by silica gel column chromatography to give the title compound 9.6g. 1 H-NMR (400MHz, DMSO-d 6) δ: 4.95 (2H, s), 7.36 (1H, dd, J = 8.0,1.6Hz), 7.47 (1H, s) , 7.70 (1H, d, J = 8.0Hz), 9.28 (1H, s).
PATENT
http://www.google.com/patents/WO2008070257A2?cl=en
5-Fluoro-l,3-dihvdro-l-hvdroxy-2, 1-benzoxaborole (Cl)
1-Hydroxy-dihydrobenzoxaboroles, such as Cl, were synthesized as shown in Scheme 1. The protected o-bromobenzyl alcohol derivative (18), prepared from 16 or 17, was converted into the corresponding phenyl boronic acid. Deprotection of the methoxymethyl ether using hydrochloric acid followed by spontaneous cyclization gave the target compounds.
Scheme 7

Conditions (a) NaBH4, MeOH, rt (when X = H ), or BH3-THF, THF, rt (when X = OH), (b) MeOCH2CI, /-Pr2NEt, CH2CI2, rt, (c) MeMgBr, THF, -78 0C to rt , (d) NBS, AIBN, CCI4, reflux, (e) NaOAc, DM F, 70 0C, (f) NaOH, MeOH, reflux, (g) n-BuLι, (/-PrO)3B, THF, -780C to rt, (h) 6N HCI, THF, rt
References
- Hui, Xiaoying; Journal of Pharmaceutical Sciences 2007, 96(10), Pg2622-2631
- Baker, Stephen J.; Journal of Medicinal Chemistry 2006, 49(15), Pg4447-4450
- Austin, Peter William; WO 9533754 A1 1995 CAPLUS
| US5880188 * | 26 May 1995 | 9 Mar 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US6083903 * | 16 May 1995 | 4 Jul 2000 | Leukosite, Inc. | Boronic ester and acid compounds, synthesis and uses |
| WO2005013892A2 | 15 Jun 2004 | 17 Feb 2005 | Tsutomu Akama | Hydrolytically-resistant boron-containing therapeutics and methods of use |
| Reference | ||
|---|---|---|
| 1 | * | Austin et al., 1996, CAS: 124:234024. |
| 2 | * | fungicide: definition from Answre.com, 1998. |
| 3 | S. J. Baker, et al., “Progress on New Therapeutics for Fungal Nail Infections,“Annual Reports in Medicinal Chemistry, 40:323-335 (2005). | |
| 4 | Sudaxshina Murdan, “Drug Delivery to the Nail Following Topical Application,” International Journal of Pharmaceutics, 236:1-26 (2002). | |
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
//////////AN-2718, Borole, PHASE 2
B1(c2ccc(cc2CO1)Cl)O
FDA Says Chinese Pfizer Plant Hid Failures, Used Old Ingredients
DRUG REGULATORY AFFAIRS INTERNATIONAL


October 31, 2015 — 2:22 AM IST,
The Pfizer Ltd. research and development plant in Dalian, China.
Bernardo De Niz/Bloomberg
A Pfizer Inc. plant in China that was being inspected by Food and Drug Administration regulators in order to ship drugs to the U.S. kept a second set of quality and manufacturing records that didn’t match official ones, according to an FDA review of the facility.
read
![]()
/////
крисаборол , كريسابورول , Crisaborole, AN 2728
Crisaborole
Treatment for Inflammatory Skin Diseases, including Atopic Dermatitis and Psoriasis
C14H10BNO3, Average mass251.045 Da
4-[(1-Hydroxy-1,3-dihydro-2,1-benzoxaborol-5-yl)oxy]benzonitrile ,
4-((1-Hydroxy-1,3-dihydrobenzo(c)(1,2)oxaborol-6-yl)oxy)benzonitrile
CAS 906673-24-3, AN-2728
Benzonitrile, 4-[(1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy]-
1,3-Dihydro-1-hydroxy-5-(4-cyanophenoxy)-2,1-benzoxaborole
5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole
crisaborol, crisaborole, Crisaborole, crisaborolum
UNII-Q2R47HGR7P
крисаборол
كريسابورول
In phase 3 for treatment of mild to moderate atopic dermatitis……Anacor Pharmaceuticals, Inc.
Psoriasis is a chronic skin disorder caused by inflammatory cell infiltration into the dermis and epidermis, and is accompanied by keratinocyte hyperproliferation. Once triggered, a strong T-cell response is mounted, and a cascade of cytokine and chemokine production is induced.
Down-regulation of certain cytokines and chemokines is considered to be a good approach to treatment, and indeed, the biologics targeting TNF-α demonstrate the effectiveness of this approach.However, biologics have intrinsic challenges, such as limited administration route, side effects, quality control and production cost.
Small molecule approaches to treat psoriasis include systemic or topical steroids, cyclosporine, psoralen plus UVA (PUVA), retinoids, methotrexete, and vitamin D3 analogs.Atopic dermatitis is an allergic skin disorder, which is typically treated with topical steroids, antihistamines, and calcineurin inhibitors.
However, there is still a need for new treatment with improved safety profile. Recently phosphodiesterase 4 (PDE4) inhibitors have been in development for such skin diseases. CC-10004 is in development as an oral treatment for psoriasis and atopic dermatitis. AWD-12-281 was, until recently, in development for the topical treatment of atopic dermatitis. In addition, roflumilast is under Phase 1 development for both diseases.

Figure 1.
PDE4 inhibitors aiming at skin inflammatory diseases.

Anacor’s lead product candidate is crisaborole, an investigational non-steroidal topical PDE-4 inhibitor in development for the potential treatment of mild-to-moderate atopic dermatitis and psoriasis
crisaborole is an investigational topical antiinflammatory drug in phase III clinical development by Anacor Pharmaceuticals for the treatment of mild to moderate atopic dermatitis and in phase II clinical trials in mild to moderate psoriasis
A novel boron-containing small molecule, Crisaborole inhibits the release of pro-inflammatory cytokines including TNF-alpha, IL-12, and IL-23, known mediators of the inflammation associated with psoriasis.
Synthesis
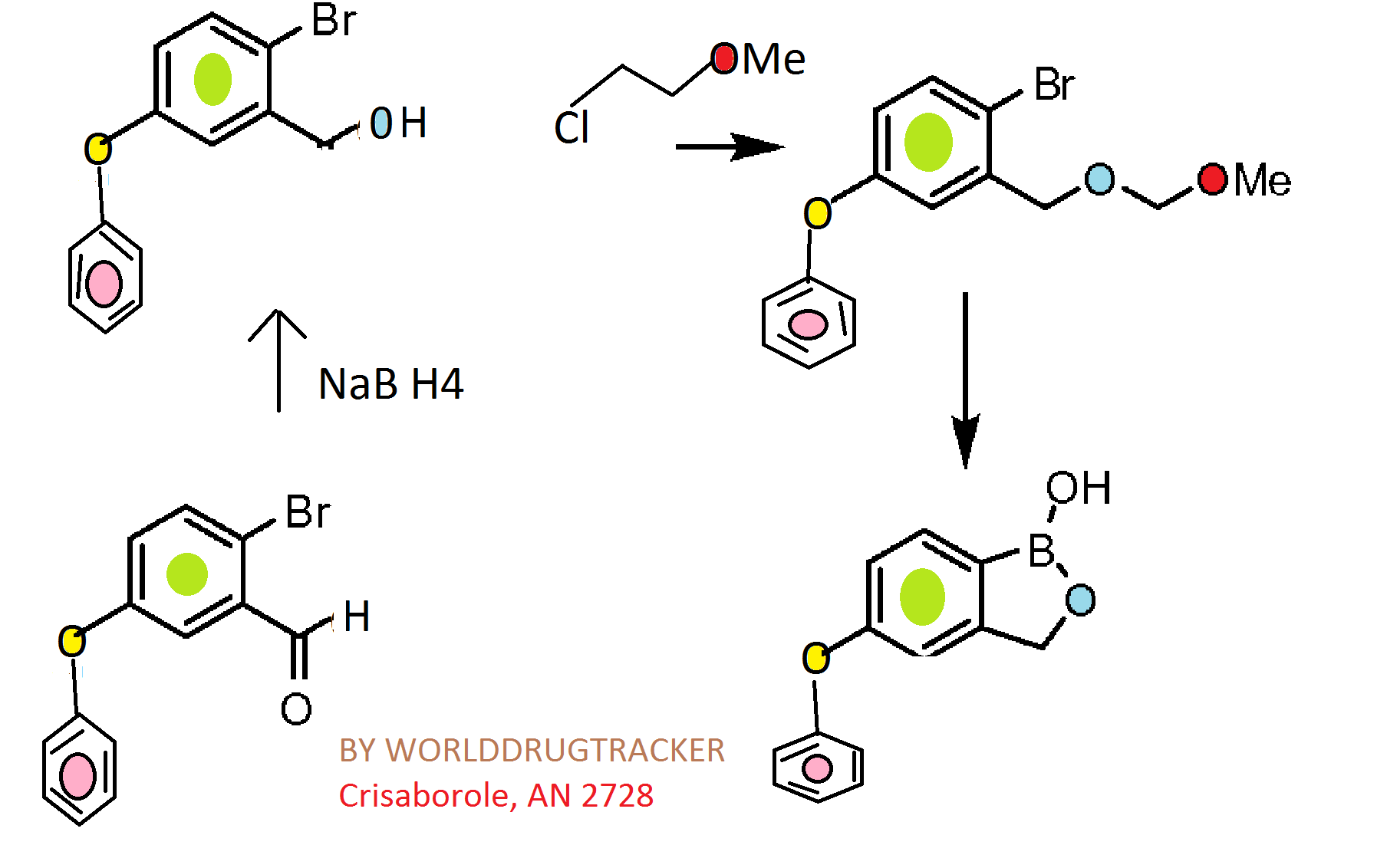
CKICK ON IMAGE FOR CLEAR VIEW
| Originator | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Therapeutic Claim | |||||||||||||||||||||
| Class | |||||||||||||||||||||
| Mechanism of action | |||||||||||||||||||||
| WHO ATC code(s) | |||||||||||||||||||||
| EPhMRA code(s) | |||||||||||||||||||||
| Clinical trial(s) |
|
PAPER
Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis
Bioorg Med Chem Lett 2009, 19(8): 2129
http://www.sciencedirect.com/science/article/pii/S0960894X09002996
- Anacor Pharmaceuticals, Inc., 1020 E. Meadow Circle, Palo Alto, CA 94303, USA
A series of phenoxy benzoxaboroles were synthesized and screened for their inhibitory activity against PDE4 and cytokine release. 5-(4-Cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2728) showed potent activity both in vitro and in vivo. This compound is now in clinical development for the topical treatment of psoriasis and being pursued for the topical treatment of atopic dermatitis


Scheme 1.
Reagents and conditions: (a) ethylene glycol, p-TsOH, toluene, reflux, 6 h (quant.); (b) K2CO3, DMF, 100 °C, overnight (82–96%); (c) 3 M HCl, THF, reflux, 2 h (80–100%); (d) NaBH4, MeOH, rt, 1 h (quant.); (e) 3,4-dihydro-2H-pyran, camphorsulfonic acid, CH2Cl2, rt, 2 h (quant.); (f) (i-PrO)3B, n-BuLi, THF, −78 °C to rt, 3 h; (g) 6 M HCl, THF, rt, 3 h (37–44%); (h) 6 M NaOH, MeOH, 1,4-dioxane, reflux, 6 days (79%); (i) diethylamine (for 5f) or morpholine (for 5g), EDCI, HOBt, DMAP, DMF, rt, overnight (41–70%).
PATENT
http://www.google.co.in/patents/WO2006089067A2?cl=en
4.2. q 5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole (C17) [0264] 1H-NMR (300 MHz,
δ ppm 4.95 (s, 2H), 7.08 (dd, J= 7.9, 2.1 Hz, IH), 7.14 (d, J= 8.8 Hz, IH), 7.15 (d, J= 2.1 Hz, IH), 7.78 (d, J= 7.9 Hz, IH), 7.85 (d, J= 9.1 Hz, 2H), 9.22 (s, IH).
PATENT
EXAMPLE 15
http://www.google.com/patents/WO2007095638A2?cl=en
4-(4-Cvanophenoxy)phenylboronic acid (C97)

(a) (4-cyanophenyl) (4-bromophenyl) ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 1000C for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with IN NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4- bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-de): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p.l94-198°C. MS: m/z = 239 (M+), 240 (M+ 1) (ESI+) and m/z = 238 (M-I) (ESI-). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6 + D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.
FURTHER METHOD

2-Bromo-5-(4-cvanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J= 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
PATENT
http://www.google.im/patents/EP1976536A2?cl=en
2.2.a 2-Bromo-5-(4-cyanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J- 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
2.2.b 2-Bromo-4-(4-cyanophenoxγ)benzyl Alcohol
1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.58 (d, IH), 7.39 (d, IH), 7.18 (dd, IH), 7.11- (d, 2H), 5.48 (t, IH) and 4.50 (d, 2H) ppm.
2.2.c 5- (4-Cyanophenoxy) -1 -Indanol
M.p.50-53°C. MS (ESI+): m/z = 252 (M+l). HPLC: 99.7% purity at 254 nm and 99.0% at 220 nm. 1H NMR (300 MHz, DMSOd6): δ 7.80 (d, 2H), 7.37 (d, IH), 7.04 (d, 2H), 6.98-6.93 (m, 2H), 5.27 (d, IH)5 5.03 (q, IH), 2.95-2.85 (m, IH), 2.75-2.64 (m, IH), 2.39-2.29 (m, IH) and 1.85-1.74 (m, IH) ppm.
2.2. d 2-Bromo-5-(tert-butyldimethylsiloxy)benzyl Alcohol [0429] 1H-NMR (300 MHz, CDCl3) δ (ppm) 0.20 (s, 6H), 0.98 (s, 9H), 4.67 (br s,lH), 6.65 (dd, J= 8.2, 2.6 Hz, IH), 6.98 (d, J= 2.9 Hz, IH), 7.36 (d, J= 8.8 Hz, IH).
3.2.k 2-Bromo-5-(2-cyanophenoχy)-l-(methoxymethoxymethyl)benzene [0443] 1H-NMR (300 MHz, CDCl3) δ (ppm) 3.41 (s, 3H), 4.64 (s, 2H), 4.76 (s, 2H), 6.8-6.9 (m, 2H), 7.16 (td, J= 7.6, 0.9 Hz, IH), 7.28 (d, J= 2.9 Hz, IH), 7.49 (ddd, J= 8.8, 7.6, 1.8 Hz, IH)5 7.56 (d, J= 8.5 Hz, IH), 7.67 (dd, J= 7.9, 1.8 Hz, IH).
EXAMPLE 32
Alternative Preparation of C17 -Intermediate

The procedure described in Example II I was followed for 1H NMR characterization of the current alcohol-borate intermediate. 1H NMR determination indicated there were 72.7 mol% of the desired alcohol-borate intermediate [2-bromo- 5-(4-cyanophenoxy)benzyl] diisopropyl borate, 20.7 mol% of an unknown intermediate and 6.5 mol% of unreacted alcohol. 1H NMR (CDCl3, 300 MHz) of [2- bromo-5-(4-cyanophenoxy)benzyl] diisopropyl borate: δ= 7.61 (d, J= 9.0 Hz, 2H), 7.52 (d, J= 8.4 Hz, IH), 7.15 (d, J= 3.0 Hz, IH), 7.03 (d, J= 8.7 Hz, 2H), 6.84 (dd, J= 8.7 Hz, J= 3.0 Hz, IH), 4.85 (s, 2H), 4.35 (septet, J= 6.1 Hz, 2H), 1.11 (d, J= 6.1 Hz, 12H) ppm.
PATENT
http://www.google.com/patents/US20090291917
- Example 154-(4-Cyanophenoxy)phenylboronic acid (C97)
-

-
(a) (4-cyanophenyl)(4-bromophenyl)ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 100° C. for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with 1N NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4-bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
-
(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p. 194-198° C. MS: m/z=239 (M+), 240 (M+1) (ESI+) and m/z=238 (M−1) (ESI−). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6+D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.

see
http://www.google.co.in/patents/WO2006089067A2?cl=en
see
http://www.google.com/patents/US20090291917
| US5688928 * | Jun 7, 1995 | Nov 18, 1997 | Prolinx, Inc. | Phenylboronic acid complexing reagents derived from aminosalicylic acid |
| US5880188 * | May 26, 1995 | Mar 9, 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US5962498 * | Dec 2, 1994 | Oct 5, 1999 | Procyon Pharmaceuticals, Inc. | Protein kinase C modulators. C. indolactam structural-types with anti-inflammatory activity |
| US6369098 * | Oct 4, 2000 | Apr 9, 2002 | Bethesda Pharmaceuticals, Inc. | Dithiolane derivatives |
| US20030032673 * | Jul 19, 2002 | Feb 13, 2003 | Isis Innovation Limited | Therapeutic strategies for prevention and treatment of alzheimer’s disease |
| US20050239170 * | Jul 16, 2001 | Oct 27, 2005 | Hedley Mary L | Alpha-MSH related compounds and methods of use |
| US20060009386 * | May 12, 2005 | Jan 12, 2006 | The Brigham And Women’s Hospital, Inc. | Use of gelsolin to treat infections |
|
Methods of treating anti-inflammatory conditions through the use of boron- containing small molecules are disclosed.
|
|
… Francisco, CA Mar. 6-10, 2009. 6, “AN2728 … Francisco, CA Mar. 6-10, 2009. 7 , “AN2728 … Kyoto, Japan, May 14-18, 2008. 10, “AN2728 …
|
|
AN2728, 5-(4-cyanophenoxy)-2,3- dihydro-1-hydroxy-2,1- …. UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
|
… Dermatology Annual Meeting, San Francisco, CA Mar. 6-10, 2009. 6, “AN2728 … 7, “AN2728 … Francisco, CA May 6-10, 2009. 10, “AN2728 …
|
|
… from the group consisting of AN-2728, AN-2898, CBS- 3595, apremilast, ELB- 353, KF-66490, K-34, LAS-37779, IBFB-211913, AWD-12-281, …
|
|
“AN2728” is the compound 4-(l-hydroxy-l,3-dihydro-2 … GSK256066, oglemilast, tetomilast, apremilast, AN2728, Compound A, Compound B, …
|
|
AN2728, 5-(4-cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1- …. UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
|
85.用于治疗疼痛的UK-500,001。 85. for the treatment of pain UK-500,001. 86.用 于治疗疼痛的AN2728。 86. for the treatment of pain AN2728.
|
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
///////////crisaborole, AN 2728, PHASE 3, Anti-inflammatory, Phosphodiesterase, Oxaborole, Psoriasis, Atopic dermatitis, borole
GSK 2251052, Epetraborole, AN3365

(S)-3-(Aminomethyl)-7-(3-hydroxypropoxy)-1-hydroxy-1,3-dihydro- 2,1-benzoxaborole (GSK2251052) is a novel boron-containing antibiotic that inhibits bacterial leucyl tRNA synthetase, and that has been in development for the treatment of serious Gramnegative infections
(S)-3-aminomethylbenzoxaborole; ABX; AN-3365; GSK ‘052; GSK-052; GSK-2251052, GSK2251052, Epetraborole
[(S)-3-(aminomethyl)-7-(3-hydroxypropoxy)-1-hydroxy- 1,3-dihydro-2,1-benzoxaborole hydrochloride],
(S)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride
1-Propanol, 3-(((3S)-3-(aminomethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaborol-7-yl)oxy)-
AN3365,
MW 237.0614,
cas 1093643-37-8
UNII: 6MC93Z2DF9
Anacor Pharmaceuticals, Inc., INNOVATOR
Glaxosmithkline Llc DEVELOPER
Biomedical Advanced Research and Development Authority (BARDA)
GSK 2251052 • $38.5M over the 1st two years; up to $94M……..http://www.idsociety.org/uploadedFiles/IDSA/Policy_and_Advocacy/Current_Topics_and_Issues/Advancing_Product_Research_and_Development/Bad_Bugs_No_Drugs/Press_Releases/FIS%20Slides.pdf

Originally came from Anacor about ten years ago, then was picked up by GlaxoSmithKline, and it’s an oxaborole heterocycle that inhibits leucyl tRNA synthetase
GlaxoSmithKline recently announced a contract with the Biomedical Advanced Research and Development
Authority (BARDA), a US government preparedness organization , The award guarantees GSK $38.5 million over 2 years towards development of GSK2251052, a molecule co-developed with Anacor Pharma a few years back, as a counter-bioterrorism agent. The full funding amount may later increase to $94 million, pending BARDA’s future option.
The goal here is to develop “GSK ‘052”, as it’s nicknamed among med-chemists, into a new antibiotic against especially vicious and virulent Gram negative bacteria, such as the classic foes plague (Yersinia pestis) or anthrax (Bacillus anthracis).
Look closely at GSK’052 (shown above): that’s a boron heterocycle there! Anacor, a company specializing in boron based lead compounds, first partnered with GSK in 2007 to develop novel benzoxaborole scaffolds. This isn’t the first company to try the boron approach to target proteins; Myogenics (which, after several acquisitions, became Millennium Pharma) first synthesizedbortezomib, a boronic acid peptide, in 1995.


Stephen Benkovic (a former Anacor scientific board member) and coworkers at Penn State first discovered Anacor’s early boron lead molecules in 2001, with a screening assay. The molecules bust bacteria by inhibiting leucyl-tRNA synthetase, an enzyme that helps bacterial cells to correctly tag tRNA with the amino acid leucine. Compounds with cyclic boronic acids “stick” to one end of the tRNA, rendering the tRNA unable to cycle through the enzyme’s editing domain. As a result, mislabeled tRNAs pile up, eventually killing the bacterial cell.
Inhibition of synthetase function turns out to be a useful mechanism to conquer all sorts of diseases. Similar benzoxaborozoles to GSK ‘052 show activity against sleeping sickness (see Trypanosoma post by fellow Haystack contributor Aaron Rowe), malaria, and various fungi.
Boron-containing molecules such as benzoxaboroles that are useful as antimicrobials have been described previously, see e.g. “Benzoxaboroles – Old compounds with new applications” Adamczyk-Wozniak, A. et al., Journal of Organometallic Chemistry Volume 694, Issue 22, 15 October 2009, Pages 3533-3541 , and U.S. Pat. Pubs. US20060234981 and US20070155699. Generally speaking, a benzoxaborole has the following structure and substituent numbering system:

Certain benzoxaboroles which are monosubstituted at the 3-, 6-, or 7-position, or disubstituted at the 3-/6- or 3-/7- positions are surprisingly effective antibacterials, and they have been found to bind to the editing domain of LeuRS in association with tRNALeu Such compounds have been described in US7, 816,344. Using combinations of certain substituted benzoxaboroles with norvaline and/or other amino acid analogs and their salts to: (a) reduce the rate of resistance that develops; and/or (b) decrease the frequency of resistance that develops; and/or (c) suppress the emergence of resistance, in bacteria exposed to compounds
Epetraborole R-mandelate
1234563-15-5
Epetraborole hydrochloride
1234563-16-6

Anacor Pharmaceuticals is out to change that. The Palo Alto, Calif.-based biotechnology company is developing a family of boron-containing small-molecule drugs. And with the assistance of Naeja Pharmaceutical, a Canadian contract research organization, Anacor has licensed one of those molecules to GlaxoSmithKline and taken another one into Phase III clinical trials.
Anacor was founded in 2002 to develop technology created by Lucy Shapiro, a Stanford University bacterial geneticist, and Stephen J. Benkovic, a Pennsylvania State University organic chemist. Through a long-standing scientific collaboration, the two researchers had discovered boron-containing compounds that inhibited specific bacterial targets.
 Lucy Shapiro is a Professor in the Department of Developmental Biology at Stanford University School of Medicine where she holds the Virginia and D. K. Ludwig Chair in Cancer Research and is the Director of the Beckman Center for Molecular and Genetic Medicine. She is a member of the Scientific Advisory Board of Ludwig Institute for Cancer Research and is a member of the Board of Directors of Pacific Biosciences, Inc. She founded the anti-infectives discovery company, Anacor Pharmaceuticals, and is a member of the Anacor Board of Directors. Professor Shapiro has been the recipient of multiple honors, including: election to the American Academy of Arts and Sciences, the US National Academy of Sciences, the US Institute of Medicine, the American Academy of Microbiology, and the American Philosophical Society. She was awarded the FASEB Excellence in Science Award, the 2005 Selman Waksman Award from the National Academy of Sciences, the Canadian International 2009 Gairdner Award, the 2009 John Scott Award, the 2010 Abbott Lifetime Achievement Award, the 2012 Horwitz Prize and President Obama awarded her the National Medal of Science in 2012. Her studies of the control of the bacterial cell cycle and the establishment of cell fate has yielded valuable paradigms for understanding the bacterial cell as an integrated system in which the transcriptional circuitry is interwoven with the three-dimensional deployment of key regulatory and morphological proteins, adding a spatial dimension to the systems biology of regulatory networks.
Lucy Shapiro is a Professor in the Department of Developmental Biology at Stanford University School of Medicine where she holds the Virginia and D. K. Ludwig Chair in Cancer Research and is the Director of the Beckman Center for Molecular and Genetic Medicine. She is a member of the Scientific Advisory Board of Ludwig Institute for Cancer Research and is a member of the Board of Directors of Pacific Biosciences, Inc. She founded the anti-infectives discovery company, Anacor Pharmaceuticals, and is a member of the Anacor Board of Directors. Professor Shapiro has been the recipient of multiple honors, including: election to the American Academy of Arts and Sciences, the US National Academy of Sciences, the US Institute of Medicine, the American Academy of Microbiology, and the American Philosophical Society. She was awarded the FASEB Excellence in Science Award, the 2005 Selman Waksman Award from the National Academy of Sciences, the Canadian International 2009 Gairdner Award, the 2009 John Scott Award, the 2010 Abbott Lifetime Achievement Award, the 2012 Horwitz Prize and President Obama awarded her the National Medal of Science in 2012. Her studies of the control of the bacterial cell cycle and the establishment of cell fate has yielded valuable paradigms for understanding the bacterial cell as an integrated system in which the transcriptional circuitry is interwoven with the three-dimensional deployment of key regulatory and morphological proteins, adding a spatial dimension to the systems biology of regulatory networks.
Stephen J. Benkovic
- Evan Pugh University Professor and Eberly Chair in Chemistry
Office:
414 Wartik Laboratory
University Park, PA 16802
Email: sjb1@psu.edu
(814) 865-2882
http://chem.psu.edu/directory/sjb1
Websites,Benkovic Research Group
Naeja was a three-year-old contract research firm run by Ronald Micetich and his son Christopher Micetich. Based in Edmonton, Alberta, the firm is staffed by chemists and biologists from a variety of nations who have found Canada welcoming to highly educated immigrants.
GSK. Last July, the British firm paid Anacor $15 million and exercised its option to take over development of AN3365. David J. Payne, vice president of GSK’s antibacterial drug discovery unit, lauded the compound, now renamed GSK2251052, as having “the potential to be the first new-class antibacterial to treat serious hospital gram-negative infections in 30 years.” GSK chemists have since developed a stereospecific synthesis for commercial-scale production
david.j.payne@gsk.com
David J. Payne, vice president of GSK’s antibacterial drug discovery unit
David J Payne Dr Payne holds a BSc in Biochemistry from Brunel University, UK, and a PhD and DSc from The Medical School, University of Edinburgh, UK. Dr Payne has 20 years of experience in antibacterial drug discovery and is currently Vice President and Head of the Antibacterial Discovery Performance Unit (DPU) within the Infectious Diseases Centre of Excellence in Drug Discovery (ID CEDD) where he is responsible for GSK’s antibacterial research effort from discovery to clinical proof of concept (up to Phase II clinical trials). At GSK, Dr Payne has played a leading role in redesigning the strategy for antibacterial research and has helped create long-term alliances with innovative biotechnology companies which has expanded GSK’s discovery pipeline. Furthermore, he has created industry-leading partnerships with the Wellcome Trust and the Defense Threat Reduction Agency (US Department of Defense) to accelerate GSK’s antibacterial programmes. To date, Dr Payne has been involved with the progression of a broad diversity of novel mechanism antibacterial agents into development. Dr Payne has authored more than 190 papers and conference presentations.
PATENT
http://www.google.com/patents/US20090227541
- General procedure for Chiral Synthesis of 3-aminomethylbenzoxaboroles
-




- 4-(3-Aminomethyl-1-hydroxy-1,3-dihydro-benzo[c][1,2]oxaborol-7-yloxy)-butyramide acetate salt (A5)

4-[2-(5,5-Dimethyl-[1,3,2]dioxaborinan-2-yl)-3-formyl-phenoxy]-butyric acid ethyl ester
-

-
A mixture of 4-(2-bromo-3-formyl-phenoxy)-butyric acid ethyl ester (5.50 g, 17.5 mmol), bis(neopentyl glucolato)diboron (6.80 g, 30.1 mmol), PdCl2(dppf).CH2Cl2 (1.30 g, 1.79 mmol), and KOAc (5.30 g, 54.1 mmol) in anhydrous THF (600 mL) was heated with stirring at 80° C. (bath temp) O/N under an atmosphere of N2. The mixture was then filtered through Celite and concentrated in vacuo to approximately one quarter of the original volume. The resulting precipitate was isolated by filtration. The precipitate was washed with THF and EtOAc and the combined filtrate was concentrated in vacuo to give an oily residue which was used directly in the next reaction without further purification.
-
1H NMR (400 MHz, CDCl3) δ (ppm): 9.95 (s, 1H), 7.47-7.39 (m, 2H), 7.09-7.07 (m, 1H), 4.14 (q, J=7.2 Hz, 2H), 4.09-4.01 (m, 2H), 3.83 (s, 3H), 3.66 (s, 3H), 2.53 (t, J=8.0 Hz, 2H), 2.19-2.07 (m, 2H), 1.32-1.22 (m, 3H), 0.98 (s, 6H).

4-(1-Hydroxy-3-nitromethyl-1,3-dihydro-benzo[c][1,2]oxaborol-7-yloxy)-butyric acid ethyl ester
-

-
MeNO2 (1.3 mL, 25 mmol) was added dropwise to a stirred solution of crude 4-[2-(5,5-dimethyl-[1,3,2]dioxaborinan-2-yl)-3-formyl-phenoxy]-butyric acid methyl ester (9.4 g), NaOH (1.0 g, 25 mmol) and H2O (35 mL) in MeCN (90 mL) at rt. The mixture was stirred at rt O/N and then acidified (pH 2) using 4 M HCl. The THF was removed in vacuo and the aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash chromatography (10% to 30% EtOAc in hexane) to give the title compound as a yellow oil: yield 2.52 g (45% over 2 steps).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.04 (s, 1H), 7.46-7.42 (m, 1H), 7.07-7.05 (m, 1H), 6.88-6.86 (m, 1H), 5.87 (d, J=8.2 Hz, 1H), 5.69 (dd, J=9.2, 2.5 Hz, 1H), 5.29 (dd, J=13.3, 2.7 Hz, 1H), 4.14-3.94 (m, 5H), 2.55-2.44 (m, 2H), 2.02-1.88 (m, 2H), 1.16 (t, J=7.2 Hz, 3H); MS (ESI) m/z=322 (M−1, negative).

4-(1-Hydroxy-3-nitromethyl-1,3-dihydro-benzo[c][1,2]oxaborol-7-yloxy)-butyric acid
-

-
A mixture of 4-(1-hydroxy-3-nitromethyl-1,3-dihydro-benzo[c][1,2]oxaborol-7-yloxy)-butyric acid ethyl ester (2.51 g, 7.78 mmol), 10% NaOH (17 mL), and 1:1 MeOH/H2O (70 mL) was stirred at rt for 5 h. The MeOH was removed in vacuo and the remaining aqueous layer was acidified to pH 1 using 2 M HCl. The aqueous layer was then extracted with EtOAc. The organic fractions were washed with brine, dried (MgSO4), and concentrated in vacuo to give the title compound as a pale yellow foam: yield 1.85 g (81%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.08 (bs, 1H), 9.01 (bs, 1H), 7.46-7.41 (m, 1H), 7.06-7.04 (m, 1H) 6.89-6.87 (m, 1H), 5.70 (dd, J=7.0, 2.3 Hz, 1H), 5.30 (dd, J=13.3, 2.3 Hz, 1H), 4.55 (dd, J=13.6, 4.2 Hz, 1H), 4.03 (t, J=6.6 Hz, 2H), 2.40 (t, J=7.5 Hz, 2H), 1.95-1.89 (m, 2H); MS (ESI) m/z=296 (M+1, positive).

- 3-Aminomethyl-6-(2-hydroxy-propoxy)-3H-Benzo[c][1,2]oxaborol-1-ol acetate salt (A31)

4-(2-Benzyloxy propoxy-2-bromobenzaldehyde
-

-
A mixture of 2-bromo-4-fluoro-benzaldehyde (30.0 g, 148 mmol), Na2CO3 (78.31 g, 738.8 mmol) and 2-benzyloxy propanol (24.56 g, 147.8 mmol) in anhydrous DMSO (300 mL) was heated with stirring at 130° C. (bath temp) for 72 h under N2. The reaction mixture was cooled to rt and diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O then brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash chromatography (hexane to 30% EtOAc in hexane) to give the title compound: yield 3.84 g (7%).
-
1H NMR (400 MHz, CDCl3) δ (ppm): 10.22 (s, 1H), 7.88 (d, J=8.6 Hz, 1H), 7.42-7.20 (m, 5H), 7.12 (d, J=2.3 Hz, 1H), 6.92 (dd, J=8.8, 2.2 Hz, 1H), 4.52 (s, 2H), 4.16 (t, J=6.2 Hz, 2H), 3.65 (t, J=6.1 Hz, 2H), 2.10 (q, J=6.2 Hz, 2H).

4-(2-Benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[/, 3, 2]dioxaborolan-2-yl)-benzaldehyde
-

-
General procedure 5: 4-(2-benzyloxy propoxy-2-bromobenzaldehyde (4.84 g, 13.9 mmol), B2pin2 (5.27 g, 20.8 mmol), KOAc (4.08 g, 41.6 mmol), PdCl2(dppf).CH2Cl2 (811 mg, 8 mol %), and 1,4-dioxane (50 mL). Purification: Biotage (gradient from 2% EtOAc/hexane to 20% EtOAc/hexane): yield 4.0 g (70%).
-
1H NMR (400 MHz, CDCl3) δ (ppm): 10.36 (s, 1H), 7.93 (d, J=8.6 Hz, 1H), 7.43-7.14 (m, 6H), 7.01 (dd, J=8.6, 2.7 Hz, 1H), 4.53 (s, 2H), 4.18 (t, J=6.2 Hz, 2H), 3.66 (t, J=6.1 Hz, 2H), 2.11 (q, J=6.1 Hz, 2H), 1.40 (s, 12H).

6-(2-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol
-

-
General procedure 8: 4-(2-benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde (3.0 g, 7.6 mmol), MeNO2 (924 mg, 15.1 mmol), NaOH (605 mg, 15.1 mmol), and H2O (10 mL). Purification: flash chromatography (10% EtOAc/hexane to 40% EtOAc): yield 820 mg (30%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.46 (bs, 1H), 7.45 (d, J=8.2 Hz, 1H), 7.41-7.18 (m, 6H), 7.09 (dd, J=8.6, 2.3 Hz, 1H), 5.71 (dd, J=9.2, 2.5 Hz, 1H), 5.31 (dd, J=13.3, 2.7 Hz, 1H), 4.58-4.40 (m, 3H), 4.08 (t, J=6.2 Hz, 2H), 3.60 (t, J=6.2 Hz, 2H), 2.08-1.94 (m, 2H).

3-Aminomethyl-6-(2-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol acetate salt (A31)
-

-
General procedure 13: 6-(2-benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (820 mg, 2.29 mmol), 20% Pd(OH)2 (850 mg, 1 equiv w/w), and AcOH (40 mL). Purification: preparative HPLC: yield 120 mg (22%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.32 (d, J=8.2 Hz, 1H), 7.22 (s, 1H), 7.02 (d, J=7.8 Hz, 1H), 4.98 (bs, 1H), 4.04 (t, J=6.2 Hz, 2H), 3.56 (t, J=6.2 Hz, 2H), 3.03-2.85 (m, 1H), 2.61 (dd, J=12.9, 7.0 Hz, 1H), 1.89 (s, 3H), 1.97-1.67 (m, 2H); MS (ESI): m/z=238 (M+1, positive); HPLC purity: 97.44% (MaxPlot 200-400 nm), 97.77% (220 nm).
-
- 7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (A47)
3-(3-Benzyloxy-propoxy)-2-hydroxy-benzaldehyde
-

-
NaH (2.95 g, 72.4 mmol) was added to an ice-cold solution of 2,3-dihydroxybenzaldehyde (5.0 g, 36 mmol) in anhydrous DMSO (45 mL). Benzyl-3-bromopropyl ether (6.45 mL, 36.2 mmol) was then added and the mixture was stirred at rt for 12 h. The mixture was neutralized using 1 N HCl and then extracted with EtOAc. The organic fraction was washed with H2O and concentrated in vacuo. The residue was purified by flash chromatography (8:2 hexane/EtOAc) to give the title compound as a brown oil: yield 8.40 g (81%).
-
[0891]1H NMR (400 MHz, CDCl3) δ (ppm): 9.93 (s, 1H), 7.36-7.23 (m, 6H), 7.20-7.16 (m, 2H), 6.98-6.91 (m, 1H), 4.53 (s, 2H), 4.19 (t, J=6.2 Hz, 2H), 3.70 (t, J=6.1 Hz, 2H), 2.19-2.16 (m, 2H).
3-(3-Benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[/, 3, 2]dioxaborolan-2-yl)-benzaldehyde
-

-
[0893]General procedure 6: 3-(3-benzyloxy-propoxy)-2-hydroxy-benzaldehyde (7.6 g, 26 mmol), pyridine (3.42 mL, 42.5 mmol), Tf2O (4.60 mL, 27.9 mmol), and CH2Cl2 (200 mL): yield 8.60 g (77%).
-
[0894]1H NMR (400 MHz, CDCl3) δ (ppm): 10.23 (s, 1H), 7.54-7.47 (m, 1H), 7.43 (t, J=8.0 Hz, 1H), 7.36-7.22 (m, 6H), 4.52 (s, 2H), 4.23 (t, J=6.3 Hz, 2H), 3.71 (t, J=6.1 Hz, 2H), 2.21-2.17 (m, 2H).
-
[0895]General procedure 5: trifluoro-methanesulfonic acid 2-(3-benzyloxy-propoxy)-6-formyl-phenyl ester (8.0 g, 19 mmol), B2pin2 (9.71 g, 38.2 mmol), KOAc (5.71 g, 57.4 mmol), PdCl2(dppf).CH2Cl2 (1.39 g, 1.89 mmol), and anhydrous dioxane (160 mL). Purification: flash chromatography (9:1 hexane/EtOAc): yield 4.80 g (43%)-some pinacol contamination, used without further purification.
-
[0896]1H NMR (400 MHz, CDCl3) δ (ppm): 9.93 (s, 1H), 7.46 (t, J=7.8 Hz, 1H), 7.41-7.36 (m, 1H), 7.35-7.24 (m, 5H), 7.08 (d, J=7.8 Hz, 1H), 4.50 (s, 2H), 4.10 (t, J=6.3 Hz, 2H), 3.67 (t, J=6.3 Hz, 2H), 2.11 (quin, J=6.2 Hz, 2H), 1.43 (s, 12H).
7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (A47)
-

-
General procedure 8: 3-(3-benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde (36 g, 91 mmol), MeNO2 (16.6 g, 273 mmol), NaOH (3.64 g, 83 mmol), H2O (180 mL), and THF (50 mL). Purification: flash chromatography (1:1 hexane/EtOAc). A47 was isolated as a light yellow oil: yield 15.9 g (50%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.05 (s, 1H), 7.44 (t, J=7.8 Hz, 1H), 7.35-7.20 (m, 5H), 7.06 (d, J=7.4 Hz, 1H), 6.88 (d, J=8.2 Hz, 1H), 5.70 (dd, J=9.4, 2.3 Hz, 1H), 5.29 (dd, J=13.7, 2.7 Hz, 1H), 4.53 (dd, J=13.3, 9.4 Hz, 1H), 4.45 (s, 2H), 4.11 (t, J=6.1 Hz, 2H), 3.60 (t, J=6.3 Hz, 2H), 2.04-1.91 (m, 2H); MS (ESI): m/z=356 (M−1, negative); HPLC purity: 99.35% (MaxPlot 200-400 nm), 97.32% (220 nm).
Alternative synthesis of 3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride (A46)
-

-
General procedure 13: A47 (0.50 g, 1.4 mmol), 20% Pd(OH)2/C (0.5 g, 1:1 w/w), AcOH (20 mL), and H2O (0.24 mL). The filtrate was concentrated and treated with 4 N HCl to give the title compound as a colorless solid: yield 0.22 g (47%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.42 (t, J=7.8 Hz, 1H), 6.97-6.90 (m, 1H), 6.86 (d, J=8.2 Hz, 1H), 5.20 (dd, J=9.2, 2.5 Hz, 1H), 4.02 (t, J=6.2 Hz, 2H), 3.54 (t, J=6.2 Hz, 2H), 3.40 (dd, J=13.3, 2.7 Hz, 1H), 2.68 (dd, J=13.1, 9.2 Hz, 1H), 1.88-1.78 (m, 2H); MS (ESI): m/z=238 (M+1, positive).
-






- 3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride (A46)

Synthesis of 3-(3-Benzyloxy-propoxy)-2-bromo-benzaldehyde (C)
-

-
To a 5° C. solution of compound A (15.0 g, 0.075 mol), B (12.0 ml, 0.075 mol) and triphenylphosphine (19.6 g, 0.075 mol) in 200 ml of anhydrous THF was added DIAD (14.8 ml, 0.075 mol) drop by drop over a period of 15 minutes. The resulting solution was warmed to room temperature over a period of 5 h and the solvent was evaporated in vacuo. The residue was dissolved in 150 ml of EtOAc and the organic layer washed with water, brine and dried over Na2SO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography (gradient of hexane to 5% EtOAc/hexane) generating 13.0 g (50% yield) of C [3-(3-benzyloxy-propoxy)-2-bromo-benzaldehyde].
-
1H NMR (400 MHz, DMSO-d6) δ (ppm) 10.41 (s, 1H), 7.49 (d, J=7.2 Hz, 1H), 7.32-7.25 (m, 6H), 7.08 (d, J=8.0 Hz, 1H), 4.54 (s, 2H), 4.16 (t, J=6.0 Hz, 2H), 3.74 (t, J=5.8 Hz, 2H), 2.19-2.14 (m, 2H).

3-(3-Benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde (D)
-

-
Compound C (8.9 g, 0.025 mol), KOAc (7.5 g, 0.076 mol), and bis(pinacolato)diboron (12.9 g, 0.051 mol) were dissolved in 50 ml of dry DMF and degassed for 30 minutes. To this was added PdCl2(dppf).DCM (0.56 g, 0.76 mmol) and the contents were again degassed for 10 minutes and then heated to 90° C. for 4 h. An additional quantity of PdCl2(dppf).DCM (0.2 g, 0.27 mmol) was added and heating was continued for an additional 2 h. The reaction was cooled to RT, filtered through celite and the solvent evaporated in vacuo. The residue was dissolved in DCM, washed with brine and the organic layer dried over Na2SO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography (gradient of hexane to 5% EtOAc/hexane) provided 5.4 g (53% yield) of D [3-(3-benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde].
-
1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.91 (s, 1H), 7.43 (t, J=7.8 Hz, 1H), 7.36 (d, J=7.2 Hz, 1H), 7.32-7.27 (m, 5H), 7.06 (d, J=8.4 Hz, 1H), 4.49 (s, 2H), 4.08 (t, J=6.0 Hz, 2H), 3.67 (t, J=6.2 Hz, 2H), 2.11-2.08 (m, 2H), 1.44 (s, 12H). ESI+MS m/z, 397 (M+H)+.

7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (E)
-

-
To an ice cold solution of NaOH (0.68 g, 0.017 mol) in 10 ml of water was added a solution of compound D (6.8 g, 0.017 mol) dissolved in 5 ml of THF. After 15 minutes, nitromethane (0.93 ml, 0.017 mol) was added drop by drop and the content stirred at RT overnight. The THF was evaporated under reduced pressure and the contents acidified to pH-3 with 2N HCl. The aqueous layer was extracted with EtOAc several times, and the combined ethyl acetate layer was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography (gradient of 10% EtOAc/hexane to 30% EtOAc/hexane) provided 3.7 g (55% yield) of E [7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol] 3.7 g.
-
1H NMR (400 MHz, DMSO-d6+D2O (0.01 ml)) δ (ppm) 7.49 (t, J=7.8 Hz, 1H), 7.34-7.25 (m, 5H), 7.08 (d, J=7.6 Hz, 1H), 6.92 (d, J=8.0 Hz, 1H), 5.71 (d, J=6.4 Hz, 1H), 5.23 (dd, J=13.2, 2.4 Hz, 1H), 4.58-4.53 (m, 1H), 4.47 (s, 2H), 4.12 (t, J=6.2 Hz, 2H), 3.63 (t, J=6.0 Hz, 2H), 2.04-2.00 (m, 2H). ESI-MS m/z, 356 [M−H]−. HPLC purity: 97.12% (MaxPlot 200-400 nm).

- 3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol (A46)
-

-
Compound E (6.0 g, 0.016 mol) was dissolved in 50 ml of glacial acetic acid and to it was added Pd(OH)2 on Carbon (20% metal content, 50% weight-wet) (5.2 g) and the content set for hydrogenation in a Parr shaker at 45 psi for 2 h. The reaction was checked for completion and the contents were filtered through Celite. The solvent was evaporated under reduced pressure at ambient temperature to yield a gummy material. To this three times was added 15 ml of dry toluene and evaporated yielding a fluffy solid. Purification was accomplished by preparative HPLC (C18 column, using acetonitrile and 0.1% AcOH/water solution) provided 1.5 g (45% yield) of compound A46 [3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol] with 0.33 mol % acetic acid (by HNMR).
-
1H NMR (400 MHz, DMSO-d6+D2O (0.01 ml)) δ (ppm) 7.52 (t, J=7.8 Hz, 1H), 7.05 (d, J=7.2 Hz, 1H), 6.95 (d, J=8.4 Hz, 1H), 5.29 (dd, J=9.2, 2.4, 1H), 4.12 (t, J=6.2 Hz, 2H), 3.62 (t, J=6.2 Hz, 2H), 3.48 (dd, J=13.2, 2.8 Hz, 1H), 2.80-2.74 (m, 1H), 1.92 (t, J=6.2 Hz, 2H). ESI+MS m/z, 238 [M+H]+. HPLC purity: 95.67% (MaxPlot 200-400 nm) and 96.22% (220 single wavelength).

- (S)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride (A49)
-

-
A solution of 1 (160 g, 962.58 mmol) and triphenylphosphine (277.72 g, 1.1 eq, 1058.83 mmol) was dissolved in dichloromethane (800 mL) and cooled to 0° C. (ice/water). A solution of carbon tetrabromide (351.16 g, 1.1 eq, 1058.83 mmol) in dichloromethane (200 mL) was added dropwise and the mixture was left to stir at rt for 18 h. The dichloromethane solvent was evaporated to obtain a white solid. The solid was treated with an excess of hexanes, stirred for 1 h, filtered off and the solvent was evaporated to yield a crude product. The crude product was purified by silica gel column chromatography using 5-10% ethyl acetate and hexane to obtain 2 (199 g, 91%) as a colorless liquid.
- (3-Benzyloxy)-1-bromo-propane (2)

3-(3-Benzyloxy-propoxy)-2-hydroxy-benzaldehyde (4)
-

-
To a solution of aldehyde 3 (27.47 g, 1 eq, 198.88 mmol) in 0.5 L of anhydrous DMSO was added sodium tertiary-butoxide (42.3 g, 2.2 eq, 440.31 mmol) portionwise. The reaction mixture was stirred at rt for 30 minutes. A brown color solution was formed. The reaction mixture was cooled to 0° C. and added bromide (56 g, 1.2 eq, 244.41 mmol) dropwise. The mixture was stirred at rt O/N. 90% of aldehyde 3 was converted to product. The reaction mixture was acidified to pH-3 and then extracted into EtOAc and washed with water. The organic layer was concentrated, the product was purified on silica gel column (EtOAc:hexane 80:20), to yield as compound 4 (48 g, 84.31% yield) (viscous oil).

Trifluoro-methanesulfonic acid 2-(3-benzyloxy-propoxy)-6-formyl-phenyl ester (5)
-

-
To an ice cold solution of 4 (48 g, 1.0 eq, 167.72 mmol) in 200 mL of dry DCM was added pyridine (22 mL, 1.62 eq, 272.11 mmol). To the reaction mixture trifluoromethanesulfonic anhydride (33 mL, 1.16 eq, 196.14 mol) was added drop by drop. The mixture was stirred for 3 h at 0° C. The mixture was quenched with 500 mL of 1N HCl. The compound was then extracted into DCM (300 mL) and passed through a small silica gel column and concentrated to give compound 5 (57 g, 82% yield) as a pale yellow thick oil.

3-(3-Benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde (6)
-

-
Compound 5 (65 g, 1.0 eq, 155.5 mmol), bis(pinacolato)diboron (86.9 g, 2.2 eq, 342.11 mmol), KOAc (45.7 g, 3.0 eq, 466.5 mmol) were mixed together and 600 mL of dioxane was added. The mixture was degassed with N2 for 30 minutes and PdCl2(dppf).DCM (5.7 g, 0.05 eq, 7.77 mmol) was added. The resulting slurry was heated to 90° C. overnight. The solvent was evaporated, EtOAc was added and then filtered through a pad of Celite. The organic layer was then washed with water (2×150 mL) and the solvent was evaporated. Column chromatography using 15% EtOAc/hexanes gave compound 6 (37.1 g, 61% yield).

7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (A47)
-

-
A solution of compound 6 (36 g, 1.0 eq, 90.91 mmol) in 50 mL of THF was cooled to 0° C. Nitromethane (16.6 g, 3.0 eq, 272.72 mmol) was added, followed by an aqueous solution of NaOH (3.64 g in 180 mL of H2O). The reaction mixture was stirred at room temperature overnight. The starting material disappeared. The cyclization was afforded by adding 1N HCl until the solution was acidified and then extracted into EtOAc. The EtOAc was evaporated and the mixture was triturated with water and decanted. Column chromatography using 50% EtOAc/hexanes gave compound A47 (15.9 g, 50% yield).

(R) and (S) 7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol
-

-
4.82 g of (A47) was resolved via chiral HPLC using CHIRALPAK ADH column and CO2:methanol (86:14) as eluent (25° C. UV detection was monitored at 230 nm. Two peaks, (S)-7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol and (R)-7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol were collected and evaporated to yellow oils. Analysis of the pooled fractions using a CHIRALPAK ADH 4.6 mm ID×250 mm analytical column and the same mobile phase provided the (S) enantiomer [0.7 g (29% yield)] with a retention time of 6.11 min and a 98.2% ee. The (R) enantiomer [1.0 g (41% yield)] had a retention time of 8.86 min and a 99.6% ee.

(S)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol (A49)
-

-
(A47) (550 mg, 1.57 mmol) was dissolved in 15 mL of glacial acetic acid. 280 mg of 20 wt % palladium hydroxide on carbon (Pearlman’s catalyst) was added and the reaction mixture was flushed with hydrogen 3× and hydrogenated at 55 psi for 3.5 hours. The mixture was filtered through Celite to remove catalyst and rinsed with methanol. Acetic acid was evaporated to obtain the crude product. HPLC purification gave 128 mg of the acetate salt of (A49). The acetate salt was treated with 10 mL of 2N HCl and stirred for 3 hours. The material was lyophilized overnight to obtain 93 mg of the hydrochloride salt of (A49) (Yield 22%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.48 (t, J=7.8 Hz, 1H), 7.05 (d, J=7.4 Hz, 1H), 6.92 (d, J=8.2 Hz, 1H), 5.27 (d, J=9.4 Hz, 1H), 4.11 (t, J=6.3 Hz, 2H), 3.58 (t, J=5.9 Hz, 2H), 2.82 (dd, J=13.3, 9.0 Hz, 1H), 1.95-1.83 (m, 2H); MS (ESI): m/z=238 (M+1, positive); HPLC purity: 98.74% (MaxPlot 200-400 nm), 98.38% (220 nm); Chiral HPLC=95.14% ee.

OTHER ISOMER
- (R)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol (A50)
-

-
(R)-7-(3-benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (0.70 g, 2.0 mmol) was dissolved in 20 mL of glacial acetic acid. 350 mg of 20 wt % palladium hydroxide on carbon (Pearlman’s catalyst) was added and the reaction mixture was flushed with hydrogen 3× and hydrogenated at 55 psi for 3.5 hours. The mixture was filtered through Celite to remove catalyst and rinsed with methanol. Acetic acid was evaporated to obtain the crude product. HPLC purification gave 65 mg of pure compound. After purification, this acetate salt was combined with material from another reaction. This product was treated with 2N HCl (10 mL) and stirred for 3 h at rt. The material was lyophilized overnight to obtain 74 mg of the hydrochloride salt of (A50) (Yield 14%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.48 (t, J=7.8 Hz, 1H), 7.05 (d, J=7.4 Hz, 1H), 6.92 (d, J=8.2 Hz, 1H), 5.27 (d, J=9.4 Hz, 1H), 4.11 (t, J=6.3 Hz, 2H), 3.58 (t, J=5.9 Hz, 2H), 2.83 (dd, J=13.3, 8.6 Hz, 1H), 1.94-1.82 (m, 2H); MS (ESI): m/z=238 (M+1, positive); HPLC purity: 99.12% (MaxPlot 200-400 nm), 98.74% (220 nm); Chiral HPLC=98.82% ee.

REFERENCES
https://pubs.acs.org/cen/coverstory/89/8912cover3.html
| US20040203094 * | Sep 20, 2002 | Oct 14, 2004 | Martinis Susan A. | Eucyl-tRNA synthetases and derivatives thereof that activate and aminoacylate non-leucine amino acids to tRNA adaptor molecules |
| US20070155699 * | Aug 16, 2006 | Jul 5, 2007 | Anacor Pharmaceuticals | Boron-containing small molecules |
| US20090227541 * | Jun 19, 2008 | Sep 10, 2009 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
 Anacor
Anacor

![]()
Dr. R. G. Micetich’s research career began in 1963 as a Research Scientist with R & L Molecular Research Ltd. (established by Dr. R. U. Lemieux). This company later became Raylo Chemicals Ltd. Dr. Micetich served as the Research Director (Pharmaceutical Research) of Raylo. During the period from 1963 to 1980 Dr. Micetich’s group was involved in pharmaceutical research and process development work in antibiotics and in NSAI’s (non-steroidal anti-inflammatory agents). This work produced a drug “Mofezolac” – a NSAI which is now marketed in Japan by the Japanese company “Yoshitomi”. Market ~ U.S. $60 million.
In 1980, Dr. R. G. Micetich joined the Faculty of Pharmacy, University of Alberta as an Adjunct Professor working on projects for international big Pharma companies. The work with Taiho Pharmaceutical Company in Japan has produced another drug – a beta-lactamase inhibitor – “TAZOBACTAM” which is now marketed worldwide. This drug now produces annual sales of over US$ 1 billion.
In 1987, Dr. Micetich established a joint venture research company with Taiho, Japan called SynPhar. SynPhar had numerous patents worldwide in various therapeutic areas and many compounds and classes of compounds at various stages of development up to late preclinical.
In view of the significant growth opportunities for SynPhar and in response to the changing international market place for pharmaceuticals, Dr. Micetich acquired and transferred all the assets including intellectual property, equipment and fixtures from SynPhar to NAEJA Pharmaceutical Inc. in 1999. NAEJA is a private Albertan company, founded by the Micetich family which from an initial staff in August 1999 of 40, has grown to 130 and is still growing. NAEJA is a completely self-supporting private company with no venture capital, nor private, nor government funding. The majority of NAEJA employees hold Ph.D.’s. NAEJA has collaborative agreements with pharmaceutical companies around the world. Based on its own intellectual property, NAEJA also has a number of co-development agreements with biotech companies worldwide. Dr. Micetich laid the seeds of foundation for NAEJA and the company continues after his passing, building his legacy.
Dr. R. G. Micetich boasted over 100 publications in well know scientific journals and composed over 100 patents taken out in many countries…………..http://www.bioalberta.com/ron-micetich
more……….
RONALD G. MICETICH (1931-2006): A Scientific Career Ronald Micetich was born in Podanur, Coimbatore (South India). Following receipt of B.Sc. Honors (Chemistry, Loyola College, Madras) and M.A. (Chemistry, Madras University) degrees in India, Ron obtained a Ph.D. (Organic Chemistry, University of Saskatchewan, Canada) in 1962. Ron initiated his interest in microbiology while he was a postdoctoral fellow at the National Research Council of Canada. During the period 1963-1980, Ron held a number of industrial appointments where he rapidly advanced his industrial scientific career (research scientist → associate research director → acting research director → director pharmaceutical / agricultural research) at Raylo Chemicals in Edmonton, Alberta. In 1981 Ron joined the Faculty of Pharmacy & Pharmaceutical Sciences at the University of Alberta as an Adjunct Professor at which time a highly successful drug development program was established with Taiho Pharmaceuticals. This joint industrial collaboration led to the birth of SynPhar with Dr. Micetich as Chairman of the Board, President, CEO and Research Director (1987-1999). Ron, again as Chairman of the Board, CEO and Research Director, established NAEJA (North America, Europe, Japan, Asia) Pharmaceuticals in 1999 with a rollover of assets, including staff, equipment and intellectual property, from SynPhar Laboratories. What began as a full fledged pharmaceutical company with an extensive intellectual property portfolio and a proven track record evolved into an internationally respected pharmaceutical outsource service provider. NAEJA has carved a unique niche in the outsource industry offering extensive discovery experience and expertise. Today, NAEJA has over 120 staff that consists of over 90% scientists holding PhD degrees.,………….see link below
[PDF]RONALD G. MICETICH – University of Alberta – Journal …



Founder, President & CEO, Board Chairman
Fedora Pharmaceuticals Inc.
January 2012 – Present (3 years 10 months)Edmonton, Alberta, Canada
See us at: http://www.fedorapharma.com
Fedora Pharmaceuticals has developed a family of beta-lactamase inhibitors designed to have activity against pathogens containing all four classes of beta-lactamases. These promising novel beta-lactamase inhibitors, used in combination with various beta-lactam antibiotics to treat those antibiotic infections currently resistant to therapy, have recently been licensed to Swiss-based pharmaceutical giant, Hoffman La Roche Ltd. in what is being touted as one of the largest biotech licensing deals ever signed in Canadian history!
Fedora Pharmaceuticals in Canada and Meiji Seika in Japan, with shared world-wide rights, have teamed and jointly entered into this significant tripartite agreement with Roche. Under the terms of the agreement, Roche will obtain exclusive rights from both companies to develop and commercialize the agent worldwide excluding Japan. Fedora and Meiji will receive from Roche; an upfront payment, development, regulatory and sales event milestone payments in addition to royalties on sales of products. While the details of the amounts have not been disclosed a total deal value of US$750 Million in addition to royalties has been announced.
Fedora was founded in 2012 and is headquartered in Edmonton, Alberta, Canada.

President & CEO, Founder
Naeja
August 1999 – Present (16 years 3 months)
NAEJA Pharmaceuticals Inc. is a privately controlled pharmaceutical CRO specializing in early stage drug discovery research with particular expertise in the area of Medicinal Chemistry. NAEJA employs highly skilled PhD scientists recruited from around the globe.
The company boasts a very long and successful track record of rapidly advancing drugs through discovery into the clinic. Several drugs in latter stages of clinical development and on the market today have originated from NAEJA. Most recently is Fedora Pharmaceuticals US $750M licensing deal to Hoffman La Roche Ltd that originated from the laboratories of NAEJA.
As a privately controlled company, NAEJA are very responsive to client’s needs offering many years of drug discovery experience to successfully find ways to advance research programs in a timely, cost effective and efficient manner. NAEJA’s longstanding track record is a testimony in itself!
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
////////////GSK 2251052, Epetraborole, Christopher Micetich, Ronald Micetich, Naeja Pharmaceuticals
B1(c2c(cccc2OCCCO)[C@H](O1)CN)O
TRAMADOL
 |
|||
|
|||


2-[(Dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol
Tramadol (marketed as Ultram, and as generics) is an opioid pain medication used to treat moderate to moderately severepain.[1] When taken as an immediate-release oral formulation, the onset of pain relief usually occurs within about an hour.[5]It has two different mechanisms. First, it binds to the μ-opioid receptor. Second, it inhibits the reuptake of serotonin andnorepinephrine.[6][7]
Serious side effects may include seizures, increased risk of serotonin syndrome, decreased alertness, and drug addiction. Common side effects include: constipation, itchiness and nausea, among others. A change in dosage may be recommended in those with kidney or liver problems. Its use is not recommended in women who are breastfeeding or those who are at risk of suicide.[1]
Tramadol is marketed as a racemic mixture of both R– and S–stereoisomers.[2] This is because the two isomers complement each other’s analgesic activity.[2] It is often combined with paracetamol (acetaminophen) as this is known to improve the efficacy of tramadol in relieving pain.[2] Tramadol is metabolised to O-desmethyltramadol, which is a more potent opioid.[8] It is of the benzenoid class.
Tramadol was launched and marketed as Tramal by the German pharmaceutical company Grünenthal GmbH in 1977 inWest Germany, and 20 years later it was launched in countries such as the UK, U.S., and Australia.[7]
Developed (from 1962) by the German company Grünenthal, and is marketed through much of the world under various trade names, including Acugersic (Malaysia), Mabron (some Eastern European countries as well as parts of the Middle and Far East), Ultram (USA), Zaldiar (France and much of Europe, as well as Russia) and Zydol (UK and Ireland).
CONFUSION ON CIS TRANS
There is some confusion within the literature as to what should be called cis and what should be called trans. For purposes of this disclosure, what is referred to herein as the trans form of Tramadol includes the R,R and S,S isomers as shown by the following two structures:
The cis form of Tramadol, as that phrase is used herein, includes the S,R and the R,S isomers which are shown by the following two structures:

Tramadol is marketed as a racemic mixture of both R and S stereoisomers. It is a μ-opioid receptor agonist, like morphine, but much less active. It inhibits reuptake of the neurotransmitters serotonin and norepinephrine, suggesting that it lifts mood and thereby may dull the brain’s perception of pain.
 |
 |
|
1R,2R-Tramadol |
1S,2S-Tramadol |
In the body, tramadol undergoes demethylation to several metabolites by a Cytochrome P450 enzyme (CYP2D6) in the liver, the most important of these products being O-desmethyltramadol. O-desmethyltramadol has a much stronger (200x) affinity for the μ-opioid receptor than tramadol, so in effect tramadol is a prodrug.
 |
 |
|
1R,2R–O-desmethyltramadol |
1S,2S–O-desmethyltramadol |
Not everyone’s liver works identically. Around 6% of the Caucasian population has a reduced CYP2D6 activity (hence reducing metabolism), so there is a reduced analgesic effect with Tramadol. These people require a dose increase of 30% to get the same pain relief as the norm. A case has been reported of a patient where, following an overdose, their ultrarapid tramadol metabolism led to excessive norepinephrine levels, with near-fatal consequences.
However, it has recently been discovered at relatively high concentrations in the roots of the African peach or pin cushion tree (Nauclea latifolia), which has a long tradition as a folk remedy. As usual, Nature got there first.

The African pin-cushion tree (Nauclea latifolia)
In the area of “legal highs”, a disturbing development is a drug blend known as “Krypton”. This isn’t the noble gas, but a mixture of O-desmethyltramadol withKratom (Mitragyna speciosa, a medicinal plant that originates in SE Asia, seemingly the local equivalent of khat), which contains an alkaloid mitragynine which is also a μ-receptor agonist. Several fatalities have been linked with its use, notably in Sweden.
SYNTHESIS
-Tramadol.svg)
-Tramadol_gespiegelt.svg)
(1R,2R)-Tramadol (1S,2S)-Tramadol
-Tramadol.svg)
-Tramadol_gespiegelt.svg)
(1R,2S)-Tramadol (1S,2R)-Tramadol
The chemical synthesis of tramadol is described in the literature.[35] Tramadol [2-(dimethylaminomethyl)-1-(3-methoxyphenyl)cyclohexanol] has two stereogenic centers at thecyclohexane ring. Thus, 2-(dimethylaminomethyl)-1-(3-methoxyphenyl)cyclohexanol may exist in four different configurational forms:
- (1R,2R)-isomer
- (1S,2S)-isomer
- (1R,2S)-isomer
- (1S,2R)-isomer
The synthetic pathway leads to the racemate (1:1 mixture) of (1R,2R)-isomer and the (1S,2S)-isomer as the main products. Minor amounts of the racemic mixture of the (1R,2S)-isomer and the (1S,2R)-isomer are formed as well. The isolation of the (1R,2R)-isomer and the (1S,2S)-isomer from the diastereomeric minor racemate [(1R,2S)-isomer and (1S,2R)-isomer] is realized by the recrystallization of the hydrochlorides. The drug tramadol is a racemate of the hydrochlorides of the (1R,2R)-(+)- and the (1S,2S)-(–)-enantiomers. The resolution of the racemate [(1R,2R)-(+)-isomer / (1S,2S)-(–)-isomer] was described[36] employing (R)-(–)- or (S)-(+)-mandelic acid. This process does not find industrial application, since tramadol is used as a racemate, despite known different physiological effects[37] of the (1R,2R)- and (1S,2S)-isomers, because the racemate showed higher analgesic activity than either enantiomer in animals[38] and in humans.[39]
Synthesised by chemists at the German company Grünenthal and brought to the market in 1977. It can readily be made by nucleophilic attack of a Grignard or RLi species upon a carbonyl group.

ALSO

Paper
http://www.jmcs.org.mx/PDFS/V49/N4/04-Alvarado.pdf
Tramadol hydrochloride (1). To a solution of 3-bromoanisol 13 (0.823 g, 4.4 mmol) in dry THF (10 mL), 1.75 M n-BuLi (2.5 mL, 4.4 mmol) was added dropwise at -78°C under argon atmosphere. The mixture was stirred at the same temperature during 45 minutes and a solution of 2-dimethylaminomethylcyclohexanone 6a (0.62 g, 4mmol) in dry THF was added dropwise. The resulting mixture was stirred at -78°C for 2 h. and the solvent was removed in vacuo. Water (30 mL) was added and the product was extracted with ethyl ether (3X30 mL). The extracts were dried over sodium sulfate, filtered and evaporated in vacuum. The residue was treated with 5mL of ethyl ether saturated with hydrogen chloride; the ethyl ether was evaporated in vacuo and the resulting solid was purified by crystallization from acetone. Tramadol hydrochloride 1 was obtained as white crystals (0.94 g, 78.6%),
MP 168- 175°C.
IR (KBr): 3410, 3185, 2935, 2826, 2782, 1601, 1249, 702 cm-1;
1H NMR (CDCl3, 300 MHz) δ 1.2-1.9 (10H, m), 2.15 (6H, s), 2.45 (1H, dd, J = 15.1, 4.4 Hz), 3.82 (3H, s), 6.76 (1H, dd, J = 8, 2.4 Hz), 7.04 (1H, d, 7.6 Hz), 7.14 (1H, s), 7.26 (1H, t, 3.9 Hz), 11.4 (1H, bs);
MS (EI): m/z 263 M+ (28), 58 (100).
PATENT
http://www.google.com.ar/patents/WO1999003820A1?cl=en
Tramadol is the compound cis(+/-)-2-[(dimethylamino)-methyl]-l-(3- methoxyphenyl) cyclohexanol which, in the form of the hydrochloride salt is widely used as an analgesic
Tramadol means the racemic mixture of cis-Tramadol as shown by the following chemical structures:

(1 R, 2R) (1S. 2S) cis-Tramadol
Step l Formation of Dimethylaminomethyl Cyclohexanone Hydrochloride

Dimethylaminomethyl Cyclohexanone Hydrochloride
Step 2 Formation of Tramadol Mannich Base
NaOH, Water
Toluene, TBME


Tramadol Mannich Base
Step 3 Formation of Tramadol Base Hydrate

Tramadol Base Hydrate (crude) Step 4 Purification of Tramadol Base Hydrate

Tramadol Base Hydrate (pure)
Step 5 Formation of Tramadol Hydrochloride

Tramadol Hydrochloride
Example 1
To produce the Tramadol base hydrate, a reaction vessel is charged successively with 69 Kg of Magnesium, 400 1 of dry Tetrahydrofuran (THF) and 15 1 of 3- bromoanisole.
With careful heating, the reactor temperature is brought up to ca. 30°C. The Grignard initiates at this point and exotherms to approximately 50°C. A further 5 1 of bromoanisole are added which maintains reflux. 400 1 of THF are then added before the remainder of the bromoanisole. This addition of the remainder of the bromoanisole is carried out slowly so as to sustain a gentle reflux. The reaction is refluxed after complete addition of 3-bromoanisole. The vessel is cooled and Mannich base is added. When addition is complete, the vessel is reheated to reflux for 30 minutes to ensure complete reaction. After cooling to ca. 10°C, 2,300 1 of water are added to quench the reaction. When complete, part of the solvents are distilled under vacuum. Approximately 260 1 of concentrated HC1 is added at a low temperature until a pH of 0 – 1 is reached. This aqueous phase is extracted with toluene. The toluene phases are discarded and ethyl acetate is added to the aqueous phase. 30% Ammonia solution is then charged to reach pH 9 – 10 and the phases are separated. The aqueous phases are extracted again with ethyl acetate and finally all ethyl acetate layers are combined and washed twice with water. Ethyl acetate is then distilled from the reaction solution at atmospheric pressure. Process water is added and the solution cooled to 20°C and seeded. After crystallisation, the vessel is cooled to -5 to 0°C and stirred for one hour.
The product is centrifuged at this temperature and washed with cold ethyl acetate 5 x 50 1. Approximately 310 – 360 Kg of moist cis-Tramadol base hydrate are obtained.
Purification
A reactor vessel is charged successively with cis-Tramadol base hydrate (crude) 200 Kg and ethyl acetate 300 1 and the contents of the vessel heated to 50°C until all solids are in solution. The vessel is then cooled to -5 to 0°C and the product crystallises. Stirring is continued for two hours and the product is then centrifuged and washed with cold ethyl acetate, 2 x 25 1. Approximately 165 – 175 Kg (moist) of cis(+/-) Tramadol base hydrate are obtained from this procedure.
The overall process produced high yields of cis-Tramadol with a trans isomer content of less than 0.03%. Analytical data of the base hydrate of cis-Tramadol
Melting point: 79 – 80°C (in comparison cis-Tramadol base anhydrous is an oil). Water content (KF) : 6.52% (= monohydrate) IR-spectrum of the base hydrate of cis-Tramadol (see Fig. 1).
IR-spectrum (=cis-Tramadol base anhydrous, see Fig. 2).
The invention provides a unique process in which a base hydrate of cis-Tramadol is selectively crystallised without impurities. The base hydrate is processed to readily form cis-Tramadol hydrochloride. The process is substantially simpler than known processes and does not require the use of potentially toxic solvents. Thus the process is environmentally friendly.
The base hydrate of cis-Tramadol prepared may also be used in various formulations.
The base hydrate of cis-Tramadol may be formulated in the form of a solid with a slow release profile. For example, slow release pellets may be prepared by coating a suitable core material with a coating, for example, of ethylcellulose/schellack solution (4:1) and suitable pharmaceutical excipients. The pellets have typical average diameter of 0.6 to 1.6 mm. The pellets may be readily converted into gelatine capsules or pressed into tablet form using well-known techniques.
Alternatively the base hydrate of cis-Tramadol may be formulated into effervescent tablets by forming granules of the base hydrate with acidity/taste modifiers and a suitable effervescent base such as sodium hydrogen carbonate /anhydrous sodium carbonate (12:1). The ingredients are typically blended in a mixer /granulator and heated until granulation occurs. The resulting granules may be pressed into tablet form, on cooling. Of particular interest is the use of the base hydrate of cis-Tramadol in a form for parenteral use/injectables. The base hydrate is typically dissolved in water together with suitable excipients (as necessary). The solution is filtered through a membrane to remove solid fibres or particles. The filtered solution may then be filled into ampoules, typically containing 10.0 mg of the active compound. Usually the formulation is prepared for intramuscular injection.
PATENT
http://www.google.com/patents/EP1346978A1?cl=en
-
Various processes for the synthesis of tramadol hydrochloride have been described in the prior art. For example, US 3 652 589 and British patent specification no. 992 399 describe the preparation of tramadol hydrochloride. In this method, Grignard reaction of 2-dimethylaminomethyl cyclohexanone (Mannich base) with metabromo-anisole gives an oily mixture of tramadol and the corresponding cis isomer, along with Grignard impurities. This oily reaction mixture is subjected to high vacuum distillation at high temperature to give both the geometric isomers of the product base as an oil. This oil, on acidification with hydrogen chloride gas, furnishes insufficiently pure tramadol hydrochloride as a solid. This must then be purified, by using a halogenated solvent and 1,4-dioxane, to give sufficiently pure tramadol hydrochloride. The main drawback of this process is the use of large quantities of 1,4-dioxane and the need for multiple crystallizations to get sufficiently pure trans isomer hydrochloride (Scheme – 1).
-
[0004]
The use of dioxane for the separation of tramadol hydrochloride from the corresponding cis isomer has many disadvantages, such as safety hazards by potentially forming explosive peroxides, and it is also a category 1 carcinogen (Kirk and Othmer, 3rd edition, 17, 48). Toxicological studies of dioxane show side effects such as CNS depression, and necrosis of the liver and kidneys. Furthermore, the content of dioxane in the final tramadol hydrochloride has been strictly limited; for example, the German Drug Codex (Deutscher Arzneimittel Codex, DAC (1991)) restricts the level of dioxane in tramadol hydrochloride to 0.5 parts per million (ppm).

-
In another process, disclosed in US patent specification no. 5 414 129, the purification and separation of tramadol hydrochloride is undertaken from a reaction mixture containing the trans and cis isomers, and Grignard reaction side products, in which the reaction mixture is diluted in isopropyl alcohol and acidified with gaseous hydrogen chloride to yield (trans) tramadol hydrochloride (97.8%) and its cis isomer (2.2%), which is itself crystallized twice with isopropyl alcohol to give pure (trans) tramadol hydrochloride (Scheme – 2). This process relies on the use of multiple solvents to separate the isomers (ie butylacetate, 1-butanol, 1-pentanol, primary amyl alcohol mixture, 1-hexanol, cyclohexanol, 1-octanol, 2-ethylhexanol and anisole). The main drawback of this process is therefore in using high boiling solvents; furthermore, the yields of tramadol hydrochloride are still relatively low and the yield of the corresponding cis hydrochloride is relatively high in most cases.

-
PCT patent specification no. WO 99/03820 describes a method of preparation of tramadol (base) monohydrate, which involves the reaction of Mannich base with metabromo-anisole (Grignard reaction) to furnish a mixture of tramadol base with its corresponding cis isomer and Grignard impurities. This, on treatment with an equimolar quantity of water and cooling to 0 to -5°C, gives a mixture of tramadol (base) monohydrate with the corresponding cis isomer (crude). It is further purified with ethyl acetate to furnish pure (trans) tramadol (base) monohydrate, which is again treated with hydrochloric acid in the presence of a suitable solvent to give its hydrochloride salt (Scheme – 2). The drawback of this method is that, to get pure (trans) tramadol hydrochloride, first is prepared pure (trans) tramadol (base) monohydrate, involving a two-step process, and this is then converted to its hydrochloride salt. The overall yield is low because of the multiple steps and tedious process involved.

-
More recently, a process for the separation of tramadol hydrochloride from a mixture with its cis isomer, using an electrophilic reagent, has been described in US patent specification no. 5 874 620. The mixture of tramadol hydrochloride with the corresponding cis isomer is reacted with an electrophilic reagent, such as acetic anhydride, thionyl chloride or sodium azide, using an appropriate solvent (dimethylformamide or chlorobenzene) to furnish a mixture of tramadol hydrochloride (93.3 to 98.6%) with the corresponding cis isomer (1.4 to 6.66%), (Scheme – 3). The product thus obtained is further purified in isopropyl alcohol to give pure (trans) tramadol hydrochloride. However, the drawback of this process is that a mixture of tramadol base with its cis isomer is first converted into the hydrochloride salts, and this is further reacted with toxic, hazardous and expensive electrophilic reagents to get semi-pure (trans) tramadol hydrochloride. The content of the cis isomer is sufficiently high to require further purification, and this therefore results in a lower overall yield.
-
Therefore, all the known methods require potentially toxic solvents and/or reagents, and multiple steps to produce the desired quality and quantity of tramadol hydrochloride. By contrast, the present invention requires a single step process (or only two steps when tramadol hydrochloride is made via the tramadol (base) monohydrate route) using a natural solvent (ie water) in the absence of carcinogenic solvents (such as the category 1 carcinogen, 1,4-dioxane) to produce pure tramadol hydrochloride, so it is ‘ecofriendly’ and easily commercialized to plant scale without any difficulties.


PATENT
http://www.google.co.in/patents/US6399829

EXAMPLE 8 Hydrochloride Formed without Improvement of the Trans:Cis Ratio
Whether a recrystallization step improves the trans:cis ratio of Tramadol depends upon the solvent composition from which the recrystallization is performed. When the hydrochloride form of Tramadol is produced and then crystallized in the presence of a solvent with a high toluene concentration, the ratio of trans:cis remains essentially unchanged. This is in contrast to the recrystallization from a solvent which has a high acetonitrile concentration as was the case in Examples 5-7.
A 21 mL solution of 1.8 g of HCl gas (bubbled at 5° C.) in acetonitrile (yielding a 2.0 M solution), was added to 10.2 g of Grignard product C (90/10 of trans/cis) in 30 mL of toluene and stirred mechanically for 3 hours. The mixture was filtered and washed with toluene. Drying in vacuo yielded 11.2 g (96% recovery). The resulting hydrochloride had a trans/cis ratio of 92:8, essentially the same trans:cis ratio as did the 10.2 g of Grignard product C.
Recrystallization from 90 mL of acetonitrile yielded 8.83 g, which was 96.6/3.4 of trans/cis by HPLC. Of this, 8.6 g was recrystallized from 75 mL of acetonitrile to give 7.44 g, trans/cis ratio of 99.6/0.4.
This example shows that the formation of the hydrochloride in the presence of a relatively large amount of toluene (here about 60%) and crystallization from toluene-acetonitrile does not improve the trans:cis ratio. As the percentage of toluene present in the mixture of toluene and acetonitrile in a crystallization step is decreased, the trans:cis ratio of the recovered product will increase. Steps in which the hydrochloride is recrystallized from acetonitrile do yield an improved trans:cis ratio.
PATENT
https://www.google.com/patents/EP0831082A1
The synthesis of Tramadol is described in U.S. Patent No. 3,652,589 and in British Patent No. 992,399. The synthesis of Tramadol consists of a Grignard reaction between 2-dimethylaminomethylcyclohexanone and 3-methoxyphenyl magnesium bromide (Equation 1). From the reaction scheme, it is clear that both isomers (RR,SS) (Structure 1) and (RS, SR) (Structure 2) are obtained in variable ratios, depending on the reaction conditions.

The original patents assigned to Gruenenthal GmbH describe the isolation of the (RR,SS) isomer, as follows:
The complex mixture of products containing both isomers of Tramadol obtained from the Grignard reaction is distilled under reduced pressure. The isomers are distilled together at 138-140°C (0.6 mm Hg). The distillate is dissolved in ether and is reacted with gaseous HCl. The resulting mixture of both isomers of Tramadol is precipitated as hydrochlorides and filtered. The resulting mixture contains about 20% of the (RS,SR) isomer. The isomer mixture is then refluxed twice with five volumes of moist dioxane, and filtered. The cake obtained consists of pure (RR,SS) isomer. The residual solution consists of “a mixture of about 20-30% of the cis (i.e. RS,SR), which cannot be further separated by boiling dioxane” [U.S. Patent 3,652,589, Example 2].
Dioxane, used in large quantities in this process, possesses many undesirable properties. It has recently been listed as a Category I carcinogen by OSHA [Kirk & Othmer, 3rd Ed., Vol. 9, p. 386], and it is known to cause CNS depression and liver necrosis [ibid., Vol. 13, p. 267]; in addition, it tends to form hazardous peroxides [ibid., Vol 17, p. 48]. As a result, the concentration of dioxane in the final product has been strictly limited to several ppb’s, and the DAC (1991) restricted the level of dioxane in Tramadol to 0.5 ppm.
A different separation method, described in Israeli Specification No. 103096, takes advantage of the fact that the precipitation of the (RR,SS) isomer of Tramadol from its solution in medium chained alcohols (C4-C8) occurs faster than the precipitation of the (RS,SR) isomer, which tends to separate later. The main disadvantage of this method is, that the time interval between the end of separation of the (RR,SS) isomer and the beginning of the (RS,SR) isomer separation is variable, and seems to depend sharply on the composition of the crude mixture. Therefore, variations in the yield and the quality of the product often occur. Furthermore, about 40% of the (RR,SS) isomer does not separate and remains in solution, along with the (RS,SR) isomer. This remaining mixture cannot be further purified by this method.
Another method, described in Israeli Specification No. 116281, relies on the fact that the (RS,SR) isomer of Tramadol undergoes dehydration much faster then the (RR,SS) isomer, when treated with 4-toluenesulfonic acid, or sulfuric acid; furthermore, when the reaction is carried out in an aqueous medium, a certain amount (up to 50%) of the (RS,SR) isomer is converted to the (RR,SS) isomer. This may, of course increase the efficiency of the process.
The unreacted (RR, SS) isomer is then separated from the dehydrated products and from other impurities by simple crystallization.
While further examining the results of the latter process, it was surprisingly found that the hydroxyl group of the (RS,SR) isomer of Tramadol reacts faster than the same group of the (RR,SS) with various reagents. A plausible explanation for this observation can be supplied by comparing the structures of both isomers, and their ability to form hydrogen bonds.
Looking closely at Fig. 1 [(RR,SS) Tramadol hydrochloride] and at Fig. 2 [(RS,SR) Tramadol hydrochloride], one can provide a plausible explanation for the difference in the OH group’s activity, as follows: The proton attached to the nitrogen atom of the protonated (RR,SS) isomer of Tramadol is capable of forming a stable hydrogen bonding with the oxygen atom of the hydroxyl group (see Fig. 1). Thus, any reaction involving protonation of the hydroxyl group (such as dehydration), or any reaction in which the hydroxyl group reacts as a nucleophile (such as a nucleophilic substitution or esterification process) is less favored to occur.
In the (RS,SR) isomer, on the other hand, there is no possible way of forming a stable intramolecular hydrogen bond, and therefore, any of the above-mentioned types of reactions can easily occur, considering the fact that this particular hydroxyl group is tertiary and benzyllic.

The general purification procedure of the present invention consists of reacting a mixture of both geometrical isomers of Tramadol hydrochloride with a potential electrophile under such conditions that the (RS,SR) isomer reacts almost exclusively, while the (RR,SS) isomer remains practically intact. The resulting mixture is evaporated and the resulting solid substance is then recrystallized from isopropanol or any other suitable solvent.
Example 1
11.1 g of a mixture consisting of 77% (RR,SS) Tramadol hydrochloride and 23% of the corresponding (RS,SR) isomer were dissolved in 30 ml DMF. 1.3 g acetic anhydride were added and the reaction mixture was stirred at room temperature for 12 hours. The solvent was partly evaporated under reduced pressure and 15 ml toluene were added. The suspension obtained was filtered and washed with 5 ml toluene. 5.8 g of crystals were obtained, in which the (RR,SS):(RS,SR) isomer ratio was 70:1. The product obtained was crystallized from 12 ml isopropanol and 4 g of pure (RR,SS) Tramadol hydrochloride were obtained.
Example 2
19.5 g of a mixture consisting of 60.5% (RR,SS) Tramadol hydrochloride and 40.5% of the corresponding (RS,SR) isomer were suspended in 55 ml chlorobenzene. A solution of 4 ml thionyl chloride in 15 ml chlorobenzene was added dropwise for two hours. The suspension was partly evaporated, the residue was filtered and rinsed with toluene, and 8.1 g of crystals were obtained, in which the (RR,SS):(RS, SR) isomer ratio was 14:1. The product obtained was recrystallized from isopropanol, and 6.7 g of pure (RR,SS) Tramadol hydrochloride were obtained.
Example 3
33.4 g of a mixture consisting of 45% (RR,SS) Tramadol hydrochloride and 55% of the corresponding (RS,SR) isomer was immersed in 50 ml trifluoroacetic acid, 5.2 g of sodium azide was added, and the reaction mixture was stirred for 24 hours. The reaction mixture was then evaporated under reduced pressure, 50 ml water was added, and the solution was brought to pH 12 with solid potassium carbonate. The suspension was extracted with 50 ml toluene, the solvent was evaporated and 25 ml hydrogen chloride solution in isopropanol were added. The solution was cooled and filtered. 9.5 g of crude (RR,SS) Tramadol were obtained, and the crude product was purified by recrystallization from isopropanol.
The hitherto unknown (RS,SR)-2-(dimethylaminomethyl)-1-azido-1-(3-methoxyphenyl)-cyclohexane hydrochloride was isolated from the reaction mixture, recrystallized from isopropanol and characterized as follows:
(RS,SR)-2-(dimethylaminomethyl)-1-azido-1-(3-methoxyphenyl)-cyclohexane hydrochloride
- ms : 288 m+
- IR : 2050 cm-1 (N3)
- 1H-NMR (DMSO): 10.42 ppm: (acidic proton); 1H; 7.40-6.90 ppm: (aromatic protons) 4H; 3.79 ppm; (OCH3 ), 3H; 2.78, 2.42 ppm: NCH2 ; 2H; 2.58, 2.37 ppm: [N(CH3 )2], 6H; 2.30-1.40 ppm: cyclohexane ring protons, 9H.
- 13C-NMR (DMSO): 159.74 ppm: C1; 144.12 ppm: C5; 130.08 ppm: C3; 117.36 ppm: C4; 112.97, 111.35 ppm: C2, C6; 69.61 ppm: C8; 59.29 ppm: C14; 55.21 ppm: C7; 44.6 ppm: C15; 40.12 ppm: C13; 35.94, 27.02, 23.56, 21.63 ppm: cyclohexane ring carbon nucleii.

AZIDE COMPD
Bibliography
Synthesis of Tramadol
http://www.nioch.nsc.ru/icnpas98/pdf/posters1/156.pdf
- http://www.opioids.com/tramadol/synthesis/index.html
- Patents to Grünenthal G.m.b.H.:- K. Flick and E. Frankus, U.S. Patent 3,652,589, March 28, 1972; Chem. Abs., 1972, 76, 153321; K. Flick and E. Frankus, U.S. Patent. 3,830,934, August 20, 1974; Chem. Abs., (1974), 82, 21817 (synth)
- C. Alvarado, Á. Guzmán, E. Díaz and R. Patiño, J. Mex. Chem. Soc., 49, (2005) 324-327 (synth)
- G. Venkanna, G. Madhusudhan, K. Mukkanti, A. Sankar, V. M. Kumar and S. A. Ali, J. Chem. Pharm. Res., 4 (2012) 4506-4513 (synth)
- A. Boumendjel, G. S.Taïwe, E. N. Bum, T. Chabrol, C. Beney, V. Sinniger, R. Haudecoeur, L. Marcourt, S. Challal, E. F. Queiroz, F. Souard, M. Le Borgne, T. Lomberget, A. Depaulis, C. Lavaud, R. Robins, J.-L. Wolfender, B. Bonaz and M. De Waard, Angew. Chem. Int. Ed., 52 (2013) 11780 –11784 (Tramadol in nature)
Action
- R. B. Raffa, E. Friderichs, W. Reimann, R. P. Shank, E. E. Codd and J. L. Vaught, J. Pharmacol. Exp. Ther., 260 (1992) 275-285 (means of action)
- P. Dayer, L. Collart and J. Desmeules, Drugs (Suppl. 1), (1994) 3-7.
- T. A. Bamigbade, C. Davidson, R. M. Langford and J. A. Stamford, Br. J. Anaesthes., 79 (1997) 352-356. (action of enantiomers)
- P. W. Keeley, G. Foster and L. Whitelaw, BMJ, 321 (2000) 1608 (auditory hallucinations with tramadol)
- U. M. Stamer, F. Musshoff, M. Kobilay, B. Madea, A. Hoeft and F. Stuber, Clin. Pharmacol. Ther., 82 (2007) 41-47 (different CYP2D6 Genotypes and metabolism)
- W. Leppert, Pharmacology, 87 (2011)274–285 (metabolism of tramadol to O-desmethyltramadol by CYP2D6)
- A. Elkalioubie, D. Allorge, L. Robriquet, J.-F. Wiart, A. Garat, F. Broly and F. Fourrier, Eur. J. Clin. Pharmacol., 67 (2011) 855–858 (ultrarapid metabolism of tramadol)
- C. F. Samer, K. I. Lorenzini, V. Rollason, Y. Daali and J. A. Desmeules, Molecular Diagnosis & Therapy, 17 (2013), 165–84 (variations in tramadol response)
Tramadol abuse
- M. K. Wedge, Can. Pharm. J., 142 (2009) 71-73. (tramadol and antidepressants)
- ACMD advice on O-desmethyltramadol
- Y. Progler, J. Res. Med. Sci., 15 (2010) 185-188 (Tramadol abuse and smuggling into Gaza)
- M. M. Fawzi, Egypt. J. Forensic Sci., 1 (2011) 99–102 (Tramadol abuse in Egypt)
- I. Giraudon, K. Lowitz, P. I. Dargan, D. M. Wood and R. C. Dart, Br. J. Clin. Pharmacol., 76, (2013) 823–824 (prescription opioid abuse in the UK)
- C. Stannard, BMJ, 347 (2013) f5108 (tramadol problem)
- N. Hawkes, BMJ, 347 (2013) f5336 (deaths from tramadol and legal highs)
- A. Winstock, J. Bell and R. Borschmann, BMJ 347 (2013) f5599 (monitoring Tramadol abuse)
- S. H. Park, R. C. Wackernah and G. L. Stimmel, J. Pharm. Pract., 27 (2014) 71-78.
- Unemployment in Gaza and Tramadol addiction.
Tramadol in “highs”
- T. Arndt, U. Claussen, B. Güssregen, S. Schröfel, B. Stürzer, A. Werle and G. Wolf, For. Sci. Int., 208 (2011) 47-52. (Kratom alkaloids and O-desmethyltramadol in urine of a “Krypton” herbal mixture consumer)
- R. Kronstrand, M. Roman, G. Thelander and A. Eriksson, J. Anal. Toxicol., 35 (2011) 242–247. (fatalities from mitragynine and O-desmethyltramadol combinations in Krypton herbal mixture)
- C. D. Rosenbaum, S. P. Carreiro and K. M. Babu, J. Med. Toxicol., 8 (2012) 15-32 (review of herbal marijuana alternatives, including Kratom)
Tramadol in cycling
- Michael Barry, “Shadows on the Road: Life at the Heart of the Peloton, from US Postal to Team Sky”, Faber 2014.
- Chris Froome, “The Climb: The Autobiography”, Viking, 2014.
- http://cyclingtips.com.au/2014/05/wada-proposes-tramadol-remains-a-monitored-rather-than-a-banned-substance-in-2015/
- Tramadol blamed for crashes
- Tramadol use for pain in cycling
| EP0778262A2 * | 19 Nov 1996 | 11 Jun 1997 | Chemagis Ltd. | Process for the purification of (RR-SS)-2-dimethyl-aminomethyl-1-(3-methoxyphenyl)cyclohexanol and its salts |
| EP0787715A1 * | 21 Dec 1996 | 6 Aug 1997 | Grünenthal GmbH | Process for the optical resolution of tramadol |
| EP0831082A1 * | 19 Aug 1997 | 25 Mar 1998 | Chemagis Ltd. | Process for the purification of (RR-SS)-2-dimethylaminomethyl-1-(3-methoxyphenyl)cyclohexanol hydrochloride |
| US5414129 * | 8 Sep 1993 | 9 May 1995 | Chemagis, Ltd. | Process for the purification of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol and its salts |
| Reference | ||
|---|---|---|
| 1 | * | CHEMICAL ABSTRACTS, vol. 126, no. 26, 30 June 1997 Columbus, Ohio, US; abstract no. 343383v, page 597; column 1; XP002079824 & IL 103 096 A (CHEMAGIS LTD) 5 December 1996 |
| 2 | * | CHEMICAL ABSTRACTS, vol. 127, no. 20, 17 November 1997 Columbus, Ohio, US; abstract no. 278028n, QIAO, BEN-ZHI ET AL.: “Synthesis and structure of 1-(m-methoxyphenyl)-2-(dimethylaminomethyl )cyclohexanol.” page 683; column 2; XP002079825 & GAODENG XUEXIAO HUAXUE XUEBAO , vol. 18, no. 6, 1997, pages 902-905, |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1999036389A1 * | 14 Jan 1999 | 22 Jul 1999 | Nicholas Archer | Purification of tramadol |
| WO2000078705A1 * | 22 Jun 1999 | 28 Dec 2000 | Bernhard Akteries | Method for separating the diastereomer bases of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)-cyclohexanol |
| WO2003078380A2 * | 20 Mar 2003 | 25 Sep 2003 | Shahid Akhtar Ansari | Process for preparing tramadol hydrochloride and/or tramadol momohydrate |
| WO2004020390A1 * | 7 Aug 2003 | 11 Mar 2004 | Bernhard Akteries | Method for the production of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol |
| EP1346978A1 * | 21 Mar 2002 | 24 Sep 2003 | Jubilant Organosys Limited | Process for preparing tramadol hydrochloride and/or tramadol monohydrate |
| US6521792 * | 21 Dec 2001 | 18 Feb 2003 | Gruenenthal Gmbh | Process for separating the diastereomeric bases of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)-cylohexanol |
| US6649783 | 3 Oct 2001 | 18 Nov 2003 | Euro-Celtique, S.A. | Synthesis of (+/-)-2-((dimethylamino)methyl)-1-(aryl)cyclohexanols |
| US6784319 | 15 Sep 2003 | 31 Aug 2004 | Euro-Celtique, S.A. | Synthesis of (±)-2-((dimethylamino)methyl)-1-(aryl)cyclohexanols |
| US7030276 | 9 Feb 2005 | 18 Apr 2006 | Gruenenthal Gmbh | Process for preparing 2-[(dimethylamino)-methyl]-1-(3-methoxyphenyl)cyclohexanol |
| US8221792 | 7 Jul 2006 | 17 Jul 2012 | Farnam Companies, Inc. | Sustained release pharmaceutical compositions for highly water soluble drugs |
| EP0940385A1 * | Mar 3, 1999 | Sep 8, 1999 | Dinamite Dipharma S.p.A. | Process for the separation of the (RR,SS)-2-(dimethylamino)methyl-1-(3-methoxyphenyl)-cyclohexanol isomer from the (RS,SR) isomer by selective precipitation |
| WO1999003820A1 * | Jun 26, 1998 | Jan 28, 1999 | Nikolopoulos Angelo | Tramadol, salts thereof and process for their preparation |
| WO1999036390A1 * | Jan 14, 1999 | Jul 22, 1999 | Nicholas Archer | Purification of tramadol |
| WO2000078705A1 * | Jun 22, 1999 | Dec 28, 2000 | Bernhard Akteries | Method for separating the diastereomer bases of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)-cyclohexanol |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7470816 | Nov 13, 2006 | Dec 30, 2008 | Ipac Laboratories Limited | Tramadol recovery process |
| US3652589 * | 27 Jul 1967 | 28 Mar 1972 | Gruenenthal Chemie | 1-(m-substituted phenyl)-2-aminomethyl cyclohexanols |
| US5414129 * | 8 Sep 1993 | 9 May 1995 | Chemagis, Ltd. | Process for the purification of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol and its salts |
| EP0778262A2 * | 19 Nov 1996 | 11 Jun 1997 | Chemagis Ltd. | Process for the purification of (RR-SS)-2-dimethyl-aminomethyl-1-(3-methoxyphenyl)cyclohexanol and its salts |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6828345 * | 31 Mar 2003 | 7 Dec 2004 | Gruenenthal Gmbh | O-substituted 6-methyltramadol derivatives |
| US7030276 | 9 Feb 2005 | 18 Apr 2006 | Gruenenthal Gmbh | Process for preparing 2-[(dimethylamino)-methyl]-1-(3-methoxyphenyl)cyclohexanol |
| US7470816 | 13 Nov 2006 | 30 Dec 2008 | Ipac Laboratories Limited | Tramadol recovery process |
| US20050215821 * | 9 Feb 2005 | 29 Sep 2005 | Gruenenthal Gmbh | Process for preparing 2-[(dimethylamino)-methyl]-1-(3-methoxyphenyl)cyclohexanol |
| US20070112074 * | 13 Nov 2006 | 17 May 2007 | Ashok Kumar | Tramadol recovery process |
| EP0778262A2 * | Nov 19, 1996 | Jun 11, 1997 | Chemagis Ltd. | Process for the purification of (RR-SS)-2-dimethyl-aminomethyl-1-(3-methoxyphenyl)cyclohexanol and its salts |
| US3652589 * | Jul 27, 1967 | Mar 28, 1972 | Gruenenthal Chemie | 1-(m-substituted phenyl)-2-aminomethyl cyclohexanols |
| US5414129 * | Sep 8, 1993 | May 9, 1995 | Chemagis, Ltd. | Process for the purification of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol and its salts |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP0940385A1 * | Mar 3, 1999 | Sep 8, 1999 | Dinamite Dipharma S.p.A. | Process for the separation of the (RR,SS)-2-(dimethylamino)methyl-1-(3-methoxyphenyl)-cyclohexanol isomer from the (RS,SR) isomer by selective precipitation |
| DE10218862A1 * | Apr 26, 2002 | Nov 6, 2003 | Gruenenthal Gmbh | Verfahren zur Chlorierung tertiärer Alkohole |
| US6169205 | Mar 4, 1999 | Jan 2, 2001 | Dipharma S.P.A. | Process for the purification of (RR,SS)-2-(dimethylamino) methyl-1-(3-methoxyphenyl)-cyclohexanol from (RS,SR)-2-(dimethylamino)methyl-1-(3-methoxyphenyl) cyclohexanol |
| US6469213 | Jan 14, 2000 | Oct 22, 2002 | Russinsky Limited | Tramadol, salts thereof and process for their preparation |
| US6649783 | Oct 3, 2001 | Nov 18, 2003 | Euro-Celtique, S.A. | Synthesis of (+/-)-2-((dimethylamino)methyl)-1-(aryl)cyclohexanols |
| US6784319 | Sep 15, 2003 | Aug 31, 2004 | Euro-Celtique, S.A. | Synthesis of (±)-2-((dimethylamino)methyl)-1-(aryl)cyclohexanols |
| US7235693 | Oct 26, 2004 | Jun 26, 2007 | Gruenenthal Gmbh | Process for chlorinating tertiary alcohols |
| US7470816 | Nov 13, 2006 | Dec 30, 2008 | Ipac Laboratories Limited | Tramadol recovery process |
| WO1999003820A1 * | Jun 26, 1998 | Jan 28, 1999 | Nikolopoulos Angelo | Tramadol, salts thereof and process for their preparation |
| Systematic (IUPAC) name | |
|---|---|
|
2-[(Dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol
|
|
| Clinical data | |
| Trade names | Ryzolt, Tramal, Ultram |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a695011 |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Dependence liability |
Present[1] |
| Routes of administration |
Oral, IV, IM, rectal |
| Pharmacokinetic data | |
| Bioavailability | 70–75% (oral), 77% (rectal), 100% (IM)[2] |
| Protein binding | 20%[3] |
| Metabolism | Liver-mediated demethylation andglucuronidation via CYP2D6 &CYP3A4[2][3] |
| Biological half-life | 6.3 ± 1.4 hr[3] |
| Excretion | Urine (95%)[4] |
| Identifiers | |
| CAS Registry Number | 27203-92-5 |
| ATC code | N02AX02 |
| PubChem | CID: 33741 |
| DrugBank | DB00193 |
| ChemSpider | 31105 |
| UNII | 39J1LGJ30J |
| KEGG | D08623 |
| ChEBI | CHEBI:9648 |
| ChEMBL | CHEMBL1066 |
| Chemical data | |
| Formula | C16H25NO2 |
| Molecular mass | 263.4 g/mol |
DMF
Tramadol hydrochloride..CEP
| Product Name | Country | Manufacture | Chemical formula | CAS # | CEP | DMF |
| Tramadol hydrochloride | India | IPCA Laboratories Ltd | C16H26ClNO2 | 22204-88-2 | R0-CEP 2008-189-Rev 00 | – – – |
| Tramadol hydrochloride | India | Dishman Pharmaceuticals and Chemicals Ltd. | C16H26ClNO2 | 22204-88-2 | R0-CEP 2003-148-Rev 01 | – – – |
| Tramadol hydrochloride | India | Sun Pharmaceutical Industries Ltd. | C16H26ClNO2 | 22204-88-2 | R1-CEP 2002-232-Rev 02 | – – – |
| Tramadol hydrochloride | China | CSPC OUYI PHARMACEUTICAL CO., LTD. | C16H26ClNO2 | 22204-88-2 | R0-CEP 2005-227-Rev 02 | – – – |
| Tramadol hydrochloride | India | JUBILANT LIFE SCIENCES LIMITED | C16H26ClNO2 | 22204-88-2 | R1-CEP 2002-199-Rev 03 | – – – |
| Tramadol hydrochloride | India | Cadila Pharmaceuticals Ltd. | C16H26ClNO2 | 22204-88-2 | R1-CEP 2004-098-Rev 01 | – – – |
| Tramadol hydrochloride | Germany | AREVIPHARMA GMBH | C16H26ClNO2 | 22204-88-2 | R0-CEP 2005-020-Rev 02 | – – – |
| Tramadol hydrochloride New process | India | Inogent Laboratories Private Limited | C16H26ClNO2 | 22204-88-2 | R0-CEP 2007-129-Rev 00 | – – – |
| Tramadol hydrochloride | India | SPIC Limited, Pharmaceuticals Division | C16H26ClNO2 | 22204-88-2 | R0-CEP 2004-245-Rev 00 | – – – |
| Tramadol hydrochloride | Switzerland | Cilag AG CH | C16H26ClNO2 | 22204-88-2 | R0-CEP 2006-262-Rev 00 | – – – |
| Tramadol hydrochloride | Italy | Dipharma Francis S.r.l. | C16H26ClNO2 | 22204-88-2 | R0-CEP 2002-105-Rev 01 | – – – |
| Tramadol hydrochloride | Israel | Chemagis Ltd | C16H26ClNO2 | 22204-88-2 | R1-CEP 2003-146-Rev 00 | – – – |
| Tramadol hydrochloride | India | Wanbury LTD | C16H26ClNO2 | 22204-88-2 | R0-CEP 2005-151-Rev 01 | – – – |
| Tramadol hydrochloride | Germany | Excella GmbH | C16H26ClNO2 | 22204-88-2 | R0-CEP 2003-137-Rev 02 | – – – |
| Tramadol hydrochloride | Switzerland | Proto Chemicals AG | C16H26ClNO2 | 22204-88-2 | R1-CEP 2002-204-Rev 01 | – – – |
| Tramadol hydrochloride | Czech Republic | ZENTIVA K.S. | C16H26ClNO2 | 22204-88-2 | R0-CEP 2009-214-Rev 00 | – – – |
| Tramadol hydrochloride | United States | Noramco, Inc. | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | Germany | GRUNENTHAL GMBH | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | Ireland | IROTEC LABORATORIES | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | Switzerland | HELSINN CHEMICALS SA | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | Israel | Chemagis Ltd | US – – | |||
| Tramadol hydrochloride | Slovakia | “ZENTIVA, A.S.” | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | United States | WYCKOFF CHEMICAL CO INC | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | Germany | Excella GmbH | US – – | |||
| Tramadol hydrochloride USP (BULK) | China | SHIJIAZHUANG PHARMACEUTICAL GROUP CO LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | Germany | EVONIK DEGUSSA GMBH | US – – | |||
| Tramadol hydrochloride | Italy | RECORDATI S.p.A. | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | India | Dishman Pharmaceuticals and Chemicals Ltd. | US – – | |||
| TRAMADOL HYDROCHLORIDE | India | Sun Pharmaceutical Industries Ltd. | US – – | |||
| TRAMADOL HYDROCHLORIDE | India | Cadila Pharmaceuticals Ltd. | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | Switzerland | PROTO CHEMICALS AG | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride EP | India | DAI ICHI KARKARIA LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | India | PEARL ORGANICS LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride EP | China | SHIJIAZHUANG PHARMACEUTICAL GROUP HUASHENG PHARMA CO LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | India | JUBILANT LIFE SCIENCES LIMITED | US – – | |||
| TRAMADOL HYDROCHLORIDE | Germany | AREVIPHARMA GMBH | US – – | |||
| Tramadol hydrochloride | India | TONIRA PHARMA LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | India | INOGENT LABORATORIES PRIVATE LIMITED | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | China | ZHEJIANG HISOAR PHARMACEUTICAL CO LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | India | IPCA Laboratories Ltd | US – – | |||
| Tramadol hydrochloride | China | HEBEI ZHONGSHENG PHARMACEUTICAL CO LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride BP | India | KAMUD DRUGS PVT LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | India | Raks Pharma Pvt Ltd. | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride USP n/sterile | India | Aurobindo Pharma Ltd. | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride (YT3 PROCESS) | India | IPCA Laboratories Ltd | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | India | PIRAMAL HEALTHCARE LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | India | Asence Pharma Private Ltd | C16H26ClNO2 | 22204-88-2 | – – – |
SYNOPSIS FOR M. PHARM DISSERTATION
Formulated and Optimized Hydrodynamically balanced Oral Controlled release Bioadhesive
Tablets of Tramadol Hydrochloride using different polymers like Carbopol 971P (CP) and …
////////TRAMADOL,
Ivermectin
IVERMECTIN
MK933
22,23-dihydroavermectin B1a + 22,23-dihydroavermectin B1b
70288-86-7 ![]() 71827-03-7
71827-03-7
| C95H146O28 | |
| Molecular Weight: | 1736.15894 g/mol |
|---|
 C48H74O14, 875.09
C48H74O14, 875.09
Ivermectin is a macrocyclic lactone derived from Streptomyces avermitilis with antiparasitic activity. Ivermectin exerts its anthelmintic effect via activating glutamate-gated chloridechannels expressed on nematode neurons and pharyngeal muscle cells. Distinct from the channel opening induced by endogenous glutamate transmitter, ivermectin-activated channels open very slowly but essentially irreversibly. As a result, neurons or muscle cells remain at either hyperpolarisation or depolarization state, thereby resulting in paralysis and death of the parasites. Ivermectin does not readily pass the mammal blood-brain barrier to the central nervous system where glutamate-gated chloride channels locate, hence the hosts are relatively resistant to the effects of this agent.
This drug, ivermectin, was developed by William C. Campbell of Drew University and Satoshi Ōmura of Japan’s Kitasato University. They were awarded the Nobel Prize in Physiology or Medicine. Originally, the drug was used to treat parasites in livestock and pets before becoming the mainstay of the global campaigns to combat lymphatic filariasis and onchocerciasis.
A workhorse of a drug that a few weeks ago earned its developers a Nobel prize for its success in treating multiple tropical diseases is showing early promise as a novel and desperately needed tool for interrupting malaria transmission, according to new findings presented today at the American Society of Tropical Medicine and Hygiene (ASTMH) Annual Meeting.
At ASTMH annual meeting, new studies explore advances in using ivermectin in ‘mass drug administration’ campaigns to reduce infections in Africa and slow spread of drug resistance in Asia…http://www.pharmpro.com/news/2015/10/nobel-prize-winning-drug-could-also-fight-malaria?et_cid=4908183&et_rid=577220619&type=cta
This new finding was presented today at the American Society of Tropical Medicine and Hygiene (ASTMH) Annual Meeting by researchers from Colorado State University.
Ivermectin has been used for decades, given once per year as a part of Mass Drug Administration (MDA) programs, to reduce the disabling worm infections onchocerciasis, which causes river blindness, and filariasis, the cause of the hugely swollen legs (elephantiasis). Merck has generously donated the entire supply of drug; other companies have followed suit with different drugs for other neglected tropical diseases.
Ivermectin (22,23-dihydroavermectin B1a + 22,23-dihydroavermectin B1b) is a broad-spectrum antiparasitic drug in theavermectin family. It is sold under brand names Heartgard, Sklice[1] and Stromectol[2] in the United States, Ivomecworldwide by Merial Animal Health, Mectizan in Canada by Merck, Iver-DT[3] in Nepal by Alive Pharmaceutical and Ivextermin Mexico by Valeant Pharmaceuticals International. In Southeast Asian countries, it is marketed by Delta Pharma Ltd. under the trade name Scabo 6. While in development, it was assigned the code MK-933 by Merck.[4]
It is taken internally or used topically, depending on the treated condition.
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basichealth system.[5]
It is a drug for the treatment of Onchocerciasis.
The disease is also known as river blindness. It is sometimes called Robles’ disease, after the Guatemalan doctor Rodolfo Robles, who first linked the blindness with an insect a century ago (1915).
Medical uses
Ivermectin is a broad-spectrum antiparasitic agent, traditionally against parasitic worms. It is mainly used in humans in the treatment of onchocerciasis (river blindness), but is also effective against other worm infestations (such as strongyloidiasis,ascariasis, trichuriasis, filariasis and enterobiasis), and some epidermal parasitic skin diseases, including scabies.
Ivermectin is currently being used to help eliminate river blindness (onchocerciasis) in the Americas, and to stop transmissionof lymphatic filariasis and onchocerciasis around the world in programs sponsored by the Carter Center using ivermectin donated by Merck.[6][7][8] The disease is endemic in 30 African countries, six Latin American countries, and Yemen, according to studies conducted by the World Health Organization.[9] The drug rapidly kills microfilariae, but not the adult worms. A single oral dose of ivermectin, taken annually for the 10- to 15-year lifespan of the adult worms, is all that is needed to protect the individual from onchocerciasis.[10]
An Ivermectin cream called Soolantra has been approved by the FDA for treatment of rosacea.[11][12]
SOOLANTRA (ivermectin) cream, 1% is a white to pale yellow hydrophilic cream. Each gram of SOOLANTRA cream contains 10 mg of ivermectin. It is intended for topical use.
Ivermectin is a semi-synthetic derivative isolated from the fermentation of Streptomyces avermitilis that belongs to the avermectin family of macrocyclic lactones.
Ivermectin is a mixture containing not less than 95.0 % and not more than 102.0 % of 5-O-demethyl-22,23-dihydroavermectin A1a plus 5-O-demethyl-25-de(1-methylpropyl)-25-(1-methylethyl)-22,23-dihydroavermectin A1a, generally referred to as 22,23-dihydroavermectin B1a and B1b or H2B1a and H2B1b, respectively; and the ratio (calculated by area percentage) of component H2B1a/(H2B1a + H2B1b)) is not less than 90.0 %.
The respective empirical formulas of H2B1a and H2B1b are C48H74O14and C47H72O14 with molecular weights of 875.10 and 861.07 respectively.
The structural formulas are:
 |
Component H2B1a: R = C2H5, Component H2B1b: R = CH3.SOOLANTRA cream contains the following inactive ingredients: carbomer copolymer type B, cetyl alcohol, citric acid monohydrate, dimethicone, edetate disodium, glycerin, isopropyl palmitate, methylparaben, oleyl alcohol, phenoxyethanol, polyoxyl 20 cetostearyl ether, propylene glycol, propylparaben, purified water, sodium hydroxide, sorbitan monostearate, and stearyl alcohol.
River blindness?
The disease is also known as river blindness. It is sometimes called Robles’ disease, after the Guatemalan doctor Rodolfo Robles, who first linked the blindness with an insect a century ago (1915).

The infection is associated with a nematode worm Onchocerca volvulus, which are transmitted by Simulium blackflies which live and breed near fast-flowing streams and rivers. The worms carry parasitic Wolbachia bacteria. The bite of the flies enables the worm larvae to enter the human’s body; after maturing into adults, followed by breeding, the larvae (microfilariae) formed move towards the skin, and release the bacteria when they die. The bacteria trigger an immune response which leads to lesions on the eye and possible blindness (the “river blindness”).


Left: the Simulium fly (from http://flipper.diff.org/app/items/6730). Right: Simplified life cycle of Onchocerciasis volvulus, modified from the original at: http://emedicine.medscape.com/article/224309-overview#a0104.
Arthropod
More recent evidence supports its use against parasitic arthropods and insects:
- Mites such as scabies:[13][14][15] It is usually limited to cases that prove to be resistant to topical treatments or that present in an advanced state (such as Norwegian scabies).[15]
- Lice:[16][17] Ivermectin lotion (0.5%) is FDA-approved for patients six months of age and older.[18] After a single, 10-minute application of this formulation on dry hair, 78% of subjects were found to be free of lice after two weeks.[19] This level of effectiveness is equivalent to other pediculicide treatments requiring two applications.[20]
- Bed bugs:[21] Early research shows that the drug kills bed bugs when taken by humans at normal doses. The drug enters the human bloodstream and if the bedbugs bite during that time, they will die in a few days.
Contraindications
Ivermectin is contraindicated in children under the age of five, or those who weigh less than 15 kg (33 lb);[22] and those who are breastfeeding, and have a hepatic or renal disease.[23]
Side effects
The main concern is neurotoxicity, which in most mammalian species may manifest as central nervous system depression, and consequent ataxia, as might be expected from potentiation of inhibitory GABA-ergic synapses.
Dogs with defects in the P-glycoprotein gene (MDR1), often collie-like herding dogs, can be severely poisoned by ivermectin.
Since drugs that inhibit CYP3A4 enzymes often also inhibit P-glycoprotein transport, the risk of increased absorption past the blood-brain barrier exists when ivermectin is administered along with other CYP3A4 inhibitors. These drugs include statins, HIV protease inhibitors, many calcium channel blockers, and glucocorticoids such as dexamethasone, lidocaine, and the benzodiazepines.[24]
For dogs, the insecticide spinosad may have the effect of increasing the potency of ivermectin.[25]
Pharmacology
Pharmacodynamics
Ivermectin and other avermectins (insecticides most frequently used in home-use ant baits) are macrocyclic lactones derived from the bacterium Streptomyces avermitilis. Ivermectin kills by interfering with nervous system and muscle function, in particular by enhancing inhibitory neurotransmission.
The drug binds and activates glutamate-gated chloride channels (GluCls).[26] GluCls are invertebrate-specific members of the Cys-loop family of ligand-gated ion channelspresent in neurons and myocytes.
Pharmacokinetics
Ivermectin can be given either by mouth or injection. It does not readily cross the blood–brain barrier of mammals due to the presence of P-glycoprotein,[27] (the MDR1 gene mutation affects function of this protein). Crossing may still become significant if ivermectin is given at high doses (in which case, brain levels peak 2–5 hr after administration). In contrast to mammals, ivermectin can cross the blood–brain barrier in tortoises, often with fatal consequences.
Ecotoxicity
Field studies have demonstrated the dung of animals treated with ivermectin supports a significantly reduced diversity of invertebrates, and the dung persists longer.[28]
History
The discovery of the avermectin family of compounds, from which ivermectin is chemically derived, was made by Satoshi Ōmura of Kitasato University, Tokyo and William C. Campbell of the Merck Institute for Therapeutic research. Ōmura identified avermectin from the bacterium Streptomyces avermitilis. Campbell purified avermectin from cultures obtained from Ōmura and led efforts leading to the discovery of ivermectin, a derivative of greater potency and lower toxicity.[29] Ivermectin was introduced in 1981.[30] Half of the 2015 Nobel Prize in Physiology or Medicine was awarded jointly to Campbell and Ōmura for discovering avermectin, “the derivatives of which have radically lowered the incidence of river blindness and lymphatic filariasis, as well as showing efficacy against an expanding number of other parasitic diseases”.[31]
It started with the avermectins. In 1974, a group of researchers headed by Professor Satoshi Ōmura of the Kitasato Institute, isolated an organism with promising antimicrobial properties in a soil sample (sample OS-3153) picked up near a golf course at Kawana, Ito City, Shizuoka Prefecture, Japan. This was passed on to researchers at the Merck, Sharpe and Dohme (MSD) research laboratories in the USA, who isolated a small family of natural products that became known as avermectins. For many years, scientists have looked in soil samples for the source of potential medicines, like the tetracyclines or streptomycin . There are 8 avermectins, molecules with closely related structures. They are made by fermentation from the bacterium Streptomyces avermitilis.
 Professor Satoshi Ōmura
Professor Satoshi Ōmura

The 8 different avermectins, with the differences between them shown in the table below.
| Name | R1 | R2 | X-Y |
|---|---|---|---|
| Avermectin A1a | Me | Et | CH=CH |
| Avermectin A1b | Me | Me | CH=CH |
| Avermectin A2a | Me | Et | CH2CH(OH) |
| Avermectin A2b | Me | Me | CH2CH(OH) |
| Avermectin B1a | H | Et | CH=CH |
| Avermectin B1b | H | Me | CH=CH |
| Avermectin B2a | H | Et | CH2CH(OH) |
| Avermectin B2b | H | Me | CH2CH(OH) |
The avermectins proved to be have biocidal activity against a wide range of parasites – such as roundworms, lungworms, mites, lice and arachnids; one of these parasites is the tick Rhipicephalus (Boophilus) microplus, one of the most important cattle parasites in tropical regions. Those with the -CH=CH- function are the more active; the most potent was Avermectin B1, occurring as an 80:20 mixture of the similar molecules B1a and B1b, particularly the B1a component. Commercially it is known as Abamectin.
Veterinary use
In veterinary medicine ivermectin is used against many intestinal worms (but not tapeworms), most mites, and some lice. Despite this, it is not effective for eliminating ticks, flies, flukes, or fleas. It is effective against larval heartworms, but not against adult heartworms, though it may shorten their lives. The dose of the medicine must be very accurately measured as it is very toxic in over-dosage. It is sometimes administered in combination with other medications to treat a broad spectrum of animal parasites. Some dog breeds (especially the Rough Collie, the Smooth Collie, the Shetland Sheepdog, and the Australian Shepherd), though, have a high incidence of a certain mutation within the MDR1 gene (coding for P-glycoprotein); affected animals are particularly sensitive to the toxic effects of ivermectin.[32][33] Clinical evidence suggests kittens are susceptible to ivermectin toxicity.[34] A 0.01% ivermectin topical preparation for treating ear mites in cats (Acarexx) is available.
Ivermectin is sometimes used as an acaricide in reptiles, both by injection and as a diluted spray. While this works well in some cases, care must be taken, as several species of reptiles are very sensitive to ivermectin. Use in turtles is particularly contraindicated.
 IVERMECTIN
IVERMECTIN
Chlorotris(triphenylphosphine)rhodium(I), [RhCl(PPh3)3]
http://www.chm.bris.ac.uk/motm/wilcat/wilcath.htm
Such selectivity found an important application in the synthesis of Ivermectin (MectizanTM). Avermectin is a naturally-occurring molecule with anthelmintic and insecticidal properties; selectively reducing one double bond using Wilkinson’s catalyst afforded Ivermectin. The resultant small change in molecular shape makes Ivermectin a much more effective drug to combat onchocerciasis (river blindness), a disease which affects many millions of people, mainly in poor African communities.

You need to add just two hydrogen atoms to reduce a C=C bond in avermectin.
Notes and references
- “SKLICE- ivermectin lotion (NDC Code(s): 49281-183-71)”. DailyMed. February 2012. Retrieved 2015-09-09.
- “STROMECTOL- ivermectin tablet (NDC Code(s): 0006-0032-20)”. DailyMed. May 2010. Retrieved 2015-09-09.
- Adhikari, Santosh (2014-05-27). “ALIVE PHARMACEUTICAL (P) LTD.: Iver-DT”. ALIVE PHARMACEUTICAL (P) LTD. Retrieved 2015-10-07.
- Pampiglione S, Majori G, Petrangeli G, Romi R (1985). “Avermectins, MK-933 and MK-936, for mosquito control”. Trans R Soc Trop Med Hyg 79 (6): 797–9. doi:10.1016/0035-9203(85)90121-X. PMID 3832491.
- “WHO Model List of Essential Medicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- The Carter Center. “River Blindness (Onchocerciasis) Program”. Retrieved2008-07-17..
- The Carter Center. “Lymphatic Filariasis Elimination Program”. Retrieved 2008-07-17..
- WHO. “African Programme for Onchocerciasis Control”. Retrieved 2009-11-12..
- United Front Against Riverblindness. “Onchocerciasis or Riverblindness”..
- United Front Against Riverblindness. “Control of Riverblindness”..
- Galderma Receives FDA Approval of Soolantra (Ivermectin) Cream for Rosacea“
- “SOOLANTRA- ivermectin cream (NDC Code(s): 0299-3823-30, 0299-3823-45, 0299-3823-60)”. DailyMed. December 2014. Retrieved 2015-09-09.
- Brooks PA, Grace RF (August 2002). “Ivermectin is better than benzyl benzoate for childhood scabies in developing countries”. J Paediatr Child Health 38 (4): 401–4.doi:10.1046/j.1440-1754.2002.00015.x. PMID 12174005.
- Victoria J, Trujillo R (2001). “Topical ivermectin: a new successful treatment for scabies”. Pediatr Dermatol 18 (1): 63–5. doi:10.1046/j.1525-1470.2001.018001063.x.PMID 11207977.
- ^ Jump up to:a b Strong M, Johnstone PW (2007). Strong, Mark, ed. “Interventions for treating scabies”. Cochrane Database of Systematic Reviews (Online) (3): CD000320.doi:10.1002/14651858.CD000320.pub2. PMID 17636630.
- Dourmishev AL, Dourmishev LA, Schwartz RA (December 2005). “Ivermectin: pharmacology and application in dermatology”. International Journal of Dermatology 44(12): 981–8. doi:10.1111/j.1365-4632.2004.02253.x. PMID 16409259.
- Strycharz JP, Yoon KS, Clark JM (January 2008). “A new ivermectin formulation topically kills permethrin-resistant human head lice (Anoplura: Pediculidae)”. Journal of Medical Entomology 45 (1): 75–81. doi:10.1603/0022-2585(2008)45[75:ANIFTK]2.0.CO;2.ISSN 0022-2585. PMID 18283945.
- “Sklice lotion”.
- David M. Pariser, M.D., Terri Lynn Meinking, Ph.D., Margie Bell, M.S., and William G. Ryan, B.V.Sc. (November 1, 2012). “Topical 0.5% Ivermectin Lotion for Treatment of Head Lice”. New England Journal of Medicine 367: 1687–1693.doi:10.1056/NEJMoa1200107.
- Study shows ivermectin ending lice problem in one treatment, Los Angeles Times, Nov 5, 2012
- DONALD G. MCNEIL JR. (2012-12-31). “Pill Could Join Arsenal Against Bedbugs”. The New York Times. Retrieved 2013-04-05.
- Jump up^ Dourmishev AL, Dourmishev LA, Schwartz RA (December 2005). “Ivermectin: pharmacology and application in dermatology”. International Journal of Dermatology 44(12): 981–988. doi:10.1111/j.1365-4632.2004.02253.x. PMID 16409259.
- Huukelbach J, Winter B, Wilcke T, et al. (August 2004). “Tratmient masivo selectivo con ivermectina contra las helmintiasis intestinales y parasitos cutáneas en una población gravemente afectada”. Bull World Health Organ 82 (7): 563–571. doi:10.1590/S0042-96862004000800005.
- Goodman and Gilman’s Pharmacological Basis of Therapeutics, 11th edition, pages 122, 1084-1087.
- Jump up^ “COMFORTIS® and ivermectin interaction Safety Warning Notification”. U.S. Food and Drug Administration (FDA) Center for Veterinary Medicine (CVM).
- Yates DM, Wolstenholme AJ (August 2004). “An ivermectin-sensitive glutamate-gated chloride channel subunit from Dirofilaria immitis”. Int. J. Parasitol. 34 (9): 1075–81.doi:10.1016/j.ijpara.2004.04.010. PMID 15313134.
- Borst P, Schinkel AH (June 1996). “What have we learnt thus far from mice with disrupted P-glycoprotein genes?”. European Journal of Cancer 32 (6): 985–990.doi:10.1016/0959-8049(96)00063-9.
- Iglesias LE, Saumell CA, Fernández AS, et al. (December 2006). “Environmental impact of ivermectin excreted by cattle treated in autumn on dung fauna and degradation of faeces on pasture”. Parasitology Research 100 (1): 93–102. doi:10.1007/s00436-006-0240-x. PMID 16821034.
- Fisher MH, Mrozik H (1992). “The chemistry and pharmacology of avermectins”. Annu. Rev. Pharmacol. Toxicol. 32: 537–53. doi:10.1146/annurev.pa.32.040192.002541.PMID 1605577.
- W. C. CAMPBELL; R. W. BURG, , M. H. FISHER, and , R. A. DYBAS (June 26, 1984).“The Discovery of Ivermectin and Other Avermectins”. American Chemical Society. pp. 5–20. ISBN 9780841210837.
|chapter=ignored (help) - “The Nobel Prize in Physiology or Medicine 2015” (PDF). Nobel Foundation. Retrieved7 October 2015.
- “MDR1 FAQs”, Australian Shepherd Health & Genetics Institute, Inc.
- “Multidrug Sensitivity in Dogs”, Washington State University’s College of Veterinary Medicine
- Frischke H, Hunt L (April 1991). “Suspected ivermectin toxicity”. Canadian Veterinary Journal 32 (4): 245. PMC 1481314. PMID 17423775.
External links
- Stromectol
- The Carter Center River Blindness (Onchocerciasis) Control Program
- Mectizan Donation Program
- American NGDO Treating River Blindness
- MERCK. 25 Years: The MECTIZAN® Donation Program
- Trinity College Dublin. Prof William Campbell – The Story of Ivermectin
- “IVERMECTIN- ivermectin tablet (NDC Code(s): 42799-806-01)”. DailyMed. November 2014. Retrieved 2015-09-09
Bibliography
- Chapman and Hall Combined Chemical Dictionary compound code number CMD99-Y (Ivermectin); CLF13-X (Avermectin).
- The World Health Organisation page on Onchocerciasis
- “Organic Chemists Fighting Blindness” – the Mectizan story from the RSC Organic Division
- A. Crump and K. Otoguro, Trends in Parasitology, 2005, 21, 126-132. (Ōmura’s work)
- Nobel committee’s announcement.
Avermectin
- R. W. Burg, B. M. Miller, E. E. Baker, J. Birnbaum, S. A. Currie, R. Hartman, Y.-L. Kong, R. L. Monaghan, G. Olson, I. Putter, J. B. Tunac, H. Wallick, E. O. Stapley, R. Oiwa, and S. Ōmura, Antimicrob. Agents Chemother., 1979, 15, 361-367 (production of avermectins)
- T. W. Miller, L. Chaiet, D. J. Cole, L. J. Cole, J. E. Flor, R. T. Goegelman, V. P. Gullo, H. Joshua, A. J. Kempf, W. R. Krellwitz, R. L. Monaghan, R. E. Ormond, K. E. Wilson, G. Albers-Schönberg and I. Putter., Antimicrob. Agents Chemother., 1979, 15, 368-371 (isolation of avermectins)
- J. R. Egerton, D. A. Ostlind, L. S. Blair, C. H. Eary, D. Suhayda, S. Cifelli, R. F. Riek and W. C. Campbell, Antimicrob. Agents Chemother., 1979, 15, 372-378 (efficacy of avermectins)
- M. H. Fisher, Pure Appl. Chem., 1990, 62, 1231-1240 (avermectin review)
- Y. J. Yoon, E.-S. Kim, Y.-S. Hwang and C.-Y. Choi, Appl. Microbiol. Biotechnol., 2004, 63, 626–634 (biosynthesis)
Ivermectin
- J. C. Chabala, H. Mrozik, R. L. Tolman, P. Eskola, A. Lusi, L. H. Peterson, M. F. Woods, M. H. Fisher and W. C. Campbell, J. Med. Chem., 1980, 23, 1134-1136 (synth)
- W. C. Campbell, M. H. Fisher, E. O. Stapley, G. Albers-Schönberg and T. A. Jacob, Science, 1983, 221, 823–828 (ivermectin as a new antiparasitic agent)
- S. Ōmura and A. Crump, Nat. Rev. Microbiol., 2004, 2, 984-989. (“The life and times of ivermectin – a success story”).
- K. Collins, Perspect. Biol. Med., 2004, 47, 100-109. (History of the Merck Mectizan donation program)
- A. D. Hopkins, Eye, 2005, 19, 1057–1066 (improvements upon ivermectin treatment)
- T. G. Geary, Trends in Parasitology, 2005, 21, 530–532 (20 years of ivermectin)
- S. Ōmura, Int. J. Antimicrob. Ag., 2008, 31, 91–98 (25 years of Ivermectin)
- A. G. Canga, A. M. S. Prieto, M. J. D. Liébana, N. F. Martínez, M. S.Vega and J. J. G. Vieitez, Vet. J., 2009, 179, 25–37 (pharmacokinetics and metabolism of ivermectin in domestic animal species)
- A. Crump and S. Ōmura, Proc. Jpn. Acad., Ser. B., 2011, 87, 13-28 (ivermectin review)
- I. Farrell, Education in Chemistry, November 2013. (“One in the eye for river blindness”), online.
- The Mectizan donation program
- Colombia eliminates river blindness
Doramectin
- K. Stutzman-Engwall, S. Conlon, R. Fedechko, H. McArthur, K. Pekrun, Y. Chen, S. Jenne, C. La, N. Trinh, S. Kim, Y.-X. Zhang, R. Fox, C. Gustafsson and A. Krebber, Metabolic Engineering, 2005, 7, 27–37 (synth.)
- J.-B. Wang, H.-X. Pan and G.-L. Tang, Bioorg. Med. Chem. Lett., 2011, 21, 3320–3323 (synth.)
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
22,23-dihydroavermectin B1a + 22,23-dihydroavermectin B1b
|
|
| Clinical data | |
| Trade names | Stromectol |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a607069 |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, topical |
| Pharmacokinetic data | |
| Protein binding | 93% |
| Metabolism | Liver (CYP450) |
| Biological half-life | 18 hours |
| Excretion | Feces; <1% urine |
| Identifiers | |
| CAS Registry Number | 70288-86-7 |
| ATC code | D11AX22 P02CF01 QP54AA01QS02QA03 |
| PubChem | CID: 9812710 |
| DrugBank | DB00602 |
| ChemSpider | 7988461 |
| UNII | 8883YP2R6D |
| KEGG | D00804 |
| ChEMBL | CHEMBL341047 |
| PDB ligand ID | IVM (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C 48H 74O 14 (22,23-dihydroavermectin B1a) C 47H 72O 14 (22,23-dihydroavermectin B1b) |
| Molecular mass | 875.10 g/mol |
SIMILAR
Doramectin is a similar molecule, used to treat parasites in animals, such as cattle, horses, sheep and pigs.

/////////////////ivermectin, MALARIA
Study Demonstrates Efficacy of New Tumor Treatment

Study Demonstrates Efficacy of New Tumor Treatment |
||||
|
The results of a study demonstrate the efficacy of this drug in treating nonfunctional neuroendocrine tumors of lung or gastrointestinal origin.
|
What are “complex manufacturing processes”? A recent reply from the EMA
DRUG REGULATORY AFFAIRS INTERNATIONAL
Sometimes a clear definition of terms is crucial in the communication between authorities and pharmaceutical companies. Find out what the European Medicines Agency EMA defines as “complex manufacturing steps” and what authorisation holders providing a variation application need to consider.
The Variations Regulation (EC) no. 1234/2008 of the European Commission defines the procedure for variations of existing marketing authorisations. The “detailed guidelines for the various categories of variations“, which were published in the consolidated version in August 2013 in the European Official Journal, explain the interpretation and application of this Variations Regulation.
Although the “detailed guidelines” describe a number of scenarios of possible variations in some detail, there are formulations in the Guideline text which require clarification due to their blur. The EMA adopted such a case in a recent update of itsquestions and answers collection “Quality of Medicines Questions and Answers: Part 1”…
View original post 258 more words
