
Home » 2014 (Page 85)
Yearly Archives: 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA publishes new Guidance on Validation of Analytical Methods
The FDA has published a new Guidance on the validation of analytical methods which shall replace the 14 years old existing Guideline on the topic. More details about the contents of this highly topical document can be found here.
A new FDA Guidance for Industry entitled “Analytical Procedures and Methods Validation for Drugs and Biologics” was published a few days ago. This Guideline replaces the Guidance for Industry “Analytical Procedures and Methods Validation” from 2000 (this document has never been finalised and has had a draft status 14 years long) and – when finalised – should also replace the “Guidelines for Submitting Samples and Analytical Data for Methods Validation” which came into force in 1987.
Unlike the previous Guideline from 2000, the new document explicitly mentions biologics in its title. The objective of the Guideline is to inform applicants about what data are expected by the FDA in marketing authorisation dossiers. The provisions of the Guideline apply to new drug applications (NDAs), abbreviated new drug applications (ANDAs), biologics license applications (BLAs), and variation applications regarding these types of application, as well as to Type II Drug Master Files. The Guideline can’t be directly used for investigational new drug applications (INDs) as the scope of data with regard to analytical procedures and methods validation varies with the development phase. Nevertheless, IND applicants should orientate themselves to the provisions of the new Guideline.
When comparing it with the former and now invalid “Methods Validation” Guidance, it is apparent that the Draft Guidance has been kept much shorter. There are no detailed descriptions available: for example the table about recommended validation parameters for different analytical tests has been deleted without substitution. Yet, new chapters have been added, like chapter “VIII. Life cycle management of analytical procedures” and its following chapter on the verification of analytical methods in FDA’s own laboratories (“IX: FDA methods verification”).
The document contains plenty of cross-references to corresponding 21 CFR paragraphs and provides – in the last chapter “X. References” – an extensive list of essential FDA Guidelines which also have to be considered in this context, as well as references to corresponding USP chapters, and technical literature on statistical topics. The fact that many aspects of methods validation are addressed in those referenced Guidelines explains the reason why the new Guidance has become shorter.
The document can be commented within 90 days.
read all at

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
http://amcrasto.bravesites.com/
http://anthonycrasto.jimdo.com/
Congratulations! Your presentation titled “Anthony Crasto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
join my process development group on google
organic-process-development group
you can post articles and will be administered by me on the google group which is very popular across the world
https://sites.google.com/site/amcrasto/
LinkedIn group
blogs are
- Green Chemistry International,
- Eurekamoments in Organic Chemistry ,
- WIX CHEMISTRY BLOG ,
- Drug regulatory affairs international
- ORGANIC CHEMISTRY SELECT
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ORGANIC CHEMISTRY INTERNATIONAL
MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ
MY CHINA AND JAPAN BLOGS
http://blog.sina.com.cn/u/5030268874
http://ameblo.jp/medchem-amcrasto/
VIETNAM
ICELAND

Tivorbex (indomethacin); Iroko Pharmaceuticals; For the treatment of acute pain,

Tivorbex (indomethacin); Iroko Pharmaceuticals; For the treatment of acute pain, Approved February of 2014
2-{1-[(4-chlorophenyl)carbonyl]-5-methoxy-2-methyl-1H-indol-3-yl}acetic acid
cas 53-86-1
PHILADELPHIA—Iroko Pharmaceuticals, LLC, a global specialty pharmaceutical company dedicated to advancing the science of analgesia, today announced that the U.S. Food and Drug Administration (FDA) has approved TIVORBEX™ (indomethacin) capsules, a nonsteroidal anti-inflammatory drug (NSAID), at 20 mg and 40 mg doses for the treatment of mild to moderate acute pain in adults1.
“TIVORBEX is the second NSAID to be approved from Iroko’s lower dose NSAID pipeline that uses proprietary SoluMatrix Fine Particle Technology™.”
TIVORBEX was approved at dosage strengths that are 20 percent lower than the 25 mg and 50 mg indomethacin products currently on the market2. FDA approval of TIVORBEX was supported by data from two Phase 3 multi-center, placebo-controlled trials that demonstrated significant improvement in pain relief in patients with post-surgical acute pain receiving TIVORBEX compared with patients receiving placebo3.
“The FDA approval of TIVORBEX is another significant milestone for Iroko as it validates our strategic approach towards developing a suite of NSAID products that offer pain management at lower doses,” said John Vavricka, President and CEO of Iroko Pharmaceuticals. “TIVORBEX is the second NSAID to be approved from Iroko’s lower dose NSAID pipeline that uses proprietary SoluMatrix Fine Particle Technology™.” read at
Indometacin (INN) or indomethacin (USAN and former BAN) is a non-steroidal anti-inflammatory drug (NSAID) commonly used as a prescriptionmedication to reduce fever, pain, stiffness, and swelling. It works by inhibiting the production of prostaglandins, molecules known to cause these symptoms. It is marketed under more than seventy different trade names.[1]

Indomethacin was discovered in 1963[8] and it was first approved for use in the U.S. by the Food and Drug Administration in 1965. Its mechanism of action, along with several other NSAIDs that inhibit COX, was described in 1971.[9]


References
- Trade names are listed on DrugBank.ca entry DB00328
- Sanders, Lisa (6 January 2012). “Think Like a Doctor: Ice Pick Pain Solved!”. The New York Times.
- Garza, I & Schwedt, TJ. “Hemicrania continua.” UpToDate.http://www.uptodate.com/contents/hemicrania-continua. Accessed 8/27/13.
- Smyth JM, Collier PS, Darwish M et al. (September 2004). “Intravenous indometacin in preterm infants with symptomatic patent ductus arteriosus. A population pharmacokinetic study”. Br J Clin Pharmacol 58 (3): 249–58. doi:10.1111/j.1365-2125.2004.02139.x.PMC 1884560. PMID 15327584.
- “INDOMETHACIN”. Hazardous Substances Data Bank (HSDB). National Library of Medicine’s TOXNET. Retrieved April 4, 2013.
- Giles W, Bisits A (October 2007). “Preterm labour. The present and future of tocolysis”. Best Pract Res Clin Obstet Gynaecol 21 (5): 857–68. doi:10.1016/j.bpobgyn.2007.03.011.PMID 17459777.
- Akbarpour F, Afrasiabi A, Vaziri N (1985). “Severe hyperkalemia caused by indomethacin and potassium supplementation”. South Med J 78 (6): 756–7. doi:10.1097/00007611-198506000-00039. PMID 4002013.
- Hart F, Boardman P (October 1963). “Indomethacin: A New Non-steroid Anti-inflammatory Agent”. Br Med J 2 (5363): 965–70. doi:10.1136/bmj.2.5363.965. PMC 1873102.PMID 14056924.
- Ferreira S, Moncada S, Vane J (Jun 23, 1971). “Indomethacin and aspirin abolish prostaglandin release from the spleen”. Nat New Biol 231 (25): 237–9.doi:10.1038/231237a0. PMID 5284362.
- Effects of Perinatal Indomethacin Treatment on Preterm Infants, academic dissertation (PDF)
- Indomethacin, from MedicineNet
- Indomethacin, from Drugs.com
- Indocin: Description, chemistry, ingredients, from RxList.com
- Indomethacin bound to proteins in the PDB
IBRUTINIB 依鲁替尼 A Btk protein inhibitor.

IBRUTINIB 依鲁替尼
A Btk protein inhibitor.
1-[(3R)-3-[4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]piperidin-1-yl]prop-2-en-1-one
1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one
| CAS number | 936563-96-1 |
|---|
- CRA-032765
- Ibrutinib
- Imbruvica
- Pc-32765
- PCI 32765
- PCI32765
- UNII-1X70OSD4VX
Company: Pharmacyclics
Approval Status: Approved February 2014US FDA:link
Treatment Area: chronic lymphocytic leukemia
Bruton’s tyrosine kinase (Btk) inhibitor
U.S. Patent No: 7,514,444 , 7,718,662
patent validity: December 2026
An orally bioavailable small-molecule inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Ibrutinib binds to and inhibits BTK activity, preventing B-cell activation and B-cell-mediated signaling and inhibiting the growth of malignant B cells that overexpress BTK. BTK, a member of the src-related BTK/Tec family of cytoplasmic tyrosine kinases, is required for B cell receptor (BCR) signaling, plays a key role in B-cell maturation, and is overexpressed in a number of B-cell malignancies.
Imbruvica (ibrutinib) is an orally available, selective inhibitor of Bruton’s tyrosine kinase (Btk), a gene that is disrupted in the human disease X-linked agammaglobulenemia (XLA). BTK is a signaling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways.
Imbruvica is specifically approved for chronic lymphocytic leukemia in patients who have received at least one prior therapy.
Imbruvica (Ibrutinib, previously known as PCI-32765) was approved as a “breakthrough therapy” on November 13, 2013 by the US Food and Drug Administration (FDA) for the treatment of mantle cell lymphoma (MCL), a rare and deadly form of blood cancer.
Ibrutinib, a first in class oral Bruton’s tyrosine kinase (Btk) inhibitor, was launched in the U.S. for the treatment of patients with mantle cell lymphoma in 2013, and for the treatment of chronic lymphocytic leukemia in 2014. In the E.U., the product candidate is awaiting registration for both indications. Additional phase III clinical trials are ongoing for the treatment of these indications in combination with bendamustine and rituximab and for the treatment of relapsed or refractory marginal zone lymphoma (MZL). Janssen and Pharmacyclics are conducting phase II clinical trials for the treatment of refractory follicular lymphoma. Early clinical development is also under way at Pharmacyclics for the treatment of recurrent B-cell lymphoma, relapsed/refractory MCL, and relapsed or relapsed and refractory multiple myeloma. The company filed an IND seeking approval to commence clinical evaluation of ibrutinib for the treatment of autoimmune disease. Preclinical studies had been under way for rheumatoid arthritis; however, no recent development has been reported. Ibrutinib is also active against Lyn and LCK tyrosine kinases.
In 2011, a codevelopment agreement was signed between the National Cancer Institute (NCI) and Pharmacyclics for the treatment of hematologic/blood cancer. Also in 2011, a worldwide codevelopment and comarketing agreement was signed by Janssen and Pharmacyclics for the treatment of cancer. In 2012, orphan drug designation was assigned in the U.S. and the E.U. for the treatment of CLL. This designation was also assigned by the FDA in 2012 for the treatment of mantle cell lymphoma. In 2013, several orphan drug designations were assigned in the U.S.; for the treatment of small lymphocytic lymphoma, for the treatment of Waldenstrom’s macroglobulinemia and for the treatment of diffuse large B-cell lymphoma. For this indication, orphan drug designation was assigned also in the E.U. the same year. In 2012, fast track designation was assigned by the FDA for the treatment of CLL. In 2013, breakthrough therapy designations were assigned to the compound in the U.S.: for the treatment (as monotherapy) of patients with chronic lymphocytic leukemia or small lymphocytic lymphoma, for the treatment of relapsed or refractory mantle cell lymphoma who have received prior therapy and for the treatment of Waldenstrom’s macroglobulinemia.
Imbruvica is supplied as a capsule for oral administration. The recommended dose is 420 mg taken orally once daily (three 140 mg capsules once daily). Capsules should be taken orally with a glass of water. Do not open, break, or chew the capsules.
The FDA approval of Imbruvica for chronic lymphocytic leukemia was based on an open-label, multi-center trial of 48 previously treated patients. Imbruvica was administered orally at 420 mg once daily until disease progression or unacceptable toxicity. The overall response rate (ORR) and duration of response (DOR) were assessed using a modified version of the International Workshop on CLL Criteria by an Independent Review Committee. The ORR was 58.3%, all partial responses. None of the patients achieved a complete response. The DOR ranged from 5.6 to 24.2+ months. The median DOR was not reached.
Imbruvica (ibrutinib) is an orally available, selective inhibitor of Bruton’s tyrosine kinase (Btk). Ibrutinib forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. BTK is a signaling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways. BTK’s crole in signaling through the B-cell surface receptors results in activation of pathways necessary for B-cell trafficking, chemotaxis, and adhesion.
Ibrutinib (USAN,[1] also known as PCI-32765 and marketed in the U.S. under the name Imbruvica) is an anticancer drug targeting B-cell malignancies. It was approved by the US FDA in November 2013 for the treatment of mantle cell lymphoma[2] and in February 2014 for the treatment ofchronic lymphocytic leukemia.[3] It is an orally-administered, selective and covalent inhibitor of the enzyme Bruton’s tyrosine kinase (BTK).[4][5][6]Ibrutinib is currently under development by Pharmacyclics, Inc and Johnson & Johnson‘s Janssen Pharmaceutical division for additional B-cell malignancies including diffuse large B-cell lymphoma and multiple myeloma.[7][8][9]
Mechanism
In preclinical studies on chronic lymphocytic leukemia (CLL) cells, ibrutinib has been reported to promote apoptosis, inhibit proliferation, and also prevent CLL cells from responding to survival stimuli provided by the microenvironment.[12] In this study, treatment of activated CLL cells with ibrutinib resulted in inhibition of Btk tyrosine phosphorylation and also effectively abrogated downstream survival pathways activated by this kinase including ERK1/2, PI3K, and NF-κB. Additionally, ibrutinib inhibited proliferation of CLL cells in vitro, effectively blocking survival signals provided externally to CLL cells from the microenvironment including soluble factors (CD40L, BAFF, IL-6, IL-4, and TNF-α), fibronectin engagement and stromal cell contact.
In early clinical studies, the activity of ibrutinib has been described to include a rapid reduction in lymphadenopathy accompanied by a transient lymphocytosis, suggesting that the drug might have direct effects on cell homing or migration to factors in tissue microenvironments.[13]
Ibrutinib has been reported to reduce CLL cell chemotaxis towards the chemokines CXCL12 and CXCL13, and inhibit cellular adhesion following stimulation at the B cell receptor.[14][15] Together, these data are consistent with a mechanistic model whereby ibrutinib blocks BCR signaling, which drives cells into apoptosis and/or disrupts cell migration and adherence to protective tumour microenvironments.
History
Ibrutinib was first designed and synthesized at Celera Genomics which reported in 2007 a structure-based approach for creating a series of small molecules that inactivate BTK through covalent binding to cysteine-481 near the ATP binding domain of BTK.[4] These small molecules irreversibly inhibited BTK by using a Michael acceptor for binding to the target cysteine. In April 2006, Pharmacyclics acquired Celera’s small molecule BTK inhibitor discovery program, which included a compound, PCI-32765 that was subsequently chosen for further preclinical development based on the discovery of anti-lymphoma properties in vivo.[16] Since 2006, Pharmacyclics’ scientists have advanced the molecule into clinical trials and identified specific clinical indications for the drug. It also has potential effects against autoimmune arthritis.[17] It was approved by the US FDA on November 13, 2013 for the treatment of mantle cell lymphoma.[2] On Feb. 12, 2014, the U.S. Food and Drug Administration expanded the approved use of the drug ibrutinib to chronic lymphocytic leukemia (CLL). [18]
Ibrutinib is an inhibitor of Bruton’s tyrosine kinase (BTK). It is a white to off-white solid with the empirical formula C25H24N6O2 and a molecular weight 440.50. Ibrutinib is freely soluble in dimethyl sulfoxide, soluble in methanol and practically insoluble in water.
The chemical name for ibrutinib is 1-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)-1Hpyrazolo[ 3,4-d]pyrimidin-1-yl]-1-piperidinyl]-2-propen-1-one and has the following structure:
 |
IMBRUVICA (ibrutinib) capsules for oral administration are supplied as white opaque capsules that contain 140 mg ibrutinib as the active ingredient. Each capsule also contains the following inactive ingredients: croscarmellose sodium, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate. The capsule shell contains gelatin, titanium dioxide and black ink. Each white opaque capsule is marked with “ibr 140 mg” in black ink.
PCI-32765 (ibrutinib) is disclose d in U.S. Patent No. 7,514,444, issued on April 7, 2009, and has the following structur

Ibrutinib is an orally available drug that targets Bruton’s tyrosine kinase (BTK).
Ibrutinib is an irreversible small molecule BTK inhibitor that is in Ph Ib/II of clinical trials in a variety of B-cell malignancies including chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), mantle cell lymphoma (MCL), diffuse large B-cell lymphoma (DLBCL) and multiple myeloma (cancer of plasma cells, a type of white blood cell present in bone marrow). At present ibrutinib is administered orally in clinical trials, via the gastrointestinal tract, at high clinical doses (420 mg/day or 840 mg/day) to patients with CLL and SLL to obtain the desired thereapeutic effect. The need for such high doses of ibrutinib may be due to low bioavailability (the oral bioavailability of ibrutinib is reported to be 22.8% in rats) and may be responsible for the adverse side effects associated with the use of ibrutinib such as nausea or emesis, dizziness and diarrhea. Moreover, low bioavailability results in more variable absorption and potential variability of the desired therapeutic response.
As stated above, at present ibrutinib is administered orally, via the gastrointestinal tract, at high clinical doses (420 mg/day or 840 mg/day) to patients to obtain the desired clinical benefit. It is presently disclosed that when ibrutinib is administered intraduodenally versus via the gastrointestinal tract in rats, the oral bioavailability of ibrutinib unexpectedly increased from 21 % to 100% as determined by AUC.
This unexpected increase in oral bioavailability of ibrutinib can translate into a number of desirable practical benefits. The increase in oral bioavailability should enable administration of ibrutinib at a significantly lower therapeutically effective dose than is currently being used. The lower variability associated with this greater bioavailability should lead to a more reliable therapeutic response as well as more predictable drug absorption.
And avoidance of exposure of Ibtrutinib to the stomach and/or use of lower therapeutically effective dose of ibrutinib can reduce or altogether eliminate potential adverse side effects of this drug such as diahrrea, nausea or emesis, and dizziness. U.S. Patent No. 7,514,444, mentioned above, discloses administration of 0.02-5000 mg/kg andl-1500 mg of ibrutinib/per day and in clinical trials 420 or 840 mg/day of ibrutinib is being administered to the patients with CLL and SLL.
There is no reasonable expectation in the art that ibrutinib can be adminstered orally at lower efficacious doses to the patients with CLL and SLL, particularly as evidenced by the 420 or 840 mg/day of ibrutinib being administered in clinical trials to those patients. Moreover, other than for active agents that are unstable in the stomach or at acidic pH delivery of any active agent with low bioavailability further along in the gastrointestinal tract reduces the path length for drug absorption and would be expected to reduce bioavailability. Therefore, it was unexpected to achieve delivery of ibruntinib directly to the small intestine with greater bioavailability.
PC1-32765 (Ibrutinib), chemical name: 1_ [(3R) _3-[4_-3 – (4 – phenoxy-phenyl)-1H-pyrazolo [3,4-d] pyrimidine – 1 – yl] – 1-piperidinyl]-2 – propen-1 – one, and its structural formula is as follows:

PC1-32765 is an oral medication that inhibits B cell as the main receptor tyrosine kinase signaling and promote cell death process, preventing cell migration and adhesion in malignant B cells.
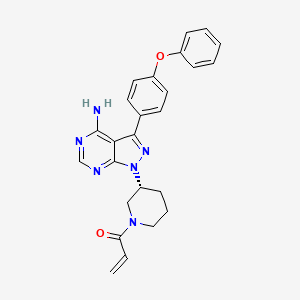
US20080108636 basic patent has been disclosed a synthetic route:
This synthetic route with 4 – phenoxy-benzoic acid as raw material, after eight-step reaction the final product, the following reaction steps:

The above method has the following disadvantages:
1, eight single-step reaction, long route, the economy is bad; i1, to use synthetic intermediates 4:00 trimethylsilyl diazomethane (TMSCHN2), this material easy to blow up, the risk coefficient is large, so large-scale production greatly reduces the possibility;
ii1, synthetic intermediates 7:00, set out to use polymer-supported triphenylphosphine, non-industrial raw materials used, the price is expensive, the cost of smell;
iv, the final step of acylation, the selectivity is poor, a large amount of negative product, purification is difficult, amplification reaction is difficult.
In summary, the route material is not common, expensive step, high costs, the reaction dangerous side reactions, purification difficult, limiting the possibility of industrial production of the route.
………………………
Polymorphs
EXAMPLES
[00438] The following ingredients, formulations, processes and procedures for practicing the methods disclosed herein correspond to that described above. Example 1; Preparation of Crystalline Forms of l-((R)-3-(4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-dlpyrimidin-l-yl)piperidin-l-yl)prop-2-en-l-one (Compound 1)
Form A – Route 1:
[00439] Amorphous Compound 1 (ca. 15 mg) was measured into a vial. Ten volumes (150 μΐ) of solvent [methyl tert-butyl ether (MTBE), diisopropyl ether (DIPE), ethyl acetate, isopropyl acetate, isopropyl alcohol, methyl isobutyl ketone (MIBK), methyl ethyl ketone (MEK), acetone, methanol, nitromethane, 10% aqueous acetone, or 10% aqueous isopropyl alcohol] were added to the vial. The vial was sealed and placed in a shaker at 50 °C for one hour. If a slurry was obtained, an additional thirty volumes (total of 600 μΐ) of solvent was added, then the slurry was returned to 50 °C for another hour. If the sample remained as a slurry at this point, no further solvent was added. The solution/slurry was stirred at 50 °C for one hour, then cooled to 0 °C at 0.1 °C/min, then held at 0 °C overnight. If a slurry was obtained, the solids were filtered under vacuum to provide Compound 1 , Form A; the solution was returned to ambient temperature for slow evaporation through a pin-hole to furnish Compound 1, Form A.
“Compound 1” or “l-((R)-3-(4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4- d]pyrimidin- 1 -yl)piperidin- 1 -yl)prop-2-en- 1 -one” or “1 – {(3i?)-3-[4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-JJpyrimidin-l-yl]piperidin-l-yl}prop-2-en-l-one” or “2-Propen- 1 -one, 1- [(3R)-3-[4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-<f]pyrimidin- 1 -yl] – 1 -piperidinyl-” or ibrutinib or any other suitable name refers to the compound with the following structure:

………….
Synthesis
Synthesis of Compound 3—Btk Activity Probe
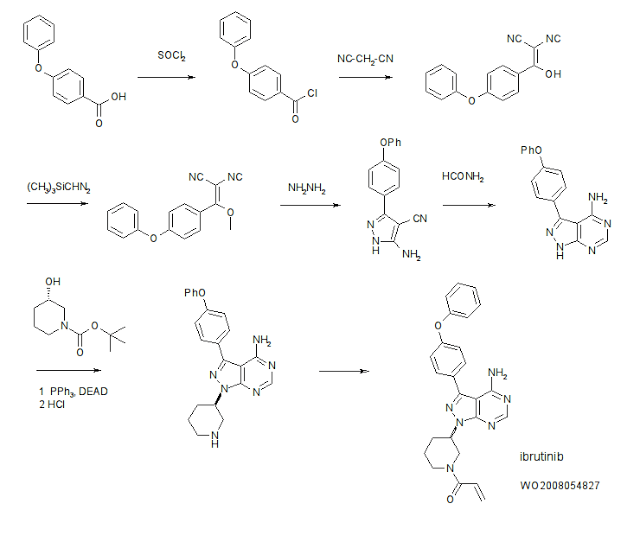
4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride was removed by distillation, the residual oil was dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride was dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) was added and the solution and stirred at −10° C. while adding diisopropylethylethylamine (57.9 g) in toluene (150 mL), while maintaining the temperature below 0° C. After 1 hour at 0° C., the mixture was stirred at 20° C. overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue was taken up in ethyl acetate and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gave a semisolid residue which was treated with a portion of ethyl acetate to give 4.1 g of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160-162° C.). The filtrate on evaporation gave 56.58 (96%) of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which was sufficiently pure for further use.
1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated. The residue is extracted with diethyl ether (3×250 mL), decanting from insoluble oil. Evaporation of the ether extracts gives 22.5 g of 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene as a pale orange solid. The insoluble oil is purified by flash chromatography to give 15.0 g of a red-orange oil.
1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2).
Synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl (Intermediate 4); a) triphenylphosphine (TPP), diisopropyl diazodicarboxylate (DIAD), tetrahydrofuran (THF); b) TFA/CH2Cl2.

To a solution of 1-boc-3-(S)-hydroxypiperidine (3.98 g, 19.8 mmol) and triphenylphosphine (5.19 g, 19.8 mmol) in THF (150 ml) was added DIAD (3.9 ml, 19.8 mmol). The yellow solution was stirred 1 minute then Intermediate 2 (4.0 g, 13.2 mmol) was added and the reaction was heated with a heat gun (3-5 minutes) until the solid had dissolved. After stirring for 1 hour at room temperature, the solvent was removed and the resulting brown oil was subjected to flash chromatography (30% then 50% THF/hexanes) to provide 4.45 g (69%) of Intermediate 3 (trace of triphenylphosphine oxide is present) as a light brown foam.
To a solution of Intermediate 3 (4.4 g, 9.0 mmol) in CH2Cl2 (20 ml) was added TFA (2.8 ml, 36.2 mmol). After stirring 2 hrs at room temperature, the solvent was removed and the residue was partitioned between ethyl acetate (250 ml) and dilute aq. K2CO3. The organic layer was dried (MgSO4), filtered and concentrated to 70 ml. The resulting solution was stirred and 4.0M HCl in dioxane (4 ml) was added to provide a thick light orange precipitate. The precipitate was collected by filtration and washed with ethyl acetate (50 ml). The material was then partitioned between ethyl acetate (300 ml) and dilute aq. K2CO3. The organic layer was dried (MgSO4), filtered and concentrated to provide 2.78 g (80%) of Intermediate 4 as a light yellow foam.
……………………
SYNTHESIS
Compounds described herein may be prepared using the synthetic methods described herein as a single isomer or a mixture of isomers.
A non-limiting example of a synthetic approach towards the preparation of compounds of any of Formula (A), (B), (C) or (D) is shown in Scheme I.

Halogenation of commercially available 1H-pyrazolo[3,4-d]pyrimidin-4-amine provides an entry into the synthesis of compounds of Formula (A), (B), (C) and/or (D). In one embodiment, 1H-pyrazolo[3,4-d]pyrimidin-4-amine is treated with N-iodosuccinamide to give 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine. Metal catalyzed cross coupling reactions are then carried out on 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine. In one embodiment, palladium mediated cross-coupling of a suitably substituted phenyl boronic acid under basic conditions constructs intermediate 2. Intermediate 2 is coupled with N-Boc-3-hydroxypiperidine (as non-limiting example) via Mitsunobu reaction to give the Boc (tert-butyloxycarbonyl) protected intermediate 3. After deprotection with acid, coupling with, but not limited to, an acid chloride, such as, but not limited to, acryloyl chloride, completes the synthesis to give compound 4.
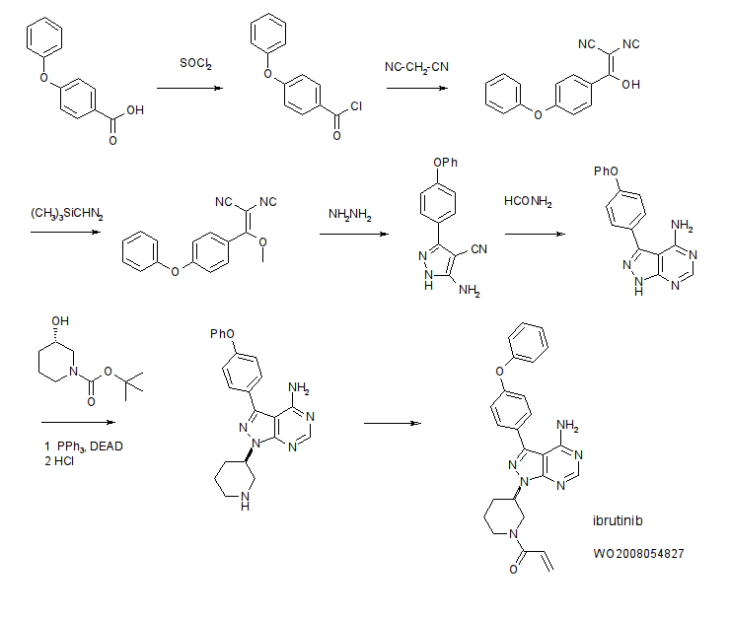
Example 1 Synthesis of Compounds Preparation of 4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2)
4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared as disclosed in International Patent Publication No. WO 01/019829. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride is removed by distillation, the residual oil dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride is dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) is added and the solution and stirred at −10° C. while adding diisopropylethylethylamine (57.9 g) in toluene (150 mL), while maintaining the temperature below 0° C. After 1 hour at 0° C., the mixture is stirred at 20° C. overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue is taken up in ethyl acetate and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gives a semisolid residue which is treated with a little ethyl acetate to give 4.1 g of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160-162° C.). The filtrate on evaporation gives 56.58 (96%) of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which is sufficiently pure for further use.
1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated. The residue is extracted with diethyl ether (3×250 mL), decanting from insoluble oil. Evaporation of the ether extracts gives 22.5 g of 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene as a pale orange solid. The insoluble oil is purified by flash chromatography to give 15.0 g of a red-orange oil.
1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine.
Example 1a Synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 4)

-
- Synthesis of compound 4; a) polymer-bound triphenylphosphine (TPP), diisopropyl diazodicarboxylate (DIAD), tetrahydrofuran (THF); b) HCl/dioxane; then acryloyl chloride, triethylamine (TEA).
Compounds described herein were synthesized by following the steps outlined in Scheme 1. A detailed illustrative example of the reaction conditions shown in Scheme 1 is described for the synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 4).
101 mg of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine and 330 mg of polymer-bound triphenylphosphine(TPP) (polymerlab) were mixed together with 5 mL of tetrahydrofuran (THF). tert-Butyl 3-hydroxypiperidine-1-carboxylate (200 mg; 2.0 equivalents) was added to the mixture followed by the addition of diisopropyl diazodicarboxylate (0.099 mL). The reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered to remove the resins and the reaction mixture was concentrated and purified by flash chromatography (pentane/ethyl acetate=1/1) to give intermediate 3 (55 mg).
Intermediate 3 (48.3 mg) was treated with 1 mL of 4N HCl in dioxane for 1 hour and then concentrated to dryness. The residue was dissolved in dichloromethane and triethylamine (0.042 mL) was added followed by acryl chloride (0.010 mL). The reaction was stopped after 2 hours. The reaction mixture washed with 5% by weight aqueous citric acid and then with brine. The organic layer was dried with MgSO4, and concentrated. Flash chromatography (with CH2Cl2/MeOH=25/1) gave 22 mg of compound 4 as a white solid. MS (M+1): 441.2; 1H-NMR (400 MHz): 8.26, s, 1H, 7.65, m, 2H, 7.42, m, 2H, 7.1-7.2, m, 5H, 6.7-6.9, m, 1H, 6.1, m, 1H, 5.5-5.7, m, 1H, 4.7, m, 1H, 4.54, m, 0.5H, 4.2, m, 1H, 4.1, m, 0.5H, 3.7, m, 0.5H, 3.2, 1,1H, 3.0, m, 0.5H, 2.3, m, 1H, 2.1, m, 1H, 1.9, m, 1H, 1.6, m, 1H.
Example 1b Synthesis of 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 13)

The synthesis of compound 13 was accomplished using a procedure analogous to that described in Example 1a. EM (calc.): 440.2; MS (ESI) m/e (M+1H)+: 441.1, (M−1H)−: 439.2.
Example 1c Synthesis of 1-((S)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 14)

The synthesis of compound 14 was accomplished using a procedure analogous to that described for Example 1a. EM (calc.): 440.2; MS (ESI) m/e (M+1H)+: 441.5, (M−1H)−: 439.2.
……………….
Synthesis of Compounds Example 1 Preparation of 4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (2a)
4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared as disclosed in International Patent Publication No. WO 01/019829. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride is removed by distillation, the residual oil dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride is dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) is added and the solution and stirred at −10° C. while adding diisopropylethylethylamine (57.9 g) in toluene (150 mL), while maintaining the temperature below 0° C. After 1 hour at 0° C., the mixture is stirred at 20° C. overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue is taken up in ethyl acetate and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gives a semisolid residue which is treated with a little ethyl acetate to give 4.1 g of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160-162° C.). The filtrate on evaporation gives 56.58 (96%) of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which is sufficiently pure for further use.
1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated. The residue is extracted with diethyl ether (3×250 mL), decanting from insoluble oil. Evaporation of the ether extracts gives 22.5 g of 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene as a pale orange solid. The insoluble oil is purified by flash chromatography to give 15.0 g of a red-orange oil.
1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine.
Example 1a Synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (4)

Synthesis of compound 4; a) polymer-bound triphenylphosphine (TPP), diisopropyl diazodicarboxylate (DIAD), tetrahydrofuran (THF); b) HCl/dioxane; then acryloyl chloride, triethylamine (TEA)
Compounds described herein were synthesized by following the steps outlined in Scheme III. A detailed illustrative example of the reaction conditions shown in Scheme II is described for the synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 4).
101 mg of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine and 330 mg of polymer-bound triphenylphosphine (TPP) (polymerlab) were mixed together with 5 mL of tetrahydrofuran (THF). tert-Butyl 3-hydroxypiperidine-1-carboxylate (200 mg; 2.0 equivalents) was added to the mixture followed by the addition of diisopropyl diazodicarboxylate (0.099 mL). The reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered to remove the resins and the reaction mixture was concentrated and purified by flash chromatography (pentane/ethyl acetate=1/1) to give intermediate 3a (55 mg).
Intermediate 3a (48.3 mg) was treated with 1 mL of 4N HCl in dioxane for 1 hour and then concentrated to dryness. The residue was dissolved in dichloromethane and triethylamine (0.042 mL) was added followed by acryl chloride (0.010 mL). The reaction was stopped after 2 hours. The reaction mixture was washed with 5% by weight aqueous citric acid and then with brine. The organic layer was dried with MgSO4, and concentrated. Flash chromatography (with CH2Cl2/MeOH=25/1) gave 22 mg of compound 4 as a white solid. MS (M+1): 441.2; 1H-NMR (400 MHz): 8.26, s, 1H, 7.65, m, 2H, 7.42, m, 2H, 7.1-7.2, m, 5H, 6.7-6.9, m, 1H, 6.1, m, 1H, 5.5-5.7, m, 1H, 4.7, m, 1H, 4.54, m, 0.5H, 4.2, m, 1H, 4.1, m, 0.5H, 3.7, m, 0.5H, 3.2, m, 1H, 3.0, m, 0.5H, 2.3, m, 1H, 2.1, m, 1H, 1.9, m, 1H, 1.6, m, 1H.
…………………….
SYNTHESIS
To solve the above problems, the present invention adopts a technical solution is: to provide a tyrosine kinase inhibitor PC1-32765 synthesis method, the reaction steps are as follows:

The beneficial effect of the present invention: The invention relates to a tyrosine kinase inhibitor synthesis of PC1-32765, as the B cell to inhibit the tyrosine kinase receptor signaling key, not only can inhibit the formation of blood cells and less side effects and mild reaction conditions, simple operation, easy purification, low cost, environmentally friendly, suitable for large-scale production.
A tyrosine kinase inhibitor PC1-32765 synthesis method comprising the steps of:
1, the compound 10 and the coupling reaction of compound 15 to give compound 6;
2, the compound 6 obtained by reacting compound 16 with compound 11 in the process, we have chosen a more perfect catalyst;
3, compound 11 to give compound 12 by protecting;
4, selective deprotection of Compound 12 Compound 13; 5, Compound 13 for Compound 17 only attack only remaining position to obtain a very pure compound 14;
6, take off the protecting group to obtain PC1-32765

Wherein the compound can 10,15,16,17 agent or industrial grade reagent compound or the use of methods and techniques related to synthesis.
Example 1 Preparation of Compound 6
Under nitrogen and the 0.1moL 1.5 equivalents of compound 10 Compound 15 and 800mL of dioxane was added to 2L reaction flask, and then 1.5 equivalents of sodium acetate was added and the catalyst PdC12 (PPh3) 2 0.2 equivalents, 50_60 ° C for 5 hours , filtered hot and the filter residue was washed three times with ethanol, the combined filtrate was concentrated to give a solid, rinsed with ethanol to give the pure product 16.2 g, yield 60%
Example 2 Preparation of Compound 6
Under nitrogen and the 0.1moL 1.5 equivalents of compound 10 Compound 15 and 2L 800mL DMF was added to the reaction flask, and then 1.5 equivalents of sodium acetate was added and the catalyst PdCl2 (PhCN) 2 0.2 equivalents, 50_60 ° C for 5 hours, hot filtered, the filter residue was washed three times with ethanol, the combined filtrate was concentrated to give a solid, which was rinsed with ethanol to give pure product 21.5 g, yield 71%.
Example 3 Preparation of Compound 11
The compound 0.1moL 1.2 equivalent of compound 6 and 16, and 2L IOOOmL THF was added to the reaction flask, 1.5 equivalents of cesium carbonate was added, refluxed for 24 hours, after the reaction, most of the solvent was concentrated and the remaining water was poured into a large, precipitated solid was filtered, washed with water to afford compound 36.9 g compound 11, yield 76%, used without further purification.
Example 4 Preparation of Compound 12
The compound will be to 0.1moL 11 and 1.2 equivalent of compound IOOOmL THF trifluoroacetyl chloride and the reaction was added to 2L flask, then triethylamine was added 2.5 ,30-40 0C for 24 hours, after the reaction, the solvent was concentrated, diluted with water, extracted with ethyl acetate, washed with water, saturated sodium chloride each time, and concentrated to obtain the product 50.1 g of ethyl acrylate, 86% yield, used directly in the next reaction.
Example 5 Preparation of Compound 13
The compound 0.1moL 12 and 500mL of methanol and 50mL 6N hydrochloric acid was added to IL reaction flask, stirred at room temperature for 3 hours to complete the reaction quickly, and a solid precipitates, filtered and the solid was washed several times with ethyl acetate, obtain 38.5 g of pure compound 13 in 80% yield.
Example 6 Preparation of Compound 14 ‘
The 0.1moL compound 13 and 1.2 equivalents of acrylic acid chloride was added to 2L of methylene chloride IL reaction flask ,20-40 ° C was added dropwise 1.2 equivalents of triethylamine was added dropwise, at room temperature for 3 hours after the reaction with two chloride extraction and concentrated to give the product 47.7 g, yield 89%. Without further purification.
Example 7 PC1-32765 Preparation
Compound 14 with the 0.1moL 160mL 800mL of methanol and a saturated solution of sodium carbonate small, 50_60 ° C for 5 hours,
After completion of the reaction was diluted with water, concentrated and then extracted with methylene chloride, concentrated to obtain crude product was recrystallized from toluene to give the final product 28.6 g, yield 65%. HPLC purity 98.6%, ee%> 98%.
The present invention relates to a tyrosine kinase inhibitor of the synthesis of PC1-32765, as the B cell to inhibit the tyrosine kinase receptor signaling key, not only can inhibit the formation of blood cells and less side effects, and the reaction conditions gentle, simple operation, easy purification, low cost, environmentally friendly, suitable for large-scale production.
…
Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase
ChemMedChem
Volume 2, Issue 1, pages 58–61, January 15, 2007
http://onlinelibrary.wiley.com/doi/10.1002/cmdc.200600221/full
http://www.wiley-vch.de/contents/jc_2452/2007/z600221_s.pdf
SYN OF COMPD 4
To 101 mg of a known intermediate 2 [WO 2001019829] and 330 mg polymer-bound Triphenylphosphine (polymerlab) in 5 ml THF, 200 mg (2.0 eq.) of 3-OH N-Boc piperidine was added followed by 0.099 ml diisopropyl diazodicarboxylate. The reaction mixture stirred at room temperature overnight. After filtered off resins, the reaction mixture was concentrated and purified with flash chromatography (pentane/ethyl acetate = 1/1) to give 55 mg of intermediate 3. This compound (48.3 mg) was treated with 1 ml of 4N HCl in dioxane for 1 hour and concentrated to dryness, which was dissolved in dichloromethane and 0.042 ml of triethylamine, followed by 0.010 ml of acryl chloride. The reaction was stopped after 2 hours. The reaction mixture was washed with 5wt% citric acid (aq.) and brine, dried with MgSO4, and concentrated. Flash chromatography with (CH2Cl2/MeOH = 25/1) gave 22 mg of compound 4 as white solids. MS (M+1): 441.2; 1H-NMR (400MHz): 8.26, s, 1H; 7.65, m, 2H; 7.42, m, 2H; 7.1-7.2, m, 5H; 6.7-6.9, m, 1H; 6.1, m, 1H; 5.5-5.7, m, 1H; 4.7, m, 1H; 4.54, m, 0.5H; 4.2, m, 1H; 4.1, m, 0.5H; 3.7, m, 0.5H; 3.2, m, 1H; 3.0, m, 0.5H; 2.3, m, 1H; 2.1, m, 1H; 1.9, m, 1H; 1.6, m, 1H
……………..
References
- Statement on a Nonproprietary Name Adopted by the USAN Council
- FDA Press Release
- Azvolinsky, PhD, Anna. “FDA Approves Ibrutinib for Chronic Lymphocytic Leukemia”. Cancer Network. Retrieved 14 February 2014.
- Pan, Z; Scheerens, H; Li, SJ; Schultz, BE; Sprengeler, PA; Burrill, LC; Mendonca, RV; Sweeney, MD; Scott, KC; Grothaus, Paul G.; Jeffery, Douglas A.; Spoerke, Jill M.; Honigberg, Lee A.; Young, Peter R.; Dalrymple, Stacie A.; Palmer, James T. (2007). “Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase”. ChemMedChem 2 (1): 58–61.doi:10.1002/cmdc.200600221. PMID 17154430.
- Celera Genomics Announces Sale of Therapeutic Programs to Pharmacyclics
- United States patent 7514444
- Janssen Biotech, Inc. Announces Collaborative Development and Worldwide License Agreement for Investigational Anti-Cancer Drug, PCI-32765
- Clinical trials involve PCI-32765
- Clinical trials involve ibrutinib
- “Imbruvica (Ibrutinib)”. Medscape Reference. WebMD. Retrieved 13 January 2014.
- “IMBRUVICA (ibrutinib) capsule [Pharmacyclics, Inc]”. DailyMed. Pharmacyclics, Inc. November 2013. Retrieved 13 January 2013.
- Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, Flynn J, Jones J, Blum KA, Buggy J.J., Hamdy A, Johnson AJ, Byrd JC, SE (2011). “Bruton’s tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765”. Blood 117 (23): 6287–6296. doi:10.1182/blood-2011-01-328484. PMC 3122947. PMID 21422473.
- The Bruton’s tyrosine kinase (BTK) inhibitor PCI-32765 (P) in treatment-naive (TN) chronic lymphocytic leukemia (CLL) patients (pts): Interim results of a phase Ib/II study” J Clin Oncol 30, 2012 (suppl; abstr 6507)
- Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, Keating MJ, O’Brien S, Chiorazzi N, Burger JA (2012). The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo 119. Blood. pp. 1182–1189.
- de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, Pals ST, Spaargaren M (2012). “The clinically active BTK inhibitor PCI-32765 targets B-cell receptor (BCR)- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia”. Blood 2012: 2590–2594.
- Honigberg, LA; Smith, AM; Sirisawad, M; Verner, E; Loury, D; Chang, B; Li, S; Pan, Z; Thamm, DH; Miller, RA; Buggy, JJ (2010). “The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy”.Proceedings of the National Academy of Sciences of the United States of America 107 (29): 13075–80. doi:10.1073/pnas.1004594107. PMC 2919935. PMID 20615965.
- Chang, BY; Huang, MM; Francesco, M; Chen, J; Sokolove, J; Magadala, P; Robinson, WH; Buggy, JJ (2011). “The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells”. Arthritis Research & Therapy 13 (4): R115.doi:10.1186/ar3400. PMC 3239353. PMID 21752263.
- http://cancer.osu.edu/mediaroom/releases/Pages/Ohio-State-Cancer-Research-Played-a-Significant-Role-in-FDA-Approval-of-Important-New-CLL-Drug.aspx#sthash.3o9uyt78.dpuf
- BTK inhibitor PCI-32765, National Cancer Institute Drug Dictionary
- Official Imbruvica patient website
MORE
1) E · Werner, L · Honeywell Berg, Z · Pan; Bruton tyrosine kinase inhibitor; drugs circulating Company; filing date: 2006.12.28, open) No. (Notice: CN101610676A CN101610676B, CN101805341A, CN101805341B, CN102746305A, CN102887900A
2) Pan, Z ; Scheerens, H; Li, SJ; Schultz, BE; Sprengeler, PA; Burrill, LC; Mendonca, RV; Sweeney, MD; Scott, KC; Grothaus, Paul G.; Jeffery, Douglas A.; Spoerke, Jill M. ..; Honigberg, Lee A.; Young, Peter R.; Dalrymple, Stacie A.; Palmer, James T. (2007) “Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase” ChemMedChem 2 (1): 58-61 ( article link )
3) Celera Genomics Announces Sale of Therapeutic Programs to Pharmacyclics , April 10, 2006
4) Honigberg; Lee, Verner; Erik, Pan; Zhengying; Inhibitors of Bruton’s tyrosine kinase, U.S. Patent 7,514,444 ; US 20080108636 A1; CA2663116A1, CN101610676B, CN101805341A, CN101805341B, CN102746305A, CN102887900A, EP2081435A2, EP2081435A4, EP2201840A1, EP2201840B1, EP2443929A1, EP2526771A1, EP2526933A2, EP2526933A3, EP2526934A2, EP2526934A3, EP2529621A1, EP2529622A1, EP2530083A1, EP2532234A1, EP2532235A1, US7514444, US7732454, US7825118, US7960396, US8008309, US8088781, US8158786, US8232280, US8236812, US8399470, US8476284, US8497277, US8501751, US8552010, US20080076921, US20080108636, US20080139582, US20090181987, US20100004270, US20100022561, US20100041677, US20100324050, US20100331350, US20110008257, US20110039868, US20110184001, US20110257203, US20110281322, US20120088912, US20120095026, US20120108612, US20120115889, US20120122894, US20120129821, US20120129873, US20120135944, US20120214826, US20120252821, US20120252822, US20120277254, US20120283276, US20120283277, US20130005745, WO2008039218A2, WO2008039218A3
5) Buggy, Joseph J. Chang, Betty Y.; Methods and Compositions . for inhibition of Bone resorption, PCT Int Appl, WO2013003629, 03 Jan 2013.
6) Wei Chen, David J. Loury, Tarak D. Mody; Preparation of pyrazolo-pyrimidine Compounds as Inhibitors of Bruton’s tyrosine kinase; U.S. Patent Number 7,718,662 , 18 May 2010; Also published as CA2776543A1, CN102656173A, EP2393816A2, EP2393816A4, EP2650294A1, US7741330, US20110086866, WO2011046964A2, WO2011046964A3
7) Wei Chen, David J. Loury, Tarak D. Mody; Inhibitors of Bruton’s tyrosine kinase; WO2013010136 A3
8) John C. Byrd, et al;. Targeting BTK with Relapsed Ibrutinib in Chronic Lymphocytic Leukemia; N Engl J Med 2013; 369:32-42
9) Michael L. Wang, MD, et al, Targeting with BTK. Ibrutinib in Refractory or Relapsed Mantle-Cell Lymphoma; N Engl J Med 2013; 369:507-516
|
8-8-2012
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
7-32-2012
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
4-18-2012
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
1-4-2012
|
INHIBITORS OF BRUTONS TYROSINE KINASE
|
|
|
11-18-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
9-16-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE FOR THE TREATMENT OF SOLID TUMORS
|
|
|
8-31-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
6-15-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
4-15-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
2-18-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
12-24-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
11-3-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
10-8-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
6-9-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
2-19-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
1-29-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
4-8-2009
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
9-5-2008
|
BRUTON’S TYROSINE KINASE ACTIVITY PROBE AND METHOD OF USING
|
| US7718662 | 16 Oct 2009 | 18 May 2010 | Pharmacyclics, Inc. | Pyrazolo-pyrimidine inhibitors of bruton’s tyrosine kinase |
| US7741330 | 16 Oct 2009 | 22 Jun 2010 | Pharmacyclics, Inc. | Pyrazolo-pyrimidine inhibitors of Bruton’s tyrosine kinase |
| US7982036 | 17 Oct 2008 | 19 Jul 2011 | Avila Therapeutics, Inc. | 4,6-disubstitued pyrimidines useful as kinase inhibitors |
| US7989465 | 20 Apr 2009 | 2 Aug 2011 | Avila Therapeutics, Inc. | 4,6-disubstituted pyrimidines useful as kinase inhibitors |
| US8008309 * | 7 Jul 2009 | 30 Aug 2011 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8088781 | 20 Jan 2009 | 3 Jan 2012 | Pharmacyclics, Inc. | Inhibitors of brutons tyrosine kinase |
| US8158786 | 15 Jun 2011 | 17 Apr 2012 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8232280 * | 21 Jan 2011 | 31 Jul 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’S tyrosine kinase |
| US8236812 | 21 Sep 2010 | 7 Aug 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8329901 | 1 Feb 2011 | 11 Dec 2012 | Celgene Avilomics Research, Inc. | 4,6-disubstitued pyrimidines useful as kinase inhibitors |
| US8338439 | 29 Dec 2009 | 25 Dec 2012 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US8377946 | 21 Jun 2012 | 19 Feb 2013 | Pharmacyclics, Inc. | Pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| US8399470 | 30 Jan 2012 | 19 Mar 2013 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8445498 | 1 Feb 2011 | 21 May 2013 | Celgene Avilomics Research, Inc. | 4,6-disubstituted pyrimidines useful as kinase inhibitors |
| US8450335 | 26 Jun 2009 | 28 May 2013 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US8476284 | 16 Dec 2011 | 2 Jul 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8481733 * | 19 May 2009 | 9 Jul 2013 | OSI Pharmaceuticals, LLC | Substituted imidazopyr- and imidazotri-azines |
| US8497277 | 6 Dec 2011 | 30 Jul 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8501724 | 30 Apr 2012 | 6 Aug 2013 | Pharmacyclics, Inc. | Purinone compounds as kinase inhibitors |
| US8501751 | 7 Jul 2009 | 6 Aug 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8552010 | 18 Jun 2012 | 8 Oct 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’S tyrosine kinase |
| US8563563 | 30 Jan 2012 | 22 Oct 2013 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8563568 | 8 Aug 2011 | 22 Oct 2013 | Celgene Avilomics Research, Inc. | Besylate salt of a BTK inhibitor |
| US8609679 | 7 Nov 2012 | 17 Dec 2013 | Celgene Avilomics Research, Inc. | 2,4-diaminopyrimidines useful as kinase inhibitors |
| US20090286768 * | 19 May 2009 | 19 Nov 2009 | Osi Pharmaceuticals, Inc. | Substituted imidazopyr- and imidazotri-azines |
| US20110184001 * | 21 Jan 2011 | 28 Jul 2011 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US20120088912 * | 29 Sep 2011 | 12 Apr 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US20120252822 * | 23 May 2012 | 4 Oct 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US20120277254 * | 5 Jul 2012 | 1 Nov 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| WO2010123870A1 * | 20 Apr 2010 | 28 Oct 2010 | Avila Therapeutics, Inc. | Heteroaryl compounds and uses thereof |
| WO2011031979A1 * | 10 Sep 2010 | 17 Mar 2011 | Cylene Pharmaceuticals Inc. | Pharmaceutically useful heterocycle-substituted lactams |
| WO2013155347A1 | 11 Apr 2013 | 17 Oct 2013 | Izumi Raquel | Bruton’s tyrosine kinase inhibitors for hematopoietic mobilization |
| WO2014004707A1 | 26 Jun 2013 | 3 Jan 2014 | Principia Biopharma Inc. | Formulations comprising ibrutinib |
FDA Approves Monovisc Injection for Knee Pain

Sodium Hyaluronate
9067-32-7 (sodium salt)
| MF: | C14H22NNaO11 |
| MW: | 403.31 |

26 feb 2014
http://www.dddmag.com/news/2014/02/fda-approves-monovisc-injection-knee-pain

Sodium hyaluronate is the sodium salt of hyaluronic acid, a glycosaminoglycan found in various connective, epithelial, and neural tissues. Sodium hyaluronate, a long-chain polymer containing repeating disaccharide units of Na-glucuronate-N-acetylglucosamine, occurs naturally on the corneal endothelium, bound to specific receptors for which it has a high affinity. The polyanionic form, commonly referred to as hyaluronan, is a visco-elasticpolymer normally found in the aqueous and vitreous humour. As a pharmaceutical, the uses of sodium hyaluronate include:

sodium hyaluronate
Sodium hyaluronate for intra-articular injection (brand names: Euflexxa, Hyalgan, Supartz, Gel-One) is used to treat knee pain in patients withosteoarthritis who have not received relief from other treatments. It is very similar to the lubricating fluid that occurs naturally in the articular capsule of the knee joint. Once injected into the joint capsule, it acts as both a shock absorber and a lubricant for the joint.[1]

Sodium hyaluronate for intraocular viscoelastic injection (brand names: Healon, Provisc, Viscoat) is used as a surgical aid in variety of surgical procedures performed on the eyeball including cataract extraction (intra- and extracapsular), intraocular lens implantation, corneal transplant,glaucoma filtration, and retina attachment surgery. In surgical procedures in the anterior segment of eyeball, instillation of sodium hyaluronate serves to maintain a deep anterior chamber during surgery, allowing for efficient manipulation with less trauma to the corneal endothelium and other surrounding tissues. Its viscoelasticity also helps to push back the vitreous face and prevent formation of a postoperative flat chamber. In posterior segment surgery, sodium hyaluronate serves as a surgical aid to gently separate, maneuver, and hold tissues. It creates a clear field of vision, facilitating intra-operative and post-operative inspection of the retina and photocoagulation.[2]
Sodium hyaluronate is used as a viscosupplement, administered through a series of injections into the knee, increasing the viscosity of the synovial fluid, which helps lubricate, cushion and reduce pain in the joint.[3] It is generally used as a last resort before surgery[4] and provides symptomatic relief, by recovering the viscoelasticity of the articular fluid, and by stimulating new production from synovial fluid.[5] Use of sodium hyaluronate may reduce the need for joint replacement.[6] Injections appear to increase in effectiveness over the course of four weeks, reaching a peak at eight weeks and retaining some effectiveness at six months, with greater benefit for osteoarthritis than oral analgesics.[7] It may also be effective when used with other joints.[8]
Sodium hyaluronate may also be used in plastic surgery to reduce wrinkles on the face or as a filler in other parts of the body.[9] It may be used in ophthalmology to assist in the extraction ofcataracts, the implantation of intraocular lenses, corneal transplants, glaucoma filtration, retinal attachment and in the treatment of dry eyes.[10]
Sodium hyaluronate is also used to coat the bladder lining in treating interstitial cystitis.
 hyaluronan
hyaluronan
cas 9004-61-9
Sodium hyaluronate functions as a tissue lubricant and is thought to play an important role in modulating the interactions between adjacent tissues. Sodium hyaluronate is a polysaccharide which is distributed widely in the extracellular matrix of connective tissue in man. It forms a viscoelastic solution in water which makes it suitable for aqueous and vitreous humor in ophthalmic surgery. Mechanical protection for tissues (iris, retina) and cell layers (corneal, endothelium, and epithelium) are provided by the high viscosity of the solution. Elasticity of the solution assists in absorbing mechanical stress and providing a protective buffer for tissues. This viscoelasticity enables maintenance of a deep chamber during surgical manipulation since the solution does not flow out of the open anterior chamber. In facilitating wound healing, it is thought that it acts as a protective transport vehicle, taking peptide growth factors and other structural proteins to a site of action. It is then enzymatically degraded and active proteins are released to promote tissue repair.[11] Sodium hyaluronate is being used intra-articularly to treat osteoarthritis.
Sodium hyaluronate is an ophthalmic agent with viscoelastic properties that is used in joints to supplement synovial fluid.
Sodium hyaluronate is absorbed and diffuses slowly out of the injection site. It is eliminated via the canal of Schlemm.
Sodium hyaluronate hyaluronan started to be in use to treat osteoarthritis of the knee in year 1986 with the product Hyalart/Hyalgan by Fidia of Italy, in intra-articular injections.
Sodium Hyaluronate
Brand names of Sodium hyaluronate in Market include (alphabetically):
- AMO Vitrax (ocular)
- AMVISIC Plus (ocular)
- CYSTISTAR, Healon (ocular)
- EYEFILL (ocular)
- HYLO-COMOD (Eye Drop)
- OLIXIA Pure (Eye Drop)
- EUFLEXXA, Bio Technology General (Israel)-Meditrina SA (Rx articular), Molecular weight: 2,400,000-3,600,000 Daltons
- GONILERT/Verisfield (UK) (Rx/articular). Molecular weight:1,800,000-2,000,000 Daltons
- HYALGAN/HYALART– Fidia (Italy)(Medical Device/Rx articular)
- MONOVISC– Anika (USA)(MedicalDevice/articular)
- OSTENIL– TRB Chemedica (Switzerland)(articular injection) [1]
- RECOSYN– Merckle Recordati (Germany) Recosyn info leaflet
- SYNOCROM– Croma Pharma (Austria) (articular injection) . Molecular weight:1,600,000 Daltons
- VISCURE– Cube (UK)(Rx/articular), Molecular weight:1,800,000-2,000,000 Daltons
- VISMED– TRB Chemedica (Switzerland)(eye drop)[2]
- YARDEL– Libytec (Impfstoffwerk Dessau-Tornau/Germany,(Rx/articular), Molecular weight:1,800,000-2,000,000 Daltons
Hyaluronic acid (HA) is a glycosaminoglycan which is present in the hyaline cartilage, synovial joint fluid and skin tissues. More particularly, HA is a linear glycosaminoglycan formed by a mixture of chains of different length constituted by the repetition of a regular disaccharide formed by a glucuronic acid unit and a N- acetyl-glucosamine unit linked beta 1-4. Disaccharides are linked beta 1-3 with an average molecular weight up to 6 Md (6×106 Da). Therefore, each chain in said mixture of chains shows the same repetitive sequence of formula (A)

the corresponding cation generally being hydrogen (hyaluronic acid) or sodium (sodium hyaluronate).
In the tissues, the function of hyaluronic acid is mainly to maintain the structural density allowing in the same time the biochemical actions of the natural products in the specific body districts. In fluids like synovia the action of HA is to keep the right viscosity by a lubricant action. To exert these actions, HA needs to be fully biocompatible including a right metabolic balance. Natural HA is continuously degraded and synthesized by the body enzymes. This homeostasis is deviated when pathological situations occur, therefore increases in the HA catabolism can results in wide range of effects from a severe pathology to simple tissue modifications. The application of HA, as sodium hyaluronate, as filler in cosmetic or in viscoelastic replacement in synovitis, requires that the employed HA polymer has enhanced viscoelastic properties. This rheology has to be balanced with an efficient capability to make the production of the injectable product.
The industrial hyaluronic acid is obtained by extraction from animal tissues or by microorganism fermentation and is commonly available as sodium hyaluronate. Concerning molecular weight, it is generally recognized that low molecular weight HA is a mixture of chains having a mean molecular weight below 250 Kd (2.5×105Da). HA is used, generally as sodium hyaluronate, in many applications in cosmetics, ophthalmology, rheumatology and tissues engineering. In particular HA with a mean molecular weight above 1 Md is used as viscosupplement in joint arthrosis or in wrinkle management. The high molecular weight is required to supplement the synovial fluid or to fill skin connective dead spaces thanks to the viscosity of the resulting solution.
Many medicaments based on the above technology are currently available on the market. They have a high biocompatibility but they are subjected to a rather rapid degradation by the body enzymes, in particular by hyaluronidase, with the consequence of a short half-life.
 sodium hyaluronate
sodium hyaluronate
References
- “Hyaluronate sodium: Indications, Side Effects, Warnings” (Web). Drugs.com. Drugs.com. 5 February 2014. Retrieved 25 February 2014.
- “Healon (Sodium Hyaluronate)” [package insert]. (2002). Kalamazo, Michigan: Pharmacia Corporation. (Web). RxList. (Updated 8 December 2004). RxList, Inc. Retrieved 25 February 2014.
- Puhl, W.; Scharf, P. (1997). “Intra-articular hyaluronan treatment for osteoarthritis”. Annals of the rheumatic diseases 56 (7): 441. doi:10.1136/ard.56.7.441. PMC 1752402.PMID 9486013. edit
- Karlsson, J.; Sjögren, L. S.; Lohmander, L. S. (2002). “Comparison of two hyaluronan drugs and placebo in patients with knee osteoarthritis. A controlled, randomized, double-blind, parallel-design multicentre study”. Rheumatology (Oxford, England) 41 (11): 1240–1248.PMID 12421996. edit
- Jubb, R. W.; Piva, S.; Beinat, L.; Dacre, J.; Gishen, P. (2003). “A one-year, randomised, placebo (saline) controlled clinical trial of 500-730 kDa sodium hyaluronate (Hyalgan) on the radiological change in osteoarthritis of the knee”. International journal of clinical practice 57 (6): 467–474. PMID 12918884. edit
- Kotz, R.; Kolarz, G. (1999). “Intra-articular hyaluronic acid: Duration of effect and results of repeated treatment cycles”. American journal of orthopedics (Belle Mead, N.J.) 28 (11 Suppl): 5–7. PMID 10587245. edit
- Bannuru, R. R.; Natov, N. S.; Dasi, U. R.; Schmid, C. H.; McAlindon, T. E. (2011). “Therapeutic trajectory following intra-articular hyaluronic acid injection in knee osteoarthritis – meta-analysis”. Osteoarthritis and Cartilage 19 (6): 611–619. doi:10.1016/j.joca.2010.09.014.PMID 21443958. edit
- Salk, R. S.; Chang, T. J.; d’Costa, W. F.; Soomekh, D. J.; Grogan, K. A. (2006). “Sodium Hyaluronate in the Treatment of Osteoarthritis of the Ankle: A Controlled, Randomized, Double-Blind Pilot Study”. The Journal of Bone and Joint Surgery 88 (2): 295–302.doi:10.2106/JBJS.E.00193. PMID 16452740. edit
- Beasley, K.; Weiss, M.; Weiss, R. (2009). “Hyaluronic Acid Fillers: A Comprehensive Review”.Facial Plastic Surgery 25 (2): 086–094. doi:10.1055/s-0029-1220647. PMID 19415575. edit
- Shimmura, S.; Ono, M.; Shinozaki, K.; Toda, I.; Takamura, E.; Mashima, Y.; Tsubota, K. (1995).“Sodium hyaluronate eyedrops in the treatment of dry eyes”. The British journal of ophthalmology 79 (11): 1007–1011. PMC 505317. PMID 8534643. edit
- Boucher, W. S.; Letourneau, R.; Huang, M.; Kempuraj, D.; Green, M.; Sant, G. R.; Theoharides, T. C. (2002). “Intravesical sodium hyaluronate inhibits the rat urinary mast cell mediator increase triggered by acute immobilization stress”. The Journal of Urology 167 (1): 380–384.doi:10.1016/S0022-5347(05)65472-9. PMID 11743360. edit
Scientists discover new drug targets for aggressive breast cancer

The image shows the aggressive growth of TNBC cells. Credit: Genome Institute of Singapore, A*STAR
Scientists at A*STAR’s Genome Institute of Singapore (GIS) led in a study that has identified genes that are potential targets for therapeutic drugs against aggressive breast cancer. These findings were reported in the July 2013 issue of PNAS.
Out of the 1.5 million women diagnosed with breast cancer in the world annually, nearly one in seven of these is classified as triple negative. Patients with triple-negative breast cancer (TNBC) have tumours that are missing three important proteins that are found in other types of breast cancer. The absence of these three proteins make TNBC patients succumb to a higher rate of relapse following treatment and have lower overall survival rates. There is currently no effective therapy for TNBC.
Using integrated genomic approaches, GIS scientists led by Dr. Qiang Yu, in collaboration with local…
View original post 374 more words
What Does 100% of Your Daily Value of Cholesterol Look Like?
Healthline just published an interesting infograph that gives a visualization of what your daily value of cholesterol looks like. In the graphic, you can see what 300 mg of cholesterol looks like for 20 high cholesterol foods: http://www.healthline.com/health/high-cholesterol/daily-value
This is a very informative resource as it helps us visualize what their cholesterol intake look like
What Does 100% of Your Daily Value of Cholesterol Look Like?
It’s no secret that eating fatty foods raises your bad cholesterol level, also known as LDL. An elevated LDL clogs up your arteries and makes it difficult for your heart to do its job. Potentially, it could lead to heart disease.
The USDA recommends consuming no more than 300 mg of cholesterol a day. While a deep-fried Twinkie at the county fair is an obvious no-no, other high cholesterol culprits may be sneaking into your diet. Check out what that number looks like in terms of everyday food items.
Warning: you may need to revise your grocery list—and your eating habits!

Fried Chicken:
4 pieces=300mg cholesterol

Croissants:
6 2/3 rolls=300mg cholesterol

Cheddar Cheese:
12 3/4 slices=300mg cholesterol

Prosciutto:
28 slices=300mg cholesterol

Corned Beef:
14 thin slices=300mg cholesterol

Butter:
1 1/5 sticks=300mg cholesterol
read at
http://www.healthline.com/health/high-cholesterol/daily-value
Bhasma : The ancient Indian nanomedicine

Bhasma : The ancient Indian nanomedicine
Dilipkumar pal
Department of Pharmaceutical Sciences, Guru Ghasidash Vishwavidyalya (A Central University), Koni, Bilaspur – 495 009, Chhattisgarh
India
Ayurveda is the science made up of Veda (knowledge) and Ayush (life) i.e. knowledge of life. An Ayurvedic system adopts a holistic approach towards health care by balancing the physical, mental and spiritual functions of the human body. Rasa–Shastra (vedic-chemistry) is one of the parts of Ayurveda, which deals with herbo-mineral/metals/non-metals preparations called Bhasmas. Rasayana (immunomodulation and anti-aging quality) and yogavahi (ability to target drugs to the site) are characteristics of a properly made herbo-mineral/metals/non-metals preparation, which is also nontoxic, gently absorbable, adaptable and digestible in the body
Pal D, Sahu CK, Haldar A. Bhasma : The ancient Indian nanomedicine. J Adv Pharm Technol Res [serial online] 2014 [cited 2014 Feb 26];5:4-12. Available from: http://www.japtr.org/text.asp?2014/5/1/4/126980
Bhasma[1] in Ayurveda has been defined as a substance obtained by calcination.
Use of both bhasma (Residue after incineration – calcined preparation) as well as in pishti (powdered gem or metal) form along with appropriate herbs for treatment of critical ailments is a medicinal preparation in Ayurveda and to some extent Unani (both Indian branches of medical science using natural curative methods. The procedures for preparing these medicines are time-consuming and complicated.
Bhasma is a calcined preparation in which the gem or metal is converted into ash. Gems or metals are purified to remove impurities and treated by triturating and macerating in herbal extracts. The dough so obtained is calcinated to obtain the ashes.[2]^
Bhasma or vibhooti is the sacred ash from the dhuni or fire of a yogi or avadhoota, or from the sacrificial fire or yajna, where special wood, ghee, herbs, grains and other auspicious and purifying items are offered in worship along with mantras. It is believed that bhasma destroys sins (paap), and that it links us with the divine. It is called ‘bhasma’ because it has the power to consume all evils. Any matter, broken up through the process of fire is reduced to its ‘bhasmic’ form, which is infinitely more refined and pure than the original matter, devoid as it is of all impurities niranjan. The grossness of matter obscures the subtle essence inherent within it, just as wood hides fire and milk conceals butter and cheese, but when it is burnt (or churned in the case of milk) only the pure essence remains. Similarly, the great heat of tapasya and the churning of the mind in meditation reveals the underlying subtle spirit or atman.
Vibhuti
In certain circumstances Bhasma, ‘Vibhuti‘ (Sanskrit) and ‘Thiruneeru’ (Tamil) are synonymous.
Bhasmikaran
Bhasmikaran is a process by which a substance which is otherwise bioincompatible is made biocompatible by certain samskaras or processes (Puranik and Dhamankar, 1964e). The objectives of samskara are :- a) elimination of harmful matters from the drug b) modification of undesirable physical properties of the drug c)conversion of some of the characteristics of the drug d) enhancement of the therapeutic action(Puranik and Dhamankar, 1964e). Various steps involved in the preparation of bhasma(or bhasmikaran) are:- 1) Shodhan -Purification, 2) Maran – Powdering, 3) Chalan- Stirring, 4) Dhavan – Washing, 5) Galan- Filtering, 6) Putan- Heating, 7) Mardan- Triturating, 8) Bhavan- Coating with herbal extract, 9) Amrutikaran – Detoxification and 10) Sandharan- Preservation (Puranik and Dhamankar, 1964e). Selection of these steps depends on the specific metal. Sometimes there is an overlapping of the steps e.g. maran is achieved by puttan. Since the present thesis work is on bhasma, Bhasmikaran process is elaborated in details in the following paragraphs.
Steps of bhasmikaran
1. Shodhan: The principle objective of shodhan is to remove unwanted part from the raw material and separate out impurities( Vaiday and Dole 1996b). Metals obtained from ores may contain several impurities, which are removed by subjecting them to Shodhan process. In context of bhasma, shodhan means purifying and making the product suitable for the next step i.e. Maran. Ayurveda classifies shodhan into a) General process and b) Specific process.
General process for shodhan:
- “The sheets of metals are heated till red hot and are successively dipped into liquids like oil, buttermilk, cow’s urine etc. The procedure is repeated seven times”.
b. Specific process for shodhan For some metals a specific process is described for shodhan e.g. for purification of Jasad, the molten mass is poured in cow’s milk 21 times (Shastri K,1979b).
2. Maran : Maran literally means killing. As the name suggests in maran process, a change is brought about in the chemical form or state of the metal. This makes it to lose its metallic characteristics and physical nature. In short, after maran, metal can be converted into powder or other form suitable for administration. To convert various metals into a form appropriate for human consumption, several techniques have been employed which ultimately gave birth to concept: “Bhasma prepared by using Rasa i.e. mercury is the best, whereas the one prepared using herbs are of better quality and those prepared using Gandhak (sulfur) are of inferior quality. Thus there are 3 methods given for maran. It is carried out by heating the metal in presence of 1) mercury 2) plants and 3 ) sulfur.
When various maran procedures for different metals were reviewed, it was found that mercury is mainly used. The unique property of mercury to amalgamate with many metals must have been the reason behind its maximum use in the process of Bhasmikaran. Ancient practitioners might have found it as the most suitable chemical and therefore probably have mentioned that bhasmas using mercury are superior. Plants used in maran process may be serving as catalyst in the process or the minerals in the plants may be forming complexes with the metals. However, no such explanation can be obtained for the use of sulfur.
3. Chalan: Process of stirring during heating the metal is chalan. Stirring is carried out either with iron rod or stick made from a specific plant. As we know today, iron serves as catalyst in many chemical reactions. The phytoconstituents of plant stick may be enhancing the therapeutic effect. For example, stick of Neem is used for chalan process of Jasad bhasma, which is used topically for ophthalmic diseases. We can interpret the significance of this process now. Neem is an antiseptic (Puranik and Dhamankar, 1964h). Zinc is antiseptic, astringent and has ulcer healing property (Block et al., 1982b). These effects of both the constituents may impart the final product better therapeutic activity.
4. Dhavan: In this process, several water washes are given to the product obtained in the previous stage. Perhaps this is to remove the excess amounts of agents used in shodhan or maran stage. Such agents may adversely affect the quality of final product. Hence intermediates are washed with water, thereby water soluble constituents are removed (Puranik and Dhamankar, 1964h).
5. Galan: The product is then sifted either through a fine cloth or through sieves of suitable mesh so as to separate residual material larger in size (Puranik and Dhamankar, 1964h).
6. Puttan: The term puttan means ignition. The general term used for heating in the process of Bhasmikaran is Puta. A special earthen pot, Sharav is generally used for the process. It has two parts, each having a shape of soccer. Sharav is used for direct heating of the material. Its shallowness is useful in heating the material faster and uniformly. After keeping the material on the shallow surface, other part is used as a lid, by placing it in an inverted position. This Puttan process can be looked upon as the key step in manufacturing of bhasma. The classification of putta is primarily done on the basic nature of the process and is as under :- (Puranik and Dhamankar, 1964f) 1)Chandraputta 2) Dhanyarashiputta 3) Suryaputta 4)Bhugarbhaputta 5) Agniptuta.
Toxicity
Modern medical science finds that mercury is inherently toxic, and that its toxicity is not due to the presence of impurities. While mercury does have anti-microbial properties, and formerly waswidely used in Western medicine, its toxicity does not warrant the risk of using it as a health product in most circumstances.[3][4] The Centers for Disease Control and Prevention have also reported a number of cases of lead and mercury poisoning associated with rasa shastra containing Ayurvedic medicines.[5]
Literal and symbolic meaning of bhasma
The Sanskrit word bhasma literally means ‘disintegration’. Bha implies bhartsanam (to destroy), while sma implies smaranam (to remember). Bhasma is thus a reminder to us of the ephemeral nature of life. Also, if we wish to unite with God (or the ‘supreme self’) and remember him constantly, our ego or ‘little self’ has first to be disintegrated or burnt to ashes. Bhasma is a symbol of this process. It is also called raksha because it protects one from all fears. When applied to the forehead before sleep, it is said to keep away spirits or ghosts, whether external or those which manifest from the depths of the mind in the form of nightmares.
Bhasma symbolises the burning of our false identification with the mortal body, and freedom from the limitations of the painfully illusive cycle of birth and death. It also reminds us of the perishable quality of the body, which will one day be reduced to mere ashes. As it says in the Bible, “Ashes to ashes; soul to soul” – the body will return to dust but the soul will continue its journey until it unites with God. All the saints and sages beseech us to remember the ephemeral nature of our earthly existence. In the Rubayyat of Omar Khyyam the poet tells us to, “ . . . make the most of what we yet may spend, before we too into the dust descend, dust into dust, and under dust to lie.” Here he calls for us to seek the eternal, not the temporal. Ash or dust, on the other hand, can be said to represent permanency (or the soul itself), because the ash, just like imperishable truth, does not itself decay. The realised soul is said to rise from the ashes (of the individual self) as the mythical phoenix. The Sufis say, “To reach the goal we have to be burned with the fire of love, so that nothing remains but ashes, and from the ashes will resurrect the new being. Only then can there be real creation!”
The power of bhasma
Bhasma is also called ‘vibhooti’, because it gives spiritual power. The Sanskrit word, vibhooti means ‘glory’, as it gives glory to one who applies it, protection (raksha) from ill health and negative forces, and attracts the higher forces of nature. Another meaning of vibhooti is ‘healing power’, and it is widely used as a medicinal treatment in both Ayurveda and Chinese and Tibetan medicine, which are all ancient and profound systems for the rejuvenation of life. Gold, silver, copper, pearls, mica and other precious stones and metals have curative properties which can quite safely and most effectively be taken into the body after being reduced to ash using great heat.
In Indian villages you will find tantric healers called ojhas who say certain mantras over the ash, which the sick person then applies to the body or eats. These healers can take some earth in their hands, hold it up to the sun, repeat some mantras, and the earth turns into the most beautifully scented ash for curative purposes. Vibhooti is also the name given to siddhis (perfections or psychic powers), as it acts as a vehicle for them. Patanjali’s Yoga Sutras devotes an entire chapter to yogic siddhis. Vibhooti also means ‘dominion’, and is the subtle power lying behind creation, from which all things manifest. From vibhooti or bhasma, anything can be created by a tantric and aghora, because the potential of creation lies within it, and he has penetrated the law and controlled the elements.
Maha Yogi Shiva, father of tantra, is usually depicted naked in sadhana, his whole body covered in bhasma. The first verse of the Shiva Panchakshara Stotram gives the following description: Naagendrahaaraaya trilochanaaya, bhasmaangaraagaaya maheshwaraaya. Nityaaya shuddhaaya digambaraaya – ‘Salutations to the mighty three-eyed Shiva, eternal and pure, wearing the king of snakes as his garland, naked and besmeared with sacred ash.’ Some other names given to Lord Shiva are Bhasmashayaaya (abode of bhasma) and Bhasmabhootaaya (covered with bhasma). Covering the body with ash is considered to be an auspicious act for discovering one’s Shiva nature. Shiva is said to be responsible for mahapralaya, the dissolution of the universe at the end of each kalpa. At this time he dances his tandava nritya, the dance of destruction.
The great tantric siddha Avadhoota Dattatreya was referred to as Bhasma Nishta – one who loves bhasma. Bhasma is generally applied on the forehead, while many sadhus also apply it on the arms, chest and stomach. Some ascetics, especially nagas (naked ascetics) rub it all over the body. While applying it, many devotees also consume a pinch. Shaivites use only bhasma from cremated bodies, which is believed to be very powerful. Bhasma has the power of fire. Agni, the inner fire, scorches and reduces all impurities in the body. It is said that one who smears ash on the body is purified as if bathed in fire. This is known as ‘the bath of fire’. After smearing the body with ash, one should reflect on and realise the highest truth.
Tripundra
Sannyasins wear three lines of bhasma on the forehead. These three lines (tripundra), with a red dot of kumkum underneath, between the eyebrows, symbolise Shiva-Shakti (the unity of energy and matter that creates the entire seen and unseen universe). The lower line represents tamoguna (the state of inertia and darkness), the middle line represents rajas (activity and dynamism) and the top line represents sattwa (balance and illumination). The red dot or tika represents the power of shakti through sadhana, which can take the sadhaka beyond the three gunas or qualities to the state of turiya, the fourth dimension of existence. This is the state of trigunatita – beyond the three gunas.
Swami Niranjanananda says, “The three stripes represent the tradition of the paramahamsas. Jignasus are one stripe sannyasins, representing the drive and motivation to overcome the tamasic tendency. Karma sannyasins are given two stripes, representing their drive to overcome the rajasic along with the tamasic tendencies. Poorna sannyasins are given three stripes, which represent their motivation to transcend the three gunas and attain inner sublimation. The red dot represents the spiritual power or energy that gives us the strength to control the three gunas. It is the awakening of that shakti which is the real aim of sannyasa.”
Bhasma and tattwa shuddhi
Consciousness manifests as energy, which then condenses into matter. In the tantric practice of tattwa shuddhi, in order to experience consciousness free from matter, we reverse the process of evolution back through more and more subtle dimensions to its original cause. Bhasma is an integral part of tattwa shuddhi sadhana, as a symbol of purification on the physical, subtle and causal realms of consciousness. The process of disintegration undergone in tattwa shuddhi is the breaking down of conscious awareness. Just as we reduce matter to its bhasmic form, the ‘fire’ of this practice leads us to the realisation of our essential essence. The stages of pratyahara (sense withdrawal) and dharana (concentration) take us through the more subtle states of consciousness, culminating in samadhi, the ultimate experience or ‘Shiva consciousness’. The journey is from gross matter to pure consciousness.
At the end of the practice of tattwa shuddhi, bhasma is applied to the forehead with the repetition of mantras. It is taken on the middle and ring fingers and wiped slowly on the forehead from left to right, repeating the mantra Om Hraum Namah Shivaya. Sannyasins use the index, middle and ring fingers, and repeat the mantra Om Hamsa. The bhasma used in tattwa shuddhi is prepared from gobar or cow dung. The word gobar literally means ‘gift from the cow’; it is also known as go-maya. The cow is a pure and sacred animal, full of auspicious qualities. It is even said to contain all the devas and devatas within it. Not only does gobar have mystical qualities, but it also contains useful hormones with germicidal properties. The word go also means ‘senses’. So bhasma is also symbolic of the disintegration of the senses which keep us trapped and bound in the gross material world. The transformation of gobar to bhasma is parallel to the transformation from the material world to cosmic consciousness that we find in tattwa shuddhi.
Panchagni bhasma
During the Sat Chandi Mahayajna, and on other auspicious occasions at Rikhia Dham, devotees receive the precious prasadam of panchagni bhasma. This is much prized by sadhakas, because as it has the power of Swami Satyananda’s sadhana behind it, it quickly helps to raise the consciousness at the time of mantra japa and other sadhana when applied to the forehead. Just keeping it in the pooja room is auspicious. This bhasma given is from the Maha Kaal Chita Dhuni, where the previously fierce fires of Sri Swamiji’s panchagni tapasya now lie smouldering quietly under ashes in their shanta roopa or peaceful form. Dhuni is the yogi’s fire, which is the witness or sakshi to his sadhana. It is also where he cooks, takes warmth, and chants the name of God. (Maha means ‘great’, kaal is ‘time’ and chita is ‘consciousness’).
This akhanda dhuni, eternal fire, has been burning in Sri Swamiji’s pooja area ever since he first came to Rikhia in 1989 and devotees come daily for its darshan. It was the centre and support of his life during his austerity, and is the very heart of the Rikhia Ashram (next to Sri Swamiji himself). Although Sri Swamiji no longer goes to this area, the fire is still tended daily. The ashes are moved to the side and the burning embers taken out. Balls of dried cow dung mixed with purifying herbs (vanaspati) are then placed inside along with fresh wood. The embers are then replaced, and the whole area is covered over once more with the ashes. From time to time the ash is removed, carefully sieved through fine cloth, and given as prasadam (that which has been blessed by a divine power.
For the panchagni sadhana itself, Sri Swamiji prepared his own bhasma to protect his body from the great heat, according to the formula prescribed in the Devi Bhagavat Purana. This special bhasma is called mahabhasma and is made from pure cow dung cakes, reeds and ghee. It is treated eleven times with many herbs, honey and other ingredients, being re-burnt each time. Bhasma is smeared on the body only during the first few days of the panchagni sadhana, and is applied in the morning. Sri Swamiji’s dog-cum-companion Bholenath, in whom he manifested the spirit of Bhairava, also took part in the tapasya. “Alsatian dogs can’t bear the heat,” commented Sri Swamiji. “I would put bhasma on him in the morning and he would sit with me.”
Tantric siddhas like Maha Yogi Shiva, Avadhoota Dattatreya and Sri Swamiji are extremely rare beings, and a gift to us beyond our understanding. They belong to a great tradition and leave behind for us a great spiritual legacy. The parampara, the line of avadhootas (those who have become immortal), continues, just as the Mahakaal Chita Dhuni continues to smoulder, unseen beneath the symbol of their glory, their bhasma – the sacred ash.
Abhrak Bhasma
Strengthens body, ligaments & Saptadhatu, effective in raktapitta, vat diseases and joint pain. Relieves problems related to chronic hyperacidity like stomache, headache etc.

Details
The Indian ayurved philosophy is founded on three basic classifications of human body known as doshasand its well-being:
Vat – related to the central nervous system and gastric tract,
Pitta –digestion and metabolism, governs movement of heat in the body and
Kapha – concerned with structure, stability and fluid balance in the body.
Indian herbal medicines are designed for the specific maintenance of these respective doshas or the aspects of the human body.
Abhrak Bhasma or Sahastraputi is an ancient Indian ayurvedic medicine which is trusted worldwide for healing Vat related issues of the human body. It is also found extremely effective for treating heart related issues.
Abhrak is known to have a high penetrative property which spreads in the body at a faster rate and impacts micro tissues quickly.That is why this herbal medicine is supremely effective in cell regeneration. When used as an alterative, it strengthens ligaments. It is known for rapidly increasing the production of T-cell phagocytes, antibacterial components, which are responsible for a strong immune system.
Kesar Herbals has been in the ayurvedic medicine manufacturing business for about 25 years. This company is one of the foremost and trusted ayurvedic medicine suppliers in India.The concept was let entire mankind benefit from the ancient herbal wealth of India.
Composition:
Purified Mica, Magnesium, Iron, Potassium, Calcium and Aluminium.
Major Benefits of Abhrak Bhasma
- It finds its application in curing diseases like Hepatitis, Tuberculosis, Asthma and Plague.
- Abhrak Bhasma is also recommended for breathlessness due to chronic Bronchitis, Asthma, and even Chest Congestion. When taken in the initial stage of TB, it can completely cure it.
- It is beneficial to all the seven dhatus (energy elements) in the body.
- It rejuvenates the human mind.
- It improves blood circulation, improves the general tone of tissues and conductivity.
- It cures erectile dysfunction and impotency in men.
- Abhrak is hematinic, which means increases the count of red blood cells. This also increases oxygen carrying ability of the cells.
- It helps in curing jaundice and anemia.
- It restores damaged tissues and maintains their health.
- It is highly effective in cases of bone marrow depletion and hepatic dysfunction.
- It is effective in curing respiratory tract infections.
- It is beneficial for curing Bells Palsy and dysentery.
- It is beneficial in curing anemia, pernicious and azoospermia.
- Also, helps in controlling many types of veneral diseases like EBV, HIV, Lupus bronchitis and pneumonia.
- It cures breast cancer.
- It is also known to cure chronic hyperacidity like stomach-ache and vomiting with blood.
Suvarna Bhasma
Boosts immunity, Acts as a nervine tonic , Indicated in old age debility, urinal disorders, anemia, Shwas, Helps to improve complexion, Kas & chronic fever etc.

Details
Swarna Bhasma is an ancient Indian ayurvedic medicine that enjoys wide popularity and application.Gold has been accepted as most useful ingredient in Indian ayurvedic medicine system. SwarnaBhasma is used for the effective treatment of Gonorrhoea and Syphilis.
Fortified with potent ayurvedic herbs it is effective for maintaining a healthy life-style and relieves stress.Bhasma denotes the metal based medicine prepared from metals after scientific process to harness the hidden richness of raw metals for therapeutic effects. Swarna Bhasmaor Gold Ash is a therapeutic form of gold metal of nano-sized particles that is one of the most treasured ayurvedic medicines in the world.
It has multi-purpose ayurvedic medicine: it can be used as an antacid, haematinic, and alterative. It is highly effective as a cardiac tonic, maintaining the vitality of the human heart. It is a general vitality booster.
Kesar Herbals firmly believes in the power of nature in healing various ailments. Ayurvedic medicines from India are a gift from nature itself, without any side-effects, and hence they are trusted world-wide.Through scientific processes, nature’s best healing powers from herbs are obtained and used for medicinal purposes, helping million people worldwide. Kesar Herbals is involved in consulting, manufacturing and supplying FDA approved ayurvedic medicines for over three decades.
Major Benefits of Swarna Bhasma
- Swarna Bhasma improves resistance power in human body.
- It is responsible for boosting immunity and strengthening the human body.
- It can be used on a regular basis as an alterative and acts as a nerve tonic.
- Swarna Bhasma is excellent in treating Anemia, and chronic fever.
- Due to its gold base, it is helpful in improving complexion.
- Swarna Bhasma eradicates all chronic disorders, when used consistently.
- It is indicated in chronic fever, cough, Asthma, urinary disorders, sleeplessness and weak digestion.
- It strengthens weak muscles, debilities associated with old-age.
- It is excellent for treating Tuberculosis.
- It increases sexual power.
- Swarna Bhasma slows down the process of degeneration of tissues thus helps to prevent premature ageing.
- It helps prevent symptoms of ageing like wrinkles, dullness of skin, dark circles around the eyes, debility, fatigue, etc.
- Swarna Bhasma strengthens tissues and ligaments of the human body.
- It is highly effective in maintaining vigor, vitality and stamina.
Hirak Bhasma
effective against immunity disorders, Bone marrow depression and arthritis, increases stamina, vitality and strength
Ayurvedic medicinal science has been at the forefront of solving most diseases and disorders of the human body because of the purity of its ingredients- this forms the core of the ancient Indian healing system. Ayurvedic medicines and herbal products are effective because of the composition or the formulation of herbs. One such effective ayurvedic medicine is the Hirak Bhasma, which contains diamond ash.
According to Ayurveda, diamond ash is very useful in cancers, immunity disorders, Crippling Rheumatoid arthritis, and even bone marrow depression. Hirak Bhasma is known for toning heart muscles and avoiding cardiovascular syndromes and diseases. It is highly effective as a tonic and alterative, which means, it can be used regularly to strengthen immunity, aid stamina and overall vitality or the functionality of the human body.
Hirak Bhasma is made with diamond- the most precious as well as the hardest gemstone known to mankind. In its healing formulation, diamond is known since ages to make the human body stronger. It is also known for sharpening the human mind by strengthening memory power.
The effectiveness of ayurvedic medicines depends on the company. So be careful in selecting the brand for these medicines and products. Kesar Herbals has been in the ayurvedic consulting space and even manufacturing and supplying of ayurvedic medicines and herbal products for over 25 years. The trust and good will it represents in the field of Ayurveda is affirmed by its customers the world over. It is important to ascertain the brand equity of the company before purchasing these medicines.
Composition:
Hirak Bhasma (diamond ash), Kumari Rasa (Aloe Vera Juice), Gulab Jala (Rose Water), Sunthi Kwath (Dry Ginger Dicoction)
Benefits:
- Hirak Bhasma is known the world over to make the human body stronger and the mind sharper.
- It is also recommended for the well-being of the human heart.
- It helps in toning the muscles of the heart.
- Its regular consumption as a tonic or an alterative, under medical prescription or supervision is known for toning the heart muscles.
- Hirak Bhasma also increases stamina, vitality and strength.
- It is highly effective against immunity disorders.
- It is known for curing Crippling Rheumatoid
- It is effective against Bone marrow depression and arthritis.
- Hirak Bhasma is also known to cure one of the most dangerous and lethal diseases known to mankind: cancer. When used in the initial stages, under medical supervision, it can cure cancerous cells.
- Ayurvedic formulary of India , 1978a
- Major Herbs of Ayurveda, Elizabeth M. Williamson. ISBN 0-443-07203-5
- http://www.atsdr.cdc.gov/toxprofiles/tp46.html
- http://toxnet.nlm.nih.gov/cgi-bin/sis/search/f?./temp/~jcaJ1Z:2
- CDC Morbidity and Mortality Weekly Report: Lead Poisoning Associated with Ayurvedic Medications — Five States, 2000–2003
FDA Approves BMS Drug for Rare Fat Disorder
1. Leptin (human), N-methionyl-
2. N-methionylleptin (human)
STRUCTURAL FORMULA
MVPIQKVQDD TKTLIKTIVT RINDISHTQS VSSKQKVTGL DFIPGLHPIL 50
TLSKMDQTLA VYQQILTSMP SRNVIQISND LENLRDLLHV LAFSKSCHLP 100
WASGLETLDS LGGVLEASGY STEVVALSRL QGSLQDMLWQ LDLSPGC 147
Disulfide bridge location
97-147
http://www.ama-assn.org/resources/doc/usan/metreleptin.pdf
MOLECULAR FORMULA C714H1167N191O221S6
MOLECULAR WEIGHT 16.16 kDa
MANUFACTURER Amylin Pharmaceuticals, Inc.
CODE DESIGNATION r-metHuLeptin
An analog of human leptin, metreleptin, has been approved in Japan and is currently under review by the FDA in the US for the treatment of diabetes and/or hypertriglyceridemia, in patients with rare forms of lipodystrophy, syndromes characterized by abnormalities in adipose tissue distribution, and severe metabolic abnormalities. Bristol-Myers Squibb has submitted a New Drug Approval (NDA) for metreleptin to the US Food and Drug Administration (FDA) Office of Orphan Products Development. In a three-year study of metreleptin in patients with lipodystrophy organized by the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health, metreleptin treatment was associated with a significant decrease in blood glucose (A1c decreased from 9.4% at baseline to 7.0% at study end) and triglyceride concentration (from 500 mg/dl at baseline to 200 mg/dl at study end). The Juvenile Diabetes Research Foundation has also partnered with Amylin Pharmaceuticals and researchers at the University of Texas Southwestern Medical Center to study whether metreleptin can be used to improve the treatment of type 1 diabetes.
N-Methionylleptin (human)
Recombinant human OB protein, purified to homogenicity as a 16-kDa monomer
Treatment of obesity and related disorders (metabolic homeostasis regulator)
- Brand name: Myalept
- Generic name: metreleptin
- Company: Amylin Pharmaceuticals, Inc.
- Treatment for: Lipodystrophy
| Feb 25, 2014 | FDA Approves Myalept to Treat Generalized Lipodystrophy |
| Dec 12, 2013 | FDA Advisory Committee Votes on Investigational Medicine Metreleptin |
| Apr 3, 2012 | Amylin Completes Biologics License Application for Metreleptin to Treat Diabetes and/or Hypertriglyceridemia in Patients With Rare Forms of Lipodystrophy |
| Dec 20, 2010 | Amylin Submits Clinical and Nonclinical Sections of Rolling Biologics License Application for Metreleptin to Treat Rare Forms of Lipodystrophy |
| Leptin | |
|---|---|

Structure of the obese protein leptin-E100.
|
“Myalept is the first approved therapy indicated for treating the complications associated with congenital or acquired generalized lipodystrophy and provides a needed treatment option for patients with this orphan disease,” said Mary Parks, M.D., deputy director of the Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research.
The safety and effectiveness of Myalept, an analog of leptin made through recombinant DNA technology, were evaluated in an open-label, single-arm study that included 48 patients with congenital or acquired generalized lipodystrophy who also had diabetes mellitus, hypertriglyceridemia, and/or elevated levels of fasting insulin. The trial showed reductions in HbA1c (a measure of blood sugar control), fasting glucose, and triglycerides.
Anti-drug antibodies with neutralizing activity to leptin and/or Myalept may develop, which could result in severe infections or loss of treatment effectiveness. T-cell lymphoma has been reported in patients with acquired generalized lipodystrophy, both treated and not treated with Myalept, so healthcare professionals should carefully consider the benefits and risks of treatment with Myalept in patients with significant hematologic abnormalities and/or acquired generalized lipodystrophy. Myalept is contraindicated in patients with general obesity. Myalept is not approved for use in patients with HIV-related lipodystrophy or in patients with metabolic disease, including diabetes mellitus and hypertriglyceridemia, without concurrent evidence of generalized lipodystrophy.
Because of the risks associated with the development of neutralizing antibodies and lymphoma, Myalept is available only through the Myalept Risk Evaluation and Mitigation Strategy (REMS) Program. Under this REMS program, prescribers must be certified with the program by enrolling in and completing training. Pharmacies must be certified with the program and only dispense Myalept after receipt of the Myalept REMS Prescription Authorization Form for each new prescription.
Myalept is also approved with a Medication Guide and instructions for use that provides patients with important information about the medication. The guide will be distributed each time a patient fills a prescription.
The FDA is requiring seven studies (post-marketing requirements) for Myalept, including a long-term prospective observational study (product exposure registry) of patients treated with Myalept, a study to assess for the immunogenicity (antibody formation) of Myalept, and an assessment and analysis of spontaneous reports of potential serious risks related to the use of Myalept. Eight additional studies are being requested as post-marketing commitments.
In clinical trials, the most common side effects observed in patients treated with Myalept were low blood sugar (hypoglycemia), headache, decreased weight, and abdominal pain.
Myalept is marketed by San Diego-based Amylin Pharmaceuticals, L.L.C.
For more information:
Metreleptin is an analogue of the human hormone leptin being developed by Amylin Pharmaceuticals (a subsidiary of Bristol-Myers Squibb) for the subcutaneous treatment of metabolic disorders including lipodystrophy. The compound is expected to improve insulin sensitivity, hypertriglyceridaemia and hyperglycaemia in patients with lipodystrophy who are unresponsive to conventional treatment.
Metreleptin has been approved in Japan as a leptin therapy for the treatment of lipodystrophy. Amylin has also completed a submission for regulatory approval to the US FDA for metreleptin in the treatment of diabetes mellitus and/or hypertriglyceridaemia in patients with rare forms of lipodystrophy.
Clinical development of the drug is also underway in the USA for the treatment of type 1 diabetes. Amgen was previously assessing the use of metreleptin as a treatment for amenorrhoea; however, it appears that development in this indication has been discontinued. This article summarizes the milestones in the development of metreleptin leading to this first approval for lipodystrophy.
Metreleptin is a leptin replacement therapy first launched in Japan in 2013 for the treatment of congenital lipodystrophy. Amylin filed for approval in the U.S. in 2010 for the treatment of diabetes and/or hypertriglyceridemia in patients with rare forms of lipodystrophy. In 2013, the Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) recommended the approval for the treatment of pediatric and adult patients with generalized lipodystrophy , but not for partial lipodystrophy.
Phase II clinical studies are also under way at Beth Israel Deaconess Medical Center for the treatment of lipodystrophy syndrome associated with AIDS. The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) is conducting phase II clinical trials for the treatment of nonalcoholic steatohepatitis. Phase II are ongoing at the National Institute for Diabetes and Digestive and Kidney Diseases for the treatment of non-alcoholic fatty liver disease (NAFLD) associated with lipodystropy. Early clinical studies had also been ongoing for the treatment of leptin deficiencies.
The University Texas Southwestern Medical Center at Dallas is evaluating metreleptin for the treatment of type 1 diabetes. Beth Israel Deaconess Medical Center is conducting phase II clinical trials for the treatment of amenorrhea. Amgen had been conducting clinical trials for this indication and for the treatment of type 1 diabetes and depression; however no recent development has been reported for this research.
In 2011, Amylin and Takeda put on hold their clinical trials with metreleptin in combination with pramlintide for the treatment of obesity in order to investigate an antibody-related laboratory finding. Amylin is currently evaluating the compound as monotherapy for the treatment of obesity. The companies had been conducting phase II clinical trials of metreleptin not in combination with pramlintide for the treatment of obesity; however, no recent development has been reported for this research.
Originally developed at the Rockefeller University, an exclusive license to metreleptin was granted to Amgen in 1995. In 2009, the drug candidate was licensed to Takeda by Amylin worldwide for the treatment of obesity. In 2010, orphan drug designation was assigned in the U.S. for the treatment of metabolic disorders secondary to lipodystrophy and for the treatment of leptin deficiency secondary to generalized lipodystrophy and partial familial lipodystrophy.
In 2012, orphan drug designation was assigned in Japan for the treatment of diabetes or hyperlipidemia due to lipoatrophy. In 2012, orphan drug designation was assigned in the E.U. for the treatment of Barraquer-Simons syndrome, Berardinelli-Seip syndrome, familial partial lipodystrophy and Lawrence syndrome. In 2014, AstraZeneca acquired the global rigths for development, manufacture and commercialization of the product.
……………
Other exemplary leptins for use in the methods and compositions described herein include, but are not limited to, the amino acid sequence for mature, recombinant methionyl human leptin (herein called rmetHu-Leptin 1-146 or Metreleptin) having the amino acid sequence:
MVPIQKVQDDTKTLIKTΓVTRINDISHTQSVSSKQKVTGLDFIPGLHPILTLSKMDQTLA VYQQILTSMPSRNVIQISNDLENLRDLLHVLAFSKSCHLPWASGLETLDSLGGVLEASG YSTEWALSRLQGSLQDMLWQLDLSPGC (SEQ ID NO:274).
Regulatory Considerations for Biosimilars

Biological medicines are already becoming an increasingly important part of health care. With patent expiries on originator biological products, biosimilars are also increasingly become a part of this future. In fact, by 2020 twelve of the top-selling biologicals will have lost patent protection, opening up an estimated US$24 billion in EU sales and US$30 billion in US sales.
Biologicals have potential to reach up to 50% share in global pharmaceutical market in the next few years.
India is one of the leading contributors in the world biosimilar market and is the third-largest in the Asia-Pacific region, after Australia and China. India has demonstrated high acceptance of biosimilars, which is reflected in the 40 biologicals marketed in India, of which 25 are biosimilars The Indian biotechnology industry is also gaining momentum, with revenues of over US$4 billion in 2011, and which are projected to reach up to US$580 million by 2012.
While small molecule drugs are ideal for generics replication, biological drugs are not so simple. Biological drugs are usually large, complex molecular structures derived from or produced through a living organism, making them very difficult to replicate
Currently there is considerable interest in the legislative debate around generic biological drugs or “biosimilars” in the EU and US due to the large, lucrative market that it offers to the industry. While some countries have issued a few regulatory guidelines as well as product specific requirements, there is no general consensus as to a single, simple mechanism similar to the bioequivalence determination that leads to approval of generic small molecules all over the world. The inherent complex nature of the molecules, along with complicated manufacturing and analytical techniques to characterize them make it difficult to rely on a single human pharmacokinetic study for assurance of safety and efficacy.
In general, the concept of comparability has been used for evaluation of the currently approved “similar” biological where a step by step assessment on the quality, preclinical and clinical aspects is made. In India, the focus is primarily on the availability and affordability of life-saving drugs. In this context every product needs to be evaluated on its own merit irrespective of the innovator brand. The formation of the National Biotechnology Regulatory Authority may provide a step in the right direction for regulation of these complex molecules. However, in order to have an efficient machinery for initial approval and ongoing oversight with a country-specific focus, cooperation with international authorities for granting approvals and continuous risk-benefit review is essential. Several steps are still needed for India to be perceived as a country that leads the world in providing quality biological products.

We are now in the twenty-fifth anniversary year of the Drug Price Competition and Patent Term Restoration Act of 1984 (better known as the Hatch Waxman Act), the landmark US regulation that jump-started the generic pharmaceutical industry. The legislation provided the required impetus to make not just cheaper price alternative medicines available to US consumers but stimulated the emergence of the Indian pharmaceutical industry which is now the dominant supplier of generic drugs to the USA.
The regulatory pathway for bringing generic drugs to market is the abbreviated new drug application (ANDA) process which relies on proving bioequivalence to the listed reference product and showing equivalent product quality. Since duplication of proof of safety and efficacy in the preclinical and clinical setting is not required, there are significant cost savings in bringing a copy of a small chemical molecule to market. This model has been so successful in economic terms that almost 7 out of 10 prescriptions in the US are now generic and for the vast majority of products there is no concern in substitution of a generic equivalent for a brand-name prescription.1

The success story of generic small molecule drugs has stimulated interest in the pharmaceutical and biotech industry for applying an analogous approach towards the highly lucrative biologics business. But biologic drugs are very different from small molecules both in their final form and in the process required to produce and control their quality. It is therefore difficult to find a simple, precise “regulatory” definition of biologics. However, biologics are generally understood to be drugs derived from an organic source. Thus, biologics may be obtained or created from living organisms, either naturally or via genetic manipulation or are manufactured from building blocks of living organisms. They demonstrate considerable molecular complexity and may comprise a diversity of molecular forms. Their larger size and heterogeneity make it difficult for complete characterization via physicochemical analysis which is possible for synthetic chemical entities. In general, biologic drugs are more expensive and the cost of a yearly treatment may run into thousands of dollars for some. They are therefore ideal targets for developing cheaper alternatives.
US FDA definition – A “biological product” means a virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product, or arsphenamine or derivative of arsphenamine (or any other trivalent organic arsenic compound), applicable to the prevention, treatment, or cure of a disease or condition of human beings (Public Health Service Act Sec. 351(i)).
Given the complexity of the final biologic product, it is clear that the nature of the manufacturing process is also complicated. In addition to aspects that are disclosed in regulatory applications, there may still be several aspects which might be held as trade secrets, thereby making it practically impossible for another company to make an identical copy of a biologic drug. While changing a host cell line or vector will definitely impact the product, effects of minor changes like temperature used in the manufacturing process may have an effect on the final characteristics of the biologic drug, including its safety and efficacy. It has been stated often that for a biologic, “the process defines the product”. Thus, while it may be possible to make a similar product, it may not be truly bioequivalent. As a result, even the term used to describe these similar biologic drugs has not been standardized globally. While the parallel term for a biologic generic may intuitively be “biogeneric”, the accepted term in Europe and Canada is “biosimilar” and the preferred term in the US is “follow-on biologic”.
Given these differences among innovator biologics and their “similar” counterparts, there is considerable hesitation on the part of the regulatory agencies to follow an abbreviated approval path similar to one widely used for generic small molecules.

Status of biosimilar regulation in Europe
EMEA Guidelines for Similar Biological Medical Products ( CHMP/437/04, 30 October 2005).2
EMEA’s Guideline on Similar Biological Medicinal Products Containing Biotechnology-derived Proteins as Active Substance: Nonclinical and Clinical Issues (EMEA/ CHMP/BMWP/42832/2005).3
EMEA’s Guideline on Similar Biological Medicinal Products Containing Biotechnology-derived Proteins as Active Substance: Quality Issues, EMEA/CHMP/49348/05.4
In Europe, the Committee for Medicinal Products for Human Use (CHMP), the European Medicines Agency (EMEA) led the way for biosimilars, by issuing its first specific regulatory guidance in October 2005. Two general guidance documents addressing quality and nonclinical and clinical perspectives (June 2006), five product-specific annexes on nonclinical and clinical issues (June-July 2006) and a manufacturing change comparability guideline (November 2007) are now available.
Biosimilars
Testing the bioequivalence of biosimilars differs from that of standard generics. Bioequivalence testing procedures for biosimilars are to be performed against the originator product as a control (reference) and include preclinical and clinical testing [2].
In the Biologics Price Competition and Innovation (BPCI) Act, a biosimilar product is defined as a product that is ‘highly similar’ to the reference product, notwithstanding minor differences in clinically inactive components and there are no clinically meaningful differences in terms of safety, purity and potency. However, little or no discussion regarding how similar is considered ‘highly similar’ is given in the BPCI Act.
For biosimilars, most of which have long half-lives, crossover study would be ineffective and unethical. This is due to the fact that a crossover study requires a wash-out period (which would be long for biosimilars with long half-lives) where the patient is not allowed to take the drug and therefore will have no treatment for their condition. On the other hand, parallel-group studies are required, but these studies do not provide an estimate of within-subject variation. For a parallel-group study, each drug is administered to a different group of subjects. Thus, we can only estimate total variance (between and within-subject variances), not individual variance components. This makes an evaluation of interchangeability difficult.
Statistical tests that may be used to asses biosimilarity are Shuirmann’s two one-sided tests procedure or the confidence interval approach.
Status of Biosimilar Regulation in US
In US, in March 2009, Representative Henry Waxman introduced H.R. 1427 to the Congress “Promoting Innovation and Access to Life-saving Medicines Act”, which authorizes FDA to approve follow-on biologics in an abbreviated manner. It has market exclusivity clauses with time frames similar to ones used currently for drugs. Other bills are expected to follow in the 2009 legislative agenda in order to establish a pathway for approval of these follow-on biologics. The contentious issues as expected, are focused around the duration of exclusivity benefits granted to innovators. The issue of substitutability of followon biologics for reimbursement is also an important one as the legislators debate the merits of each bill.
Korea and Singapore have released draft guidelines on biosimilars in 2009. The Singapore guideline is derived mainly from the EMEA guidelines and defines a similar biological/ biosimilar product as “a biological medicinal product referring to an existing registered product, submitted for medicinal product registration by an independent applicant, and is subject to all applicable data protection periods and/or intellectual property rights for the original product”. In addition to specifying the requirements for biosimilars, the guidance requires that the product have prior approval in countries such as Australia, Canada, EMEA or US.
Indian scenario
The Indian biotech industry is a thriving industry which got its start from vaccine manufacturing. In addition to meeting domestic demands, the Indian vaccine industry also fulfils export requirements to a large extent. Therefore it is evident that manufacturing expertise in producing biologic products of required export quality already exists in the country. What is not readily evident is whether these products can prove to be “comparable” to innovator products when we look into all categories of biologics.
The evolution of regulations governing pharmaceuticals in India has historically been driven by the need to make essential medicines accessible to patients. Access encompasses availability and affordability. It applies to medicines for all indications, acute and chronic illness, small molecules and biologics alike. The absence of product patent regulations for drugs marked a period in the country’s history where it was imperative to make inexpensive medicinal products available to the masses – it did not matter whether these products were innovator-made or copies thereof. In the post-TRIPS era however, there is need to offer and enforce adequate protections for patentable drugs, particularly biologics that inherently involve huge investments in R & D, manufacture and clinical development.

Today, several biologics have been approved in India , including recombinant human insulin, recombinant human erythropoietin (EPO), interferon (IFN), granulocyte colony stimulating factor (GCSF). The versions of biologics available in India are typically products whose patents have expired or do not exist in India. Therefore, from a technical standpoint, there is no concern about patent infringement regarding these (there are no patents in India for these products). If a biosimilar results in a price drop of 30%, it is a significant improvement to patients who may now be able to afford this generic version of a life-saving drug. In many ways, the debate about biosimilars that rages across the developed world and regulated markets is irrelevant to India where the central concern revolves around access.
Partly due to the dearth of appropriate resources and experience, Indian regulators have sought to mimic regulations already in use in the developed world without much customization. A host of agencies have been created to address the issues brought forth by biologics.
Basic facts about biosimilars.
Biotechnological drugs have become an essential part of modern pharmacotherapy and are expected to reach a 50% share in the pharmaceutical market in the next few years. The expiry of patent protection for many original biotechnological medicines has led to the development of what are called biosimilars or follow-on biologics. Biosimilars attempt to copy the original technology leading to the production of innovative biotechnological medicines to obtain a product which is similar to the original one. The first two biosimilars have recently been approved in the European Union and one application was rejected. Many more biosimilars will likely see approval in the near future. Our experience with biosimilars has been very limited to date and long-term safety data including immunogenicity are not available. Although biosimilars will likely lower the cost of modern therapies there are issues which have to be discussed at this stage among physicians regarding in particular the differences between biosimilars and generics of the classical chemical drugs, need for appropriate regulations as well as identification of potential problems with biosimilars. Other specific problems which will also be addressed in this review are safety of biosimilars, pharmacovigilance, automatic substitution, naming and labeling/prescription rules. 7
List of agencies
- Indian Council for Medical Research (ICMR)
- Central Drugs Standard Control Organisation (CDSCO)
- Department of Biotechnology (DBT)
- Genetic Engineering Approval Committee (GEAC)
- Recombinant DNA advisory Committee (RDAC)
- Review Committee on Genetic Manipulation (RCGM)
- Institutional Biosafety Committee (IBSC)
- National Centre for Biological Sciences
- National Control Laboratory for Biologicals
Notwithstanding the above, there is clarity on the fact that biologics and drugs need to be scrutinized differently. With this in mind, the DBT has been given the mandate to set up the National Biotechnology Regulatory Authority (NBRA). This is envisaged as an independent, autonomous and professionally led body to provide a single window mechanism for biosafety clearance of genetically modified products and processes.
Before such an organization can be effectively implemented, it will be necessary to put in place appropriate new legislation, namely the “National Biotechnology Regulatory Act” or the NBR Act. Draft establishment plan and “Draft National Biotechnology Regulatory Bill, 2008” are currently available on the DBT website for comments. The responsibility of consolidating the feedback has been entrusted to Biotech Consortium India Limited (BCIL). The draft bill envisions the scope of this authority to encompass research, manufacture, import and use of genetically engineered organisms and products derived thereof.
Biosimilars: how similar or dissimilar are they?
The imminent expiry of patents on biological medicinal products, such as epoetin alfa in 2006, has significant implications for nephrology in Australia. The purpose of this review is to examine the differences between biosimilars (similar biological medicinal products) and generic low molecular weight (chemical) drugs. The approach that regulatory agencies, including the European Medicines Agency (EMEA) and the Therapeutic Goods Administration (TGA), are taking towards biosimilars is also discussed. Biosimilars differ from generic chemical drugs in many important ways, including the size and complexity of the active substance, the nature of the starting materials (cell banks, tissues and other biological products), and the complexity of the manufacturing processes. Therefore, it has been acknowledged by the EMEA that established legal and regulatory principles of ‘essential similarity’ that are applied to standard chemical generics cannot be readily applied to biosimilars. One of the key areas of concern with the introduction of biosimilars into the field of nephrology will be guaranteeing the safety and efficacy of biosimilars. New manufacturers will need to ensure that their biopharmaceutical has a similar efficacy and safety profile to the innovator product through more extensive clinical trials than the limited testing done for generic versions of low molecular weight chemical medicines. 6
Safety
The primary importance of the manufacturing process was highlighted when a slight change in the production process of an originator recombinant erythropoietin resulted in patients developing pure red cell aplasia.
To try to address this possible safety issue, guidelines from EMA on comparability of biosimilars state that preclinical data must be insufficient to demonstrate the immunological safety of some biosimilars. This means that safety must be demonstrated in cohorts of patients enrolled in clinical trials and using post marketing surveillance.
-
The challenge of biosimilars.
The purpose of this report was to review issues associated with the introduction of alternative versions of biosimilars used in the oncology setting.
Data were obtained by searches of MEDLINE, PubMed, references from relevant English-language articles, and guidelines from the European Medicines Agency.
When biosimilars are approved in EU, they will be considered ‘comparable’ to the reference product, but this does not ensure therapeutic equivalence. Inherent differences between biosimilars may produce dissimilarities in clinical efficacy, safety, and immunogenicity. Switching biosimilars should be considered a change in clinical management. Regulatory guidelines have been established for some biosimilar categories but, because of the limited clinical experience with biosimilars at approval, pharmacovigilance programs will be important to establish clinical databases. Guidelines also provide a mechanism for the extrapolation of clinical indications (approved indications for which the biosimilar has not been studied). This may be of concern where differences in biological activity can result in adverse outcomes or when safety is paramount (e.g. stem cell mobilization in healthy donors). These issues should be addressed in biosimilar labeling.
Biosimilars should provide cost savings and greater accessibility to biopharmaceuticals. A thorough knowledge surrounding biosimilars will ensure the appropriate use of biopharmaceuticals.
Pharmacovigilance
Due to the limited clinical database at the time of approval of a biosimilar, vigorous pharmacovigilance is required. EMA guidelines require pharmacovigilance programmes to monitor the safety of biosimilar products post-approval.Substitution
For small molecule generics the issue of substitution is easy, since they are considered identical to the originator molecule. This, however, is not the case for biosimilars, which are large complex molecules prone to heterogeneity.In the US, the BPCI Act gives FDA the authority to designate a biosimilar as interchangeable with its reference product. This means that the biosimilar may be substituted for the originator product by the pharmacist without reference to the prescribing physician. This is not the case, however, in the EU, where decisions on interchangeability are not made by EMA, but at a national level.
Global concerns regarding product safety and quality
Every drug/biologic manufacturer needs to own the responsibility for putting a high quality, safe drug on the market, after appropriate review and approval by the concerned regulatory authority. While the safety of original biologics products is assured by the innovator by adherence to rigorous standards required for approval, the resistance towards biosimilars on the part of regulators, stems from the concern that an abbreviated approval process may not be adequate to ensure safe performance of the product in the market. For a manufacturer looking to get into the biosimilar market, he needs to overcome major challenges in making a complex product, getting regulatory approval by satisfying stringent criteria and then selling it in the market. Typically, facilities required for manufacture of biologicals are very expensive and the kind of infrastructure required to meet high regulatory expectations is limited to only a few companies. Clinical trial expenditure and ongoing analysis requires compliance to pharmacopoeial monographs when available and access to reference standards, which are not always available.
The cornerstone of generic drug approvals has been the concept of bioequivalence, using equivalence of pharmacokinetic parameters as surrogates for clinical efficacy. But in the context of biosimilars, the concept of comparability is the one used to make such an evaluation. Comparability protocols are used for chemistry, manufacturing and controls (CMC) sections to make the case on the quality aspects of the product. Preclinical testing requires knowledge of study designs used by innovator in order to truly compare performance of the biosimilar. For clinical evaluation, at least one clinical comparability trial is required to demonstrate comparability (non-inferiority in terms of efficacy to innovator and comparable safety profile). But long term safety issues remain unaddressed for biosimilars, requiring thorough postmarketing studies and pharmacovigilance and adequate risk management plans.
In terms of preclinical studies, for biologics, pharmacodynamic endpoints are more relevant than pharmacokinetics, which is the key measure with small molecules. For animal safety studies, choice of appropriate animal species and duration of studies are important criteria for proving comparability. Clinically, comparative PK/PD study is required to compare the reference and biosimilar product. However, clinical trial design selection and a thorough understanding and a priori statement of margins chosen for comparability must be stated for meaningful evaluation of data.
Follow-on biologics: challenges of the “next generation”.
The imminent patent expiration of many biopharmaceutical products will produce the possibility for generic versions of these therapeutic agents (i.e. biosimilars). However, there are a number of issues that will make approval of biosimilars much more complicated than the approval of generic equivalents of conventional pharmaceuticals. These issues centre on the intrinsic complexity of biopharmaceutical agents, which are recombinant proteins in most cases, and the heterogeneity of proteins produced by different manufacturing processes (i.e. differences in host cells, purification and processing, formulation and packaging). The increased occurrence of antibody (Ab)-mediated pure red cell aplasia (PRCA) associated with a change in the formulation of one particular epoetin-alpha product highlights the potential for increased immunogenicity of recombinant proteins with different formulations, or those manufactured by different processes. Thus, verification of the similarity to or substitutability of biosimilars with reference innovator biopharmaceutical products will require much more than a demonstration of pharmacokinetic similarity, which is sufficient for conventional, small molecule generic agents. Regulatory requirements for the approval of biosimilars have not yet been fully established, but preliminary guidelines from the European Agency for the Evaluation of Medicinal Products (EMEA) state that the complexity of the product, the types of changes in the manufacturing process, and differences in quality, safety and efficacy must be taken into account when evaluating biosimilars. For most products, results of clinical trials demonstrating safety and efficacy are likely to be required. In addition, because of the unpredictability of the onset and incidence of immunogenicity, extended post-marketing surveillance is also important and may be required. 10
Statistical assessment of biosimilar products.
Biological products or medicines are therapeutic agents that are produced using a living system or organism. Access to these life-saving biological products is limited because of their expensive costs. Patents on the early biological products will soon expire in the next few years. This allows other biopharmaceutical/biotech companies to manufacture the generic versions of the biological products, which are referred to as follow-on biological products by the U.S. Food and Drug Administration (FDA) or as biosimilar medicinal products by the European Medicine Agency (EMEA) of the European Union (EU). Competition of cost-effective follow-on biological products with equivalent efficacy and safety can cut down the costs and hence increase patients’ access to the much-needed biological pharmaceuticals. Unlike for the conventional pharmaceuticals of small molecules, the complexity and heterogeneity of the molecular structure, complicated manufacturing process, different analytical methods, and possibility of severe immunogenicity reactions make evaluation of equivalence (similarity) between the biosimilar products and their corresponding innovator product a great challenge for both the scientific community and regulatory agencies. In this paper, we provide an overview of the current regulatory requirements for approval of biosimilar products. A review of current criteria for evaluation of bioequivalence for the traditional chemical generic products is provided. A detailed description of the differences between the biosimilar and chemical generic products is given with respect to size and structure, immunogenicity, product quality attributed, and manufacturing processes. In addition, statistical considerations including design criteria, fundamental biosimilar assumptions, and statistical methods are proposed. The possibility of using genomic data in evaluation of biosimilar products is also explored.15
A way forward for India
In today’s scenario, India needs to focus on quality of each biological product per se, whether that is demonstrated through comparability or by its own merit; and assurance of safety through appropriate regulatory review and approval of available data.
Irrespective of the authority entrusted to oversight of biologics, the debate on appropriate level of regulatory scrutiny for biologics will continue to focus on requiring adequate characterization while balancing cost, with the overall goal of having a much needed product on the market with reasonable assurance of efficacy and safety. Intense discussion on publication of appropriate monographs in the Indian Pharmacopeia and availability of reference standards continues amidst regulatory circles. Indian manufacturers have always sought to enter new markets and have voluntarily raised the bar in order to secure approvals for their products in the regulated markets where profit margins are high. From a facility infrastructure and systems point of view, most companies eyeing the regulated markets for their products will most likely fulfil expectations. State-of-the art analytical techniques are available within the industry. Therefore from a quality standpoint, biologic products made in India should not have any trouble in meeting market expectations. However, physicochemical characterization of a biologic product and compliant facilities form only one part of the evaluation required to demonstrate product comparability.
The practical way forward for approval of biosimilar products in India would have to be unique to the Indian context while staying rooted to scientific basics and keeping in mind the needs and limitations of the country. The large majority of biosimilars introduced in India would be products whose patents have expired and where the “original innovator” product may not be approved in the country. It is also possible that no patent exists in India for some products and therefore , originator and similars coexist. For all products, the question of available reference standards and monographs would continue to remain. The next wave of biologics of commercial interest to the industry will become a burning issue where the regulator cannot expect to wait to see how the legislation is crafted in the US or elsewhere before making a move.
In my opinion, it seems that India, having the benefit of in-house (in-country) expertise in the area, should utilize the various agencies currently entrusted with splintered tasks and responsibilities to come up with working group or taskforce whose goal is to develop product-specific guidelines for approval. These can be developed using available worldwide regulatory knowledge by signing appropriate MOUs if necessary, studying the scientific literature and current industry standards and practice with respect to characterization, focusing on specific areas of unique concern for each product and proposing an approval path. These guidelines can be widely disseminated in the community. There will still be grey areas that need clarification and in such cases, a system for formal meeting with members of the working group/taskforce can be instituted, similar to the scientific advice that is currently available through the EMEA or individual European country competent authorities.
As a nation that takes pride in being the “exporter to the world” in the arena of pharmaceuticals, it behooves not just the regulators but all those in the regulatory affairs profession in India to support such initiatives to make life-saving products available to our countrymen that are unquestionably of the highest standards in terms of quality, safety and efficacy such that we become the supplier of choice when it comes to exporting biosimilars to markets in every corner of the world.
Biosimilar therapeutics-what do we need to consider?
Patents for the first generation of approved biopharmaceuticals have either expired or are about to expire. Thus the market is opening for generic versions, referred to as ‘biosimilars’ (European Union) or ‘follow-on protein products’ (United States). Healthcare professionals need to understand the critical issues surrounding the use of biosimilars to make informed treatment decisions.The complex high-molecular-weight three-dimensional structures of biopharmaceuticals, their heterogeneity and dependence on production in living cells makes them different from classical chemical drugs. Current analytical methods cannot characterize these complex molecules sufficiently to confirm structural equivalence with reference molecules. Verification of the similarity of biosimilars to innovator biopharmaceuticals remains a key challenge. Furthermore, a critical safety issue, the immunogenicity of biopharmaceuticals, has been highlighted in recent years, confirming a need for comprehensive immunogenicity testing prior to approval and extended post-marketing surveillance.Biosimilars present a new set of challenges for regulatory authorities when compared with conventional generics. While the demonstration of a pharmacokinetic similarity is sufficient for conventional, small-molecule generic agents, a number of issues will make the approval of biosimilars more complicated. Documents recently published by the European Medicines Agency (EMEA) outlining requirements for the market approval of biosimilars provide much-needed guidance. The EMEA has approved a number of biosimilar products in a scientifically rigorous and balanced process. Outstanding issues include the interchangeability of biosimilars and innovator products, the possible need for unique naming to differentiate the various biopharmaceutical products, and more comprehensive labelling for biosimilars to include relevant clinical data. 5
Biosimilars: policy, clinical, and regulatory considerations.
The regulatory background surrounding biosimilars (biopharmaceuticals that are considered similar in composition to an innovator product, but not necessarily clinically interchangeable); equivalence, interchangeability, and unique considerations associated with biopharmaceuticals; the biopharmaceutical protein production process; scientific facts for use in the policy discussion about biosimilars; the European Union system for biosimilars; and the current status of biosimilars legislation in the United States are described.
An abbreviated regulatory pathway for the approval of biosimilars, and a process for safely demonstrating the therapeutic interchangeability of these proteins, has the potential to provide meaningful cost savings. This economic advantage to patients can translate into important public health benefits. But to date, no formal regulatory process exists in the United States for bringing these drugs to market. In addition, the current tools for fully characterizing biopharmaceuticals are not–in certain cases–well developed, especially for proteins that have complex structures or are heavily glycosylated. In addition, using “similar” but not completely “identical” proteins interchangeably raises concerns about potentiating immunogenicity. The bottom line is that demonstrating therapeutic equivalence and interchangeability for biosimilars is not a straightforward matter–it cannot be based on the same criteria as for conventional small-molecule drugs. The science, while obtainable, is more complex. For example, it is assumed that showing that a biosimilar protein can be safely used interchangeably with an innovator protein would require, at the least, some limited clinical data and interchangeability studies. Notwithstanding the more complex scientific and clinical issues particular to protein products, most believe that a process for enabling the approval of safe and effective biosimilar proteins is not only possible, but an important public health goal. The European Union system for biosimilars may provide a model for anticipating and resolving the scientific and policy issues related to biosimilars in the U.S.
The legal and regulatory status of biosimilars remains to be resolved in the United States as policymakers address the scientific and policy issues surrounding product manufacturing, patent terms, and clinical use.
Biosimilars: it’s not as simple as cost alone.
Biosimilars or follow-on biologics (FoB) are biopharmaceuticals that, unlike small molecule generic products, are copies of larger, much more complex proteins. As such, data generated from one biopharmaceutical cannot be extrapolated to another. Unlike small molecule generics, FoB require a full developmental programme, albeit smaller than for an originator product. This has been recognized by European regulatory authorities and it is becoming clear that accelerated processes for FoB marketing approval are not feasible.
To determine the balance between costs surrounding FoB (including relatively extensive developmental programmes and subsequent price to the market) and the necessity to ensure efficacy and safety.
It is important that FoB are sufficiently tested to ensure patient safety is not compromised. Conducting such a development programme followed by sound pharmacovigilance is very challenging and costly.
Cost-savings associated with FoB may be limited. 10
Recommendations regarding technical standards for follow-on biologics: comparability, similarity, interchangeability.
Policy makers around the world are currently considering the creation of a regulatory pathway for follow-on biologics (FOB), which will have to account for the substantial technical challenges associated with FOB development. These challenges will likely involve more complexity than comparability assessments of process changes made by the same manufacturer. The history of industry-regulator comparability discussions helps explain why the same degree of testing and flexibility now applied to change-control within a manufacturer’s own process, at this time, cannot be extrapolated to the observed and possibly unknown differences between two manufacturing processes that are independently developed by different (non-collaborating) parties.
This commentary provides recommendations on the technical aspects that should be considered in the creation of an approval pathway for FOB products.
In the authors’ view, analytical methodology in its current state cannot alone provide full assurance that the FOB is sufficiently similar to the innovator product. Moreover, the FOB manufacturer will not have access to the extensive knowledge accumulated by the innovator manufacturer from early development through marketing. Thus, extensive clinical evaluation will likely be necessary to provide assurance that the FOB is safe and efficacious. If such testing demonstrates the FOB is safe and efficacious per existing regulatory standards, the product should receive marketing approval as a ‘similar’ product. Since ‘similarity’ is a fundamentally different determination than establishing interchangeability between the two products, an interchangeability determination must be based on additional testing and market experience to ensure patient safety. Post-marketing surveillance of the FOB should be conducted to ensure that the approved molecule has similar clinical safety and efficacy as the innovator product, prior to any consideration of interchangeability 11
European regulatory guidelines for biosimilars.
The impending arrival en masse of biosimilars on Western markets is placing drug regulatory agencies under pressure to realign their policies. Biosimilars require more rigorous assessments than traditional chemical generics. This is because of the molecular complexity of recombinant proteins, and the complexity of biological manufacturing processes. Small differences can arise in a recombinant protein product which are hard or impossible to detect with even state-of-the-art analytical techniques. Yet, these differences can have significant impact on the safety and efficacy of the drug. The European Medicines Agency (EMEA) has taken the lead in issuing guidelines, most of which are still under review. The guidelines advocate pre-clinical and clinical testing of biosimilars prior to market authorization, complemented by tailored pharmacovigilance plans. These guidelines provide a valuable base from which to develop in this evolving regulatory environment.12
Legislative initiatives in Europe, Canada and the US for market authorization of follow-on biologics.
The formulation and application of legal and regulatory requirements for the market authorization of follow-on versions of biological drugs present challenges. This review discusses relevant regulatory guidelines and legislative initiatives related to market authorization for follow-on biologics in Europe, Canada and the US. The respective positions of these three markets is analyzed with regard to several factors: criteria for the choice of reference products; requirements for the comparability exercise between a candidate follow-on biologic and the selected reference product, with an emphasis on considerations of quality, safety and efficacy data; the interchangeability of a reference product with related follow-on drugs; data exclusivity provisions; and the application of specialized patent enforcement mechanisms to follow-on biologics.13
Quality, safety and efficacy of follow-on biologics in Japan.
Recently, WHO, EU, Japan and Canada have published guidelines on biosimilar/follow-on biologics. While there seems to be no significant difference in the general concept in these guidelines, the data to be submitted for product approval are partially different. Differences have been noted in the requirements for comparability studies on stability, prerequisites for reference product, or for the need of comparability exercise for determination of process-related impurities. In Japan, there have been many discussions about the amount and extent of data for approval of follow-on biologics. We try to clarify the scientific background and rational for regulatory pathway of biosimilar/follow-on biologics in Japan in comparison with the guidelines available from WHO, EU and Canada. In this article, we address and discuss the scientific background underlying these differences to facilitate the harmonization of follow-on biologic principles in the guidelines in future.14
References
RAMELTEON, TAK 375 ..Melatonin MT1/MT2 receptor agonist

RAMELTEON
- HSDB 7787
- Ramelteon
- Rozerem
- TAK-375
- UNII-901AS54I69
| CAS number | 196597-26-9 |
|---|
| (S)-N-[2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)ethyl]propionamide |
(5)-N-[2-(l,6,7,8-tetrahydro-2H-indeno-[5,4-ό]furan-8- yl)ethyl]propionamide
| United States | US 6034239 | 1999-07-22 | expiry 2019-07-22 |
EP885210B1 , EP1792899A1 and J. Med Chem. 2002, 45, 4222-4239
read all at
http://www.allfordrugs.com/2014/02/23/ramelteon-tak-375-melatonin-mt1mt2-receptor-agonist/
