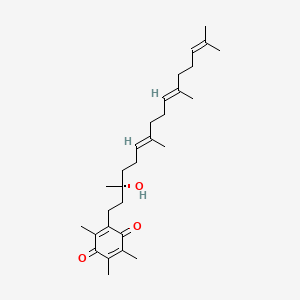

Vatiquinone
バチキノン
Vatiquinone; Alpha-Tocotrienol quinone; EPI-743; UNII-6O85FK9I0X; 1213269-98-7; Vincerenone
| Molecular Formula: |
C29H44O3 |
| Molecular Weight: |
440.668 g/mol |
2-[(3R,6E,10E)-3-hydroxy-3,7,11,15-tetramethylhexadeca-6,10,14-trienyl]-3,5,6-trimethylcyclohexa-2,5-diene-1,4-dione
2-((R,6E,10E)-3-hydroxy-3,7,11,15-tetramethylhexadeca-6,10,14-trien-1-yl)-3,5,6-trimethylcyclohexa-2,5-diene-1,4-dione
2-[(3R,6E,10E)-3-hydroxy-3,7,11,15-tetramethylhexadeca-6,10,14-trien-1-yl]-3,5,6-trimethylcyclohexa-2,5-diene-1,4-dione
6O85FK9I0X
9604
Research Code:EPI-743; ATQ-3, BioE-743
MOA:Mitochondria
- Originator Edison Pharmaceuticals
- Developer Edison Pharmaceuticals; Sumitomo Dainippon Pharma; University of Florida; Yale University
- Class Alkadienes; Benzoquinones; Cyclohexenes; Small molecules
- Mechanism of Action Antioxidants; NQO1 modulators
- Orphan Drug Status Yes – Mitochondrial disorders; Leigh disease; Friedreich’s ataxia
- New Molecular Entity Yes
Highest Development Phases
- Phase III Leigh disease
- Phase II Friedreich’s ataxia; Methylmalonic acidaemia; Mitochondrial disorders; Noise-induced hearing loss; Parkinson’s disease; Rett syndrome
- No development reported Gilles de la Tourette’s syndrome
Most Recent Events
- 04 Nov 2017 No recent reports of development identified for phase-I development in Gilles-de-la-Tourette’s-syndrome in USA (PO)
- 01 Apr 2017 Efficacy data from a phase II trial in Friedreich’s ataxia presented at the 69th Annual Meeting of the American Academy of Neurology (AAN- 2017)
- 16 Apr 2016 Initial efficacy and safety data from a phase IIa trial in Parkinson’s disease presented at the 68th Annual Meeting of the American Academy of Neurology (AAN – 2016)
Vatiquinone is in phase II/III clinical trials for the treatment of leigh syndrome in JP. Phase II clinical trials is also ongoing for Friedreich’s ataxia, Parkinson’s disease, Pearson syndrome, cobalamin C deficiency syndrome, hearing loss and Rett’s syndrome.
Vatiquinone was originally developed by Edison Pharmaceuticals, then licensed to Sumitomo Dainippon Pharma in Japan in 2013.
Orphan drug designations for the treatment of Friedreich’s, Leigh syndrome and Rett’s syndrome were granted to the compound by FDA in 2014.
In 2013, the compound was licensed to Sumitomo Dainippon Pharma by Edison Pharmaceuticals in Japan for development and commercialization for the treatment of pediatric orphan inherited mitochondrial and adult central nervous system diseases.
EU
On 17 January 2018, orphan designation (EU/3/17/1971) was granted by the European Commission to Edison Orphan Pharma BV, The Netherlands, for vatiquinone (also known as alpha-tocotrienol quinone) for the treatment of RARS2 syndrome.
http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/orphans/2018/03/human_orphan_002075.jsp&mid=WC0b01ac058001d12b
Vatiquinone, also known as EPI 743, is an orally bioavailable para-benzoquinone being developed for inherited mitochondrial diseases. The mechanism of action of EPI-743 involves augmenting the synthesis of glutathione, optimizing metabolic control, enhancing the expression of genetic elements critical for cellular management of oxidative stress, and acting at the mitochondria to regulate electron transport.
Vatiquinone has been investigated for the treatment and prevention of Retinopathy, Rett Syndrome, Genetic Disease, Noise-induced Hearing Loss, and Methylmalonic Aciduria and Homocystinuria,Cblc Type.
EPI-743 (vatiquinone) is a compound being developed by BioElectron (previously known as Edison Pharmaceuticals) to treat Friedreich’s ataxia (FA), a rare, autosomal recessive genetic disorder. The disorder is caused by mutations in the FXN gene, which encodes for a protein called frataxin. Frataxin is required for the normal functioning of mitochondria, or the energy factories of the cells. Decreased levels of frataxin, as observed in patients with FA, disrupts the normal function of mitochondria and leads to the gradual development of symptoms associated with the disease: impairment of muscle coordination, loss of muscle strength and sensation, and impaired speech, vision, and hearing.
Currently, there are no drugs available that could cure or help to effectively manage the condition, although a large number of potential treatments are in the pipeline.
How EPI-743 works
EPI-743 is a drug belonging to the class of para-benzoquinones, a group of potent antioxidants. The regulation of oxidative stress is disturbed in people with FA. EPI-743 targets an enzyme called NADPH quinone oxidoreductase 1 (NQO1), helping to increase the biosynthesis of glutathione, a compound essential for the control of oxidative stress. The drug does not target any FA-specific biochemical pathways directly, but helps to improve the regulation of cellular energy metabolism in general. Due to its non-specific mechanism, the drug can be used in a variety of disorders where mitochondrial function is affected.
EPI-743 in clinical trials
In December 2012, Edison Pharmaceuticals started a placebo-controlled Phase 2 study (NCT01728064) to examine the safety and efficacy of EPI-743 on visual and neurological function in FA patients. The study was completed in February 2016. The results indicated no significant differences in visual function at six months between patients treated with EPI-743 and those who received a placebo. However, researchers reported a trend toward improvement in neurological function.
In October 2013, the University of South Florida started a small Phase 2 study (NCT01962363) to evaluate the effects of EPI-743 in patients with rare point mutations leading to FA. The study investigated whether treatment with EPI-743 has a discernible impact on neurological function. The results announced in April 2016 demonstrated significant improvements in neurological functions over 18 months. However, the trial only included three participants.
Currently, no further trials testing EPI-743 in FA patients is taking place. However, the drug is in clinical trials for several other disorders that affect the functions of mitochondria, including Leigh syndrome, mitochondrial respiratory chain disease, Pearson syndrome, and others.
Other information
In February 2014, the U.S. Food and Drug Administration (FDA) granted orphan drug status to EPI-743, which allows a more expedited drug approval process. The FDA also granted fast track status to EPI-743 for the treatment of FA in March 2014.
ADDITIONAL INFORMATION
Edison Pharmaceuticals is developing vatiquinone, which was awarded Fast Track status for Friedreich’s ataxia in March 2014.
Reference
Bioorg. Med. Chem. Lett. 2011, 21, 3693-3698.
https://www.sciencedirect.com/science/article/pii/S0960894X11005440

Reference
WO2013041676A1 / US9045402B2.

It is known that a-tocotrienol quinones are pharmaceutically active.
US 201 1 /0172312 A1 discloses that tocotrienol quinones are used in treating Leight Syndrome. WO 2010/126909 A1 and US 2006/0281809 A1 disclose that tocotrienol quinones can be used for treating ophthalmic diseases and mitochondrial diseases. US 5,318,993 discloses the activity of tocotrienol quinones as cholesterol suppression. W.D. Shrader et al., Bioorganic & Medical Chemistry Letters 21 (201 1 ), 3693-3698 disclose that the R-isomer of a-tocotrienol quinone is a metabolite of α-tocotrienol and is a potent cellular protectant against oxidative stress and ageing. The R-isomer of α-tocotrienol used for this study has been extracted from Elaeis guineensis. All these documents either use tocotrienol from natural sources or do not disclose the source of tocotrienol respectively tocotrienol quinones or disclose very specific complex synthesis thereof. These methods are very expensive and limited in producing industrial amounts of the desired products.
It is well known that from vitamin E the tocopherols and tocotrienols having the R-configuration have a significantly higher bioactivity (biopotency) than the corresponding S-isomer. This is also the case for the corresponding R-isomers of tocotrienol quinones.
Synthetic pathways to produce the R-isomer of tocotrienol quinones in a stereospecific way are very expensive and therefore only of limited interest.
The synthesis of a-tocotrienol is known from Kabbe and Heitzer, Synthesis 1978, 888-889, however, no indication of chirality whatsoever is indicated.
The synthesis of tocotrienol from the corresponding 4-oxo-chromanol-derivative is known from US 6,096,907, however, no indication of chirality is indicated.
J. Org. Chem. 1981 , 46, 2445-2450 and CH 356754 disclose the chemical transformation of a-tocopherol to a-tocopheryl quinone and to a-tocopherylhydro-quinone, however, neither tocotrienols nor tocotrienol quinones are mentioned.
Separation of chiral compounds by chromatography is principally known. However, it is also known that the quantitative separation is very often very difficult to achieve.
Due to the importance of these substances, there exists a high interest in a process which would produce R-tocotrienol quinones in a large scale in an easy and economic way.
Examples
The present invention is further illustrated by the following experiments.
1 . Chromatographic separation
Starting materials:
Solvents and reagents used as received were heptane (Fluka, 51750), ethanol (Merck, 1 .00983), isopropanol (Sigma-Aldrich, 59300) and acetic acid (Fluka, 45730).
Chromatography:
Preparative separations were performed on an Agilent 1 100 series hplc system consisting of an Agilent 1 100 degasser, Agilent 1 100 preparative pump, Agilent 1 100 diode array detector, Agilent 1 100 MPS G2250A autosampler/fraction collector controlled by chemstation/CC-mode software package.
HPLC conditions for preparative separation:
Column: Daicel Chiracel® OD-H, 250 mm x 20 mm; eluent 0.5% isopropanol, 0.2 % acetic acid in n-heptane; flow 13 ml/min; detection 220 nm, 400 μΙ injection.
Separation of (R)-6-hydroxy-2,5,7,8-tetramethyl-2-((3E,7E)-4,8, 12-trimethyl-trideca-3,7, 11-trienyl) chroman-4-one and (S)-6-hydroxy-2,5,7,8-tetramethyl-2-((3E, 7E)-4,8, 12-trimethyltrideca-3, 7, 11-trienyl) chroman-4-one
Example 1 :
6-Hydroxy-2,5,7,8-tetramethyl-2-((3E,7E)-4,8,12-trimethyltrideca-3,7,1 1 -trienyl) chroman-4-one was prepared according to the example 6a in Kabbe and Heitzer, Synthesis 1978, 888-889.
The product was analyzed by HPLC (Column: Daicel Chiracel® OD-H, 250 mm x 4.6 mm; eluent 1 % ethanol in n-hexane; flow 1 ml/min; detection 220 nm, 2 μΙ injection). Figure 9 b) shows this chromatogram. It shows that the product is a 49.5 : 50.5 mixture (Retention time 13.2 and 14.2 min.)
87.5 mg of this product in heptane was injected and the two peaks with retention time at maximum 35.4 min. (1 ) (50.9%) resp. 43.5 min. (2) (49.1 %) were se-parated by the preparative HPLC separation. Figure 9 a) shows the chromatogram of the preparative HPLC separation.
After evaporation to dryness and dissolution the two collected fractions have been reanalysis on an analytical column (Daicel Chiracel® OD-H, 250 mm x 4.6 mm; eluent 1 % ethanol in n-hexane; flow 1 ml/min; detection 220 nm, 2 μΙ injection). Figure 9 c), respectively Figure 9 d), show the chromatogram of the first fraction, respectively the second fraction. The separation of the two isomers (Retention time 13.2 min, resp. 14.2 min) in the two fraction shows to be 94.9 : 5.1 (Figure 9 c)) resp. 7.1 : 92.9 (Figure 9 d)). Hence, the two isomers have been separation by preparative chromatography almost completely.
Patent
WO2010126909
The active component of the formulation of the present invention is selected from alpha- tocotrienol quinone, beta-tocotrienol quinone, gamma-tocotrienol quinone, delta-tocotrienol quinone, and mixtures thereof. In one embodiment, the formulation of the present invention comprises alpha-tocotrienol quinone as the active component. In other embodiments, the formulations of the present invention comprise one or more tocotrienol quinones of Formula I or mixtures thereof, in a pharmaceutically acceptable vehicle, and in other embodiments, the formulations of the present invention comprise alpha-tocotrienol quinone in a pharmaceutically acceptable vehicle. In other particular embodiments, the formulations are administered orally. In other embodiments, the formulations of the present invention comprise one or more tocotrienol quinones of Formula I or mixtures thereof, in an ophthalmically acceptable vehicle for topical, periocular, or intraocular administration, and in other embodiments, the formulations of the present invention comprise alpha-tocotrienol quinone in an ophthalmically acceptable vehicle.
[0120] The formulations of the present invention comprise tocotrienol quinones which can be produced synthetically from the respective tocotrienol by oxidation with suitable oxidizing agents, as for example eerie ammonium nitrate (CAN). Particularly, the formulations of the present invention comprise alpha-tocotrienol quinone (CAS Reg. No. 1401-66-7) produced by oxidation of alpha-tocotrienol. A preferred process for the production of alpha-tocotrienol has been described in co-owned US provisional application USAN 61/197,585 titled “Process for Enrichment and Isolation of alpha-Tocotrienol from Natural Extracts”.
[0121] Syntheses of various members of the tocotrienol family in the d,l- or (RS)-form have been published, see for example Schudel et al, HeIv. Chim. Acta (1963) 46, 2517-2526; H. Mayer et al, HeIv. Chim. Acta (1967) 50, 1376-11393; H.-J. Kabbe et al, Synthesis (1978), 888-889; M. Kajiwara et al, Heterocycles (1980) 14, 1995-1998; S. Urano et al, Chem. Pharm. Bull. (1983) 31, 4341-4345, Pearce et al, J. Med Chem. (1992), 35, 3595-3606 and Pearce et al, J. Med. Chem. (1994). 37, 526-541. None of these reported processes lead to the natural form of the tocotrienols, but rather produces racemic mixtures. Syntheses of natural form d-tocotrienols have been published. See for example. J. Scott et al, HeIv. CMm. Acta (1976) 59, 290-306, Sato et al. (Japanese Patent 63063674); Sato et al. (Japanese Patent NoJP 01233278) and Couladouros et al. (US Patent No. 7,038,067).
[0122] While synthetic and natural tocopherols are readily available in the market, the natural tocotrienol supply is limited, and generally comprises a mixture of tocotrienols. Crude palm oil which is rich in tocotrienols (800-1500 ppm) offers a potential source of natural tocotrienols. Carotech, Malaysia is able to extract and concentrate tocotrienols from crude palm oil, by a process patented in U.S. Pat. No. 5,157,132. Tocomin®-50 typically comprises about 25.32% mixed tocotrienols (7.00% alpha-tocotrienol, 14.42% gamma-tocotrienol, 3.30% delta-tocotrienol and 0.6% beta-tocotrienol ), 6.90% alpha-tocopherol and other phytonutrients such as plant squalene, phytosterols, co-enzyme QlO and mixed carotenoids.
[0123] Other methods for isolation or enrichment of tocotrienol from certain plant oils and plant oil by-products have been described in the literature. For some examples of such isolation and purification processes, see for instance Top A. G. et al, U.S. Pat. No. 5,190,618; Lane R et al, U.S. Pat No. 6,239,171; Bellafiore, L. et al. U.S. Pat. No.6,395,915; May, CY et al, U.S. Pat. No.6,656,358; Jacobs, L et al, U.S. Pat. No. 6,838,104; Sumner, C et al. Int. Pat. Pub. WO 99/38860, or Jacobs, L, Int. Pat. Pub. WO 02/500054. The compounds for use in the present invention and the other therapeutically active agents can be administered at the recommended maximum clinical dosage or at lower doses. Dosage levels of the active compounds in the compositions for use in the present invention may be varied so as to obtain a desired therapeutic response depending on the route of administration, severity of the disease and the response of the patient. When administered in combination with other therapeutic agents, the therapeutic agents can be formulated as separate compositions that are given at the same time or different times, or the therapeutic agents can be given as a single composition.
REFERENCES
1: Peragallo JH, Newman NJ. Is there treatment for Leber hereditary optic neuropathy? Curr Opin Ophthalmol. 2015 Nov;26(6):450-7. doi: 10.1097/ICU.0000000000000212. PubMed PMID: 26448041; PubMed Central PMCID: PMC4618295.
2: Miller DK, Menezes MJ, Simons C, Riley LG, Cooper ST, Grimmond SM, Thorburn DR, Christodoulou J, Taft RJ. Rapid identification of a novel complex I MT-ND3 m.10134C>A mutation in a Leigh syndrome patient. PLoS One. 2014 Aug 12;9(8):e104879. doi: 10.1371/journal.pone.0104879. eCollection 2014. PubMed PMID: 25118196; PubMed Central PMCID: PMC4130626.
3: Strawser CJ, Schadt KA, Lynch DR. Therapeutic approaches for the treatment of Friedreich’s ataxia. Expert Rev Neurother. 2014 Aug;14(8):949-57. doi: 10.1586/14737175.2014.939173. Epub 2014 Jul 18. PubMed PMID: 25034024.
4: Enns GM. Treatment of mitochondrial disorders: antioxidants and beyond. J Child Neurol. 2014 Sep;29(9):1235-40. doi: 10.1177/0883073814538509. Epub 2014 Jun 30. PubMed PMID: 24985754.
5: Avula S, Parikh S, Demarest S, Kurz J, Gropman A. Treatment of mitochondrial disorders. Curr Treat Options Neurol. 2014 Jun;16(6):292. doi: 10.1007/s11940-014-0292-7. PubMed PMID: 24700433; PubMed Central PMCID: PMC4067597.
6: Hargreaves IP. Coenzyme Q10 as a therapy for mitochondrial disease. Int J Biochem Cell Biol. 2014 Apr;49:105-11. doi: 10.1016/j.biocel.2014.01.020. Epub 2014 Feb 2. Review. PubMed PMID: 24495877.
7: Chicani CF, Chu ER, Miller G, Kelman SE, Sadun AA. Comparing EPI-743 treatment in siblings with Leber’s hereditary optic neuropathy mt14484 mutation. Can J Ophthalmol. 2013 Oct;48(5):e130-3. doi: 10.1016/j.jcjo.2013.05.011. PubMed PMID: 24093206.
8: Pastore A, Petrillo S, Tozzi G, Carrozzo R, Martinelli D, Dionisi-Vici C, Di Giovamberardino G, Ceravolo F, Klein MB, Miller G, Enns GM, Bertini E, Piemonte F. Glutathione: a redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol Genet Metab. 2013 Jun;109(2):208-14. doi: 10.1016/j.ymgme.2013.03.011. Epub 2013 Mar 24. PubMed PMID: 23583222.
9: Sadun AA, La Morgia C, Carelli V. Mitochondrial optic neuropathies: our travels from bench to bedside and back again. Clin Experiment Ophthalmol. 2013 Sep-Oct;41(7):702-12. doi: 10.1111/ceo.12086. Epub 2013 Apr 11. Review. PubMed PMID: 23433229.
10: Kerr DS. Review of clinical trials for mitochondrial disorders: 1997-2012. Neurotherapeutics. 2013 Apr;10(2):307-19. doi: 10.1007/s13311-013-0176-7. Review. PubMed PMID: 23361264; PubMed Central PMCID: PMC3625388.
11: Blankenberg FG, Kinsman SL, Cohen BH, Goris ML, Spicer KM, Perlman SL, Krane EJ, Kheifets V, Thoolen M, Miller G, Enns GM. Brain uptake of Tc99m-HMPAO correlates with clinical response to the novel redox modulating agent EPI-743 in patients with mitochondrial disease. Mol Genet Metab. 2012 Dec;107(4):690-9. doi: 10.1016/j.ymgme.2012.09.023. Epub 2012 Sep 28. PubMed PMID: 23084792.
12: Martinelli D, Catteruccia M, Piemonte F, Pastore A, Tozzi G, Dionisi-Vici C, Pontrelli G, Corsetti T, Livadiotti S, Kheifets V, Hinman A, Shrader WD, Thoolen M, Klein MB, Bertini E, Miller G. EPI-743 reverses the progression of the pediatric mitochondrial disease–genetically defined Leigh Syndrome. Mol Genet Metab. 2012 Nov;107(3):383-8. doi: 10.1016/j.ymgme.2012.09.007. Epub 2012 Sep 10. PubMed PMID: 23010433.
13: Büsing A, Drotleff AM, Ternes W. Identification of α-tocotrienolquinone epoxides and development of an efficient molecular distillation procedure for quantitation of α-tocotrienol oxidation products in food matrices by high-performance liquid chromatography with diode array and fluorescence detection. J Agric Food Chem. 2012 Aug 29;60(34):8302-13. doi: 10.1021/jf301137b. Epub 2012 Aug 16. PubMed PMID: 22747466.
14: Sadun AA, Chicani CF, Ross-Cisneros FN, Barboni P, Thoolen M, Shrader WD, Kubis K, Carelli V, Miller G. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol. 2012 Mar;69(3):331-8. doi: 10.1001/archneurol.2011.2972. PubMed PMID: 22410442.
15: Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, Amagata A, Barnes A, Kheifets V, Shrader WD, Thoolen M, Blankenberg F, Miller G. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. 2012 Jan;105(1):91-102. doi: 10.1016/j.ymgme.2011.10.009. Epub 2011 Oct 21. PubMed PMID: 22115768.
16: Shrader WD, Amagata A, Barnes A, Enns GM, Hinman A, Jankowski O, Kheifets V, Komatsuzaki R, Lee E, Mollard P, Murase K, Sadun AA, Thoolen M, Wesson K, Miller G. α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg Med Chem Lett. 2011 Jun 15;21(12):3693-8. doi: 10.1016/j.bmcl.2011.04.085. Epub 2011 Apr 24. PubMed PMID: 21600768.
17: Gagnon KT. HD Therapeutics – CHDI Fifth Annual Conference. IDrugs. 2010 Apr;13(4):219-23. PubMed PMID: 20373247.
18: Bidichandani SI, Delatycki MB. Friedreich Ataxia. 1998 Dec 18 [updated 2014 Jul 24]. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. Available from http://www.ncbi.nlm.nih.gov/books/NBK1281/ PubMed PMID: 20301458.
19: Yu-Wai-Man P, Chinnery PF. Leber Hereditary Optic Neuropathy. 2000 Oct 26 [updated 2013 Sep 19]. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. Available from http://www.ncbi.nlm.nih.gov/books/NBK1174/ PubMed PMID: 20301353.
////////////orphan drug status, EPI-743, fast track, EPI743, EPI-743, EPI 743, Vatiquinone; alpha-Tocotrienol quinone, Vincerenone, バチキノン , BioE-743
CC1=C(C(=O)C(=C(C1=O)C)CCC(C)(CCC=C(C)CCC=C(C)CCC=C(C)C)O)C
Biogen Idec, Atlas Venture Pump $17M into Ataxion
-
Biogen Idec and Atlas Venture have agreed to invest a combined $17 million of Series A
financing in a nearly-year-old drug developer focused on hereditary ataxias. Biogen Idec is separately providing R&D and other funding to the company, called Ataxion. The biotech giant has the option to acquire Ataxion to continue development of the program upon completion of a Phase I multiple ascending dose (MAD) study at pre-negotiated terms, including undisclosed upfront and milestone payments. Earlier this month, Edison Pharmaceuticals won FDA “fast-track” designation for its own Fredrich’s ataxia drug, the company’s lead drug candidate EPI-743, now in Phase II trials. And on February 12, the developer of a preclinical gene therapy for Friedrich’s ataxia, Voyager Therapeutics, was launched by Third Rock Ventures with $45 million in Series A financing. read at
http://www.genengnews.com/gen-news-highlights/biogen-idec-atlas-venture-pump-17m-into-ataxion/81249632/
-
EPI-743 is being developed at Edison Pharmaceuticals in phase II clinical trials for several indications; Leigh syndrome, Friedreich’s ataxia, Parkinson’s disease, Pearson syndrome, cobalamin C deficiency syndrome and Rett’s syndrome. The licensee, Dainippon Sumitomo is developing the product in phase II/III study for the treatment of Leigh syndrome in children. Preclinical studies are also underway for the treatment of Huntington’s disease. In 2011, an orphan drug designation was assigned by the FDA for the treatment of inherited mitochondrial respiratory chain diseases and by the EMA for the treatment of Leigh syndrome, and in 2014, the FDA assigned another orphan drug for the treatment of Friedreich’s ataxia. In 2014, the product was granted fast track designation for this indication. In 2013, the compound was licensed to Dainippon Sumitomo Pharma by Edison Pharmaceuticals in Japan for development and commercialization for the treatment of pediatric orphan inherited mitochondrial and adult central nervous system diseases.
-
OLD ARTICLE

19 February 2013 EPI-743 Vatiquinone is a new drug that is based on vitamin E. Tests have shown that it can help improve the function of cells with mitochondrial problems. It may be able to treat people with genetic disorders that affect metabolism and mitochondria Edison Pharmaceuticals and Bambino Gesu Children’s Hospital have announced the commencement of EPI-743 Phase 2 cobalamin C deficiency syndrome trial. EPI-743 is an orally bioavailable small molecule and a member of the para-benzoquinone class of drugs. The trial’s principal investigator, Bambino Gesu Children’s Hospital, division of metabolism Professor Carlo Dionisi-Vici said, “Given the central role of glutathione in cellular redox balance and antioxidant defense systems, we are eager to explore whether a therapeutic that increases glutathione such as EPI-743 will provide clinical benefit.” Improvement in visual function is the primary endpoint of the placebo-controlled study while secondary outcome measurements assess neurologic and neuromuscular function, glutathione biomarkers, quality of life, in addition to safety parameters. The investigation is aimed at assessing the efficacy of EPI-743 in disorders of intermediary metabolism that also result in redox disturbances. EPI-743 is an orally absorbed small molecule that readily crosses into the central nervous system. It works by targeting the enzyme NADPH quinone oxidoreductase 1 (NQO1). Its mode of action is to synchronize energy generation in mitochondria with the need to counter cellular redox stress Friedreich’s ataxia (FRDA) is an autosomal recessive neurodegenerative and cardiodegenerative disorder caused by decreased levels of the protein frataxin. The disease causes the progressive loss of voluntary motor coordination (ataxia) and cardiac complications. Symptoms typically begin in childhood, and the disease progressively worsens as the patient grows older; patients eventually become wheelchair-bound due to motor disabilities. Patients with Friedreich’s ataxia develop loss of visual acuity or changes in color vision. Most have jerky eye movements (nystagmus), but these movements by themselves do not necessarily interfere with vision. ……………… Bioorg Med Chem Lett 2011, 21(12): 3693 http://www.sciencedirect.com/science/article/pii/S0960894X11005440We report that α-tocotrienol quinone (ATQ3) is a metabolite of α-tocotrienol, and that ATQ3 is a potent cellular protectant against oxidative stress and aging. ATQ3 is orally bioavailable, crosses the blood–brain barrier, and has demonstrated clinical response in inherited mitochondrial disease in open label studies. ATQ3 activity is dependent upon reversible 2e-redox-cycling. ATQ3 may represent a broader class of unappreciated dietary-derived phytomolecular redox motifs that digitally encode biochemical data using redox state as a means to sense and transfer information essential for cellular function. 
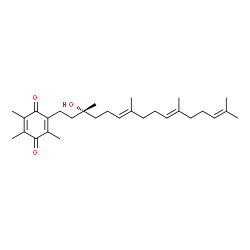
Figure 1.
The conversion of α-tocotrienol to α-tocotrienol quinone.

Figure 1.
The conversion of α-tocotrienol to α-tocotrienol quinone.


 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....






















































 f the general formula I belonging to the class of phospholipids (X is O and R2 is a group of formula II), e.g. alkyloxy phospholipids (Y is O) and the corresponding alkylthio derivatives (Y is S), can be prepared as described in the literature (Bittman, R.; J. Med. Chem. 1997, 40, 1391-1395; Reddy, K. C.; Tetrahedron Lett. 1994, 35, 2679-2682; Guivisdalsky, P. N.; J. Med. Chem. 1990, 33, 2614-2621 and references cited therein) or by standard variations of the procedures described therein. Synthesis of the corresponding ester and thioester analogues (Y is OCO and SCO, respectively) can be accomplished by standard acylation of the hydroxy or thio precursor materials.
f the general formula I belonging to the class of phospholipids (X is O and R2 is a group of formula II), e.g. alkyloxy phospholipids (Y is O) and the corresponding alkylthio derivatives (Y is S), can be prepared as described in the literature (Bittman, R.; J. Med. Chem. 1997, 40, 1391-1395; Reddy, K. C.; Tetrahedron Lett. 1994, 35, 2679-2682; Guivisdalsky, P. N.; J. Med. Chem. 1990, 33, 2614-2621 and references cited therein) or by standard variations of the procedures described therein. Synthesis of the corresponding ester and thioester analogues (Y is OCO and SCO, respectively) can be accomplished by standard acylation of the hydroxy or thio precursor materials. an animation to soothe ones eye
an animation to soothe ones eye










 Amgen announced that the Phase 3 TESLA (Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities) trial evaluating evolocumab met its primary endpoint of the percent reduction from baseline at week 12 in low-density lipoprotein cholesterol (LDL-C). The percent reduction in LDL-C, or “bad” cholesterol, was clinically meaningful and statistically significant………….read at
Amgen announced that the Phase 3 TESLA (Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities) trial evaluating evolocumab met its primary endpoint of the percent reduction from baseline at week 12 in low-density lipoprotein cholesterol (LDL-C). The percent reduction in LDL-C, or “bad” cholesterol, was clinically meaningful and statistically significant………….read at









 mifepristone
mifepristone



 mifepristone
mifepristone
