
Home » 2014 (Page 70)
Yearly Archives: 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sotagliflozin, LX 4211 in phase 2 For type 1, 2 diabetes
LX 4211, Sotagliflozin, LP-802034 , lex 1287
UNII-6B4ZBS263Y
Methyl (5S)-5-[4-chloro-3-(4-ethoxybenzyl)phenyl]-1-thio-beta-L-xylopyranoside
β-L-Xylopyranoside, methyl 5-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-thio-, (5S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4- ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol,
(5S)-Methyl 5-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-thio-beta-L-xylopyranoside
1018899-04-1
C21H25ClO5S, 424.94, LP-802034
LX-4211 is a dual SGLT2/1 inhibitor; Antidiabetic agents.
LX-4211 is a SGLT-2 inhibitor being evaluated in phase II clinical studies at Lexicon Pharmaceuticals for the oral treatment of type 2 diabetes.



Summary
- Co-administration of LX4211 led to a nearly one-third reduction in mealtime insulin for Type 1 diabetics.
- Although there was no reduction in basal insulin use, the LX4211 group saw better glucose control, lower HbA1c, and weight loss.
- Partnering LX4211 is still management’s top priority but independent development in Type 1 diabetes is at least an option.
Lexicon Pharmaceuticals (LXRX) continues to generate data on its SGLT-1/2 inhibitor LX4211 that suggest this is an effective and promising medication for treating not only Type 2 diabetes (the common target for non-insulin medications for diabetes), but also Type 1 as well. Lexicon’s most recent update, a small short-term Phase II study in Type 1 diabetics is certainly a positive update, but it’s not what investors really want to see. Lexicon still needs to find a development partner for LX4211 and the ongoing delays don’t help sentiment or the long-term prospects for the drug.
A Potentially Meaningful Addition To Type 1 Care
On Monday morning, Lexicon released top-line data from a small (33-patient) Phase II study of LX4211 in Type 1 diabetics on insulin. The results support the notion that SGLT inhibition can play a valuable role in improving glucose control for Type 1 diabetics.
This small study enrolled generally well-controlled patients (HbA1c levels ranging from 7 to 9, with an average of 7.9) and the addition of LX4211 led to 32% reduction in bolus (mealtime) insulin versus a 6% reduction in the placebo group. Even with the lower bolus insulin, patients in the LX4211 group showed a 0.55% reduction in HbA1c versus a 0.06% reduction in the placebo group. Patients taking LX4211 demonstrated better glucose control (more time spent in the target range of 70-180 mg/dL) and saw a 1.7kg weight loss versus a 0.5kg weight gain in the placebo group

……………………..
Scheme 1 :




3(a) 3(b)


4(a) 4(b)

Scheme 3:

…………………
http://www.google.com/patents/EP2332947A1?cl=en
EXAMPLES
-
Aspects of this invention can be understood from the following examples.
6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2.3-d][13]dioxol-5-yl)(morpholino)methanone
-
To a 12L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(-)-xylose (504.40 g, 3.360 mol), acetone (5L, reagent grade) and anhydrous MgSO4 powder (811.23g, 6.740 mol / 2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol / 0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24°C over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH = 8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50% w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wt% water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23g, 86% yield, 96.7 area% pure by GC). 1H NMR (400 MHz, DMSO-d6)δ1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93 – 4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 13C NMR (101MHz, DMSO-d6) δ26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73.
-
To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0g, 131 mmol) in acetone (375 mL, 15X) and H2O (125 mL, 5X) was added NaHC03 (33.0g, 3.0 equiv), NaBr (2.8g, 20 mol%) and TEMPO (0.40g, 2 mol%) at 20°C. The mixture was cooled to 0-5°C and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20°C for 24h. Methanol (20 mL) was added and the mixture was stirred at 20°C for 1h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2X). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12X x3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5X) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0g, 79%). 1H NMR (methanol-d4), δ 6.00 (d, J= 3.2 Hz, 1H), 4.72 d, J= 3.2 Hz, 1H), 4.53 (d, J= 3.2 Hz, 1H), 4.38 (d, J= 3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H).
-
To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0g, 24.5 mmol) in THF (100 mL, 20X) was added TBTU (11.8g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20°C for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20°C for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2X x2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4:1) to afford 4.3 g of the desired morpholine amide (64%) as a white solid. 1H NMR (CDCl3), 8 6.02 (d, J= 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J= 3.2 Hz, 1H), 4.58 (d, J= 3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
6.2. Alternative synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahvdrofuro[2.3-d][1,3]dioxol-5-yl)(morpholino)methanone
-
A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2 solution (1.6 L) and bleach/K2HPO4 solution (400 mL),∼1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25°C. Additional charges of TEMPO (14.3g, 0.5 mol%) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to < 10°C and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15°C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L x 3). The combined organic layer was concentrated under vacuo (∼100-120 torr) at < 35°C (28-32°C vapor, 45-50°C bath) to low volume (- 6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached < 1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5°C and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) δ5.96 (d, J = 3.6 Hz, 1H), 4.5 8 (d, J = 3.6 Hz, 1H), 4.53 (d, J =3.2Hz,1H), 4.30 (d, J= 3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) 8 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25. 1.
-
The morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (7.85 kg, 26.9 mol), morpholine (2.40 L, 27.5 mol) and boric acid (340 g, 5.49 mol, 0.2 eq) were added to toluene (31 L). The resulting slurry was degassed and heated at reflux with a Dean-Stark trap under nitrogen for 12 h and then cooled to room temperature. The mixture was filtered to remove insolubles and the filter cake washed with toluene (5 L). The filtrate was concentrated to about 14 L and flushed with toluene (-80 L) to remove excess morpholine. When final volume reached -12 L, heptane (14 L) was added slowly at 60-70°C. The resulting slurry was cooled gradually to room temperature and aged for 3 h. It was then filtered and washed with heptane (12 L) and dry under nitrogen gave a slightly pink solid (6.26 kg, 97% pure, 98% yield). m.p.: 136°C (DSC). 1H NMR (CDCl3), δ 6.02 (d, J = 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H). 13C NMR (methanol-d4) δ 26.84, 27.61, 44.24, 47.45, 68.16, 77.14, 81.14, 86.80, 106.87, 113.68, 169.05.
1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene:

6.3. Synthesis of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene
-
A 2L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with 2-chloro-5-iodobenzoic acid (199.41 g, 0.706 mol), dichloromethane (1.2L, KF = 0.003 wt% water) and the suspension was set stirring at ambient temperature. Then N,N-dimethylformamide (0.6 mL, 1.1 mol %) was added followed by oxalyl chloride (63 mL, 0.722 mol, 1.02 equiv) which was added over 11 min. The reaction was allowed to stir at ambient overnight and became a solution. After 18.75hours, additional oxalyl chloride (6 mL, 0.069 mol, 0.10 equiv) was added to consume unreacted starting material. After 2 hours, the reaction mixture was concentrated in vacuo to afford crude 2-chloro-5-iodobenzoyl chloride as a pale yellow foam which will be carried forward to the next step.
-
A jacketed 2L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with aluminum chloride (97.68 g, 0.733 mol, 1.04 equiv), dichloromethane (0.65 L, KF = 0.003 wt% water) and the suspension was set stirring under nitrogen and was cooled to about 6°C. Then ethoxybenzene (90 mL, 0.712 mol, 1.01 equiv) was added over 7 minutes keeping internal temperature below 9°C. The resulting orange solution was diluted with dichloromethane (75mL) and was cooled to -7°C. Then a solution of 2-chloro-5-iodobenzoyl chloride (≤ 0.706 mol) in 350 mL dichloromethane was added over 13 minutes keeping the internal temperature below +3°C. The reaction mixture was warmed slightly and held at +5°C for 2 hours. HPLC analysis suggested the reaction was complete and the reaction was quenched into 450mL pre-cooled (∼5°C) 2N aq. HCl with stirring in a jacketed round bottom flask. This quench was done in portions over 10min with internal temperature remaining below 28°C. The quenched biphasic mixture was stirred at 20°C for 45min and the lower organic phase was washed with 1N aq. HCl (200mL), twice with saturated aq sodium bicarbonate (200mL per wash), and with saturated aq sodium chloride (200mL). The washed extract was concentrated on a rotary evaporator to afford crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone as an off-white solid (268.93g, 99.0 area% by HPLC at 220nm, 1.0 area% regioisomer at 200nm, 98.5 % “as-is” yield).
-
A jacketed 1 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged with crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone (30.13 g, 77.93 mmol), acetonitrile (300mL, KF = 0.004 wt% water) and the suspension was set stirring under nitrogen and was cooled to about 5°C.Then triethylsilane (28mL, 175.30 mmol, 2.25 equiv) was added followed by boron trifluoride-diethyletherate (24mL, 194.46mmo1,2.50 equiv) which was added over about 30 seconds. The reaction was warmed to ambient over 30min and was stirred for 17 hours. The reaction was diluted with methyl tert-butyl ether (150mL) followed by saturated aq sodium bicarbonate (150mL) which was added over about 1 minutes. Mild gas evolution was noticed and the biphasic solution was stirred at ambient for 45 minutes. The upper organic phase was washed with saturated aq sodium bicarbonate (100 mL), and with saturated aq sodium chloride (50mL). The washed extract was concentrated on a rotary evaporator to about one half of its original volume and was diluted with water (70 mL). Further concentration in vacuo at 45°C was done until white prills formed which were allowed to cool to ambient while stirring. After about 30 minutes at ambient, the suspended solids were isolated by filtration, washed with water (30 mL), and were dried in vacuo at 45°C. After about 2.5 hours, this afforded 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene as a slightly waxy white granular powder (28.28 g, 98.2 area % by HPLC at 220nm, 97.4 % “as-is” yield).
6.4. Synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanone
-
To a solution of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene (500mag, 1.34 mmol) in THF (5.0 mL) was added i-PrMgCl (2.0M in THF, 1.0 mL, 2.00 mmol) at 0-5°C, and the mixture was stirred for 1.5 h at 0-5°C. A solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone (146.5 mg, 0.536 mmol) in THF (1.0 mL) was added dropwise at 0-5°C and the mixture was kept stirring for 1h, warmed to 20°C and stirred at 20°C for 2 hours. The reaction was quenched with saturated aq NH4CI, extracted with MTBE, washed with brine. The organic layer was concentrated and the residue was purified by silica gel column chromatography to afford the desired ketone (178 mg, 76%) as a white solid. 1H NMR (CDCl3) δ 7. 88 (dd, J= 8.4, 2.0 Hz, 1H), 7.82 (d, J= 2.0 Hz, 1H), 7.50 (d, J= 8.4 Hz, 1H), 7.12 (d, J= 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 6.07 (d, J = 3.2 Hz, 1H), 5.21 (d, J = 3.2 Hz, 1H), 4.58 (d, J = 3.2 Hz, 1H), 4.56 (d, J = 3.2 Hz, 1H), 4.16 (d, J = 7.2 Hz, 2H), 4.03 (q, J = 7.2 Hz, 2H), 1.54 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H), 1.37 (s, 3H).
6.5. Alternative synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone
-
To a 20 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet was charged with the iodide (3.00 kg, 8.05 mol) and THF (8 L, 4X to the morpholinoamide) at room temperature and cooled to -5°C. To the above solution was added dropwise a solution of i-PrMgCl in THF (Aldrich 2 M, 4.39 L, 8.82 mol) at -5°C over 3 hours. This Grignard solution was used in the ketone formation below.
-
[0055]To a 50 L reactor equipped with a mechanical stirrer, a temperature controller, and a nitrogen inlet was charged the morpholinoamide (HPLC purity = 97 wt%, 2.01 kg, 7.34 mol) and THF (11 L, 5.5X) at room temperature and stirred for 45 minutes at room temperature and for 15 minutes at 30°C. The homogeneous solution was then cooled to – 25°C. To this solution was added a solution of t-BuMgCl in THF (Aldrich 1 M, 7.32 L, 7.91 mol) at -25°C over 3 hours. Then the above Grignard solution was added to this solution at -20 over 41 minutes. The resulting solution was further stirred at -20°C before quench. The reaction mixture was added to 10 wt% aqueous NH4Cl (10 L, 5X) at 0°C with vigorous stirring, and stirred for 30 minutes at 0°C. To this mixture was added slowly 6 N HCl (4 L, 2X) at 0°C to obtain a clear solution and stirred for 30 minutes at 10°C. After phase split, the organic layer was washed with 25 wt% aq NaCl (5 L, 2.5X). Then the organic layer was concentrated to a 3X solution under the conditions (200 mbar, bath temp 50°C). EtOAc (24 L, 12X) was added, and evaporated to a 3X solution under the conditions (150 mbar, bath temp 50°C). After removed solids by a polish filtration, EtOAc (4 L, 2X) was added and concentrated to dryness (150 mbar, bath temp 50°C). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70°C to obtain a 2.5X homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5X) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5X) was added slowly to a little cloudy solution at 70°C. After stirred for 0.5 h at 70°C, the suspension was slowly cooled to 60°C and stirred for 1 h at 60°C. The suspension was then slowly cool to room temperature and stirred for 14 h at the same temperature. The crystals were collected and washed with heptane (8 L, 4X), dried under vacuum at 45°C to give the desired ketone as fluffy solids (2.57 kg, 100 wt% by HPLC, purity-adjusted yield: 81%).
(2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate:

6.6. Synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
-
To a solution of the ketone (4-chloro-3-(4-ethoxybenzyl)phenyl)-((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone (114.7 g, 0.265 mol) in MeOH (2 L, 17X) was added CeCl3.7H2O (118.5g, 1.2 equiv) and the mixture was stirred at 20°C until all solids were dissolved. The mixture was then cooled to -78°C and NaBH4 (12.03g, 1.2 equiv) was added in portions so that the temperature of the reaction did not exceed -70°C. The mixture was stirred at – 78°C for 1 hour, slowly warmed to 0°C and quenched with saturated aq NH4Cl (550 mL, 5X). The mixture was concentrated under vacuum to remove MeOH and then extracted with EtOAc (1.1L, 10X x2) and washed with brine (550 mL, 5X). The combined organics were concentrated under vacuum to afford the desired alcohol as a colorless oil (crude, 115g). To this colorless oil was added AcOH (650 mL) and H2O (450 mL) and the mixture was heated to 100°C and stirred for 15 hours. The mixture was then cooled to room temperature (20°C) and concentrated under vacuum to give a yellow oil (crude, ∼118 g). To this crude oil was added pyridine (500 mL) and the mixture was cooled to 0°C. Then, Ac2O (195 mL, -8.0 equiv) was added and the mixture was warmed to 20°C and stirred at 20°C for 2h. The reaction was quenched with H2O (500 mL) and diluted with EtOAc (1000 mL). The organic layer was separated and concentrated under vacuum to remove EtOAc and pyridine. The residue was diluted with EtOAc (1000 mL) and washed with aq NaHSO4 (1N, 500 mL, x2) and brine (300 mL). The organic layer was concentrated to afford the desired tetraacetate intermediate as a yellow foam (-133g).
-
To a solution of tetraacetate (133 g, 0.237 mol assuming pure) and thiourea (36.1, 2.0 equiv) in dioxane (530 mL, 4X) was added trimethylsilyl trifluoromethanesulfonate (TMSOTf) (64.5 mL, 1.5 equiv) and the reaction mixture was heated to 80°C for 3.5 hours. The mixture was cooled to 20°C and Mel (37 mL, 2.5 equiv) and N,N-diisopropylethylamine (DiPEA) (207 mL, 5.0 equiv) was added and the mixture was stirred at 20°C for 3h. The mixture was then diluted with methyl tertiary-butyl ether (MTBE) (1.3 L, 10X) and washed with H2O (650 mL, 5X x2). The organic layer was separated and concentrated under vacuum to give a yellow solid. To this yellow solid was added MeOH (650 mL, 5X) and the mixture was reslurried at 60°C for 2h and then cooled to 0°C and stirred at 0°C for 1 hour. The mixture was filtered and the cake was washed with MeOH (0°C, 70 mL, x3). The cake was dried under vacuum at 45°C overnight to afford the desired triacetate (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (88 g, 60% over 4 steps) as a pale yellow solid. 1H NMR (CDCl3) δ 7.37 (d, J= 8.0 Hz, 1H), 7.20 (dd, J= 8.0, 2.0 Hz, 1H), 7.07 (m, 2H), 6.85 (m, 2H), 5.32 (t, J = 9.6 Hz, 1H), 5.20 (t, J = 9.6 Hz, 1H), 5.05 (t, J= 9.6 Hz, 1H), 4.51 (d, J=9.6Hz, 1H), 4.38 (d, J= 9.6Hz, 1h), 4.04 (m, 2H), 2.17 (s, 3H), 2.11 (s, 3H), 2.02 (s, 3H), 1.73 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H).
6.7. Alternative synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
-
To a 50 L reactor under nitrogen atmosphere, 40 L MeOH was charged, followed with the ketone (2.50 kg, 5.78 mol) and CeCl3.7H2O (2.16 kg, 1.0 equiv). Methanol (7.5 L) was added as rinse (totally 47.5 L, 19X). A freshly prepared solution of NaBH4 (87.5 g, 0.4 equiv) in aqueous 1 N NaOH (250 mL) was added slowly (35 min) at 15-25°C. The mixture was then stirred for 15 min. HPLC analysis of the reaction mixture showed approximately 90:10 diastereomeric ratio. The reaction was quenched with 10 wt% aq NH4Cl (2.5 L, 1X) and the mixture was concentrated under vacuum to 5X, diluted with water (10 L, 4X) and MTBE (12.5L, 5X). The mixture was cooled to 10°C and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H2O (5L, 2X). The combined aqueous layer was extracted with MTBE (12.5 L, 5X). The combined organic layers were washed with brine (2.5 L, 1X) and concentrated under vacuum to 3X. MeCN (15 L, 6X) was added. The mixture was concentrated again to 10 L (4X) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN.
-
The organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol% aqueous H2SO4 solution (61.8 mL 98% concentrated H2SO4 and 5 L H2O) was added. The mixture was heated to 80°C for 2 hours and then cooled to 20°C. The reaction was quenched with a solution of saturated aqueous K2CO3 (5 L, 2X) and diluted with MTBE (15 L, 6X). The organic layer was separated, washed with brine (5 L, 2X) and concentrated under vacuum to 5 L (2X). MeCN (12.5 L, 5X) was added and the mixture was concentrated to 7.5 L (3X).
-
The above MeCN solution of (3S,4R,SR,6S)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl)tetrahydro-2H-pyran-2,3,4,5-tetraol was cooled to 10°C, added with dimethylaminopyridine (17.53 g, 2.5 mol%), followed by slow addition of acetic anhydride (3.23 L, 6.0 equiv) and triethylamine (5 L, 2X, 6.0 equiv) so that the temperature of the mixture was kept below 20°C. The reaction was then warmed to 20°C and stirred for 1 hour and diluted with MTBE (15 L, 6X). The mixture was slowly quenched with water (7.5 L, 3X). The organic layer was separated and washed with saturated aqueous KHCO3 (5L, 2X), 1 N NaHSO4 (5 L, 2X), and brine (5 L, 2X) in sequence.
-
The organic layer was then concentrated under vacuum to 5 L (2X). MeCN (12.5 L, 5X) was added and the solution was concentrated to 7.5 L (3X) (KF = 0.08%). Dioxane (12.5 L, 5X) was added and the solution was concentrated to 7.50 L (3X) (KF = 0.02%). Any residual solid was removed by a polish filtration and the cake was washed with minimal amount of dioxane (500 mL).
-
To the above filtrate was added thiourea (880 g, 2.0 equiv) and TMSOTf (1.57 L, 1.5 equiv). The reaction mixture was heated to 80°C for 3 hours (>97% conversion). The mixture was cooled to 20°C and methyl iodide (541 mL, 1.5 equiv) and diethylisopropylamine (3.02 L, 3.0 equiv) were added and the mixture was stirred at 20°C for 18 hours. An extra methyl iodide charge (90 mL, 0.25 equiv) was added and the mixture was stirred at 20°C for 1 hours. The mixture was then diluted with MTBE (25 L, 10X) and washed with water (12.5 L, 5X x2). The organic layer was separated and concentrated under vacuum to -5 L (2X). MeOH (12.5 L, 5X) was added and the mixture was concentrated to 5X to afford a slurry. The mixture was then heated at 60°C for 1 hour and cooled to 0°C and stirred at 0°C for 1 hour. The mixture was filtered and the cake was washed with MeOH (0°C, 2.5 L, 1X x2, 1.0 L, 0.4X). The cake was dried under vacuum at 45°C overnight to afford the desired triacetate (1.49 kg, 47% over 4 steps) as a pale yellow/off-white solid.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol
-
To a slurry of (2S,3S,4R,SS,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0.164mo1) in MeOH (900 mL, 10X) was added NaOMe in MeOH (25 wt%, 18 mL, 0.2X) at 20°C and the mixture was stirred at 20°C for 2 hours until all solids disappeared. The mixture was then concentrated to 300 mL, added to H2O (1L) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, x3) and the cake was dried under vacuum at 45°C overnight to afford the desired methyl thiolate (67.0g, 95%). 1H NMR (CDCl3) δ 7.38 (d, J = 8.4 Hz, 1H), 7.22 (m, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.35 (d, J = 9.6 Hz, 1H), 4.15 (d, J = 9.6 Hz, 1H), 4.10-3.95 (m, 3H), 3.64 (t, J = 8.8 Hz, 1H), 3.50 (m, 2H), 3.42 (br s, 1H), 2.95 (br s, 1H), 2.57 (br s, 1H), 2.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H).
…………
http://www.google.com/patents/WO2010009197A1?cl=en
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H- pyran-3,4,5-triol:

LEX-1287 The compound is an inhibitor of the sodium glucose co-transporter 2, and may be useful in the treatment of diabetes and a variety of other diseases and conditions. See U.S. patent application no. 11/862,690, filed September 28, 2007.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4- ethoxybenzyl)phenyl)-6-fmethylthio)tetrahydro-2H-pyran-3,4,5-triol To a slurry of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-
(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0.164mol) in MeOH (900 mL, 10X) was added NaOMe in MeOH (25 wt%, 18 mL, 0.2X) at 200C and the mixture was stirred at 200C for 2 hours until all solids disappeared. The mixture was then
18
LEX-1287 concentrated to 300 mL, added to H2O (IL) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, x3) and the cake was dried under vacuum at 45°C overnight to afford the desired methyl thiolate (67.Og, 95%). IH NMR (CDC13) δ 7.38 (d, J = 8.4 Hz, IH), 7.22 (m, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.35 (d, J = 9.6 Hz, IH), 4.15 (d, J = 9.6 Hz, IH), 4.10-3.95 (m, 3H), 3.64 (t, J = 8.8 Hz, IH), 3.50 (m, 2H), 3.42 (br s, IH), 2.95 (br s, IH), 2.57 (br s, IH), 2.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H).
6.9. Preparation of Crystalline Anhydrous (2S,3R,4R,5S,6R)-2-(4-chloro-
3-f4-ethoxybenzyl)phenyl)-6-fmethylthio)tetrahydro-2H-pyran- 3,4,5-triol Form 1
Under slightly positive nitrogen pressure, to a 50 L reactor was charged MeOH (12 L) and the triacetate (1.70 Kg, 3.09 mol). Methanol (5L) was added as a rinse. The slurry was then added NaOMe in MeOH (25 wt%, 340 mL, 0.2X) in 15 minutes at 200C and the mixture was stirred at 200C for 2 hours until all solids disappeared. To the mixture was added slowly water (25.5 L, 15X) in 45 minutes with 5 g seeding (DSC123°C). Solids crashed out and the mixture was stirred at 200C for 1 hour, cooled to 00C and stirred for 30 minutes. The solid was filtered and washed with water (1.7 L, IX, x2) and the cake was dried under vacuum at 45°C overnight to afford the title compound (m.p. ~ 123 0C by DSC peak; 1.28 Kg, 97.7% yield).
…………..
http://www.google.com/patents/US20090030198

EXAMPLES
Aspects of this invention can be understood from the following examples, which do not limit its scope.
6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone

To a 12 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(−)-xylose (504.40 g, 3.360 mol), acetone (5 L, reagent grade) and anhydrous MgSO4 powder (811.23 g, 6.740 mol/2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol/0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24° C. over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH =8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142 mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5 S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50% w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25 L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wt % water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23 g, 86% yield, 96.7 area % pure by GC). 1H NMR (400 MHz, DMSO-d6) δ 1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93-4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 3C NMR (101 MHz, DMSO-d6) δ 26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73. To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0 g, 131 mmol) in acetone (375 mL, 15×) and H2O (125 mL, 5×) was added NaHCO3 (33.0 g, 3.0 equiv), NaBr (2.8 g, 20 mol %) and TEMPO (0.40 g, 2 mol %) at 20° C. The mixture was cooled to 0-5° C. and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20° C. for 24h. Methanol (20 mL) was added and the mixture was stirred at 20° C. for 1 h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2×). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12× ×3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5×) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0 g, 79%).1H NMR (methanol-d4), δ 6.00 (d, J=3.2 Hz, 1H), 4.72 d, J=3.2 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.38 (d, J=3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H). To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0 g, 24.5 mmol) in THF (100 ML, 20×) was added TBTU (11.8 g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20° C. for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20° C. for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2× ×2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4: 1) to afford 4.3 g of the desired morpholine amide (64%) as a white solid. 1H NMR (CDCl3), δ 6.02 (d, J=3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
6.2. Alternative synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone
A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0 L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2solution (1.6 L) and bleach/K2HPO4 solution (400 mL, 1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25° C. Additional charges of TEMPO (14.3 g, 0.5 mol %) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to <10° C. and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15° C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L ×3). The combined organic layer was concentrated under vacuo (˜100-120 torr) at <35° C. (28-32° C. vapor, 45-50° C. bath) to low volume (˜6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached <1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5° C. and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) δ 5.96 (d, J=3.6 Hz, 1H), 4.58 (d, J=3.6 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.30 (d, J=3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) δ 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25.1. The morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (7.85 kg, 26.9 mol), morpholine (2.40 L, 27.5 mol) and boric acid (340 g, 5.49 mol, 0.2 eq) were added to toluene (31 L). The resulting slurry was degassed and heated at reflux with a Dean-Stark trap under nitrogen for 12 h and then cooled to room temperature. The mixture was filtered to remove insolubles and the filter cake washed with toluene (5 L). The filtrate was concentrated to about 14 L and flushed with toluene (˜80 L) to remove excess morpholine. When final volume reached 12 L, heptane (14 L) was added slowly at 60-70° C. The resulting slurry was cooled gradually to room temperature and aged for 3 h. It was then filtered and washed with heptane (12 L) and dry under nitrogen gave a slightly pink solid (6.26 kg, 97% pure, 98% yield). m.p.: 136° C. (DSC). 1H NMR (CDCl3), δ 6.02 (d, J=3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H). 13C NMR (methanol-d4) δ 26.84, 27.61, 44.24, 47.45, 68.16, 77.14, 81.14, 86.80, 106.87, 113.68, 169.05.
6.3. Synthesis of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene

A 2 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with 2-chloro-5-iodobenzoic acid (199.41 g, 0.706 mol), dichloromethane (1.2L, KF=0.003 wt % water) and the suspension was set stirring at ambient temperature. Then N,N-dimethylformamide (0.6 mL, 1.1 mol %) was added followed by oxalyl chloride (63 mL, 0.722 mol, 1.02 equiv) which was added over 11 min. The reaction was allowed to stir at ambient overnight and became a solution. After 18.75hours, additional oxalyl chloride (6 mL, 0.069 mol, 0.10 equiv) was added to consume unreacted starting material. After 2 hours, the reaction mixture was concentrated in vacuo to afford crude 2-chloro-5-iodobenzoyl chloride as a pale yellow foam which will be carried forward to the next step. A jacketed 2 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with aluminum chloride (97.68 g, 0.733 mol, 1.04 equiv), dichloromethane (0.65 L, KF=0.003 wt % water) and the suspension was set stirring under nitrogen and was cooled to about 6° C. Then ethoxybenzene (90 mL, 0.712 mol, 1.01 equiv) was added over 7 minutes keeping internal temperature below 9° C. The resulting orange solution was diluted with dichloromethane (75 mL) and was cooled to −7° C. Then a solution of 2-chloro-5-iodobenzoyl chloride (<0.706 mol) in 350 mL dichloromethane was added over 13 minutes keeping the internal temperature below +3° C. The reaction mixture was warmed slightly and held at +5° C. for 2 hours. HPLC analysis suggested the reaction was complete and the reaction was quenched into 450 mL pre-cooled (˜5° C.) 2N aq. HCl with stirring in a jacketed round bottom flask. This quench was done in portions over 10 min with internal temperature remaining below 28° C. The quenched biphasic mixture was stirred at 20° C. for 45 min and the lower organic phase was washed with 1N aq. HCl (200 mL), twice with saturated aq. sodium bicarbonate (200 mL per wash), and with saturated aq. sodium chloride (200 mL). The washed extract was concentrated on a rotary evaporator to afford crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone as an off-white solid (268.93 g, 99.0 area % by HPLC at 220 nm, 1.0 area % regioisomer at 200 nm, 98.5 % “as-is” yield). A jacketed 1 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged with crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone (30.13 g, 77.93 mmol), acetonitrile (300 mL, KF=0.004 wt % water) and the suspension was set stirring under nitrogen and was cooled to about 5° C. Then triethylsilane (28 mL, 175.30 mmol, 2.25 equiv) was added followed by boron trifluoride-diethyletherate (24 mL, 194.46 mmol, 2.50 equiv) which was added over about 30 seconds. The reaction was warmed to ambient over 30 min and was stirred for 17 hours. The reaction was diluted with methyl tert-butyl ether (150 mL) followed by saturated aq sodium bicarbonate (150 mL) which was added over about 1 minutes. Mild gas evolution was noticed and the biphasic solution was stirred at ambient for 45 minutes. The upper organic phase was washed with saturated aq. sodium bicarbonate (100 mL), and with saturated aq. sodium chloride (50 mL). The washed extract was concentrated on a rotary evaporator to about one half of its original volume and was diluted with water (70 mL). Further concentration in vacuo at 45° C. was done until white prills formed which were allowed to cool to ambient while stirring. After about 30 minutes at ambient, the suspended solids were isolated by filtration, washed with water (30 mL), and were dried in vacuo at 45° C. After about 2.5 hours, this afforded 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene as a slightly waxy white granular powder (28.28 g, 98.2 area % by HPLC at 220 nm, 97.4 % “as-is” yield).
6.4. Synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanone

To a solution of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene (500 mg, 1.34 mmol) in THF (5.0 mL) was added i-PrMgCl (2.0M in THF, 1.0 mL, 2.00 mmol) at 0-5° C., and the mixture was stirred for 1.5 h at 0-5° C. A solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone (146.5 mg, 0.536 mmol) in THF (1.0 mL) was added dropwise at 0-5° C. and the mixture was kept stirring for 1 h, warmed to 20° C. and stirred at 20° C. for 2 hours. The reaction was quenched with saturated aq NH4Cl, extracted with MTBE, washed with brine. The organic layer was concentrated and the residue was purified by silica gel column chromatography to afford the desired ketone (178 mg, 76%) as a white solid. 1H NMR (CDCl3) δ 7.88 (dd, J=8.4, 2.0 Hz, 1H), 7.82 (d, J=2.0 Hz, 1H), 7.50 (d, J=8.4 Hz, 1H), 7.12 (d, J=8.4 Hz, 2H), 6.86 (d, J=8.4 Hz, 2H), 6.07 (d, J=3.2 Hz, 1H), 5.21 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 4.56 (d, J=3.2 Hz, 1H), 4.16 (d, J=7.2 Hz, 2H), 4.03 (q, J=7.2 Hz, 2H), 1.54 (s, 3H), 1.42 (t, J=7.2 Hz, 3H), 1.37 (s, 3H).
6.5. Alternative synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d]1,3]dioxol-5-yl)methanone
To a 20 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet was charged with the iodide (3.00 kg, 8.05 mol) and THF (8 L, 4× to the morpholinoamide) at room temperature and cooled to −5° C. To the above solution was added dropwise a solution of i-PrMgCl in THF (Aldrich 2 M, 4.39 L, 8.82 mol) at −5° C. over 3 hours. This Grignard solution was used in the ketone formation below. To a 50 L reactor equipped with a mechanical stirrer, a temperature controller, and a nitrogen inlet was charged the morpholinoamide (HPLC purity=97 wt %, 2.01 kg, 7.34 mol) and THF (11 L, 5.5×) at room temperature and stirred for 45 minutes at room temperature and for 15 minutes at 30° C. The homogeneous solution was then cooled to −25° C. To this solution was added a solution of t-BuMgCl in THF (Aldrich 1 M, 7.32 L, 7.91 mol) at −25° C. over 3 hours. Then the above Grignard solution was added to this solution at −20 over 41 minutes. The resulting solution was further stirred at −20° C. before quench. The reaction mixture was added to 10 wt % aqueous NH4Cl (10 L, 5×) at 0° C. with vigorous stirring, and stirred for 30 minutes at 0° C. To this mixture was added slowly 6 N HCl (4 L, 2×) at 0° C. to obtain a clear solution and stirred for 30 minutes at 10° C. After phase split, the organic layer was washed with 25 wt % aq NaCl (5 L, 2.5×). Then the organic layer was concentrated to a 3× solution under the conditions (200 mbar, bath temp 50° C.). EtOAc (24 L, 12×) was added, and evaporated to a 3× solution under the conditions (150 mbar, bath temp 50° C.). After removed solids by a polish filtration, EtOAc (4 L, 2×) was added and concentrated to dryness (150 mbar, bath temp 50° C.). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70° C. to obtain a 2.5× homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5×) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5×) was added slowly to a little cloudy solution at 70° C. After stirred for 0.5 h at 70° C., the suspension was slowly cooled to 60° C. and stirred for 1 h at 60° C. The suspension was then slowly cool to room temperature and stirred for 14 h at the same temperature. The crystals were collected and washed with heptane (8 L, 4×), dried under vacuum at 45° C. to give the desired ketone as fluffy solids (2.57 kg, 100 wt % by HPLC, purity-adjusted yield: 81%).
6.6. Synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate

To a solution of the ketone (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone (114.7 g, 0.265 mol) in MeOH (2 L, 17×) was added CeCl3.7H2O (118.5 g, 1.2 equiv) and the mixture was stirred at 20° C. until all solids were dissolved. The mixture was then cooled to −78° C. and NaBH4 (12.03 g, 1.2 equiv) was added in portions so that the temperature of the reaction did not exceed −70° C. The mixture was stirred at −78° C. for 1 hour, slowly warmed to 0° C. and quenched with saturated aq NH4Cl (550 mL, 5×). The mixture was concentrated under vacuum to remove MeOH and then extracted with EtOAc (1.1 L, 10× ×2) and washed with brine (550 mL, 5×). The combined organics were concentrated under vacuum to afford the desired alcohol as a colorless oil (crude, 115 g). To this colorless oil was added AcOH (650 mL) and H2O (450 mL) and the mixture was heated to 100° C. and stirred for 15 hours. The mixture was then cooled to room temperature (20° C.) and concentrated under vacuum to give a yellow oil (crude, 118 g). To this crude oil was added pyridine (500 mL) and the mixture was cooled to 0° C. Then, Ac2O (195 mL, ˜8.0 equiv) was added and the mixture was warmed to 20° C. and stirred at 20° C. for 2 h. The reaction was quenched with H2O (500 mL) and diluted with EtOAc (1000 mL). The organic layer was separated and concentrated under vacuum to remove EtOAc and pyridine. The residue was diluted with EtOAc (1000 mL) and washed with aq NaHSO4 (1N, 500 mL, ×2) and brine (300 mL). The organic layer was concentrated to afford the desired tetraacetate intermediate as a yellow foam (˜133 g). To a solution of tetraacetate (133 g, 0.237 mol assuming pure) and thiourea (36.1, 2.0 equiv) in dioxane (530 mL, 4×) was added trimethylsilyl trifluoromethanesulfonate (TMSOTf) (64.5 mL, 1.5 equiv) and the reaction mixture was heated to 80° C. for 3.5 hours. The mixture was cooled to 20° C. and MeI (37 mL, 2.5 equiv) and N,N-diisopropylethylamine (DiPEA) (207 mL, 5.0 equiv) was added and the mixture was stirred at 20° C. for 3 h. The mixture was then diluted with methyl tertiary-butyl ether (MTBE) (1. 3 L, 10×) and washed with H2O (650 mL, 5× ×2). The organic layer was separated and concentrated under vacuum to give a yellow solid. To this yellow solid was added MeOH (650 mL, 5×) and the mixture was reslurried at 60° C. for 2 h and then cooled to 0C and stirred at 0° C. for 1 hour. The mixture was filtered and the cake was washed with MeOH (0° C., 70 mL, ×3). The cake was dried under vacuum at 45° C. overnight to afford the desired triacetate (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (88 g, 60% over 4 steps) as a pale yellow solid. 1H NMR (CDCl3) 6 7.37 (d, J=8.0 Hz, 1H), 7.20 (dd, J=8.0, 2.0 Hz, 1H), 7.07 (m, 2H), 6.85 (m, 2H), 5.32 (t, J=9.6 Hz, 1H), 5.20 (t, J=9.6 Hz, 1H), 5.05 (t, J =9.6 Hz, 1H), 4.51 (d, J =9.6 Hz, 1H), 4.38 (d, J=9.6 Hz, 1h), 4.04 (m, 2H), 2.17 (s, 3H), 2. 11 (s, 3H), 2.02 (s, 3H), 1.73 (s, 3H), 1.42 (t, J=7.2 Hz, 3H).
6.7. Alternative synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a 50 L reactor under nitrogen atmosphere, 40 L MeOH was charged, followed with the ketone (2.50 kg, 5.78 mol) and CeCl3.7H2O (2.16 kg, 1.0 equiv). Methanol (7.5 L) was added as rinse (totally 47.5 L, 19×). A freshly prepared solution of NaBH4 (87.5 g, 0.4 equiv) in aqueous 1 N NaOH (250 mL) was added slowly (35 min) at 15-25° C. The mixture was then stirred for 15 min. HPLC analysis of the reaction mixture showed approximately 90:10 diastereomeric ratio. The reaction was quenched with 10 wt % aq NH4Cl (2.5 L, 1×) and the mixture was concentrated under vacuum to 5×, diluted with water (10 L, 4×) and MTBE (12.5 L, 5×). The mixture was cooled to 10° C. and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H2O (5L, 2×). The combined aqueous layer was extracted with MTBE (12.5 L, 5×). The combined organic layers were washed with brine (2.5 L, 1×) and concentrated under vacuum to 3×. MeCN (15 L, 6×) was added. The mixture was concentrated again to 10 L (4×) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN. The organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol % aqueous H2SO4 solution (61.8 mL 98% concentrated H2SO4 and 5 L H2O) was added. The mixture was heated to 80° C. for 2 hours and then cooled to 20° C. The reaction was quenched with a solution of saturated aqueous K2CO3 (5 L, 2×) and diluted with MTBE (15 L, 6×). The organic layer was separated, washed with brine (5 L, 2×) and concentrated under vacuum to 5 L (2×). MeCN (12.5 L, 5×) was added and the mixture was concentrated to 7.5 L (3×). The above MeCN solution of (3S,4R,5R,6S)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl)tetrahydro-2H-pyran-2,3,4,5-tetraol was cooled to 10° C., added with dimethylaminopyridine (17.53 g, 2.5 mol %), followed by slow addition of acetic anhydride (3.23 L, 6.0 equiv) and triethylamine (5 L, 2×, 6.0 equiv) so that the temperature of the mixture was kept below 20° C. The reaction was then warmed to 20° C. and stirred for 1 hour and diluted with MTBE (15 L, 6×). The mixture was slowly quenched with water (7.5 L, 3×). The organic layer was separated and washed with saturated aqueous KHCO3 (5L, 2×), 1 N NaHSO4 (5 L, 2×), and brine (5 L, 2×) in sequence. The organic layer was then concentrated under vacuum to 5 L (2×). MeCN (12.5 L, 5×) was added and the solution was concentrated to 7.5 L (3×) (KF=0.08%). Dioxane (12.5 L, 5×) was added and the solution was concentrated to 7.50 L (3×) (KF=0.02%). Any residual solid was removed by a polish filtration and the cake was washed with minimal amount of dioxane (500 mL). To the above filtrate was added thiourea (880 g, 2.0 equiv) and TMSOTf (1.57 L, 1.5 equiv). The reaction mixture was heated to 80° C. for 3 hours (>97% conversion). The mixture was cooled to 20° C. and methyl iodide (541 mL, 1.5 equiv) and diethylisopropylamine (3.02 L, 3.0 equiv) were added and the mixture was stirred at 20° C. for 18 hours. An extra methyl iodide charge (90 mL, 0.25 equiv) was added and the mixture was stirred at 20° C. for 1 hours. The mixture was then diluted with MTBE (25 L, 10×) and washed with water (12.5 L, 5× ×2). The organic layer was separated and concentrated under vacuum to ˜5 L (2×). MeOH (12.5 L, 5×) was added and the mixture was concentrated to 5× to afford a slurry. The mixture was then heated at 60° C. for 1 hour and cooled to 0° C. and stirred at 0° C. for 1 hour. The mixture was filtered and the cake was washed with MeOH (0° C., 2.5 L, 1× ×2, 1.0 L, 0.4×). The cake was dried under vacuum at 45° C. overnight to afford the desired triacetate (1.49 kg, 47% over 4 steps) as a pale yellow/off-white solid.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol

To a slurry of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0. 164 mol) in MeOH (900 mL, 10×) was added NaOMe in MeOH (25 wt %, 18 mL, 0.2×) at 20° C. and the mixture was stirred at 20° C. for 2 hours until all solids disappeared. The mixture was then concentrated to 300 mL, added to H2O (1 L) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, ×3) and the cake was dried under vacuum at 45° C. overnight to afford the desired methyl thiolate (67.0 g, 95%). 1H NMR (CDCl3) 6 7.38 (d, J=8.4 Hz, 1H), 7.22 (m, 2H), 7.11 (d, J=8.8 Hz, 2H), 6.83 (d, J=8.8 Hz, 2H), 4.35 (d, J=9.6 Hz, 1H), 4.15 (d, J=9.6 Hz, 1H), 4.10-3.95 (m, 3H), 3.64 (t, J=8.8 Hz, 1H), 3.50 (m, 2H), 2.73 (br s, 3H), 2.17 (s, 3H), 1.40 (t, J=7.2 Hz, 3H).
…………..
SGLT inhibitors: a novel target for diabetes.
Kanwal A, Banerjee SK.
Pharm Pat Anal. 2013 Jan;2(1):77-91. doi: 10.4155/ppa.12.78.
clinical trials………..http://clinicaltrials.gov/search/intervention=LX-4211+OR+LX4211
On the importance of synthetic organic chemistry in drug discovery: reflections on the discovery of antidiabetic agent ertugliflozin. , Med. Chem. Commun., 2013, 4, 101
Carbohydrate Derivatives and Glycomimetic Compounds in Established and Investigational Therapies of Type 2 Diabetes Mellitus

A PART IS PASTED
Carbohydrate Derivatives and Glycomimetic Compounds in Established and Investigational Therapies of Type 2 Diabetes Mellitus
László Somsák, Éva Bokor, Katalin Czifrák, László Juhász and Marietta Tóth (2011). Carbohydrate Derivatives and Glycomimetic Compounds in Established and Investigational Therapies of Type 2 Diabetes Mellitus, Topics in the Prevention, Treatment and Complications of Type 2 Diabetes, Prof. Mark Zimering (Ed.), ISBN: 978-953-307-590-7, InTech, DOI: 10.5772/23463. Available from: http://www.intechopen.com/books/topics-in-the-prevention-treatment-and-complications-of-type-2-diabetes/carbohydrate-derivatives-and-glycomimetic-compounds-in-established-and-investigational-therapies-of-
1. Introduction
Diabetes mellitus is characterized by chronically elevated serum glucose levels resulting in damage of several tissues (e. g. retina, kidney, nerves) due to higher protein glycation, retardation of wound healing, impaired insulin secretion, enhanced insulin resistance, cell apoptosis, and increased oxidative stress. Type 2 diabetes (T2DM), representing 90-95 % of all diabetic cases, is a multifactorial disease where impaired insulin secretion and the development of insulin resistance ultimately leads to hyperglycemia (Hengesh, 1995). The end of the 20th century has witnessed a dramatic increase in the number of patients diagnosed with diabetes worldwide. The predicted number for the year 2025 is well over 300 million representing a 4-5 % yearly increase of the population above 20 years of age (Treadway et al., 2001). This striking prevalence can even be an underestimate due to methodological uncertainties as well as undiagnosed cases (Green et al., 2003). The highest increases are expected in the developing countries of Africa, Asia, and South America, while European populations seem to be less affected (Diamond, 2003). T2DM has been considered as the adult- or late-onset variant, however, the recent decade has seen the appearance and spreading of the disease among young people including children: this forecasts severe economic and health service burdens in the near future (Alberti et al., 2004; Ehtisham & Barrett, 2004).
The epidemic of T2DM is in conjunction with genetic susceptibility: evidence for a genetic component to the disease are accumulating, and the potential of these factors in the treatment and prevention of diabetes has been reviewed (Barroso, 2005; Bonnefond et al., 2010; Sladek et al., 2007; Toye & Gauguier, 2003). A similarly high contribution to this epidemic may originate from behavioral factors such as sedentary lifestyle, overly rich nutrition, and obesity (Bloomgarden, 2004).
Especially due to its long term complications (Brownlee, 2001) like retinopathy, neuropathy, nephropathy, and in particular cardiovascular diseases, as well as significantly higher risk of myocardial infarction, stroke, gangrene, and limb amputation diabetes has become one of the largest contributors to disability and mortality. Although several pathomechanisms (Lowell & Shulman, 2005;Panunti et al., 2004; Stumvoll et al., 2005) are under investigation, no firm understanding of the molecular origins (Ross et al., 2004) of the disease exists. Thereby, all available and investigational treatments are symptomatic. As the complications can first of all be attributed to the high blood glucose levels, current antidiabetic therapies (Table 1) aim at reaching normoglycemia. However, most of the applied oral hypoglycemic agents (Cheng & Fantus, 2005; Krentz & Bailey, 2005; Mizuno et al., 2008; Padwal et al., 2005; Rendell, 2004; Uwaifo & Ratner, 2005) have several side effects and are inadequate for 30-40 % of the patients (Wagman & Nuss, 2001). On the other hand, their efficacy is lost over the time, and several concerns exist regarding their safety (Israili, 2011).
TABLE 1.
Main types of current therapeutic agents for T2DM and their major side effects (Israili, 2011;Moller, 2001)
The complexity of T2DM offers many potential points of intervention for pharmacotherapy for which the main molecular targets and strategies such as insulin secretagogues, insulin sensitizers, hormones, inhibitors of PTP-1B, GSK3, and hepatic glucose production, methods for altering lipid metabolism, combination therapies, etc. have been reviewed in details (Israili, 2011; Morral, 2003; Nourparvar et al., 2004; Wagman et al., 2004).
Among the numerous methods used to treat type 2 diabetes and investigated to find new therapeutic possibilities there are several approaches which apply carbohydrate (especially glucose) derivatives as well as compounds mimicking the properties of sugars. Based on our experience in the chemistry of carbohydrates and glycomimetics, in this survey we summarize the roles of such compounds in combatting type 2 diabetes relying on the review literature and very recent primary scientific papers.
2. Inhibitors of α-glucosidase enzymes
Starch and sucrose are the most important dietary carbohydrates but they are not directly available for the cells. They are digested in the gastrointestinal tract to monosaccharides which can be absorbed to the circulation to raise the serum concentration (Hanhineva et al., 2010). The normal blood glucose level (3.6–5.8 mM) fluctuates throughout the day, is usually lowest in the morning, before the first meal of the day, and rises after meals for an hour or two.
A medically applied treatment of diabetes is to retard the absorption of glucose by inhibition of the carbohydrate hydrolyzing enzymes α-amylase and α-glucosidase in the digestive tract. In humans the digestion of starch, maltodextrins, and maltooligosaccharides includes several stages: degradation of the polymeric substrates results in shorter oligomers which are than cleaved by α-amylase into smaller oligosaccharides. This mixture is broken down to monosaccharides by α-glucosidase from the non-reducing end of the oligosaccharides. By inhibition of these enzymes the rate of glucose production can be reduced that contributes to diminishing the blood glucose levels, too (Tundis et al., 2010). Such inhibitors decrease postprandial hyperglycaemia and hyperinsulinaemia, thereby may improve sensitivity to insulin and release the stress on β-cells (Scheen, 2003).
Glycosidases are a long known and studied class of glycoenzymes for which an enormous number of compounds have been tested as inhibitors (El Ashry et al., 2000a; El Ashry et al., 2000b; El Ashry et al., 2000c; Lillelund et al., 2002). Analogues of monosaccharides in which the ring oxygen is replaced by a nitrogen atom are known as iminosugars (or less properly azasugars) comprising both natural and synthetic molecules (Table 2) which, as the most potent inhibitors of glycosidases, have high pharmacological potential not only in the context of T2DM (Asano, 2009; Compain & Martin, 2007).
The naturally occurring salacinol and analogous sugar mimics with a 4-thiofuranoid type ring (Table 2) belong to a growing class of zwitterionic glycosidase inhibitors, which attract great interest both as synthetic targets and applications for α-glucosidase inhibition (Praly & Vidal, 2010).
The positive charge on the sulfur atom in the thiosugar derivatives and in the iminosugar-based glycosidase inhibitors at physiological pH is facilitating the binding in the active sites of glycosidase enzymes as a mimicry of the charge of the oxocarbeniumion-like transition state formed during hydrolysis of the natural enzyme substrate (Zechel & Withers, 2000). The stabilizing electrostatic interactions between the ammonium (protonated nitrogen) or sulfonium (positively charged sulfur) moieties and an active-site carboxylate residue are considered to be a possible mechanism of action of these inhibitors (Mohan & Pinto, 2007).
Three competitive inhibitors of α-glucosidases: acarbose, miglitol, and voglibose (de Melo et al., 2006) (Table 3) are used as drugs in the treatment of T2DM under various brand names. These compounds are known to inhibit a wide range of glycosidases. In the absence of specificity and because of the known serious side effects, the applications of these first generation iminosugar drugs are limited. Current investigations aim at discovering safer, more specific, and effective iminosugar based derivatives not only as hypoglycemic agents but for several other purposes among others in oncology, as antivirals, and against cystic fibrosis as reviewed in (Home et al., 2011).

Select iminosugar and thiosugar type inhibitors and their effect againstα-glucosidases originating from mammalian gastrointestinal tract



TABLE 3.
α-Glucosidase inhibitors in the clinical practice against T2DM
CONTD……………………
Merck & Co gets FDA OK for allergy treatment Grastek

Merck & Co and ALK-Abello are celebrating the US green light for their grass pollen allergy immunotherapy Grastek.![]()
The US Food and Drug Administration has approved Grastek, an allergen extract in a sublingual tablet, for the treatment of Timothy grass pollen-induced allergic rhinitis with or without conjunctivitis. The thumbs-up was expected given that the FDA’s Allergenic Products Advisory Committee voted unanimously to recommend approval at the end of 2013 but it does come with a boxed warning regarding severe allergic reactions.
Read more at: http://www.pharmatimes.com/Article/14-04-15/Merck_Co_gets_FDA_OK_for_allergy_treatment_Grastek.aspx#ixzz2z11s8ydQ

Telapristone acetate

Telapristone acetate
[(8S,11R,13S,14S,17R)-11-[4-(Dimethylamino)phenyl]-17-(2-methoxyacetyl)-13-methyl-3-oxo-1,2,6,7,8,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-17-yl] acetate
17-acetoxy- 11 β-[4-(dimethylamino)-ρhenyl]-21-methoxy-19-noφregna-4,9-dien-3,20-dione
17-Acetoxy-llβ-f4-(dimethylamino)-phenyl)1-21-methoxy-19-norpregna-4,9-dien-3,20- dione
17α-acetoxy-llβ-[4-(N,N-dimethylamino)phenyl]-21-methoxy- 19-norpregna-4, 9-diene-3,20-dione
CDB-4124; 17α-Acetoxy-21-methoxy-11β-[4-N,N-dimethylaminophenyl]-19-norpregna-4,9-diene-3,20-dione)
Telapristone (proposed trade names Proellex and Progenta) is an investigational selective progesterone receptor modulator, tested for treatment of progesterone sensitive myomata.[1] CDB-4124 was originally developed the National Institutes of Health, and as of 2012 is in Phase II clinical trials for uterine fibroids and endometriosis.[2] It also has some antiglucocorticoidactivity

17α-acetoxy-21-methoxy-11β-[4-N,N-dimethylaminophenyl]-19-norpregna-4,9-diene-3,20-dione) is a selective progesterone receptor modulator, it is being tested for treatment of progesterone sensitive myomata.
International patent application WO 97/41145 disclosed for the first time the preparation of 17α-acetoxy-21-methoxy-11β-[4-N,N-dimethylaminophenyl]-19-norpregna-4,9-diene-3,20-dione). In example 9 it is characterized as light-yellow powder with a melting point of 116° C. (purity: 98.06%, characteristic FT-IR absorption bands at: 1124, 1235, 1370, 1446, 1518, 1612, 1663, 1734, 2940 cm−1).
According to the published international patent applications of WO 01/47945 and WO 01/74840 the obtained 17α-acetoxy-21-methoxy-11β-[4-N,N-dimethylaminophenyl]-19-norpregna-4,9-diene-3,20-dione) was light-yellow powder as well having a melting point of 116° C. (purity: 98.87%, 98.06%, characteristic FT-IR absorption bands at: 1124, 1235, 1370, 1446, 1518, 1612, 1662, 1734, 2940 cm−1)
………………
http://www.google.com/patents/WO2001047945A1?cl=en
Preparation of 17α-hydroxy-llβ-[4-(N,N-dimethylamino)phenyl]-21-methoxy- 19-norpregna-4,9-diene-3,20-dione (10) :
A suspension of 2-iodoxybenzoic acid (IBX, 599 mg, 2.14 mmol) in anhydrous dimethylsulfoxide (DMSO) (5.0 mL; Aldrich, Sure-Seal) was stirred magnetically under nitrogen and warmed in an oil bath at 55 – 60°C. After several minutes, all of the IBX was solubilized. To the IBX solution was added a solution of the 20-alcohol (18, 500 mg, 1.07 mmol) in DMSO (5 mL). Additional DMSO (3 mL) was used to rinse in residual 18. After a period V2 hr of reaction, approximately 70% of the 20-alcohol (18) had been converted to the 20-ketone (10), as evidenced by TLC (15% acetone in methylene chloride; aliquot was diluted in water and extracted by EtOAc). After 3 hr, there was no observable change in the conversion. The reaction mixture was transferred to a separatory funnel, diluted with water, and extracted by EtOAc (3x). The EtOAc extracts were washed with additional water (2x) and brine (lx). The combined extracts were dried by filtration through sodium sulfate, evaporated in vacuo, and dried overnight under high vacuum to recover 600 mg of a brown film. The film product was taken up in EtOAc and filtered through silica on a sintered glass funnel to remove residual DMSO and highly polar impurities. Evaporation of EtOAc afforded 450 mg of a yellow film. Repeated trituration with hexane, with scratching and sonicating, produced a solid. The solid was dried overnight under high vacuum to give 349 mg of 10 as a yellow powder in 70.1% yield. The product was carried directly to the next reaction without further purification. NMR (300 MHz, CDCI3) : δ 0.408 (s, 3 H, C18-CH3),2.906 (s, 6 H, -N(CH3)2), 3.454 (s, 3 H, C21-OCH3), 4.245 and 4.388 (AB, 2 H, C21-CH2, JAB = 17.41 Hz), 4.378 (d, 1 H, Cllβ-CH, J = 7.50), 5.758 (s, 1 H, C4-CH), 6.638 (d, 2 H, 3′,5′-aromatic CH, J = 8.55 Hz) and 6.975 (d, 2 H, 2′,6′-aromatic CH, J = 8.55 Hz).
Preparation of 17α-acetoxy-llβ-[4-(N,N-dimethylamino)phenyl]-21-methoxy- 19-norpregna-4, 9-diene-3,20-dione (11) :
A mixture of trifluoroacetic anhydride (47 mL) and glacial acetic acid (19.1 mL) in methylene chloride (300 mL) was allowed to stir at room temperature under nitrogen. After 1/2 hr of stirring, the mixture was cooled to 0°C in an ice water bath and tosic acid (2.85 g, 14.98 mmol) was added. A solution of the 17α-hydroxy compound (10, 6.18 g, 13.33 mmol) was added in 50 mL of methylene chloride and rinsed in with additional CH2CI2 (50 mL). After stirring for a period of 2 hr at 0°C, examination by TLC (silica; 10% acetone in methylene chloride; neutralized with NH4OH before developing) indicated that the reaction was >95% complete. The reaction mixture was diluted with water (300 mL) and neutralized by careful addition of concentrated NH4OH (75 mL).
More NH4OH was added to a pH of 7 as indicated by a pH paper. The product obtained was extracted by CH2CI2 (3x) and the organic extracts were washed with water (2x) and brine (lx). The combined organic extracts were dried by filtration through Na2SO4 and evaporated in vacuo to give 7.13 g of the crude product (11). A pure material was obtained by flash column chromatography (silica; 10% acetone in methylene chloride). The impure fractions were combined and chromatographed a second time. The pure fractions from both chromatographic runs were combined and evaporated in vacuo, then evaporated from ether, and further dried under high vacuum to produce a pale yellow foam. Treatment with pentane followed by scratching and sonicating produced 4.13 g of 11 as a fine yellow powder in 61.3% yield; m.p. softens at 116°C.
Analysis by a reverse phase HPLC on a NOVAPAK™ Cι8 column eluted with 70% CH3OH in water with 0.03% Et3N at a flow rate of 1 mL per min and at λ = 302 indicated 98.87 % purity of 11 with retention time tR = 6.45 min.
FTIR (KBr, diffuse reflectance) : vmax 2940, 1734, 1662, 1612, 1518, 1446, 1370, 1235 and 1124 cm“1.
NMR (300 MHZ, CDCI3) : δ 0.38 (s, 3 H, C18-CH3), 2.08 (s, 3H, C17α-0Ac), 2.90 (s, 6 H, -N(CH3)2), 3.42 (s, 3 H, C21-OCH3), 4.07 and 4.33 (AB, 2 H, C21-CH2, JAB= 18 Hz), 4.37 (s, 1 H, Cllβ-CH), 5.80 (s, 1 H, C4-CH), 6.67 (d, 2 H, 3′,5′-aromatic CH, J = 9 Hz) and 7.0 (d, 2 H, 2′, 6′- aromatic CH, J = 9 Hz).
MS (El) m/z (relative intensity) : 505 (M+, 13.5), 445 (1.1), 372 (2.7), 134 (16.2) and 121 (100).
Anal. Calcd for C31H39NO5: C, 73.64; H, 7.77; N, 2.77 Found : C, 73.34; H, 7.74; N, 2.70.
…………….
synthesis
http://www.google.com/patents/WO2009001148A2?cl=en
According to the above mentioned facts, there is no such known process, which is suitable for the realization of the synthesis of CDB-4124 on industrial scale using simple reaction conditions. Our aim was to elaborate a process, which is easy to scale-up, the industrial realization of which is safe, economical and the purity of the active ingredient fulfils the requirements of the pharmacopoeia.
Surprisingly it was found, that the following process fulfils the above mentioned requirements: i) epoxide formation on the double bond in position 5(10) of 3,3-[l,2-ethandiyl- bis(oxy)]-oestr-5(10),9(l l)-dien-17-one of formula (II)

with hydrogen peroxide; ii) addition of hydrogen cyanide formed in situ on position 17 of the obtained 5,1 Oa- epoxy-3,3-[l,2-ethandiyl-bis(oxy)]-5α-oestr-9(l l)-en-17-one of formula (III)

iii) silylation of the hydroxyl group in position 17 of the formed 5,10α-epoxy-3,3-[l,2- ethandiyl-bis(oxy)]-17α-hydroxy-5α-oestr-9(l l)-en-17β-carbonitrile of formula (IV)

with trimethyl chlorosilane; iv) reacting the obtained 5,10α-epoxy-3,3-[l,2-ethandiyl-bis(oxy)]-17-[trimethyl-silyl- oxy]-5α-oestr-9(ll)-en-17β-carbonitrile of formula (V)

with 4-(dimethylamino)-phenyl magnesium bromide Grignard reagent in the presence of CuCl
(Teutsch reaction); v) silylation of the hydroxyl group in position 5 of the formed 1 lβ-[4-(dimethyl-amino)- phenyl]-3 ,3-[ 1 ,2-ethandiyl-bis(oxy)] -5-hydroxy- 17α-[trimethylsilyl-(oxy)] -5α-oestr-9-en- 17β- carbonitrile of formula (VI)

with trimethyl chlorosilane; vi) reacting the obtained llβ-[4-(dimethylamino)-phenyl]-3,3-[l,2-ethandiyl-bis(oxy)]- 5,17α-bis-[trimethyl-silyl-(oxy)]-5α-oestr-9-en-l 7β-carbonitrile of formula (VII)

with diisobutyl aluminum hydride and after addition of acid to the reaction mixture vii) methoxy-methylation of the obtained llβ-[4-(dimethylamino)-phenyl]-3,3-[l,2- ethandiyl-bis(oxy)]-5,17α-bis-[trimethyl-silyl-(oxy)]-5α-oestr-9-en-17β-carbaldehide of formula (VIII)

with methoxy-methyl Grignard reagent formed in situ, while hydrolyzing the trimethylsilyl protective groups; viii) oxidation of the hydroxy! group in position 20 of the obtained 17,20ξ-dihydroxy-
3-[4-(dimethylamino)-phenyl]-21 -methoxy- 19-norpregna-4,9-dien-3-one of formula (IX)

with dicyclohexyl carbodiimide in the presence of dimethyl sulfoxide and a strong organic acid (Swern oxidation), and in given case after purification by chromatography ix) acetylation of the hydroxyl group in position 17 of the obtained l lβ-[4- (dimethylamino)-phenyl]- 17-hydroxy-21 -methoxy- 19-norpregna-4,9-dien-3 ,20-dione of formula (X)

with acetic anhydride in the presence of perchloric acid, and in given case the obtained 7- acetoxy-11 β-[4-(dimethylamino)-phenyl)]-21-methoxy-19-norpregna-4,9-dien-3 ,20-dione of formula (I) is purified by chromatography.

Example 11
17-Acetoxy-llβ-f4-(dimethylamino)-phenyl)1-21-methoxy-19-norpregna-4,9-dien-3,20- dione [compound of formula (Dl 70 % Perchloric acid (6 ml) was added to stirred and cooled ((-20) – (-25) 0C) acetic anhydride (45 ml) at such a rate to keep the temperature below (-15) °C. Then a solution of l lβ-[4-(dimethylamino)-phenyl)]-17-hydroxy-21-methoxy-19-norpregna-4,9-dien-3,20-dione (15.5 g) in dichloromethane (60 ml) was added at (-20) – (-25) 0C. After completion of the reaction – followed by thin layer chromatography – the reaction mixture was diluted with dichloromethane (50 ml), cooled to (-10) 0C and ion exchanged water (52 ml) was added to decompose the acetic anhydride. After stirring for 10 min 25 % ammonium hydroxide solution (77 ml) was added at such rate to keep the temperature below 25 0C (pH=7-8). Then the precipitated carbamide by-product was filtered off, the aqueous phase was separated, extracted with dichloromethane (2×30 ml) and the combined organic layers were concentrated to yield 16.2 g (95.8 %) of the title compound, which was purified by HPLC according to method described in the next example. NMR: 1H NMR C500 MHz. CDCl1 (TMS), δ (ppmT): 0.40 (3H, s, 18-CH3); 2.10 (3H5 s, O-CO- CH3); 2.90 (6H, s, N-CH3); 3.41 (3H, s, 0-CH3); 4.09 (IH, d, Hx-21); 4.38 (IH, m, H-Il); 4.29 (IH, d, Hy-21); 5.77 (IH, br, H-4); 6.62 (2H5 m, H-3′ & H-5′); 6.96 (2H, m, H-2′ & H-6′) 13C NMR (125 MHz. CDCU (TMS), δ fppmϊ): 15.6 (C-18); 21.1 (0-CO-CH3); (39.3 (C-Il); 40.6 (N-CH3); 59.4 (0-CH3); 76.0 (C-21); 93.9 (C-17); 112.8 (C-3′ & C-5′); 123.0 (C-4); 127.3 (C-2′ & C-6′); 129.4 (C-IO); 131.3 (C-I’); 145.5 (C-9); 148.7 (C-4′); 156.4 (C-5); 170.7 (0-CO-CH3); 199.4 (C-3); 202.7 (C-20)
Example 12 Purification of crude CDB-4124 by HPLC (eluent: cyclohexanermethyl-tert-butyl- ether;acetone = 60:30:10) (laboratory scale) [compound of formula (DI
Silicagel (51O g, ZEOPREP C-GEL C-490L, 15-35 μm of particle size; bed length about 60 cm) was filled to an axial bed compression HPLC column of 5 cm of diameter with slurry packing method and the column was equilibrated with a 60:30:10 mixture of cyclohexane – methyl-tert-butyl ether – acetone eluent. 5.1 g of the crude compound of formula (I) (CDB-4124) obtained in the previous example (content of impurities: less than 4 %) was dissolved in the eluent (100 ml), filtered and injected on the column. The product was eluted with 85 ml/min flow rate and UV detection was used. The first fraction was about 40 ml, the main fraction containing the pure CDB-4124 was about 560 ml. The solid title compound was obtained by concentration of the eluted main fraction. Yield: 4.25 g (83.33 %), content of impurities: less than 0.5 %. Melting point: 1180C.
[a^ = +127.2 ° (c=l %, chloroform)
NMR: 1H NMR (500 MHz. CDCh (TMS). δ fppmV): 0.40 (3H, s, 18-CH3); 2.10 (3H, s, O-CO-
CH3); 2.90 (6H, s, N-CH3); 3.41 (3H, s, 0-CH3); 4.09 (IH, d, Hx-21); 4.38 (IH, m, H-I l); 4.29 (IH, d, Hy-21); 5.77 (IH, br, H-4); 6.62 (2H, m, H-3′ & H-5′); 6.96 (2H, m, H-2′ & H-6′)
13C NMR (125 MHz. CDCh (TMS), δ (ppm)): 15.6 (C-18); 21.1 (0-CO-CH3); (39.3 (C-Il);
40.6 (N-CH3); 59.4 (0-CH3); 76.0 (C-21); 93.9 (C-17); 112.8 (C-3′ & C-5′); 123.0 (C-4);
127.3 (C-2′ & C-6′); 129.4 (C-IO); 131.3 (C-I’); 145.5 (C-9); 148.7 (C-4′); 156.4 (C-5); 170.7
(0-CO-CH3); 199.4 (C-3); 202.7 (C-20)
References
- Attardi BJ, Burgenson J, Hild SA, Reel JR (2004). “In vitro antiprogestational/antiglucocorticoid activity and progestin and glucocorticoid receptor binding of the putative metabolites and synthetic derivatives of CDB-2914, CDB-4124, and mifepristone”. J Steroid Biochem Mol Biol 88 (3): 277–88. doi:10.1016/j.jsbmb.2003.12.004. PMID 15120421.
- ClinicalTrials.gov
|
5-23-2012
|
Industrial method for the synthesis of 17-acetoxy-11[beta][4-(dimethylamino)-phenyl]-21-methoxy-19-norpregna-4,9-dien-3,20-dione and the key intermediates of the process
|
|
|
6-11-2010
|
Treatment of Macular Degeneratio
|
| ATTARDI BARBARA J ET AL: “CDB-4124 and its putative monodemethylated metabolite, CDB-4453, are potent antiprogestins with reduced antiglucocorticoid activity: In vitro comparison to mifepristone and CDB-2914” MOLECULAR AND CELLULAR ENDOCRINOLOGY, ELSEVIER IRELAND LTD, IE, vol. 188, no. 1-2, 25 February 2002 (2002-02-25), pages 111-123, XP002496575 ISSN: 0303-7207 | ||
| 2 | * | MEALY N E ET AL: “CDB-4124” DRUGS OF THE FUTURE 200411 ES, vol. 29, no. 11, November 2004 (2004-11), page 1133, XP009118559 ISSN: 0377-8282 |
| WO2010106383A1 * | Mar 22, 2010 | Sep 23, 2010 | Richter Gedeon Nyrt | Novel crystalline form of antiprogestin cdb-4124 |
| WO2011015892A2 * | Aug 5, 2010 | Feb 10, 2011 | Richter Gedeon Nyrt. | Novel crystal form of an organic compound and process for the preparation thereof |
| US8513228 | Mar 22, 2010 | Aug 20, 2013 | Richter Gedeon Nyrt. | Crystalline form of antiprogestin CDB-4124 |
Arno Therapeutics (ARNI) in a Phase I/II trial assessing its oral, anti-progestin hormone blocker ‘onapristone’ in men with advanced castration-resistant prostate cancer (CRPC) after failure of abiraterone or enzalutamide.

| CAS No. | 96346-61-1 |
| AU 8817602; EP 0299209; JP 1989045385; US 4925849; US 4994453; US 5087629; US 5102878; US 5179103; US 5296490 | |
| Synonyms: | ZK-299;ZK-98299;ONAPRISTONE;ONAPRINSTONE;(13α)-11β-[4-(Dimethylamino)phenyl]-17α-hydroxy-17β-(3-hydroxypropyl)estra-4,9-dien-3-one;(11R)-11-[4-(dimethylamino)phenyl]-17-hydroxy-17-(3-hydroxypropyl)-13-methyl-1,2,6,7,8,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-3-one |
Schering AG (Originator) |
|
| Molecular Formula: | C29H39NO3 |
| Formula Weight: | 449.62 |


Mifepristone Onapristone Asoprisnil(ZK-98299)
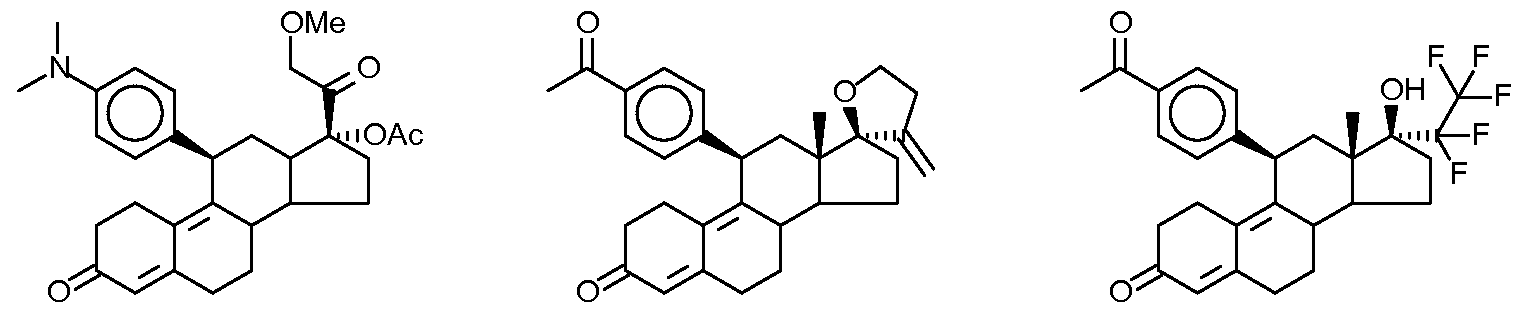
Proellex ORG-33628 Lonaprisan


US-based clinical stage biopharmaceutical firm Arno Therapeutics (ARNI) has started enrolling patients in a Phase I/II trial (NCT02049190) assessing its oral, anti-progestin hormone blocker ‘onapristone’ in men with advanced castration-resistant prostate cancer (CRPC) after failure of abiraterone or enzalutamide.
In previous Phase II clinical trials, onapristone has shown to exhibit anti-tumour activity in patients with breast cancer.
The pre-clinical testing has showed that onapristone had blocked the activation of the progesterone receptor (PR), which is believed to be a mechanism that inhibits the growth of APR-driven breast, endometrial and other tumours.
The company said that tests for the activated form of the progesterone receptor (APR) have the potential to function as a biomarker of anti-progestin activity, as detected by a companion diagnostic under development.
Enrolment of patients in the randomised, open-label Phase I/II trial follows approval of an Investigational Medicinal Product Dossier from the UK Health Authority, Medicines and Healthcare products Regulatory Agency (MHRA), ethics committee authorisation and subsequent site authorisation.
Arno Therapeutics president and chief executive officer Glenn Mattes said globally, prostate cancer is the second most common cancer in men, and the fifth leading cause of death from cancer in men, with an estimated 1.1 million new cases diagnosed and 307,000 deaths during 2012 alone, according to the International Agency for Research on Cancer.
“These numbers are staggering, and our ultimate goal is to evaluate onapristone in the subset of advanced CRPC patients who are more likely to respond to this personalised treatment, for which there is an immense unmet medical need,” Mattes said.
“The trial marks Arno’s second Phase I study actively enrolling this year and we are excited by the momentum generated thus far.”
The Phase I/II trial, designed to assess the safety and anti-cancer activity of onapristone in the select patient population, is being carried out at the Institute of Cancer Research, London, and the Royal Marsden NHS Foundation Trust in the UK.
A total of 60 patients will be enrolled in the trial, which additional sites are planned for in the UK.
The company has engaged Biotrial, a drug evaluation and pharmacology research company, as its contract research organisation (CRO) for the Phase I/II trial.
The trial will evaluate onapristone in extended-release tablet formulations in up to five dose levels (10mg-50mg BID) in patients with advanced CRPC where PR may be contributing to tumour progression.
Patients in the trial will be evaluated for whether their tumours express APR, which may help identify patients who are more likely to respond to onapristone.
A second group of patients will be included at the recommended Phase II dose to gain additional understanding of the onapristone safety profile and potential anti-cancer activity.
J Steroid Biochem1987,27,(4-6):851
Steroids1984,44,(4):349-72
| ATTARDI BARBARA J ET AL: “CDB-4124 and its putative monodemethylated metabolite, CDB-4453, are potent antiprogestins with reduced antiglucocorticoid activity: In vitro comparison to mifepristone and CDB-2914” MOLECULAR AND CELLULAR ENDOCRINOLOGY, ELSEVIER IRELAND LTD, IE, vol. 188, no. 1-2, 25 February 2002 (2002-02-25), pages 111-123, XP002496575 ISSN: 0303-7207 | ||
| 2 | * | MEALY N E ET AL: “CDB-4124” DRUGS OF THE FUTURE 200411 ES, vol. 29, no. 11, November 2004 (2004-11), page 1133, XP009118559 ISSN: 0377-8282 |
| WO2010106383A1 * | Mar 22, 2010 | Sep 23, 2010 | Richter Gedeon Nyrt | Novel crystalline form of antiprogestin cdb-4124 |
| WO2011015892A2 * | Aug 5, 2010 | Feb 10, 2011 | Richter Gedeon Nyrt. | Novel crystal form of an organic compound and process for the preparation thereof |
| US8513228 | Mar 22, 2010 | Aug 20, 2013 | Richter Gedeon Nyrt. | Crystalline form of antiprogestin CDB-4124 |
Glenmark Pharmaceuticals Ltd. through its Swiss Subsidiary receives USD 5 Mn. as milestone fee payment from Sanofi

Glenmark Pharmaceuticals Ltd. through its Swiss Subsidiary receives USD 5 Mn. as milestone fee payment from Sanofi
Total Payment received for GBR 500 monoclonal antibody programme from Sanofi is USD 55 Mn
MUMBAI, April 15, 2014: Glenmark Pharmaceuticals Ltd. has informed the Stock Exchange today that the company through its Swiss subsidiary has received USD 5 million as
milestone payment from Sanofi on a collaboration of its VLA2 (alpha2-beta1) integrin monoclonal antibody. GBR 500 is a first-in-class therapeutic monoclonal antibody for chronicautoimmune disorders.
Glenmark has received from Sanofi already USD 50 Mn as an upfront payment in FY2011-12. Hence, the total amount received by Glenmark from Sanofi for its first in class VLA-2monoclonal antibody is USD 55 million
read at
http://www.moneycontrol.com/stocks/stock_market/corp_notices.php?autono=790416
(copy paste on browser)
MD and CEO Mr Glenn Saldanha
old updates
Glenmark GBR 500 enters into Phase II clinical development for ulcerative colitis
17 September 2012
Glenmark Pharmaceuticals, a wholly-owned subsidiary of Glenmark Pharmaceuticals, has commenced the Phase II study of GBR 500 for ulcerative colitis.
GBR 500, an antagonist of the VLA2 (alpha2-beta1) integrin, is a first-in-class therapeutic monoclonal antibody for chronic autoimmune disorders.
The randomised, double-blind, placebo-controlled study will investigate the efficacy and safety of GBR 500 in patients with moderate to severe ulcerative colitis (UC).
Glenmark Pharmaceuticals chief scientific officer Dr Michael Buschle said that UC represents an area of substantial unmet medical need, despite treatment advances in recent years.
“We’re pleased with the continued progress of our partnership with Sanofi and excited about the commencement of this trial,” Buschle said.
The trial, which will be conducted at multiple clinical sites in North America and Europe, is expected to involve approximately 84 patients.
Patients participating in the study will receive multiple doses of either GBR 500 or placebo, administered over a period of several weeks.
Glenmark has completed Phase I of GBR 500 in the US, won licensing rights to all therapeutic indications from Sanofi and is conducting the clinical development programme.
The trial is part of a strategic global collaboration between Glenmark and Sanofi to investigate GBR 500 for the treatment of chronic inflammatory disorders.
MUMBAI, India, May 16, 2011
Glenmark Pharmaceuticals Out-Licenses Novel Monoclonal Antibody, GBR 500, to Sanofi
Combined Upfront and Potential Development, Regulatory and Commercial Milestone Payments Could Total US$613 Mn
MUMBAI, India, May 16, 2011 /PRNewswire-FirstCall/ — Glenmark Pharmaceuticals S.A (GPSA), a wholly owned subsidiary of Glenmark Pharmaceuticals Limited India (GPL), announced today that it has entered into an agreement with Sanofi to grant Sanofi a license for the development and commercialization of GBR 500, a novel monoclonal antibody for the treatment of Crohn’s Disease and other inflammatory conditions. The transaction is expected to close in the coming month subject to customary closing conditions, including the expiration or early termination of the waiting period under the HSR Antitrust Improvements Act.
Under the terms of the agreement, Glenmark will receive an upfront payment of US$ 50 million, of which US$ 25 million will be paid upon closing of the transaction and US$ 25 million, which is contingent upon Sanofi’s positive assessment of certain data to be provided by Glenmark. In addition, Glenmark could receive potential success-based development, regulatory and commercial milestone payments. The total of these payments could reach US$613 Mn. In addition, Glenmark is eligible to receive tiered double-digit royalties on sales of products commercialized under the license.
GBR 500 is an antagonist of the VLA-2 (alpha2-beta1) integrin. It is a first-in-class therapeutic monoclonal antibody and has established proof of concept in animal models across a range of anti-inflammatory conditions. Glenmark has completed Phase I dosing of GBR 500 in the US and the drug has been well tolerated with a good pharmacokinetic profile. Plans are in place to initiate clinical proof of concept studies in Crohn’s Disease. Sanofi has licensed the rights to all therapeutic indications.
“There continues to be a strong medical need for safer and more efficacious products for the treatment of Inflammatory Diseases,” said Elias Zerhouni, M.D., President, Global Research & Development, Sanofi. “GBR500 brings an innovative approach to Sanofi’s Immuno-Inflammation portfolio, which we believe may address a significant gap in treating Inflammatory Diseases which would be of huge benefit to patients”.
Glenn Saldanha MD and CEO of GPL, “This collaboration on a novel first-in-class monoclonal antibody validates Glenmark’s world-class innovative R&D capabilities in the drug discovery arena. We are pleased to have this second licensing collaboration with Sanofi, one of the largest pharmaceutical companies in the world and the first one from Glenmark in the field of novel biologics”.
Carcerand for Molecular encapsulation …Drug Delivery
.jpg)
Crystal structure of a nitrobenzene bound within a hemicarcerand reported by Cramand coworkers in Chem. Commun., 1997, 1303-1304.

A carcerand is a host molecule that completely entraps its guest so that it will not escape even at high temperatures.[1] This type of molecule was first described by Donald J. Cram in 1985 and is derived from the Latin carcer, or prison. The complexes formed by a carcerand with permanently imprisoned guests are called carceplexes.
In contrast hemicarcerands allow guests to enter and exit the cavity at high temperatures but will form stable complexes at ambient temperatures.[2] The complexes formed by a hemicarcerand and a guests are called hemicarceplexes.

Reactivity of bound guests
Cram described the interior of the container compound as the inner phase in which radically different reactivity was observed.[3] He used a hemicarcerand to isolate highly unstable, antiaromatic cylobutadiene at room temperature. The hemicarcerand stabilizes guests within its cavity by preventing their reaction with other molecules.
Synthesis

Synthesis of a carcerand from two calixarenes.
The first generation carcerands are based on calixarene hemicarcerands with 4 alkyl substituents on the upper rim and 4 reactive substituents on the lower rim. The coupling of both hemicarcerands takes place through a spacer group. In the original 1985 publication two different hemicarcerands react, one with chloromethyl reactive groups and one with thiomethyl reactive groups in a nucleophilic displacement and the resulting the spacer group is a dimethylsulfide (CH2SCH2). In this experiment the guests were the molecules already present in the reaction medium such as argon and dimethylformamide.
In another configuration the 4 lower rim functional groups are aldehydes which condense with O-Phenylenediamine to the corresponding di-imines. The 4 spacer groups connecting the two spheres are now much longer and consequently the internal cavity is much larger. Compounds trapped in the cavity are said to be held there by constrictive binding.[4] They can be introduced by simply heating in neat solvent like hexachlorobutadiene (a fungicide). The half-life of the reverse process is 3.2 hours at 25 °C in CDCl3 by NMR analysis. Ferrocene can be introduced by heating with the hemicarcerand in a large bulky solvent such as tripiperidylphosphine oxide. The half-life for ferrocene liberation is 19.6 hours at 112 °C.
Large Carcerands

Octahedral Nanocontainer
The internal cavity of a carcerand can be as large as 1700 Å3 (1.7 nm3) when six hemicarcerands form a single octahedral compound.[5] This is accomplished by dynamic covalent chemistry in a one-pot condensation of 6 equivalents of a tetraformyl calixarene and 12 equivalents of ethylene diamine withtrifluoroacetic acid catalyst in chloroform at room temperature followed by reduction of the imine bonds with sodium borohydride.









References
- Shell closure of two cavitands forms carcerand complexes with components of the medium as permanent guests Donald J. Cram, Stefan Karbach, Young Hwan Kim, Lubomir Baczynskyj, Gregory W. KallemeynJ. Am. Chem. Soc.; 1985; 107(8); 2575-2576. Abstract
- Recent Highlights in Hemicarcerand Chemistry Ralf Warmuth and Juyoung Yoon, Accounts of Chemical Research Volume 4, Issue 2, Pages 95-105, 2001.
- The Inner Phase of Molecular Container Compounds as a Novel Reaction Environment Ralf Warmuth Journal of Inclusion Phenomena and Macrocyclic Chemistry 37: 1–38, 2000.
- Constrictive binding of large guests by a hemicarcerand containing four portals Mimi L. C. Quan, Donald J. Cram J. Am. Chem. Soc.; 1991; 113(7); 2754-2755. Abstract
- One-Pot, 18-Component Synthesis of an Octahedral Nanocontainer Molecule Xuejun Liu, Yong Liu, Gina Li, Ralf Warmuth, Angewandte Chemie International Edition Volume 45, Issue 6 , Pages 901 – 904 2006 Abstract
extra info
This review focuses on how self-assembly can form hosts capable of binding large guests. Its sister article, ”Guests within Large Synthetic Hydrophobic Pockets Synthesized Using Polymer and Conventional Techniques,” reviews like-minded work using either polymers or hosts synthesized by traditional synthetic approaches. As described in more detail in that paper, the focus here is on hosts capable of binding organic molecules of more than seven nonhydrogen atoms. Likewise, a similar definition of a ”pocket” is retained, with the focus on hosts possessing well-defined, highly concave or enclosed surfaces. An arbitrary value of greater than approximately 50% encapsulation has been set. ”Comprehensive Supramo-lecular Chemistry” covers much of our discussion topic up to 1995.[1] This review is therefore primarily interested with the literature since that time.
Motivators for supramolecular chemists include molecular storage/delivery, the detection of substances, and the conversion of one substance into another via catalytic processes. All these processes include at some point the binding of a guest to a host. The hard part in these endeavors is the synthesis of the host, with all the required functionality gathered in a converging array. One approach uses self-assembly, whereby molecular subunits are designed to merge in a specific pattern that possesses a hydrophobic pocket. In this regard, both self-assembly and self-assembly with covalent modification have been used. As with the polymer and traditional synthetic strategies, the self-assembly approach has pros and cons. Normally, relatively rigid subunits are used and so a common worry in cavity design—hydrophobic pocket collapse—is generally avoided. On the other hand, at our current level of understanding we are limited to relatively symmetrical subunits and assembled structures. Nevertheless, testimony to the power of this approach is found in the large cavities formed, some in the order of thousands of cubic Angstroms.
The bulk of recent self-assembly research has focused on understanding the rules that govern how one product can arise out of a reaction mixture that, if all things were equal, would lead to a highly complex mixture. Hence in a manner analogous to contemporary polymer research, the emphasis is on understanding how the structure of the product is reached, rather than on understanding the properties of any cavity in the product. As a result, many of the very large cavities created by self-assembly are ”simply” filled with a large number of (small) solvent molecules. Such examples are not dealt with explicitly here but can be found in citations throughout the text.
HOSTS BASED ON RESORCINARENES
In the last seven or so years it has become apparent that resorcinarenes, 1 (n=4), a family of molecules[2-5] held in much regard for their hosting properties and their use in the synthesis of a plethora of cavitands, also possess a spectacular flair for self-assembly. Two general assembly products have been identified (Fig. 1). Either two molecules can come together in apseudo C4h symmetric host, or six assemble into a pseudo-octahedral array. A second component, usually solvent, is necessary to ”glue” the subunits together. The first hint of this supramolecular chemistry was pinpointed by MacGillivray and Atwood, who identified the pseudo-octahedral complex both in the solid and the solution state.[6] The total assembly consists of six resorcinarenes, eight water molecules and has a solvent-filled cavity. Shortly thereafter, the dimer, again in part held together by 2-propoanol/water molecules, was identified independently by the Rose and Aoki groups.[7,8] The latter identified a tetraethylammonium ion within the cavity. A third such structure, this time hosting triethyl-ammonium, was identified shortly thereafter.1-9-1 Recently, Atwood et al. identified a more robust pseudo-spherical hexamer derived from pyrogallol[4]arenes 2,[10] while Shivanyuk and Rebek have determined that the corresponding dimeric assembly also forms and encapsulates large guests.[11] Thus, in deuterated methanol or aqueous acetonitrile, NMR evidence suggests that 2 (R=Pr) is monomeric. However, the addition of tropylium tetra-fluoroborate results in an intense red color indicating a charge transfer complex between 2 (R=Pr) and guest. Furthermore, at host/guest ratios larger than two, the 1H NMR at 233 K demonstrates that a 2:1 complex of 2 (R=Pr) and tropylium tetrafluoroborate exists. At this temperature, the kinetics of guest exchange is slow on the (600 MHz) NMR time-scale. Further experiments revealed that 1) at higher equivalents of guest a 1:1 complex was formed, and 2) a protic solvent was necessary for assembly of the dimer host. This latter point was noted when more lipophilic 2 (R=CnH23) was shown to only assemble in deuterated chloroform when a trace of methanol was present. Interestingly, resorcin-arenes 1 (R=Me, or Et) did not undergo this encapsulation of the tropylium ion.

Fig. 1 Dimeric and hexameric assemblies of resorcinarenes.
The octahedral assemblies of 1 (n=4)[12,13] have demonstrated some fascinating encapsulation properties. For example, in water saturated, deuterated chloroform, the hexamer of 1 encapsulates a range of ammonium ions in a manner that is intimately tied to the size and concentration of the guest.[14] Tetrahexylammonium bromide is an ideal guest and exchanges slowly, relative to the 600-MHz NMR time-scale, between cavity and free solution. Nuclear magnetic resonance evidence suggests that the larger guest tetraheptylammonium bromide is cramped within the confines of the assembly, while the still larger tetra-hexadecylammonium bromide was not complexed. With smaller guests, things became even more interesting. Thus, in the case of Bu4N+BF4~, the small counter ion is coencapsulated along with cation. In such cases, solvent is also present in the cavity but leaves when a more suitable space-filler is present. Hence, at the expense of bound water 4-phenyltoluene is also encapsulated along with Bu4N+BF4~. Still smaller ammonium ions instead template the assembly of the dimeric host. Bu4SbBr, along with a variety of co-encapsulated aromatic guests, is also seen within the hexameric assembly.[15] Co-guests included are benzene, p-xylene, 4-phenyltoluene, and naphthalene. In contrast, 4,4′-dimethylbiphenyl was not encapsulated with the antimony guest, suggesting that adding an extra methyl group to 4-phenyltoluene was enough to destabilize the complex. Size, however, is not the only important factor for encapsulation. Neither hexa-fluorobenzene, cyclohexane, nor pentane was coencapsu-lated. Rather, the antimony guest and presumably some solvent molecules occupied the cavity.

As the basis of cavitands, resorcinarenes have also been instrumental in the synthesis of carceplexes such as 3 (Scheme 1).[16-18] As defined by their inventor, Donald Cram, carceplexes are closed surface compounds that permanently entrap guest molecules or ions within their shell, such that guest escape can only occur by rupture of covalent bonds. Since their initial synthesis,[19] their self-assembly (with covalent modification) has been intensively investigated; as has the relationship between host shell (the carcerand) and guest. As it transpires, these two facets are intimately tied. Early work focusing on small guests established that templation[20] is essential for successful synthesis. The best yields arise when a template stabilizes the transition state of the rate-determining step (rds) for the synthesis. Furthermore, the transition state at the rds has a similar cavity to the product.[21-29] Hence good templates for assembly make good guests for the carcerands. In these initial studies, the best guest identified was pyrazine, while the worst guest, and hence the solvent of choice for many of these studies, was N-methylpyrrolidinone (NMP).

Scheme 1 Synthesis of carceplex 3.

Carceplexes can be increased in size to allow the encapsulation of large guests. One way is to use wide-bodied cavitands—cavitands derived from resorcin [5]arenes—in the carceplex reaction.[30] Another is to join several ”normal”-sized cavitands together.[31-33] To date, only relatively small molecules have been observed within hosts synthesized by these methods. A more common approach is to increase the size of the linker groups that join the two ”hemispheres” of the shell, i.e., replace bromochloromethane in the synthesis shown in Scheme 1 with a compound with two separate electro-philic centers.[34] The resulting products, such as 4 (R=-(CH2)4-),[35] are called hemicarceplexes; so named because the portals in the shell, or hemicarcerand, are large enough for guests to exit or enter the cavity without covalent bond rupturing.[36] The ability of over 68 molecules to template the formation of 4 (R=-(CH2)4-) has been recently studied.[37] Of these, 30 proved capable of templation, with the best template p-xylene proving to be 3600 times better than the worst N-formylpiperidine. However, in contrast to carceplex 3, the final host structure 4 did not appear to be a reasonable model of the transition state of the rds in its synthesis.
Although many hemicarceplexes can be assembled via templation, most have been synthesized by inserting the desired guest post-assembly. This has provided information about how solvent and the shape of the portals or guest influence thermodynamic stability, and complexa-tion and decomplexation rates. For example, after synthesizing hemicarceplex 4 (R=-(CH2)4-, guest= solvent), the guest solvent can be exchanged using the law of mass action. Either heating the hemicarceplex in the presence of 100 equivalents of new guest in a solvent too large to enter the cavity, or more simply heating the complex in neat guest, leads to exchange. Using this technique, it is possible to synthesize a range of hemicarceplexes.[38] In general, long, thin guests complexed the fastest, while for disubstituted aromatic guests a general order of com-plexation was demonstrated by the xylene isomers; p-xylene ^ m-xylene> o-xylene. Computational studies on a related hemicarceplex indicated that two type of gating processes, involving conformational changes in the intra-hemispherical linker groups, affect guest entry or egres-sion.[39,40]
How do guests inside the cavity interact with species in free solution? Host 4 (R=-(CH2)4-) was used to study the first Sn2 reactions of inner-phase guests with outer-phase reactants.[41] For example, when the complexes 4 (R= -(CH2)4-, guest=either 4-HOC6H4OH or 3-HOC6H4OH or 2-HOC6H4OH) were exposed to THF/NaH/CH3I, three different reaction outcomes were noted. The encapsulated para-isomer gave no reaction, the meta-isomer was doubly methylated, while the ortho-isomer gave a mixture of mono-and dimethylated guest. Different reactivity profiles were noted because each guest prefers a different orientation within the cavity. In the case of the 1,4-isomer for example, the two OH groups can reside deep within the ”poles” of the host and cannot be readily alkylated. A similar rationale explains the observed alkyllithium additions and borane reductions of 4 (R=-(CH2)4-, guest =benzaldehyde, benzocyclobutenone, and benzocyclobutenedione).[42] If a reactive species is generated within the cavity, it can sometimes react with the hemicarcerand shell. For example, reaction at 0°C between 4 (R=-(CH2)4-, guest =-benzocyclobutenedione) and an excess of MeLi gave diol 5 (Scheme 2). Experiments revealed that a possible mechanism for this conversion begins with the addition of one equivalent of MeLi to a C=0 group of the guest. The basic lithiate complex 6 can then induce a p-elimination in one of the linkers. This produces a butene ether derivative 7 that can then undergo a further elimination to yield the bis-phenoxide. Workup yields 5. The tetramethylene linkers are not the sole reactive sites in this hemicarcerand. Thus the methylene bridges in each hemisphere undergo reaction with the guest derived from methyl lithium addition to encapsulated N-methyl-2-pyrrolidinone.[42]

Scheme 2 Synthesis of diol 5 via an inner-molecular reaction.
By carrying out a two-step (hemi)carceplex reaction, lower symmetry hydrophobic pockets can be synthe-sized.[43,44] To point out just two examples, hosts such as 4 (R=-CH2-, or R=-(CH2)6-) were synthesized and their corresponding hemicarceplexes examined by X-ray crystallography, NMR, and computational studies.[45] As anticipated, introducing a dissimilar linker between the hemispheres altered the shape of the pocket, the orientation of the guest, and hence the thermodynamic and kinetic stability of the corresponding complexes. The kinetics of exchange were measured at different temperatures for 1,4-dimethoxybenzene vacating 4 (R= 1,3-(CH2)2C6H4), to show how AHz, ASz, and hence AGz varied as a function of solvent.[43] These studies revealed that solvation plays an important role in decomplexation. Hence decomplexa-tion rates in CDCl3 were 800 times faster than in deuterated 1,1,2,2-tetrachloroethane. An examination of different hemicarcerands showed that the structure of the unique linker also has a considerable effect on egression rates. For example, changing the dissimilar linker R in 4 from pentamethylene to hexamethylene increased the rate constant for egression by a factor of 177. Lower symmetry cavities can also be made by using two different cavitands to construct a hemicarceplex. For example, in hosts 8 and 9 methylene and dimethylene bridges link the phenol oxygens of the former, while dimethylene and trimethylene linkers are used in the latter.[46,47] Nuclear magnetic resonance evidence demonstrates that these subtle changes in the host are manifest in how the guest moves within the pocket. Hence when the guest in 8 is 1,2,3-trimethoxybenzene three signals are observed for the methoxy groups. The 1- and 3-methoxy groups reside within different hemispheres, and the movement of the guest that allows them to exchange is slow on the (500 MHz) NMR time-scale; in contrast, the slightly bigger cavity of 9 means that the corresponding process is fast on the NMR time-scale. Although no simple rules were discernable, the size of the bridges between the phenolic O-atoms undoubtedly influences guest movement and decomplexation rates.

![tmp1CF-162_thumb[1]](http://what-when-how.com/wp-content/uploads/2011/03/tmp1CF162_thumb1.jpg "tmp1CF-162_thumb[1]")
Building on these developments, Cram moved the carceplex and hemicarceplex field into the realms of aqueous solution, while at the same time synthesizing chiral hosts. The first water soluble hemicarceplexes were isolated from hemicarcerand 10.[48] In aqueous solution, this host is capable of sequestering a number of guests including naphthalene and 1,3-dimethoxybenzene. Guests such as alkyl ammonium salts that are well solvated by water did not bind. Chiral hemicarceplexes can be synthesized by using the two-step process discussed above. Hosts 11 and 12 are two examples.[49] Introducing the chiral linker group of 11 in the presence of a racemic mixture of selected chiral guests resulted in ratios of the diastereomeric complexes of up to 1:1.5. Alternatively, higher diastereomeric ratios could be attained if the chiral hemicarceplex 11 containing chloroform was heated either in the presence of pure, racemic guest, or in diphenylether with an excess of racemic guest. By this approach the highest diastereomeric ratio observed was >20:1 in favor of the R-isomer of 4-MeC6H4S(O)Me binding to 11. In contrast, the diastereomeric ratio observed for complexing racemate C6H5S(O)Me was only 1.6:1 in favor of the R-isomer. Overall, the hemicarcerand 12 (guest=chloroform) was less discriminating than 11. This was attributed to the two ”nonchiral,” 26-membered ring portals in 12 being less encumbering than its two chiral portals.

As a rule, the shape and functionality of the guest can be transferred through a hemicarceplex shell to the external environment. A simple thin layer chromatography experiment is usually sufficient to demonstrate this point. Furthermore, hemicarcerand shells allow the transfer of triplet energy from aryl ketone guests to free naphthalene.1-50-1 These results notwithstanding, guests in carce-plexes or hemicarceplexes are in relatively sheltered waters, and this has allowed these unique hosts to be used as storage containers for highly reactive guests.[34,51,52] The first example, involving a small guest, was carried out over 12 years ago. Nevertheless, the trapping and room temperature analysis of cyclobutadiene 13 (Fig. 2), the Mona Lisa of organic chemistry as Cram described it, is always worth mentioning.[34] Equally as exciting was the trapping by Warmuth of o-benzyne 14 inside the cavity of 4 (R=-(CH2)4-).[51] This remarkable feat was accomplished by the photolysis of 4 (R=-(CH2)4-, guest=benzocyclo-butenedione) and allowed its :H and 13C NMR analysis. The former suggested that ”free” benzyne would possess :H NMR chemical shifts of d=7.0 and 7.6 ppm, while 13C-13C coupling in the latter suggests that 14 is best described as a cumulene. Interestingly, when warmed up to room temperature, the guest reacted with the hemi-carcerand in an inner-molecular Diels-Alder reaction.[53] Following on from this work, Warmuth and Marvel successfully trapped an enantiomeric mixture of 1,2,4,6-cycloheptatriene 15 inside hemicarceplex 4 (R=-(CH2)4), by first encapsulating phenyldiazirine and then irradiating the resulting hemicarceplex.[54] Protected by the host shell, 15 could not dimerize and was stable for weeks at ambient temperature. By carrying out the same chemistry within chiral host 12, a 3:2 ratio of the two resulting diastereomeric complexes was formed.[55] No coalescence of NMR signals could be observed when heating these complexes up to 100°C, which puts a lower limit to the enantiomerization barrier of > 19.6 kcal mol—1 This value was collaborated with a parallel experiment using the diastereomeric complex of 12 (guest= 16). Thus using line broadening analysis of the :H NMR signals from the methyl groups of the two complexes (d = — 1.47 and — 1.57 ppm), a similar isomerization barrier was determined.[56] Although tetraenes 15 and 16 are stabilized by the protective shell, the hemicarcerand cannot stop oxygen and other small reagents from entering the cavity. Hence when a solution of 4 (guest = 15) is exposed to oxygen, the spirodioxirane 17 forms, which upon heating evolves CO2 to leave an entrapped benzene guest. Furthermore, when heated entrapped 16 rearranges to the corresponding p-tolylcarbene, which goes on and reacts with the hemi-carcerand shell in a number of different ways.


Fig. 2 Reactive species trapped within hemicarceplexes.
Animation of nanoparticles for drug delivery in cancer treatment

Animation of nanoparticles for drug delivery in cancer treatment
read at
Immunotherapy could help tackle tough liver cancer
Significant new data presented today at the International Liver Congress 2014 indicate that liver cancer (Hepatocellular Carcinoma (HCC)) may be treated by adoptive T-cell therapy.
This new therapeutic approach in the treatment of HCC could be very important as without treatment the 5 year survival rate is just 5%. Globally, HCC accounts for 746,000 deaths, and in the UK alone is responsible for over 4,000 deaths per year.
Glypican-3 (GPC3) is a tumour associated antigen expressed in up to 70% of HCC but not in healthy human tissue. Isolating GPC3-specific T-cell receptors and expressing them on patient’s T-cells can help treat HCC, as these T cells can recognise and eliminate GPC3-postive HCC.
The study detected and expanded MHC-multimer-positive CD8+ T-cells specific for targeted GPC3 epitopes and grew T-cell clones. From these clones, the most specific and active T-cell receptor was isolated. When this T-cell receptor was expressed on donor T…
View original post 131 more words
BI 224436 an investigational new drug under development for the treatment of HIV infection


(2S)-2-tert-butoxy-2-(4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-2- methylquinolin-3-yl)acetic acid
BI 224436
1155419-89-8 cas no
mw
| 442.51 |
3-Quinolineacetic acid, 4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-α-(1,1-dimethylethoxy)-2-methyl-, (αS,4R)-
hemi-succinate of (2S)-2-tert-butoxy-2-(4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-2-methylquinolin-3-yl)acetic acid)
BI 224436 is an investigational new drug under development for the treatment of HIV infection. BI 224436 is the first non-catalytic site integrase inhibitor (NCINI). It inhibits HIV replication via binding to a conserved allosteric pocket of the HIV integrase enzyme. This makes the drug distinct in mechanism of action compared to raltegravir and elvitegravir, which bind at the catalytic site.[2] In October 2011, Gilead Sciences purchased exclusive rights to develop BI 224436 and several related compounds under investigation in Boehringer Ingelheim’s noncatalytic site integrase inhibitor program.[3][4]
Novel hemi-succinate salt form of (2S)-2-tert-butoxy-2-(4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-2-methylquinolin-3-yl)acetic acid (presumed to be BI-224436) and its crystalline forms is desc in WO-2014055618.
Gilead, under license from BI, was developing BI-224436 for the oral treatment of HIV infection. In September 2011, this drug had entered phase 1 trials. Picks up from WO2012138670, claiming a process for the preparation of the same drug. Also see the concurrently published WO2014055603. This compound is claimed specifically in WO2009062285 and generically in WO2007131350.
BI 224436 has antiviral EC50 values ranging between 4 and 15 nM against different HIV-1 laboratory strains and CC50 values >90 μM in different cells, including peripheral blood mononuclear cells. BI 224436 also has a low, 2.2-fold shift in antiviral potency in the presence of 50% human serum and by virtue of a steep dose-response curve slope, BI 224436 exhibits serum-shifted EC95 values ranging between 22 and 75 nM. Drug combination studies performed in cell-based antiviral assays have shown that BI 224436 displays, at the least, an additive effect in combination with any of the marketed antiviral classes including INSTIs. BI 224436 has drug-like ADME properties including a Caco-2 cell permeability of 14 .10-6 cm/sec, solubility > 24 mg/ml in the pH range 2.0-6.8 and low cytochrome P450 inhibition. Moreover BI 224436 shows excellent PK profiles in rat (CL=0.7% QH; F= 54%), monkey (CL= 23% QH; F= 82%) and dog (CL= 8%QH; F= 81%).

http://www.natap.org/2011/ICAAC/ICAAC_32.htm
……………………
Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1
ACS Med. Chem. Lett., 2014, 5 (4), pp 422–427
DOI: 10.1021/ml500002n
http://pubs.acs.org/doi/abs/10.1021/ml500002n

1H NMR: 12.4 (br, 1H), 8.52 (d, 1H, J = 4.4Hz), 7.94 (d, 1H, J = 7.9 Hz),7.65-7.61 (m, 1H), 7.45 (d,
1H, J = 8.2 Hz), 7.31-7.24 (m, 2H), 7.12 (d, 1H, J = 7.9 Hz), 6.94-6.92 (m, 1H), 4.99 (s, 1H), 4.57-4.47
(m, 2H), 3.37-3.30 (m, 2H), 2.86 (s, 3H), 0.82 (s, 9H).
13C NMR: 172.2, 158.4, 153.1, 150.1, 146.6,
146.1, 145.0, 141.0, 130.8 (br), 130.6 (br), 128.9, 128.0, 127.2, 127.1 (br) 126.4, 125.6, 118.0, 116.7,
109.1, 75.2, 70.8, 65.6, 27.7, 27.5, 24.9.
HRMS: m/z calc. for C27H26N2O4 + H+: 443.1965, m/z found:
443.1951 (-3.2 ppm).
UPLC-MS: rt = 0.68 min, m/z 443.3 [M + H]+, purity: >99.9% @ 254 nm.
http://pubs.acs.org/doi/suppl/10.1021/ml500002n/suppl_file/ml500002n_si_001.pdf
………………………….
http://www.google.com/patents/WO2012138670A1?cl=en
General Scheme IA:

G1 1001
wherein Y is I, Br or CI;
General Scheme 11 A:

wherein:
Example 1

1 a 1 b
1a (600 g, 4.1 mol) was charged into a dry reactor under nitrogen followed by addition of Ac20 (1257.5 g, 12.3 mol, 3 eq.). The resulting mixture was heated at 40 °C at least for 2 hours. The batch was then cooled to 30 °C over 30 minutes. A suspension of 1b in toluene was added to seed the batch if no solid was observed. After toluene (600 ml_) was added over 30 minutes, the batch was cooled to -5— 10 °C and was held at this temperature for at least 30 minutes. The solid was collected by filtration under nitrogen and rinsed with heptanes (1200 ml_). After being dried under vacuum at room temperature, the solid was stored under nitrogen at least below 20 °C. The product 1 b was obtained with 77% yield. 1H NMR (500 MHz, CDCI3): δ = 6.36 (s, 1 H), 3.68 (s, 2H), 2.30 (s, 3H). Example 2

2a 2b
2a (100g, 531 mmol) and 1b (95 g, 558 mmol) were charged into a clean and dry reactor under nitrogen followed by addition of fluorobenzene (1000 mL). After being heated at 35-37 °C for 4 hours, the batch was cooled to 23 °C. Concentrated H2S04 (260.82 g, 2659.3 mmol, 5 eq.) was added while maintaining the batch temperature below 35 °C. The batch was first heated at 30-35 °C for 30 minutes and then at 40- 45 °C for 2 hours. 4-Methyl morpholine (215.19 g, 2127 mmol, 4 eq.) was added to the batch while maintaining the temperature below 50 °C. Then the batch was agitated for 30 minutes at 40-50 °C. eOH (100 mL) was then added while maintaining the temperature below 55 °C. After the batch was held at 50-55 °C for 2 hours, another portion of MeOH (100 mL) was added. The batch was agitated for another 2 hours at 50-55 °C. After fluorobenzene was distilled to a minimum amount, water (1000 mL) was added. Further distillation was performed to remove any remaining fluorobenzene. After the batch was cooled to 30 °C, the solid was collected by filtration with cloth and rinsed with water (400 mL) and heptane (200 mL). The solid was dried under vacuum below 50 °C to reach KF < 0.1%. Typically, the product 2b was obtained in 90% yield with 98 wt%. 1H NMR (500 MHz, DMSO- d6): δ = 10.83 (s, 1 H), 9.85 (s, bs, 1 H), 7.6 (d, 1 H, J
Hz), 6.40 (s, 1 H), 4.00 (s, 2 H), 3.61 (s, 3 H). Example 3

2b 3a
2b (20 g, 64 mmol) was charged into a clean and dry reactor followed by addition of THF (140 mL). After the resulting mixture was cooled to 0 °C, Vitride® (Red-AI, 47.84 g, 65 wt%, 154 mmol) in toluene was added while maintaining an internal temperature at 0-5 °C. After the batch was agitated at 5-10 °C for 4 hours, IPA (9.24 g, 153.8 mmol) was added while maintaining the temperature below 10 °C. Then the batch was agitated at least for 30 minutes below 25 °C. A solution of HCI in IPA (84.73 g, 5.5 M, 512 mmol) was added into the reactor while maintaining the temperature below 40 °C. After about 160 mL of the solvent was distilled under vacuum below 40 °C, the batch was cooled to 20-25 °C and then aqueous 6M HCI (60 mL) was added while maintaining the temperature below 40 °C. The batch was cooled to 25 °C and agitated for at least 30 minutes. The solid was collected by filtration, washed with 40 mL of IPA and water (1V/1V), 40 mL of water and 40 mL of heptanes. The solid was dried below 60 °C in a vacuum oven to reach KF < 0.5%. Typically, the product 3a was obtained in 90-95% yield with 95 wt%. 1H NMR (400 MHz, DMSO-d6): δ = 10.7 (s, 1 H), 9.68 (s, 1 H), 7.59 (d, 1 H, J = 8.7 Hz), 6.64 (, 1 H, J = 8.7 Hz), 6.27 (s, 1 H), 4.62 (bs, 1 H), 3.69 (t, 2H, J = 6.3 Hz), 3.21 (t, 2H, J = 6.3 Hz).
Example 4

3a (50 g, 174.756 mmol) and acetonitrile (200 mL) were charged into a dry and clean reactor. After the resulting mixture was heated to 65 °C, POCI3 (107.18 g, 699 mmol, 4 eq.) was added while maintaining the internal temperature below 75 °C. The batch was then heated at 70-75 °C for 5-6 hours. The batch was cooled to 20 °C. Water (400 mL) was added at least over 30 minutes while maintaining the internal temperature below 50 °C. After the batch was cooled to 20-25 °C over 30 minutes, the solid was collected by filtration and washed with water (100 mL). The wet cake was charged back into the reactor followed by addition of 1 M NaOH (150 mL). After the batch was agitated at least for 30 minutes at 25-35 °C, it was verified that the pH was greater than 12. Otherwise, more 6M NaOH was needed to adjust the pH >12. After the batch was agitated for 30 minutes at 25-35 °C, the solid was collected by filtration, washed with water (200 mL) and heptanes (200 mL). The solid was dried in a vacuum oven below 50 °C to reach KF < 2%. Typically, the product 4a was obtained at about 75-80% yield. H NMR (400 MHz, CDCI3): δ = 7.90 (d, 1 H, J = 8.4 Hz), 7.16 (s, 1 H), 6.89 (d, 1 H, J = 8.4 Hz), 4.44 (t, 2 H, J = 5.9 Hz), 3.23 (t, 2 H, J = 5.9 Hz). 13C NMR (100 MHz, CDCI3): δ = 152.9, 151.9, 144.9, 144.1 , 134.6, 1 19.1 , 1 17.0, 1 13.3, 1 1 1.9, 65.6, 28.3.
Example 5

4a 5a
Zn powder (54 g, 825 mmol, 2.5 eq.) and TFA (100 mL) were charged into a dry and clean reactor. The resulting mixture was heated to 60-65 °C. A suspension of 4a (100 g, 330 mmol) in 150 mL of TFA was added to the reactor while maintaining the temperature below 70 °C. The charge line was rinsed with TFA (50 mL) into the reactor. After 1 hour at 65±5 °C, the batch was cooled to 25-30 °C. Zn powder was filtered off by passing the batch through a Celite pad and washing with methanol (200 mL). About 400 mL of solvent was distilled off under vacuum. After the batch was cooled to 20-25 °C, 20% NaOAc (ca. 300 mL) was added at least over 30 minutes to reach pH 5-6. The solid was collected by filtration, washed with water (200 mL) and heptane (200 mL), and dried under vacuum below 45 °C to reach KF ≤ 2%. The solid was charged into a dry reactor followed by addition of loose carbon (10 wt%) and toluene (1000 mL). The batch was heated at least for 30 minutes at 45-50 °C. The carbon was filtered off above 35 °C and rinsed with toluene (200 mL). The filtrate was charged into a clean and dry reactor. After about 1000 mL of toluene was distilled off under vacuum below 50 °C, 1000 mL of heptane was added over 30 minutes at 40-50 °C. Then the batch was cooled to 0±5 °C over 30 minutes. After 30 minutes, the solid was collected and rinsed with 200 mL of heptane. The solid was dried under vacuum below 45 °C to reach KF≤ 500 ppm. Typically, the product 5a was obtained in about 90-95 % yield. 1H NMR (400 MHz, CDCI3): δ = 8.93 (m, 1 H), 7.91 (dd, 1 H, J = 1.5, 8 Hz), 7.17 (m 1 H), 6.90 (dd, 1 H, J = 1 .6, 8.0 Hz), 4.46-4.43 (m, 2 H), 3.28-3.23 (m, 2 H). 13C NMR (100 MHz, CDCI3): δ = 152.8, 151 .2, 145.1 , 141.0, 133.3, 1 18.5, 1 18.2, 1 14.5, 1 1 1.1 , 65.8, 28.4.
Example 6

5a 6a
5a (1.04 kg, 4.16 mol) and toluene (8 L) were charged into the reactor. The batch was agitated and cooled to -50 to -55 °C. BuLi solution (2.5 M in hexanes, 1.69 L, 4.23 mol) was charged slowly while maintaining the internal temperature between – 45 to -50 °C. The batch was agitated at -45 °C for 1 hour after addition. A solution of triisopropyl borate (0.85 kg, 4.5 mol) in MTBE (1 .48 kg) was charged. The batch was warmed to 10 °C over 30 minutes. A solution of 5 N HCI in I PA (1 .54 L) was charged slowly at 10 °C, and the batch was warmed to 20 °C and stirred for 30 minutes. It was seeded with 6a crystal (10 g). A solution of aqueous concentrated HCI (0.16 L) in IPA (0.16 L) was charged slowly at 20 °C in three portions at 20 minute intervals, and the batch was agitated for 1 hour at 20 °C. The solid was collected by filtration, rinsed with MTBE (1 kg), and dried to provide 6a (943 g, 88.7 % purity, 80% yield). 1H NMR (400 MHz, D20): δ 8.84 (d, 1 H, J = 4 Hz)
1 H), 7.68 (d, 1 H, J = 6 Hz), 7.09 (m, 1 H), 4.52 (m, 2H), 3.47 (m, 2H).
Example 7

Iodine stock solution was prepared by mixing iodine (57.4 g, 0.23 mol) and sodium iodide (73.4 g, 0.49 mol) in water (270 mL). Sodium hydroxide (28.6 g, 0.715 mol) was charged into 220 mL of water. 4-Hydroxy-2 methylquinoline 7a (30 g, 0.19 mol) was charged, followed by acetonitrile (250 mL). The mixture was cooled to 10 °C with agitation. The above iodine stock solution was charged slowly over 30 minutes. The reaction was quenched by addition of sodium bisulfite (6.0 g) in water (60 mL). Acetic acid (23 mL) was charged over a period of 1 hour to adjust the pH of the reaction mixture between 6 and 7. The product was collected by filtration, washed with water and acetonitrile, and dried to give 7b (53 g, 98%). MS 286 [M + 1].
Example 8

7b 8a
4-Hydroxy-3-iodo-2-methylquinoline 7b (25 g, 0.09 mol) was charged to a 1-L reactor. Ethyl acetate (250 mL) was charged, followed by triethylamine (2.45 mL, 0.02 mol) and phosphorus oxychloride (12 mL, 0.13 mol). The reaction mixture was heated to reflux until complete conversion (~1 hour), then the mixture was cooled to 22 °C. A solution of sodium carbonate (3 .6 g, 0.3 mol) in water (500 mL) was charged. The mixture was stirred for 20 minutes. The aqueous layer was extracted with ethyl acetate (120 mL). The organic layers were combined and concentrated under vacuum to dryness. Acetone (50 mL) was charged. The solution was heated to 60 °C. Water (100 mL) was charged, and the mixture was cooled to 22 °C. The product was collected by filtration and dried to give 8a (25 g, 97.3 % pure, 91.4 % yield). MS 304 [M + 1].
(Note: 8a is a known compound with CAS # 1033931-93-9. See references: (a) J. Org Chem. 2008, 73, 4644-4649. (b) Molecules 2010, 15, 3171 -3178. (c) Indian J. Chem. Sec B: Org. Chem. Including Med Chem. 2009, 488(5), 692-696.)
Example 9

8a 9a
8a (100 g, 0.33 mol) was charged to the reactor, followed by copper (I) bromide dimethyl sulfide complex (3.4 g, 0.017 mol) and dry THF (450 mL). The batch was cooled to -15 to -12 °C. i-PrMgCI (2.0 M in THF, 173 mL, 0.346 mol) was charged into the reactor at the rate which maintained the batch temperature < -10 °C. In a 2nd reactor, methyl chlorooxoacetate (33 mL, 0.36 mol) and dry THF (150 mL) were charged. The solution was cooled to -15 to -10 °C. The content of the 1 st reactor (Grignard/cuprate) was charged into the 2nd reactor at the rate which maintained the batch temperature < -10 °C. The batch was agitated for 30 minutes at -10 °C. Aqueous ammonium chloride solution ( 0%, 300 mL) was charged. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Aqueous ammonium chloride solution (10%, 90 mL) and sodium carbonate solution (10%, 135 mL) were charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Brine (10%, 240 mL) was charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes. The aqueous layer was separated. The batch was concentrated under vacuum to -1/4 of the volume (about 80 mL left). 2-Propanol was charged (300 mL). The batch was concentrated under vacuum to -1/3 of the volume (about 140 mL left), and heated to 50 °C.
Water (70 mL) was charged. The batch was cooled to 20 – 25 °C, stirred for 2 hours, cooled to – 0 °C and stirred for another 2 hours. The solid was collected by filtration, washed with cold 2-propanol and water to provide 58.9 g of 9a obtained after drying (67.8 % yield). 1H NMR (400 MHz, CDCI3): δ 8.08 (d, 1 H, J = 12 Hz), 7.97 (d, 1 H, J = 12 Hz), 7.13 (t, 1 H, J = 8 Hz), 7.55 (t, 1 H, J= 8 Hz), 3.92 (s, 3H), 2.63 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 186.6, 161.1 , 155.3, 148.2, 140.9, 132.0, 129.0, 128.8, 127.8, 123.8, 123.7, 53.7, 23.6.

Catalyst preparation: To a suitable sized, clean and dry reactor was charged dichloro(pentamethylcyclopentadienyl)rhodium (III) dimer (800 ppm relative to 9a, 188.5 mg) and the ligand (2000 ppm relative to 9a, 306.1 mg). The system was purged with nitrogen and then 3 ml. of acetonitrile and 0.3 ml_ of triethylamine was charged to the system. The resulting solution was agitated at room temperature for not less than 45 minutes and not more than 6 hours. Reaction: To a suitable sized, clean and dry reactor was charged 9a (1.00 equiv, 100.0 g (99.5 wt%), 377.4 mmol). The reaction was purged with nitrogen. To the reactor was charged acetonitrile (ACS grade, 4 L/Kg of 9a, 400 mL) and
triethylamine (2.50 equiv, 132.8 mL, 943 mmol). Agitation was initiated. The 9a solution was cooled to Tint= -5 to 0 °C and then formic acid (3.00 equiv, 45.2 mL, 1 132 mmol) was charged to the solution at a rate to maintain Tint not more than 20 °C. The batch temperature was then adjusted to Tint= -5 to -0 °C. Nitrogen was bubbled through the batch through a porous gas dispersion unit (Wiimad-LabGlass No. LG-8680-1 0, VWR catalog number 14202-962) until a fine stream of bubbles was obtained. To the stirring solution at Tint= -5 to 0 °C was charged the prepared catalyst solution from the catalyst preparation above. The solution was agitated at Tint= -5 to 0 °C with the bubbling of nitrogen through the batch until HPLC analysis of the batch indicated no less than 98 A% conversion (as recorded at 220 nm, 10-14 h). To the reactor was charged isopropylacetate (6.7 L/Kg of 9a, 670 ml_). The batch temperature was adjusted to Tint= 18 to 23 °C. To the solution was charged water (10 L/Kg of 9a, 1000 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. To the solution was charged water (7.5 L/Kg of 9a, 750 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. The batch was then reduced to 300 mL (3 L/Kg of 9a) via distillation while maintaining Text no more than 65 °C. The batch was cooled to Tint= 35 to 45 °C and the batch was seeded (10 mg). To the batch at Tint= 35 to 45 °C was charged heptane (16.7 L/Kg of 9a, 1670 mL) over no less than 1.5 hours. The batch temperature was adjusted to Tint= -2 to 3 °C over no less than 1 hour, and the batch was agitated at Tint= -2 to 3 °C for no less than 1 hour. The solids were collected by filtration. The filtrate was used to rinse the reactor (Filtrate is cooled to Tint= -2 to 3 °C before filtration) and the solids were suction dried for no less than 2 hours. The solids were dried until the LOD is no more than 4 % to obtain 82.7 g of 10a (99.6- 100 wt%, 98.5% ee, 82.5% yield). 1H-NMR (CDCI3, 400 MHz) δ: 8.20 (d, J= 8.4 Hz, 1 H), 8.01 (d, J= 8.4 Hz, 1 H), 7.73 (t, J= 7.4 Hz, 1 H), 7.59 (t, J= 7.7 Hz, H), 6.03 (s, 1 H), 3.93 (s, 1 H), 3.79 (s, 3H), 2.77 (s, 3H). 13C-NMR (CDCI3, 100 MHz) δ: 173.5, 158.3, 147.5, 142.9, 130.7, 128.8, 127.7, 127.1 , 125.1 , 124.6, 69.2, 53.4, 24.0.
Example 11

10a 6a
10a (2.45 kg, 96.8% purity, 8.9 mol), 6a (2.5 kg, 88.7% purity, 8.82 mol), tris(dibenzylideneacetone)dipalladium(0) (Pd2dba3, 40 g, 0.044 mol), (S)-3-ieri-butyl- 4-(2,6-dimethoxypheny1 )-2,3-dihydrobenzo[d][1 ,3]oxaphosphole (32 g, 0.01 1 mol), sodium carbonate (1.12 kg, 10.58 mol), 1-pentanol (16.69 L), and water (8.35 L) were charged to the reactor. The mixture was de-gassed by sparging with argon for 10-15 minutes, was heated to 60-63 °C, and was agitated until HPLC analysis of the reaction shows <1 A% (220 nm) of the 6a relative to the combined two atropisomer products (-15 hours). The batch was cooled to 18-23 °C. Water (5 L) and heptane (21 L) were charged. The slurry was agitated for 3 – 5 hours. The solids were collected by filtration, washed with water (4 L) and heptane/toluene mixed solvent (2.5 L toluene/5 L heptane), and dried. The solids were dissolved in methanol (25 L) and the resulting solution was heated to 50 °C and circulated through a CUNO carbon stack filter. The solution was distilled under vacuum to ~ 5 L. Toluene (12 L) was charged. The mixture was distilled under vacuum to ~ 5 L and cooled to 22 °C. Heptane (13 L) was charged to the contents over 1 hour and the resulting slurry was agitated at 20-25 °C for 3 – 4 hours. The solids were collected by filtration and washed with heptanes to provide 2.58 kg of 11a obtained after drying (73% yield). 1H NMR (400 MHz, CDCI3): δ 8.63 (d, 1 H, J = 8 Hz), 8.03 (d, 1 H, J = 12 Hz), 7.56 (t, 1 H, J = 8 Hz), 7.41 (d, 1 H, J = 8 Hz), 7.19 (t, 1 H, J = 8 Hz), 7.09 (m, 2H), 7.04 (d, 1 H, J = 8 Hz), 5.38 (d, 1 H, J = 8 Hz), 5.14 (d, 1 H, J = 8 Hz), 4.50 (t, 2H, J = 4 Hz), 3.40 (s, 3H), 3.25 (t, 2H, J = 4 Hz), 2.91 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 173.6, 158.2, 154.0, 150.9, 147.3, 147.2, 145.7, 141.3, 132.9, 123.0, 129.4, 128.6, 127.8, 126.7, 126.4, 125.8, 1 18.1 , 1 17.3, 109.9, 70.3, 65.8, 52.3, 28.5, 24.0.
Example 12

11a 12a
To a suitable clean and dry reactor under a nitrogen atmosphere was charged 11a (5.47 Kg, 93.4 wt%, 1 .00 equiv, 12.8 mol) and fluorobenzene (10 vols, 51.1 kg) following by trifluoromethanesulfonimide (4 mol%, 143 g, 0.51 mol) as a 0.5 M solution in DCM (1.0 Kg). The batch temperature was adjusted to 35-41 °C and agitated to form a fine slurry. To the mixture was slowly charged i-butyl-2,2,2- trichloroacetimidate 12b as a 50 wt% solution (26.0 Kg of f-butyl-2,2,2- trichloroacetimidate (1 19.0 mol, 9.3 equiv), the reagent was -48-51 wt% with the remainder 52-49 wt% of the solution being – 1.8:1 wt:wt heptane: fluorobenzene) over no less than 4 hours at Tint= 35-41 °C. The batch was agitated at Tint= 35-41 °C until HPLC conversion (308 nm) was >96 A%, then cooled to Tint= 20-25 °C and then triethylamine (0.14 equiv, 181 g, 1 .79 mol) was charged followed by heptane (12.9 Kg) over no less than 30 minutes. The batch was agitated at Tint= 20-25 °C for no less than 1 hour. The solids were collected by filtration. The reactor was rinsed with the filtrate to collect all solids. The collected solids in the filter were rinsed with heptane (1 1 .7 Kg). The solids were charged into the reactor along with 54.1 Kg of DM Ac and the batch temperature adjusted to Tint= 70-75 °C. Water ( .2 Kg) was charged over no less than 30 minutes while the batch temperature was maintained at Tint= 65-75 °C. 12a seed crystals (34 g) in water (680 g) was charged to the batch at Tlnt= 65-75 °C. Additional water (46.0 Kg) was charged over no less than 2 hours while maintaining the batch temperature at Tint= 65-75 °C. The batch temperature was adjusted to Tint= 18-25 °C over no less than 2 hours and agitated for no less than 1 hour. The solids were collected by filtration and the filtrate used to rinse the reactor. The solids were washed with water (30 Kg) and dried under vacuum at no more than 45 °C until the LOD < 4% to obtain 12a (5.275 Kg, 99.9 A% at 220 nm, 99.9 wt% via HPLC wt% assay, 90.5% yield). 1H-NMR (CDCI3, 400
MHz) δ: 8.66-8.65 (m, 1 H), 8.05 (d, J= 8.3 Hz, 1 H), 7.59 (t, J= 7.3 Hz, 1 H), 7.45 (d, J= 7.8 Hz, 1 H), 7.21 (t, J= 7.6 Hz, 1 H), 7.13-7.08 (m, 3H), 5.05 (s, 1 H), 4.63-4.52 (m, 2H), 3.49 (s, 3H), 3.41 -3.27 (m, 2H), 3.00 (s, 3H), 0.97 (s, 9H). 13C-NMR (CDCI3, 100 MHz) δ: 172.1 , 159.5, 153.5, 150.2, 147.4, 146.9, 145.4, 140.2, 131.1 , 130.1 , 128.9, 128,6, 128.0, 127.3, 126.7, 125.4, 117.7, 117.2, 109.4, 76.1 , 71.6, 65.8, 51 .9, 28.6, 28.0, 25.4. Example 13

To a suitable clean and dry reactor under a nitrogen atmosphere was charged 12a (9.69 Kg, 21.2 mol) and ethanol (23.0 Kg). The mixture was agitated and the batch temperature was maintained at Τίηί= 20 to 25 °C. 2 M sodium hydroxide (17.2 Kg) was charged at Tint= 20 to 25 °C and the batch temperature was adjusted to Tint= 60- 65°C over no less than 30 minutes. The batch was agitated at Tint= 60-65°C for 2-3 hours until HPLC conversion was >99.5% area (12a is <0.5 area%). The batch temperature was adjuted to Tlnt= 50 to 55°C and 2M aqueous HCI (14.54 Kg) was charged. The pH of the batch was adjusted to pH 5.0 to 5.5 (target pH 5.2 to 5.3) via the slow charge of 2M aqueous HCI (0.46 Kg) at Tint= 50 to 55°C. Acetonitrile was charged to the batch (4.46 Kg) at Tint= 50 to 55°C. A slurry of seed crystals (1001 , 20 g in 155 g of acetonitrile) was charged to the batch at Tint= 50 to 55°C. The batch was agitated at Tint= 50 to 55°C for no less than 1 hour (1-2 hours). The contents were vacuum distilled to -3.4 vol (32 L) while maintaining the internal temperature at 45-55°C. A sample of the batch was removed and the ethanol content was determined by GC analysis; the criterion was no more than 10 wt% ethanol. If the ethanol wt% was over 10%, an additional 10% of the original volume was distilled and sampled for ethanol wt%. The batch temperature was adjusted to Tint= 18-22°C over no less than 1 hour. The pH of the batch was verified to be pH= 5 – 5.5 and the pH was adjusted, if necessary, with the slow addition of 2 M HCI or 2 M NaOH aqueous solutions. The batch was agitated at Tint= 18-22°C for no less than 6 hours and the solids were collected by filtration. The filtrate/mother liquid was used to remove all solids from reactor. The cake with was washed with water (19.4 Kg) (water temperature was no more than 20 °C). The cake was dried under vacuum at no more than 60 °C for 12 hours or until the LOD was no more than 4% to obtain 1001 (9.52 Kg, 99.6 A% 220 nm, 97.6 wt% as determined by HPLC wt% assay, 99.0% yield).
…………………
compd 1144
http://www.google.com/patents/WO2009062285A1?cl=en


……………………
http://www.google.com/patents/WO2012138669A1?cl=en
Compound (I), (2S)-2-tert-butoxy-2-(4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-2- methylquinolin-3-yl)acetic acid, is an HIV non-catalytic site integrase inhibitor.

Compound (I) falls within the scope of the HIV inhibitors disclosed in WO
2007/131350. Compound (I) is disclosed specifically as compound no. 1144 in WO 2009/062285. Compound (I) can be prepared according to the general procedures found in WO 2007/13 350 and WO 2009/062285, which are hereby incorporated by reference.
Example 1

1 a 1b
1a (600 g, 4.1 mol) was charged into a dry reactor under nitrogen followed by addition of Ac20 (1257.5 g, 12.3 mol, 3 eq.). The resulting mixture was heated at 40 °C at least for 2 hours. The batch was then cooled to 30 °C over 30 minutes. A suspension of 1b in toluene was added to seed the batch if no solid was observed. After toluene (600 mL) was added over 30 minutes, the batch was cooled to -5 ~ -10 °C and was held at this temperature for at least 30 minutes. The solid was collected by filtration under nitrogen and rinsed with heptanes (1200 mL). After being dried under vacuum at room temperature, the solid was stored under nitrogen at least below 20 °C. The product 1b was obtained with 77% yield. 1H NMR (500 MHz, CDCI3): δ = 6.36 (s, 1 H), 3.68 (s, 2H), 2.30 (s, 3H).
Example 2

2a (100 g, 531 mmol) and 1 b (95 g, 558 mmol) were charged into a clean and dry reactor under nitrogen followed by addition of fluorobenzene ( 000 mL). After being heated at 35-37 °C for 4 hours, the batch was cooled to 23 °C. Concentrated H2S04 (260.82 g, 2659.3 mmol, 5 eq.) was added while maintaining the batch temperature below 35 °C. The batch was first heated at 30-35 °C for 30 minutes and then at 40- 45 °C for 2 hours. 4-Methyl morpholine (215.19 g, 2127 mmol, 4 eq.) was added to the batch while maintaining the temperature below 50 °C. Then the batch was agitated for 30 minutes at 40-50 °C. MeOH ( 00 mL) was then added while maintaining the temperature below 55 °C. After the batch was held at 50-55 °Cfor 2 hours, another portion of MeOH (100 mL) was added. The batch was agitated for another 2 hours at 50-55 °C. After fluorobenzene was distilled to a minimum amount, water (1000 mL) was added. Further distillation was performed to remove any remaining fluorobenzene. After the batch was cooled to 30 °C, the solid was collected by filtration with cloth and rinsed with water (400 mL) and heptane (200 mL). The solid was dried under vacuum below 50 °C to reach KF < 0.1 %. Typically, the product 2b was obtained in 90% yield with 98 wt%. 1H NMR (500 MHz, DMSO- cfe): δ = 10.83 (s, 1 H), 9.85 (s, bs, 1 H), 7.6 (d, 1 H, J = 8.7 Hz), 6.55 (d, 1 H, J = 8.7 Hz), 6.40 (s, 1 H), 4.00 (s, 2 H), 3.61 (s, 3 H).
Example 3

2b 3a
2b (20 g, 64 mmol) was charged into a clean and dry reactor followed by addition of THF (140 mL). After the resulting mixture was cooled to 0 °C, Vitride® (Red-AI, 47.84 g, 65 wt%, 154 mmol) in toluene was added while maintaining an internal temperature at 0-5 °C. After the batch was agitated at 5-10 °C for 4 hours, IPA (9.24 g, 153.8 mmol) was added while maintaining the temperature below 10 °C. Then the batch was agitated at least for 30 minutes below 25 °C. A solution of HCI in IPA (84.73 g, 5.5 M, 512 mmol) was added into the reactor while maintaining the temperature below 40 °C. After about 160 mL of the solvent was distilled under vacuum below 40 °C, the batch was cooled to 20-25 °C and then aqueous 6M HCI (60 mL) was added while maintaining the temperature below 40 °C. The batch was cooled to 25 °C and agitated for at least 30 minutes. The solid was collected by filtration, washed with 40 mL of IPA and water (1 V/1 V), 40 mL of water and 40 mL of heptanes. The solid was dried below 60 °C in a vacuum oven to reach KF < 0.5%. Typically, the product 3a was obtained in 90-95% yield with 95 wt%. 1H NMR (400 MHz, DMSO-c/e): 5 = 10.7 (s, 1 H), 9.68 (s, 1 H), 7.59 (d, 1 H, J = 8.7 Hz), 6.64 (, 1 H, J = 8.7 Hz), 6.27 (s, 1 H), 4.62 (bs, 1 H), 3.69 (t, 2H, J = 6.3 Hz), 3.21 (t, 2H, J = 6.3 Hz).
Example 4

3a 4a
3a (50 g, 174.756 mmol) and acetonitrile (200 mL) were charged into a dry and clean reactor. After the resulting mixture was heated to 65 °C, POC13 (107.18 g, 699 mmol, 4 eq.) was added while maintaining the internal temperature below 75 °C. The batch was then heated at 70-75 °C for 5-6 h. The batch was cooled to 20 °C. Water (400 mL) was added at least over 30 minutes while maintaining the internal temperature below 50 °C. After the batch was cooled to 20-25 °C over 30 minutes, the solid was collected by filtration and washed with water (100 mL). The wet cake was charged back into the reactor followed by addition of 1 M NaOH (150 mL). After the batch was agitated at least for 30 minutes at 25-35 °C, verify that the pH was greater than 12. Otherwise, more 6M NaOH was needed to adjust the pH >12. After the batch was agitated for 30 minutes at 25-35 °C, the solid was collected by filtration, washed with water (200 mL) and heptanes (200 mL). The solid was dried in a vacuum oven below 50 °C to reach KF < 2%. Typically, the product 4a was obtained at about 75-80% yield. 1H NMR (400 MHz, CDCI3): δ = 7.90 (d, 1 H, J = 8.4 Hz), 7.16 (s, 1 H), 6.89 (d, 1 H, J = 8.4 Hz), 4.44 (t, 2 H, J = 5.9 Hz), 3.23 (t, 2 H, J = 5.9 Hz). 13C NMR (100 MHz, CDCI3): δ = 152.9, 151.9, 144.9, 144.1 , 134.6, 119.1 , 1 17.0, 1 13.3, 1 1 1.9, 65.6, 28.3.
Example 5

4a 5a
Zn powder (54 g, 825 mmol, 2.5 eq.) and TFA (100 mL) were charged into a dry and clean reactor. The resulting mixture was heated to 60-65 °C. A suspension of 4a (100 g, 330 mmol) in 150 mL of TFA was added to the reactor while maintaining the temperature below 70 °C. The charge line was rinsed with TFA (50 mL) into the reactor. After 1 hour at 65±5 °C, the batch was cooled to 25-30 °C. Zn powder was filtered off by passing the batch through a Celite pad and washing with methanol (200 mL). About 400 mL of solvent was distilled off under vacuum. After the batch was cooled to 20-25 °C, 20% NaOAc (ca. 300 mL) was added at least over 30 minutes to reach pH 5-6. The solid was collected by filtration, washed with water (200 mL) and heptane (200 mL), and dried under vacuum below 45 °C to reach KF ≤ 2%. The solid was charged into a dry reactor followed by addition of loose carbon (10 wt%) and toluene (1000 mL). The batch was heated at least for 30 minutes at 45-50 °C. The carbon was filtered off above 35 °C and rinsed with toluene (200 mL). The filtrate was charged into a clean and dry reactor. After about 1000 mL of toluene was distilled off under vacuum below 50 °C, 1000 mL of heptane was added over 30 minutes at 40-50 °C. Then the batch was cooled to 0±5 °C over 30 minutes. After 30 minutes, the solid was collected and rinsed with 200 mL of heptane. The solid was dried under vacuum below 45 °C to reach KF≤ 500 ppm. Typically, the product 5a was obtained in about 90-95 % yield. 1H NMR (400 MHz, CDCI3): δ = 8.93 (m, 1 H), 7.91 (dd, 1 H, J = 1.5, 8 Hz), 7.17 (m 1 H), 6.90 (dd, 1 H, J = 1.6, 8.0 Hz), 4.46-4.43 (m, 2 H), 3.28-3.23 (m, 2 H). 13C NMR (100 MHz, CDCI3): δ = 152.8, 151 .2, 145.1 , 141.0, 133.3, 1 18.5, 1 18.2, 1 14.5, 1 1 1 .1 , 65.8, 28.4.
Example 6

5a (1.04 kg, 4.16 mol) and toluene (8 L) were charged into the reactor. The batch was agitated and cooled to -50 to -55 °C. BuLi solution (2.5 M in hexanes, 1.69 L, 4.23 mol) was charged slowly while maintaining the internal temperature between – 45 to -50 °C. The batch was agitated at -45 °C for 1 hour after addition. A solution of triisopropyl borate (0.85 kg, 4.5 mol) in MTBE (1.48 kg) was charged. The batch was warmed to 10 °C over 30 minutes. A solution of 5 N HCI in IPA (1.54 L) was charged slowly at 10 °C, and the batch was warmed to 20 °C and stirred for 30 minutes. It was seeded with 6a crystal (10 g). A solution of aqueous concentrated HCI (0.16 L) in IPA (0.16 L) was charged slowly at 20 °C in three portions at 20 minute intervals, and the batch was agitated for 1 hour at 20 °C. The solid was collected by filtration, rinsed with MTBE (1 kg), and dried to provide 6a (943 g, 88.7 % purity, 80% yield). 1H NMR (400 MHz, D20): δ 8.84 (d, 1 H, J = 4 Hz), 8.10 (m, 1 H), 7.68 (d, 1 H, J = 6 Hz), 7.09 (m, 1 H), 4.52 (m, 2H), 3.47 (m, 2H).
Example 7

7a 7b
Iodine stock solution was prepared by mixing iodine (57.4 g, 0.23 mol) and sodium iodide (73.4 g, 0.49 mol) in water (270 mL). Sodium hydroxide (28.6 g, 0.715 mol) was charged into 220 mL of water. 4-Hydroxy-2 methylquinoline 7a (30 g, 0.19 mol) was charged, followed by acetonitrile (250 mL). The mixture was cooled to 10 °C with agitation. The above iodine stock solution was charged slowly over 30 minutes. The reaction was quenched by addition of sodium bisulfite (6.0 g) in water (60 mL). Acetic acid (23 mL) was charged over a period of 1 hour to adjust the pH of the reaction mixture between 6 and 7. The product was collected by filtration, washed with water and acetonitrile, and dried to give 7b (53 g, 98%). MS 286 [M + 1].

7b 8a
4-Hydroxy-3-iodo-2-methylquinoline 7b (25 g, 0.09 mol) was charged to a 1 -L reactor. Ethyl acetate (250 mL) was charged, followed by triethylamine (2.45 mL, 0.02 mol) and phosphorus oxychloride (12 mL, 0.13 mol). The reaction mixture was heated to reflux until complete conversion (~1 hour), then the mixture was cooled to 22 °C. A solution of sodium carbonate (31.6 g, 0.3 mol) in water (500 mL) was charged. The mixture was stirred for 20 minutes. The aqueous layer was extracted with ethyl acetate (120 mL). The organic layers were combined and concentrated under vacuum to dryness. Acetone (50 mL) was charged. The solution was heated to 60 °C. Water (100 mL) was charged, and the mixture was cooled to 22 °C. The product was collected by filtration and dried to give 8a (25 g, 97.3 % pure, 91.4 % yield). MS 304 [M + 1].
(Note: 8a is a known compound with CAS # 1033931-93-9. See references: (a) J. Org Chem. 2008, 73, 4644-4649. (b) Molcules 2010, 15, 3171-3178. (c) Indian J. Chem. Sec B: Org. Chem. Including Med Chem. 2009, 48B(5), 692-696.)

8a (100 g, 0.33 mol) was charged to the reactor, followed by copper (I) bromide dimethyl sulfide complex (3.4 g, 0.017 mol) and dry THF (450 mL). The batch was cooled to – 5 to – 2 °C. i-PrMgCI (2.0 M in THF, 173 mL, 0.346 mol) was charged into the reactor at the rate which maintains the batch temperature < -10 °C.
In a 2nd reactor, methyl chlorooxoacetate (33 mL, 0.36 mol) and dry THF (150 mL) was charged. The solution was cooled to -15 to -10 °C. The content of the 1 st reactor (Grignard/cuprate) was charged into the 2nd reactor at the rate which maintained the batch temperature < -10 °C. The batch was agitated for 30 minutes at -10 °C. Aqueous ammonium chloride solution (10%, 300 mL) was charged. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Aqueous ammonium chloride solution (10%, 90 mL) and sodium carbonate solution (10%, 135 mL) were charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Brine (10%, 240 mL) was charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes. The aqueous layer was separated. The batch was concentrated under vacuum to -1/4 of the volume (about 80 mL left). 2-Propanol was charged (300 mL). The batch was concentrated under vacuum to -1/3 of the volume (about 140 mL left), and heated to 50 °C. Water (70 mL) was charged. The batch was cooled to 20 – 25 °C, stirred for 2 hours, cooled to -10 °C and stirred for another 2 hours. The solid was collected by filtration, washed with cold 2-propanol and water to provide 58.9 g of 9a obtained after drying (67.8 % yield). 1H NMR (400 MHz, CDCI3): δ 8.08 (d, 1 H, J = 12 Hz), 7.97 (d, 1 H, J = 12 Hz), 7.13 (t, 1 H, J = 8 Hz), 7.55 (t, 1 H, J = 8 Hz), 3.92 (s, 3H), 2.63 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 186.6, 161.1 , 155.3, 148.2, 140.9, 132.0, 129.0, 128.8, 127.8, 123.8, 123.7, 53.7, 23.6.
Example 10

Catalyst preparation: To a suitable sized, clean and dry reactor was charged dichloro(pentamethylcyclopentadienyl)rhodium(lll) dimer (800 ppm relative to 9a, 188.5 mg) and the ligand (2000 ppm relative to 9a, 306.1 mg). The system was purged with nitrogen and then 3 ml_ of acetonitrile and 0.3 ml_ of triethylamine was charged to the system. The resulting solution was agitated at RT for not less than 45 minutes and not more than 6 hours.
Reaction: To a suitable sized, clean and dry reactor was charged 9a (1.00 equiv, 100.0 g (99.5 wt%), 377.4 mmol). The reaction was purged with nitrogen. To the reactor was charged acetonitrile (ACS grade, 4 L/Kg of 9a, 400 ml_) and
triethylamine (2.50 equiv, 132.8 ml_, 943 mmol). Agitation was initiated. The 9a solution was cooled to Tint= -5 to 0 °C and then formic acid (3.00 equiv, 45.2 ml_, 1 132 mmol) was charged to the solution at a rate to maintain Tint not more than 20 °C. The batch temperature was then adjusted to Tlnt= -5 to -0 °C. Nitrogen was bubbled through the batch through a porous gas dispersion unit (Wilmad-LabGlass No. LG-8680-1 10, VWR catalog number 14202-962) until a fine stream of bubbles was obtained. To the stirring solution at Jml= -5 to 0 °C was charged the prepared catalyst solution from the catalyst preparation above. The solution was agitated at Tint= -5 to 0 °C with the bubbling of nitrogen through the batch until HPLC analysis of the batch indicated no less than 98 A% conversion (as recorded at 220 nm, 10-14 h). To the reactor was charged isopropylacetate (6.7 L/Kg of 9a, 670 mL). The batch temperature was adjusted to Tint= 18 to 23 °C. To the solution was charged water (10 L/Kg of 9a, 1000 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. To the solution was charged water (7.5 L/Kg of 9a, 750 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. The batch was then reduced to 300 mL (3 L/Kg of 9a) via distillation while maintaining Text no more than 65 °C. The batch was cooled to Tint= 35 to 45 °C and the batch was seeded ( 0 mg). To the batch at Tint= 35 to 45 °C charged heptane (16.7 L/Kg of 9a, 1670 mL) over no less than 1.5 hours. Adjusted the batch temperature to Tint= -2 to 3 °C over no less than 1 hour, and agitated the batch at Tint= -2 to 3 °C for no less than 1 hour. Collected the solids by filtration. Used the filtrate to rinse the reactor (Filtrate is cooled to
-2 to 3 °C before filtration) and the solids were suction dried for no less than 2 hours. The solids were dried until the LOD was no more than 4 % to obtain 82.7 g of 10a (99.6-100 wt%, 98.5% ee, 82.5% yield). 1H- NMR (CDCI3, 400 MHz) δ: 8.20 (d, J= 8.4 Hz, 1 H), 8.01 (d, J= 8.4 Hz, 1 H), 7.73 (t, J= 7.4 Hz, 1 H), 7.59 (t, J= 7.7 Hz, 1 H), 6.03 (s, 1 H), 3.93 (s, 1 H), 3.79 (s, 3H), 2.77 (s, 3H). 13C-NMR (CDCI3, 100 MHz) δ: 173.5, 158.3, 147.5, 142.9, 130.7, 128.8, 127.7, 127.1 , 125.1 , 124.6, 69.2, 53.4, 24.0.
Example 11

10a 6a 11a
10a (2.45 kg, 96.8% purity, 8.9 mol), 6a (2.5 kg, 88.7% purity, 8.82 mol), tris(dibenzylideneacetone)dipalladium(0) (Pd2dba3, 40 g, 0.044 mol), (S)-3-iert-butyl-4-(2,6-dimethoxyphenyl)-2,3-dihydrobenzo[d][1 ,3]oxaphosphole (32 g, 0.01 1 mol), sodium carbonate (1.12 kg, 10.58 mol), 1 -pentanol (16.69 L), and water (8.35 L) were charged to the reactor. The mixture was de-gassed by sparging with argon for 10-15 minutes, was heated to 60-63 °C, and was agitated until HPLC analysis of the reaction shows <1 A% (220 nm) of the 6a relative to the combined two atropisomer products (-15 hours). The batch was cooled to 8-23 °C. Water (5 L) and heptane (21 L) were charged. The slurry was agitated for 3 – 5 hours. The solids were collected by filtration, washed with water (4 L) and heptane/toluene mixed solvent (2.5 L toluene/5 L heptane), and dried. The solids were dissolved in methanol (25 L) and the resulting solution was heated to 50 °C and circulated through a CUNO carbon stack filter. The solution was distilled under vacuum to ~ 5 L. Toluene (12 L) was charged. The mixture was distilled under vacuum to – 5 L and cooled to 22 °C. Heptane (13 L) was charged to the contents over 1 hour and the resulting slurry was agitated at 20-25 °C for 3 – 4 hours. The solids were collected by filtration and washed with heptanes to provide 2.58 kg of 11a obtained after drying (73% yield). 1H NMR (400 MHz, CDCI3): δ 8.63 (d, 1 H, J = 8 Hz), 8.03 (d, 1 H, J = 12 Hz), 7.56 (t, 1 H, J = 8 Hz), 7.41 (d, 1 H, J = 8 Hz), 7.19 (t, 1 H, J = 8 Hz), 7.09 (m, 2H), 7.04 (d, 1 H, J = 8 Hz), 5.38 (d, 1 H, J = 8 Hz), 5.14 (d, 1 H, J = 8 Hz), 4.50 (t, 2H, J = 4 Hz), 3.40 (s, 3H), 3.25 (t, 2H, J = 4 Hz), 2.91 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 173.6, 158.2, 154.0, 150.9, 147.3, 147.2, 145.7, 141.3, 132.9, 123.0, 129.4, 128.6, 127.8, 126.7, 126.4, 125.8, 1 18.1 , 1 17.3, 109.9, 70.3, 65.8, 52.3, 28.5, 24.0.

To a suitable clean and dry reactor under a nitrogen atmosphere was charged 1a (5.47 Kg, 93.4 wt%, 1 .00 equiv, 12.8 mol) and fluorobenzene (10 vols, 51.1 kg) following by trifluoromethanesulfonimide (4 mol%, 143 g, 0.51 mol) as a 0.5 M solution in DCM (1.0 Kg). The batch temperature was adjusted to 35-41 °C and agitated to form a fine slurry. To the mixture was slowly charged i-butyt-2,2,2- trichloroacetimidate 12b as a 50 wt% solution (26.0 Kg of f-butyl-2,2,2- trichloroacetimidate (119.0 mol, 9.3 equiv), the reagent was -48-51 wt% with the remainder 52-49 wt% of the solution being ~ 1.8:1 wt:wt heptane: fluorobenzene) over no less than 4 hours at Tint= 35-41 °C. The batch was agitated at Tint= 35-41 °C until HPLC conversion (308 nm) was >96 A%, then cooled to Tlnt= 20-25 °C and then triethylamine (0.14 equiv, 181 g, 1.79 mol) was charged followed by heptane (12.9 Kg) over no less than 30 minutes. The batch was agitated at Tint= 20-25 °C for no less than 1 hour. The solids were collected by filtration. The reactor was rinsed with the filtrate to collect all solids. The collected solids in the filter were rinsed with heptane (1 1.7 Kg). The solids were charged into the reactor along with 54.1 Kg of DM Ac and the batch temperature adjusted to Tint= 70-75 °C. Water (1 1.2 Kg) was charged over no less than 30 minutes while the batch temperature was maintained at Tint= 65-75 °C. 12a seed crystals (34 g) in water (680 g) was charged to the batch at Tint= 65-75 °C. Additional water (46.0 Kg) was charged over no less than 2 hours while maintaining the batch temperature at Tint= 65-75 °C. The batch temperature was adjusted to Tint= 18-25 °C over no less than 2 hours and agitated for no less than 1 hour. The solids were collected by filtration and the filtrate used to rinse the reactor. The solids were washed with water (30 Kg) and dried under vacuum at no more than 45 °C until the LOD < 4% to obtain 12a (5.275 Kg, 99.9 A% at 220 nm, 99.9 wt% via HPLC wt% assay, 90.5% yield). H-NMR (CDCI3l 400 MHz) δ: 8.66-8.65 (m, 1 H), 8.05 (d, J= 8.3 Hz, 1 H), 7.59 (t, J= 7.3 Hz, 1 H), 7.45 (d, J= 7.8 Hz, 1 H), 7.21 (t, J= 7.6 Hz, 1 H), 7.13-7.08 (m, 3H), 5.05 (s, H), 4.63-4.52 (m, 2H), 3.49 (s, 3H), 3.41 -3.27 (m, 2H), 3.00 (s, 3H), 0.97 (s, 9H). 13C-NMR (CDCI3, 100 MHz) δ: 172.1 , 159.5, 153.5, 150.2, 147.4, 146.9, 145.4, 140.2, 131.1 , 130.1 , 128.9, 128.6, 128.0, 127.3, 126.7, 125.4, 1 17.7, 1 17.2, 109.4, 76.1 , 71.6, 65.8, 51.9, 28.6, 28.0, 25.4.
Example 13

To a suitable clean and dry reactor under a nitrogen atmosphere was charged 12a (9.69 Kg, 21.2 mol) and ethanol (23.0 Kg). The mixture was agitated and the batch temperature was maintained at Tjnt= 20 to 25 °C. 2 M sodium hydroxide (17.2 Kg) was charged at Tint= 20 to 25 °C and the batch temperature was adjusted to Tlnt= 60- 65°C over no less than 30 minutes. The batch was agitated at Tint= 60-65°C for 2-3 hours until HPLC conversion was >99.5% area (12a is <0.5 area%). The batch temperature was adjuted to Tint= 50 to 55°C and 2M aqueous HCI (14.54 Kg) was charged. The pH of the batch was adjusted to pH 5.0 to 5.5 (target pH 5.2 to 5.3) via the slow charge of 2M aqueous HCI (0.46 Kg) at Tint= 50 to 55°C. Acetonitrile was charged to the batch (4.46 Kg) at Τ,ηί= 50 to 55°C. A slurry of seed crystals (1001 , 20 g in 155 g of acetonitrile) was charged to the batch at Tint= 50 to 55°C. The batch was agitated at Tint= 50 to 55°C for no less than 1 hour (1-2 hours). The contents were vacuum distilled to -3.4 vol (32 L) while maintaining the internal temperature at 45-55°C. A sample of the batch was removed and the ethanol content was determined by GC analysis; the criterion was no more than 10 wt% ethanol. If the ethanol wt% was over 10%, an additional 10% of the original volume was distilled and sampled for ethanol wt%. The batch temperature was adjusted to Tint= 8-22°C over no less than 1 hour. The pH of the batch was verified to be pH= 5 – 5.5 and the pH was adjusted, if necessary, with the slow addition of 2 M HCI or 2 M NaOH aqueous solutions. The batch was agitated at Tint= 18-22°C for no less than 6 hours and the solids were collected by filtration. The filtrate/mother liquid was used to remove all solids from reactor. The cake with was washed with water (19.4 Kg) (water temperature was no more than 20 °C). The cake was dried under vacuum at no more than 60 °C for 12 hours or until the LOD was no more than 4% to obtain 1001 (9.52 Kg, 99.6 A% 220 nm, 97.6 wt% as determined by HPLC wt% assay, 99.0% yield). Example 14
Hydrochloride salt of Compound (I), Type A
Compound (I) (263 mg) was added to a vial of ethanol (1.5 ml_), and then 36.5% HCL aqueous solution (59 mg) was added. The mixture was heated to 70 °C; and stirred at this temperature until solid material was obtained. The mixture was cooled to 20 °C over a period of 10 hours. After cooling, isopropanol (400 μΙ_) was added over a period of 3 hours. The resulting solids were collected and characterized as the hydrochloride salt of Compound (I), Type A.
The hydrochloride salt of Compound (I), Type A was prepared analogously to the aforementioned procedure using methyl ethyl ketone, tetrahydrofuran, acetonitrile, ethyl acetate, dichloroethane and methyl-t-buyl ether instead of ethanol.

References
- Pharmacodynamics of BI 224436 for HIV-1 in an in vitro hollow fiber infection model system
- Levin, Jules. BI 224436, a Non-Catalytic Site Integrase Inhibitor, is a potent inhibitor of the replication of treatment-naïve and raltegravir-resistant clinical isolates of HIV-1. Conference Reports for NATAP. ICAAC Chicago Sept 17-20 2011.
- Gilead Negotiates Worldwide License to BI’s Early Clinical Stage HIV Program. Genetic Engineering and Biotechnology News. 6 Oct 2011.
- Highleyman, Liz. ICAAC: New Integrase Inhibitor BI 224436 Active against Raltegravir-Resistant HIV. HIVandHepatitis.com. 7 Oct 2011.
- WO-2014055618

