
Home » 2014 (Page 7)
Yearly Archives: 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ragaglitazar ……..Dr. Reddy’s Research Foundation
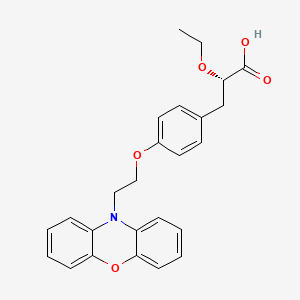
Ragaglitazar
NNC-61-0029, (-) – DRF-2725, NN-622,
(−)DRF 2725
cas 222834-30-2
222834-21-1 (racemate)
Hyperlipidemia; Hypertriglyceridemia; Lipid metabolism disorder; Non-insulin dependent diabetes
PPAR alpha agonist; PPAR gamma agonist
(2S)-2-ETHOXY-3-{4-[2-(10H-PHENOXAZIN-10-YL)ETHOXY]PHENYL}PROPANOIC ACID,
(2S)-2-ethoxy-3-[4-(2-phenoxazin-10-ylethoxy)phenyl]propanoic acid, DRF, 1nyx





……………………………………………………………..

J Med Chem 2001,44(16),2675
http://pubs.acs.org/doi/abs/10.1021/jm010143b

(−)DRF 2725 (6) is a phenoxazine analogue of phenyl propanoic acid. Compound 6 showed interesting dual activation of PPARα and PPARγ. In insulin resistant db/db mice, 6 showed better reduction of plasma glucose and triglyceride levels as compared to rosiglitazone. Compound 6 has also shown good oral bioavailability and impressive pharmacokinetic characteristics. Our study indicates that 6 has great potential as a drug for diabetes and dyslipidemia.

Scheme 1 a
a (a) NaH, DMF, 0−25 °C, 12 h; (b) triethyl 2-ethoxy phosphosphonoacetate, NaH, THF, 0−25 °C, 12 h; (c) Mg/CH3OH, 25 °C, 12 h; (d) 10% aq NaOH, CH3OH, 25 °C, 6 h; (e) (1) pivaloyl chloride, Et3N, DCM, 0 °C, (2) (S)-2-phenyl glycinol/Et3N; (f) 1 M H2SO4, dioxane/water, 90−100 °C, 80 h.
Compound 6 is prepared from phenoxazine using a synthetic route shown in Scheme 1. Phenoxazine 7 upon reaction with p-bromoethoxy benzaldehyde 89 gave benzaldehyde derivative 9. Reacting 9 with triethyl 2-ethoxy phosphonoacetate afforded propenoate 10 as a mixture of geometric isomers. Reduction of 10 using magnesium methanol gave propanoate 11, which on hydrolysis using aqueous sodium hydroxide gave propanoic acid 12 in racemic form. Resolution of 12 using (S)(+)-2-phenyl glycinol followed by hydrolysis using sulfuric acid afforded the propanoic acid 6 in (−) form.
Nate, H.; Matsuki, K.; Tsunashima, A.; Ohtsuka, H.; Sekine, Y. Synthesis of 2-phenylthiazolidine derivatives as cardiotonic agents. II. 2-(phenylpiperazinoalkoxyphenyl)thiazolidine-3-thiocarboxyamides and corresponding carboxamides. Chem. Pharm. Bull. 1987, 35, 2394−2411
(S)-3-[4-[2-(Phenoxazin-10-yl)ethoxy]phenyl]-2-eth-oxypropanoic Acid (6). as a white solid, mp: 89−90 °C.
[α]D 25 = − 12.6 (c = 1.0%, CHCl3).
1H NMR (CDCl3, 200 MHz): δ 1.16 (t, J = 7.0 Hz, 3H), 1.42−1.91 (bs, 1H, D2O exchangeable), 2.94−3.15 (m, 2H), 3.40−3.65 (m, 2H), 3.86−4.06 (m, 3H), 4.15 (t, J = 6.6 Hz, 2H), 6.63−6.83 (m, 10H), 7.13 (d, J = 8.5 Hz, 2H). Mass m/z (relative intensity): 419 (M+, 41), 197 (15), 196 (100), 182 (35), 167 (7), 127 (6), 107 (19).
Purity by HPLC: chemical purity: 99.5%; chiral purity: 94.6% (RT 27.5).

…………………………………
http://www.google.com/patents/US6608194?cl=zh
EXAMPLE 23 (−) 3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxypropanoic acid:

The title compound (0.19 g, 54%) was prepared as a white solid from diastereomer [(2S-N(1S)]-3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxy-N-(2-hydroxy-1-phenyl)ethylpropanamide (0.45 g, 0.84 mmol) obtained in example 21b by an analogous procedure to that described in example 22. mp: 89-90° C.
[α]D 25=−12.6 (c=1.0% CHCl3)
1H NMR (CDCl3, 200 MHz): δ 1.16 (t, J=7.02 Hz, 3H), 1.42-1.91 (bs, 1H, D2O exchangeable), 2.94-3.15 (complex, 2H), 3.40-3.65 (complex, 2H), 3.86-4.06 (complex, 3H), 4.15 (t, J=6.65 Hz, 2H), 6.63-6.83 (complex, 10H), 7.13 (d, J=8.54 Hz, 2H).
………………………..
http://www.google.com/patents/EP1049684A1?cl=en
Example 23
(S)-3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxypropanoic acid :

The title compound (0.19 g, 54 %) was prepared as a white solid from diastereomer [(2S- N(lS)]-3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxy-N-(2-hydroxy-l- phenyl)propanamide (0.45 g, 0.84 mmol) obtained in example 21b by an analogous procedure to that described in example 22. mp : 89 – 90 °C. [α]D 25 = – 12.6 (c = 1.0 %, CHC13)
*H NMR (CDC13, 200 MHz) : δ 1.16 (t, J = 7.02 Hz, 3H), 1.42 – 1.91 (bs, IH, D20 exchangeable), 2.94 – 3.15 (complex, 2H), 3.40 – 3.65 (complex, 2H), 3.86 – 4.06 (complex, 3H), 4.15 (t, J = 6.65 Hz, 2H), 6.63 – 6.83 (complex, 10H), 7.13 (d, J = 8.54 Hz, 2H).
| Patent | Submitted | Granted |
|---|---|---|
| Benzamides as ppar modulators [US2006160894] | 2006-07-20 | |
| Novel tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US2002077320] | 2002-06-20 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US7119198] | 2006-07-06 | 2006-10-10 |
| Tricyclic compounds and their use in medicine: process for their preparation and pharmaceutical compositions containing them [US6440961] | 2002-08-27 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6548666] | 2003-04-15 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6608194] | 2003-08-19 | |
| CRYSTALLINE R- GUANIDINES, ARGININE OR (L) -ARGININE (2S) -2- ETHOXY -3-{4- [2-(10H -PHENOXAZIN -10-YL)ETHOXY]PHENYL}PROPANOATE [WO0063189] | 2000-10-26 | |
| Pharmaceutically acceptable salts of phenoxazine and phenothiazine compounds [US6897199] | 2002-11-14 | 2005-05-24 |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6939988] | 2005-09-06 |
WO-2014181362
-
wo/2014/181362 a process for the preparation of 3 … – WIPO
patentscope.wipo.int/search/en/WO2014181362Nov 13, 2014 – (WO2014181362) A PROCESS FOR THE PREPARATION OF 3-ARYL-2-HYDROXY PROPANOIC ACID COMPOUNDS …
A process for the preparation of 3-aryl-2-hydroxy propanoic acid compounds
ragaglitazar; saroglitazar
Council of Scientific and Industrial Research (India)
Process for preparing enantiomerically pure 3-aryl-2-hydroxy propanoic acid derivatives (eg ethyl-(S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate), using S-benzyl glycidyl ether as a starting material. Useful as intermediates in the synthesis of peroxisome proliferator activated receptor agonist such as glitazars (eg ragaglitazar or saroglitazar). Appears to be the first filing on these derivatives by the inventors; however see WO2014181359 (for a concurrently published filing) and US8748660 (for a prior filing), claiming synthesis of enantiomerically pure compounds.
- Dolling, U. H.; Davis, P.; Grabowski, E. J. J. Efficient Catalytic Asymmetric Alkylations. 1. Enantioselective Synthesis of (+)-Indacrinone via Chiral Phase-Transfer Catalysis. J. Am. Chem. Soc. 1984, 106, 446–447.
- Andrus, M. B.; Hicken, E. J.; Stephens, J. C. Phase-Transfer Catalyzed Asymmetric Glycolate Alkylation. Org. Lett. 2004, 6, 2289–2292.
- Andrus, M. B.; Hicken, E. J.; Stephens, J. C.; Bedke, D. K. Asymmetric Phase-Transfer Catalyzed Glycolate Alkylation, Investigation of the Scope, and Application to the Synthesis of (-)-Ragaglitazar. J. Org. Chem. 2005, ASAP.
- Henke, B. R. Peroxisome Proliferator-Activated Receptor Dual Agonists for the Treatment of Type 2 Diabetes. J. Med. Chem. 2004, 47, 4118–4127.
- Wilson, T. M.; Brown, P. J.; Sternbach, D. D.; Henke, B. R. The PPARs: from orphan receptors to drug discovery. J. Med. Chem. 2000, 46, 1306–1317.
- Uchida, R.; Shiomi, K.; Inokoshi, J.; Masuma, R.; Kawakubo, T.; Tanaka, H.; Iwai, Y.; Omura, A. Kurasoins A and B, New Protein Farnesyltrasferase Inhibitors Produced by Paecilomyces sp. FO-3684. J. Antibio. 1996, 49, 932–934.
TAK-733……. clinical studies for cancer treatment.

TAK-733
CAS: 1035555-63-5
Synonym: TAK-733; TAK 733; TAK733.
IUPAC/Chemical name:
(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
Chemical Formula: C17H15F2IN4O4
Exact Mass: 504.01060
Molecular Weight: 504.23
Elemental Analysis: C, 40.49; H, 3.00; F, 7.54; I, 25.17; N, 11.11; O, 12.69
Phase I clinical studies for cancer treatment.Takeda Pharmaceutical,
Solid Tumors Therapy
Description of TAK-733: TAK-733 is an orally bioavailable small-molecule inhibitor of MEK1 and MEK2 (MEK1/2) with potential antineoplastic activity. MEK inhibitor TAK-733 selectively binds to and inhibits the activity of MEK1/2, preventing the activation of MEK1/2-dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling and tumor cell proliferation. MEK1/2 (MAP2K1/K2) are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway and are often upregulated in a variety of tumor cell types.
Current developer: Millennium Pharmaceuticals, Inc./Takeda Pharmaceutical Company Limited.
TAK-733 is being developed at Millennium Pharmaceuticals for the treatment of adult patients with advanced non-hematological malignancies. Phase I clinical trials are ongoing for the treatment of advanced metastatic melanoma. In preclinical studies, the compound has been shown to bind to and potently inhibit MEK.
………………………………….
Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer
- Takeda San Diego;10410 Science Center Drive, San Diego, CA 92121, United States
http://www.sciencedirect.com/science/article/pii/S0960894X11000941

Synthesis of compounds 26 and 27 (Route 4). Reagents and conditions: (a) 1-chloro-2,4-dinitrobenzene, K2CO3, DMF; (b) (R)-O-((2,2-dimethyl-1,3-dioxolan-4-yl)methyl)hydroxylamine or 2,2-dimethyl-1,3-dioxan-5-amine, K2CO3 or Cs2CO3, DMF; (c) HCl, THF; (d) Selectfluor, CH3CN,DMF.
TAK-733 exhibited potent enzymatic and cell activity with an IC50 of 3.2 nM against constitutively active MEK enzyme and an EC50 of 1.9 nM against ERK phosphorylation in cells. TAK-733 did not inhibit any other kinases, receptors or ion channels that were tested with inhibitor concentrations up to 10 μM. TAK-733 was found to bind plasma protein moderately (ca. 97% for human and 96% for mouse), and exhibit high permeability and high microsomal stability across species. It did not inhibit P450s up to 30 μM.
The co-crystal structure of TAK-733 in the MEK1 allosteric site has been solved (Fig. 3). As predicted, the pyridone oxygen makes a hydrogen bond with the backbone NH of Ser212. The 2-fluoro-4-iodoaniline moeity sits in the deep lipophilic pocket. The pyrimidinone oxygen makes a hydrogen bond with Lys97, and the propanediol terminal hydroxyl interacts with both Lys97 and the ADP phosphate.

-
Figure 3.
The X-ray co-crystal structure of TAK-733 in the MEK1 allosteric site.

(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
| Molecular Weight: | 504.23 |
| TAK-733 Formula: | C17H15F2IN4O4 |
| CAS Number: | 1035555-63-5 |
TAK-733 is an orally bioavailable small-molecule inhibitor of MEK1 and MEK2 (MEK1/2) with potential antineoplastic activity. MEK inhibitor TAK-733 selectively binds to and inhibits the activity of MEK1/2, preventing the activation of MEK1/2-dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling and tumor cell proliferation. MEK1/2 (MAP2K1/K2) are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway and are often upregulated in a variety of tumor cell types.
BRAF L597 mutations in melanoma are associated with sensitivity to MEK inhibitors.
Dahlman et al. Cancer Discov. 2012 Jul 13. PMID: 22798288.Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer.
Dong et al. Bioorg Med Chem Lett. 2011 Mar 1;21(5):1315-9. PMID: 21310613.

16: 1652-1659


MEK kinases regulate the pathway that mediates proliferative and anti-apoptotic signaling factors that promote tumor growth and metastasis. TAK-733 is an MEK kinase inhibitor that entered phase I clinical trials for the treatment of cancer. A noteworthy feature of this short synthesis (25% yield overall) is the one-pot, three-step synthesis of the fluoropyridone D, in which the fluorine atom is present at the outset.
The reaction of F with the nosylate G gave a mixture of N- and O-alkylation products (8:1) from which the desired N-alkylation product was isolated by crystallization. The mixture of N-methyl pyrrolidine (NMP) and methanol used in the final deprotection step, helped to ensure formation of the desired polymorph. The nine-step discovery synthesis (3% overall yield) is also presented.
…………………………….
http://www.google.com/patents/WO2008079814A2?cl=en
Example 18: (i?)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione

[0364] To a solution of (i?)-3-(2,3-dihydroxypropyl)-5-(2-fluoro-4-iodophenylamino)- 8-methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione (example 6) (1 g, 2.06 mmol, 1 eq) in DMF (19 mL) was added dropwise a mixture of Selectfluor (801 mg, 2.26 mmol, 1.1 eq) in acetonitrile (9 niL) and DMF (5 niL) at ambient temperature while stirring under nitrogen. The reaction mixture was stirred for 10 minutes at ambient temperature and filtered. The filtrate was purified by preparatory LC/MS (30-55% CH3CN in H2O) to give (i?)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (example 18) as an off-white solid. The recovered starting material was subjected to the same reaction conditions to produce a second crop of product. The total yield of product was 347 mg (33% yield). 1H NMR (400 MHz, DMSO-J6) δ ppm 3.36 – 3.40 (m, 1 H) 3.43 – 3.50 (m, 1 H) 3.58 (s, 3 H) 3.61 – 3.72 (m, 1 H) 3.72 – 3.82 (m, 1 H) 4.27 – 4.37 (m, 1 H) 4.78 – 4.87 (m, 1 H) 5.14 (d, J=5.81 Hz, 1 H) 6.93 – 7.03 (m, 1 H) 7.53 (d, J=8.84 Hz, 1 H) 7.65 – 7.74 (m, 1 H) 8.52 (s, 1 H) 10.25 (d, J=LOl Hz, 1 H). [M+H] calc’d for Ci7Hi5F2IN4O4, 505; found, 505.
OTHER ISOMER
Example 19: (5)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione
 CAREFUL PLEASE
CAREFUL PLEASE[0365] To a solution of (5)-3-(2,3-dihydroxypropyl)-5-(2-fluoro-4-iodophenylamino)- 8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (example 5) (1.25 g, 2.58 mmol, 1 eq) in DMF (26 mL) was added a mixture of Selectfluor (1.187 g, 3.35 mmol, 1.3 eq) in acetonitrile (26 mL). The reaction mixture was irradiated with microwave at 82 0C for 10 minutes and filtered. The filtrate was purified by preparatory LC/MS (30-55% CH3CN in H2O) to give (5)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione (example 19) as an off-white solid (331 mg, 25% yield). 1H NMR (400 MHz, DMSO-J6) δ ppm 3.36 – 3.42 (m, 1 H) 3.42 – 3.51 (m, 1 H) 3.59 (s, 3 H) 3.63 – 3.72 (m, 1 H) 3.73 – 3.81 (m, 1 H) 4.32 (dd, J=13.14, 3.03 Hz, 1 H) 4.79 (br. s., 1 H) 5.10 (br. s., 1 H) 6.98 (td, J=8.46, 5.31 Hz, 1 H) 7.52 (dd, J=8.46, 1.14 Hz, 1 H) 7.68 (dd, J=10.36, 1.77 Hz, 1 H) 8.51 (s, 1 H) 10.24 (d, J=2.27 Hz, 1 H). [M+H] calc’d for Ci7Hi5F2IN4O4, 505; found, 505.
……………………………………….
http://www.google.com/patents/US8436200?cl=en

The synthetic process of the present invention allows for the preparation of (R)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione while avoiding the use of a fluorinating reagent in the last step. That is, the present invention provides a valuable process for making (R)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione characterized by the reaction of a compound of formula (8) with 2-fluoro-4-iodoaniline to give a compound of formula (9). Such a process avoids the use of costly and possible hazardous fluorinating regents in later steps which has significant advantages in large-scale manufacture.
Example 8.1(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
Combine (R)-3-((2,2-dimethyl-1,3-dioxolan-4-yl)methyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (24.75 g, 45.58 mmol) and ethanol (250 mL). Add aqueous 9N sulfuric acid (50 mL) over 5 minutes. Heat to 75° C. After 2 hour, cool to ambient temperature and then cool in an ice bath to give a solid. Collect the solid by filtration, rinse with ethanol (3×30 mL), and dry to give the title compound. 1H NMR (400 MHz, DMSO-d6) δ10.24 (s, 1H), 8.52 (s, 1H), 7.69 (dd, 1H, J=10.4, 1.8 Hz), 7.52 (d, 1H, J=8.6 Hz), 6.98 (m, 1H), 5.14 (brs, 1H), 4.83 (brs, 1H), 4.32 (dd, 1H, J=12.9, 2.5 Hz), 3.76 (m, 1H), 3.67 (dd, 1H, J=13.1, 12.9 Hz), 3.58 (s, 3H), 3.46 (ddd, 1H, J=10.9, 5.3, 5.1 Hz), 3.38 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ161.3 (d, J=4.0 Hz), 155.6 (d, J=22.8 Hz), 154.6 (d, J=250 Hz), 152.0, 150.6, 134.3 (d, J=231 Hz), 133.8 (d, J=7.1 Hz), 133.1 (d, J=3.0 Hz), 127.8 (d, J=10.3 Hz), 125.3 (d, J=7.0 Hz), 123.9 (d, J=21.5 Hz), 95.0 (d, J=4.0 Hz), 87.1 (d, J=7.8 Hz), 68.0, 63.8, 50.1, 28.8; 19F NMR (376 MHz, DMSO-d6) δ-124.4, −149.8; MS (M+H)+m/z calcd 505.0. found 505.0.
Google+
|
Information about this agent |
TAK-733 is currently in Phase I clinical trials and is being developed by Millennium Pharmaceuticals, Inc. (a part of Takeda Pharmaceutical Company Limited).
|
References |
1: Acquaviva J, Smith DL, Jimenez JP, Zhang C, Sequeira M, He S, Sang J, Bates RC, Proia DA. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol Cancer Ther. 2014 Feb;13(2):353-63. doi: 10.1158/1535-7163.MCT-13-0481. Epub 2014 Jan 7. PubMed PMID: 24398428.
2: Zhang Y, Xue D, Wang X, Lu M, Gao B, Qiao X. Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways. Mol Med Rep. 2014 Jan;9(1):83-90. doi: 10.3892/mmr.2013.1781. Epub 2013 Nov 7. PubMed PMID: 24213221.
3: Nakamura A, Arita T, Tsuchiya S, Donelan J, Chouitar J, Carideo E, Galvin K, Okaniwa M, Ishikawa T, Yoshida S. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013 Dec 1;73(23):7043-55. doi: 10.1158/0008-5472.CAN-13-1825. Epub 2013 Oct 11. PubMed PMID: 24121489.
4: Garraway LA, Baselga J. Whole-genome sequencing and cancer therapy: is too much ever enough? Cancer Discov. 2012 Sep;2(9):766-8. doi: 10.1158/2159-8290.CD-12-0359. PubMed PMID: 22969114.
5: Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P, Atefi M, Su Z, Branch S, Lyle PL, Hicks DJ, Bozon V, Glaspy JA, Rosen N, Solit DB, Netterville JL, Vnencak-Jones CL, Sosman JA, Ribas A, Zhao Z, Pao W. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012 Sep;2(9):791-7. Epub 2012 Jul 13. PubMed PMID: 22798288; PubMed Central PMCID: PMC3449158.
6: von Euw E, Atefi M, Attar N, Chu C, Zachariah S, Burgess BL, Mok S, Ng C, Wong DJ, Chmielowski B, Lichter DI, Koya RC, McCannel TA, Izmailova E, Ribas A. Antitumor effects of the investigational selective MEK inhibitor TAK733 against cutaneous and uveal melanoma cell lines. Mol Cancer. 2012 Apr 19;11:22. PubMed PMID: 22515704; PubMed Central PMCID: PMC3444881.
7: Dong Q, Dougan DR, Gong X, Halkowycz P, Jin B, Kanouni T, O’Connell SM, Scorah N, Shi L, Wallace MB, Zhou F. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg Med Chem Lett. 2011 Mar 1;21(5):1315-9. doi: 10.1016/j.bmcl.2011.01.071. Epub 2011 Jan 22. PubMed PMID: 21310613.
| US8030317 | Dec 18, 2007 | Oct 4, 2011 | Takeda Pharmaceutical Company Limited | MAPK/ERK kinase inhibitors |
| US20080255160 | Dec 18, 2007 | Oct 16, 2008 | Qing Dong | Mapk/erk kinase inhibitors |
| WO2008000020A1 | Jun 27, 2007 | Jan 3, 2008 | Gary L Corino | Improved process |
| EP1894932A1 | Jun 10, 2005 | Mar 5, 2008 | Japan Tobacco, Inc. | 5-amino-2,4,7-trioxo-3,4,7,8-tetrahydro-2H-pyrido[2,3-d]pyrimidine derivatives and related compounds for the treatment of cancer |
| US20050222177 * | Jul 29, 2004 | Oct 6, 2005 | Irm Llc | Diseases with abnormal activation of the Abl, BCR-Abl, Bmx, CSK, TrkB, FGFR3, Fes, Lck, B-RAF, C-RAF, MKK6, alpha and beta SAPK2 kinases; antiproliferative; pyrrolo[2,3-d]pyrimidine-7-carboxylic acid [3-phenylcarbamoyl-phenyl]-amides and pyrrolo[3,2-c]pyridine analogs |
EMA Guideline on similar Biological Medicinal Products adopted
![]()

EMA Guideline on similar Biological Medicinal Products adopted
On 23 October, the CHMP adopted the revised Guideline on similar biological medicinal products. Get more details here.

Last year the “Draft Guideline on Similar Biological Medicinal Products” was published by EMA.
After agreement of the revised draft by the Biosimilar Medicinal Products Working Party and Biologics Working Party in July, the CHMP adopted and published the final Guideline on 23 October 2014. They summarized the outline of the document as follows:
“This Guideline outlines the general principles to be applied for similar biological medicinal products (also known as biosimilars) as referred to in Directive 2001/83/EC, as amended, where it is stated that ‘the general principles to be applied [for similar biological medicinal products] are addressed in a guideline taking into account the characteristics of the concerned biological medicinal product published by the Agency’.
This Guideline describes and addresses the application of the biosimilar approach, the choice of the reference product and the principles for establishing biosimilarity.

The scope of the guideline is to fulfil the requirement of section 4, Part II, Annex I to Directive 2001/83/EC, as amended, which states that ‘the general principles to be applied [for similar biological medicinal products] are addressed in a guideline taking into account the characteristics of the concerned biological medicinal product published by the Agency’.”
The date for coming into effect is 30 April 2015 (with the advice: After adoption by CHMP applicants may apply some or all provisions of this guideline in advance of this date.). The document replaces the Guideline on similar biological medicinal products (CHMP/437/04).
For further infromation please see the complete “Guideline on similar biological medicinal products“.
Aplaviroc, AK602, GSK-873140

Aplaviroc
4-(4-{[(3R)-1-butyl-3-[(R)-cyclohexylhydroxymethyl]-2,5-dioxo- 1,4,9-triazaspiro[5.5]undecan-9-yl]methyl}phenoxy)benzoic acid
for the treatment of HIV infection
461023-63-2 of hydrochloride
461443-59-4 (free base)
873140
AK-602
GW-873140
ONO-4128
ono…….innovator
| Ono Pharmaceutical Co., Ltd. |
| Identifiers | |
|---|---|
| CAS number | 461023-63-2 |
| ATC code | None |
| PubChem | CID 3001322 |
| ChemSpider | 2272720 |
| UNII | 98B425P30V |
| KEGG | D06557 |
| ChEMBL | CHEMBL1255794 |
| Chemical data | |
| Formula | C33H43N3O6 |
| Mol. mass | 577.711 g/mol |
Aplaviroc (INN, codenamed AK602 and GSK-873140) is a CCR5 entry inhibitor developed for the treatment of HIV infection.[1][2] It is developed by GlaxoSmithKline.
In October 2005, all studies of aplaviroc were discontinued due to liver toxicity concerns.[3][4] Some authors have claimed that evidence of poor efficacy may have contributed to termination of the drug’s development;[5] the ASCENT study, one of the discontinued trials, showed aplaviroc to be under-effective in many patients even at high concentrations.[6]
Aplaviroc hydrochloride, an orally-effective, long-acting chemokine CCR5 receptor antagonist, had been under development by Ono and GlaxoSmithKline for the treatment of HIV infection. In early 2006, the companies discontinued development of the antagonist based on reports of elevated liver function test values from clinical studies.
Originally developed at Ono, aplaviroc was licensed to GlaxoSmithKline in 2003 for development, manufacturing and marketing. GlaxoSmithKline also obtained rights to evaluate the agent in non-HIV conditions worldwide with the exception of Japan, South Korea and Taiwan.
A low-molecular-weight compound, aplaviroc prevents HIV viral infection by blocking the binding of the virus to the CCR5 receptor
……………….
WO 2002074770
0r
http://www.google.com/patents/EP1378510A1?cl=en
Reference example 3(3)
- (3R)-1-butyl-2,5-dioxo-3-((1R)-1-hydroxy-1-cyclohexyl)-1,4,9-triazaspiro[5.5]undecane • hydrochloride
-
[0136]

TLC:Rf 0.32 (butanol:acetic acid:water = 4:2:1);
NMR (CD3OD): δ 4.16 (d, J = 2.0 Hz, 1H), 3.95 (m, 1H), 3.70 (m, 1H), 3.52 (m, 1H), 3.37 (m, 1H), 3.28 (m, 1H), 3.22-3.13 (m, 2H), 2.46-1.93 (m, 6H), 1.80-1.64 (m, 5H), 1.48-1.15 (m, 6H), 1.02-0.87 (m, 5H);
Optical rotation:[α]D +1.22 (c 1.04, methanol, 26°C).

Example 9(54)
- (3R)-1-butyl-2,5-dioxo-3-((1R)-1-hydroxy-1-cyclohexylmethyl)-9-(4-(4-carboxyphenyloxy)phenylmethyl)-1,4,9-triazaspiro[5.5]undecane • hydrochloride
-
[0359]

TLC:Rf 0.43(chloroform:methanol = 5:1);
NMR (CD3OD):δ 8.05 (d, J = 9.0 Hz, 2H), 7.61 (d, J = 9.0 Hz, 2H), 7.19 (d, J = 9.0 Hz, 2H), 7.08 (d, J = 9.0 Hz, 2H), 4.38 (s, 2H), 4.17 (d, J = 2.1 Hz, 1H), 4.02 (m, 1H), 3.78 (m, 1H), 3.60-3.40 (m, 3H), 3.30-3.10 (m, 2H), 2.56-1.86 (m, 6H), 1.82-1.60 (m, 5H), 1.52-1.16 (m, 6H), 1.06-0.82 (m, 2H), 0.97 (t, J = 7.2 Hz, 3H).

………………….
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
Owing to the special properties of piperazines (increased solubility and H-bond acceptor capability etc.) it is often considered to be a privileged structure and therefore occurs widely, for instance in GlaxoSmithKlines investigational anti-HIV drug aplaviroc (4.37) which, despite being a promising CCR5 receptor antagonist, was discontinued due to hepatotoxicity concerns. In this compound the spirodiketopiperazine unit (4.35) was designed to mimic a type-1 β-turn (4.36) as present in G-protein coupled receptors (Figure 14) [117].
![[1860-5397-9-265-14]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-14.png?scale=2.0&max-width=1024&background=FFFFFF)
The synthesis of aplaviroc and its analogues can be accomplished via the use of an Ugi multicomponent reaction (Ugi-MCR) [118]. The procedure involved the condensation of piperidone 4.38 and butylamine (4.39) followed by reaction of the resulting imine with isocyanide 4.41 and interception of the nitrilium intermediate with the amino acid4.40 (Scheme 47) [119]. This sequence was completed by structural rearrangement and acid-mediated ring closure to produce the spirocyclic diketopiperazine 4.43. Following debenzylation this material was subjected to a reductive amination finally affording aplaviroc analogues (Scheme 47).
![[1860-5397-9-265-i47]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i47.png?scale=2.0&max-width=1024&background=FFFFFF)
- 117 Habashita, H.; Kokubo, M.; Hamano, S.; Hamanaka, N.; Toda, M.; Shibayama, S.; Tada, H.; Sagawa, K.; Fukushima, D.; Maeda, K.; Mitsuya, H. J. Med. Chem. 2006, 49, 4140–4152. doi:10.1021/jm060051s
- Dömling, A.; Huang, Y. Synthesis 2008, 2859–2883. doi:10.1055/s-0030-1257906
ref 118 - Nishizawa, R.; Nishiyama, T.; Hisaichi, K.; Matsunaga, N.; Minamoto, C.; Habashita, H.; Takaoka, Y.; Toda, M.; Shibayama, S.; Tada, H.; Sagawa, K.; Fukushima, D.; Maeda, K.; Mitsuya, H.Bioorg. Med. Chem. Lett. 2007, 17, 727–731. doi:10.1016/j.bmcl.2006.10.084
ref 119
| Patent | Submitted | Granted |
|---|---|---|
| Triazaspiro[5.5]undecane derivative and pharmaceutical composition comprising the same as active ingredient [US7262193] | 2005-09-29 | 2007-08-28 |
| Drugs containing triazaspiro[5.5]undecane derivatives as the active ingredient [US7285552] | 2004-06-03 | 2007-10-23 |
| Triazaspiro[5.5]undecane derivatives and drugs containing the same as the active ingredient [US7053090] | 2004-04-29 | 2006-05-30 |
| WO1998031364A1 * | Jan 20, 1998 | Jul 23, 1998 | Timothy Harrison | 3,3-disubstituted piperidines as modulators of chemokine receptor activity |
| WO2000014086A1 * | Jan 21, 1999 | Mar 16, 2000 | Kyowa Hakko Kogyo Kk | Chemokine receptor antagonists and methods of use therefor |
| WO2002074769A1 * | Mar 18, 2002 | Sep 26, 2002 | Kenji Maeda | Drugs containing triazaspiro[5.5]undecane derivatives as the active ingredient |
References
- Maeda, Kenji; Ogata, Hiromi; Harada, Shigeyoshi et al. (2004). “Determination of binding sites of a unique CCR5 inhibitor AK602 / ONO-4128/ GW873140 on human CCR5” (PDF). Conference on Retroviruses and Opportunistic Infections. Archived from the original on November 3, 2005.
- Nakata, Hirotomo; Maeda, Kenji; Miyakawa, Toshikazu et al. (February 2005). “Potent Anti-R5 Human Immunodeficiency Virus Type 1 Effects of a CCR5 Antagonist, AK602/ONO4128/GW873140, in a Novel Human Peripheral Blood Mononuclear Cell Nonobese Diabetic-SCID, Interleukin-2 Receptor γ-Chain-Knocked-Out AIDS Mouse Model”. Journal of Virology 79 (4): 2087–96.doi:10.1128/jvi.79.4.2087-2096.2005.
- “Aplaviroc (GSK-873,140)”. AIDSmeds.com. October 25, 2005. Retrieved September 5, 2008.[dead link]
- Nichols WG, Steel HM, Bonny T et al. (March 2008). “Hepatotoxicity Observed in Clinical Trials of Aplaviroc (GW873140)”.Antimicrobial Agents and Chemotherapy 52 (3): 858–65. doi:10.1128/aac.00821-07. PMC 2258506. PMID 18070967.
- Moyle, Graeme (December 19, 2006). “The Last Word on Aplaviroc: A CCR5 Antagonist With Poor Efficacy”. The Body.Archived from the original on 6 October 2008. Retrieved September 5, 2008.
- Currier, Judith; Lazzarin, Adriano; Sloan, Louis et al. (2008). “Antiviral activity and safety of aplaviroc with lamivudine/zidovudine in HIV-infected, therapy-naive patients: the ASCENT (CCR102881) study”. Antiviral Therapy (Lond.) 13 (2): 297–306.PMID 18505181.
Further reading
- Horster, S; Goebel, FD (April 2006). “Serious doubts on safety and efficacy of CCR5 antagonists: CCR5 antagonists teeter on a knife-edge”. Infection 34 (2): 110–13. doi:10.1007/s15010-006-6206-1. PMID 16703305.
FAMOTIDINE


FAMOTIDINE
76824-35-6
3-(2-Guanidinothiazol-4-ylmethylthio)-N-sulfamoylpropanamidine
MK-208
YM-11170
YM-1170
Histamine H2 Receptor Antagonists
Gastroesophageal Reflux Disease,
Agents forGastric Antisecretory Drugs (GERD)
Astellas Pharma (Innovaator)Launched – 1985

| Systematic (IUPAC) name | |
|---|---|
| 3-[({2-[(diaminomethylidene)amino]-1,3-thiazol-4-yl}methyl)sulfanyl]-N’-sulfamoylpropanimidamide | |
| Clinical data | |
| Trade names | Pepcid |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a687011 |
| Licence data | US FDA:link |
| Pregnancy cat. | |
| Legal status | |
| Routes | Oral (tablet form) |
| Pharmacokinetic data | |
| Bioavailability | 40-45% (Oral)[1] |
| Protein binding | 15-20%[1] |
| Metabolism | hepatic |
| Half-life | 2.5-3.5 hours[1] |
| Excretion | Renal (25-30% unchanged [Oral])[1] |
| Identifiers | |
| CAS number | 76824-35-6 |
| ATC code | A02BA03 |
| PubChem | CID 3325 |
| DrugBank | DB00927 |
| ChemSpider | 3208 |
| UNII | 5QZO15J2Z8 |
| Chemical data | |
| Formula | C8H15N7O2S3 |
| Mol. mass | 337.449 g/mol |
Famotidine is an H2-histamine antagonist that was first launched by Astellas Pharma (formerly Yamanouchi) in Japan in 1985 as an injectable for the treatment of upper gastrointestinal hemorrhage and for the treatment of Zollinger-Ellison syndrome. In 1986, the drug was launched pursuant to a collaboration between Merck Sharp & Dohme and Sigma-Tau for the oral prevention and treatment of gastroesophageal reflux disease (GERD).
Famotidine (INN) /fəˈmɒtɪdiːn/ is a histamine H2-receptor antagonist that inhibits stomach acid production, and it is commonly used in the treatment of peptic ulcer disease (PUD) and gastroesophageal reflux disease (GERD/GORD). It is commonly marketed byJohnson & Johnson/Merck under the trade names Pepcidine and Pepcid and by Astellas under the trade name Gaster. Unlikecimetidine, the first H2 antagonist, famotidine has no effect on the cytochrome P450 enzyme system, and does not appear to interact with other drugs.[2]
Medical use
Certain preparations of famotidine are available over the counter (OTC) in various countries. In the US 20 or more mg, sometimes in combination with a more traditional antacid, are available OTC. Larger doses still require a prescription.

Famotidine is given to surgery patients before operations to prevent postoperative nausea and to reduce the risk of aspiration pneumonitis. Famotidine is also given to some patients taking NSAIDs, to prevent peptic ulcers.[3] It serves as an alternative toproton-pump inhibitors.[4]
It is also given to dogs and cats with acid reflux.
Famotidine has also been used in combination with an H1 antagonist to treat and prevent urticaria caused by an acute allergic reaction.[5]
Side-effects
Side-effects are associated with famotidine use. In clinical trials, the most common adverse effects were headache, dizziness, andconstipation or diarrhea.[6]
History
Famotidine was developed by Yamanouchi Pharmaceutical Co.[7] It was licensed in the mid-80s by Merck & Co.[8] and is marketed by a joint venture between Merck and Johnson & Johnson. The imidazole-ring of cimetidine was replaced with a 2-guanidinothiazole ring. Famotidine proved to be 30 times more active than cimetidine.[citation needed]
It was first marketed in 1981. Pepcid RPD orally-disintegrating tablets were released in 1999. Generic preparations became available in 2001, e.g.Fluxid (Schwarz) or Quamatel (Gedeon Richter Ltd.).
In the United States and Canada, a product called Pepcid Complete, which combines famotidine with an antacid in a chewable tablet to quickly relieve the symptoms of excess stomach acid, is available. In the UK, this product is known as Pepcidtwo.
Famotidine suffers from poor bioavailability (50%), as it is poorly soluble in the low pH of the stomach. Famotidine used in combination with antacids promotes local delivery of these drugs to the receptor of the parietal cell wall. Therefore, researchers are developing innovative formulations of tablets, such as gastroretentive drug delivery systems. Such tablets are retained in the stomach for a longer period of time, thereby improving the bioavailability of drugs. Local delivery also increases bioavailability at the stomach wall receptor site and increases the efficacy of drugs to reduce acid secretion.[9]
Research
Famotidine has been investigated as an adjunct in treatment-resistant schizophrenia. In one trial it caused a 10% reduction in schizophrenic symptom severity in treatment-resistant patients.[10]
Famotidine is also indicated in the treatment of duodenal and benign gastric ulcers, for the prevention of relapse of duodenal ulceration, for the treatment of gastric mucosal lesions associated with acute gastritis and acute exacerbation of chronic gastritis, for the treatment of heartburn associated with acid indigestion and sour stomach, for the prevention of meal-induced heartburn, and for the treatment of reflux esophagitis due to GERD, including ulcerative disease as diagnosed by endoscopy. The drug is currently marketed in tablet, film-coated tablet, orally-disintegrating tablet, powder, lyophilized powder for injection solution and injectable formulations. The compound had been in development for the treatment of non-erosive reflux disease (NERD), however, Astellas Pharma discontinued development for this indication in 2007.

Famotidine was developed by replacing the imidazole ring of GlaxoSmithKline’s cimetidine with a 2-guanidinothiazole ring, a modification proven to increase the activity of the drug 30-fold. Famotidine competitively inhibits the action of histamine at the histamine H2 receptors of the parietal cells. It suppresses the normal secretion of acid by parietal cells and the meal-stimulated secretion of acid by two mechanisms: by blocking histamine released by enterochromaffin-like (ECL) cells in the stomach from binding to H2 receptors and stimulating acid secretion, and by reducing the effect that other compounds (such as gastrin, pentagastrin, caffeine, insulin and acetylcholine) have on the promotion of acid secretion due to H2 receptor blockade.
Famotidine was originally developed at Astellas Pharma. It was subsequently licensed in the U.S. to Merck & Co., known outside the U.S. and Canada as Merck Sharp & Dohme. In 2007, Salix acquired the U.S. rights to famotidine oral solution (Pepcid[R]) for the treatment of GERD and peptic ulcer. Sigma-Tau holds rights to the drug and is responsible for marketing activities in Italy. Famotidine is sold in over 110 countries worldwide, including France, Germany, Italy, Japan, the U.S. and the U.K.

……………………………..
US 4283408
http://www.google.co.in/patents/US4283408
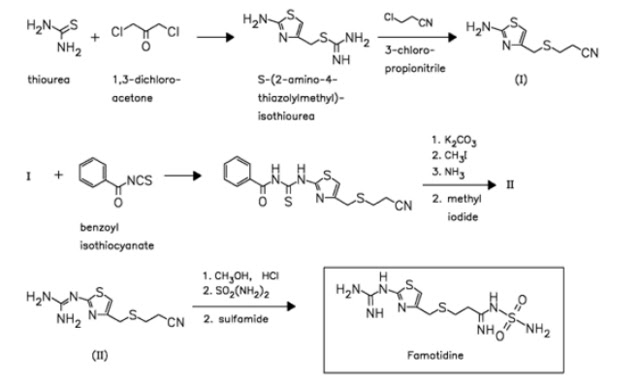
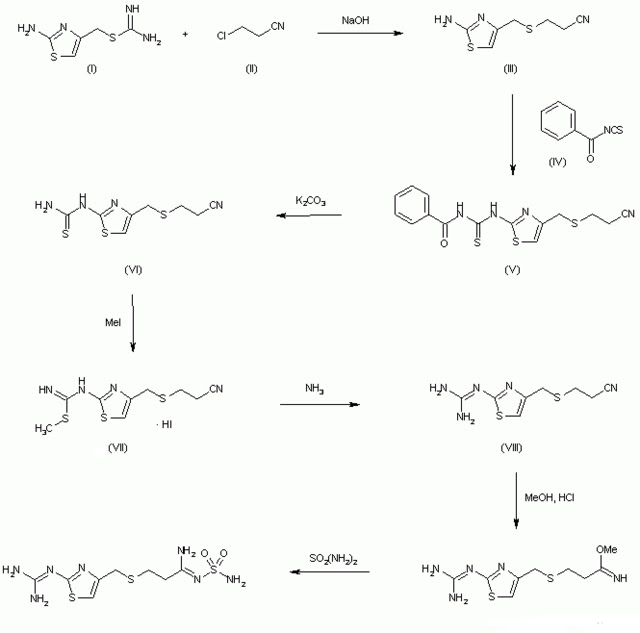
The reaction ot S-(2-aminothiazol-4-ylmethyl)isothiourea (I) with 3-chloropropionitrile (II) by means of NaOH in ethanol – water gives 3-(2-aminothiazol-4-ylmethylthio)propionitrile (III), which is condensed with benzoyl isothiocyanate (IV) in refluxing acetone to afford 3-[2-(3-benzoylthioureido)thiazol-4-ylmethylthio]propionitrile (V). The hydrolysis of (V) with K2CO3 in acetone – methanol – water yields 3-(2-thioureidothiazonl-4-ylmethylthio)propionitrile (VI), which by methylation with methyl iodide in refluxing ethanol is converted into 3-[2-(S-methylisothioureido)thiazol-4-ylmethylthio]propionitrile hydroiodide (VII). The reaction of (VII) with NH3 and NH4Cl in methanol at 90 C in a pressure vessel affords 3-(2-guanidinothiazol-4-ylmethylthio)propionitrile (VIII), which by partial alcoholysis with methanol by means of dry HCl in CHCl3 is converted into methyl 3-(2-guanidinothiazol-4-ylmethylthio)propionimidate (IX). Finally, this compound is treated with sulfamide in refluxing methanol.
Drugs Fut 1983, 8, 1, 14
US 4283408
DOS 2 951 675 (Yamanouchi; appl. 21.12.1979; J-prior. 2.8.1979).
DOS 3 008 056 (Yamanouchi; appl. 3.3.1980; J-prior. 6.3.1979, 23.6.1979).
GB 2 052 478 (Yamanouchi; appl. 6.3.1980; J-prior. 6.3.1979, 23.6.1979).
GB 2 055 800 (Yamanouchi; appl. 20.12.1979; J-prior. 2.8.1979).
synthesis of S-[2-aminothiazol-4-ylmethyl]isothiourea:
Spragne, J.M.; Lund, A.H.; Ziegler, C.: J. Am. Chem. Soc. (JACSAT) 68, 2155 (1946).


FT IR OF FAMOTIDINE

http://link.springer.com/article/10.1007%2Fs00216-011-5599-6

[1H,13C] 2D NMR Spectrum
………………………..
DSC OF FAMOTIDINE
WILL BE ADDED
……………………………
…………………..
UV – range
 |
||||
| Conditions : Concentration – 1 mg / 100 ml | ||||
| The solvent designation graphics | Methanol |
Water |
0.1М HCl |
0.1M NaOH |
|---|---|---|---|---|
| Maximum absorption | 287 nm | – | 265 nm | 286 nm |
 |
465 | – | 309 | 440 |
| e | 15700 | – | 10400 | 14850 |
FIG WILL BE ADDED
IR – spectrum
| Wavelength (μm) |
|---|
 |
| Wave number (cm -1 ) |
…………..
Synthesis pathway
| Synthesis of a) |
|---|
   |
Trade names
| Country | Trade name | Manufacturer |
|---|---|---|
| Germany | Fadul | Hexal |
| Famobeta | betapharm | |
| Famonerton | Dolorgiet | |
| Pepdul | TEOFARMA | |
| various generic drugs | ||
| France | Peptsidak | McNeil |
| Peptsidduo | McNeil | |
| Pepdin | Merck Sharp & Dohme-Chibret | |
| Great Britain | Peptsid | Merck Sharp & Dohme |
| Italy | Famoudou | Sigma-Tau |
| Gastridin | Merck Sharp & Dohme | |
| Motiaks | Neopharmed | |
| Japan | Gaster | Astellas |
| United States | Peptsid | Merck, 1986 |
| – “- | Johnson & Johnson; Merck | |
| Ukraine | Ulfamid | Krka, dd, Novo mesto, Slovenia |
| Kvamatel | JSC “Gedeon Richter”, Hungary | |
| Famasan | ABM. MED. CA AT Prague, Czech Republic | |
| FamodinGeksal | Salyutas Pharma GmbH, Germany, venture hexane AG, Germany | |
| various generic drugs | ||
Formulations
-
ampoule 10 mg, 20 mg;
-
Tablets coated with 10 mg, 20 mg, 40 mg;
-
oral suspension, 40 mg / 5 ml;
-
2% powder, 10%;
-
10 mg tablets, 20 mg;
-
vials (lyophilisate) 20 mg
Reference for above
-
DOS 2,951,675 (Yamanouchi; appl. 21.12.1979; J-prior. 2.8.1979).
-
DOS 3,008,056 (Yamanouchi; appl. 3.3.1980; J-prior. 6.3.1979, 23.6.1979).
-
GB 2052478 (Yamanouchi; appl. 6.3.1980; J-prior. 6.3.1979, 23.6.1979).
-
GB 2055800 (Yamanouchi; appl. 20.12.1979; J-prior. 2.8.1979).
-
US 4,283,408 (Yamanouchi; 11.8.1981; J-prior. 2.8.1979).
References
- Truven Health Analytics, Inc. DRUGDEX® System (Internet) [cited 2013 Oct 10]. Greenwood Village, CO: Thomsen Healthcare; 2013.
- Humphries TJ, Merritt GJ (August 1999). “Review article: drug interactions with agents used to treat acid-related diseases” (pdf). Aliment. Pharmacol. Ther. 13 (Suppl 3): 18–26.doi:10.1046/j.1365-2036.1999.00021.x. PMID 10491725.
- “Horizon Pharma, Inc. Announces FDA Approval of DUEXIS(R) for the Relief of the Signs and Symptoms of Rheumatoid Arthritis and Osteoarthritis and to Decrease the Risk of Developing Upper Gastrointestinal Ulcers” (Press release). Horizon Pharma. 2011-04-25.
- Brauser D (Jul 13, 2009). “Famotidine May Prevent Peptic Ulcers, Esophagitis in Patients Taking Low-Dose Aspirin”. Medscape.
- Fogg TB, Semple D (29 November 2007). “Combination therapy with H2 and H1 antihistamines in acute, non compromising allergic reactions”. BestBets. Manchester, England: Manchester Royal Infirmary. Retrieved 26 April 2011.
- “Pepcid Side Effects & Drug Interactions”. RxList.com. 2008. Retrieved 2008-07-31.
- US patent 4283408, HIRATA YASUFUMI; YANAGISAWA ISAO; ISHII YOSHIO; TSUKAMOTO SHINICHI; ITO NORIKI; ISOMURA YASUO; TAKEDA MASAAKI, “Guanidinothiazole compounds, process for preparation and gastric inhibiting compositions containing them”, issued 1981-08-11
- “Sankyo Pharma”. Skyscape Mediwire. 2002. Retrieved 2009-10-30.[dead link]
- “Formulation and Evaluation of Gastroretentive Floating Tablets of Famotidine”. Farmavita.Net. 2008. Retrieved 2009-01-30.
- Meskanen, K; Ekelund, H; Laitinen, J; Neuvonen, PJ; Haukka, J; Panula, P; Ekelund, J (August 2013). “A randomized clinical trial of histamine 2 receptor antagonism in treatment-resistant schizophrenia.”. Journal of Clinical Psychopharmacology 33 (4): 472–478. doi:10.1097/JCP.0b013e3182970490. PMID 23764683.
Esoxybutynin, (S)-Oxybutynin

![Chemical structure for Esoxybutynin [INN]](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?t=l&deposited=t&sid=135253519)
Drug name, 药物名称….. Esoxybutynin, (S)-Oxybutynin

| CAS No. | 119618-22-3 |
| Chemical Name: | (S)-Oxybutynin |
| Synonyms: | Esoxybutynin;(S)-Oxybutynin;(S)-OXYBUTYNIN HCL;(S)-OXYBUTYNIN CHLORIDE;(S)-OXYBUTYNIN HYDROCHLORIDE;(S)-Hydroxycyclohexylphenylacetic acid 4-(diethylamino)-2-butynyl ester;(S)-CYCLOHEXYL-HYDROXY-PHENYL-ACETIC ACID 4-DIETHYLAMINO-BUT-2-YNYL ESTER;(αS)-α-Cyclohexyl-α-hydroxybenzeneacetic acid 4-(diethylamino)-2-butin-1-yl ester;Benzeneacetic acid, a-cyclohexyl-a-hydroxy-, 4-(diethylamino)-2-butynyl ester, (S)-;(S)-α-Phenylcyclohexaneglycolic Acid 4-(Diethylamino)-2-butynyl Ester, Hydrochloride |
| CBNumber: | CB1746039 |
| Molecular Formula: | C22H31NO3 |
| Formula Weight: | 357.49 |
Oxybutynin and its derivatives are applicable as a bronchodilator or a remedy for pollakisuria. Also, oxybutynin exerts a direct antispasmodic effect on various forms of smooth muscle, mainly by inhibiting the action of acetylcholine on smooth muscle as an anti-cholinergic drug and the like. Oxybutynin is marketed in hydrochloride form. Oxybutynin known as [α-cyclohexyl-hydroxy-benzeneaceticacid- 4-(diethyl amino)-2-butynyl ester] he US Patent No. 3,176,019 (‘019) discloses about 4-amino-2-butynol esters and their derivatives, particularly about oxybutynin hydrochloride. It also reveals about the synthesis of oxybutynin, wherein, the methyl phenyl cyclohexyl glycolate is reacted with 4-diethylamino-2-butynylacetate in presence of base to yield oxybutynin followed by further workup. Further, it is treated with 2N HCl solution to form hydrochloride salt. It is recrystallised by employing ethyl acetate or water to obtain pure oxybutynin hydrochloride. Further, the US Patent ‘019 unveils about the reaction of propargyl-2-cyclohexyl-2-hydroxy-2-phenyl acetate, /^-formaldehyde and diethyl amine in dry dioxane to obtain crude product of oxybutynin. The dry hydrogen chloride gas is passed through the ether solution of oxybutynin to yield the oxybutynin chloride as precipitate.
According to the prior art process oxybutynin is obtained as oil, which contains lot of impurities, therefore, it needs to purify high vacuum distillation. Also, the resultant oxybutynin base is having a low melting point, which may decompose during high vacuum distillation. Further, the existence of any polymorphism in oxybutynin is not disclosed in prior arts. In light of the foregoing, a need exists in the art for inventing a new form and the process thereof. Objects and Summary of the Invention
It is a principal object of the present invention is to provide a novel crystalline oxybutynin base in a solid state having improved quality.
Another object of the present invention is to provide a process for the preparation of novel crystalline oxybutynin base as a solid state. Further, object of the present invention is to provide a process for preparing an acid addition salt of oxybutynin employing crystalline oxybutynin base
In accordance with one preferred embodiment of the present invention, there is provided a crystalline oxybutynin base characterized by using different analytical tools including X-ray powder diffraction pattern, Thermo Gravimetric Analysis (TGA), and Differential Scanning Calorimetry (DSC).
Oxybutynin is used therapeutically in the treatment of intestinal hypermotility and in the treatment of urinary incontinence due to detrusor instability. Oxybutynin is sold for this purpose under the trade name of Ditropan®. Chemical names for oxybutynin are 4- (diethylamino)-2-butynyl-α-cyclohexyl-α-hydroxy benzeneacetate, and 4-(diethylamino)-2- butynylphenylcyclohexyl-glycolate. It is a racemic mixture of the R-enantiomer, R- oxybutynin, and the S-enantiomer, S-oxybutynin.
Use of the S-enantiomer of oxybutynin, S-oxybutynin, for the treatment of urinary incontinence has been described in U.S. Patent Numbers 5,532,278, and 5,736,577. The structure of S-oxybutynin (Registry Number 1 19618-22-3) is shown in formula I. S- oxybutynin is not commercially available at the present time.
Administration of racemic oxybutynin may result in a number of adverse effects. These adverse effects include, but are not limited to, xerostomia, mydriasis, drowsiness, nausea, constipation, palpitations and tachycardia. The amelioration of cardiovascular side effects of racemic oxybutynin, such as tachycardia and palpitations, is of particular therapeutic value.
The synthesis of S-oxybutynin has been described in the literature by Kacher et al, J. Pharmacol. Exp. Ther., 247, 867-872 (1988). An improved synthetic method is disclosed in copending U.S. patent application, serial number 09/21 1,646, the contents of which are incorporated in their entirety. In this method, an activated derivative of cyclohexylphenylglycolic acid (CHPGA), the mixed anhydride I, is prepared.
isobutylchloroforrnate
The mixed anhydride I is coupled with the propargyl alcohol derivative 4-N,N-diethylamino butynol (4-N,N-DEB)( III where R1 is -CH2R2; R2 is -ΝR3R4; and R3 and R4 are each ethyl.) Reaction of the optically active mixed anhydride with 4-NN-DEB produces a single enantiomer of oxybutynin, in this case, (S)-4-diethylamino-2- butynylphenylcyclohexylglycolate.
Improved syntheses of starting material CHPGA have been described in two copending U.S. Patent Applications, Serial Numbers 09/050,825 and 09/050,832. The contents of both are incorporated by reference in their entirety. In the first (09/050,825), phenylglyoxylic acid or cyclohexylglyoxylic acid is condensed with a single enantiomer of a cyclic vicinal aminoalcohol to form an ester of the phenylglyoxylic acid or the cyclohexylglyoxylic acid. The ester is reacted with an appropriate Grignard reagent to provide an α-cyclohexylphenylglycolate ester. A single diastereomer of the product ester is separated from the reaction mixture, and hydrolyzed to provide S-α- cyclohexylphenylglycolic acid (S-CHPGA). The second (09/050,832) discloses an alternate stereoselective process for preparing CHPGA. A substituted acetaldehyde is condensed with mandelic acid to provide a 5-phenyl-l,3-dioxolan-4-one, which is subsequently reacted with cyclohexanone to provide a 5-(l-hydroxy cyclohexyl)-5-phenyl-l,3-dioxolan-4-one. The product is dehydrated to a 5-(l-cyclohexenyl)-5-phenyl-l,3-dioxolan-4-one, hydrolyzed and reduced to CHPGA.
……………………………..
SYNTHESIS

Racemic cyclohexylphenyl glycolic acid (CHPGA) (I) is dissolved with (L)-tyrosine methyl ester (II) in refluxing acetonitrile/water to yield a mixture of diastereomeric salts, which is resolved by crystallization to afford the desired diastereomeric salt [(S)-CHPGA-(L)-TME] (III). Finally, the hydrolysis of salt (III) with HCl or H2SO4 at 40-50篊 in toluene yields the enantiomer (IV). Alternatively intermediate (IV) can be obtained as follows: acetalization of (S)-mandelic acid (V) with pivaldehyde (VI) in pentane and catalytic TfOH provides derivative (VII), which is then treated with LHMDS and then condensed with cyclohexanone (VIII) in THF to furnish aldol adduct (IX). Elimination of tertiary alcohol in (IX) with SOCl2 and pyridine in THF gives derivative (X), which is then converted into intermediate (IV) either by first hydrolysis of lactone (X) with KOH in MeOH and subsequent hydrogenation of the obtained derivative (XI) over Pd/C in MeOH, or by first hydrogenation of (X) over Pd/C in MeOH to give (XII), followed by hydrolysis with KOH in MeOH. On turn, derivative (XII) can alternatively be synthesized by treatment of derivative (VII) with LHMDS, followed by reaction with 3-bromocyclohexene (XIII) in THF to provide derivative (XIV), which is then hydrogenated over Pd/C.
US 5973182; US 6140529; WO 0023414
……………………………………………………………

The desired product is finally obtained by first formation of a mixed anhydride (XVI) by reaction of the cyclohexylphenyl glycolic acid (IV) with isobutylchloroformate (XV) in cyclohexane in the presence of Et3N, followed by treatment with 4-N,N-diethylamino butynol (XVII) (obtained on turn from reaction of propargyl alcohol (XVIII) with diethylamine (XIX) in the presence of paraformaldehyde and CuCl.
J Org Chem 2000,65(19),6283
Racemic cyclohexylphenyl glycolic acid (CHPGA) (I) is dissolved with (L)-tyrosine methyl ester (II) in refluxing acetonitrile/water to yield a mixture of diastereomeric salts, which is resolved by crystallization to afford the desired diastereomeric salt [(S)-CHPGA-(L)-TME] (III). Finally, the hydrolysis of salt (III) with HCl or H2SO4 at 40-50篊 in toluene yields the enantiomer (IV). Alternatively intermediate (IV) can be obtained as follows: acetalization of (S)-mandelic acid (V) with pivaldehyde (VI) in pentane and catalytic TfOH provides derivative (VII), which is then treated with LHMDS and then condensed with cyclohexanone (VIII) in THF to furnish aldol adduct (IX). Elimination of tertiary alcohol in (IX) with SOCl2 and pyridine in THF gives derivative (X), which is then converted into intermediate (IV) either by first hydrolysis of lactone (X) with KOH in MeOH and subsequent hydrogenation of the obtained derivative (XI) over Pd/C in MeOH, or by first hydrogenation of (X) over Pd/C in MeOH to give (XII), followed by hydrolysis with KOH in MeOH. On turn, derivative (XII) can alternatively be synthesized by treatment of derivative (VII) with LHMDS, followed by reaction with 3-bromocyclohexene (XIII) in THF to provide derivative (XIV), which is then hydrogenated over Pd/C.
CLIP

Tetrahedron Lett 2002,43(48),8647
The catalytic enantioselective cyanosilylation of the ketone (I) by means of Tms-CN catalyzed by gadolinium isopropoxide and the chiral ligand (II) in THF/propionitrile gives the silylated cyanohydrin (III), which is reduced by means of DIBAL in toluene to yield the carbaldehyde (IV). The desilylation of (IV) by means of HCl in aqueous THF affords the hydroxyaldehyde (V), which is finally oxidized by means of NaClO2 in tert-butanol/water to provide the target (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid intermediate (VI) (see Scheme no. 23604001a, intermediate (IV)).
PATENT
http://www.google.com/patents/WO2009122429A2?cl=en
Example-1 Preparation of 4-diethylamino-2-butyne-ol
A mixture of para formaldehyde (105.Og), N,N-diethyl amine(300g) and copper(II) acetate (7.5g) in 1,4 dioxane (900ml) was heated to 60-65° C. After 1.5 h, 2-propyne-l-ol (150g, 2.7 moles) was added and the mixture was heated at 90-95° C. after 2 hrs; excess solvent, 1,4 dioxane, evaporated at reduced pressure to afford 315g
(84%) of the product as an oil. Example-2
Preparation of diethylamino-2-butvnylacetate
A mixture of 4-diethylamino-2-butyne-l-ol (30Og), acetic acid (600ml); acetic anhydride (300ml) and con.sulphuric acid (15ml) was heated to 65-70° C. After 2hrs.of maintenance excess solvent mixture was evaporated at reduced pressure. The residue was cooled and poured in a mixture of dichloromethane (1800ml) and DM water (3000ml).The reaction mass was saturated with sodium bicarbonate (300g) solid slowly controlling effervescences. The organic layer was separated and washed with 2% sodium bicarbonate and 1% EDTA solution to afford 318g (81%) of product as oil.
Example-3
Preparation of 4-diethylamino-2-butvnyl phenyl cvclohexyl alveolate hydrochloride (Oxybutynin Hydrochloride)
A mixture of 150g of methyl phenyl cyclohexyl glycolate, 133g of 4- diethylamino-2-butynyl acetate was dissolved in 1.8 ltr of n-heptane. The solution was added with 1.2 g of sodium methoxide. The solution was heated with stirring to a temperature of 95-100° C and distillate was collected. After 30min of maintenance at 95-100° C, the solution was cooled to 65-70° C under nitrogen. The solution was added with 3.24 g of sodium methoxide. The solution was heated with stirring to a temperature of 95-100° C and distillate was collected. After 1 hr. maintenance at 95- 100° C, reaction mass cooled to room temperature, washed with water. n-Heptane layer was separated and added 300 ml of 2N Hydrochloric acid to give oxybutynin hydrochloride. The crude was recrystallised from ethyl acetate.
Example-4 Preparation of Oxybutvnin base
A mixture of 150g of methyl phenyl cyclohexyl glycolate, 133g of 4- diethylamino-2-butynyl acetate was dissolved in 1.8 ltr of n-heptane. The solution was added with 1.2 g of sodium methoxide. The solution was heated with stirring to a temperature of 95-100° C and distillate was collected. After 30min of maintenance at 95-100° C, the solution was cooled to 65-70° C under nitrogen. The solution was added with 3.24 g of sodium methoxide. The solution was heated with stirring to a temperature of 95-100° C and distillate was collected. After 1 hr. maintenance at 95-
100° C, reaction mass cooled to room temperature, washed with ‘water. n-Heptane layer was separated, concentrated under reduced pressure to give residue. n-Pentane (250ml) was added to the residue and stirred under nitrogen atmosphere at 25-30° C. The solid product was filtered and washed with chilled n-pentane. Wet cake was dried at 40-42° C. Dry weight = 160.O g
Example-5 Preparation of Oxybutvnin (Base)
Oxybutynin chloride (lOOgm) was treated with DM water (500ml) at 25-30° C and heated to 40-45° C to observe clear solution. n-Heptane (500ml) was added to the solution and adjusted the pH of the mass to 10.0-11.0 using 5% sodium hydroxide solution at 20-25° C. Layers obtained were separated and aqueous layer was extracted with heptane. Organic layers were combined and concentrated under vacuum at 40- 45° C to, give residue. n-Pentane (250ml) was added to the residue and stirred under nitrogen atmosphere at 25-30° C. The solid product was filtered and washed with chilled n-pentane. Wet cake was dried at 40-42° C. Dry weight = 85.0 gm
PATENT
http://www.google.com/patents/US3176019
Example XIX 4-diethylamino-2-butynyl phenylcyclohexylglycolate hydrochl0ride.-A mixture of 394.2 g. of methyl phenylcyclohexylglycolate, 293.1 g. of 4-diethylamino-2-butynyl acetate was dissolved with Warming in 2.6 l. of n-heptane. The solution was heated with stirring to a temperature of 60-70 C. and 8.0 g. of sodium methoxide were added. The temperature of the mixture was then raised until the solvent began to distill. Distillation was continued at a gradual rate and aliquots of the distillate were successively collected and analyzed for the presence of methyl acetate by measurement of the refractive index. The reaction was completed when methyl acetate no longer distilled, and the refractive index observed was that of pure heptane (11 1.3855). About three and one-half hours were required for the reaction to be completed. The reaction mixture was then allowed to cool to room temperature, washed with Water, and extracted with four ml. portions of 2 N hydrochloric acid. The aqueous extracts Were combined and stirred at room temperature to permit crystallization of the hydrochloride salt of the desired product. Crystallization was completed by cooling the slurry in an ice bath, and the product was collected by filtration, pressed dry, and recrystallized from 750 ml. of water. Yield of pure crystalline material, 323 g.
PATENT
http://www.google.com/patents/EP1185498A2?cl=en
Background of the Invention Cyclohexylphenyl glycolic acid (also referred to herein as “CHPGA”) is used as a starting material for manufacturing compounds that have important biological and therapeutic activities. Such compounds include, for example, oxphencyclimine, oxyphenonium bromide, oxypyrronium bromide, oxysonium iodide, oxybutynin (4- diethylamino-2-butynyl phenylcyclohexylglycolate) and its metabolites, such as desethyloxybutynin (4-ethylamino-2-butynyl phenylcyclohexylglycolate). The important relation between stereochemistry and biological activity is well known. For example, the (S)-enantiomers of oxybutynin and desethyloxybutynin have been shown to provide a superior therapy in treating urinary incontinence, as disclosed in U.S. Patent Nos. 5,532,278 and 5,677,346. The (R) enantiomer of oxybutynin has also been suggested to be a useful drug candidate. [Noronha-Blob et al., J. Pharmacol. Exp. Ther. 256, 562-567 (1991)]. Racemic CHPGA is generally prepared by one of two methods: (1) selective hydrogenation of phenyl mandelic acid or of phenyl mandelate esters, as shown in Scheme 1; or (2) cyclohexyl magnesium halide addition to phenylglyoxylate as shown in Scheme 2. Scheme 1.
R is hydrogen or lower alkyl.
Scheme 2.
Asymmetric synthesis of individual enantiomers of CHPGA has been approached along the lines of Scheme 2, by Grignard addition to a chiral auxiliary ester of glyoxylic acid to give a diastereomeric mixture of esters. In addition, multiple step asymmetric synthesis of (R)-CHPGA from fDJ-arabinose using Grignard reagents has been reported. In general, simple primary alkyl or phenyl Grignard (or alkyllithium) reagents are used for the addition, and the addition of inorganic salts (e.g. ZnCl2) appears to increase the diastereoselectivity of the products.
As outlined in Scheme 3 below, the simple chiral ester wherein R* is the residue of a chiral alcohol, can be directly converted to chiral drugs or drug candidates by trans-esterification (R’=acetate), or hydrolyzed to yield chiral CHPGA (R’=H).
Scheme 3
esterification
(S) or (R)-Oxybutynin
(S) or (R)-CYLOHEXYLPHENYL GLYCOLIC ACID VIA RESOLUTION The resolution process of the present invention provides an inexpensive and efficient method for preparing a single enantiomer from racemic CHPGA via the formation of the diastereomeric salt with (L) or (D) -tyrosine methyl ester, also referred to herein as “(Z) or (D)-TME”. The process consists of three parts, which are depicted and described below: Part 1: Preparation of (S)-CHPGA-(Z)-TME diastereomeric salt or (R)-CHPGA-(D)-TME diastereomeric salt; Part 2:
Preparation of (S) or (R) CHPGA; and Part 3 – Recovery of (L) or (D)-tyrosine methyl ester. The ability to recover the resolving agent in high yield is an advantageous feature of the process of the invention. It greatly reduces cost by allowing recycling of the resolving agent. For ease in understanding, the diastereomeric salt, (<S)-CHPGA-(E)-TME, and the pure enantiomer (S)-CHPGA are depicted in the reactions below. However, the (R) enantiomeric series could instead be depicted and is similarly produced using the opposite enantiomer of TME.
Part 1 : Preparation of (5VCHPGA-(XVTyrosine Methyl Ester Diastereomer Salt
* ( )-TME
(S, R)-CHPGA (S)-CHPGA – (J)-TME (MW= 234.3) (MW = 429.5)
For use in the process of Part 1, the racemic starting material, (S, R)- cyclohexylphenyl glycolic acid (CHPGA) can be prepared by the process described above, i.e. (1) selective hydrogenation of phenyl mandelic acid or of phenyl mandelate esters or (2) cyclohexyl magnesium halide addition to phenylglyoxylate. Mandelic acid and phenylglyoxylic acid, also known as benzoylformic acid, are commercially available. Phenyl mandelic acid may be prepared by Grignard addition of phenyl magnesium bromide to diethyl oxalate followed by hydrolysis. The (L) enantiomer of tyrosine methyl ester is also readily available from commercial sources, as is (Z))-tyrosine, which can then be esterified to produce (_9)-tyrosine methyl ester using conventional techniques, such as acid-catalyzed esterification with methanol. The diastereomer of the present process is produced by dissolving racemic
CHPGA and an appropriate amount of an enantiomer of tyrosine methyl ester in a suitable solvent and then bringing about the insolubilization of one diastereomer. For example, racemic CHPGA and about 0.5 molar equivalents of (Z)-tyrosine methyl ester or (Z))-tyrosine methyl ester can be dissolved in a mixture of acetonitrile and water. When the solvent is about 10 wt % water in acetonitrile, solution may be achieved by heating, preferably by heating to reflux (approximately 78° C). After heating the solution for a sufficient time to achieve complete dissolution, usually about 5 minutes at reflux, followed by cooling, preferably to about 0-5° C, the diastereomeric salt (S)-CHPGA – (E)-TME or (R)-CHPGA – (£>)- TME, depending on the TME enantiomer used, crystallizes from solution. Better yields are obtained when the cooling temperature is maintained until crystallization of the diastereomer salt is complete, typically a period of about four hours. The salt crystals are then separated from the solution, for example by filtration. The crystalline product may be washed with solvent and dried. When the solvent is water/acetonitrile, drying under vacuum at about 40-50° C is effective. The mother liquor stream may be saved for later racemization and recovery of residual CHPGA. Racemization may be effected with aqueous mineral acids, particularly aqueous sulfuric acid in ethanol. Part 2: Preparation of (S.-CHPGA
(S)-CHPGA – (Z)-TME (S)-CHPGA
(MW = 429.5) (MW= 234.3)
In Part 2, the CHPGA enantiomer produced, (S) or (R)-CHPGA, is liberated from the diastereomeric salt. For the preparation of (S)-CHPGA, the (S)-CHPGA-(E)- TME salt from Part 1 is added to and dissolved to form a solution which is about 15 wt % substrate in toluene. The solution is treated with an excess of dilute mineral acid, such as 1.1 equivalents of 0.5 M HC1 or H2SO4. Upon dissolution of the diastereomeric salt, essentially all the TME enantiomer is converted to the hydrochloride salt. The diastereomeric salt mixture may be heated to about 40-50° C for about 10 minutes to facilitate dissolution of the solids. A phase split yields an aqueous solution containing (Z)-TME-HCl and an organic solution of (S)-CHPGA in toluene. The aqueous phase is separated from the organic solution and saved for recovery of the tryrosine methyl ester in Step 3 below. A common method of separation, which may be used throughout the processes described herein, is gravitational settling followed by drainage of the aqueous phase through a tap in the bottom of the reaction vessel.
The toluene organic phase containing (S)-CHPGA may be washed a second time with mineral acid, as specified above, and heated. The organic phase and aqueous phase are then separated, and the aqueous phase is discarded along with the rag layer, i.e. the layer separating the two phases. The retained toluene organic phase is then preferably concentrated, typically by vacuum distillation, to a weight that is about 2.1 to 2.3 times the weight of the diastereomeric salt originally present, followed by gradual cooling to 0-5° C to initiate crystallization of the single (S) enantiomer of CHPGA, as indicated by the formation of a thick slurry. The slurry is cooled for at least an hour to ensure that crystallization is complete, then filtered to isolate (S)-CHPGA. The (S)-CHPGA cake is then dried under vacuum while heating to a temperature of about (40-45° C).
Part 3 : Recovery of (X -Tyrosine Methyl Ester The aqueous phase containing (Z)-TME-HCl or (D)-TME-HCl saved from
Part 2 is cooled, preferably to about 0-5° C. While maintaining the cooling temperature, the aqueous solution is titrated with 0.5M NaOH to a pH of approximately 9.0. Typically, a thin slurry will form as the TME enantiomer precipitates. The TME enantiomer is isolated by filtration, washing with deionized water, and drying under vacuum at a temperature of about (40-50° C).
The resolution process of the present invention set forth above is illustrated by, but not limited to, the following example:
Example 1 Part 1 : Preparation of (S)-CHPGA-(E -Tyrosine Methyl Ester Diastereomer Salt A 2-liter reactor was charged with 100.0 g racemic CHPGA, 41.7 g (L)-
TME (0.5 equiv.), 549.2 g CH3CN, and 54.8 g deionized water. The reaction mixture was heated to reflux at approximately 78° C for about 5 min. The solution was then cooled to a temperature between 0-5° C over a period of 2 hours and remained cooling (0-5 ° C) for about 2 hours. The solution was filtered to isolate the (S)-CHPGA-(Z)-TME diastereomeric salt, and the salt cake was washed with 130 g chilled ( 0-5° C) CH3CN. The salt cake was dried in vacuo at 40-50° C , and the residual solvent remaining in the cake was < 0.5%. Yield = 77.1 g (42.1 mole %); ee > 99.0% (S).
Part 2: Preparation of .S.-CHPGA A 1000 mL reactor was charged with 77. 1 g (S)-CHPGA-(E)-TME from
Part 1, 447.0 g toluene, 339.2 g 0.5M HC1 (1.1 equiv.) and heated to 40-50°C while stirring until the solids dissolved (about 10 min). While maintaining the temperature at 40-50° C, the organic and aqueous phases separated after about 10 minutes. The phases were divided, and the aqueous (bottom) phase containing (L)- TME-HC1 was saved for recovery in Part 3 below. Approximately 370 g aqueous phase was recovered.
To the toluene organic phase an additional 169.6 g 0.5M HC1 (0.6 equiv.) were added, and the solution was heated to a temperature between 40-50° C while stirring for about 10 minutes. The toluene and aqueous phases were allowed to separate (~ 10 min.), while maintaining the temperature between 40-50° C. The phases were divided, and the aqueous (bottom) phase and rag layer were discarded. The organic phase was concentrated by vacuum distillation to a final weight of 168.0 g, then cooled to 0-5 °C over a period of about one hour during which time a thick slurry formed spontaneously. Agitation was adjusted as necessary. The slurry was cooled at 0-5 °C for an additional one hour. The slurry was filtered to recover the (S)-CHPGA. The (S)-CHPGA filter cake was dried in vacuo at 40-45° C , and the residual solvent remaining in the cake was < 0.2%. Yield = 35.8 g (85 mole %); ee > 99.0%; chemical purity (% HPLC area) > 99.0%.
Part 3: Recovery of (Z)-Tyrosine Methyl Ester
A 2-liter vessel was charged with the aqueous phase saved from Part 2 (370 g). The solution was cooled to 0-5 °C, and the cooling temperature was maintained while titrating with 0.5 M NaOH to a pH of 9.0 ±0.5 over approximately 30 min. A thin slurry formed as (Z)-TME precipitated. The slurry was filtered, and the (L)-
TME cake was washed with 154 g deionized water. The cake was dried in vacuo at 40-50°C , and the residual solvent remaining in the cake was < 1.0%. Yield = 30.5 g (E)-TME (87 mole %).
(S) OR fRVOXYBUTYNIN AND RELATED COMPOUNDS VIA DIRECT COUPLING
The synthesis of a single enantiomer of oxybutynin and oxybutynin analogs according to the present invention comprises coupling an enantiomer of cyclohexylphenyl glycolic acid with a propargyl alcohol derivative utilizing carboxylic acid activation. Optically active CHPGA may be prepared either by the resolution process described above or by asymmetric methods. The present invention also provides a process for converting the aforementioned enantiomers of oxybutynin and oxybutynin analogs to their corresponding hydrochloride salts. The synthetic process consists of two reactions, which are depicted and described below: Part 1: Formation of the Mixed Anhydride; Part 2: Formation of (S) or (R) oxybutynin and its related compounds. Again for ease in understanding, the (S) enantiomeric series is depicted, although the (R) series is produced similarly.
Part 1 : Formation of the Mixed Anhydride
isobut lchloroformate
(S)-CHPGA Mixed Anhydride MW=234.29
In Part 1, (S) or (R) cyclohexylphenyl glycolic acid (CHPGA) is reacted with an alkyl chloroformate in an organic solvent to form a mixed anhydride enantiomer, as shown above, which can then react to form the desired chiral product in Part 2 below.
It should be noted that, while mixed anhydrides are often employed for the synthesis of amides, their use for ester synthesis is quite unusual. It should also be noted that a surprising and unexpected aspect of the present process is that the mixed anhydride intermediate proceeds to a chiral product without affecting the tertiary carbinol of CHPGA, which would lead to impurity formation or racemization. One would expect reaction with an acyl halide at the benzylic hydroxyl resulting in the formation of a stable, but undesired compound, such as an ester. Alternatively, if the hydroxyl were activated (unintentionally) to form a good leaving group, as, for example, under acidic conditions, the dissociation of the leaving group would form a benzylic carbonium ion, leading to racemization. One would therefore expect a loss in optical activity of the oxybutynin or the extensive production of by-products. Surprisingly, the present process produces a high purity product, and no racemization is observed.
In the preparation of the mixed anhydride, two intermediates, in addition to the mixed anhydride shown above, were detected. The two were isolated and their structures were determined by NMR to be
carbonate-anhydride A carbonate-acid B wherein R5 was isobutyl. Both intermediates were smoothly converted to oxybutynin upon treatment with 4-N,N-DEB.
The reaction is preferably carried out in an inert atmosphere, such as nitrogen or argon, and the reaction solution is stirred using conventional techniques. In the depiction above, isobutyl chloroformate (IBCF) is shown as the preferred alkyl chloroformate for reaction with (S)-CHPGA forming the isobutyloxy mixed anhydride. However, other alkyl chloroformates, such as isopropenylchloroformate and 2-ethylhexylchloroformate, for example, may instead be used. The amount of alkyl chloroformate used in the reaction is preferably about 1.2 equivalents with respect to the CHPGA enantiomer.
Preferably, the reaction proceeds in the presence of a tertiary amine (2.5 equiv.), such as triethylamine (TEA), 4-N,N-dimethylaminopyridine (DMAP), pyridine, diisopropylethylamine, diethylmethylamine, Ν-methylpiperidine or Ν- methylmorpholine, which scavenges the HC1 produced. Organic solvents that may be used include, but are not limited to cyclohexane, heptane, toluene, tetrahydrofuran (THF), ethylene glycol dimethyl ether (DME), diethoxy methane (DEM), and methyl t-butyl ether (MTBE). Part 2: Formation of (S) or (R -Oxybutynin and its Analogs
Mixed Anhydride (S)-Oxybutynin or Analog
A sidechain propargyl alcohol derivative of formula (III), wherein R1 is as previously defined, is added to the mixed anhydride contained in the reaction mixture to produce the single enantiomer of oxybutynin or analog thereof (II). About 1.3 equivalents of the formula (III) compound relative to (S) or (R)-CHPGA is sufficient. Typically, the reaction mixture is heated to reflux at a temperature of about 65-80° C, but more preferably about 70-75° C, until the reaction is complete, as determined by HPLC.
Most preferably, the propargyl alcohol derivative of formula (III) is a 4- amino propargyl alcohol derivative, wherein R1 is represented as -CH2R2; R2 is – NR3R4; and R3 and R4 are each independently lower alkyl, benzyl or methoxybenzyl. For example, the compound of formula (III) is most preferably 4-N,N-diethylamino butynol (4-N,N-DEB), where R3 and R4 are each ethyl. Reaction of the mixed anhydride with 4-N.N-DEB produces the single enantiomer of oxybutynin, i.e. (S) or (R)-4-diethylamino-2-butynyl phenylcyclohexylglycolate. Another preferred embodiment is the reaction using an Ν-protected 4-N-ethylamino butynol, such as Ν-ethyl-Ν-(4-methoxybenzyl)butynol, as the propargyl alcohol derivative and then cleaving the protecting group (by methods well known in the art) to produce (S) or (R)-4-ethylamino-2-butynyl phenylcyclohexylglycolate, also known as desethyloxybutynin. In that case, R3 is ethyl, and R4 is converted to hydrogen in formula (III). Suitable protecting groups are described in Greene and Wuts Protecting Groups in Organic Synthesis. Second Edition Wiley, New York 1991, p. 362-371, which is incorporated herein by reference. In another preferred embodiment, the propargyl alcohol derivative of formula (III) is 4-N,N- ethylmethylamino butynol, which results in the formation of (Sf) or (R)-4- ethylmethylamino-2-butynyl phenylcyclohexylglycolate. In this case, R3 is ethyl, and R4 is methyl.
Other useful sidechain propargyl alcohol compounds in which R1 is -CH2R2 are those wherein R2 is azide, hydroxy, or halo. In addition, propargyl alcohol itself, also known as 2-propyn-l-ol, may be reacted with the mixed anhydride. In this case, R1 is hydrogen in formula (III). 4-N,N-Diethylamino butynol for use as the sidechain propargyl alcohol in the present invention may be prepared by reacting propargyl alcohol, paraformaldehyde, and diethylamine under standard Mannich conditions. Other amino and alkyl amino propargyl alcohol derivatives of structure (III) can be formed by the process disclosed in U.S. Patent No. 5,677,346. Briefly, a secondary amine, in which one or more substituents may be a protecting group, such as N-ethyl-4- methoxybenzenemethanamine for example, is reacted with propargyl alcohol and paraformaldehyde in the presence of cuprous chloride. After condensation with the activated CHPGA, the addition of α-chloroethyl carbonochloridate removes the protecting group. In this example, the 4-N-ethylaminobutynyl ester is the ultimate product. The remaining propargyl alcohol derivatives for use in the present invention are commercially available or can be synthesized by methods known in the art.
As stated above, the progress of the condensation of the mixed anhydride with the propargyl alcohol may be conveniently monitored by periodic HPLC analyses of the reaction mixture until the desired extent of conversion is reached. At >80% conversion, the reaction is preferably quenched by washing with 10-12% aqueous monobasic sodium phosphate and water. About 8.5 g of the phosphate per gram of enantiomeric CHPGA used is typical. After separation of the organic phase, the aqueous washes are then discarded. A final wash using deionized water may then be performed, after which the bottom aqueous phase is discarded. The retained organic phase containing the enantiomer of structure (II) in solution with the organic solvent can then be concentrated to remove most of the solvent, typically by vacuum distillation.
Formation of the Hvdrochloride Salt
(S)-Oxybutynin or Analog (S)-Oxybutynin or Analog-HCl
To promote crystallization, the organic solvent containing the enantiomer of oxybutynin or one of its analogs (II) produced by the process outlined above (also referred to herein as “free base enantiomer (II)”) is exchanged with ethyl acetate (EtOAc). Typically, the organic solvent is removed by vacuum distillation to contain about 20-25 wt % (S) or (R) enantiomer of structure (II), which is based on the theoretical amount of free base (II) formed from the coupling process above. Ethyl acetate is then added to obtain the original solution volume or weight. This step may be repeated substituting the removal of EtOAc for the organic solvent. The EtOAc solution may be filtered through a filtering agent, such as diatomaceous earth. The filter cake is washed with EtOAc as needed.
The filtrate is then concentrated by vacuum distillation, for example, to contain about 20-25 wt % (theoretical) of free base enantiomer (II) and < 0.3 wt % water. To maximize product yield and purity and to encourage crystallization, most of the water should be removed from the solution. If the foregoing concentration processes are insufficient to reduce the water to < 0.3%, the vacuum distillation may be repeated with fresh solvent or a drying agent, such as magnesium sulfate may be employed. Water content can be determined by KF (Karl Fisher method). Methyl t-butyl ethyl (MTBE) is then added to the concentrated EtOAc solution to a volume that reduces the concentration by weight of the free base enantiomer (II) by about one third, or optimally to between about 6.5 and 8.5 wt %. The hydrochloride salt is then formed by the addition of HC1, while stirring. A slight excess of HC1 in ethanol, for example about 1.1 equivalents of 35-40 wt % HC1, is generally sufficient. The temperature may be increased to 35-45° C. To initiate recrystallization, the solution may be seeded with the hydrochloride salt of the enantiomer of structure (II). After about an hour of stirring, which may be done at 35-45° C, a slurry forms. If the slurry is cooled to about 0-5° C and this temperature maintained for about two hours, filtration provides a very good recovery of the hydrochloride salt of the enantiomer of structure (II). The filter cake is typically a white to off-white crystalline solid, which can then be washed with ambient temperature methyl t-butyl ether (at least 2.2 g MTBE per gram free base enantiomer (II)), followed by vacuum drying at 40- 50° C.
The following example is illustrative, but the present invention is not limited to the embodiment described therein:
Example 2 Preparation of (S)-Oxybutvnin-HCl
A 3-neck round bottomed flask was charged with 50.0 g (S)-CHPGA (213.0 mmol) and 780 g (1000 mL) cyclohexane under nitrogen. While stirring, 54 g triethylamine (2.5 equiv.) and 35 g isobutyl chloroformate (IBCF)(1.2 equiv.) were slowly added while maintaining the temperature at 20-30° C. After about 0.5 hour, while continuing to stir the reaction mixture, 39.15 g 4-N,N-DEB (1.3 equiv.) were added, and the mixture was heated to 65 °C to reflux . Mixing continued at reflux until the formation of (S)-oxybutynin was complete by HPLC area normalization. Heating was discontinued, and the reaction mixture was cooled to between
20-30° C. At this time, 425 g of 11.5% ΝaH2PO4Η20 aqueous solution were added to the mixture, and the mixture was stirred for 10 min. Stirring was discontinued, and the organic and aqueous phases separated after about 15 minutes. The aqueous (bottom) phase was discarded. 425 g of 11.5% NaH2PO4Η20 aqueous solution were again added to the retained organic phase, and the mixture was stirred for about 10 min. The phases were then permitted to separate, which took about 15 minutes. The aqueous (bottom) phase was again discarded. To the remaining organic phase, deionized water (400 g) was added. The mixture was stirred for about 10 min, followed by phase separation after about 15 minutes. The aqueous (bottom) phase was discarded.
Cyclohexane was removed from the organic phase by vacuum distillation to about 350 g (~ 22 wt % (S)-oxybutynin based on the theoretical amount (76.29 g) of (S)-oxybutynin free base formed). Ethyl acetate (EtOAc) was added to obtain the original solution volume of about 1000 mL (or about 83 Og), followed by vacuum distillation to 20-25 wt % (S)-oxybutynin. EtOAc was then added a second time to a volume of about 1000 mL (or about 830g). The batch was then polish filtered through about 5.0 g CELITE® while washing the filter cake with EtOAc as needed. The filtered mixture was concentrated and dried by vacuum distillation to 339 g (~ 22.5 wt % (S)-oxybutynin) and < 03 wt % water, as measured by KF. Based on the theoretical amount of (S)-oxybutynin free base (76.29 g), methyl t-butyl ether was added to adjust the (S)-oxybutynin free base concentration to 8.0 wt % (953 g). With agitation, 23 g of 37 wt % HC1 in EtOH (1.1 equiv.) were slowly added to the solution, while maintaining the temperature between 20 and 45° C. The temperature of the solution was then adjusted to 35-45° C, and the solution was seeded with about 500 mg (S)-oxybutynin-HCl crystals (approximately 10 mg of seeds per g (S)-CHPGA ). The temperature was maintained, and the solution was stirred for about one hour. A slurry formed, which was then cooled to 0-5 °C over a minimum of 1 hour and held for 2 hours. The slurry was then filtered to recover the (S)-oxybutynin-HCl. The filter cake was a white to off-white crystalline solid. After washing with MTBE (a minimum of 167.84 g MTBE (2.2 g MTBE/g (S)-oxybutynin free base), the cake was dried in vacuo at 40-45 °C. The residual solvent remaining in the cake was < 0.5%. Dry weight = 57.9 g. Overall yield = 68.9%.
Example 3 Isolation of the two carbonate intermediates A and B To a racemic mixture of cyclohexylphenylglycolic acid [CHPGA] (5.0 g, 0.0213mol) in cyclohexane (100 mL) was added triethylamine (7.4 mL, 0.053 mol) and isobutylchloroformate (5.5mL, 0.0426mol). The slurry was allowed to stir at ambient temperature for approximately 0.5 h, at which time the reaction was quenched with a 10% aq. NaH2PO4 (50 ml). The organic phase was separated from the aqueous phase and washed with 10% aq. NaH2PO4 (50mL) followed by DI water (50mL). The organic phase was dried over anhydrous MgSO4 and concentrated in vacuo to afford a colorless oil. The product was purified by flash chromatography eluting with 95:5 hexane-EtOAc [Rf = 0.2] to afford pure carbonate-anhydride A. The structure was confirmed by H and 13C NMR, IR, in sttw IR and MS.
To a racemic mixture of cyclohexylphenylglycolic acid [CHPGA] (5.0 g, 0.0213 mol) in cyclohexane (100 mL) was added triethylamine (7.4 mL, 0.053mol) or preferably 1-methyl piperidine (O.053mol), and isobutylchloroformate (3.3 mL, 0.026mol). The slurry was allowed to stir at ambient temperature for approximately 0.5 h, at which time the reaction was quenched with a 10% aq. solution of NaH2PO4 (50 mL) followed by DI water (50mL). The organic phase was dried over anhydrous MgSO4 and concentrated in vacuo to afford a colorless oil as a 4:1 mixture of A and B by HPLC. The crude product was purified by passing the mixture through a plug of neutral alumina. Compound A was eluted first using
CHClj B was then washed off the alumina with acetone and concentrated in vacuo to afford pure carbonate-acid B. The structure was confirmed by H and 13C NMR, IR, in situ IR and MS.
PATENT
http://www.google.com/patents/US6294582
The synthesis of S-oxybutynin has been described in the literature by Kacher et al., J. Pharmacol. Exp. Ther., 247, 867-872 (1988). An improved synthetic method is disclosed in copending U.S. patent application, Ser. No. 09/211,646, now U.S. Pat. No. 6,140,529, the contents of which are incorporated in their entirety. In this method, an activated derivative of cyclohexylphenylglycolic acid (CHPGA), the mixed anhydride I, is prepared.
The mixed anhydride I is coupled with the propargyl alcohol derivative 4-N,N-diethylamino butynol (4-N,N-DEB)(III where R1 is —CH2R2; R2 is —NR3R4; and R3 and R4 are each ethyl.) Reaction of the optically active mixed anhydride with 4-N,N-DEB produces a single enantiomer of oxybutynin, in this case, (S)-4-diethylamino-2-butynylphenylcyclohexylglycolate.
Improved syntheses of starting material CHPGA have been described in two copending U.S. patent applications, Ser. No. 09/050,825, now U.S. Pat. No. 6,013,830, and 09/050,832. The contents of both are incorporated by reference in their entirety. In the first (09/050,825, now U.S. Pat. No. 6,013,830), phenylglyoxylic acid or cyclohexylglyoxylic acid is condensed with a single enantiomer of a cyclic vicinal aminoalcohol to form an ester of the phenylglyoxylic acid or the cyclohexylglyoxylic acid. The ester is reacted with an appropriate Grignard reagent to provide an a-cyclohexylphenylglycolate ester. A single diastereomer of the product ester is separated from the reaction mixture, and hydrolyzed to provide S-α-cyclohexylphenylglycolic acid (S-CHPGA). The second (09/050,832) discloses an alternate stereoselective process for preparing CHPGA. A substituted acetaldehyde is condensed with mandelic acid to provide a 5-phenyl-1,3-dioxolan-4-one, which is subsequently reacted with cyclohexanone to provide a 5-(1-hydroxy cyclohexyl)-5-phenyl-1,3-dioxolan-4-one. The product is dehydrated to a 5-(1-cyclohexenyl)-5-phenyl-1,3-dioxolan-4-one, hydrolyzed and reduced to CHPGA.
The magnitude of a prophylactic or therapeutic dose of S-oxybutynin in the acute or chronic management of disease will vary with the severity of the condition to be treated, and the route of administration. The dose, and perhaps the dose frequency will also vary according to the age, body weight, and the response of the individual patient. In general, the daily dose ranges when administered by inhalation, for the conditions described herein, are from about 0.1 mg to about 100 mg in single or divided dosages. Preferably a daily dose range should be between about 10 mg to about 25 mg, in single or divided dosages, preferably in from 2-4 divided dosages. In managing the patient the therapy should be initiated at a lower dose, perhaps from 5 mg to about 10 mg, and increased up to about 2×20 mg or higher depending on the patient’s global response. When administered orally, preferably as a soft elastic gelatin capsule, the preferred dose range is from about 1 mg to about 1 g per day, more preferably, from about 25 mg to about 700 mg per day, and most preferably, from about 100 mg to about 400 mg per day. It is further recommended that children and patients over 65 years and those with apaired renal, or hepatic function, initially receive low dosages and that they be titrated based on individual responses and blood levels. It may be necessary to use dosages outside these ranges in some cases, as will be apparent to those skilled in the art. Further, it is noted that the clinician or treating physician possesses knowledge of how and when to interrupt, adjust, or terminate therapy in conjunction with individual patient response. The terms “a therapeutically effective quantity”, and “a quantity sufficient to alleviate bronchospasms” are encompassed by the above described dosage amounts and dose frequency schedule.
The methods of the present invention utilize S-oxybutynin, or a pharmaceutically acceptable salt thereof. The term “pharmaceutically acceptable salt” or “a pharmaceutically acceptable salt thereof” refer to salts prepared from pharmaceutically acceptable nontoxic acids including both inorganic and organic acids. Suitable pharmaceutically acceptable acid addition salts for the compound of the present invention include acetic, benzenesulfonic (besylate), benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, and p-toluene sulfonic. The hydrochloride has particular utility.
Preferred unit dosage formulations are those containing an effective dose, as recited, or an appropriate fraction thereof, of S-oxybutynin or pharmaceutically acceptable salts thereof. The formulations of this invention may include other agents conventional in the art having regard to the type of formulation in question. For example, formulations for oral administration may include carriers such as starches, sugars, microcystalline cellulose, diluents, granulating agents, flavoring agents and the like. Formulations suitable for oral, rectal and parenteral administration (including subcutaneous, transdermal, intramuscular, and intravenous) and inhalation may be used for treatment according to the present invention.
Any suitable route of administration may be employed for providing the patient with an effective dosage of S-oxybutynin. For example, oral, rectal, parenteral (subcutaneous, intramuscular, intravenous), transdermal, and like forms of administration may be employed. Transdermal administration may be improved by the inclusion of a permeation enhancer in the transdermal delivery device, for example as described in PCT application WO 92/20377. Dosage forms include troches, dispersions, suspensions, solutions, aerosols, patches, syrups, tablets and capsules, including soft elastic gelatin capsules. Oral and parenteral sustained release dosage forms may also be used.
Because of their ease of administration, tablets and capsules represent one of the more advantageous oral dosage unit forms, in which case solid pharmaceutical carriers are employed. If desired, tablets may be coated by standard aqueous or nonaqueous techniques. Soft elastic gel capsules are a preferred form of administration of S-oxybutynin.
Soft elastic gelatin capsules may be prepared by mixing S-oxybutynin with a digestible oil such as soybean oil, lecithin, cottonseed oil, or olive oil. The mixture is then injected into gelatin by means of a positive pressure pump, such that each dosage unit contains an effective dose of S-oxybutynin. The capsules are subsequently washed and dried.
Oral syrups, as well as other oral liquid formulations, are well known to those skilled in the art, and general methods for preparing them are found in most standard pharmacy school textbooks. An exemplary source is Remington: The Science and Practice of Pharmacy. Chapter 86 of the 19th edition of Remington entitled “Solutions, Emulsions, Suspensions and Extracts” describes in complete detail the preparation of syrups (pages 1503-1505) and other oral liquids. Similarly, sustained release formulation is well known in the art, and Chapter 94 of the same reference, entitled “Sustained-Release Drug Delivery Systems”, describes the more common types of oral and parenteral sustained-release dosage forms (pages 1660-1675.) The relevant disclosure, Chapters 86 and 94, is incorporated herein by reference.
Controlled release means and delivery devices are also described in U.S. Pat. Nos. 3,845,770; 3,916,899; 3,536,809; 3,598,123; and 4,008,719, and in PCT application WO 92/20377. Because they reduce peak plasma concentrations, controlled release dosage forms are particularly useful for providing a therapeutic plasma concentration of S-oxybutynin while avoiding the side effects associated with peak plasma concentrations.
Formulations suitable for inhalation include sterile solutions for nebulization comprising a therapeutically effective amount of S-oxybutynin or a pharmaceutically acceptable salt thereof, dissolved in aqueous saline solution and optionally containing a preservative such as benzalkonium chloride or chlorobutanol, and aerosol formulations comprising a therapeutically effective amount of S-oxybutynin, or a pharmaceutically acceptable salt thereof, dissolved or suspended in an appropriate propellant (e.g., HFA-134a, HFA-227, or a mixture thereof, or a chlorofluorocarbon propellant such as a mixture of Propellants 11, 12 and/or 114) optionally containing a surfactant. Aerosols may be conveniently presented in unit dosage form and prepared by any of the methods well-known in the art of pharmacy. The preparation of a particularly desirable aerosol formulation is described in European Patent No. 556239, the disclosure of which is incorporated herein by reference. Also suitable are dry powder formulations comprising a therapeutically effective amount of S-oxybutynin or a pharmaceutically acceptable salt thereof, blended with an appropriate carrier and adapted for use in connection with a dry-powder inhaler.
……………………….
| CZ20013826A3 * | Title not available | |||
| US3176019 * | Jun 20, 1961 | Mar 30, 1965 | Mead Johnson & Co | Substituted aminobutynyl acetates |
| Reference | ||
|---|---|---|
| 1 | * | DATABASE CAPLUS [Online] 13 November 2010 STN Database accession no. 2006:220682 & CZ 20 013 826 A3 18 June 2003 |
| 2 | * | DATABASE CAPLUS 13 January 2010 STN: ‘Syntheses of oxybutynin hydrochloride‘ Database accession no. 1997:395370 & ZHONGGUO YIYAO GONGYE ZAZHI vol. 27, no. 9, pages 387 – 389 |
![]()
CLIP
Racemic
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 4-Diethylaminobut-2-ynyl 2-cyclohexyl-2-hydroxy-2-phenylethanoate | |
| Clinical data | |
| Trade names | Ditropan |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a682141 |
| Pregnancy cat. | |
| Legal status | |
| Routes | oral, transdermal gel, transdermal patch |
| Pharmacokinetic data | |
| Protein binding | 91%-93% |
| Half-life | 12.4-13.2 hours |
| Identifiers | |
| CAS number | 5633-20-5 |
| ATC code | G04BD04 |
| PubChem | CID 4634 |
| IUPHAR ligand | 359 |
| DrugBank | DB01062 |
| ChemSpider | 4473 |
| UNII | K9P6MC7092 |
| KEGG | D00465 |
| ChEBI | CHEBI:7856 |
| ChEMBL | CHEMBL1231 |
| Chemical data | |
| Formula | C22H31NO3 |
| Mol. mass | 357.486 g/mol |
Oxybutynin (Ditropan, Lyrinel XL, Lenditro (South Africa)) is an anticholinergic medication used to relieve urinary and bladder difficulties, including frequent urination and inability to control urination (urge incontinence), by decreasing muscle spasms of the bladder.[1]
It competitively antagonizes the M1, M2, and M3 subtypes of the muscarinic acetylcholine receptor. It also has direct spasmolytic effects on bladder smooth muscle as a calcium antagonist and local anesthetic, but at concentrations far above those used clinically.
Oxybutynin is also a possible treatment of hyperhidrosis (hyper-active sweating).[2][3][4]
Chemistry
Oxybutynin contains one stereocenter. Commercial formulations are sold as the racemate. The (R)-enantiomer is a more potent anticholinergic than either the racemate or the (S)-enantiomer, which is essentially without anticholinergic activity at doses used in clinical practice.[5][6] However, (R)-oxybutynin administered alone offers little or no clinical benefit above and beyond the racemic mixture. The other actions (calcium antagonism, local anesthesia) of oxybutynin are not stereospecific. (S)-Oxybutynin has not been clinically tested for its spasmolytic effects, but may be clinically useful for the same indications as the racemate, without the unpleasant anticholinergic side effects.
Clinical efficacy
In two trials of patients with overactive bladder, transdermal oxybutynin 3.9 mg/day decreased the number of incontinence episodes and increased average voided volume to a significantly greater extent than placebo. There was no difference in transdermal oxybutynin and extended-release oral tolterodine.[7]
Adverse effects
Common adverse effects associated with oxybutynin and other anticholinergics include: dry mouth, difficulty in urination, constipation, blurred vision, drowsiness, and dizziness.[8] Anticholinergics have also been known to induce delirium.[9]
These are dose-related and sometimes severe. In one population studied—after six months, more than half of the patients had stopped taking the medication because of side effects and calcium defects. An intake of calcium of 800 to 1000 mg is suggested.Dry mouth may be particularly severe; one estimate is that over a quarter of patients who begin oxybutynin treatment may have to stop because of dry mouth.
N-Desethyloxybutynin is an active metabolite of oxybutynin that is thought responsible for much of the adverse effects associated with the use of oxybutynin.[10] N-Desethyloxybutynin plasma levels may reach as much as six times that of the parent drug after administration of the immediate-release oral formulation.[11] Alternative dosage forms have been developed in an effort to reduce blood levels of N-desethyloxybutynin and achieve a steadier concentration of oxybutynin than is possible with the immediate release form. The long-acting formulations also allow once-daily administration instead of the twice-daily dosage required with the immediate-release form. The transdermal patch, in addition to the benefits of the extended-release oral formulations, bypasses the first-pass hepatic effect that the oral formulations are subject to.[12] In those with overflow incontinence because of diabetes or neurological diseases like multiple sclerosis or spinal cord trauma, oxybutynin can worsen overflow incontinence since the fundamental problem is that the bladder is not contracting.
Clinical pharmacology
Oxybutynin chloride exerts direct antispasmodic effect on smooth muscle and inhibits the muscarinic action of acetylcholine on smooth muscle. It exhibits one-fifth of the anticholinergic activity of atropine on the rabbit detrusor muscle, but four to ten times the antispasmodic activity. No blocking effects occur at skeletal neuromuscular junctions or autonomic ganglia (antinicotinic effects).
Sources say the drug is absorbed within one hour and has an elimination half-life of 2 to 5 hours.[13][14][15] There is a wide variation among individuals in the drug’s concentration in blood. This, and its low concentration in urine, suggest that it is eliminated through the liver.[14]
Contraindications
Oxybutynin chloride is contraindicated in patients with untreated angle closure glaucoma, and in patients with untreated narrow anterior chamber angles—since anticholinergic drugs may aggravate these conditions. It is also contraindicated in partial or complete obstruction of the gastrointestinal tract, hiatal hernia, gastroesophageal reflux disease, paralytic ileus, intestinal atony of the elderly or debilitated patient, megacolon, toxic megacolon complicating ulcerative colitis, severe colitis, and myasthenia gravis. It is contraindicated in patients with obstructive uropathy and in patients with unstable cardiovascular status in acute hemorrhage. Oxybutynin chloride is contraindicated in patients who have demonstrated hypersensitivity to the product.
Formulations
It is available orally in generic formulation or as the brand-names Ditropan, Lyrinel XL, or Ditrospam, as a transdermal patch under the brand name Oxytrol, and as a topical gel under the brand name Gelnique.
A 2009 Weill Cornell Medical College study concluded that patients switched to generic oxybutynin experienced a degradation in therapeutic value: “In women, there was a doubling of daytime frequency of urination, a slight 20% increase in nocturia, and a 46.3% increase in urge incontinence. In men, there was a 2.4-fold increase in daytime frequency, a 40% increase in nocturia, and a 40.6% increase in urge incontinence”.[16]
PATENT
http://www.google.com/patents/US3176019
Example XIX 4-diethylamino-2-butynyl phenylcyclohexylglycolate hydrochl0ride.-A mixture of 394.2 g. of methyl phenylcyclohexylglycolate, 293.1 g. of 4-diethylamino-2-butynyl acetate was dissolved with Warming in 2.6 l. of n-heptane. The solution was heated with stirring to a temperature of 60-70 C. and 8.0 g. of sodium methoxide were added. The temperature of the mixture was then raised until the solvent began to distill. Distillation was continued at a gradual rate and aliquots of the distillate were successively collected and analyzed for the presence of methyl acetate by measurement of the refractive index. The reaction was completed when methyl acetate no longer distilled, and the refractive index observed was that of pure heptane (11 1.3855). About three and one-half hours were required for the reaction to be completed. The reaction mixture was then allowed to cool to room temperature, washed with Water, and extracted with four ml. portions of 2 N hydrochloric acid. The aqueous extracts Were combined and stirred at room temperature to permit crystallization of the hydrochloride salt of the desired product. Crystallization was completed by cooling the slurry in an ice bath, and the product was collected by filtration, pressed dry, and recrystallized from 750 ml. of water. Yield of pure crystalline material, 323 g.


References
- Chapple CR. “Muscarinic receptor antagonists in the treatment of overactive bladder”. Urology (55)5, Supp. 1:33-46, 2000.
- Tupker RA, Harmsze AM, Deneer VH (2006). “Oxybutynin therapy for generalized hyperhidrosis.”. Arch Dermatol 142 (8): 1065–6. doi:10.1001/archderm.142.8.1065. PMID 16924061.
- Mijnhout GS, Kloosterman H, Simsek S, Strack van Schijndel RJ, Netelenbos JC. (2006). “Oxybutynin: dry days for patients with hyperhidrosis.”. Neth J Med 64 (9): 326–8. PMID 17057269.
- Schollhammer M, Misery L. (2007). “Treatment of hyperhidrosis with oxybutynin.”. Arch Dermatol. 143 (4): 544–5. doi:10.1001/archderm.143.4.544. PMID 17438194.
- Kachur JF, et al. “R and S enantiomers of oxybutynin: pharmacological effects in guinea pig bladder and intestine.” Journal of Pharmacology and Experimental Therapeutics 247:867-72, 1988.
- Noronha-Blob L, Kachur JF. “Enantiomers of oxybutynin: in vitro pharmacological characterization at M1, M2 and M3 muscarinic receptors and in vivo effects on urinary bladder contraction, mydriasis and salivary secretion in guinea pigs.” Journal of Pharmacology and Experimental Therapeutics 256:562-7, 1991.
- Baldwin C, Keating GM.[1].Drugs 2009;69 (3):327-337. doi:10.2165/00003495-200969030-00008.
- Mehta D (Ed.) 2006. British National Formulary 51. Pharmaceutical Press. ISBN 0-85369-668-3
- Andreasen NC and Black DW, “Introductory Textbook of Psychiatry.” American Psychiatric Publishing Inc. 2006
- Allen B. Reitz, Suneel K. Gupta, Yifang Huang, Michael H. Parker, and Richard R. Ryan (2007). “The preparation and human muscarinic receptor profiling of oxybutynin and N-desethyloxybutynin enantiomers”. Med Chem 3 (6): 543–5. doi:10.2174/157340607782360353. PMID 18045203.
- Zobrist RH, et al. “Pharmacokinetics of the R- and S-Enantiomers of Oxybutynin and N-Desethyloxybutynin Following Oral and Transdermal Administration of the Racemate in Healthy Volunteers”. Pharmaceutical Research 18:1029-1034, 2001.
- Oki T, et al. “Advantages for Transdermal over Oral Oxybutynin to Treat Overactive Bladder: Muscarinic Receptor Binding, Plasma Drug Concentration, and Salivary Secretion”. Journal of Pharmacology and Experimental Therapeutics Fast Forward 316:1137-1145, 2006.
- [2] “Oxybutynin” Retrieved on 30 August 2012.
- [3] “The pharmacokinetics of oxybutynin in man. (Abstract)” Retrieved on 30 August 2012.
- [4] “Oxybutynin” Retrieved on 30 August 2012.
- Kerr, Martha (2009-05-03). “AUA 2009: Generics Not Equal to Brand-Name Drugs for Overactive Bladder”. American Urological Association (AUA) 104th Annual Scientific Meeting (Medscape). Retrieved 2013-04-20.
External links
- http://health.yahoo.com/urinary-medications/oxybutynin-oral/healthwise–d00328a1.html
- http://www.mayoclinic.com/health/drug-information/DR601047
- http://www.medicinenet.com/oxybutynin_er-oral/article.htm
| Title: OxybutyninCAS Registry Number: 5633-20-5CAS Name: a-Cyclohexyl-a-hydroxybenzeneacetic acid 4-(diethylamino)-2-butynyl esterAdditional Names: a-phenylcyclohexaneglycolic acid 4-(diethylamino)-2-butynyl ester; 4-diethylamino-2-butynyl phenylcyclohexylglycolate; oxibutininaMolecular Formula: C22H31NO3Molecular Weight: 357.49Percent Composition: C 73.91%, H 8.74%, N 3.92%, O 13.43%Literature References: Muscarinic receptor antagonist. Prepn: GB 940540 (1963 to Mead Johnson). Physico-chemical properties: E. Miyamoto et al., Analyst 119, 1489 (1994). GC-MS determn in plasma: K. S. Patrick et al., J. Chromatogr. 487, 91 (1989). Toxicity: E. I. Goldenthal, Toxicol. Appl. Pharmacol. 18, 185 (1971). Review of pharmacodynamics and therapeutic use: Y. E. Yarker et al., Drugs Aging 6, 243-262 (1995).Properties: pKa 8.04. Log P (n-octanol/water): 2.9 (pH 6). Soly in water (mg/ml): 77 (pH 1); 0.8 (pH 6); 0.012 (pH >9.6).pKa: pKa 8.04Log P: Log P (n-octanol/water): 2.9 (pH 6)Derivative Type: HydrochlorideCAS Registry Number: 1508-65-2
Additional Names: Oxybutynin chloride Manufacturers’ Codes: MJ-4309-1 Trademarks: Cystrin (Sanofi-Synthelabo); Ditropan (Sanofi-Synthelabo); Dridase (Sanofi-Synthelabo); Kentera (UCB); Pollakisu (Kodama); Tropax (BMS) Molecular Formula: C22H31NO3.HCl Molecular Weight: 393.95 Percent Composition: C 67.07%, H 8.19%, N 3.56%, O 12.18%, Cl 9.00% Properties: Crystals, mp 129-130°. Sol in water, acids. Practically insol in alkali. LD50 orally in rats: 1220 mg/kg (Goldenthal). Melting point: mp 129-130° Toxicity data: LD50 orally in rats: 1220 mg/kg (Goldenthal)
Therap-Cat: In treatment of urinary incontinence. Keywords: Antimuscarinic. |
Launching new Blog……DRUG PATENTS INTERNATIONAL

Beta carotene may protect people with common genetic risk factor for type-2 diabetes
25 JAN 2013
STANFORD, Calif. — Stanford University School of Medicine investigators have found that for people harboring a genetic predisposition that is prevalent among Americans, beta carotene, which the body converts to a close cousin of vitamin A, may lower the risk for the most common form of diabetes, while gamma tocopherol, the major form of vitamin E in the American diet, may increase risk for the disease.
The scientists used a “big data” approach to hunt down interactions between gene variants previously associated with increased risk for type-2 diabetes and blood levels of substances previously implicated in type-2 diabetes risk. In people carrying a double dose of one such predisposing gene variant, the researchers pinpointed a highly statistically significant inverse association of beta carotene blood levels with type-2 diabetes risk, along with a suspiciously high positive association of gamma tocopherol with risk for the disease.
“Type-2 diabetes affects…
View original post 1,081 more words
Tagged Phosphine Reagents to Assist Reaction Work-up by Phase-Switched Scavenging Using a Modular Flow Reactor Process

The use of three orthogonally tagged phosphine reagents to assist chemical work-up via phase-switch scavenging in conjunction with a modular flow reactor is described. These techniques (acidic, basic and Click chemistry) are used to prepare various amides and tri-substitutedguanidines from in situ generated iminophosphoranes.

Tagged Phosphine Reagents to Assist Reaction Work-up by Phase-Switched Scavenging Using a Modular Flow Reactor Process
C.D. Smith, I.R. Baxendale, G.K. Tranmer, M. Baumann, S.C. Smith, R.A. Lewthwaite and S.V. Ley, Org. Biomol. Chem., 2007, 5, 1562-1568.
http://pubs.rsc.org/en/content/articlelanding/2007/ob/b703033a/unauth#!divAbstract
New USP Requirements on Plastic Packaging Systems
DRUG REGULATORY AFFAIRS INTERNATIONAL

New USP Requirements on Plastic Packaging Systems |
| The USP describes in an article of the Pharmacopeial Forum the future requirements for plastic packaging systems. Here, the importance is laid on the selection of suitable, safe plastic materials and the verification of potential interactions. More information can be found here in the News. |
GMP News: New USP Requirements on Plastic Packaging Systems
An interesting article from the USP on the future requirements for plastic packaging systems has been published in the Pharmacopoeial Forum 39(6).
In this article, the USP’s experts group provides an overview of the already existing and also the planned General Chapters on pharmaceutical plastic packaging systems. Together both chapters aim to describe a general and chemistry-based approach for the quality and safety of packaging systems and their starting materials for the construction of these packaging systems.
Among the key topics which are discussed, you can find:
- a. The…
View original post 160 more words
