MK-0822; Odanacatib.
603139-19-1
Formula: C25H27F4N3O3S
Mass: 525.17093
Merck Frosst Canada Ltd. phase 3
(2S)-N-(1-Cyanocyclopropyl)-4-fluoro-4-methyl-2-({(1S)-2,2,2-trifluoro-1-[4′-(methylsulfonyl)biphenyl-4-yl]ethyl}amino)pentanamide
(S)-N-(1-cyanocyclopropyl)-4-fluoro-4-methyl-2-(((S)-2,2,2-trifluoro-1-(4′-(methylsulfonyl)-[1,1′-biphenyl]-4-yl)ethyl)amino)pentanamide
N1-(1-Cyanocyclopropyl)-4-fluoro-N2-[2,2,2-trifluoro-1(S)-[4′-(methylsulfonyl)biphenyl-4-yl]ethyl]-L-leucinamide
Odanacatib (pINN; codenamed MK-0822) is an investigational treatment for osteoporosis and bone metastasis. It is an inhibitor of cathepsin K, an enzyme involved in bone resorption. It is being developed by Merck & Co. As of November 2009, Merck is conducting phase III clinical trials.
Odanacatib, also known as MK-0822, is an inhibitor of cathepsin K with potential anti-osteoporotic activity. Odanacatib selectively binds to and inhibits the activity of cathepsin K, which may result in a reduction in bone resorption, improvement of bone mineral density, and a reversal in osteoporotic changes. Cathepsin K, a tissue-specific cysteine protease that catalyzes degradation of bone matrix proteins such as collagen I/II, elastin, and osteonectin plays an important role in osteoclast function and bone resorption
Osteoporosis is a disease characterized by excessive bone loss causing skeletal fragility and an increased risk of fracture. One in two women and one in eight men over the age of 50 will have an osteoporotic fracture. Cathepsin K is a recently discovered member of the papain superfamily of cysteine proteases that is abundantly expressed in osteoclasts, the cells responsible for bone resorption.
MK-0822 is in phase III clinical trials at Merck & Co. for the treatment of postmenopausal osteoporosis. Several phase II trials had been ongoing for the treatment of cancer, specifically for the treatment of women with breast cancer and metastatic bone disease and also for the treatment of osteoarthritis in the knee and for the treatment of arthritis; however, no recent development has been reported for these indications. MSD KK (formed in 2010 following the merger of Banyu and Schering-Plough KK) is developing the compound for the treatment of osteoporosis in Japan.
Bone is a living tissue that is remodeled every five to seven years in a dynamic process governed by the balance between bone formation and resorption in which osteoblasts and osteoclasts play a pivotal role. The abundant and selective expression of Cathepsin K in osteoclasts has made it an attractive therapeutic target for the treatment of osteoporosis.
Odanacatib (MK-0822) 1 has been identified as a potent and selective inhibitor of Cathepsin K.
A variety of disorders in humans and other mammals involve or are associated with abnormal bone resorption. Such disorders include, but are not limited to, osteoporosis, glucocorticoid induced osteoporosis, Paget’s disease, abnormally increased bone turnover, periodontal disease, tooth loss, bone fractures, rheumatoid arthritis, osteoarthritis, periprosthetic osteolysis, osteogenesis imperfecta, metastatic bone disease, hypercalcemia of malignancy, and multiple myeloma. One of the most common of these disorders is osteoporosis, which in its most frequent manifestation occurs in postmenopausal women. Osteoporosis is a systemic skeletal disease characterized by a low bone mass and microarchitectural deterioration of bone tissue, with a consequent increase in bone fragility and susceptibility to fracture. Osteoporotic fractures are a major cause of morbidity and mortality in the elderly population. As many as 50% of women and a third of men will experience an osteoporotic fracture. A large segment of the older population already has low bone density and a high risk of fractures. There is a significant need to both prevent and treat osteoporosis and other conditions associated with bone resorption. Because osteoporosis, as well as other disorders associated with bone loss, are generally chronic conditions, it is believed that appropriate therapy will typically require chronic treatment.
Osteoporosis is characterized by progressive loss of bone architecture and mineralization leading to the loss in bone strength and an increased fracture rate. The skeleton is constantly being remodeled by a balance between osteoblasts that lay down new bone and osteoclasts that breakdown, or resorb, bone. In some disease conditions and advancing age the balance between bone formation and resorption is disrupted; bone is removed at a faster rate. Such a prolonged imbalance of resorption over formation leads to weaker bone structure and a higher risk of fractures. Bone resorption is primarily performed by osteoclasts, which are multinuclear giant cells. Osteoclasts resorb bone by forming an initial cellular attachment to bone tissue, followed by the formation of an extracellular compartment or lacunae. The lacunae are maintained at a low pH by a proton-ATP pump. The acidified environment in the lacunae allows for initial demineralization of bone followed by the degradation of bone proteins or collagen by proteases such as cysteine proteases. See Delaisse, J. M. et al, 1980, Biochem J 192:365-368; Delaisse, J. et ah, 1984, Biochem Biophys Res Commun:44l-447; Delaisse, J. M. et α/., 1987, Bone 8^305-313, which are hereby incorporated by reference in their entirety. Collagen constitutes 95 % of the organic matrix of bone. Therefore, proteases involved in collagen degradation are an essential component of bone turnover, and as a consequence, the development and progression of osteoporosis.
Cathepsins belong to the papain superfamily of cysteine proteases. These proteases function in the normal physiological as well as pathological degradation of connective tissue. Cathepsins play a major role in intracellular protein degradation and turnover and remodeling. To date, a number of cathepsin have been identified and sequenced from a number of sources. These cathepsins are naturally found in a wide variety of tissues. For example, cathepsin B, F, H, L, K, S, W, and Z have been cloned. Cathepsin K (which is also known by the abbreviation cat K) is also known as cathepsin O and cathepsin O2. See PCT Application WO 96/13523, Khepri Pharmaceuticals, Inc., published May 9, 1996, which is hereby incorporated by reference in its entirety. Cathepsin L is implicated in normal lysosomal proteolysis as well as several diseases states, including, but not limited to, metastasis of melanomas. Cathepsin S is implicated in Alzheimer’s disease and certain autoimmune disorders, including, but not limited to juvenile onset diabetes, multiple sclerosis, pemphigus vulgaris, Graves’ disease, myasthenia gravis, systemic lupus erythemotasus, rheumatoid arthritis and Hashimoto’s thyroiditis; allergic disorders, including, but not limited to asthma; and allogenic immunbe responses, including, but not limited to, rejection of organ transplants or tissue grafts. Increased Cathepsin B levels and redistribution of the enzyme are found in tumors, suggesting a role in tumor invasion and matastasis. In addition, aberrant Cathpsin B activity is implicated in such disease states as rheumatoid arthritis, osteoarthritis, pneumocystisis carinii, acute pancreatitis, inflammatory airway disease and bone and joint disorders.
Cysteine protease inhibitors such as E-64 (trαns-epoxysuccinyl-L- leucylamide-(4-guanidino) butane) are known to be effective in inhibiting bone resorption. See Delaisse, J. M. et al., 1987, Bone 8:305-313, which is hereby incorporated by reference in its entirety. Recently, cathepsin K was cloned and found specifically expressed in osteoclasts See Tezuka, K. et al., 1994, J Biol Chem 269:1106-1109; Shi, G. P. et αZ.,1995, EEES Lett 357: 129-134; Bromme, D. and Okamoto, K., 1995, Biol Chem Hoppe Seyler 376:379-384; Bromme, D. et al, 1996, J Biol Chem 271:2126-2132: Drake, F. H. et al, 1996, J Biol Chem 271:12511- 12516, which are hereby incorporated by reference in their entirety. Concurrent to the cloning, the autosomal recessive disorder, pycnodysostosis, characterized by an osteopetrotic phenotype with a decrease in bone resorption, was mapped to mutations present in the cathepsin K gene. To date, all mutations identified in the cathepsin K gene are known to result in inactive protein. See Gelb, B. D. et al., 1996, Science 273:1236-1238; Johnson, M. R. et al., 1996, Genome Res 6:1050-1055, which are hereby incorporated by reference in their entirety. Therefore, it appears that cathepsin K is involved in osteoclast mediated bone resorption.
Cathepsin K is synthesized as a 37 kDa pre-pro enzyme, which is localized to the lysosomal compartment and where it is presumably autoactivated to the mature 27 kDa enzyme at low pH. See McQueney, M. S. et al., 1997, J Biol Chem 272:13955-13960; Littlewood-Evans, A. et al, 1997, Bone 20:81-86, which are hereby incorporated by reference in their entirety. Cathepsin K is most closely related to cathepsin S having 56 % sequence identity at the amino acid level. The S2P2 substrate specificity of cathepsin K is similar to that of cathepsin S with a preference in the PI and P2 positions for a positively charged residue such as arginine, and a hydrophobic residue such as phenylalanine or leucine, respectively. See Bromme, D. et al., 1996, J Biol Chem 271: 2126-2132; Bossard, M. J. et al, 1996, J Biol Chem 271:12517-12524, which are hereby incorporated by reference in their entirety. Cathepsin K is active at a broad pH range with significant activity between pH 4-8, thus allowing for good catalytic activity in the resorption lacunae of osteoclasts where the pH is about 4-5.
Human type I collagen, the major collagen in bone is a good substrate for cathepsin K. See Kafienah, W., et al, 1998, Biochem J 331:727-732, which is hereby incorporated by reference in its entirety. In vitro experiments using antisense oligonucleotides to cathepsin K, have shown diminished bone resorption in vitro, which is probably due to a reduction in translation of cathepsin K mRNA. See Inui, T., et al, 1997, Biol Chem 272:8109-8112, which is hereby incorporated by reference in its entirety. The crystal structure of cathepsin K has been resolved. See McGrath, M. E., et al, 1997, Nat Struct Biol 4:105-109; Zhao, B., et al, 1997, Nat Struct Biol 4: 109-11, which are hereby incorporated by reference in their entirety. Also, selective peptide based inhibitors of cathepsin K have been developed See Bromme, D., et al, 1996, Biochem 315:85-89; Thompson, S. K., et al, 1997, Proc Natl Acad Sci U S A 94: 14249-14254, which are hereby incorporated by reference in their entirety. Accordingly, inhibitors of Cathepsin K can reduce bone resorption. Such inhibitors would be useful in treating disorders involving bone resorption, such as osteoporosis.
……………….
The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K
Bioorg Med Chem Lett 2008, 18(3): 923
http://www.sciencedirect.com/science/article/pii/S0960894X07015065


Scheme 2.
Reagents and conditions: (a) ClCOOiBu, NMM, NaBH4, DME, 85%; (b) Ts2O, pyr, dichloroethane, 83%; (c) MeMgBr, toluene/THF, 85%; (d) DAST, CH2Cl2, 60%; (e) Ba(OH)2, EtOH/H2O, 100%; (f) TBSCl, Et3N; (g) CF3C(OH)OEt, PhH, 88% (two steps); (h) BrPhLi, THF; (i) TBAF, THF, 75% (two steps); (j) H5IO6, CrO3, CH3CN, 60%; (k) 1-amino-1-cyanocyclopropane hydrochloride, i-Pr2NEt, HATU, DMF, 80%; (l) MeSPhB(OH)2, PdCl2dppf, Na2CO3, DMF, 70%; (m) H2O2, Na2WO42H2O, Bu4NHSO4, EtOAc, 97%. see Supplementary data.
………………
WO 2003075836 or http://www.google.com/patents/EP1482924A2?cl=en
EXAMPLE 10
Synthesis of N l-cyanocyclopropyl)-N2{(lS)-2,2,2-trifluoro-l-[4′-(m^
1 , 1 -biphenyl-4-yl]ethyl ) -L-leucinamide
To a mixture of Ν-{(lS)-2,2,2-trifluoro-l-[4′-(methylsulfonyl)-l,l’- biphenyl-4-yl]ethyl} -L-leucine from Example 8 (0.83 g), O-(7-azabenzotriazol-l-yl)- N, N, N\ N’-tetramethyluronium hexafluorophosphate (0.78 g), cyclopropylamine hydrochloride (0.466 g) in DMF (18 mL) at 0 °C was added triethylamine (0.9 mL). The mixture was kept at room temperature for 48 hours and then poured into dilute aqueous ammonium cholride and diethyl ether. The ether layer was separated and the aqueous further extracted with diethylether. The combined ether extracts were washed with brine, dried with magnesium sulfate and the solvent was removed in vacuo. The residue was purified in SiO2 using ethyl acetate and hexanes (1:1) as eluant, followed by a swish in diethyl ether to yield the title compound.
H NMR (CD3COCD3) δ 8.15(1H, bs), 8.05(2H, d), 8.0(2H, d), 7.8(2H, d), 7.65(2H, d), 4.35-4.45(lH, m), 3.35-3.45(lH, m), 3.2(3H, s), 2.65-2.7(lH, m), 1.85-1.95(1H, m), 1.3-1.6(5H, m), 1.05-1.15(1H, m), 0.85-0.95(6H, m).
……….
WO 2008119176

http://www.google.com/patents/WO2008119176A1?cl=en
EXAMPLE 1
4-FLUORO-iV- {(1 S)-2,2,2-TRIFLUORO- 1 -[4′-(METHYLSULFONYL)BIPHENYL^- YL]ETHYL}-L-LEUCINE DICYCLOHEXYLAMINE SALT

Biphenyl acid (20.74 g) was dissolved in 2-propanol (186 mL) / water (20.7 mL). A solution of iV,jV-dicyclohexylamine (9.82 mL) in 2-propanol (21 mL) / water (2 mL) was added (-10% of volume) and the solution was seeded with DCHA salt (10 mg). A heavy seed bed formed and the slurry was let stir at rt for 30 min. Addition of DCHA was continued over 20-30 min. The slurry was let stir at rt overnight and filtered. The filter cake was washed with 2-propanol / water (2 x 30 mL, 10:1) and MTBE (2 x 30 mL). DCHA salt was obtained as a white solid, 24.4 g, 84% yield. 1H NMR (CD3OD) δ 8.07 (d, 2H, J- 8.0), 7.94 (d, 2H, J= 8.0), 7.75 (d, 2H, J= 8.0), 7.61 (d, 2H, J= 8.0), 4.31 (m, IH), 3.46 (bq, IH, J= 4), 3.22 (m, 2H), 3.19 (s, 3H), 2.11 (bm, 5H), 1.91 (bm, 5H), 1.75 (bm, 2H), 1.49 (d, 3H, J= 21.6), 1.48 (d, 3H, J= 21.6), 1.35 (m, 9H); 19F NMR (CD3OD) δ – 72.9, – 129.4; mp 209-211°C, [α]D 20 + 18.7 (c = 0.29, MeOH).
EXAMPLE 2
N-(I -CYANOCYCLOPROPYL)-4-FLUORO-N2– {(1 S)-2,2,2-TRIFLUORO- 1 -[4′- (METHYLSULFOΝYL)BIPHEΝYL-4- YL]ETHYL}-L-LEUCINAMIDE
Acid (1.9 g) was dissolved in DMAc (10 mL) and cooled to 0°C. 1 –
Aminocyclopropane carbonitrile hydrochloride (0.57 g) and HATU (1.85 g) were added. The resulting slurry was stirred for 15 min and DIEA (2.12 mL) was added over 1.5 h. The reaction was aged for 1 h. Water (11.2 mL) was added via dropping funnel over 70 min and the slurry was aged for Ih at 2O0C. The mixture was filtered and the filter cake was washed with a solution of DMAc:water (9.4 mL, 1 : 1.2), water (18.7 mL), 2-propanol (9.3 mL) The batch was dried to yield 1.67 g, 79% yield of the corresponding amide.
Amide (2.56 g), was dissolved in THF (30.7 mL) at 30°C. Water (19 mL) was added via dropping funnel. The batch was seeded and aged for Ih at 2O0C. Additional water (40.9 mL) was added over 1.5 h and the batch was aged for 16 h. The batch was filtered and washed with water (15 mL). The solids were dried to a constant weight to yield 2.50 g, 97% yield of pure amide. 1H NMR (CD3OD) δ 8.17 (bs, IH), 8.05 (d, 2H, J= 8.5), 7.96 (d, 2H, J= 8.5), 7.80 (d, 2H, J= 8.0), 7.64 (d, 2H, J= 8.0), 4.43 (m, IH), 3.55 (ddd, IH, J= 5.0, 8.5, 8.0), 3.18 (s, 3H), 2.84 (bm, IH), 2.02 (m, 2H), 1.46 (d, 3H, J= 21.5), 1.43 (d, 3H, J= 22.0), 1.36 (m, 2H), 1.07 (m, IH), 0.94 (m, IH); 13C NMR (CD3OD) δ; 19F NMR (CD3OD) δ -73.2, -136.8; IR (cm“1) 3331, 2244, 1687, 1304, 1152; mp 223-224 0C, [α]D 20 + 23.3 (c = 0.53, MeOH).
EXAMPLE 3
N-(l-CYANOCYCLOPROPYL)-4-FLUORO-iV2-{(l1S)-2,2,2-TRrFLUORO-l-[4′- (METHYLSULFONYL)BIPHENYL^-YL]ETHYL) -L-LEUCINAMIDE

A round-bottom flask was charged with biphenyl acid’DCHA salt (76.6 g, 99.2% ee, diastereomeric ratio 342:1) and DMF (590 g). Solid aminocyclopropane carbonitrile-HCl (15.2 g), HOBt-H2O (17.9 g), and EDCΗC1 (29.1 g) were all charged forming a white slurry. The batch was then heated to 38-42°C and aged for 5 hours. The batch was then cooled to 20- 250C and held overnight. HPLC analysis showed 99.4% conversion. The batch was heated to 38-42°C and water (375 g) was charged to batch over 2 hours. The batch remained as a slurry throughout the water addition. The batch was then heated to 58-620C and aged for 1 hour. Following age, water (375 g) was charged over 3 hours, at a rate of 2.1 g/min. The batch was then cooled to 15-25°C and aged overnight. The batch was filtered and washed with 39% DMF in water (2 x 300 g) and 2-propanol (180 g). The solids were dried in the filter at 40-600C for 24 hours. The desired crude product was isolated as a white solid (57g, 92% yield, 99.4 wt%). A round-bottom flask was charged with crude solid (57 g) and acetone/water solution (324 g, 88/12). The slurry was then heated to 400C, at which point the batch was in solution, and aged for an hour. Water (46 g) was then charged over 30 minutes. The batch was then seeded (1.7 g, 3.0 wt%), and the batch was aged at 40°C for an hour prior to proceeding with the crystallization. Water (255 g) was charged over 4.5 h. The batch was then cooled to 230C over 1.5 h, aged for 4 h and filtered. The solids were washed with acetone/water (158 g, 45/55) and water (176 g). The filter cake was dried with nitrogen sweep / vacuum at 55°C. The desired product (57.2 g , 99.9wt%, 99.8A% (enantiomer ND), was obtained in 94.9% yield. 1H NMR (CD3OD) δ 8.17 (bs, IH), 8.05 (d, 2H, J= 8.5), 7.96 (d, 2H, J= 8.5), 7.80 (d, 2H, J= 8.0), 7.64 (d, 2H, J= 8.0), 4.43 (m, IH), 3.55 (ddd, IH, J= 5.0, 8.5, 8.0), 3.18 (s, 3H), 2.84 (bm, IH), 2.02 (m, 2H), 1.46 (d, 3H, J= 21.5), 1.43 (d, 3H, J= 22.0), 1.36 (m, 2H), 1.07 (m, IH), 0.94 (m, IH); 13C NMR
(CD3OD) δ; 19F NMR (CD3OD) δ -73.2, -136.8; IR (cm“1) 3331, 2244, 1687, 1304, 1152; mp 223-224 0C, [α]D 20 + 23.3 (c = 0.53, MeOH).
……………..
J. Org. Chem., 2009, 74 (4), pp 1605–1610
DOI: 10.1021/jo802031
JOC 2009 74(4): 1605-1610
http://pubs.acs.org/doi/abs/10.1021/jo8020314

An enantioselective synthesis of the Cathepsin K inhibitor odanacatib (MK-0822) 1 is described. The key step involves the novel stereospecific SN2 triflate displacement of a chiral α-trifluoromethylbenzyl triflate 9a with (S)-γ-fluoroleucine ethyl ester 3 to generate the required α-trifluoromethylbenzyl amino stereocenter. The triflate displacement is achieved in high yield (95%) and minimal loss of stereochemistry. The overall synthesis of 1 is completed in 6 steps in 61% overall yield.
(2S)-N-(1-Cyanocyclopropyl)-4-fluoro-4-methyl-2-({(1S)-2,2,2-trifluoro-1-[4′-(methylsulfonyl)biphenyl-4-yl]ethyl}amino)pentanamide (1)
To a visually clean 5-necked 50-L round-bottomed flask equipped with a mechanical stirrer, a thermocouple, a dropping funnel, and a nitrogen inlet was added biaryl acid 12a (1.87 kg, 4.0 mol) and DMAc (9.3 L)…………………………………….deleted……………………………………………………. and dried under vacuum at 35 °C to yield 1 as a white solid (2.50 kg, 97% yield, 99.7 area %, 99.9% de by HPLC):
mp 223−224 °C;
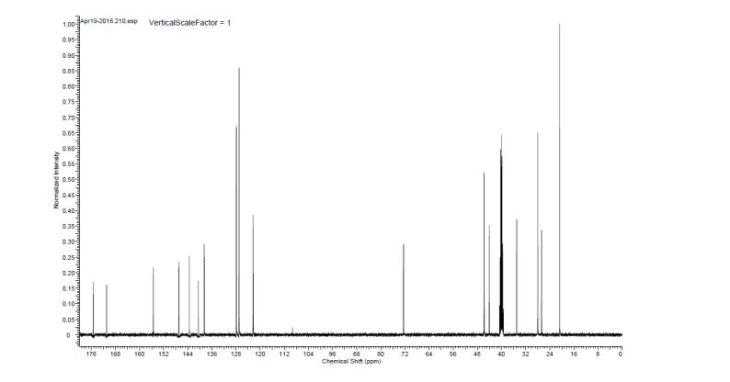
1H NMR (CD3OD) δ 8.17 (br s, 1H), 8.05 (d, 2H, J = 8.5 Hz), 7.96 (d, 2H, J = 8.5 Hz), 7.80 (d, 2H, J = 8.0 Hz), 7.64 (d, 2H, J = 8.0 Hz), 4.43 (m, 1H), 3.55 (ddd, 1H, J = 5.0, 8.5, 8.0 Hz), 3.18 (s, 3H), 2.84 (br m, 1H), 2.02 (m, 2H), 1.46 (d, 3H, J = 21.5 Hz), 1.43 (d, 3H, J = 22.0 Hz), 1.36 (m, 2H), 1.07 (m, 1H), 0.94 (m, 1H); 13C NMR (125 MHz, acetone-d6) δ 175.2, 146.0, 141.2, 140.6, 136.1, 130.3, 128.9 (q, J = 282.8 Hz), 128.7, 128.6, 128.4, 120.9, 95.9 (d, J = 164.3 Hz), 63.5 (q, J = 30.0 Hz), 59.2 (d, J = 3.5 Hz), 44.8 (d, J = 23.1 Hz), 44.3, 27.5 (d, J = 23.9 Hz), 27.1 (d, J = 24.9 Hz), 20.7, 16.5;
19F NMR (CD3OD) δ −73.2, −136.8; IR (cm−1) 3331, 2244, 1687, 1304, 1152; [α]20D + 23.3 (c 0.53, MeOH);
HRMS calcd for C25H28F4N3O3S [MH]+ 526.1782; found 526.1781;
HPLC Phenomenex Spherisorb 4.6 mm × 25 cm column; eluants (A) 0.1% aqueous H3PO4 and (B) acetonitrile; 1 mL/min; gradient A/B 60:40 to 30:70 over 30 min; λ = 265 nm; temperature 45 °C; tR(1 (major diastereoisomer)) = 15.8 min, tR(1 (minor diastereoisomer)) = 16.4 min; HPLC (chiral) Chiralpak AD 4.6 mm × 15 cm column; eluants (A) hexanes, (B) ethanol, and (C) methanol; 1 mL/min; isocratic A/B/C 80:10:10 for 60 min; λ = 265 nm; temperature 40 °C; tR((S,S)-1) = 14.5 min, tR((R,S)-1) = 11.9 min, tR((S,R)-1) = 18.2 min, tR((R,R)-1) = 25.3 min, >99.5% (S,S).
…………..
| In vitro protocol: |
XXX |
| In vivo protocol: |
bone marrow of CatK(-/-) mice: Bone. 2011 Oct;49(4):623-35Pharmacokinetics and metabolism in rats, dogs, and monkeys: Drug Metab Dispos. 2011 Jun;39(6):1079-87. |
in Ovariectomized Rabbits. Calcif Tissue Int. 2013 Oct 2. [Epub ahead of print]Clinical study:Int J Clin Pharmacol Ther. 2013 Aug;51(8):688-92.J Clin Endocrinol Metab. 2013 Feb;98(2):571-80.
Br J Clin Pharmacol. 2013 May;75(5):1240-54.
J Bone Miner Res. 2010 May;25(5):937-47.
Clin Pharmacol Ther. 2009 Aug;86(2):175-82.Review papers:Clin Interv Aging. 2012;7:235-47.Clin Calcium. 2011 Jan;21(1):59-62.
IDrugs. 2009 Dec;12(12):799-809.
Ther Adv Musculoskelet Dis. 2013 Aug;5(4):199-209.
1: Chapurlat RD. Treatment of postmenopausal osteoporosis with odanacatib. Expert Opin Pharmacother. 2014 Jan 24. [Epub ahead of print] PubMed PMID: 24456412.
2: Odanacatib, a cathepsin K inhibitor, superior to alendronate. Bonekey Rep. 2013 Sep 4;2:426. doi: 10.1038/bonekey.2013.160. eCollection 2013. PubMed PMID: 24422127; PubMed Central PMCID: PMC3789222.
3: Retraction notice: Odanacatib for the treatment of postmenopausal osteoporosis. Expert Opin Pharmacother. 2014 Jan;15(1):151. doi: 10.1517/14656566.2014.868399. Epub 2013 Nov 30. PubMed PMID: 24289716.
4: Anderson MS, Gendrano IN, Liu C, Jeffers S, Mahon C, Mehta A, Mostoller K, Zajic S, Morris D, Lee J, Stoch SA. Odanacatib, a selective cathepsin K inhibitor, demonstrates comparable pharmacodynamics and pharmacokinetics in older men and postmenopausal women. J Clin Endocrinol Metab. 2013 Dec 20:jc20131688. [Epub ahead of print] PubMed PMID: 24276460.
5: Chapurlat RD. Odanacatib for the treatment of postmenopausal osteoporosis. Expert Opin Pharmacother. 2014 Jan;15(1):97-102. doi: 10.1517/14656566.2014.853038. Epub 2013 Oct 25. Retraction in: Expert Opin Pharmacother. 2014 Jan;15(1):151. PubMed PMID: 24156249.
6: Stoch SA, Witter R, Hrenuik D, Liu C, Zajic S, Mehta A, Chandler P, Morris D, Xue H, Denker A, Wagner JA. Odanacatib does not influence the single dose pharmacokinetics and pharmacodynamics of warfarin. J Popul Ther Clin Pharmacol. 2013;20(3):e312-20. Epub 2013 Oct 2. PubMed PMID: 24142206.
7: Jensen PR, Andersen TL, Pennypacker BL, Duong le T, Delaissé JM. The bone resorption inhibitors odanacatib and alendronate affect post-osteoclastic events differently in ovariectomized rabbits. Calcif Tissue Int. 2014 Feb;94(2):212-22. doi: 10.1007/s00223-013-9800-0. Epub 2013 Oct 2. PubMed PMID: 24085265.
8: Bonnick S, De Villiers T, Odio A, Palacios S, Chapurlat R, Dasilva C, Scott BB, Le Bailly De Tilleghem C, Leung AT, Gurner D. Effects of Odanacatib on BMD and Safety in the Treatment of Osteoporosis in Postmenopausal Women Previously Treated With Alendronate: A Randomized Placebo-Controlled Trial. J Clin Endocrinol Metab. 2013 Dec;98(12):4727-35. doi: 10.1210/jc.2013-2020. Epub 2013 Sep 24. PubMed PMID: 24064689.
9: Zerbini CA, McClung MR. Odanacatib in postmenopausal women with low bone mineral density: a review of current clinical evidence. Ther Adv Musculoskelet Dis. 2013 Aug;5(4):199-209. doi: 10.1177/1759720X13490860. PubMed PMID: 23904864; PubMed Central PMCID: PMC3728981.
10: Williams DS, McCracken PJ, Purcell M, Pickarski M, Mathers PD, Savitz AT, Szumiloski J, Jayakar RY, Somayajula S, Krause S, Brown K, Winkelmann CT, Scott BB, Cook L, Motzel SL, Hargreaves R, Evelhoch JL, Cabal A, Dardzinski BJ, Hangartner TN, Duong le T. Effect of odanacatib on bone turnover markers, bone density and geometry of the spine and hip of ovariectomized monkeys: a head-to-head comparison with alendronate. Bone. 2013 Oct;56(2):489-96. doi: 10.1016/j.bone.2013.06.008. Epub 2013 Jun 24. PubMed PMID: 23806798.
11: Cabal A, Jayakar RY, Sardesai S, Phillips EA, Szumiloski J, Posavec DJ, Mathers PD, Savitz AT, Scott BB, Winkelmann CT, Motzel S, Cook L, Hargreaves R, Evelhoch JL, Dardzinski BJ, Hangartner TN, McCracken PJ, Duong le T, Williams DS. High-resolution peripheral quantitative computed tomography and finite element analysis of bone strength at the distal radius in ovariectomized adult rhesus monkey demonstrate efficacy of odanacatib and differentiation from alendronate. Bone. 2013 Oct;56(2):497-505. doi: 10.1016/j.bone.2013.06.011. Epub 2013 Jun 20. PubMed PMID: 23791777.
12: Stoch SA, Witter R, Hreniuk D, Liu C, Zajic S, Mehta A, Brandquist C, Dempsey C, Degroot B, Stypinski D, Denker A, Wagner JA. Absence of clinically relevant drug-drug interaction between odanacatib and digoxin after concomitant administration. Int J Clin Pharmacol Ther. 2013 Aug;51(8):688-92. doi: 10.5414/CP201864. PubMed PMID: 23782582.
13: Nakamura T, Shiraki M, Fukunaga M, Tomomitsu T, Santora AC, Tsai R, Fujimoto G, Nakagomi M, Tsubouchi H, Rosenberg E, Uchida S. Effect of the cathepsin K inhibitor odanacatib administered once weekly on bone mineral density in Japanese patients with osteoporosis-a double-blind, randomized, dose-finding study. Osteoporos Int. 2014 Jan;25(1):367-76. doi: 10.1007/s00198-013-2398-2. Epub 2013 May 29. PubMed PMID: 23716037.
14: Brixen K, Chapurlat R, Cheung AM, Keaveny TM, Fuerst T, Engelke K, Recker R, Dardzinski B, Verbruggen N, Ather S, Rosenberg E, de Papp AE. Bone density, turnover, and estimated strength in postmenopausal women treated with odanacatib: a randomized trial. J Clin Endocrinol Metab. 2013 Feb;98(2):571-80. doi: 10.1210/jc.2012-2972. Epub 2013 Jan 21. PubMed PMID: 23337728.
15: Fratzl-Zelman N, Roschger P, Fisher JE, Duong le T, Klaushofer K. Effects of Odanacatib on bone mineralization density distribution in thoracic spine and femora of ovariectomized adult rhesus monkeys: a quantitative backscattered electron imaging study. Calcif Tissue Int. 2013 Mar;92(3):261-9. doi: 10.1007/s00223-012-9673-7. Epub 2012 Nov 23. PubMed PMID: 23179105.
16: Stoch SA, Zajic S, Stone JA, Miller DL, van Bortel L, Lasseter KC, Pramanik B, Cilissen C, Liu Q, Liu L, Scott BB, Panebianco D, Ding Y, Gottesdiener K, Wagner JA. Odanacatib, a selective cathepsin K inhibitor to treat osteoporosis: safety, tolerability, pharmacokinetics and pharmacodynamics–results from single oral dose studies in healthy volunteers. Br J Clin Pharmacol. 2013 May;75(5):1240-54. doi: 10.1111/j.1365-2125.2012.04471.x. PubMed PMID: 23013236; PubMed Central PMCID: PMC3635595.
17: Ng KW. Potential role of odanacatib in the treatment of osteoporosis. Clin Interv Aging. 2012;7:235-47. doi: 10.2147/CIA.S26729. Epub 2012 Jul 12. Review. PubMed PMID: 22866001; PubMed Central PMCID: PMC3410681.
18: Langdahl B, Binkley N, Bone H, Gilchrist N, Resch H, Rodriguez Portales J, Denker A, Lombardi A, Le Bailly De Tilleghem C, Dasilva C, Rosenberg E, Leung A. Odanacatib in the treatment of postmenopausal women with low bone mineral density: five years of continued therapy in a phase 2 study. J Bone Miner Res. 2012 Nov;27(11):2251-8. doi: 10.1002/jbmr.1695. PubMed PMID: 22777865.
19: Jayakar RY, Cabal A, Szumiloski J, Sardesai S, Phillips EA, Laib A, Scott BB, Pickarski M, Duong le T, Winkelmann CT, McCracken PJ, Hargreaves R, Hangartner TN, Williams DS. Evaluation of high-resolution peripheral quantitative computed tomography, finite element analysis and biomechanical testing in a pre-clinical model of osteoporosis: a study with odanacatib treatment in the ovariectomized adult rhesus monkey. Bone. 2012 Jun;50(6):1379-88. doi: 10.1016/j.bone.2012.03.017. Epub 2012 Mar 24. PubMed PMID: 22469953.
20: Khosla S. Odanacatib: location and timing are everything. J Bone Miner Res. 2012 Mar;27(3):506-8. doi: 10.1002/jbmr.1541. PubMed PMID: 22354850.
21 nmr……..http://www.medkoo.com/Product-Data/Odanacatib/Odanacatib-QC-BBC20130906Web.pdf
http://www.medkoo.com/Product-Data/Odanacatib/JOC2009p1605-NMR-Data.pdf

| WO2003075836A2 |
Feb 28, 2003 |
Sep 18, 2003 |
Axys Pharm Inc |
Cathepsin cysteine protease inhibitors |
| WO2005019161A1 |
Aug 19, 2004 |
Mar 3, 2005 |
Merck Frosst Canada Inc |
Cathepsin cysteine protease inhibitors |
| WO2005021487A1 |
Aug 23, 2004 |
Mar 10, 2005 |
Christopher Bayly |
Cathepsin inhibitors |
| WO2006034004A2 |
Sep 16, 2005 |
Mar 30, 2006 |
Axys Pharm Inc |
Processes and intermediates for preparing cysteine protease inhibitors |
| CA2477657A1 * |
Feb 28, 2003 |
Sep 18, 2003 |
Axys Pharmaceuticals, Inc. |
Cathepsin cysteine protease inhibitors |
Lems, Willem F.; Geusens, Piet. Established and forthcoming drugs for the treatment of osteoporosis. Current Opinion in Rheumatology (2014), 26(3), 245-251.
Schwarz, Peter; Jorgensen, Niklas Rye; Abrahamsen, Bo. Status of drug development for the prevention and treatment of osteoporosis. Expert Opinion on Drug Discovery (2014), 9(3), 245-253.
Anderson, Matt S.; Gendrano, Isaias Noel; Liu, Chengcheng; Jeffers, Steven; Mahon, Chantal; Mehta, Anish; Mostoller, Kate; Zajic, Stefan; Morris, Denise; Lee, Jessie; et al. Odanacatib, a selective cathepsin K inhibitor, demonstrates comparable pharmacodynamics and pharmacokinetics in older men and postmenopausal women.Journal of Clinical Endocrinology and Metabolism (2014), 99(2), 552-560.
Nelo Rivera, Yadagiri R. Pendri, Sreenivas MENDE, Bramhananda N. REDDY. Process for preparing fluoroleucine alkyl esters. PCT Int. Appl., WO2013148554 A1,Oct 3, 2013
Humphrey, Guy and Yong, Kelvin. Improved amidation process for the preparation of dipeptide nitriles from fluorinated amino acids in the absence of HOBt coupling agent. PCT Int. Appl., WO2012148555, 01 Nov 2012
Kassahun, Kelem et al. Cathepsin cysteine protease inhibitors. PCT Int. Appl., WO2012112363, 23 Aug 2012
Qiu, Xiao-Long; Yue, Xuyi; Qing, Feng-Ling. Fluorine-containing chiral drugs. Chiral Drugs (2011), 195-251.
O’Shea, Paul D. et al. A Practical Enantioselective Synthesis of Odanacatib, a Potent Cathepsin K Inhibitor, via Triflate Displacement of an α-Trifluoromethylbenzyl Triflate. Journal of Organic Chemistry, 74(4), 1605-1610; 2009
O’Shea, Paul and Gosselin, Francis. Amidation process for the preparation of cathepsin K inhibitor 4-fluoro-N-[(S)-2,2,2-trifluoro-1-[4′-(methylsulfonyl)-1,1′-biphenyl-4-yl]ethyl]-L-leucine 1-cyanocyclopropylamide. PCT Int. Appl., WO2008119176, 09 Oct 2008
Gauthier, Jacques Yves et al. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorganic & Medicinal Chemistry Letters, 18(3), 923-928; 2008
Sarah J. Dolman, Francis Gosselin, Paul D. O’Shea, Ian W. Davies. Selective metal-halogen exchange of 4,4′-dibromobiphenyl mediated by lithium tributylmagnesiate. Tetrahedron, 2006, 62, 5092–5098
Gauthier, Jacques Yves and Truong, Vouy Linh. Preparation of amino acid derivatives as cathepsin cysteine protease inhibitors. PCT Int. Appl., WO2005019161, 03 Mar 2005
Bayly, Christopher et al. Preparation of amino acid derivatives as cathepsin inhibitors. PCT Int. Appl., WO2005021487, 10 Mar 2005
Limanto, John et al. An Efficient Chemoenzymatic Approach to (S)-γ-Fluoroleucine Ethyl Ester. Journal of Organic Chemistry, 70(6), 2372-2375; 2005
Devine, Paul et al. Process for preparing fluoroleucine alkyl esters. U.S. Pat. Appl. Publ., US20050234128, 20 Oct 2005
Bayly, Christopher I. et al. Cathepsin cysteine protease inhibitors and their therapeutic use. PCT Int. Appl., WO2003075836, 18 Sep 2003
updated


 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....