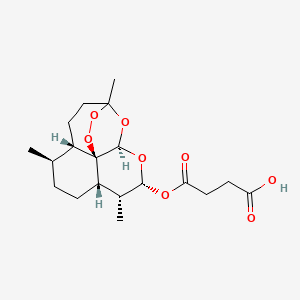
ARTESUNATE
Butanedioic acid mono[(3R,5aS,6R,8aS,9R,10R,12R,12aR)-decahydro-3,6,9-trimethyl-3,12-epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin-10-yl] ester
(3R,5aS,6R,8aS,9R,10S,12R,12aR)-Decahydro-3,6,9-trimethyl-3,12-epoxy-12H-pyrano(4,3-j)-1,2-benzodioxepin-10-ol, hydrogen succinate
Butanedioic acid, mono((3R,5aS,6R,8aS,9R,10S,12R,12aR)-decahydro-3,6,9-trimethyl-3,12-epoxy-12H-pyrano(4,3-j)-1,2-benzodioxepin-10-yl) ester
Additional Names: artesunic acid; dihydroqinghaosu hemisuccinate, Succinic ester of artemether.
Molecular Formula: C19H28O8
Molecular Weight: 384.42
Percent Composition: C 59.36%, H 7.34%, O 33.30%
Artesunate (superseded RN); Dihydroartemisinine-12-alpha-succinate; Succinyl dihydroartemisinin; Quinghaosu reduced succinate ester
Therap-Cat: Antimalarial.
Artesunate (INN) is part of the artemisinin group of drugs that treat malaria. It is a semi-synthetic derivative of artemisinin that is water-soluble and may therefore be given by injection. It is sometimes abbreviated AS.
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basic health system.[1]



- Arinate
- Armax 200
- Arsuamoon
- Arsumax
- Artesunata
- Artesunate
- Artesunato
- Artesunato [INN-Spanish]
- Artesunatum
- Artesunatum [INN-Latin]
- Artesunic acid
- Asumax
- Cosinate
- Dihydroqinghasu hemsuccinate
- Gsunate Forte
- HSDB 7458
- Plasmotrin
- Qinghaozhi
- Quinghaosu reduced succinate ester
- Saphnate
- SM 804
- Succinyl dihydroartemisinin
- UNII-60W3249T9M
- WR 256283
- Zysunate
SODIUM ARTESUNATE;
Dihydroartemisinin alpha-hemisuccinate sodium salt;
Sodium dihydroarteannuin hydrogen succinate monoester;
Butanedioic acid 1-[(3R,5aα,8aα,12aR)-decahydro-3,6α,9β-trimethyl-3β,12α-epoxypyrano[4,3-j]-1,2-benzodioxepin-10α-yl]4-sodium salt;
Butanedioic acid, mono(decahydro-3,6,9-trimethyl-3,12-epoxy-12H-pyrano(4,3-J)-1,2-benzodioxepin-10-yl)ester, sodium salt, (3R-(3-alpha,5A-beta,6-beta,8A-beta,9-alpha,10-alpha,12-beta,12ar*))-
CAS 82864-68-4
Derivative Type: Sodium salt
Manufacturers’ Codes: SM-804
Molecular Formula: C19H27NaO8
Molecular Weight: 406.40
Percent Composition: C 56.15%, H 6.70%, Na 5.66%, O 31.49%
Properties: Poor stability in aqueous solutions. LD50 in mice (mg/kg): 520 i.v.; 475 i.m. (China Cooperative Research Group); also reported as 699 ± 58.5 i.v. (Zhao, 1985).
Toxicity data: LD50 in mice (mg/kg): 520 i.v.; 475 i.m. (China Cooperative Research Group); also reported as 699 ± 58.5 i.v. (Zhao, 1985)
Medical uses
The World Health Organization recommends intramuscular or intravenous artesunate as the first line treatment for severe malaria.[2]Artesunate was shown to prevent more deaths from severe malaria than quinine in two large multicentre randomized controlled trials from Africa[3] and Asia.[4] A subsequent systematic review of seven randomized controlled trials found this beneficial effect to be consistent across all trials.[5]
For severe malaria during pregnancy, there is less certainty about the safety of artesunate during the first trimester but artesunate is recommended as first-line therapy during the second and third trimesters.[6]
Artesunate is also used to treat less severe forms of malaria when it can be given orally, but should always be taken with a second antimalarial such as mefloquine or amodiaquine to avoid the development of resistance.[2]
While artesunate is used primarily as treatment for malaria, there is some evidence that it may also have some beneficial effects inSchistosoma haematobium infection,[7] but this needs confirming in large randomized trials.
Adverse effects
Artesunate is generally safe and well-tolerated. The best recognised side effect of the artemesinins that they lower reticulocyte counts.[8] This is not usually of clinical relevance.
Delayed haemolysis (occurring around two weeks after treatment) has been observed in patients treated with artesunate for severe malaria.[9] Whether or not this haemolysis is due to artesunate, or to the malaria itself is unclear.[10]
The safety of artesunate in pregnancy is unclear. There is evidence of embryotoxicity in animal models (defects in long bones and ventricular septal defects in the heart in rates and monkeys). However, observational evidence from 123 human first-trimester pregnancies showed no evidence of damage to the fetus.[11]
Synthesis
Artesunate is prepared from dihydroartemisinin (DHA) by reacting it with succinic acid anhydride in basic medium. Pyridine as base/solvent, sodium bicarbonate in chloroform and catalyst DMAP (N,N-dimethylaminopyridine) and triethylamine in 1,2-dichloroethane have been used, with yields of up to 100%. A large scale process involves treatment of DHA indichloromethane with a mixture of pyridine, a catalytic amount of DMAP and succinic anhydride. The dichloromethane mixture is stirred for 6–9 h to get artesunate in quantitative yield. The product is further re-crystallized from dichloromethane. alpha-Artesunate is exclusively formed (m.p 135–137˚C).
Artemisinin and its ether and ester derivatives show antimalarial activity against multidrug resistant strains. Ether derivatives like arteether and artemether shows better activity but they suffer from some limitation like solubility, short half life. Unlike ether derivatives, ester derivatives like artesunate has increased solubility and improved pharmacokinetic properties. The water insoluble dihydroartimisinin hemisuccinate is given orally in tablet form and water soluble artesunate sodium is given as LV.
Artesunate was first prepared by Chinese scientists, using different methods. One of them describes acylation of dihydroartemisinin by succinic anhydride in pyridine at 300C for 24 hr with yield of 60%. In another method, described in Acta. Chim. Sinica 40(6), 557-561., ester derivatives of dihydroartemisinin was prepared in presence of 4- (N, N-dimethylamino) pyridine and triethylamine as basic catalyst and 1 ,2 dichloroethane as solvent. The reaction is continued until complete conversion of dihydroartemisinin and product is isolated and purified by silica gel column giving overall yield 60-90%.
Another improved method disclosed in US patent 5654446, describes preparation of artesunate from dihydroartemisinin and succinic anhydride in presence of triethylamine as basic catalyst and in low boiling water miscible dry solvent like acetone. After completion of reaction, mixture is acidified and diluted with water to get artesunate. The yield of esterification is 96%.
U.S. patent 6677463 discloses one pot method for preparation of artesunate from artemisinin. Method describes reduction of artermisinin to dihydroartemisinin in presence of polyhydroxy compound and sodium borohydride. After completion of reaction succinic anhydride and anion exchange resin was added to reaction mass and stirred for 2 hrs. Then cold water was added and product was extracted with ethylacetate hexane mixture in pH range of 6-7. Distilling off the solvent yields the crude artesunate which on silica gel column purification gives 96 % of pure artesunate. The process is complex and time consuming as it involves chromatographic purification step.
……………………………..
http://www.google.com/patents/WO2008087667A1?cl=en

Example 1 discloses the process for obtaining artesunate. The process involves reducing artemisinin to dihydroartemisinin in presence of 1, 2-propanediol and sodium borohydride in a solvent mixture of hexane and isopropanol to give dihydroartemisinin in a yield of 92%. The ratio of artemisinin to 1 , 2-propanediol is 1 :0.66 w/w and the ratio of artemisinin to sodium borohydride is 1 :0.33 w/w. The high yield is attributed to the combination of 1 , 2-propanediol and sodium borohydride in a solvent mixture of hexane and isopropanol that could not be derived from prior art. The dihydroartemisinin is esterified using succinic anhydride and imidazole to give the artesunate in a yield of 100% in 40 min. The ratio of artemisinin to succinic anhydride is 1 :0.52 w/w and that of artemisinin to imidazole is 1 :0.2 w/w. Further, high yield of artesunate obtained in less time was due to imidazole catalyst that accelerates the rate of reaction. Moreover, the process of the present disclosure does not employ purification over silica gel as is in the prior art, but the pure compound is obtained by simple crystallization using suitable solvent.
Example 2 describes the process for obtaining artesunate. The process involves reducing artemisinin to dihydroartemisinin as in example 1. The dihydroartemisinin is esterified using succinic anhydride and imidazole to give the artesunate in a yield of
100% in 25 min. The ratio of artemisinin to succinic anhydride is 1 :0.52 w/w and that of artemisinin to imidazole is 1 :0.3 w/w.
Example 3 describes the process for obtaining artesunate involving reducing artemisinin to dihydroartemisinin in presence of 1, 2-propanediol and sodium borohydride in a solvent mixture of hexane and isopropanol to give dihydroartemisinin in a yield of 88% in 40 min. The ratio of artemisinin to 1, 2-propanediol is 1 :0.8 w/w and the ratio of artemisinin to sodium borohydride is 1 :0.4 w/w. The dihydroartemisinin is esterified using succinic anhydride and imidazole to give the artesunate in a yield of 86%. The ratio of artemisinin to succinic anhydride is 1 :0.52 w/w and that of artemisinin to imidazole is 1 :0.2 w/w.
Example 4 describes the process for obtaining artesunate involving reducing artemisinin to dihydroartemisinin as in example 1. The dihydroartemisinin is esterified using succinic anhydride and imidazole to give the artesunate in a yield of 90% in 210 min. The ratio of artemisinin to succinic anhydride is 1 :0.52 w/w and that of artemisinin to imidazole is 1 :0.1 w/w.
Example 5 describes the process for obtaining artesunate involving reducing artemisinin to dihydroartemisinin as in example 1. The dihydroartemisinin is esterified using succinic anhydride and imidazole in dichloromethane to give the artesunate in a yield of 92% in 60 min. The ratio of artemisinin to succinic anhydride is 1:0.44 w/w and the ratio of artemisinin to imidazole is 1 :0.2 w/w.
Example 6 describes the process for obtaining artesunate involving reducing artemisinin to dihydroartemisinin as in example 1. The dihydroartemisinin is esterified using succinic anhydride and imidazole in acetonitrile to give the artesunate in a yield of 92% in 180 min. The ratio of artemisinin to succinic anhydride is 1:0.52 w/w and that of artemisinin to imidazole is 1 :0.2 w/w.
Example 1 Artemisinin (1.0 g) and 1, 2-propanediol (0.66 g) was added to a mixture of isopropanol (3.5 ml) and hexane (10 ml) and the suspension was stirred for 2 minutes at 2O0C followed by the addition of Sodium borohydride (0.33 gm). After 2 minutes of stirring, dihydroartemisinin started precipitating and the reaction mixture was further stirred for about 8 minutes at 2O0C. Water (10 ml) was added to the reaction mixture and stirred for 10 minutes at 100C. Solid was filtered, washed with hexane (2 * 20 ml) and dried to yield 0.92 g (92% w/w) dihydroartemisinin.
Dihydroartemisinin (0.92 g) was stirred in dichloromethane (10 ml) for 2 minutes at room temperature. Succinic anhydride (0.52 g) and imidazole (0.2 g) were added to this solution and stirred for 40 minutes. The pH of reaction mixture was adjusted to 5-6 and organic layer was washed with water, dried and concentrated to oily mass. The oily mass was dissolved in methanol (1.5 ml) and stirred for 2 min to obtain a clear solution. Water (ImI) was added dropwise to this solution to start the precipitation of artesunate and the suspension was stirred for 5 minutes. The solid was filtered, washed with cold water (2 x 2ml) and dried to yield 1.0 g of artesunate. The overall yield of artesunate was 100 % w/w.
Example 2
Reduction of artemisinin to dihydroartemisinin was carried out as described in Example 1. Dihydroartemisinin (0.92 g) was stirred in dichloromethane (10 ml) for 2 minutes at room temperature. Succinic anhydride (0.52 g) and imidazole (0.3 g) were added to this solution and stirred for 25 minutes. The pH of reaction mixture was adjusted to 5-6 and organic layer was washed with water, dried and concentrated to oily mass. The oily mass was dissolved in methanol (1.5 ml) and stirred for 2 min to obtain a clear solution. Water (ImI) was added dropwise to this solution to start the precipitation of artesunate and the suspension was stirred for 5 minutes. The solid was filtered, washed with cold water (2 χ 2 ml) and dried to yield 1.0 g of artesunate. The overall yield of artesunate was 100 % w/w.
Example 3
Artemisinin (1.0 g) and 1, 2-propanediol (0.8 g) was added to a mixture of isopropanol (3.5 ml) and hexane (10 ml) and the suspension was stirred for 2 minutes at 2O0C followed by the addition of Sodium borohydride (0.4 g). After 2 minutes of stirring, dihydroartemisinin started precipitating and the reaction mixture was further stirred for about 8 minutes at 200C. Water (7.5 ml) was added to the reaction mixture and stirred for 10 minutes at 100C. Solid was filtered, washed with hexane (2 ^ 2 ml) and dried to yield 0.88 g (88% w/w) dihydroartemisinin.
Dihydroartemisinin (0.88 g) was stirred in dichloromethane (10 ml) for 2 minutes at room temperature. Succinic anhydride (0.52 g) and imidazole (0.2 g) were added to this solution and stirred for 40 minutes. The pH of reaction mixture was adjusted to 5-6 and organic layer was washed with water, dried and concentrated to oily mass. The oily mass was dissolved in methanol (1.5 ml) and stirred for 2 min to obtain a clear solution. Water (ImI) was added dropwise to this solution to start the precipitation of artesunate and the suspension was stirred for 5 minutes. The solid was filtered, washed with cold water (2 x 2ml) and dried to yield 0.86 g of artesunate. The overall yield of artesunate was 86 % w/w.
Example 4
Reduction of artemisinin to dihydroartemisinin was carried out as described in Example 1. Dihydroartemisinin (0.92 g) was stirred in dichloromethane (10 ml) for 2 minutes at room temperature. Succinic anhydride (0.52 g) and imidazole (0.1 g) were added to this solution and stirred for 210 minutes. The pH of reaction mixture was adjusted to 5-6 and organic layer was washed with water, dried and concentrated to oily mass. The oily mass was dissolved in methanol (1.5 ml) and stirred for 2 min to obtain a clear solution. Water (ImI) was added dropwise to this solution to start the precipitation of artesunate and the suspension was stirred for 5 minutes. The solid was filtered, washed with cold water (2 x 2 ml) and dried to yield 0.9 g of artesunate. The overall yield of artesunate was 90 % w/w.
Example 5
Reduction of artemisinin to dihydroartemisinin was carried out as described in Example 1. Dihydroartemisinin (0.92 g) was stirred in dichloromethane (10 ml) for 2 minutes at room temperature. Succinic anhydride (0.44 g) and imidazole (0.2 g) were added to this solution and stirred for 60 minutes. The pH of reaction mixture was adjusted to 5-6 and organic layer was washed with water, dried and concentrated to oily mass. The oily mass was dissolved in methanol (1.5 ml) and stirred for 2 min to obtain a clear solution. Water (ImI) was added dropwise to this solution to start the precipitation of artesunate and the suspension was stirred for 5 minutes. The solid was filtered, washed with cold water (2 x 2 ml) and dried to yield 0.92 g of artesunate. The overall yield of artesunate was 92 % w/w. Example 6
Reduction of artemisinin to dihydroartemisinin was carried out as described in
Example 1. Dihydroartemisinin (0.92 g) was stirred in acetonitrile (10 ml) for 2 minutes at room temperature. Succinic anhydride (0.52 g) and imidazole (0.2gm) were added to this solution and stirred for 180 minutes. The pH of reaction mixture was adjusted to 5-6 and it was extracted with dichloromethane (10 ml). The organic layer was washed with water (20 ml), dried and concentrated to oily mass. The oily mass was dissolved in methanol (1.5 ml) and stirred for 2 min to obtain a clear solution. Water (ImI) was added dropwise to this solution to start the precipitation of artesunate and the suspension was stirred for 5 minutes. The solid was filtered, washed with cold water (2 x 2 ml) and dried to yield 0.92 g of artesunate. The overall yield of artesunate was 92 % w/w.
Mechanisms of action
In a hematin dependent manner, artesunate has been shown to potently inhibit the essential Plasmodium falciparum exported protein 1 (EXP1), a membrane glutathione S-transferase.[12]
Drug resistance
Clinical evidence of drug resistance has appeared in Western Cambodia, where artemisinin monotherapy is common.[13] There are as yet no reports of resistance emerging elsewhere.

…………………………………
http://www.google.com/patents/WO2004050661A1?cl=en
Malaria is caused by protozoan parasites, notably Plasmodium falciparum. The range of drugs available in the market for prevention and treatment of malaria is limited, and there are problems of drag resistance. Artemisinin and its derivatives: artemether and arteether (oil soluble), artelinate and artesunate (water soluble), are a class of anti-malarial compounds derived from Artemisia annua which are now proving their promising activity and are being used for the treatment of uncomplicated/severe complicated/cerebral and multi drug resistant malaria. The chemistry and the anti-protozoal action of these compounds, described in the publications are listed as references cited.
The water-insoluble artesunic acid is customarily administered orally in the form of tablets or rectally in the form of suppositories, while the water- soluble artesunate is administered intravenously.
Artesunic acid together with a number of other Cio-ester and CiQ-ether” derivatives of dihydroartemisinin, were prepared for the first time by Chinese scientists at the end of 1979 to the beginning of 1980. Shaofeng et al., H Labeling of QHS Derivatives, Bull. Chin. Materia Medica 6 (4), 25-27 (1981) and Li et al, Synthesis of Ethers. Carboxylic esters and carbonates of Dihydroartemisinin, Acta Pharm. Sin 16(6), 429-39, 1981) describe the preparation of artesunic acid by acylation of dihydroartemisinin with succinic anhydride in pyridine. The above mentioned publications describe a general method for preparing various dihydroartemisinin Cι0-esters and also provide a process for preparing artesunic acid in a yield of 60% by means of warming dihydroartemisinin and succinic anhydride in pyridine at 30° C for 24 hours.
Ying et al. in the Synthesis of some carboxylic esters and carbonates of Dihydroartemisinin by using 4-(N, N-Dimethylamino) pyridine as an active acylation catalyst, Acta Chim Sinica 40 (6), 557-561 982) proposed an improved version of the acylation of dihydroartemisinin. The said publication described in detail with the aid of the preparation of dihydroartemisinin – 10-valerate the aforesaid process. In this process dihydroartimisinin was dissolved in 1,2-dichloroethane and treated with valeric anhydride, 4-(N, N-dimethylamino) pyridine and triethylamine, and the mixture was stirred at room temperature until dihydroartemisinin had been used up. The reaction mixture was then acidified with dilute hydrochloric acid and the aqueous phase was separated off. The oily residue, obtained after washing and drying the organic phase and distilling off the solvent, was purified by chromatography on silica gel using petroleum ether 60-80° C degree/ethyl acetate (10:1) as an eluent. The use of this procedure for the preparation of the artesunic acid from dihydroartemisinin with succinic anhydride and 4-(N, N-dimethylamino) pyridine afforded artesunic acid in a yield of 65% in 5 hours.
U.S. Patent No. 5,654,446 granted to Ognyanov et al. titled “Process for preparation of Dihydroartemisinin Hemisuccinate (artesunic acid)”, dated August 5, 1997 teaches a process for preparing o α-artesunic acid by acylation of dihydroartemisinin with succinic anhydride, in the presence of trialkylamines and their mixture in a low boiling, neutral water miscible, inert organic solvent or solvent mixture at 20-60°C in 0.5 hours and the artesunic acid is then isolated directly at pH 5 to 8 in 91.8 to 97.2% yield.
The above mentioned methods carry some disadvantages being less cost effective and more time consuming as compared to the present invention it should be noted that all the above referenced methods require two separate steps to convert artemisinin into 10-esters of dihydroartemisinin i.e. (a) reduction of artemisinin into dihydroartemisinin in the first pot following by isolation of dihydroartemisinin, and (b) esterification of dihydroartemisinin into different esters in the second pot.
Further, solvent pyridine or 1,2 dichloroethane and catalyst, 4 (N, N-dimethylamino) pyridine used in these processes are not acceptable according to the health standard. Hence there is a need to provide a single step process that overcomes the above-mentioned disadvantages.
EXAMPLE 1
Artemisinin (500mg) and polyhydroxy compound (dextrose, 2.5g) are stirred in 1,4-dioxan (15ml) at room temperature for 5 minutes. Sodium borohydride (2.5g) is added slowly for 10 minutes and the reaction mixture is stirred for about 2 hours at room temperature (20- 30° C). After completion of the reaction (Checked by TLC), succinic anhydride (250 mg) and anion exchange (basic) resin (1.5g) are added at room temperature and the reaction mixture is stirred further for 2 hours at room temperature. Cold water (50 ml) is added to the reaction mixture and pH is adjusted between 6-7 with dilute acetic acid and extracted with 40% ethyl acetate in hexane (3 x 25 ml). The combined extract is washed with water (50 ml). The ethyl acetate π-hexane extract is dried over anhydrous sodium sulphate and evaporation of the solvent yield 655 mg of crude artesunic acid which upon purification over silica gel (1:5 ratio) with 20-30% ethyl acetate in hexane, furnish pure artesunic acid in 93% w/w (465 mg) yield (according to CO-TLC). After drying the pure α-artesunic acid, mp 140-142° C is characterized by spectral analysis.
EXAMPLE 2
Artemisinin (500 mg), polyhydroxy compound (dextrose, 2.0g) are stirred in 1,4-dixan (10 ml). Sodium borohydride (2.5g) is added slowly for 10 minutes and the reaction mixture is stirred for about 2 hours at room temperature (20-30° C). After completion of the reduction step, succinic anhydride (250 mg) and triethylamine (1ml) are added and the reaction mixture is further stirred for 2 hours at room temperature (20-30 degree C). After usual work up and purification of crude product (690mg) through column chromatography (1:4 ratio) 91.2%) pure artesunic acid is obtained.
EXAMPLE 3 Artemisinin (500 mg), polyhydroxy compound (dextrose, 2.0g) are stirred in tetrahydrofuran (10 ml). Sodium borohydride (2.5g) is added slowly for 10 minutes and the reaction mixture is stirred for about 2 hours at room temperature. After completion of the reduction step succinic anhydride (250 mg) and triethylamine (1ml) are added and the reaction mixture is further stirred for 2 hours at room temperature. After usual work up and purification of the crude product (615mg) through column chromatography 87.4% pure artesunic acid is obtained.
EXAMPLE 4
Artemisinin (500 mg) and polyhydroxy compound (dextrose, 2g) are stirred in dioxan (15 ml) for 5 minutes. Sodium borohydride (2.4gm) is added slowly and the reaction mixture is stirred for 2 hours at room temperature (20-30 degree C). After completion of the reduction step succinic anhydride (250 mg) and sodium bicarbonate (3.5g) are added and the reaction mixture is further stirred for 2 hours. After usual workup and purification of impure reaction product (650 mg), 89.6%w/w pure artesunic acid is obtained.
EXAMPLE 5
Artemisinin (500mg) and cation exchange resin (lg) are stirred in tetrahydrofuran (10ml) at room temperature for 5 minutes. Sodium borohydride (250mg) is added slowly for 10 minutes and the reaction mixture is stirred for about 30 minutes at room temperature (20- 35 degree C). After completion of the reaction succinic anhydride (250mg) and triethylamine (0.7ml) are added at room temperature and the reaction mixture is stirred further for 1 hours at room temperature. The resin is filtered. After usual workup and column chromatography of the crude product (710mg), 480mg of pure artesunic acid (yield
= 96%w/w) is obtained.
EXAMPLE 6
Artemisinin (500mg) and cation exchange resin (lg) are stirred in 1,4 dioxan (10ml) at room temperature for 5 minutes. Sodium borohydride (250mg) is added slowly for 10 minutes and the reaction mixture is stirred for about 30 minutes at room temperature (20-35 degree C). After completion of the reaction succinic anhydride (250mg) and triethylamine (0.7ml) are added slowly at room temperature and the reaction mixture is stirred further for 1.25 hours at room temperature. After usual work up and purification of the crude artesunic acid (680mg) pure product in 91.7% w/w is obtained.
EXAMPLE 7
Artemisinin (500 mg), cation exchange resin (lOg) are stirred in 1,4 dioxan (10 ml). Sodium borohydride (250mg) is added slowly for 10 minutes and the reaction mixture is stirred for about 45minutes at room temperature (20-35 degree C). After completion of the reduction step succinic anhydride (250 mg) and sodium bicarbonate (2.5g) are added and the reaction mixture is further stirred for 1.5 hours at room temperature (20-35 degree C). After usual work up and purification of the crude artesunic acid (630mg) pure product in 85%o w/w yield is obtained.
EXAMPLE 8 Artemisinin (500 mg) and cation exchange resin (lg) are stirred in tetrahydrofuran (15 ml) for 5 minutes. Sodium borohydride (2.4gm) is added slowly and the reaction mixture is stirred for 45 minutes at room temperature (20-35 degree C). After completion of the reduction reaction, succinic anhydride (245 mg) and sodium bicarbonate (3.5g) are added and the reaction mixture is further stirred for 1.25 hours. After usual workup and purification of impure reaction product (650 mg), pure artesunic acid in 93%w/w yield is obtained.
EXAMPLE 9
Artemisinin (lOOmg) and cation exchange resin (200mg) are stirred in tetrahydrofuran (3ml) at room temperature for 5 minutes. Sodium borohydride (50mg) is added slowly for 10 minutes and the reaction mixture is stirred for about 30 minutes at room temperature (20-35 degree C). After completion of the reaction propionic anhydride (0.5ml) and triethylamine (0.2ml) are added at room temperature and the reaction mixture is stirred further for 1.5 hours at room temperature. After usual workup and purification of the crude products through preparative TLC 44 mg of pure dihydroartemisinin 10- propionate characterized by its spectral analysis is obtained.
EXAMPLE 10
Artemisinin (lOOmg) and cation exchange resin (200mg) are stirred in tetrahydrofuran (3ml) at room temperature for 5 minutes. Sodium borohydride (50mg) is added slowly for
10 minutes and the reaction mixture is stirred for about 30 minutes at room temperature (20-35 degree C). After completion of the reaction chloroacetic anhydride (50mg) and triethylamine (0.2ml) are added at room temperature and the reaction mixture is stirred further for 1.5 hours at room temperature. After usual workup and purification of the crude products through preparative TLC 35mg of pure dihydroartemisinin 10- chloroacetate characterized by its spectral analysis is obtained.
EXAMPLE 11
Artemisinin (lOOmg) and cation exchange resin (200mg) are stirred in tetrahydrofuran (3ml) at room temperature for 5 minutes. Sodium borohydride (50mg) is added slowly for 10 minutes and the reaction mixture is stirred for about 30 minutes at room temperature (20-35 degree C). After completion of the reaction acetic anhydride (50mg) and triethylamine (0.2ml) are added at room temperature and the reaction mixture is stirred further for 1.5 hours at room temperature. After usual workup and purification of the crude products through preparative TLC 42mg of pure dihydroartemisinin 10-acetate identified by its spectral analysis is obtained.
EXAMPLE 12
Artemisinin (5g) and cation exchange resin (lOg) are stirred in tetrahydrofuran (60ml) at room temperature for 5 minutes. Sodium borohydride (2.5g) is added slowly for 20 minutes and the reaction mixture is stirred for about 1 hour at room temperature (20-35 degree C). After completion of the reaction succinic anhydride (2.5g) and triethylamine (6ml) are added at room temperature and the reaction mixture is stirred further for 1.5 hours at room temperature. After usual workup and purification of the crude product
(6.92g) through CC pure artesunic acid in 94.6%w/w yield is obtained.
ADVANTAGES OF THE PRESENT INVENTION
1. The two pot reactions: reduction of artemisinin into dihydroartemismin and esterification of dihydroartemisinin to artesunic acid carried out in one pot avoids the process of isolation of dihydroartemisinin is avoided which saves chemicals, labour and losses of dihydroartemisinin in isolating it.
2. Conversion of artemisinin into artesunic acid in one pot takes place in about 2-5 hours and is a less time consuming method as compared to previously reported methods in which conversion of artemisinin into dihydroartemisinin in first pot followed by isolation of dihydroartemisinin and its esterification into artesunic acid in the second pot is also a long process. 3. The conversion of artemisinin into artesunic acid in one pot is carried out at room temperature (20-35 degree C) and thereby avoids use of cooling unit.
4. The solvent used to carry out the reduction reaction is also being used in esterification and thus enabling the process cost effective.
5. The catalysts, polyhydroxy compound or cation exchange resin used to carry out the reduction of artemisinin into dihydroartemisinin at room temperature (20-35°C) are cost effective.
6. The conversion of artemisinin into crude artesunic acid followed by workup and purification to yield pure product takes 6-10 hours as compared to previously reported methods (about 20-40 hours) and thus the process is less time consuming.
7. The yield of final product in the present invention i.e. pure artesunic acid is upto 96%, w/w.
8. Thus, this improved process which avoids the disadvantages of previously known process is suitable for the preparation of artesunic acid in large scale.
References
- “WHO Model List of EssentialMedicines”. World Health Organization. October 2013. Retrieved 22 April 2014.
- World Health Organization. “Guidelines for the treatment of malaria; Second edition 2010”. World Health Organization. Retrieved 10 January 2014.
- Dondorp AL, et al. (2010). “Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial”.The Lancet 376 (9753): 1647–1657. doi:10.1016/S0140-6736(10)61924-1.PMC 3033534. PMID 21062666.
- South East Asian Quinine Artesunate Malaria Trial (SEAQUAMAT) (2005). “Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial”. The Lancet 366 (9487): 717–725. doi:10.1016/S0140-6736(05)67176-0. PMID 16125588.
- Jump up^ Sinclair, D; Donegan, S; Isba, R; Lalloo, DG (Jun 13, 2012). “Artesunate versus quinine for treating severe malaria.”. The Cochrane database of systematic reviews 6: CD005967.doi:10.1002/14651858.CD005967.pub4. PMID 22696354.
- Jump up^ WHO (2007). Assessment of the safety of artemisinin compounds in pregnancy. World Health Organization, Geneva.
- Jump up^ Boulangier D, Dieng Y, Cisse B, et al. (2007). “Antischistosomal efficacy of artesunate combination therapies administered as curative treatments for malaria attacks”. Trans R Soc Trop Med Hyg 101 (2): 113–16. doi:10.1016/j.trstmh.2006.03.003.PMID 16765398.
- Jump up^ Clark RL (2012). “Effects of artemisinins on reticulocyte count and relationship to possible embryotoxicity in confirmed and unconfirmed malarial patients”. Birth defects research. Part A, Clinical and molecular teratology 94 (2): 61–75.doi:10.1002/bdra.22868.
- Rolling T, Agbenyega T, Issifou S, et al. (2013). “Delayed hemolysis after treatment with parenteral artesunate in African children with severe malaria—a double-center prospective study.”. J Infect Dis 209 (12): 1921–8. doi:10.1093/infdis/jit841.PMID 24376273.
- Clark RL (2013). “Hypothesized cause of delayed hemolysis associated with intravenous artesunate.”. Med Hypotheses 82 (2): 167–70.doi:10.1016/j.mehy.2013.11.027. PMID 24370269.
- Clark RL (2009). “Embryotoxicity of the artemisinin antimalarials and potential consequences for use in women in the first trimester.”. Reprod Toxicol 28 (3): 285–96.doi:10.1016/j.reprotox.2009.05.002. PMID 19447170.
- Lisewski, A. M.; Quiros, J. P.; Ng, C. L.; Adikesavan, A. K.; Miura, K; Putluri, N; Eastman, R. T.; Scanfeld, D; Regenbogen, S. J.; Altenhofen, L; Llinás, M; Sreekumar, A; Long, C; Fidock, D. A.; Lichtarge, O (2014). “Supergenomic Network Compression and the Discovery of EXP1 as a Glutathione Transferase Inhibited by Artesunate”. Cell 158(4): 916–28. doi:10.1016/j.cell.2014.07.011. PMID 25126794. edit
- White NJ (2008). “Qinghaosu (Artemisinin): The price of success”. Science 320 (5874): 330–334. doi:10.1126/science.1155165. PMID 18420924.
Literature References:
Derivative of artemisinin, q.v. Prepn: China Cooperative Research Group on Qinghaosu, J. Tradit. Chin. Med. 2, 9 (1982).
Absolute configuration: X.-D. Luo et al., Helv. Chim. Acta 67, 1515 (1984).
GC/MS determn.: A. D. Theoharideset al., Anal. Chem. 60, 115 (1988);
HPLC determn in plasma: H. Naik et al., J. Chromatogr. B 816, 233 (2005).
Pharmacology: Y. Zhao, J. Trop. Med. Hyg. 88, 391 (1985). Antimalarial activity: W. Peters et al., Ann. Trop. Med. Parasitol. 80, 483 (1986); A. J. Linet al., J. Med. Chem. 30, 2147 (1987).
Inhibition of cytochrome oxidase: Y. Zhao et al., J. Nat. Prod. 49, 139 (1986).
Toxicology: China Cooperative Research Group on Qinghaosu, J. Tradit. Chin. Med. 2, 31 (1982).
Series of articles on chemistry, pharmacology, and antimalarial efficacy: ibid. 3-50.
Clinical trial as add-on therapy in pediatric malaria: L. von Seidlein et al.,Lancet 355, 352 (2000).
Review: R. N. Price, Expert Opin. Invest. Drugs 9, 1815-1827 (2000).
| “THE CHEMISTRY AND SYNTHESIS OF QINGHAOSU DERIVATIVES” JOURNAL OF TRADITIONAL CHINESE MEDICINE, BEIJING, CN, vol. 2, no. 1, 1982, pages 9-16, XP008019918 ISSN: 0255-2922 |
| 2 |
* |
DATABASE CAPLUS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; PHAN DINH CHAU ET AL: “Semisynthesis of an antimalarial artesunate” retrieved from STN Database accession no. 119:249761 XP002250029 -& TAP CHI DUOC HOC (1992), (5), 10-12 , XP001162710 |
| 3 |
* |
HAYNES, RICHARD K. ET AL: “C-10 ester and ether derivatives of dihydroartemisinin – 10-.alpha. artesunate, preparation of authentic 10-.beta. artesunate, and of other ester and ether derivatives bearing potential aromatic intercalating groups at C-10” EUROPEAN JOURNAL OF ORGANIC CHEMISTRY (2002), (1), 113-132 , XP002250027 |
| 4 |
* |
LI Y ET AL: “Studies on artemisinine analogs. I. Synthesis of ethers, carboxylates and carbonates of dihydroartemisinine” YAO HSUEH HSUEH PAO – ACTA PHARMACEUTICA SINICA, BEIJING, CN, vol. 16, no. 6, June 1981 (1981-06), pages 429-439, XP002119789 ISSN: 0513-4870 |
Artesunate

Artesunate is a drug that was initially designed for combating malaria, however, recently it has shown great promise as a cancer therapy1,2,3. It has been used in combination with some chemotherapies to improve outcomes in advanced cancer patients5. When fighting cancer it is important to use every tool at your disposal to weaken the cancer and strengthen your own cells. Artesunate is another weapon in the arsenal of natural remedies that can make a significant difference in the fight against cancer.
The mechanism of action for artesunate in the context of cancer therapy is very well defined. Cancer cells have a tendency to absorb iron at high levels and this is thought to accelerate the mutation rate within these cells. Iron reacts with oxygen to form free radicals, which are reactive molecules that damage DNA. In normal cells this reaction is a problem; in cancer cells it allows them to mutate and develop resistance to therapies. Artesunate activates mitochondrial apoptosis by iron catalyzed lysosomal reactive oxygen species production4. In other words, this drug will use the iron within the cancer cells against them.
http://yaletownnaturopathic.com/how-does-artesunate-kill-cancer/
Dr. Adam McLeod is a Naturopathic Doctor (ND), BSc. (Hon) Molecular biology, First Nations Healer, Motivational Speaker and International Best Selling Author. He currently practices at his clinic in Vancouver, British Columbia where he focuses on integrative oncology. http://www.yaletownnaturopathic.com
References:
1) MIYACHI, HAYATO, and CHRISTOPHER R. CHITAMBAR. “The anti-malarial artesunate is also active against cancer.” International journal of oncology 18 (2001): 767-773.
2) Michaelis, Martin, et al. “Anti-cancer effects of artesunate in a panel of chemoresistant neuroblastoma cell lines.” Biochemical pharmacology 79.2 (2010): 130-136.
3) Du, Ji-Hui, et al. “Artesunate induces oncosis-like cell death in vitro and has antitumor activity against pancreatic cancer xenografts in vivo.” Cancer chemotherapy and pharmacology65.5 (2010): 895-902.
4) Efferth, Thomas, et al. “Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron.” Free Radical Biology and Medicine 37.7 (2004): 998-1009.
5) Zhang, Z. Y., et al. “[Artesunate combined with vinorelbine plus cisplatin in treatment of advanced non-small cell lung cancer: a randomized controlled trial].” Zhong xi yi jie he xue bao= Journal of Chinese integrative medicine 6.2 (2008): 134-138.
 Artesunate is…
Artesunate is…
a water-soluble ‘artemesinin’ drug derived from the ‘sweet wormwood’ plant, Artemsia annua, an herb used to treat infections and other illnesses in China for centuries. Interestingly, according to Wikipedia artemsia was lost as an herbal remedy in China until 1970 when an ancient Chinese medical manual dating back to 340 AD was found. The active ingredient in the plant – artemesinin – was isolated by scientists and it anti-malarial properties were quickly noted (1972). It is now used to treat malaria and schistosoma infections.
Artesunate also reduces anti-oxidant activity in the red blood cell thus exposing the cell to high free radical levels. Artesunate is currently being studied as an adjunct to chemotherapeutic agents because of its ability to induce cancerous cells to commit suicide (apoptosis) by inducing high rates of oxidative stress. The ability of the antioxidant NAC to thwart Artesunate’s effects in one study substantiated the important role increased free radical production plays in the drugs effect.
Malaria – Artemesia annua is native to China but has become naturalized around the world including the eastern United States. Artesunate was recently approved for emergency use in patients with severe malaria in the United States.
In April 2009 the FDA approved CoArtem which contains a derivative of artemesinin and a broad spectrum antibiotic called lumefantrine. Upon binding to infected red blood cells artesunate triggers the release of oxygen and carbon-based free radicals that attack proteins in the parasites.
Herpesviruses – Recent culture cell experiments indicated Artesunate was effective at significantly reducing viral protein production in HHV-6A infected cells. A 2005 in vitro study suggested Artesunate significantly reduced cytomegalovirus replication in cells. Because Artesunate effects HHV-6 early in its life cycle it may hold special promise in the kind of smoldering infections that may occur in chronic fatigue syndrome (ME/CFS).
Artesunate’s effects on herpesviruses, however, have not been well studied with just five studies published to date. Interest in this drug appears to be increasing, however, three of the five studies were published in 2008.
Artesunate May Work in Chronic Fatigue Syndrome (ME/CFS) Because..
it may be able to reduce herpesvirus activity in some patients. It’s use, however, is highly experimental.


 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....






































 The National Chemical Laboratory is located in the state of Maharashtra in India. Maharashtra state is the largest contributor to India’s GDP. The National Chemical Laboratory is located in Pune city, and is the cultural capital of Maharashtra. Pune city is second only to Mumbai (the business capital of India) in size and industrial strength. Pune points of interest include: The tourist places in Pune include: Lal Deval Synagogue, Bund Garden, Osho Ashram, Shindyanchi Chhatri and Pataleshwar Cave Temple.
The National Chemical Laboratory is located in the state of Maharashtra in India. Maharashtra state is the largest contributor to India’s GDP. The National Chemical Laboratory is located in Pune city, and is the cultural capital of Maharashtra. Pune city is second only to Mumbai (the business capital of India) in size and industrial strength. Pune points of interest include: The tourist places in Pune include: Lal Deval Synagogue, Bund Garden, Osho Ashram, Shindyanchi Chhatri and Pataleshwar Cave Temple.